The Obstetric Hematology Manual. Manual
User Manual: manual pdf -FilePursuit
Open the PDF directly: View PDF ![]() .
.
Page Count: 278 [warning: Documents this large are best viewed by clicking the View PDF Link!]
- Cover
- Half-title
- Title
- Copyright
- Contents
- Contributors
- Preface
- Acknowledgments
- Section 1 Cellular changes
- Chapter 1 Normal hematological changes during pregnancy and the puerperium
- Chapter 2 Hematinic deficiencies
- Introduction
- Iron deficiency
- Epidemiology
- Pathogenesis
- Iron homeostasis
- Diagnosis of iron deficiency
- Decrease in storage iron
- Decrease in iron for erythropoiesis
- Decrease in peripheral hemoglobin
- Clinical signs and symptoms
- Management options
- Iron
- Parenteral iron
- Erythropoietin
- Blood transfusion
- Prevention strategies
- Screening for iron deficiency
- Summary – key points
- Folate deficiency
- Epidemiology
- Diagnosis
- Management
- Prevention strategies
- Summary
- B12 deficiency
- Epidemiology
- Diagnosis
- Management
- Dilemmas
- Summary
- References
- Chapter 3 Inherited red cell disorders
- Introduction
- Ante-and neonatal screening for hemoglobin disorders
- NHS sickle and thalassemia screening – an example of a linked ante-natal and neonatal program
- Sickling disorders in pregnancy
- Pathogenesis
- Contraception
- Maternal and fetal complications of pregnancy
- General management of sickle cell pregnancy
- Management
- Management of chest crises in pregnancy
- Postpartum
- Dilemmas
- Summary
- Thalassemia and pregnancy
- Pathogenesis
- Preconception
- Medical problems in pregnancy
- Dilemmas
- Summary
- Red cellmembrane disorders
- Glucose-6-phosphate dehydrogenase deficiency
- Acknowledgment
- Chapter 4 Maternal autoimmune cytopenias
- Introduction
- Idiopathic/immune thrombocytopenic purpura (ITP)
- Planning for delivery
- Management of labor when platelet count has not been corrected
- Prenatal counseling
- Autoimmune neutropenia (AIN)
- Incidence and pathogeneisis
- Diagnosis
- Management
- Autoimmune hemolytic anemia (AIHA)
- References
- Section 2 Feto-maternal alloimmune syndromes
- Chapter 5 Fetal/neonatal alloimmune thrombocytopenia
- Introduction
- Epidemiology
- Incidence
- Clinical diagnosis
- Laboratory diagnosis
- Clinical significance of FNAIT
- Prediction of the severity of FNAIT in subsequent pregnancies
- Consideration of ante-natal screening for FNAIT
- Management of FNAIT
- Summary
- References
- Chapter 6 Red cell alloimmunization
- Chapter 5 Fetal/neonatal alloimmune thrombocytopenia
- Section 3 Thromboembolism and anticoagulation
- Chapter 7 Acutemanagement of suspected thromboembolic disease in pregnancy
- Introduction
- Epidemiology of VTE during pregnancy
- Assessment and diagnosis of acute VTE in pregnancy
- Initial treatment of VTE in pregnancy
- Maintenance treatment of VTE
- Management at the time of delivery
- Postpartum anticoagulation and duration of anticoagulation therapy
- Management of massive, life-threatening PE
- References
- Chapter 8 Thromboprophylaxis
- Chapter 9 Prosthetic heart valves
- Introduction
- Indication for valve replacement
- Consideration of valve type in women of child bearing age
- Prevention of thromboembolism
- Anticoagulant management during pregnancy
- Anticoagulation with warfarin during pregnancy
- Anticoagulation with heparin during pregnancy
- Role of aspirin
- Other management issues
- Management of labor and delivery
- Women on oral anticoagulants
- Prevention of infective endocarditis
- Management of valve thrombosis
- Summary
- References
- Chapter 10 Management of anticoagulants at delivery
- Chapter 7 Acutemanagement of suspected thromboembolic disease in pregnancy
- Section 4 Thrombophilia and fetal loss
- Chapter 11 Antiphospholipid syndrome
- Introduction
- Pathophysiology and etiology
- Clinical features
- Laboratory evaluation
- Principles of management
- Background
- Pre-pregnancy management
- Management of thrombosis
- Management of women with antiphospholipid antibodies and a previous thrombotic event
- Management of women with multiple previous venous events, or venous plus arterial events
- Management of women with APS pregnancy morbidity
- Management of thrombocytopenia associated with APS in pregnancy
- Management dilemmas
- Neonatal issues
- References
- Chapter 12 Thrombophilia and pregnancy loss
- Introduction
- Epidemiology
- Pathogenesis
- Thrombophilia
- Placentation in normal pregnancy
- Placental pathology in pregnancy loss
- Is heritable thrombophilia associated with pregnancy loss?
- Does maternal heritable thrombophilia cause pregnancy loss?
- Diagnosis
- Thrombophilia testing
- Management
- General measures
- Are antithrombotics useful?
- Purist vs. pragmatic management
- Dilemmas
- Lack of evidence
- Randomized trials are needed urgently
- Summary
- References
- Chapter 11 Antiphospholipid syndrome
- Section 5 Hemorrhagic disorders
- Chapter 13a Management of obstetric hemorrhage: obstetric management
- Chapter 13b Management of obstetric hemorrhage: anestheticmanagement
- Introduction
- Communication
- Access
- Monitoring
- Oxytocics (see also Chapter 13a)
- Fluids
- Preventing the “lethal triad” of hypothermia, acidosis and coagulopathy
- Blood and blood component therapy (see Chapter 13c)
- Cell salvage
- Investigations
- Regional Vs general anesthesia
- Post-hemorrhage care
- Documentation
- Drills/protocols
- Debriefing and counseling
- References
- Chapter 13c Management of obstetric hemorrhage: hemostatic management
- Chapter 13d Management of obstetric hemorrhage: radiologicalmanagement
- Chapter 14 Inherited disorders of primary hemostasis
- Introduction
- Von Willebrand disease
- VWF activity
- Clinical features
- Disease prevalence
- Classification of VWD
- Laboratory evaluation
- Hormonal influences on levels in pregnancy
- Obstetric complications
- Maternal bleeding
- Pregnancy outcomes
- Pregnancy management for women with VWD
- Ante-natal management
- DDAVP in pregnancy
- Coagulation factor replacement
- Intrapartum management
- Analgesia
- Postpartum management (Table 14.6)
- Tranexamic acid
- DDAVP and plasma products
- Potential complications of factor replacement
- Neonatal management
- Inherited disorders of platelet function
- References
- Chapter 15 Inherited coagulopathies
- Hemophilia
- Introduction
- Disease incidence
- Clinical features
- Hormonal influences on levels in pregnancy
- Obstetric complications
- Neonatal risk
- Pre-pregnancy management
- Ante-natal management (Table 15.2)
- Intrapartum management (Table 15.3)
- Postpartum
- Neonatal management
- Factor XI deficiency
- Introduction
- Disease incidence
- Clinical features
- Hormonal influences on factor levels
- Obstetric complications
- Ante-natal management
- Treatment options to cover delivery and ante-natal procedures
- Intrapartum management
- Regional anesthesia
- Postpartum management
- Neonatal management
- Rare coagulation factor deficiences
- Hormonal influences on factor levels
- Pre-pregnancy management
- Ante-natal management
- Treatment options
- Early pregnancy failure
- Thrombosis
- Neonatal management
- References
- Chapter 16 Genetic counseling and pre-natal diagnosis in hemophilia
- Section 6 Microangiopathies
- Chapter 17 Pre-eclampsia
- Introduction
- Epidemiology
- Diagnosis of PET
- Current concepts on the pathogenesis of PET
- Management of pre-eclampsia
- Hematological complications of PET
- Differential diagnosis of PET and HELLP by microangiopathic hemolytic anemias (MAHA)
- Acute fatty liver of pregnancy (AFLP)
- Summary
- References
- Chapter 18 Thrombotic thrombocytopenic purpura and other microangiopathies
- Introduction
- Moderate to severe thrombocytopenia presenting during pregnancy
- Placental profiles in high risk pregnancies
- Thrombotic thrombocytopenic purpura (TTP)
- Hemostatic changes of normal pregnancy-Factor VIII, VonWillebrand Factor (VWF), and ADAMTS 13
- Women presenting with acute TTP during pregnancy
- Risk associated with pregnancy in women with previous acquired idiopathic (non-pregnancy associated) TTP
- Treatment of TTP in pregnancy
- Liver disease in pregnancy
- Intrahepatic cholestasis of pregnancy (ICP)
- Acute fatty liver of pregnancy (AFLP)
- Hemolysis, elevated liver enzymes and low platelets (HELLP)
- Pre-eclampsia (PET)
- Hemolytic uremic syndrome (HUS)
- Treatment
- Exacerbation of systemic lupus erythematosis (SLE)
- Disseminated intravascular coagulation (DIC)
- References
- Chapter 17 Pre-eclampsia
- Section 7 Malignant conditions
- Chapter 19 Myeloproliferative disorders
- Introduction
- Epidemiology
- Pathogenesis
- Diagnosis
- Treatment options
- Recommendations for management of MPDs in pregnancy
- Dilemmas
- References
- Chapter 20 Effects of chemoradiotherapy for hematological malignancy on fertility and pregnancy
- Chapter 19 Myeloproliferative disorders
- Index

This page intentionally left blank

The Obstetric Hematology
Manual

The Obstetric Hematology
Manual
Edited by
Sue Pavord
University Hospitals of Leicester NHS Trust
Beverley Hunt
Guy’s and St. Thomas’ NHS Foundation Trust and King’s College, London

CAMBRIDGE UNIVERSITY PRESS
Cambridge, New York, Melbourne, Madrid, Cape Town, Singapore,
São Paulo, Delhi, Dubai, Tokyo
Cambridge University Press
The Edinburgh Building, Cambridge CB2 8RU, UK
First published in print format
ISBN-13 978-0-521-86564-7
ISBN-13 978-0-511-67748-9
© Cambridge University Press 2010
2010
Information on this title: www.cambrid
g
e.or
g
/9780521865647
This publication is in copyright. Subject to statutory exception and to the
provision of relevant collective licensing agreements, no reproduction of any part
may take place without the written permission of Cambridge University Press.
Cambridge University Press has no responsibility for the persistence or accuracy
of urls for external or third-party internet websites referred to in this publication,
and does not guarantee that any content on such websites is, or will remain,
accurate or appropriate.
Published in the United States of America by Cambridge University Press, New York
www.cambridge.org
eBook
(
NetLibrar
y)
Hardback
Contents
List of contributors page vii
Preface ix
Acknowledgments x
Section 1. Cellular changes
1. Normal hematological changes
during pregnancy and the puerperium 3
Margaret Ramsay
2. Hematinic deficiencies 13
Jane Strong
3. Inherited red cell disorders 28
Emma Welch and Josh Wright
4. Maternal autoimmune cytopenias 45
Hamish Lyall and Bethan Myers
Section 2. Feto-maternal
alloimmune syndromes
5. Fetal/neonatal alloimmune
thrombocytopenia 63
Michael F. Murphy
6. Red cell alloimmunization 73
Alec McEwan
Section 3. Thromboembolism and
anticoagulation
7. Acute management of suspected
thromboembolic disease in pregnancy 91
Andrew J. omson and Ian A. Greer
8. Thromboprophylaxis 99
Sarah Germain and Catherine Nelson-Piercy
9. Prosthetic heart valves 109
Claire McLintock
10. Management of anticoagulants
at delivery 120
Christina Oppenheimer and Paul Sharpe
Section 4. Thrombophilia and
fetal loss
11. Antiphospholipid syndrome 131
Sue Pavord, Bethan Myers, and Beverley Hunt
12. Thrombophilia and pregnancy loss 141
Isobel D. Walker
Section 5. Hemorrhagic disorders
13a. Management of obstetric
hemorrhage: obstetric management 151
Annette Briley and Susan Bewley
13b. Management of obstetric
hemorrhage: anesthetic management 158
Vivek Kakar and Geraldine O’Sullivan
13c. Management of obstetric
hemorrhage: hemostatic
management 166
Eleheria Leou and Beverley Hunt
13d. Management of obstetric
hemorrhage: radiological
management 171
Ash Saini and John F. Reidy
14. Inherited disorders of primary
hemostasis 176
Sue Pavord
15. Inherited coagulopathies 186
Sue Pavord v

Contents
16. Genetic counseling and pre-natal
diagnosis in hemophilia 194
Andrew Mumford
Section 6. Microangiopathies
17. Pre-eclampsia 203
Eleheria Leou and Beverley Hunt
18. Thrombotic thrombocytopenic
purpura and other microangiopathies 218
Marie Scully and Pat O’Brien
Section 7. Malignant conditions
19. Myeloproliferative disorders 229
Claire Harrison and Susan E. Robinson
20. Effects of chemoradiotherapy for
hematological malignancy on fertility
and pregnancy 243
Seonaid Pye and Nina Salooja
Index 253
vi

Contributors
Susan Bewley
Women’s Services, Guy’s and St. omas’ NHS
Foundation Trust, London, UK
Annette Briley
Maternal and Fetal Research, Guy’s and St. omas’
NHS Foundation Trust, London, UK
Sarah Germain
Diabetes and Endocrine Centre, Guy’s and
St. omas’ NHS Foundation Trust, London, UK
Ian A. Greer
Hull York Medical Centre, University of York,
Heslington, York, UK
Claire Harrison
Department of Haematology, Guy’s and St. omas’
NHS Foundation Trust, London, UK
Beverley Hunt
Department of Haematology, Guy’s and St. omas’
NHS Foundation Trust and King’s College,
London, UK
Eleftheria Lefkou
Department of Haematology, Guy’s and St. omas’
NHS Foundation Trust, Lambeth Palace Road,
London, UK
Vivek Kakar
Department of Anaesthesia and Intensive Care, Guy’s
and St. omas’, NHS Foundation Trust, London, UK
Hamish Lyall
Department of Haematology, Norfolk and Norwich
University, Norwich, UK
Alec McEwan
Department of Obstetrics and Gynaecology, Queen’s
Medical Centre, Nottingham, UK
Claire McLintock
Natural Women’s Health, Auckland City Hospital,
Auckland, New Zealand
Andrew Mumford
Bristol Haemophilia Centre, Bristol Haematology and
Oncology, Bristol, UK
Michael Murphy
National Blood Service, John Radclie Hospital,
Headington, Oxford, UK
Bethan Myers
Department of Haematology, Queen’s Medical
Centre, Nottingham, UK
Catherine Nelson-Piercy
Department of Obstetrics, Guy’s and St. omas’
NHS Foundation Trust, London, UK
Pat O’Brien
Department of Obstetrics and Gynaecology,
University College London Hospitals, London, UK
Christina Oppenheimer
Department of Obstetrics and Gynaecology, Leicester
Royal Inrmary, Leicester, UK
Geraldine O’Sullivan
Department of Anaesthetics, Guy’s and St. omas’
NHS Foundation Trust, London, UK
Sue Pavord
Department of Haematology, Leicester Royal
Inrmary,Leicester,UK
Seonaid Pye
Department of Haematology, Charing Cross Hospital,
London, UK
Margaret Ramsay
Department of Obstetrics and Gynaecology, Queen’s
Medical Centre, Nottingham, UK vii

List of contributors
John F. Reidy
Department of Radiology, Guy’s and St. omas’
NHS Foundation Trust, London, UK
Susan E. Robinson
Department of Haematology, Guy’s and St. omas’
NHS Foundation Trust, London, UK
Nina Salooja
Division of Investigating, Imperial College London,
London, UK
Marie Scully
Department of Haematology, University College
London, London, UK
Paul Sharpe
Department of Anaesthesia, Leicester Royal
Inrmary,Leicester,UK
Jane Strong
Department of Haematology, Leicester Royal
Inrmary,Leicester,UK
Isobel D. Walker
Department of Haematology, Glasgow Royal
Inrmary, Glasgow, UK
Emma Welch
Department of Haematology, Royal Hallamshire
Hospital,Sheeld,UK
Josh Wright
Department of Haematology, Royal Hallamshire
Hospital,Sheeld,UK
viii

Preface
is book aims to appeal to both those who have
already submersed themselves in the eld of obstet-
ric haematology and new-comers to the area. Many
have already discovered the numerous challenges and
dilemmas involved but also have found this area of
medicine to be both stimulating and rewarding. Oth-
ers may be new to the eld or have unwittingly found
themselves regularly involved in the care of these
women. We hope that all will benet from this man-
ual, which reects up-to-date clinical management of
this complex group of patients as they present in clin-
ical practice.
e impact of haematological disease on fertility,
pregnancy and the puerperium can be consider-
able. rombosis and haemorrhage are the leading
causes of maternal mortality and a large number of
haematological conditions are associated with fetal
loss. Advances in fetal maternal medicine and obstet-
ric care has enabled high expectations of fetal sur-
vival and maternal wellbeing. However the stakes
are high, management can be complex and good
outcomes require excellent multidisciplinary team
work.
New challenges arise in the light of changing cos-
mopolitan populations, including rising birth rates
and improved survival and fertility from chronic ill-
nesses and life-threatening conditions. us in-depth
understanding is required to deal with this broad
rangeofdisease.Wearefortunatetohavesuchadis-
tinguished group of contributors, whose knowledge,
experience and opinions are invaluable, particularly in
an area where randomised clinical trials are scant and
good quality evidence hard to nd.
is branch of medicine is gaining increasing
recognition as a subspecialist area, with the growth
of national and international specialist groups and
development of educational courses in the area.
Clinical problems have become an important fea-
ture in postgraduate examinations, both in hematol-
ogy and obstetrics. is book is therefore not only
an important guide for practitioners in haematol-
ogy, obstetrics, midwifery, and obstetric anaesthesia
butisinvaluableforthosestudyingforpostgraduate
examinations.
Obstetric haematology is immensely rewarding,
andwehopethisbookprovidesencouragement,
particularlyforthosewhoarenewtothespe-
ciality, to view it as both thought-provoking and
enjoyable.
Sue Pavord
Beverley Hunt
ix

Acknowledgments
anks to our families for tolerating our time away
in writing and editing, and to the Sta of Cambridge
University Press, who guided us.
x

Section
1
Cellular changes

Section 1 Cellular changes
Chapter
1Normal hematological changes during
pregnancy and the puerperium
Margaret Ramsay
Introduction
ere are both subtle and substantial changes in
hematological parameters during pregnancy and
the puerperium, orchestrated by changes in the
hormonal milieu. A thorough understanding of these
is important to avoid both over and under-diagnosing
abnormalities. Appreciation of the time frame for
some of the changes allows sensible planning; this
is particularly true when considering thrombo-
prophylaxis.
Some of the quoted reference ranges may dier
between centers, depending on laboratory techniques.
However, the principles of recognizing physiological
changes can still be applied.
Red cells
During pregnancy, the total blood volume increases by
about 1.5 l, mainly to supply the needs of the new vas-
cular bed. Almost 1 liter of blood is contained within
the uterus and maternal blood spaces of the placenta.
Expansion of plasma volume by 25%–80% is one of
the most marked changes, reaching its maximum by
mid pregnancy. Red cell mass also increases by 10%–
20% but the net result is that hemoglobin (Hb) con-
centration falls.1Typically, this is by 1–2 g/dL by the
late second trimester and stabilizes thereaer. Women
whotakeironsupplementshavelesspronouncedHb
changes, as they increase their red cell mass propor-
tionately more than those without dietary supplements
(the increase is approximately 30% over pre-pregnancy
values).1
It is hard to dene a normal reference range for
Hb during pregnancy and the limit for diagnosing
anemia. e World Health Organization has sug-
gested that anemia is present in pregnancy when Hb
concentration is ⬍11 g/dL. However, large studies
in healthy Caucasian women taking iron supplements
frommidpregnancyfoundHbvaluesintheearly
third trimester to be 10.4–13.5 g/dL (2.5th–97.5th cen-
tiles)2. Studies from other ethnic populations have
documented lower third trimester Hb concentrations,
which may be attributable to the women entering preg-
nancy with poor iron stores or with dietary deciencies
of iron and folic acid.
Red cell count and hematocrit (Hct) values are like-
wise lower in pregnancy, but the other red cell indices
change little (Table 1.1), although red cells show more
variation in size and shape than in the non-pregnant
state. ere is a small increase in mean cell volume
(MCV), of on average 4 fL for iron-replete women,
which reaches a maximum at 30–35 weeks gestation
and occurs independently of any deciency of B12 and
folate.2
Hemoglobinandhematocritincreaseaerdeliv-
ery. Signicant increases have been documented
between measurements taken at 6–8 weeks postpar-
tum and those at 4–6 months postpartum, demonstrat-
ing that this length of time is needed to restore them to
non-pregnant values.1
Summary points
rHb concentrations decrease in pregnancy.
rHb ⬍10.4 g/dL suggests anemia.
rHb ⬎13.5g/dLisunusualandsuggests
inadequate plasma volume expansion (which can
be associated with pregnancy problems including
pre-eclampsia and poor fetal growth).
rMCV is normally slightly increased.
rMCH and MCHC are normally unchanged in
pregnancy and do not change with gestation.
3
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 1. Cellular changes
Table 1.1 Red cell indices during pregnancy and the puerperium
Gestation
Red cell indices 18 weeks 32 weeks 39 weeks 8 weeks postpartum
Hemoglobin (Hb) g/dL 11.9 (10.6–13.3) 11.9 (10.4–13.5) 12.5 (10.9–14.2) 13.3 (11.9–14.8)
Red cell count ×1012/L 3.93 (3.43–4.49) 3.86 (3.38–4.43) 4.05 (3.54–4.64) 4.44 (3.93–5.00)
Mean cell volume (MCV) fL 89 (83–96) 91 (85–97) 91 (84–98) 88 (82–94)
Mean cell hemoglobin (MCH) pg 30 (27–33) 30 (28–33) 30 (28–33) 30 (27–32)
Mean cell hemoglobin concentration
(MCHC) g/dL
34 (33–36) 34 (33–36) 34 (33–36) 34 (33–36)
Hematocrit 0.35 (0.31–0.39) 0.35 (0.31–0.40) 0.37 (0.32–0.42) 0.39 (0.35–0.44)
Mean and reference ranges (2.5th–97.5th centiles). Samples were collected longitudinally from 434 women.
Adapted from Ref 2.
White cells
White cell count (WBC) is increased in pregnancy2
with a typical reference range of 6 ×109–16 ×109/L.
In the hours aer delivery3, healthy women have been
documented as having WBC 9 ×109–25 ×109/L. By
4 weeks post-delivery, typical WBC ranges are similar
to those in healthy non-pregnant women (4 ×109–10
×109/L).
ere has been much discussion about the nor-
mal ranges for the dierent types of white cells.4Neu-
trophils contribute most to the overall higher WBC.
ere is an increase in immature forms and the cyto-
plasm shows toxic granulation. e count3,4 is rela-
tively constant throughout gestation (3 ×109–10 ×
109/L), markedly elevated in the hours aer deliv-
ery (up to 23 ×109/L) and back to non-pregnant
values by 4 weeks post-partum (1.5 ×109–6 ×
109/L). Neutrophil chemotaxis and phagocytic activ-
ity are depressed, the latter being inhibited by factors
present in pregnancy serum. ere is also evidence of
increased oxidative metabolism in neutrophils during
pregnancy.
Lymphocyte count3,4 decreases during pregnancy
through rst and second trimesters, increases during
the third trimester, but remains low in the early puer-
perium as compared to normal non-pregnant values.
Typical pregnancy range for lymphocyte count is 1.1
×109–2.8 ×109/L, compared with the non-pregnant
reference range 0.8 ×109–4.0 ×109/L. Lymphocyte
count is restored to normal range by 4 weeks aer
delivery. Detailed studies of T and B lymphocyte sub-
sets in peripheral blood and the proliferative responses
of these cells to mitogens found more helper and sup-
pressor cells and less killer cells during pregnancy.
Lymphocyte proliferation in response to a variety of
agents was found to be impaired in pregnancy, suggest-
ing that there is an immunosuppressant factor present
in the serum.
Monocyte count is higher in pregnancy, espe-
cially in the rst trimester, but decreases as gestation
advances.4Typical values3,4 in the third trimester are
0.2 ×109–1.0 ×109/L,ascomparedtonon-pregnant
values 0.1 ×109–0.9 ×109/L.emonocytetolym-
phocyte ratio is markedly increased in pregnancy.
Eosinophil and basophil counts do not change sig-
nicantly during pregnancy.3
Myelocytes and metamyelocytes may be found in
the peripheral blood lm of healthy women during
pregnancy and do not have any pathological signi-
cance.
Summary points
rWBC is elevated in pregnancy, mostly due to
neutrophilia.
rLymphocyte count is lower and monocyte count
higher.
rDuring pregnancy, only WBC ⬎16 ×109/L is
considered abnormal.
rSoon aer delivery, only WBC ⬎25 ×109/L is
considered abnormal.
rEosinophil and basophil counts do not change in
pregnancy.
4

Chapter 1. Normal changes
Platelets
Large cross-sectional studies in pregnancy of healthy
women (specically excluding any with hypertension)
have shown that the platelet count decreases during
pregnancy, particularly in the third trimester.5is is
termed “gestational thrombocytopenia.” Almost 12%
of women in one study5were found to have a platelet
count of ⬍150 ×109/L late in pregnancy. Of these
women, 79% had platelet counts 116 ×109–149 ×
109/L; none had complications related to thrombocy-
topenia and none of their babies had severe throm-
bocytopenia (platelet count ⬍20 ×109/L). us, it
has been recommended that the lower limit of platelet
count in late pregnancy should be considered as 115 ×
109/L. Only 1% of healthy women have platelet counts
⬍100 ×109/L.
Platelet size is an indicator of the age of the
platelets; young ones are large and they become pro-
gressively smaller with age. Platelet volume has a
skewed distribution, tailing o at larger volumes. e
platelet volume distribution width increases signi-
cantly and continuously as gestation advances and the
mean platelet volume becomes an insensitive measure
of platelet size. Studies suggest that platelet lifespan is
shorter in pregnancy. e decrease in platelet count
andincreaseinplateletsizeinpregnancysuggeststhat
there is hyperdestruction of platelets.
Platelet function, as assessed by the time required
for whole blood to occlude a membrane impregnated
with either epinephrine or adenosine 5’diphosphate
(ADP), has been studied in late pregnancy.7,8 No cor-
relation was found between platelet count and the “clo-
sure times” over a range of platelet counts 44 ×109–
471 ×109/L in healthy women.8Another study found
that the closure times were increased in women with
severe pre-eclampsia, although they did not correlate
with clinical bleeding problems in these women.9In
women with gestational thrombocytopenia, platelet
closure times are inuenced by hemoglobin level,
being prolonged when there is both thrombocytopenia
and anemia.7is is perhaps not surprising, given the
contribution of red cells to the hemostatic process, in
part due to ADP donation. e increase in brinogen
during pregnancy helps to maintain platelet function.
Summary points
rPlatelet count decreases during pregnancy in
some patients.
re lower limit of normal platelet count at term is
115 ×109/L.
rere is evidence of platelet hyperdestruction in
pregnancy.
rPlatelet closure times are not aected by absolute
platelet count in healthy women during
pregnancy.
rPlatelet closure times are prolonged when there is
anemia in addition to a low platelet count.
re increase in brinogen during pregnancy more
than compensates for the fall in platelet count.
Coagulation factors
Screening tests used to assess the coagulation path-
ways include the activated partial thromboplastin time
(APTT), which measures the intrinsic pathway, the
prothrombin time (PT), which measures the extrin-
sic pathway, and the thrombin time (TT) which meas-
uresthenalcommonpathway.Inpregnancy,the
APTT is usually shortened, by up to 4 seconds in the
third trimester, largely due to the hormonally inu-
enced increase in factor VIII. No marked changes in
PT or TT occur.
Many coagulation factors are increased in preg-
nancy (Table 1.2). Von Willebrand Factor and Factors
VII, VIII, X, and brinogen increase substantially as
gestation advances. In one longitudinal study,10 Fac-
tor VII activity increased from the range 60%–206%
(compared to standard) at the end of the rst trimester
to 87%–336% by term. e same study, found Fac-
tors II and V increased in early pregnancy, but then
reduced in the third trimester. Another cross-sectional
study found a 29% rise in Factor V from 6–11 weeks’ to
36–40 weeks’ gestation.11 Increased levels of coagula-
tion factors are mediated by rising estrogen levels and
thought to be due to both increased protein synthe-
sis and enhanced activation by thrombin. Coagulation
factors remain elevated in the early puerperium and
for assessment of true non-pregnant levels, it is best to
sample 8–12 weeks aer delivery.
Summary points
rAPTT is usually shortened in pregnancy.
rVon Willebrand factor and factors VII, VIII, X,
and brinogen increase.
rere is a variable change in factor XI levels.
rCoagulation factor levels remain high in the early
postpartum period. 5

Section 1. Cellular changes
Table 1.2 Coagulation factors during pregnancy and the early puerperium
6–11
weeks
N=41
12–16
weeks
N=28
17–23
weeks
N=10
24–28
weeks
N=19
29–35
weeks
N=36
36–40
weeks
N=23
3 days
post-natal
N=87
Prothrombin fragments 1 + 2 nmol/l 1.1
⬍2.9
1.1
⬍1.5
1.3
⬍2.1
1.8
⬍3.4
2.0
⬍3.9
1.9
⬍3.5
2.2
⬍4.9
Fibrinogen activity
g/l
3.6
2.5–4.8
3.8
2.5–5.1
3.6
2.6–4.7
4.4
2.9–5.9
4.1
2.5–5.8
4.2
3.2–5.3
4.5
3.1–5.8
Prothrombin activity
iu/dl
153
107–200
160
111–209
153
41–265
172
92–252
153
100–211
162
107–217
169
108–231
Factor V activity
u/dL
99
39–159
101
39–162
111
47–175
108
50–166
111
43–179
129
65–194
141
71–211
Factor VIII activity
iu/dl
107
62–220
129
82–130
189
59–159
187
71–341
180
31–328
176
50–302
192
54–331
Factor IX activity
iu/dl
100
49–151
106
82–130
96
74–118
121
59–183
109
65–154
114
79–150
136
65–207
Factor X activity
iu/dl
125
88–162
129
78–180
128
50–206
159
52–263
146
81–212
152
113–191
162
69–254
Factor XI activity
iu/dl
102
50–154
103
58–147
86
58–114
102
45–162
100
31–169
92
36–181
96
46–146
Factor XII activity
iu/dl
137
70–204
160
52–268
186
64–247
170
54–286
178
78–278
179
62–296
174
86–262
Von Willebrand Antigen iu/dl 137
70–204
160
52–268
186
64–247
170
54–286
178
78–278
179
62–296
174
86–262
RCo
iu/dl
117
47–258
132
55–298
128
50–206
204
68–360
169
86–466
240
100–544
247
97–630
Mean and 2 standard deviation normal ranges. From a cross sectional study of 239 women, each of whom was only sampled once.
Adapted from ref. 11.
RCo: Ristocetin cofactor activity.
Table 1.3 Natural anticoagulant factors during pregnancy and the early puerperium
6–11
weeks
N=41
12–16
weeks
N=28
17–23
weeks
N=10
24–28
weeks
N=19
29–35
weeks
N=36
36–40
weeks
N=23
3 days
post-natal
N=87
Total Protein S
u/dl
80
34–126
77
45–109
66
40–92
68
38–98
67
27–106
58
27–90
69
37–85
Free Protein S
u/dl
81
47–115
72
44–101
64
38–90
60
34–86
54
32–76
57
15–95
58
29–87
Protein C activity
u/dl
95
65–125
94
62–125
101
63–139
105
73–137
99
60–137
94
52–136
118
78–157
Antithrombin activity
u/dl
96
70–122
100
72–128
100
74–126
104
70–138
104
68–140
102
70–133
108
77–137
Mean and 2 standard deviation normal ranges. From a cross sectional study of 239 women, each of whom was only sampled once.
Adapted from ref. 11.
Natural anticoagulants
ere are changes in the balance of the natural anti-
coagulants during pregnancy and the puerperium
(Table 1.3). Levels and activity of Protein C do not
change and remain within the same ranges as for non-
pregnant women of similar age.11 ere are increased
levels and activity of Protein C in the early puer-
perium. Total and free (i.e. biologically available) Pro-
tein S levels decrease progressively through gestation.
Ranges for total and free Protein S are lower in the
6

Chapter 1. Normal changes
Table 1.4 Natural anticoagulants and markers of fibrinolysis
Number of patients
Weeks
41
11–15
48
16–20
47
21–25
66
26–30
62
31–35
48
36–40
61 Post-
delivery
61 Post-
natal
Fibrin degradation
Products g/ml
Mean 1.07 1.06 1.09 1.13 1.28 1.32 1.66 1.04
Fibrinolytic activity
(100/Lysis time)
Mean 7.6 7.4 7.3 5.5 4.5 5.6 6.75 5.75
Lysis time in hours Mean 13.25 13.5 13.75 18.25 22.25 17.8 14.8 17.4
Antithrombin III:C Mean
Range
85
49–120
90
46–133
87
42–132
94
47–141
87
42–132
86
40–132
87
48–127
92
38–147
Antithrombin III:Ag Mean
Range
93
60–126
94
56–131
93
56–130
97
56–138
96
59–132
93
50–136
95
58–133
100
64–134
␣1Antitrypsin Mean
Range
124
66–234
136
86–214
125
53–295
146
85–249
149
89–250
154
91–260
172
84–352
77
44–135
␣2Macroglobulin Mean
Range
176
100–309
178
98–323
170
92–312
160
88–294
157
85–292
153
85–277
146
81–265
142
82–245
Where no units are shown, values are expressed as per cent of standard. Where shown, range is 2.5th–97.5th centile. Samples were collected
longitudinally from 72 women. Post-natal samples were collected 2 weeks-12 months following delivery. The post-natal values were found
to be similar to those obtained from healthy pre-menopausal women who were not using oral contraceptives.
Adapted from ref. 10.
rst trimester (34–126 and 47–115 iu/dL, respectively)
than in women of similar age, not using oral con-
traceptives (64–154 and 54–154 iu/dL, respectively).11
is makes it dicult to diagnose Protein S deciency
in pregnancy. Antithrombin levels and activity are usu-
ally stable during pregnancy, fall during labor and rise
soon aer delivery (Tables 1.3 and 1.4).
Acquired activated Protein C (APC) resistance
has been found in pregnancy, in the absence of Fac-
tor V Leiden, antiphospholipid antibodies or a pro-
longed APTT.11 is has been attributed to high
Factor VIII activity and may also be inuenced by
high Factor V activity and low free Protein S lev-
els. Similar acquired APC resistance has been found
in women using oral contraceptives and in associa-
tion with inammatory disorders. e changes in APC
resistance with gestation preclude use of APC sensitiv-
ity ratios as a screening test for Factor V Leiden during
pregnancy.
Summary points
rProteinCisunchangedinpregnancy.
rProteinSdecreasesinpregnancy.
rAntithrombin levels decrease during labor.
rere is acquired APC resistance during
pregnancy.
Fig. 1.1 Thromboelastograph analyzer.
Thromboelastography
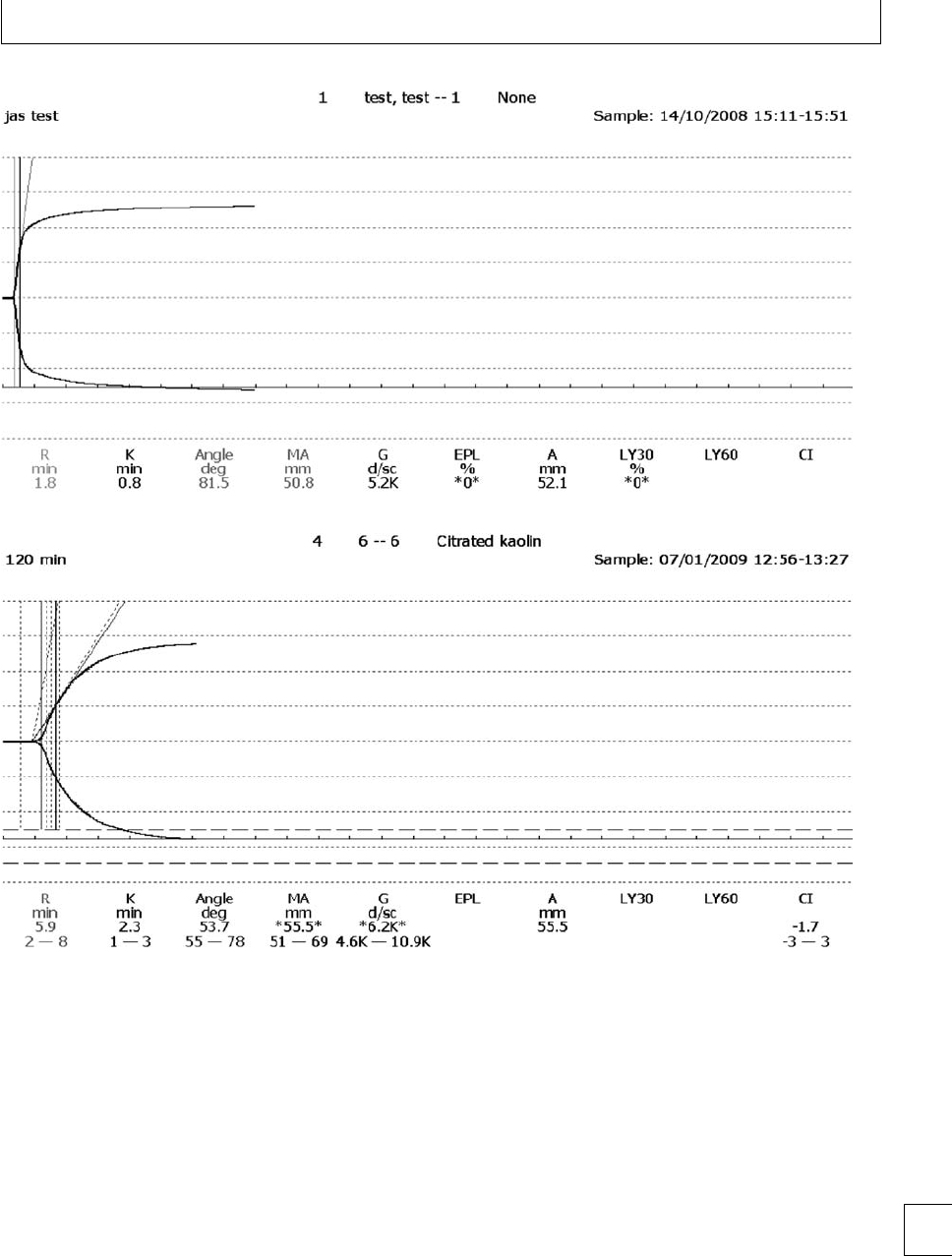
romboelastography (TEG)(Fig. 1.1) provides an
overall assessment of coagulation by measuring the 7

Section 1. Cellular changes
viscoelastic properties of whole blood as it is induced
to clot in a low-shear environment. e parameters
derived from the automated TEG equipment dene
the reaction time to initiation of a clot (R), the clot
formation rate (␣) and time (K), the clot strength or
maximum amplitude (MA) and clot lysis (reduction
in maximum amplitude aer 60 minutes, LY60) (Fig.
1.2). e various parameters are correlated and are
aected by the availability of brinogen and platelet
function. e TEG coagulation index (TEG CI) is
derivedfromR,K,MA,and␣, which has a normal
range of −3 (hypocoagulability) to +3(hypercoagu-
lability).
In healthy late pregnancy, there is increasing hyper-
coagulability and the TEG CI has been measured in
the range −0.6 to +4.3. Within the rst 24 hours of
delivery, TEG CI values of −0.5 to +3.9havebeen
found.12 e highest TEG CI values have been found
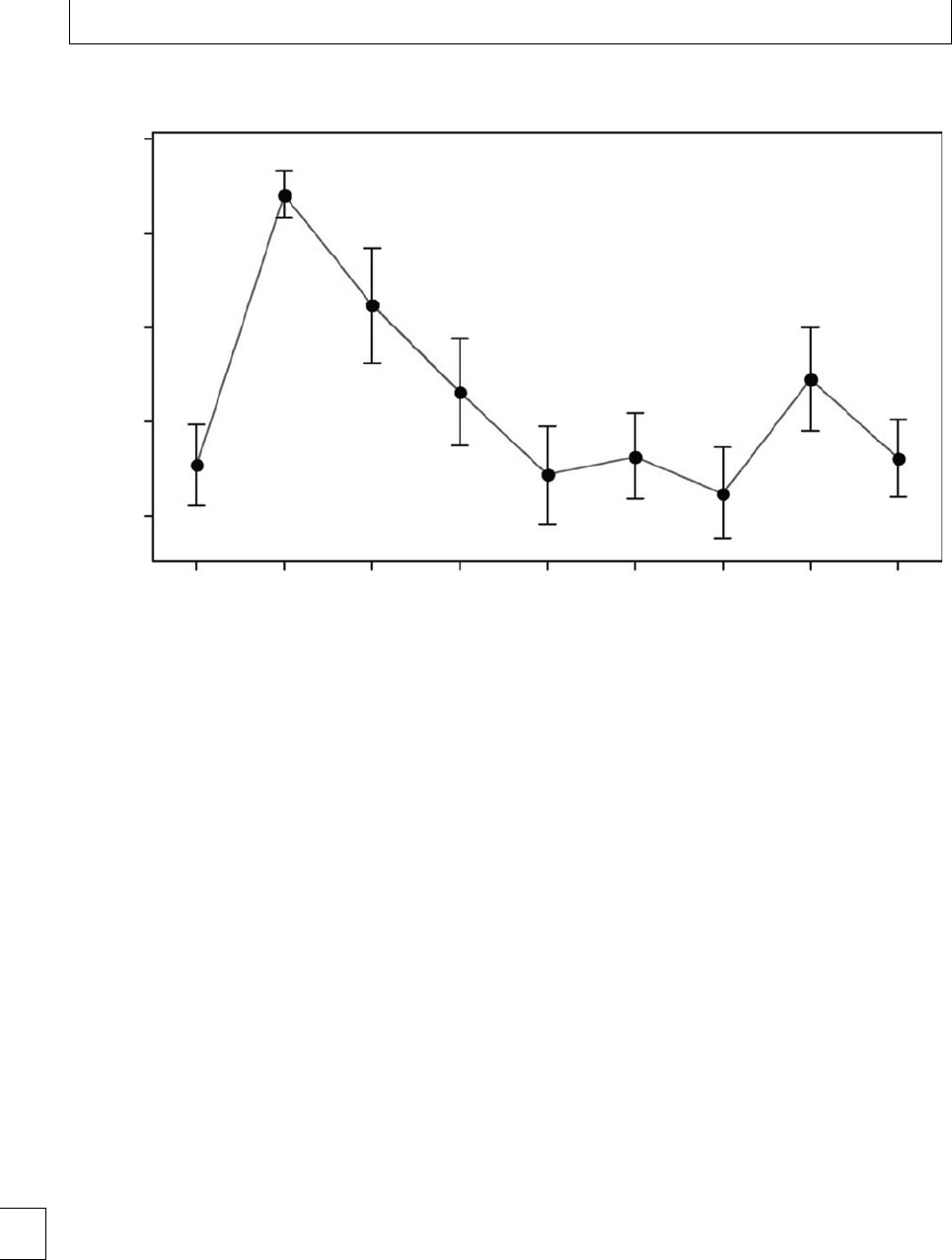
during active labor. Parameters return to baseline by 4
weeks postpartum13(Fig 1.3). No dierences have been
found in TEG parameters during pregnancy between
smokers and non-smokers. Signicantly lower TEG
CI values were found in a large study of women who
took folic acid supplements14 during the rst trimester
(−1.22 to +2.87), indicating that they were less
hypercoagulable than those who did not take supple-
ments (−1.52 to +2.60).
Studies of TEG in pregnant women with thrombo-
cytopenia are inconclusive to date. e TEG MA corre-
lates with platelet count as well as brinogen, but it is as
yet unclear whether TEG parameters can be used clin-
ically to predict the safety of regional anesthetic tech-
niques in women with low platelet counts, especially
those with pre-eclampsia.8,9
Summary points
rTEG gives a global assessment of coagulation
status.
rTEG CI measurement demonstrates the tendency
to hypercoagulability in pregnancy.
rere is insucient experience with TEG in
pregnant women with thrombocytopenia or
pre-eclampsia to judge its clinical usefulness.
Markers of hemostatic activity
Hemostatic activity can be assessed by measuring
markers of both clot formation and clot destruc-
tion.15 Many have been used in research settings,
but the ones that have clinical applications are
thrombin–antithrombin complexes (TAT) and pro-
thrombin fragments (F 1+2), which reect in vivo
thrombin formation, plus tests that demonstrate plas-
min degradation of brin polymer to yield fragments,
namely D-dimers and brin degradation products
(FDP). Exact reference ranges depend on the reagents
and testing kits used for the assays. Increased lev-
els of F 1+2 are shown in Table 1.2; by term, lev-
els are approximately four times higher than those
from a healthy adult population. Likewise, TAT lev-
els15 increase with gestation; in early pregnancy the
upper limit of normal is similar to the adult range of
2.63 g/L, whereas by term, the upper limit of normal
is 18.03 g/L.
D-dimer levels are very markedly increased in
pregnancy, with typical ranges tenfold higher in
late pregnancy than in early pregnancy or the non-
pregnant state. In one study,15 where the healthy
adult range for D-dimers was ⬍433 g/L, by mid
pregnancy the range was ⬍3000 g/L and by
late pregnancy ⬍5300 g/L. It is thought that the
increase in D-dimers reects the increase in brin
during pregnancy, rather than increased brinolytic
activity.
Summary points
rMarkers of thrombin production (TAT and F1+2)
are elevated in pregnancy.
rD-dimers are tenfold higher in late normal
pregnancy than typical levels from healthy
non-pregnant women.
Fibrinolysis
ere is additional hemostatic control exerted by
lysis of the brin clot. is is achieved by plas-
min, created from plasminogen by activators. e
brin mesh is lyzed to brin degradation prod-
ucts, including D-dimers. Tissue plasminogen acti-
vator is the most important endothelial cell derived
plasminogenactivator.ereisreductioninthe
activity of the brinolytic system during pregnancy,
mostly due to increased levels of plasminogen acti-
vator inhibitors (PAI-1 and PAI-2), which are pro-
duced by the placenta. PAI-1 is also produced by
platelets and endothelium. ere is an exponential
8
Chapter 1. Normal changes
(a)
(b)
Fig. 1.2 Thromboelastograph trace (a) pregnant (b) non-pregnant, showing shortened R and K times and increased maximum amplitude in
pregnancy.
increase in PAI-1 with gestation, from typical val-
ues ⬍50 g/Linearlypregnancyandthenon-
pregnant state, to values 50–300 g/L at term.15 Old
studies of brinolytic mechanisms in pregnancy and
the puerperium demonstrated that levels of plas-
minogen activator decline through pregnancy, reach
their lowest levels during labor and increase soon
aer delivery.16 e discovery of PAI-1 and PAI-2
provides the explanation for these changes, which
lead to maximum suppression of brinolysis during
labor.
ere are a number of inhibitors of plasmin,
including ␣2antiplasmin, antithrombin, ␣1antitryp-
sin, ␣2macroglobulin and C1-esterase inhibitor. Levels
of ␣1antitrypsin and ␣2macroglobulin increase aer
delivery (Table 1.4), as do Factor VIII and brinogen 9
Section 1. Cellular changes
70
95% CI for the mean
P<0.0001
P<0.0001
P<0.05
65
60
MA (mm)
55
50
Control 1234
Weeks’ postpartum
567-910-12
Fig. 1.3 Interval plot of maximum amplitude vs. weeks’ postpartum after normal delivery.
activities (Table 1.2); this is an acute phase reaction,
similar to that seen aer surgery. ere are also
increased levels of thrombin activatable brinoly-
sis inhibitor (TAFI) in pregnancy, which inhibits
brinolysis by various mechanisms.17 Overall,
although brinolytic activity increases aer delivery,
it takes at least 6 weeks to be completely restored to
normal non-pregnant levels.
Clotlysistimeisprolongedinpregnancy
(Table 1.4), particularly in the third trimester. In
one study,17 the median and interquartile range
for clot lysis time was 98 (90–111) minutes in the
rst trimester, 110 (99–124) minutes in the second
trimester and 127 (107–171) minutes in the third
trimester, but 92 (80–99) minutes in the rst 24 hours
aer delivery of the placenta.
Increased circulating FDP levels (Table 1.4) and
D-dimers15 are found during pregnancy despite sys-
temic suppression of brinolysis. It is thought that
there is increased brin generation and degradation
locally in the placental circulation. Dierences have
been found in hemostatic and brinolytic processes
in blood samples from venous placental blood and
from forearm blood10. It is also possible that clearance
of FDP and D-dimers may be altered in pregnancy.
Overall, there is a low level of intravascular coagu-
lation, demonstrable from as early as 11–15 weeks
gestation.10 Levels of FDP, D-dimers and soluble b-
rin remain high aer delivery for at least the rst
week.
Summary points
rFibrinolysis is suppressed during pregnancy and
especially during labor.
rPAI-1fromendothelialcellsisincreasedin
pregnancy.
rPAI-2isproducedintheplacenta.
rVarious factors continue to suppress brinolysis
soon aer delivery.
rRaised FDP and D-dimers indicate clot formation
and destruction, possibly locally in the placental
circulation.
10

Chapter 1. Normal changes
Homocysteine
Homocysteine levels fall in early pregnancy and are
signicantly reduced compared to the non-pregnant
state, in all three trimesters.18 is appears to be
multifactorial and related to the hormonal changes
in pregnancy, physiological hemodilution, increased
renal clearance of homocysteine, folic acid supple-
mentation and enhanced remethylation of homocys-
teine due to increased demands for methionine by the
fetus.
11

Section 1. Cellular changes
References
1. Taylor DJ, Lind T. Red cell mass during and aer
normal pregnancy. British Journal of Obstetrics and
Gynaecology 1979; 86: 364–370.
2. Milman N, Bergholt T, Byg K-E et al. Reference
intervals for haematological variables during normal
pregnancy and postpartum in 434 healthy Danish
women. European Journal of Haematology 2007; 79:
39–46.
3. Edlestam G, Lowbeer C, Kral G et al. New reference
values for routine blood samples and human
neutrophilic lipocalin during third trimester
pregnancy. Scandinavian Journal of Clinical
Laboratory Investigation 2001; 61: 583–592.
4. Valdimarsson H, Mulholland C, Fridriksdottir V
et al. A longitudinal study of leucocyte blood counts
and lymphocyte responses in pregnancy: a marked
early increase of monocyte-lymphocyte ratio.
Clinical and Experimental Immunology 1983; 53:
437–443.
5. Boehlen F, Hohfeld P, Extermann P et al. Platelet
count at term pregnancy: a reappraisal of the
threshold. Obstetrics and Gynecology 2000; 95:
29–33.
6. Fay RA, Hughes AO, Farron NT. Platelets in
pregnancy: hyperdestruction in pregnancy. Obstetrics
and Gynecology 1983; 61: 238–240.
7. Vincelot A, Nathan N, Collert D et al. Platelet function
during pregnancy: an evaluation using the PFA-100
analyser. British Journal of Anaesthesia 2001; 87:
890–893.
8. Beilin Y, Arnold I, Hossain S. Evaluation of the platelet
function analyzer (PFA-100 R
)vs.the
thromboelastogram (TEG) in the parturient.
International Journal of Obstetric Anesthesia 2006; 15:
7–12.
9. DaviesJR,RoshanF,HallworthSP.Hemostatic
function in healthy pregnant and preeclamptic
women: an assessment using the platelet function
analyzer (PFA-100 R
) and romboelastograph R
.
Anesthesia and Analgesia 2007; 104: 416–420.
10. Stirling Y, Woolf L, North WRS et al. Haemostasis in
normal pregnancy. rombosis and Haemostasis 1984;
52: 176–182.
11. Clark P, Brennand J, Conkie JA et al.Activatedprotein
C sensitivity, protein C, protein S and coagulation in
normal pregnancy. rombosis and Haemostasis 1998;
79: 1166–1170.
12. Sharma SK, Philip J, Wiley J. romboelastographic
changes in healthy parturients and postpartum
women. Anesthesia and Analgesia 1997; 85: 94–98.
13. Maybury HJ, Waugh JJS, Gornall A, Pavord S. ere is
a return to non-pregnant coagulation parameters aer
four not six weeks postpartum following spontaneous
vaginal delivery. Obstetric Medicine 2008; 1: 92–94.
14. Deol PS, Barnes TA, Dampier K, Pasi KJ,
Oppenheimer C, Pavord SR. e eects of folic acid
supplements on coagulation status in pregnancy.
British Journal of Haematology 2004; 127: 204–
208.
15. Cadroy Y, Grandjean H, Pichon J et al. Evaluation of
six markers of haemostatic system in normal
pregnancy and pregnancy complicated by
hypertension or pre-eclampsia. British Journal of
Obstetrics and Gynaecology 1993;100: 416–420.
16. Bonnar J, McNicol GP, Douglas AS. Fibrinolytic
enzyme system and pregnancy. British Medical Journal
1969; iii: 387–389.
17. Mousa HA, Downey C, Alrevic Z, Toh C-H.
rombin activatable brinolysis inhibitor and its
brinolytic eect in normal pregnancy. rombosis
and Haemostasis 2004; 92: 1025–1031.
18. Walker MC, Smith GN, Perkins SL et al. Changes in
homocysteine levels during normal pregnancy.
American Journal of Obstetrics and Gynecology
1999;180: 660–4.
12

Section 1 Cellular changes
Chapter
2Hematinic deciencies
Jane Strong
Introduction
Deciency of any of the vitamins and minerals essen-
tial for normal erythropoiesis (hematinics) may be
associated with defective erythropoiesis and anemia.
Hematinics include iron, copper, cobalt, vitamins A,
B12,B
6, C, E, folic acid, riboavin, and nicotinic acid.
Iron, folate, and vitamin B12 deciency are the most
common hematinic deciencies. ese are the focus of
this chapter.
Iron deciency
Epidemiology
Iron deciency anemia is the most common health
problem that women face worldwide. It aects about
20% of the world’s population and is a signicant cause
of morbidity and mortality. Of anemias diagnosed in
pregnancy, 75% are due to iron deciency.
On a worldwide perspective, the deciency in iron
reects poor nutrition resulting from widespread eco-
nomic and social deprivation. Many women have
depleted or borderline iron stores due to menstrua-
tion and the demands of previous pregnancies, and
few women enter into pregnancy with sucient iron
stores. Combined with the increased iron demands in
pregnancy due to the expansion in red cell mass and
the requirements of the developing fetus, many women
become iron decient.
Worldwide, iron deciency anemia in pregnancy
aects about 50% of women. In developing countries
the prevalence is 56% and in developed countries 18%.
e majority of these women are already anemic prior
to pregnancy. Prevalence studies in the United States
reveal iron store depletion in about 10% of women of
reproductive age, with anemia present in 5%.
e iron deciency anemia rates in pregnancy
increase with each trimester – starting with 9% in
the rst trimester, 14% in the second, and 37% in the
third.
It is of note that it takes 2 years of normal dietary
iron to replace the iron lost with each pregnancy. More
than 500 mg of storage iron are required to avoid iron
deciency in pregnancy. is amount of storage iron
is present in only 20% of women with 40% having no
storage iron at the start of pregnancy.
Pathogenesis
Iron homeostasis
Dietary elemental iron is absorbed from the duode-
num and jejunum. e typical western diet will contain
15 mg/day iron. e recommended daily allowance of
iron for pregnancy is 30 mg/day.
e dietary bioavailability of iron depends on the
iron content of the food and its form. Heme iron,
derived from meat is more readily absorbed than
non-heme iron. Absorption is facilitated by reducing
agents such as vitamin C, hence the recommendation
to take iron supplements with orange juice or ascor-
bic acid tablets. Absorption is inhibited by phytates
in cereals, tannins in tea and polyphenols in some
vegetables.
Only approximately 10% of dietary iron is
absorbed. is increases in pregnancy and triples
from the rst to the third trimester peaking aer 30
weeks.
e iron requirements of a pregnancy, labor, and
delivery are approximately 1240 mg (see Table 2.1).
Iron requirements in pregnancy rise sharply from
1–2 mg/day in the rst trimester to 4 mg/day in the sec-
ond trimester and peaking at 6 mg a day in the third
trimester. Lactation requires 0.5–1.0 mg/day of iron.
13
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 1. Cellular changes
Table 2.1 Iron requirements for pregnancy, labor, and delivery
Source of increased iron requirement Iron demand
Increase in red cell mass 450 mg
Fetus and placenta 300 mg
Increase in basal maternal requirements 240 mg
Blood loss at delivery (normal vaginal
delivery)
250 mg
Iron requirements for pregnancy, labor,
and delivery
1240 mg
Absorption is regulated by the gastrointestinal
tract and is dependent on iron stores. In normal preg-
nancy a physiological hypervolemia occurs and this
resultsinamodiedresponsetobloodloss.e
plasma volume increases from 6 weeks gestation by
50%. e red cell mass has a slower rate of expansion.
By term, it has increased by 25%, but this is dependent
on iron status.
Iron is required for the red cell expansion and fer-
ritin levels show a marked decline between 12 and
25 weeks. is results in a physiological reduction
in hemoglobin concentration that is maximal at 32
weeks. Hemoglobin concentrations return to normal
within 1 week in the postpartum period in iron-replete
women.
e increase in blood volume helps to compensate
for blood loss at delivery. A blood loss of 1000 ml can
be tolerated without a signicant drop in hemoglobin.
Provided the blood loss at delivery does not exceed
25% of the pre-delivery blood volume, there is no
further increase in blood volume. e plasma vol-
ume decreases as a result of diuresis, the hemato-
crit increases, and the blood volume returns to non-
pregnant values.
The placental regulation of iron transfer
to the fetus
e apical surface of the placental syncytiotrophoblast
has transferrin receptors that trap maternal transfer-
rin by endocytosis, and the iron is bound to halotrans-
ferrin within the placental cell. Iron is released, bound
to ferritin within the placenta, and then actively trans-
ported to the fetus initially as fetal apotransferrin and
then as holotransferrin in the fetal circulation.
If maternal iron decreases, the placental tranfer-
rin receptors increase and conversely placental iron
uptake is inhibited by placental synthesis of ferritin.
Transfer of iron to the fetus occurs predominantly in
the last 4 weeks of pregnancy. Two-thirds of fetal iron
isfoundinthefetalhemoglobin,therestinthefetal
liver.
Maternal iron deciency anemia aects both
mother and fetus. Iron-dependent enzymes in every
cell are aected and there are neuromuscular, gastroin-
testinal, and epithelial consequences that can inuence
fetal mortality, growth, and programing.
Diagnosis of iron deciency
Iron deciency develops sequentially, with storage iron
becoming depleted initially. is is followed by a fall in
the amount of iron available for erythropoiesis. Subse-
quently, the peripheral blood hemoglobin drops and,
with that, there is a fall in the delivery of oxygen to
peripheral tissues, and the patient develops clinical
symptoms and signs.
Each phase in the development of symptomatic
iron deciency anemia has various hallmarks outlined
below.
Decrease in storage iron
Tissue and bone marrow iron become deplete rst.
Bone marrow samples can be specically stained to
look for iron. Without iron supplementation, 80% of
women are deplete of iron stores at term with no stain-
able iron in their bone marrow samples. Although this
is a rapid and reliable method of assessing iron stores,
theinvasivenatureofthetestmeansitisrarelydone
to diagnose iron deciency as there are several reliable
non-invasive tests. Bone marrow examination is gen-
erally reserved for severe anemias when the cause can-
not be determined by other means and when there is
evidence of marrow failure.
Serum ferritin levels fall early in the development
of iron deciency. is is one of the rst abnormal la-
boratory tests. Ferritin levels are not aected by recent
ingestion of iron, but they are an acute phase reactant
rising if there is active infection or inammation.
Transferrin levels increase early in the develop-
ment of iron deciency, but are rarely available as a la-
boratory measure. is transporter protein increases
in an attempt to deliver more iron to the tissues.
Decrease in iron for erythropoiesis
Serum transferrin receptors are transmembrane pro-
teins present in all cells. ey bind transferrin-bound
iron and transport it to the cell interior. Receptors
increase as the iron supply decreases. Small amounts
14

Chapter 2. Hematinic deficiencies
of transferrin receptors circulate in the plasma in
amounts proportional to the total. ese soluble trans-
ferrin receptors can be measured by immunological
techniques. is test is reported as being 100% specic
in identifying iron deciency in pregnancy and has sig-
nicant advantages over ferritin and transferrin satu-
ration.
Once tissue iron deciency is established, serum
transferrin receptors increase in proportion to the
degree of iron deciency. Serum transferrin receptor
level changes occur before a reduction in the mean
corpuscular volume (MCV) and mean corpuscular
hemoglobin concentration (MCHC) in red cells and
also before the rise in free erythrocyte protoporphyrin.
A reduction in MCV and MCHC are seen at an
early stage in the development of iron deciency in
the non-pregnant state, but these are a poor indicator
of iron deciency that develops during pregnancy. e
increased drive to erythropoiesis resulting in the phys-
iological increase in red cell mass means that there are
a higher proportion of young large red cells and this
can mask the eect of iron deciency on red cell MCV.
A normal MCV does not exclude iron deciency and
the red cell indices in established iron decient women
in pregnancy may be normochromic normocytic.
Iron replete pregnancies are associated with a phys-
iological increase in red cell size – usually around 4fL
(femtoliters – 10−15L).
Free erythrocyte protoporphyrin increases as iron
for erythropoiesis reduces. Iron addition to the por-
phyrin ring is the last step in heme biosynthesis. When
iron is low, free protoporphyrin increases. Zinc com-
petes with iron and, if iron is unavailable, zinc proto-
porphyrin levels increase and these can also be meas-
ured. Both free erythrocyte and zinc protoporphyrin
increase in situations of acute infection or inamma-
tion. ese measurements are also elevated in lead poi-
soning.
Decrease in peripheral hemoglobin
Anemia is dened as a hemoglobin level at least two
standard deviations below the median value for a
healthy matched population. e World Health organ-
ization denes anemia in pregnancy as a hemoglobin
below 11 g/dL. Some dene a dierent cut-o in the
second trimester – the United States Centers for Dis-
ease Control (US CDC) use a value of 10.5 g/dL.
e maternal blood volume expands in the rst and
second trimesters – the plasma volume expansion is
increased by 50% and the red cell mass by 18%–25%
depending on iron status. ese physiological changes
cause a dilutional decrease in hemoglobin and hema-
tocrit. Increased hemoglobin in the second trimester
may represent poor maternal blood volume expansion
andisassociatedwithmaternalandfetalmorbidity.A
hematocrit above 43% has been associated with a four-
fold increased risk of fetal growth retardation.
Iron deciency is oen diagnosed retrospectively
aeragoodhemoglobinresponsetoatherapeutictrial
of iron supplements. In populations where there is a
possibility of thalassemia that can present with full
blood count features similar to iron deciency, iron
therapy should only be started aer iron deciency
is conrmed with a measure of iron stores such as
ferritin.
Clinical signs and symptoms
Patients with iron deciency are oen asymptomatic,
but symptoms may occur without an anemia. Iron-
dependent enzymes in every cell are aected and there
are neuromuscular, gastrointestinal and epithelial con-
sequences. Prior to the development of an anemia, the
signs and symptoms of iron deciency are non-specic
and include reduced exercise tolerance and tiredness.
Severe iron deciency is associated with pallor,
glossitis, angular chelitis, nail ridging, and when severe
nail spooning – koilonychia. Dsyphagia can develop
if a post-cricoid web occurs. Iron deciency can
also aect cellular immunity and phagocytosis, with
women being increasingly susceptible to infection.
Pica can occur in as many as 50% of patients as a
symptom of severe iron deciency and can take dier-
entforms–cravingforearth,clay,starch,andice.It
improves with iron replacement (Tables 2.2, 2.3, 2.4).
Table 2.2 Clinical signs and symptoms of iron deficiency
Symptoms Signs
Iron
deficiency
without
anemia
Irritability
Poor concentration
Tiredness and fatigue
Reduced exercise tolerance
None
Iron
deficiency
with anemia
Tiredness and fatigue
Reduced exercise tolerance
Shortness of breath on exercise
Palpitations
Headache
Dysphagia
Pica
Pallor
Glossitis
Angular chelitis
Koilonychia
15

Section 1. Cellular changes
Table 2.3 Effects of iron deficiency
Mother Fetus and pregnancy outcome Neonate, infant, and older
Effects of iron
deficiency
See Table 2.2 above
Decreased cognitive function
Tissue enzyme malfunction
Effects on neuromuscular
transmission
Hb≤9 g/dL – increased risk of:
•Prematurity (doubles risk)
•Small for gestational age
•Spontaneous abortion
Lower Apgar scores
Low iron stores in newborn
associated with growth
restriction, neurological
and mental impairment
Low ferritin:
Placental hypertrophy – increase in
angiogenesis
Increased placenta:fetal ratio is
a predictor of cardiovascular
disease and diabetes in adult life
Table 2.4 Laboratory investigations in iron deficiency
Laboratory test and normal
non-pregnant female
range Normal – pregnancy
Iron deficiency without
anemia – iron store
depletion
Iron deficiency with
anemia –
Mild – severe
Bone marrow reticuloendothelial
iron 2+ – 3+
2+–3+
Difficult to maintain by third trimester
without iron
None None
Serum iron
60–150 mcg/dL
⬎60 mcg/dL progressive fall over
pre-pregnancy values
Borderline low low
Transferrin
200–400 mg/dL
Progressive rise over pre-pregnancy
values – within normal range
Borderline high Raised
Saturation
SI/TIBC: 20%–50%
Progressive fall within normal range Normal Low
Plasma or serum ferritin
40–200 g/L
Decreases within normal range between
12th and 25th week (hemodilution)
⬍40 ⬍20 (mild)–⬍10 (severe)
Soluble transferrin receptors
2.9–8.3 mg/L
First trimester –2.6–6.7 mg/L
Second and third trimester – 25%
increase (increased erythropoiesis)
Increased Increased
Red cell indices and red cell
morphology
MCV can rise: average 4–6 fL Normal Mild hypochromia and
microcytosis
Erythrocyte protoporphyrin
30–70 ng/mL
Progressive rise, usually within normal
range
30–70 ⬎100 – 200
Hemoglobin 12–15 g/dL ⬎11 first and third trimesters
⬎10.5 second trimester
Normal 9–12 (mild), 6–7(severe)
Other tissue changes None None Nail/epithelial changes
Management options
Iron
Iron is available in a variety of forms – dietary, tablet,
and liquid, intravenous and intramuscular.
Dietary iron
In pregnancy it is recommended that iron consump-
tion is increased by 15 mg/day to a daily recom-
mended allowance of 30 mg/day. Women will oen
nd it dicult to increase dietary iron suciently, but
these recommended amounts are met by most pre-
natal vitamin formulations.
Dietary iron is predominantly in the reduced fer-
ric form (Fe3+) and this is poorly soluble above a pH
of 3. It is poorly absorbed at the duodenal pH of 7–
8. e oxidized ferrous form of iron (Fe2+)ismore
soluble at the duodenal pH and hence more easily
absorbed.
Heme dietary sources of iron – meat, sh, and
poultry have a much greater bioavailability than non-
heme vegetable sources. Iron bioavailability from
heme sources is approximately 30% vs. 10% for non-
heme sources.
16

Chapter 2. Hematinic deficiencies
Table 2.5 Iron absorption
Enhanced iron
absorption Reduced iron iron absorption
Ascorbic acid Phytates in bran, oats, rye, and fiber
Heme iron Tannins in tea
Oxidized, ferrous form of
iron (Fe2+)
Polyphenols in some vegetables
High dietary calcium content
Intraluminal factors in the gastrointestinal tract
also aect absorption (Table 2.5).
Tablet and liquid iron
Iron can be given to supplement dietary iron and
maintain iron stores at a time of marked increased
iron demand. Most studies report that this approach
decreases the prevalence of iron deciency anemia
at delivery. is may help anemia in infancy, but
it is unclear whether iron supplementation in well-
nourished non-anemic women improves birth out-
come.
It can be given selectively based on a measure of
iron stores or routinely. e need for iron supplemen-
tation in Western countries is debatable, but the prac-
tice is recommended in the developing world. e
World Health Organization (WHO) recommend uni-
versal oral iron supplementation with 60 mg elemental
iron daily for 6 months in pregnancy in areas where
the prevalence of iron deciency is less than 40%. e
supplementation is continued for 3 months postpar-
tum in areas where the prevalence is greater than 40%.
e Center for Disease Control and prevention rec-
ommends supplementation with 30 mg elemental iron
daily as does the American College of Obstetricians
and Gynecologists.
Universal supplementation is considered practical
and cost eective by some. e debate is ongoing. A
recent Cochrane database library review demonstrated
no denite advantage to mother or fetus with routine
iron or iron and folate supplementation.1
Womenwithirondeciencyanemiashouldreceive
iron supplements of 30–120 mg elemental iron until
theanemiaiscorrectedandtherehasbeentimefor
iron stores to replenish. Oral iron is an eective, cheap,
and safe way of replacing iron, provided there is com-
pliance.
ere are a large number of oral iron-containing
preparations and they oen come combined with other
vitamins and minerals. As a general principle, enteric
coated or slow release formulations should be avoided
as the iron is released beyond the duodenum and prox-
imal jejunum where it is maximally absorbed. Women
should be counseled regarding diet and the factors that
can inhibit iron absorption. Iron salts should ideally
not be given with food because the phytates, tannins,
andphosphateswithinthedietcanbindironprevent-
ing its absorption. Antacids should also be avoided
around the ingestion of iron and ideally ascorbic acid
shouldbetakentoenhanceabsorption.
e iron preparation of choice is based on eective-
ness and minimal side eects. e three ferrous salts
available are ferrous fumurate, ferrous gluconate, and
ferrous sulphate. ey each contain diering quanti-
ties of elemental iron:
rferrous fumarate – 65 mg elemental (ferrous) iron
per 200 mg tablet
rferrous sulphate – 60 mg elemental iron per
300 mg tablet
rferrous sulphate, dried – 65 mg elemental iron per
200 mg tablet
rferrous gluconate – 35 mg elemental iron per
300 mg tablet.
e recommended oral dose of elemental iron for
the treatment of iron deciency is 100–200 mg daily.
Ferrous sulphate 200 mg three times daily provides
195 mg elemental iron and, on this treatment, regimen
the hemoglobin should rise 2 g/dL over 3–4 weeks.
Once the hemoglobin has normalized, the treatment
should be continued for a further 3 months to replen-
ish the iron stores.
Side eects are experienced in 10%–20% of patients
at treatment doses. Iron salts irritate the gastrointes-
tinal tract and can cause nausea, vomiting, epigastric
discomfort, and altered bowel habit (constipation or
diarrhea). ere appears to be a clear dose relationship
with the upper gastrointestinal symptoms, but this is
less clear with the altered bowel habit.
If side eects occur, an iron preparation containing
a smaller dose of iron can be tried. Liquid preparations
can be useful, allowing patients to titrate their dose to
a level where side eects are acceptable. Iron can be
taken with meals, but this will decrease the amount
absorbed.
Parenteral iron
Parenteral iron therapy is available as iron dextran or
sucrose. It is reserved for patients unable to tolerate 17

Section 1. Cellular changes
oral iron or where compliance is in doubt or in patients
where there is a level of bleeding that exceeds the abil-
ityoftheGItracttoabsorbironorthereismalabsorp-
tion. It should be noted that parenteral administration
does not produce a faster response than correctly taken
oral iron that is absorbed adequately. It merely ensures
compliance. First trimester administration is not rec-
ommended.
ere are currently two well-established prepara-
tions approved for use in the UK:
riron dextran (Cosmofer R
) – a complex of ferric
hydroxide with dextran containing 50 mg of
elemental iron/ml that can be given either
intramuscularly or intravenously;
riron sucrose (Venofer R
) – a complex of ferric
hydroxide with sucrose containing 20 mg of
elemental iron/ml that is approved for intravenous
use.
Dose is calculated according to body weight and
iron decit. Cosmofer R
has the advantage of being
licensed for administration as a single total dose infu-
sion. Anaphylactoid reactions can occur with parental
iron preparations and a test dose is recommended
prior to the rst dose. Cardiopulmonary resusci-
tation facilities should be available with injectable
1:1000 adrenaline solution, antihistamines, and corti-
costeroids.
Irondextranhassafetyissuesrelatedtoanaphy-
laxis. e high molecular weight dextran moiety is
thought to share antigens with gastrointestinal organ-
isms. Much of the reported experience with this drug
is in hemodialysis patients. e safety of intravenous
iron dextran has been reviewed in 573 hemodialysis
patients:2
r4.7% had an adverse reaction.
rTen patients (1.7%) had reactions classied as
anaphylactoidincludingcardiacarrestin0.2%,
chest pain1%, and hypotension 0.5%.
rere were no deaths.
rOnly in 4 of the 10 with anaphylactoid reactions
did these occur during the test dose
administration, emphasizing the need for
vigilance.
rDrug allergies were strong predictors for
reactions.
e iron dextran SPC report severe anaphylactoid
reactionsasbeingveryrare⬍1/10 000.
Iron sucrose appears to be safe even amongst
those with a prior history of sensitivity to iron dex-
tran. Again, the experience comes from hemodialy-
sis patients. A group of 665 patients including 80 with
previous iron preparation intolerance experienced no
adverse reactions to iron sucrose.3
e next generation of parenteral iron has recently
become available:
rFerric carboxymaltose (Ferinject R
)–contains
50 mg of elemental iron/ml that can be given
intravenously.
Dose is also calculated according to body weight
and iron decit. It is contraindicated in the rst
trimester of pregnancy. Clinical data on pregnant
women are not currently available and the SPC
advises a careful risk/benet evaluation prior to
use in pregnancy. e appeal of this new product
includes signicantly reduced infusion times and no
requirement for a test dose. Adverse events from
pooled data from 10 multicenter trials involving 2800
patients reported no serious or life-threatening hyper-
sensitivity (anaphylactic) events, but as with other
parenteral iron preparations the SPC warns that
facilities for cardiopulmonary resuscitation must be
available.
Intramuscular iron
Iron administration given by deep intramuscular
injection into the gluteal muscle. is route is oen
painful, can stain the skin and the mobilization of iron
from intramuscular sites is slow and oen incomplete.
ere have been diculties in sourcing the intramus-
cular preparation and its administration has become
less popular.
Erythropoietin
Recombinant human erythropoietin is widely used for
anemia associated with chronic renal failure, malig-
nancy, and cytotoxic chemotherapy. It has been used in
dicult anemia cases in pregnancy. e widest expe-
rience is with Jehovah’s witnesses. It does not cross
the placenta, but carries a risk of hypertension and
thrombosis. Currently, its role in the treatment of
maternal anemia or to increase the yield in autolo-
gous or salvage techniques in pregnancy is not well
established. e usual dose is of 50–200 IU/kg sub-
cutaneously two to three times weekly, usually along
with supplemental iron. Trials of intravenous iron with
18

Chapter 2. Hematinic deficiencies
Table 2.6 Summary of treatment options in iron deficiency
Treatment
option Indication Dose Advantages Disadvantages
Oral iron Standard
treatment
30–120 mg elemental
iron/day until anemia
corrected and stores
replenished
Cheap
Easy to administer
Low bioavailability
Poorly tolerated
Frequent side effects
Often poor compliance
Intravenous
iron
Non-compliance
or intolerance
Calculated according to
body weight and iron deficit
Fast Efficient
Ensures compliance
Reduced need for blood transfusions
Anaphylactoid reactions (see
text)
Intramuscular
iron
Non-compliance
or intolerance
Calculated according to
body weight and iron deficit
Ensures compliance
Reduced need for blood transfusions
Pain, abscess, skin
pigmentation at injection site
Difficult to source product
Erythropoietin Specialist
sub-groups of
patients
50–200 IU/kg sc 2–3
times/week
Useful adjunct in difficult cases and
where blood transfusion prohibited
(e.g., Jehovah’s witnesses)
Hypertension
Pure red cell aplasia
Clinical benefit and
cost-effectiveness not well
established
Blood
transfusion
Emergency
treatment in acute
hemorrhage
Assessment based on
volume lost and
hemoglobin
Fast rise in hemoglobin Risks of infection,
contamination, reaction,
antibody formation
or without recombinant erythropoietin conclude that
intravenous iron therapy is the rst-line treatment
in resistant iron deciency anemia but that erythro-
poietin may be considered in severe anemia requir-
ing rapid correction in patients who do not respond
to intravenous iron alone. e hemoglobin rise with
combination therapy is quicker than with parenteral
iron alone. Median duration of therapy in the com-
bined treatment group was 18 days vs. 25 days in
the group treated with i.v. iron alone. It has also
been used eectively in the setting of postpartum ane-
mia.4,5 Further assessment of clinical benet and cost-
eectiveness is required.
Blood transfusion
Blood transfusion should be avoided if possible. Trans-
fusion should be reserved for acute hemorrhage. In
chronic iron deciency, transfusion is not indicated.
ere are circumstances when women with severe iron
deciency are not detected until just prior to delivery
and there is not enough time for iron in any form to
raise the hemoglobin. Transfusion may be required in
these circumstances and this is regrettable as it reects
lack of antenatal surveillance and action.
Transfusionhasmanyknownrisks,including
transmission of viruses and bacteria, immunomodu-
lation and increase of post-operative infections, mor-
bidity, and mortality. e possibility of transmitting
prions is an increasing concern. Furthermore, con-
dential reporting systems indicate that human error
resulting in incorrect blood being transfused is still the
commonest serious hazard of transfusion.
Top-up transfusions are inappropriate especially
in a patient group that are young, essentially well
and undergoing a physiological process known to be
demanding on iron stores. Transfusion remains the
emergency treatment for acute hemorrhage.
To reduce transfusion in this group of patients,
anemia prevention strategies during pregnancy and
peripartum are required, including antepartum
screening, monitoring, and iron supplementation and
treatment when necessary, to optimize pre-delivery
hemoglobins. ere should also be strategies to
minimize parturition hemorrhage.
Despite the high incidence and burden of disease
associated with iron deciency, good-quality studies
evaluating clinical, maternal, and neonatal eects of
iron administration in pregnant women with anemia
are lacking.6(Tables 2.6, 2.7).
Prevention strategies
Iron supplementation for all
Iron supplementation is a controversial issue in preg-
nancy. Despite the precarious and oen depleted
iron stores during and aer pregnancy, iron 19
Section 1. Cellular changes
Table 2.7 Summary of the iron formulations available
Formulations available Elemental iron content
Prophylactic
dose Treatment dose
Oral iron – tablets
Oral iron – syrups
Oral iron – over-the-
counter
preparations
Ferrous fumarate
Ferrous sulphate
Ferrous sulphate, dried
Ferrous gluconate
Ferrous fumurate –
ferasamal
Sodium Feredetate – sytron
Spatone – iron rich water
straight from source
65 mg/200 mg tablet
60 mg /300 mg tablet
65 mg /200 mg tablet
35 mg /300 mg tablet
45 mg/5 mL
27.5 mg/5 mL
5mg/10mLsachet
26% to 40% bioavailability
compared to an average
5%–20% from food and 3%
–10% from iron pills
1 tablet daily
1 tablet daily
1 tablet daily
2 tablets daily
5mL3xdaily
5mL3xdaily
1 sachet daily
1 tablet twice daily
1 tablet 2–3 x daily
1 tablet 2–3 x daily
4–6 tablets daily in
divided doses
10–20 mL twice daily
10 mL 3 x daily
2 sachets daily
Intravenous iron Iron dextran (Cosmofer R
)–
a complex of ferric
hydroxide with dextran
Iron sucrose (Venofer R
)–a
complex of ferric hydroxide
with sucrose
50 mg of elemental iron/ml
20 mg of elemental iron/ml
N/A Calculated according to
body weight and iron
deficit: consult
product literature
Intramuscular iron Iron dextran (Cosmofer R
)–
a complex of ferric
hydroxide with dextran
50 mg of elemental iron/ml N/A Calculated according to
body weight and iron
deficit: consult
product literature
supplementation in non-anemic women has not
been shown to improve pregnancy outcome.
e Cochrane library database has reviewed the
eects of routine oral iron supplementation with or
without folic acid during pregnancy. Forty trials with
a total of 12 706 women were reviewed. Iron supple-
mentation with or without folic acid does reduce the
number of women with a hemoglobin less than 10 g/dL
in late pregnancy, at delivery and 6 weeks postpartum,
but there are no clear conclusions regarding clinical
outcomes for mother, fetus, or neonate.1
Routine iron and folic acid supplementation is
recommended by international organizations in areas
where there is a high prevalence of anemia.
Selective iron supplementation is the approach
adoptedinmostindustrializedcountries.Assessment
of iron stores, usually with a ferritin level in the rst
trimester, identies women with low or depleted iron
stores and these women are the ones given iron sup-
plements. In the United States the Centers for Dis-
easesControlandPreventionandtheAmericanCol-
lege of Obstetricians and Gynecologists recommend
routine iron supplementation with lower doses of
elemental iron (30 mg/day) as a primary prevention
intervention.
ere are several concerns about iron supplemen-
tation in iron-replete women. ese include hemo-
concentration leading to impaired placental circula-
tion and fetal growth, the production of free radicals,
and oxidative damage and the risk of iron overload
in women with hemochromatosis. Iron-replete women
givenirondonotincreasetheirhemoglobinlevels,and
hemoglobin concentration in these women is a reec-
tion on the degree of plasma volume increase. Inef-
fective plasma volume expansion is predictive of poor
pregnancy outcome.
Claims that iron causes a variety of chronic diseases
and birth defects have not been substantiated. Acci-
dental ingestion of iron supplements by children is a
potential hazard, however. Although most fatal cases
involve ingestion of 2–10 g of iron, as little as 1–2 g
can cause death in young children. Women should be
made aware of this and advised to keep iron tablets well
outofthereachofyoungchildren.
Screening for iron deciency
In industrialized countries screening of women for
iron deciency anemia is done by measuring the
hemoglobin concentration at booking, 28 weeks and
again at 36 weeks if the 28-week blood result is abnor-
mal. Practice does vary in dierent countries.
For many clinicians it is dicult to accept that
in well-nourished populations the extra requirements
of pregnancy are not met by a normal mixed diet.
e hemodilution, which occurs in healthy pregnancy,
20

Chapter 2. Hematinic deficiencies
has encouraged the acceptance of abnormally low
hemoglobin levels as being physiological.
It is my personal opinion, based on experience in
the UK, that iron store depletion without anemia is
not well identied. Women rarely have routine rst
trimester ferritin measurements, which are then acted
on to prevent the development of anemia. By the time
an anemia is established in pregnancy, it is more di-
cult to correct and replenish stores as the iron require-
ments continue to escalate. Either ferritin screen-
ing and selective supplementation needs improve-
ment or universal supplementation should be adopted
as a practical and cost eective approach. Con-
trolled trials do not demonstrate any obvious bene-
t of iron supplementation, but indirect associations
such as general maternal wellbeing, reduced fetal pla-
cental ratio, preterm deliveries, postpartum hemor-
rhage, and recovery from blood loss at delivery have
notbeenlookedatinanobjectivemanner.
Postpartum anemia
Postpartum anemia is dened as a Hb value less than
10 g/dL and an acute or severe anemia corresponds to
Hb less than 8 g/dL. e prevalence of postpartum ane-
miavariesfrom4%to27%.Inindustrializedcountries,
iron stores are depleted in approximately one-third to
one-half of parturients.
e physiological hemodilution that occurs in
pregnancy protects the mother from blood loss at
delivery, but the 5% of deliveries that have a blood loss
greater than 1 litre can result in a symptomatic anemia
andanincreasedriskofbloodtransfusion.
At the present time, there is no consensus on the
management of postpartum anemia, and clinical prac-
tice varies. Treatment currently consists of oral iron
therapy and blood transfusions.
e Cochrane library database has reviewed the
treatment for women with postpartum iron deciency
and concluded that there is some limited evidence
of favorable outcomes for treatment of postpartum
anemia with erythropoietin. Some of the studies sug-
gest improved lactation with this approach. Labora-
tory hematological parameters improve, but it is not
clear how this relates to clinical outcomes. Six ran-
domizedcontrolledtrialswerereviewedinvolving411
women. All the trials involved erythropoietin. e
authors state that further high-quality trials assessing
the treatment of post partum anemia with iron supple-
mentation (e.g., intravenous administration of iron)
and blood transfusions are needed.4
Summary – key points
Reference ranges in pregnancy are dierent from the
non-pregnant adult female population, but are rarely
quoted on laboratory reports.
Hemoglobin levels fall in pregnancy as a result
of a physiological increase in plasma volume that is
greater than the pregnancy-associated increase in red
cell mass.
Iron stores are exhausted by the end of pregnancy
in the majority of women unless iron is given.
Iron deciency accounts for over 90% of anemia
during pregnancy, therefore iron should be the main-
stay of therapy.
Anemia aects quality of life and virtually all
organs.
Maternal anemia inuences mortality, fetal
growth, premature death in utero, and fetal program-
ing.
Anemia is screened for in pregnancy at booking,
28weeks and possibly 36 weeks (if the 28-week test
result is low). e frequency of testing is dependent on
the country of care – France, for example, carries out a
hemoglobin level at every pregnancy visit.
Anemia screening is primarily done by measuring
the hemoglobin. Further investigation is usually a fer-
ritin level or a trial of iron.
All tests assessing iron status have to be assessed in
the light of gestational changes.
Oral iron is the usual rst-line treatment in iron
deciency.
Parenteral forms of iron are of use if there is non-
tolerance or non-compliance of oral iron.
Blood transfusion should not be given as top-ups
for iron deciency anemia.
Peripartum transfusions are oen inappropriate
(in 2004 one series put the rate at 32%).
e prevalence of postpartum anemia varies
between 4% and 27%.
Iron stores are depleted in 33%–50% of parturients
in industrialized countries (Fig. 2.1)
Folate deciency
Epidemiology
Folate deciency has a prevalence of less than 5% in
developed countries and a very low prevalence where 21
Section 1. Cellular changes
Normal result
Abnormal result
Normal Abnormal
Normal Abnormal
BOOKING First trimester FBC
Hb ≥ 11
Repeat Hb
At 28/40
Hb ≥ 10.5 Hb <10.5
Repeat Hb
At 36/40
Hb ≥ 10.5 Hb < 10.5
↓Hb only
Abnormalities of white cells
and/or platelets in addition to
low Hb or in isolation
Check hemoglobinopathy
screen
If positive check ferritin
prior to treatment with iron
Discuss with senior
obstetric/hematology staff
Consider referral to
hematology/obstetric clinic
Hb < 9 Hb < 11
Hb > 9
Send ferritin
Start treatment
doses of iron
whilst awaiting
results, give
dietary advice
Start iron and
folate
supplements, e.g.
Pregaday, give
dietary advice
Women at risk of
anemia
Multiple pregnancies
Multiparity and
pregnancy recurring
after a short interval
Previous iron deficiency
anemia
Repeat approximately 2−4
weekly depending on
hemoglobin and timing in
pregnancy
•Check compliance
•Investigate and treat causes
•Refer to consultant for individual
management plan
Continue
NB ? require permission fig
4.1 hematology in Pregnancy
chptMidwife’s guide to
antenatal investigations A
Sullivan, L Kean, A Cryer
Response Non-response
Fig. 2.1 Algorithm summarizing the management of ante-natal hemoglobin level – haematology in pregnancy, chapter in
Midwife’s Guide
to Antenatal Investigations
, A Sullivan, L Kean, A Cryer.
22

Chapter 2. Hematinic deficiencies
there is food fortication with folic acid. Worldwide
folate deciency is far more common and may compli-
cate one-third of pregnancies. It is a reection of nutri-
tional status.
Pathogenesis
Folateisawater-solubleBvitamin.Itcannotbesynthe-
sized by humans, but is found in a wide variety of food
sources, including leafy green vegetables, liver, citrus
fruits, nuts, bread, and dairy produce. Folate is heat
labile and it is oen lost in the cooking process. It is
absorbed mainly in the jejunum and then taken up by
the liver. Folate stores last several months.
ere are various disorders and factors that cause
or exacerbate folate deciency including malabsorp-
tion, hemolysis (particularly congenital red cell disor-
ders and hemoglobinopathies), myeloproliferative dis-
orders, and anticonvulsants.
Diagnosis
Full blood count and blood lm
B12 and/or folate deciency cause a megaloblastic
anemia. is is usually suspected by the presence
of macrocytic red cells. Megaloblastic erythropoiesis
requires a bone marrow to demonstrate large develop-
ing red cells with nuclear cytoplasmic asynchrony and
giant metamyelocytes. In practice, this is rarely neces-
sary.Inpregnancy,interpretationofMCVcanbemore
dicult due to the physiological increase in red cell
size and the increased likelihood of an additional iron
deciency anemia that may reduce the MCV. Blood
lm examination can provide useful diagnostic clues.
Features suggestive of a megaloblastic anemia include
hyper-segmented neutrophil nuclei (more than ve
segments), oval macrocytes and mild leukopenia, and
thrombocytopenia in severe cases. If iron deciency
co-exists with a megaloblastic anemia, the blood lm
will be dimorphic with a mixture of large and small red
cells (macrocytic and microcytic cells).
Hematinic assays
Redcellfolateassaysgiveanindicationofoverallbody
tissue levels and are better than serum folate levels
that are aected by recent diet and uctuate signi-
cantlyfromdaytoday.Evenso,redcellfolatedoes
not have good sensitivity or specicity in pregnancy.
Serum and red cell folate levels are lower in smokers.
Redcellfolatelevelsmayshowaslightdownwardtrend
in pregnancy, but recover by 6 weeks postpartum. Nor-
mal references in pregnancy have not been established
and standard adult reference ranges quoted on labora-
tory reports are not applicable in pregnancy. 7
Homocysteine levels
Homocysteine is the precursor to methionine in the
remethylation cycle and increases in B12 or folate de-
ciency, as both are required as cofactors. is indirect
measurement is a sensitive marker for folate deciency.
roughout pregnancy, plasma homocysteine levels
are lower than non-pregnant controls. e lowest lev-
els are in the second trimester but may rise slightly
in the third trimester. Normal references in pregnancy
have not been established and standard adult reference
ranges quoted on laboratory reports are not applicable
in pregnancy. 7
Bone marrow
Megaloblastic erythropoiesis is demonstrated by the
nding of large erythroblasts and giant abnormally
shaped metamyelocytes. Although this is a rapid and
reliable method of assessment, the invasive nature of
the test means it is rarely done to diagnose folate de-
ciency as there are several reliable non invasive tests.
Bone marrow examination is generally reserved for
patients with pancytopenia.
Management
Prophylaxis of folate deciency
Mild folate deciency can be associated with neural
tube defects. Folic acid supplementation at a dose of
400 g/day is recommended 3 months prior to con-
ception and throughout the rst trimester. Pericon-
ceptual folic acid reduces the incidence of neural tube
defects by 70%. If women have had a child aected by a
neural tube defect, the recommendation is for a higher
dose of folic acid periconceptually – at least 5 mg/day.
Folate deciency is also an independent risk factor
for thrombosis.
Folate prophylaxis is required in those with an
increased red cell turnover seen in inherited and
acquired red cell disorders and also for those on anti-
convulsants. Currently, doses of 5 mg/day are used for
these indications (Table 2.8). 23
Section 1. Cellular changes
Table 2.8 Folate requirements
Folate supply
Folate demand/
requirement
Typical Western
diet
250 g/day
Average daily
requirements
(non-pregnant)
100 g/day
Average daily
requirements
(pregnant)
400 g/day
increased folate
metabolism
transfer of folate from the
mother to the fetus
(approximately 800 gat
term).
Puerperium 100 g/day from 6 weeks
postpartum
25 g/day in breast milk7
Treatment of folate deciency
Proven folate deciency should be treated with folic
acid 5 mg three times a day. Dietary history should be
taken and advice given. Deciency of B12 should be
excluded as folic acid in these doses can improve the
anemia of B12 deciency, masking the underlying B12
deciency and thereby potentially exacerbating a neu-
rological deterioration.
Treatment of hyperhomocysteinemia
is is based on increasing folate, vitamin B6, and vita-
min B12 levels. Doses used in treatment are usually
1–5 mg folic acid, 10 mg vitamin B6, and 0.4–1 mg
vitamin B12.
Prevention strategies
Folate food fortication
e addition of folic acid to bread has been considered
in government committees for years. e fortication
of the nation’s food supply with vitamins and miner-
als dates back to the post-second world war era. e
British population at this time had signicantly poor
nutrition and, as a consequence, diseases due to vita-
min deciencies such as rickets.
In the US, the Food and Drug Administration have
made folic acid food fortication mandatory since
1998. All enriched our, pasta, rice, and other grain
products contain 140 g of folic acid per 100 grams.
is strategy has reduced the incidence of neural tube
defects by 20% from 37.8 per100 000 live births in
1998 to 30.5 per 100 000 live births currently.8ere
are concerns that, in the eort to reduce the neural
tube defects, other patient groups may have suered
from the increased levels of folate in the diet. Folic
acid supplementation is potentially harmful in B12
deciency, where it can mask the anemia leading to
delayed treatment and risk of neuropsychiatric symp-
toms such as peripheral neuropathy, mood changes,
dementia type syndromes, and posterolateral spinal
cord demyelination (subacute combined degeneration
of the cord). ere are also concerns that high intakes
of folic acid may speed up the progression of certain
cancers.
A major objection against folic acid fortication
in the UK is that it requires mass supplementing the
population at large to treat a relatively small target
group of young mothers and that this group should be
targeted by other means. It is estimated that, by adding
folic acid to bread, spina bida is prevented in 120
babies in the UK every year. For every baby saved, half
a million people, male and female, will have to take the
added folic acid.
Implementing periconceptual folic acid
supplementation
Neural tube closure is complete 4 weeks aer concep-
tion, when many women are not aware that they are
pregnant and will not have initiated folic acid supple-
ments. Women need to be made aware of the recom-
mendation for folic acid so that they may start it early
when attempting to conceive.
Summary
Periconceptual folic acid is advised to reduce the inci-
dence of neural tube defects. Ideally, it should be
started 3 months prior to conception and continued
throughout the rst trimester. e dose is 400 gdaily
unless there has been a previously aected child in
which case the dose should be at least 5 mg/daily.
Folate prophylaxis should be considered in at risk
groups such as those on anticonvulsants and with
chronic hereditary or acquired red cell disorders.
Folate stores can be depleted within months and
women need education on diet to ensure recom-
mended folate consumption.
24

Chapter 2. Hematinic deficiencies
B12 deciency
Epidemiology
Deciency of B12 in pregnancy is rare. It is usually
associated with infertility. B12 plays a key role in the
development of new tissue; thus women who are de-
cient may not ovulate, or a fertilized egg may not
develop, resulting in miscarriage
e most common cause of B12 deciency in the
general population is pernicious anemia and this is
rare in women of childbearing years. Pernicious ane-
mia usually begins aer the age of 40 years. Pernicious
anemia is due to lack of intrinsic factor that is required
to bind B12 in the stomach prior to absorption in the
terminal ileum. Other causes of B12 deciency include
ileal resection, partial gastric resection, Crohn’s dis-
ease, tropical sprue.
Dietary deciency can occur and is most oen seen
inveganswhodonoteatanimalproducts.Evenveg-
ans, however, obtain B12 from bacteria synthesis in the
gastrointestinal tract or on legumes and in marmite.
Maternal cobalamin stores are around 3 mg and the
daily dietary requirement is approximately 3 ug/day.
e developing fetus requires 50 ug/day. It takes about
5 years for a deciency of B12 to manifest itself clin-
ically because of the stores.9
Pathogenesis
Vitamin B12, also known as cobalamin, is present in
animal-derived foodstus such as meat, milk and eggs.
It is required for methionine synthesis and the con-
version of methylmalonyl CoA to succinyl CoA. It is
involved in myelin synthesis, protein and DNA synthe-
sis, and fatty acid degradation.
Inadequate B12 levels leads to hyperhomocysteine-
mia and this in itself can be associated with obstetric
complications. Small subsets of women with recurrent
miscarriages have been found to have elevated homo-
cysteine and is hoped that treatment with vitamins will
reduce levels and prevent pregnancy loss. Low levels
of vitamin B12 have also been found in women with
children with neural tube defects. It is unknown, how-
ever, whether vitamin B12 status aects the incidence
of neural tube defects. Meta-analyses have suggested
an association, but methodological dierences in the
studies mean it is dicult to draw this conclusion.
ere is a correlation between maternal and neona-
tal B12 levels. Whilst persistent deciency can lead to
infertility, mild B12 deciency can be compatible with
Table 2.9 Features of B12 and folate deficiency
Folate deficiency B12 deficiency
Symptoms Anemia
Very occasionally
neuropsychiatric
symptoms
Gradually progressive
symptoms and signs
of anemia
Neuropsychiatric
symptoms inc.
dementia
Signs Neural tube defects
Vascular disease
(associated with high
levels of homocysteine)
Mild jaundice may be
present
Glossitis
Angular stomatitis
purpura due to
thrombocytopenia
Subacute combined
degeneration of the
cord
Rarely optic atrophy
a normal pregnancy outcome but a low B12 level in
the baby especially if the baby is breastfed. is usually
becomes apparent at about the age of 6 months when
the infant fails to thrive, has regression of development
and anemia. Prompt recognition and treatment with
B12 will limit neurological damage.
Diagnosis
See Table 2.9 for clinical signs and symptoms of B12
deciency.
Full blood count and blood lm
Megaloblastic anemia is the hallmark of B12 de-
ciency. Blood lm examination can be useful. See
above, under diagnosis of folate deciency.
Hematinic assays
B12 assays give an indication of overall body tissue lev-
els. B12 levels fall in pregnancy, but this is not thought
to represent a true tissue deciency. It is likely to be
a consequence of increasing maternal plasma volume
and transfer to the fetus. e physiological reduction
can be 30%–50% during pregnancy. Levels tend to be
lowerinsmokers.elevelsreturntonormalrapidly
aer delivery without supplementation. Levels greater
than 130 ng/ml may be considered normal but lev-
els with less than 130 ng/ml with macrocytosis and/or
neurological symptoms should be considered for B12
treatment.
B12 levels can be falsely lowered by folate de-
ciency that resolves with folate treatment. 25

Section 1. Cellular changes
Homocysteine and methylmalonic
acid levels
Homocysteine is the precursor to methionine in the
remethylationcycleandincreasesifB12and/orfolate
are decient as both are required as cofactors. Methyl-
malonic acid is the precursor for the conversion of
methylmalonyl-CoA to succinyl CoA. It increases if
there is a deciency of cobalamin, but it is not aected
by folate stores.
is indirect measurement is a sensitive marker for
B12 deciency in the non-pregnant setting. In preg-
nancy, however, there is a poor correlation between
serum B12 and no correlation between urinary methy-
malonic acid and serum B12. Normal references in
pregnancy have not been established and standard
adult reference ranges quoted on laboratory reports are
not applicable in pregnancy.
Auto-antibodies
Intrinsic factor antibodies can be helpful in the diag-
nosis of pernicious anemia if the results are positive.
Antibodies to intrinsic factor are found in 70% of
patients with pernicious anemia. ese antibodies can
cross the placenta and cause intrinsic factor deciency
in the fetus. Antiparietal antibodies are non-specic
and not very sensitive in diagnosing pernicious ane-
mia. ey are no longer recommended.
Schilling test
is test has been used classically to diagnose perni-
cious anemia. It is contradicted in pregnancy because
of the radiation risks.
Trial of B12
A therapeutic trial of B12 can conrm the diagno-
sis. A reticulocytosis occurs within 3–4 days and
peaks at day 6–7. e hemoglobin concentration rises
within 10 days and usually returns to normal within
8 weeks. Hyper-segmented neutrophils disappear at
around 10–14 days.
In patients with severe anemia, hypokalemia can
occur as potassium is used in the production of new
red cells. is requires monitoring and potassium sup-
plementation if necessary.
Neurological abnormalities are slower to improve
and can take months.
Bone marrow
Megaloblastic erythropoiesis is demonstrated by the
nding of large erythroblasts and giant abnormally
shaped metamyelocytes. Although this is a rapid and
reliable method of assessment, the invasive nature of
the test means it rarely done to diagnose B12 deciency
as there are several reliable non-invasive tests. Bone
marrow examination is generally reserved for patients
with pancytopenia.
Management
Most mechanisms of B12 deciency are absorptive and
treatment is generally parenteral. Hydroxycobalamin
or cyanocobalamin 1 mg is given three times a week
for 2 weeks and then every 3 months. Neurological
involvement may require higher doses.
Oral B12 can be given if dietary deciency is the
etiology. ere is literature supporting the use of high
dose cobalamin 1–2 mg/day in patients with impaired
intrinsic factor function. ere is a second less e-
cient cobalamin transport system that does not require
intrinsic factor. is type of treatment requires very
good patient compliance and monitoring of cobalamin
levels.
Dilemmas
B12 assays are often coupled with
folate assays
Oen unrequested B12 results are generated because
of the coupling of these tests. is can lead to dicul-
ties as the B12 level is almost always low in the preg-
nant population but the quoted reference range on the
laboratory report is that of a non pregnant popula-
tion. is can lead to many phone calls, referrals, and
concerns. Ideally, B12 assays should not be carried out
unless a specic request, based on clinical grounds, has
been made.
Summary
B12 deciency is rare in pregnancy and vitamin B12
levels should be interpreted with caution. B12 levels
fall in pregnancy by up to 50% in the third trimester.
e reference ranges quoted on reports are for non-
pregnant populations.
26

Chapter 2. Hematinic deficiencies
References
1. Pena Rossa JP, Viteri FE. Eects of routine oral iron
supplementation with or without folic acid for women
during pregnancy. Cochrane Database of Systematic
Reviews 2006; 3: CD004736.
2. Fishbane S, Ungureanu VD, Maesaka JK et al.e
safety of intravenous iron dextran in hemodialysis
patients. American Journal of Kidney Disease 1996; 28:
529–534.
3. Arono GR, Bennett WM, Blumenthal S et al.Iron
sucrose in hemodialysis patients: safety of replacement
and maintenance regimens. Kidney International 2004;
66: 1193–1198.
4. Dodd J, Dare MR, Middleton P. Treatment for women
with postpartum iron deciency anaemia. Cochrane
Database of Systematic Reviews 2004; 4: CD004222.
5. Sifakis S, Angelakis E, Vardaki E et al. Erythropoietin
in the treatment of iron deciency anemia during
pregnancy. Gynecological Obstetric Investment 2001;
51: 150–156.
6. Reveiz L, Gyte GML, Cuervo LG. Treatments for
iron-deciency anaemia in pregnancy. Cochrane
Database of Systematic Reviews 2001; 2:
CD003094.
7. Magda M Megahed, IM Taher. Folate and
homocysteine levels in pregnancy. British Journal of
Biomedical Science 2004; 62: 84–86.
8 Honein MA, Paulozzi LJ, Mathews TJ et al.Impactof
folic acid food fortication of the food supply on the
occurrence of neural tube defects. Journal of the
American Medical Association, 2001; 285: 2981–
2986.
9 Murphy MM, Molloy A, Ueland PM et al. Longitudinal
studyoftheeectofpregnancyonmaternalandfetal
cobalaminstatusinhealthywomenandtheirospring.
e Journal of Nutrition 2007; 137: 1863–1866.
27

Section 1 Cellular changes
Chapter
3Inherited red cell disorders
Emma Welch and Josh Wright
Introduction
e hemoglobinopathies are common genetic disor-
ders. ey may result in signicant morbidity and
mortality, aecting all age groups and genders. is
chapter will concentrate on sickle cell disease and thal-
assemia. e abnormalities of hemoglobin can be of
two kinds.
Structural: such as in sickle cell disease, where a sin-
gle nucleotide change in the -globin gene leads to the
substitution of valine for glutamine at position 6 on the
-globin chain.
Or
Disorders resulting from unbalanced globin chain pro-
duction: the thalassemias, the globin chains produced
are structurally normal, but reduced in quantity.
Ante- and neonatal screening for
hemoglobin disorders
Rationale
Ante-natal screening aims to allow informed repro-
ductive choice by identifying couples, at risk of an
aected infant, at an early stage in pregnancy. Options
include pre-natal diagnosis with either termination or
continuation of aected pregnancies.
It has long been known that morbidity and mor-
tality in children with sickle cell disease is high in the
rst5yearsoflife.eprotectiveeectsofhighlev-
els of HbF in the newborn decline over the rst 4–6
months of life, thereaer much of the mortality is due
to pneumococcal septicemia and acute splenic seques-
tration. Successful antibiotic prophylaxis, vaccination
and education programs have all but eliminated these
problems2and are perhaps the single most important
step in the improved survival of sickle cell disease.
Since these severe complications are oen the pre-
senting features of sickle cell disease, a screening pro-
gram is required to identify at-risk couples and/or
aected newborns.
In thalassemia major the failure of globin chain
production results in a severe transfusion-dependent
anemia, which is manifest as HbF levels reduce in
the rst few months of life. From this point on, the
management of thalassemia is based upon regular
transfusion and iron chelation to reduce the risk of
organ damage, particularly cardiac. Care of the patient
with thalassemia involves collaboration of hematolo-
gists, endocrinologists, diabetologists, cardiologists,
with occasional input from other specialities such as
hepatology. With appropriate care and good compli-
ance, life expectancy may be normal; however, early
cardiac death is common in those who do not comply
with iron chelation.
Neonatal screening for inherited disease is only
undertaken if:
1. it is common in a particular population;
2. there is a cost-eective reliable screening strategy;
3. detection of disease leads to improvements in
care/survival.
NHS sickle and thalassemia
screening – an example of a linked
ante-natal and neonatal program
e newborn program screens all births in England,
with samples collected by heel prick onto a Guthrie
card. e regional screening laboratories generally use
high performance liquid chromatography (HPLC) to
detect the presence of signicant variant hemoglobins;
second-line conrmation is performed by iso-electric
focusing. e program has close links with Child
28 e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.
Chapter 3. Inherited red cell disorders
MED
5−15%
α 3.7 I
αTα
SEA
α 3.7 I
α 4.2
α 3.7 III
5%−15%
5%−80%
α 4.2
αTα
αTα
(−α 3.7 1)
5%−40%
αTa
(−α 3.7)
(−α 3.7 I)
(−α 3.7 II)
(−α 4.2)
α+ thalassemia
α0 thalassemia
15−80%
60%
αTα
Fig. 3.1 Distribution of ␣thalassemia
(taken from Barbara Bain,
Hemoglobinopathy Diagnosis
).
Reproduced with permission.
Table 3.1 Outcomes for neonatal screening
Hemoglobin results Diagnostic possibilities
Sickle disorders
FS Sickle cell disease 81%
Sickle cell 0 thalassemia 17%
Sickle HPFH 2%
FSC Hemoglobin SC disease
FSA
HbS⬎HbA
Sickle + thalassemia
?transfusion
Other significant disorders
F only Possible thalassemia major
Prematurity
Homozygous HPFH
FE Homozygous hemoglobin E
Hemoglobin E thalassemia
Hemoglobin E HPFH
FA plus Hb Bart’s
Barts⬎20% A
Hemoglobin H disease
␣thalassemia carrier
HPFH, Hereditary persistence of fetal hemoglobin.
Health, to allow appropriate referral of those requir-
ing further follow-up, and the antenatal laboratories
to highlight mothers at risk of an aected child. e
main aim of the program is the detection of children
with sickling disorders, those at risk of thalassemia
major will be highlighted for further investigation (see
Table 3.1).
With the aim of the antenatal program being
choice, there is considerable time pressure to obtain
results of the patient and partner and to counsel and
arrange antenatal diagnostic procedure if required.
Since termination is one option, early diagnosis is cru-
cial and a target for identication of at risk couples is
set at 10 weeks.
All couples at risk of having an aected child
should be oered pre-natal diagnosis, although many
will decline. Prenatal diagnosis is usually by chorionic
villous sampling between 10 and 12 weeks’ gestation.
efetallossrateisapproximately1%.Alternatively,
amniocentesis may be performed at 15 weeks or more
with a miscarriage rate 0.5%–1%.
If prenatal testing results conrm a fetus aected
with a major hemoglobin disorder, then a couple
need counseling about living with a child aected
by hemoglobinopathy. e earlier a diagnosis of a
hemoglobinopathy is made, the higher the likelihood
that termination is acceptable. In a study examining
at prenatal testing in thalassemia amongst British
Pakistanis, 70% accepted prenatal diagnosis if oered
in the rst trimester, with over 90% of pregnancies
being terminated. However, if testing was oered in
the second trimester, only 40% of couples accepted
prenatal testing with fewer aected pregnancies
terminated.129

Section 1. Cellular changes
Antenatal screening for hemoglobin disorders is
universal in areas of high prevalence and, where preva-
lence is low, the selection for screening is on the basis
of family origin using an ethnicity questionnaire and
red cell indices (see screening algorithms: Figs. 3.2 and
3.3).
Sickling disorders in pregnancy
e sickling disorders are a group of inherited chronic
hemolytic anemias with clinical manifestations occur-
ring as a result of the polymerization of hemoglobin S.
e disorders in which sickling occurs are:
rHomozygous sickle cell disease – HbSS. e most
common and generally the most severe.
Compound heterozygous states
rHemoglobin SC disease
rHemoglobin Salassemia
rHemoglobin SD Punjab
rHemoglobin SO Arab
rHemoglobin SLepore Boston.
CarriageofhemoglobinSisnotassociatedwith
signicant disease and its only signicance in preg-
nancy is in terms of genetic counseling and the need
for partner testing.
Pathogenesis
e clinical manifestations in sickle cell disease are
as a result of many interacting pathological processes
including:
rpolymerization of HbS;
rhemolysis and nitric oxide depletion;
rvaso-occlusion.
Polymerization of HbS
HbS forms insoluble polymers at low oxygen tensions.
e polymers interact with red cell membrane proteins
causing progressive damage ultimately leading to the
formation of the typical sickled blood cell.
Hemolysis
Sickle cell disease is characterized by chronic intravas-
cular and extravascular hemolysis, red cell lifespan is
shortened from 120 days to 16–20 days. is chronic
hemolysisleadstotheliberationoffreehemoglobin
which mops up nitric oxide released from the vas-
cular endothelium. is, in turn, leads to endothelial
activation and vasoconstriction, providing ideal con-
ditions for adherence of cellular blood components.
Vaso-occlusion
e combination of poorly deformable red blood
cells, increased viscosity, endothelial activation, and
vasoconstriction causes ongoing vaso-occlusion in the
microvasculature. e process is further exacerbated
by leukocytosis, platelet activation, and increased lev-
els of pro-inammatory cytokines. Vaso-occlusion
leads to both the acute complications of sickle cell dis-
ease such as painful crises as well as chronic organ
damage, including cardiac and renal impairment seen
in older patients.2,3
Contraception
ere are few data to guide contraceptive choice for
womenwithsicklecelldisorders.Whatiscertainis
that the risks of pregnancy in sickle cell disease far
outweigh the risks of contraception. e condition is
listed as a relative contraindication for some combined
oral contraceptive preparations based upon the theo-
retically greater risk of thromboembolism in sickle cell
disease. ere is little evidence to support this, partic-
ularly with the lower dose pills, which are commonly
prescribed. Progesterone-only contraceptives are also
safe, indeed limited data suggests they are associated
with a favorable change in hematological parameters
such as reduction in hemolytic rate and increased HbF.
Levonorgestrol implants and intrauterine systems are
safe and have a low failure rate. Copper-containing
intrauterine devices have been felt to be contraindi-
cated because of infection, and possibly heavier men-
strual loss.
In general, sickle patients should be oered the full
range of contraceptives available and counseled about
the risks and benets of each method.
Maternal and fetal complications
of pregnancy
Much of the published information on pregnancy in
sickle cell disorders relates to homozygous (SS) sickle
cell disease. is, and S0thalassemia, are, in general,
the most severe forms. Patients with milder sickle con-
ditions such as SC disease and S+thalassemia can
also have complicated pregnancies though the risks are
lower. All patients with sickling disorders should be
30
FBC
Test baby’s
father
High-risk
family origin
Refer to
Consultant
hematologist
Test baby’s
father
Test baby’s
father
No further
action
No further
action
Test baby’s
father
Refer to
Consultant
hematologist
Test baby’s
father
High risk of
alphao
thalassemia
Low risk of
alphao
thalassemia
HbS, HbC,
HbDPunjab, HbE,
HbOArab,
HbLepore
Other
variant
Hb variant
No further
action HPLC
Low-risk
family origin
No further
action
No Hb variant
HbA2< 4%
HbF < 5%
HbA24.0%
or
HbF > 5%
HbA2> 3.5%
beta thal trait
Consider family
origin
MCH < 25 pg
Iron deficiency
alpha thal
HbA2< 3.5% HbF > 5%
Refer to
Consultant
hematologist
Consider
family origin
MCH > 27 pg
MCH > 25 pg
HbS, HbC,
HbDPunjab, HbE,
HbOArab,
HbLepore
Other variant
Hb variant
HPLC
MCV < 27 pg
Fig. 3.2 Testing algorithm for laboratory screening in low prevalence areas.
Test baby’s
father
Refer to
Consultant
hematologist
Test baby’s
father
Test baby’s
father
No further
action
No further
action
Test baby’s
father
Refer to
Consultant
hematologist
No further
action
High risk of
alphao
thalassemia
Low risk of
alphao
thalassemia
Consider family
origin
MCH < 25 pg MCH > 25 pg
Iron deficiency
alpha thal
HbA2< 3/5%
MCH < 27 pg
HbF > 5%
HbA2> 4.0%
or
HbF > 5%
HbA2< 4.0%
HbF < 5%
MCH > 27 pg
No variant
HbS, HbC,
HbDPunjab, HbE,
HBOArab,
H
b
L
epo
r
e
Other variant HbA2> 3.5%
beta thal trait
Hb variant
FBC
HPLC
Fig. 3.3 Testing algorithm for laboratory screening in high prevalence areas.

Chapter 3. Inherited red cell disorders
jointly managed by an obstetrician and a hematologist
with interest and experience in these diseases. Since
these pregnancies are high risk, patients will require
frequent review by the multidisciplinary team.
Twin and multiple birth pregnancies are associated
with a higher rate of serious complications.
Problem-free pregnancies
Despite the potential complications, more than one-
quarter of these pregnancies occur without problems.
Table 3.2
Maternal risks
Increased mortality
Painful crisis
Infection
Chest syndrome
Hypertension & pre eclampsis
Worsening anaemia
Increased cesarian rate
Thrombosis
Table 3.3
Fetal/neonatal risks
Miscarriage
Increased perinatal mortality
Intrauterine growth retardation and low birth weight
Premature delivery
Increased cesarean rate
Maternal mortality
Maternal mortality rates are known to be increased
in sickling disorders. Prior to the 1970s, 30%–40% of
women with sickle cell disease did not survive preg-
nancy, prompting obstetricians to question whether
the maternal risks of pregnancy were justied. Recent
decades have seen a marked improvement, currently
mortality has been shown to be 1%–2% in studies from
USA and Europe.4,5 In Africa, maternal mortality rates
are between 7% and 12%, probably as a result of a lack
of ante-natal care. In Benin, one of the least devel-
oped countries in Africa, an active pre-natal program
reduced mortality to 1.8%, comparable to the West.6
Mortality and morbidity rates have been found to be
similar in both HbSS and HbSC pregnancies.
In the triennial “Condential Enquiries into
Maternal Deaths in the UK” there were ve deaths
between 1982 to 1999 associated with sickling con-
ditions. ese were due to pneumonia, multi-organ
failure following placental abruption in SS disease,
acute chest crisis in SS disease, septicemia in S
thalassemia and sickle crisis with multi-organ failure
in SC disease. From 1999 to 2005 there were four
deaths in women with sickling disorders, but not all
directly associated with their hemoglobinopathy. One
woman with SC disease died of thromboembolism,
another with SS disease and myocardial brosis
diedwhilsthavingatandapainfulcrisis,another
woman also died during an epileptic t, and nally
a woman with SC disease died of an amniotic uid
embolism.
e recent NCEPOD report (“A Sickle Crisis?”
July 2008) highlights diculties with death certica-
tion and autopsy in sickle cell disorders. Few patholo-
gists have signicant experience and non-specialist
sickle clinicians are in a similar position. It is recom-
mended that pathologists with appropriate experience
perform such autopsies, though there are now national
guidelines for autopsy in sickle cell disease. Clinico-
pathological correlation is crucial, for example, in dif-
ferentiating sickle chest from pneumonia or whether
thrombosis is likely to have been in situ or embolic.
Notwithstanding this proviso, the reports into mater-
nal death illustrate the importance of multidisciplinary
management and, in several cases, suggest a lack of
awareness of the nature and diculty of sickle cell
pregnancy.
ese women may have complex co-existing medi-
cal problems which can make the management of their
pregnancy even more challenging.
Perinatal mortality
e last 30 years or so have seen marked improvements
in fetal outcomes as a consequence of joint obstetric/
hematology care. Peri-natal mortality was reported to
be as high as 50%–80% prior to the 1970s. More recent
studies in USA and Europe have reported a peri-natal
mortality rate of between 1–8%,5even in Benin rates
are between 12% and 19%.6Howard et al. reported a
peri-natal death rate of 60 per 1000 in the period 1991–
1993 in UK centers, ve times higher than the general
obstetricpopulationatthistime.
533

Section 1. Cellular changes
Miscarriage
ere is known to be an increased risk of miscar-
riage in the sickling disorders. is has previously been
reported at between 19% and 24%. A recent study in
Jamaica found a miscarriage rate of 36% in sickle preg-
nancies and 10% in controls. is is higher than previ-
ously documented. 7
Premature deliveries
Since the 1970s it has been known that women with
sickling disorders are more likely to have premature
deliveries. is has been reported at an average of
between 34.1 to 38.5 weeks’ gestation. In a recent
Jamaican study the mean gestational age was found to
be 37.0 weeks compared with 38.7 weeks in controls.
In African Americans the mean gestational age was 37
weeks. Infants born to SS mothers are twice as likely to
bepretermcomparedtoHbSCmothers.
7
Fetal intrauterine growth retardation
Intrauterine growth retardation is a well-documented
complication of sickle cell pregnancy. is is thought
to arise as a consequence of maternal anemia
and impaired placental function resulting from
vaso-occlusion in uteroplacental circulation. Histo-
logical studies have shown placental infarction with
abruptions and villous edema.
Of infants born to mothers with sickle cell anemia,
77% have a birth weight below the 50th centile, with
21% below the 10th centile. Neonates born to mothers
with Hb SS disease are signicantly smaller than babies
borntomotherswithHbSCdisease.
4–7
Infections
Patients with sickle cell disease have a complex
immune defect. In addition to hyposplenism, there are
data suggesting subtle changes in leukocyte function,
opsonization and complement pathways. Urinary tract
infections are increased in normal pregnancies and can
lead to pyelonephritis and premature labor. ere may
be a further increase in risk in sickle pregnancy.
Other common sites of infection include chest
and bone. Common pathogens include Pneumococ-
cus,Salmonella,E.ColiandMycoplasma.Infectionis
a common precipitant of painful crises.
Hypertension
Pregnancy-induced hypertension and pre-eclampsia
complicate one-third of pregnancies in sickle cell dis-
ease. ere is an association between hypertension
with proteinuria and simultaneous sickling complica-
tions.4
Thrombotic risk
Pregnancy labor and the puerperium are associated
with complex changes of the hemostatic enzyme sys-
tems. rombotic risk is increased in normal preg-
nancy. To further complicate this situation, it has long
been recognized that steady state sickle cell disease
is associated with evidence of platelet and coagula-
tion activation. Furthermore, changes in the levels of
the naturally occurring anticoagulants and endothelial
activation also have the potential to increase the risk of
thrombosis in sickle cell pregnancy.
Despite these biochemical changes, the role of
thrombosis in sickle cell disease has been dicult to
establish. e pregnant patient with sickle cell disease
should be regarded as at high risk of venous thrombo-
embolism. Pulmonary embolism is dicult to diag-
nose in this setting, but should be considered within
the dierential of a patient presenting with dyspnea
and chest pain.
General management of sickle cell
pregnancy
Preconception
rDiscuss maternal and fetal risks of pregnancy and
counsel about availability of pre-natal diagnosis.
rPartner screening.
rFolic acid supplements.
rReview medications, with assessment of risks vs.
benets for individual drugs. Stop
hydroxycarbamide 3 months before conception
and discuss potential need for transfusion.
At booking
rDiscussion of pregnancy and associated risks.
rEarly involvement of a hematologist with
expertise in the hemoglobinopathies.
rReview by an obstetrician experienced in the care
of women with hemoglobinopathies.
34

Chapter 3. Inherited red cell disorders
rEarly booking appointment and establishment of
a planned schedule of care between obstetrician
and hematologist.
rFBC, Hb electrophoresis/ HPLC, U&E plus full
red cell phenotype. Check ferritin and folate
status.
rEnsure partner screening.
rDiscussion of pre-natal diagnosis, if appropriate
rFolic acid 5 mg daily, continued throughout
pregnancy.
rTakefullhistoryparticularlyfrequencyand
management of crises, transfusions, previous
pregnancies, evidence of chronic organ damage,
which may contribute to risk.
rReview medication – penicillin, folic acid,
hydroxycarbamide, iron chelators, analgesic usage.
rStress the importance of early presentation if
unwell.
rEducation about the signs and symptoms of
infection.
rAnte-natal screening for Hepatitis B, C and HIV,
given likely transfusion history.
rEchocardiogram to assess le ventricular function
and pulmonary pressures if evidence of iron
overload or cardiorespiratory symptoms/signs.
rUltrasound to assess viability and conrm
gestation.
Throughout pregnancy
rContinued health education.
rContinue folic acid 5 mg.
rIron supplementation if ferritin low.
rRegular FBC checks every 4 weeks and U&E every
8weeks.
rSerialultrasoundscansfrom20weekstoassess
fetal growth/placental function.
rMonthly mid-stream urine culture.
rLow threshold for admission especially if limb,
bone, abdominal, chest pain aer 28 weeks.
r24-hour admission policy and contact numbers.
rAppropriate plan for use of analgesia in
pregnancy. Avoid non-steroidal
anti-inammatory drugs aer 34 weeks.
rInvolve obstetric anesthetist to discuss
management in labor.
rPrompt treatment of emesis to avoid dehydration.
rTransfuse only aer discussion with a
hematologist.
rWatch closely for features of acute chest
syndrome. Seek advice from obstetrician,
hematologist and anesthetist. Chest crises are
most likely to occur during late third trimester
and postpartum.
rIf admitted during pregnancy, use low molecular
weight heparin for thrombopropylaxis and
compression stockings.
Painful crisis in pregnancy
re majority of severe crises occur in the third
trimester oen, at the time of delivery, oen the
complications of sickle cell disease precipitate
labor rather than labor precipitating sickling
complications.
r30%–80% of women with Hb SS pregnancies have
crises.
r30% of women with HbSC have crises in
pregnancy. SC disease is generally a milder
condition when not pregnant but patients may
present with pain and other sickle complications
in the third trimester.
rLabor and early puerperium are risk periods for
development of pain. is becomes more likely in
the presence of infection, dehydration or acidosis.
rSickle patients have a renal concentrating defect
from early childhood and pass large volumes of
dilute urine. Attention to hydration status is
therefore crucial.
rCrises in pregnancy may present as abdominal
pain which can be dicult to distinguish from
obstetric complications.
re risk of thromboembolism increases in
pregnancy.
Management
rAdmit to obstetric or hematology ward as per
local protocol. In the nal trimester with the high
risk of obstetric problems, the obstetric setting is
most appropriate.
rInform relevant sta (hematologist/obstetrician).
rEnsure rest and warmth.
rGive oxygen if hypoxic on monitoring of O2
saturation.
rEnsure adequate hydration – oral or intravenous
uids 3–4 liters. Strict uid balance essential.
rPain relief – take account of previous analgesic
history. Use paracetamol, non-steroidals if 35

Section 1. Cellular changes
pregnancy less than 34 weeks but subcutaneous
opiates are oen necessary.
rPethidine is not recommended for the treatment
of sickle pain. Morpine, diamorphine or
oxycodone are appropriate but intravenous use
should be discouraged.
rUse linear analog scale to assess pain control.
Patient-controlled analgesia or subcutaneous
pumps are occasionally required.
rRegular assessment of sedation and conscious
level if on strong opiates. e recent NCEPOD
report highlights deciencies in the care and
monitoring of patients on opiate analgesics.
rInvestigations – FBC, reticulocytes, U&Es, group
and screen, pulse oximetry and arterial blood
gases if appropriate.
rMicrobiology – urine culture, blood cultures and
throat swabs.
rConsider chest X-ray if chest involvement.
rAntibiotics are not routinely required unless
evidence of infection, low grade fever ⬍38 ◦Cis
common in painful crisis even in the absence of
infection.
rLow molecular weight heparin
thromboprophylaxis and compression stockings.
rDiscuss indication for transfusion or exchange
transfusion with hematologist.
rChest physiotherapy including incentive
spirometry will reduce the risk of a subsequent
chest syndrome in patients with rib pain.
Acute chest syndrome (ACS)
is condition remains one of the most common
causes of death in sickle cell disease. It is characterized
by pulmonary inltrates on the chest X-ray, chest pain,
shortness of breath and fever. Not surprisingly, those
unfamiliar with sickle cell disease frequently diagnose
a chest infection and manage with antibiotics alone.
Despite the radiological appearances (which may lag
behind clinical signs), this is predominantly a vascular
event and responds well to blood transfusion.
Management of chest crises
in pregnancy
rInform consultant obstetric and hematology sta
on admission.
rContinuous monitoring of O2saturation and
supplemental oxygen.
rInvestigations – CXR, blood gases on air, pulse
oximetry, FBC, reticulocytes.
rBroad spectrum antibiotics – should include a
macrolide.
rBronchodilators.
riv uids.
rTransfusion, either exchange or top up, should be
considered in hypoxemia (SaO2⬍5% lower than
patient’s steady-state level), deteriorating clinical
status or progressive multi-lobe involvement.
re timing of transfusion rather than the volume
is critical (i.e. early in disease course).
ekeytoappropriatetransfusioninACSisthe
timing rather than the volume of blood used or the
target %HbS. In most cases early top-up or partial
exchange transfusion is the optimal approach. In the
United States, e National ACS study group showed
simple top-up transfusion was performed in 68% of
patients using an average of 3.2 units of packed cells.
isappearedtobeaseectiveasanexchangetrans-
fusion. In the absence of a randomized controlled trial
a sensible approach is to use simple top-up transfu-
sion, aiming for a hemoglobin of no more than 9–10
g/dL, in patients with relatively mild episodes or those
with severe anemia, e.g. ⬍5g/dLandtouseexchange
transfusioninthemoreseverecases.Again,thetim-
ing of exchange transfusion is crucial. It is preferable
to perform a limited manual partial exchange urgently
rather than waiting for several hours or overnight
until sta are available to perform an automated
exchange.
Labor and delivery
rAim to achieve a vaginal delivery, no need to
schedule delivery.
rKeep warm.
rMaintain good hydration – commence iv uids at
time of admission in labor at rate 1 L/8 hours to
maintain good urine output. Strict uid balance.
rCheck full blood count, blood group, and
antibody screen.
rContinuous pulse oximetry. May need
supplemental oxygen.
rContinuous CTG monitoring throughout labor
rEpidural analgesia is pain relief of choice.
rAvoid prolonged labor, not more than 12 hours
and prolonged rupture of membranes, which
increase the risk of infection and dehydration.
36

Chapter 3. Inherited red cell disorders
rIf operative delivery necessary, discuss with
hematologist. Regional (rather than general)
anesthesia reduces the likelihood of sickle crisis
and post-op acute chest syndrome.
rromboprophylaxis with low molecular weight
heparin and graduated compression stockings.
rAlert pediatricians.
Postpartum
Baby
rMonitor for signs of respiratory depression if
opiateshavebeenusedintrapartum.
Mother
rMaintain hydration and oxygenation. Watch for
signs of painful or chest crises.
r4-hourly observations for 24 hours post-delivery.
rLow threshold for the use of antibiotics
particularly aer operative delivery.
rCheck FBC day 1 post-delivery.
rMobilize early and continue thromboprophylaxis
until discharge.
rNo contraindication to breastfeeding.
rGive appropriate contraceptive advice prior to
discharge.
rEnsure patient has follow-up both for post-natal
check and with hemoglobinopathy team.
Dilemmas
Operative deliveries
To section or not? e management of labor in patients
with sickling disorders varies widely from unit to unit.
ere are risks and benets of planned vs. sponta-
neous labor. Many units oer a planned induction at
38 weeks. ere is, however, no evidence to support
this approach and in general spontaneous labor is pre-
ferred. Induction leads to a higher cesarean section
rate, with its own complications plus the implication
thatfuturepregnancieswillneedatrialofscarandbe
associated with a risk of subsequent operative deliv-
ery.ElectiveCesareanisnotusuallyadvisedinsick-
ling disorders. ey are associated with a 30% increase
in maternal morbidity, signicantly higher than when
emergency section is performed in spontaneous labor
for obstetric reasons.
If operative delivery is felt necessary then
the patient’s condition should be optimized pre-
anesthetic. Particular attention needs to be paid to
hydration and oxygenation. e procedure may be
undertaken without transfusion support. However,
if felt necessary, then simple top-up transfusion is
adequate.
Post-operative chest physiotherapy including
incentive spirometry may reduce the risk of chest
syndrome.
Hydroxycarbamide
Many patients with sickle cell disease are routinely
managed with hydroxycarbamide. is agent induces
hemoglobin F and also acts as a nitrous oxide donor.
Hydroxycarbamide has been found to reduce the
occurrence of painful and chest crises and may also
prolong life. It has been found to be teratogenic in ani-
mal studies. Males and females on hydroxycarbamide
should therefore be counseled about the importance of
using contraception whilst on the drug. ey should
be asked to stop hydroxycarbamide at least 3 months
before trying to conceive. However, case series have
been published showing that hydroxycarbamide can
be taken throughout pregnancy without complication.
If conception occurs accidentally whilst on hydroxy-
carbamide, the drug should be stopped.
Prophylactic transfusion
e role of transfusion in sickle cell disease in preg-
nancy is controversial though it is generally accepted
that transfusion is not required as part of the manage-
ment of uncomplicated sickle pregnancy.
e rationale is to reduce the amount of circulat-
ing hemoglobin S thereby improving oxygenation and
placental function. A single randomized control trial
in 1980s concluded that routine prophylactic transfu-
sion from the onset of pregnancy does not alter the
outcome for the fetus; however, the numbers involved
in this study are small and it should therefore be inter-
preted with caution.8A retrospective study of the use
of red cell transfusion in the UK noted a trend towards
fewer sickling complications in third trimester and
puerperium.4ere was no evidence that transfusion
improved fetal growth or outcome. A further study
compared a restricted transfusion policy (not trans-
fusing blood unless the hemoglobin fell below 6 g/dl)
vs. a prophylactic transfusion policy (transfusing if 37

Section 1. Cellular changes
hemoglobin fell below 10 g/dl). ey found similar
rates of crises and other complications in both groups.
e risk of alloimmunization was found to be 10%–
20%, this rate can be reduced but not completely abol-
ished by the use of phenotypically matched blood.4
ese antibodies have potential to produce hemolytic
disease of the newborn and may cause diculty in
provision of compatible units for future transfusions.
All women should have a group and full phenotype at
booking visit to screen for antibodies present.
In conclusion, transfusion should be reserved for
high-risk pregnancies. is would include twin preg-
nancies, women with previous poor obstetric history,
chest crises, recurrent pain, and severe anemia.
Summary
e key to successful outcome of sickle pregnancy lies
in the close interaction between obstetric teams and
hematologists. Close monitoring, awareness of risks
and complications is essential. e majority of preg-
nancies have a successful outcome. Where possible,
pregnancy should be allowed to proceed with mini-
mal intervention there being little evidence that trans-
fusion or operative delivery are of any benet in the
majority of cases.
Thalassemia and pregnancy
In the past, thalassemia major was associated with a
high mortality rate in the rst decade of life. Over
recent years outcomes have improved, with children
surviving into adult life in good health, leading normal
lives, and able to have families of their own.
e mainstay of management is regular transfu-
sions with concurrent iron chelation to reduce iron
overload. e most common cause of death is cardiac
failure due to siderosis, although iron overload can
also occur in the endocrine glands, pancreas, and liver.
Many patients develop growth failure, central hypogo-
nadism, and diabetes.
Pathogenesis
e thalassemias are almost always autosomal reces-
sive disorders caused by mutations or deletions in the
␣or globin genes leading to diminished or absent
production of one or more globin chains. e other
globin chain is produced in relative excess and pre-
cipitates within erythroid precursors causing chronic
hemolysis and ineective erythropoiesis.
␣thalassemia
Four ␣globin genes are inherited as a pair from
each parent. A normal individual is annotated thus
(␣␣/␣␣). e more ␣genes deleted, the more severe
the condition (Table 3.4).
Alpha thalassemias are the commonest single gene
disorders worldwide. Approximate frequencies and
types of carriage are illustrated in Fig. 3.1.
␣thalassemia carrier (␣␣/–), (␣-/␣-) or
(-␣/␣␣)
Carriers of ␣thalassemia are asymptomatic and are
usually rst detected at ante natal screening. eir
hemoglobin is in the normal range or minimally
decreased with low mean cell volume (MCV) and
mean cell hemoglobin (MCH).
Hemoglobin H disease (–/-␣)
oseaectedbyhemoglobinHdiseasehavethree
non-functioning alpha genes. e hemoglobin is
commonly in the range 8–9 g/dl with microcytic,
hypochromic red cell indices, and splenomegaly. HbH
disease is a mild form of thalassemia intermedia,
those aected rarely need transfusion. e anemia may
worsen in pregnancy and with infection. e condition
is diagnosed by the presence of an HbH peak on the
HPLC trace and typical “Golf ball” cells on supravital
staining.
Hemoglobin Bart’s hydrops (–/–)
A complete absence of ␣chains is incompatible with
life and results in the unopposed ␥chains forming
tetramers called hemoglobin Bart’s. is is a common
causeofstillbirthinareaswithahighfrequencyof
(–/␣␣)suchasSEAsiaandtheEasternMediterranean.
efetusisstillbornat34–40weeksordiessoon
aer birth. e Hb Bart’s binds oxygen poorly impair-
ing tissue oxygenation. e fetus appears edematous
andjaundicedwithmassivehepatosplenomegalyand
ascites.
Couples at risk of a child with Bart’s Hydrops
should be picked up by ante-natal screening programs
and oered ante-natal diagnosis. If found to have an
aected infant, termination should be oered.
38

Chapter 3. Inherited red cell disorders
Table 3.4 Effects of alpha gene deletion
Genotpye Outcome Effect
␣␣/␣␣Normal Normal
-␣/␣␣ Heterozygous ␣+
thalassemia trait
Frequently silent or
slight decrease in
MCV/MCH
-␣/-␣Homozygous ␣+MCH⬍25 pg
–/␣␣Heterozygous ␣0
thalassemia trait
MCH⬍25 pg
–/-␣Hemoglobin H disease Hb 8–9 g/dL
–/– Hemoglobin Bart’s
Hydrops
Death in utero
MCV, mean cell volume, MCH, mean cell hemoglobin.
Thalassemia carrier
Asymptomatic and diagnosed at ante-natal screening
or during investigation of microcytic, hypochromic
indices. e hemoglobin is rarely less than 10 g/dl.
Hemoglobin A2 is raised. Iron replacement need not
be given unless a deciency state is proven by reduced
serum ferritin.
Thalassemia intermedia
A range of interacting genetic lesions may lead to a
thalassemic phenotype of varying severity. Some will
be asymptomatic whilst others require intermittent
transfusion. e hemoglobin is usually 10–12 g/dl,
but can be as low as 5–6 g/dl in severe forms. Hep-
atosplenomegaly may be present.
Thalassemia major
is is the inheritance of severe abnormalities in
both globin genes. Onset of symptoms of anemia
occurs as fetal hemoglobin levels decline in the rst
few months of life. Patients are transfusion depend-
ent. If not treated with transfusion, extramedullary
hematopoeisis occurs leading to characteristic skeletal
deformities and hepatosplenomegaly. Morbidity and
mortality in this condition is now caused by transfu-
sional iron overload (Table 3.5).
Management
Carriers of ␣and thalassemia and those with
hemoglobin H disease or other mild forms of tha-
lassemia intermedia can be managed as a normal preg-
nancy. Anemia may worsen during pregnancy because
of the normal physiological changes. Oral iron sup-
plements should be given where there is a reduced
Table 3.5 Effects of iron overload
Common problems due to iron overload with
relevance to pregnancy
rCentral hypogonadism- may require referral to assisted
conception unit
rDiabetes or impaired glucose tolerance
rCardiac siderosis
rSmall stature
rendocrine dysfunction, for example, hypothyroidism
ferritin, but not for microcytosis and hypochromia
alone.
It is important to identify couples at risk of a baby
aected by hemoglobin Bart’s. is should be picked
up by the ante-natal screening program and parents
oered counseling, education and pre-natal diagno-
sis. e mother may also develop “mirror syndrome” a
severe pre-eclampsia, and delivery of a hydropic fetus
and placenta can cause obstetric diculties.
thalassemia major and severe forms of interme-
dia are clinically signicant in pregnancy and require
careful multidisciplinary management.
Fertility
Because of the eects of iron overload, transfused
patients oen have hypogonadotrophic hypogo-
nadism, many patients are on hormone replacement
therapies but this does not restore fertility. e Stan-
dards for the Clinical Care of Children and Adults
with alassemia in the UK 9,10 state that:
rIron chelation should be optimized from
childhood to reduce the risk of infertility.
rWhere there is clinical or biochemical evidence of
pubertal or hormone disturbance, management
by an endocrinologist is required.
rEarly referral for discussion of fertility issues
should be oered. is should be to a clinic
experienced in treating patients with thalassemia.
rCouples may be infertile for a number of reasons
including those unrelated to thalassemia and a
range of investigations may be necessary.
rInduction of ovulation or spermatogenesis may be
required for patients with central hypogonadism.
is needs to be done in a center with experience
of such patients to minimize the risk of
hyperstimulation syndrome and multiple
births.
rIt is imperative that a couple are given the
opportunity to discuss the risk of having a child 39
Section 1. Cellular changes
Risks to women with thalassemia in pregnancy
rPregnancy causes a 30%–50% increase in cardiac output,
thus patients with significant cardiac siderosis are at risk of
decompensation and death
rTransfusion requirements increase in pregnancy
rRisk of accelerating pre-existing diabetic retinopathy or
nephropathy
rWorsening osteoporosis
rHigh incidence of gestational diabetes
rHigh incidence of operative delivery
Table 3.6 Risks to women of thalassemia in pregnancy
with thalassemia or other major hemoglobin
disorder, e.g. sickle cell conditions if partner is a
sickle carrier. e partner must be tested and if
they carry thalassemia or variant hemoglobin
counseled about options and oered pre-natal
diagnosis.
Preconception
Careful preassessment of a woman with thalassemia
considering pregnancy is required.
rFull cardiology assessment including
echocardiogram, T2∗MRI quantication of
cardiac iron (where available) as well as
assessment by a cardiologist
rEndocrinological assessment including glucose
tolerance test. Optimize diabetic control if known
to be diabetic
rIron chelation should be optimized before
pregnancy considered. For well-controlled
patients with evidence of normal pituitary
function, it may be reasonable to stop chelation
for natural conception.
rFolic acid should be started prior to conception
until the end of pregnancy.
rReview rubella status, HIV, Hepatitis C status
prior to pregnancy.
rDiscuss smoking and alcohol consumption.
rPartner screening and risk assessment for
thalassemia
rReview medication – ACE inhibitors should be
changed (Tables 3.6, 3.7).
Management of pregnancy
rEarly booking appointment.
rFBC, group and save and full antibody screen at
booking.
rU&Es and LFTS at booking.
Risks to the baby
rPossibility of a major hemoglobin disorder (depending on
partner carrier status)
rDiabetes is associated with a four fold increased risk of fetal
anomaly and threefold increased risk of peri-natal mortality
rIncreased risk of chromosomal non-dysjunction, related to
maternal iron overload
rIncreased risks of multiple pregnancies secondary to
fertility procedures
rSudden maternal death in late pregnancy
Table 3.7 Risks to the baby of thalassemia
rRegular FBCs throughout pregnancy. Transfusion
requirements are likely to increase.
rClose involvement by obstetrician (experienced in
hemoglobinopathy), consultant hematologist and
cardiologist.
rReview all medications.
rStart folic acid before pregnancy and continue
throughout.
rContinue penicillin prophylaxis (if
splenectomized) throughout pregnancy.
rCalcium and vitamin D supplements are advisable
if bone density already reduced prior to
pregnancy.
rStop ACE inhibitors and bisphosphonates.
rStop iron chelators prior to ovarian stimulation
and pregnancy. Rate of iron accumulation during
pregnancy is surprisingly low.
rIncreased risk of thrombosis in splenectomized
patients.
rromboprophylaxis whilst an inpatient and
during labor and puerperium.
rDiscussmodeofdeliveryinadvance–consider
cardiac problems and possible bony abnormalities
of pelvis to assess suitability for vaginal delivery.
rDiscuss contraception post-delivery.
Medical problems in pregnancy
Bone problems
rTransfusion-dependent thalassemics show very
high rates of osteoporosis and osteopenia which
may be exacerbated by pregnancy.
rDuring pregnancy bisphosphonates need to be
stoppedbutvitaminDandcalciumsupplements
may be continued.
rPatients should be advised against smoking and
alcohol and encouraged to take regular exercise.
40

Chapter 3. Inherited red cell disorders
rPatients with back pain should be told this may
worsen in pregnancy and appropriate analgesia
discussed.
Liver complications
rCommon problem in thalassemia due to viral
infections, iron overload, biliary problems
secondary to gallstones, and drug toxicity.
rIn North America 14% of the thalassemic
population are hepatitis C RNA positive.
rVertical transmission of hepatitis C does occur but
is rare – upper estimates are 6%, but this increases
to 14%–17% where there is co-infection with HIV.
Endocrine problems
re incidence of Type 1 Diabetes Mellitus in
thalassemia major is 6%–8% – these patients need
to be managed as per standard recommendations
for diabetes in pregnancy.
rGlucose tolerance should be assessed throughout
pregnancy.
rTreated hypothyroidism is present in 9% but up to
75% have evidence of thyroid dysfunction.
rAny patient with endocrine dysfunction should be
regularly assessed by a consultant endocrinologist.
Dilemmas
Iron chelation during pregnancy
rIron chelation should be maximized prior to
pregnancy. Where possible, a low cardiac iron
load should be shown by T2∗MR.
rIt is advised that chelation agents are withheld
during pregnancy.
rere are case reports of women receiving iron
chelators throughout pregnancy without
teratogenic eects. Recommencement of chelation
could be considered for patients felt to be at high
risk of cardiac death.
rVitamin C should also be stopped due to a risk of
precipitating cardiac damage.
rSerum ferritin levels may remain stable in
pregnancy, with no more than a 10% increase aer
delivery despite cessation of iron chelation. is
may be due to the hemodilution eect or fetal
consumption of iron.
rWomenshouldbeencouragedtoresumeiron
chelation aer delivery.
rDesferrioxamine is safe to use whilst breast
feeding. Deferiprone and deferasirox should not
be used until breastfeeding ceases.
Transfusion
rTransfusion requirements will increase in
pregnancy.
rPatients who are not normally transfusion
dependent, e.g. thalassemiaintermediaor
hemoglobin H disease may require transfusion in
pregnancy or post-delivery.
rMaintain hemoglobin over 10 g/dl in thalassemia
major.
rIt is reasonable to observe patients with
thalassemia intermedia, provided there is no
cardiac dysfunction and serial ultrasound shows
normal fetal growth, transfusion may be avoided.
rAlloimmunization to minor blood antigens,
which may lead to increased diculties in
cross-matching blood and risk of hemolytic
disease of the newborn in the fetus.
rRisk of transmission of blood-borne viral
infections via transfusion.
Delivery
rMode of delivery needs to take account of
pre-existing cardiac problems.
rere is a high rate of Cesarean section in
thalassemic patients. In the majority of patients
this is due to cephalo-pelvic disproportion
resulting from the small stature of thalassemic
patients and normal growth of the fetus.
rIn the absence of contraindications, labor may
proceed normally.
Cardiac problems
re most common cause of death in thalassemic
patients is cardiac failure secondary to iron
deposition in the myocardium.
rPatients with poor compliance with iron chelators
and a ferritin above 2500 g/l are more likely to
develop cardiac problems, pregnancy should be
delayed in such patients until chelation status is
acceptable.
rCardiac arrythmias, cardiac failure and sudden
death can occur in a previously well patient – and
those without grossly elevated ferritins.
rCardiac T2∗MRI is the investigation of choice to
quantify cardiac iron and assess myocardial 41

Section 1. Cellular changes
function, though is only available in a few UK
centers. Cardiac T2∗levels less than 20
milliseconds correlate with le ventricular
dysfunction. Further aggressive chelation prior to
pregnancy should be undertaken in such cases.
rCardiovascular changes in pregnancy, anemia,
increase in plasma volume and increased cardiac
output can aggravate or precipitate cardiac failure.
rSeverely impaired le ventricular function during
periods of stress maybe evident long before the
onset of cardiac failure and is a contraindication
to pregnancy.
Summary
Multidisciplinary care is essential to the management
of this complex group of patients.
Prior to conception eorts need to be made to max-
imize chelation and assess organ and endocrine func-
tion so that the patient can be counseled accurately.
From assessment of risk (e.g. cardiac, endocrine)
through to induction of ovulation and management of
established pregnancy it is vital to maintain good com-
munication between the various specialist teams.
Red cell membrane disorders
Hereditary spherocytosis refers to a group of disorders
characterized by spherical erythrocytes of increased
osmotic fragility. ere are a variety of molecular
lesions which are typically inherited in an autosomal
dominant manner and result in defects in the pro-
tein structure and interaction between various red
cell membrane components, leading to loss of mem-
brane surface area and reduced deformability. ese
cells have a reduced lifespan, resulting in a hemolytic
anemia. Hereditary spherocytosis occurs in all ethnic
and racial groups and there is considerable hetero-
geneity reecting the wide range of molecular lesions.
Diagnosis is made by the typical blood lm appear-
ances, most patients have anemia, with hemoglobin
between 9–12 g/dL associated with a reticulocytosis
and other biochemical evidence of hemolysis, such
as reduced haptoglobin, raised LDH and bilirubin.
Approximately 10% of patients may have a more severe
anemia (6–8 g/dL). e diagnosis can be conrmed by
an incubated osmotic fragility test or ow cytometry.
Many patients lead normal lives and indeed the diag-
nosis may be an incidental nding.
For the most part, there are few implications for
pregnancy and the outcome is good. Some experience
anemia greater would be expected from the expanded
plasma volume due to higher hemolytic rate. Folate
requirements are increased in any hemolytic anemia
and patients known to have HS should be encour-
aged to take pre-conception folic acid supplements and
continue these through their pregnancy. In the more
severe cases transfusion may be required on an inter-
mittent basis.
Acordsampleshouldbetakenforhemoglobin
and bilirubin levels. Neonates who have inherited HS
themselves may require transfusion, but it is worthy of
note that the degree of anemia at this stage does not
correlate with the hemoglobin level in later life.
Elliptocytosis has no signicant implications for
pregnancy, though folate supplementation throughout
is prudent. Hereditary pyropoikilocytosis is a related
condition and is associated with typical blood lm
appearances and a more severe degree of anemia. In
addition to folate supplementation, such patients may
require transfusion. e need for intervention with
transfusioninallredcellmembranedisordersshould
be judged individually and based upon hemoglobin
level, symptoms and assessments of fetal wellbeing.
Glucose-6-phosphate dehydrogenase
deciency
Deciencies in red cell enzymes oen lead to short-
ened red cell lifespan. G6PD deciency was the rst
of such abnormalities to be discovered and is the most
common. e presence of G6PD is crucial to protect
the red cell from oxidative damage. e deciency is
X linked. Despite the mode of inheritance, females
mayhaveclinicalmanifestationsandbesusceptible
to hemolysis. Because of X chromosome inactivation,
heterozygotes have two populations of red cells, one
normal and one G6PD decient.
e prevalence of G6PD deciency varies consid-
erablybeingrareinNorthernEuropeanpopulations
to frequencies of 20% in parts of Southern Europe,
Africa, and Asia. A large number of mutations within
the gene for G6PD may result in a decient pheno-
type. e majority cause mild deciency and only
result in signicant hemolysis in “stress” situations
such as infection and as a complication of certain
drugs. Rarely individuals have a more severe chronic
non-spherocytic hemolytic anemia. Hemolysis is char-
acterized by the presence of denatured hemoglobin
within the red cell, which can be seen on supravital
staining (Heinz bodies). Diagnosis may be made using
42

Chapter 3. Inherited red cell disorders
G6PD deciency screening tests available in the major-
ity of hematology laboratories or by direct quantica-
tion, which is available in certain centers and used to
conrm positive screens. For the most part, mild de-
ciency has little eect on the pregnancy.
Antenatal management
rDetermine history of hemolytic episodes and
precipitating factors.
rFBC, blood lm for characteristic red cell changes,
serum folate, G6PD assay if not previously tested.
Reticulocyte count, LDH, and bilirubin. Heinz
body preparation is helpful during active
hemolysis.
rAdvise against oxidant drugs (see BNF) and
consumption of fresh or lightly cooked broad
(fava)beans.Ifadrugisfelttobeindicatedand
there is no alternative, then the risks and benets
must be taken into account. G6PD deciency is
heterogenous, patients with a signicant history of
hemolytic crises or chronic hemolysis are more
likely to react adversely than those with a milder
phenotype.
rCheck folate status and prescribe folic acid 5 mg
daily for all patients with chronic hemolysis.
rPatients should be made aware of the symptoms
and signs of acute hemolytic anemia. Hemolysis is
usually self-limiting, as reticulocytes have higher
enzyme activity. However, red cell transfusion
may be required in severe cases. Occasionally,
renal failure can complicate acute severe
intravascular hemolysis and should be treated as
required.
rCaution with all drugs prescribed to the mother to
ensure there is no associated risk of hemolysis.
Management of the neonate
Neonatal erythrocytes have an increased suscepti-
bility to oxidative hemolysis. Immaturity of hepatic
enzyme systems may enhance the risk of jaundice,
G6PD deciency has rarely been described as a cause
of Kernicterus. Hemolysis is usually self limiting but
exchange transfusion may be required for those cases
with severe jaundice.
rA cord sample should be taken at birth for
hemoglobin and bilirubin. G6PD assays should
also be performed, although this may be dicult
to interpret.
rPhytomenadione (a fat soluble preparation of
vitamin K) can be administered to the baby in
accordance with normal procedures. (Water
soluble preparations of the vitamin K should be
avoidedinviewofthepossibleriskofhemolysis
in newborns, though the evidence for this is
conicting.)
rObserve over the rst 4 days of life for jaundice.
Hemolysis is usually self limiting but exchange
transfusion using G6PD screened blood may be
required in selected cases.
Breast feeding
re mother should be advised that certain drugs
may be excreted in breast milk and may trigger
hemolysis in a G6PD decient baby.
Acknowledgment
e authors are grateful for the review and construct-
ivecommentsofDr.DFothergillConsultantObstetri-
cian, Jessops Hospital for Women, Sheeld.
43

Section 1. Cellular changes
References/suggested reading
1. Modell B, Harris R, Lane B et al. Informed consent in
genetic screening for thalassemia during pregnancy:
audit from a national condental inquiry. British
Medical Journal 2000; 320: 337–341.
2. Powars DR. Natural history of sickle cell disease: the
rst 10 years. Seminars in Haematology 1975; 12:
267–285.
3. Serjeant GR. Sickle Cell Disease.Oxford:Oxford
University Press, 1992.
4. Howard RJ, Tuck SM, Pearson TC. Pregnancy in sickle
cell disease in the UK: results of a multicentre survey
of the eect of prophylactic blood transfusion on
maternal and fetal outcome. British Journal of
Obstetrics and Gynaecology 1995; 102: 947–951.
5. Smith JA, Espeland M, Bellevue R et al. Pregnancy in
sickle cell disease: experience of the cooperative study
of sickle cell disease. Obstetrics and Gynaecology 1996;
87: 199–204.
6. Rahimy MC, Gango A, Adjou R et al. Eect of active
prenatal management on pregnancy outcome in sickle
cell disease in African setting. Blood 2000; 96:
1685–1689.
7. Serjeant GR, Loy LL, Crowther M et al. Outcome of
pregnancy in homozygous sickle cell disease.
Obstetrics and Gynaecology 2004; 103: 1278–1285.
8. Koshy M, Burd L, Wallace D et al. Prophylactic red cell
transfusion in pregnant patients with sickle cell
disease. A randomized cooperative study. New
England Journal of Medicine 1988; 319: 1447–1452.
9. United Kingdom alassemia Society Standards for
the Clinical Care of Children and Adults with
alassemia in the UK, 2005.
10. JensenCE,TuckSM,WonkeB.Fertilityin
alassemia major: a report of 16 pregnancies,
preconceptual evaluation and a review of the literature.
British Journal of Obstetrics and Gynaecology 1995;
102: 625–629.
44

Section 1 Cellular changes
Chapter
4Maternal autoimmune cytopenias
Hamish Lyall and Bethan Myers
Introduction
Autoimmune conditions are characterized by the pro-
duction of antibodies against self-antigens (autoanti-
bodies). Since these conditions oen occur during the
second and third decades of life, they may occur dur-
ing, or predating pregnancy. In these circumstances,
the additional considerations of both the eect of
pregnancy on the disease, and the disease (and its
treatment) on the pregnancy need to be taken into
account.
It is recognized that pregnancy may inuence
thecourseofmaternalautoimmunediseases.is
can result in remissions, relapses, or new presenta-
tions of these disorders. e pathogenesis of this phe-
nomenon is likely to be related to the hormonal and
complex immunological changes that occur during
pregnancy. Immunological changes in pregnancy are
necessary to prevent rejection of the fetus, which
expresses both paternal as well as maternal anti-
gens. Placental immunology, and modulation of the
systemic immune response, have been identied as
importantmechanismsofthisimmunetolerance.Itis
probable that these features have a signicant inuence
on autoimmune hematological disorders that occur
during pregnancy.
In this chapter, three auto-immune hemato-
logical conditions that may complicate pregnancy:
immune/idiopathic thrombocytopenic purpura (ITP),
autoimmune hemolytic anemia (AIHA), and autoim-
mune neutropenia (AIN) are discussed. ey are char-
acterized by the development of an autoantibody spe-
cic for a surface antigen on the platelet, erythrocyte,
or neutrophil. Premature cellular destruction occurs,
by reticuloendothelial phagocytosis, T lymphocyte
cytotoxicity, or complement mediated cell lysis.
To date, the relationship between these immune
mechanisms and the immunological changes in
pregnancyisnotfullyunderstood.
Cytopenias occur when the enhanced clearance of
theplatelet,erythrocyteorneutrophilfromtheperiph-
eral blood is greater than the bone marrow’s ability to
produce new cells. ITP is by far the most frequently
seen condition. AIN and AIHA rarely occur in preg-
nancy and few cases are reported in published liter-
ature. e three conditions usually occur in isolation
but occasionally may be seen together, e.g. Evans syn-
drome (ITP and AIHA).
About two-thirds of cases present prior to preg-
nancy with the diagnosis already established, but the
remaining third present during pregnancy, either as
an incidental nding or less commonly in the symp-
tomatic state. For many women, pregnancy is the rst
time that a full blood count (FBC) is performed. Care-
ful evaluation of any abnormal result is required before
an immune cytopenia can be diagnosed.
e majority of autoantibodies implicated in these
disorders are of the IgG subtype, and hence are able
to cross the placenta. Consideration therefore, needs
to be given not just to the implications for the mother,
but also for the developing fetus, and aer delivery, the
neonate.
Management is dicult because, where treatment
is required, there are no agents which are univer-
sally ecacious and all carry the potential for adverse
eects. As with all therapies in pregnancy, the bene-
ts of treatment compared with the relative risks to
mother and baby have to be considered. A multidisci-
plinary approach, combining expertise from obstetri-
cians, hematologists, anesthetists, and neonatologists
is required for optimal care.
45
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 1. Cellular changes
Idiopathic/immune thrombocytopenic
purpura (ITP)
Introduction
ITP is usually a chronic condition in adults, oen
occurring in young women, and can be challenging
to diagnose and manage in pregnancy. Although it
is principally mediated by autoantibodies, the devel-
opment of specic assays as a diagnostic tool has, to
date, proved unsuccessful. erefore, the diagnosis is
predominantly one of exclusion with frequent di-
culty in excluding alternative causes of thrombocy-
topenia. Fortuitously, the risk of major hemorrhagic
complications is low. Successful management requires
maintaining adequate platelet counts for pregnancy
and delivery whilst minimizing the risks of treatment-
related side eects for mother and baby. Potential risks
of fetal thrombocytopenia need to be appreciated and
measures taken to prevent hemorrhagic complications
at delivery.
Epidemiology
e annual incidence, of acute and chronic ITP in
adults, from population-based studies is estimated as
2–4 per 100 000, when dened using a platelet count of
less than 100 ×109/L. ese incidence gures are simi-
lar for Europe and the USA.1Inkeepingwithother
immune disorders, it is more common in women than
men (F:M 1.7–1.9:1), and frequently occurs during
the reproductive years, occurring in all ethnic groups.
e incidence in pregnancy has been estimated at 0.1–
1 per 1000 pregnancies,1,2 accounting for about 3%3
of cases of thrombocytopenia in pregnancy. Approxi-
mately two-thirds of cases of ITP already have an
established diagnosis prior to pregnancy, allowing the
opportunity for pre-pregnancy counseling and plan-
ning for a future pregnancy.
Pathogenesis
rombocytopenia is predominantly caused by
autoantibodies specic for platelet glycoproteins
binding to platelets in the maternal circulation. is
results in immune mediated platelet destruction. e
immune dysregulation which permits autoantibody
formation is still the subject of much research. More
recently it has been found that, in addition to increased
destruction of platelets, there is also suppression of
megakaryopoiesis in the bone marrow. erapeutic
agents targeting this phenomenon are now licensed
foruseinthenon-pregnantsetting.ereisusuallyno
apparent stimulus for the autoantibody production;
however, occasionally a history of recent viral illness
or drug exposure can be implicated. ITP usually
occurs in isolation but may occur with other immune
cytopenias or be secondary to a systemic autoimmune
condition, e.g. SLE. e spleen has an important
role in ITP, being both a major source of antibody
production and the predominant site for destruction
of antibody-bound platelets. e antibodies are of
the IgG subtype and therefore able to cross the pla-
centa and potentially cause thrombocytopenia in the
fetus/neonate.
Diagnosis
Thrombocytopenia in pregnancy
e reference range for platelet counts outwith preg-
nancy is 150–400 ×109/L. During pregnancy there
is a general trend downwards in platelet count, espe-
cially in the last trimester, resulting in a fall of around
10% from the pre-pregnancy level.4,5 is is thought
to be due to accelerated destruction of platelets and
normal physiological dilutional eects. For the major-
ity of women this will not result in the platelet count
falling below the normal laboratory range. However,
if the pre-pregnancy platelet count lies at the lower
end of the normal range, or if there is a more severe
drop in counts, thrombocytopenia occurs. e nd-
ing of mild thrombocytopenia in pregnancy is com-
mon, with approximately 8%–10% of women having a
platelet count below the laboratory normal range.4
Since the diagnosis of ITP is one of exclusion (when
presenting during pregnancy), alternative diagnoses
must be considered and excluded where possible. e
principal dierential diagnoses of thrombocytopenia
in pregnancy are discussed below and are summarized
in Table 4.1.
Gestational thrombocytopenia
e majority of cases of thrombocytopenia in preg-
nancy (74%) are attributable to gestational throm-
bocytopenia (incidental thrombocytopenia) of preg-
nancy.5is is a benign condition and represents
no bleeding risk to mother or fetus. It proba-
bly reects the extreme end of the normal physio-
logical eect described above. It typically occurs in
the third trimester and usually results in a mild
thrombocytopenia. Platelet counts below 70 ×109/L
46

Chapter 4. Maternal autoimmune cytopenias
Table 4.1 Causes of thrombocytopenia in pregnancy
Thrombocytopenic condition Pathogenesis of thrombocytopenia Diagnostic characteristics
Gestational thrombocytopenia Physiological dilution
Accelerated destruction
Third trimester, plts ⬎70 ×109/L
Incidental finding,
no features of other disease
HIP/ Pre-eclampsia/eclampsia Peripheral consumption Unwell patient
clinical features – hypertension, proteinuria,
neurological signs/symptoms
Micoangiopathic hemolytic anemias
(MAHA) – TTP, HUS, HELLP syndrome
Mechanical destruction and peripheral
consumption (accumulation of
micro-thrombi in small vessels)
Unwell patient.
Clinical features – neurological signs, fever,
renal impairment, deranged LFTs, hemolysis
ITP Immune mediated peripheral consumption
and occasional bone marrow suppression
Absence of other causes of thrombocytopenia
Diagnosis of exclusion
Hereditary thrombocytopenia Bone marrow underproduction Family history
Somatic abnormalities
Abnormal blood film
Leukemia/lymphoma Bone marrow infiltration Lymphadenopathy, hepatosplenomegaly,
Other FBC abnormalities
Pseudothrombocytopenia EDTA artefact Platelet clumping seen on blood film
Viral infection Multifactorial Recent viral illness. Risk factors
Drugs Multifactorial Timing of drug exposure
HIP: Hypertension in pregnancy, TTP: Thrombotic Thrombocytopenic purpura, HUS: Hemolytic uremic syndrome, HELLP: Hemolysis with
elevated liver enzymes and low platelets, ITP: Immune thrombocytopenic purpura
should alert the physician to consider alternative diag-
noses, although in rare cases the diagnosis has been
subsequently conrmed in women with counts as
low as 50 ×109/L.6Gestational thrombocytopenia
is not immune mediated and therefore poses no risk
to the fetus. A platelet count that has been normal
before pregnancy, and normal in the rst and second
trimesters is useful in helping make the diagnosis. e
FBC returns to normal within a few weeks of delivery.
It may cause diagnostic diculty with ITP when there
are no pre-pregnancy counts.
Hypertensive disorders
Hypertensive disorders of pregnancy complicate
between 12%–22% of pregnancies, and are a common
cause of thrombocytopenia in pregnancy, accounting
for approximately 20% of cases. “Gestational hyper-
tension,” which includes “hypertension in pregnancy”
(HIP), pre-eclampsia and eclampsia, is responsible for
the vast majority of these. rombotic thrombocy-
topenic purpura (TTP), hemolytic uremic syndrome
(HUS) and Hemolysis, Elevated Liver Enzymes and
Low Platelets (HELLP) syndrome can share similar
features with pre-eclampsia, and distinguishing
between these conditions is sometimes problematic.
Together, these rarer microangiopathic hemolytic
anemia (MAHA) conditions cause less than 1% of
pregnancy-related thrombocytopenia. Management
of these conditions is described in Chapters 17 and
18. Hypertensive disorders may be associated with the
disseminated intravascular coagulation (DIC), which
will contribute to further platelet reduction.
Constitutional thrombocytopenia
Hereditary causes of thrombocytopenia are rare,
accounting for less than 1% of cases. is includes
MYH-9 disorders characterized by giant platelets on
the blood lm and Dohle body inclusions in neu-
trophils. Of these, the May Hegglin anomaly is the
most likely to be encountered in pregnancy. Rarely,
hereditary bone marrow failure syndromes such as
Fanconi’s anemia may present with an isolated throm-
bocytopenia in pregnancy. ese diagnoses may be
suspected if there is a family history of thrombocy-
topenia, unexplained thrombocytopenia in more than
two rst-degree relatives, or physical abnormalities
suggestive of the disorder.
Drugs and infections
rombocytopenia is a frequently occurring side
eect of many medications. Heparin induced throm-
bocytopenia, a potentially life-threatening condition, 47

Section 1. Cellular changes
may occur rarely with unfractionated heparin use in
pregnancy, but to date has not been described with
low molecular weight heparin therapy in pregnancy.
As with the non-pregnant setting, viral infection is
an important cause of thrombocytopenia. Whilst this
occurs as a transient phenomenon with many viruses,
specic consideration should be given to hepatitis and
HIV infection, particularly if risk factors are present.
rombocytopenia in this setting is likely to be mul-
tifactorial with both an immune and non-immune
pathogenesis. Diagnosing these infections early in
pregnancy may allow treatment to be initiated, reduc-
ing the risk of related complications and vertical trans-
mission.
Others
Hematological malignancies can present in pregnancy
and the initial feature may be isolated thrombocytope-
nia. Occasionally, a bone marrow examination may be
required to exclude these.
Laboratory artifact from EDTA present in the sam-
ple tubes may account for some cases of apparent
thrombocytopenia. Examination of the blood lm is
essential to exclude this possibility.
ere are no specic diagnostic tests for ITP.
Although platelet glycoprotein specic antibodies can
be detected in the majority of cases, this test lacks the
sensitivity and specicity to be of clinical use. Diag-
nostic parameters for ITP in pregnancy are: thrombo-
cytopenia with a past history of ITP, or a platelet count
during pregnancy of less than 70 ×109/L with other
causes excluded. Mild thrombocytopenia presenting
in the rst or second trimesters may also represent
ITP, but this is not clinically signicant for the mother
since no treatment is required. In all cases, a careful
history and examination of the blood lm are critical
to the evaluation of thrombocytopenia and diagnosing
ITPinpregnancy.eimportantclinicalandlabora-
tory points for diagnosis are discussed below and listed
inTable 4.2.
History
rWhere there is a preceding history of ITP, check
diagnosis for accuracy.
rDocumented response to corticosteroids or
intravenous immunoglobulin (IVIG) is usually
diagnostic of ITP. In addition, this information is
valuable for deciding on future treatment.
rPrevious pregnancy experience and any
documented blood counts both during and
outside of pregnancy are very useful.
rNeonatal platelet counts from previous successful
pregnancies should be noted.
rNote any illnesses associated with ITP (e.g. SLE),
or the occurrence of other autoimmune disorders
in the patient.
rA family history of thrombocytopenia may
suggest a hereditary disorder.
rIdentify any risk factors for HIV and viral
hepatitis, and include the relevant tests.
rAny current medications should be considered for
the possibility of drug-induced
thrombocytopenia.
Clinical examination
rClinical examination is occasionally of value.
re presence of purpura or mucosal bleeding
should be sought.
rSplenomegaly and/or lymphadenopathy are not
characteristic of ITP.
rPhysical abnormalities may suggest a hereditary
disorder.
Laboratory assessment
rFBC: Incidental nding of thrombocytopenia
should prompt a recheck of the FBC.
rBlood lm is essential to exclude alternative
diagnoses:
(a) Spurious thrombocytopenia is caused either by
EDTA artifact (causing platelet clumps – if
present, repeat count in citrate sample) or platelet
satellitism. Both are readily seen on the lm.
(b) Check erythroid and leukocyte morphology and
conrm within normal limits. Abnormal red cell
(e.g. fragmentation) or white cell morphology
suggests alternative diagnosis.
(c) Check platelet morphology. Giant platelets may be
seen in ITP but, if this is the dominant nding,
consider a MYH-9 disorder and examine the
neutrophils for D¨
ohle bodies. Giant platelets and
thrombocytopeniamayalsobeseenwithBernard
Soulier disease, but a lifelong history of abnormal
bleeding would be expected. Abnormally small
platelets may be seen with hereditary
thrombocytopenia and bone marrow failure
48

Chapter 4. Maternal autoimmune cytopenias
Table 4.2 Evaluation of suspected ITP
Specific point to elicit Relevance
Current history Is patient hemorrhagic?
Viral illness/ risk factors for HIV or hepatitis
Thrombocytopenia genuine. Indication for treatment
Viral cause for thrombocytopenia;
Check serology
Family history Family history of unexplained
thrombocytopenia
Consider hereditary causes
Past medical history Known ITP ITP likely cause thrombocytopenia.
? previous response to steroids or immunoglobulin
SLE, thyroid or other autoimmune
disorders
ITP likely cause thrombocytopenia. Possibility of SLE related
complications
Past obstetric history History of pre-eclampsia or previous
thrombocytopenia in pregnancy
Previous baby with neonatal
thrombocytopenia
Increased likelihood of recurrence
Establish cause
ITP likely. If not, consider possibility of neonatal alloimmune
thrombocytopenia (NAIT)(see chapter 5A)
May predict risk future neonatal thrombocytopenia
Clinical examination Mucocutaneous bleeding
Lymphadenopathy, hepatosplenomegaly
Thrombocytopenia genuine; indication for treatment
Possible leukemia/lymphoma
Not consistent with ITP
Laboratory assessment Platelets: clumping present?
Giant platelets
Normal red cell and white cell numbers
and normal morphology
Schistocytes/red cell fragments
LFTs abnormal
coagulation screen abnormal
Pseudothrombocytopenia; repeat FBC in citrate
Check platelet count by alternative method. Consider MYH-9
disorders
Consistent with gestational thrombocytopenia or ITP
Consider MAHA
Consistent with HELLP syndrome
Consider DIC, hypertensive disorders of pregnancy
syndromes. Automated platelet counts may be
erroneous if they are performed on an analyzer
that relies on impedence counting and if the
plateletsareverylarge.isshouldbesuspectedif
the lm appearances dier signicantly from the
analyzer result. An alternative method of platelet
measurement available on some analyzers (e.g.
ow cytometry platelet count) may give a more
accurate measurement.
Some analyzers are able to measure reticulated
platelets and this percentage increases signicantly in
ITP.
rOther routine investigations which should be
performed are listed in Table 4.2.
Bone marrow examination
rA bone marrow examination can conrm the
presence of normal megakaryocytes, normal
hematopoiesis, and the absence of bone marrow
inltration.
ris is not necessary for younger patients where
there are no other clinical or laboratory features to
suggest bone marrow failure or inltration (BSCH
guidelines 2003).
rConsider performing a bone marrow examination
in cases that do not respond to standard
treatments.
rNB: bone marrow examination will not
dierentiate between ITP and gestational
thrombocytopenia or other consumptive causes,
which constitute the main dierential diagnoses,
only conrming that thrombocytopenia is due to
peripheral consumption.
Management
e aim of management of ITP in pregnancy is not
to achieve a sustained normal platelet count but sim-
ply to maintain a platelet count which is adequate to
avoid hemorrhagic complications during pregnancy,
delivery and immediately postpartum. is conser-
vative approach minimizes the risks of maternal and
fetal exposure to therapeutic agents. ere are no
universally accepted criteria for “safe” platelet counts
in pregnancy. It is advisable that members of the
team involved in managing these cases (obstetricians, 49
Section 1. Cellular changes
Table 4.3 Suggested platelet thresholds for intervention
Intervention Platelet count
Ante-natal, no invasive procedure planned ⬎20 ×109/L
Vaginal delivery ⬎40 ×109/L
Operative or instrumental delivery ⬎50 ×109/L
Epidural anesthesia ⬎80 ×109/L
hematologists, anesthetists) agree a consensus for min-
imum accepted platelet thresholds. Generally, these
can be low in the antenatal period if the patient is not
hemorrhagic. resholds typically need to be higher
for delivery. Suggested platelet thresholds for ITP are
stipulated in Table 4.3.
Monitoring during pregnancy
Platelet counts in women with ITP need to be closely
monitored through pregnancy: in general, monthly in
the rst and second trimesters, 2-weekly in the third,
and weekly near term, although the frequency of mon-
itoring will depend on the rate of change as well as
absolute values.
Treatment
erearetwodecisionstobemadeintreatingITP
in pregnancy: when to treat and what treatment to
give. e majority of women will not require ther-
apy throughout the whole duration of the ante-natal
period.7,8 Only women with very low platelet counts
(⬍20 ×109/L) or who are hemorrhagic will require
treatment at this stage. By contrast, treatment is
oen required to raise the platelet count prior to
delivery. e two treatment options for the initial
management of ITP usually considered are corticos-
teroids and intraveous immunoglobulin (IVIG); anti-
D immunoglobulin appears to have equivalent ecacy
toIVIG,andcouldbeconsideredasanalternative
in non-splenectomized Rhesus positive patients.9e
characteristics of these agents are summarized in Table
4.4a, 4.4b and 4.4c. e choice of which agent to use
requires discussion with the individual about the rela-
tive risks and benets of each treatment. Some author-
ities advocate rst-line therapy with IVIG rather than
corticosteroids. Currently, there is little experience of
using Anti D in pregnancy for ITP; however, it is
widely used in Rhesus D negative women for the pre-
vention of hemolytic disease of the newborn (HDN).
It should be noted that the dose for ITP is substan-
tially higher than for HDN and this may result in an
Table 4.4a Corticosteroids
Advantage Disadvantage
rOral therapy
rMost experience
rCan be used for extended
periods if prolonged
platelet count rise is
required
rDose can be tapered to
minimum required for
desired effect.
rNot a blood product
rInexpensive
rRisk of gestational
diabetes mellitus
rImmunosuppressive
rSlow response 3–7 days
for first response, maximal
response 2–3 weeks
rRisk of osteoporosis with
prolonged therapy
rRisk of hypertension
rPossible adverse effects
on fetus at high doses
(but 90% metabolized)
Table 4.4b Intravenous immunoglobulin (IVIG)
Advantage Disadvantage
rEstablished therapy
rResponse to
treatment is rapid
(6–72 hours)
rNo corticosteroid
side effects
rLow risk to fetus
rIntravenous therapy with long
duration of administration
rPooled plasma product
therefore potential risk of
pathogen transmission for
mother and fetus
rTransient response (⬍1 month)
rRisk of infusional reactions
rRisk of aseptic meningitis
rHeadache common
rExpensive
Table 4.4c Anti D immunoglobulin
Advantage Disadvantage
rShort administration
period (3–15 min)
rGood reported efficacy
rNon-immunosuppressive
rNo corticosteroid side
effects
rLimited experience in
pregnancy
rTransient response ⬍1
month
rOccasionally may induce
significant hemolysis
rPooled plasma product
therefore potential risk of
pathogen transmission for
mother and fetus
rCrosses the placenta.
Fetus may be at risk of
hemolysis
rOnly available to patients
who are Rh positive
(approx. 90% of
individuals)
rNo efficacy if prior
splenectomy
increased risk of neonatal hemolysis. Currently, there
are no anti D preparations licensed in the UK for
the treatment of ITP. Patients with contraindications
to corticosteroids (diabetes mellitus, concurrent infec-
tions, history of steroid psychosis) should be managed
with IVIG or Anti D alone.
50
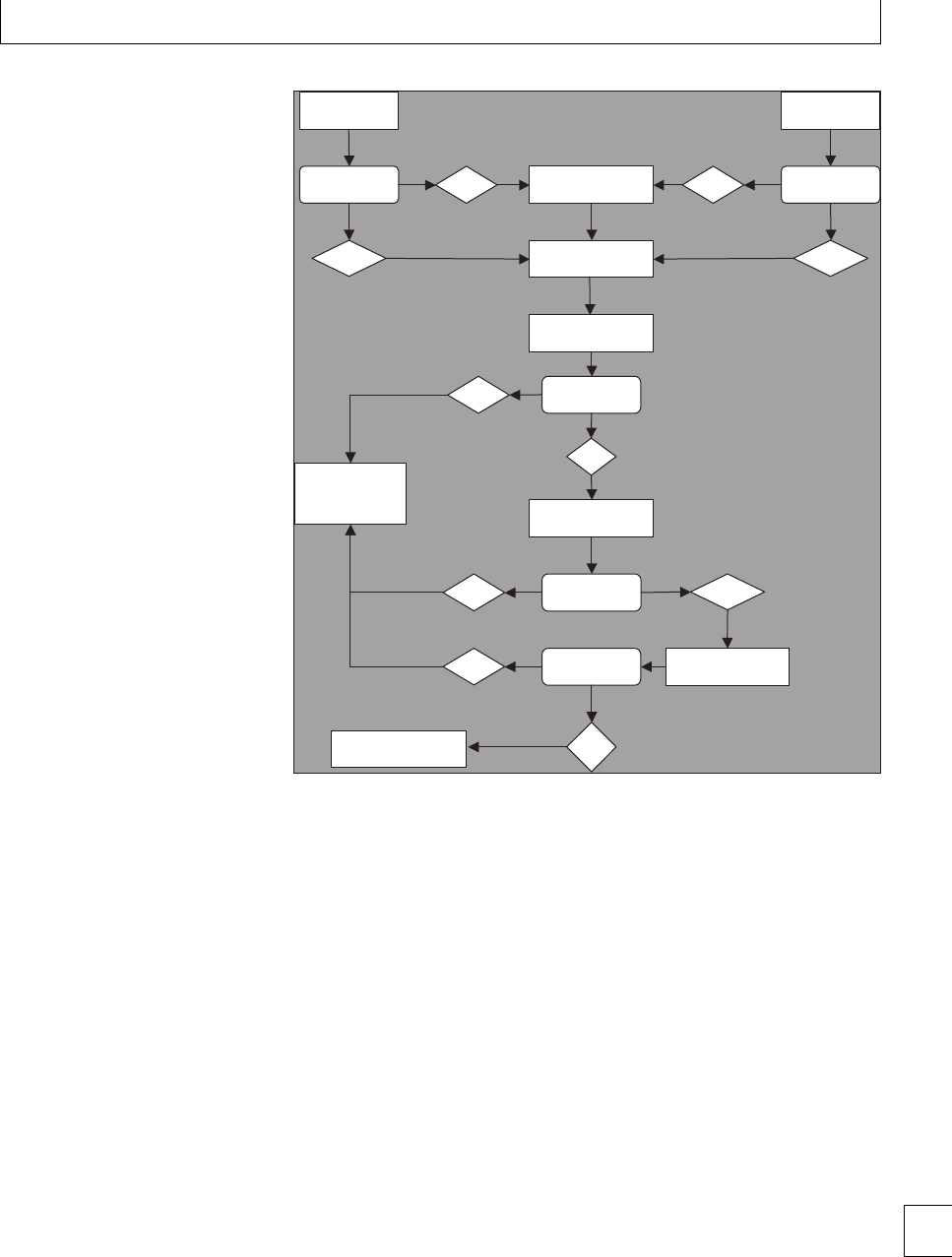
Chapter 4. Maternal autoimmune cytopenias
<36 weeks Assessment at 36
weeks **
Yes
No
Platelets > 50
and stable
Yes
No
Yes
Taper
prednisolone to
lowest effective
dose Prednisolone 60 mg
OD for 1 week
Monitor FBC until
treatment required
Treatment required
Prednisolone 20 mg
OD for 1 week
Response to
treatment?
No
Response to
treatment?
Yes No
Continue Prednisolone.
Add IVIG or Anti D*
Response to
treatment?
Yes
No
Consider alternative
therapies
Platelets >20 and
asymptomatic?
Fig. 4.1 Algorithm for initial ITP therapy
∗IVIG 0.4 g/kg for 5 days or 1 g/kg for 2
days
Anti D 50–70 mcg/kg single dose
Consider methylprednisolone 1 g IV in
addition to IVIG or Anti D.
∗∗ Reassess earlier if obstetric indications
that might need early delivery
e choice of therapy depends on the following factors:
rthe speed with which a platelet increment is
required;
rthe length of time for which a rise needs to be
sustained;
rwhich therapy carries the least potential risk for a
given individual.
A suggested algorithm for initial therapy is shown in
Fig. 4.1. is algorithm is only suitable for uncompli-
cated cases. Suggested management for various scen-
arios are listed below.
Patients with moderate/severe thrombocytopenia (⬍20 ×109/L)
rPrednisolone 20 mg/day10 (it is common practice
to use less than the 1 mg/kg to avoid adverse
eects).
Patients with very severe thrombocytopenia (≤10×109/L)
signicant major bleeding:
rrequires treatment to raise the platelet count
urgently;
rIVIG +/−high dose corticosteroids (usually
60 mg daily);
rconsider platelet transfusions if signicant
bleeding.
Patients with life-threatening bleeding
rplatelet transfusion +;
rIVIG +IV methyl-prednisolone;
Where possible, the dose of prednisolone should be
promptly reduced to the minimum eective dose.
Unless hemorrhage is a major feature, prolonged
therapy (⬎6weeks)withhighdosesofprednisolone
is considered to carry too high a risk of adverse
events for the mother. e response to IVIG or anti 51

Section 1. Cellular changes
D is oen transient. Patients receiving these treat-
ments may require repeat infusions. e addition of
methylprednisolone 1 g intravenously is used to speed
up/improve the response as compared to standard
prednisolone in dicult or refractory cases.
It is not always possible to achieve the desired
platelet count in individuals with ITP. Many patients
(35% in one series) diagnosed with ITP in pregnancy
will not respond to corticosteroids or IVIG. In addi-
tion, response to platelet transfusions are transient
with poor increments as circulating antibody rapidly
clears transfused platelets.
Management of refractory cases
rIn considering other therapeutic options the
balance of risks need to be considered between
treatment-related toxic eects vs. risk of major
bleeding with prolonged severe
thrombocytopenia. In many circumstances it may
be preferable or necessary to accept the increased
hemorrhagic risk of signicant thrombocytopenia
ratherthanusemoreaggressivetherapies.
rSplenectomy: this procedure has a well
established, though diminishing, role in ITP. It
can generally be performed safely in pregnancy
but carries the risks of general surgery and of fetal
loss. Where possible, it should be performed in
the second trimester. is avoids the risks of
teratogenicity associated with drugs in the rst
trimester. In the third trimester the gravid uterus
may make splenectomy technically more
demanding, although laparoscopic splenectomy
maymaketheproceduremorefeasible.
rTranexamic acid: this is an antibrinolytic,
normallyavoidedinpregnancybecauseof
concerns that it may increase thrombotic risk.
Reproductive animal studies do not indicate risk
to the fetus, but there are no adequate and
well-controlled studies done on pregnant women
(category B). It could be considered in the
refractory patient with ongoing symptoms, aer
the rst trimester.
rAzathioprine is used as a second-line agent, and
has been given safely in pregnancy, but there is
insucient evidence to currently advocate its
routine use in this setting. It has a slow onset of
action (about 8 weeks), which also reduces its
utility.
rRituximab: this agent is an anti-CD20
monoclonal antibody, which is increasingly used
to treat non-pregnancy related ITP. However,
there is insucient evidence regarding safety and
ecacy to advocate its use during pregnancy. e
manufacturer currently recommends avoiding
pregnancy for 1 year following treatment.
Other agents, which are useful outside pregnancy,
such as androgen analogs (e.g. danazol), and cytotoxic
agents such as cyclophosphamide or vinca alkaloids,
are contraindicated in pregnancy.
General measures – the following should be avoided:
raspirin and non-steroidal medication;
rintramuscular injections;
rstrenuous activity.
Planning for delivery
Consideration of potential maternal and neonatal
thrombocytopenia is required in addition to any
obstetric factors that may be present when planning
delivery.
Maternal considerations
e principal concern is hemorrhage. is may be dur-
ing delivery or postpartum. Postpartum hemorrhage is
of particular concern due to the sharp fall in procoagu-
lant factors that occurs at this time. As discussed above,
there is no universally agreed safe platelet count; how-
ever, hemorrhage caused by thrombocytopenia occur-
ring at a platelet count ⬎50 ×109/L would be consid-
ered unusual.
Epidural analgesia is of particular concern, as even
a small increase in venous hemorrhage could have the
potential for spinal cord compression. e risk is con-
sidered to be greatest at the time of insertion and with-
drawal of the catheter. ere is controversy over the
safe threshold for epidural anesthesia; there is some
evidence to suggest that a platelet count of 50 ×109/L
is adequate (based on British Society of Haematol-
ogy guidelines), however anesthetic practice is to use a
threshold of at least 80 ×109/L, in experienced hands
(based on BCSH and anesthetic guidelines). A pre-
delivery anesthetic consultation is helpful to discuss
alternative analgesia during labor. e role of spinal
anesthetic is more dicult. is procedure may allow a
cesarean section to be performed without the need for
52

Chapter 4. Maternal autoimmune cytopenias
a general anesthetic. A decision may be taken that the
risksofasinglepassspinalneedlecouldbelessthan
those of a general anesthetic in some situations and, if
an experienced obstetric anesthetist is available, a cut-
o of 50 ×109/L is suggested.
Chronic immunosuppression antenatally for ITP
may increase the risks of postpartum sepsis.
Neonatal considerations
e principal neonatal risk is intracranial hemorrhage
due to severe thrombocytopenia and birth trauma.
is is rare (⬍1% of ITP cases), although potentially
devastating when it occurs. e overall incidence of
thrombocytopenia in neonates born to mothers with
ITP is reported in various studies as 14%–37.5%.8,11,12
However, only approximately 5% of babies born to
mothers with ITP will have platelet counts ⬍20 ×
109/L, with a further 5% having counts between 20 ×
109–50 ×109/L.8,13
Unfortunately, predicting which babies may be
aected or directly assessing the fetal platelet count is
dicult. No correlation has been established with the
severity of maternal ITP or levels of circulating anti-
body. Although there are no reliable predictors of its
occurrence or severity, neonatal thrombocytopenia is
more likely if:
rthere is a previous sibling with
thrombocytopenia.13
rthe mother has had a splenectomy prior to this
pregnancy (although not all studies conrmed
this nding).
rsevere maternal ITP.13,14
Wherebabieshavebeenbornpreviouslywithsevere
thrombocytopenia, testing for paternal platelet anti-
gen incompatibility to exclude Neonatal alloimmune
thrombocytopenia (NAIT) is required.
ere is currently little role for the routine measur-
ing of fetal platelet counts by percutaneous umbilical
blood sampling (PUBS) in ITP. Studies evaluating this
technique have estimated the procedure-related risk to
be greater than the risk of preventing neonatal hem-
orrhage. Platelet counts taken from fetal scalp sam-
ples are prone to erroneously low results, and carry
the risk of scalp hematoma, and are therefore best
avoided.
Mode of delivery
Concerns regarding potential neonatal thrombocy-
topenia and birth trauma have previously led some
clinicians to recommend cesarean section. ere is
currently no evidence that cesarean section reduces
the incidence of intracranial hemorrhage in suscep-
tiblebabiescomparedwithanuncomplicatedvagi-
nal delivery. is is true for congenital bleeding disor-
ders as well as ITP. For this reason it is recommended
that the mode of delivery is determined by obstetric
indications rather than ITP. However, vaginal deliv-
ery that is augmented by ventouse or rotational for-
ceps does carry an increased risk of head trauma to
the neonate and where possible should be avoided.
Induction of labor at the time of maximal platelet
count may be required if platelet count rises are very
transient with therapy. e exact mode and timing of
delivery has many patient-specic variables and there-
fore an individualized plan with multidisciplinary
input is advised.
Management of labor when platelet count
has not been corrected
In these cases a pragmatic approach needs to be taken.
Experiences suggest that normal delivery can occur
without excess hemorrhage, reassuringly, even at very
low platelet counts. It is advisable to have platelet trans-
fusions available on standby and to proceed with deliv-
ery. If time allows, high dose IVIG (1 g/kg) may be
used. Epidural anesthesia should be avoided as should
non-steroidal anti-inammatory (NSAIDs) drugs for
postpartum pain relief.
Postpartum – neonatal care
e neonatal team should be alerted prior to deliv-
ery. A cord platelet count should be measured at
birth. If the platelet count is normal, further neona-
tal platelet counts are not required. If thrombocytope-
niaispresent,thisshouldbeconrmedonacapillary
or venous sample. Intramuscular injections are best
avoided, if severe thrombocytopenia is present, and
vitaminKgivenorally.
Further alternate-daily FBC measurements over
the next week are required to ensure that the neonate
is not at risk of hemorrhage. e nadir platelet count
is usually between days 2 and 5.
Babies with severe thrombocytopenia of ⬍20 ×
109/L or clinical hemorrhage require treatment
with IVIG. Life-threatening complications should
be treated with immediate platelet transfusions and
IVIG. Consideration should be given to using HPA 53

Section 1. Cellular changes
1a 5b negative platelets if available until NAIT is
excluded.
Babies with severe thrombocytopenia should have
a cranial ultrasound to assess for evidence of intracra-
nial hemorrhage.
Prenatal counseling
Women who have an established diagnosis of ITP may
request pre-natal counseling before deciding whether
to embark on a pregnancy. ere are few predictors of
outcome that can be used to assess risk. While preg-
nancy should not be discouraged, it is suggested that
the following points should be discussed:
rCirculating antiplatelet antibodies may still be
present in the maternal blood. is is particularly
relevant for women who have had a splenectomy.
In these circumstances the ITP may appear in
remission with normal platelet counts. However,
this is primarily due to an inability to clear
platelet–antibody complexes rather than a
cessation of antibody production. ese women
willstillbeatriskofneonatalthrombocytopenia
or hemorrhagic complications in utero.
rITPmayrelapseorworsenduringpregnancy.
rIf treatment of ITP is required it will carry both
maternal and fetal risks.
rere is an increased risk of hemorrhage at
delivery, but the risk is small even if the platelet
count is low.
rEpidural anesthesia may not be possible.
rAlthough it is not possible to accurately predict if
a neonate will be aected, the risk is high if a
sibling had thrombocytopenia, or mother had
undergone splenectomy.
rMaternal death or serious adverse outcomes for
mothers with ITP are rare.
re risk of intracranial hemorrhage for the
fetus/neonate is very low.
Autoimmune neutropenia (AIN)
Introduction
Neutropenia is a common nding in routine FBC test-
ing, and is dened as an absolute neutrophil count
(ANC) of ⬍1.5 ×109/L (or ⬍1.2 for some ethnic
groups – see below). e majority of cases are mild,
transient, and no specic etiology is determined. By
contrast, AIN is a rare disorder and can cause severe
neutropenia associated with recurrent infection.15 It
may occur in isolation or in conjunction with ITP
or AIHA. Many cases in adults are secondary, asso-
ciated with collagen vascular disorders, rheumatoid
conditions, and SLE. Primary AIN is predominantly
a disease of childhood. e main complication of this
condition is recurrent infection, which occurs if the
neutropenia is severe (ANC ⬍0.5 ×109/L). Diagno-
sis can be problematic as laboratory investigation of
neutropenia is limited, and usually restricted to spe-
cialist centers. Pregnancy poses an additional prob-
lem, as autoantibodies may cross the placenta result-
ing in neonatal neutropenia aer delivery. Currently,
published evidence on management of these cases is
lacking.
Incidence and pathogeneisis
e true incidence of AIN is not known. Persistent
neutropenia in adults is a common nding and is
frequently not investigated if asymptomatic and mild
(ANC 1.0 ×109–2.0 ×109/L). Cases are oen labeled
as chronic idiopathic neutropenia (CIN). It is prob-
able that some cases with a presumptive diagnosis
of CIN are immune mediated. e benign nature of
asymptomatic CIN means that specialist investiga-
tion is oen of little value and immunological stud-
ies are therefore not pursued. is may not be the
case for women of childbearing age, as identication
of immune-mediated cases may help with neonatal
assessment.
e pathogenesis of AIN is similar to that of other
immune cytopenias. It is an acquired disorder in which
autoantibodies specic for neutrophil surface glyco-
proteins result in reduced neutrophil survival and
neutropenia.
Diagnosis
Patients with symptomatic neutropenia (recurrent
infections, severe neutropenia) are likely to present
outside of pregnancy and have an established diag-
nosis. Diculty occurs in the asymptomatic patient
if an incidental nding of neutropenia is made fol-
lowing FBC testing during routine ante-natal care.
Assessment involves a careful history, examination of
the other FBC indices and inspection of the blood
lm.
e dierential diagnosis includes: drugs, viral
infections, immune mediated disorders, large granu-
lar lymphocyte (LGL) disease (oen associated with
54

Chapter 4. Maternal autoimmune cytopenias
Table 4.5 Severity of neutropenia according to the ANC
ANC Severity Clinical effect
⬎1.0 ×109/L Mild Usually asymptomatic
0.5 ×109–1.0 ×109/L Moderate Usually asymptomatic
0.2 ×109–0.5 ×109/L Severe Infections possible
⬍0.2 ×109/L Very Severe High risk of infection
rheumatoid arthritis), benign ethnic neutropenia, and
CIN. Important clinical and laboratory aids to diagno-
sis are listed below.
History
rHistory of SLE, rheumatoid arthritis or other
autoimmune disease suggests secondary immune
neutropenia. (more common than primary AIN
in adults.)
rEthnic origin (ANC ⬎1.2 ×109/L may be
considered within normal limits for some African,
Middle Eastern and Yemenite Jew populations).
rAsk about any recent viral illness.
rAssess risk factors for HIV.
rAssess for evidence of recurrent infections,
particularly unusual infections or mouth ulcers (if
there is a temporal pattern – consider cyclical
neutropenia).
rTake a careful drug history (especially antithyroid
drugs, phenothiazines, and NSAIDs), which are
known to cause neutropenia.
rIs there a known family history of neutropenia?
rIs there a history of ITP or AIHA?
Laboratory assessment
rA blood lm should be examined to conrm
neutropenia. Severity may be graded using the
criteria in Table 4.5.
rIncrease in LGLs on the blood lm should be
noted.
re presence of abnormalities other than
neutropenia suggests an alternative diagnosis to
AIN.
rAsymptomatic cases with ANC ⬎0.5 ×109/L and
where there is no apparent cause are best managed
by repeating the test aer 4 weeks. Further
investigation during pregnancy is warranted if the
neutropenia persists, or if the patient is
symptomatic.
rAnti-neutrophil antibody results also produce
frequent false negatives and positives, similar to
anti-platelet antibodies, making the test of little
use. Repeat samples may help diagnosis in some
cases.
rA bone marrow examination is of value in cases of
severe neutropenia. e bone marrow
appearances in AIN may show normal
hematopoiesis or an apparent arrest at the
metamyelocyte stage with a reduction in the
number of mature neutrophils and band forms.
Management
ere are two main risks during pregnancy – the
maternal risk of sepsis and the risk of neonatal neu-
tropenia. Sepsis in pregnancy may provoke miscar-
riage or premature labor and is the main concern,
for example, a normally benign urinary infection may
progress to pyelonephritis and septicemia.
Information on neonatal outcomes in women
with AIN is limited. Neutropenia from all causes
in neonates is common. Information from neonates
aected by neonatal alloimmune neutropenia (NAIN)
suggests that infections are, in the main, mild and
death or serious morbidity from sepsis is very rare.
As with ITP, steroids are the usual rst line of treat-
ment, if required. IVIG may be given if no response.
Sepsis
Sepsis in individuals with severe neutropenia is an
emergency. Untreated sepsis in the setting carries a
signicant mortality for both mother and baby. Blood
cultures should be taken and broad-spectrum intra-
venous antibiotics commenced promptly according to
local protocols. Fetuses tolerate pyrexia poorly, and
neurological damage may occur if the baby suers pro-
longed fever.
Granulocyte colony stimulating factor (GCSF)
GCSF has signicantly changed the management of
severe chronic neutropenia. For many individuals the
administration of low doses of GCSF 2–3 times per
week substantially reduces the incidence of infection.
GCSF has replaced traditional therapies such as IVIG,
corticosteroids or splenectomy as rst line therapy
outside of pregnancy. Long-term follow-up to date
has suggested that this is a safe treatment and there-
fore patients with symptomatic neutropenia are oen
on regular therapy.15 ItisnotyetclearthatGCSF
is safe for use in pregnancy. Studies investigating
prematurity have noted a potential association with 55

Section 1. Cellular changes
spontaneous preterm birth and elevated cytokines
including endogenous GCSF. In addition, GCSF car-
ries a small risk of venous thrombo-embolism, which
may constitute a signicant risk factor for some preg-
nancies. It is known that GCSF crosses the placenta.
Despite these reservations, it is likely that GCSF is rel-
atively safe in pregnancy. Several cases of successful
pregnancywithcontinuationofGCSFinpregnancy
are documented in the published international severe
chronic neutropenia registry and this is supported by
individual case reports.
Postpartum
Women with proven AIN or where AIN is strongly
suspected are at risk of delivering a neutropenic baby.
e neutrophil count at birth should be measured and
subsequent measurements performed according to the
degree of neutropenia and the infection risk. Immune
neutropenia may take several weeks to resolve.
Practical approach to pregnant patients
with diagnosed AIN
rIndividuals who are asymptomatic are unlikely to
benet from specic therapy.
rIf the ANC is ⬍0.5 ×109/L, advice on treating
sepsis promptly with intravenous antibiotics is
required.
rIndividuals who are symptomatic and already on
GCSF may benet from continuing therapy but a
careful discussion of the risks of therapy is
necessary. Consideration can be given to stopping
GCSF, particularly for the rst trimester.
rMonitoring FBC to tailor GCSF dose may be
required.
re neonatal team should be alerted prior to
delivery.
rA cord blood sample should be taken.
rA postpartum FBC should be sent.
Management of a newly presenting case of neutropenia
in pregnancy
rExclude other causes of neutropenia.
rCheck hematinics – (ferritin, B12 and folate – see
Chapter 2).
rAssess for evidence of associated auto-immune
conditions.
rIf severe neutropenia, warn patient of risk of
life-threatening infection – ensure they
understand that prompt treatment is necessary,
and have clear, ecient self-referral route.
rTreatment options should be discussed: steroids
are rst-line choice in pregnancy, with IVIG and
GCSF as second- and third-line options if no
response.
Autoimmune hemolytic anemia (AIHA)
Introduction
Hemolysisisdenedasshortenedredcellsurvival,
the average lifespan of an erythrocyte being 120 days.
Mild hemolysis is compensated for by an increase in
bone marrow erythropoeisis and may not aect the
hemoglobin concentration. Anemia occurs when red
cell survival is suciently shortened to exceed this
increase in erythropoetic activity. Causes of hemoly-
sis are listed in Table 4.6. AIHA is a common cause
of hemolysis but rarely complicates pregnancy. Non-
immune hemolysis occurs more frequently in preg-
nancy and is mostly associated with pre-eclampsia or
other hypertension-related disorders. It is essential to
distinguish between these types of hemolysis as the
management is very dierent. AIHA may be further
divided into “warm” and “cold” types. Warm AIHA is
usuallyIgGmediated.ColdAIHAismostlyIgMand
complement mediated. e blood lm appearances
and direct antiglobulin test (DAT) are characteristic.
TreatmentofAIHAinpregnancyissimilartooutside
pregnancy. Transplacental passage of IgG antibodies
may occur, but neonatal hemolysis is rarely severe.
Epidemiology and pathogenesis
AIHA in pregnancy is a rare disorder with an esti-
mated incidence of 1:50 000 pregnancies.16 Pregnancy
appears to be a stimulus for AIHA with a 4 ×higher
incidence than outside pregnancy. Cases of AIHA may
predate conception and relapse in pregnancy or occur
as a new presentation. Secondary causes include lym-
phoproliferative disorders, infections (mycoplasma,
Epstein–Barr virus) and connective tissue disorders.
AIHA is caused by the production of autoantibodies
directed against a red cell surface antigen, which on
binding results in premature destruction of the ery-
throcyte. is is usually extravascular in the spleen or
liver but occasionally may be intravascular. e anti-
bodies are most frequently IgG followed by the IgM
subtype. A spectrum of severity exists. In mild cases
a positive direct antiglobulin test (DAT) is the only
56

Chapter 4. Maternal autoimmune cytopenias
Table 4.6 Causes of hemolysis
Immune Autoimmune warm type IgG mediated
•Autoimmune cold type
•Autoimmune mixed type
•Alloimmune
IgM mediated
IgG and IgM mediated
Reaction to blood transfusion,
Hereditary •Disorder of hemoglobin synthesis
•Disorder of red cell enzymes
•Disorder of red cell membrane
e.g. sickle cell anemia
e.g. G6PD deficiency
e.g. hereditary spherocytosis
Mechanical •Red cell fragmentation •Mechanical heart valve
•MAHA (TTP, HUS, HELLP syndrome, pre-eclampsia)
Paroxysmal nocturnal
Hemoglobinuria
•Clonal stem cell disorder Increased susceptibility to complement lysis
Drugs •Oxidative stress, immune e.g. Dapsone
Infections •Bacterial enzymes e.g.
Clostridium perfringens
abnormality found. More severe cases have evidence of
compensated hemolysis with the most severe resulting
in signicant anemia.
Diagnosis
Anemia during pregnancy is a common nding. For
patients presenting during pregnancy, the diagnosis of
AIHA requires careful exclusion of other causes of ane-
mia, biochemical evidence of hemolysis and serologi-
cal evidence that the hemolysis is immune mediated.
Important clinical and laboratory features for diagno-
sis are summarized below.
History
rIs the patient symptomatic of anemia?
rIs there a history of cardiovascular or pulmonary
problems which may impair ability to cope with
anemia?
rIsthereevidenceofasecondarycause,e.g.recent
chest infection (mycoplasma) or autoimmune
disorders?
rIs the patient on any drugs known to cause
hemolysis (especially penicillins, methyldopa,
NSAIDs)?
rIdentify other potential causes of anemia
(hematinic deciency, hereditary disorders, etc).
Examination
rClinical examination may demonstrate evidence
of a secondary disorder.
rCasesofchronichemolysis(e.g.hereditary
spherocytosis) can have mild splenomegaly
present.
Laboratory
Hemolysis is characterized by:
r↑bilirubin, ↑LDH, ↑reticulocytes, ↓haptoglobins;
rblood lm – polychromasia, spherocytes, red cell
agglutination (Cold AIHA);
rimmune-mediated hemolysis – characterized by
positive DAT (Coombs test);
rIntravascular hemolysis – characterized by
urinary hemosiderin, hemoglobinuria.
Management
e principal risk is of a sudden fall in hemoglobin
resulting in symptomatic anemia and spontaneous
abortion. Successful management requires maintain-
inganadequatehemoglobinlevelwithredcelltransfu-
sion and giving specic therapy (usually prednisolone)
to arrest the hemolysis. Although transplacental pas-
sage of antibodies occurs, the risk of developing ane-
mia in utero or signicant neonatal anemia with asso-
ciated hyperbilirubinemia is small. Published experi-
ence of AIHA in pregnancy is limited, but the majority
of reports are favorable using this approach.
Blood transfusion
e presence of autoantibodies can cause diculty in
identifying suitable units for transfusion. Autoanti-
bodies may mask alloantibodies present in the
maternal serum, with the possibility of causing a
hemolytic transfusion reaction. Specialist investiga-
tion is required to exclude an alloantibody or identify
the specicity of an alloantibody if present. is may 57

Section 1. Cellular changes
delay the provision of suitable units. Many hospital
transfusion laboratories refer this work to specialist
transfusion centers, and the following points should
be considered.
rEnsure close liaison with the transfusion
laboratory to ensure that adequate samples have
been provided for testing.
rEnsure that the time within which blood is
required is clearly agreed with the transfusion
laboratory.
rIn cases requiring emergency transfusion, the
risks of issuing blood without compatibility being
fully determined should be discussed between the
hematologist and obstetrician.
rPatients with cold hemaglutinin disease (CHAD)
may benet from receiving transfusions via a
blood warmer.
Treatment of hemolysis
Corticosteroids may be eective in reducing hemoly-
sis. e risks of corticosteroid use are listed in Table
4.4a on ITP. Patients with warm AIHA are more likely
to respond than those with cold AIHA. A similar treat-
mentpatterntothatforITPmaybeused,andaswith
ITP the minimum dose possible to control hemolysis
should be used.
Experience with other agents in pregnancy is lim-
ited. IVIG can be eective and its use may be justi-
ed in pregnancy if corticosteroids are ineective or
contraindicated. Rituximab is increasingly used out-
side of pregnancy but there are insucient data cur-
rently available in pregnancy to advise its use.
Additional measures
rFolic acid 5 mg daily should be given. is
prevents folate deciency occurring as a result of
increased erythropoiesis. Increased dosage may
occasionally be necessary.
rromboprophylaxis should be considered.
Hemolysis is a prothrombotic condition and there
is an increased risk of venous thromboembolism
(VTE). Individual assessment of the degree of risk
is necessary, and should include assessment of
other risk factors for VTE. General measures
should be emphasized, such as ensuring adequate
hydration, and re-evaluation of degree of risk
should continue through the pregnancy. e
puerperium is a peak time for thrombotic events,
and pharmacological thromboprophylaxis
during the rst 6 weeks postpartum is
recommended.
Considerations for fetus and at birth
ere is the potential for in utero hemolysis if transpla-
cental passage of antibodies occurs. is applies only
to cases of IgG mediated hemolysis. Unlike hemolytic
disease of the newborn, the role of monitoring mater-
nal antibody titers has not been established. Non-
invasive monitoring for anemia using ultrasonography
may be of value.
e neonatal team should be alerted prior to deliv-
ery and neonates should have a hemoglobin and biliru-
bin measured at birth. Neonates born to mothers
with AIHA frequently have a positive DAT; however,
hemolysis is usually mild if present. Signicant ane-
mia or elevated bilirubin levels requiring treatment
is unusual. is is in contrast to hemolytic disease of
the newborn (HDN), which may result in very severe
hemolysis requiring in utero transfusion or neonatal
exchange transfusion.
58

Chapter 4. Maternal autoimmune cytopenias
References
1. Segal JB, Powe NR. Prevalence of immune
thrombocytopenia: analyses of administrative data.
JournalofrombosisandHaemostasis2006; 4:
2377–2383.
2. Sainio S, Kekomaki R, Riikonen S, Teramo K. Maternal
thrombocytopenia at term: a population-based study.
Acta Obstetrica Gynecologica Scandinavica 2000;
79:744–749.
3. Gill KK, Kelton JG. Management of idiopathic
thrombocytopenic purpura in pregnancy. Seminars in
Hematology 2000; 37: 275–289.
4. Boehlen F, Hohlfeld P, Extermann P et al. Platelet
count at term pregnancy: a reappraisal of the
threshold. Obstetrics and Gynecology 2000; 95: 29–33.
5. Verdy E, Bessous V, Dreyfus M et al. Longitudinal
analysis of platelet count and volume in normal
pregnancy. rombosis and Haemostasis 1997; 77:
806–807.
6. Win N, Rowley M, Pollard C et al. Severe gestational
(incidental) thrombocytopenia: to treat or not to treat.
Haematology 2005; 10: 69–72.
7. British Committee for Standards in Haematology
General Haematology task force. Guidelines for
investigation and management of idiopathic
thrombocytopenic purpura in adults, children and in
pregnancy. British Journal of Haematology 2003; 120:
574–596.
8. Webert KE, Mittal R, Sigouin C et al.Aretrospective
11-year analysis of obstetric patients with idiopathic
thrombocytopenic purpura. Blood 2003; 102:
4306–4311.
9. Michel M, Novoa MV Bussel JB. Intravenous anti-D as
a treatment for immune thrombocytopenic purpura
(ITP) during pregnancy. British Journal of
Haematology 2003; 123: 142–146.
10. Provan D, Stasi R, Newland AC. International
consensus report on the investigation and
management of primary immune thrombocytopenia.
Blood 2010, 115: 168–186.
11. Veneri D, Franchini M, Raaelli R et al. Idiopathic
thrombocytopenic purpura in pregnancy: analysis of
43 consecutive cases followed at a single Italian
institution. Annals of Hematology 2006; 85: 552–554.
12. Yamada H, Kato E, Kobashi G et al.Passiveimmune
thrombocytopenia in neonates of mothers with
idiopathic thrombocytopenic purpura: incidence and
risk factors. Seminars in rombosis Hemostasis. 1999;
25: 491–496.
13. Christiaens GC, Niewenhuis HK, Bussel JB.
Comparison of platelet counts in rst and second
newborns of mothers with immune thrombocytopenic
purpura. Obstetrics and Gynecology 1997; 90: 546–552.
14. Burrows, R, Kelton J. Pregnancy in patients with
idiopathic thrombocytopenic purpura: assessing the
risksfortheinfantatdelivery.Obstetrical and
Gynecological Survey 1993; 48: 781–788.
15. Dale DC, Cottle TE, Fier CJ et al. Severe chronic
neutropenia: treatment and follow-up of patients in
the severe chronic neutropenia international registry.
American Journal of Hematology 2003; 72: 82–93.
16. Sokol RJ, Hewitt S, Stamps BK. Erythrocyte
autoantibodies, autoimmune haemolysis and
pregnancy. Vox S anguinis 1982; 43: 169–176.
59

Section
2
Feto-maternal alloimmune
syndromes

Section 2 Feto-maternal alloimmune syndromes
Chapter
5Fetal/neonatal alloimmune
thrombocytopenia
Michael F. Murphy
Introduction
Fetal and neonatal alloimmune thrombocytope-
nia (FNAIT) is the commonest cause of severe
neonatal thrombocytopenia, and is analogous to
the fetal/neonatal anemia caused by hemolytic dis-
ease of the fetus and newborn (HDFN).1,2 Fetal
platelet antigens are expressed on platelets in normal
amounts from as early as the 16th week of preg-
nancy. Feto-maternal incompatibility for human
platelet alloantigens (HPAs) may cause maternal
alloimmunization, and fetal and neonatal thrombo-
cytopenia may result from placental transfer of IgG
antibodies. Many HPA systems have been described3.
e majority of HPA antigens such as HPA-1a are
locatedonthe3subunitofthe␣IIb3integrin
(GPIIb/IIIa,CD41/CD61) which is present at high
density on the platelet membrane. Others such as
HPA-5b are on ␣21 (GPIa/IIa, CD49b). However,
the antigen incompatibility HPA-1a is found in
about 80% of cases of FNAIT in Caucasians and, in
contrast to HDFN, FNAIT frequently occurs in rst
pregnancies.
Considerable progress has been made in the la-
boratory investigation of FNAIT since it was rst rec-
ognized in the 1950s.1ere have also been improve-
ments in its management, particularly in the ante-natal
management of women with a history of one or more
pregnancies aected by FNAIT, resulting from a bet-
ter understanding of the risk of severe hemorrhage and
advances in fetal and transfusion medicine.
Epidemiology
e normal platelet count in the fetus and the neonate
isthesameasinadults.Neonatalthrombocytopenia
has many causes, and is the commonest hematologi-
cal problem in the newborn infant. A platelet count
of ⬍150 ×109/L occurs in about 1% of unselected
neonates, and is ⬍50 ×109/L in about 0.2%. FNAIT is
the most important cause of severe fetal and neona-
tal thrombocytopenia, both because of its frequency
and the severity of the bleeding associated with it.
For example, FNAIT is associated with more severe
fetal/neonatal bleeding than with maternal autoim-
mune thrombocytopenic purpura for reasons which
are not entirely clear but could be due to associated
platelet and/or endothelial dysfunction.
A fetal or neonatal platelet count of ⬍20 ×109/L
is usually caused by FNAIT due to anti-HPA-1a as are
approximately half of the cases in which the neonatal
platelet count is ⬍50 ×109/L.
e most common entities in the dierential diag-
nosis of severe fetal and neonatal thrombocytopenia
are:
rcongenital infections such as toxoplasmosis,
rubella, and cytomegalovirus;
rmaternal autoimmune thrombocytopenic
purpura;
rchromosomal abnormalities;
rcongenital heart disease;
rdisseminated intravascular coagulation (DIC).
Incidence
Prospective studies in Caucasian populations for
FNAIT due to anti-HPA-1a indicate that about 2% of
women are HPA-1a negative, and that about 10% of
HPA-1a negative women develop anti-HPA-1a.4
Alloimmunization to HPA-1a is HLA class
II restricted. ere is a strong association with
HLADRB3∗0101 (HLADRw52a), which is present in
1 in 3 of Caucasian women, and HPA-1a alloimmu-
nization is rare in HPA-1a negative women who lack
this antigen.
63
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 2. Feto-maternal alloimmune syndromes
Using data from prospective studies, the overall
incidence of FNAIT due to anti-HPA-1a is estimated to
be 1 in 1163 live births (86 per 100 000), and the inci-
denceofseverethrombocytopenia(plateletcount⬍50
×109/L) to be 1 in 1695 (or 59 per 100 000).4
FNAIT is under-diagnosed in routine clinical prac-
tice. e evidence for this is the mismatch in the inci-
dence of FNAIT between prospective studies involving
laboratory screening for HPA antibodies and the iden-
tication of clinically diagnosed cases. It is estimated
that only 7%–23% of cases of FNAIT, and only 37% of
severe cases, are detected clinically.
Clinical diagnosis
FNAIT is usually suspected in neonates with bleed-
ing or severe, unexplained, and/or isolated post-
natal thrombocytopenia. e clinical diagnosis is one
of exclusion.
re infant has no signs of DIC, infection or
congenital anomalies known to be associated with
thrombocytopenia.
re mother has had a normal pregnancy with no
history of autoimmune disease,
thrombocytopenia, or drugs that may cause
thrombocytopenia.
Specic criteria which distinguish cases of FNAIT
from other causes of unexplained thrombocytopenia
include:
rsevere thrombocytopenia (platelet count ⬍50 ×
109/L);
rno additional, non-hemorrhagic neonatal medical
problems;
rintracranial hemorrhage (ICH) associated with
one or more of:
Apgar score at 1 minute ⬎5;
birthweight ⬎2.2 kg;
documented ante-natal or post-natal bleeding.
Laboratory diagnosis
Detailed laboratory investigations are required for
conrmation of a provisional clinical diagnosis, and
should be performed by an experienced reference lab-
oratory. e diagnosis is based on:
rdetection and identication of the maternal HPA
antibody;
rdetermination of the HPA genotype of mother,
father and, if needed, the child (or fetus).
In the past, it was dicult to dierentiate between
HLA and HPA antibodies in standard serological
assays. e description of the monoclonal antibody-
specic immobilization of platelet antigens (MAIPA)
assay overcame this problem. Rather than working
with intact platelets, the assay involves capture of spe-
cic GPs using monoclonal antibodies enabling ana-
lysis of complex mixtures of platelet antibodies. How-
ever, it requires considerable operator expertise in
order to ensure maximum sensivity and specicity, and
the selection of appropriate screening cells is critical.
Immunization against HPA-1a and HPA-5b are
responsible for up to 95% of cases of FNAIT. Antibod-
ies against other HPAs are more frequently detected
in recent large series of FNAIT. In some of these
cases, testing against standard donor platelet pan-
els may be negative. To pursue further investigation
requires strong clinical suspicion of FNAIT. Possible
approaches include:
rcross-match of maternal serum and paternal
platelets using MAIPA;
ridentication of a mismatch between maternal
and paternal (or neonatal) genotypes for low
frequency HPA antigens, and then screen
maternal serum for the corresponding HPA
antibodies.
Clinical signicance of FNAIT
ICHisthemajorcauseofmortalityandlong-term
morbidity in FNAIT. e long-term outcome may be
devastating with blindness and major physical and
mental disability (Fig. 5.1). ICH was reported in a large
review of the literature to occur in 74/281 (26%) of
cases of FNAIT due to anti-HPA-1a with a mortality
of 7%.5
Although there is a risk of hemorrhage due to
severe thrombocytopenia at the time of delivery,
80% of ICH associated with FNAIT occur in utero,
with 14% occurring before 20 weeks and a fur-
ther 28% occurring before 30 weeks.5ere may
also be unusual presentations such as isolated fetal
hydrocephalus, unexplained fetal anemia, or recurrent
miscarriages.
Bleeding is more severe with FNAIT due to anti-
HPA-1a than for example anti-HPA-5b, possibly due to
the higher density of HPA-1a antigen sites on platelets.
64

Chapter 5. Fetal/neonatal alloimmune thrombocytopenia
Fig. 5.1 Intracranial hemorrhage in FNAIT: MRI scan showing
subacute hematoma (black arrow) and chronic hematoma (open
arrow). Reproduced from De Vries
et al. Br J Obstet Gynaecol
;95:
299–302.
Prediction of the severity of FNAIT in
subsequent pregnancies
Laboratory testing
Unfortunately, there is no reliable laboratory method
to predict severe clinical disease, which might be used
to identify pregnancies at risk of severe thrombocy-
topenia and ICH. Some studies have observed an asso-
ciation between high levels of maternal anti-HPA-1a
and the severity of neonatal thrombocytopenia, but
this is not a suciently reliable association to be clini-
cally useful. Reliable methods for quantifying the other
antibodies are not yet available. e lack of laboratory
parameters predictive of severe disease remains one of
the major barriers to optimizing ante-natal manage-
ment for FNAIT, and is an important area for future
research.
History of FNAIT in previous pregnancies
Subsequent pregnancies of HPA-1a alloimunized
women with a history of a previously aected infant
with FNAIT are well recognized to be associated with
a high risk of recurrence of FNAIT and poor outcome.
A detailed literature search found that the recurrence
rate of ICH in the subsequent pregnancies of women
withahistoryofFNAITwithICHwas72%(con-
dence interval 46%–98%) without the inclusion of
fetal deaths, and 79% (condence interval 61%–97%)
with their inclusion.6e risk of ICH following a
previous history of FNAIT without ICH was estimated
to be 7% (condence interval 0.5%–13%).
ese data provide the justication for ante-natal
intervention in women with a past history of pregnan-
cies aected with FNAIT, particularly where there has
been fetal or neonatal ICH in a previous pregnancy, to
reduce the risk of morbidity and mortality from severe
hemorrhage.
If there is paternal heterozygosity for the rele-
vant HPA, fetal platelet genotyping should be con-
sidered, for example, by obtaining a sample using
amniocentesis.
Consideration of ante-natal
screening for FNAIT
Advances in the laboratory diagnosis and ante-
natal management of FNAIT have drawn attention to
the fact that the rst aected fetus/neonate is usu-
ally only recognized aer bleeding has occurred or
severe thrombocytopenia detected by chance. is
raises the question of whether routine screening for
FNAIT should be considered. It is recognized that
there are signicant shortcomings in the knowledge
about FNAIT necessary for the introduction of an
antenatal screening program.7
More research is required, for example, on the clin-
ical outcome of rst aected pregnancies, the identi-
cation of laboratory measures predictive of severe dis-
ease where ante-natal intervention might be justied,
and the optimal approach for the ante-natal manage-
ment of pregnant women with HPA antibodies, but
with no previous history of aected pregnancies, as
ante-natal treatment carries signicant risks and costs.
Management of FNAIT
Post-natal
e thrombocytopenia in FNAIT usually resolves
within 2 weeks, although it may last as long as 6 weeks.
A cerebral ultrasound should be carried out to deter-
mine if ICH has occurred because of the changes in
management that would occur if there had been a
hemorrhage. 65

Section 2. Feto-maternal alloimmune syndromes
e optimal post-natal management of FNAIT
depends on its rapid recognition, and prompt correc-
tion by transfusion of platelet concentrates to neonates
who are severely thrombocytopenic (platelet count
⬍30 ×109/L) or bleeding. It is not appropriate to wait
for the laboratory conrmation of the diagnosis in sus-
pected cases.
Whiletherehasbeendebateaboutthevalueof
random donor platelets in the immediate post-natal
management of FNAIT, two recent studies reported
that random donor (i.e. not HPA-matched) platelets
were oen eective in increasing the platelet count
in FNAIT. However, in some of the cases, sponta-
neous recovery of the neonatal platelet count may have
been the reason for the apparent response to random
donor platelet transfusions. Compatible platelet con-
centrates were shown in another study to produce a
larger increase in platelet count and twice the length
of survival of the transfused platelets compared to ran-
dom donor platelets.8
Compatible platelet concentrates, for example,
from HPA-1a and 5b negative donors, should be used
initially, if they are available, on the basis of the cer-
tainty of their eectiveness in the more than 90% of
cases of FNAIT which are due to anti-HPA-1a or anti-
HPA-5b. Unfortunately, the routine availability of such
HPA-1a and 5b-negative platelets for immediate use in
suspected cases of FNAIT is limited to only a minority
of countries, including England.
Although intravenous immunoglobulin (IVIG) is
eective in at least 75% of cases, the platelet count does
not increase in responders for 24–72 hours so it should
not be used for the initial therapy of FNAIT. Its role in
the management of post-natal FNAIT should be lim-
ited to those few cases with very prolonged and severe
thrombocytopenia.
Provision of information to the mother
e parents should be provided with information
about FNAIT once the platelet antigen typing and anti-
body results are complete, specically to provide:
1. an explanation of the cause of FNAIT;
2. the risk of recurrence in subsequent pregnancies;
3. the options for ante-natal management as well as
the fact that this is an evolving eld;
4. a request that the mother should notify the fetal
medicine center as soon as she becomes pregnant;
5. her risk for the future of transfusion reactions,
and potentially post-transfusion purpura (PTP),
although it appears that the risk of PTP is very low
with leukocyte-reduced blood components which
are now standard in the UK;
6. Testing of female relatives of the mother should be
suggested.
Ante-natal
e traditional management of subsequent pregnan-
cies in women with a previous history of FNAIT
consisted of performing early elective Cesarean sec-
tion, and then transfusing compatible platelets aer
birth. Major advances in the ante-natal management
of FNAIT have been made in the last 25 years.1,2,9
Early ante-natal treatment strategies
In 1984, the use of ultrasound-guided fetal blood sam-
pling (FBS) was described to obtain the fetal platelet
count at 32 weeks’ gestation in the second pregnancy of
a woman whose rst child had ICH due to FNAIT; the
fetal platelet count was 15 ×109/L. ere was no ultra-
sound evidence of ICH by 37 weeks, and an in utero
transfusion of maternal platelets was given 6 hours
priortodeliverybyCesareansection.Asaresult,the
cord platelet count was 95 ×109/L and there were no
signs of bleeding.
e use of in utero platelet transfusion (see Fig. 5.2)
immediately before delivery was described in greater
detail in a series of 9 cases, where FBS was carried
outat21weeks’gestationtoconrmthediagnosis
of FNAIT.1FBS was repeated at 37 weeks with an in
utero platelet transfusion if the fetal platelet count was
⬍50 ×109/L followed by delivery 6–36 hours later.
However, over the next 10 years, it became clearer that
an aected fetus is at risk of ICH in utero,evenbefore
20 weeks’ gestation, indicating that earlier ante-natal
intervention is required in cases likely to be severely
aected. During this period, dierent groups began
to explore alternative approaches to ante-natal man-
agement, one based around serial weekly fetal platelet
transfusion, and the other around medical treatment
ofthemotherwithIVIGand/orsteroids.
Serial fetal platelet transfusions
Early studies with fetal platelet transfusions high-
lighted the short survival of transfused platelets, and
the diculty of maintaining the fetal platelet count
at a “safe” level. Further experience indicated that it
was possible to maintain the count above 30 ×109/L
using transfusions at weekly intervals (Fig. 5.3). is
66
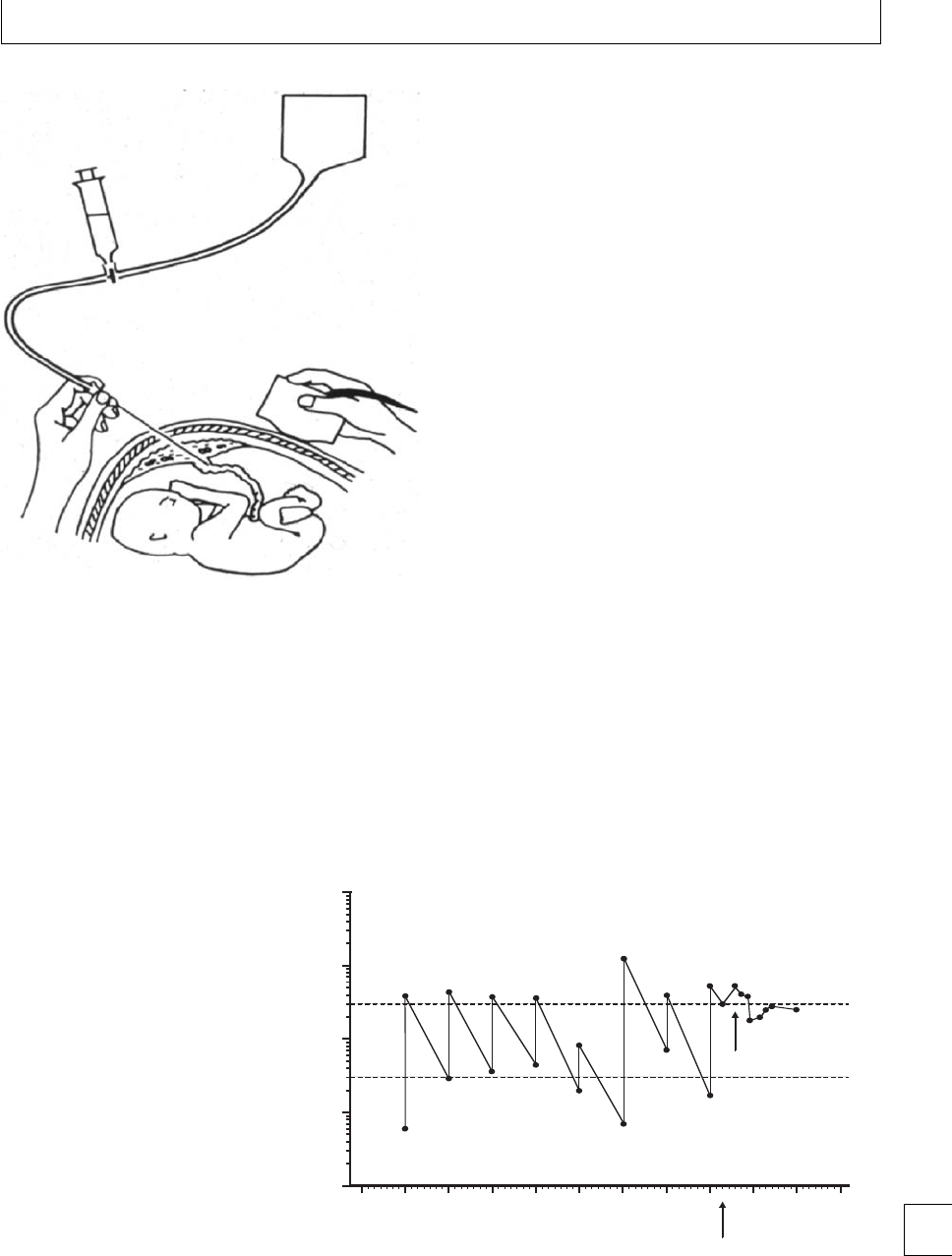
Chapter 5. Fetal/neonatal alloimmune thrombocytopenia
Donor
platelet
transfusion
3-way tap
Fig. 5.2 Schematic diagram of ultrasound-guided fetal blood
sampling and platelet transfusion.
was achieved by increasing the dose of platelets, whilst
avoiding an unacceptable increase in the transfused
volume, by concentrating the platelet collection by
centrifugation and removal of plasma. Later improve-
ments in apheresis technology allowed the prepara-
tion of leukocyte-depleted concentrated platelets suit-
able for fetal transfusion without the need for further
processing.
Technical aspects and complications of FBS
e technique employed for trans-abdominal
ultrasound-guided FBS and intravascular transfusion
is the same as for red cell alloimmunization. Unlike
HDFN, where the needle may be removed while
the hematocrit is estimated before transfusion is
commenced, removal of the needle from the umbilical
cord in the presence of a very low platelet count can
result in rapid exsanguination of the fetus. Very few
operators check the platelet count during the proce-
dure and it is standard practice to transfuse platelets
to the fetus following FBS even if the procedure is
undertaken for diagnosis or monitoring of FNAIT
rather than part of serial fetal transfusions.
e main risks of FBS are severe cord bleeding,
cardiac arrhythmias, and miscarriage. Pooling data
fromseveralstudiesindicatesafetallossrateof3/223
(1.3%)/procedure and 3/55 (5.5%)/pregnancy.
From 26 weeks’ gestation, FBS and platelet trans-
fusion should be performed in the operating the-
ater where facilities are available to perform an emer-
gency Cesarean section, should there be signs of fetal
distress or bleeding from the sampling site. Unpub-
lished data from the Oxford Rhesus erapy Unit
indicate that there is approximately a 4% chance of
rapid delivery being required at the time of each
transfusion.
e volume of platelet hyperconcentrate to be
transfused is calculated from a formula:
Volume of concentrate =desired platelet incre-
ment ×feto-placental blood volume for gestational
age ×R÷platelet count of the concentrate
1
10
100
1000
10000
25 26 27 28 29 30 31 32 33 5 12
Weeks’ gestation Days’ post-natal
CS
30
300
Platelet
transfusion
Platelets × 109/L
Fig. 5.3 Pre- and post-transfusion
platelet counts following serial FBS and
platelet transfusions. The fetal platelet
count was ⬍10 ×109/L at 26 weeks. The
aim was to maintain the fetal platelet
count above 30 ×109/L by raising the
immediate post-transfusion platelet count
to above 300 ×109/L after each
transfusion. The fetal platelet count fell
below 10 ×109/L on one occasion when
there were problems in preparing the fetal
platelet concentrate and the dose of
platelets was inadequate. CS =Cesarean
section. Reproduced from
Practical
Transfusion Medicine
, 3rd edn. Murphy MF
& Pamphilon D. Wiley-Blackwell
Publishing, 2009.
67
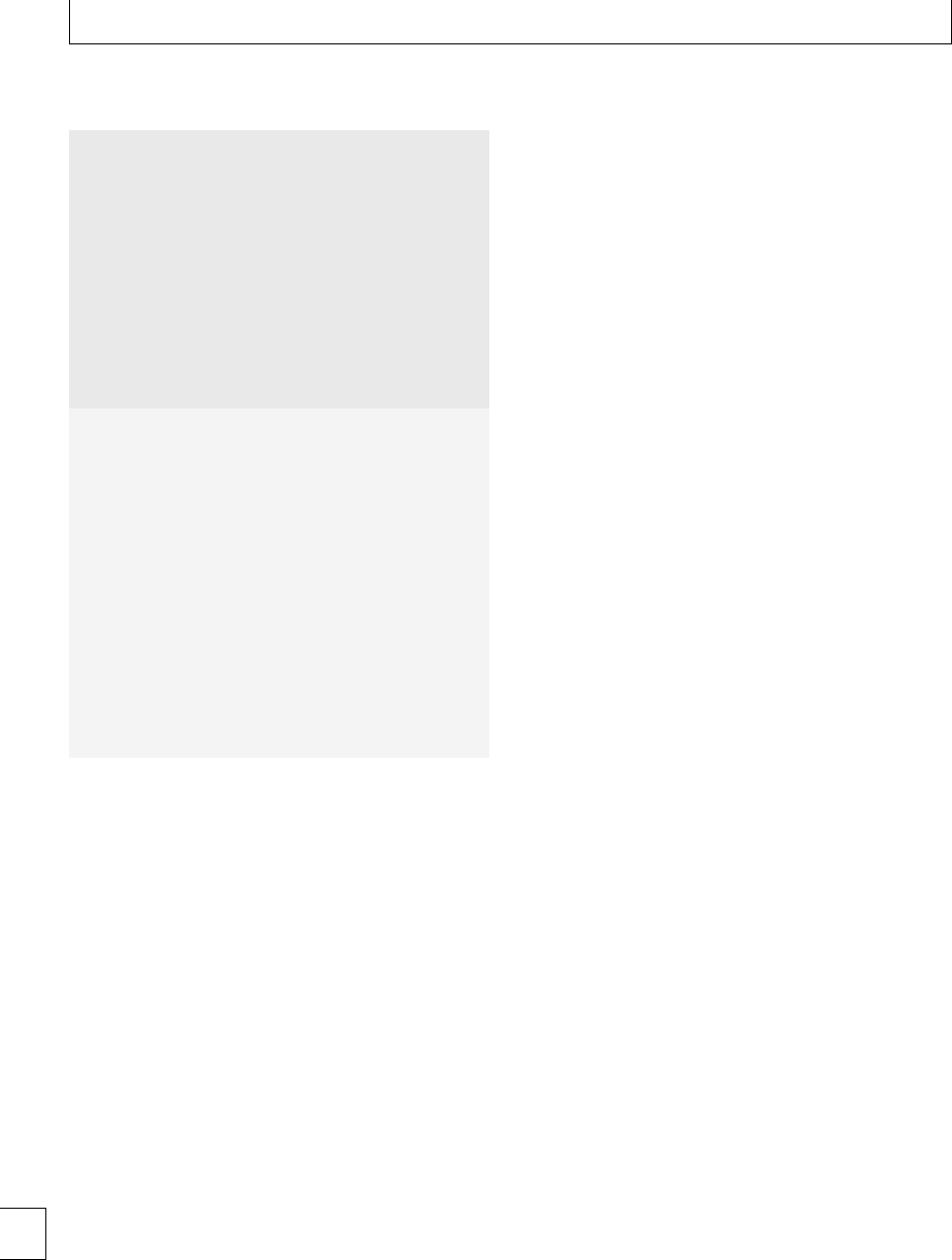
Section 2. Feto-maternal alloimmune syndromes
Table 5.1 Specification of the platelet product for
intra-uterine transfusion
Donor
rHPA type compatible with maternal
antibodies, usually HPA-1a negative
rGroup O RhD negative for the first transfusion
(for subsequent transfusions, the ABO and RhD
group of the donor should be compatible with
the fetal blood group which should be
determined from a sample taken at the first
FBS)
rNoHPAorHLAantibodies
rNo high titer ABO antibodies
Platelet concentrates
rHigh concentration of platelets (usually in the
range 2.5 ×109–3.0 ×1012/L compared to
1.4 ×1012/L for standard platelet concentrates
for use in neonates or adults) to reduce the
volume of the transfusion. The
hyperconcentrates
can be prepared using a
modification of the procedure for collection of
platelet concentrates by apheresis.
rGamma-irradiated to prevent
transfusion-associated graft-vs.-host disease
rCMV-seronegative
rLeukocyte-reduced
rTransfuse within 24 hours after collection
e feto-placental volume for gestational age is cal-
culatedfromstandardcharts.Inearlyfetalplatelet
transfusion studies, the immediate post-transfusion
platelet increment was found to be 50% of that
expected, i.e. 50% platelet recovery, probably because
of pooling in the feto-placental circulation. e vol-
ume calculation takes account of this by introducing
the factor R=2, thus doubling the volume of platelets
transfused.
e specication of the platelet product for intra-
uterine transfusion is provided in Table 5.1.
Maternal treatment
One of the main drivers for the development of mater-
nally directed ante-natal treatment for FNAIT was
concern about the risks of FBS and platelet transfusion.
Steroids
ere is considerable experience from North Amer-
ica with the combined use of steroids and IVIG.10
Although low dose steroids did not add signicantly
to the eect of IVIG, high dose steroids (prednisolone
60 mg and later 1 mg/kg) added substantially to the
eect of IVIG. e use of 0.5 mg/kg prednisolone in
the lowest risk cases (no previous sibling ICH, initial
fetal count ⬎20 ×109/L) demonstrated ecacy com-
parable to that of IVIG in this group of patients.
Intravenous immunogloblin (IVIG)
e rst protocol involving maternal administration
of IVIG was described in 1988. Initial FBS was car-
ried out at 20–22 weeks’ gestation to conrm the diag-
nosisofFNAITanditsseverity.IVIG(dose1g/kg
body weight/week) was administered to the mother,
and FBS was repeated 4–6 weeks later to assess the
eect of IVIG. None had ICH in contrast to three of
their respective untreated siblings, two of whom had
antenatal ICH, and there were no serious complica-
tions of treatment.Overall, there was an increase of
36 ×109/LbetweentherstandsecondFBS,andan
increase of 69 ×109/L between the rst FBS and birth.
Of fetuses 62%–85% responded to therapy depend-
ing on the denition of response used, and there were
no cases with ICH. However, other reports described
cases in which IVIG was ineective in raising the fetal
platelet count, and ante-natal ICH was reported during
maternal treatment with IVIG.
Complications of maternal teatment
e use of IVIG is expensive, and both IVIG and
prednisolone can cause adverse maternal eects. IVIG
appears to be a safe blood product when adminis-
tered to otherwise healthy young women. e risks
of renal disease, hemolysis, uid overload, and trans-
mission of infection are extremely low, and none of
these have been reported in a patient undergoing ante-
natal treatment for FNAIT. Headaches occur but usu-
ally lessen with time. Prednisolone has been widely
used in pregnancy, and is known to cause uid over-
load, high blood pressure, diabetes mellitus, irritabil-
ity, and osteoporosis.
Recent studies of maternal treatment
A collaborative study in European centers reported
in 2003 on the ante-natal management of FNAIT
in 56 fetuses managed with either maternal treat-
ment or platelet transfusions. Maternal therapy,
predominantly IVIG, resulted in a platelet count
exceeding 50 ×109/L in 67%. e most serious
complications encountered were associated with FBS
68

Chapter 5. Fetal/neonatal alloimmune thrombocytopenia
and platelet transfusion, and the results support the
use of maternal therapy as rst-line treatment for the
ante-natal management of FNAIT. e association of
lower pre-treatment platelet counts in cases with a
sibling history of ante-natal ICH or severe thrombocy-
topenia favors stratication of ante-natal management
on the basis of the history of FNAIT in previous
pregnancies.
In 2006, the North American team reported
two randomized controlled trials of maternal treat-
ment stratied according to the previous history of
FNAIT.11
(1) “High risk” patients had either a sibling with
peripartum ICH or one with an initial fetal
platelet count ⬍20 ×109/L. Patients underwent
FBS at 20 weeks or later, and were randomized to
receive IVIG alone (1 g/kg/week) or in
combination with prednisolone 1 mg/kg/day.
ere was a satisfactory increase in the fetal
platelet count in 89% of pregnancies receiving
combination treatment compared to 35%
receiving IVIG alone (P=⬍0.05). In those
with initial fetal platelet counts ⬍10 ×109/L,
82% had a satisfactory response to IVIG and
prednisolone compared to only 18% treated with
IVIG alone (P=⬍0.03). ere was one ICH; this
occurred in a pregnancy managed with IVIG
alone.
(2) “Standard” risk patients were those with a sibling
who had not had an ICH and a fetal platelet count
between 20 and 100 ×109/L. ese patients
underwent FBS near to 20 weeks, and were
randomized to receive IVIG (1 g/kg/week) or
prednisolone 0.5 mg/kg/day. Subsequent FBS was
carried out in all patients at 3–8 weekly intervals.
ere were no signicant dierences in the
responses to the two treatments. ere were two
ICHs; one in a fetus born at 38 weeks’ gestation
withaplateletcountof172×109/L, and one in an
infant with a birth platelet count of 68 ×109/L
deliveredat28weeksbecauseofbradycardia
following FBS.
ere were 11 serious complications out of a total
of 175 (6%) FBS conrming the dangers of FBS and
platelettransfusioninFNAIT.isstudydemonstrates
that eective ante-natal treatment can be stratied
according to the previous history of FNAIT.
The search for less invasive strategies for the ante-natal
management of FNAIT
Concern regarding the safety of FBS and platelet trans-
fusion has led to a search to develop less invasive
treatment strategies involving maternal administra-
tion of IVIG while reducing or even avoiding FBS for
monitoring the fetal platelet count and administering
platelet transfusions.
Some studies suggested that the pre-treatment
platelet count had predictive value for the response
to maternal treatment. A review of patients treated in
North America found that the response rate in fetuses
with a pre-treatment platelet count of ⬎20 ×109/L
was 89%, but was only 51% in those with an initial fetal
platelet count ⬍20 ×109/L. e authors suggested
thatadditionalFBSmightnotbewarrantedinthose
cases with an initial fetal platelet count ⬎20 ×109/L;
any gain from identifying and intensifying treatment
in “poor responders” would be oset by the complica-
tions of additional FBS.
e Leiden group have evaluated less intensive
ante-natal treatment strategies over a number of years
and found that that a non-invasive strategy based on
treatment with IVIG without FBS appears to be eect-
ive when there is no history of ICH in a previous preg-
nancy.12 e same group extended this approach to
the management of seven high risk pregnancies where
there had been a previous sibling history of ICH. IVIG
was administered from 16–19 weeks’ gestation in the
six pregnancies where there had been previous ante-
natal ICH, and from 28–29 weeks in the case where
ICH was post-natal. e total number of weekly IVIG
infusions ranged from 8 to 21. e platelet count at
birth ranged from 10 ×10949 ×109/L. No ICH was
seen on ante-natal or post-natal ultrasound examina-
tions, and all infants were doing well at follow-up at
3months.
ese recent studies indicating success with less
invasive strategies suggest that further work is ne-
cessary to determine the optimal ante-natal manage-
ment for FNAIT. An alternative to the “empirical” (no
FBS) and “invasive” (FBS before and during treat-
ment) approaches is to initiate maternal treatment
(type and timing determined by consideration of the
previoushistoryofFNAIT)withoutperformingFBS,
and then to carry out FBS 4–8 weeks aer the initia-
tion of treatment to identify the non-responding cases
which may benet from a change in treatment. is
is the approach being followed by some UK referral 69

Section 2. Feto-maternal alloimmune syndromes
Table 5.2 Suggested ante-natal management depending on
previous history of FNAIT ∗
(1) Ante-natal ICH in previous sibling:
ICH in second trimester
rAt 12 weeks, IVIG 2 g/kg/week (given as 1
g/kg/twice a week)
rFBS at week 20–22
ICH in third trimester
rAt 16 weeks, IVIG 1 g/kg of IVIG
rFBS at week 20–22
If fetal platelet count at rst FBS ⬎30 ×109/l:
rContinue current treatment
rFurther FBS at 28 weeks at 34–36 weeks and/or
pre-delivery
If fetal platelet count at rst FBS ⬍30 ×109/l:
rAdd prednisolone 1 mg/kg/day
rRepeat FBS 2 weeks later. If no response, where
relevant increase IVIG to 2 g/kg/week (given as
1 g/kg/twice a week) and repeat FBS at 2 weeks
rIf no response to maximal combination
therapy, proceed to weekly IUT and discontinue
medical treatment
rIf response to maximal combination therapy
repeat FBS at 2–4 weekly intervals
(2) Neonatal ICH or platelet count ≤50 ×109/l
in previous sibling:
rIVIG 1 g/kg/week at 20 weeks
rFBS at 28–32 weeks
If fetal platelet count ⬎30 ×109/l:
rContinue current treatment
rFurther FBS at 34–36 weeks
If fetal platelet count at rst FBS ⬍30 ×109/l:
rAdd prednisolone 1 mg/kg/day
rRepeat FBS 2 weeks later. If no response, where
relevant increase IVIG to 2 g/kg/week (given as
1 g/kg/twice a week) and repeat FBS at 2 weeks
rIf no response to maximal combination
therapy, proceed to weekly IUT and discontinue
medical treatment
rIf response to maximal combination therapy,
repeat FBS at 2–4 weekly intervals
Table 5.2 (
cont.
)
Mode of delivery:
Based on FBS at 30–32 weeks:
rIf fetal platelet count ≥100 ×109/L, proceed to
spontaneous vaginal delivery with no further
fetal blood sampling
rIf fetal platelet count ≤100 ×109/L, continue
with treatment and perform repeat sampling at
35–37 weeks, with transfusion of platelets
rIf fetal platelet count ≥50 at 35–37 weeks
(prior to platelet transfusion), allow
spontaneous vaginal delivery
rIf platelet count ⬍50 ×109/L at 35–37 weeks,
discuss options:
– Induction of labor within 5 days of IUT
– Weekly IUT until either spontaneous labor,
induction of labour or planned Cesarean
section
There is no evidence to suggest that elective Cesarean section
is safer than vaginal delivery, if the platelet count is above 50 ×
109/L.
∗developed by Rachel Rayment, Mike Murphy and Jim Bussel
(unpublished data).
centers including our own (Table 5.2). Recommenda-
tions about the mode of delivery are also provided in
Table 5.2.
How to manage the ‘non-responders’ to initial
maternal therapy
e options are to increase the dose of IVIG, add pred-
nisolone, switch to serial platelet transfusions and/or
consider early delivery. e North American group
have developed this concept of “salvage” or “intensi-
cation” therapy. Only about 25% of “high risk” or
“standard risk” patients required more intensive treat-
ment because of a lack of response to their initial
therapy. “Intensication” therapy comprised adding
IVIG or prednisolone if not being used already, or
increasing the dose of IVIG, and all but six had platelet
counts at birth ⬎50 ×109/L.
e ability to modify ante-natal treatment in an
individual case does depend on the use of FBS to mon-
itor the fetal platelet count. Although empirical treat-
ment without knowledge of the fetal platelet count
before or during treatment avoids the risks of FBS, it
has the drawbacks of the administration of potentially
unnecessary or inadequate treatment.
70

Chapter 5. Fetal/neonatal alloimmune thrombocytopenia
Optimal approach for the modern ante-natal
management of FNAIT
ere has been huge progress in the ante-natal man-
agement of FNAIT over the last 20 years. However, the
ideal eective treatment without signicant side eects
to the mother or fetus has yet to be determined.
ere are some basic principles to consider in the
management of an individual case.2
1. Obtain as much information as possible about the
clinical history of previously aected pregnancies
with FNAIT focusing on the neonatal
thrombocytopenia to exclude other causes of
thrombocytopenia. It is important to determine as
conclusively as possible if an ICH has occurred
and if so, when.
2. Ensure that comprehensive laboratory
investigations have been carried out in a reference
laboratory, including testing for HPA antibodies
and the identication of their specicity, and HPA
genotyping of the mother and her partner. If the
partner is heterozygous for the relevant HPA, the
fetal HPA genotype should be established.
3. Aected fetuses should be managed in referral
centers with experience in the ante-natal
management of FNAIT. Close collaboration is
required between specialists in fetal medicine,
obstetrics, hematology/transfusion medicine, and
pediatrics.
4. e mother and her partner should be provided
with detailed information about FNAIT and its
potential clinical consequences, and the benets
and risks of dierent approaches to ante-natal
management.
5. Maternally administered therapy should be the
rst-line approach in all cases. is is based on
data describing the eectiveness and safety of
maternal treatment in contrast to the toxicity of
serial FBS to deliver weekly fetal platelet
transfusions.
6. An important goal is to minimize the number of
FBS. However, the debate between empirical
treatment and treatment guided by measurement
of the fetal platelet count using FBS is not yet
resolved. Either approach is acceptable until the
issue is resolved by further clinical trials. It is to be
hoped that there will be developments in
laboratory testing allowing non-invasive
assessment of the likely severity of FNAIT in
individual cases.
7. Dierent centers currently have dierent
strategiesbasedontheirownexperienceand
those of published studies. Stratication of
ante-natal treatment based on the history of
FNAIT in previous pregnancies is common (and
appropriate) to both empirical and “invasive”
approaches to treatment.
8. Further progress is only likely to be achieved by
conducting randomized controlled trials to
resolve outstanding management issues. Patients
should be entered into trials, wherever possible.
Even referral centers see relatively small numbers
of patients, and to obtain sucient patient
numbers for adequately powered trials,
collaboration will be required between referral
centers.
Summary
ere have been considerable advances in the clini-
cal and laboratory diagnosis of FNAIT, and its postna-
tal and ante-natal management. e ante-natal man-
agement of FNAIT has been particularly problematic,
becauseseverehemorrhageoccursasearlyas16weeks’
gestationandthereisnonon-invasiveinvestigation
which reliably predicts the severity of FNAIT in utero.
e strategies for ante-natal treatment have included
the use of serial platelet transfusions, which while
eective are invasive and associated with signicant
morbidity and mortality. Maternal therapy involving
the administration of intravenous immunoglobulin
and/or steroids is also eective and associated with
fewer risks to the fetus. Signicant recent progress has
involved renement of maternal treatment, stratify-
ing it according to the likely severity of FNAIT based
on the history in previous pregnancies. However, the
ideal ante-natal treatment, which is eective without
causing signicant side-eects to the mother or fetus,
has yet to be determined, and further clinical trials are
needed.
71

Section 2. Feto-maternal alloimmune syndromes
References
1. Kaplan C. Neonatal alloimmune thrombocytopenia: a
50 year story. Immunohematology 2007; 23: 9–13.
2. Murphy MF, Bussel JB. Advances in the management
of alloimmune thrombocytopenia. British Journal of
Haematology 2007; 136: 366–378.
3. Ouwehand WH, Staord P, Ghevaert C et al. Platelet
immunology, present and future. ISBT Science Series
2006; 1: 96–102.
4. Turner ML, Bessos H, Fagge T et al. Prospective
epidemiologic study of the outcome and
cost-eectiveness of antenatal screening to detect
neonatal alloimmune thrombocytopenia due to
anti-HPA-1a. Transfusion 2005; 45: 1945–
1956.
5. Spencer JA, Burrows RF. Feto-maternal alloimmune
thrombocytopenia: a literature review and statistical
analysis. Australia and New Zealand Journal of
Obstetrics and Gynaecology 2001; 41: 45–55.
6. Radder CM, Brand A, Kanhai HH. Will it ever be
possible to balance the risk of intracranial
haemorrhage in fetal or neonatal alloimmune
thrombocytopenia against the risk of treatment
strategies to prevent it? Vox Sanguinis 2003; 84:
318–325.
7. Murphy MF, Williamson LM, Urbaniak SJ. Antenatal
screening for fetomaternal alloimmune
thrombocytopenia: should we be doing it? Vox
Sanguinis 2002; 83: 409–16.
8. Allen D, Verjee S, Rees S et al. Platelet transfusion in
neonatal alloimmune thrombocytopenia. Blood 2007;
109: 388–389.
9. Rayment R, Brunskill SJ, Stanworth S et al.Antenatal
interventions for fetomaternal alloimmune
thrombocytopenia. e Cochrane Library,Issue1,
2005. Chichester, UK: John Wiley & Sons, Ltd.
10. Bussel JB, Berkowitz RL, Lynch L et al. Antenatal
management of alloimmune thrombocytopenia with
intravenous gammaglobulin: a randomized trial of the
addition of low dose steroid to IVIg in y-ve
maternal–fetal pairs. American Journal of Obstetrics
and Gynecology 1996; 174: 1414–1423.
11. Berkowitz RL, Kolb EA, McFarland JG et al. Parallel
randomized trials of risk-based therapy for fetal
alloimmune thrombocytopenia. Obstetrics and
Gynecology 2006; 107: 91–96.
12. Radder CM, Brand A, Kanhai HHH. A less invasive
treatment strategy to prevent intracranial hemorrhage
in fetal and neonatal alloimmune thrombocytopenia.
American Journal of Obstetrics and Gynecology 2001;
185: 683–688.
72

Section 2 Feto-maternal alloimmune syndromes
Chapter
6Red cell alloimmunization
Alec McEwan
Introduction
Hemolytic disease of the newborn (HDN) describes a
process of rapid red blood cell breakdown, which puts
the baby at risk of anemia and kernicterus (bilirubin
induced cerebral damage) within the rst few days of
life. A variety of etiologies are recognized; however,
this chapter focuses on red cell alloimmunization, i.e.
the immune-mediated destruction of erythrocytes ini-
tiated by maternal red cell antibodies which reach the
fetal circulation by transportation across the placenta,
onwards from approximately 12 weeks’ gestation.
Pathogenesis
Antibodies recognizing red cell surface antigens usu-
ally arise secondary to a blood transfusion, or fol-
lowingthebirthofababywithadierentblood
group to the mother. Fetal red blood cells “track”
into the maternal circulation throughout pregnancy,
but “isoimmunization” against foreign antigens occurs
most frequently around the time of delivery when
the size of feto-maternal hemorrhage (FMH) tends
to be greatest. Other events associated with FMH are
listed in Table 6.1. ese red cell antibodies can, in a
subsequent pregnancy, reach the fetal circulation and
cause immune mediated destruction of fetal red blood
cells. is transplacental transportation of mater-
nal immunoglobulin G begins in the early second
trimester and red cell antibodies recognizing certain
erythrocyte antigens may bind and bring about pre-
mature destruction of the fetal red cells by the reticu-
loendothelial system. One of the breakdown products
of heme is bilirubin, and levels rise within the fetus
and amniotic uid, although placental transfer limits
this accumulation. Progressive anemia initially stimu-
lates the bone marrow rst but, as its capacity to main-
tain the hemoglobin levels is exceeded, extramedullary
hematopoiesis becomes increasingly important. is
hyperactivity of the reticuloendothelial system results
in fetal hepatosplenomegaly. A degree of portal hyper-
tension and hypoalbuminaemia secondary to liver
dysfunction may contribute to extracellular uid accu-
mulation within the fetus (hydrops fetalis); however,
cardiac dysfunction is more likely to be the main
explanation for hydropic change. Fetal anemia induces
a high-output cardiac state and a degree of hypoxia
may directly impair myocardial contractility. Hydrops
is characterized by skin edema, pleural and pericardial
eusions, cardiomegaly, atrioventicular valve dysfunc-
tion, ascites, polyhydramnios, and placentomegaly, all
of which can be detected by ultrasound scanning
(Fig. 6.1–6.3). ese changes are seen only when fetal
hemoglobin levels decline well below the normal range
and are a late feature of erythroblastosis fetalis.Intra-
uterine death will ensue in severe cases if the problem
isnottreated,orthebabydelivered.
HDN describes the consequences of this ante-
natal pathogenic process which continues on into the
newborn period. Maternal immunoglobulin G (IgG)
remains with the baby for 4–6 months aer birth
and top-up blood transfusions may be needed by the
infant whilst hemolysis continues. Far more concern-
ing than this semi-chronic post-natal anemia, how-
ever, is the risk of kernicterus which occurs within
therstfewdaysoflife.eimmaturefetalliveris
unable to conjugate the excessive circulating biliru-
bin and, as serum levels rise, it permeates the blood–
brain barrier. e globus pallidus of the basal ganglia
and the brain stem nuclei are the structures most at
risk of damage from the unconjugated bilirubin, which
is thought to uncouple phosphorylation from oxida-
tion, resulting in reduced ATP synthesis and impair-
ment of energy-dependent metabolism. Athetoid cere-
bral palsy, other movement disorders, deafness and
73
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.
Section 2. Feto-maternal alloimmune Syndromes
Table 6.1 Clinical scenarios associated with FMH and risk of
isoimmunization (adapted from RCOG Green top guideline
(No. 22))
Any birth (including by cesarean section)
Manual removal of retained placenta
Stillbirths and intrauterine deaths
Abdominal trauma in the third trimester
Delivery of twins
Unexplained hydrops fetalis
Invasive pre-natal diagnostic procedures such as amniocentesis
or CVS
Antepartum hemorrhage
External cephalic version
Hydatidiform mole
Termination of pregnancy (prophylaxis is recommended at all
gestations and with all methods)
Ectopic pregnancy (regardless of mode of treatment)
Spontaneous miscarriage ≥12 weeks (see below)
Fig. 6.1 Ante-natal ultrasound showing a transverse section
through the upper fetal abdomen at the level of the stomach and
liver. The calipers are measuring a 10 mm rim of ascites. There are
numerous etiologies for fetal ascites, but fetal anemia (from any
cause) is one of the more common explanations.
impaired eye movements may all be long-term seque-
lae of kernicterus.
Repeated exposure of an isoimmunized woman
to the same red cell antigen, as occurs in successive
pregnancies, will further stimulate antibody produc-
tion. Subsequent pregnancies, which express the blood
group in question, have a tendency to show more
severe hemolysis, and at earlier gestations.
Fig. 6.2 Ante-natal ultrasound showing a transverse section
through the fetal cranium. The calipers are measuring 9 mm of scalp
edema. Edema can collect throughout the skin of the fetus in severe
anemia. This results from a combination of high output cardiac
failure and also possible hepatic dysfunction and hypoproteinemia.
Fig. 6.3 Ante-natal ultrasound showing a transverse section
through the fetal chest. A slender fetal pericardial effusion and a
small left sided pleural effusion behind the heart can be seen. The
heart is also subjectively enlarged. These features are all consistent
with, but are non-specific signs of, fetal anemia.
Genotype and phenotype
ere are almost 30 dierent blood grouping systems,
but the ABO and Rhesus groups are arguably the most
important clinically. e Rhesus D (RhD) antigen was
discovered in 1939, but the full complexity of this
74

Chapter 6. Red cell alloimmunization
blood group system has only become evident much
more recently with the advent of molecular biology.
Of white Europeans, 16% are RhD negative, 5% of
West Africans, and virtually no Chinese. Of all deliv-
eries in the UK, 10% are of RhD positive babies born
to RhD negative women. In the absence of preventive
measures, 1 in 6 RhD negative women will isoimmun-
ize if they deliver a term RhD positive baby, and in the
1950s 1 in 2000 babies died of HDN, principally due to
RhD isoimmunization.
e Rhesus proteins are coded for by two genes,
which share a major degree of homology. RHD and
RHCE lie very close to one another on chromosome
1, back-to-back, and are thought to have arisen from
a duplication event involving the original ancestral
Rhesus gene, which can still be found in rodents and
most other mammals. e Rhesus proteins are char-
acterized by 12 intramembranous segments and 6
extra cellular “surface” loops. eir function remains
unclear, although ammonium ion transportation and
gas exchange across the erythrocyte cell membrane
have been postulated.
eRhDnegativephenotypeisrecognizedinthe
laboratory by failure of red cells to agglutinate with
standard anti-D reagents (antibodies). e underlying
genetic explanation for this phenotype is more com-
plex. In Europeans, 90% of RhD negative individuals
have a complete deletion of RHD, with the remain-
ing cases being explained by nonsense and frameshi
mutations which truncate the protein. However, in the
majority of African individuals typed as RhD nega-
tive the genotype is very dierent. e two common
RHD variants resulting in the D negative phenotype
are the RHD pseudogene, RHD,whichcodesfora
non-functional protein, and the Cdesallele which con-
tains segments from both the RHD and the RHCE
genes.
e situation is confused even further by alleles
of RHD, which cause subtle qualitative changes in
the extracellular surface loops of the RhD protein,
meaning that serological tests are only weakly positive
with standard anti-D reagents. Furthermore, missense
mutations causing single amino acid substitutions in
the intramembranous or cytoplasmic portions of the
RhD protein may impair integration of the protein
into the membrane, so bringing about a quantitative
reduction in the number of cell surface antigen sites
per red blood cell. is too may reduce the agglutina-
tion response of these cells to standard laboratory anti-
Dantibodies.ese“partialD”and“weakD”pheno-
Table 6.2 Key events in the history of prevention of RhD
isoimmunization
1938 Darrow concludes that “erythroblastosis fetalis” results
from the formation of a maternal antibody against some
component of fetal blood
1939 Levine and Stetson postulate that maternal
immunization is caused by a fetal antigen inherited from
the father which is lacking in the mother
1940 Landsteiner and Wiener discover the Rhesus antigen
1948 Wiener suggests that the initiating process is occult
placental hemorrhage
1957 Kleihauer devises a test able to detect fetal cells in the
maternal circulation
1961 Stern gives RhD positive red blood cells to RhD negative
volunteers, both with and without anti-D, and shows
that alloimmunization can be prevented
1966 Freda demonstrates that isoimmunization can be
prevented by giving anti-D to recently delivered RhD
negative women
1969 Widespread introduction of routine post-natal
prophylaxis with anti-D following multicenter trials
types, as they are respectively known, can be import-
ant from a clinical perspective and will be discussed in
greater detail later.
e DNA sequence of the RHCE gene shows far less
variation, and dierences at just ve amino acid posi-
tions result in the four dierent antigens C, c, E, and e.
Each allele expresses only C or c, in combination with
Eore,and,amongstEuropeans,theCehaplotypeis
most common.
Prevention of RhD isoimmunization
Antibodies against all the Rhesus proteins, and other
red cell antigens, can cause erythroblastosis and HDN;
however, anti-D has historically been of greatest sig-
nicance. Prevention of RhD isoimmunization, and
improvements in the ante-natal and neonatal care of
isoimmunized women and their babies, has all but
eradicated serious morbidity and mortality associated
with this condition. Some of the key landmarks in the
evolution of this success story are listed in Table 6.2. By
the early 1960s Stern had demonstrated that exogen-
ous anti-D given to RhD negative individuals could
prevent immunization occurring when RhD positive
blood was transfused into them.
Exogenous anti-D is produced by exposing RhD
negative volunteers to the RhD antigen. ese individ-
ualsareeithermale,orarewomenwhohavecompleted
their families. ey regularly donate their blood, and
cold-ethanol precipitation is used to separate the 75

Section 2. Feto-maternal alloimmune Syndromes
Table 6.3 Tests used to quantify the size of a FMH
Kleihauer : Fetal hemoglobin (HbF) is more resistant to acid or
alkaline elution than adult hemoglobin. After treatment, any
erythrocytes containing HbF retain their hemoglobin and can
be stained and recognized. Unfortunately, some adults have
persistent HbF production, and this can confuse matters.
Furthermore, quantification is less precise with bigger bleeds.
Flow cytometry: This uses immunofluorescently stained
antibodies to recognize fetal erythrocytes, which can then be
flow-sorted and quantified. This method is often preferred for
larger bleeds.
immunoglobulins from their hyperimmune plasma.
Following the emergence of variant Creutzfeldt–Jakob
disease in the UK, only plasma from US volunteers
hasbeenusedmorerecently,althoughitisnotknown
for certain if prions can be transmitted via trans-
fused immunoglobulins. A solvent/detergent treat-
ment inactivates HIV, hepatitis B and hepatitis C. BPL,
one of the major manufacturers of anti-D, estimates
a risk of viral infection of 1 in 10 000 billion doses of
their product and, to date, there have been no recorded
cases.
ere were theoretical concerns that passive anti-
D might itself cause haemolysis within the fetus. ere
is certainly no doubt that it can cross the placenta.
Although a small number of babies were born in the
anti-D trials with a weakly positive direct antiglobu-
lin test (DAT), the reaction was insuciently strong to
cause signicant hemolysis or anemia.
Delivery was recognized to be the time of great-
est risk for FMH and by the end of the 1960s
widespread post-natal prophylaxis had been intro-
duced. A Cochrane review of six eligible trials of rou-
tine postpartum anti-D prophylaxis gives a relative risk
of 0.12 for RhD alloimmunization in the subsequent
pregnancy, i.e. a tenfold reduction in the incidence of
isoimmunization.1Various doses of anti-D have been
tried, and indeed protocols still vary around the world
today. Doses of less than 500 iu are associated with a
greaterriskofisoimmunization;however,higherdoses
do not seem to confer any obvious benet. 125 iu anti-
D, is able to neutralize 1 ml of fetal red blood cells.
Feto-maternal hemorrhage (FMH) of ≥30 ml occurs
in only 0.6% of all deliveries. A dose of 1500 iu has
beenadoptedintheUSAtocoverthepossibilityof
larger hemorrhages. In the UK and France, a smaller
dose of 500 iu is routinely used; however, a test is also
performed to quantify the size of the FMH (Table 6.3).
Occasional bleeds exceeding 4 ml are recognized and a
higher dose of anti-D is administered.
e anti-D is usually given by intramuscular injec-
tion (although intravenous preparations are available)
and ideally should be given within 72 hours of delivery
(or any other possible sensitizing event). ere may,
however, be benet in giving anti-D as much as 9–10
days following potential isoimmunizing events.
Later came the recognition that a variety of events
during pregnancy might cause or be associated with
FMH, other than delivery, and that these might subse-
quently also lead to isoimmunization (Table 6.1). e
RCOG Green-Top Guideline (No. 22) lists these situ-
ations and recommends the use of anti-D prophylaxis
in these scenarios also.2e RhD antigen is thought to
be expressed as early as 7–8 weeks gestation and there
is no doubt that FMH can be demonstrated during the
rst trimester. As little as 0.25 ml of fetal RhD posi-
tive blood may be sucient to cause isoimmunization
and older studies have shown that this value is oen
exceeded with FMH occurring aer 8 weeks. e stud-
ies examining the risk of rst trimester isoimmuniza-
tion are old, and few in number.3e risk probably lies
between0and3%,butdoesseemtobehigherwhenthe
uterus is instrumented.
e RCOG have recommended anti-D only for
miscarriages prior to 12 weeks if the uterus is instru-
mented. Aer 12 weeks, and before 20 weeks, 250 iu
of anti-D should be given for all threatened and actual
miscarriages. Miscarriages and other potential sensi-
tizing events aer 20 weeks should be covered by 500 iu
of anti-D and a Kleihauer should be taken to identify
those cases where the size of the FMH exceeds 4 ml.2
Routine antenatal prophylaxis
Even in the absence of dened events known to
be associated with FMH, leakage of fetal red blood
cells into the maternal circulation is known to occur
throughout pregnancy. Beyond 28 weeks’ gestation the
quantity of tracked cells can be great enough to
bring about alloimmunization. Indeed, “silent” FMH
will cause RhD isoimmunization in 1%–2% of all RhD
negative women with RhD positive pregnancies.
ere is good-quality evidence supporting the use
of routine ante-natal anti-D prophylaxis (RAADP) to
prevent these isoimmunizations. A consensus confer-
ence hosted by the RCOG and RCP in 1997 came
out strongly in favor of routine ante-natal prophylaxis.
Crowther subsequently published a systematic review
in the Cochrane database, although only two trials
were deemed of high enough quality to be included.
76

Chapter 6. Red cell alloimmunization
is review reported a relative risk of isoimmunization
of 0.4 in the women receiving RAADP. More recently,
a Technology Appraisal Guidance (No. 41), produced
by NICE,4has reviewed the wider evidence from nine
trials. Although the trials varied in design and
methodology, they gave remarkably consistent results.
Without RAADP the isoimmunization rate ranged
from 0.9%–1.6%. is fell to approximately 0.3% in the
groups receiving RAADP.
A number of attempts at estimating the cost eec-
tiveness of this intervention have been made. e
number of HDN related deaths would be reduced
fromapproximately30to10peryearintheUKif
all women received RAADP. e cost–benet seems
clearforwomenintheirrstpregnancy,butlesssofor
parous women. Ultimately, however, both the RCOG
and NICE have recommended RAADP for all RhD
negative women, irrespective of parity.
e following dosage schedules are currently in use
in the UK:
A 500 iu at 28 weeks and 34 weeks’ gestation
B A single dose of 1500 iu at 28 weeks’ gestation
Schedule A was used in the only randomized con-
trolled trial of RAADP (Hutchet) and was most widely
adopted in the UK.4e half-life of anti-D is 24
days and theoretically there is less circulating anti-
D le at 40 weeks’ gestation with schedule B than
with A. e trials using this regime however did
not show signicantly poorer results. Commercially
available preparations of 1500 iu anti-D have recently
become available in the UK and there is a move
toward schedule B, mostly for reasons of convenience
and patient preference (one injection rather than
two).
Refusal of anti-D prophylaxis
A small minority of women will refuse anti-D, either
as part of RAADP or following potentially sensitiz-
ing events (including delivery), perhaps due to safety
fears or “needle phobia.” e woman should be pro-
vided with good-quality information to ensure that
this choice is truly informed; however, the nal deci-
sion of course must lie with her. Declining anti-D pro-
phylaxis carries no risk when;
1. the woman is condent she is not going to have
further children (e.g. requesting sterilization); or
2. when the father of the baby, or the fetus itself, is
known with certainty to be RhD negative.
Widespread non-invasive pre-natal fetal RhD test-
ingispossible(seelater)and,ifadopted,willmean
that RAADP and the use of anti-D following sen-
sitizing events will be reserved for women carry-
ingafetuswhichisRhDpositive,orofunknown
status.5
Traditional management of
isoimmunization
Despiteeectiveprophylaxisprograms,newcasesof
RhD isoimmunization do arise, either because guide-
lines are not followed appropriately, women fail to seek
medical advice around the time of potentially sensi-
tizing events, or because of “silent” isoimmunizations,
perhaps occurring prior to 28 weeks’ gestation. Man-
agement of these pregnancies has become limited to a
relatively small number of centers. Preventing morbid-
ity and mortality in these cases necessitates the iden-
tication of pregnancies at risk, subsequent monitor-
ing of disease severity, and timely intervention in the
form of intrauterine transfusion and/or delivery of the
baby. Modern management is quite dierent to that of
even just 10 years ago and, to best appreciate the recent
advances made, a rapid review of traditional methods
is included here.
Historical perspectives
Routine maternal blood typing and serological test-
ing was introduced in the 1950s. RhD negative women
with anti-D antibodies were recognized as being at risk
of having their pregnancies complicated by hydrops,
stillbirth, and hemolytic disease of the newborn.
Approximately 85% of the white European and North
American population is RhD positive, and just over
half are heterozygous. e ospring of RhD heterozy-
gous males and RhD negative women are at 50% risk
of being RhD positive themselves, and 50% will be
RhD negative. e RhD negative fetus is at no risk
of hemolysis, whatever the levels of maternal anti-D.
Although RhD negativity in male partners could be
determined with certainty, predicting whether a RhD
positive man was homo- or heterozygous was impre-
cise prior to the advent of molecular biology and relied
on the results of serological testing with anti-sera to the
D, C, c, E and e antigens and racially specic incidence
charts. However, this prediction was inexact and, when
a male partner was thought to be heterozygous, the 77

Section 2. Feto-maternal alloimmune Syndromes
status of the fetus remained unclear. A RhD negative
pregnancy could be exposed to serial invasive testing
when there was no actual risk.
With the development of molecular genetic tech-
niques, and improved understanding of the Rhesus
gene cluster, it became possible to determine RhD
status precisely using DNA amplication techniques.6
ese are able to sensitively distinguish between
homozygotes and heterozygotes and can be applied to
DNA from amniocytes to precisely assign RhD posi-
tive or negative status to the fetus of a couple where
themalepartnerisheterozygous.Asingleamniocente-
sis meant that further testing could be avoided in 50%
of cases (those found to be RhD negative). Surveil-
lance and invasive testing could then be appropriately
focusedontheRhDpositivepregnancies.
A number of dierent factors have been used to
time interventions in Rhesus disease. e simplest and
least sensitive of these is previous obstetric history.
Walker showed how, in a RhD isoimmunized preg-
nancy, the risk of stillbirth was 8% if there was no
previous history of HDN. is rose to 18% if a pre-
vious child had been moderately aected and to 58%
if there was a previous history of stillbirth caused
byhemolyticdisease.etendencyforthedisease
to become more severe, and at progressively earlier
gestations, was well recognized. Recent retrospective
reviews of isoimmunized pregnancies have conrmed
these historical conclusions. However, relying on pre-
vious history to guide intervention was imprecise and
hazardous.
Coombs demonstrated that the strength of anti-D
isoimmunization could be measured by serially dilut-
ing maternal serum until agglutination of RhD posi-
tive red blood cells no longer occurred. e more
doubling dilutions were required to lose this reaction,
the more anti-D must have been there to begin with.
Serial dilutions of 1/2, 1/4, 1/8, 1/16, 1/32, 1/64, 1/128
indicated progressively higher starting levels of anti-
D. More recently, levels of anti-D have been quan-
tied more precisely in “international units per ml”
(IU/ml) using dierent techniques. Signicant hemol-
ysis is unlikely at levels below 4 IU/ml and is unlikely
to be severe at levels below 10–15 IU/ml. However,
this threshold too is insensitive and the relationship
between absolute anti-D levels and disease severity
weakens in pregnancies beyond the rst where anti-
bodies are detected.
Nevertheless, these factors have been used (and
still are to some degree) to decide when to investigate
further with amniocentesis, perform fetal blood sam-
pling, or indeed deliver.
Amniocentesis
Immune-mediated hemolysis within the fetus gen-
erates bilirubin, which is excreted by the fetal kid-
neys into the amniotic uid. Ballantyne, at the end of
the nineteenth century, recognized that yellow stain-
ing of amniotic uid was associated with the subse-
quent development of severe jaundice in the newborn.
Bevis recognized that the degree of yellow pigmen-
tation of amniotic uid samples taken during preg-
nancy oered a guide to the nal outcome; how-
ever, reliable measurement of bilirubin concentrations
proved dicult and the alternative technique of meas-
uring the optical density shi caused by the biliru-
bin was adopted. Using a spectrophotometer, the opti-
cal density of amniotic uid is assessed across a wide
spectrum of wavelengths. Bilirubin causes a shi in
absorption at the 450 nm wavelength and the degree
of this shi (⌬OD450) is proportional to the con-
centration of bilirubin. In the early 1960s, Liley pub-
lished a chart which could be used to estimate the
risk of severe anemia in an isoimmunized pregnancy
based on the ⌬OD450 of amniotic uid collected by
amniocentesis aer 27 weeks’ gestation; the higher the
⌬OD450, the greater the chance of severe fetal ane-
mia. Results falling above a certain threshold (“Zone
3”) would prompt intrauterine blood sampling and a
subsequent transfusion if the fetus was found to be sig-
nicantly anemic. When managing a RhD isoimmu-
nized pregnancy, the timing of the rst amniocentesis
was decided by a number of factors, including previ-
ous history and anti-D titer (or concentration). If the
⌬OD450 fell below these thresholds, repeated amnio-
centeses were subsequently required at intervals of 1
to 4 weeks, depending on the initial result, the rate of
rise between successive samplings, the Rhesus history
and the anti-D level. In a group of pregnancies with a
high incidence of fetal anemia, the sensitivity for detec-
tion of severe anemia (Hb of less than 5 SD below the
mean) was found to be approximately 80%,7meaning
that 1 in 5 severely anemic fetuses would be missed
by the screening test. Reducing the ⌬OD450 threshold
abovewhichfetalbloodsamplingwouldbeperformed
did improve the sensitivity to nearly 100% but went
hand-in-hand with a drop in the specicity (below
50%) and positive predictive value, meaning that a sig-
nicant number of fetal blood samplings were being
78
Chapter 6. Red cell alloimmunization
Table 6.4 The disadvantages of amniocentesis and ⌬OD450
measurements in the assessment of immune-mediated fetal
anemia
r0.5–1.0% risk of miscarriage/preterm
delivery/chorioamnionitis/peri-natal loss with each
procedure
rLimited performance as a screening test, particularly at
gestations <28 weeks
rCause of fetomaternal hemorrhage and subsequent rise in
antibody levels in 50% of cases (Bowell
et al.
)
rA surrogate marker for fetal anemia. Particularly
problematic in Kell isoimmunized pregnancies (see later)
rUnpleasant for the woman
prompted by the ⌬OD450 when, in fact, the fetus was
not severely aected (false positives). Other studies
quote somewhat dierent sensitivities and specicities
but the overall message remains the same. Later, the
Liley charts were extrapolated backwards to 20 weeks’
gestation but this too was associated with a reduc-
tion in the sensitivity, as demonstrated by Nicolaides.
ese were not the only weaknesses of amniocentesis
used in this way (Table 6.4).
Nevertheless, when the procedure-related risks
associated with amniocentesis (0.5%–1.0%) were com-
pared with those of fetal blood sampling (1%–4%)
the benet in “screening” by ⌬OD450 prior to fetal
blood sampling seemed clear. e optimum timing of
rst, and repeated, amniocenteses required signicant
experience but it soon became adopted as standard
practice in most centers. Some questioned whether,
with improvements in fetal blood transfusion tech-
niques, it should be abandoned; however, the practice
continued until newer non-invasive methods became
more widespread at the beginning of the new millen-
nium. It is rare now for amniocentesis to be performed
in the management of RhD isoimmunization.
Fetal blood transfusion
Pre-natal treatment options for RhD hemolytic dis-
ease, other than preterm delivery, really began in the
1960s when Liley showed that the fetus could be trans-
fused in utero by injection of blood into the fetal peri-
toneal cavity under X-ray guidance. A radio-opaque
dye was injected into the amniotic cavity and taken
up by the fetus. On reaching the bowel, it outlined the
peritoneal cavity, into which the blood was injected.
Erythrocytes were then absorbed directly across the
bowelwallintothefetalintravascularcompartment.
is hazardous procedure was later superseded by
ultrasound guided transfusions into the peritoneal
Table 6.5 Historical landmarks in the development of fetal
blood transfusion
1963 (Liley) X-ray guided intraperitoneal IUT
1964 (Freda and
Adamsons)
Direct IUT through hysterotomy
incision
1981 (Rodeck) Fetoscopically guided intravascular IUT
1981 (Berkowitz and
Hobbins)
Ultrasound guided intraperitoneal IUT
1986 (Nicolaides and
Rodeck)
Ultrasound guided intravascular IUT
cavity and then directly into the fetal circulation. e
timeline of the evolution of this remarkable fetal ther-
apy is detailed in Table 6.5.
Intraperitoneal transfusion (IPT) had a num-
ber of drawbacks. No pre- or post-transfusion fetal
hemoglobin level or hematocrit was available, so the
volume to be transfused was, at best, an educated
guess. Absorption of the donated red cells across the
bowelwallwasslowandthiswasofparticularcon-
cern for the very anemic fetus where immediate and
rapid correction was needed. e presence of edema
in the bowel wall of a hydropic fetus could interfere
with absorption completely. Furthermore, overdisten-
sion of the fetal abdomen by the donated blood pos-
siblyendangeredthefetusbyinterferingwithvenous
return and cardiac output. As intravascular transfu-
sion (IVT) became more popular, comparative studies
suggested a sixfold greater risk of fetal death following
IPT.
Ultrasound guided direct intravascular transfu-
sions are performed via a number of dierent routes.
Direct intracardiac transfusion is possible but, for
understandable reasons, is not ideal. e majority of
fetal IVT are performed into the umbilical vein at the
insertion of the umbilical cord into the placenta or
percutaneously into the intrahepatic portion of the
umbilical vein.8A free loop of cord can be used if the
placental insertion cannot be accessed. ere is lit-
tle evidence to suggest any one method is superior
to the others. Whichever route is favored, the uter-
ine wall, amniotic membrane, and sometimes the pla-
centa, must be breached by the needle. e risk of fetal
loss, membrane rupture, bradycardia, or fetal bleed-
ing requiring delivery is quoted between 2% and 4%
per procedure, but this depends on gestation, opera-
tor experience, and fetal condition prior to the transfu-
sion. Transfusions below 20 weeks gestation are a par-
ticular challenge and fetal loss rate is at least 10% if
there is hydrops. 79

Section 2. Feto-maternal alloimmune Syndromes
e blood used for a fetal transfusion is cross-
matched against the maternal blood and should
be fresh, CMV negative, and irradiated. e cross-
matched unit ideally has a hematocrit of 75%–80%, so
as to minimize the volume load of the transfusion on
the fetus. A degree of “over transfusion” to a hematocrit
of 40%–50% will prolong the interval between subse-
quent transfusions. However, the transfused blood is
acidotic and this and the volume load can be hazardous
to the very compromised hydropic fetus. In these cases,
a hematocrit of 25% should be aimed for with the rst
transfusionwhichcanbefollowed2or3dayslater
with a second “top-up.” e total volume (in ml) to
be transfused (VT) is determined by the fetoplacen-
tal blood volume (VFet), which increases with gesta-
tion, the hematocrit of the fetal and donated blood
(HctFet and HctDon), and the target “desired” hemat-
ocrit (HctDes ) according to the equation:
VT(ml) =VFet(ml) ×HctDes −HctFet
HctDon −HctDes
ere is a steady increase in the total blood volume of
the fetoplacental circuit (VFet ) with advancing gesta-
tion, from approximately 25 ml at 20 weeks’ gestation
to 100 ml at 28 weeks’, and 210 ml at 34 weeks’ gesta-
tion.
Maternal sedation is usually employed because the
procedure can be uncomfortable and may last more
than 30 minutes. ese sedative drugs may also help
to reduce fetal movement. Antibiotics and oral tocolyt-
ics are used by some operators, but there is no evi-
dence to support or refute this practice.e maternal
abdomen is sterilized and draped and strict aseptic
techniqueisfollowed.Localanestheticisinjectedinto
the maternal abdominal wall. Under ultrasound guid-
ance, a 20 Gauge needle is inserted through the uter-
ine wall into the umbilical cord, or the vessels within
the fetal liver. A small sample of blood is taken to
conrm correct positioning and the hematocrit meas-
ured immediately, so that the total volume required
can be calculated rapidly. Transfusion of blood at 5 ml
per minute is usually tolerated well by the fetus. A
post-transfusion hematocrit is taken to help guide the
interval between transfusions. e hematocrit drops
by approximately 1% per day, but this rate may decline
as subsequent transfusions replace the fetal RhD pos-
itive blood with donated RhD negative blood, which
survives longer. e interval is usually 2–3 weeks and
newer scanning techniques may help to ne-tune this
(see later). Serial transfusions are usually performed
until 34–36 weeks’ gestation, aer which delivery is
organized. Severe, early onset hemolytic disease may
necessitate more than ve transfusions in a single preg-
nancy. A vaginal birth is aimed for unless there are
other obstetric factors, or the fetus is sick.
Recent advances in management
Despite the declining incidence of RhD isoimmuniza-
tion, the management of aected pregnancies has not
stood still. e need for invasive assessment of the
pregnancy at risk can be directed now with greater
precision, and newer treatment options have shown
promise in helping to avoid the need for transfusions
at very early gestations.
Non-invasive testing for fetal
RhD status
e plasma of pregnant women contains free (i.e. non-
cell associated) fetal DNA (DNA) in signicant quan-
tities, from the early rst trimester. e main source of
this DNA is debated, but it probably originates from
trophoblastic cells at the maternal–fetal interface. is
DNA is fragmented and is degraded rapidly. Mater-
nal free DNA is present in much larger quantities.
Lo, in 1997, demonstrated how DNA could be used
for the non-invasive pre-natal diagnosis of fetal RhD
status9and, since then, the same principles have been
applied to non-invasive fetal sexing and the inherit-
ance of paternally derived disease causing mutations.
istechnologyisstronglypromotedby“SAFE”(e
Special Non-invasive Advances in Fetal and Neonatal
Evaluation Network), a multinational European group
established in 2004 and funded by monies from the
European Union.10
A RhD negative woman should have no RhD DNA
sequences in her plasma because her negative status
is usually caused by a deletion of the RhD gene (but
see later). If DNA probes designed to recognize RhD
gene exons are added to her plasma, along with DNA
polymerase,thennoproductshouldformifthefetus
is also RhD negative, because there are no binding
sites for the probes. If the fetus is RhD positive, then
theprobeswillbindtotheDNAandaPCRproduct
will be produced, which can be detected easily using
standard molecular techniques. ree separate exons
from RHD are amplied, and if only one or two of the
PCR products is generated then the result is considered
80
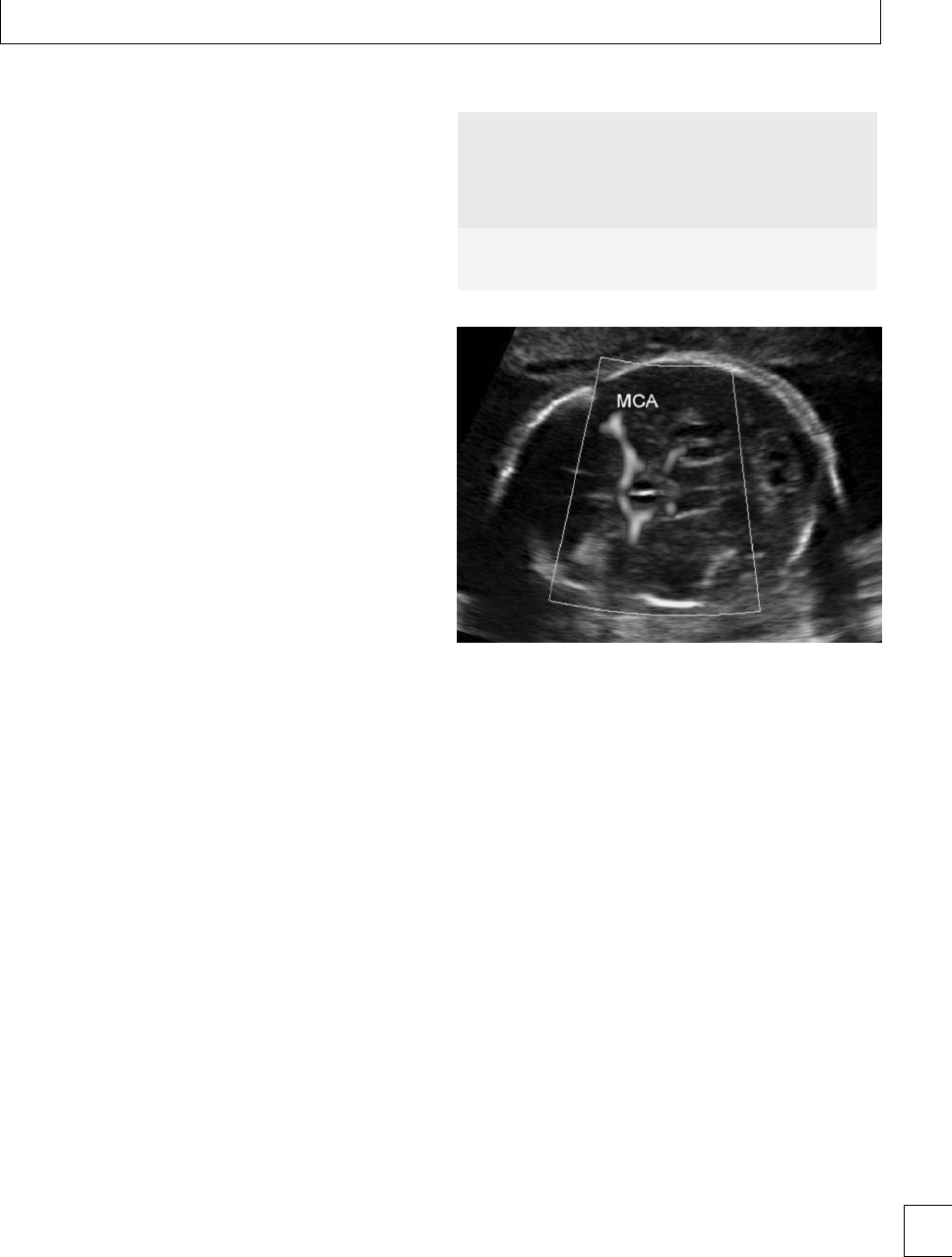
Chapter 6. Red cell alloimmunization
equivocal and further investigation is required before
aresultcanbegiven.
Use of this technology has become almost rou-
tine practice in the UK, and in parts of Europe, where
the father of the baby is a RhD positive heterozygote,
where his status is unknown, or if paternity is uncer-
tain. e team performing this work in the UK is based
at the International Blood Group Reference Labora-
tory in Bristol, and their results are impressive.11 Over
a thousand samples have so far been tested and there
have only been two false positive and two false neg-
ative results. e accuracy of the test is independent
of gestation, however the rate of inconclusive results is
higher at gestations below 16 weeks and the test must
be repeated in a h of cases (personal communica-
tion March 2008). A simple maternal blood test eect-
ively avoids the need for amniocentesis to determine
fetal RhD status when the father is heterozygous, or
hisbloodgroupisunknown.is,inturn,bypasses
the 1% risk of miscarriage associated with the amnio-
centesis, and the likely fetomaternal transfusion of red
cells which may cause a subsequent rise in anti-D lev-
els in already isoimmunized women. A RhD negative
result provides welcome relief and reassurance without
having put the pregnancy under any risk at all.
Middle cerebral artery blood flow
Since the late 1980s the relationship between fetal ane-
mia and the velocity of blood ow in the middle cere-
bralartery(MCA)hasbeenclearlydocumented.is
can be measured using ultrasound, and a collaborative
group, led by Mari,12 are usually credited with enhanc-
ing the prole of this technique so that today it has
eectively replaced amniocentesis in the monitoring of
pregnancies complicated by red cell antibodies.
A decrease in total red cell mass results in a reduc-
tion in blood viscosity and an increase in cardiac out-
put. e eect on viscosity is thought to be the prin-
cipal mechanism causing an increase in fetal peak sys-
tolic blood ow velocities (PSV). e vessel where this
can be measured most easily and reliably is the middle
cerebral artery.
e technique is not dicult; however, a number
of factors inuence the MCA PSV (see Table 6.6) and
guidelines must be adhered to strictly. e angle of
insonation of the pulse wave Doppler must be as close
to 0 degrees as possible, or angle correction must be
used.evesselmustbeinsonatedwithintherst
2 mm of the proximal portion of the vessel as it arises
Table 6.6 Factors influencing the MCA PSV value
Fetal
Gestational age
Fetal activity
Cardiac status
Gender
Uterine contractions
Technical
Angle of insonation of Doppler wave
Positioning of the Doppler gate along the MCA
Fig. 6.4 Power Doppler study showing the fetal Circle-of-Willis and
the near-field middle cerebral artery (MCA)
from the Circle of Willis (see Figs. 6.4 and 6.5). Meas-
urements taken at a distal point in the vessel may be
6–10 cm s−1less than values taken proximally. Usually
the MCA closest to the transducer is chosen, for ease,
but the far-eld vessel gives similar results. e fetus
must be quiescent as fetal movement and breathing can
have a signicant impact on the values obtained.
Normal ranges and charts (Fig. 6.6) are available
to plot the values onto and these show a normal grad-
ual increase in the MCA PSV as gestation advances.
Although the trend in values is important in any par-
ticular case, the most valuable indicator of signicant
fetal anemia has proven to be a threshold of 1.5 MoM
(multiples of the median). Below this level, it is highly
unlikely that the fetus will be more than just mildly
anemic. e higher the value lies above this line, the
more likely moderate or severe anemia becomes. If the
MCA PSV in an “at-risk” pregnancy is found to fall
above this threshold, the study will usually be repeated
within 24 hours. If the nding is persistent, then fetal
blood sampling (with or without transfusion) will usu-
ally be performed soon aer. MCA PSV studies are
usually performed at intervals varying from 3 days to 81
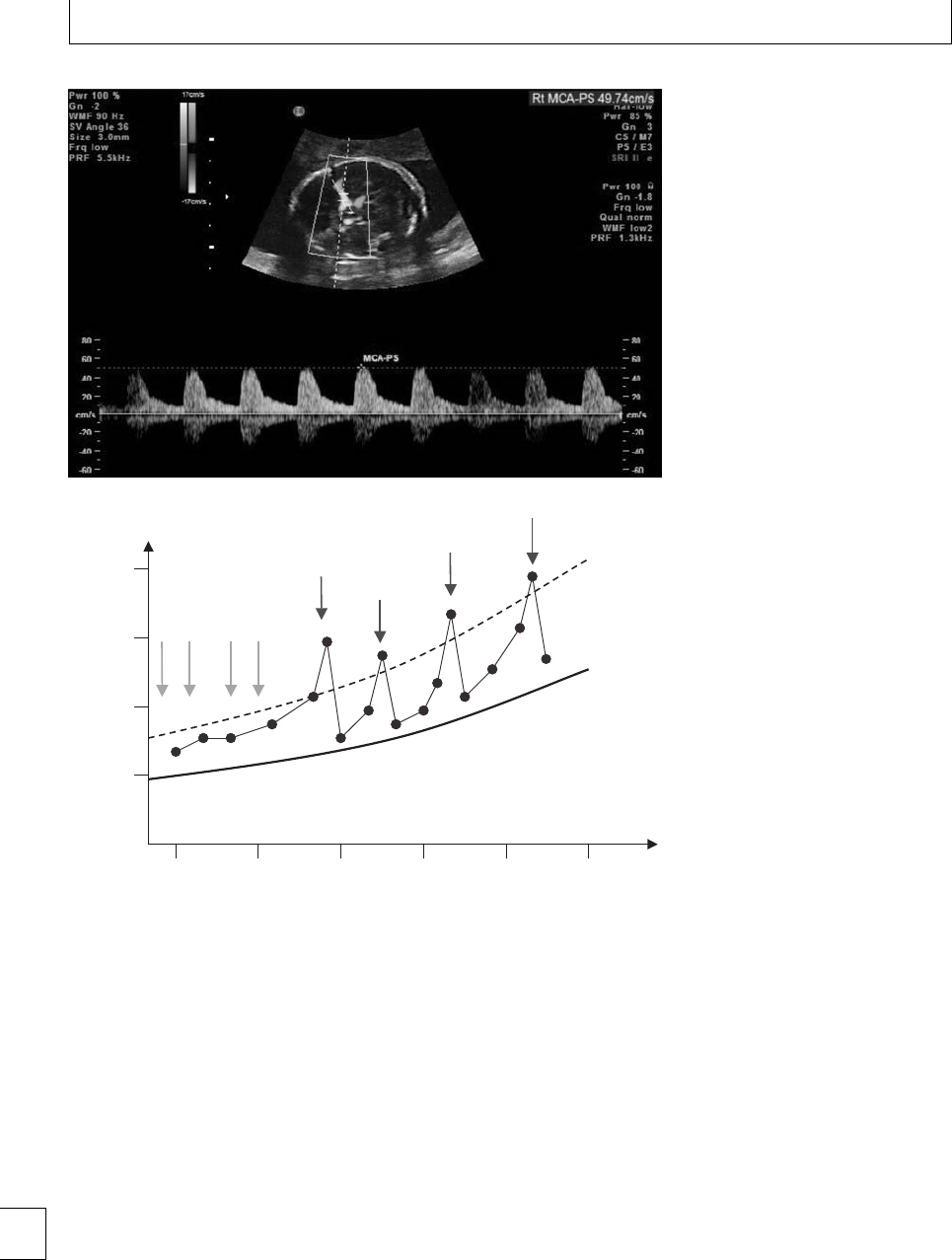
Section 2. Feto-maternal alloimmune Syndromes
Fig. 6.5 Color Doppler measurement of
middle cerebral artery peak systolic
velocity (MCA PSV). This study was
performed at 22 weeks’ gestation. A PSV
of almost 50 cm s−1in the MCA at this
gestation is well above 1.5 multiples of
the median and did indeed indicate fetal
anemia in this case. Although the
circulation is hyperdynamic in fetal
anemia, the actual cause of the rise in
blood velocity is thought to be a
reduction in blood viscosity secondary to
a falling fetal hematocrit. The fetal
hemoglobin was found to be 4 g/dl on
subsequent testing.
20
40
60
80
16 20 24 28 32 36
MCA PSV (cm s−1)
Gestational age (weeks)
Median
1.5 MoM
Fig. 6.6 A chart showing median fetal
middle cerebral artery peak systolic
velocities (MCA PSV) throughout the
second and third trimesters. An imaginary
line has been drawn at 1.5 multiples of
this median value and acts as an action
line to prompt fetal blood sampling +/−
transfusion. Superimposed is a fictitious
case of severe RhD isoimmunization with
a RhD positive fetus. The blue arrows
show where maternal IVIG was given, and
the red arrows represent fetal blood
transfusions. The MCA PSV can be seen to
fall immediately following a fetal blood
transfusion, but gradually increases again
as hemolysis continues. As the fetal RhD
positive blood becomes replaced by
successive donations of RhD negative
blood, the interval between transfusions
increases. However, this donated blood
also has a limited lifespan, even though it
is not subject to the antibody mediated
hemolysis.
4 weeks, depending on the perceived degree of risk,
and previous values.
e collaborative group found a 100% sensitivity
for the prediction of moderate to severe anemia, with a
false positive rate of 28%.13 iscomparesveryfavor-
ably with amniocentesis. Other studies have failed to
achieve quite such impressive results, although those
of Zimmermann are typical and are at least as good
as amniocentesis. ey found a sensitivity of 88% and
a positive predictive value of 53%, i.e. approximately
1 in 10 cases of moderate-severe anemia were missed
andhalfofallcaseswithaPSVgreaterthan1.5
MoM required transfusion. ey recommended that
this technique should not be used aer 35 weeks’ ges-
tation, when the false positive rate is higher. Other
studies have suggested that the MCA PSV operates
well in the second trimester, in contrast to ⌬OD450.13
Even if MCA PSV only matches the predictive abili-
ties of liquor ⌬OD450 measurements, the advantage
remains clear; the technique is non-invasive. Indeed,
by adopting MCA PSV measurements as the method
of determining when fetal blood sampling is required,
the number of invasive procedures can be reduced by
two-thirds. Furthermore, reducing the time interval
between MCA studies should improve the sensitiv-
ity of the test. Recent studies show that it remains a
82

Chapter 6. Red cell alloimmunization
Table 6.7 Possible mechanism of action of IVIG in Rhesus D
isoimmunization
Increased catabolism of maternal IgG
Competitive blockade of placental IgG transport mechanisms
Fc receptor blockade within the fetal reticuloendothelial system
Precipitation of immune complexes by excess antibody
Antigen neutralization
Binding of exogenous anti-idiotypic Ab to endogenous Ab
useful tool for timing second, third, and subsequent
fetal blood transfusions.
Adjunctive ante-natal treatments
Targeting the maternal immune response is a tempt-
ing strategy for tackling severe cases of isoimmuniza-
tion. e knowledge that disease severity is, at least in
part, related to absolute anti-D levels led to the pro-
posal that plasmapheresis might help ameliorate the
disease. Anti-D levels can be kept under control with
this technique, but it is not without maternal risk,
causes a rebound of antibody levels when treatment
comes to an end, and was never convincingly shown
to make a dierence in erythroblastosis fetalis. For
thesereasons,itsuseasanadjuncttowell-established
management techniques had fallen out of favor until
more recently when it has been used in combination
with a second form of immunomodulation which itself
has shown greater promise. e use of intravenous
immunoglobulin (IVIG) to prevent/treat fetal and
neonatal alloimmune thrombocytopenia is described
in Chapter 5 and there is more evidence of its value
in this condition than there is for Rhesus isoimmun-
ization. Nevertheless, non-randomized studies pro-
vide support for its use in cases of severe RhD
isoimmunization. 1 g/kg is administered on a weekly
or fortnightly basis from 13 to 20 weeks’ gestation,
the aim being to delay the onset of moderate-severe
hemolysis to a point in the pregnancy where IVT can
be more readily and reliably performed. e num-
ber of cases of hydrops can be reduced, as can the
number of fetal blood transfusions needing to be per-
formed. Table 6.7 lists some of the possible mech-
anisms by which IVIG might work. A more recent
study has reported on a combination of IVIG with
serial plasmapheresis from 12 weeks’ gestation for
women with the most severe histories, with impres-
sive outcomes.14 e contribution of the two dier-
ent methods of immunomodulation is not possible to
Table 6.8 Management strategies where moderate to severe
HDN is expected
Close communication with neonatal team
Planned delivery (induction or Cesarean section)
Cord blood analysis for bilirubin, Hb, group and DCT
Drainage of effusions and ascites in the hydropic infant
Phototherapy
Intravenous immunoglobulin
Exchange transfusion
Recombinant erythropoietin
Top-up blood transfusions
assess. IVIG is expensive, and prepared from multiple
donors. Rarely, it may cause unpleasant and potentially
serious side eects, including;
rpyrexia and rigors;
rheadache, backache, and myalgia;
rhypotension and tachycardia;
rtachypnea and chest tightness;
ralopecia;
rhemolytic anemia;
rrenal impairment.
Figure 6.6 illustrates how serial MCA PSV moni-
toring, early IVIG administration and multiple fetal
blood transfusions can support a pregnancy at risk of
early and severe erythroblastosis fetalis through to a
gestation where induction can be expected to bring
about the normal birth of a non-hydropic baby with
adequate hemoglobin levels.
Pediatric management
ItisrarenowforababytobebornatriskofHDNwith-
out the prior knowledge of maternity and pediatric
sta. A multidisciplinary approach is vital for optimiz-
ing outcomes. Infants born with only a low risk of sig-
nicanthemolysisshould,attheveryleast,havecord
blood sent for Coombs test (DCT), blood group,
hemoglobin and bilirubin levels. Close observation
over the next 2–3 days is necessary and repeat bilirubin
estimations may be required, as may phototherapy.
Management strategies for more signicant cases
of HDN are listed in Table 6.8.
It is far preferable to treat a hydropic fetus in
utero than it is to deliver the baby in such poor con-
dition. However, complications from an intrauterine
transfusion may precipitate the unplanned delivery
of such a baby in which case intubation, ventilation, 83

Section 2. Feto-maternal alloimmune Syndromes
and drainage of pleural eusions and ascites will be
required. ese babies are volume overloaded, making
transfusion hazardous (although still necessary). ey
are at risk of hypoglycemia, hypocalcemia, hypona-
tremia, hyperkalemia, hyperbilirubinemia, acidosis,
and renal failure. Mortality rates are high.
With modern ante-natal and fetal management,
this situation is fortunately rare. Nevertheless, plan-
ning delivery is important for the cases of moderate
or severe erythroblastosis fetalis, even if intrauterine
transfusions have minimized the risk. Bilirubin lev-
els will rise sharply aer birth and phototherapy must
begin immediately.
Light from the blue–green region of the spec-
trum (425–490 nm) is most eective at convert-
ing non-polar bilirubin to water-soluble photoiso-
mers and uorescent tubes producing irradiance of
⬎30 W/cm2/nm are optimal. e surface area of the
baby exposed to the light is crucial, and ber-optic
pads placed under the neonate, or the use of specif-
ically designed “bili-beds,” ensure that this is maxi-
mized. Bilirubin levels must be measured regularly and
phototherapy may need to continue for a number of
days. Gestation specic charts are available for biliru-
bin levels and thresholds for exchange transfusion are
recognised. A rise in serum bilirubin beyond these lev-
els puts the newborn at increasing risk of kernicterus.
Severe anemia, high absolute bilirubin levels,
excessive rise in bilirubin concentration, and unsafe
bilirubin-to-albumin ratios are all indicators for
exchange transfusion. An intravenous catheter is
placed into the inferior vena cava via the umbilical vein
through the cord stump and the entire blood volume of
the neonate is usually replaced twice (“double-volume”
exchange) by removing neonatal blood and replacing it
with RhD negative blood in 5–10 ml aliquots. is pro-
cess removes bilirubin and antibody-coated red blood
cells, and at the same time provides new albumin with
unoccupied bilirubin binding sites and RhD negative
erythrocytes. Between 70% and 90% of all fetal red
blood cells are removed, but because most of the biliru-
bin is in the extravascular compartment, 75% of total
body bilirubin remains and can cause a rebound rise
in serum levels soon aer the exchange, necessitat-
ing a repeat procedure. e inherent risks of exchange
transfusion are substantial, however (Table 6.9),15 and
experience with the technique is declining. As many as
1-in-20 infants undergoing exchange transfusion may
die and 1-in-4 suer non-fatal complications. Much
of this morbidity and mortality is found in preterm
Table 6.9 Potential complications of exchange transfusion
Haematological : Over-anticoagulation with hemorrhage, ane-
mia, neutropenia, thrombocytopenia
Cardiac : Volume overload, congestive heart failure, hyperten-
sion, arrhythmia, arrest
Metabolic : Acidosis, hypocalcemia, hypoglycemia, hyper-
kalemia, hypernatremia,
Vascular : Thromboembolic events, necrotizing enterocolitis,
vessel perforation
Infectious : Bacterial, viral, malarial
Other : Hypothermia, apnea, bowel perforation
babies, emphasizing again the massive impact that
ante-natal management has had on this condition by
delaying the gestation at which the baby needs to be
born. Furthermore, intra-uterine transfusions provide
the fetus with red blood cells not at risk of immune-
mediated hemolysis. By the third IUT, the fetal blood
will be almost entirely RhD negative. At birth there-
fore, these babies paradoxically are less likely to need
exchange transfusion.
Avoiding exchange transfusion is clearly benecial.
e use of intravenous immunoglobulin is well estab-
lished now in the treatment of neonatal alloimmune
thrombocytopenia and HDN. Although a number of
mechanisms are possible, the main action is thought
tobeablockadeofFcreceptorsinthereticuloendothe-
lial system. IVIG reduces carboxyhemoglobin levels, a
sensitive indicator of hemolysis. Although IVIG is pre-
pared from multiple donors, and is extremely expen-
sive, its use as an adjunct in moderate-to-severe HDN
seems justied. A Cochrane systematic review in 2002
concluded that IVIG signicantly reduces the need for
exchange transfusion (RR =0.28), and reduces the
number of exchanges needed when they cannot be
avoided.16 However, better quality studies are few in
number and there has been a call for larger random-
ized trials. It should be used only as an adjunct to pho-
totherapy and 0.5–1.0 g/kg is usually given as a single
dose soon aer delivery.
e baby remains at risk of developing anemia for
some months, for two reasons. Firstly, maternal anti-
bodies circulate for 4–6 months and continue to cause
low-grade hemolysis. Secondly, intrauterine and new-
born transfusions may suppress normal erythropoiesis
and it may be a number of months before reticulo-
cytes appear. During this time, “top-up” blood trans-
fusions may be required, although these carry mini-
mal risk in comparison with exchange transfusion.
84

Chapter 6. Red cell alloimmunization
Regular recombinant erythropoietin (EPO) injections
can be used during this time to limit the number of
top-up transfusions required.
Rhesus D variants
RHD is a complex gene and much variation exists
within it, particularly between racial groups. Under-
standing this is crucially important for a number of
reasons. Phenotypic tests of RhD status examine how
blood from an individual behaves when it is added
to serum containing anti-D antibodies. Agglutination
indicates that the individual is RhD positive. Geno-
typic tests of RhD status look for key DNA sequences
from RHD.eentiregenecannotbeexamined,so
sections from a variety of coding exons are chosen for
multiplication, using the polymerase chain reaction.
If a PCR product is produced, then the assumption is
made that the individual is phenotypically RhD posi-
tive.
RhD variants can confuse this. e common cause
for RhD negativity in Africans is an allele called the
RHD pseudogene (RHD)whichcontainsa37base
pair insert in exon 4 and a nonsense mutation in exon
6, which eectively make the protein non-functional.
Phenotypically, these individuals are RhD negative,
but genetic tests might, for example, amplify exon 7
successfully and give a false positive result for RhD
status. Amplifying more exons, such as exon 5, would
allow clarication because this exon is amplied from
the normal RHD but not RHD.ishasparticu-
lar relevance to non-invasive pre-natal RhD testing.
Knowledge of the racial origin of the woman’s part-
ner is clearly important. A second common African
variant is the RHD/CE hybrid allele, of which there
are more than 20. In these alleles, entire segments
of the RHCE gene have been substituted into RHD.
ese red blood cells will agglutinate with polyclonal
serum, but fail to react with monoclonal antibodies
raised specically against the extracellular loops coded
by the missing exons. Although certain RHD exons
will amplify with standard RHD probes, not all will
(because they are missing), and this allows them to
be distinguished from true RhD positive individuals
using genetic rather than serological tests.
ese RHD/CE hybrids are usually known as “par-
tial D” alleles. e changes usually aect a long string
of amino acids which is always located on the ery-
throcyte surface. e protein is altered so dramati-
cally in these external “antigenic” portions that it is
not recognized by anti-D and these individuals are
prone to true RhD isoimmunization if exposed to nor-
mal RhD positive red cells. In the majority of cases,
women with these alleles should be treated like RhD
negative women and oered RhD negative blood if it
is required and anti-D if the fetus is possibly or de-
nitely RhD positive. It is very important that genetic
testsinthisgroupdonotfalselyclassifythemasRhD
positive and a variety of strategies are in place in most
laboratories to prevent this happening. e most com-
mon European partial D variant is DNB, caused by a
missense mutation, which alters one amino acid in the
sixth extracellular loop of the protein.
e second group of RHD variants is known as
“weak D.” e changes within these alleles substi-
tute amino acids in the transmembranous portions
of the protein. e surface antigenic sites are unal-
tered; however, integration of the weak D protein into
thecellmembraneishinderedorrenderedunsta-
ble, eectively reducing the number of RHD antigenic
sites expressed per red cell. e eect is quantitative
rather than qualitative. Blood from these individuals
will eventually show agglutination with anti-D if given
more time and assisted by the addition of anti-human
globulin reagent. Weak D type 1 is the most common
European weak D variant and is caused by a single mis-
sense mutation at amino acid 270. Most women with
weakDvariants(type1–3)canbegivenRhDpositive
bloodanddonotneedprophylacticanti-D,although
there are a few exceptions.
e term “Du” was previously applied to variants
of RHD. In view of the complexity of the situation,
and the consequences of treating women as RhD posi-
tive when in fact they carry a variant which puts them
at risk of isoimmunization against RhD, it is recom-
mended that advice is taken from the laboratory per-
forming the serological and molecular tests in each
case where a variant is identied.
Other red cell antibodies
Table 6.10 lists some of the other red cell antigens,
which have been documented to be the target of mater-
nal antibodies, resulting in hemolytic disease. ose
highlighted are the most signicant from a clinical
perspective. Certain red cell antibodies never cause
hemolysis. Regional blood transfusion services will
advise where rare antibodies are discovered on ante-
natal screening. 85
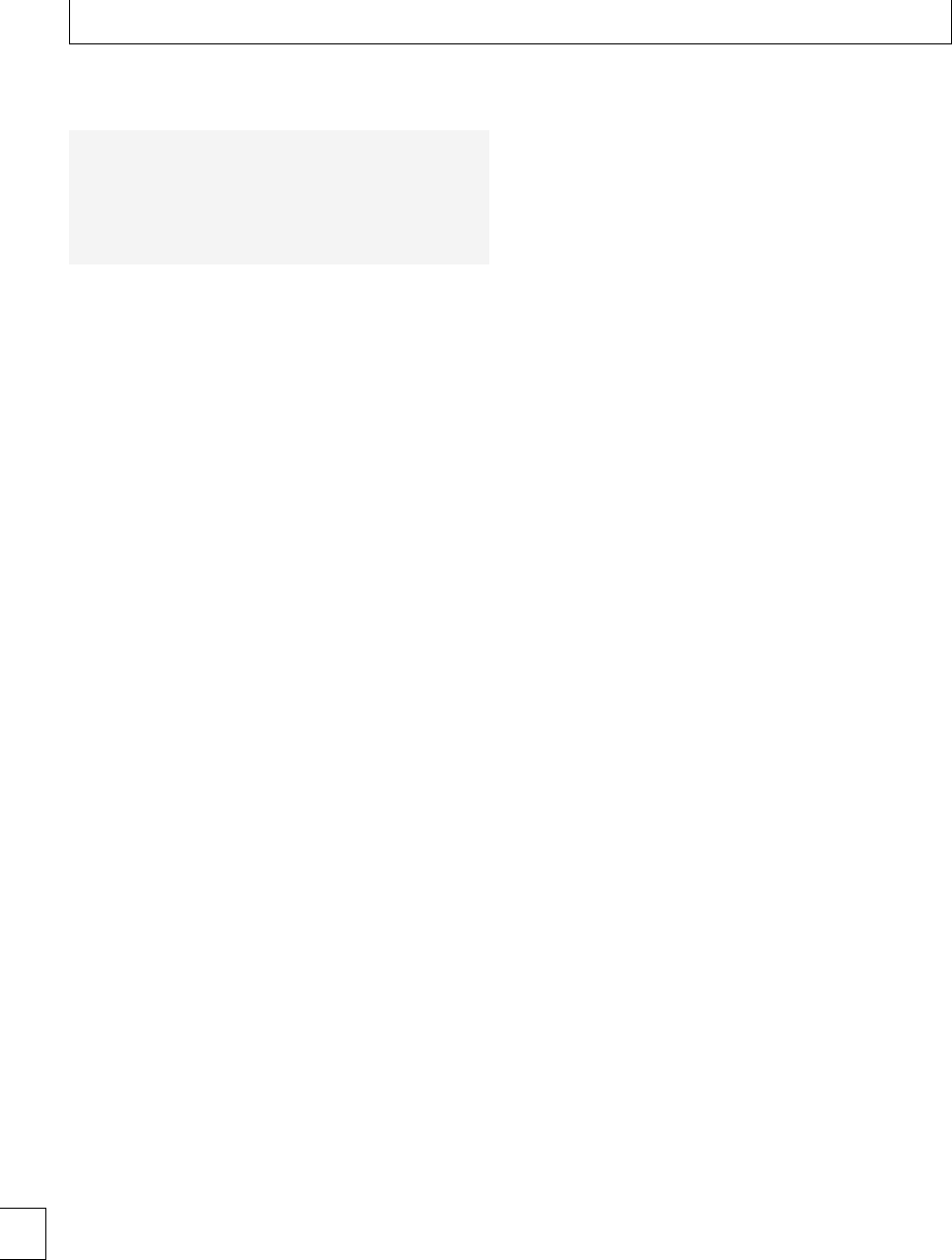
Section 2. Feto-maternal alloimmune Syndromes
Table 6.10 Other red cell antigens implicated in
fetal/neonatal hemolytic disease
Kell
Rhesus c,CandE
AandB
Fya(Duffy) and Fyb
Jka(Kidd) and Jkb
S, M and U
Lua
Isoimmunization against the Kell antigen deserves
special mention. Although there are four separate Kell
antigens, Kell 1causes most concern. Anti-Kell1anti-
bodies are the second most common cause of fetal
immune mediated hemolysis and early onset anemia
and hydrops have been well documented. Nine out of
10 of the general population are Kell1negative and
only1in20babiesofKell
1negative women are Kell1
positive. e Kell antigen is expressed on red cell
progenitorsinthebonemarrowanditisviathese
cells that anti-Kell1antibodies are able to suppress
hematopoiesis, as well as causing hemolysis. is made
ante-natal surveillance with amniocentesis unreliable
because ⌬OD450 of amniotic uid acts only as a surro-
gate for hemolysis and cannot estimate the impact that
the antibodies have on erythropoeisis. Fortunately, this
problem is not shared by MCA PSV, which is used in
exactly the same way as in RhD isoimmunization. In
approximately half of all cases of Kell isoimmuniza-
tion the cause is a previous blood transfusion where
cross-matching did not take account of Kell status
of the woman, or donor. e remainder result from
FMH occurring at the delivery of a previous Kell posi-
tive baby. Absolute levels of anti-Kell antibodies are
less useful in the prediction of disease severity. Non-
invasive pre-natal diagnosis (NIPD) for fetal Kell sta-
tus is available through the BTS laboratory in Bristol
and is particularly helpful because most Kell positive
individuals are heterozygous. NIPD is also possible for
the Rhesus c and E antigens, although currently rela-
tively few tests have been performed, when compared
with RhD, meaning that the degree of diagnostic cer-
tainty is less. Anything more than mild disease is very
unlikely with RhE antibodies.
Women who have the blood group O quite com-
monlyhaveantibodiestotheA-andB-antigens,
although these are more likely to be of the IgM class
which does not cross the placenta. Anti-A and anti-
B IgGs can reach the fetal circulation, but only mild
hemolysisisthegeneralrulefortworeasons.Firstly,
these antigens are expressed on a wide variety of cell
types, eectively diluting their eect on red blood
cells. Secondly, cell-surface expression is incomplete
during gestation and develops gradually, thus limit-
ing the risk before birth. Jaundice caused by ABO
incompatibility is usually mild and readily treated with
phototherapy.
The future
e successes of recent years have not brought research
and progress in the prevention and management of
RhD hemolytic disease to a close. Although polyclonal
anti-D is a safe product, it is pooled from various
donors and anxieties about viral and prion disease
transfection continue. e infection of hundreds of
Irish women with hepatitis C in 1977–78 following the
administration of contaminated anti-D illustrates this
pointalltoowellandcurrently4outof10womenin
theUKreceivinganti-DwillinfactbecarryingaRhD
negative fetus. e anti-D for these women is unneces-
sary, unpleasant, expensive, and not without a degree
of risk.
Limiting the administration of anti-D to only those
RhD negative women carrying a RhD positive fetus is
a worthy goal. e techniques used for non-invasive
pre-natal fetus RhD testing (see above) are time con-
suming and expensive, although very accurate. Appli-
cation of this technology to all pregnant RhD nega-
tive women is impractical. Mass screening requires an
automated test, and robotic systems have been devel-
oped and tested recently with very promising results.
Results from a Bristol study have given a detection
rate for fetal RhD positive status of 99.7% for a false
positive rate of 2%.5ese false positives represent a
group of RhD negative women who would continue to
receive anti-D unnecessarily, but 2% nevertheless is a
signicant improvement on 38%. Of greater concern
are the false negative results (i.e. failure to detect fetal
RhD positivity) of which there were only 3 cases from
nearly 1200 RhD positive pregnancies. Although these
women would be at a three fold greater risk of isoim-
munization because they would not receive ante-natal
prophylactic anti-D (they would still receive post-natal
anti-D), mathematical modeling shows that this actu-
ally equates to only one extra case of hemolytic disease
in 86 000 future pregnancies.
An alternative approach to improve on the
safety of anti-D prophylaxis is to use recombin-
ant monoclonal antibodies (mAb) produced from
hybridoma or human B-cell lines, instead of polyclonal
86

Chapter 6. Red cell alloimmunization
antibodies collected from human serum. A number of
these cell lines exist and progress to date has recently
been summarized by Kumpel.17 Results with some of
the mAb are encouraging and D-immunization can
be prevented in RhD negative volunteers transfused
with RhD positive cells. A “clean” and eective
recombinant anti-D mAb may be on the horizon, but
hurdles still exist, not least obtaining ethical approval
for large-scale trials.
More distant are further exciting possibilities.18
Mutated recombinant anti-D monoclonal antibodies
have been designed and produced, which are able to
bind to the RhD antigen but have a much lower an-
ity for the Fc␥receptor on macrophages than nor-
mal anti-D.19 ese mutated antibodies would displace
endogenous anti-D from its binding sites on the RhD
antigen. Complement mediated lysis, hemolysis and
phagocytosis could all be reduced. ere are many dif-
culties to overcome. e lifespan of these antibodies
islimitedandveryhighandfrequentmaternaladmin-
istrations might be needed for transplacental transfer
to maintain sucient levels in the fetus.
A welcome move in neonatal care would be the
avoidancealtogetherofexchangetransfusion.e
use of IVIG seems to have made some headway
with this however a further option is close at hand.
Competitive heme oxygenase inhibitors, such as tin-
mesoporphyrin, have recently undergone phase III
trials and are already available for certain conditions.
is structural analog of heme competitively blocks
heme-oxygenase, a rate limiting enzyme in bilirubin
production. Heme is le unaltered to be excreted in
bile. It does not pass through the blood–brain barrier
and does not accumulate in tissues. Several random-
ized trials have conrmed that these substances can
prevent and block jaundice progression in the new-
born. In the future these drugs may result in a reduc-
tion in the need for phototherapy, and exchange trans-
fusion for HDN may become a procedure conned to
the history books.
87

Section 2. Feto-maternal alloimmune Syndromes
References
1. Crowther C, Middleton P. Anti-D administration aer
childbirth for preventing Rhesus alloimmunisation.
Cochrane Database of Systematic Reviews 1997 (2)
CD000021.
2. Royal Institute for Clinical Excellence. Guidance on
the use of routine antenatal anti-D prophylaxis.
Guideline no. 22. RCOG Press, 2002.
3. Jabara S, Barnhart KT. Is Rh immune globulin needed
in early rst-trimester abortion? A review. American
Journal of Obstetrics and Gynecology 2003; 188:
623–627.
4. National Institute for Clinical Excellence. Guidance on
the use of routine antenatal anti-D prophylaxis for
RhD-negative women. Technology Appraisal
Guidance no. 41. www.nice.org.uk, 2002.
5. Finning K, Martin P, Summers J et al.Eectof
high-throughput RHD typing of fetal DNA in maternal
plasma: on use of anti-RHD immunoglobulin in RHD
negative pregnant women: prospective feasibility
study. British Medical Journal 2008; 336: 816–818.
6. Bennett PR, Le Van Kim C, Colin Y et al. Prenatal
determination of fetal RhD type by DNA
amplication. New England Journal of Medicine 1993;
329: 607–610.
7. Sikkel E, Vandenbussche FPHA, Oepkes D et al.
Amniotic uid ⌬OD450 values accurately predict
severe fetal anaemia in D-alloimmunisation. Obstetrics
and Gynecology 2002; 100: 51–57.
8. Nicolaides KH, Soothill PW, Clewell W, Rodeck CH.
Rh disease: intravascular fetal blood transfusion by
cordocentesis. Fetal erapy 1986; 1: 185–192.
9. Lo YMD, Corbetta N, Chamberlain PF et al. Presence
of fetal DNA in maternal plasma and serum. Lancet
1997; 350: 485–487.
10. Chitty LS, van der Schoot, Hahn S, Avent ND. SAFE –
e special non-invasive advances in fetal and
neonatal evaluation network: aims and achievements.
Prenatal Diagnosis 2008; 28: 83–88.
11. Daniels G, Finning K, Martin P, Summers J. Fetal
blood group genotyping. Present and future. Annals
of e New York Academy Science 2006; 1075:
88–95.
12. Mari G, Deter RL Carpenter RL et al.Forthe
collaborative group for the assessment of the blood
velocity in anaemic fetuses. Non-invasive diagnosis by
Doppler ultrasonography of fetal anaemia due to
maternal red-cell alloimmunisation. New England
Journal of Medicine 2000; 342: 9–14.
13. Pereira L, Jenkins TM, Berghella V. Conventional
management of maternal red cell alloimmunisation
compared with management by Doppler assessment of
middle cerebral artery peak systolic velocity. American
Journal of Obstetrics and Gynecology 2003; 189:
1002–1006.
14. RumaMS,MoiseKJ,KimEet al. Combined
plasmapheresis and intravenous immune globulin for
the treatment of severe maternal red cell
alloimmunisation. American Journal of Obstetrics and
Gynecology, 2007; 196: 138.e1–138.e6.
15. Jackson JC. Adverse events associated with exchange
transfusion in healthy and ill newborns. Paediatrics
1997; 99:E7.
16. Alcock GS, Liley H. Immunoglobulin infusion for
isoimmune haemolytic jaundice in neonates.
Cochrane Database of Systematic Reviews 2002; (3):
CD003313.
17. Kumpel BM. Ecacy of RhD monoclonal antibodies
in clinical trails as replacement therapy for
prophylactic anti-D immunoglobulin: more questions
than answers. Vox Sanguinis 2007; 93: 99–111.
18. Urbaniak SJ. Noninvasive approaches to the
management of RhD haemolytic disease of the fetus
and newborn. Transfusion 2008; 48:2–5.
19. Nielson LK, Green TH, Sandlie I et al.Invitro
assessment of recombinant, mutant anti-D
immunoglobulin G devoid of haemolytic activity for
treatment of on-going haemolytic disease of the fetus
and newborn, Transfusion 2008; 48: 12–19.
88

Section
3
Thromboembolism and
anticoagulation

Section 3 Thromboembolism and anticoagulation
Chapter
7Acute management of suspected
thromboembolic disease in pregnancy
Andrew J. Thomson and Ian A. Greer
Introduction
Antenatal and postnatal venous thromboembolism
(VTE)isaround10and25timesmorecommon
respectively, than in non-pregnant women of the
same age and is the major cause of direct maternal
mortality in the developed world. European studies
have consistently found the pregnancy-related VTE
mortality to be 8.5 – 14 per million live births.1,2
In the United Kingdom, sequential reports from
Condential Enquiries into Maternal Deaths have
demonstrated that VTE remains the main direct cause
of maternal death and have highlighted failures in
obtaining objective diagnoses and employing adequate
treatment.3Fatal pulmonary embolism (PE) arises
from deep venous thrombosis (DVT), many cases of
which are not recognized clinically and are only iden-
tied at post-mortem following a maternal death.3
e subjective, clinical assessment of DVT and PE is
particularly unreliable in pregnancy and a minority of
women with clinically suspected VTE has the diag-
nosisconrmedwhenobjectivetestingisemployed.
4
Acute VTE should be suspected during pregnancy
in women with symptoms and signs consistent with
possible VTE,4–6 particularly if there are other risk
factors for VTE (see Tables 8.1 and 8.2).7–11 e symp-
toms and signs of VTE include leg pain and swelling
(usually unilateral), lower abdominal pain, low grade
pyrexia, dyspnoea, chest pain, haemoptysis and
collapse.
Epidemiology of VTE during pregnancy
Virchow’s triad for VTE consists of alterations in nor-
mal blood ow (stasis), trauma or damage to the vas-
cular endothelium and alterations in the constitution
of blood (hypercoagulability), and describes the three
broad categories of factors that contribute to throm-
bosis. During normal pregnancy, hypercoagulability
results from increases in the levels of factor VIII and
brinogen, reduction in protein S levels, a resistance
to activated protein C and impaired brinolysis. Stud-
ies assessing blood ow velocity in the lower limbs in
pregnancy have shown an extensive reduction in ow
of up to 50% by 29 weeks’ gestation, reaching its nadir
at 36 weeks. e changes in both blood ow velocity
and coagulation factors may persist for up to 6 weeks
aer delivery. e third component of Virchow’s triad,
damage to the vascular endothelium, arises during the
course of vaginal or abdominal delivery – whilst VTE
can occur at any stage of pregnancy, the puerperium is
the time of greatest risk.
Almost 90% of cases of DVT occur in the le leg
in pregnancy, in contrast to the non-pregnant situ-
ation, where only 55% occur on the le.4is may
reect compression of the le iliac vein by the right
iliac artery and the ovarian artery, which cross the vein
only on the le side. Over 70% of DVTs in pregnancy
arise in the iliac and femoral veins rather than the calf
veins, whereas in non-pregnant patients only about 9%
arise in the ilio-femoral area. is is of importance
since ileo-femoral DVTs are more likely to result in PE
than are calf vein thromboses.
Assessment and diagnosis of acute
VTE in pregnancy
Clinical diagnosis of both DVT and PE is unreliable.
In non-pregnant patients where DVT is suspected, the
diagnosis is conrmed in about 20–30% of cases when
objective testing is performed. During pregnancy, clin-
ical assessment is even more unreliable since many of
the symptoms and signs of VTE, such as leg swelling,
91
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 3. Thromboembolism and anticoagulation
Table 7.1 – Symptoms and signs of VTE in pregnancy
Deep venous thrombosis
rleg pain or discomfort
rtenderness
rswelling
rlower abdominal pain
rincreased temperature and oedema
relevated white cell count
Pulmonary thromboembolism
rchest pain
rdyspnoea
rhaemoptysis
rtachycardia
rfocal signs in the chest
rraised jugular venous pressure
rcollapse
rabnormalities on the chest X-ray
rsymptoms and signs associated with DVT
chest pain and dyspnea, are commonly found in nor-
mal pregnancy (Table 7.1). As a consequence, the accu-
racy of clinical diagnosis falls to about 8% for DVT and
to less than 5% for suspected PE.4–6
It is therefore essential that objective testing is per-
formed in women with suspected VTE. Failure to iden-
tifyVTEwillplacethemother’slifeatrisk,whilst
unnecessary treatment is associated with risks, incon-
venience and costs during the pregnancy and may also
have implications for her future health care (including
future use of oral contraception and hormone replace-
menttherapy,andthromboprophylaxisinfuturepreg-
nancies). ere are no well-designed large clinical tri-
als to support the management of suspected VTE in
pregnancy and guidelines are therefore empirical and
based on extrapolation from studies performed in
non-pregnant patients.1,2,4
If there is a delay in obtaining objective tests, the
woman should be commenced on anticoagulant ther-
apy, unless contraindicated, until testing can be per-
formed.
Diagnosis of DVT
Compression Duplex ultrasound of the entire prox-
imal venous system is the optimal initial diagnostic
test for DVT in pregnancy. If the initial ultrasound
shows an abnormality in the popliteal or femoral veins,
the diagnosis of proximal DVT is conrmed and anti-
coagulant treatment should be commenced and con-
tinued. A normal ultrasound does not exclude a calf
DVT and therefore, if ultrasound is negative and a high
level of clinical suspicion exists, the patient should
remain anticoagulated and the ultrasound repeated in
one week or an alternative diagnostic test employed. If
repeat testing is negative, anticoagulant treatment can
be discontinued.4
For the diagnosis of iliac vein thrombosis, which
may present with back pain and/or swelling of
the entire limb, pulsed Doppler, magnetic reso-
nance venography or conventional contrast venogra-
phy should be considered.
Diagnosis of PE
When a woman with suspected PE is haemodynamic-
ally stable, a chest X-ray (CXR) should be performed.
CXR may identify other pulmonary disease such as
pneumonia, pneumothorax or lobar collapse. Whilst
the CXR is normal in over half of pregnant patients
with objectively proven PE, abnormal features caused
by PE include atelectasis, eusion, focal opacities,
regional oligaemia or pulmonary edema. e radia-
tion dose to the fetus from a CXR performed at any
stageofpregnancyisnegligible.IftheCXRisabnor-
mal, ventilation perfusion (V/Q) scanning is unreli-
able and CT pulmonary angiography (CTPA) should
be performed.4–6
If the CXR is normal, the authors recommend that
bilateral Doppler ultrasound leg studies should be per-
formed. A diagnosis of DVT may indirectly conrm a
diagnosis of PE and since anticoagulant therapy is the
same for both conditions, further investigation is not
usually necessary. is would limit the radiation doses,
particularly associated with CTPA, given to the mother
and her fetus.4
e choice of technique for denitive diagnosis
(V/Q scan or CTPA) may depend on factors such as
local availability and guidelines, and should usually
be made aer discussion with a radiologist. During
pregnancy the ventilation component of the V/Q scan
can oen be omitted, thereby minimizing the radia-
tion dose for the fetus. In the United Kingdom, the
British oracic Society5recommends CTPA as rst-
line investigation for non-massive PE in non-pregnant
patients. is technique has potential advantages over
radionuclide (V/Q) imaging including better sensitiv-
ity and specicity, (at least in non-pregnant patients)
and a lower radiation dose to the fetus (see section
below). In addition it can identify other pathology
such as aortic dissection. e main disadvantage of
CTPAisthehighradiationdosetothematernalbreasts
associated with an increased lifetime risk of develop-
ing breast cancer. is is particularly relevant when it is
92

Chapter 7. Acute venous thromboembolism
known that only around 5% of such investigations will
have a positive result. In addition, conventional CTPA
may not identify small peripheral PEs, although this
is overcome by the latest multidetector row spiral CT
techniques. In contrast to CTPA, V/Q scanning may
be delayed because of availability of isotope. Despite
these potential advantages of CTPA, many authorities,
including the authors, continue to recommend V/Q
scanning, where possible, as rst-line investigation in
pregnancy because of its high negative predictive value
in this situation, its substantially lower radiation dose
to pregnant breast tissue, and because most pregnant
womenintheUKwillnothaveco-morbidpulmonary
pathology.4,12
Radiation exposure associated with
diagnostic tests
CTPA delivers less radiation to the fetus than V/Q
scanning during all trimesters of pregnancy. It has
been estimated that the risk of fatal cancer to the age
of 15 years is ⬍1/1,000,000 aer in utero exposure
to CTPA and 1/280 000 following a perfusion scan.
While CTPA is associated with a lower risk of radia-
tion for the fetus, this must be oset by the relatively
high radiation dose (20 mGy) to the mother’s thorax
and in particular breast tissue. e delivery of 10 mGy
of radiation to a woman’s breast increases her life-
time risk of developing breast cancer. It has been esti-
mated that the increased risk is 13.6% (background
risk 1/200), a gure that has been cited widely (4,
12). More recently, authorities have suggested that this
risk is an overestimate. Nevertheless, breast tissue is
especially sensitive to radiation exposure during preg-
nancy, and it therefore seems sensible to recommend
that lung perfusion scans should be considered the
investigation of rst choice for young women, espe-
cially if there is a family history of breast cancer or
the patient has had a previous chest CT scan. Radia-
tion exposure from pulmonary angiography is approx-
imately0.5mSvtofetus,and5to30mSvtomother.
D-dimer testing in pregnancy
Outwith pregnancy, a normal plasma D-dimer level
has been shown to have excellent negative predic-
tive value in patients with a low clinical probability
score for VTE. However levels increase physiologically
throughout pregnancy, becoming elevated at term and
in the post-natal period in most healthy pregnant
women. Furthermore, D-dimer levels are increased if
there is a concomitant problem such as pre-eclampsia,
preterm labour, and placental abruption. us the
probability of a negative result is lower and objec-
tive testing is more oen required. For this reason
guidelines produced by the Royal College of Obste-
tricians and Gynaecologists in the United Kingdom,4
do not recommend that D-dimer levels are evaluated
in pregnant women with suspected VTE. In contrast,
the European Society of Cardiology13 recommend that
D-dimer levels should be measured, as a proportion of
patients will have a normal result and be able to avoid
unnecessaryimaging.Itshouldbenoted,however,that
although the SimpliRED test has been reported to have
a negative predictive value of 100% in pregnancy, false
negative results have been reported.
Thrombophilia testing in acute VTE
pregnancy
Almost half of all women who have an episode of
VTEinpregnancy,willhaveanunderlyingherita-
ble or acquired thrombophilia.14 e prevalence rates
forthrombophiliasinEuropeanpopulationsisshown
in Table 12.2 and the relative risk of each condition,
shown by metaanalysis, is shown in Table 7.2. Per-
forming a thrombophilia screen in the acute stages of
thrombosis may give misleading results and is not rou-
tinely recommended. Levels of antithrombin, protein
C and protein S may fall, particularly if thrombus is
extensive. In addition, protein S levels fall in normal
pregnancy and an acquired activated protein C resis-
tance is found with the APC sensitivity ratio test in
around 40% of pregnancies, due to the physiological
changes in the coagulation system. Clearly, genotyp-
ing for factor V Leiden and prothrombin G20210A will
not be aected by pregnancy or thrombus. Whilst the
results of a thrombophilia screen will not inuence the
immediate management of acute VTE, they might pro-
vide information that can inuence the duration and
intensity of anticoagulation, such as when antithrom-
bin deciency or antiphospholipid syndrome is iden-
tied.
Initial treatment of VTE in pregnancy
Before anticoagulant therapy is initiated, blood should
be taken for a full blood count and coagulation screen.
Urea, electrolytes and liver function tests should also 93
Section 3. Thromboembolism and anticoagulation
Table 7.2. Risk of pregnancy associated VTE in women with
underlying thrombophilia (14)
Relative Risk of
Thrombophilic defect thrombosis
Factor V Leiden (heterozygote)8.32
Factor V Leiden (homozygote)34.4
Prothrombin 20210A (heterozygote)6.8
Prothrombin 20210A (homozygote)26.4
Protein C deficiency 4.76
Protein S deficiency 3.19
Antithrombin deficiency 4.69
be checked to exclude renal or hepatic dysfunction,
which are cautions for anticoagulant therapy.
e treatment of VTE in pregnancy is heparin.
Vitamin K antagonists are rarely employed in this
setting as they cross the placenta and are associated
with increased pregnancy loss, a specic embryopa-
thy and other abnormalities in the rst trimester, as
well as fetal haemorrhagic complications and cen-
tral nervous system anomalies at any stage of preg-
nancy. Although for many years, unfractionated hep-
arin (UFH) was the standard anticoagulant used dur-
ing and outwith pregnancy, it has now largely been
replaced by low molecular weight heparin (LMWH).
Meta-analyses of randomised controlled trials (RCTs)
in non-pregnant patients indicate that LMWHs are
more eective and are associated with a lower risk
of haemorrhagic complications and lower mortality
than unfractionated heparin in the initial treatment
of DVT. A meta-analysis of RCTs has shown equiva-
lent ecacy of LMWH to unfractionated heparin in
the initial treatment of PE. A systematic review of
LMWH in pregnancy has conrmed its ecacy and
safety in the management of acute thrombosis and
in the provision of thromboprophylaxis. Furthermore,
compared with UFH, LMWH is associated with a sub-
stantially lower risk of heparin-induced thrombocy-
topenia, haemorrhage and osteoporosis.1,2,4,15–17 Nei-
ther UFH nor LMWH cross the placenta and both are
safe for breast feeding.
Whilst several LMWH preparations are available,
most experience currently exists with enoxaparin, dal-
teparin and tinzaparin. In non-pregnant patients with
acute VTE, LMWH is usually administered in a once
daily dose. In view of recognised alterations in the
pharmacokinetics of dalteparin and enoxaparin dur-
ing pregnancy, a twice daily dosage regimen is recom-
mended for these LMWHs in the treatment of VTE
in pregnancy, (enoxaparin 1mg/kg twice daily; dal-
teparin 100 units/kg twice daily). Preliminary bio-
chemical data suggest that once daily administration
of tinzaparin (175 units/kg) may be appropriate in the
treatment of VTE in pregnancy. Whichever, prepara-
tion of LMWH is employed, the woman should be
taught to self-administer the drug by subcutaneous
injection, allowing further management on an out-
patient basis until delivery.4
In the very early management of DVT, the leg
should be elevated and a graduated elastic compres-
sion stocking applied to reduce oedema. Once full anti-
coagulation has been commenced, the woman should
be encouraged to mobilize whilst wearing compres-
sion hosiery as this has been shown to reduce pain and
swelling in the aected leg. Studies in non-pregnant
patients have shown that early mobilization, with com-
pression therapy, does not increase the likelihood of
developing PE and helps prevent the development of
post-thrombotic syndrome. For patients with persist-
ing leg oedema aer DVT, class II compression hosiery
is more eective than class I stockings. Where DVT
threatens leg viability through venous gangrene, the
leg should be elevated, anticoagulation given and con-
sideration given to surgical embolectomy or throm-
bolytic therapy.4
Monitoring of LMWH therapy
Experience indicates that satisfactory anti-Xa levels
(peak anti-Xa activity, 3 hours post-injection, of 0.5–
1.2 u/ml) are obtained using a weight-based regimen
and monitoring of anti-Xa is not routinely required
in patients with VTE on therapeutic doses of LMWH,
particularly as there are concerns over the standardiza-
tion and accuracy of anti-Xa monitoring. ere may
be a case for monitoring levels at extremes of body
weight (⬍50 kg and ≥90 kg), and women with other
complicating factors including renal disease and recur-
rent VTE.4,16
Guideline documents from North America2rec-
ommend that routine platelet count monitoring is
not required in obstetric patients who have received
only LMWH as heparin induced thrombocytopenic
thrombosis is not a feature in pregnancies managed
exclusively with LMWH. If unfractionated heparin
is employed, or if the obstetric patient is receiving
LMWH aer rst receiving unfractionated heparin, or
if she has received unfractionated heparin in the past,
94

Chapter 7. Acute venous thromboembolism
theplateletcountshouldideallybemonitoredevery2–
3 days from day 4 to day 14, or until heparin is stopped,
whichever occurs rst.
Maintenance treatment of VTE
Women with ante-natal VTE can be managed with
subcutaneous LMWH for the remainder of the preg-
nancy using LMWH administered subcutaneously. It
is our practice to continue with the initial dose regi-
men throughout pregnancy despite the pregnancy-
associated weight gain, (since LMWH does not cross
the placenta and therefore the weight of the feto-
placental unit is not relevant). If LMWH therapy
requires monitoring, for example in extremes of body
weightorrenalimpairment,theaimistoachievea
peak anti-Xa activity, three hours post-injection of
0.5 –1.2 u/ml.
It is not yet established whether the initial dose of
LMWHcanbereducedtoanintermediatedoseaer
an initial period of several weeks of therapeutic anti-
coagulation, although this practice has been success-
fully employed in some centres. Outwith pregnancy in
patients with underlying malignancy, a reduction in
dosehasbeenshowntobesafeaer4weeksofther-
apeutic anticoagulation. Although there have been no
studies directly comparing these two types of dosing
strategies in pregnant women, this type of modied
dosing regimen may be useful in pregnant women at
increased risk of bleeding or osteoporosis.18
Management at the time of delivery
For women on therapeutic anticoagulation, a planned
delivery,eitherthroughinductionoflabourorelec-
tive caesarean section, allows accurate timing of events
and minimizes the risk of the woman having to deliver
on full anticoagulation. e dose of LMWH should
be reduced to a once daily thromboprophylactic dose
on the day before induction of labour or caesarean
section. When a woman presents whilst on a thera-
peutic, twice daily regimen of LMWH, regional tech-
niques should not usually be employed for at least 24
hours aer the last dose of LMWH. LMWH should
not be given for at least four hours aer the epidu-
ral catheter has been removed and the cannula should
not be removed within 12 hours of the most recent
injection.4
Fordeliverybyelectivecaesareansection,thetreat-
mentdosesofLMWHshouldbeomittedfor24
hours prior to surgery. A thromboprophylactic dose of
LMWH (enoxaparin 40mg; dalteparin 5000iu; tinza-
parin 75 iu/kg) should be given by three hours post-
operatively (⬎4 hours aer removal of the epidu-
ral catheter, if appropriate), and the treatment dose
recommenced that evening. ere is an increased risk
of wound haematoma following caesarean section with
both unfractionated heparin and LMWH of around
2%. For this reason, wound drains should be consid-
ered at caesarean section, and the skin incision should
ideally be closed with staples or interrupted sutures to
allow easy drainage of any haematoma.
If the thrombosis occurred in the last week of preg-
nancy, consideration should be given to the use of
unfractionated heparin (since it can be relatively eas-
ily reversed using protamine sulfate and has a short
duration of action). If spontaneous labour occurs in
women receiving therapeutic doses of subcutaneous
unfractionated heparin, careful monitoring of the anti-
coagulant eect by measuring the activated partial
thromboplastin time (APTT) is required. Subcuta-
neous unfractionated heparin should be discontinued
12 hours before and intravenous unfractionated hep-
arin stopped 6 hours before induction of labour or
regional anaesthesia. It should be noted however that
theaPTTislessreliableinpregnancyduetoincreased
levels of FVIII and heparin binding proteins, which
can lead to an apparent heparin resistance.4,18
Postpartum anticoagulation and
duration of anticoagulation therapy
In the United Kingdom, it is recommended that in
non-pregnant patients, anticoagulant therapy should
be continued for 6 weeks for calf vein thrombosis
and 3 months for proximal DVT or PE when VTE
has occurred in relation to a temporary risk factor,
and 6 months for a rst episode of idiopathic VTE.4
e presence of ongoing risk factors and the safety of
LWMH have led authorities to propose that anticoag-
ulant therapy should be continued for the duration of
the pregnancy and until at least six weeks postpartum
and to allow a total duration of treatment of at least
3 months. Both heparin and warfarin are satisfactory
for use postpartum – in our experience most women
prefer to use LMWH (which can be used with once
daily dosing postpartum) because they have become
accustomed to its administration, and they appreciate
the convenience of not having to attend clinics to have
their INR checked. Before discontinuing treatment
the ongoing risk of thrombosis should be assessed 95
Section 3. Thromboembolism and anticoagulation
Transfer to intensive therapy area
•Emergency call to multi-disciplinary resuscitation team
•Oxygen administered
•Heparinise with intravenous unfractionated heparin
•IV fluids and inotropic support
•Inform on-call obstetric team immediately for consideration of early delivery
If persistent hypotension (SBP < 90mmHg), consider:-
•Thrombolysis
If thrombolysis is contraindicated, consider
•Percutaneous catheter fragmentation
•Surgical embolectomy
Diagnosis made by emergency CTPA or echocardiogram
If the patient becomes periarrest at any stage, consider thrombolysis
without imaging.
CTPA confirms significant PE
or
Cardiac echo confirms RV dilatation/dysfunction
Negative investigations:
Search for other diagnosis
Figure 7.1. Management of women
with clinically suspected massive PTE
including a review of personal and family history of
VTE and any thrombophilia screen results. rom-
bophilia screening should be discussed and arranged
if required.
Neither heparin (unfractionated or LMWH) nor
warfarin is contraindicated in breastfeeding. ere are
little published data on whether LMWHs are secreted
in breast milk, although extensive experience of enoxa-
parin in the puerperium reports no problems during
breastfeeding and other heparins are known not to
cross the breast. Furthermore, neither unfractionated
heparin nor LMWH are orally active and no eect
would therefore be anticipated in the fetus.
e post-thrombotic syndrome is a common com-
plication following DVT.18 It is found in over 60%
of cases followed up over a median of 4.5 years. It is
characterized by chronic persistent leg swelling, pain,
a feeling of heaviness, dependant cyanosis, telangiec-
tasis, chronic pigmentation, eczema, associated vari-
cose veins and in some cases lipodermatosclerosis,
and chronic ulceration. Symptoms are made worse
by standing or walking and improve with rest and
recumbancy. e syndrome is more common where
there is a recurrent DVT, with obesity and where
there has been inadequate anticoagulation. It is rec-
ommended that graduated elastic compression stock-
ings (class II) should be worn on the aected leg for
two years aer the acute event to reduce the risk of
post-thrombotic syndrome. is recommendation is
based upon studies in non-pregnant patients where
such therapy reduces the incidence of post throm-
botic syndrome from 23% to 11% over 2 years. Grad-
uated elastic compression stockings will improve the
microcirculation by assisting the calf muscle pump,
reducing swelling and reux, and reducing venous
hypertension.4
96

Chapter 7. Acute venous thromboembolism
Management of massive,
life-threatening PE
Massive, life-threatening PE may be dened as embo-
lus associated with haemodynamic compromise (a sys-
tolic blood pressure ⬍90 mmHg or a drop in sys-
tolic blood pressure of ≥40mmHg from baseline
for a period ⬎15 minutes), not otherwise explained
by hypovolaemia, sepsis or new arrhythmia. is is
an obstetric and medical emergency and hospitals
should have in place guidelines for the management
of non-haemorrhagic obstetric shock (see gure 7.1).
e collapsed, shocked pregnant woman needs to be
assessed by a multi-disciplinary resuscitation team of
experienced clinicians including senior obstetricians,
physicians and radiologists, who should decide on
an individual basis whether a woman receives intra-
venous unfractionated heparin, thrombolytic therapy
or thoracotomy and surgical embolectomy.
Oxygen should be administered and the circula-
tion supported using intravenous uids and inotropic
agents if required. Intravenous unfractionated heparin
is the traditional method of heparin administration
in acute VTE and remains the preferred treatment in
massive PE because of its rapid eect and extensive
experience of its use in this situation. e diagnosis
should be established using either portable echocar-
diogramorCTPAperformedwithin1hourofpresen-
tation.
In massive life-threatening PE with haemody-
namic compromise there is a case for considering
thrombolytic therapy as anticoagulant therapy will
not reduce the obstruction of the pulmonary cir-
culation. Aer thrombolytic therapy has been given
an infusion of unfractionated heparin can be given.
ere are now a large number of published case
reports on the use of thrombolytic therapy in preg-
nancy, streptokinase being the agent most frequently
employed. Streptokinase, and probably other throm-
bolytic agents, do not cross the placenta. No mater-
naldeathsassociatedwiththrombolytictherapyhave
been reported, and the maternal bleeding complica-
tion rate is approximately 6%, which is consistent with
that in non-pregnant patients, receiving thrombolytic
therapy. Most bleeding events occur around catheter
and puncture sites, and, in pregnant women, from the
genital tract. If the patient is not suitable for thrombol-
ysis or moribund, a discussion with the cardiothoracic
surgeons with a view to urgent thoracotomy should be
had.4
97

Section 3. Thromboembolism and anticoagulation
References
1. Bates SM, Ginsberg JS. How we manage venous
thromboembolism during pregnancy. Blood 2002; 100:
3470–8.
2. Bates SM, Greer IA, Pabinger I et al.Venous
thromboembolism, thrombophilia, antithrombotic
therapy, and pregnancy: American College of Chest
Physicians Evidence-Based Clinical Practice
Guidelines (8th Edition). Chest 2008; 133: 844S–
886S.
3. Lewis, G. ed. e Condential Enquiry into Maternal
and Child Health. Saving mothers’ lives: reviewing
maternal deaths to make motherhood safer 2003–2005.
e seventh report on Condential Enquiries into
Maternal Deaths in the United Kingdom. London:
CEMACH, 2007.
4. Royal College of Obstetricians and Gynaecologists.
romboembolic disease in pregnancy and the
puerperium: acute management. RCOG Green Top
Guideline 2007, No 28.
5. British oracic Society Standards of Care Committee
Pulmonary Embolism Guideline Development Group.
British oracic Society guidelines for the
management of suspected acute pulmonary embolism.
orax 2003; 58: 470–84.
6. Scarsbrook AF, Evans AL, Owen AR, Gleeson FV.
Diagnosis of suspected venous thromboembolic
disease in pregnancy. Clinical Radiology 2006; 61:
1–12.
7. Royal College of Obstetricians and Gynaecologists.
Reducing the risk of thrombosis and embolism during
pregnancy and the puerperium. RCOG Green Top
Guideline 2009, No 37.
8. Jacobsen AF, Skjeldestad FE, Sandset PM. Ante- and
postnatal risk factors of venous thrombosis: a
hospital-based case-control study. Journal of
rombosis and Haemostasis 2008; 6: 905–12.
9. Lindqvist P, Dahlb¨
ack B, Marˆ
s´
al K. rombotic risk
during pregnancy: a population study. Obstetrics and
Gynecology 1999; 94: 595–9.
10. James AH, Jamison MG, Brancazio LR, Myers ER.
Venous thromboembolism during pregnancy and the
postpartum period: incidence, risk factors, and
mortality. American Journal of Obstetrics and
Gynecology 2006; 194: 1311–5.
11. Knight M; on behalf of UKOSS. Antenatal pulmonary
embolism: risk factors, management and outcomes.
BJOG: An International Journal of Obstetrics &
Gynaecology 2008; 115: 453–461.
12. Cook JV, Kyriou J. Radiation from CT and perfusion
scanning in pregnancy. British Medical Journal 2005;
331: 350.
13. e Task Force for the Diagnosis and Management of
Acute Pulmonary Embolism of the European Society
of Cardiology. Guidelines on the diagnosis and
management of acute pulmonary embolism. European
Heart Journal 2008; 29: 2276–2315.
14. Robertson, L. and Wu, O. and Langhorne, P. and
Twaddle, S. and Clark, P. and Lowe, G.D.O. and
Walker,I.D.andGreaves,M.andBrenkel,I.and
Regan, L. and Greer, I.A. rombophilia in pregnancy:
asystematicreview.British Journal of Haematology
2006; 132: 171–196.
15. Greer IA, Nelson-Piercy C. Low-molecular-weight
heparins for thromboprophylaxis and treatment of
venous thromboembolism in pregnancy: a systematic
review of safety and ecacy. Blood 2005; 106: 401–7.
16. omson AJ, Walker ID, Greer IA. Low molecular
weight heparin for the immediate management of
thromboembolic disease in pregnancy. e Lancet
1998; 352: 1904.
17. James AH. Prevention and management of venous
thromboembolism in pregnancy. American Journal of
Medicine 2007; 120: S26–S34.
18. Greer I, Hunt BJ. Low molecular weight heparin in
pregnancy: current issues. British Journal of
Haematology 2005; 128: 593–60.
19. McColl D, Ellison J, Greer IA. et al. Prevalence of the
post thrombotic syndrome in young women with
previous venous thromboembolism. British Journal of
Haematology 2000; 108: 272–274.
98

Section 3 Thromboembolism and anticoagulation
Chapter
8Thromboprophylaxis
Sarah Germain and Catherine Nelson-Piercy
Introduction and epidemiology
Venous thromboembolism (VTE) remains the leading
direct cause of maternal death in the UK. In the lat-
est CEMACH report “Saving Mothers Lives: Review-
ing maternal deaths to make motherhood safer, 2003–
5”1there were 33 deaths from VTE and eight from
cerebral vein thrombosis. Although the absolute num-
bers of both fatal and non-fatal VTE in pregnancy
and the puerperium are small, with an overall inci-
dence of approximately 2 per 1000 births,2,3 many
are preventable. Successive enquiry reports have high-
lighted the need to identify risk factors for VTE early
in pregnancy and ensure adequate thromboprophy-
laxis is employed. e Royal College of Obstetricians
and Gynaecologists have published guidelines regard-
ing thromboprophylaxis covering both the ante-natal
and post-natal periods4,5 and these have recently been
updated.6Despite this, a recent case control study
of 143 cases of ante-natal pulmonary embolism (PE)
in the UK, by the UK Obstetric Surveillance System
(UKOSS), demonstrated that, although nine of the
womenshouldhavereceivedante-natalthrombopro-
phylaxis according to national guidelines, only 33%
had actually done so, and 50% of the six women who
had a PE while on prophylactic low molecular weight
heparin (LMWH) were receiving lower than recom-
mended doses.7
Traditionally, VTE has been considered a compli-
cation of late pregnancy and Post-Cesarean section.
e Condential Enquiries into Maternal Deaths have
shown that this is by no means the case, with two-
thirds of ante-natal fatal pulmonary VTE in 2003–
2005 occurring in the rst trimester, and just over
half of the post-natal deaths from pulmonary VTE
aer vaginal delivery.1AstudyfromtheUSAfound
that 44% of deep vein thromboses DVTs in pregnancy
occurred in the rst trimester,8and a more recent
Spanish study similarly found that 40% of ante-natal
VTE were in the rst trimester.9ese all emphasize
the need for risk assessment pre-pregnancy and insti-
tution of prophylaxis if appropriate in early pregnancy.
Although numerically most VTE occurs ante-natally,
the highest risk per day is during the immediate post-
partum period, and this is demonstrated by a cohort
study from the USA, which showed that the annual
incidence of VTE was ve times higher among post-
partum compared to pregnant women.3
As clinicians we therefore ideally need to iden-
tifywomenatriskpriortoconception,oratleast
early in pregnancy, then establish their level of risk,
and nally initiate an appropriate thromboprophylaxis
regimen, which extends into the puerperium. We also
need to be aware that a woman who starts pregnancy
as “low risk” of VTE may develop or acquire risk
factors as pregnancy progresses, and thromboprophy-
laxis may need to be introduced at that point or aer
delivery.
Pathogenesis and risk factor
assessment
Pregnancyitselfputsallwomenathigherriskof
VTE, with a four to ten-fold increase compared to
an age-matched non-pregnant female population.3,5
is is primarily related to the pro-coagulant changes
that occur during pregnancy to promote hemostasis
post-delivery,2and are evident from early in the rst
trimester. Other components of Virchow’s triad are
also present, namely increased venous stasis and vas-
cular trauma, the latter particularly around the time of
delivery. Superimposed on this background risk, are a
range of additional risk factors which may either pre-
date the pregnancy or develop during the pregnancy or
puerperium, and can be persistent or transient.
99
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 3. Thromboembolism and anticoagulation
Table 8.1 Additional risk factors for VTE in pregnancy and the
puerperium∗
Pre-existing
Age ⬎35 years
Obesity
(BMI
⬎
30 kg/m
2
, either pre-pregnancy or in early
pregnancy)
Parity ≥3
Gross varicose veins
Paraplegia
Sickle cell disease
Smoking
ART
Inflammatory disorders
(e.g. inflammatory bowel disease)
Some medical disorders
(e.g. nephrotic syndrome, certain
cardiac diseases)
Myeloproliferative disorders
(e.g. essential thrombocythemia,
polycythemia vera)
New onset or transient
Ovarian hyperstimulation syndrome
Hyperemesis
Dehydration
Severe infection
(e.g. pyelonephritis)
Long-distance travel
Immobility
(
⬎
4daysbedrest)
Pre-eclampsia
Excessive blood loss/blood transfusion
Surgical procedure in pregnancy or puerperium
(e.g. ERPC,
postpartum sterilization)
Specic to postpartum VTE only
Cesarean section (especially in labor)
Prolonged labor
Midcavity instrumental delivery
Immobility after delivery
∗Adapted from ref. 6
VTE =venous thromboembolism; BMI =body mass index;
ERPC =evacuation of retained products of conception; ART =
Assisted Reproductive therapy.6
e major additional risk factors are a previous
VTE and/or a documented thrombophilia.2Ahistory
of thrombosis increases the risk of pregnancy-related
VTE to 2%–12%.2De Stefano et al. found recurrence
ratesfollowingasinglepreviousDVTorPEof5.8%for
ante-natal and 8.3% for post-natal VTE.10
rombophilias may either be heritable
(antithrombin deciency, Protein C deciency,
Protein S deciency, Factor V Leiden, or Prothrom-
bin gene variant) or acquired (antiphospholipid
syndrome, including lupus anticoagulant and anticar-
diolipinantibodies).Upto50%ofwomenwhodevelop
VTE during pregnancy or the postpartum period have
an underlying thrombophilia.11 e relative risk of
VTEinpregnancyvariesdependingonthethrom-
bophilia, but can be as high as ten fold with antithrom-
bin deciency. e most important determinant of
Table 8.2 Quoted adjusted odds ratios for individual risk
factors
Risk factor for VTE aOR 95% CI
Previous VTE 11 24.8 17.1–36
Age ⬎35 14 1.3 1.0–1.7
BMI ⬎30 12
11
5.3
4.4
2.1–13.5
3.4–5.7
Smoking 13 2.7 1.5–4.9
Parity ≥314 2.4 1.8–3.1
Medical conditions11 2.0–8.7
Sickle cell disease, SLE, heart
disease, anemia, infection,
2.0–3.2
Hyperemesis 2.51
Immobility 13 7.7 (an)
10.8 (pn)
3.2–19
4–28.8
Pre–eclampsia13 3.1 1.8–5.3
+Fetal growth restriction 5.8 2.1–16
ART 13 4.3 2.0–9.4
Twins13 2.6 1.1–6.2
APH 13 2.3 1.8–2.8
PPH 13 4.1 2.3–7.3
Cesarean section 14 3.6 3.0–4.3
Varicose veins 2.4 1.04–5.4
Transfusion11 7.6 6.2–9.4
VTEriskinapregnantwomanwithathrombophilia
is a personal or family history of VTE.
Additional risk factors are detailed in Table 8.1,
adapted from the Royal College of Obstetricians Green
Top Guideline on Reducing the risk of rombosis and
Embolism during pregnancy and the puerperium.6
All are accepted VTE risk factors, but the degree of
increased VTE risk associated with them varies, as
indicated in Table 8.2. Of particular importance are
obesity and increasing maternal age.
e growing problem of maternal obesity in asso-
ciation with VTE is highlighted in the ndings from
both the most recent Condential Enquiry into Mater-
nal Deaths (2003–5)1and the UKOSS study of ante-
natal PE,7that have already been discussed. e lat-
ter demonstrated that one of the main risk factors was
aBMI⬎30 with an adjusted odds ratio (OR) of 2.65
(95% condence interval (CI) 1.09–6.45). Similarly, a
recent case control study from Denmark of 129 cases
of VTE in pregnancy or the puerperium demonstrated
an adjusted OR of 5.3 (95% CI 2.1–13.5) for obesity
(BMI ⬎30), with a higher risk of PE (adjusted OR 14.9,
100

Chapter 8. Thromboprophylaxis
95% CI 3.0–74.8) than of DVT (adjusted OR 4.4, 95%
CI 1.6–11.9)12 e only other risk factors that reached
statistical signicance in these two studies were mul-
tiparity (adjusted OR 4.03, 95% CI 1.60–9.84)7and
current smoking (adjusted OR 2.7, 95% CI 1.5–4.9),12
although Knight et al. highlight the diculty of obtain-
ing sucient power to show other associations, even in
a large national study such as theirs, as ante-natal PE is
still a relatively rare condition.7
Management
Management strategy
e management strategy for thromboprophylaxis
during pregnancy and the postpartum period is
detailed in Table 8.3. All women should have an assess-
ment of risk factors for VTE, in early pregnancy, or
ideally pre-pregnancy. It is particularly important to
identify those with a previous VTE and/or known
thrombophilia. If there is a past history of VTE, then
the details of presentation, means of diagnosis, and
drug treatment and length of course should be deter-
mined. If deemed appropriate, women with a previ-
ous VTE should be screened for both heritable and
acquired thrombophilia prior to pregnancy, as inter-
pretation of some of the tests (especially protein S) is
unreliable in pregnancy. e other risk factors detailed
in Table 8.1 should also be considered, as well as any
family history of VTE in a rst-degree relative.
Following discussion with the woman, a written
plan can then be made for ante-natal thrombopro-
phylaxis, if required, and prescribed if the woman is
already pregnant. Women should be taught how to
self-administer subcutaneous LMWH. If the woman is
not yet pregnant, the prescription can still be given, so
LMWH can be started as soon as a pregnancy test is
positive. is is particularly for women in the very high
and high risk groups, as the pregnancy-related increase
in VTE risk starts from the beginning of the rst
trimester.8,9 e recommendations should be detailed
in a letter copied to both the GP and patient, so pro-
phylaxis can be started without the woman needing to
come back to the hospital clinic rst.
It is important to remember that the VTE risk
may change for a particular woman as pregnancy pro-
gresses, for example, if she develops pre-eclampsia,
and the risk factor assessment should be repeated if
there is any change in circumstances, and LMWH ini-
tiated as appropriate. It may be that the additional
risk factor is only temporary, for example hyperemesis
gravidarum, and the original regimen can be returned
to once the condition or situation has resolved.
Issues regarding anticoagulant use peri-delivery
should be discussed, including epidural timing, and
again documented in the ante-natal notes (discussed
further in Chapter 10). Assessment by an obstetric
anesthetist in the ante-natal period if the woman
is using ante-natal LMWH, especially if at high
prophylactic or therapeutic dose, should be part of
this process.
A clear plan for postpartum prophylaxis should
also be documented in the obstetric notes. e level
of risk will need to be reassessed post-natally, as it
may be increased depending on the mode of delivery
and any associated complications. Women on long-
term warfarin are usually managed with high-dose
prophylactic LMWH for the rst week post-natally
and then converted back to warfarin. Follow-up
should be arranged for those women with a previous
VTE who have not had a thrombophilia screen, once
they have completed the 6-week post-natal anticoagu-
lant course, so they can be investigated for an underly-
ing thrombophilia.
Non-pharmacological and pharmacological
measures used
romboprophylaxis involves both non-
pharmacological and pharmacological measures,
and the various modalities and drugs that can be used
are discussed below.
Non-pharmacological
Non-pharmacological measures include appropriate
hydration, early mobilization aer surgery or delivery,
graduated compression stockings (TEDS), and pneu-
matic compression boots. e aim is to improve blood
ow and decrease stasis in the femoral and popliteal
vessels.
No randomized controlled trials have been car-
ried out in pregnancy to study the ecacy of TEDS
or pneumatic compression for thromboprophylaxis,
but the latter have been shown to be cost-eective
when used at Cesarean section,16 and trials outside
of pregnancy have demonstrated signicant reduc-
tion in incidence of VTE when used peri-operatively.
ese measures are also not associated with the poten-
tial hemorrhagic side eects of the pharmacological 101
Section 3. Thromboembolism and anticoagulation
Table 8.3 Management strategy for thromboprophylaxis in pregnancy and the postpartum period
Pre-pregnancy Assess women with prior history of VTE and/or known thrombophilia
Consider VTE risk factors when giving pre-pregnancy counseling to women with other medical problems
Make plan for future pregnancy regarding thromboprophylaxis and document in letter to GP and patient
Consider giving high risk women prescription for LMWH to start with positive pregnancy test
Ante-natal Assess all women for VTE risk factors at booking
Determine need for ante-natal thromboprophylaxis and prescribe if required
Liaise with obstetric hematologist and/or obstetric physician
Discuss implications of anticoagulants peri-delivery
Arrange assessment by obstetric anesthetist if using ante-natal LMWH
Document plan for peri-delivery and postpartum periods in obstetric notes
Reassess woman’s risk status, need for LMWH, and LMWH dose if events change in pregnancy (e.g. acquire additional
risk factors detailed in Table 8.1)
Peri-delivery Liaise with obstetric anesthetist regarding timing of regional analgesia and anesthesia.
Postpartum Reassess woman’s risk status depending on mode of delivery and any obstetric complications
Prescribe postpartum prophylaxis if required
Arrange follow-up for thrombophilia testing once off anticoagulants if previous VTE and not already tested
VTE =venous thromboembolism; LMWH =low molecular weight heparin.
agents discussed below. ey can be of particular use
in patients requiring Cesarean section or having pro-
longed bedrest.
Pharmacological
Aspirin, heparin, and warfarin are the main pharma-
cological agents to be used in thromboprophylaxis,
and each will be discussed in turn. Although they dif-
fer in their ability to cross the placenta, all are safe to
use in breast-feeding mothers.17
Aspirin
Aspirin inhibits the enzyme cyclo-oxygenase in
platelets, thus reducing thromboxane production and
platelet aggregation. Aspirin is known to be eective
in reducing the risk of VTE in both surgical and
medical patients but no randomized controlled trials
have been carried out looking at the use of aspirin as
thromboprophylaxis in pregnancy. However studies
ofitsuseforarangeofotherindicationsinpregnancy
haveshownittobesafeatlowdose(75mg).
17 It may
therefore be reasonable to consider low dose aspirin
for women who have an increased risk of VTE, but not
high enough to warrant LMWH, although its use for
this indication is controversial, and recent guidelines
do not recommend it.6,17
Heparin
Neither unfractionated (UH) nor low molecular
weight (LMWH) heparin cross the placenta, so there
are no adverse eects on the fetus.
LMWHs are now the anticoagulants of choice in
the UK for prophylaxis and treatment of VTE in preg-
nancy, for the vast majority of cases. LMWH have
a longer half-life and increased bioavailability, which
allows once daily dosing for prophylaxis. UH is usually
only used for thromboprophylaxis if there is an allergy
to LMWH or in renal failure.
Hospitals dier in the LMWH used, and Table 8.4
details the recommended, prophylactic and therapeu-
tic doses for the dierent LMWHs available. Doses
need to be adjusted according to maternal weight in
early pregnancy.18
UH is a heterogenous mixture of high molecu-
lar weight molecules (3 000–30 000 daltons), whereas
LMWHs are a derivative of UH with molecular weights
of 4 000–5 000 daltons. is aords LMWH a num-
ber of advantages over UH, including predictable and
reliable pharmacokinetics, a higher ratio of anti-Xa
to anti-IIa activity, providing good antithrombotic
eect with possibly a lower risk of bleeding. ey also
have less of an eect on platelet aggregation, func-
tionandactivation,andbindplateletfactor4lesswell,
hence reducing the risk of both early and late heparin-
induced thrombocytopenia (HIT).19
Heparin-induced osteoporosis is an important
risk, especially if heparin is required throughout the
ante-natal period, but the risk is much lower with
LMWH (0.04%) than UH (2%).19 Also, even if there
is loss in bone density during pregnancy (up to
5%), this usually improves on stopping breast feed-
ing postpartum. LMWHs in prophylactic doses have
notbeenassociatedwithareductioninbonedensity
102

Chapter 8. Thromboprophylaxis
Table 8.4 Ante-natal prophylactic and therapeutic doses of different low molecular weight heparins∗
Dose
(based on early pregnancy
maternal weight) Enoxaparin∗∗ Dalteparin Tinzaparin
Standard prophylactic
⬍
50 kg
50–90 kg
91–130 kg
131–170 kg
20 mg od
40 mg od
60 mg od or 40 mg bd
80 mg od or 40 mg bd
2500 units od
5000 units od
7500 units od or 5000 bd
10,000 units od or 5000 units bd
3500 units od
4500 units od
7000 units od
High prophylactic
40 mg bd 5000 units bd 4500 units bd
Treatment/therapeutic
⬍
50 kg
50–90 kg
⬎
90 kg
1mg/kgbd
40 mg bd
60–80 mg bd
100 mg bd
100 units/kg bd
5000 units bd
6000–8000 units bd
10 000 units bd
175 units/kg od
∗Adapted from4,14;∗∗ 100 units/mg
od =once daily; bd =twice daily.
overandabovewhatwouldbeexpectedinnormal
pregnancy.20
Of women, 1%–2% may develop a local aller-
gic skin rash with LMWH. e rst-line pragmatic
approach is to substitute with an alternative LMWH,
but there may be cross-reactivity, as also occurs with
UH.19 If this is the case, or if other complications such
as HIT develop, then consideration of alternative hep-
arinoids such as danaparoid or the synthetic pentasac-
charidefondaparinuxisappropriate.However,HIT
has never been described with LMWH in a pregnant
woman.2,17
Warfarin
Warfarin crosses the placenta and is teratogenic.17
e characteristic “warfarin embryopathy” includes
chrondrodysplasia punctata, nasal hypoplasia, and
short proximal limbs. e period of greatest risk is
between 6 and 12 weeks. Warfarin is also associated
with an increased rate of miscarriage and stillbirth,
andinthethirdtrimesterasignicantriskoffetal
intracerebral hemorrhage and maternal retroplacental
bleeding, especially aer 36 weeks’ gestation. Use in
the second trimester has been linked to microcephaly
and neurological abnormalities, which may be due to
over-anticoagulation of the fetus.
Because of these eects, warfarin is not used rou-
tinely during pregnancy, and most women requir-
ing thromboprophylaxis or treatment can be managed
with LMWH or unfractionated heparin. ere are
some women though, who that require continued full
anticoagulation with warfarin throughout pregnancy
until planned delivery. e only non-controversial
indication for use of warfarin in pregnancy is for
women with a metal prosthetic heart valves (particu-
larly of the older type and those in the mitral posi-
tion), whose thrombotic risk is extremely high, and in
whom valve thrombosis carries a high mortality rate
(see Chapter 9).
In other high-risk women the decision is more dif-
cult, and the risk/benet balance for the particular
individual has to be considered. is would include
those with a known thrombophilia and VTE or cere-
bral arterial thrombosis despite full dose LMWH. A
compromise option here is to use LMWH for the high-
est risk periods for warfarin side eects, namely, the
rst trimester and aer 36 weeks’ gestation, and con-
vert back to warfarin for the second and early third
trimesters. is obviously requires very close supervi-
sion and thorough discussion with the woman.
Recommendations
Although VTE in pregnancy and postpartum is a
major cause of maternal morbidity and mortality, the
absolute risk for most women is low and there are few
randomized controlled trials of thromboprophylaxis
in the pregnancy-related setting on which to base rec-
ommendations. A Cochrane review published in 2002,
looking at trials of thromboprophylaxis during preg-
nancy and the early puerperium, concluded there was
insucient evidence on which to base recommenda-
tions, as the number of trials and sample sizes were too
small.21A number of professional bodies have drawn 103

Section 3. Thromboembolism and anticoagulation
Table 8.5 Ante-natal VTE risk factor assessment and thromboprophylaxis management
Risk level Risk factors Recommended thromboprophylaxis
Very high Previous VTE on long-term warfarin +/– thrombophilia
antithrombin deficiency or APS
High prophylactic or therapeutic dose LMWH
Requires specialist management by experts in hemostasis
and pregnancy
High Previous recurrent VTE
Previous VTE:
bplus thrombophilia
bplus family history of VTE
bplus other risk factor(s)∗
bon COCP or during pregnancy
bat unusual site
Asymptomatic thrombophilia:
bAntithrombin deficiency
bCombined defects
bHomozygous factor V Leiden
bHomozygous prothrombin gene defect
bCompound heterozygote
Three or more other risk factors∗
Three or more risk factors plus admission to hospital
Prophylactic dose LMWH
Intermediate Previous single VTE without family history,
thrombophilia or other risk factor(s)∗
Other asymptomatic thrombophilias including APS
(not covered above)
Close surveillance
Advise to keep mobile and avoid dehydration consider
TEDS
Lower less than 3 other risk factors∗Advise to keep mobile and avoid dehydration
Consider TEDS
∗See Table 8.1
VTE =venous thromboembolism; COCP =combined oral contraceptive pill; LMWH =low molecular weight heparin;
APS =anti-phospholipid syndrome; TEDS =graduated compression stockings.
up guidelines,4–6,17 many recommendations are based
on low grade evidence. However, recent data show
that risk assessment and allocation of thrombo-
prophylaxis according to such guidelines is eca-
cious and cost eective, with few clinically signi-
cant adverse events.22,23
Ante-natal
Recommendations for ante-natal assessment and
management of VTE risk are given in Table 8.5. It
is important to remember that there are probably
many heritable thrombophilias as yet undiscovered
and therefore unable to be conrmed with in vitro
testing. So, if the history is suspicious, for example,
previous VTE in an unusual site or previous recurrent
VTE especially associated with as family history of
VTE, it is sensible to treat these women as high risk,
even without an identiable thrombophilia, and give
both ante-natal and post-natal LMWH prophylaxis.
In comparison, women with a single previous VTE
relatedtoatemporarybutnon-estrogenrelated
(pregnancy or the combined oral contraceptive pill)
risk factor, who have no identiable thrombophilia
or additional current risk factor, do not require ante-
natal LMWH.24 ArecentstudyfromItalyof88women
who became pregnant aer a single previous episode
of VTE, with no ante-natal thromboprophylaxis,
demonstrated no recurrence of VTE in pregnancy if
the initial VTE was related to transient risk factors
other than pregnancy or oral contraceptive use.10 is
contrasted with a 7.5% recurrence rate if the rst VTE
was unprovoked, or estrogen related.
Women who are on long-term warfarin outside
pregnancy because of previous VTE or stroke associ-
ated with APS are at very high risk of recurrence, and
shouldbemanagedwithhighprophylacticorsome-
times full therapeutic LMWH ante-natally, depending
ontheclinicalhistory,underthecareofanexpertin
hemostasis and pregnancy.
Even women without a previous VTE or identied
thrombophilia may require ante-natal thrombopro-
phylaxis due to other risk factors. ose with at least
three of the risk factors detailed in Table 8.1 should
be considered for LMWH ante-natally and those
with two risk factors require LMWH during hospital
104

Chapter 8. Thromboprophylaxis
Table 8.6 Post-natal VTE risk factor assessment and thromboprophylaxis management
Risk level Risk factors Recommended prophylaxis
Very high Previous VTE on long-term warfarin +/–
thrombophilia antithrombin deficiency or APS
LMWH until re-established on warfarin
High Any other previous VTE
Asymptomatic thrombophilia:
bAntithrombin deficiency
bCombined defects
bHomozygous factor V Leiden
bHomozygous prothrombin gene defect
bCompound heterozygote
Extended major pelvic or abdominal surgery
(e.g. CS hysterectomy)
Paralysis of lower limbs
6 weeks prophylactic dose LMWH
Intermediate Emergency CS in labor
Any CS plus any other risk factor
Three or more other risk factors∗
Asymptomatic thrombophilia:
bHeterozygous factor V Leiden
bHeterozygous prothrombin gene defect
7 days prophylactic dose LMWH∗∗
Consider extending if other risk factors or
positive family history
Lower CS not in labor no other risk factors
Less than 3 other risk factors∗
Early mobilization and avoidance of dehydration
Consider TEDS
∗See Table 8.1; ∗∗ See text – there is minimal evidence to determine optimum duration of postpartum thromboprophylaxis.
VTE =venous thromboembolism; LMWH =low molecular weight heparin; CS =Cesarean section; TEDS =graduated compression
stockings.
admissions. is stresses the importance of repeated
risk assessment as pregnancy progresses. It also high-
lights the need to assess all pregnant women for their
thromboembolic risk, not just women under the care
of hematologists with previous VTE and/or throm-
bophilia.
Interestingly, the UKOSS7study demonstrated that
approximately one-third of women with an ante-natal
PE had no classical risk factors for VTE disease (apart
from pregnancy), and only 9 of the 99 who did would
have been eligible for thromboprophylaxis under the
2004 RCOG guidelines.5Knight et al. discuss inclu-
sion of women with at least two risk factors being eli-
gible, but also acknowledge that this requires an esti-
mated 9% of maternities to receive LMWH, or 5.5% if
just multiparity was added as a risk factor. ey suggest
further studies are required to assess the cost benet of
this inclusion.7
Post-natal
Table 8.6 details recommendations for post-natal
assessment and management of VTE risk. e most
recent RCOG guidelines supersede an order guideline
regarding women who have had a Cesarean section.4,6
ere is signicant variation between units as to how
aggressively these are applied, with some prescribing
LMWH to all who have undergone Cesarean section,
while others just for emergency Cesarean section in
labor.
It is important to remember that thromboprophy-
laxis should not be limited to those who have delivered
by Cesarean section, as women die from VTE even
aer a normal vaginal delivery. Women with at least
two persisting risk factors from Table 8.1 should be
considered for postpartum LMWH.
e immediate postpartum period is the time of
highest risk for VTE, and post-natal LMWH should be
continued for at least 7 days. Recently published data,
assessing changes in thromboelastography parameters
in the postpartum period suggest the risk is high for 7
days.25 ere is also emerging evidence that it may take
several weeks for the hypercoagulable state of preg-
nancy to return to non-pregnant levels,24 therefore for
high risk patients, for example, those with a previous
VTE or thrombophilia, it is standard practice to con-
tinue LMWH for six weeks.
Dilemmas – current research and
future direction
rIdentication of other thrombophilias
rCost–benet analysis of extending ante-natal
thromboprophylaxis 105

Section 3. Thromboembolism and anticoagulation
rInvestigation of the optimal duration of
postpartum thromboprophylaxis in women with
risk factors other than previous VTE or
thrombophilia.
Summary
rVenous thromboembolism (VTE) is the
leading direct cause of maternal mortality
in the UK, but many cases are potentially
preventable.
rRisk factors for VTE should be identied
pre-pregnancy, or at least early in
pregnancy, and reassessed throughout
pregnancy and the puerperium, as level of risk
may change.
rPregnancy itself is a risk factor for VTE, and
additional risk factors include previous VTE,
thrombophilia, and obesity.
rromboprophylaxis should be introduced
depending on the level of risk. Guidelines are
given for both ante-natal and post-natal
management, and in particular for the highest risk
period immediately postpartum.
rromboprophylaxis includes both
non-pharmacological and pharmacological
measures, mainly low molecular weight heparin.
rFurther research is required to identify additional
thrombophilias, assess whether
thromboprophylaxis should be extended, and
determine optimal duration for postpartum
thromboprophylaxis.
106

Chapter 8. Thromboprophylaxis
References
1. Condential Enquiry into Maternal and Child Health.
Saving Mothers’ Lives: reviewing maternal deaths to
make motherhood safer, 2003–5. e Seventh Report of
the Condential Enquiries into Maternal Deaths in the
United Kingdom. London, RCOG Press, 2007.
2. James AH. Prevention and management of venous
thromboembolisminpregnancy.American Journal of
Medicine 2007; 120: S26–S34.
3. Heit JA, Kobbervig CE, James AH, Petterson TM,
Bailey KR, Melton LJ 3rd. Trends in the incidence of
venous thromboembolism during pregnancy or
postpartum: a 30-year population-based study. Annals
of Internal Medicine 2005; 143: 697–706.
4. Royal College of Obstetricians and Gynaecologists.
Report of the RCOG Working Party on prophylaxis
against thromboembolism in gynaecology and obstetrics.
London, RCOG Press, 1995.
5. Royal College of Obstetricians and Gynaecologists.
romboprophylaxis during pregnancy, labour and aer
normal vaginal delivery. Guideline no.37. London,
RCOG Press, 2004.
6. Royal College of Obstetricians and Gynaecologists.
Reducing the risk of thrombosis and embolism during
pregnancy and the puerperium.Guideline no.37.
London, RCOG Press, 2009.
7. Knight M on behalf of UKOSS. Antenatal pulmonary
embolism: risk factors, management and outcomes.
British Journal of Obstetrics and Gynecology 2008; 115:
453–461.
8. James AH, Tapson VF, Goldhaber SZ. rombosis
during pregnancy and the postpartum period.
American Journal of Obstetrics and Gynecology 2005;
193: 216–219.
9. Blanco-Molina A, Trujillo-Santos J, Criado J
et al. Venous thromboembolism during pregnancy or
postpartum: ndings from the RIETE Registry.
rombosis and Haemostasis 2007; 97: 186–
190.
10. De Stefano V, Martinelli I, Rossi E et al. e risk
of recurrent venous thromboembolism in pregnancy
and puerperium without antithrombotic
prophylaxis. British Journal of Haematology 2006; 135:
386–391.
11. NelsonSM,GreerIA.rombophiliaandtheriskfor
venous thromboembolism during pregnancy, delivery,
and puerperium. Obstetric Gynecologic Clinics of North
America 2006; 33: 413–427.
12. James AH, Jamison MG, Brancazio LR, Myers ER.
Venous thromboembolism during pregnancy and the
postpartum period: incidence, risk factors, and
mortality. American Journal of Obstetrics and
Gynecology 2006; 194: 1311–5.
13. Larsen TB, Sorensen HT, Gislum M, Johnsen SP.
Maternal smoking, obesity, and risk of venous
thromboembolism during pregnancy and the
puerperium: a population-based nested case-control
study. rombosis Research 2007; 120: 505–
509.
14. Jacobsen AF, Skjeldestad FE, Sandset PM. Ante- and
postnatal risk factors of venous thrombosis: a
hospital-based case-control study. Journal of
rombosis and Haemostasis 2008; 6: 905–912.
15. Lindqvist P, Dahlb¨
ack B, Marˆ
s´
al K. rombotic risk
during pregnancy: a population study. Obstetrics and
Gynecology. 1999; 94: 595–599.
16. Casele H, Grobman WA. Cost-eectiveness of
thromboprophylaxis with intermittent pneumatic
compression at Cesarean delivery. Obstetrics and
Gynecology 2006; 108:535–540.
17. Bates SM, Greer IA, Pabinger I et al.Venous
thromboembolism, thrombophilia, antithrombotic
therapy, and pregnancy. American College of Chest
Physicians Evidence-Based Clinical Practice
Guidelines (8th edn.). Chest 2008; 133: 844S–886S.
18. British National Formulary 55. London, BMJ Group
and RPS Publishing, 2008.
19. Greer IA, Nelson-Piercy C. Low-molecular-weight
heparins for thromboprophylaxis and treatment of
venous thromboembolism in pregnancy: a systematic
review of safety and ecacy. Blood 2005; 106:
401–407.
20. Carlin AJ, Farquharson RG, Quenby SM et al.
Prospective observational study of bone mineral
density during pregnancy: low molecular weight
heparin versus control. Human Reproduction 2004; 19:
1211–1214.
21. Gates S, Brocklehurst P, Davis LJ. Prophylaxis for
venous thromboembolic disease in pregnancy and the
early postnatal period. Cochrane Database Systematic
Review 2002; (2):CD001689.
22. Johnston JA, Brill-Edwards P, Ginsberg JS et al.
Cost-eectiveness of prophylactic low molecular
weightheparininpregnantwomenwithaprior
history of venous thromboembolism. American
Journal of Medicine 2005; 118: 503–514.
23. Bauersachs RM, Dudenhausen J, Faridi A et al. Risk
stratication and heparin prophylaxis to prevent
venous thromboembolism in pregnant women.
rombosis and Haemostasis 2007; 98: 1237–1245.
107

Section 3. Thromboembolism and anticoagulation
24. Brill-Edwards P, Ginsberg JS, Gent M et al. Safety of
withholding heparin in pregnant women with a
history of venous thromboembolism. Recurrence of
Clot in this Pregnancy Study Group. New England
Journal of Medicine 2000; 343: 1439–1444.
25. Maybury H J, Waugh J J S, Gornall A, Pavord S. ere
is a return to non-pregnant coagulation parameters
aer four not six weeks postpartum following
spontaneous vaginal delivery. Obstetric Medicine 2008;
1: 92–94.
108
Section 3 Thromboembolism and anticoagulation
Chapter
9Prosthetic heart valves
Claire McLintock
Introduction
Patients with mechanical heart valves require long-
term anticoagulation, but the choice of anticoagulant
for these women during pregnancy presents a major
challenge. Oral anticoagulants such as warfarin and
coumadin are the most eective agents for preven-
tion of maternal thromboembolism, but freely cross
the placenta and are teratogenic. ey also cause late
fetal loss in as many as one in ten pregnancies. Anti-
coagulation with unfractionated heparin (UFH) and
low molecular weight heparin (LMWH), which do not
cross the placenta will reduce the risk of these adverse
fetal outcomes. However, there is concern that these
drugs are less eective at preventing maternal valve
thrombosis and systemic thromboembolism. erein
lies the challenge: the anticoagulant that is safest for
the mother’s physical health carries the greatest poten-
tial risk for her infant. Many women will choose a
treatment that is safest for her baby, even if her own
health may be compromised. Despite being advised
that thrombotic complications may necessitate urgent
valve-replacement surgery or lead to major neuro-
logical sequelae, many women are reluctant to take
oral anticoagulants during pregnancy when informed
about fetal risks. At worst, some women are non-
compliant with all therapy, causing even greater mater-
nal risk. is chapter addresses the management of
pregnancy in women with mechanical heart valves and
discusses the maternal and fetal risks associated with
the dierent anticoagulant options, to enable clinicians
and women to make the most informed choice in this
challenging clinical situation.
Indication for valve replacement
e most common indications for replacement of a
native heart valve are congenital valvular disease and
rheumatic heart disease. While the incidence of con-
genital valvular disease is relatively stable at around
0.25% of births, there is marked variation in the rates
of rheumatic fever and rheumatic heart disease (RHD)
across dierent countries. Rheumatic fever is certainly
more common in resource-poor countries and com-
munities, but it is also prevalent in countries such
as New Zealand and Australia with high rates in
the indigenous Aboriginal and Maori people in these
countries and in Pacic Island people in New Zealand
and the Pacic region (Table 9.1).1–3
Table 9.1 Rates of acute rheumatic fever in different regions of
the world
Country Ethnicity
Rate of acute
rheumatic fever
in children aged
5–14 y (rate per
100 000)
Sub-Saharan
Africa1
13.4
South-central
Asia1
54.0
New Zealand2Maori
Pacific Island peoples
European
30.4
77.7
1.7
Australia3Aboriginal and
Torres Strait
Islander peoples
Other Australian
people
162–375
1.0
China121.2
Consideration of valve type in women
of child bearing age
For patients who require heart valve replacement,
the alternatives include bioprosthetic valves – either
homogra (human tissue) or heterogra (porcine or
109
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 3. Thromboembolism and anticoagulation
Table 9.2 Mechanical valve types
Valve type
Ball and cage Starr-Edwards R
Tilting disc Bjork-Shiley R
Medtronic-Hall R
Bileaflet St Judes R
CarboMedics R
On-X R
ATS valve R
bovine tissue) – or mechanical valves (Table 9.2). e
major advantage of bioprosthetic valves for women
of child-bearing age is that, in the absence of other
thromboembolic risk factors, oral anticoagulant ther-
apy is not required. e disadvantage is the high
rate of structural valve deterioration, the common-
est reason for replacement of bioprosthetic valves.
Structural deterioration of bioprosthetic valves in the
mitral position occurs more commonly than of those
in the aortic or tricuspid position and was reported
in 84% of bioprosthetic mitral valves by 10 years.
Although valve deterioration occurred more rapidly
in younger patients, it was not further accelerated
by pregnancy.4In contrast, structural valve failure is
extremely uncommon with mechanical heart valves,
buttherisksofvalvethrombosisandsystemicthrom-
boembolism mean that patients must take long-term
oral anticoagulant therapy. When considering which
type of valve to use in young women, clinicians should
take into account the impact of the decision on man-
agement and outcome of future pregnancies.5
Prevention of thromboembolism
An overall approach to management of women with
prosthetic heart valves is summarized in Figure 9.1.
romboembolic complications of mechanical valves
include valve thrombosis, causing valve obstruc-
tion or systemic embolization, mainly cerebrovascu-
lar accidents (CVA) but also myocardial infarction or
embolization into peripheral arteries. Systemic throm-
boembolism can develop from either obstructed or
non-obstructed valves. Prevention of these complica-
tions is the main indication for long-term anticoag-
ulation. Outside of pregnancy, the rate of major sys-
temic embolization in patients with mechanical valves
isaround1%peryearinpatientstakingwarfarin,2%
per year in patients taking aspirin, and 4% per year in
patients on no anticoagulation.6Additional risk fac-
tors for thromboembolism are shown in Table 9.3. e
Table 9.3 Risk factors for thromboembolism in patients with
prosthetic valves
Valve related factors Older type ball-cage valves, i.e.
Starr–Edwards valve
Valve in mitral position
≥2 prosthetic valves
Clinical risk factors Previous thromboembolism
Atrial fibrillation
Dilated left atrium
Heart failure
prothrombotic changes of pregnancy further increase
theriskofthromboembolismwitheventsreportedin
4% of women taking warfarin during pregnancy.7
Anticoagulant management
during pregnancy
Available guidelines accurately state that continuation
of oral anticoagulants (OAC) is the safest option for
the mother for prevention of valve thrombosis and rec-
ommend that they should be used during pregnancy
in spite of the known fetal risks (Table 9.4). Most sug-
gest continuation of OAC throughout pregnancy, per-
haps substituting adjusted-dose UFH or LMWH dur-
ing the rst trimester, the teratogenic risk-period.8–10
However, ongoing use of OAC is associated with a sig-
nicant risk of late fetal loss, as high as 10%, mainly
as a result of complications from fetal anticoagulation.
Maternal concern relating to these fetal risks and the
desireforahealthybabyoenmeansthatwomenopt
for the most dangerous option of all, taking no anti-
coagulation during pregnancy. e recent American
College of Chest Physicians Guidelines on Anticoagu-
lant erapy 8is the rst published guideline to rec-
ommend either adjusted dose LMWH or unfraction-
ated heparin (UFH) throughout pregnancy or until the
beginning of the 13th week of pregnancy, switching to
warfarin until close to delivery.
Unfractionated heparin and LMWH are recom-
mended for prevention and treatment of thrombosis
during pregnancy in women who are at risk of throm-
boembolism from other causes.8ese agents do not
cross the placenta, are not teratogenic and have no
fetal anticoagulant eect. LMWH with its more pre-
dictable anticoagulant eect and better side eect pro-
le has supplanted UFH for treatment and preven-
tion of venous thromboembolism during pregnancy.
Data relating to the safety and ecacy of LMWH in
prevention of thromboembolic complications in preg-
nant women with mechanical heart valves are limited,
110
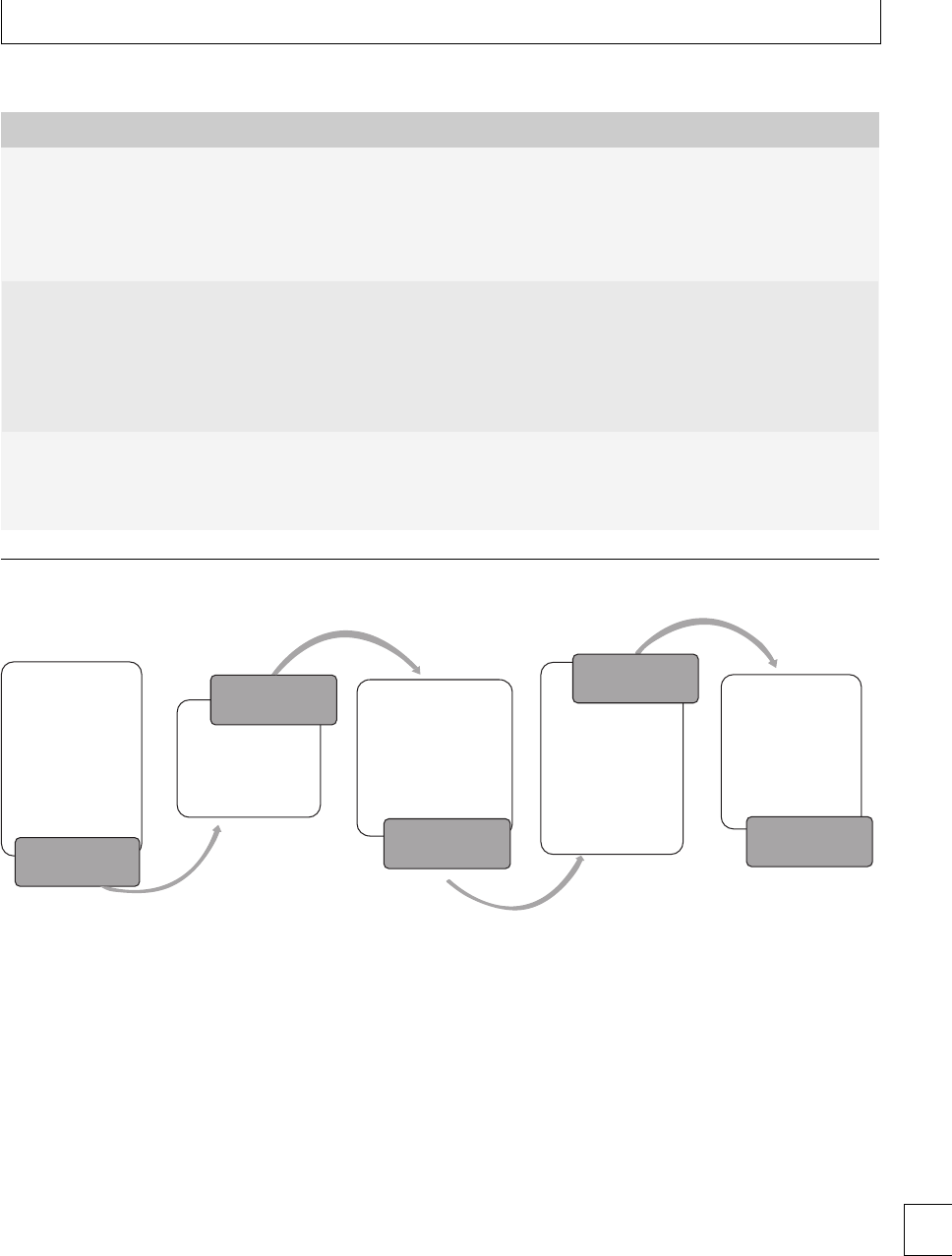
Chapter 9. Prosthetic heart valves
Table 9.4 Summary of published guidelines of management of anticoagulation in pregnant women with mechanical heart valves
Warfarin LMWH UFH Aspirin
American College of
Chest Physicians
20088
Only for women at very high
risk thromboembolism, i.e.
history thromboembolism,
older type valve in mitral
position
Can be used throughout
pregnancy. Twice daily
dose-adjusted,
manufacturers peak
anti-Xa level (1.0 U/ml)
4h post-dose
Can be used throughout
pregnancy. Initial dose
17 500–20 000 U every
12 h. 6 hour
post-injection aPTT 2x
baseline or anti-Xa
levels 0.35–0.70 U/mL
75–100 mg
throughout
pregnancy for
high risk
women
American Heart
Association 20089
Reasonable to avoid 6–12
weeks’ gestation.
Embryopathy risk 4–10%.
INR target 3.0 (range
2.5–3.5). Discontinue 2–3
weeks before planned
delivery
Can be used 6–12 weeks’
gestation. Twice daily,
dose-adjusted with 4 h
post-dose target
anti-Xa levels 0.7–1.2
U/ml
Can be used 6–12 weeks’
gestation. Continuous
iv UFH or
dose-adjusted s/c UFH
bid. Starting dose s/c
17 500–20 000 U every
12h. Target aPTT 2x
baseline
75–100 mg
during second
and third
trimesters
European Society of
Cardiology 200710
Favored in first trimester if
dose ≤5 mg. Favored
anticoagulant during the
second and third trimester
until week 36.
Currently not
recommended –
insufficient safety and
efficacy data
Close monitoring is
required when used
Not discussed
All guidelines recommend full discussion of the risks and benefits of anticoagulant regimens.
Assess thrombotic risk: valve
type, position, previous
thromboembolism, cardiac
rhythm, left atrial size
Discuss anticoagulant
regimens
Option 1: therapeutic dose
LMWH throughout, i.e.
enoxaparin 1 mg / kg bid
Option 2: therapeutic dose
LMWH weeks 6−12, warfarin
from week 13
Option 3: warfarin
throughout
Pre- pregnancy
counseling
Early clinical review
when pregancy
confirmed
1st trimester
Option 1 or 2
Ongoing management
for option 2 (and 3)
All groups
Decide on anticoagulant
regimen
Option 1, 2 or 3
Start low dose aspirin: 100−
150 mg
Option 1 or 2: stop warfarin
before 6 weeks
Switch LMWH to warfarin at
week 13 (option 2)
Planned delivery at 38−39
weeks
Mode of delivery detrmined
by obstetric indications
Last dose LMWH 36 hours
pre-delivery
Start intravenous UFH 24
hours prior to delivery
INR
At least monthly INR
measurement: target range
determined by valve type
Switch to therapeutic dose
enoxaparin at week 34−36
Start therapeutic dose LMWH
Measure anti-Xa levels day 3−5
Target anti-Xa: trough (pre-
dose) 0.4−0.7 IU and peak
(4 hours postdose) 0.7−1.2 IU
Monthly monitoring of anti-Xa
levels
Further discussion of anticoagulant
regimen at week 13.
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
*
Fig. 9.1 Approach to management of women with mechanical heart valves in pregnancy.
butsomestudiessuggestthatitislesseectivethan
warfarin at preventing maternal thromboembolism.
A major systematic review by Chan and co-workers7
compared the maternal and fetal outcome of dier-
ent anticoagulant regimens in 1234 pregnancies in 976
womeninwomenwithmechanicalheartvalvesfrom
28 studies of women conducted between 1966 and
1997. Comparisons were made across four broad cate-
gories of anticoagulant regimens:
roral anticoagulants throughout pregnancy (792
pregnancies);
runfractionated heparin in the rst trimester
followed by oral anticoagulants (230 pregnancies);
runfractionated heparin throughout pregnancy (21
pregnancies);
rantiplatelet agents or no anticoagulant therapy
(102 pregnancies).
Since that time, a number of further studies have been
published,11–13 including reviews of pregnancy out-
comes in women taking LMWH at some stage during
pregnancy.14,15 Asummaryofpublisheddataofmater-
nalandfetaloutcomesinpregnancieswherewomen 111

Section 3. Thromboembolism and anticoagulation
Table 9.5 Maternal and fetal outcomes in pregnancies in women with mechanical heart valves related to anticoagulant
management approach
Anticoagulant regimen Pregnancies
Miscarriage
N (%)
Stillbirth∗
(Σ)
Warfarin
embryopathy
Maternal
thromboembolic
complications
Warfarin throughout pregnancy7,12,13,27 983 253
(25.7%)
13/127§
(10.2%)
39/740
(5.3%)
36
(3.7%)
Warfarin-UFH-warfarin7,11,13 285 66
(23.4%)
3/43§
(7.0%)
6/229
(2.6%)
28
(9.9%)
Warfarin-LMWH-warfarin15 56 4
(7.1%)
1/51
(1.9%)
05
(8.9%)
LMWH throughout11,13,15 29 2
(8.3%)
00 1
(4.2%)
∗pregnancies included in Chan review excluded as stillbirths not reported as a separate group.
§excluding spontaneous miscarriages and terminations of pregnancy.
have received dierent anticoagulant regimens is pre-
sented in Table 9.5.
Anticoagulation with warfarin
during pregnancy
Oral anticoagulants, such as warfarin and aceno-
coumarol, are the most eective agents for prevention
of valve thrombosis and systemic thromboembolism
during pregnancy in women with mechanical heart
valves. Disadvantages of oral anticoagulants include
teratogenicity, high rates of spontaneous abortion and
latefetallossaswellasneurologicalabnormalitiesin
surviving infants.
Fetal eects of warfarin
Exposure to warfarin in the rst trimester causes war-
farin embryopathy in as many as 6% of infants. Hall
and co-workers16 recommended that nasal hypopla-
sia and stippled epiphyses should be the minimal fea-
turesrequiredtoclassifyacaseaswarfarinembry-
opathy. ese features are not described in women
who substitute heparin for warfarin between 6–12
weeks of pregnancy or who are exposed to warfarin
from the second trimester onwards. A recent review of
63 published cases of warfarin-related abnormalities17
described skeletal anomalies in 81% of cases (n=51)
with mid facial hypoplasia described in 47 infants and
epiphyseal calcic stippling of long bones, vertebrae,
calcanei, or phalanges in 32 infants. Breathing and
feeding problems were present in 24 of 47 infants who
had severe midfacial hypoplasia. e period of expo-
sure to warfarin, common to infants who developed
embryopathy, was between 6 and 9 weeks’ gestation.
Long-term follow-up information was available on 20
of 46 children in this cohort who survived the neona-
tal period with abnormalities persisting in about half
of the children with midline hypoplasia and spinal
deformities. ese teratogenic eects are unlikely to
be caused by inhibition of Vitamin K-dependent clot-
ting proteins by warfarin, as these are not produced
by the fetal liver until aer 12–14 weeks’ gestation.
Vitamin K-dependent proteins are also important in
development of bone and cartilage and inhibition of
these proteins may account for this teratogenic eect of
warfarin.
Microcephaly, cerebral atrophy, hydrocephalus,
optic atrophy, and intracranial hemorrhage are among
the central nervous system abnormalities described
in 1% of liveborn infants exposed to warfarin during
pregnancy. e prevalence of long-term neurological
problems, such as developmental delay and low IQ, in
infants who appear normal aer in-utero exposure to
warfarin is still debated.
Warfarin and pregnancy outcome
Warfarin exposure during pregnancy is associated
with increased rates of spontaneous miscarriage and
a 10% rate of late fetal loss, Table 9.5. e majority
of late losses are thought to be due to fetal intracra-
nial hemorrhage (ICH) as a result of anticoagulation
of the fetus. Warfarin freely crosses the placenta so
it will inhibit fetal vitamin K-dependent clotting pro-
teins that are produced from 12–14 weeks’ gestation.
As vitamin K levels in the fetus are one-tenth of the
levels in the mother, the dose of warfarin taken by the
112

Chapter 9. Prosthetic heart valves
mother to achieve a therapeutic INR will cause severe
over-anticoagulation in the fetus, possibly with INR
levels as high as those that develop in elderly patients
on warfarin who become vitamin K decient when
they are unwell, stop eating, and take antibiotics.
Eect of warfarin dose on
pregnancy outcome
Vitale and co-workers18 described increased fetal com-
plication rates in women with mechanical heart valves
taking sodium warfarin doses of ⬎5 mg during preg-
nancy compared to women on lower doses. is dose-
dependent relationship was further examined in an
updated publication of their pregnancy cohort includ-
ing 71 pregnancies in 52 consecutive patients.19 Fetal
losses were reported in 78.8% of 33 pregnancies in
women taking ⬎5 mg warfarin, including 21 sponta-
neous abortions between 12 and 20 weeks’ gestation
and ve stillbirths aer 20 weeks’ gestation. Of 38 preg-
nancies in women taking ≤5 mg of warfarin daily only
two spontaneous abortions (5.3%) were reported. e
cause of spontaneous abortion or stillbirth was not
reported. Rates of classical embryopathy did not dif-
fer between groups with skeletal anomalies reported in
three infants; two with nasal hypoplasia (one in a term
infant in the ≤5 mg warfarin group and one in a spon-
taneously aborted fetus in the ⬎5mggroup)andcer-
vical spine abnormalities reported in a spontaneously
aborted fetus from the higher dose group. is dose-
dependent relationship with adverse fetal outcome is
not described in all studies4and the clinical impact
of such an association is uncertain as OAC dosage is
determined by maternal INR.
Anticoagulation with heparin
during pregnancy
Unfractionated heparin and
antiplatelet agents
Unfractionated heparin and antiplatelet agents such
as aspirin were used as alternatives to warfarin for
anticoagulation of pregnant women with mechani-
cal heart valves prior to the development of LMWH.
Although use of antiplatelet agents avoided the risk
of congenital anomalies the thromboembolic compli-
cations were reported in 29% of pregnancies where
women took antiplatelet agents alone.7Similarly, no
fetal anomalies were reported in pregnancies where
UFH was substituted for warfarin prior to six weeks
gestation and continued until completed 12 weeks’ ges-
tation. Variation in rates of thromboembolic compli-
cations with UFH are likely to be confounded by dif-
ferences in heparin dosing, anticoagulant monitoring,
and duration of treatment. romboembolic compli-
cations occurred in 9.9% of these pregnancies where
women received UFH (Table 9.5) including events in
the two recent studies11,13 where the dose of UFH was
adjusted to keep the activated partial thromboplas-
tin time (aPTT) at two to three times baseline lev-
els. In 21 pregnancies where UFH was used through-
out, thromboembolic complications were reported in
33%.7Concerns with maternal safety with UFH pro-
vided the impetus to explore the ecacy of LMWH
with its more predictable anticoagulant eect as an
alternative anticoagulant for women with mechanical
heart valves who do not wish to take warfarin during
pregnancy. Long-term use of therapeutic dose UFH
also increases the risk of osteoporosis and heparin-
induced thrombocytopenia.
Maternal and fetal outcomes in women
receiving LMWH during pregnancy
Limited data are available of pregnancy outcome in
women with mechanical heart valves treated with
LMWH during pregnancy; outcomes in women tak-
ing either LMWH during the rst trimester only or
throughout pregnancy account for only 4.1% and
2.1% of published reports, respectively (Table 9.5). In
their review of published studies of pregnancy out-
come in women with mechanical heart valves who
received LMWH during pregnancy, Oran and co-
workers reported seven episodes of valve thrombosis
and two CVAs in 81 pregnancies, a complication rate
of 11.1%.15 Single case reports and small case series
accounted for six of the thromboembolic events in this
review. Limiting analysis to studies including ≥5preg-
nancies the rate of valve thrombosis was 4.8%. Dose
adjustment of LMWH in response to anti-Xa levels
appears to be a key factor in management of anticoagu-
lation with LMWH in pregnant women with mechan-
ical heart valves: six of the seven valve thromboses in
Oran’s review occurred in women who did not have
anti-Xa levels measured.
Another consideration with heparin use is the
risk of heparin-induced thrombocytopenia and bone 113

Section 3. Thromboembolism and anticoagulation
Table 9.6 Quick-reference guide: estimates of maternal and fetal risks with different anticoagulant regimens for pregnant women with
mechanical heart valves
Anticoagulant regimen
Maternal valve
thrombosis and systemic
thromboembolism
Spontaneous
abortion
Late fetal
loss
Warfarin
embryopathy
Warfarin continued throughout pregnancy Low High Moderate Moderate
Dose-adjusted UFH weeks 6–12, then warfarin Moderate High Moderate No risk
Therapeutic-dose LMWH weeks’ 6–12 with
anti-Xa monitoring, then warfarin
? Low Moderate Low No risk
Therapeutic-dose LMWH throughout
pregnancy started ⬍6 weeks’ gestation
with anti-Xa monitoring
? Low Moderate Low No risk
Low risk ⬍5% Moderate risk =5%–10% High
risk ⬎10%
mineral density loss. is is less common with LMWH
than with UFH.
Role of aspirin
Outside of pregnancy, low dose aspirin (100–150 mg)
is recommended in addition to warfarin for patients
with mechanical heart valves with other thromboem-
bolic risk factors (Table 9.3). Low dose aspirin has been
shown to reduce the risk of major thromboembolic
events such as valve thrombosis and CVA in patients
at the expense of an increase in minor, but not major
bleeding.20 e prothrombotic changes of pregnancy
could also be considered as an additional risk factor
for thromboembolism supporting the use of low dose
aspirin during pregnancy in addition to warfarin or
heparin.
A simplied summary of risks and benets asso-
ciated with UFH, LMWH and warfarin is provided in
Table 9.6.
Although there are limited published data relating
to use of LMWH for anticoagulation during pregnancy
in women with mechanical heart valves, it is pos-
sible that thromboembolic complications may in part
relate to suboptimal LMWH doses. Regular mon-
itoring of anti-Xa levels with dose-adjustment of
LMWH may provide more eective thrombopro-
phylaxis. e high risk ante-natal medical clinic at
National Women’s Health, Auckland City Hospital has
published one of the largest single case series of women
with mechanical heart valves treated with enoxaparin
and aspirin during pregnancy.21 Women attending this
clinic are informed that the safest option for them is
nottobecomepregnantbutthosewhochoosetopro-
ceed with pregnancy have in-depth counseling of the
Table 9.7 Anticoagulant options offered to women with
mechanical heart valves attending high-risk medical ante-natal
clinic, National Women’s Health, Auckland City Hospital,
New Zealand
Option 1. Substitution of warfarin with therapeutic dose
enoxaparin (1 mg/kg bd) and aspirin 150 mg before 6
weeks’ gestation, continued until planned delivery
Option 2. Substitution of warfarin with therapeutic dose
enoxaparin before 6 weeks until 12 completed weeks,
reverting to warfarin until 34–36 weeks, then therapeutic
dose enoxaparin until planned delivery. Aspirin 150 mg
throughout pregnancy.
Option 3. Warfarin and aspirin 150 mg throughout pregnancy,
switching to therapeutic enoxaparin and aspirin at 34–36
weeks’ gestation until planned delivery.
Low-dose aspirin (100–150 mg) recommended for all women
maternal and fetal risks and benets of dierent anti-
coagulant agents and provide written consent for their
choice of one of three anticoagulant regimens (Table
9.7). Regular anticoagulant monitoring is carried out
with target therapeutic ranges of INR and anti-Xa lev-
els as listed in Table 9.8. Low-dose aspirin is recom-
mended for all women.
e thromboembolic complications reported with
dose-adjusted UFH and the variable dose–response
rateswiththisagentsuggestthatitmaybealessreliable
alternative to LMWH. Given the signicant advan-
tages in terms of fetal outcome, perhaps monitored
therapeutic-dose LMWH for prevention of mater-
nal valve thrombosis and systemic thromboembolism
could be an attractive alternative to warfarin for use in
this clinical setting.22 Certainly, it would seem prema-
ture to contraindicate use of therapeutic dose LMWH
during pregnancy for these women23 given the lack
of an acceptable alternative anticoagulant and also the
114
Chapter 9. Prosthetic heart valves
Table 9.8 Anticoagulant monitoring for women with mechanical heart valves attending high-risk medical ante-natal clinic, National
Women’s Health, Auckland City Hospital, New Zealand
Drug Laboratory test Valve type
Anticoagulant target
range
Enoxaparin (1 mg/kg bd) Anti-Xa levels – 3–5 days after first
dose then monthly
All valves Trough (pre-dose) 0.4–0.7 IU
Peak (4 h post-dose) 0.7–1.2 IU
Warfarin Monthly INR Starr–Edwards valves
Bileaflet and tilting disc valves
3.0–4.5
2.5–3.5
continued emergence of safety and ecacy data of its
useinthisclinicalsetting.Intheabsenceofaran-
domized clinical trial, clinicians must rely on best-
practice guidelines based on existing evidence and
experience.
Other critical issues to consider when deciding on
anticoagulant management in pregnant women with
mechanical heart valves include the cost of LMWH
and access to laboratory testing for anti-Xa levels. In
countries with limited resources it may be more appro-
priate to treat women with warfarin but these decisions
will rest with individual clinicians.
Other management issues
Regular follow-up during pregnancy is essential to
make sure frequent monitoring of anticoagulation is
done, and careful clinical assessment to detect any car-
diac complications. Table 9.9 lists important aspects of
care. e infants of mothers with congenital heart dis-
ease have a ve- to tenfold increased risk of congenital
heart defects themselves and careful anatomy scan or
fetal echocardiogram is required at 20–24 weeks’ ges-
tation.
Table 9.9 Summary of ante-natal care required for women
with mechanical heart valves: other maternal and fetal issues
Management of pregnancies in women with mechanical heart
valves: other issues
(1) Regular patient review: every 4 weeks until 28 weeks’
gestation, every two weeks until 34 weeks’ gestation then
weekly until delivery
(2) Urgent assessment if patient concerns about possible
thromboembolism, change in cardiac symptoms,
bleeding, preterm labor
(3) Clinical assessment to include enquiry about symptoms of
systemic thromboembolism, heart failure, cardiac rhythm,
maternal hypertension.
(4) Careful auscultation of maternal heart at each visit: new
murmurs.
(5) Maternal echocardiogram every trimester or more
frequently if clinical concern
(6) Fetal surveillance: regular scans for fetal growth, evidence
of intracranial hemorrhage
Management of labor and delivery
Management of women in the peri-delivery period
requires close clinical monitoring, given the bleed-
ing risks associated with therapeutic anticoagulation.
e mode of delivery should be determined by obstet-
ric indications but vaginal delivery is preferable to
Cesarean section as it minimizes time o therapeutic-
dose anticoagulation – the increased risk of bleeding
from the operative site necessitates a delay in restart-
ing anticoagulation postpartum. A planned delivery
allows for better control and adjustment of anticoagu-
lation.
Women on oral anticoagulants
Women should stop warfarin by 34–36 weeks’ gesta-
tion to allow normalization of the infant’s INR and
minimize the risk of fetal intracranial hemorrhage at
delivery. Although cessation of OAC leads to normal-
ization of the woman’s INR within 3–4 days, the pro-
cess oen takes much longer in the infant as it is likely
to be over-anticoagulated given the immaturity of its
coagulation system. Anticoagulant options for women
who have taken warfarin until the peri-delivery period
include:
rcontinuous intravenous unfractionated heparin
aiming for aPTT 2–3x baseline;
rdose-adjusted subcutaneous UFH bid aiming for
aPTT 2–3x baseline;
rtherapeutic dose LMWH with monitoring of
anti-Xa levels (see Table 9.8).
Tables 9.10 and 9.11 summarize peri-delivery man-
agement of anticoagulation for planned vaginal deliv-
ery or cesarean section, respectively. Gradual re-
introduction of intravenous UFH, as outlined in Tables
9.10 and 9.11, is the preferred option for anticoagu-
lation in the immediate postpartum period. Intra-
venous UFH has the advantage over LMWH of allow-
ing more exible dose adjustment including more 115

Section 3. Thromboembolism and anticoagulation
Table 9.10 Management of anticoagulation prior to induction
of labor for planned vaginal delivery in women on
subcutaneous LMWH (National Women’s Health, Auckland City
Hospital, New Zealand)
(1) 36 hours prior to planned CS – last dose s/c LMWH
(2) 24 hours prior to induction – start IV UFH infusion 5000 U
bolus dose then 1200 U/h
(3) Check aPTT 6-hourly – target aPTT 2–3 ×baseline∗
(4) Discontinue iv UFH when woman in established labor
(5) If regional analgesia required stop iv UFH 4 hr prior to
epidural catheter placement, check aPTT back to baseline
prior to placement, restart iv UFH 3 hours after catheter
placement
(6) 4–6 h postpartum, restart iv UFH at 500 U/h (no bolus dose)
if no bleeding concerns then increase dose by 250 U every
4–6 hours until aPTT 2–3 ×baseline∗
(7) Start oral anticoagulant on first postpartum day if
uncomplicated vaginal delivery or day 2–3 if Cesarean
section or other bleeding complications
∗therapeutic range may vary between centers, being dependent
on the sensitivity of the aPTT reagent used. It should also be
noted that the aPTT can be less reliable in pregnancy due to
increased levels of factor VIII and heparin binding proteins.
Table 9.11 Management of anticoagulation prior to elective
Cesarean section in women on subcutaneous LMWH (National
Women’s Health, Auckland City Hospital, New Zealand)
(1) 36 hours prior to planned CS – last dose s/c LMWH
(2) 24 hours prior to planned CS – start iv UFH infusion 5000 U
bolus dose then 1200 U/h
(3) Check aPTT 6-hourly – target aPTT 2–3 ×baseline∗
(4) Stop iv UFH 4 hr prior to epidural catheter placement for
regional anesthesia, check aPTT back to baseline prior to
placement.
(5) 6–12 h post-delivery, restart iv UFH at 500 U/h (no bolus
dose) if no bleeding concerns, then increase dose by 250 U
every 4–6 hours until aPTT 2–3 ×baseline∗
(6) Delay starting oral anticoagulant until epidural catheter
removed if remains in situ for post-operative pain
management
∗therapeutic range may vary between centers, being dependent
on the sensitivity of the aPTT reagent used. It should also be
noted that the aPTT can be less reliable in pregnancy due to
increased levels of factor VIII and heparin binding proteins.
rapid and complete reversal of its anticoagulant eect
in the event of clinically signicant bleeding in the
postpartum period, especially as oral anticoagulants
are restarted. Some centers may lack the experience of
managing intravenous UFH infusions so an alternative
may be to use intermediate doses of LMWH i.e. enoxa-
parin 40mg daily then twice daily until the rst few
days postpartum until the INR is therapeutic. How-
ever, not only is there the concern of a higher risk of
valve thrombosis with low doses of LMWH, if bleed-
ing does occur, the anticoagulant eect cannot be com-
pletely reversed.
In the case of an obstetric emergency such as
preterm labor or placental abruption, rapid rever-
sal of anticoagulation is required. Management
of heparin reversal with protamine is outlined in
Chapter 10. Eective reversal of oral anticoagulation
requires administration of 5.0–10.0 mg vitamin K1
intravenously, as well as prothrombin concentrate or
fresh frozen plasma.
Prevention of infective endocarditis
Antibiotic prophylaxis against infective endocarditis is
recommended for women with prosthetic heart valves
following vaginal delivery or Cesarean section, given
thehighriskofadverseoutcomeshouldthiscom-
plication occur.24 Women who have prosthetic heart
valves as a result of rheumatic heart disease require
secondary prevention of acute rheumatic fever and
rheumatic heart disease for a minimum of 10 years
aer the most recent episode or until 30–40 years
of age (whichever is longer). Benzathine penicillin-G,
benzylpenicillin, or erthyromycin are considered safe
in pregnancy.
Management of valve thrombosis
Development of neurological symptoms, chest pain or
symptoms of heart failure, or detection of a new car-
diac murmur warrants exclusion of valve thrombosis
using transthoracic or transesophageal echocardiog-
raphy. Minor valve thromboses can oen be man-
aged by increasing the intensity of anticoagulation,
but for more severe thromboses valve replacement
surgery is usually required, although some centers
have reported success with thrombolysis using rtPA
or streptokinase.25 Women with valve thrombosis
shouldbemanagedjointlybytheobstetricandcar-
diothoracic surgical team. Readers are referred to
recent review articles discussing management issues in
pregnancy.26–28
Summary
e clinical challenge of managing anticoagulation in
pregnancy in women with mechanical heart valves is
set to continue. ese women and the clinicians caring
for them are faced with a true dichotomy: the choice
of warfarin or heparin. Taking warfarin in pregnancy
minimizes the risk of thrombotic complications but
carries a high-risk of adverse fetal outcome; LMWH
116

Chapter 9. Prosthetic heart valves
has an uncertain maternal risk but the fetal compli-
cations are very low. Experience suggests that, given
the choice, many women will opt for what is safer
forthebaby–especiallyifitistheirrstpregnancy
or they have had a bad fetal outcome with warfarin
previously. However, women who develop complica-
tions with valve thrombosis or stroke due to subop-
timal anticoagulation also place their babies at risk.
Data relating to the safety and ecacy of LMWH con-
tinues to emerge and in the absence of a random-
ized trial, data from registries such as the International
Registry of Pregnancies in Women with Mechanical
HeartValvesdevelopedbytheISTHSubcommitteeon
Women’sHealthIssuesinrombosisandHaemosta-
sis may help provide clearer guidance for management
in the future.
117

Section 3. Thromboembolism and anticoagulation
References
1. Carapetis JR, Steer AC, Mulholland EK, Weber M. e
global burden of group A streptococcal diseases. e
Lancet Infectious Diseases. 2005; 5: 685–694.
2. Atatoa-Carr P, Lennon D, Wilson N. Rheumatic fever
diagnosis, management, and secondary prevention: a
New Zealand guideline. New Zealand Medical Journal
2008; 121: 59–69.
3. Australian Institute of Health and Welfare. Rheumatic
heart disease: all but forgotten in Australia except
among Aboriginal and Torres Strait Islander peoples.
Australian Institute of Health and Welfare: Australian
Government 2004.
4. Sadler L, McCowan L, White H. Pregnancy outcomes
and cardiac complications in women with mechanical,
bioprosthetic and homogra valves. BJOG: An
International Journal of Obstetrics and Gynaecology
2000; 107: 245–253.
5. Bach DS. Choice of prosthetic heart valves:update for
the next generation. Journal of the American College of
Cardiology 2003; 19; (10): 1717–1719.
6. Cannegieter SC, Rosendaal FR, Briet E.
romboembolic and bleeding complications in
patients with mechanical heart valve prostheses.
Circulation 1994; 89: 635–641.
7. Chan WS, Anand S, Ginsberg JS. Anticoagulation of
pregnant women with mechanical heart valves: a
systematic review of the literature. Archives of Internal
Medicine 2000; 160: 191–196.
8. Bates SM, Greer IA, Pabinger I et al.Venous
thromboembolism, thrombophilia, antithrombotic
therapy, and pregnancy: American College of Chest
Physicians Evidence-Based Clinical Practice
Guidelines (8th edn). Chest 2008; 133: 844S–886S.
9. Bonow RO, Carabello BA, Chatterjee K et al. focused
update incorporated into the ACC/AHA 2006
guidelines for the management of patients with
valvular heart disease: a report of the American
College of Cardiology/American Heart Association
Task Force on Practice Guidelines. 2008; 52: e1–142.
10. De Caterina R, Husted S, Wallentin L et al.
Anticoagulants in heart disease: current status and
perspectives [see comment]. European Heart Journal
2007; 28: 880–913.
11. Kawamata K, Neki R, Yamanaka K et al. Risks and
pregnancy outcome in women with prosthetic
mechanical heart valve replacement. Circulation
Journal 2007; 71: 211–213.
12. Lee JH, Park NH, Keum DY et al. Low molecular
weight heparin treatment in pregnant women with a
mechanical heart valve prosthesis. Journal of Korean
Medical Science 2007; 22: 258–261.
13. Meschengieser SS, Fondevila CG, Santarelli MT,
Lazzari MA. Anticoagulation in pregnant women with
mechanical heart valve prostheses. Heart 1999; 82:
23–26.
14. James AH, Brancazio LR, Gehrig TR et al.
Low-molecular-weight heparin for
thromboprophylaxis in pregnant women with
mechanical heart valves. Journal of Maternal–Fetal and
Neonatal Medicine 2006; 19:543–549.
15. Oran B, Lee-Parritz A, Ansell J. Low molecular weight
heparin for the prophylaxis of thromboembolism in
women with prosthetic mechanical heart valves during
pregnancy. rombosis and Haemostasis 2004; 92:
747–751.
16. Hall JG, Pauli RM, Wilson KM. Maternal and fetal
sequelae of anticoagulation during pregnancy.
American Journal of Medicine 1980; 68: 122–138.
17. Van Driel D, Wesseling J, Sauer PJJ et al. Teratogen
update: feta; eects oer in utero exposure to
coumarins overview of cases, follow-up ndings, and
pathogenesis. Teratolog y 2002; 66: 127–140.
18. Vitale N, De Feo M, Cotrufo M. Anticoagulation for
prosthetic heart valves during pregnancy: the
importance of warfarin daily dose. European Journal of
oracic and Cardiovascular Surgery 2002; 22: 656.
19. Cotrufo M, De Feo M, De Santo LS et al. Risk of
warfarin during pregnancy with mechanical valve
prostheses. Obstetrics and Gynecology 2002; 99:
35–40.
20. Turpie AGG, Gent M, Laupacis A et al. Acomparison
of aspirin with placebo in patients treated with
warfarin aer heart-valve replacement. New England
Journal of Medicine 1994; 329: 524–529.
21. Rowan JA, McCowan LM, Raudkivi PJ, North RA.
Enoxaparin treatment in women with mechanical
heart valves during pregnancy. American Journal of
Obstetrics Gynecology 2001; 185: 633–637.
22. McLintockC,NorthRA,WhiteHD.Prostheticheart
valves and pregnancy. Circulation 2003; 108: e159–
60.
23. American College of Obstetricians and Gynecologists
Committee on Obstetric Practice. ACOG committee
opinion. Safety of Lovenox in pregnancy. Number 276,
October 2002. Committee on Obstetric Practice.
International Journal of Gynaecology and Obstetrics
2002 ; 79: 299–300.
24. Wilson W, Taubert KA, Gewitz M et al. Prevention of
infective endocarditis: guidelines from the American
Heart Association: a guideline from the American
Heart Association Rheumatic Fever, Endocarditis, and
Kawasaki Disease Committee, Council on
Cardiovascular Disease in the Young, and the Council
118

Chapter 9. Prosthetic heart valves
on Clinical Cardiology, Council on Cardiovascular
Surgery and Anesthesia, and the Quality of Care and
Outcomes Research Interdisciplinary Working Group.
Circulation 2007; 116: 1736–1754.
25. Leonhardt G, Gaul C, Nietsch HH et al. rombolytic
therapy in pregnancy. Journal of rombosis and
rombolysis 2006; 21: 271–276.
26. Dieter RS, Dieter RA, Jr., Dieter RA, 3rd et al.
Prosthetic heart valve thrombosis: an overview.
Wisconsin Medical Journal 2002; 101: 67–69.
27. Lengyel M, Horstkotte D, Voller H, Mistiaen WP,
Working Group Infection rombosis, Embolism and
Bleeding of the Society of Heart Valve Disease.
Recommendations for the management of prosthetic
valve thrombosis. Journal of Heart Valve Disease 2005;
14: 567–575.
28. Nassar AH, Hobeika EM, Abd Essamad HM et al. Usta
IM. Pregnancy outcome in women with prosthetic
heart valves. American Journal of Obstetrics and
Gynecology 2004; 191: 1009–1013.
119

Section 3 Thromboembolism and anticoagulation
Chapter
10 Management of anticoagulants at delivery
Christina Oppenheimer and Paul Sharpe
Introduction
is chapter will address the practical obstetric and
anesthetic management of women on prophylactic
heparin and therapeutic anticoagulation in the peri-
partum period, and the dilemmas for obstetricians,
anesthetists, and hematologists. Also considered will
be issues surrounding use of thrombolytic agents in
pregnancy and unusual but complex situations such as
cardiopulmonary bypass in pregnancy.
Increasing use of prophylactic anticoagulants in
pregnancy, both for venous thromboprophylaxis and
to modify fetal risk, as in antiphospholipid syn-
drome, means that more women are now reaching the
peri-partum period on anticoagulants, usually a low
molecular weight heparin. erapeutic doses are used
for treatment of acute venous thromboembolic events,
prevention of thromboembolism in women with car-
diac disease including mechanical heart valves, acute
cardiac events and cardiomyopathy, and those on long-
term anticoagulation outside of pregnancy for a variety
of other indications. is situation necessitates careful
assessment of risks, close multidisciplinary discussion
and planning, and expert management by the medi-
cal and midwifery teams during labor or Cesarean sec-
tion. Careful discussion of risks and therapeutic deci-
sions with the patient and her partner are also essen-
tial.
Thromboprophylaxis
e use of a variety of anticoagulants in obstetric prac-
tice has been increasing steadily over the last 15 years,
bringing with it increasing awareness of the need for
attentiontothromboprophylaxis,butalsotheneed
to adapt and plan for the risks associated with this.
romboprophylaxis guidance from the Royal Col-
lege of Obstetricians and Gynaecologists (RCOG)1
and the recent report from the Condential Enquiry
into Maternal Health (CEMACH)2have both signif-
icantly raised awareness of the importance of risk
assessment for venous thromboembolism, and hence
increased use of low molecular weight heparins in par-
ticular (Chapter 8). e recent and rapid rise in preva-
lence of obesity in women of child-bearing age also
brings many women into a high risk category neces-
sitating use of general as well as pharmacological anti-
thrombotic measures.
Antiplatelet agents
Increasing use of low-dose aspirin, rstly following the
publication of the CLASP trial in 1994,3then the work
on anti-phospholipid syndrome in recurrent miscar-
riage by Regan and colleagues, as well as a gradual
increase in numbers of women on long-term aspirin
for medical conditions, such as previous stroke, has led
to guidance on aspirin use around the time of delivery.
e use of aspirin 75 mg up to the time of labor and
delivery is not contraindicated on either obstetric or
anesthetic grounds. At this dose there is no increased
risk of bleeding either at vaginal or Cesarean deliv-
ery, nor is there evidence of any increase in the risk
of vertebral canal hematoma aer spinal or epidural
block insertion. However, it must be remembered that,
if used with either a heparin or warfarin in the postpar-
tum period, there may be an additive eect and par-
ticular care should be taken with timing of dose
administration and epidural catheter removal. ere
is no contraindication to breastfeeding on aspirin at
75 mg daily, in particular, there is no evidence of risk
of Reye’s syndrome at this dose.
e safety in pregnancy of other antiplatelet agents
such as clopidogrel or ticlopidine at usual therapeu-
tic doses has not been established and they are rarely
used. Both the indication for use and a clear plan of
120 e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Chapter 10. Management of anticoagulants at delivery
management to minimize risk at the time of delivery,
including whether to interrupt the treatment, should
be made on an individual case basis with consultation
among responsible obstetrician, physician, and anes-
thetist.
Non-steroidal anti-inammatory drugs are largely
contra-indicated in pregnancy and should not be used
aroundthetimeofdelivery.
Low molecular weight
heparins (LMWH)
Obstetric aspects
Most of the women requiring prophylactic doses of
anticoagulant will be given one of the low molecu-
lar weight heparins. is is usually given once daily,
although those with a particularly high thrombotic
risk, obesity, or mechanical heart valves may be on
twice daily doses.
While increasing numbers of women being pre-
scribed LMWH for thromboprophylaxis leads to
increased experience for sta in managing such preg-
nancies, it also necessitates sucient knowledge for
safe practice, particularly around the time of labor and
delivery. is would take into account the half-life,
increased clearance in pregnancy and dierent thera-
peutic index of dierent heparins.4Underpinning all
of the clinical management described below must be
collaboration, clear local guidelines, knowledge and
education of medical and midwifery sta, as well as
written information for patients and individual care
plans based on a woman’s particular risk factors.
Labor and delivery
In the presence of increased risk of venous throm-
boembolism, the optimum management will be to aim
for a spontaneous onset of labor as these are on aver-
age shorter and have a lower risk of operative deliv-
ery. Clearly, this advantage needs to be weighed against
other obstetric and medical risks if present.
AstandardapproachistoomittheLMWHat
the onset of labor and ensure general antithrombotic
measures, including adequate hydration, with early
recourse to intravenous uids if necessary, mobiliza-
tion, with passive movements or massage if mobility
restricted by epidural, and the wearing of graduated
compression stockings. If a woman has no additional
complications other than a need for LMWH thrombo-
prophylaxis, there is no contraindication to delivering
in a midwifery-led setting provided at least 8 hours has
elapsed since the last dose by delivery. If women in this
situation request a home connement, this should be
discussed on an individual basis.
Induction of labor
If an induction of labor is planned, the LMWH should
beomittedatthestartoftheprocess.Ifamorn-
ing dose is usual, the dose on the day of induction
should be omitted, or if an evening dose, that on the
evening before should be taken as usual, but none fur-
ther until aer delivery. If an omission of more than
48 hours is thought to be contraindicated, intermittent
doses of subcutaneous UFH, 5000u, could be consid-
ered 8 hourly until articial rupture of the membranes
(ARM) is possible, although this is rarely needed. UFH
has a half-life of about 3 hours, so that an anticoagulant
eect at a level suitable for regional anesthetic block-
ade is possible around that point. With prophylactic
LMWH this delay needs to be 8–12 hours depending
on dose as discussed below. Attention should be paid to
general antithrombotic measures including compres-
sion hose, hydration, and mobility.
A prolonged induction process is more likely in
primigravida and those with an unfavorable cervix at
the start of the induction. us, where possible, care-
ful assessment should be made to try to delay induc-
tion until the cervix is more favorable. If this is not
possible, there will then be the dual additional risk of
a prolonged period where the woman is only covered
by general antithrombotic measures and an increased
risk of instrumental delivery or Cesarean section (CS)
inherent in an induced labor.
Practical issues
At prophylactic doses of LMWH or UFH, there is no
contraindication to intramuscular analgesics during
laborortointramuscularsyntometrineatdelivery.e
LMWH should be recommenced 3–6 hours aer deliv-
ery, providing hemostasis is secured and the timing
around removal of epidural catheter is considered, as
described below.
It should also be emphasized that, for standard pro-
phylactic doses of LMWH, no signicant increase in
the risk of intra- or postpartum bleeding, paravagi-
nal hematoma or prolonged lochia has been shown;
however, it is good practice to ensure adequate and
timely use of uterotonics and early suturing of any tear
or episiotomy. ere is no contraindication to puden-
dal or perineal block for analgesia. Also, there is no 121

Section 3. Thromboembolism and anticoagulation
increased risk of excess surgical bleeding at Cesarean
section, although risk of wound hematoma may be
slightly increased.
Breastfeeding
ere should be a clear plan of the length of time
to continue the LMWH postpartum. e woman
should be reassured that there is no contraindication
to breastfeeding since, although the LMWH will be
present in breastmilk, it will be broken down in the
gastric acid before absorption can occur.
Anesthetic aspects
Oneofthemajorconcernsabouttheuseofhep-
arins in the peripartum period in relation to anes-
thesia is the risk of vertebral canal hematoma and
its severe sequelae. is was rst raised as a signi-
cant issue in publications from the USA,5and subse-
quently there has been much study and debate lead-
ing to some standardization and guidance.6,7 Over
40 cases of vertebral canal hematoma were reported
in the American literature in 1997–1998, from a 5-
year observation period, in patients given enoxaparin,
mostly following epidural, spinal, or lumbar punc-
ture needle insertion. However, subsequent European
reports include only two such cases with vertebral
canal hematoma. e incidence has been estimated at
1 in over 2 million in a European study, but about
1 in 15 000 in American studies. Numerous factors
including timing of LMWH in relation to needle
insertion or epidural catheter removal, and dosing
schedules have been implicated. Other studies have
suggested that technical diculties with needle and
particularly epidural catheter insertion may also be
associated with increased risk of hematoma, particu-
larly multiple insertion attempts or blood-stained tap.8
(Table 10.1)
Table 10.1 Estimated incidence for vertebral canal
hematoma in spinal and epidural anesthesia
Epidural Spinal
Without heparin
Atraumatic
Traumatic
1/220 000
1/20 000
1/320 000
1/29 000
Heparin given after procedure
Atraumatic
Traumatic
UFH more than 1 hr after puncture
UFH less than 1 hr after puncture
1/70 000
1/2 000
1/100 000
1/8 700
1/100 000
1/2 900
1/150 000
1/13 000
Thromboprophylaxis for elective operative procedures
Single shot spinal versus epidural catheter techniques
Single shot subarachnoid anesthesia remains a popular
choice amongst obstetric anesthetists to provide oper-
ative conditions for elective Cesarean section. is is
primarily because of the superior nature of the quality
of anesthesia produced when compared with epidural
anesthesia. Because there is no catheter to be removed,
this technique facilitates the use of a single agent in the
post-operative period. A minimum of 2 hours should
be allowed to elapse between completing the subarach-
noid injection and the administration of a LMWH.
e use of an epidural catheter in isolation, for
example to provide more controlled onset of anesthe-
sia in patients with cardiovascular instability, is less
common in the elective setting. e introduction of
the combined spinal epidural (CSE) has allowed a ex-
ible approach. e epidural space is located with a
Tuohy needle using a loss of resistance technique, usu-
ally with a 16- or 18-gauge needle. A spinal needle, of
smaller gauge such as 26, is then introduced through
the Tuohy needle to pierce the dura and enter the
subarachnoid space. Once this thinner spinal needle
has been removed, the epidural catheter can then be
threaded through the Tuohy needle into the epidu-
ral space. is technique has provided great exibil-
ity. Administration of reduced doses of local anes-
thetic into the subarachnoid space, with further doses
administered via the epidural catheter allows excel-
lent control over the cardiovascular system. e pres-
ence of an epidural catheter also allows the anesthetist
to provide additional doses of anesthetic in cases of
inadequate anesthesia or prolonged operative deliv-
ery. However, as this technique involves a catheter in
situ at the end of surgery, the operative team are pre-
sented with two options in women requiring thrombo-
prophylaxis: (1) administration of a single anticoagu-
lant, most commonly a LMWH or (2) a combination
approach.
IfaLMWHisgivenasasoleagent,itispreferable
to wait 2–4 hours aer removal of the Tuohy needle.
e epidural catheter should be le in situ until the
drug levels have reached a safe trough (see Table 10.2).
A combination of unfractionated heparin followed by
LMWHallowstheepiduralcathetertoberemovedina
highly monitored environment in the Recovery Ward
and also permits more controlled reversal of heparin
eects if required in cases of massive post partum
hemorrhage. When unfractionated heparin is given at
the end of Cesarean section, local data suggest that
122

Chapter 10. Management of anticoagulants at delivery
coagulation proles are equal to pre-operative values
at 4 hours post-operation, providing there has been no
signicant peripartum bleed.
Labor analgesia
It is quite common for a “safe window” to be present
to administer epidural analgesia as a result of ante-
natal assessment and a plan documented to omit or
appropriately manage heparin once labor starts. For
prophylactic dosing, there is current guidance on tim-
ings for insertion of epidural blockade. is should
be utilized regularly as part of ante-natal planning,
and agreed between obstetric, anesthetic, and hema-
tology teams. e demonstrated increase in clearance
of LMWH in the pregnant woman allows a slightly dif-
ferent regime to surgical patients in general. A scheme
allowing neuraxial analgesia 8–10 hours following the
administration of 2500 units and 12 hours following
a dose of 5000 units of dalteparin for example, would
be appropriate (see Table 10.2). e ability to pre-
dict an appropriate time is harder when higher doses
of LMWH have been used, or in situations where a
woman presents on full anticoagulant therapy, but in
general this needs to be at least 24 hours aer the last
dose. An assessment based on previous anti-Xa lev-
els if available, and on a risk–benet analysis for each
patient and situation is needed.
If anticoagulation is to commence aer delivery,
an adequate gap should be le aer the removal of
the epidural catheter before the heparin is adminis-
tered, usually around 4 hours for LMWH. Whilst most
anesthetists accept a 2-hour interval between catheter
Table 10.2 Guidance for relative timings of heparin and
epidural or spinal block
A. Subcutaneous prophylactic dose
unfractionated heparin
Catheter placement or removal ⬎2–4 h after injection
Delay next dose until ⬎2 h after catheter insertion
or ⬎4 h after removal
B. Intravenous infusion of unfractionated heparin
Catheter placement ⬎4 h after stopping infusion, when aPTT
back to baseline
Restart infusion ⬎2 h after catheter insertion
or ⬎4 h after removal
C. Low molecular weight heparin
Spinal or epidural catheter insertion
⬎8 h after last injection – low dose
⬎12 h after last injection – intermediate dose
⬎24 h after last injection – full anticoagulation
Remove epidural catheter
12hafteranydose
Delay next dose until ⬎2 h after catheter insertion or
subarachnoid injection
or ⬎4 h after catheter removal
insertion and heparin injection, as this can easily be
lengthened if the procedure is dicult or traumatic,
4 hours is preferred aer removal as the catheter can
pull on established clot and stir up bleeding, even when
insertion had been apparently uneventful.
Full anticoagulation
Womenwhoarefullyanticoagulatedatthetimeof
labor and delivery include those with signicant risks
of morbidity – a recent venous thromboembolic event,
those normally on long-term warfarin for a variety
of conditions, cardiac disease including mechanical
valves, ischemic heart disease and cardiomyopathy,
and symptomatic homozygous or combination herit-
able thrombophilias.9–11
e management of these women with a need
for therapeutic levels of anticoagulation in the peri-
partum period involves balancing the risks of exces-
sive bleeding and diculties with analgesia and
anesthesia against the risk of thrombosis associ-
ated with the underlying condition. is requires
a careful and individualized approach and thor-
ough forward planning, by a team including obste-
trician, midwife, anesthetist, and hematologist, and
in full consultation with the patient (table 10.3).
is is particularly important for the small number
ofwomeninwhomtheusuallyrecommendedtem-
porary peripartum reduction in level of anticoagu-
lation may be considered unsafe.
If it has been necessary to use warfarin during the
pregnancy, this should be stopped by 34–36 weeks’
gestation, to allow correction of the fetal coagulopa-
thy, which takes longer than that of the mother,
and minimize the risk of intracranial hemorrhage at
delivery. e most common practice is to replace it
with therapeutic dose LMWH and monitor with anti-
Xa levels as well as clinically. e anti Xa level is
checked at 3–5 days following the rst dose and, if in
the desired therapeutic range does not usually need
repeating.
Ifvaginaldeliveryisintended,plannedinduc-
tion of labor once the cervix is suciently favorable
should be considered. is allows more accurate tim-
ing of events and minimizes the risk of delivery whilst
fully anticoagulated. e LMWH should be omitted
on the morning of induction. If prophylaxis needs to
be continued for the day of labor, 5000iu UFH can
be given subcutaneously 8 hourly. If treatment doses
are necessary, an infusion of UFH at 1200 u/h should
be started. APTT should be checked at 4 hours aer the
123

Section 3. Thromboembolism and anticoagulation
infusion was commenced, aiming for the therapeutic
range as determined by the local laboratory. It should
be noted that the aPTT is less reliable in pregnancy due
to increased levels of factor VIII and heparin binding
proteins.
is regime has been shown to be useful when
stopped 1–6 hours (typically 4) pre-labor, with mini-
mal obstetric or anesthetic complication.12 However,
if this interruption prior to labor is not appropriate,
the UFH infusion should be stopped when cervical
dilatation reaches 5 cm dilatation in primipara, or an
appropriate pre-planned dilatation depending on pre-
vious labor experience in multipara. It should also be
stopped if increased unscheduled blood loss is noted
or if urgent Cesarean section is required.13
Protamine sulphate must be immediately available
asshouldtheregimeforitsuse,and4unitsofcross-
matched red cells should be available. A protamine sul-
phate regime is described in Table 10.4.
Graduated compression stockings should be worn
throughout induction and labor and throughout the
inpatient stay, mobility should be encouraged and
hydration ensured.
Aer delivery, the third stage of labor should be
actively managed with oxytocin, given by intravenous
bolus 10 u followed by an infusion of 40 u over 4 hours.
Perineal tears or episiotomy should be repaired imme-
diately with careful attention to hemostasis and hep-
arin restarted as soon as hemostasis has been secured.
A high index of suspicion must always be main-
tained when caring for these women with regard
to hemorrhagic complications during pregnancy. e
incidence of antepartum hemorrhage and abruption
is not increased, but is more likely to be signicant.
A careful plan for investigation and management of
unexplained abdominal pain or unscheduled bleeding
must be available in the notes.
Other issues which must be considered are the
inadvisability of pudendal block, the potential mater-
nal risks of instrumental delivery if labor occurs
spontaneously and without the lapse of time since
thelastdoseofheparin,andtheimportanceof
careful postpartum observations for development of
hematomata.
If the woman is on warfarin when labor starts, the
INR must be checked urgently and if greater than 3 or
if a Cesarean section is required, reversed with vita-
min K and if more urgent, prothrombin complex con-
centrate as well. In this instance, a cord blood clot-
ting screen must be taken aer delivery as intravenous
Table 10.3 Recommended thromboprophylactic and
anti-hemorrhagic measures
General measures: Optimize anticoagulation ante-natally
Formulate intrapartum care plan
Multidisciplinary intrapartum care
Senior staff involvement in care
throughout
Aim for vaginal delivery whenever
possible
Specific
thromboprophylactic
measures:
Mobilize during labor
Maintain hydration
Graduated compression stockings
throughout labor and postpartum
Specific
anti-hemorrhagic
measures:
Minimize maternal soft tissue trauma
(e.g. difficult instrumental delivery,
perineal trauma)
Active third stage with intravenous
oxytocics
Prompt management of
complications (e.g. retained
placenta, perineal trauma)
Vitamin K may be required for the neonate. Early
involvement of the neonatologists is essential.
Anesthetic considerations
Analgesia and anesthesia in this group of women is a
major challenge and early involvement of senior anes-
thetic sta is essential in planning intrapartum care.
Regional analgesia is clearly contraindicated because
of the risk of vertebral canal hematoma. A review
by Loo et al. documented an overall incidence of
0.2–0.3/100 000 following obstetric epidural analge-
sia, with coagulation abnormalities being identied as
a major risk factor.14
Analgesia for labor
e role of a consistent intrapartum care provider, par-
ticularly midwifery, is of high importance – one-to-
one care from a single carer has been shown to reduce
analgesia requirements and will also reduce the many
anxieties associated with labor in such a situation.13
Other guidance aids decision making, concerning the
need for continuity of anticoagulation, or a window to
reducetheriskofhemorrhageandaltertherangeof
analgesics available.15
Pharmacological intervention includes inhaled
analgesia in the form of entonox, or administration
of systemic opioids. In a fully anticoagulated woman
intramuscular injection is contraindicated, so bolus or
patient-controlled intravenous administration of opi-
oid analgesia is a suitable option.
124

Chapter 10. Management of anticoagulants at delivery
Traditionally, opioid therapy used by most deliv-
ery suites across the United Kingdom was intermit-
tent bolus administration of intramuscular drugs such
as pethidine. e desire to match the pharmacokinet-
ics of opioids to the time course of the cyclical pain
associated with labor has led to the investigation of
shorter-acting opioids administered in small repeated
doses. Patient-controlled analgesia pumps deliver a
small pre-set dose of opioid via an intravenous can-
nula.
Fentanyl is a synthetic phenylpiperidine derivative
andisahighlyselectivemuopioidagonist.Whengiven
by the intravenous route, the dose is eective within
2 to 5 minutes. Fentanyl is highly lipid soluble and
therefore the drug in the plasma rapidly redistributes
to fat-rich areas. is accounts for the short duration
of fentanyl in clinical practice. If large doses are given,
in, for example, a prolonged labor, then the reservoir
for redistribution becomes full. e duration of action
therefore becomes exaggerated from each subsequent
dose, behaving more like morphine. e time to clin-
icalactionmeansthatitcanbedicultforthelaboring
woman to coincide the analgesic action with the peak
of each contraction.
Remifentanil is another synthetic opioid of the
anilidopiperidine group. It has an ultra-short dura-
tion of action due to unique metabolism by plasma
esterases. It has a peak onset of 1 to 3 minutes, thus
making the timing relative to contractions easier to
manage. Remifentanil can be given by bolus dose,
or a combination of background infusion with sup-
plemental bolus doses. e pharmacokinetic prole
of remifentanil means that, of all the opioids avail-
able, it should most closely match the time prole
ofacontraction.Followingarecentsurveyofopi-
oid use in labor, remifentanil is now the most com-
monly used opioid in the UK for women labor-
ing with a live fetus. e recommended dose varies
between studies, and whilst the presence of a back-
ground infusion seems to increase analgesia, it is
oen at the expense of increased adverse events.
e narrow therapeutic window means respiratory
depression, and indeed apnea, are signicant risks.
e degree of monitoring required is oen well in
excess of that which can be oered on a delivery
suite.
Anesthesia for operative delivery
General anesthesia is associated with signicant mor-
bidity and even mortality, for example, the risk of fail-
ing to intubate and protect the trachea is increased by
a factor of ten during pregnancy. is must be consid-
eredinarisk–benetassessmentforeachindividual
woman presenting for delivery.
Advice regarding timing of regional anesthesia rel-
ative to anticoagulant dose is the same as for regional
analgesia (see Table 10.2), although the use of a single
shot spinal anesthetic technique with a ne gauge nee-
dle may be considered earlier than standard guidance
if the woman has other signicant anesthetic risk fac-
tors.
e anesthetist must be aware of the increased risk
of bleeding at Cesarean section in those women taking
higher doses of LMWH. e introduction of cell sal-
vage machines in obstetric practice to collect autolo-
gous blood may help in the management of these
patients.
Other potential complications of regional anesthe-
sia include :
Problems with blood patches
Accidental puncture of the dura mater occurs in 0.5%
to 2.0% of all epidural procedures. Of these women
70% will develop a post-dural puncture headache. If
this headache is severe, it is oen treated with an epidu-
ral blood patch. Maternal blood is withdrawn asepti-
cally from a suitable vein and introduced via a Tuohy
needle into the epidural space. As this requires fur-
ther passage of a Tuohy needle, the same time delay
should be introduced aer LMWH administration to
avoid bleeding risk. If the woman is fully anticoagu-
lated in the post-delivery period, it should not be done.
If headache is severe, then a hiatus in anticoagulant
therapy may be considered; however, it is unlikely that
the risks of coming o anticoagulant therapy will be
outweighed by the treatment of the headache. Review
of the current literature would suggest that continuing
on prophylactic doses of LMWH does not aect the
ability of the epidural blood patch to treat the headache
eectively.
Cauda equina syndrome
e cauda equina is formed from the terminal nerve
roots of the spinal cord, aer the spinal cord has for-
mally terminated around the L2 lumbar disc space.
Compression of the cauda equina presents in a com-
monpattern.Table10.5showsthesymptomsandsigns
classically displayed by patients with a cauda equina
syndrome. Patients on anticoagulant therapy whom
have had regional analgesia or anaesthesia should be 125
Section 3. Thromboembolism and anticoagulation
Table 10.4 Treatment of severe heparin overdosage
Protamine sulphate regime (1) heparin infusion
25 mg–50 mg after stopping heparin infusion
(1 mg of protamine sulphate neutralizes 80–100 units of heparin)
(2) heparin bolus:
1.00–1.5 mg/100 IU Heparin can be given if 30 minutes have elapsed
0.5–0.7 mg/100 IU Heparin can be given if 30–60 minutes have elapsed
0.25 mg–0.375 mg IU Heparin can be given if 2 hours have elapsed
(3) subcutaneous heparin injection:
1–1.5 mg/100 IU heparin. 25–50 mg by slow IV injection and the remainder by slow IV infusion
over 8–16 hours (or the expected duration of absorption of heparin), or 2 hourly divided doses.
(4) heparin during extracorporeal circulation:
1.5 mg per 100 IU heparin. Sequential APTTs may be needed to calculate correct dosage
Dose in renal/hepatic impairment. For hepatic impairment, seek further advice
No dose adjustment necessary for renal impairment
Note Excessive doses of protamine can have an anticoagulant effect
Table 10.5 The cauda equina syndrome
Low back pain
Bilateral, occasionally unilateral, sciatica
Perineal numbness (saddle numbness)
Bladder dysfunction
Bowel dysfunction
Variable lower limb weakness and sensory loss
carefully monitored in the post delivery period. Imag-
ing of a potential lesion is usually undertaken with
MRI scanning. Cauda equina syndrome is a medi-
calemergency,andinthecaseofhematoma,requires
urgent surgical opinion to schedule evacuation of the
clot, as delay is likely to increase the risk of residual
neurological dysfunction.
Thrombolysis and bypass
It has been traditional to consider pregnancy and the
puerperium as contraindications to the use of throm-
bolytic agents. However, in the situations where they
areneeded,thelifeofthemotherislikelytobeat
high risk, as in cardiac compromise following massive
central pulmonary embolus (PE), or acute myocardial
infarction. In this situation, the balance of risks needs
careful consideration, but if thrombolysis is likely to be
life-saving, it should not be delayed as if hemorrhage
occurs it can be managed with a combination of surgi-
cal and hematological techniques. Clearly for massive
central PE targeted injection via catheter is the ideal,
if appropriate stas are available, but there are reports
of successful thrombolysis without this.16 Life-saving
treatmentshouldnotbedelayedorwithheldforthe-
oretical risk which cannot be substantiated. Senior and
experienced decision making is essential – for example
if a Cesarean section is required as part of resuscitation
in a woman collapsed from PE or MI, the timing and
management of thrombolytic agents would need to be
discussed carefully but swily.
e risk–benet balance when cardio-pulmonary
bypass is contemplated in pregnancy is dierent.
Clearly, the seriousness of the situation giving rise
to the need for bypass, usually associated with car-
diac surgery, combined with prolonged and signicant
anticoagulation means that to facilitate optimization
of both maternal condition for anesthesia and surgery
and post-operative recovery, emptying of the uterus
prior to bypass is advisable. ere are case reports
of successful prolongation of pregnancy in this situa-
tion, but also numerous reports published and unpub-
lished of signicant retroplacental bleeding, inability
to maintain maternal blood pressure or oxygenation,
and diculty in maintaining good peri-operative con-
ditions associated with attempting to continue preg-
nancy. Clearly, in a viable fetus these risks are unac-
ceptable and delivery should be expedited. Prior to
this, serious consideration should be given to medical
or surgical termination.
Summary
Management of women with any degree of anticoagu-
lation in the peripartum period is oen challeng-
ing. However, attention to detail, careful planning and
documentation, departmental education and involv-
ing both appropriate disciplines as well as the patient,
should allow optimal management to be achieved.
126

Chapter 10. Management of anticoagulants at delivery
References
1. Royal College of Obstetricians and Gynaecologists.
Reducing the risk of rombosis and Embolism
during pregnancy and the puerperium. Guideline
no.37. London, RCOG Press, 2009.
2. Lewis G (Ed.). Saving Mothers’ Lives : reviewing
maternal deaths to make motherhood safer
2003–2005. e Seventh Report on Condential
Enquiries into Maternal Deaths in the United
Kingdom. London, CEMACH, 2007.
3. CLASP (Collaborative Low dose Aspirin Study in
Pregnancy) Collaborative Group. CLASP: a
randomised trial of low-dose aspirin for the
prevention and treatment of pre-eclampsia among
9364 pregnant women. Lancet 1994; 343: 619–629.
4. James AH, Abel DE, Braucazio LR. Anticoagulants in
pregnancy. Obstetrical and Gynaecological Survey
2005; 61: 59–61.
5. WysowskiDK,TalaricoL,BacsanyiJ,BotsteinP.
Spinal and epidural haematoma and low molecular
weight heparin. New England Journal of Medicine 1998;
338: 1774–1775.
6. Scottish Intercollegiate Guidelines Network. Section 7
Spinal and epidural blocks; Section 9 Pregnancy and
puerperium. In: Prophylaxis of Venous
romboembolism. Publication No. 62. Edinburgh,
SIGN 2002; 24–26; 30–35.
7. Horlocker TT, Wedel DJ, Benzon H et al. Regional
anesthesia in the anticoagulated patient: dening the
risks. (e Second ASRA Consensus Conference on
Neuraxial Anesthesia and Anticoagulation.) Regional
Anesthesia and Pain Medicine 2003; 23: 172–197
8. Staord Smith M. Impaired haemostasis and regional
anaesthesia. Canadian Journal of Anesthesia 1996; 43:
R129–R141.
9. Royal College of Obstetricians and Gynaecologists.
romboembolic disease in pregnancy and the
puerperium : acute management. Green-top guideline
28, 2007.
10. Asghar F, Bowman P. A Clinical Approach to the
Management of rombosis in Obstetrics Part 2 :
diagnosis and management of venous
thromboembolism. e Obstetrician and Gynaecologist
2007; 9:3–8.
11. Gelson E, Johnson M, Gatzoulis M, Uebing A.
Cardiac Disease in Pregnancy Part 2 : Acquired Heart
Disease. e Obstetrician and Gynaecologist 2007; 9:
83–87.
12. Austin SK, Lambert J, Peebles D, Cohen H. Managing
peri-delivery anticoagulation in women on therapeutic
dose low molecular weight heparin : a role for
unfractionated heparin? Journal of rombosis and
Haemostasis 2007; 5: P-S-622.
13. Akkad A, Oppenheimer C, Mushambi M, Pavord S.
Intrapartum care for women on full anticoagulation.
International Journal of Obstetric Anaesthesia 2003; 12:
188–192.
14. Loo CC, Dahlgren G, Irestedt L. Neurological
complications of obstetric regional analgesia.
International Journal of Obstetric Anesthesia 2000; 9:
99–124.
15. Bates SM, Greer IA, Pabinger I, Sofaer S, Hirsh J.
Venous romboembolism, rombophilias,
Anti-thrombotic therapy and Pregnancy. American
College of Chest Physicians Evidence-Based Clinical
Practice Guidelines (8th edn). Chest 2008; 133:
844S–886S.
16. Leonhardt G, Gaul C, Nietsch HH et al. rombolytic
therapy in pregnancy. Journal of rombosis and
rombolysis 2006; 21: 271–276.
127

Section
4
Thrombophilia and fetal loss

Section 4 Thrombophilia and fetal loss
Chapter
11 Antiphospholipid syndrome
Sue Pavord, Bethan Myers, and Beverley Hunt
Introduction
Antiphospholipid syndrome (APS) is an autoim-
mune disorder characterized by vascular thrombosis
and/or obstetric morbidity in the presence of persist-
ent antiphospholipid antibodies (aPL)1namely, lupus
anticoagulant antibodies (LAC), anti-cardiolipin anti-
bodies (aCL) and/or anti-2-glycoprotein I antibod-
ies. ere are numerous potential complications for
pregnancy but with optimal management, good mater-
nal health and a live birth rate of 80%–90% can be
achieved.
e syndrome produces a spectrum of disease,
both in terms of clinical manifestations and the
presence of other autoimmune conditions. Arterial,
venous, or small vessel thrombosis may occur, there is
an array of adverse obstetric outcomes and a number of
additional clinical features may be present, involving
organs such as the heart, skin, and central nervous sys-
tem. e disease is classied as primary (PAPS) when
it occurs in the absence of any features of other autoim-
mune disease, and secondary where other autoim-
mune disease is present (SAPS). Predominantly, this
is systemic lupus erythematosus (SLE), but other con-
ditions such as inammatory bowel disease may be
involved.
Prevalence
e syndrome occurs most commonly in young to
middle-aged adults, with a mean age of onset of
31years. Women are more frequently aected, with a
female to male ratio of 5:1 which is even higher in
SAPS associated with SLE. ere is no dened racial
predominance for APS, although an increased inci-
dence of SLE occurs in African Americans and the His-
panic population. Among patients with SLE, the preva-
lence of aPL is 15% to 35%, but only around half of
these cases will have clinical features of antiphospho-
lipid syndrome.
Pathophysiology and etiology
Dierent pathological mechanisms may be responsible
for the varying clinical manifestations. Recurrence of
complications oen follows a similar pattern of disease
and recurrent thrombosis usually occurs in the same
vascular eld, although this is not always the rule.
It was originally thought that aPL were directed
against negatively charged phospholipid, but it is now
clear that they target plasma proteins with anity
for these anionic phospholipids. ere is concordance
between the LAC, aCL, and anti-2-GP I, antibodies;
however, they are not identical and some LAC antibod-
ies react with phospholipids other than cardiolipin and
proteins other than 2-GP I, whereas some aCL and
anti-2-GP I antibodies have no LAC activity. In gen-
eral, LAC antibodies are more specic for the diagno-
sis of APS, but there is no association with particular
clinical manifestations and antibody type.
e main antigens involved are 2-glycoprotein 1
(2GPI) and prothrombin, although many more anti-
genic targets have been described. Antibodies to the
2GPI are persistent and associated with thrombotic
complications, whereas those independent of 2GPI
tend to be present only transiently, in association with
infectious diseases or drugs.
Procoagulant eects
2GPI is a multifunctional apolipoprotein, which
contributes to the regulation of hemostasis as well
as other physiological processes. Not surprisingly
therefore, 2GPI-dependent antibodies have been
associated with a number of dierent biological
eects (Table 11.1). ese include direct cellular eects
131
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 4. Thrombophilia and fetal loss
Table 11.1 Procoagulant effects of 2GPI-dependent aPL
Up-regulation of the tissue factor pathway
Inhibition of the activated protein C pathway
Inhibition of antithrombin activity
Inhibition of fibrinolysis
Activation of endothelial cells
Enhanced expression of adhesion molecules by
endothelial cells with increased binding of
leukocytes
Activation and degranulation of neutrophilis
Potentiation of platelet activation
Enhanced platelet aggregation
Displacement of annexin V from cell membranes
caused by bound 2GPI –antibody complexes, with
anity for both anionic phospholipid expressed on
the surface of activated cells and heparin sulphate-
containing structures on non-activated cells. e bind-
ing of 2GPI to anionic structures, through domain
5, induces the expression of new cryptic epitopes in
domain 1 and may increase the antigenic density,
twoeventsthatseemtobepivotalfortheantibody
binding. Studies show that dimerization of 2GPI
by anti-2GPI antibodies causes a conformational
change in the molecule increasing its anity for phos-
pholipids by 100-fold. 2GPI can bind to the low-
density lipoprotein receptor, ApoER2, on the surface
of platelets and thus mediate platelet activation, with
increased thromboxane synthesis and platelet aggrega-
tion.Invitroendothelialcellsandmonocytescanalso
be activated by aPL and 2GPI binding, resulting in
tissue factor expression. In addition, in vitro studies
haveshownsomeaPLcauseinterferencewithhemo-
static factors such as IX, X, and XII, resistance to acti-
vated protein C, and a reduction in brinolysis from
antiplasmin or anti-tissue-type plasminogen activator
(tPA) activity. At this time, there is no clarity as to how
aPL cause thrombosis, and it is the subject of much
research.
Obstetric morbidity
e pathophysiological mechanisms underlying fetal
loss or morbidity also appear to be multiple. Due to
the wide spectrum of manifestations and heteroge-
neous ndings in placental tissue in these patients, it
is unclear whether one or several aPL subgroups are
responsible for the varying phenotypes and whether
concurrent, aPL-independent genetic and environ-
mental factors aecting the maternal–fetal inter-
face, inuence the potential pathogenicity of these
antibodies.
Early histological studies demonstrated decidual
vasculopathy and placental thrombosis. Displacement
of annexin V from trophoblasts contributes to a proco-
agulant state through acceleration of coagulation reac-
tions. An elegant mouse model has demonstrated acti-
vation of complement through the classical pathway,
with consequent inux of inammatory cells into tis-
sues, mediating placental injury, and leading to fetal
loss and growth restriction. In this model, heparin pre-
vented pregnancy loss by blocking activation of com-
plement, rather than primarily via an anticoagulant
eect.2
Direct trophoblastic damage by aPL, independent
of mechanisms involving thrombosis and comple-
ment activation, has also been demonstrated recently.
Interaction of aPL with 2GPI, exposed during tro-
phoblast syncytium formation, has been shown to
cause inhibition of the intercytotrophoblast fusion
process, gonadotrophin secretion, and trophoblast
invasiveness. is mechanism has been hypothesized
to contribute to early pregnancy loss. ere is evidence
of a signicant reduction in intradecidual endovascu-
lar trophoblast invasion on analysis of the products of
conception (rst-trimester failure) from APS patients.
e factors that determine whether aPL induce a
thrombotic or non-thrombotic disease phenotype in
the placenta are not known. It is likely that interplay
between patient background traits and distinct aPL
subgroups determines disease manifestation.
Clinical features
International consensus criteria for the classication
of denite APS were initially published in 19993and
updated in 20061(Table 11.2).
Thrombosis
rombosis is the most common presenting feature of
APS.4rombosis may occur in both the venous and
arterial circulation as well as the microvasculature. It
can involve vascular beds that are infrequently aected
by other prothrombotic states and is independent of
atherosclerotic vascular disease.
132

Chapter 11. Antiphospholipid syndrome
Table 11.2 Summary of the revised classification criteria for the Antiphospholipid Syndrome (APS)1
APS is diagnosed if at least one clinical and one laboratory criteria are met (although not if there is less than 12
weeks or more than 5 years between the positive aPL test and the clinical manifestation).
Clinical criteria
1. Vascular thrombosis
One or more clinical episodes of arterial, venous, or microvascular thrombosis occurring in any tissue or
organ (superficial venous thrombosis is not included)
2. Pregnancy morbidity
(a) One or more unexplained deaths of a morphologically normal fetus at, or beyond, 10 weeks’ gestation
or
(b) One or more premature births of a morphologically normal neonate before 34 weeks’ gestation
because of eclampsia, severe pre-eclampsia or recognized features of placental insufficiency∗.
(c) Three or more unexplained consecutive spontaneous abortions before the 10th week of gestation,
with maternal anatomic or hormonal abnormalities and paternal and maternal chromosomal causes
excluded.
Laboratory criteria
1. LAC present on two or more occasions at least 12 weeks apart detected according to the guidelines of the
International Society on Thrombosis and Haemostasis (10)
2. aCL of IgG and/or IgM isotype in serum or plasma, present in medium or high titer (i.e. ⬎40GPLorMPL,or
⬎the 99th percentile), on two or more occasions, at least 12 weeks apart, measured by a standardized
ELISA
3. Anti-2GP-I antibody of IgG and/or IgM isotype in serum or plasma (in titer ⬎the 99th percentile), present
on two or more occasions, at least 12 weeks apart, measured by standardized ELISA
∗Generally accepted features of placental insufficiency include: (i) abnormal or non-reassuring fetal surveillance test(s), e.g. a non-reactive
non-stress test, suggestive of fetal hypoxemia, (ii) abnormal Doppler flow velocimetry waveform analysis suggestive of fetal hypoxemia,
e.g. absent end-diastolic flow in the umbilical artery, (iii) oligohydramnios, e.g. an amniotic fluid index of 5 cm or less, or (iv) a post-natal
birth weight less than the 10th percentile for the gestational age.
Venous thrombosis
aPL are found in approximately 2% of patients present-
ingwithacutevenousthromboembolism.Oen,the
venous thrombosis occurs in an unusual site such as
the cerebral, retinal, splanchnic or axillary, and sub-
clavian veins and APS can account for up to 70% of
such presentations. Venous thromboembolism, espe-
cially deep venous thrombosis of the legs, has been
shown to occur in around 30%–50% of patients with
APS during an average follow-up of less than 6 years.5
Following a rst episode, the risk for future venous
thrombosis increases signicantly.
Arterial thrombosis
e most common site of arterial thrombosis is the
central nervous system, with strokes and transient
ischemic attacks accounting for 50% of the arterial
events seen with APS. Myocardial infarction accounts
for around 20% and other vascular beds may be
involved including those of the lungs, retina, gastroin-
testinal tract, spleen, and extremities. In many cases
the event is otherwise unexplained, with no other iden-
tiable risk factors for arterial disease, such as smok-
ing, diabetes, or hypertension.
Obstetric complications
Complications during pregnancy, in addition to
maternal thrombosis, include recurrent spontaneous
abortions in the rst trimester as well as adverse
outcomes occurring late in pregnancy. However, there
are women with aPL who have no problems at all in
pregnancy.
Early pregnancy loss
Pregnancy loss is one of the leading problems in
women’s health issues. Approximately one-third of all 133

Section 4. Thrombophilia and fetal loss
conceptions and 15% of clinically recognized pregnan-
cies (⬍6 wk of gestation) fail to result in a live birth.
Of women, 5% experience two or more losses and 1%–
2%suerwiththreeormore.Uptohalfofthecases
remain unexplained aer gynecological, hormonal,
and karyotypic analyses. Of the women who have
recurrent pregnancy loss, dened as three or more rst
trimester miscarriages, 10% to 20% have detectable
aPL.6ese women potentially have a 90% risk of fur-
ther fetal loss if le untreated.7e diagnostic cri-
teria for APS suggest that evaluation should begin
aer the third consecutive early miscarriage, dened
by less than 10 weeks’ gestation (Table 11.2). However,
in practice, evaluation aer two early miscarriages is
oen initiated at the discretion of the physician.
Late complications of aPL in pregnancy
Complications occurring late in pregnancy relate to
placental dysfunction caused by aPL. e manifes-
tations include pre-eclampsia, prematurity, fetal dis-
tress, intrauterine growth restriction, and fetal death.
Preterm delivery is associated with premature rupture
of the membranes or pre-eclampsia. e median rate
of gestational hypertension or pre-eclampsia is 30%–
50% in untreated women with previously diagnosed
APS but falls to 10% with eective management. In
contrast, aPL are not found in a signicantly higher
proportion of general obstetric patients present-
ing with pre-eclampsia.8HELLP syndrome (hemoly-
sis, elevated liver enzymes, and low platelets) may
occur, being associated with pre-eclampsia/eclampsia
in most cases and seems to occur earlier than in
women without APS, oen in the second trimester.
Other clinical manifestations
In addition to thrombosis and obstetric morbidity,
there are a number of additional clinical manifesta-
tions which are not included in the ocial denition of
APS. ese include abnormalities of skin (particularly
livedo reticularis), cardiac valves, central nervous sys-
tem, kidneys, and hematological disturbances such as
thrombocytopenia and a positive direct Coombs test
with occasional cases of clinical hemolytic anemia.
Thrombocytopenia
Many patients with APS have thrombocytopenia
(platelets⬍100 ×109/L). e pathogenic antibodies
are directed towards epitopes on platelet membrane
glycoproteinsandaredistinctfromantiphospholipid
antibodies. Conversely, aPL are found in approxi-
mately 25% of patients with chronic autoimmune
thrombocytopenia. ey do not confer a dierent clin-
ical phenotype initially, but the persistence of lupus
anticoagulant in these patients has been found to be
an important risk factor for subsequent development
of APS.9
CNS aects
A variety of neurological manifestations may occur,
mostly secondary to cerebrovascular infarcts. e clin-
ical features depend upon the caliber and location of
the vessels occluded and include multi-infarct demen-
tia, psychomotor agitation and insomnia, movement
disorders such as chorea, dystonia, oral dyskinesias
and speech impairment, transverse myelitis, seizures,
migraine, psychosis, and optic neuritis.
Valve defects
Up to 30% of patients with APS have minor valvu-
lar abnormalities, which usually do not cause hemo-
dynamic disturbance. Non-bacterial thrombotic endo-
carditis (Libman–Sacks endocarditis) is a rare disorder
characterized by sterile, thrombotic vegetations of the
heart valves and can occur rarely in APS. ese throm-
botic lesions carry signicant embolic potential.
Catastrophic antiphospholipid
syndrome (CAPS)
is term denes a severe accelerated form of APS
that results in multi-organ failure from widespread
thromboses, which are usually microvascular rather
than large vessel occlusions. e pathogenesis
appears dependent on a multi-hit phenomenon, with
infection, trauma or surgery, drug administration,
or warfarin withdrawal exacerbating an already
procoagulable state. In 50% of cases no triggering
factor is identied and in some it may be relatively
minor such as a biopsy. Around one-quarter suer
with disseminated intravascular coagulation (DIC),
contributing to “thrombotic storm” and end organ
damage. Severe thrombocytopenia is common. Acute
adult respiratory distress syndrome (ARDS) occurs in
one-third of patients and death in around 50%, mainly
from cardiac or respiratory failure, despite treatment
with anticoagulation and plasma exchange.
134

Chapter 11. Antiphospholipid syndrome
Laboratory evaluation
Limitations to laboratory testing include lack of la-
boratory standardization for aPL and the heteroge-
nous nature of the antibodies resulting in low speci-
city of the assays. Many healthy individuals can have
aPL without thrombosis or obstetric morbidity; indeed
aPL are found in 3%–5% of the normal population.
ey are oen transient and associated with infection
or drugs. e clinical importance of these aPL is uncer-
tain.TosatisfytheAPSlaboratoryclassicationcrite-
ria, a patient has to be persistently positive for either
one of the assays – anticardiolipin antibody, lupus anti-
coagulant, or 2GPI antibody, for at least 12 weeks.
Testing should occur away from the acute event.
Lupus anticoagulant (LAC)
ese antibodies are detected by their ability to pro-
long phospholipid-dependent coagulation reactions,
not corrected by mixing patient and normal plasmas.
As aPL showing LAC activity are heterogeneous, it
is recommended that at least two methods are per-
formed;APTTandadirectantiXaassaysuchasthe
dilute Russell viper venom assay (dRVVT). It should
be conrmed that the anticoagulant is directed against
protein bound to negatively charged phospholipids.
e International Society for rombosis and
Haemostasis has identied the following criteria for
the conrmation of a LAC.10
(1) Prolongation of a phospholipid-dependent
clotting assay
(2) Evidence of an inhibitor demonstrated by mixing
studies
(3) Conrmation of the phospholipid-dependent
nature of the inhibitor (platelet or other
phospholipid neutralization procedure).
Most LACs are directed against either 2GPI or pro-
thrombin and recently methods to distinguish those
associated with anti 2GPI have been developed but
whilst these are highly specic, their sensitivity is low.
LAC has been shown to be the most relevant assay
in relation to vascular events and obstetric morbid-
ity. e odds ratios for thrombotic risk ranges from 5
to 16.
Anticardiolipin antibodies (aCL)
Anticardiolipin antibodies are measured by ELISA,
although, again, concordance amongst laboratories is
poor and it is dicult to distinguish between the sig-
nicant antibodies associated with 2GPI from those
bound to other plasma proteins or directly bound
to cardiolipin. e correlation between aCL titer and
thrombotic risk is well established, with the IgG sub-
type having stronger association than IgM or IgA.11
e revised classication criteria for APS uses an IgG
aCL cut o of 40 U. Low levels of aCL, although statis-
tically abnormal, may not be associated with a signi-
cant risk of thrombosis and in a systematic review of
the literature, Galli et al. observed no correlation with
venous thrombosis and only a weak correlation with
arterial thrombosis.12
Anti-beta 2 glycoprotein 1 antibodies
Anti-2GPI antibodies are now included in the revised
criteria for the diagnosis of APS1(Table 11.2). ey
show better correlation with thrombosis than aCL
but there is a high false-positive rate. Recently, new
guidelines have been published for the performance of
an anti-2GPIantibodyELISA,whichmightimprove
standardization of the assay. However, the specicity
remains low as there are non-pathogenic antibod-
ies that bind 2GPI. Indeed, of the ve domains of
2GPI involved, only those antibodies directed against
domain 1 correlate with thromboembolic complica-
tions, with an odds ratio of around 18.
Principles of management
Individual treatment strategies for the management of
the antiphospholipid syndrome in pregnancy in part
depends on the assessment of a number of dierent
factors. ese include:
rhistory of prior thrombosis;
rwhether the thrombotic event was provoked or
spontaneous;
rwhether the thrombotic event was venous or
arterial;
rhistory of obstetric morbidity alone;
revidence of any organ damage;
rthe presence of SLE or other autoimmune disease;
rother maternal risk factors, such as obesity and
maternal age.
Background
e rst treatment used and studied for pregnant
patients with APS, was a combination of cortico-
steroids and low dose aspirin. Low-dose aspirin is 135

Section 4. Thrombophilia and fetal loss
knowntobesafeinpregnancy,intherstandsecond
trimesters, and also recognized to reduce risk of pre-
eclampsia.
Corticosteroids were an obvious choice as an
immunosuppressant to suppress the antibodies
present. Small, early studies were encouraging. Sub-
sequent studies comparing heparin and prednisolone
concluded that low dose heparin was preferable,
since, although eective, steroids induced signicant
maternal morbidity, and more premature deliveries.13
Heparin (either unfractionated or low molecular
weight) is the standard anticoagulant in pregnancy for
prophylaxis and treatment of VTE. With improved
understanding of the mechanisms of action of
heparins and the pathophysiology of APS, it is a
logical drug of choice in this condition. In addition
to anticoagulant activity, it has anti-inammatory
and anti-complement eects, both of which may
be involved in APS pathogenesis. In vitro heparin
also appears to enhance trophoblast development,
apparently limiting aPL attack on trophoblasts. Two
systematic reviews of small studies recommended
a combination of aspirin and heparin to reduce
fetal loss, concluding that this regime may reduce
pregnancy loss by 54%.14,15 Some authors, however,
question the role of pharmacological treatment in
improved live birth rate, as some studies showed no
dierence between treatment and placebo arms, in
low-risk patients. Problems in interpretation are due
to the small size of studies, variable entry criteria, lack
of placebo arms, and absence of blinding.
Pre-pregnancy management
Pre-pregnancy counseling should be oered, taking all
factors into consideration. Where necessary, recom-
mendations should be made to improve general health
and reduce risk before a pregnancy is undertaken, such
as the need for weight loss, to wait at least 6 months fol-
lowing an acute thrombotic event or until SLE has been
quiescent for 3 months. ere may be circumstances
where pregnancy should be actively discouraged, for
example, if pulmonary hypertension is present the
risk of maternal death is estimated at greater than
35%.
During this review, a clear proposed plan for preg-
nancy management should be outlined, both ver-
bally and in writing. e following issues need to be
addressed:
rReview detailed medical and obstetric history.
rDocument and conrm persistant aPL, assess
renal function and presence of thrombocytopenia
and/or anemia.
rOptimize the patient’s clinical state and
pharmacological treatment before pregnancy.
Advise postponing pregnancy if a thrombotic
event has occurred within the last 6 months, SLE
has been active or hypertension uncontrolled.
rAssess individual additional risk factors such as
obesity and maternal age and give a clear
indication to the patient regarding the degree of
risk for both thrombosis and obstetric
complications.
rAssess for the presence of anti-Ro or La
antibodies, even if no evidence of SLE. ese
antibodies are associated with a 2% risk of
complete heart-block in the fetus and up to a 10%
risk of neonatal lupus. If found, fetal cardiology
assessment should be oered and any pregnancy
aected by complete heart block should be
managed by a specialized centre where there is a
pediatric cardiologist to manage the neonate.
rProvide contact information for prompt, early
referral at the onset of pregnancy and ensure a
clear understanding of the need to substitute
heparin for warfarin at the time of the rst missed
period. Ideally, the woman can be provided with a
supply of LMW heparin and lessons in
self-injection so should they get pregnant, they
can switch quickly from warfarin.
Management of thrombosis
e immediate management of thrombosis should be
the same in those with or without the antiphospho-
lipid syndrome. It may, indeed, be the rst indication
of underlying APS. Samples should not be sent for
thrombophilia testing at the time of an acute event. e
results may be misleading for several reasons, particu-
larly during pregnancy, and the laboratory results do
not change initial management.
Management of women with
antiphospholipid antibodies and a
previous thrombotic event
ese women may be on long-term anticoagulants,
although this depends on the circumstances of the
thrombotic event; if it was a single venous event with
136

Chapter 11. Antiphospholipid syndrome
a clear temporary provoking factor, a limited duration
of anticoagulation may have been given.
In the former case, a change from warfarin to hep-
arincanbemadeonceapregnancytestispositive.is
requires the woman to be well motivated to check care-
fully in order to ensure that the substitution occurs
before 6 weeks’ gestation, to minimize risk of terato-
genicity of warfarin. In the UK, it is usual to recom-
mend a pregnancy test on the rst or second day of
a missed period and then to switch to low molecu-
lar weight heparin (LMWH) either at intermediate
or full therapeutic doses, depending on the extent of
risk when all factors are taken into account. Low dose
aspirin, at 75 mg daily, is also given.
e patient requires regular, frequent review
throughout the pregnancy, and scans to assess fetal
growth should be performed throughout the second
half (monthly or more oen as indicated). Uterine
artery Doppler ow measurements (between 20 and
24 weeks onwards) are a further useful tool to assess
platelet dysfunction. e absence of bilateral notching
is a good prognostic sign for fetal outcome.
Post-natally, the patient may be re-established on
warfarin or continued on LMWH for at least 6 weeks.
Management of women with multiple
previous venous events, or venous plus
arterial events
isgroupofwomenwilllikelybeonlong-termwar-
farin, possibly at a higher INR range (3–4s), and are
at very high risk during pregnancy. In this group,
pre-pregnancy counseling is particularly important, to
assess the extent of risk on an individual basis, and this
risk clearly conveyed to the patient and partner prior to
embarking on pregnancy. Low molecular weight hep-
arin should be substituted for warfarin before 6 weeks’
gestation, in intermediate or full dose. Some groups
monitor anti-Xa levels, although they are not clearly
predictive of anti-thrombotic eect. Other manage-
ment is as above, i.e. low dose aspirin, fetal growth
assessment by regular ultrasound scanning, and war-
farin reintroduced post-natally.
Management of women with APS and
pregnancy morbidity
A Cochrane systematic review in 2005 assessed 13 tri-
als published (between 1991 and 1999) on recurrent
pregnancy loss associated with aPL. ey commented
that the quality of the studies was not good, which lim-
ited useful conclusions.
Aspirin
In obstetric patients, low dose aspirin has been used
to improve pregnancy outcome in those with hyper-
tension, pre-eclampsia, preterm birth, and intrauter-
ine growth restriction. e Cochrane review and
meta-analysis summarized the studies with aspirin.16
ree trials with aspirin alone showed no signi-
cant reduction in pregnancy loss; two studies using
unfractionated heparin and aspirin showed a signif-
icant improvement in fetal outcome compared with
aspirin alone; but in a further randomised controlled
trial, whilst high success rates were achieved with low
dose aspirin, the addition of LMWH did not pro-
vide further benet.17 is latter study has been crit-
icized as the laboratory criteria for APS were not
met.
However, these studies were done at a time when
LMWH was just being introduced, and it is dicult
to draw rm conclusions from this collection of data.
Subsequently obstetric hematology groups, have accu-
mulated a large volume of experience with LMWH,
and it is considered safer and as eective as unfrac-
tionated heparin, and more convenient with a once-
daily dosing regimen. e risk of osteoporosis which
is as high as 2% with unfractionated heparin, is rarely
described with LMWH and prophylactic dose LMWH
and low dose aspirin has become standard practice.
Improved outcomes for women with previous late fetal
loss or early delivery due to placental insuciency
have been conrmed and a recent metaanalysis sup-
ports the ecacy of this approach for recurrent preg-
nancy loss.18
Ultrasonography
In patients with poor obstetric history, pre-eclampsia
or evidence of fetal growth restriction, fetal growth
scansevery4weeks,from20weeksisrecom-
mended, in addition to pharmacological treatments.
Studies have shown uterine artery Doppler to be
valuable in predicting placental dysfunction, i.e.
pre-eclampsia and intrauterine growth restriction
and the discovery of bilateral uterine artery notching
can allow the obstetrician to monitor the pregnancy
more closely. Multivariate analysis of data on 100
pregnancies demonstrated that a notched uterine
artery at the second trimester was the only predictor 137

Section 4. Thrombophilia and fetal loss
for adverse pregnancy outcome. Uterine artery
Doppler assessment should be performed with the
fetalscanat20weeksand24weeks.LeiHuong
et al.19 showedthepredictivevalueoftheumbilical
artery Doppler ultrasound examination for late preg-
nancy outcome, together with clinical examination
and laboratory tests in women with SLE and/or
APS.
Table 11.3 Summary of pharmacological management of APS
in pregnancy
Clinical feature Management
APS with prior fetal
death or recurrent
pregnancy loss
Aspirin 75 mg od
LMWH prophylactic
dosage
Doppler ultrasound
for fetal assessment
APS with prior venous
or arterial thrombosis
Low molecular weight
heparin at
intermediate or
therapeutic dosage,
plus aspirin
Antiphospholipid
antibodies without
clinical features and
healthy previous
pregnancies
No treatment, or low
dose aspirin
Primigravida with
isolated aPL
Low dose aspirin and
fetal monitoring
APS with recurrent
thrombotic events
Full therapeutic dose
LMWH; consider
warfarin
Other treatment modalities
A summary of pharmacological management of APS
in pregnancy is provided in Table 11.3.
If there is continued pregnancy loss despite
prophylactic-dose heparin and low dose aspirin, the
following options should be considered.
(a) Increasing the dose of heparin to therapeutic
levels may be eective, although there are no trials
that have demonstrated this.
(b)ereareanecdotalcasereportsofuseof
intravenous immunoglobulin in refractory cases
with success, although several studies have not
demonstrated its benet.20 It may also be useful in
APS cases who have additional indications for its
use, for example, in auto-immune
thrombocytopenia or Guillain–Barr´
esyndrome.
(c) Successful use of plasma exchange to remove
antibodies temporarily has been reported, in high
risk pregnancies where plasma exchange was
administered.
Management of thrombocytopenia
associated with APS in pregnancy
is may be due to pre-eclampsia, HELLP syndrome
or worsening maternal idiopathic thrombocytopenic
purpura (ITP). is should be managed in the same
way as those complications occurring without APS
(see Chapters 4,17, and 18).
Management dilemmas
(1) Management of women who have isolated aPL,
with no prior pregnancy loss or thrombo-embolic
phenomena, do not generally merit ante-natal
pharmacological treatment, although low dose
aspirin is oen used. e best predictor of
maternal and fetal outcome in APS pregnancies is
the previous obstetric history. Mothers with a
previously normal obstetric history despite aPL
can be reassured that any future pregnancy has a
low risk of complications.
(2)Forwomeninanultra-highriskgroup,suchas
those with multiple unprovoked venous and
arterial event, or patients who develop a new
thrombotic event during pregnancy despite
anticoagulation, consideration should be given for
warfarin usage from the second trimester, or even
all trimesters. e risks of warfarin to the fetus
must be explained – including the teratogenic risk
in the rst trimester and the ongoing risk of fetal
loss, hemorrhage and subtle neurological changes,
which have been described aer its use in the
second and third trimester.
(3) Where in vitro fertilization (IVF) or other assisted
reproductive techniques are planned, LMWH
should be substituted for warfarin at the time at
which assisted reproductive procedure is
performed,i.e.atthetimeofeggtransfer;ifthe
woman is not on anticoagulation prior to IVF,
138

Chapter 11. Antiphospholipid syndrome
then prophylactic LMWH and aspirin should be
used.
(4) For the rare seronegative APS, or SNAPS, where
typical clinical features occur in the absence of
measurable standard antibodies – expert clinical
judgment is required to make this diagnosis and
to determine need for treatment.
(5) Management of women with SLE in pregnancy is
one of the few indications for the use of
glucocorticoids during the pregnancy. SLE may
are during pregnancy and increasing or starting
small doses of prednisolone is appropriate.
Hydroxychloroquine and azathioprine, standard
drugs for the management of SLE, are safe in
pregnancy and should be continued if the disease
is stable. Stopping such medications may lead to a
are of SLE which could be harmful to mother
and fetus.
(6) Chorea gravidarum is a rare complication in
pregnancy and may be associated with primary or
secondary APS. It is thought to be due to
development of antibodies against components of
the basal ganglia, or rarely due to infarction of this
area. It is usually self-limiting and resolves
following pregnancy, although may recur in
subsequent pregnancies. When severe, a variety of
treatments have been described, including low
dose haloperidol, steroids, anticoagulants,
antiplatelet medication, or a combination of
treatments.21
Neonatal issues
Neonatal APS has been described, although the exist-
ence of this syndrome has not been fully accepted.
It is a rare occurrence, characterized by neonatal
thrombosis thought to be due to the transplacental
passage of maternal aPL.22 Ischemic stroke is the main
event described. In comparison to a high incidence
of thrombotic and obstetric complications in women
with APS, the aPL-associated thrombotic events in
neonates are extremely rare. ere is no benet from
routine screening for aPL in neonates born to mothers
with APS.
139

Section 4. Thrombophilia and fetal loss
References
1. Miyakis S, Lockshin MD, Atsumi T et al. International
consensus statement on an update of the classication
criteria for denite antiphospholipid syndrome (APS).
JournalofrombosisandHaemostasis2006; 4:
295–306.
2. Girardi G, Redecha P, Salmon JE. Heparin prevents
antiphospholipid antibody-induced fetal loss by
inhibiting complement activation. Nature Med. 2004;
10: 1222–1226.
3. Wilson WA, Gharavi AE, Koike T et al. International
consensus statement on preliminary classication
criteria for denite antiphospholipid syndrome: report
of an international workshop. Arthritis and
Rheumatism 1999; 42: 1309–1311.
4. Cervera R, Piette JC, Font J et al. Euro-Phospholipids
Project Group. Antiphospholipid syndrome: clinical
and immunologic manifestations and patterns of
disease expression in a cohort of 1,000 patients.
Arthritis and Rheumatism 2002; 46: 1019–1027.
5. Asherson RA, Khamashta MA, Ordi-Ros J et al.e
“primary” antiphospholipid syndrome: major clinical
and serological features. Medicine (Baltimore) 1989;
68: 366–374.
6. Yetman DL, Kutteh WH. Antiphospholipid antibody
panels and recurrent pregnancy loss: prevalence of
anticardiolipin antibodies compared with other
antiphospholipid antibodies. Fertility and Sterility
1996; 66: 540–546.
7. Rai RS, Cliord K, Cohen H, Regan L. High
prospective fetal loss rate in untreated pregnancies of
women with recurrent miscarriage and
antiphospholipid antibodies. Human Reproduction
1995; 10: 3301–3304.
8. Dreyfus M, Hedelin G, Kutnahorsky R et al. Antipho-
spholipid antibodies and preeclampsia: a case-control
study. Obstetrics and Gynecology 2001; 97: 29–34.
9. Diz-K¨
uc¸¨
ukkaya R, Hacihaneoglu A, Yenerel M et al.
Antiphospholipid antibodies and antiphospholipid
syndrome in patients presenting with immune
thrombocytopenic purpura: a prospective cohort
study. Blood 2001; 98: 1760–1764.
10. Brandt JT, Triplett DA, Alving B, Scharrer I. Criteria
for the diagnosis of lupus anticoagulants: an update.
On behalf of the Subcommittee on Lupus
Anticoagulant/Antiphospholipid Antibody of the
Scientic and Standardisation Committee of the ISTH.
rombosis and Haemostasis 1995; 74: 1185–1190.
11. Harris EN, Pierangeli SS. Revisiting the anticardiolipin
test and its standardization. Lupus 2002; 11: 269–275.
12. Galli M, Luciani D, Bertolini G, Barbui T. Lupus
anticoagulants are stronger risk factors for thrombosis
than anticardiolipin antibodies in the
antiphospholipid syndrome: a systematic review of the
literature. Blood 2003; 101: 1827–1832.
13. Cowchock FS, Reece EA, Balaban D et al. Repeated
fetallossesassociatedwithantiphospholipid
antibodies: a collaborative randomised trial comparing
prednisone with low-dose heparin treatment.
American Journal of Obstetrics and Gynaeology 2003
101; 1319–1332.
14. Rai R, Cohen H, Dave M, Regan L. Randomised
controlled trial of aspirin and aspirin plus heparin in
pregnant women with recurrent miscarriage
associated with phospholipid antibodies. British
Medical Journal 1997; 314: 253–257.
15. Kutteh WH. Antiphospholipid antibody associated
recurrent pregnancy loss: treatment with heparin and
low-dose aspirin is superior to low dose aspirin alone.
American Journal of Obstetrics and Gynaecology 1996;
174: 1584–1589.
16. Empson M, Lassere M, Craig J, Scott J. Prevention of
recurrent miscarriage for women with
antiphospholipid antibody or lupus anticoagulant.
Cochrane Database of Systematic Reviews 2005; 002859.
17. Farquharson RG, Quenby S, Greaves M.
Antiphospholipid syndrome in pregnancy: a
randomised controlled trial of treatment. Obstetrics
and Gynecology 2002; 100: 408–413.
18. Mak A, Cheung MWL, Cheak AA, Ho RC.
Combination of heparin and aspirin is superior to
aspirin alone in enhancing live births in patients with
recurrent pregnancy loss and positive
anti-phospholipid antibodies: a meta-analysis of
randomized controlled trials and meta-regression.
Rheumatology 2010; 49: 281–288.
19. Le i Huong D, Weschler B, Vauthier-Brouzes et al.
e second trimester Doppler ultrasound examination
isthebestpredictoroflatepregnancyoutcomein
systemic lupus erythematosis and/or the
antiphospholipid syndrome. Rheumatology 2006; 45:
332–338.
20. Triolo G, Ferrante A, Ciccia F et al. Randomised study
of subcutaneous low molecular weight heparin plus
aspirin versus intravenous immunoglobulin in the
treatment of recurrent fetal loss. Arthritis and
Rheumatism 2003; 48: 728–731.
21. Cervera R, Asherson RA, Font J et al. Chorea in the
antiphospholipid syndrome. Clinical, radiologic and
immmunolgic characteristics of 50 patients from our
clinics and recent literature. Medicine 1997; 76(3):
203–212.
22. Boa MC, Lachassine E. Infant perinatal thrombosis
and antiphospholipid antibodies: a review. Lupus 2007;
16: 634–641.
140

Section 4 Thrombophilia and fetal loss
Chapter
12 Thrombophilia and pregnancy loss
Isobel D. Walker
Introduction
Pregnancy loss is psychologically and emotionally
extremely dicult for the mother, her partner and
wider family. Couples who have had such an event,
have many questions including, what caused the preg-
nancy to fail, will it happen again, and what can be
done to minimize the risk of a recurrence?
It has been postulated that, in some cases, preg-
nancy failure may be, at least in part, due to inade-
quate placental circulation and that thrombophilia, by
increasing the risk of brin deposition or thrombo-
sis within the placental circulation, may increase the
risk of pregnancy loss. is postulate has led to the
hypothesis that, for women with a history of pregnancy
losswithnoidentiablecauseotherthananunderly-
ing thrombophilia, intervention with antithrombotics
may improve the outcome in subsequent pregnancies.
Epidemiology
Sporadicpregnancylossisverycommon.Ithasbeen
estimated that 30%–50% of fertilized ova are sponta-
neously aborted with around 15% of clinically recog-
nized pregnancies being lost before 24 weeks’ gesta-
tion. Around one in 20 women will suer two or more
consecutive pregnancy losses and 1% suers the loss
of three or more consecutive pregnancies (Table 12.1).
e observed incidence of recurrent pregnancy loss
is greater than the 0.34%, which may be expected by
chance, suggesting that some women are predisposed
to pregnancy loss.
e World Health Organization’s denition of mis-
carriage is a pregnancy which fails to progress, result-
inginthedeathandexpulsionofanembryoorfetus
weighing no more than 500 g (which corresponds to
a gestational age of 20 weeks or less). Unfortunately,
this denition is not used consistently and a huge range
of “denitions” have been employed in the many stud-
ies examining potential associations between throm-
bophilia and pregnancy loss. Recurrent pregnancy loss
is dened as the occurrence of three or more con-
secutive miscarriages. is includes women with pri-
mary recurrent pregnancy loss (with ≥3consecu-
tive pregnancy losses and no pregnancy proceeding
beyond 20 weeks’ gestation) and women with sec-
ondary recurrent pregnancy loss (with ≥3consecu-
tive pregnancy losses following a pregnancy which
proceeded beyond 20 weeks’ gestation and resulted in
a live birth, stillbirth, or neonatal death). Some investi-
gators include within the denition of recurrent preg-
nancy loss, women who have had ≥3 non-consecutive
pregnancy losses. To add further confusion, because
reproductive practice has changed and many women
nowadays do not embark on their rst pregnancy
until they are in their mid to late 30s, there has
been an increasing tendency to consider intervention
for women who have a history of only two consecu-
tive miscarriages and to “label” these women as hav-
ing a history of recurrent pregnancy loss.
Pathogenesis
Thrombophilia
By denition, thrombophilias are disorders of
hemostasis which predispose to thrombosis. Included
are heritable deciencies of the natural anticoagulants
antithrombin, protein C, and protein S and common
mutations in the genes encoding clotting factor V,
factor V Leiden which results in increased resistance
to activated protein C and clotting factor II, the
prothrombin G20210A mutation. Also included are
acquired abnormalities such as antiphospholipid
antibodies and some disorders of mixed genetic and
141
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 4. Thrombophilia and fetal loss
Table 12.1 Incidence of pregnancy loss
Spontaneous abortion of fertilized ova 30%–50%
Spontaneous loss of clinically recognized
pregnancy before 24 weeks
15%
Spontaneous loss of two or more consecutive
pregnancies
5%
Three or more consecutive pregnancy losses
before 20 weeks
1%
Table 12.2 The prevalences of heritable thrombophilias in
European populations
Thrombophilia Prevalence (%)
Antithrombin deficiency 0.25–0.55
Protein C deficiency 0.20–0.33
Protein S deficiency 0.03–0.13
Factor V Leiden (heterozygous) 2–7
Prothrombin G20210A (heterozygous). 2
environmental etiology such as hyperhomocysteine-
mia. Around 10% of Caucasians carry an identiable
heritable thrombophilia (Table 12.2).
Placentation in normal pregnancy
Successful pregnancy requires trophoblast invasion
into the maternal uterine spiral arteries converting
them into large dilated vessels, which lack a func-
tioning contractile smooth muscle wall. Prior to this
remodeling, the spiral arterioles are occluded by
endovascular trophoblasts. It is postulated that this
plugging protects the early intervillous spaces from
maternal systemic arterial pressure and protects the
developing intervillous trophoblasts from high oxy-
gentensionandoxidativedamage.Althoughtheblood
owing through the spiral arteries and the placental
intervillous spaces is maternal, the cells lining these
spaces are embryonic trophoblasts. us the hemo-
static balance within the placenta may be disturbed as
a result of hypercoagulability of the maternal blood or
as a result of abnormality of cellular regulatory mech-
anisms of fetal origin operating at the feto-maternal
interface. Since the fetal blood is separated from the
trophoblasts by fetal endothelial cells, the fetal hemo-
static balance is inuenced only by components of fetal
origin.
Placental pathology in
pregnancy loss
rombi in the spiral arteries or brin deposition in
the intervillous spaces on the maternal side of the pla-
centa may result in inadequate placental perfusion.
Microthrombi are frequently found in the vessels of the
placentas from women who have experienced preg-
nancy loss and placental infarction has been described
intheplacentasofsome,butnotall,womenwhohavea
pregnancy loss and who have thrombophilia. Placental
thrombosis and infarction are, however, not uncom-
moninfetallosscasesintheabsenceofanyidenti-
able thrombophilia and no placental lesion is spe-
cic for thrombophilia. Most of the studies that have
reported on the placental pathology in women with
thrombophilia and a history of pregnancy loss have
concentrated on women with antiphospholipid syn-
drome. ere is limited information about the pla-
cental pathology in women with pregnancy loss and
an underlying heritable thrombophilia. Furthermore,
there are methodological problems with many of the
published studies. Some studies have compared the
placentas from women with thrombophilia and preg-
nancy loss with the placentas from non-thrombophilic
women with normal gestations – others with placentas
from non-thrombophilic women with pregnancy loss.
Othershaveincludednocontrolgroupatall.
Is heritable thrombophilia associated
with pregnancy loss?
Observational studies
It has long been accepted that antiphospholipids in
maternal plasma increase the risk of both early and late
pregnancy loss. More recently, attention has turned to
the question of the potential role of heritable throm-
bophiliasinthecausationofpregnancyloss.Asso-
ciations between heritable thrombophilias and preg-
nancy loss were rst noted in families in which the
probands had presented with venous thrombosis. One
such study of family members, the European Prospect-
ive Cohort on rombophilia (EPCOT), reported the
incidence of pregnancy loss in a cohort of 571 women
with heritable thrombophilia who had experienced
1524 pregnancies and 395 age-matched controls who
had had 1019 pregnancies and reported a signicantly
greater percentage of women with thrombophilia had
a history of pregnancy loss (29.4% vs. 23.5%).1e
142

Chapter 12. Thrombophilia and pregnancy loss
Table 12.3 Pregnancy loss in women with heritable
thrombophilia (identified because of a family history of venous
thrombosis). Data from EPCOT Study1
Pregnancy loss
at <28 weeks
Odds Ratio
(95% CI)
Pregnancy loss
at ≥28 weeks
Odds Ratio
(95% CI)
Antithrombin
deciency
1.7 (1.0–2.8) 5.2 (1.5–18.1)
Protein C deciency 1.4 (0.9–2.2) 2.3 (0.6–8.3)
Protein S deciency 1.2 (0.7–1.9) 3.3 (1.0–11.3)
Factor V Leiden 0.9 (0.5–1.5) 2.0 (0.5–7.7)
Combined defects 0.8 (0.2–3.6) 14.3 (2.4–86.0)
Odds Ratio (OR) for fetal loss associated with throm-
bophilia was 1.35, 95% Condence Interval (CI) 1.01–
1.82. When the data for all thrombophilic women and
the controls were stratied according to the stage of
gestation at which the pregnancy losses occurred, the
odds ratio was statistically signicant only for fetal
losses aer 28 weeks’ gestation (OR 3.6, 95% CI 1.4–
9.4). For pregnancy loss at, or before, 28 weeks the
odds ratio was 1.27 (95% CI 0.94–1.71). When the data
for fetal losses were stratied according to the specic
thrombophilic defects, the odds ratios for individual
thrombophilias were signicant only for antithrombin
deciency and protein S deciency for pregnancy loss
aer 28 weeks’ gestation (Table 12.3). e odds ratio
for pregnancy loss aer 28 weeks’ gestation in women
with more than a single identiable heritable throm-
bophilia was 14.3 (95% CI 2.4–86.0), suggesting a pos-
sible dose–response eect.
Meta-analyses
Following the early studies of families, numerous stud-
ies have examined possible associations between her-
itable thrombophilias and pregnancy loss. Within the
past 5 years, two meta-analyses have been published.
Both noted signicant heterogeneity between studies.
Rey et al., following analysis of 31 case-control, cohort,
and cross-sectional studies, reported that factor V Lei-
den and the prothrombin G20210A mutation are sig-
nicantly associated with recurrent early fetal loss and
with non-recurrent late fetal loss, and that protein S
deciency was associated with late non-recurrent fetal
loss but not with recurrent fetal loss (Table 12.4).2Rey
also found that maternal factor V Leiden was associ-
ated with recurrent late pregnancy loss – odds ratio
7.83 (95% CI 2.83–21.7). e meta-analysis by Robert-
son et al. conrmed3that factor V Leiden and the
prothrombin G20210A mutation are associated with
recurrent early fetal loss and non-recurrent late fetal
loss, and reported that both factor V Leiden and the
prothrombin G20210A mutation are associated with
non-recurrent second trimester loss (Table 12.4). As
in the Rey meta-analysis, Robertson et al. found that
protein S deciency is associated with an increased
risk of late pregnancy loss. is meta-analysis also
reported an association between hyperhomocysteine-
mia and early pregnancy loss (odds ratio 6.25, 95% CI
1.37–28.42).
Very early pregnancy loss – embryo loss
Most reports do not separate very early pregnancy
losses (before 10 weeks’ gestation) from later rst
trimester losses. In a cohort study, 491 patients with
a history of adverse pregnancy outcome, maternal
thrombophilia was associated with an increased risk
of pregnancy loss aer 10 weeks’ gestation (odds
ratio 1.76, 95% CI 1.05–2.94 for women with one
thrombophilia and odds ratio 1.66, 95% CI 1.03–2.68
for women with more than one identiable throm-
bophilia) (Table 12.5).4Paradoxically, the presence of
Table 12.4 Heritable thrombophilia and pregnancy loss. Data from two meta analyses∗2and∗∗3
Factor V Leiden Prothrombin G20210A Protein S deficiency
Odds Ratio (95% CI) Odds Ratio (95% CI) Odds Ratio (95% CI)
Rey∗Robertson∗∗ Rey∗Robertson∗∗ Rey∗Robertson∗∗
Recurrent 1st
trimester loss
2.01
(1.13–3.58)
1.91
(1.01–3.61)
2.56
(1.04–6.29)
2.70
(1.37–5.34)
14.7
(0.99–218)
-
Non recurrent
2nd trimester loss
-4.12
(1.93–8.81)
-8.60
(2.18–33.95)
- -
Late pregnancy
loss
3.26
(1.82–5.83)
2.06
(1.10–3.86)
2.30
(1.09–4.87)
2.66
(1.28–5.53)
7.39
(1.28–42.6)
20.09
(3.70–109.15) 143

Section 4. Thrombophilia and fetal loss
Table 12.5 Heritable thrombophilia and embryo or fetal loss4
1 thrombophilia
Odds Ratio (95% CI)
>1 thrombophilia
Odds ratio (95% CI)
Embryo loss
⬍10 weeks’
gestation
0.55 (0.33–0.92) 0.48 (0.29–0.78)
Fetal loss
⬎10 weeks’
gestation
⬎14 weeks’
gestation
1.76 (1.05–2.94)
3.41 (1.9–6.1)
1.66 (1.03–2.68)
3.86 (2.26–6.59)
one or more maternal thrombophilias seemed to be
protective of recurrent very early (less than 10 weeks’
gestation) pregnancy loss (odds ratio 0.55, 95% CI
0.33–0.92 for one and odds ratio 0.48, 95% CI 0.29–
0.78 for multiple thrombophilias).
Fetal thrombophilia
It has been suggested that fetal carriage of throm-
bophilic mutations may have adverse clinical conse-
quences. In one case control study a twofold increase
in factor V Leiden carrier frequency was noted in abor-
tuses compared with unselected pregnant women, but
most studies have not shown a signicant associa-
tion between fetal carriage of the most prevalent herit-
able thrombophilias (factor V Leiden and prothrom-
bin G20210A) and feto-placental thrombosis.
Does maternal heritable
thrombophilia cause pregnancy loss?
e majority of published studies have been too small
and therefore inadequately powered to detect odds
ratios of 2 or more for heritable thrombophilias which
usually have prevalences of less than 5% in the general
population. Meta-analyses support the hypothesis that
at least some heritable thrombophilias are associated
with pregnancy loss but, where the data are mature
and the condence intervals are narrow (i.e. for factor
V Leiden and prothrombin G20210A), the point esti-
mates of the odds ratios are small, suggesting that the
associations, if they truly exist, are weak.
Even if an association exists, it may not be a causal
association. However, a few studies have shown that,
compared with heterozygotes carrying a single throm-
bophilic variant, homozygous patients or patients
with combinations of thrombophilic variants have
increased odds ratios for pregnancy loss. is appar-
ent dose eect would support the hypothesis of causal-
ity but needs further evidence. Evidence that preg-
nancy outcome could be improved in thrombophilic
women with a history of pregnancy loss by reducing
the hypercoagulability with anticoagulant treatment
wouldoerindirectsupporttothehypothesisthat
maternal thrombophilia may cause pregnancy loss.
Many factors including chromosomal abnormal-
ities, endocrine disorders, anatomical aberrations,
and infections have been shown to cause preg-
nancy loss, but around 40% of cases of recurrent
pregnancy losses are unexplained aer gynecologi-
cal, hormonal, immunological, microbiological, and
karyotypic investigations. ere is an increasing risk
of pregnancy loss as the number of previous losses
increases. Recurrent pregnancy loss is recognized to
be a multi-causal disorder. Whilst heritable throm-
bophilias may not alone cause pregnancy loss, it is pos-
siblethatcarriageofathrombophilicvariantmaycon-
tribute to a complex network of factors, which together
result in pregnancy failure.
It is generally assumed that the mechanism of preg-
nancy failure associated with maternal thrombophilia
involves brin deposition or thrombosis secondary to
hypercoagulability but, although it is biologically plau-
siblethatplacentalthrombosismayhavearolein
the causation of fetal loss aer 10 weeks’ gestation,
it is not plausible that this mechanism would cause
embryo loss (before 10 weeks) prior to development of
the placental vasculature. e vast majority of recur-
rent pregnancy losses occur early in pregnancy. In
women with antiphospholipid syndrome in addition
to hypercoagulability, a non-prothrombotic mecha-
nism has been postulated. Antiphospholipid antibod-
ies have been shown to inhibit extravillous trophoblast
dierentiation and subsequent placentation. Studies of
trophoblast dierentiation and early placental devel-
opment are lacking in heritable thrombophilias, but
experiments in mice have shown that maternal pro-
tein C is activated following binding to thrombomod-
ulin on the trophoblast surface. Activated protein C
then binds to endothelial protein C receptor also on
the trophoblast surface and with protein S as a cofac-
tor down-regulates local coagulation activation. Tight
regulation of thrombin generation is essential for the
regulation of trophoblast cell growth and limits the
production of brin degradation products, which trig-
ger trophoblast apoptosis. It is therefore possible that,
in early pregnancy in humans, some maternal herit-
able thrombophilias may exert an adverse eect on
normal trophoblast development.
144

Chapter 12. Thrombophilia and pregnancy loss
Diagnosis
Thrombophilia testing
Who should be tested?
Routine testing for thrombophilias in unselected pop-
ulations is not recommended. ere are important
issues relevant to the clinical utility and cost eective-
ness of testing that must be addressed in consider-
ing who should be tested. Positive tests are not sensi-
tive predictors of poor pregnancy outcome in women
with no history of pregnancy complications. ere
are a number of published guidelines which suggest
thatwomenwithahistoryofrecurrentpregnancyloss
and women with a history of unexplained late preg-
nancy loss be tested for antiphospholipids, testing for
both lupus anticoagulant activity and elevated anticar-
diolipins.5Some authors have extended this guidance
andhavesuggestedthatwomenwithahistoryofrecur-
rent early pregnancy loss or an unexplained late preg-
nancy loss should, in addition, be tested for heritable
thrombophilia.5Others take an opposing view sug-
gesting testing for heritable thrombophilia is at present
not indicated, since there is insucient evidence on
which to base any intervention in women with a his-
tory of pregnancy loss with no other identied abnor-
mality apart from a heritable thrombophilia.6
What tests?
Currently,notonlyistherealackofconsensusabout
which individuals (if any) merit thrombophilia test-
ing, but there is also no universal agreement regarding
which tests should be included in the “thrombophilia
screen.” Most diagnostic laboratories would include
functional assays of antithrombin, protein C and an
immunologic assay of free protein S along with tests to
detect factor V Leiden and the prothrombin G20210A
mutation (Table 12.6). A few centers include an assay
of homocysteine in the panel of tests they oer for
women with a history of pregnancy loss.
Pitfalls
If testing for heritable thrombophilia is pursued, man-
aging clinicians should be aware that there are numer-
ous potential pitfalls in the interpretation of “throm-
bophilia screens” particularly in pregnant or recently
pregnant women. Antithrombin activity falls slightly
towards the end of a normal pregnancy, but usually
Table 12.6 Thrombophilia screening tests
Heritable
thrombophilias Preferred test method
Antithrombin deficiency Heparin co-factor activity
Protein C deficiency Chromogenic activity
Protein S deficiency Free protein S antigen
Factor V Leiden Activated protein C resistance after
predilution of test plasma in factor V
depleted plasma or DNA based
Prothrombin G20210A DNA based
Acquired
thrombophilia Preferred test method
Lupus anticoagulant Clotting based tests∗
Anticardiolipins Immunologic assays of IgG and IgM
anticardiolipin
∗See Chapter 11
levels remain within the reference range for non preg-
nant subjects. Protein C activity is unaected by ges-
tation, although an elevation of protein C activity
occurs in the early puerperium. Even in non-pregnant
women there is considerable overlap of protein S lev-
els between “normals” and subjects with heritable pro-
tein S variants. e levels of both free and total pro-
tein S are reduced by 60%–70% in uncomplicated preg-
nancy. A diagnosis of possible protein S deciency
made on a sample collected during pregnancy or the
puerperium requires conrmation when the woman
is no longer pregnant, puerperal or using hormonal
contraception. Pregnancy is also associated with a pro-
gressive increase in resistance to activated protein C
(APC) due to the physiological rise in clotting fac-
tor VIII levels and fall in protein S levels. Using the
original APC resistance test, around 40% of pregnant
women in their third trimester have an APC sensitiv-
ity ratio below the general population reference range.
Testing for factor V Leiden therefore requires the use of
a modied APC resistance test with predilution of the
test sample in factor V decient plasma or genetic test-
ing. rombophilia test results should always be inter-
preted by sta experienced in the reporting of throm-
bophiliatestsandinthelightofclearclinicalinforma-
tion about each particular patient.
Management
General measures
First, primary prevention of vascular placental com-
plications using antithrombotics is not indicated, so 145

Section 4. Thrombophilia and fetal loss
routine screening of asymptomatic women cannot be
justied on this basis. Second, the prognosis for future
pregnancies in women with a heritable thrombophilia
whohaveahistoryofrecurrentpregnancymaybebet-
ter than generally expected. e EPCOT investigators
reported that the prognosis in subsequent pregnancies
of women with recurrent pregnancy loss and under-
lying heritable thrombophilia was a live birth rate of
63%.1
In women with a history of previous pregnancy
loss,ante-natalsurveillancetoassessplacentalfunc-
tion is useful. Given the possible association between
hyperhomocysteinemia and pregnancy loss, it would
seem prudent to suggest that women with a history of
pregnancy loss take prophylactic doses of folic acid in
subsequent pregnancies
Are antithrombotics useful?
Studies published in the 1990s reported improved
pregnancy outcome in women with antiphospholipid
syndrome and a history of recurrent pregnancy loss,
given prophylactic doses of heparin combined with
low dose aspirin compared with those given aspirin
alone, but there is a paucity of data to indicate whether
antithrombotic therapy is benecial in women with
heritable thrombophilia and pregnancy loss.
In a prospective study, 131 women with heritable
thrombophilia(identiedbecauseofafamilyhistory
of venous thrombosis) were followed through their
rst pregnancy. Only 7 of 83 (8%) given thrombo-
prophylaxis to prevent venous thrombosis had a preg-
nancylosscomparedwith10of48(21%)whoreceived
no thromboprophylaxis (relative risk 0.3; 95% CI 0.1–
1.0).7
Open, non-controlled studies
In women with heritable thrombophilia and a previ-
ous history of pregnancy loss, low molecular weight
heparin has been tested in open, non-controlled stud-
ies in which outcomes were compared with the out-
come of the subjects’ previous pregnancies or with out-
comesincontrolswhowereeitheruntreatedortreated
dierently. In studies in which pregnancy outcome in
womenwiththrombophiliaandahistoryofpregnancy
loss treated with once daily prophylactic doses of a low
molecular weight heparin was compared with their
past obstetric history, low molecular weight heparin
use was associated with an increase in the live birth rate
from 20% to 75%.8
Randomized studies
Although some randomized controlled studies have
been reported, they lack a no treatment or placebo
group. In one, women with factor V Leiden, prothrom-
bin G20210A or protein S deciency and a history of
unexplained pregnancy loss aer 10 weeks’ gestation,
live births were recorded in 86% of 80 women given
daily prophylactic doses of a low molecular weight
heparin from 8 to 37 weeks’ gestation and in only 29%
ofthe80womengivendailylowdoseaspirin(odds
ratio 15.5, 95% CI 7–34; P⬍0.0001).9e randomiza-
tion and “blinding” in this trial have been criticized.
ere is no evidence that aspirin improves fetal out-
come in women with heritable thrombophilias and a
history of fetal loss.8
Purist vs. pragmatic management
From a purists’ standpoint there is currently insu-
cient evidence on which to base antithrombotic inter-
vention in women with a history of pregnancy loss
with no other identied abnormality apart from a her-
itable thrombophilia. is is the position adopted by
many authors, by the British Committee for Standards
in Haematology6and by the authors of a recently pub-
lished Cochrane Review.10 In support of this position
it has to be reiterated that the use of antithrombotic
drugs during pregnancy is not without risk for mother
and fetus and, in general, empirical intervention dur-
ing pregnancy should be discouraged.
Pragmatists, on the other hand, argue that there is
at least some observational evidence that women with
heritable thrombophilia who have suered pregnancy
loss may benet from intervention with antithrom-
botic drugs in future pregnancies and they point out
that low molecular weight heparins are used increas-
inglyinpregnantwomenandare,ingeneral,consid-
ered safe. Based on extrapolation from the evidence of
benet from intervention with heparin and low dose
aspirin in women with antiphospholipid syndrome
and recurrent pregnancy loss, the limited evidence in
women with heritable thrombophilia, and the relative
safety of prophylactic doses of low molecular weight
heparininpregnancy,anincreasingnumberofclin-
icians are willing to prescribe antithrombotic agents
to women with heritable thrombophilia and a history
of two or more otherwise unexplained miscarriages
or one unexplained later intra-uterine fetal death.
e American Consensus of Chest Physicians sug-
gested for women with heritable thrombophilia and
146

Chapter 12. Thrombophilia and pregnancy loss
recurrent miscarriage or a second-trimester or later
loss, prophylactic doses of low molecular weight hep-
arin (or minidose unfractionated heparin) with low
dose aspirin therapy during pregnancy and following
delivery.5
Dilemmas
Women who have suered pregnancy loss have many
questions and will seek information about the possible
cause, the likelihood of recurrence, and the possibil-
ity of intervention to try to reduce the chances of fur-
ther pregnancy loss. At present, however, there is a lack
ofsolidevidenceonwhichtobaseadviceaboutthe
appropriateness or otherwise of testing these women
for heritable thrombophilias or on the management of
those who may be found to have a heritable throm-
bophilia.
Lack of evidence
Studies on the management of pregnancy loss are fre-
quently awed. e subjects included are oen poorly
selected and form a heterogeneous group lacking strat-
ication for important factors such as maternal age,
past obstetric history, and stage of gestation. Some
studies have compared pregnancy outcome in patients
subjected to a new intervention with the outcome in
their own previous pregnancies. is strategy ignores
the fact that some of these women will have suf-
fered previous pregnancy loss merely by chance and
will, as a result of the phenomenon of “regression to
mean,” have a high chance of having a successful preg-
nancy outcome without any additional intervention.
Reports of studies in which the pregnancy outcome
in women with a history of pregnancy loss subjected
to some experimental intervention is compared with
pregnancy outcome in historical controls who were
either untreated or treated dierently are subject not
only to the phenomenon of “regression to mean” in the
treated patient group but also to problems ascertaining
the control information.
Randomized trials are needed urgently
Proper evaluation of interventions in women with
a history of pregnancy loss requires randomized,
double-blind, controlled trials in which patients are
carefully selected to ensure that the treated and con-
trol groups are similar with respect to all of the import-
ant determinants of pregnancy outcome. Random-
ized, controlled trials have proven dicult to complete
because many women do not wish to risk being
randomized to the control group. A number of ran-
domized double-blind studies are, however, nearing
completion. It has to be hoped that these studies will
provide more solid evidence on which to base infor-
mation and advice for women with a history of unex-
plained late pregnancy loss or recurrent early loss.
In the meantime, many clinicians choose to treat
patients on an individual and pragmatic basis with
prophylactic daily doses of low molecular weight
heparin (e.g. enoxaparin 40 mg daily or dalteparin
5000 units daily) throughout pregnancy and the puer-
perium. Some also advocate the addition of low dose
aspirin (⬍150 mg) daily. e pros and cons of inter-
vention should be discussed with the patient and the
lack of proof of ecacy of thromboprophylaxis made
clear. Ideally, this discussion should take place during
preconception counseling.
Summary
rMany studies have examined the association
between heritable thrombophilias and fetal loss,
but the results are frequently contradictory,
populations heterogeneous, and the absolute risk
(if any) small.
rPublished meta-analyses suggest that factor V
Leiden, prothrombin G20210A, and protein S
deciency are associated with an increased risk of
recurrent early pregnancy loss and non-recurrent
late pregnancy loss.
rWomen with a history of pregnancy loss merit
increased surveillance in subsequent pregnancies
and should be given folic acid during pregnancy.
rCurrently, there is a lack of evidence on which to
base any pharmacologic intervention in women
with a history of pregnancy loss with no other
identied abnormality apart from a heritable
thrombophilia.
rDespite the lack of evidence from randomized,
double-blind, placebo-controlled trials, many
clinicians are oering women with a history of
pregnancy loss found to have a heritable
thrombophilia self-administered prophylactic
doses of low molecular weight heparin +/−daily
low dose aspirin in subsequent pregnancies. 147

Section 4. Thrombophilia and fetal loss
References
1. Preston FE, Rosendaal FR, Walker ID et al.
Increased fetal loss in women with heritable
thrombophilia.[comment]. e Lancet 1996; 348:
913–916.
2. Rey E, Kahn SR, David M, Shrier I. rombophilic
disorders and fetal loss: a meta-analysis. e Lancet
2003; 361: 901–908.
3. RobertsonL,WuO,LanghornePet al. rombophilia
in pregnancy: a systematic review. British Journal of
Haematology 2006; 132: 171–196.
4. Roque H, Paidas MJ, Funai EF et al. Maternal
thrombophilias are not associated with early
pregnancy loss. rombosis and Haemostasis 2004; 91:
290–295.
5. Bates SM, Greer IA, Hirsh J, Ginsberg JS. Use of
antithrombotic agents during pregnancy: the Seventh
ACCP Conference on Antithrombotic and
rombolytic erapy. Chest 2004; 126: 627S–
644S.
6. Walker ID, Greaves M, Preston FE. Investigation and
management of heritable thrombophilia. British
Journal of Haematology 2001; 114: 512–528.
7. VossenCY,PrestonFE,ConardJet al. Hereditary
thrombophilia and fetal loss: a prospective follow-up
study. JournalofrombosisandHaemostasis2004; 2:
592–596.
8. Brenner B, Homan R, Blumenfeld Z, Weiner Z,
Younis JS. Gestational outcome in thrombophilic
women with recurrent pregnancy loss treated by
enoxaparin. rombosis and Haemostasis 2000; 83:
693–697.
9. Gris JC, Mercier E, Quere I et al.
Low-molecular-weight heparin versus low-dose
aspirin in women with one fetal loss and a
constitutional thrombophilic disorder. Blood 2004;
103: 3695–3699.
10. Di Nisio M, Peters L, Middeldorp S. Anticoagulants
for the treatment of recurrent pregnancy loss in
women without antiphospholipid syndrome. Cochrane
Database of Systematic Reviews 2005;(2):CD004734.
148

Section
5
Hemorrhagic disorders

Section 5 Hemorrhagic disorders
Chapter
13a Management of obstetric hemorrhage:
obstetric management
Annette Briley and Susan Bewley
Introduction and epidemiology
Obstetric hemorrhage (OH) is the leading cause of
maternal mortality worldwide. In the UK, mortality
rates are relatively low, with 17 deaths per 100 000
maternities recorded in the Condential Enquiry into
Maternal and Child Health (CEMACH) report 2000–
2002.1However, morbidity remains high, and timely
recognition and management is of the utmost import-
ance.
Antepartum hemorrhage (APH) is dened as
bleeding from the genital tract aer 24 weeks’ gestation
and aects approximately 3%–4% of all pregnancies.
e most common cause of APH is due to the pres-
ence of placenta previa, where the placenta is abnor-
mally located in the lower uterine segment, covering
or partially covering the internal os. As pregnancy pro-
gresses, especially as the lower segment forms or the
cervix dilates, the woman is prone to episodes of bleed-
ing that may be profuse. Another common cause of
APH is placental abruption, when the placenta prema-
turely separates either partially or totally. It may be a
single episode or recurrent, small or large, and the fea-
tures may be typical and multiple (bleeding and pain,
tender and woody Couvelaire uterus with stillbirth)
or atypical and isolated (bleeding, premature labor,
fetal growth restriction, abnormal CTG). However, the
cause of many cases of APH is oen unknown.
Primary postpartum hemorrhage (PPH) is the
most common obstetric hemorrhage and is dened
by the World Health Organization (WHO) as the loss
of blood estimated to be ⬎500 ml from the geni-
tal tract within 24 hours of delivery. Aer this,
and until 6 weeks’ postpartum, abnormal bleeding
from the genital tract is dened as secondary PPH.
Hemorrhage is considered severe when blood loss
exceeds 1000 ml.2e major cause of postpartum
hemorrhage is uterine atony, when the uterus fails to
contract fully aer delivery of the placenta. PPH com-
plicates 11% of deliveries worldwide, and is annually
responsible for 132 000 maternal deaths.3.Evenwith
appropriate active management, around 3% of women
will experience a PPH2following vaginal delivery and
a recent study in low risk Australian women suggested
it was as high as 12%.4Hemorrhage is a direct cause
of around 30% of all maternal deaths worldwide, the
majority occurring in the poorest countries.5Substan-
dard care has been highlighted as a factor in 60% of
maternal deaths in the UK Condential Enquiry into
Maternal Deaths 2003–2005 report.6
Table 13a.1 shows the estimated time to death for
obstetric emergencies, highlighting APH and PPH, in
particular, and revealing obstetric hemorrhage as the
most dangerous complication of pregnancy for the
mother.7
Prevention
Prevention of PPH is via the recognition of any risk
factors present either ante-natally or during the intra-
partum period, and the subsequent implementation of
preventative management/strategies.
Although there are a host of risk factors (see Table
13a.2 below), postpartum hemorrhage oen occurs in
womenwithnoidentiablepredictorsandtherefore
clinicians must be prepared for this eventuality at each
and every delivery.8
e degree of risk will inuence the management
of these women from place of birth to mode of deliv-
eryandpost-natalcare.Womenathigherriskofhem-
orrhage should be advised to have their babies in
consultant-led units with an on-site blood bank. It is
important to involve the woman and her family in the
multi-disciplinary plan for her delivery, which must
151
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 5. Hemorrhagic disorders
Table 13a.1 Estimated time to death for
obstetric emergencies
Cause Time to death
Postpartum hemorrhage 2 hours
Antepartum hemorrhage 12 hours
Uterine rupture 1 day
Eclampsia/severe PET 2 days
Obstructed labor 3 days
Infection 6 days
be well documented and reviewed as the pregnancy
progresses and risk factors change. CEMACH recom-
mends planned management, particularly in cases of
placenta percreta.6
Ultrasound localization of the placenta in all
women, especially those who have had previous
Cesarean section,9should be reported and documen-
ted clearly in the handheld notes. Ante-natal assess-
ment of full blood count and treatment of anemia is
essential.
e importance of communication with all mem-
bers of the multi-disciplinary team, and early involve-
ment of senior medical and midwifery sta, have been
highlighted in successive Condential Enquiries and
CEMACH Reports to improve prognosis. e Scot-
tish Audit has found that reporting morbidities and
the resultant review of management has consistently
reduced the incidence of substandard care.10
If a woman is at risk of PPH, there are preventative
and predictive measures which can be implemented
in the intrapartum period. Such interventions include
giving oral ranitidine (150 mg), gaining intravenous
access with two large bore cannulae and taking blood
to send for a full blood count, group, and save.
Active management of the third stage of labor is
recommended for any woman at increased risk of
PPH. is shortens the time between delivery of the
baby and the placenta and membranes with no signi-
cant increase in retained placenta.11,12 Active manage-
ment involves the administration of oxytocin (or other
uterotonic drug) with, or shortly following, delivery
Table 13a.2 Risk factors for postpartum hemorrhage4,7, 20
Factors Risk (if known)
Pre-pregnancy Maternal age ⬎35 years
Nulliparity ×3
Grand multiparity
Asian ethnicity ×2
Obesity ×2
Previous Cesarean section
Previous PPH ×3
Uterine fibroids
Factor VIII deficiency – hemophilia A carrier
Factor IX deficiency – hemophilia B carrier
Pregnancy acquired Multiple pregnancy
Placenta praevia
Abnormal placental implantation – accreta, increta, and percreta
APH in current pregnancy
Polyhydramnios
Pre-eclampsia or pregnancy induced hypertension
Sepsis (including chorioamnionitis and/or endometritis)
×5
×15
×4
Delivery acquired Elective cesarean section ×4
Precipitate labor
Maternal pyrexia in labor ×2
Oxytocin administration for induction or augmentation of labor
Labor lasting ⬎12 hours ×2
Operative vaginal delivery ×2
Emergency Cesarean section ×9
Fetal macrosomia (Baby weight ⬎4kg) ×2
Ruptured uterus
Third stage Tissue – Retained placenta (causes 10% of PPH)
Tone – Uterine atony (causes 70% of PPH)
Trauma – laceration to perineum, vagina, or cervix (causes 20% of PPH)
Thrombin – coagulopathies (causes 1% of PPH)
Infection
152

Chapter 13a. Obstetric management
Increta:
placenta invades
myometrium
Percreta:
placenta
penetrates
through
myometrium and
serosa
Accreta:
placenta
adherent
Normal
Implantation:
note cleavage plane
Fig. 13a.1 Abnormal uteroplacental
implantation.
of the anterior shoulder of the baby. Controlled cord
traction may reduce the risk of retained placenta and
subsequent need for medical intervention. ere have
been no reported adverse eects of controlled cord
traction.13–15 More recently, international guidelines
include uterine massage following delivery of the pla-
centa as the last part of active management of the third
stage,16 although there is little evidence of the eective-
ness of this.17
Pathogenesis of PPH
e most common cause of PPH, accounting for
approximately 70% of occurrences is uterine atony.
ere are numerous reasons for the uterus failing
to contract eectively; including exhaustion, sepsis,
and retained products. Other causes of PPH include
perineal trauma, uterine inversion, clotting disorders,
pelvic hematomas, and cervical tears. An abnormally
implanted placenta (see Fig 13a.1) (placenta accreta,
increta or percreta) can remain in situ and hence pre-
vent the uterus from contracting properly. Placenta
previa and accreta are becoming an increasing prob-
lem, attributed to abnormal adherence of the pla-
centa in subsequent pregnancies following Cesarean
section.
If obstetric hemorrhage is not managed eciently
and eectively, this will lead to shock, hemostatic
failure from disseminated intravascular coagulation
(DIC), and ultimately death.
Diagnosis
Diagnosis of obstetric hemorrhage is typically by the
visualization of blood loss from the genital tract. In the
case of APH, bleeding can be concealed, and the only
signmaybeevidenceofmaternalcompromiseand/or
fetal distress. With PPH, the volume of blood loss
is usually estimated visually, although this is notori-
ously inaccurate. In the acute situation before hemod-
ilution, hemoglobin will not represent the amount of
blood lost and t, young women may appear to com-
pensate and maintain vital signs until a late stage.
Alertness and attention to clinical symptoms and signs
arevital.Someunitsattempttomeasurebloodloss
by weighing blood-soaked items, for example, sani-
tary pads and sheets. CEMACH suggest that an Early
Warning Score chart be used to assess maternal com-
promise and so give a more accurate representation
of maternal condition compared with visual estima-
tion of blood loss, to prevent delay in emergency
management.6
Obstetric management
In the case of APH, management will depend on
the amount of bleeding, maternal compromise and/or
degree of fetal distress. A concealed APH large enough
to cause intra-uterine death is probably at least
1.5 liters and DIC and PPH should both be assumed
and anticipated.
Immediate management of PPH
Once PPH has been diagnosed, action must be rapid.
Figure 13a.2 is an eective tool in identifying what
needs to be done by whom, as oen in the case of a
PPH several actions need to be taken simultaneously.
Whilst constantly assessing maternal resuscitation
requirements (pulse, blood pressure, respiration, tem-
perature),theuterusshouldbemassagedtostimu-
late a contraction, which may assist stemming of the 153

Section 5. Hemorrhagic disorders
HEAD
•Check airway
•Check breathing
•Administer O2
•Lie flat
•Record time of relevant events ARMS
Check pulse and BP
Establish LARGE BORE IV access x 2
Check FBC, clotting and Xmatch 4–6 units
Start FLUID RESUSCITATION
2 liters crystalloid
Drugs:
Ergometrine 0.5 mg IV (IM)
Syntocinon infusion (10 u/hr)
Prostaglandin F2α 0.25 mg IM
Consider moving to theater if >2 doses required
Consider misoprostol 800 mcg PR
UTERUS
START HERE- CALL FOR HELP
•Massage uterus to stimulate contraction
•Deliver placenta if still in situ
•CO-ORDINATE:
o Assistant 1 at “HEAD”
o Assistants 2 and 3 at “ARMS”
•Empty bladder- insert catheter
•If atony persists apply bimanual compression
•Review other causes; 4 T’s (Tone, Trauma, Tissue, Thrombin)
•Move to theater early if bleeding persists
Fig. 13a.2 Management of PPH: organizing the team (adapted from PersePhone/Pingirl).2
bleeding should the cause be atonic. is mas-
saging action also helps expel retained products
orbloodclots.Afullbladdercouldpreventthe
uterus from contracting properly by impeding on the
space, and therefore catheterization is recommended
(Table 13a.3).
Other uterotonics have been suggested with lim-
ited anecdotal evidence. ese include:
rDinoprostone; however, this is not suitable in
hypovolemic situations.
rGemeprost (cervogem)
rSulproston – a Prostaglandin E2widely used in
France as a second line drug aer oxytocin (before
ergometrine). Can cause coronary spasm,
hypertension, pyrexia, nausea, and vomiting.
rVas opressin R
5iuin19mlnormalsalinegiven
by subendothelial inltration. Avoid intravenous
administration as it causes severe hypertension.
rTranexamic acid – a lysine derivative, which
appears well tolerated. ere is no evidence of
increased thrombosis and this drug is probably
underused. I g intravenously with, if necessary,
repeatdose4hourslater.
rMethotrexate – prevents DNA replication and
may be useful in conservative management of
placenta accreta.
Volume maintenance in PPH
Initially, while blood is being cross-matched,
volume replacement with crystalloid should
be instituted. Close attention to uid balance is
required to avoid the perils of hypoxia-hypovolemia,
on the one hand, and cardio-pulmonary overload
on the other. In massive hemorrhage, uid replace-
ment can be controlled with central venous and
arterial lines and anesthetic and hematology input
is vital both during the event and subsequently
on high-dependency or intensive care units (see
Chapter 13b).
154
Chapter 13a. Obstetric management
Table 13a.3 Drug Management of PPH
Name of drug How it works Administration Side effects
Oxytocin
Prevention
Stimulates rhythmic upper uterine
segment contractions
IM as part of syntometrine (acts in 2–3
minutes, lasts up to 60 mins)
IV bolus 5 i.u. (acts in 1 min, has half life of
3mins)
IV 5 i.u. bolus can be repeated
Infusion- 40 units/500 mls over 4 hours
Hypotension, due to vasodi-
latation, especially in cardiac
patients
rAdminister slowly
rIn cardiac patients infuse
10 units over 30 mins
Anti-diuretic hormone effect
rFluid overload – can lead
to pulmonary edema
rHyponatremia
Ergometrine
Recognition
Sustains uterine contraction via alpha
receptors in the upper and lower
uterine segment of the uterus
Combined with oxytocin as
Syntometrine
Acts in 2–5 mins lasts up to 3 hours
If atony persists give 0.5mg IV
Potent agonist causes blood
vessels to constrict
Vomiting+++most women
WILL vomit
Hypertension- do not give to
women with pre-existing
high BP, Pre-eclampsia or PIH
or cardiac disease
Carboprost
(Hemobate R
)
Treatment
This is a prostaglandin F2 alpha Not for IV administration
Intramyometrially into the fundus of the
uterus to avoid blood vessels (but there
are very large vessels in the uterus)
Intramuscularly, 250 g (microgram) every
15 mins (maximum of 2mg)
In practice most women are not given
more ⬎2doses
85% women respond to the first dose
Has predelection for smooth
muscle of the bronchi and
therefore caution is
required with asthmatics
Bronchospasm
Significant intrapulmonary
shunt
Hypoxia
Microprostol
(Cytotec R
)
Treatment
This is a prostaglandin E1analog
It is thermostable and does not
require refrigeration.
Multiple routes of administration
Orally, sublingually, rectally
800–1000 mcg (effective within 3 mins)
Shivering
Pyrexia
This drug is cheap and therefore
useful in resource limited
countries.
Fig. 13a.3 Bimanual uterine compression.
Surgical management of PPH
rManual removal of placenta – is is an
emergency procedure to separate the placenta
manually that should be considered if it has not
deliveredwithinanhourofbirth.Partial
separation and delays can be associated with very
heavy bleeding.
rBimanual uterine compression is is an eective
way of stemming bleeding by compressing the
uterus with both hands (Fig 13a.3)
rExamination under anesthesia (EUA) and
evacuation of retained products of conception
(ERPC) is should be performed in theater with
appropriate conditions, personnel, and
instruments and in preparation for further
procedures. It is important to explore the whole of
the uterus, cervix, vagina, and perineum in a
rigorous way even if one cause is found or
excluded. e aim of EUA is to assess the cause of
bleeding and take action accordingly. e cause
may be found, for example, retained cotyledon,
pelvic hematoma (common aer a normal vaginal 155

Section 5. Hemorrhagic disorders
delivery and oen requiring surgery) or cervical
tears. ERPC for secondary PPH, especially if
associated with sepsis, must be performed with
greatcareasitcanleadtoperforationofthe
uterus.
rBalloon tamponade can be used. is involves
placing a balloon in the uterus and inating it.
Most commonly around 800–1000 ml are used to
ensure the balloon does not fall out aer a vaginal
delivery, but less is required if only compressing
the lower segment aer elective Cesarean; the
balloon is then le in situ for 24 hours aer which
time it is gradually deated. is is a cost-eective
method. In an emergency a condom could be
lledwithuidandinsertedintotheuterusto
apply pressure to stop the bleeding.
rPacking with surgical gauze. is is a traditional
and eective way to stop surgical bleeding and
ooze from a raw or sutured surface, although the
disadvantage is that a second procedure may be
required for removal. Packs have to be placed
under pressure, and can be le in the uterus, the
vagina, or in the abdomen.
re B-Lynch brace suture –Althoughnotpopular
in some units, this has revolutionized practice in
recent years. e brace suture has not been
evaluated in a RCT, but the case history evidence
is compelling, and any speculated eect on
fertility has to be compared with hysterectomy,
which would otherwise be the next surgical
option. It has been suggested that the B-Lynch
bracesuturemayindenttheuterusorcause
necrotic uterus. Prophylactic brace sutures can be
advocatedinJehovah’sWitnessesandinothers
who will refuse transfusion who require Cesarean
section and are assessed as at increased risk of
PPH (although risk assessment is dicult).
rInternal iliac vessel ligation –thisisan
old-fashioned technique and less familiar to
obstetricians nowadays, though more oen used
by gynecological oncologists. e principle has
evolved into an interventional radiology
technique of uterine artery embolization to stop
bleeding. e collateral circulation is adequate to
protect the uterus, but the equipment is not
availableinallunits,andthereisarisktofuture
fertility.
rHysterectomy – Women may die if the decision to
do a hysterectomy is not made or made too late,
but practitioners must be prepared to defend their
decision making in the legal process, as
unnecessary loss of fertility is devastating. is
must be the treatment of last resort, having
attempted conservative measures rst. e
UKOSS study of hysterectomy did show a failure
rate both for brace suture and interventional
radiology embolization.17
All women, and their companions, deserve a con-
temporaneous explanation, and a discussion aer-
wards about what happened, so that any questions
can be answered. Aer a PPH, women can make a
remarkable physical recovery. However, unrecognized
or undocumented PPH may lead to dizziness, fainting,
or collapse in the immediate postpartum period. All
women with symptoms or a recognized PPH should
have a postpartum or day 2–3 hemoglobin in case iron
supplements should be prescribed. A prolonged recov-
ery may be associated with fatigue, exhaustion, and
interference with breast feeding and bonding. Massive
hemorrhage can be very traumatic, for women, their
families, and for sta, and the need for explanation and
reection must not be underestimated. Sta skills must
be constantly updated.
156

Chapter 13a. Obstetric management
References
1. Lewis G. e Condential Enquiry into Maternal and
ChildHealth(CEMACH).Why mothers die
(2000–2002), 2005; London: CEMACH.
2. American Academy of Family Practitioners (AAFP)
Advanced Life Support in Obstetrics. Syllabus Updates
2008. www.aafp.org/online/en/home/cme/
aafpcourses/clinicalcourses/also/syllabus.html
#Parsys0003 downloaded 21/05/2008
3. WorldHealthReport.Makeeverymotherandchild
count. http://who.int/whr/2005 en.pdf 2005; accessed
19th May 2008.
4. Ford JB, Roberts CL, Simpson JM, Vaughan J,
Cameron CA. Increased postpartum hemorrhage rates
in Australia. International Journal of Gynaecology and
Obstetrics (the ocial organ of the International
Federation of Gynaecology and Obstetrics) 2007; 98,
237–243.
5. Khan KS, Wojdyla D, Say L, Gulmezoglu AM, van
Look PF. WHO analysis of causes of maternal death: a
systematic review. e Lancet 2006; 367: 1066–1074.
6. CEMACH. Saving Mother’s Lives: Reviewing maternal
deaths to make motherhood safer – 2003–2005. e
Seventh Report of the Condential Enquiries into
Maternal Deaths in the United Kingdom DoH 2007.
7. Magann EF, Evans S, Chauhan SP, Lanneua G, Fisk
AD, Morrison JC. e length of the third stage of labor
and risk of postpartum hemorrhage. Obstetrics and
Gynecology 2005; 105(2): 290–293.
8. DoH. Why Mothers Die, 1997–1999. e Fih Annual
Report of the Condential Enquiries in to Maternal
Deaths in the United Kingdom.DoHRCOGPress
London.
9. Magann EF, Evans S, Hutchinson M et al. Postpartum
hemorrhage aer cesarean delivery; an analysis of risk
factors. South Medical Journal 2005; 98: 681–685.
10. Penney G, Adamson L. Scottish Condential Audit of
Severe Maternal Morbidity 4th Annual Report 2006
SPCERH.
11. Bais JM, Eskes M, Pel M, Bonsel GJ, Bleker OP.
Postpartum haemorrhage in nulliparous women:
incidence and risk factors in low and high risk women.
A Dutch population-based cohort study on standard
(⬎or =500 ml) and severe (⬎or =1000 ml)
postpartum haemorrhage. European Journal of
Obstetrics Gynecology Reproduction Biology 2004; 115:
166–172.
12. Prendiville WJ, Elbourne D, McDonald S. Active
versus expectant management of the third stage of
labour. Cochrane Database System Review 2000(2):
CD00007.
13. Khan GQ, John IS, Wani S et al. Controlled cord
traction versus minimal intervention techniques in
delivery of the placenta: a randomised controlled trial.
American Journal of Obstetrics and Gynecology 1997;
177: 770–774.
14. Zhao S, Xiaofeng S. Clinical study in curing
postpartum haemorrhage in the third stage of labor.
JournalofPracticalObstetricsandGynecology2003; 19:
278–280.
15. Giacalone PL, Vignal J, Daures JP et al. A randomised
evaluation of two techniques of management of the
third stage of labour in women at low risk of
postpartum haemorrhage. British Journal of Obstetrics
and Gynaecology 2000; 107:396–400.
16. Lalonde A, Daviss BA, Acosta A, Herschderfer K.
Postpartum hemorrhage today: ICM/FIGO initiative
2004–2006. International Journal of Gynaecology and
Obstetrics 2006; 94: 243–253.
17. Chelmow D. Postpartum haemorrhage: prevention.
British Medical Journal Clinical Evidence 2007; 2:
1410.
18. Knight M, UKOSS. Peripartum hysterectomy in the
UK: management and outcomes of the associated
haemorrhage. British Journal of Obstetrics and
Gynecology: an International Journal of Obstetrics and
Gynaecology, 2007; 114, 1380–1387.
157

Section 5 Hemorrhagic disorders
Chapter
13b Management of obstetric hemorrhage:
anesthetic management
Vivek Kakar and Geraldine O’Sullivan
Introduction
Once the diagnosis of obstetric hemorrhage has been
made, early senior anesthetic involvement (experi-
enced registrar or a consultant) is vital. e UK Con-
dential Enquiry into Maternal and Child Health 2000–
2002 (Why Mothers Die1) and 2003–2005 (Saving
Mothers Lives2) show that hemorrhage is still one of
the commonest causes of direct maternal deaths. In
Why Mothers Die 1999–2002, 17 maternal deaths were
caused by hemorrhage; care was considered subopti-
mal in 5 of these 17 cases. In the Saving Mothers Lives
2003–2005, hemorrhage caused 14 deaths and was a
complicatingfactorin9others.Inasmanyas10of
these 14 deaths, the patient received suboptimal care.
Communication
Early and clear communication between obstetricians,
midwives, anesthetists, hematologists, and the porters
is essential. A “leader” should coordinate the ongoing
management of the hemorrhage. Arguably, this leader
should be the senior anesthetist. Extra help, surgical,
and/or anesthetic, should also be summoned.
Access
Several large bore intravenous (IV) cannulae
(14G/16G) should be sited. Central venous catheter-
ization may be needed at a later stage, but should
not delay resuscitation in emergent situations, in
otherwise healthy patients. Ultrasound guidance is
recommended by NICE3for the insertion of a central
venous catheter in the internal jugular vein. Subcla-
vian vein cannulation should specically be avoided
in established or suspected coagulopathy, as occurs in
concealed abruption, sepsis, severe pre-eclampsia and
massive transfusion.
Monitoring
Blood pressure, oxygen saturation, and electrocardio-
gram (ECG) should be continuously monitored. ere
should be a relatively low threshold to insert an arter-
ial line in bleeding patients. A central venous pres-
sure (CVP) monitor may be required in cases of
massive hemorrhage, although not as a part of the
initial resuscitation. A urinary catheter should be
inserted in all cases to measure the hourly urine
output. Resuscitation should be guided by clinical
parameters, arterial blood gases, lactates, CVP, and
urinary output. In cases with cardiovascular and/or
renal problems, some form of cardiac output mon-
itoring (LiDCO, PiCCO, or esophageal Doppler in
intubated patients) can provide useful information.
Accurate assessment of blood loss is essential and
can be achieved by weighing the surgical swabs
andmeasuringthevolumeofbloodinthesurgi-
cal suction. e Association of Anaesthetists of Great
Britain and Ireland (AAGBI) guidelines4for moni-
toring should be followed during surgery, recovery
and transfer of these patients, should that be required.
Other recommendations2,5–6 are that some form of
track and trigger scoring system (such as Modi-
ed Early Warning Scores (Table 13b.1)) should be
used in high risk patients monitored on the labor ward
to facilitate early identication of patients with ongo-
ing hemorrhage.
Oxytocics (see also Chapter 13a)
(a) Oxytocin:Fivetotenunitsshouldbeadministered
as a slow IV bolus. Rapid IV administration can
cause profound hypotension and tachycardia.
Cardiac arrest has also been reported. It works
within 2–3 minutes but due to its short half-life it
needs to be administered as an infusion, e.g.
40 IU in 40ml of normal saline over 4 hours.
158 e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Chapter 13b. Anesthetic management
Table 13b.1 Modified early warning scoring system5
32 1 0 1 2 3
HR
(bpm)
⬍40 40–50 51–100 101–110 111–129 ≥130
BP ⬎45%↓30%↓15%↓Normal 15%↑30%↑⬎45%↑
RR
(/min)
≤89–14 15–20 21–29 ≥30
Temp
(◦C)
⬍35.0 35.0–38.4 ⬎38.5
CNS A V P U
Urine
Output
Nil ⬍1ml/kg/2h ⬍1ml/kg/h ⬎3ml/kg/2h
A=Alert V =Responds to Verbal commands P =Responds to Pain U =Unresponsive.
(b) Ergometrine:Itisanextremelyeective
second-line drug for an atonic uterus. e IV dose
is 100–300 mcg. Uterine contraction occurs
within 5 min of an intramuscular (IM) injection
and 1 minute aer an IV injection. Its eects last
at least 1 hour.
(c) Carboprost (methyl prostaglandin F2␣):Itisan
extremely potent drug and is administered by IM
or intramyometrial injection. It should be
administered in 250 g increments, repeated at
15-minute intervals up to a maximum of 2 mg.
e majority (85%) of patients will usually
respond to the rst or second dose, and in practice
the full 2 mg dose will rarely be employed as
ongoing severe hemorrhage usually necessitates
further surgical/radiological intervention.
(d) Misoprostol: is is a prostaglandin E1analog. It
is supplied as 200 g tablets and 800–1000 g
should be administered rectally.
Fluids
Immediate resuscitation should begin with crystal-
loids and colloids. ere is no evidence of superi-
ority of one over the other in non-septic obstetric
patients.7In an emergency, the choice of uid is imma-
terial. Hartmann’s solution is the most physiologically
balanced solution; normal saline can also be used,
although it can itself cause metabolic acidosis aer sev-
eral liters have been used.
Commonly available colloids include starches
(Voluven), gelatins (Haemaccel, Gelofusine), and albu-
min. ere have been concerns about the eect of
starches on platelet function and renal function. A
recent large study8found signicantly increased renal
failure and blood transfusion requirements in sep-
tic patients who required more than 22 ml/kg of 10%
hetastarch (0.5/200). is study did not include the
obstetric population. Gelatins can interfere with blood
grouping, cross-matching, and cause allergic reac-
tions. A recent Cochrane review failed to establish any
dierence between dierent colloids in terms of out-
come.9
Blood pressure, heart rate, and urine output are
good endpoints in assessing adequate resuscitation. In
t and healthy young patients, tachycardia will usu-
ally represent uncorrected hypovolemia. Blood pres-
sure will generally not fall until 30% of the blood vol-
ume (∼1500 ml) has been lost (Table 13b.2).
Preventing the “lethal triad” of
hypothermia, acidosis and
coagulopathy
It has been demonstrated in trauma patients with
massive bleeding, that if they are allowed to become
hypothermic and acidotic, their coagulopathy wors-
ensorisrefractorytocorrection.
10 is has also been
shown to be true in other cases of hemorrhagic shock
(Fig. 13b.1).
Evidence suggests that a fall in temperature from
37 ◦Cto33◦C reduces rFVIIa activity by 20%, whilst
afallinpHfrom7.4to7.0reducesrFVIIaactiv-
ity by 90%. Platelet function is also inhibited to a
varying degree. It seems that acidosis alone does not
seem to aect the clotting, but increases the eect of
hypothermia on clotting.11 erefore, the prevention 159

Section 5. Hemorrhagic disorders
Table 13b.2 Classification of hemorrhagic shock
Class 1
(Compensated)
Class 2
(Mild)
Class 3
(Moderate)
Class 4
(Severe)
Blood loss
(% Circulating blood
volume)
750 ml
(⬍15%)
800 – 1500 ml
(15%–30%)
1500 – 2000 ml
(30%–40%)
⬎2000 ml
(⬎40%)
Systolic blood pressure No change Orthostatic Fall Low Very low
Diastolic blood pressure No change Raised Reduced Very low
Pulse rate ⬍100 ⬎100 ⬎120 (weak) ⬎140 (very weak)
Capillary rell Normal Slow (⬎2s) Slow(⬎2s) Prolonged (⬎5s)
Respiratory rate Normal Normal Raised (⬎20/min) Raised (⬎20/min)
Urine output ⬎30 ml/h 20 – 30 ml/h 10 – 20 ml/h 0 – 10 ml/h
Extremities Normal Pale Pale Pale and cold
Complexion Normal Pale Pale Ashen
Mental state Alert, thirsty Anxious, thirsty Anxious, aggressive or
drowsy
Drowsy, confused
or unconscious
Adapted from ATLS manual.
Hypothermia
and
acidosis
Coagulopathy
Hemorrhage
The lethal triad
Fig. 13b.1 The lethal triad
of hypothermia and acidosis is an essential component
in the successful management of massive hemorrhage.
Patients should be kept warm using forced air
warming blankets. All uids should be warmed using
any of the commonly available uid warmers (Fig.
13b.2). When the uid needs to be infused rapidly, nor-
mal uid warming devices may not be able to perform
eciently, when pressurized uid is run through them.
erefore, every obstetric unit should have access to a
Level 1 infusor (Fig. 13b.3) which can both pressurize
and warm blood and uids rapidly.
Fig. 13b.2 Fluid warmer.
Blood and blood component therapy12
(see Chapter 13c)
Blood replacement should be guided by bedside
and/or laboratory hemoglobin testing. When hemor-
rhage is rst diagnosed, blood should be sent to the
laboratory for a group and cross match, full blood
count (FBC) and coagulation screen (INR, aPTT, b-
rinogen). e hemoglobin, coagulation screen, and
platelet count will need to be repeated at regular
160

Chapter 13b. Anesthetic management
Fig. 13b.3 Level 1 Infusor.
intervals in order to facilitate the logical use of coagu-
lation factors. Alternatively, if available, a thrombo-
elastogram (TEG) can be used to guide the replace-
ment of coagulation factors.
Cell salvage13–18
Intraoperative cell salvage and auto transfusion has
been available for many years and has been very useful
in cardiac surgery, major vascular surgery, orthopedic
surgery, and trauma. Cell salvage is now increasingly
considered in massive obstetric hemorrhage as recent
research has shown it to be safe in obstetrics.
Indications
Cell salvage is a technique for re-cycling operative
blood loss. It is particularly appropriate for elective
surgery where massive blood loss is anticipated, e.g.
placenta previa/acreta/percreta and for mothers who
refuse blood and blood products, e.g. Jehovah’s wit-
nesses. Once skill has been acquired with the tech-
nique, it can be rapidly set up, even in an emergency.
Principles of cell salvage (Figs 13b.4
and 13b.5)
Blood is aspirated from the surgical site through hep-
arinized tubing and a lter into a collecting reservoir.
e cells are separated by hemoconcentration and dif-
ferential centrifugation in 0.9% saline, and washed in
1–2 L of 0.9% saline. is process removes circulating
brin, debris, plasma, micro-aggregates, complement,
platelets, free hemoglobin, circulating pro-coagulants,
and most of the heparin. At the end of the salvaged
process, the hematocrit of the salvaged blood is usu-
ally between 55% and 60%.
Problems
Cell salvage was until recently considered to be contra-
indicated in obstetrics because of the following two
theoretical risks:
rAmniotic uid embolism:Intheliterature,only
onedeathhassofarbeenreportedaerthe
use of salvaged blood. However, the patient was
aJehovah’sWitnesswithseverepre-eclampsia
and HELLP syndrome (Hemolysis, elevated liver
enzymes and low platelet count), and a leuko-
cyte depletion lter was not used. It has now
been shown that use of a 40 leukocyte deple-
tion lter (Fig. 13b.6), on the return limb to the
patient, eectively depletes or entirely removes
fetal squames, white blood cells, and platelets from
the salvaged blood. To date, no case of amni-
otic uid embolism has been reported in patients
who received salvaged blood during Cesarean sec-
tion where a leukocyte lter was used. In addi-
tion, amniotic uid embolism is now considered to
be more of an immunological phenomenon rather
than actual physical embolism. Nevertheless, any
contamination of the salvaged blood with amniotic
uid should be avoided as far as possible.
rAllo-immunization: Despite the use of several
wash cycles and lters, it is still not possible to
avoid contamination of the salvaged blood with
fetalredbloodcells.isisbecausethemachine
cannot distinguish between fetal and maternal red
blood cells. e amount can vary between 2 ml and
19 ml and Kleihauer counts should be routinely
performed in the postpartum period. All rhesus
negative mothers should be immunized with anti-
D. A second dose may be required if the Kleihauer
suggests heavy contamination with fetal cells.
Cost
Although the machines can be very expensive, most
hospitals lease them from the manufacturer. Typically, 161
Section 5. Hemorrhagic disorders
Suction from Double lumen aspiration
+ anticoagulent assembly
Saline +
anticoagulant
Reservoir
Saline
wash
Pump
Centrifuge
bowl
Return to patient
Waste bag
operative slte
Retransfusion
bag
Fig. 13b.4 Cell salvage schematic diagram.17
it costs approximately £100–170 per patient (towards
disposables) to setup and use the cell salvage machine.
So, the cost of disposables is covered as soon as you
need to transfuse more than one unit. A systematic
review of over 600 studies comparing various trans-
fusion strategies to reduce allogenic blood transfusion
found that the relative risk of requiring allogenic blood
transfusion with cell salvage was 0.59 and it was more
cost eective than all other strategies except acute nor-
movolemic hemodilution. Every unit with cell salvage
facilities should have a protocol for the use of cell sal-
vage in obstetrics.
Investigations
FBC and clotting should be checked frequently. In
cases of ongoing blood loss, resuscitation should
be guided by bedside hemoglobin estimation (e.g.
HemoCue, Fig. 13b.7) and/or arterial blood gas
estimation. Liver and renal function should also
be assessed at baseline and, once the patient has
been stabilized, especially in patients with com-
plex co-morbidities, multiple medications, massive
transfusion, or prolonged period of intraoperative
hypotension.
Regional Vs general anesthesia
In an elective situation, where signicant blood loss
is anticipated, such as with anterior placenta praevia,
regional anesthesia can still be considered, although
thepatientsshouldbewarnedoftheoccasionalneedto
convert to general anesthesia (GA) intra-operatively.
Baseline hemoglobin, venous access, invasive moni-
toringandacellsalvageunitshouldbeestablished
prior to starting such cases. Two to four units of blood
should also be cross-matched.
In an emergency situation,anestheticmanage-
ment will be determined by both fetal and mater-
nal considerations. GA is usually considered in cases
of severe hemodynamic instability, sepsis, and sus-
pected or conrmed coagulopathy. If GA is used in
severely hypovolemic patients, the anesthetic induc-
tion agents, ketamine or etomidate should be used
instead of thiopentone or propofol, as they do not
cause the profound hypotension commonly seen when
the latter two agents are used in hypovolemic patients.
If a bleeding patient needs to be transferred to the
interventional radiology suite, it might be worth secur-
ing the airway prior to transfer, especially if the radiol-
ogy department is not very close to the obstetric unit.
In addition, adequate anesthetic facilities and assis-
tanceshouldbeavailableintheradiologysuite.
162

Chapter 13b. Anesthetic management
Fig. 13b.5 Cell salvage machine.
Post-hemorrhage care
Post-operative care will usually be on a high depend-
ency unit, but transfer to an intensive care unit
may be necessary particularly if the patient requires
mechanical ventilation. If possible, cardiovascular and
metabolic parameters should be stabilized prior to
transfer. Acceptable standards of monitoring should be
maintained during the transfer as mentioned earlier.
Once the bleeding has been controlled and the
patient is stable, regular thromboprophylaxis should
be commenced.
Documentation
Accurate and complete documentation of the sequence
of events is very important. In cases of poor outcome,
poor documentation is indefensible even if excellent
care was provided. One person can be assigned the
job of keeping a record of all the drugs and uids
Fig. 13b.6 Leukocyte filters.
Fig. 13b.7 Hemocue.TM 163

Section 5. Hemorrhagic disorders
administeredandofthepersonnelinvolvedinthe
resuscitation.
Drills/protocols
A multidisciplinary massive hemorrhage protocol
must be available in all units and should be updated
and rehearsed regularly. Women known to be at high
risk of bleeding should be seen by a consultant anes-
thetist in the ante-natal period. ese patients should
ideally be delivered in centers with facilities for blood
transfusion, cell salvage, intensive care, and inter-
ventional radiology, and plans for their management
should be made in advance.
Debrieng and counseling
Supportive counseling of all the team members
involved is vital, should the hemorrhage result in
maternal death. Such an event represents a tragedy
not only for the woman’s family, but also for the
carers. Debrieng aer such episodes can be a very
good opportunity to reinforce learning points and seek
improvements for future.
164

Chapter 13b. Anesthetic management
References
1. CEMACH Report 2000–2002(Why Mothers Die).
(www.cemach.org.uk)
2. CEMACH Report 2003–2005(Saving Mothers’ Lives).
(www.cemach.org.uk)
3. National Institute for Clinical Excellence. Ultrasound
Locating Devices for Placing Central Venous Catheters.
Guideline number 49. London: NICE; 2002.
4. Association of Anaesthetists of Great Britain and
Ireland. Recommendations for Standards of Monitoring
During Anaesthesia and Recovery. London: Association
of Anaesthetists; 2007.
5. IntensiveCareSociety.GuidelinesfortheIntroductionof
Outreach Services. Intensive Care Society; 2002.
6. NICE clinical guideline 50. Acutely ill patients in
hospital. July 2007.
7. Perel P, Roberts I. Colloids versus crystalloids for uid
resuscitation in critically ill patients. Cochrane
Database Systematics Reviews 2007; 17: CD000567.
8. Brunkhorst FM, Engel C, Bloos F et al.Intensive
insulin therapy and pentastarch resuscitation in severe
sepsis. New England Journal of Medicine 2008; 358:
125–139.
9. Bunn F, Trivedi D, Ashraf S. Colloid solutions for uid
resuscitation. Cochrane Database System Review 2008
4: CD001319.
10. Tsuei BJ, Kearney PA. Hypothermia in the trauma
patient. Injury 2004; 35: 7–15.
11. Dirkmann D, Hanke AA, Gorlinger K, Peters J.
Hypothermia and acidosis synergistically impair
coagulation in human whole blood. Anesthesia and
Analgesia 2008; 106: 1627–1632.
12. Association of Anaesthetists of Great Britain and
Ireland. BloodTransfusionandtheAnaesthetist.
London: Association of Anaesthetists; 2008.
13. Allam J, Cox M, Yentis S M. Cell salvage in obstetrics.
International Journal of Obstetric Anesthesia 2008; 17:
37–45.
14. Sullivan I, Faulds J, Ralph C. Contamination of
salvaged maternal blood by amniotic uid and fetal
red cells during elective Caesarean section. British
Journal of Anaesthesia 2008; 101: 225–229.
15. Catling SJ, Williams S, Fielding A. Cell salvage in
obstetrics: an evaluation of ability of cell salvage
combined with leucocyte depletion lter to remove
amniotic uid from operative blood loss at caesarean
section. International Journal of Obstetric Anesthesia
1999; 8: 79–84.
16. Waters JH, Biscotti C, Potter PS, Phillipson E.
Amniotic uid removal during cell salvage in
caesarean section patients. Anesthesiology 2000; 92:
1531–1536
17. UK National Institute for Health and Clinical
Excellence. Intraoperative blood cell salvage in
obstetrics. IP Guidance Number: IPG144. Available
from URL: http://www.nice.org.uk/guidance
18. Davies L, Brown TJ, Haynes S, Payne K, Elliott RA,
McCollum C. Cost eectiveness of cell salvage and
alternative methods of minimising perioperative
allogeneic blood transfusion: a systematic review and
economic model. Health Technology Assessment 2006;
10: 1–228.
165

Section 5 Hemorrhagic disorders
Chapter
13c Management of obstetric hemorrhage:
hemostatic management
Eleftheria Lefkou and Beverley Hunt
Blood loss
Obstetric hemorrhage (OH) is dened by the World
Health Organization (WHO)1as a blood loss of more
than 500 ml in the rst 24 hours aer birth, or of
more than 1000 ml when Cesarean section has been
performed. A more comprehensive denition could
be any blood loss which can provoke a physiologi-
cal change threatening the woman’s life. According to
the American College of Obstetrics and Gynecology,
OH is dened as either a 10% change in hematocrit
between admission and postpartum, or the need for a
blood transfusion.2
Currentbestpracticeforthehematologicalman-
agement of obstetric hemorrhage (OH) emphasizes
the need for speedy and appropriate use of blood com-
ponents with close monitoring of blood loss. However,
best practice is not always followed. is seems, in part,
to be due to poor understanding in the appropriate use
of blood components and pharmacological agents to
reduce bleeding.
In this chapter we give practical guidelines for the
hematological management of OH. Table 13c.1 shows
the available blood components and their derivatives
used in hemostatic replacement therapy.
Hemostatic replacement therapy
Red cell products
Red blood cell (RBC) transfusion is a rst-line inter-
vention to treat the inadequate oxygen delivery (but
not the volume loss) seen in OH. In the UK whole
blood is not usually available.
One unit of packed red cells increases the
hemoglobin by approximately one g/dl and the
hematocrit by 3%. ere are no evidence-based guide-
lines for transfusion of RBC into hemodynamically
unstable women with OH. According to the British
Committee for Standards in Hematology guidelines
on the management of massive blood loss, red cell
transfusion is likely to be required when 30%–40%
blood volume is lost; when 40% blood volume loss
is immediately life-threatening.3As a general rule,
thetargethemoglobinlevelsshouldbegreaterthan
8g/dL.
Ideally, all pregnant women should be transfused
withredcellsofthesameABOandRhesusgroup.
4In
an urgent situation where blood is required immedi-
ately, with unknown patient’s blood group, all women
under 50 years must be given group O Rhesus negative
red cells, in order to avoid sensitization and hemolytic
disease of the newborn in subsequent pregnancies.
Every obstetric unit should have 2 units of O-Negative
blood in the fridge for emergency use. However, ante-
natalABOandrhesusgroupingandsendingasam-
ple to the blood transfusion laboratory to conrm
ABO grouping allows the release of matched blood.
e physicians should be mindful that blood group-
ing takes less than 10 min and so ABO group-specic
red cells should be administrated as soon as possible.
Platelet transfusion
IntheUKplateletsareusuallyobtainedbyplatelet-
pheresis from one donor- single donor plateletphere-
sis and stored in polyolen packs with a viability of
about 5 days; or they are removed from a unit of blood
andbaggedtogethersknownaspooledrandomdonor
platelets.
In massive OH aer a 1.5–2 ×blood volume
replacement, a platelet count ⬍50 ×109/l should be
anticipated. e target of platelet transfusion is to
maintain platelet count ⬎50 ×109/l (70–110 ×109/l).
In cases with qualitative platelet abnormalities, as in
166 e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Chapter 13c. Hemostatic management
Table 13c.1 Blood components and their derivatives used in
hemostatic replacement therapy
rRed cells
rPlatelet pools
rFresh frozen plasma
rCryoprecipitate or fibrinogen concentrates
rRecombinant Factor VIIa
rAntithrombin, Protein C and activated Protein C
concentrates
rProthrombinase concentrates (II, VII, IX and X)
rPlasma-derived and Recombinant Factor VIII and IX
rvon Willebrand Factor concentrates
some inherited diseases such as Glanzmann’s throm-
basthenia or Bernard–Soulier syndrome, or acquired
disorders such as liver or kidney disease, or drug-
induced platelet dysfunction, the trigger for platelet
transfusion should be higher, depending not on the
number but on the function of platelets. In the UK one
platelet apheresis concentrate will increase the platelet
count by 50 ×109/L in most adult patients. Ideally, the
platelet count should be checked 10–15 minutes aer
platelet infusion to ensure the adequacy of therapy. A
poor increment of less than 20 ×109/L aer 15 min-
utes in a patient without ongoing bleeding to suggest
thepresenceofantiplateletantibodies,usuallyhuman
leukocyte antigen (HLA) antibodies.
Fresh frozen plasma (FFP)
Fresh frozen plasma (FFP) is separated within 6–8
hours of whole blood collection, frozen at −18 ◦Cand
stored for up to 1 year. e volume of a typical unit is
200 to 250 ml. FFP contains normal levels of all coagu-
lation factors, except FVIII, which rapidly decays, leav-
ing around 60% levels. e indications for use of FFP
in massive transfusion and disseminated intravascular
coagulation with signicant bleeding is PT or APTT
ratio ⬎1.5. ere is no evidence base for the dose that
should be used, however 15 mL/kg is widely accepted.
Solvent detergent prepared FFP has a lower risk of
transfusion transmitted infection but has reduced lev-
els of macromolecular von Willebrand factor (VWF),
which is of little concern in the management of
bleeding.
Fibrinogen
ere are two sources of brinogen available in
the UK: cryoprecipitate and brinogen concentrate.
Cryoprecipitate is made from donor plasma by plac-
ing the plasma in a fridge at 4 ◦C. is allows all the
large molecules such as brinogen, von Willebrand
factor, and Factor VIII to precipitate out. ese are sep-
arated o and they are redissolved in a small residue
ofplasma,asitiswarmed.Atypicaladultdoseistwo
ve-donor pools (equivalent to 10 single donor units)
containing 3–6 g brinogen in a volume of 200 to 500
ml. As a rule of thumb, 10 bags of cryoprecipitate will
increase a normal adult’s brinogen by 1 g/L.
Fibrinogen concentrate is available but not licensed
for use in massive transfusion in the UK. Its poten-
tial side eects include hypertension, anaphylaxis, and
arterial thrombosis. It is licensed for use in patients
with congenital a- or hypo-brinogenemia.
In normal pregnancy brinogen levels are elevated
as part of the hemostatic response to pregnancy, with
levelsattermbetween5and7g/L.Soevennormal
non-pregnant levels (range: 1.5–4.0 g/L) of brinogen
mean that signicant consumption of brinogen has
occurred.
In OH brinogen levels are oen very low. With a
poorly contractile uterus or intra-abdominal bleeding,
large volumes of clot form and rapidly consume all the
availablebrinogen.Oen,brinogenlevelsareaslow
as 0.1 g/L. In this situation there is not enough brino-
gen for normal coagulation to occur. Infusion of fresh
frozen plasma is not enough to replete the deciency
andso20bagsofcryoprecipitateshouldbeusedas
soon as possible to elevate brinogen by approximately
2g/dL.
Pharmacological agents that
reduce bleeding
Antibrinolytics
e two agents which have been used in the UK are
tranexamic acid and aprotinin. Aprotinin has been
suspended from marketing in the UK with concerns
about its safety.5
Tranexamic acid binds to plasminogen and thus
inhibits its binding to brin. It has a plasma half-life
of 2 hours. It is contraindicated in renal tract bleeding
andinrenalfailure.
Tranexamic acid has been used extensively to
reduce perioperative bleeding, whether or not there
is evidence of hyperbrinolysis. A recent Cochrane
review6showsitissafeinthatitisnotassociated
with an increased risk of venous thromboembolism
with short-term use. However, there are no studies
of the ecacy and use of antibrinolytics in OH,
but theoretically the hemostatic changes of obstetric 167

Section 5. Hemorrhagic disorders
hemorrhage should be little dierent from surgical
bleeding and trauma. We know that massive bleed-
ing will stimulate epinephrine, which will cause release
of brinolytic activators. If there is a low brinogen,
clot formation will be defective and the clot is open
and more liable to being penetrated by brinolytic
activators. e use of tranexamic acid in traumatic
bleeding is being investigated in the CRASH-2 (clin-
ical randomization of antibrinolytic therapy in sig-
nicant hemorrhage), which aims to randomize 20 000
patients to tranexamic acid vs. placebo and will report
in 2010. e same group plan to do a similar random-
ized controlled trial of tranexamic acid in obstetric
hemorrhage (the WOMAN study).
e authors suggest in the interim that tranexamic
acid in a 1–2 g bolus should be strongly considered in
the management of OH in view of its safety and ecacy
in other settings.
Recombinant VIIa
Recombinant FVIIa is not adequately studied in OH.
It is licensed in Europe for treatment of hemophilia
patients with inhibitors to factors VIII and IX, and
for patients with Glanzmann’s thrombasthenia, and
FVII deciency. It has no other licensed indication for
any other group of patients but it has been used “o
license” in the management of bleeding.
ere are no prospective, randomized placebo-
controlled studies in the use of rFVIIa in OH, but many
case reports. Unfortunately, case reports can lead to
considerable reporting bias with a tendency towards
reporting only positive outcomes. ere are, however,
three major studies reporting data in the use of rFVIIa
in this setting. e rst study comes from the North-
ern European Registry 2000–2004, and gives data from
the use of rFVIIa in primary postpartum hemorrhage
(PH), from nine European countries.7A total of 113
individual cases are presented and the authors con-
clude that there was some improvement in more than
80% of women and few adverse eects. But it is not
clear that best practice for blood components was
applied prior to use of rFVIIa, i.e that the use of blood
components appropriately would not have resulted in
thesameimprovedoutcome.esecondstudy,from
Finland, reports retrospectively the one-center experi-
ence on the administration of rFVIIa to 38 parturi-
ents.8e authors conclude that there is no evidence
that the use of rFVIIa was better than standard man-
agement with blood components. e last study from
Ireland, reports massive OH in 28 cases, with rFVIIa
use in six patients, in a 3-year period at one insti-
tution.9e authors concluded that there is a need
for resuscitation, surgical intervention and appropri-
ate use of blood products and no place for the routine
use of rFVIIa. Haynes et al.10 summarizes 44 reported
cases with rFVIIa in OH and added four cases from
their experience. Data from this study showed that,
despite the administration of rFVIIa, invasive surgery
or procedures, such as hysterectomy or emboliza-
tion, remained necessary. From the 48 patients, seven
responded only partially to treatment and three died
despite treatment. A relatively recent systematic review
on the ecacy and safety of rFVIIa for treatment of
severe bleeding conclude that more randomized con-
trolled trials are required to assess the use of rFVIIa for
patients without a pre-existent coagulation disorder
and with severe bleeding.11 ArecentCochranereview
did not nd real evidence of its o license use but there
was a trend towards reduced mortality and increased
thromboembolic events.12 e review did not include
any studies of obstetric hemorrhage.
ere is current concern about the safety of rFVIIa
in “o licence” indications. A recent meta-analysis
showed an arterial thrombosis rate of 5.6% in those
receiving rVIIa compared with 3% in the placebo-
treated patients.
us the use of rVIIa in OH should ideally be lim-
ited to clinical trials or in intractable hemorrhage in
carefully selected patients, where there are adequate
levels of platelets and coagulation factors and bleed-
ing has not resolved despite optimal management and
good transfusion practice.
e current recommended dose is 90 g/kg
repeated up to every 2 hours. Currently, no monitor-
ing is available for rFVIIa therapy. It is important to
remember that the success of rFVIIa is dependent on
several pre-conditions that include:
(a) the presence of adequate platelets (⬎50 ×109/L)
and coagulation factors (brinogen levels ⬎1g/L),
and
(b) the absence of acidemia and hypothermia.
Other products
Prothrombinase complexes contain Factors II, VII, IX,
and X isolated from thousands of units of blood and
stored as a powder that requires rehydration for use.
ey are used for the emergency reversal of vitamin K
antagonists, an unlikely prospect in pregnant women.
168

Chapter 13c. Hemostatic management
eir use in OH has been restricted to cases with inher-
ited or acquired deciency of coagulation factors.
DDAVP (1-deamino-8-d-arginine vasopressin,
desmopressin) is a non-blood-derived alternative
(a synthetic analog of vasopressin) that retains the
antidiuretic action of the natural hormone and also
stimulates the release of tissue plasminogen factor
(tPA). ese eects are used to elevate the plasma
factorVIIIandvWFleveltwo-tofourfoldabove
the baseline, by its release from storage sites. It can
correct the hemostatic defect in mild hemophilia
A or von Willebrand disease (VWD) suciently
to cover minor surgery or at a minor bleeding
episode.DDAVPshouldbeusedwithcautionin
women with pre-eclampsia, due to the antidiuretic
eect and to the potential risk of hyponatremia
that can lead to convulsions. erefore, restriction
of uid intake is required to accompany its use.
Other side eects comprise mild facial ushing and
headache.
ere are insucient data about the ecacy and
safety of DDAVP for prophylaxis and treatment of OH.
It has been used safely during pregnancy in women for
other indications (see Chapter 14). DDAVP does not
pass into breast milk in signicant amounts and so it
maybeusedinlaborandinthepostpartumperiod.
Hematological monitoring
and management
Regular full blood counts (FBCs) and coagulation
screens should be used to guide therapy, with regu-
lar (up to hourly) requests with massive loss. e
turnaround time in an average hospital makes near–
patient testing an attractive option. A thromboelas-
togram (TEG) is an alternative, but is poorly validated
in this setting.
In general, if the bleeding continues and no bleed-
ing point can be found:
rKeep hemostatic monitoring going.
rConsider an antibrinolytic agent.
rConsider the bleeding history and the possibility
of undiagnosed von Willebrands disease or other
bleeding disorder.
Speedy responses are required to prevent a cycle of
failure to catch up e.g. excessive hemorrhage →deple-
tion of hemostatic factors →further bleeding →,etc.
Table 13c.2 Use of Blood components in OH
Red blood cells:
rMaintain Hb ⬎8g/dL
rOne unit of packed red cells increases the hemoglobulin by
1 g/dL and the hematocrit by 3%.
Platelet transfusion:
rMaintain platelet count ⬎50 ×109/L (70 ×109–110 ×
109/L).
rIf platelet count ⬍50 ×109/L give one adult therapeutic
dose of platelets
FFP:
rIf INR/APTT ratios ⬎1.5 give FFP 15ml/kg
Cryoprecipitate:
rMaintain fibrinogen ⬎1.0 g/L
r10 bags of cryoprecipitate will increase a normal adult’s
fibrinogen by 1.0 g/L
Ideally, a blood warmer should be used to help prevent
hypothermia, and regular blood gases should be per-
formed (see Chapter 13b).
Disseminated intravascular coagulation
DIC is not a usual direct consequence of OH but
rather a complication of appropriate, delayed or inad-
equate treatment. Delayed and inappropriate treat-
ment will lead to prolonged hypoxia, hypovolemia,
hypothermia or extensive muscle damage and thus
to a DIC-like syndrome. A prolonged PT and aPTT,
thrombocytopenia and low brinogen levels (⬍1.0
g/l), are highly suggestive of a developing DIC-like
syndrome.
Post-hemorrhage care
Once bleeding has stopped, it should be remembered
that, in the UK, the most common cause of maternal
direct deaths in UK is thromboembolism.
Mothers that hemorrhage and have operative inter-
vention will have excellent acute phase responses post-
partum that will make the blood more prothrom-
botic and the patients at high risk of venous throm-
boembolism. erefore, thromboprophylaxis with a
low molecular weight heparin should be started as
soon as possible postpartum, according to current
guidelines.
Table 13c.2 summarizes the suggested guidelines
for the management of OH. Current best practice
emphasizes the need for speedy and appropriate use of
blood components with monitoring if bleeding contin-
ues.
169

Section 5. Hemorrhagic disorders
References
1. Ronsmans C, Graham WJ; Maternal mortality: who,
when, where, and why? Lancet Maternal Survival
Series steering group. Lancet 2006; 368: 1189–1200.
2. ACOG Practice Bulletin: Clinical Management
Guidelines for Obstetrician-Gynecologists Number 76,
October 2006: postpartum hemorrhage. Obstetrics and
Gynecology 2006; 108: 1039–1047.
3. Stainsby D, MacLennan S, omas D et al. Guidelines
on the management of massive blood loss. British
Journal of Haematology 2006; 135: 634–641.
4. Handbook of Transfusion Medicine,edDBL
McClelland, United Kingdom Blood Services, 4th edn.
5. Ferguson A.D., Hebert C.P.A, Mazer C.D et al.A
comparison of aprotinin and lysine analogues in
high-risk cardiac surgery. e BART study. New
England Journal of Medicine 2008; 358: 2319–2331.
6. Henry DA, Carless PA, Moxey AJ et al.
Anti-brinolytic use for minimising perioperative
allogeneic blood transfusion. Cochrane Database of
Systematic Reviews 2007; 17: CD001886.
7. Alrevic Z, Elbourne D, Pavord S et al.Useof
recombinant activated factor VII in primary
postpartum hemorrhage: the Northern European
registry 2000–2004. Obstetrics and Gynecology 2007;
110: 1270–1278.
8. Ahonen J, Jokela R, Kortila K. An open
non-randomized study of recombinant activated factor
VIIa in major postpartum haemorrhage. Acta
Anaesthesiologica Scandinavica 2007; 51: 929–936.
9. McMorrow RC, Ryan SM, Blunnie WP et al.Useof
recombinant factor VIIa in massive post-partum
haemorrhage. European Journal of Anaesthesiology
2008; 7:1–6.
10. Haynes J, Laan M, Plaat F. Use of recombinant
activated factor VII in massive obstetric haemorrhage.
International Journal of Obstetrics and Anesthesia 2007;
16: 40–49.
11. Levi M, Peters M, Buller HR. Ecacy and safety of
recombinant factor VIIa for treatment of severe
bleeding: a systematic review. Critical Care Medicine
2005; 33: 883–890.
12. Stanworth S, Birchall J, Doree C, Hyde C. Recombinant
factor VIIa for the prevention and treatment of
bleeding in patients without haemophilia. Cochrane
Database of Systematic Reviews 2007; 18: CD005011.
170

Section 5 Hemorrhagic disorders
Chapter
13d Management of obstetric hemorrhage:
radiological management
Ash Saini and John F. Reidy
Introduction
ose rare cases of obstetric hemorrhage that do
not respond to conservative measures such as the
use of uterotonics, correction of coagulopathies, lac-
eration repair, evacuation of retained products, and
uterine packing have traditionally been treated by
more invasive surgical methods. ese include: appli-
cation of a uterine compression suture, ligation of
the arterial supply to the uterus and, if all else fails,
hysterectomy.
e use of interventional radiology (IR) to treat
postpartum hemorrhage (PPH) was initially described
in 1979.1Since then, uterine artery embolization
(UAE) to treat broids has become common prac-
tice and interventional radiologists have been able
to apply the technique to the treatment of obstetric
hemorrhage.UAEplaysavitalroleonceconserva-
tive treatments have failed, avoiding the trauma of
major surgery, reducing transfusion requirements and
importantly preserving fertility. Although there are no
randomized controlled trials, there are a large num-
ber of case series which have established arteriogra-
phy and embolization for PPH to be safe and eective,
with a success rate of 90% and very low complications.2
In the United Kingdom recommendations have been
made that all hospitals with delivery units should have
access to an emergency IR service and to consider early
or prophylactic embolization as an important tool in
the prevention and management of obstetric hemor-
rhage.3,4
e main indications for embolization are in
the emergency treatment of PPH and electively, in
high risk cases. Embolization has also successfully
been used to treat hemorrhage secondary to ectopic
pregnancy, gestational trophoblastic disease, and
acquired uterine arterio-venous malformations.5–9
Contraindications are all relative and include
coagulopathy, renal failure, and severe contrast
allergy.
The role of interventional
radiology in the management
of emergency postpartum
hemorrhage
ere are a variety of indications for the use of
embolization, but it is most commonly used follow-
ing a vaginal delivery to treat primary PPH secondary
to uterine atony (Table 13d.1). Obstetric units should
have protocols to guide referral (Fig. 13d.1).
Although embolization should ideally be per-
formed on a hemodynamically stable patient, some
leeway exists depending on anesthetic support and the
speed and experience of the local IR service. Early
liaison with interventional radiology and referral for
embolization is critical. In this respect obstetricians
should be aware of the potential delay in treatment
whilstthepatientistransferredtoanangiographic
suite and plan accordingly. It is oen the case that, if
conservative treatments are unsuccessfully employed
for such prolonged periods, the patient is then too
unstable to be transferred to the radiology department
for embolization.
It is essential that the procedure is performed
by an experienced interventional radiologist. As with
other more invasive surgical procedures, it is impera-
tive that the patient is resuscitated by another prac-
titioner, preferably an anesthetist. e procedure is
minimally invasive, carried out under local anesthetic.
Standard transfemoral arterial access is obtained
and specialized catheters inserted into the anterior
171
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.
Section 5. Hemorrhagic disorders
Table 13d.1 Indications for the use of interventional radiology
in the treatment of emergency postpartum hemorrhage
Primary postpartum hemorrhage due to:
– atonic uterus post-vaginal delivery (most common, usually
following prolonged labor)
– uterine, cervical, or vaginal tears
– hemorrhage post-Cesarean delivery
– pelvic bleeding in a surgically challenging location, e.g.
broad ligament, pelvic, or vulval hematoma
– undiagnosed placenta previa or accreta
Other indications for the use of emergent embolization:
– hemorrhage following therapeutic or accidental abortion or
interstitial ectopic pregnancy
– secondary post-operative PPH
– acquired uterine arterio-venous malformations caused by
instrumentation after delivery
division of each internal iliac artery. Arteriography
isperformed,butitisnotessentialthatbleeding
is demonstrated. If the source of bleeding is identi-
ed (usually the uterine or vaginal arteries), selective
catheterization is performed and the vessel embolized
with small particles (1–2 mm) of Gelfoam (Upjohn)
until the antegrade ow of contrast stops (Fig. 13d.2).
Gelfoam is the embolic agent of choice as it creates a
temporary occlusion and is small enough to stop ow
in distal branches but unlikely to reach capillary level
and cause uterine necrosis. If the source of bleeding
remains undetected, or if time and patient anatomy
does not allow selective catheterization, then empiric
embolization of the anterior division of the internal
iliac artery is performed.
The role of interventional
radiology in the elective or
prophylactic management of
postpartum hemorrhage
Even rarer than those cases of unexpected emergency
obstetric hemorrhage treated by embolization, are a
high risk group of patients with abnormalities of pla-
centation in whom massive bleeding at the time of
delivery may occur. In these patients the elective use
of embolization has been advocated.
Placenta accreta occurs when there is abnormally
rm attachment of the placental villi to the uter-
ine wall and remains a formidable clinical challenge,
with patients most at risk of emergency hysterectomy.
Although previously extremely rare, there has been
a tenfold increase in cases, thought to be secondary
to the rise in Cesarean deliveries, in which the uter-
ine incision acts as the nidus for abnormal placen-
tation.10,11 Placenta accreta most commonly occurs
in patients with placenta previa. ree variants are
BEST MEDICAL THERAPY
Continued hemorrhage
Hemorrhage stopped
UTERINE
PACKING/TAMPONADE
CRITICAL
EARLY LIAISON WITH
INTERVENTIONAL
RADIOLOGY
Stable patient – fit
for transfer to
angiographic suite
Unstable patient – unfit
for transfer
UTERINE ARTERY
EMBOLIZATION
SURGICAL THERAPY
•Uterine compression suture
•Hysterectomy
Continued hemorrhage
Obstetric and anesthetic review
Hemorrhage stopped
Fig. 13d.1 Example of a protocol for the
management of emergency postpartum
hemorrhage.
172
Chapter 13d. Radiological management
Fig. 13d.2 Persistent massive primary PPH following Cesarean
section and uterine packing (∗). Right transfemoral arterial access
has been obtained and a catheter placed in the origin of the right
internal iliac artery. Angiography demonstrates active pelvic
hemorrhage
(arrow)
, however the precise source is not
demonstrated (a). Selective arteriography identifies hemorrhage
arising from a vasoconstricted right uterine artery (b), (c) which was
successfully embolized with particles of Gelfoam.
recognized, depending on the depth to which the pla-
centa extends through the myometrium (Fig 13a.1).
In its most severe form the placenta extends beyond
the serosal surface of the uterus to invade neighboring
structures, usually the bladder (placenta percreta).
Eective management starts with a high index
of clinical suspicion and accurate pre-natal diagno-
sis. Pelvic ultrasound by a skilled ultrasonographer is
reliable in excluding a diagnosis of placenta accreta.
In cases with suspicious but inconclusive ultrasono-
graphic ndings, magnetic resonance imaging (MRI)
may be used to optimize diagnostic accuracy.12 Once
diagnosed, it is vital that a multi-disciplinary approach
is adopted, with appropriate anesthetic, hematologi-
Fig. 13d.3
cal, IR, and urological support as necessary. Patients
are treated either by elective Cesarean hysterectomy
or, if possible, by Cesarean delivery, for example, in
cases of placenta percreta where attempts to per-
form a hysterectomy would lead to further blood
loss. Although based on very small numbers of cases,
IR has an important role in helping to minimize
hemorrhage.5,13–16
Bilateral transfemoral arterial access is obtained
and pre-delivery catheterization of the anterior divi-
sion of both internal iliac arteries with occlusion bal-
loons performed (Fig. 13d.3). e balloons are deated
throughout the delivery and then inated immedi-
ately aerward, thus allowing time to better con-
trolthehemorrhagesurgically.Alternatively,Gelfoam
embolizationcanthenbeperformednon-selectivelyor
selectively through a micro-catheter passed co-axially
through the lumen of the occlusion balloon catheter
and into the uterine arteries. Although the ecacy of
this technique remains to be fully determined, it is
likely to represent the safest form of combined man-
agement in a group of patients that have a higher risk of
hemorrhage and are more likely to undergo an emer-
gency hysterectomy.17 173

Section 5. Hemorrhagic disorders
Complications of
embolization and eects
on fertility
Minor complications related to the puncture site
include hematoma or pseudoaneurysm formation, but
are uncommon since patients are usually young and
free of vascular disease. Major complications are even
rarer with anecdotal reports of pelvic sepsis, uter-
ine necrosis, bladder necrosis, and transient buttock
ischemia.2
A prime advantage of embolization is that it
avoids the need for hysterectomy. Although no large
prospective studies have been completed, several case
series have reported a return to normal menses with
no signicant adverse eects on future fertility.18–20
Summary
e techniques of selective arterial embolization to
treat emergency obstetric hemorrhage, and balloon
occlusion used electively in high risk patients are
proven safe and eective methods of treatment for
obstetric hemorrhage. ey can reduce transfusion
requirements, preserve fertility and thus have the
potential to reduce maternal morbidity and mortality.
Obstetric departments should have protocols for man-
agement that include early referral to IR and consider-
ation of embolization prior to surgery.
174

Chapter 13d. Radiological management
References
1. BrownBJ,HeastonDK,PoulsonAMet al.
Uncontrollable postpartum bleeding: a new approach
to hemostasis through angiographic arterial
embolisation. Obstetrics and Gynaecology 1979; 54:
361–365.
2. Doumouchtis SK, Papageorgiou AT, Arulkumaran S.
Systematic review of conservative management of
postpartum haemorrhage: what to do when medical
treatment fails. Obstetrical and Gynaecological Survey
2007; 68: 540–547.
3. Investigation into 10 maternal deaths at, or following
delivery at, Northwick Park Hospital, North West
London NHS Trust, between April 2002 and April
2005. London: Healthcare Commission, 2006.
4. e role of emergency and elective interventional
radiology in postpartum haemorrhage – good practice
No. 6 Royal College of Obstetricians and
Gynaecologists, June 2007.
5. Mitty HA, Sterling KM, Alvarez M et al.Obstetric
hemorrhage: prophylactic and emergency arterial
catheterisation and embolotherapy. Radiology 1993;
188: 183–187.
6. Kerr A, Trambert J, Mikhail M et al.Preoperative
transcatheter embolization of abdominal pregnancy:
report of three cases. Journal of Vascular and
Interventional Radiology 1993; 4: 733–735.
7. Lobel SM, Meyetovitz MF, Benson CC et al.
Preoperative angiographic uterine artery embolization
in the management of cervical pregnancy. Obstetrics
and Gynaecology 1990; 76: 938–941.
8. Pearl ML, Braga CA. Percutaneous transcatheter
embolization for control of life-threatening pelvic
hemorrhage from gestational trophoblastic disease.
Obstetrics and Gynaecology 1992; 80: 571–574.
9. Badawy SZ, Etman A, Singh M et al. Uterine artery
embolization: the role in obstetrics and gynaecology.
Clinical Imaging 2001; 25: 288–295.
10. Wu S, Kocherginsky M, Hibbard JU. Abnormal
placentation: twenty-year analysis. American Journal of
Obstetrics and Gynaecology 2005; 192: 1458–1461.
11. Silver RM, Landon MB, Rouse DT et al. Maternal
morbidity associated with multiple repeat cesarean
delivery. Obstetrics and Gynaecology 2006; 107:
1226–1236.
12. Warshak CR, Eskander R, Hull AD et al. Accuracy of
ultrasonography and magnetic resonance imaging in
the diagnosis of placenta accreta. Obstetrics and
Gynaecology 2006; 108: 573–582.
13. Weeks SM, Stroud TH, Sandhu J et al.Temporary
balloon occlusion of the internal iliac arteries for
control of hemorrhage during caesarean hysterectomy
in a patient with placenta previa and placenta increta.
Journal of Vascular and Interventional Radiology 2000;
11:622–624.
14. Levine AB, Agarwal R, Seckl MJ et al. Placenta accreta:
comparison of cases managed with and without pelvic
artery balloon catheters. Journal of Maternal and Fetal
Medicine 1999; 8: 173–176.
15. Dubois J, Garel L, Grignon A et al. Placenta percreta:
balloon occlusion and embolization of the internal
iliac arteries to reduce intra-operative blood losses.
American Journal of Obstetrics and Gynaecology 1997;
176: 723–726.
16. Hansch E, Chitkara U, McAlpine J et al.Pelvicarterial
embolization for control of obstetric hemorrhage: a
ve year experience. American Journal of Obstetrics
and Gynaecology 1999; 180:1454–1460.
17. Zaki ZM, Bahar AM, Ali ME et al.Riskfactorsand
morbidity in patients with placenta previa accreta
compared to placenta previa non-accreta. Acta
Obstetrics and Gynaecologica Scandinavica 1998; 77:
391–394.
18. Ornan D, White R, Pollak J et al. Pelvic embolization
for intractable postpartum hemorrhage: long-term
follow-up and implications for fertility. Obstetrics and
Gynecology 2003; 102: 904–910.
19. Salomon LJ, deTayrac R, Castaigne-Meary V et al.
Fertility and pregnancy outcome following pelvic
arterial embolization for severe post-partum
haemorrhage. A cohort study. Human Reproduction
2003; 18: 849–852.
20. Descargues G, Mauger Tinlot F et al.Menses,fertility
and pregnancy aer arterial embolization for the
control of postpartum haemorrhage. Human
Reproduction 2004; 19: 339–343.
175

Section 5 Hemorrhagic disorders
Chapter
14 Inherited disorders of primary hemostasis
Sue Pavord
Introduction
e management of inherited bleeding disorders dur-
ing pregnancy, delivery, and the postpartum period
is particularly challenging. Consideration should be
given to the inheritance risk to the fetus and the bleed-
ing risk to the mother, with appropriate multidisci-
plinary management plans to minimize complications
for both. Good communication among the haematol-
ogists, obstetricians, anesthetists, neonatologists, and
laborwardstaisrequired,aswellasfullinformation
for the patient. is should begin prior to conception
and be reviewed as pregnancy advances. Guidelines for
management have been provided by a task force of the
UK Haemophilia Centre Doctors’ Organization.1
Von Willebrand disease
Von Willebrand disease (VWD) is the most com-
mon of the inherited bleeding disorders. It is char-
acterized by a deciency of Von Willebrand factor
(VWF), a large multimeric glycoprotein, which plays
a crucial role in the rst steps of thrombus formation.
Pregnancy, delivery, and the postpartum period pose
signicant challenges to the hemostatic system, and
women with VWD need to be carefully managed dur-
ing these at-risk times.
VWF activity
e functions of VWF are twofold:
rIt mediates platelet adhesion and aggregation at
sitesofvasculardamage,initiallybyforminga
bridge between the platelet Gp1b receptor and the
subendothelial collagen bers, exposed by the
injured vessel.
rIt acts as a carrier for FVIII, protecting it from
proteolysis and facilitating its cofactor activity by
transportation to the site of vascular injury.
e synthesis by endothelial cells and subsequent
secretion from its storage site in the Wiebel Palade
bodies determines the plasma concentration of VWF.
A separate supply is synthesized by megakaryocytes
andstoredinthealphagranulesofplatelets,from
where it is released during platelet activation, provid-
ing a rapid and local increase to levels during ves-
sel repair. ese actions are particularly important in
sites of fast owing blood and high shear forces, as
occurs in the arterial circulation and the microvascula-
ture. In these conditions, the globular VWF molecule
is dragged into an elongated shape, exposing its platelet
binding sites, whilst in the venous system where blood
ows more slowly, brinogen-dependent clot forma-
tion predominates.
Clinical features
e principal clinical manifestations of VWD reect
the dual function of VWF. Reduced activity of VWF,
leading to impaired platelet plug formation gives rise
to bleeding from mucosal surfaces, typical of the
thrombocytopathies, whilst the rapid clearance of the
unprotected factor VIII impedes brin clot forma-
tion, causing symptoms characteristic of the coagu-
lopathies, such as prolonged bleeding aer surgery.
Both these eects have potentially serious implications
for women in pregnancy.
Disease prevalence
VWF is encoded by a gene spanning 178 kb of genomic
DNA on the short arm of chromosome 12. Numerous
genetic mutations, aecting VWF production, have
been described2andVWDhasanestimatedfrequency
176 e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Chapter 14. Inherited disorders of primary hemostasis
Table 14.1 Subtypes of von Willebrand disease
Subclassification Multimeric pattern VWF function Specific characteristics
Type 1 Normal Normal Quantitative deficiency of VWF.
Accounts for about 70% of all cases.
Type 2a Absent HMW multimers Abnormal Impaired platelet adhesion
2b Loss of HMW multimers through
increased platelet binding
Abnormal Increased affinity for platelets causing
thrombocytopenia
2M Normal Abnormal Impaired platelet adhesion
2N Normal Abnormal FVIII binding Reduced affinity for FVIII with low FVIII levels due
to short half-life
Type 3 Absent Absent Severe bleeding phenotype
HMW =high molecular weight.
in the population of around 1%, based on the num-
ber of people with bleeding symptoms, low VWF, +/−
a positive family history. Clinical penetrance of the
genetic abnormalities is variable;3in some cases they
are fully penetrant, accounting for the low VWF lev-
els and bleeding phenotype, but in others the VWF
mutation may simply act as a risk factor for bleeding
in combination with other modifying factors, such as
platelet dysfunction or the presence of blood group O,
which is typically associated with 25% lower levels than
the other blood groups.4In other cases, classic VWD
mutations have not been identied but VWF may still
play a role. us, those with a clinically signicant
bleeding phenotype amount to only about 0.02% of
the population. ese patients are usually diagnosed in
childhood, whereas the milder forms may not present
until aer signicant hemostatic challenges, such as
menstruation and childbirth. is probably explains
the misconception in the original reports by Erik von
Willebrand, describing women as twice as likely to be
aected than men.
Classication of VWD
VWF is present in the plasma as a series of multimers,
assembled from varying amounts of identical subunits.
e composition of multimers, which range from 150
000 to 20 000 000 Daltons, has been used to classify
VWF into its dierent subtypes (Table 14.1). Its adhe-
sive function is largely dependent on the high molecu-
lar weight (HMW) multimers, which are released dur-
ing platelet and endothelial cell activation.
Type 3 disease is characterized by unmeasurable
VWF levels and consequently, severely low FVIII lev-
els, with median FVIII levels being around 4%. us,
in addition to clinical features of impaired primary
hemostasis, these patients behave like those with mod-
erate hemophilia, with potential for spontaneous joint
and muscle bleeds. Transmission is autosomal reces-
sive with patients being homozygous or double het-
erozygous for the abnormal VWF gene, inherited from
asymptomatic parents. Prevalence in the UK is around
1 per million, being most frequent in communities
where consanguinous marriages are common.
Laboratory evaluation
Routine coagulation screening tests including the pro-
thrombin time (PT) and activated partial thrombo-
plastin time (APTT) do not detect VWD unless the
factor VIII level is below normal, prolonging the
APTT. Specic assays for FVIII activity and VWF anti-
gen and activity are available, the latter including Ris-
tocetin cofactor activity and collagen binding assay.
Platelet function can be assessed by PFA-100, which
measures the time taken for platelets to close over
a hole in a collagen membrane coated with ADP or
epinephrine. However, all assays are limited in their
specicity and sensitivity and none show good corre-
lation with the severity of bleeding. Furthermore, lev-
els can be inuenced by external factors such as physi-
cal and mental stress. us, despite the numerous tests
available, the diagnosis of VWD and its subclassica-
tion is oen dicult.
Hormonal inuences on levels in pregnancy
Levels of von Willebrand factor and factor VIII start to
rise from 6 weeks’ gestation, increasing progressively
throughout pregnancy to three to ve times baseline
levels by delivery.5is is due to increased synthesis 177

Section 5. Hemorrhagic disorders
of VWD, although the cause for the increase in FVIII
is not entirely clear but in part reects improved stabil-
ization by VWF. is rise is benecial to many patients
with type 1 VWD, in whom normal levels are oen
reached by delivery. However, those with starting base-
line levels of ⬍15 IU/dL may fail to reach normal val-
ues6and in patients with type 2 disease, where the
molecule functions abnormally, the condition may not
improve and may even deteriorate.7is is particu-
larly evident in type 2b where the rise in dysfunctional
VWF protein enhances abnormal platelet binding and
exacerbates thrombocytopenia. In patients with type
2N disease, factor VIII levels tend to remain low
because of impaired binding by the abnormal VWF
and patients with type 3 disease show little or no rise
in VWF.8
A study of VWF levels in 248 healthy women,
showed that they remained elevated for 1–3 days post-
partum and then returned to baseline by day 7–21.9
Factor VIII and VWF levels are also inuenced
by thyroxine, which shows a physiological rise in its
bound form during pregnancy. Hypothyroidism may
be associated with clinical and laboratory features of
VWD which corrects with thyroid replacement. 10
Obstetric complications
Maternal bleeding
Women with VWD have an increased risk of bleeding
events and even death during childbirth.11 Although
the physiological rise in VWF and FVIII protects many
women with mild type 1 disease during delivery, they
remain vulnerable in early pregnancy and in the post-
partum period. Studies have found:
rOne-third of women with VWD have bleeding
during their rst trimester.
r15%–30% of women with VWD have primary
postpartum hemorrhage.
rDelayed postpartum bleeding occurs in 20%–25%
of women with VWD.
rere is a relatively high frequency of perineal
hematoma, a normally rare complication of
vaginal birth.
re risk of receiving a blood transfusion is
increased vefold
rMaternal mortality rate is ten times higher than
that for women without the condition.
Table 14.2 Pre-pregnancy management of VWD
rReassess severity of clinical bleeding tendency
including previous responses to hemostatic
challenges.
rCheck baseline investigations if not already
known
rEstablish response to DDAVP
rObtain consent for use of plasma products
after full counseling of risks
rWhere plasma-derived products have been
received in the past, the presence of
transfusion transmitted infection should be
excluded.
rVaccinate against hepatitis A and B if not
already immune
rCheck hemoglobin and serum ferritin and give
oral supplements as necessary
rAll should receive counseling about risks of
increased bleeding, particularly in the
postpartum period and particularly for women
with type 2 or 3 VWD
rA management plan should be discussed with
all patients.
rAll patients should be offered genetic
counseling as they are at risk of delivering an
affected child
rAll should receive explanation regarding
evaluation of the infant after delivery
Pregnancy outcomes
Women with VWD are no more likely to experience
premature labor, placental abruption, fetal growth
restriction or intrauterine fetal death.11 Early miscar-
riage has not been shown to be any more frequent than
in the general population, but can be complicated by
signicant bleeding.8,12
Pregnancy management for women
with VWD
e safe management of women with VWD requires
good communication between the hematologist,
obstetricians, anesthetists, neonatologists, and labor
ward sta. e patient should be fully informed of
potential bleeding risks and the plan for management
of pregnancy, delivery, and the postpartum period.
is should begin prior to conception and should be
reviewed as pregnancy advances (Table 14.2).
178

Chapter 14. Inherited disorders of primary hemostasis
Ante-natal management
In view of the physiological rise in factor VIII and
VWF during pregnancy, most women with mild type
1 VWD achieve levels above 50 IU/dL, the lower limit
of the normal range outside of pregnancy, and can be
safelymanagedinstandardobstetricunitsincollabor-
ation with hemophilia center sta. Women with types
2and3VWD,ormoderateorseveretype1,orahis-
tory of severe bleeding, should be referred for pre-natal
care and delivery to a center where there are specialists
in high risk obstetrics, as well as a Hemophilia Center.
Laboratory, pharmacy, and blood bank support is also
essential.
For all types of VWD, levels should be checked rou-
tinely at booking, 28 weeks and if still abnormal, 34
weeks’ gestation. If an adequate rise is demonstrated,
only a third trimester sample may be necessary for sub-
sequent pregnancies, unless earlier interventions are
required.
Levels are always needed prior to invasive proce-
dures such as chorionic villus sampling, amniocen-
tesis, or cervical cerclage. If VWF activity or FVIII
levels are ⬍50 IU/dL, women should receive prophy-
laxis. DDAVP should be used in preference to plasma
derivedproductsintype1VWD,toavoidpotentialfor
transfusion transmitted infections (Table 14.3).
DDAVP in pregnancy
DDAVP (1-deamino-8-D-arginine vasopressin) a syn-
thetic derivative of antidiuretic hormone that acts
specically through type 2 vasopressin receptors,
stimulates release of ultralarge VWF multimers from
storage in the Wiebel Palade bodies of the endothelial
cells. is is not a direct stimulatory eect, but is medi-
ated through intracellular calcium mobilization and
cyclic adenosine monophosphate. Administration has
traditionally been by slow intravenous infusion, over
20 minutes, of 0.3 g per kilogram of body weight,
although a subcutaneous preparations are now com-
monlyused,withsimilarecacyandfewersideeects.
Intranasal preparations are also available. Adminis-
tration results in a three to vefold increase in both
VWF and factor VIII, within 30–60 minutes, lasting
for 8–10 hours. To assess the response to DDAVP,
VWF activity levels and factor VIII should be mea-
sured before administration and again at 1 and 4
hours aer, to determine peak levels and clearance rate,
respectively.
Table 14.3 Ante-natal management of VWD
rCheck VWF antigen and activity and FVIII levels
at booking, 28 and if still abnormal, 34 weeks’
gestation and prior to any invasive procedure.
rIn patients with type 2B VWD, the platelet
count should also be monitored. Platelet
transfusions, as well as VWF factor replacement
may sometimes be required for bleeding or to
cover surgical procedures and spontaneous
miscarriage.
rAim for FVIII and VWF:RCo activity levels of ≥
50% to cover surgical procedures or
spontaneous miscarriage.
rTreat with desmopressin in preference to
coagulation factor concentrates whenever
possible, checking pre- and post-treatment
VWF activity levels and factor VIII.
rDistribute action plan for acute bleeding
events, to hematology and obstetric staff and
ensure patient is given an emergency number
for contact.
DDAVP is generally thought to be safe for mother
and fetus13 and previous concerns regarding the
potential risk of uterine contractions or neonatal
hyponatremia have diminished, in view of its select-
ive eect on V2R receptors. Fluid intake should
be restricted to 1 liter for the following 24 hours,
to prevent maternal hyponatremia caused by water
retention from the antidiuretic eect. An in vitro
placenta model showed that DDAVP, at therapeu-
tic dose, did not cross the placenta in detectable
amounts.14
e advantages of DDAVP are its low cost, unlim-
ited availability, and most importantly, the avoidance
of blood products. However, there are many situations
where DDAVP will be contraindicated or ineective
and plasma products necessary (Table 14.4). In these
patients, prophylactic treatment with a clotting con-
centrate containing Factor VIII and von Willebrand
Factor should be considered to raise levels ⬎50 IU/dL
for ante-natal procedures and childbirth. Patients with
type 2B disease may also require platelet transfusion if
thrombocytopenia is severe.
Coagulation factor replacement
ere are several licensed plasma-derived VWF/FVIII
products available. e spectrum of VWF HMW 179

Section 5. Hemorrhagic disorders
Table 14.4 Situations where DDAVP may not be suitable
Patients with insufficient baseline
levels
Patients with baseline VWF or FVIII levels of less than 15 IU/dl may not
achieve post-infusion levels, which are sufficient to control or
prevent bleeding.
Some subtypes of Type 1, including
Vicenza subtype
Some subtypes of type 1 VWD show decreased survival of
endogenously produced VWF following DDAVP compared with
normal survival of exogenously administered VWF.
Previous intolerance or severe
adverse effects
During intravenous infusion, hypotension, headache and facial
flushing are common but generally mild. Blood pressure should be
monitored during and after infusion.
Known cardiovascular disease,
pre-eclampsia or unstable blood
pressure
There are anecdotal reports of myocardial and cerebral infarction and
DDAVP should be avoided in patients known to have arterial disease.
Tachyphylaxis after repeated doses The response to DDAVP diminishes after repeated doses.
Type 2 VWD (except some cases of
2M)
DDAVP is less likely to correct the abnormality in type 2 VWD,
although there may be a transient shortening of the bleeding time.
Because the molecule is abnormal, the usual increase in the amount
of VWF protein following DDAVP is unlikely to improve function.
Type 2B The heightened and spontaneous binding of the abnormal VWF
molecule to normal platelets may be aggravated by the rise in VWF
levels after DDAVP, increasing platelet clearance from the circulation
and exacerbating thrombocytopenia.
Type 2N DDAVP also causes release of Factor VIII from stores. However, due to
the abnormal VWF : FVIII binding, the response to DDAVP is
shortened to a median of 3 hours.
Type 3 VWD These patients lack releasable stores of VWF and do not respond to
DDAVP.
multimers and ratio of VWF:RCo/FVIII activity dif-
fers between them, but this does not appear to
cause a dierence in ecacy.15 ey are available as
lyophilized powders and, aer reconstitution in water,
can be administered by slow bolus intravenous injec-
tion. Treatment is usually given 1 hour pre-operatively.
Pre-and post-levels should be checked and therapeutic
levels of FVIII and VWF:RCo ⬎50 IU/dL maintained
until hemostasis is secure. For most ante-natal proce-
dures, a single pre-operative treatment is sucient, but
in some cases a second dose may be required at 12–24
hours, depending on the nature of the procedure and
the measured levels.
Intrapartum management
Although there are no large prospective studies that
correlate VWF:RCo and FVIII levels with the risk
of bleeding at the time of childbirth, the opin-
ion of experts is that levels above 50 IU/dL should
be achieved before vaginal delivery or Cesarean
section.1,16
Neonates are at risk of intracranial hemorrhage
and cephalhematomas during labor and delivery. e
increase in FVIII and VWF, induced by the stress of
labor, provides some protection for babies with mild
type 1 disease but in more severe types, trauma to the
baby should be minimized by avoiding extracephalic
180

Chapter 14. Inherited disorders of primary hemostasis
Table 14.5 Intrapartum management
rAllow spontaneous labor and normal vaginal
delivery, if no other obstetric concerns, to
minimize risk of intervention.
rIf FVIII or VWF:RCo activity levels ⬍50 IU/dl at
the last check, the test needs to be repeated.
rIf levels ⬍50 IU/dL, treat with DDAVP if known
responder, otherwise plasma-derived factor
concentrate. Treatment should be given at the
onset of established labor and pre- and
post-treatment levels should be obtained.
rAvoid prolonged second stage of labor, with
early recourse to Cesarean section if necessary,
to reduce risk of trauma to mother and baby
and risk of uterine atony.
rFor fetuses at risk of having type 2 or 3 disease
or moderately severe type 1, avoid fetal blood
sampling, fetal scalp monitoring, Ventouse
delivery and mid-cavity or rotational forceps.
rAvoid aspirin and consider alternatives for
NSAIDs. Intramuscular injections may be
suitable if FVIII and VWF activity and PFA-100
are in the normal range.
rActive management of the third stage of labor
and early suturing of episiotomy and
lacerations.
version, Ventouse delivery, fetal blood sampling, scalp
electrodes, and rotational forceps.
Analgesia
ere is no consensus on the levels that are safe for
regional anesthesia but if FVIII and VWF activity lev-
els are ⬎50 IU/dL, regional anesthesia may be under-
taken. Consideration should also be given to the levels
at the time of catheter removal and repeat treatment
givenbeforehandifnecessary.Intramuscularinjec-
tions are not contraindicated if FVIII and Von Wille-
brand activity are shown to be normal, but attention
shouldbegiventotheprolongedantiplateleteect
of non-steroidal anti-inammatory drugs, being 2–3
days for indomethacin and diclofenac. Aspirin has an
irreversible eect on platelets, but following ibuprofen
normalization of platelet aggregation occurs within 24
hours. Before delivery, all women with VWD should
have the opportunity to discuss analgesia with an anes-
thetist.
Table 14.6 Postpartum management
rEnsure careful surgical hemostasis and effective
uterine contraction in all cases.
rRepeat VWF activity levels and Hb prior to
discharge.
rGive prophylactic tranexamic acid.
rFor patients with significantly low
pre-pregnancy levels, consider DDAVP if known
responder.
rFor types 2 and 3 disease or severe type 1,
ensure VWF activity levels are maintained at
⬎50 IU/dL for 3 days following vaginal delivery
or 5 days if Cesarean section has been
performed.
rMaintain regular contact with the patient after
discharge and encourage them to report
excessive blood loss.
rConsider use of combined oral contraceptive
pill if excessive bleeding is ongoing despite
prophylaxis. This is particularly beneficial to
patients with type 1 disease due to the
associated increase in functional VWF protein.
Postpartum management (Table 14.6)
e postpartum fall in FVIII and VWF levels is vari-
able, occurring between 24 hours and 2 weeks aer
birth. In normal pregnancies, the median duration of
bleedingaerchildbirthis21to27days,withdelayed
or secondary postpartum hemorrhage occurring in
fewer than 1% of cases. In women with VWD this is
much more common, aecting 20%–25% of cases. In
addition, there are multiple cases of postpartum hem-
orrhage that have occurred despite prophylaxis. e
average time of presentation of postpartum hemor-
rhage in women with VWD is 10–20 days aer deliv-
ery.17 Women with mild type 1 disease should be
encouraged to report excessive bleeding but, for more
severe cases, hemoglobin should be monitored and
regular contact with the patient maintained for several
weeks.
Tranexamic acid
Patients with mild type 1 disease can usually be
safely managed with tranexamic acid alone. is is
a lysine analog, which saturates lysine binding sites
on plasminogen and prevents them from interacting
with the brin surface, thus inhibiting brinolysis. 181

Section 5. Hemorrhagic disorders
It has proven ecacy in reducing blood loss, with-
out increasing thrombotic risk. It is contraindicated in
patients with hematuria and doses should be reduced
in renal failure.
Tranexamic acid crosses the placenta and should
generally be avoided during pregnancy, although it
has been used to treat ante-natal bleeding in a limited
number of cases without adverse fetal eects reported.
Traces have been found in breast milk but this has not
been associated with changes to neonatal brinolytic
activity.
DDAVP and plasma products
For patients with signicantly low pre-pregnancy lev-
els, DDAVP can be given at the time of cord clamp-
ing, although as the peak eect is 40 to 60 minutes aer
administration, it may be more benecial if adminis-
tered during the second stage of labor or immediately
before Cesarean section. DDAVP may be used to raise
factor levels in responders, but care must be taken in
its administration at the time of childbirth and extra
uids should be avoided. Tranexamic acid is a useful
adjunct to desmopressin, particularly as it counteracts
the mild brinolytic eect of DDAVP related to the
associated rise in tissue plasminogen activator.
All patients with type 3 and most with type 2
disease require plasma derived VWF concentrates, to
maintain levels ⬎50 IU/dl for at least 3 days aer vagi-
naldeliveryand5daysfollowingCesareansection.
1
ese patients also usually require prolonged admin-
istration of tranexamic acid and close monitoring
(Table 14.6).
Potential complications
of factor replacement
Transfusion transmitted infections
To minimize risk of viral transmission, two independ-
ent and eective steps which complement each other
in their mode of action, are incorporated into the
plasma product manufacturing process. ese include
dryheattreatmentat80
◦C for 72 h, pasteurization
at 60 ◦C for 10 h, or solvent detergent (SD) treat-
ment with tri(n-butyl) phosphate and Tween-80 or Tri-
ton X. A third step of nanoltration has been intro-
duced for some products. No cases of HIV, hepatitis
B, and hepatitis C have occurred with products inacti-
vated by the currently used processes; however, some
viruses, such as parvovirus B19, are relatively resist-
ant to all these inactivation techniques. Parvovirus
infection can have serious consequences in pregnancy,
being associated with hydrops fetalis and intrauterine
fetal death. In addition, new emerging infections as
wellasthosesuchasvCJD,capableofcrossingbetween
species, will remain potential infective risks.
Inhibitor formation
Alloantibodies to exogenous VWF are a rare complica-
tion of treatment and more likely to occur in patients
with type 3 disease, associated with large or complete
VWF gene deletions or stop codons. e prevalence
in these patients is around 8%. e antibodies render
replacement therapy ineective and can cause severe
anaphylactic reactions.
Thrombosis
Excessive accumulation of FVIII may arise aer
repeated administration of Von Willebrand factor con-
taining concentrates. Resulting thrombosis has been
reported but mostly these cases were peri-operative
without use of monitoring.
It is advised that when using VWF containing con-
centrates peri-operatively, monitoring of FVIII:C and
VWF:RCo should be used in deciding dosing of ther-
apy and excessive FVIII levels avoided. Mobilization
and hydration should be encouraged and anti-embolic
or stockings considered. Pharmacological thrombo-
prophylaxis should generally be avoided, particularly
with type 3 disease.
Neonatal management
Being an autosomal dominant condition in most cases,
the risk of transmission is 50%. However, the variable
penetrance of type 1 VWD results in only around one
third being clinically aected. Type 3 disease is auto-
somal recessive giving a 25% risk if a previous sibling
has been aected. e risk of peri-natal intracranial
hemorrhage is low, even in neonates with VWD type 3.
Nevertheless newborns at risk of moderate and severe
types need to be tested for VWD using cord blood and
assessed to exclude intracranial hemorrhage. For diag-
nostic purposes, however, levels are unreliable in most
cases, being articially low due to the gestational age
or increased from the stress of labor and delivery and
need repeating at 6–12 months when adult values are
reached (Table 14.7).
182

Chapter 14. Inherited disorders of primary hemostasis
Table 14.7 Neonatal management
rIf severe disease phenotype is expected, a cord
sample should be tested for FVIII level and VWF
activity. The limitations of testing at this stage
should be understood.
rBabies with type 2B disease may require
platelet transfusion if there is severe
thrombocytopenia or bruising/ bleeding
manifestations.
rIntramuscular vitamin K should be avoided until
results are known and given orally if necessary.
Any heel prick tests should have pressure
applied afterwards for 5 minutes.
Inherited disorders of platelet
function
ere are a number of platelet function disorders and
formostcasesmanagementrequiresassessmentof
maternal bleeding phenotype with consideration given
to use of platelets to cover ante-natal procedures and
delivery, DDAVP may be used if response has been
previously demonstrated. Patients with bleeding his-
tories should be given tranexamic acid for 5–14 days
postpartum. If there is a neonatal risk of platelet dys-
function, traumatic delivery should be avoided and if
thrombocytopenia is a feature of the condition, a cord
sample should be taken at birth. Special mention is
given to Glanzmann’s thrombasthenia and Bernard–
Soulier syndrome.
Glanzmann’s thrombasthenia
Glanzmann’s thrombasthenia is a congenitally
acquired platelet disorder with an autosomal recessive
mode of inheritance. Platelets are normal in number,
but their ability to aggregate is reduced due to loss of
the surface receptor glycoprotein IIbIIIa. Pregnancy
and delivery are rare in these patients but have been
associated with a high risk of severe postpartum
hemorrhage.
Recombinant activated factor VIIa is licensed for
use in patients with this disorder. In pharmacologi-
cal concentrations, FVIIa is capable of binding to the
surface of activated platelets and improving throm-
bin generation to enhance adhesion and aggregation
of platelets lacking GPIIb/IIIa. e usual dose given
is 90 mcg/kg 2–3 hourly. Bleeding can also be suc-
cessfully prevented by transfusion of platelets before
and aer delivery. However, platelet transfusion can
stimulate isoantibody formation against glycoprotein
IIb–IIIa, resulting in a decreased ecacy of subse-
quent transfusions. A single donor platelet preparation
should be used in preference to pooled platelet trans-
fusion to reduce this risk and where possible should be
HLA matched.
Delayed bleeding up to 2–3 weeks postpartum has
been reported and in these circumstances, DDAVP
and tranexamic acid are useful to reduce platelet trans-
fusion requirements.
Neonatal management
Unless the father has the same condition, the fetus is
heterozygous, with platelets carrying specic paternal
antigens that are not present on the maternal platelets
and thus are capable of causing maternal alloim-
munization. Transplacental transfer of the maternal
antiplatelet immunoglobulin G antibodies can lead to
severe isoimmune neonatal thrombocytopenia and a
risk of intracranial hemorrhage in the fetus. Women
should be monitored for the development of platelet
specic antibodies.
Bernard–Soulier Syndrome
e Bernard–Soulier syndrome (BSS) is a rare auto-
somal recessive bleeding disorder, characterized by
impaired platelet aggregation with ristocetin and
a normal to decreased number of unusually large
platelets whose membranes lack glycoprotein complex
GP Ib/IX/V. In some patients the disease can go unrec-
ognized until the third or fourth decade.
Four dierent features of BSS may contribute to the
hemorrhagic diathesis: thrombocytopenia, abnormal
platelet interaction with vWF, impaired platelet inter-
action with thrombin, and abnormal platelet coagulant
activity. BSS is caused by genetic defects in the genes
of GPIb␣,GPIb,GPIXorGPV.isvarietyofmuta-
tions could explain the heterogeneity of the syndrome;
however, the clinical manifestation may even dier in
consecutive pregnancies of the same patient.
e main complications encountered in reported
cases have been antepartum hemorrhage excessive
intra-operative bleeding and immediate and delayed
postpartum hemorrhage, development of maternal
antiplatelet antibodies leading to fetal intracranial
hemorrhageandneonatalalloimmunethrombocy-
topenia. 183

Section 5. Hemorrhagic disorders
Management
Management is similar to that for Glanzmanns
thrombasthenia and includes the judicious and
timely use of platelet transfusions to prevent bleeding
whilst minimizing the risk of platelet refractoriness.
Regional anesthesia should be avoided and postpar-
tum tranexamic acid and DDAVP prescribed as nece-
ssary.
Neonatal management
e risk to the fetus is unpredictable but thrombocy-
topenia can occur due to heterozygosity of the platelet
function disorder and, more signicantly, fetoma-
ternal alloimmunization, which may be encountered
even in the absence of demonstrable antibodies. us
the management may follow that for fetal/neonatal
alloimmune thrombocytopenia. (See Chapter 5.)
184

Chapter 14. Inherited disorders of primary hemostasis
References
1. Lee CA, Chi C, Pavord SR et al. UK haemophilia
Center Doctors’ Organization. e obstetric and
gynaecological management of women with inherited
bleeding disorders – review with guidelines produced
by a taskforce of UK Haemophilia Center Doctors’
Organisation. Haemophilia 2006; 12: 301–336.
2. ISTH. SSC VWF Database [Database on the
Internet]. Sheeld, UK: University of Sheeld.
http://www.vwf.group.shef.ac.uk/index.html
(Accessed 23.01.2008).
3. Goodeve A, Eikenboom J, Castaman G et al.
Phenotype and genotype of a cohort of families
historically diagnosed with type 1 von Willebrand
disease in the European study, Molecular and Clinical
Markers for the Diagnosis and Management of Type 1
von Willebrand Disease (MCMDM-1VWD). Blood
2007; 109: 112–121.
4. Gill JC, Endres-Brooks J, Bauer PJ et al.eeectof
ABO blood group on the diagnosis of von Willebrand
disease. Blood 1987; 69: 1691–1695.
5. Stirling Y, Woolf L, North WR, Seghatchian et al.
Haemostasis in normal pregnancy. rombosis and
Haemostasis 1984; 52: 176–182.
6. Ramsahoye BH, Davies SV, Dasani H, Person JF.
Obstetric management in von Willebrand’s disease: a
report of 24 pregnancies and a review of the literature.
Haemophilia 1885; 1: 140–144.
7. Conti M, Mari D, Conti E et al.Pregnancyin
women with dierent types of von Willebrand
disease. Obstetrics and Gynaecology 1986; 68:
282–285.
8. Kadir RA, Lee CA, Sabin CA et al.Pregnancyin
women with von Willebrand’s disease or factor XI
deciency. British Journal of Obstetrics and
Gynaecology 1998; 105: 314–321.
9. Sanchez-Luceros A, Meschengieser SS, Marchese C.
et al. Factor VIII and von Willebrand factor changes
during normal pregnancy and puerperium. Blood
Coagulation and Fibrinolysis 2003; 14: 647–651.
10. Manfredi E, van Zaane B, Gerdes VE et al.
Hypothyroidism and acquired von Willebrand’s
syndrome: a systematic review. Haemophilia 2008; 14:
423–433.
11. James AH, Jamison MG. Bleeding events and other
complications during pregnancy and childbirth in
women with von Willebrand disease. Journal of
rombosis and Haemostasis 2007; 5: 1165–1169.
12. Foster PA. e reproductive health of women with von
Willebrand Disease unresponsive to DDAVP: results of
an international survey. On behalf of the
Subcommittee on von Willebrand Factor of the
Scientic and Standardization Committee of the ISTH.
rombosis and Haemostasis 1995; 74: 784–790.
13. Mannucci PM. Use of desmopressin (DDAVP) during
early pregnancy in factor VIII-decient women. Blood
2005; 105: 3382.
14. JG Ray, R Boskovic, B Knie et al.Invitroanalysisof
human transplacental transport of desmopressin.
Clinical Biochemistry 2004; 37: 10–13.
15. Mannucci PM, Tenconi PM, Castaman G, Rodeghiero
F. Comparison of four virus-inactivated plasma
concentrates for treatment of severe von Willebrand
disease: a cross-over randomized trial. Blood 1992; 79:
3130–3137.
16. Pasi KJ, Collins PW, Keeling DM et al. Management of
von Willebrand disease: a guideline from the UK
Haemophilia Center Doctors’ Organization.
Haemophilia 2004; 10: 218–231.
17. RoqueH,FunaiE,LockwoodCJ.vonWillebrand
disease and pregnancy. e Journal of Maternal–Fetal
Medicine. 2000; 9: 257–266.
185

Section 5 Hemorrhagic disorders
Chapter
15 Inherited coagulopathies
Sue Pavord
Hemophilia
Introduction
Hemophilia is characterized by a deciency of factor
VIII (hemophilia A) or factor IX (hemophilia B), both
key components of the intrinsic pathway of the coagu-
lation cascade. e gene is carried on the long arm
of the X chromosome, so males are clinically aected
and females are carriers. Female carriers may also have
low factor levels due to skewed X chromosome inacti-
vation, giving rise to an increased tendency to bleed.
us management of pregnancy requires the assess-
ment of bleeding risk for both mother and baby, with
particular attention given to multidisciplinary plan-
ning and co-ordination of healthcare professionals at
the time of, and aer, delivery.
Disease incidence
Hemophilia A and B occur with an incidence of
around 1:5000 and 1:10 000 male births, respectively.
e severity of the disease runs true in families and, if
the family history is known, the bleeding risk to male
ospringcanbelargelypredicted(Chapter16).How-
ever, 40%–50% of cases are sporadic and unexpected
with no family history of the condition.
Clinical features
e hallmark of the condition is hemarthrosis, result-
ing in progressive arthropathies requiring joint fusions
or joint replacement to alleviate pain. e bleeding risk
correlates with the level of coagulation factor (Table
15.1). Patients with severe hemophilia or those with
recurrent joint bleeds require prophylaxis with factor
concentrate twice (factor IX) or thrice (factor VIII)
weekly with an aim to keep trough levels at, or above,
5% and avoid spontaneous bleeds. Acute bleeds require
immediate treatment to minimize joint and so tis-
sue damage. Aer a period of training and assess-
ment of competency, factor concentrate can be self-
administered using home stocks, although many chil-
dren on prophylaxis have diculty with venous access
and require insertion of portacaths, which are oen
complicated by recurrent infections.
Hormonal inuences on levels
in pregnancy
Levels of factor VIII increase from 6 weeks’ gestation,
totwotothreetimesbaselinebyterm.FactorIXlevels
are relatively unaltered.1
Obstetric complications
Maternal bleeding
Female carriers of hemophilia typically have half levels
of factor VIII/IX. Unbalanced lyonization, where there
is uneven X chromosome inactivation, may result in
signicantly lower levels. For carriers of hemophilia
A, the pregnancy-induced rise in factor VIII level alle-
viates any potential problems for childbirth, although
they remain vulnerable in early pregnancy and those
with baseline levels ⬍15 IU/dL may not achieve nor-
mal levels by delivery. Women with low factor IX levels
remain at risk of bleeding throughout pregnancy.
Pregnancy outcomes
Miscarriage and placental insuciency syndromes are
not increased. e main risk is to the neonate at the
timeofdelivery,aswellasthematernalbleedingrisk,
particularly postpartum, for those mothers with low
factor levels.
186 e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.
Chapter 15. Inherited coagulopathies
Table 15.1 Severity of hemophilia according to factor level
Factor level
(% activity)
Severity of
clinical condition Bleeding risk
⬍1 Severe Spontaneous joint and
muscle bleeds
1–5 Moderate Joint and muscle bleeds
mainly after trauma.
Occasional spontaneous
bleeds
⬎5 Mild Trauma/surgery induced
bleeding
Neonatal risk
e most signicant potential complication for the
neonateisintracranialhemorrhage(ICH),particularly
following instrumental or traumatic birth. e risk is
approximately 50 times greater than for the general
populationandaectsaround4%ofallhemophilia
boys,2although it is clearly highest in those with severe
hemophilia or where the disease is unexpected and no
preventative strategies, neonatal surveillance, or con-
sidered management plan are in place.
ICH is most oen associated with extracranial
hemorrhage (ECH) aer trauma and any signicant
ECH in a newborn should raise the suspicion of under-
lying coagulopathy and ICH. Common complications
are cephalhematomas and abnormal bleeding aer
injection or venepuncture. Other reported events are
umbilical bleeding, hematuria and retro-orbital bleed-
ing.
Pre-pregnancy management
All women with a family history of hemophilia should
be assessed for carrier status, including pedigree pro-
le and calculation of statistical risk, baseline factor
levels and genetic mutation analysis where possible.
(See Chapter 16.)
rCarriers should receive eective counseling
regarding their risk of
(a) bleeding, particularly in the postpartum period;
(b) delivering an aected male.
ese risks need to be determined and fully dis-
cussed with the patient, including options for pre-natal
diagnosis. Appropriate multidisciplinary management
plans should be agreed to minimize complications for
both mother and baby.
Table 15.2 Ante-natal management of hemophilia carriers
rCheck factor levels at booking, 28 and if still abnormal, 34
weeks’ gestation or prior to any invasive procedure.
rAim for FVIII /FIX levels of ≥50 U/dL to cover surgical
procedures or spontaneous miscarriage.
rFor carriers with low factor VIII levels DDAVP may be used
but recombinant factor concentrate is required to raise
factor IX levels.
Ante-natal management (Table 15.2)
rAll women should be oered pre-natal diagnosis
(Chapter 16), but women who do not wish for this
should to have the fetal sex determined by
ultrasound when the anomaly scan is
performed.
rFactor levels should be checked at booking, 28 and
if still abnormal, at 34 weeks’ gestation. Factor
VIII levels usually rise in pregnancy, but factor IX
tends to remain constant. If an adequate rise in
Factor VIII is demonstrated, only a third trimester
sample may be necessary for subsequent
pregnancies, unless earlier interventions are
required.
rFactor levels should also be checked prior to
potentially hemorrhagic events such as invasive
diagnostic procedures, spontaneous abortion, or
termination of pregnancy. If levels are ⬍50 IU/dL,
women should receive prophylaxis.
rDDAVP can be used to raise factor VIII levels by
around three times, but recombinant clotting
factor concentrate is needed for factor IX decient
women and may be required for those with factor
VIII levels below 15 IU/dL, as the response to
DDAVP may be insucient. Pre- and
post-treatment levels should be checked and
therapeutic levels maintained for a suitable time
period depending on the procedure.
Intrapartum management (Table 15.3)
Although there are no large prospective studies that
correlate FVIII or IX levels with the risk of bleeding at
thetimeofchildbirth,theopinionofexpertsisthatlev-
els should be above 50 IU/dL. If treatment is required,
the level should be brought to 100 U/dL pre-delivery
and maintained at ⬎50 U/dL for at least 3–5 days.
Excessive treatment should be avoided due to the risk
of thrombosis and thus careful titration and monitor-
ing of levels is required. 187

Section 5. Hemorrhagic disorders
Table 15.3 Intrapartum management of hemophilia carriers
rIf FVIII/IX levels ⬍50 IU/dL at the last check, the
test needs to be repeated on arrival in labor
rRecombinant factor concentrate is required to
raise factor IX levels. Treatment should be given
at the onset of established labor and pre and
post treatment levels should be obtained.
rAllow spontaneous labor and normal vaginal
delivery, if no other obstetric concerns, to
minimize risk of intervention.
rAvoid prolonged second stage of labor, with
early recourse to Cesarean section if necessary,
to reduce risk of trauma to the baby.
rAvoid fetal blood sampling, fetal scalp
monitoring, Ventouse delivery, and mid-cavity
forceps, or forceps involving rotation of the
head.
rActive management of the third stage of labor
and early suturing of episiotomy and
lacerations for patients with low factor levels.
rRegional anesthesia has been shown to be safe
if the coagulation screen is normal and factor
levels are ⬎50 IU/dL treatment is required but
levels must be checked prior to removal of the
catheter as they may fall rapidly in the
postpartum period.
rIf maternal FVIII/IX levels ⬍50 IU/dL, caution
with non-steroidal anti-inflammatory drugs and
intramuscular injections.
Neonates are at risk of intracranial hemorrhage
and cephalhematomas during labor and delivery. e
risk is not increased by vaginal delivery but the sec-
ond stage of labor should not be prolonged and early
recourse to Cesarean section may be required. Trauma
should be minimized by avoiding extra cephalic ver-
sion, Ventouse delivery, fetal blood sampling, scalp
electrodes, and rotational forceps. e cut-o value
for predicted factor VIII or IX, above which no
birth restrictions are necessary, has not been dened,
although mild hemophilia is unlikely to be associ-
ated with severe bleeding at birth. Furthermore, the
increase in FVIII, induced by the stress of labor, pro-
vides some protection for babies with mild hemophilia
A. Female carriers have a small risk of extreme lyoniza-
tion and low factor levels, but this needs to be weighed
up against the possibly greater risks of withholding
instrumental delivery and invasive fetal monitoring.
As Factor IX levels are lower at birth and are not
increased by the stress of delivery, female carriers of
severe Hemophilia B are theoretically at higher risk.
Analgesia
ere is no consensus on the levels required for
regional anesthesia but this is generally considered to
be safe if FVIII /IX levels are ⬎50 IU/dL.3,4 Consider-
ation should also be given to the levels at the time of
catheter removal and repeat treatment given before-
hand if necessary. Intramuscular injections and non-
steroidal anti-inammatory drugs are not contraindi-
cated if factor levels are normal. All women with low
factor levels should have the opportunity to discuss
analgesia with an anesthetist prior to delivery.
Postpartum
Postpartum blood loss should be assessed as factor
levels may fall rapidly aer delivery. Levels should be
maintained at ⬎50 IU/dL for at least 3 days, or for 5
days if Cesarean section has been performed.5
DDAVP and/or tranexamic acid may be useful to
prevent excessive postpartum bleeding. ese agents
are described in Chapter 14.
Neonatal management
Aected babies may suer bruising and bleeding at
venepuncture and heel prick sites and even sponta-
neous organ or joint bleeding. To identify neonates at
risk, a cord sample should be taken for coagulation
factor assay and the result must be known before the
patient leaves hospital. Female babies at risk of being
carriers for severe hemophilia may also require a cord
sample, as very low levels may occur due to severely
unbalanced lyonization. However factors VIII and IX
from the newborn do not reect the true baseline level
and may need repeating at 6 months of age, when adult
values are reached.
Venepunctures and intramuscular injections,
including vitamin K should be avoided until the cord
factor level is known. Vitamin K could be given orally
if the results are delayed. Severely aected babies
should receive an ultrasound scan of the head to
assess for signs of intracranial hemorrhage (ICH),
particularly if delivery was traumatic or labor pro-
longed. is investigation is non-invasive, but lacks
sensitivity, particularly for subdural bleeds, which is
the commonest site of ICH in the neonate.
188
Chapter 15. Inherited coagulopathies
e mean age for occurrence of ICH is 4.5 days,2
whenthebabyislikelytobeathome.Henceparents
andmidwivesshouldbeinformedoftheearlysignsof
ICH; poor feeding, listlessness, vomiting, and seizures,
so that treatment can be administered without delay.
To date, there is no evidence for the benet of pro-
phylactic factor concentrate, about which the risk of
inhibitor development is debated. However, it may be
justied in selected cases, such as prematurity or trau-
matic delivery, where the risk of ICH is greater.
Factor XI deciency
Introduction
Factor XI is an important component of the intrinsic
coagulation pathway, playing a key role in the ampli-
cation of initial thrombin production, via activa-
tion of factor IX. e additional amount of thrombin
activates thrombin-activatable brinolysis inhibitor
(TAFI), which consolidates the brin clot and pro-
tects it from degradation by brinolysis. us de-
ciency of Factor XI is manifest mostly by injury or
surgery-related bleeding at sites which are prone to
local brinolysis, such as the nose and genitouri-
nary tract. Women with factor XI deciency are
at risk of menorrhagia and bleeding in relation to
childbirth.
Disease incidence
e inheritance of Factor XI deciency is autosomal. It
is most common amongst Ashkenazi Jews, where the
estimated heterozygosity rate is as high as 8%.6e
incidence in the non-Jewish population is reported to
be around 1:100 000, although this is likely to be an
underestimate, as it may frequently remain undetected
as routine coagulation assays may be normal in het-
erozygotes and there may be no bleeding history.
e predominant mutations in Ashkenazi Jews are
a Glu117stop codon in exon 5 designated type II, and
a Phe283Leu mutation in exon 9 designated type III.
Homozygotes for type II and type III mutation have
factor XI activities ⬍1 U/dL and 8–15 U/dL, respect-
ively, with compound heterozygotes for type II and
III having factor XI levels between these values.7In
non-Jewish populations, rapidly increasing numbers
of mutations and polymorphisms have been reported,
now reaching over 80. For the majority of these, the
level of FXI antigen has not been reported.
Table 15.4 Pre-pregnancy management of women with
Factor X1 deficiency
rAssess clinical bleeding tendency and
coexistence of confounding factors such as
VWD or platelet dysfunction.
rOffer pre-natal diagnosis where there is a risk of
severe deficiency and the mutation is known.
rDiscuss potential maternal bleeding risk and
options for management.
rConsent for use of blood products if necessary
and ensure hepatitis A and B immunity.
Clinical features
e bleeding tendency in FXI-decient individuals is
highly variable.8Factor XI activities ⬍15 U/dL have
been designated as severe deciency, although bleed-
ing is not closely correlated with factor levels9as it is
with hemophilia A and B. Neither does the abnormal
genotype causing the condition seem to bear any rela-
tionship to bleeding tendency, which is inconsistent
amongstfamilymembers.Indeed,inmostpatients
spontaneous bleeding, as well as bleeding aer hemo-
static challenge, does not occur and phenotype may
depend on other associated factors, such as coexistence
of mild von Willebrand disease.
Hormonal inuences on factor levels
Factor XI levels usually remain constant in pregnancy,
but studies have shown inconsistencies in levels with
increases or decreases as pregnancy advances.10
Obstetric complications
e main risk in pregnancy is of uterine hemorrhage
during invasive procedures, miscarriage, or postpar-
tum. Patients with FXI levels ⬍15 IU dL-1 have a 16%–
30% risk of peripartum bleeding11 and this has been
conrmed to almost exclusively aect those with a pre-
determined bleeding phenotype.10 usitisimportant
to attempt to ascertain, by thorough history taking,
which patients are at risk of bleeding, so that ante-natal
procedures, childbirth, and the postpartum period can
be managed appropriately (Table 15.4).
Ante-natal management
As it is oen not feasible to check levels in an acute
situation, routine monitoring should be carried out 189
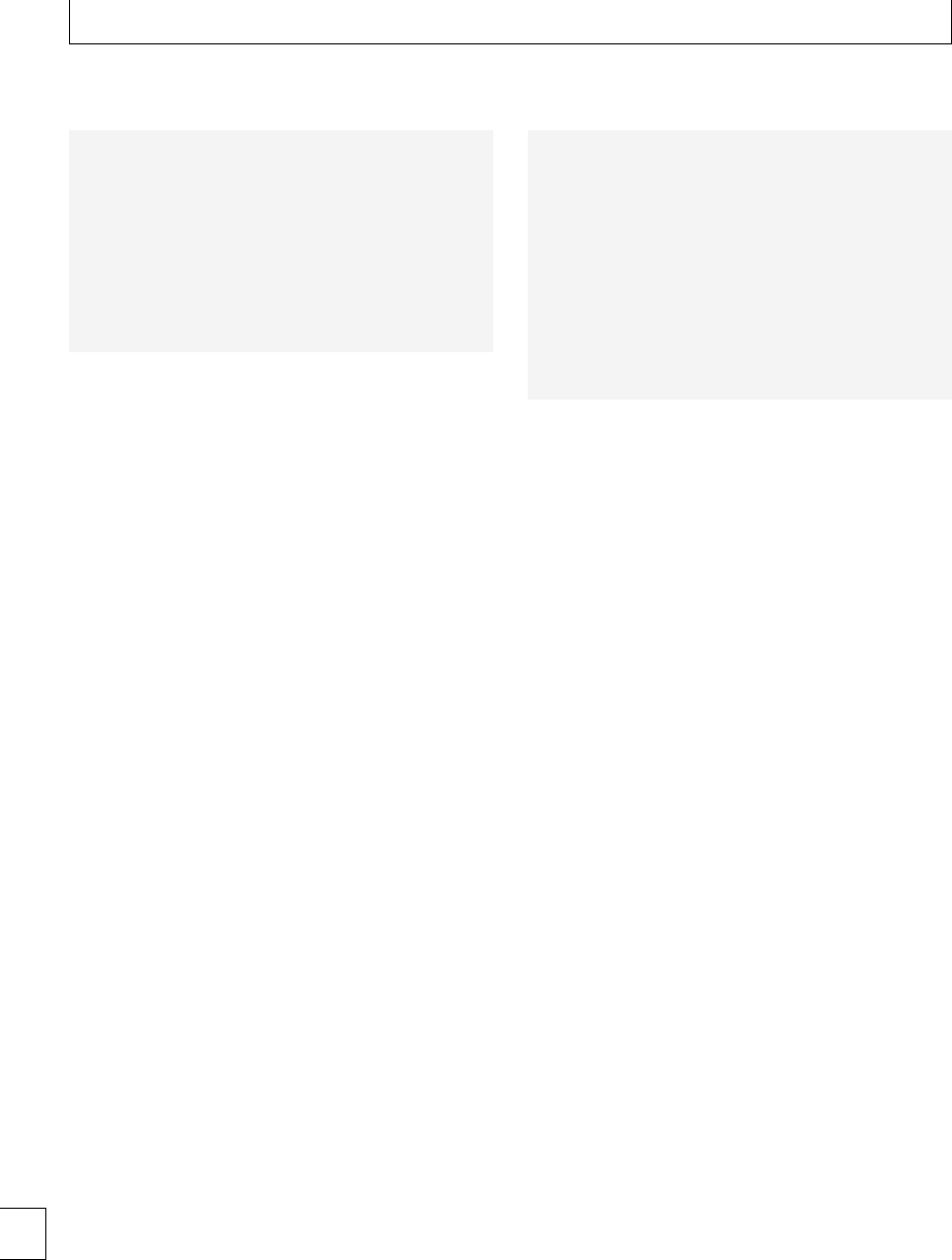
Section 5. Hemorrhagic disorders
Table 15.5 Ante-natal management of women with Factor X1
deficiency
rCheck levels at booking, 28 and 34 weeks’
gestation and prior to invasive procedures.
rPatients with severely low levels or a positive
bleeding history should be given prophylaxis to
cover invasive procedures.
rOther patients can be managed expectantly
with close observation and treatment available
on standby should bleeding occur.
at booking, 28, and 34 weeks. Whilst low factor lev-
els cause prolongation of the APTT, test reagents vary
in their sensitivity to factor XI levels and a normal
APTT does not exclude mild deciency. is is par-
ticularly so in pregnancy where the increase in factor
VIII may normalize the APTT even when factor XI
is reduced. us a specic coagulation factor assay is
required (Table 15.5).
Treatment options to cover delivery
and ante-natal procedures
Most patients can be managed expectantly,12 but those
with severely reduced levels or a positive bleeding his-
tory require prophylaxis for invasive ante-natal proce-
dures, miscarriage, and delivery.
Factor XI concentrate provides eective cover and
has a mean half-life of 52 hours, so a single dose is usu-
allysucient.However,itisassociatedwithapoten-
tial risk of transfusion transmitted infections, com-
mon to all plasma products, as well as an increased
risk of thrombosis due to coagulation activation.13
e increased thrombotic risk may be further exagger-
ated in pregnancy where there is already activation of
coagulation and increased thrombin generation.
Fresh frozen plasma contains variable amounts
of factor XI and patients with severe deciency are
unlikely to achieve levels above 30 IU dL−1.However,
it is helpful for milder cases and involves less donor
exposure than factor XI concentrate. A dose of 15–
20 mL/kg is eective, but the risk of uid overload
must be considered.
Monitoring of the response to FFP or factor XI con-
centrate is important and, due to the thrombogenic-
ity of the latter, levels should not be allowed to exceed
70 IU dL−1. A recent study found inhibitor develop-
ment, aer transfusion of plasma derived factor XI, in
Table 15.6 Intrapartum management of women with Factor
X1 deficiency
rAn on-demand policy can be advocated,
including for those with severely low levels,
during and after vaginal delivery.
rMost patients undergoing Cesarean Section
can be managed expectantly, but those with
severe deficiency should be given prophylaxis.
rMeasures should be taken to avoid unnecessary
trauma to the baby during delivery.
rThe third stage of labor should be actively
managed.
33% of patients with severe factor XI deciency due
to homozygous type II mutation (which accounts for
approximately 25% of Jewish patients with severe fac-
tor XI deciency).
Recombinant factor VIIa (rFVIIa, NovoSeven R
,
Novo Nordisk Ltd, Bagsvaerd, Denmark) is currently
being assessed as a possible alternative to plasma-
derived FXI replacement and avoids the risk of bacter-
ial or viral infections, transfusion-related lung injury
and development of inhibitors to factor XI. It is as
yet unlicensed for use in this setting and the opti-
mal dose has not been ascertained. A suggested dose
for minor procedures is 90 g/kg administered intra-
venously before surgery and 4 h later. For major
surgery, 2 hourly infusions are necessary due to the
short half-life of the product.
Intrapartum management
Around 70% of patients do not experience bleeding
problems at delivery. is may be due to increased lev-
els of coagulation factors, including factor VIII and
brinogen, at term. Also, the pregnancy-associated
reduction in brinolytic activity, due to decreased lev-
els of tissue plasminogen activator and urokinase and
an increased level of plasminogen activator inhibitor-
2, contributes to hemostasis. us, even for those with
severe factor XI deciency an on-demand policy can
usually be adopted for vaginal delivery.14 However, it
isimportantthatthepatientiscloselyobservedand
that all relevant sta is aware of the management plan.
It may be that a similar policy can be adopted for
patients with severe deciency undergoing Cesarean
section,butuntilfurtherstudiesaredone,these
patients should probably receive prophylaxis with one
of the agents described above (Table 15.6).
190

Chapter 15. Inherited coagulopathies
Regional anesthesia
Epidural anesthesia should be avoided in patients with
low factor XI levels. If the procedure is necessary, it
should be covered with factor XI concentrate and an
adequate response demonstrated. FFP is not recom-
mended due to the variable levels of FXI. Recombi-
nantfactorVIIamayprovideeectivecover,butfur-
ther evaluation is required in this area.
Postpartum management
e incidence of primary and secondary postpartum
hemorrhage, in patients with untreated factor XI de-
ciency, has been reported to be 16% and 24%, respect-
ively. Tranexamic acid is eective, although its use with
factor XI concentrate should be avoided. e standard
dose is 1g 6–8 hourly for 3–5 days, with the rst dose
being administered in labor.
Neonatal management
Neonatal hemorrhage due to peripartum events is
rare but nevertheless care should be taken during
delivery to avoid unnecessary trauma to the baby,
including avoidance of Ventouse extraction, rotational
forceps, and invasive monitoring techniques. Sponta-
neous bleeding or intracranial hemorrhage has not yet
been reported in neonates, but a cord blood sample
should be taken to determine the potential for bleed-
ing during high risk procedures such as circumcision.
Neonatal levels are approximately half that of adults
and repeat testing aer 6 months of age is required to
provide an accurate baseline level.
Rare coagulation factor deciences
e rare coagulation disorders include deciencies of
coagulation factors II, V, VII, X, V, and VIII, combined
vitamin K-dependent factors, FXIII, and disorders of
brinogen. e spectrum of bleeding manifestations
in individuals with these disorders is variable, but
some may present with severe bleeds, including
intracranial hemorrhage and hemarthroses. With the
exception of dysbrinogenemia, these disorders have
autosomal recessive inheritance and their prevalence,
in the severe form, varies between 1:500 000 and
1:2,000 000. Pregnancy in women with these disorders
or couples at risk of having an aected child, should
bemanagedinanobstetricunitwithcloselinkstoa
Hemophilia Center.
Hormonal inuences on factor levels
Factors II, V, and XIII tend to remain constant
throughout pregnancy or show a slight increase but
thereisaprogressiveriseinfactorsVII,X,andb-
rinogen, particularly in the third trimester. is is ben-
ecial to heterozygous women with mild or moder-
ate factor deciency, but in homozygous women, with
severe deciency, levels remain low.
Pre-pregnancy management
e clinical bleeding tendency and response to hemo-
static challenges should be ascertained. Women should
be counseled about their potential bleeding risk in
relation to pregnancy, ante-natal procedures, delivery,
and the postpartum period. Consent for use of blood
products should be obtained and immunity to hep-
atitis A and B ensured. Genetic counseling should be
given and pre-natal diagnosis oered where possible.
Ante-natal management
Levels should be checked at booking and repeated at
28 and 34 weeks’ gestation. Depending on the fac-
tor level and clinical bleeding tendency, prophylaxis
may be required for ante-natal procedures and deliv-
ery. ere is little evidence to guide therapeutic deci-
sions but in general, relatively low levels of factors
II, V, VII, and X, of around 20 IU/dL, are sucient
for normal hemostasis.12,15 erefore, patients with
partial deciencies and no history of bleeding can be
managed expectantly. Otherwise, replacement therapy
should start at the onset of established labor and the
factor half-life should be considered to determine the
need for, and timing of, repeat doses. Pre- and post-
treatment factor levels should be obtained and eective
levels maintained for 3–5 days aer delivery.
Treatment options
Prothrombin complex concentrates can be used for
patients with factors II or X deciency. ese are
pooled plasma-derived products containing known
quantities of factors II, IX, and X, with or without fac-
tor VII. e strength of the concentrate is expressed in
termsofunitsofFIX,butthisisapproximatelyequalto
the units of prothrombin. Concomitant use of tranex-
amic acid should be avoided because of the risk of
thrombosis.
FFP is the only available product for FV deciency
and may also be used for patients with prothrombin 191

Section 5. Hemorrhagic disorders
and FX deciency. A virally inactivated product should
beused.Aninitialdoseof15mL/kgshouldbegiven,
with repeat doses dictated by factor levels and clin-
ical response. Women with factor V deciency failing
to respond to FFP may benet from platelet transfu-
sions, which provide a concentrated supply of platelet
factor V.
Recombinant FVIIa is the treatment of choice for
surgery or childbirth in women with FVII deciency,
at a dose of 20–25 mcg/kg administered every 4–6
hours.
Tranexamic acid is useful in preventing postpar-
tum bleeding, although should not be used in conjunc-
tion with prothrombin complex concentrates.
Patients with combined vitamin K-dependent fac-
tors can be treated with daily vitamin K, although FFP
may be needed in the event of bleeding.
Early pregnancy failure
Maternal FXIII plays a critical role in uterine hemosta-
sis and maintenance of the placenta during gestation.
e risk of miscarriage in women with severe factor
XIII deciency is around 50%, depending on the sub-
type. ese women should receive prophylactic infu-
sionsofFXIIIatmonthlyintervals,aimingforatrough
level of ⬎3 U/dL, although higher Factor XIII levels
may be needed for delivery. 16
Fibrinogen is important for implantation and
patients with abrinogenemia or hypobrinogenemia
have a high rate of early miscarriage occurring at 6–8
weeks’ gestation. Regular infusions of brinogen con-
centrate, to maintain trough levels ⬎0.6 g/L, should
be started as soon as pregnancy is conrmed and
continued throughout pregnancy and the peripartum
period.17 Fibrinogen consumption tends to increase as
pregnancy advances. Repeated ultrasounds should be
carried out to detect concealed placental bleeding and
monitor fetal growth.
Dysbrinogenemia has been associated with a high
incidence of miscarriage and stillbirth18 but clinical
phenotypes vary and management should be indi-
vidualized, depending on the brinogen level and
the clinical presentation of the disorder in the fam-
ily. romboprophylaxis with low molecular weight
heparin is required for those with personal or fam-
ily history of thrombosis and brinogen replacement
for bleeding phenotypes, but many cases are asymp-
tomatic without the need for specic treatment.
Thrombosis
e potential for thrombosis following factor replace-
ment must be considered and attention given to sim-
ple thromboprophylactic measures such as adequate
hydration, compression stockings, and early mobil-
ization. Patients with abrinogenemia- or dysbrino-
genemia are at particular risk of thrombosis due to
impaired regulation of thrombin generation. Loose
platelet thrombi form and are susceptible to emboliza-
tion, therefore careful consideration should be given
to the balance of bleeding and thrombotic risk. For
these patients, a continual infusion of brinogen con-
centrate to maintain levels above 1.5 g/L during the
peripartum period allows for ne control.
Neonatal management
PerinataltraumasuchasVentousedelivery,rotational
forceps, and fetal blood sampling should be avoided.
Severe and moderate deciencies can be diagnosed on
a cord blood sample. Severely aected babies require
cranial ultrasound to detect any ICH.
192

Chapter 15. Inherited coagulopathies
References
1. ChiC,LeeCA,ShitaghNet al. Pregnancy in carriers
of haemophilia. Haemophilia 2008; 14: 56–64.
2. Kulkarni, Roshni and Lusher, Jeanne M. Intracranial
and extracranial hemorrhages in newborns with
hemophilia: a review of the literature. Journal of
Pediatric Hematology/Oncology 1999; 21: 289–
295.
3. Kadir RA, Economides DL, Braithwaite J et al.e
obstetric experience of carriers of haemophilia. British
Journal of Obstetrcs and Gynaecology 1997; 104:
803–810.
4. EA., Letsky. Haemostasis and epidural anaesthesia.
International Journal of Obstetric Anesthesia. 1991; 1:
51–54.
5. Lee CA, Chi C, Pavord SR et al. UK haemophilia
Centre Doctors’ Organization. e obstetric and
gynaecological management of women with inherited
bleeding disorders – review with guidelines produced
by a taskforce of UK Haemophilia Centre Doctors’
organization. Haemophilia 2006; 12: 301–336.
6. Asakai R, Chung DW, Davie EW, Seligsohn U. Factor
XI deciency in Ashkenazi Jews in Israel. New England
Journal of Medicine 1991; 325: 153–158.
7. Hancock JF, Wieland K, Pugh RE et al. A molecular
genetic study of factor XI deciency. Blood 1991; 77:
1942–1948.
8. Bolton-Maggs PH, Patterson DA, Wensley RT,
Tuddenham EG. Denition of the bleeding tendency in
factor XI-decient kindred – a clinical and laboratory
study. rombosis and Haemostasis 1995; 73: 194–
202.
9. Bolton-Maggs PH, Wan-Yin BY, McCraw AH et al.
Inheritance and bleeding in factor XI deciency.
British Journal of Haematology 2008; 69: 521–528.
10. Myers B, Pavord S, Kean L et al.Pregnancyoutcomein
Factor XI deciency: incidence of miscarriage,
antenatal and postnatal haemorrhage in 33 women
with Factor XI deciency. British Journal of Obstetrics
and Gynaecology: an International Journal of Obstetrics
and Gynaecology 2007; 114: 643–646.
11. Kadir RA, Lee CA, Sabin CA, Pollard D, Economides
DL. Pregnancy in women with von Willebrand’s
disease or factor XI deciency. British Journal of
Obstetrics and Gynaecology 1998; 105: 314–321.
12. Bolton-Maggs PH, Perry DJ, Chalmers EA et al.e
rare coagulation disorders–review with guidelines for
management from the United Kingdom Haemophilia
Centre Doctors’ Organisation. Haemophilia 2004; 10:
593–628.
13. Bolton-Maggs PH, Colvin BT, Satchi BT et al.
rombogenic potential of factor XI concentrate.
Lancet 1994; 344: 748–749.
14. Salomon O, Steinberg DM, Tamarin I et al.Plasma
replacement therapy during labor is not mandatory for
women with severe factor XI deciency. Blood
Coagulation and Fibrinolysis 2005; 16: 37–41.
15. Kadir R, Chi C, Bolton-Maggs P. Pregnancy and rare
bleeding disorders. Haemophilia 2009; 15: 990–1005.
16. Asahina T, Kobayashi T, Takeuchi K et al. Blood
coagulation factor XIII deciency and successful
deliveries: a review of the literature. Obstetrical and
Gynecological Survey 2007; 62: 255–260.
17. Kobayashi T, Kanayama N, Tokunaga N et al. Prenatal
and peripartum management of congenital
abrinogenaemia. British Journal of Haematology
2000; 109: 364–366.
18. Haverkate F, Samama M. Familial dysbrinogenemia
and thrombophilia. Report on a study of the SSC
Subcommittee on Fibrinogen. rombosis and
Haemostasis 1995; 73: 151–161.
193

Section 5 Hemorrhagic disorders
Chapter
16 Genetic counseling and pre-natal diagnosis
in hemophilia
Andrew Mumford
Introduction
Hemophilia is the most common severe genetic bleed-
ing disorder and presents signicant risk to the fetus
at delivery. Hemophilia is also associated with signif-
icant morbidity later in life and may require inten-
sive long-term treatment, which may be a considerable
burden to aected families. High quality obstetric care
of women with a family history of hemophilia is there-
fore paramount but presents particular management
challenges.
Genetic counseling in hemophilia
Genetic counseling refers to the process of communi-
cation of information to women and families to enable
informed decision making about the consequences of
carrying a fetus with hemophilia. Genetic counseling
for hemophilia should encompass the issues of carrier
testing and pre-natal diagnosis.
Successful genetic counseling should be support-
ive and requires careful two-way discussion between
families and healthcare professionals who are famil-
iar with hemophilia management and with the tech-
niques available for carrier testing and pre-natal diag-
nosis. Since genetic counseling oen raises complex
ethicalandmoralissues,thisprocessmayrequiremul-
tiple face-to-face consultations supported by clear and
objective written information. Ideally, genetic counsel-
ing should be initiated before pregnancy is planned.1
Genetic counseling is a step-wise process and may
require discussion about the following issues:
rfamily diagnosis of hemophilia and clinical
severity;
rinheritance pattern of hemophilia within the
family to exclude carriership or to identify
“possible” and “obligate” carriers;
rpattern of transmission and consequences of
hemophilia in future ospring;
rbenets and hazards of carrier detection
techniques;
rOptions available for management of pregnancy,
including pre-natal diagnosis.
Heritability of hemophilia
As hemophilia A and B are sex-linked disorders,
aected families may contain males with hemophilia
and female hemophilia carriers who usually do not
have abnormal bleeding, but may transmit hemophilia
to males in the next generation (Fig. 16.1). Analysis
of an accurate family pedigree is essential to establish
the probability of hemophilia carriership and trans-
mission risk.
rSons of female hemophilia carriers have a 50%
chance of having hemophilia.
rDaughters of female hemophilia carriers have a
50% chance of being carriers.
rSons of males with hemophilia will not inherit
hemophilia.
rDaughters of males with hemophilia will always
inherit hemophilia and will therefore be obligate
hemophilia carriers.
rApproximately 50% of individuals newly
diagnosed with hemophilia have no family history
of hemophilia.
Some women can be excluded as being hemophilia
carriers by analysis of the family pedigree. Although
these women do not have the gene change responsible
for hemophilia elsewhere in their families, they retain
a small risk, as in the general population, of being car-
riers of a dierent hemophilia gene change that has
arisen by spontaneous mutation. For hemophilia A,
194 e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.
Chapter 16. Genetic counseling and pre-natal diagnosis in hemophilia
1
1
1
2
2
2
34
I
II
III
Fig. 16.1 Pedigree of a family with hemophilia showing possible
patterns of transmission of hemophilia over three generations. The
offspring of a female hemophilia carrier (I.2) can include males with
hemophilia (II.1), males without hemophilia (II.2), female hemophilia
carriers (II.3), and females who are not hemophilia carriers (II.4). The
offspring of males with hemophilia (II.1) can either be males without
hemophilia (III.1) or female obligate hemophilia carriers (III.2).
this background risk of carriership is approximately 1
in 20 000 women.
Prediction of carrier status by
pedigree analysis
For women from families with hemophilia, the proba-
bility that a pregnancy will yield a fetus that is a male
with hemophilia can be calculated from the family
pedigree using simple rules of Mendelian inheritance
(Fig. 16.2).
Forfamiliesinwhichthereishemophiliainone
individual but no antecedent history of hemophilia
(sporadic hemophilia), calculating the risk of
hemophilia in subsequent members of the same
generation is more dicult (Fig. 16.3). Sporadic
hemophilia usually arises because of new mutations
in the F8 or F9 genes (hemophilia A and B, respect-
ively) occurring during gametogenesis in either the
mother or a maternal ancestor of an aected male.
However, spontaneous mutations occur more readily
during spermatogenesis than oogenesis. erefore, the
causative mutation in a male with sporadic hemophilia
is more likely to have arisen during spermatogenesis
in the maternal grandfather than in oogenesis in the
mother. It follows that mothers of males with sporadic
hemophilia are likely to be constitutional hemophilia
carriers. Observational population studies conrm
this prediction and show that approximately 90% of
mothers of males with sporadic hemophilia are car-
riers and therefore, have signicant risk transmitting
hemophilia to future male ospring.2
1
1
2
2
2
1
I
II
III
IV
50% chance of being
hemophilia carrier
12.5% chance of being
male with hemophilia
Obligate hemophilia
carrier
1
Fig. 16.2 Example pedigree allowing calculation of the probability
of carriership and transmission of hemophilia. The female proband
(III.2-arrowed) has a maternal grandfather (I.1) with hemophilia.
Since I.1 is a male with hemophilia, the mother of the proband (II.2)
is an obligate carrier of hemophilia. The proband III.2 therefore has a
50% chance of hemophilia carriership. Since the probability that a
hemophilia carrier will carry a fetus that is a male with hemophilia at
each pregnancy is 25%, the absolute probability that the unborn
fetus IV.1 will be affected with hemophilia is 12.5%.
1
1
1
2
2
2
I
II
III
4
3
90% chance of being
hemophilia carrier
23% chance of being
male with hemophilia
Fig. 16.3 Example pedigree showing estimated probability of
hemophilia carriership and transmission in a family with sporadic
hemophilia The female proband (II.2-arrowed) already has a son
who is affected with hemophilia (III.3) but has no other family
history. The mutation responsible for hemophilia in III.1 is most likely
to have occurred during spermatogenesis in individual I.3 and so
the proband II.2 is likely to be a carrier of hemophilia. The estimated
probability that II.2 is a carrier of hemophilia is approximately 90%
and so the probability that each subsequent pregnancy will yield a
male with hemophilia is approximately 23%. 195

Section 5. Hemorrhagic disorders
Laboratory detection of hemophilia
carriership
Determining the probability of hemophilia carriership
by pedigree analysis is essential for the genetic coun-
seling process. However, all women who are potential
hemophilia carriers should also be oered laboratory
carriership detection. Two complementary approaches
are available; coagulation factor activity assays and
mutation analysis.
Coagulation factor activity assays
Female carriers of hemophilia usually show reduced
activities of coagulation factor VIII or IX (hemophilia
A and B, respectively) to levels of 40%–80% that of
unaected individuals.3However, there is wide varia-
tion in factor activity between carriers and there is sig-
nicant overlap with women who are not hemophilia
carriers. Measurement of coagulation factor activity
may therefore guide identication of hemophilia carri-
ers but is insucient for denitive diagnosis. e ratio
of FVIII to Von Willebrand factor (VWF) may be help-
ful, as these two molecules normally circulate in the
plasma with 1:1 stoichiometry. us carrier status may
be suspected if the ratio falls below 0.7, despite the
absolute FVIII level being normal.4
Genetic detection of carriership
Female hemophilia carriers are heterozygous for
mutations in F8 or F9 and demonstration of a
hemophilia – associated mutation in these genes is suf-
cient to diagnose carriership. It is good practice to
conrm hemophilia carriership with genetic testing
even in women identied as obligate carriers by pedi-
gree analysis. Denitive exclusion of hemophilia car-
riership in potential carriers requires demonstration
that the hemophilia mutation in the family is absent.
In this circumstance, prior knowledge of the causative
mutationinamalewithhemophiliaoranobligate
female carrier from the family is essential.
Testing the potential for transmission of
hemophilia in asymptomatic women raises com-
plex moral issues for the individual and families
undergoing testing. e full implications of genetic
testing should therefore be discussed during coun-
seling and informed written consent is mandatory.
Counseling should include specic discussion about
the limitations of F8 and F9 genetic analysis.
Mutations associated with hemophilia
Although more than 1800 F8 mutations have now been
identied in individuals with hemophilia A, many
defects are recurrent and have been recognized in mul-
tiple aected families. A major structural rearrange-
mentoftheF8generesultingfromaninversioninvolv-
ing intron 22 accounts for approximately 50% of cases
of severe hemophilia A. Other recurrent mutations
associated with severe hemophilia A include point
mutations, non-sense mutations, deletions, or other
major structural changes in F8 that prevent expression
of the gene. Mild hemophilia A and hemophilia B are
usually associated with point mutations in F8 and F9,
respectively, although heterogeneity between aected
families means that previously unreported mutations
are common.
e mutation databases for hemophilia A (http://
europium.csc.mrc.ac.uk/WebPages/Main/main.htm)
and hemophilia B (http://www.kcl.ac.uk/ip/
petergreen/haemBdatabase.html) contain biblio-
graphic references and phenotypic data from pre-
viously reported families with hemophilia. ese
resources are valuable for conrming that a newly
identied mutation in a hemophilia family is causative
and in predicting the future clinical phenotype of
aected males.
Limitations and hazards of genetic diagnosis
of carriership
1. Failure to detect causative mutations.
Approximately 5% of hemophilia mutations are
not detected by analysis of the coding sequence of
F8 or F9. Similarly, some mutations such as large
deletions may be readily detected in males but not
in heterozygous female carriers. In these
circumstances, detection of carriers currently
requires techniques such as linkage analysis. is
may not be informative in all families and,
because of genetic recombination events, has
lower diagnostic accuracy than direct mutation
detection by sequencing.
2. False-negative carrier detection because of
somatic mosaicism. An individual is a somatic
mosaic for hemophilia when a spontaneous
hemophilia mutation occurs in a somatic cell
during early embryogenesis rather than during
gametogenesis in one or other parent. In an
aected embryo, the hemophilia mutation is
196

Chapter 16. Genetic counseling and pre-natal diagnosis in hemophilia
therefore present in some, but not all, cells
including the germ cells. ose germ cells which
contain the hemophilia mutation may then go on
to form gametes. For women who are somatic
mosaics, this population of gametes is then
capable of transmitting hemophilia to the
subsequent generation.
Somatic mosaicism was identied in a female
proband in more than 10% of families with severe
hemophilia and, in some cases, the hemophilia muta-
tion was present in up to 25% of maternal cells.5In
this circumstance, standard genetic testing of DNA
obtained from peripheral blood cells may not detect
ahemophiliamutationandsomaticmosaicmothers
may therefore be mis-classied as “not hemophilia car-
riers.” Somatic mosaicism should be considered in all
women who have a son with hemophilia but no other
family history and who have been classied as “not a
hemophilia carrier” by standard genetic testing. For
families with sporadic hemophilia B, one study has
estimated that women with this background have a
risk of hemophilia B in a second fetus of ⬍6%.6is
very low probability may be similar for all forms of
hemophilia and should be discussed during genetic
counseling for all potential carrier women.
Pre-natal diagnosis of hemophilia
Women who have been identied as hemophilia
carriers by pedigree analysis and laboratory investi-
gation may be oered several dierent options for
pre-natal diagnosis. Involvement of healthcare profes-
sionals with expertise in fetal medicine is essential.
Pre-natal diagnosis in hemophilia may be per-
formed currently for two dierent reasons:
rto oer a more accurate probability of whether a
fetus will be aected with hemophilia by fetal
sexing to assist management of delivery;
rto oer early denitive diagnosis of a hemophilia
in male fetus by rst trimester pre-natal genetic
diagnosis to enable the option of termination of
an aected pregnancy.
Pre-natal fetal sexing
rFetal sexing may be performed by non-invasive
(ultrasound or free fetal DNA (DNA) analysis)
or invasive (chorionic villus sampling (CVS) or
amniocentesis) techniques.
rIf a female fetus is identied, hemophilia is
excluded and hemostatic precautions at delivery
are unnecessary.
rFetal ultrasound allows gender to be identied
reliably in most pregnancies from about 18–20
weeks’ gestation.
rPCR detection of the fetal SRY locus in DNA
circulating in maternal plasma is highly specic
for a male fetus. is assay requires a maternal
venous blood specimen of ⬍20 mL and has ⬎99%
diagnostic accuracy at 10–12 weeks’ gestation in
expert centers.7Arecentstudyevaluatingthe
method in 196 women, including hemophilia
carriers, showed 100% accuracy as early as 7
weeks’ gestation.8
First trimester pre-natal genetic diagnosis
rFirst trimester pre-natal genetic diagnosis allows
denitive diagnosis of hemophilia in a fetus but
requires invasive testing by CVS.
rCVS is an important option for conrmed
hemophilia carriers who are considering
termination of a male fetus with hemophilia. e
uptake of this approach is very low in most
reported series of pregnancies in hemophilia
carriers.
rCVS for pre-natal genetic diagnosis is performed
at 11–14 weeks’ gestation and carries a
miscarriagerateofapproximately1%.Earlier
procedures have resulted in fetal limb reduction
defects, particularly if performed before 10 weeks’
gestation. Hemophilia carrier mothers with low
coagulation factor levels may need treatment to
cover the procedure. (See Chapter 15.)
rPlacental cells obtained by CVS are rst used to
determine fetal sex. Detection of the hemophilia
mutation present in the family is then performed
on male fetuses.
rAdvances in very early fetal sexing by DNA
analysis may enable female fetuses to be identied
before 11–14 weeks so that CVS is then
unnecessary. Conrmation of gender by fetal
ultrasound at 18 weeks’ gestation is then
recommended.
rAmniocentesis is an alternative technique for
pre-natal genetic diagnosis in hemophilia and can
safely be performed from 15 weeks’ gestation. is
technique is therefore less suitable for women
contemplating termination of pregnancy. 197

Section 5. Hemorrhagic disorders
Amniocentesisisassociatedwithmiscarriagerates
of 0.5%–1%, but higher miscarriage rates and fetal
talipes have been associated with amniocentesis
performed before 15 weeks. Cord blood sampling
is unsuitable for pre-natal diagnosis of hemophilia
because of bleeding risk in an aected
fetus.
Future techniques for pre-natal diagnosis
Third trimester amniocentesis and mutation detection
Amniocentesis performed at around 36 weeks enables
genetic diagnosis of hemophilia in male fetuses and
carries a risk of preterm labor of approximately 1%
in experienced centers. is approach allows hemo-
static precautions to be applied only to male fetuses
with hemophilia, so that, unaected male fetuses can
be delivered without these constraints. e risk to the
fetusofdeliveryprecipitatedbytheamniocentesisis
very small at this late gestation. e potential clinical
benets of this approach in hemophilia are currently
under evaluation.
Pre-implantation diagnosis
Pre-implantation sexing with re-implantation of
female or unaected male embryos requires standard
in vitro fertilization techniques, with harvesting of
cells from embryos at the 8-cell stage for analysis.
Single-cell PCR enables detection of specic mutations
in male embryos.9ese approaches are technically
feasible in hemophilia and have now been performed
in small numbers of successful pregnancies.
Mutation detection using DNA or fetal cells
in maternal blood
Detection of hemophilia mutations in DNA or in fetal
cells in maternal blood in pregnancy potentially oers
non-invasive pre-natal diagnosis of hemophilia. is
approach requires highly ecient purication of fetal
material from maternal blood and may only be feas-
ible in the third trimester when DNA and fetal cells
aremostabundant.isapproachiscurrentlyatearly
development stage.
Genetic counseling for other heritable
bleeding disorders
Genetic counseling, carrier detection and pre-natal
diagnosis should also be considered in families with
heritable bleeding disorders other than hemophilia,
which may also present bleeding risk to an aected
fetus. Most of these rare bleeding disorders show auto-
somalrecessiveinheritance(e.g.severeFactorXde-
ciency, severe Factor V deciency, Type III VWD) and
genetic counseling requires discussion about the trans-
mission of homozygous or compound heterozygous
mutations from both parents. Aected fetuses are usu-
ally sporadic and arise in families with no bleeding his-
tory in heterozygous “carrier” ancestors. For mothers
who are known heterozygous “carriers” or who them-
selves are homozygous or compound heterozygous for
a recessive bleeding disorder, accurate prediction of
fetal bleeding risk may require partner testing. is
is particularly important in consanguineous partner-
shipswheretheriskoftransmissionofhomozygous
recessive mutations is high.
Genetic counseling for the rare bleeding disorders
should reect that the relationship between plasma
coagulation factor activity and bleeding risk in aected
individuals is less predictable than in hemophilia and
that some disorders show variable penetrance. Since
the range of reported mutations in the rare bleed-
ing disorders is less than for hemophilia, detection of
previously undescribed mutations in aected families
is common. Uncertainty about whether a candidate
mutation is the true disease-associated mutation may
hamper genetic carrier detection and pre-natal diag-
nosis in some families.
198

Chapter 16. Genetic counseling and pre-natal diagnosis in hemophilia
References
1. Ludlam CA, Pasi KJ, Bolton-Maggs P et al.A
framework for genetic service provision for
haemophilia and other inherited bleeding disorders.
Haemophilia 2005; 11: 145–163.
2. Kasper CK, Lin JC. Prevalence of sporadic and familial
haemophilia. Haemophilia 2007; 13: 90–92.
3. Plug I, Mauser-Bunschoten EP, Broker-Vriends AH
et al. Bleeding in carriers of haemophilia. Blood 2006;
108: 52–56.
4. Shetty S, Ghosh K, Pathare A, Mohanty D. Carrier
detection in haemophilia A families: comparison of
conventional coagulation parameters with DNA
polymorphism analysis – rst report from India.
Haemophilia 2001; 5: 243–246.
5. Leuger M, Oldenburg J Lavergne J-M et al.Somatic
mosaicism in Haemophilia A: a fairly common event.
American Journal of Human Genetics 2001; 69: 75–87.
6. Green PM, Saad S, Lewis CM, Gianelli F. Mutation
rates in humans I: overall and sex-specic rates
obtained from a population study of haemophilia B.
American Journal of Human Genetics 1999; 65:
1572–1579.
7. Avent N, Chitty LS. Non-invasive diagnosis of fetal
sex; utilisation of free fetal DNA in maternal plasma
and ultrasound. Prenatal Diagnosis 2006; 26: 598–
603.
8. Bustamente-Aragones A, Rodrguez de Alba M,
Gonzalez-Gonzalez C et al. Foetal sex determination
in maternal blood from the seventh week of gestation
and its role in diagnosing haemophilia in the foetuses
of female carriers. Haemophilia 2008; 14: 593–598.
9. Michaelidis K, Tuddenham EG, Turner C et al.Live
birth following the rst mutation specic
pre-implantation genetic diagnosis for haemophilia A.
rombosis and Haemostasis 2006; 95: 373–379.
199

Section
6
Microangiopathies

Section 6 Microangiopathies
Chapter
17 Pre-eclampsia
Eleftheria Lefkou and Beverley Hunt
Introduction
Pre-eclampsia (PET) is a pregnancy-related mul-
tisystem syndrome, that is characterized by new-
onset of hypertension (blood pressure greater than
140/90 mmHg) aer 20 weeks of gestation and pro-
teinuria (greater than 1+, or urinary excretion of pro-
tein ≥300 mg/24 hours) resolving aer delivery. PET
is also termed toxemia, pregnancy-induced hyperten-
sion, and pre-eclamptic toxemia. Symptoms can occur
any time aer 20 weeks of gestation or even start in the
rst few days aer delivery, and always resolve within
a few days to weeks aer delivery of the placenta. Early
onset PET is when it develops before the 34th week
of gestation and late onset PET when it presents aer
the34thweekofpregnancy.Predisposingfactorsare
shown in Table 17.1. It is not known why some women
develop pre-eclampsia, while others with the same risk
factors do not.
Eclampsia occurs when PET is complicated by
seizures.
Chronic hypertension is dened as systolic pres-
sure ≥140 mmHg and/or diastolic pressure ≥90
mmHg that antedates pregnancy, that is present before
the 20th week of pregnancy, or persists longer than 12
weeks’ postpartum.
PET with chronic hypertension is diagnosed when
a pregnant woman has a history of chronic hyperten-
sion and then develops features suggestive of PET aer
the20thweekofpregnancy.
Gestational hypertension,ortransienthyperten-
sion of pregnancy refers to the situation that is charac-
terized by elevated blood pressure (⬎140/90 mmHg)
aer the 20th week of gestation, but without pro-
teinuria, that occurs uniquely during pregnancy and
resolves aer birth.
e aim of this chapter is to provide a basic under-
standing of PET and a detailed understanding of
hematological complications and their management.
Epidemiology
Pre-eclampsia (PET), the commonest medical compli-
cation of pregnancy, aecting approximately 2%–14%
of all pregnancies, remains a major cause of maternal
and fetal morbidity and mortality worldwide. It is esti-
mated that 50 000 women die annually worldwide due
to PET and eclampsia. In the United States the inci-
dence of PET is approximately 5%–8%, with 75% of
cases being mild and 10% of cases due to early onset
PET. According to the latest report from the Conden-
tialEnquiryintoMaternalandChildHealthitremains
the second major cause of maternal mortality and mor-
bidity in the UK aer venous thromboembolism.1e
incidence of PET in the UK is reported as 2%–8%, with
a fatality rate of 18/ 100 000 pregnancies. Mild PET is
under-reported and so the true incidence is potentially
much higher.
PET is associated with intrauterine growth restric-
tion (IUGR), in one-third of cases. Premature deliv-
ery to prevent the progression of PET is responsible for
15% of all preterm births. Infants of women with PET
have a vefold increase in mortality compared with
infants of mothers without the disorder.
e recurrence likelihood for PET is reported as
60% if it had occurred ⬍34 weeks’ gestation and 10%–
20% if occurred near term. e key to appropriate
management is early clinical recognition.
Diagnosis of PET
e diagnosis of PET is based on the maternal history,
signs, and symptoms (Table 17.2). e current aim of
203
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 6. Microangiopathies
Table 17.1 Risk factors for pre-eclampsia (PET)
Nulliparity
High body mass index (BMI) (⬎35 at booking)
Multiple gestation (twins, triplet pregnancies)
Chronic hypertension
Diastolic pressure ⬎89 mmHg at booking
Proteinuria at booking
Previous pregnancy with PET or IUGR child
Family history of PET (in mother or sisters)
Black race
Maternal age under 20 and possibly maternal age over 35 to 40
Diabetes mellitus or insulin resistance
Renal disease
Thrombophilias and hyperviscosity syndromes
Underlying maternal collagen vascular disease
Presence of antiphospholipid syndrome, or antibodies
Increased circulating testosterone
Protein D deficiency in mother during pregnancy
Trisomy 13
High altitude
Mirror syndrome
Fetal (genetic) factors from donor eggs
Father of Hispanic origin
Parental specific genes
ante-natal care is to monitor for signs of PET at each
clinic visit, with assessment of blood pressure, urinaly-
sisandthepresenceofedema.esevisitsoccurmore
frequently in the third trimester of pregnancy, espe-
cially in women with risk factors. Most women with
PET experience only mildly increased blood pressure
and small amounts of proteinuria. Edema, especially
in the face and hands, is a frequent sign of PET, but
is not pathognomonic, for many women without PET
also develop edema during pregnancy. Other forms of
hypertensive disorders also occur in pregnancy and
should be considered in the dierential diagnosis of
PET.
e maternal manifestations of PET can aect
almost every organ, depending on severity. Possible
complications of PET in the mother and in the fetus
are listed in Table 17.3.
Table 17.2 Signs and symptoms of severe pre-eclampsia
Blood pressure greater than 160/110 mm Hg
Impaired kidney function (serum creatinine concentration
⬎110 mol/L, urine protein greater than 5 grams in a 24-hour
urine collection) or low urine production (less than 500 mL in
24 hours)
Persistent severe headache
Papilledema and/or visual disturbances (blurred vision, diplopia,
blind spots, flashes of light, or squiggly lines).
Hyperreflexia, brisk tendon reflexes (3+)
Pulmonary edema, shortness of breath
Nausea, vomiting
Abdominal pain, persistent new epigastric pain or tenderness
Impaired functional liver tests (elevated alanine
aminotransferase, aspartate aminotransferase)
Thrombocytopenia (⬍100×109/L)
Microangiopathic hemolytic anemia
Table 17.3 Complications of PET
(A) Maternal
Central nervous system
rEclampsia
rCerebral edema
rCerebral hemorrhage
rRetinal edema
rRetinal blindness
rCortical blindness
Liver
rHELLP syndrome
rAcute liver failure
rHepatic rupture
Renal system
rAcute renal failure
rRenal cortical necrosis
rRenal tubular necrosis
Respiratory
rPulmonary edema
rLaryngeal edema
Hemostatic system
rThrombocytopenia
rDIC
rMicroangiopathic hemolytic anemias
Cardiovascular system
rRisk factor for later cardiovascular disease
Labor
rPlacental infarction
rPlacental abruption
rPreterm delivery
(B) Fetal–Neonatal
rIUGR
rPrematurity
rDeath
rNeurological complications
rLater cardiovascular disease
204

Chapter 17. Pre-eclampsia
Current concepts on the pathogenesis
of PET
Placental dysfunction is the central feature in the
development of PET. In 1939 Ernest Page introduced
the concept that PET may be due to the reduced per-
fusion of the placenta.
The two stage model of PET
Currently, the development of PET is hypothesized,
to be in two stages, according to a theory introduced
by the Oxford Group in 1991, and supported and
expanded by Roberts.2e rst stage is reduced pla-
cental perfusion and the second the maternal response
to this – maternal endothelial cell activation. Failure of
endovascular trophoblast invasion is thought to lead to
relative under perfusion of the placenta. Under condi-
tions of hypoperfusion, the placenta probably releases
factors into the circulation, which then trigger mater-
nal endothelial dysfunction.
Early in normal gestation, cytotrophoblast cells
invade the decidua and myometrium. ese cells also
invade endovascularly, replacing rst the endothelium
and then the media of the spiral arteries. is cre-
ates a system of accid, low resistance, large diameter,
unresponsive arterioles, that increase placental perfu-
sion. e outcome is an increment in blood ow to the
fetus and lack of adrenergic vasomotor control. e
endothelial lining is replaced by the cytotrophoblast
cells, which adapt to mimick an endothelial pattern of
adhesion molecule expression.
In PET, this vascular phenotype is not expressed
and the pattern of invasion is much more super-
cial. ere is a restriction of trophoblast invasion
into the spiral arteries, particularly those within the
myometrium. ese decidual vessels may later show
atherosis, and superimposed thrombosis augments
hypoperfusion. It seems plausible that, consequent to
these changes, placental hypoperfusion causes a state
of relative hypoxia.
Various factors have been postulated as the sub-
stance produced from the placenta that aects blood
ow, arterial pressure, and maternal endothelial cell
activation (ECA). ese factors include oxidative
stress, cytokines such as tumor necrosis factor ␣(TNF-
a) and IL-6, insulin-like growth factors, nitric oxide
(NO), heparin-binding endothelial growth factor-like
growth factor, endothelin-1, arachidonic acid metabo-
lites, angiotensin II type-1 receptor autoantibody
(AT1-AA), and angiogenic factors.
Recently, the focus has been on angiogenic fac-
tors. It has been proposed that PET could be related
to an imbalance between proangiogenic (as vascular
endothelial growth factor, VEGF) and antiangiogenic
factors as Fms-like tyrosine kinase (sFlt-1) and soluble
endoglin (sEng).
sFlt-1 is an endogenous inhibitor of both VEGF
and PGF and may regulate placental angiogenesis by
preventing the interaction between circulating VEGF
and PGF with their proangiogenic receptors. Level
of sFlt-1 in the plasma of women with PET are ele-
vated compared with normal pregnancies. When sFlt-
1 is exogenously administered via adenovirus medi-
ated gene transfer in pregnant rats and mice, there
are increases in arterial blood pressure and protein-
uria,aswellasdecreasedlevelsofVEGFandPIGF,
similar to these observed in PET.3,4 Another observa-
tion was that VEGF infusion attenuates the increased
blood pressure and renal dysfunction observed in
pregnant rats overexpressies sFlt-1.5Also uteroplacen-
tal ischemia has been shown to increase plasma and
placental sFlt-1, and to decrease the levels of VEGF and
PIGF in late gestation of rats and baboons.6Endoglin
(Eng) is a component of the transforming growth
factor (TGF)-beta receptor complex and a hypoxia
inducible protein, and is related to cellular prolifera-
tion and NO signaling. Soluble Eng (sEng) has been
shown to act as an anti-angiogenic factor, possibly via
the inhibition of TGF-beta of binding to cell surface
receptors.7Recent works showed that sEng inhibits in
vitro endothelial cell tube formation and that aden-
ovirus mediated increase of both s-t-1 and sEng in
pregnant rats resulted in IUGR and in a syndrome
resembling PET.8Recently, sEng levels have been pro-
posed as a predictor of PET.9
Whichever factor provokes maternal ECA, when
the latter is established, it leads to upregulation
of a number of inammatory molecules, including
adhesion molecules. ese procedures change the
endothelium phenotype from antithrombotic to pro-
thrombotic, with a decrease in the formation of the
vasodilator and antiplatelet agents prostacyclin and
nitric oxide, the production of endothelin, and nally
the downregulation of anticoagulant systems.
Redman’sresearchgroupinOxfordproposedthat
the endothelial dysfunction seen in PET is part of
a wider inammatory response and that placental
hypoperfusion is not necessarily the sole primary 205
Section 6. Microangiopathies
Reduced perfusion of
the
p
lacenta
Why? Unknown. Proposed factors:
Hypoxia, ischemia, oxidative stress, altered
NK cell signaling, syncytial debris, altered
hemeoxygenase expression, etc.
Oxidative stress, cytokines (TNF-a, IL-6), insulin-like growth factors, NO, heparin-binding endothelial growth factor-
like growth factor, endothelin-1, arachidonic acid metabolites, angiotensin II type-1 receptor autoantibody (AT1-AA)
and an
g
io
g
enic factors
(
sFlt-1, sEn
g)
↑ Blood flow and arterial pressure
Maternal endothelial cell activation (ECA)
Endothelin, reactive oxygen species (ROS), thromboxane, 10-HETE, ↑ on vascular sensitivity to
angiotensin II, ↓vasolidators (as nitric acid (NO) and prostacyclin)
Generalized dysfunction of the maternal vascular endothelium +Maternal constitutional factors
First stage: reduced placental perfusion, abnormal implantation/vascular remodeling
Second stage: maternal syndrome
Fig. 17.1 Summary of current concepts on the pathogenesis of PET. The two stages model of PET.
event. ey argue that pregnancy normally elicits an
inammatory response.10 isisevidencedbychanges
in granulocytes and monocytes such as increased
intracellular production of reactive oxygen species
and upregulation of surface molecules such as CD11b
and CD64, as well as release of L-selectin, which is
related to granulocyte activation. During PET there
is increased activation of platelets, neutrophils, and
monocytes and an increase in the release of micropar-
ticles when compared with normal pregnancy. Per-
haps these inammatory changes are a response to
the presence of fetal (or paternal) antigens. If so, then
abnormalities of the normal immunomodulation seen
at the feto-placental interface could act to trigger PET.
HLA-G is important in the prevention of recognition
of the placenta as “non-self” and there is a reduction
of expression of HLA-G in PET along with abnormal
responsiveness of maternal lymphocytes towards fetal
cells.
Microparticles are fragments of cell membranes
released into the circulation as a result of cellular acti-
vation or apoptosis and can have a procoagulant eect.
Microparticles in pregnancy are derived from a num-
ber of cells, but the predominant population is platelet
derived. Vesicles prepared from syncytiotrophoblast
microvillous membranes (STBM) have been shown to
suppress the proliferation of endothelial cells in vitro.
ey also aected an in vitro model of endothelial cell-
dependent arterial relaxation. e numbers of STBM
detected in the circulation of pre-eclamptic women
have been shown to be signicantly elevated compared
with those with normal pregnancies.
A summary of the currently favored pathogenesis
of PET is shown in Fig. 17.1.
206

Chapter 17. Pre-eclampsia
Relation between PET and IUGR/ fetal
growth restriction (FGR)
e consequences of placental dysfunction can be
twofold – intra-uterine growth restriction and the
maternal symptoms and signs of PET. What is not
understood is why some women only have FGR, while
others have both FGR and PET. It has been sug-
gested that the maternal syndrome of PET may only
occur in women with “constitutional factors” (genet-
ics, environmental, dietary, behavior, etc.) that render
the mother sensitive to the eects of reduced placental
perfusion.
Constitutional factors that have been proposed to
actastheinductorsofthematernalsyndromeof
PET, include several dietary factors, metabolic condi-
tions such as diabetes, insulin resistance and uric acid,
low melatonin levels, obesity, metabolic syndrome,
folic acid and hyperhomocysteinemia, hyperlipidemia
with elevated triglycerides, free fatty acids and LDL
cholesterol and reduced HDL, maternal vitamin D
deciency, and thrombophilia. e factors that cause
ECA may contribute to the development or severity
of PET.
Relation of PET with later cardiovascular risk
in women and their babies
Despite PET and FGR occurring only in pregnancy,
they have been shown to have long-term conse-
quences. Mothers, who have had PET or have deliv-
ered a baby with FGR, experience a 2–8-fold increased
risk of atherosclerotic cardiovascular disease (CAD)
in later life.11 It is unclear whether PET causes CAD
or whether these two entities share the same causal
origin. It has also been shown that the earlier PET
presents in pregnancy, the more severe the maternal
CAD is. Women with PET before 37 weeks of gesta-
tion had eight times more cardiovascular deaths than
woman with normal pregnancy 14 years later.
ere is a large body of epidemiological studies
showing that the long-term consequences of FGR in
thebabylastwellintoadulthood.eseindividu-
als have a predisposition to develop a metabolic syn-
drome later in life, manifesting as obesity, hyper-
tension, hypercholesterolemia, cardiovascular disease,
and type 2 diabetes, in agreement with the theory
of early origin of CAD, also known as “the Barker
hypothesis.”12
A recent study showed positive associations
between maternal pre-pregnancy levels of trigly-
cerides, cholesterol, low-density lipoprotein, and
baseline systolic blood pressure and subsequent
development of PET.13 e authors concluded that
thepresenceofcardiovascularriskfactorspriorto
pregnancy, are predisposing to PET. e prevalence
of chronic hypertension is signicantly higher among
women with a history of PET (46.7%) as well as those
with previous IUGR (8.9%).14 Women w it h PET and
FGR with chronic hypertension on follow-up had
increased carotid intimal-media thickness, suggesting
a predisposition to atherosclerosis. Women with
previous PET have signicantly higher fasting glucose
levels, waist circumference, body mass index, and
higher prevalence of metabolic syndrome compared
to normal women
New modications of current theories
on the pathogenesis of PET
As long as the initial causative factor for PET remains
unrecognized, dierent theories continue to be gener-
ated, some of them challenging the currently accepted
origins of PET.
It has been suggested that early (before 34 weeks)
and late onset (aer 34 weeks) PET are two dier-
ent clinical entities with dierent pathogenesis, ori-
gins, etiology, severity, and clinical expression. Cer-
tainly, FGR is more strongly associated with severe
rather than with milder pregnancy-induced hyperten-
sion.15 According to this theory, early PET is associated
with reduced perfusion but PET at term may not, sug-
gesting dierent genetic origins for early and late PET.
Huppetz, in a recent paper, has challenged the
placental origins of PET and proposed that PET is
a syndrome of early placental formation.16 He sug-
gested that an insult results in aberrant development
and dierentiation of the villous syncitiotrophoblast
causing impaired maintenance of the placental bar-
rier. is subsequently leads to the release of necrotic
and aponecrotic fragments culminating in a systemic
inammatory response of the mother. According to
this theory FGR is due, in contrast, to a failure of
extravillous trophoblast invasion. is new concept
clearly separates the origins of PET and FGR, and pro-
poses alterations in dierent trophoblast dierentia-
tion pathways as origins of both syndromes.
Genome-wide expression analysis in rodents
showed that spontaneous dierentiation of 207

Section 6. Microangiopathies
trophoblast stem cells is associated with the acqui-
sition of an endothelial-cell like thromboregulatory
gene expression program.17 is program is develop-
mentally regulated and conserved between mice and
humans. ey further showed that trophoblast cells
sense, via the expression of protease activated recep-
tors, the presence of activated coagulation factors.
Engagement of these receptors results in cell-type
specic changes. ese observations dene candidate
fetal genes that are potential risk modiers of PET
and suggest that hemostasis can aect trophoblast
physiology and thus aect placental function in the
absence of frank thrombosis. It is postulated that PET
is not only due to a maternal cause, but also that fetal
genes could contribute to the development of the
disease.
Thrombophilia and PET
Acquired thrombophilia
Mothers with antiphospholipid antibodies have a pre-
disposition to PET and FGR. Indeed, the develop-
ment of these conditions before 34 weeks in a woman
with antiphospholipid antibodies has now become
dening criteria for obstetric antiphospholipid syn-
drome. is is discussed in more depth in Chapter 11.
Other acquired conditions that predispose to throm-
bosis such as myeloproliferative disease would also be
expected to predispose to PET (see Chapter 19).
Genetic thrombophilias
An association between PET and inherited throm-
bophilias was rst reported by Dekker et al. in 1995,
who proposed that maternal thrombophilia could act
as a genetic constitutional factor for the development
of PET.18 Since then, a large number of retrospective
and case-controlled studies have examined the associ-
ation between dierent types of thrombophilic muta-
tions and PET. e results of published reports have
been inconsistent. Meta-analysis of all case-control
studies suggests that only FVL mutation is associ-
ated with a minor increased risk of PET (odds ratio,
l.18; 95% condence interval, 1.14 to 2.87). Over-
all, studies suggest that women with genetic throm-
bophilia have more severe PET than those with-
out, but thrombophilia itself does not precipitate the
condition.
Prediction of PET
As the exact causative factor that provokes PET is not
yetknown,atpresentthereisnoclearstrategyforits
prevention and so the clinical and research focus has
been on early detection and prediction.
Hyperuricemia is an established marker of severe
PET, correlating with the histological severity of renal
lesions, and clinically with adverse fetal outcomes, but
has a low negative predictive value.
Uterine artery Doppler screening between 20 and
24 weeks identies mothers at high risk for developing
adverse pregnancy outcomes. e correlation between
elevated uterine artery resistance and a high risk of
PETand/orFGRwasrstdemonstratedattheendof
second trimester, probably reecting the ongoing pro-
cess of trophoblast invasion into the spinal arteries.
Bilateral notching at 20–24 weeks identies the preg-
nancies that will have FGR and PET, although there is
ahighfalsepositiverate.
19
An algorithm of placental and endothelial mark-
ers between 20 and 24 weeks’ gestation was developed
and showed good prediction of the later development
of PET.20 isstudyproposedsixmarkersaspoten-
tial predictive indicators: HDL cholesterol, PAI-1/PAI-
2 ratio, leptin, and PIGF. At 20 weeks’ of gestation, an
algorithmofthesemarkersdistinguishedPETfrom
the low risk group. At 24 weeks’ of gestation the posi-
tive predictive value was even better. Increased levels of
soluble fms-like tyrosine kinase 1 (sFlt-1) and reduced
levels of soluble placental growth factor (PIGF) have
been shown to predict the subsequent development
of PET, as early as 5 weeks before the onset of PET.
Human cancer patients treated with anti-VEGF anti-
body developed hypertension and proteinuria.
In association with increased levels of sFlt-1,
symptoms were dramatically worse, and typical of
HELLP syndrome, leading the authors to postulate
that increased levels of sFlt-1 were responsible for PET,
but the combination of increased sFlt-1 and sEng led
to HELLP syndrome. In a longitudinal analysis, the
rise in soluble endoglin concentrations occurred ear-
lier and was more marked in pregnancies with subse-
quent pre-eclampsia.
Soluble endoglin (sEng) is a co-receptor for
transforming growth factor 1and3, expressed
on trophoblasts. Its levels are increased in pre-
eclampsia15 and in pregnant rats this has been
associated with increased vascular permeability and
hypertension. Other serum markers that have been
208

Chapter 17. Pre-eclampsia
proposed to predict PET as early as the rst trimester,
are placental protein 13 (PP13), placenta associated
plasma protein A (PAPP-A), and long pentraxin 3
(PTX3).Allofthosemarkersstillneedfurtherevalu-
ation in larger multicenter trials.
Management of pre-eclampsia
At present, the sole eective therapy for pre-eclampsia
is delivery and removal of the placenta. Symptoms usu-
ally improve within days. erefore, early diagnosis
and timely delivery are imperative for maternal and
peri-natal survival.
Prevention of PET
Several drugs have been tried for the prevention of
PET. Despite the rst promising publications, it has
been shown later that there is a lack of evidence for cal-
cium, vitamin C, and E in PET’s prevention. e main
drugs that are used for the prevention of PET are anti-
hypertensives and antithrombotics.
Prevention of PET with antihypertensives
Antihypertensive drugs are used for secondary pre-
ventionofPET,inwomenwithmildtomoderate
hypertension developing or pre-existing to pregnancy.
Data from several studies showed that, although there
was a reduction in hypertension, it was unlikely that
this had a major impact on the progression to PET.
Furthermore, it has been argued that the impact to
the fetus of lowering maternal blood pressure could
provoke FGR. Although there is no big randomized
trial, beta-blockers are more likely to have such an
impact (eight trials, 810 women; relative risk 1.56,
1.10 to 2.22). e antihypertensive drug methyldopa
has oen been used in gestational hypertension. Side
eects include depression and drowsiness. Other drugs
that can be used are labetalol and calcium channel
blockers.Atenololisrelativelycontraindicatedinpreg-
nancy due to possible FGR; absolutely contraindi-
cated are angiotensin converting enzyme inhibitors
and angiotensin receptor antagonists due to possible
teratogenicity. Diuretics should be avoided in general,
andshouldbekeptonlyforspecialindicationssuchas
renal or cardiac diseases.
Prevention of PET with antithrombotics
Antiplatelet agents
e Collaborative low-dose Aspirin Study in Preg-
nancy (CLASP study), was a randomized trial of low
dose aspirin for the prevention and treatment of PET
among 9364 pregnant women.21 e women were
randomly assigned 60 mg aspirin daily or matching
placebo. To simulate real obstetric practice, the entry
criteria were broad and embraced women thought to
beatriskofPETandFGRfrom12to32weeks’ges-
tation. Primiparous women, women with pre-existing
hypertension or a history of FGR, PET, or stillbirth
and women with established PET could all be entered
in the study: 74% were entered for prophylaxis of
PET, 12% for prophylaxis of FGR, 12% for treatment
of PET, and 3% for treatment of FGR. Overall, the
use of aspirin was associated with a reduction of only
12% in the incidence of proteinuric PET, which was
not signicant. Nor was there any signicant eect on
the incidence of IUGR or of stillbirth and neonatal
death. Aspirin did, however, signicantly reduce the
likelihood of preterm delivery (7% aspirin vs. 2% con-
trol); absolute reduction of 5 per 100 women treated.
ere was a signicant trend towards progressively
greater reductions in proteinuric pre-eclampsia, the
more preterm the delivery. Aspirin was not associated
with a signicant increase in placental hemorrhage or
in bleeding during preparation for epidural anesthe-
sia, but there was a slight increase in use of blood
transfusion aer delivery. Low dose aspirin appeared
safe for the fetus and newborn infant, with no evi-
dence of an increased likelihood of bleeding. e rate
of stillbirth, neonatal death, or fetal growth retarda-
tion occurring before 32 weeks was 5.3% in the aspirin
groupascomparedwith10.6%intheplacebogroup.
ese ndings do not support routine prophylactic or
therapeutic administration of aspirin in pregnancy to
all women at increased risk of pre-eclampsia or IUGR.
Low dose aspirin may be justied in women judged to
be especially liable to early-onset PET severe enough
to need very preterm delivery. In such women it seems
appropriate to start low dose aspirin prophylactically
early in the second trimester.
e Cochrane Library Update summarizing data
from 37 560 women for 59 trials of aspirin to pre-
vent PET showed that the use of aspirin is associ-
ated with a 17% reduction in the risk of pre-eclampsia
(46 trials, 32 891 women, relative risk (RR) 0.83, 95%
condence interval (CI) 0.77 to 0.89), an 8% reduc-
tion in the relative risk of preterm birth (29 trials,
31 151 women, RR 0.92, 95% CI 0.88 to 0.97); NNT
72 (52 119)), and a 14% reduction in fetal or neonatal
deaths (40 trials, 33 098 women, RR 0.86, 95% CI 0.76
to 0.98); NNT 243 (131, 1 666) and a 10% reduction in 209

Section 6. Microangiopathies
small-for-gestational age babies (36 trials, 23 638
women, RR 0.90, 95% CI0.83 to 0.98). e authors con-
cluded that antiplatelet agents, largely low dose aspirin,
have moderate benets when used for prevention of PE
and its consequences.22
e Perinatal Antiplatelet Review of Interna-
tional Studies (PARIS) Collaborative Group published
a meta-analysis, included 31 randomized trials of
PET primary prevention enrolling a total of 32. 217
women and their 32.819 infants.23 According to their
results antiplatelet agents, particularly aspirin, moder-
ately reduce the relative risk for PET, preterm births
before 34 weeks’ gestation, and serious adverse preg-
nancy outcomes. For women randomized to receive
antiplatelet agents, the relative risk of developing PET,
comparedwithwomenincontrolgroups,was0.90
(95% condence interval (CI) 084–0.97). e risk of
delivering before 34 weeks’ gestation was 0.90 (95% CI,
0.83–0.98) and of having a pregnancy with a serious
adverse outcome was 0.90 (95% CI, 0.85–0.96). Use of
antiplatelet agents was not associated with any signi-
cant eect on the risk for death of the fetus or new-
born,riskofhavinganinfantbornsmallforgestational
age, or risk for bleeding events for either the women or
their babies. No subgroups of women who were sub-
stantially more or less likely to benet from antiplatelet
agents than any other were identied.23 Despite these
two large meta-analyses, further studies are required
to assess which women are most likely to benet, when
treatment is best started, and at what dose.
Heparin and antithrombin concentrates
Heparin as monotherapy or in combination with
aspirin has also been suggested for the prevention of
PET in women with high risk pregnancies, but data are
not yet sucient for a nal conclusion. For example,
a recent study investigated the eect of low molecular
weightheparin(LMWH)onpregnancyoutcome,on
the maternal blood pressure values, and on uteropla-
cental ow in angiotensin-converting enzyme (ACE)
non-thrombophilic women, with insertion/deletion
(I/D) polymorphism, with history of PET.24 e study
included 80 women, 41 treated with dalteparin 5000
IU/day, and 39 untreated (control group). is study
suggests that LMWH may reduce the recurrence of
PET, of negative outcomes, and the resistance of utero-
placental ow, and also prevents maternal blood pres-
sure increase in ACE DD homozygote women with a
previous history of PET.
Antithrombin (AT) levels are reduced in PET. Pre-
vious randomized controlled trials of AT therapy in
PET between 24–35 weeks’ gestation have shown sig-
nicantly improved maternal symptoms and birth
weight.25 A further trial examined AT therapy in
severe PET in women presenting before 32 weeks’ ges-
tation.42patientswereenrolledandeachreceived
3000 IU per day for 7 days compared to albumin
582 mg/day for 7 days. An equal number of women
discontinued the intervention in the AT and placebo
(albumin) groups. AT treatment improved or at least
preserved fetal biophysical status. It prolonged the
pregnancy to reach 34 weeks and fetal growth rate
was preserved. However, AT treatment of PET is still
largely conned to research settings.
Planning for the optimal timing of delivery
One can justify PET, of any severity, presenting aer
34 weeks as an indication for delivery. If earlier than
34 weeks, the balance of expectant management is set
against risk to the mother, but potentially benets the
child in terms of risks of prematurity. Generally, hemo-
dynamic instability, fetal distress, and rapid disease
progression are indications for delivery. ere is no
evidence base to support these decisions, as only small
trials of expectant management prior to 34 weeks vs.
delivery have been carried out.
If an induced pre-term delivery is contemplated,
it may be necessary to give prostaglandins to ripen
the cervix. Steroid therapy to improve fetal lung matu-
rity should also be considered, in discussion with the
pediatric team. In general, a vaginal delivery is consid-
ered safer than Cesarean section for those with com-
plications of PET. For both forms of delivery, a platelet
count of greater than 50 ×109/l is recommended, and
platelet transfusions may be necessary to achieve this.
Regional anesthesia is also generally preferred, but
depends on the platelet count, and guidelines recom-
mend a count of greater than 80 ×109/L, in the set-
ting of normal platelet function. Coagulation param-
eters should also be checked prior to delivery because
oftheriskofDICinPET.
It should be emphasized that the disease does not
abate immediately post-delivery and that seizures can
occur up to a week later. Hence, seizure prophylaxis,
anti-hypertensive therapy, and frequent monitoring
should be continued for an appropriate period, e.g. 12–
48 hours for seizure prophylaxis and close monitoring,
210

Chapter 17. Pre-eclampsia
up to 12–16 weeks or indenitely for anti-hypertensive
therapy.
Other pharmaceutical management of PET
Anti-hypertensive drugs for the management of PET
e most used anti-hypertensive drugs in the manage-
ment of PET are methyldopa, labetalol, and nifedip-
ine.25,26 Labetalol is quite safe and eective, decreas-
ing heart rate and having fewer side eects than
other drugs (lack of reex tachycardia, hypotension, or
increased intracranial pressure).25 Best avoided drugs
are high dose diazoxide, due to increased risk for
hypotension and Cesarean section, and the serotonin
receptor antagonist kentaserin.25
In general, angiotensin converting enzyme (ACE)
inhibitors, angiotensin receptor-blocking drugs
(ARB), and diuretics should be avoided. Nifedipine
should be given orally and not sublingually.
Concern has been about hydralazine as rst-line
treatment (due to the potential unpredictable hypoten-
sion) and the combination of nifedipine and magne-
sium sulfate.
Magnesium sulfate
Magnesiumsulfateisthedrugofchoiceforthepreven-
tion and treatment of pre-eclampsia. e epidemio-
logical and basic science evidence suggesting that mag-
nesium sulphate when given to early pregnancy in
womenconsideredatriskofpretermbirthmaybeneu-
roprotective for the fetus, has now being conrmed
by a recent Cochrane systematic review.26 It acts by
causing cerebral vasodilation, thereby reversing the
ischemia produced by cerebral vasospasm during an
eclamptic episode. Data suggest that women receiving
magnesium sulfate therapy have a 58% lower risk of
eclampsia than placebo and that also reduces the risk
for maternal death.26 Apossiblesideeectisushing,
which occurs in one-quarter of women.
Suggesting guidelines for the management
of established PET
rClose in or outpatient monitoring of vital signs,
deep tendon reexes, neurological examination.
rBed rest and relaxation.
rFetal monitoring: external fetal monitor,
oxcytocin challenge test, biophysical prole.
rGive steroids to accelerate fetal lung maturation
when ⬍34 weeks of gestation; betamethasone
12 mg IM/day for 2 doses, or dexamethasone 6 mg
IM/ 12 hours ×4 doses.
rCareful uid restriction to reduce the risk of uid
overload. Total uid intake should be limited to
80 mL/h (max 150 mL/h), or 1 mL/kg/h, urine
output can be tolerated as low as 10 mL/h.
rGive supplemental oxygen.
rMaintain diastolic blood pressure ⬍110 mmHg/
and systolic ⬍160 mmHg with anti-hypertensive
drugs.
rGive prophylactic intravenous magnesium sulfate
for the prevention of eclampsia during labor and
the postpartum.
rLaboratory monitoring; complete blood count,
platelets count; coagulation studies in severe PET
(PT, PTT, brinogen, FDP) urea, ser um
creatinine, uric acid, serum electrolytes, liver
functional tests, lactate dehydrogenase.
Suggesting guidelines for the management
of eclampsia
rClose monitoring.
rGive oxygen.
rFluid restriction is advisable to reduce the risk of
uid overload. Total uid should be limited to
80 mL/h, or 1 mL/kg/h.
rGive magnesium sulfate. Alternative drugs
include diazepam, phenytoin.
rGive steroids if ⬍34 weeks’ gestation.
rUrgent delivery.
Hematological complications of PET
All the changes taking place during PET due to
endothelial cell activation can produce hematological
complications.
Frequent (at least every 8 hours) full blood count
and coagulation screen should be performed in case of
severe PET, or where there is suspicion of subsequent
development of hematological complications.
Thrombocytopenia
e most common hematological complication of
PET is thrombocytopenia, occurring in 18% of pre-
eclamptic women. is is probably due to platelet
and endothelial activation generating thrombin and 211

Section 6. Microangiopathies
causing platelet consumption. In general, the severity
of thrombocytopenia is related to the severity of PET.
If the platelet count is greater than 40 000 ×109/L,
the risk of bleeding is small. In the majority of cases
thrombocytopenia resolves aer delivery, but rarely
maycontinuetofallaerbirth.Severethrombocy-
topenia persisting aer delivery could be a possible
indicator of developing microangiopathic hemolytic
anemia.
Management of thrombocytopenia in PET
Platelet counts of ⬎50 ×109/L in patients with other-
wise normal coagulation are regarded as safe for nor-
mal vaginal delivery and Cesarean section. Concerns
over the risk of hematoma formation and neurologi-
cal damage have led to the use of regional anesthesia
not being recommended unless the platelet count is
⬍75×109/L with a normal coagulation screen. is
recommendation is based on consensus rather than on
evidence.
If platelet count ⬍50 ×109/L and there is no bleed-
ing, then no treatment is necessary unless there is
active bleeding, when it is appropriate to transfuse
platelets.
Disseminated intravascular
coagulation (DIC)
DIC is a clinicopathological syndrome characterized
by a systemic activation of coagulation leading to
microvascular deposition of brin, and thus to con-
sumption of coagulation factors, platelets and physio-
logical anticoagulants. is produces a reduction in
platelet count, a fall in brinogen, and a prolongation
of the activated partial thromboplastin time (APTT)
and international normalized ratio (INR).
Prolongation of PT and APTT with severe throm-
bocytopenia and low brinogen levels (⬍1.0 g/L) are
signs of a developing DIC-like state and hence fre-
quent estimation of platelet count, brinogen (using
Clauss method), prothrombin time (PT), and APTT
is strongly recommended. Laboratory evidence of a
consumptive coagulopathy should be sought before
microvascular bleeding becomes evident, so that
appropriate and aggressive action can be taken to
address the underlying cause.
DIC occurs in about 10%–12% of all cases of PET
andin7%ofseverePET.eetiologyofDICin
pre-eclampsia is not well understood, but is probably
a consequence of endothelial cell activation. In only
10%–15% of DIC cases in PET, it can become more
systematic and even lethal. In PET there is a low grade
brin deposition in the renal and placental microcir-
culation.
DIC in obstetric patients could be a complication
of other obstetric conditions or of none related directly
with pregnancy. e most common causes of DIC in
obstetrics, besides PET, are abruption placentae and
amniotic-uid embolism (occurring in more than 50%
of obstetric cases), and retained dead fetus, sepsis, and
septic abortion.
Management of DIC
Management of DIC involves (1) treating the cause and
(2) replacement of missing hemostatic components
with blood products. Rarely, chronic DIC requires low
dose anticoagulation to “switch o” the stimulus to
DIC.
Hematological treatment consists of platelets, FFP,
and cryoprecipitate (see Table 17.3, Chapter 13c),
but avoiding circulatory overload. Novel therapeutic
strategies are based on current insights into the patho-
genesis of DIC, and include anticoagulant strategies
(e.g. directed at switching o coagulation stimulus)
and strategies to restore physiological anticoagulant
pathways (such as activated protein C concentrate).
ese have not been evaluated adequately in the man-
agement of DIC in pregnancy and postpartum.
HELLP syndrome
Denition
HELLP syndrome (hemolysis, elevated liver enzymes,
low platelets) occurs in the second and third trimester
of pregnancy and presents occasionally postpartum.
ere are no clear denition criteria for HELLP.
Epidemiology of HELLP
is disorder complicates between 0.5% and 1% of
pregnancies and is associated with a maternal morbid-
ity ranging between 1% and 4%. HELLP syndrome is
reported in PET with an incidence ranging between
2% and 50% (5% and 15%), depending on the popu-
lation studied and the diagnostic criteria used: 70%
of cases occur ante-natally and 30% occur within the
rst 48 hours’ to 7 days’ postpartum. 20% of women
who develop HELLP post-labor had no evidence
of PET before delivery. e incidence of HELLP is
212

Chapter 17. Pre-eclampsia
Table 17.4 Differential diagnosis of HELLP syndrome
Acute fatty liver of pregnancy
Gal bladder disease
Gastroenteritis
Appendicitis
Diabetes insipidus
Hemolytic uremic syndrome
Thrombotic thrombocytopenic purpura
Idiopathic thrombocytopenic purpura
Acute renal failure
Pyelonephritis
Glomerulonephritis
Peptic ulcer
Flair of systemic lupus erythematosus
Viral hepatitis
signicantly increased among white middle-class and
older multiparous women. DIC is founded in approxi-
mately 20%–30% of women with HELLP. Recurrence
rates in subsequent pregnancies is 3% for HELLP,
10%–14% for IUGR and 18%–20% for PET.
Clinical presentation of HELLP
e clinical presentation is with fatigue and malaise
for a few days, followed by nausea, vomiting, shoul-
der, neck, epigastric or right upper quadrant pain,
headache, and visual disturbances. Right upper-
quadrant or epigastric pain is thought to be due to
obstruction of blood ow in the hepatic sinusoids,
which are blocked by intravascular brin deposits.
Usually, the patients present with signicant weight
gain, due to the associated generalized edema, and
with proteinuria greater than 1+(in 90% of cases).
Severe hypertension is not a constant or a frequent
nding in HELLP syndrome. at is why it can usu-
ally be misdiagnosed as having another disease (listed
in Table 17.4).
Pathophysiology of HELLP syndrome
e pathophysiology is not clear, but it is helpful to
consider that it represents PET conned to the liver,
which may result in necrosis of areas of the liver.
According to one theory, pre-eclamptic patients are
already prone to spontaneous hemorrhages. e liver
is thought to be particularly prone because brin split
products can deposit in the reticuloendothelial sys-
tem of the liver. Multiple previous subclinical sponta-
neous hemorrhages within the small hepatic sinusoids
and arterioles may go unnoticed symptomatically and
leave the liver in a fragile state. Fibrin thrombi may be
le uncleared in the liver. Occasionally, a trigger (such
as DIC) may cause extreme hypoperfusion of the liver,
leading to infarction.
As the liver is the primary site of plasma protein
production and pregnancy is a hypermetabolic con-
dition, a specic plasma protein prole was noted in
women with HELLP syndrome compared with nor-
mal control cases. e primary candidate identied
was serum amyloid A (SAA), which was signicantly
dierent between the HELLP cases and controls. How-
ever, further work is needed to determine if this is truly
a predictive marker for the development of HELLP or
merely a surrogate of liver impairment.
Complications of HELLP
Possible complications of HELLP syndrome include
subcapsular hematoma of the liver, liver rupture,
excessive bleeding, DIC, pulmonary edema, acute
renal failure, abruptio placentae, peri-natal asphyxia,
fetal death, and maternal death.
Diagnosis of HELLP syndrome
ediagnosisismadebythendingsoffragmenta-
tion on the blood lm, low platelets and abnormal liver
function tests, and with abdominal ultrasound. e
patient may or may not have signs of PET.
Management of HELLP syndrome
Stabilization of hypertension, if present, and other
manifestations of HELLP, such as seizures or DIC are
required as well as fetal monitoring. e only cer-
tain therapeutic measure is prompt delivery, and in
the majority of cases women have complete recovery
within 24–48 hours aer labor, although some women
may continue to have symptoms for up to 14 days.
In the majority of patients, normalization of platelet
count and resolution of HELLP occurs 5 days post-
partum.Ifthesesignsofdiseasepersistbeyond5
days postpartum (and indeed if they don’t begin to
improvewithin48hoursofdelivery),thediagnosis
of HELLP should be reconsidered. Ideally, all women
with HELLP should be referred to a tertiary hos-
pital. Anti-hypertensive drugs, steroids, and plasma
exchange/plasmapheresis have also been used with
variable results.
A Cochrane review summarized the evidence on
the eects of corticosteroids on maternal and neonatal 213

Section 6. Microangiopathies
mortality and morbidity in women with HELLP syn-
drome.28 From the ve studies reviewed (n=170),
threewereconductedantepartumandtwopostpar-
tum. Four of the studies randomized participants to
standard therapy, or to the administration of dexa-
methasone. One study compared dexamethasone with
betamethasone. e conclusions were that there is
insucient evidence to determine whether steroid
use in HELLP decreases the major maternal and
peri-natal morbidity and the maternal and peri-natal
mortality.
Platelet transfusions and HELLP syndrome
A randomized trial of women with class 1 HELLP
syndrome received either dexamethasone (n=26) or
dexamethasone and platelet transfusions (n=20).
Liver function tests were signicantly higher in the
steroid plus platelets group. Platelet count normal-
ized signicantly faster in the dexamethasone only
group, and the postpartum stay was more prolonged
in the dexamethasone and platelet group. e group
that received platelets reported complications such as
wound dehiscence, wound infection and pulmonary
edema.11 Apreviousreportofintrapartumuseof
platelets when platelet count was ⬍40 ×109/L did
not nd a signicantly lower incidence of hemorrhagic
complications. As a result, platelet transfusion is not
oen used in the management of HELLP.
Massive bleeding secondary to
placental abruption
Placental abruption is dened as the premature sep-
aration of a normally located placenta. Patients with
defective placentation and abnormal placental vascu-
lature, such as in PET, are predisposed to ischemia
and rupture of these placental vessels, which is thought
to lead to placental abruption. Other risk factors
include smoking and cocaine use. Presenting fea-
tures include mild vaginal bleeding, signs of hypo-
volemia, fetal compromise, uterine contractions or
hypertonicity, DIC, and renal failure. Ultrasonogra-
phy may be useful to conrm the position of the pla-
centa, or the presence of a large hemorrhage, but is
insensitive.
e management of placental abruption, whether
expectant or with delivery depends on the extent of the
abruption, the gestational age of the fetus, and the pres-
ence of fetal or maternal compromise. A full review is
beyond the scope of this chapter and is covered in other
sources. In general terms, however, delivery may be
vaginal (usually due to the stimulation of rapid labor in
response to the abruption), or by Cesarean section. e
latter scenario may occur in the case of failed progres-
sionoflabororinmaternalorfetalinstability.Expec-
tant management with or without the use of tocolytics
may be possible if the presentation of bleeding is less
acute and earlier in the pregnancy.
DIC oen occurs in association with abruption,
particularly with a complete abruption, and may fol-
low within hours. e specic management of DIC has
already been mentioned. e hemostatic management
of massive bleeding is presented in Chapter 13c.
e maternal complications of placental abruption
include massive hemorrhage, DIC, renal failure, and
amniotic uid embolism. Fetal complications relate
primarily to premature delivery, i.e. stillbirth (adjusted
relative risk of 8.9), growth restriction (adjusted rela-
tive risk of 2.0), and complications of prematurity.
Dierential diagnosis of PET and HELLP
by microangiopathic hemolytic
anemias (MAHA)
e dierential diagnosis of thrombotic thrombocy-
topenicpurpura(TTP)andhemolyticuremicsyn-
drome (HUS) from PET and HELLP may be dicult
(see Chapter 18). TTP is diagnosed during pregnancy
or postpartum, with 75% of episodes occurring around
the time of delivery.
Postpartum HUS is a rare syndrome of unknown
cause, not related to E. coli (D-). e prognosis is poor
for both the mother and the fetus. It is recognized that
HUS recurs in subsequent pregnancies, although the
reasonforthatisnotknown.Manypregnantwomen
who survive aer HUS develop chronic hypertension
and chronic renal failure later in life. Plasma exchange
(PE) has low response rates.
Acute fatty liver of pregnancy (AFLP)
HELLP syndrome should be distinguished from AFLP,
a rare condition, also associated also thrombocytope-
nia, but without microangiopathic hemolytic anemia.
Clinical presentation is similar with HELLP, occurring
almost always in the third trimester. DIC accompanies
AFLP in 90% of cases. Maternal mortality is approxi-
mately 15% and fetal mortality ⬍5%.
214

Chapter 17. Pre-eclampsia
Summary
rPre-eclampsia (PET), the new onset of
hypertension aer 20 weeks’ of gestation and
proteinuria, resolving aer delivery, aects
approximately 2%–14% of all pregnancies and
remains a major cause of maternal and fetal
morbidity and mortality worldwide.
rPlacental dysfunction is considered to be the
central feature in the development of PET.
rCurrent hypothesis is that PET is a two-stage
disease: the rst stage is reduced placental
perfusion and the second stage is the maternal
response to this with endothelial cell activation.
rProposed placental factors produced from the
placenta that aect blood ow, arterial
pressure and maternal endothelial cell activation
(ECA) include oxidative stress, cytokines
(TNF-a,IL-6), and angiogenic factors (VEGF,
s-FLT-1, sEng).
rMaternal constitutional factors that have been
proposed to act as inductors of the maternal
syndrome of PET, include several dietary factors,
metabolic conditions (diabetes, insulin resistance,
anduricacid),obesity,metabolicsyndrome,folic
acid and hyperhomocysteinemia, hyperlipidemia,
maternal vitamin D deciency, and
thrombophilia.
rPETisassociatedwithfetalgrowthrestriction
(FGR), in one-third of cases.
rDespite PET and FGR occurring only in
pregnancy, they have been shown to have
long-term consequences for both mother and
fetus. Mothers who have had PET or who have
delivered a baby with FGR, experience a 2–8-fold
increased risk of atherosclerotic cardiovascular
disease (CAD) in later life.
re key to good management is early detection
and secondary prevention with anti-hypertensive
and antithrombotic drugs (aspirin, heparin).
rHematological complications of PET include
thrombocytopenia, disseminated intravascular
coagulation (DIC), HELLP syndrome, and
massive bleeding aer placental abruption.
rDierential diagnosis includes microangiopathic
hemolytic anemias: (thrombotic
thrombocytopenic purpura, TTP, hemolytic
uremic syndrome, HUS), and acute fatty liver of
pregnancy.
215

Section 6. Microangiopathies
References
1. Lewis, G (ed). e Condential Enquiry into Maternal
and Child Health. (CEMACH). Saving Mothers; Lives:
reviewing maternal deaths to make motherhood safer
2003–2005. eSeventhReportonCondential
Enquiries into Maternal Deaths in the United Kingdom,
2007.
2. Roberts JM, Gammill HS. Preeclampsia: recent
insights. Hypertension 2005; 46: 1243–1249.
3. Maynard SE, Min JY, Mercham J et al. Excess placental
solublefms-liketyrosinekinase1(sFlt-1)may
contribute to endothelial dysfunction, hypertension,
and proteinuria in preeclampsia. Journal of Clinical
Investigations 2003; 111: 649–658.
4. LuF,LongoM,TamayoEet al. e eect of
over-expression of sFlt-1 on blood pressure and the
occurrence of other manifestations of preeclampsia in
unrestrained conscious pregnant mice. American
Journal of Obstetrics and Gynecology 2007; 196:
396.
5. Li B, Ogasawara AK, Yang R et al. KDR (VEGF
receptor2)isthemajormediatorforthehypotensive
eect of VEGF. Hypertension 2002; 39: 1095–1100.
6. Gilbert JS, Babcock SA, Granger JP. Hypertension
produced by reduced uterine perfusion in pregnant
rats is associated with increased soluble fms-like
tyrosine kinase-1 expression. Hypertension 2007; 50:
1142–1147.
7. Levine RJ, Lam C, Qian C et al. Soluble endoglin and
other circulating antiangiogenic factors in
preeclampsia. New England and Journal of Medicine
2006; 355: 992–1005.
8. Venkatesha S, Toporsian M, Lam C et al. Soluble
endoglin contributes to the pathogenesis of
preeclampsia. Nature Medicine 2006; 12:
642–649.
9. Masuyama H, Nakatsukasa H, Takamoto N,
Hiramatsu Y. Correlation between soluble endoglin,
vascular endothelial growth factor receptor-1 and
adipocytokines in preeclampsia. Journal of Clinical
and Endocrinological Metabolism 2007; 92:
2672–2679.
10. Redman CW, Sacks GP, Sargent IL. Preeclampsia: an
excessive maternal inammatory response to
pregnancy. American Journal of Obstetrics and
Gynecology 1999; 180: 499–506.
11. Irgens HU, Reisaeter L, Irgens LM, Lie RT. Long term
mortality of mothers and fathers aer pre-eclampsia:
population based cohort study. British Medical Journal
2001; 323: 1213–1217.
12. Barker DJB (ed.) Foetal and Infant Origins of Adult
Disease. London: BMJ Publishing Group, 1992.
13. Magnussen EB, Vatlen LJ, Lund-Nilsen TI, Salvesen
KA, Smith GD, Romundstad PR. Pregnancy
cardiovascular risk factors as predictors of
pre-eclampsia: population based cohort study. British
Medical Journal 2007; 225: 978–981.
14. Berends AL, de Groot CJM, Sijbrands EJ et al. Shared
constitutional risks for maternal vascular-related
pregnancy complications and future cardiovascular
disease. Hypertension 2008; 51: 1034–1041.
15. Rasmussen S, Irgens LM. History of fetal growth
restriction is more strongly associated with severe
rather than milder pregnancy-induced hypertension.
Hypertension 2008; 51: 1231–1238.
16. Huppetz B. Placental origins of preeclampsia:
challenging the current hypothesis. Hypertension 2008;
51: 970–975.
17. Sood R, Kalloway S, Mast AE, Hilard CJ, Weiler H.
Fetomaternal cross talk in the placental vascular bed:
control of coagulation by trophoblast cells. Blood 2006;
107(8): 3173–3181.
18. Papageorgiou AT, Yu CK, Bindra R, Pandis G,
Nicolaides KH. Multicenter screening for
pre-eclampsia and fetal growth restriction by
transvaginal uterine artery Doppler at 23 weeks’ of
gestation. Ultrasound Obstetrics and Gynecology 2001;
18: 441–449.
19. ChappellLC,SeedPT,BrileyAet al. A longitudinal
study of biochemical variables in women at risk of
preeclampsia. American Journal of Obstetrics and
Gynecology 2002; 187:127–136.
20. CLASP: a randomised trial of low dose aspirin for the
prevention and treatment of pre-eclampsia among
9364 pregnant women. CLASP (Collaborative low
dose Aspirin Study in Pregnancy) Collaborative
Group. e Lancet 1994; 343: 619–629.
21. Askie LM, Duley L, Henderson-Smart DJ, Stewart LA.
PARis Collaborative Group. Antiplatelet agents for
prevention of pre-eclampsia: a meta-analysis of
individual patient data. Lancet 2007; 369: 1791–
1798.
22. Askie L, Duley L, Henderson-Smart D, Stewart L.
Antiplatelet agents for prevention of pre-eclampsia: a
meta-analysis of individual patient data. e Lancet
2007; 369: 1791–1798.
23. MelloG,ParrettiE,FatiniCet al.
Low-molecular-weight heparin lowers the recurrence
rate of preeclampsia and restores the physiological
vascular changes in angiotensin-converting enzyme
DD women. Hypertension 2005; 45: 86–91.
24. Maki M, Kobayashi T, Terao T et al. Antithrombin
therapy for severe preeclampsia: results of a
double-blind, randomized, placebo-controlled trial.
216

Chapter 17. Pre-eclampsia
BI51.017 Study Group. rombosis and Haemostasis
2000; 84: 583–590.
25. Doyle LW, Crowther CA, Middleton P et al.
Magnesium sulphate for women at risk of preterm
birth for neuroprotection of the fetus. Cochrane
Database Systems Review 2009; 1: CD004661.
26. McCoy S, Baldwin K. Pharmacotherapeutic options
for the treatment of preeclampsia. American Journal
Health Systems Pharm. 2009; 66: 337–344.
27. Matchaba P, Moodley J. Corticosteroids for HELLP
syndrome in pregnancy. Cochrane Database Syst. Rev.
2004; (1): CD 002076.
28. Fonseca JE, Mendez F, Catano C, Arias F.
Dexamethasone treatment does not improve the
outcome of women with HELLP syndrome: a
double-blind, placebo-controlled, randomized clinical
trial. American Journal of Obstetrics and Gynecology
2005; 193: 1591–1598.
217

Section 6 Microangiopathies
Chapter
18 Thrombotic thrombocytopenic purpura
and other microangiopathies
Marie Scully and Pat O’Brien
Introduction
rombotic microangiopathies (TMAs) describe the
clinical and pathohistological eects of thrombosis in
small vessels. ere is usually thrombocytopenia and
anemia and review of the blood lm conrms the
microangiopathic process, with evidence of red cell
fragmentation and oen polychromasia. One of the
earliest diagnoses was by Moschowitz in 1924, who
described a young woman with anemia and thrombo-
cytopenia, neurological and renal symptoms, and signs
with fever. is described the typical pentad of fea-
tures of acute thrombotic thrombocytopenic purpura
(TTP). However, in pregnancy, the dierential diagno-
sis may be very dicult and oen clinical suspicion in
conjunction with laboratory parameters requires dif-
ferentiation from other TMAs, which are specic to
this period. e diagnostic challenge is the dierenti-
ation from acute fatty liver of pregnancy (AFLP), pre-
eclampsia (PET) or eclampsia, HELLP (hemolysis, ele-
vated liver enzymes, low platelets), antiphospholipid
syndrome (APS), systemic lupus erythematosus (SLE),
hemolyticuremicsyndrome(HUS),anddisseminated
intravascular coagulation (DIC) (See Table 18.1).
Moderate to severe thrombocytopenia
presenting during pregnancy
rombocytopenia is dened by a platelet count
⬍150 ×109/L. It results from increased destruction
and/or decreased production and can aect 10%
of pregnancies. e most common is gestational
thrombocytopenia, accounting for 75% of all cases.
Rarely, the count is below 70 ×109/L, typically in
the third trimester and it returns to normal within 12
weeks postpartum. It is thought to result from a hemo-
dilutional eect in pregnancy and placental platelet
destruction. ere is very little risk of hemorrhage to
themotherorthefetus.
ITP (immune thrombocytopenic purpura) occurs
in 5% of pregnancies with thrombocytopenia and is
a result of immunological peripheral platelet destruc-
tion. Maternal treatment and precautions during deliv-
ery may be required, but rarely does it have an eect on
the fetus (Chapter 4).
PET and HELLP account for 21% of all cases of
thrombocytopenia in pregnancy; the platelet count
(and other pathological features) usually return to nor-
mal within 3–5 days aer delivery.
Placental proles in high risk
pregnancies
Abnormal uterine artery blood ow in the second
trimester is indicative of an increased risk of placen-
tal pathology later in the pregnancy, including intra-
uterine growth restriction (IUGR) and PET. Uterine
artery Doppler examination is oen carried out at
around 24 weeks’ gestation in women considered to be
at increased risk of these disorders. Increased resist-
ance in the uterine arteries (indicated by increased
pulsatility index or “notched” waveforms) are associ-
ated with a sixfold increased risk of thrombotic placen-
tal injury, leading to IUGR and/or PET, compared with
normal uterine artery Dopplers. However, the sensitiv-
ityofthistestispoor,soitsuseisusuallyrestricted
to high risk women. In early pregnancy, increased
levels of biochemical markers such as alpha fetopro-
tein (AFP), beta-human chorionic gonadotrophin (-
HCG), and decreased levels of placental protein 13
(PP–13),intheabsenceofDownsyndromeandspina
bida, are associated with an increased risk of PET,
IUGR, placental abruption, and intra-uterine fetal
death. ese biochemical markers can improve the
218 e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Chapter 18. Thrombotic thrombocytopenic purpura and other microangiopathies
Table 18.1 Typical features in pregnancy associated microangiopathies
MAHA Thrombocytopenia Coagulopathy HBP
Abdominal
symptoms
Renal
impairment
Neurological
symptoms
PET ++ ± +++± ± ++
HELLP ++ ± ++++ + ±
TTP ++ + + + − ± + ++ + + +
HUS +++ ± +++ +++ ±
AFLP ±+ ++ +++ + ±
SLE ++ ± +± ++ +
APS +++ ± ++− ++ ++
MAHA: microangiopathic hemolytic anemia, HBP: High blood pressure.
PET: pre-eclampsia, HELLP: hemolysis, elevated liver enzymes and low platelets, TTP: thrombotic thrombocytopenia HUS: hemolytic
uremic syndrome AFLP: acute fatty liver of pregnancy SLE: systemic lupus erythematosis APS:
±: possibly occurs.
+++: definitive feature.
predictive value of uterine artery Doppler imaging for
the prediction of the smaller subset of women with a
high risk of later developing serious problems related
to placental disease.
Thrombotic thrombocytopenic
purpura (TTP)
TTP is an acute life-threatening disorder associated
with thrombocytopenia, microangiopathic hemolytic
anemia, and symptoms related to microvascular
thrombosis. Clinically, in addition to a low platelet
count (below 150 ×109/L, but more usually ⬍50 ×
109/L), patients are anemic secondary to fragmenta-
tion hemolysis with an associated acute consumption
of folate. Corresponding blood lm changes include
polychromasia, anemia, reduced platelets, and frag-
mented red blood cells. Bilirubin is oen raised, but
the direct antiglobulin test is negative and the clot-
ting screen is normal. Lactate dehydrogenase (LDH)
is increased, oen out of proportion to the degree of
hemolysis, due to associated tissue ischemia.
Von Willebrand factor (VWF), a plasma glycopro-
tein synthesized by megakaryocytes and endothelial
cells, normally circulates as multimers of 500–20 000
kDa. Ultra-large VWF multimers (ULVWFM), which
have a molecular weight greater than 20 000 kDa, and
arenotnormallydetectedinplasma,wereinitially
detected in patients with chronic relapsing TTP. Sub-
sequently, a deciency of VWF-cleaving protease in
patients with TTP, was dened in 2001 as “a disinte-
grin and metalloprotease with thrombospondin type
1 motif, member 13” or ADAMTS 13.1is enzyme is
required to break down ULVWFMs. Failure to do so,
due to an inherited deciency or acquired reduction
of ADAMTS 13, or due to antibodies to ADAMTS 13,
for example, leads to platelet adhesion and aggregation
of UL VWFMs and resulting microvascular throm-
bosis. Hence, platelet transfusions are relatively con-
traindicated in TTP, as infusions potentiate the eects
of platelet aggregation on UL VWFMs.
Pregnancy is a precipitating cause of acute TTP,
accounting for approximately 10%–25% of all cases of
TTP in women. From the Oklahoma registry, 19 of the
61 women of child-bearing age presented with TTP
during pregnancy or postpartum.2TTP is more com-
mon in women (3:2), and 45% of all cases of TTP occur
in women of child-bearing age. ere is also a risk
of relapse of TTP during subsequent pregnancies in
women diagnosed with TTP.
Other pregnancy-related thrombotic microan-
giopathies, such as pre-eclampsia / HELLP and
hemolytic–uremic syndrome may further complicate
the diagnosis of TTP. Gestational thrombocytopenia,
which occurs in around 7% of pregnancies and is a
diagnosis of exclusion, may explain a reduction in
platelet counts, when all other laboratory parameters
are normal. Management approaches dier for these
conditions, although dierentiation may be clinically
challenging.
Hemostatic changes of normal
pregnancy-Factor VIII, Von Willebrand
Factor (VWF), and ADAMTS 13
Normal pregnancy is associated with marked changes
in hemostasis, which are hormonally mediated and 219

Section 6. Microangiopathies
protect against severe hemorrhage at the time of
delivery, but ultimately result in a hypercoagulable
state. Factor VIII and VWF increase in parallel in the
rst half of pregnancy; thereaer, the increase in VWF
is greater throughout the remainder of pregnancy,
returning to normal levels over the 6 weeks’ postpar-
tum. Reciprocal changes of VWF and ADAMTS 13
have been documented. erefore, with the increased
VWF in pregnancy, ADAMTS 13 would be expected
to decrease. A review of ADAMTS 13 in normal
women with no history of TTP documented a reduc-
tion in ADAMTS 13 activity in the second and third
trimesters of pregnancy. A further study in healthy
women conrmed a reduction in ADAMTS 13 activity
aer the rst trimester (weeks 12–16) up until the end
of the post-natal period when the levels normalized to
pre-pregnancy levels. ADAMTS 13 activity was lower
in non-pregnant nulliparous women (mean 65%) com-
pared with parous women (mean 83%). In pregnancy
andpost-delivery,meanADAMTS13activitywas
slightly, but non-signicantly, lower in primigravidae
than in multigravidae (68% vs. 74%). ADAMTS 13 was
unaected by platelet count, but was higher in smokers
than in non-smokers during pregnancy (mean 79% vs.
70%, respectively). ere was a signicant correlation
between higher VWF:Ag levels and lower ADAMTS
13 activity.3e reason for the decrease in ADAMTS
13 during pregnancy may be twofold. First, enzyme
levels decrease with excess substrate, VWF. Second,
a hormonal inuence, possibly estrogen, may lower
ADAMTS13 levels. A role for the eect of estrogen on
parity, and ADAMTS 13 levels, are in line with estro-
diol levels in the pregnant and non-pregnant state.
Women presenting with acute TTP
during pregnancy
Women presenting with T TP during pregnanc y
appear to fall into two groups: those with congenital
TTP and those with acquired, antibody mediated
TTP. Congenital TTP may rst present during preg-
nancy and these women are more likely to relapse
in subsequent pregnancies. Diagnosis is conrmed
with ADAMTS 13 activity ⬍5%,noevidenceofan
inhibitor, and conrmation by mutational analysis
of the ADAMTS 13 gene, revealing a homozygous
or compound heterozygous abnormality. To date,
the published literature includes 14 patients, eight of
whom received plasma during pregnancy.
In women who present with acquired TTP related
to pregnancy, the literature presents varying outcomes.
Successful pregnancy outcome can be achieved in
women with an initial episode of TTP.4In the Okla-
homa registry,5therewere11womenwhohadatotal
of 17 pregnancies subsequent to a diagnosis of acute
TTP in pregnancy. Two of these pregnancies were
associated with TTP recurrence and neither infant sur-
vived. In women with no TTP in a subsequent preg-
nancy (15/17), infant survival was 80%. However, it
appears from the remaining published literature, with
the proviso that these are small case series and there is
likely to be some reporting bias, that the risk of recur-
rence in subsequent pregnancies is approximately 50%,
and infant survival rates are around 67%.
Risk associated with pregnancy
in women with previous acquired
idiopathic (non-pregnancy associated)
TTP
A particular concern in women who have had acute
TTP unrelated to pregnancy is the risk of relapse from
TTP during a subsequent pregnancy. From the Okla-
homa Registry,5of 7 women with idiopathic TTP, 3
had recurrent relapsing TTP. In the 12 subsequent
pregnancies following a diagnosis of TTP, 3 devel-
oped TTP in pregnancy and infant survival was 67%.
Interestingly, in women who did not relapse from
TTP during pregnancy (9/12 pregnancies, 75%), infant
survival was only 33% (3/9). From the literature to
date, including 20 women who had a total of 26
pregnancies following the diagnosis of acute TTP,
17/26 had a relapse of TTP during pregnancy and
infant survival was 15/26. In those patients in whom
ADAMTS 13 testing was available, normal levels pre-
pregnancy/onset of pregnancy were associated with a
lower likelihood of relapse. Another important fea-
ture of women reported in the literature is the number
of complications documented associated with throm-
botic microangiopathies, such as pre-eclampsia and
HELLP syndrome, as well as reduced fetal survival (see
Table 18.2). It could be hypothesized that women with
TTP are at increased risk of prothrombotic complica-
tions and increased risk of placental infarction, despite
normal routine TTP-based laboratory parameters.
rombotic microangiopathies during pregnancy
may be clinically indistinguishable and very dicult
to treat. With the normal reduction in ADAMTS 13
220

Chapter 18. Thrombotic thrombocytopenic purpura and other microangiopathies
Table 18.2 Complications in pregnancy in women with a history of TTP
Reference
Number of
pregnancies
In utero fetal
death
Maternal
death
Pre-eclampsia/
HELLP Other
10 16 4 1
11 6 1 – – –
529
∗8 – 11/2 11x first trimester spontaneous abortions
12 4 3 (set of twins) 1 1 –
13 10 2 1 – 1x fetal distress, 1x placental abruption
4 5 2 – – 1x Hypertension, 1x first trimester
spontaneous abortion
∗: Includes patients with HUS, but excludes those presenting with bloody diarrhea, therefore 29 pregnancies in 18 women.
HELLP: hemolysis with elevated liver enzymes and low platelets.
Table 18.3 Thrombotic Thrombocytopenic Purpura Presenting During Pregnancy
Trimester
First Second Third/postpartum
Case series
References
Number of women
diagnosed with TTP
during pregnancy
14 25 4 6 15
15 9 0 0 9
16 4 0 0 4
17 9 2 1 6
10 5 3 1 1
519 1315
11 3 1 1 1
12 4 1 1 2
13 9 1 3 5
43 120
Total 90 14 18 58
from the onset of the second trimester, it had originally
been proposed that this was the time of increased
presentation of acute TTP. However, it now appears
that the greatest risk is in the third trimester or post-
partum (see Table 18.3).
Treatment of TTP in pregnancy
e combination of thrombocytopenia and MAHA
encompasses a number of diagnoses in pregnancy and
it is oen dicult to dierentiate TTP from these. e
primary decision is whether delivery will be associ-
ated with remission of the TMA (as in PET or HELLP)
or whether plasma exchange should be instigated, as
recovery following delivery is unlikely and there is
a risk of multi-organ dysfunction/ death. A further
complicating issue is the development of HELLP/PET
following delivery, which may occur in 20%–30% of
cases of TTP in pregnancy.
IfTTPdevelopsinthersttrimester,plasma
exchange (PEX) may allow continuation of pregnancy
with delivery of a live infant. However, as HELLP/pre-
eclampsia or TTP can present in the post-natal period
or there may be progression of symptoms despite deliv-
ery, PEX is the most appropriate option. With the
availability of ADAMTS 13 activity measurement and
detection of inhibitors to ADAMTS 13 (or more specif-
ically IgG antibodies), it may be possible to distinguish
TTP from other pregnancy associated TMAs, speci-
cally if ADAMTS 13 activity is ⬍5% and/or if IgG anti-
bodies are present. In HELLP syndrome, ADAMTS
13 activity is reduced (median 31%, range 12%–43%)
but with no inhibitor/antibodies to ADAMTS 13 and
higher VWF levels. 221

Section 6. Microangiopathies
Steroids may be useful in HELLP syndrome and
in TTP, but for dierent reasons. ey have been used
empirically in TTP because of the underlying autoim-
mune basis of the disorder, and in HELLP may accel-
erate recovery from delivery.
However, women presenting with thrombo-
cytopenia, MAHA, neurological features (such as
stroke/TIAs, seizures, encephalopathy), and renal
impairment, should be treated with PEX until the
diagnosis of TTP is excluded. In women with congen-
italTTP,theriskofrelapseinasubsequentpregnancy
is such that elective plasma therapy during pregnancy
is warranted. Plasma infusions may be satisfactory;
however, to deliver sucient volumes, PEX may be
required. e optimal frequency of plasma replace-
ment is unknown; the half-life of ADAMTS 13 is
2–3 days and plasma therapy every 2 weeks appears
satisfactory.4
In women with acquired TTP, it is not as easy to
predict who are likely to relapse and the literature is
sparse in this area. e previous history of TTP and
theADAMTS13activityattheonsetofpregnancy
may be helpful in dierentiating patients most likely
to relapse. A normal ADAMTS 13 at the onset of preg-
nancy appears to predict women at reduced risk of sub-
sequent relapse.4However, if there is low ADAMTS 13
activity (⬍5%) at the onset of pregnancy, consideration
shouldbegiventoelectivetherapytopreventrelapse.
In contrast, women with normal ADAMTS 13 activity
at the onset of pregnancy, who maintain normal rou-
tine laboratory parameters, ADAMTS 13 activity, and
antibody/inhibitor levels throughout pregnancy, do
not usually require intervention for TTP. A reduction
in ADAMTS 13 activity (⬍10%) may be the trigger for
elective therapy to prevent microvascular thrombosis
during pregnancy.
Supportive therapy during pregnancy has not
been addressed in the literature; specically, low dose
aspirin (LDA) and/or prophylactic low molecular
weight heparin (LMWH). All patients in our cohort
are maintained on LDA throughout pregnancy and
womenwithadocumentedthrombophiliaorapast
history of venous thromboembolism (VTE) associated
withTTParestartedonprophylacticLMWH.e
aim is to optimize implantation and preserve placental
function as abnormalities of the utero-placental circu-
lation, resulting in insuciency are established in the
rst trimester. LDA/LMWH may be benecial in other
thrombophilic disorders during pregnancy, reducing
the risk of placental abnormalities secondary to infarc-
Table 18.4 Physiological changes during normal pregnancy
Test Change in pregnancy
Bilirubin Unchanged
Aminotransferases Unchanged
Alkaline phosphatase Increase two to fourfold
Cholesterol Increase twofold
Prothrombin time Unchanged
Fibrinogen 50% increase
Hemoglobin Decrease in later pregnancy
White cells Increase
tion. However, this therapy has not been formally
evaluated in pregnancy associated TTP. ere are no
data on the microvascular eects of “subacute” TTP
before presentation with thrombocytopenia. ere-
fore, women with a previous pregnancy loss due to
TTP or low ADAMTS 13 activity at the onset of preg-
nancy can be assumed to be at increased risk of further
episodes of placental disorders in subsequent pregnan-
cies. Interestingly, especially as reported in the Okla-
homa registry data, there were a large number of rst
trimester losses in such women. is may be due to the
underlying TTP risk, but there is no conclusive histo-
logical conrmation.
erefore, women with congenital TTP require
therapy with plasma, either as infusions or as
PEX. In women with acquired, previous acute TTP
episodes, the baseline ADAMTS 13 activity, and
inhibitor/antibody status at the onset of pregnancy
maybeusefulintheidenticationofthosemostlikely
to relapse. Monitoring of enzyme activity in those
with normal early pregnancy levels may be useful,
but in women with low (⬍5%) ADAMTS 13 activity
and/or raised IgG antibody levels, which appear to
be at increased risk of relapse, elective PEX may be
useful. Adjunctive therapy with LDA in all women
+/−prophylacticLMWH,shouldbeaddedtohelp
prevent complications related to placental thrombosis.
Liver disease in pregnancy
ere are some changes in liver function in normal
pregnancy (see Table 18.4), but clinically abnormal
liver function can be detected in 3%–5% of all preg-
nancies. e cause may be coincidental to pregnancy
or pre-existing chronic liver disease may be docu-
mented. However, in the majority of cases, pregnancy
itself is the precipitant. Hyperemesis gravidarum
222

Chapter 18. Thrombotic thrombocytopenic purpura and other microangiopathies
typically occurs in the rst trimester and intrahepatic
cholestasis of pregnancy (ICP) in the second or third
trimesters. PET, HELLP and acute fatty liver of preg-
nancy (AFLP) are also associated with abnormal liver
function.
Intrahepatic cholestasis
of pregnancy (ICP)
ICP has been associated with impaired sulphation
and abnormalities of progesterone metabolism. Clin-
ically, initially there is pruritus, which in 10%–25%
progresses to jaundice associated with 10–20-fold
increases in aminotransferases, but a less marked rise
in bilirubin. e diagnosis is helped by measuring bile
acid levels. Treatment is supportive and ursodeoxy-
cholic acid (UDCA) is used. Steroids, although use-
ful for fetal lung maturation pre-delivery have not
been shown to be benecial compared with UDCA
therapy. e main risk of raised bile acid levels is
to the fetus; there is an increased risk of placental
insuciency but more importantly an association with
sudden intrauterine fetal death, the precise cause of
which is not clear. Resolution of the condition occurs
with delivery. However, recurrence occurs in 45%–
70% of subsequent pregnancies or with use of the com-
bined oral contraceptive pill, the progesterone only pill
(mini-pill) appears not to increase the risk of recur-
rence.
Acute fatty liver of pregnancy (AFLP)
is is a rare disorder (incidence estimated at 1/13 000
deliveries), but is an acute life-threatening illness asso-
ciated with signicant maternal and peri-natal mor-
tality.6Typically, it presents in the third trimester,
between the 30th and 38th weeks of pregnancy,
although it has been rarely described in the rst
and second trimesters. It usually aects primigravid
women, although reports of recurrence in subsequent
pregnancies have been documented.
Clinically, presentation is non-specic with
headache, fatigue, nausea, vomiting (70%), and right
upper quadrant or epigastic pain (50%). Progression
of the illness is oen rapid and, early in the presen-
tation, there may be gastrointestinal hemorrhage,
coagulation abnormalities, acute renal failure, infec-
tion, pancreatitis, and hypoglycemia. Later in the
disease process, liver failure and encephalopathy may
occur. Early delivery is imperative and improvement
occurs over 1–4 weeks’ postpartum, although an
improvement in liver function is usually seen within a
few days of delivery.
Diagnosis is suggested by the clinical features and
may be conrmed by liver biopsy. Histologically, there
is characteristic microvesicular steatosis and with Oil
Red O staining, cytoplasmic vesiculation as a result
of microvesicular fat. However, because of the acute
presentation and laboratory features including coag-
ulopathy, it is usually not possible to undertake liver
biopsy, and the diagnosis is made by a combination of
clinical and biochemical features.
In routine laboratory tests, there may be a raised
white cell count and thrombocytopenia with nor-
moblasts on the blood lm. ere is DIC (with pro-
longed PT, APPT, and reduced brinogen). Urea,
creatinine, and uric acid levels are raised, there are
elevated ammonia levels and hypoglycemia. Serum
aminotransferases are markedly raised and alkaline
phosphatase are three to four times the normal level
(althoughthisisraisedinnormalpregnancybecause
of placental production).
e primary dierential diagnoses are acute fulmi-
nant hepatitis and severe HELLP, although the latter
arelesslikelytobeassociatedwithhypoglycemiaand
prolonged PT. e histological features of liver biopsy
are described above.
Pathogenesis: with advances in molecular biology,
ithasbecomeevidentthatAFLPmayresultfrommito-
chondrial dysfunction. ere is a strong association
between AFLP and a deciency of the enzyme long
chain 3-hydroxyacyl-CoA dehydrogenase (LCHAD)
in the fetus, a disorder of mitochondrial fatty acid
beta-oxidation. -oxidation of fatty acids is a major
source of energy for skeletal muscle and the heart,
while the liver oxidizes fatty acids under conditions
of prolonged fasting, during illness, and at peri-
ods of increased muscular activity. Mitochondrial -
oxidation of fatty acids is a complex process. LCHAD
is part of an enzyme complex, the mitochondrial tri-
functional protein (MTP), associated with the inner
mitochondrial membrane. MTP contains four ␣and
four subunits. A hydratase enzyme is located in the
amino-terminal domain and LCHAD is located in the
carboxy-terminal region of the ␣subunit. e sub-
unit contains thiolase enzymatic activity. Defects in the
MTP complex are recessively inherited and are due
to an isolated LCHAD deciency, specically associ-
ated with G1548C mutation, with relatively normal
hydratase and thiolase activities. In complete MTP 223

Section 6. Microangiopathies
deciency, there is a marked reduction in all three
enzymes. A few hours aer birth, children with these
disorders, which are primarily LCHAD, present with
non-ketotic hypoglycemia and hepatic encephalopa-
thy, progressing to coma or death if untreated.
Studies suggest an association between fetal MTP
defects and AFLP. In one study, in every pregnancy
in which the fetus had an LCHAD deciency, the
mother developed AFLP or HELLP syndrome. Sub-
sequent work in pregnancies without a LCHAD de-
cient fetus found that the pregnancy progressed nor-
mally, with no liver dysfunction. In another study of
prospectively screened mothers who developed AFLP
(27 pregnancies) or HELLP (81 pregnancies), 5 fetuses
in the AFLP group, but none in the HELLP group, had
an MTP mutation.
e precise mechanism by which a LCHAD-
decientfetuscausesAFLPinaheterozygotemother
remains unclear. However, there are several hypothe-
ses. e mother who is heterozygote for an MTP
defect has reduced capacity to oxidize long chain fatty
acids. e stress of pregnancy associated with altered
metabolism, increased lipolysis, and decreased oxi-
dation, and the hepatotoxic LCHAD produced by the
fetus or placenta may accumulate in the maternal cir-
culation. erefore, approximately one in ve women
who develop AFLP may carry an LCHAD-decient
fetus. Screening of newborn infants at birth for this
disorder of fatty acid oxidation can be lifesaving and
allows for genetic counseling in subsequent pregnan-
cies.
Hemolysis, elevated liver enzymes
and low platelets (HELLP)
is is a microangiopathy associated with endothe-
lial cell injury, brin deposition, platelet activation
and consumption, and areas of hepatic hemorrhage
and necrosis. e underlying precipitating cause is
unknown but it occurs only in pregnancy and the
incidence is between 0.17% and 0.85% of all live
births. Maternal mortality is 3%–4%, with fetal mor-
tality reaching approximately 25%, mainly due to pre-
maturity. Diagnostically, there is considerable overlap
with other TMAs especially PET, and they may repre-
sent dierent points on a single pathological spectrum
(see Chapter 17). ere are no obvious precipitating
factors associated with development of HELLP and
it typically presents between the second and third
trimesters, although approximately a quarter of all
cases are postpartum.7Typical presenting symptoms
include upper abdominal pain and tenderness, nausea,
vomiting, malaise, headache, and rarely jaundice.
ere are no clinical or laboratory factors that are
diagnostic, but bilirubin is not usually raised. Amino-
transferases can be marginally increased or up to
20-fold. HELLP syndrome may be classied according
to the degree of thrombocytopenia: HELLP 1 (≤50 ×
109/L), HELLP 2 (between 50 ×109and 100 ×109/L)
and HELLP 3 (between 100 ×109and 150 ×109/L).
Serious maternal complications include DIC, pla-
cental abruption, acute renal failure, pulmonary
edema, and hepatic failure, occasionally requiring liver
transplantation. Hepatic rupture is a further rare,
acute, life-threatening complication.
Pre-eclampsia (PET)
is is classically dened as the triad of hypertension,
proteinuria, and edema, but is best thought of as a mul-
tisystem disorder resulting from endothelial damage.
It is a leading cause of maternal and neonatal mor-
bidity and mortality, aecting 5%–10% of all preg-
nancies. It is more common in primigravid women. It
rarely occurs before 24 weeks of gestation and the inci-
dence rises as pregnancy advances, being most com-
mon in the third trimester. Liver involvement is com-
mon although rarely severe and is the most common
cause of hepatic tenderness and liver dysfunction in
pregnancy. It is an indicator for delivery because of
the increased risk of severe eclampsia, hepatic rupture,
DIC, and necrosis. e high peri-natal morbidity and
mortality are partly due to the association with placen-
tal insuciency and IUGR, but partly due to prema-
ture delivery for maternal indications. Severe PET is
complicated in 2%–12% of cases by HELLP syndrome,
consistent with the idea that they lie on a spectrum of a
single disorder. Renal impairment, eclampsia (convul-
sions), and abnormalities of the coagulation system are
further complications.
Hemolytic uremic syndrome (HUS)
D+(diarrhea positive) HUS is typically preceded by
an illness with a verotoxin-producing bacteria, usually
E.coli 0157:H7. Atypical, D– (diarrhea negative) HUS,
is rare, with an incidence of 1/25 000 pregnancies, and
in nearly all documented cases associated with preg-
nancy, occurs postpartum. Atypical HUS (aHUS) may
be familial and has a poorer prognosis, with a mor-
tality of 25% acutely and 50% requiring chronic renal
224

Chapter 18. Thrombotic thrombocytopenic purpura and other microangiopathies
therapy. Like all TMAs, it is a disease of microvascular
endothelial activation, cell injury, and thrombosis, but
associated with complement deregulation, leading to
an increase in activity in the alternative pathway. Muta-
tions within the complement regulatory proteins and
activating components are found.
Typically, the presentation in HUS is of MAHA,
thrombocytopenia, and renal impairment. e pri-
mary pathology is in the renal arterioles and interlob-
ular arteries, with widespread endothelial cell swelling,
leading to exposure of the underlying basement mem-
brane. e vessel lumens are occluded by red cells and
platelet brin thrombi. e pre-glomerular pathol-
ogy distinguishes it from D+HUS and TTP. ere is
consequently excess complement activation particu-
larly along glomeruli, arteriolar endothelium, and
basement membranes. More than 50% of cases result
from mutations in complement genes controlling the
alternative complement pathway. Mutations may aect
complement regulatory genes, such as Factor H, I or
MCP, or complement activating genes, Factor B (CBF),
or C3 (C3). Single nucleotide polymorphisms and anti-
bodies, such as to Factor H, have also been found
to play a role. Factor H mutations, mostly heterozy-
gote, account for 15%–30% of all cases of aHUS.8MCP
mutations account for 10%–13% of aHUS patients, the
majority being heterozygote, with approximately 25%
homozygous/compound heterozygote.
Treatment
isisprimarilysupportive,includingredcelltrans-
fusion, blood pressure control, and renal dialysis. e
role of plasma therapy remains undetermined, but has
been successful in some cases.
Exacerbation of systemic lupus
erythematosis (SLE)
SLE is an autoimmune disease, the active phase of
which may be associated with thrombocytopenia,
hemolytic anemia, pancytopenia, and an increase in
double-stranded DNA. e disorder is multisystem
and, typically, there are associated skin and joint
symptoms. Serum complement levels may be nor-
mal or decreased. An acute exarcerbation occurs in
25%–30% of women during pregnancy, but it may
occur for the rst time during pregnancy9or in
the postpartum period. An acute episode of lupus
nephritis, associated with hypertension and protein-
uria, may be dicult to dierentiate from HELLP or
pre-eclampsia.
Antiphospholipid antibodies (aPL) may be present
in 30%–49% of women with lupus and further increase
the risk of thrombotic events, the risk of tissue
ischemia and TMA. rombocytopenia is present in
aminority.
Disseminated intravascular
coagulation (DIC)
In pregnancy, DIC must not be forgotten as a cause
of MAHA with an abnormal clotting screen. Usually,
there is an underlying precipitating cause that must be
treated and it can be a complication of any of the above
TMAs in severe cases. Treatment of DIC requires
platelet transfusions to maintain a count ⬎50 ×109/L,
fresh frozen plasma, and cryoprecipitate, depend-
ing on the level of abnormality of the coagulation
parameters.
225

Section 6. Microangiopathies
References
1. Sadler JE. Von Willebrand factor, ADAMTS13, and
thrombotic thrombocytopenic purpura. Blood 2008;
112: 11–18.
2. Vesely SK, George JN, Lammle B et al. ADAMTS13
activity in thrombotic thrombocytopenic
purpura–hemolytic uremic syndrome: relation to
presenting features and clinical outcomes in a
prospective cohort of 142 patients. Blood 2003; 102:
60–68.
3. Sanchez-Luceros A, Farias CE, Amaral MM et al.von
Willebrand factor-cleaving protease (ADAMTS13)
activity in normal non-pregnant women, pregnant and
post-delivery women. rombosis and Haemostasis
2004; 92: 1320–1326.
4. Scully M, Starke R, Lee R et al. Successful management
of pregnancy in women with a history of thrombotic
thrombocytopaenic purpura. Blood Coagulation and
Fibrinolysis 2006; 17: 459–463.
5. Vesely SK, Li X, McMinn JR, Terrell DR, George JN.
Pregnancy outcomes aer recovery from thrombotic
thrombocytopenic purpura–hemolytic uremic
syndrome. Transfusion 2004; 44: 1149–1158.
6. Riely CA. Acute fatty liver of pregnancy. Seminars
Liver Disease 1987; 7: 47–54.
7. Rath W, Faridi A, Dudenhausen JW. HELLP
syndrome. Journal of Perinatal Medicine 2000; 28:
249–260.
8. Caprioli J, Noris M, Brioschi S et al. Genetics of HUS:
the impact of MCP, CFH, and IF mutations on clinical
presentation, response to treatment, and outcome.
Blood 2006; 108: 1267–1279.
9. Cortes-Hernandez J, Ordi-Ros J, Paredes F et al.
Clinical predictors of fetal and maternal outcome in
systemic lupus erythematosus: a prospective study of
103 pregnancies. Rheumatology (Oxford). 2002; 41:
643–650.
10. Ezra Y, Rose M, Eldor A. erapy and prevention
of thrombotic thrombocytopenic purpura during
pregnancy: a clinical study of 16 pregnancies.
American Journal of Hematology 1996; 51:
1–6.
11. Ducloy-Bouthors AS, Caron C, Subtil D et al.
rombotic thrombocytopenic purpura: medical and
biological monitoring of six pregnancies. European
Journal of Obstetrics and Gynecology Reproduction
Biology 2003; 111: 146–152.
12. Shamseddine A, Chehal A, Usta I et al.rombotic
thrombocytopenic purpura and pregnancy: report of
four cases and literature review. Journal of Clinical
Apheresis, 2004; 19: 5–10.
13. Castell´
aM,PujolM,Juli
´
aAet al. rombotic
thrombocytopenic purpura and pregnancy: a
review of ten cases. Vox Sang uinis 2004; 87: 287–
290.
14. Ridol RL, Bell WR. rombotic thrombocytopenic
purpura. Report of 25 cases and review of the
literature. Medicine (Baltimore) 1981; 60: 413–
428.
15. BellWR,BraineHG,NessPM,KicklerTS.Improved
survival in thrombotic thrombocytopenic
purpura-hemolytic uremic syndrome. Clinical
experience in 108 patients. New England Journal of
Medicine 1991; 325: 398–403.
16. ompson CE, Damon LE, Ries CA, Linker CA.
rombotic microangiopathies in the 1980s: clinical
features, response to treatment, and the impact of the
human immunodeciency virus epidemic. Blood 1992;
80: 1890–1895.
17. Hayward CP, Sutton DM, Carter WH, Jr. et al.
Treatment outcomes in patients with adult thrombotic
thrombocytopenic purpura-hemolytic uremic
syndrome. Archives in Internal Medicine 1994; 154:
982–987.
226

Section
7
Malignant conditions

Section 7 Malignant conditions
Chapter
19 Myeloproliferative disorders
Claire Harrison and Susan E. Robinson
Introduction
e myeloproliferative disorders (MPDs) encompass
chronic myelogenous leukemia (CML), polycythemia
vera (PV), myelobrosis (PMF), primary thrombo-
cythemia (PT also known as essential thrombo-
cythemia or ET), rarer entities such as chronic neu-
trophilic leukemia, chronic eosinophilic leukemia,
chronic myeloproliferative disease unclassiable, and
the mast cell diseases. is chapter will concentrate
upon the management of the more common classi-
cal Philadelphia negative MPDs; PT, PV, and PMF in
pregnancy.
Epidemiology
e incidence of the classical Philadelphia nega-
tive MPDs combined is approximately 6/100 000–
9/100 000, with a peak in frequency between 50 and 70
years of age; they are less frequent in women of repro-
ductive age.
rombosis and hemorrhage are a major cause of
morbidity in MPD patients; progression to myelo-
brosis or an acute leukemia occur less frequently. His-
toricalcasereportsofpregnancyinMPDshavesug-
gested signicant maternal morbidity and poor fetal
outcome. An increase in awareness of MPDs, advanced
maternal age, and automation of blood counts to
include a platelet count has led to an increase in the
diagnosesofMPDsinwomenofareproductiveage.
Hence issues concerning the management of these dis-
orders in pregnancy are a real clinical challenge to
hematologists and obstetricians that is compounded
by a lack of clinical data and evidence-based guidance.
is chapter provides a summary of the epidemiology,
pathogenesis, and diagnosis of the MPDs in pregnancy
and a management strategy developed from current
experience attained in a tertiary referral center.
Previous reports of MPD in pregnancy
A recent meta-analysis reported the outcome of 461
pregnancies in women diagnosed with PT.1e mean
age was 29 years and the mean platelet count at the
beginning of pregnancy was 1000 ×109/L declining to
599 ×109/Linthesecondtrimester.elivebirth
rate was 50%–70%, rst trimester loss occurred in
25%–40%, and late pregnancy losses in 10%. Rates of
placental abruption (3.6%) and intrauterine growth
restriction (IUGR) (4.5%) were higher than in the gen-
eral population. Postpartum thrombotic episodes were
reported in 5.2% of pregnancies and pre/postpartum
hemorrhage in 5.2%. A summary of 208 historical
cases of PT collated from case series that included
greater than six pregnancies produced comparable
data (presented in Table 19.1). e literature for preg-
nancies aected by PV is sparse; pregnancy outcome
in a case series of 18 pregnancies in PV combined with
20 historical reports was concordant with the preg-
nancyoutcomesinPT(andissummarizedinTable
19.2).2In PV rst trimester loss was the most frequent
complication (21%), followed by late pregnancy loss
(18%), IUGR (15%) and premature delivery (13%),
which included three neonatal deaths resulting in a
50% survival rate. Maternal morbidity was also sig-
nicant including three thromboses, one large post-
partum hemorrhage, four cases of pre-eclampsia and
one maternal death associated with evidence of a deep
vein thrombosis, pulmonary emboli, sagittal sinus
thrombosis and disseminated intravascular coagula-
tion. Lastly, PMF is the least prevalent MPD in women
of child-bearing age. A report of four pregnancies in
PMF combined with four historical cases suggested a
50% risk of fetal loss; however, no maternal complica-
tions of thrombosis or disease progression were noted
but the numbers are probably too small to draw any
rm conclusions (summarized in Table 19.3).3
229
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Table 19.1 Summary of reported pregnancies affected by PT
Reference
Number
of pts
Number of
Pregnancies
Previous
thrombosis
Previous
hemorrhage
Maternal
outcome
Live
birth
total
Pregnancy
loss total
Loss
<12/40
Loss
>12/40 IUGR
Placental
abruption
Live birth
premature
delivery
<37/40
Live
birth
FTD
9 3 11 Detail not
available
Detail not
available
Detail not available 4 7 6 1 Detail not
available
12 2
10 6 9 0 0 1 Phlebitis 1 Leg ulcer
1 PPH
8 1 1 0 0 0 1 7
11 8 10 Detail not
available
Detail not
available
Detail not available 7 3 0 3 Detail not
available
00 7
12 915 (1 TOP) 1VTE
2 TIA
02 VTE 2TIA 1
Hemorrhage
9 6 3 2 2 0 4 5
13 13 16 1 VTE 0 3VTE 13 3 3 0 0 0 3 10
14 12 30 (1 Ectopic) Detail not
available
Detail not
available
1PE 17 13 4 8 2 5 5 12
15 9 17 1 CVA
1VTE
1 Epistaxis 1 TIA 2 Acquired vWD
3 Vaginal bleeds 2
Epistaxis
11 6 6 0 0 0 3 8
16 20 43 (2 TOP, 1
Ectopic)
Detail not
available
Detail not
available
Detail not available 22 21 16 2 Detail not
available
1 1 21
17 12 17 0 0 3 Vaginal bleeds 7 10 8 2 0 0 0 7
18 16 40 Detail not
available
Detail not
available
1 Eclampsia 2
Pre-eclampsia 1
Vaginal bleed
26 (1
Twin)
15 13 2 1 0 2 23
Total 108 208 124
(60%)
85 (41%) 60 (29%) 20 (10%) 5 (2%) 7 (3%) 22 (11%) 102
(49%)
FTD: full term delivery; IUGR: intrauterine growth restriction; TOP: elective termination of pregnancy
Adapted from refs. 4 and 5.

Table 19.2 Summary of literature regarding pregnancy in PV
Author
Number
of Pts
No. of
Pregnancies
Previous
thrombosis
Previous
hemorrhage
Treatment
during
pregnancy
High
risk
Maternal
outcome
Live
birth
total
Pregnancy
loss total FTM
Stillbirth
(gestation) IUGR
Placental
abruption
Live birth
premature
delivery
<37/40
Live
birth
FTD
19 1 1 No No Aspirin +
dipyrimadole
No Death∗01 1TOP0 00 0 0
20 1
1
3
2
No
No
No
No
Nil
Nil
No
No
Alive
Alive PET
1
2
2
0
2
0
0
0
0
0
0
0
0
0
1
2
PET
21 1 2 Superficial
thrombo
phlebitis
No None No Alive PET 1 1 0 1 (35/40) PET 1 0 0 1
22 1 3 No No Aspirin,
heparin∗∗
venesection
No Alive PE
postpartum
1 2 0 2 (24/40 and
28/40)∗∗∗
2 0 1 (32/40) 0
23 1 4 Yes, CVA No Nil Yes Alive PET 2 2 0 2 (5 + 7
months) PET
0 0 1 (7 months,
PET), 1
(8 months)
0
24 2 2 No No Nil No Alive PPH 2 0 0 0 0 0 0 2
25 1 2 No No Heparin 3/52
postpartum
No Alive, PE
24/7
postpartum
11 10 00 0 1
26 1 1 No No Hydroxyurea
9/40 then nil
No Alive 1 0 0 0 0 0 0 1
27 8 18 (1 twin) Yes (1 patient) No Varied:
Venesection,
Aspirin,
Interferon
LMWH,
vitamin C+E
No Alive PET in 1 11 7 4 2 3 0 1 (34/40,
IUGR), 1
(36/40), 1
26/40) (NND)
9
Total 18 38 1 CVA 1
thrombo
phlebitis
None 1yes 1death4
PET 2 PE 1
PPH
22
3 NNDs
16 8 7 6 0 6 17
FTM: first trimester miscarriage, IUGR: intrauterine growth restriction, FTD: full term delivery, NND: Neonatal death, TOP: termination of pregnancy, PE: pulmonary embolism, PET: pre-eclampsia,
PPH: postpartum hemorrhage, LMWH: low molecular weight heparin
∗the patient died with evidence of deep vein thrombosis, pulmonary embolism, sagittal Sinus thrombosis and disseminated intravascular coagulation
∗∗ postpartum heparin after 2nd pregnancy, LMWH throughout third pregnancy, aspirin throughout both pregnancies
∗∗∗multiple placental infarcts in first and abnormal uterine artery Doppler waveforms and Severe IUGR in third pregnancy
Adapted from ref. 2.

Table 19.3 Summary of literature regarding PMF in pregnancy
Author Patient Pregnancy
Previous
thrombosis
Previous
hemorrhage
Treatment
Pre
pregnancy
Treatment
during
pregnancy
Maternal
outcome
First
trimester
miscarriage
Stillbirth
(gestation) IUGR
Placental
abruption
Live birth
premature
delivery
<37 wks
1 1 No No Supportive Supportive No
complications
0 0 0 0 1 Elective
induction at 36
wks
1 1 No No None None No
complications
0 30 (placental
infarctions)
00 0
2 Placental
infarctions
No None None No
complications
0 27 (placental
infarctions)
00 0
3 Placental
infarctions
No Interferon ␣Interferon ␣No
complications
0 0 1 0 1 Elective
delivery at 34
wks due to
IUGR birth
weight 2000 g
30 A 3 (nb. 2
preceeding
PMF
diagnoses)
No No Aspirin Aspirin Disseminated
TB
00001FTND
B 1 Digital
ischemia
No Aspirin Aspirin, LMWH Postpartum
hemorrhage
00001FTND
B 2 Digital
ischemia
No Aspirin Aspirin, LMWH No
complications
0 24/40 cardiac
malformation
00 0
B 3 Digital
ischemia
No Aspirin Aspirin, LMWH No
complications
10000
Total 4 8 3 0 3 3 0 3 1 0 4
Adapted from ref. 3.
IUGR: Intrauterine growth restriction, FTND: full term normal delivery.

Chapter 19. Myeloproliferative disorders
Historical reports of pregnancy in MPD are likely
to be subject to selection bias, favoring cases associ-
ated with a poor outcome; prospective surveillance of
pregnancies in MPDs and the development of struc-
tured evidence-based guidance would be of benet in
this eld.
Pathogenesis
MPDs result from the transformation of a hematopoi-
etic progenitor cell and are characterized by over-
production of mature blood cells. e proliferation
of one single cell type predominates, resulting in
increased numbers of granulocytes (CML), erythro-
cytes (PV), platelets (PT), or broblasts (PMF). A
single, acquired point mutation in the Janus kinase
2 (JAK2) gene occurring in the majority of patients
with PV and almost half of those with ET and PMF
was discovered in 2005. e mutation is a guanine to
thymidine substitution that substitutes phenylalanine
for valine at position 617 (V617F) of the JAK2 pro-
tein.isresidueislocatedwithintheJH2pseudo-
kinase domain, which negatively regulates the JH1 cat-
alytically active kinase domain. e wild-type JAK 2
protein binds to multiple cytokine receptors including
the erythropoietin, thrombopoietin, and granulocyte-
colony stimulating factor receptors that are essen-
tial for hemopoietic stem cell biology and dieren-
tiation. e JAK2 protein with the V617F mutation
enables constitutive, cytokine independent activation
of the JAK-STAT, PI3K, and MAPK signal transduc-
tion pathways at various stages of development and in
various lineages of hemopoietic cells (Fig. 19.1). Four
further mutations aecting the JAK2 exon 12 that were
recently identied dene a distinctive myeloprolifer-
ative syndrome that aects patients who previously
received a diagnosis of PV or idiopathic erythrocyto-
sis. Finally, two further mutations in the thrombopoi-
etin receptor MPL W515L/K have been described in
patients with PMF(5%) and PT(1%). e reported
mutations have been shown to produce an MPD-like
phenotype in various murine models.
Pathogenesis of placental infarction
and thrombosis
rombosisisconsistentlyidentiedastheleading
cause of maternal mortality in apparently healthy nor-
mal pregnancies. rombotic occlusion of the placen-
tal circulation may be a late manifestation of placental
dysfunction or an independent mechanism of preg-
nancy morbidity. In women with PT, placental throm-
bosis was reported in pregnancies which resulted in
late fetal loss, pre-term delivery, and IUGR. IUGR,
which is associated with utero-placental dysfunction,
is known to occur in other acquired and inherited
causes of thrombophilia.
Multiple factors are likely to contribute to the
pathogenesis of thrombosis in MPDs, including the
degree of thrombocytosis, leukocytosis, raised hema-
tocrit, activation of platelets and leukocytes, the for-
mation of platelet leukocyte aggregates, circulating
prothrombotic and endothelial factors, and their inter-
actions.6It is of interest to our understanding of how
the MPD phenotype contributes to placental dysfunc-
tion that independently, studies of MPDs and pre-
eclampsia both report increased platelet activation,
platelet monocyte aggregate formation, and micropar-
ticle formation. It is currently unclear whether the
presence of the JAK2 V617F and MPL W515L/K muta-
tions increase the risk of thrombosis in MPDs and,
if so, by what mechanism. However, recently a study
of 103 pregnancies in 62 women with PT, identied
the JAK2 V617F mutation as an independent predic-
tor of pregnancy complications (P =0.01). A total of
17 (71%) of 24 women carrying the JAK2 mutation had
complicationsatrstpregnancy.estudyconcluded
that women with PT with the JAK2 V617F mutation
had a twofold higher risk of developing complications
than those without the mutation. A recent matched
case control study of unexplained rst pregnancy loss
involving 32 683 patients is in support of these nd-
ings. In 3496 pairs of women the JAK2 V617F muta-
tion occurred more frequently in patients with preg-
nancy loss (1.06%) than in control subjects (0.20%).
Diagnosis
e diagnosis of MPDs in pregnancy requires an
increased awareness of these disorders occurring in
pregnant women. Suspicion may be secondary to an
abnormal full blood count, a thrombotic, or hemor-
rhagic event and should prompt referral to a hema-
tologist. In view of pregnancy morbidities and the
likelihood of improved outcome with intervention,
these women benet from a diagnosis being made
pre-conceptually, during pregnancy or the postpar-
tum period. e following section details local policy
including adaptation for diagnostic investigations in
pregnancy. 233
Section 7. Malignant conditions
Intracellular domain
Extracellular domain
Wild-type JAK2 without
erythropoeitin
Wild-type JAK2 with
erythropoeitin
JAK2 with V617F mutation
without erythropoeitin
STAT dimer
STAT
EPO
JAK JAK
V617F
P
PP
PP
PPP
P
PP
PP
P
STAT dimer
P13K
Nucleus
Enhanced DNA transcription
P13K
RAS-MAPK RAS-MAPK
Fig. 19.1 Jak-Stat pathways.
Primary thrombocythemia
ere is no diagnostic hallmark for this condition.
ediagnosisismadebyexcludingotherMPDs
and a reactive or secondary cause of a thrombocy-
tosis. Causes of a reactive thrombocytosis include
iron deciency anemia, chronic inammation (e.g.
rheumatoid arthritis, or inammatory bowel disease),
splenectomy, acute hemorrhage, and malignant dis-
ease. Where conditions co-exist that may cause a
reactive thrombocytosis, this may make the diagno-
sismoredicult.Inpregnancytheplateletcountmay
fall especially during the second and third trimesters,
thereby masking the diagnosis.
Historically, the diagnostic criteria for PT were
those of the polycythemia vera study group; 40 years
on, continual development of the diagnostic criteria
for MPDs set the stage for the World Health Orga-
nization diagnostic criteria 2001, modied in 2007.
e revised WHO criteria require characteristic bone
marrowmorphology(thisisacontroversialaspectnot
universally accepted), a platelet threshold of 450 ×
109/L and molecular analysis for the JAK 2 V617F
mutation and other clonal markers. Investigations
should include a blood count, blood lm, hema-
tinics, renal, and liver prole, CRP, ANA and RhF,
genetic screen for the JAK2 V617F, MPL W515L/K,
and bcr/abl mutation and abdominal ultrasound scan.
Polycythemia vera
An erythrocytosis requiring investigation is dened
as a packed cell volume (PCV) greater than 0.48 in
234

Chapter 19. Myeloproliferative disorders
non-pregnant women; in pregnancy this threshold has
not formally been dened. To determine whether there
is an absolute increase in PCV or an erythrocytosis
or an apparent increase due to reduced plasma vol-
ume has traditionally required a red cell mass study,
which would be contraindicated in pregnancy. Red
cell mass scans have been largely superseded by test-
ing for the presence of the JAK2 V617F mutation,
which indicates the presence of the majority of PV
cases. e JAK2 V617F mutation negative erythrocy-
tosis cases may still be PV without a genetic marker or
with a JAK2 exon 12 mutation; alternatives include a
pseudo/apparent, primary congenital, secondary con-
genital or acquired, or an idiopathic erythrocytosis, all
of which require denition.
e current British Committee for Standards in
Haematology guideline for investigation and manage-
ment of erythrocytosis7suggest a staged approach
to investigation as the dierential diagnosis is broad
and secondary causes must be excluded. is is fol-
lowed by investigations to conrm or refute a diag-
nosis of a JAK2 V617F positive PV. e majority of
patients (excluding borderline erythrocytosis) and all
ex- and current smokers will require a chest X-ray.
is should be avoided in pregnancy unless there is
a strong suspicion of a causative lung pathology, in
which case appropriate screening should be used. Uri-
nalysisisasimpleeectivescreenforrenaldisease,
which should be performed in all patients at the ini-
tial visit. Patients may present with co-morbidity, thus
regardlessofadiagnosisofPVareviewofsecondary
causes is pertinent. Additional investigation of possi-
ble secondary causes will vary according to symptoms
or signs present.
Myelobrosis
Myelobrosis is very rare indeed in women of child-
bearingage.Toachievethisdiagnosisitisnecessary
to exclude other MPDs (PV, PT and CML) as well as
disordersinwhichmarrowbrosiscandevelopasa
secondary feature such as metastatic carcinoma, lym-
phoma, irradiation, TB, and leishmaniasis. e fol-
lowing features are generally necessary to conrm a
diagnosis of MF: splenomegaly, increased bone mar-
row brosis (coarse reticulin bers arranged in parallel
in trephine biopsy), a leukoerythroblastic blood lm
(immature red cells and myeloid precursors with tear
drop-shaped red cells) and the exclusion of secondary
causes of myelobrosis (see above). In all suspected
cases of MF a bone marrow aspirate and trephine are
required.
Treatment options
WomenofreproductiveagewithadiagnosisofMPD
should receive information and assurance regarding
management and outcome of future pregnancies. If
fertility issues arise, optimal disease management may
need to be re-addressed prior to a timely referral
for standard fertility investigation. A risk assessment
according to disease status, concomitant illnesses, and
prior obstetric history forms the basis for a discussion
of the risks and benets of therapeutic options in preg-
nancy. According to perceived risk, the therapeutic
options include aspirin, heparin, venesection, cytore-
ductive agents, and thromboembolic deterrent stock-
ings. From pre-conceptual planning to the postpartum
period, access to joint care from an obstetrician with
experience of high risk pregnancies and a hematolo-
gist in a multidisciplinary setting is paramount.
e pre-conception to postpartum management
plan should include:
rinformed multidisciplinary care and education;
rrisk assessment and discussion of therapeutic
options and implementation of an appropriate
management plan;
radditional monitoring during pregnancy;
rfurther optimization of disease control, if fertility
is an issue, prior to timely referral for standard
investigation;
rA comprehensive delivery and postpartum plan.
is approach enables optimal disease control with
the aim of increasing the possibility of conception,
implantation, and maintenance of placental function.
us reducing complications secondary to placental
dysfunction, such as IUGR and pregnancy losses. An
emphasis upon the prevention of thrombosis and hem-
orrhage and management of events pre- and postpar-
tum is also required.
Two key treatment aims, in high risk non-pregnant
patients with MPDs, are to attain a platelet count less
than 400 ×109/L and a PCV less than 0.45 and possibly
less than 0.42 in women who remain symptomatic. In
pregnancy an increase in the plasma volume reduces
both the platelet count and PCV, which is likely to
further alter blood cell rheology. Interestingly, it has
been suggested that the decrease in platelet count is
greater than that expected in a normal pregnancy. One 235

Section 7. Malignant conditions
theory is whether the placenta produces an interferon-
like substance. An understanding of this physiological
dilutional eect and the brisk return to pre-pregnancy
levels in the postpartum period in MPD pregnan-
cies, are important when considering optimal moni-
toring intervals and suitable treatment targets. e tar-
get PCV and platelet count in pregnant women with
MPDs are ongoing debates, but appropriate targets are
probably a platelet count of less than 400 ×109/L and
a hematocrit certainly less than 0.45 and probably in
the mid gestation appropriate range.
Aspirin
Low dose aspirin is considered safe in pregnancy in
accordance with the Collaborative Low dose Aspirin
Study in Pregnancy (CLASP), although its use for
thromboprophylaxis in MPDs has never been assessed
by a controlled trial. e European Collaborative Low-
dose Aspirin in Polycythaemia Vera (ECLAP) study
supports the use of low dose aspirin in non-pregnant
patients with PV. Aspirin has been the most widely
used therapy (in at least half of published pregnan-
cies) for pregnancies aected by PT. Although the evi-
dence is both retrospective and based on small num-
bers, the use of low dose aspirin in pregnancy in myelo-
proliferative disorders seems advantageous, and a low
risk strategy for the pregnancy. A recent update of
the largest case series of pregnant women with PT to
date provided analysis of pregnancy outcomes treated
with aspirin vs. those managed by observation alone.
ere was no evidence that therapy with aspirin pos-
itively inuenced pregnancy outcome in women with
PT; however, interpretation should be cautious as with
all retrospective reports.
Low molecular weight heparin
A study of women with the Factor V Leiden, pro-
thrombin gene mutation or protein S deciency, and
one fetal loss demonstrated aspirin to be inferior to
low molecular weight heparin (LMWH) in terms of
live birth rate and birth weight. e successful use of
LMWH in other pregnancies at high risk of thrombo-
sis and in reducing fetal morbidity has drawn attention
to the possibility of its use, in addition to aspirin, dur-
ing pregnancy in women with MPDs with a previous
thrombosis or pregnancy-related events.
Our regime for LMWH use, if necessary, in
pregnant patients with MPDs is:
rante-natal dose of LMWH, e.g. 40 mg enoxaparin
daily or 5000 IU dalteparin daily, increasing to 12
hourly from 16 weeks onwards;
rat low body weight (e.g. ⬍50 kg), lower doses of
LMWH may be required, e.g. 20 mg enoxaparin
daily or 2500 IU dalteparin daily;
rIn obese patients (e.g. BMI ⬎30 in early
pregnancy), higher doses of LMWH may be
required, e.g. 40 mg enoxaparin 12 hourly or 5000
IU dalteparin 12 hourly;
rpostpartum dose of LMWH, e.g. 40 mg
enoxaparin daily or 5000 IU dalteparin daily for 6
weeks;
rif uterine artery Dopplers are repeatedly
abnormal, increase to a therapeutic dose of
LMWH.
In women with previous arterial thrombotic events
or in those with recurrent thrombosis on warfarin
prior to pregnancy, therapeutic doses of subcutaneous
LMWH may be required. Some patients may require
monitoring of anti-Xa levels.
Graduated elastic compression
stockings (GECS)
GECSmaybeusedante-natallyandduringthepost-
partum period. ere are no trials to support such
practice, but the British Society for Haematology
(BSH) guidelines suggest that all women with previ-
ous venous thrombosis or a thrombophilia should be
encouraged to wear GECS throughout their pregnancy
and for 6–12 weeks aer delivery.
Cytoreductive therapy
Cytoreduction is used where necessary to reduce the
plateletcountoraraisedPCVthatisresistanttovene-
section, but these agents should preferably be avoided
in pregnancy, particularly in the rst trimester. None
of the cytoreductive drugs mentioned in this chap-
ter have a product licence for use in pregnancy. e
expected natural fall of the platelet count and hema-
tocrit during pregnancy may reduce the need for
cytoreduction or venesection. However, in high risk
situations where cytoreduction is deemed necessary
(see below), interferon ␣(IFN-␣) is the drug of choice.
ere are no reports of teratogenic eects in animals
or adverse eects in the admittedly small numbers of
pregnancies exposed to this drug. However, some evi-
dence suggests that IFN-␣may decrease fertility and
236
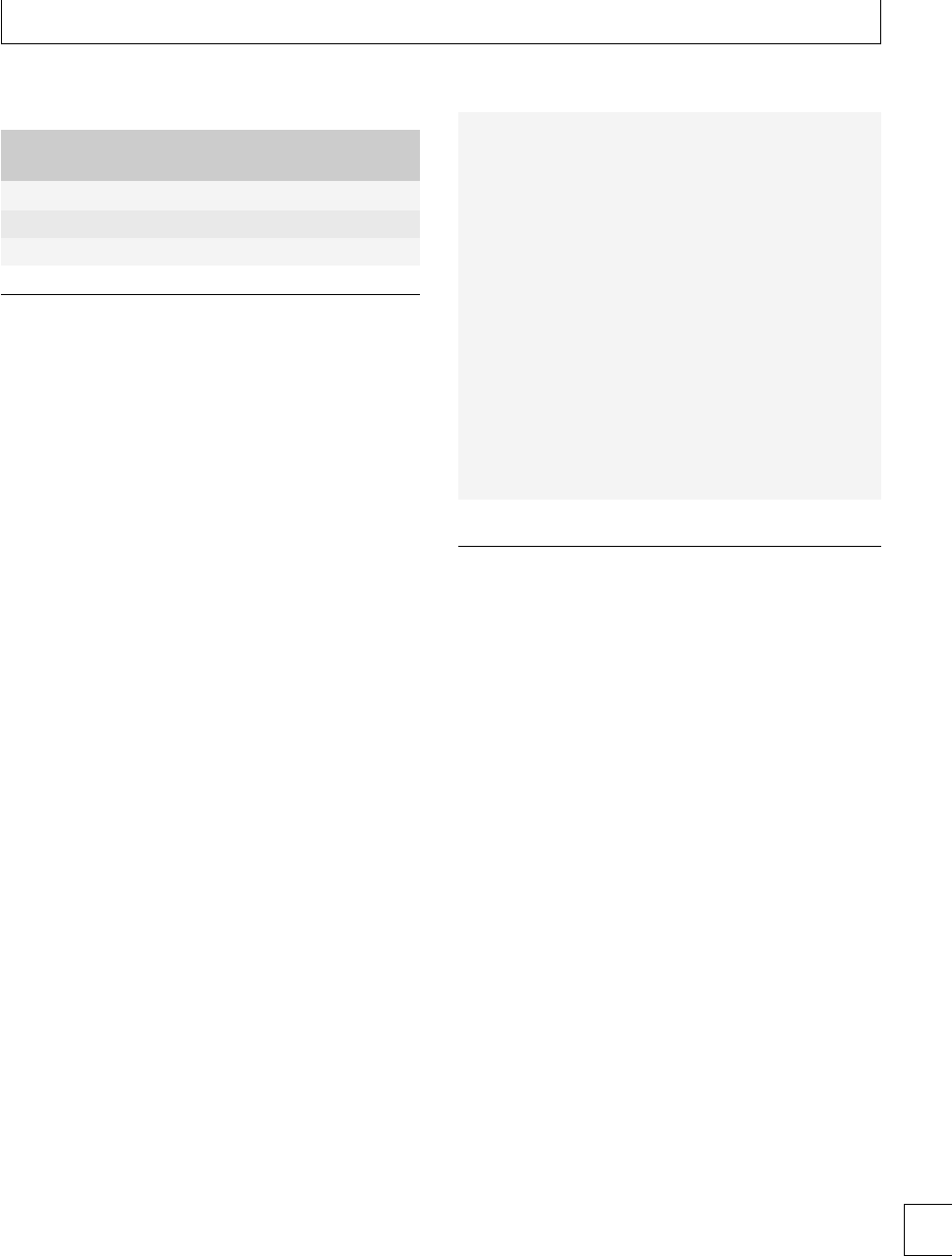
Chapter 19. Myeloproliferative disorders
Table 19.4 95% ranges for hematological variables during
pregnancy
Gestation
First
trimester
Second
trimester
Third
trimester
Hb (g/dL) 11–14.3 10–13.7 9.8–13.7
PCV (l/L) 0.31–0.41 0.30–0.38 0.28–0.39
Platelet count (109/L) 174–391 171–409 155–429
Adapted from ref. 8.
so it is best avoided in women with diculty conceiv-
ing. In relation to hydroxyurea, the outcomes of small
numbers of pregnancies have been published and
these are mainly without fetal complications, although
one stillbirth and one malformed infant have been
reported aer exposure to hydroxyurea. Teratogenic-
ity in animals has also been reported. us the use of
hydroxyurea is probably contraindicated at the time
of conception and during pregnancy. e use of ana-
grelide in pregnancy is similarly not recommended
because of insucient documentation of its use in this
situation and because of the possibility of thrombocy-
topenia in the fetus.
Venesection
Although the natural fall in the hematocrit or PCV
in pregnancy may obviate the need for venesection, it
is an option in resistant cases. If venesection fails to
control the PCV, then cytoreduction should be consid-
ered. e target PCV for a pregnant woman has yet to
be determined. A reasonable target PCV would be the
middle of the gestation appropriate range (Table 19.4).
ere is currently no evidence for maintaining it lower
than this in pregnancy, although this has been an area
of controversy.
Recommendations for management
of MPDs in pregnancy
An overview of the small groups of MPD patients and
individual cases described in the literature does not
enable condent management guidelines to be drawn
up. e following are recommendations based on cur-
rent knowledge of this and other thrombophilic states
and upon personal experience in a tertiary referral
unit. Good communication between consultant obste-
trician and hematologist is essential.
Table 19.5 High risk MPD pregnancy criteria
rPrevious venous or arterial thrombosis in mother (whether
pregnant or not)
rPrevious hemorrhage attributed to MPD (whether pregnant
or not)∗
rPrevious pregnancy complication that may have been
caused by a MPD:
–
Three pregnancy losses
⬍
10 weeks
–
One or more pregnancy losses
⬎
24 weeks
–
Intra-uterine growth restriction or other evidence of
placental dysfunction
–
Intra-uterine death or stillbirth (with no obvious other
cause)
–
A significant ante- or postpartum hemorrhage
(requiring red cell transfusion)
–
Severe pre-eclampsia (necessitating preterm delivery
⬍
37 weeks)
rPlatelet count rising to ⬎1500 ×109/L prior to pregnancy
or during pregnancy∗
rDiabetes mellitus or hypertension requiring treatment
rJAK2 V6127F mutation, the status of this as a risk marker is
currently unclear
Note ∗these criteria would be an indication for cytoreductive
treatment but
not
LMWH.
Pre-conceptual meeting
e patient should ideally have a pre-conceptual meet-
ing with both an obstetrician and a hematologist to
discuss a plan of management for a future pregnancy,
including the necessity for cytoreductive therapy. Ide-
ally, this should be written out and copied to the
patient.
Control of platelet count and haematocrit
If a patient is already taking hydroxyurea or anagrelide,
this should be gradually withdrawn before conception,
followed ideally by a wash-out period of 3 months for
hydroxyurea following the last dose. e platelet count
and PCV must be closely monitored thereaer. Care-
ful venesection should be commenced if the hema-
tocrit rises above the gestational appropriate range.
Cytoreduction with IFN-␣may be necessary in cases
with a raised PCV resistant to venesection or persistent
thrombocytosis. Most patients with a clear indication
for cytoreductive therapy pre-pregnancy will require
cytoreduction during pregnancy.
Cytoreduction with IFN-␣may also be necessary
if any of the factors are present, or if they develop
in the index pregnancy, which in our experience
denes a high risk MPD pregnancy (Table 19.5). Treat-
ment should be guided by monitoring the full blood
count and by maintaining the platelet count less than 237

Section 7. Malignant conditions
400 ×109/L and the PCV in the appropriate gesta-
tional range.
Management of thrombotic risk
Assessment of need for antithrombotic medication
e assessment of the need for antithrombotic medi-
cation should preferably be done in the pre-conceptual
meeting, but on-going individual risk assessment
should occur and may warrant commencing or
increasing thromboprophylaxis.
Aspirin
In the absence of clear contraindications, i.e. asthma,
history of peptic ulceration, or current hemorrhage,
all patients should be on aspirin (initially 75 mg once
daily) throughout the pregnancy and for at least 6
weeks aer delivery. In the event of a platelet count in
excess of 1000 ×109/L, acquired von Willebrand dis-
ease should be excluded prior to commencing aspirin.
Low molecular weight heparin
Consider the use of subcutaneous LMWH during
pregnancy in addition to cytoreduction in patients
with any of the high risk MPD pregnancy factors
listed in Table 19.5 with the exception of hemorrhage
and extreme thrombocytosis. LMWH is an option to
be introduced in patients with persistently abnormal
uterine artery Dopplers. Once adequate hemostasis
has been achieved postpartum, all women should be
oered 6 weeks of LMWH thromboprophylaxis in the
absenceofapriorhistoryofasignicanthemorrhage.
Caution should be applied to cases where women have
aprevioushistoryofasignicanthemorrhagewith
aplateletcount⬍1000 ×109/L and no other obvi-
ous cause except for platelet dysfunction secondary to
aMPD.
Graduated elastic compression stockings
Consider the use of GECS as a supplementary therapy
throughout pregnancy and for 6–12 weeks aer deliv-
ery in accordance with current BSH guidelines.
Maternal and fetal monitoring
Maternal monitoring
Full blood count monitoring, blood pressure, and
urine testing should be performed 4 weekly until 24
weeks and thereaer at 2-weekly intervals.
Fetal monitoring
e local protocol for fetal monitoring includes scans
at 12 and 20 weeks. If the uterine artery Doppler ultra-
soundat20weeksisabnormal,itshouldberepeated
at 24 weeks; if abnormal then, consideration should
be given to increasing or escalating therapy. Regular
growth scans should also be performed.
Uterine artery doppler scanning at 20 and 24 weeks
will reveal whether the woman has bilateral notch-
ing. e persistence of bilateral notching indicates
increased resistance to ow and possible placental dys-
function. e presence of persistent bilateral notching
at 24 weeks should prompt commencing or increasing
LMWH to 40 mg s/c twice daily, and further treatment
escalation including interferon ␣and the possibility of
an early delivery may need to be considered.
Delivery
Prior to labor or Cesarean section, it is important to
discuss the implications of the use of thromboprophy-
laxis for epidural or spinal anesthesia with the woman
and obstetric anesthetist following locally agreed pro-
tocols. If a woman develops a hemorrhagic problem
while on LMWH, the treatment should be stopped
and hematological advice sought; a platelet transfu-
sion may be useful in patients with MPDs. It should be
remembered that excess blood loss and blood transfu-
sion are risk factors for VTE, so thromboprophylaxis
should be begun or reinstituted as soon as the imme-
diate risk of hemorrhage is reduced. e third stage of
labor should be managed actively.
Postpartum thromboprophylaxis
e time of greatest risk for VTE associated with
pregnancy is the immediate puerperium period. e
prothrombotic pregnancy phenotype does not revert
back to normal until 6 weeks aer delivery. All MPD
patients should receive 6 weeks of postpartum LMWH
thromboprophylaxis unless contraindicated. Aspirin
should also be continued for at least 6 weeks. As dis-
cussed above, where women have a history of hemor-
rhage, the addition of postpartum heparin should be
cautiousandconsideredonanindividualcasebasis.
Postpartum assessment
e platelet count and PCV may rise dramatically
postpartum, but can usually be controlled with cytore-
ductive therapy or venesection. Cytoreductive therapy
238

Chapter 19. Myeloproliferative disorders
suitable immediately post-delivery if required include
hydroxyurea, IFN-␣and anagrelide, the choice and
dose depending on previous experience in that patient.
Hydroxyurea, IFN-␣, and possibly anagrelide are
excreted in breast milk, so breast feeding is then con-
traindicated. In the current literature there is no evi-
dence that pregnancy predisposes MPD patients to
acceleration of their disease to PMF or acute leukemia,
nor would this be anticipated. e most signicant
risk is of thrombosis in the mother and adverse fetal
outcome. A frequent question asked by these patients
is what is the chance of their children being aected
by MPD. Until recently, it was believed that famil-
ialMPDwasrelativelyrareand,whilstthisistrue
for large kindreds with many aected individuals, it
has become apparent that up to 8% of MPD patients
may have an aected relative, usually a cousin, aunt,
uncle, etc. Parent:child combinations are extremely
uncommon and routine testing of children is not
recommended.
Dilemmas
e following section includes a series of challenging
cases, which enable discussion of management options
accordingly in these women with complex pregnan-
cies.
Case 1
A 37-year-old lady with a diagnosis of PT is referred
for a tertiary opinion regarding conception and
pregnancy management. The referral to a
hematologist and initial diagnosis of PT followed a
full blood count screen by her GP. The obstetric
history includes one full-term spontaneous vaginal
delivery 12 years previously. Following remarriage
3 years ago, she has undergone two spontaneous
miscarriages ⬍10 weeks. The current platelet count
is 1700 ×109/L, current medication includes aspirin
75 mg daily.
In an attempt to establish when the patient devel-
oped PT prior to the ocial diagnosis, any previous
full blood counts could be reviewed. However, this is
unlikely to change the management in this case. e
diagnosisofPTandobstetrichistoryaresuggestiveof
poor pregnancy outcome secondary to PT. e platelet
count enables us to stratify the patient as being at high
risk of a vaso-occlusive event and outside of pregnancy
would suggest benet from commencing cytoreduct-
ive therapy. With the stated aim to conceive in the near
future, the appropriate cytoreductive agent would be
interferon ␣as this could be continued throughout
pregnancy. A screen for an acquired von Willebrand’s
disease should be completed prior to continuation of
aspirin. e woman should be monitored in her local
clinic and the dose of interferon adjusted to main-
tain a platelet count ⬍400 ×109/L.Ifapregnancy
is conrmed, monitoring according to the treatment
algorhythm should commence and follow-up could be
shared between the local hospital and tertiary refer-
ral unit. e patient does not currently meet the cri-
teria to commence LMWH ante-natally, although may
benet from heparin and aspirin for 6 weeks post-
partum. However, a discussion within the multidisci-
plinary pre-conceptual meeting in view of patients age
and prior obstetric history may conclude the addition
of LMWH from conception as suitable on an individ-
ual basis regardless that the history is of two not three
miscarriages ⬍10 weeks. Clearly, these complex cases
needtobemanageduponanindividualbasisandman-
agement plans may need to encompass aspects which
are outside of general guidance. Close collaboration
between the local hematology and obstetric unit and
the tertiary center may enable delivery outside of the
tertiary center dependent upon the progress of the
individual pregnancy.
Case 2
A pregnant 26-year-old woman diagnosed with PV
following investigation of menorrhagia and
epistaxis 4 years previously is referred for an opinion
at 9 weeks’ gestation. There is no prior obstetric
history, and the current management is low dose
aspirin and venesection. Her current blood count
reveals Hb 15 g/dL, HCT 0.47, plt 567 ×109/L.
e history of hemorrhage and or epistaxis may
be attributed to MPD in this case and, if so, the
woman would meet the criteria to consider interferon
in this pregnancy, but this is a relatively so indica-
tion and the authors would not use this treatment in
this setting. However, this history should be exam-
inedcarefullyashemorrhageisrareinthesecon-
ditions and, even though the platelet count is not
markedly abnormal, it would be wise to screen for
vonWillebrand’sdisease.Withnohistoryofthrom-
botic events or pregnancy complications, there is no
indication for ante-natal heparin prophylaxis unless 239
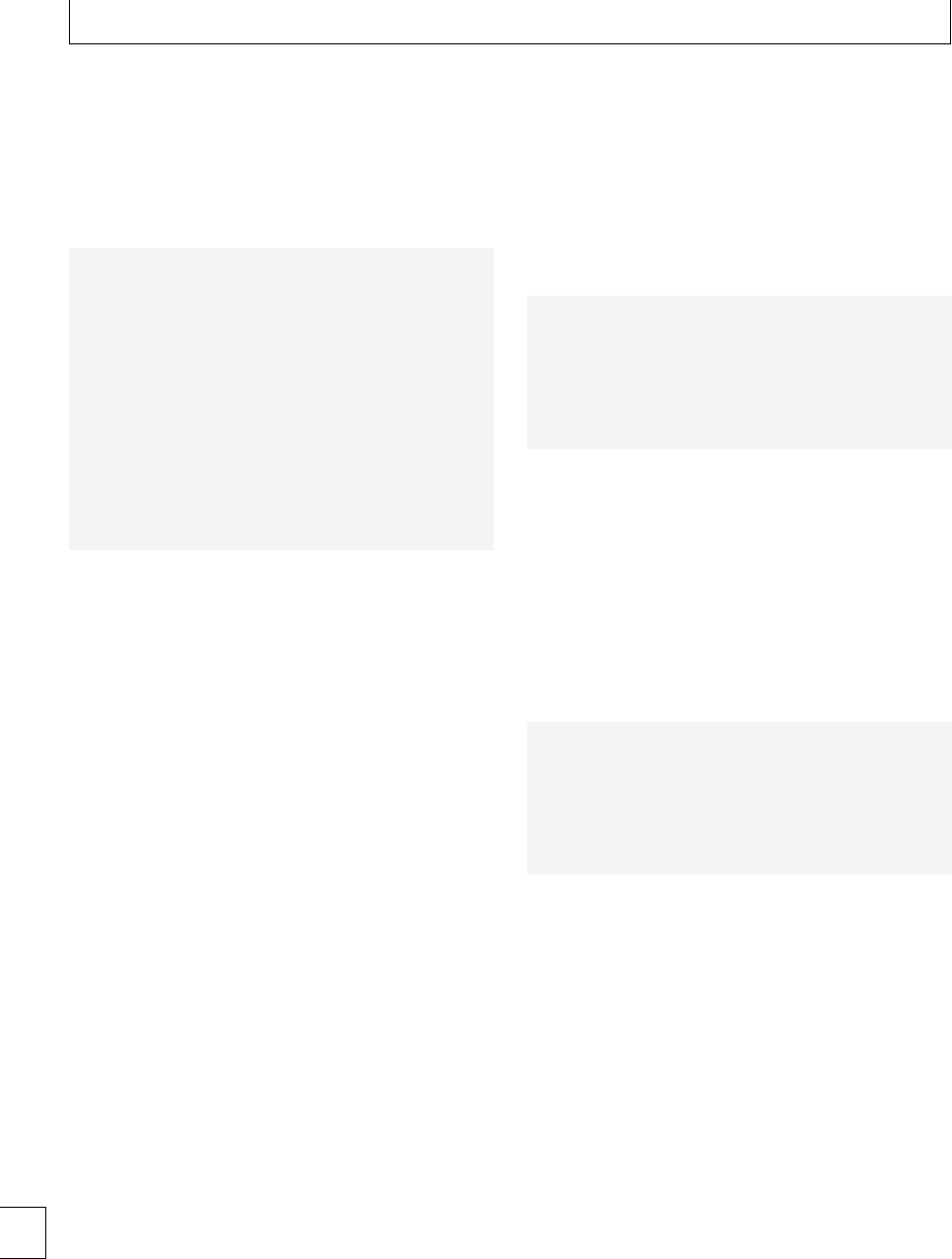
Section 7. Malignant conditions
uterine artery dopplers subsequently suggest impaired
placental function; heparin should be given for 6 weeks
postpartum. Aspirin should be continued throughout
pregnancy. e current PCV is outside the appropriate
range for the rst trimester and venesection should be
considered if tolerated.
Case 3
A 32-year-old woman diagnosed with PMF 3 years
ago following a recent hepatic vein thrombosis
attends clinic to discuss treatment options
regarding future pregnancies. She has one healthy
daughter of 5 years delivered by Cesarean section
following a trial of labor which failed to progress.
Current medication includes aspirin, warfarin, and
hydroxyurea. The question of future cord blood
stem cell storage is also raised. Her current blood
count in Hb 11 g/dL, PCV 0.35, plt 147, Wbc 7.6 ×
109/L.
In the pre-conception planning, a management
plan regarding anticoagulation, cytoreductive therapy,
and review of concomitant liver disease is required.
e issues of stem cell storage needs to be addressed
regarding reasoning and practicalities. Clearly, the
woman may have a personal interest in cord stem
cell storage and this needs to be addressed pre-
conceptually. is complex case would benet from
follow-up and delivery in a tertiary referral center.
e aspirin should be continued throughout preg-
nancy; testing should be performed in the fortnight
following a possible conception in order to stop the
warfarin as early as possible and commence LMWH.
In view of the history of a hepatic vein thrombo-
sis, a therapeutic dose of LMWH should be used
and switched back to warfarin postpartum. ree
months prior to conception the hydroxyurea needs
to be stopped and interferon commenced. Optimiza-
tion of any concomitant liver pathology and portal
hypertension secondary to the previous hepatic vein
thrombosis is important. e obstetric history needs
to be reviewed in light of whether portal hypertension
and varices are present. e presence of varices may
require banding and additional medication, which
should be instigated and followed up by the gastroen-
terology team. e planned mode of delivery accord-
ing to concomitant pathology needs to be addressed in
a multidisciplinary meeting.
Case 4
A pregnant 31-year-old female with diabetes is
referred at 10/40 with a platelet count of 759 ×
109/L from the combined endocrine obstetric clinic
and is subsequently diagnosed with PT.
Althoughthereisnopreviousobstetrichistory,a
diagnosis of diabetes suggests a high risk pregnancy.
Outside of pregnancy, the patient would be in the high
risk of vaso-occlusive event group secondary to the
diagnosis of diabetes. Low dose aspirin should be com-
menced and continued postpartum. A prophylactic
dose of LMWH once daily increased to twice daily at
16 weeks followed by 6 weeks’ postpartum prophylac-
tic once daily LMWH should be considered. Interferon
wouldalsobeconsideredinthiscase.
Case 5
A 29-year-old pregnant woman with a diagnosis of
PT with no prior obstetric history or thrombotic
events attends clinic at 15/40.
Hb 12 g/dL, HCT 0.34, Plt 580 ×109/L.
Low dose aspirin throughout pregnancy continued
indenitely postpartum, combined with 6 weeks post-
partum heparin prophylaxis would be appropriate in
this case. Additional treatment will depend upon how
the pregnancy progresses.
240

Chapter 19. Myeloproliferative disorders
References
1. Barbui T, Finazzi G. Myeloproliferative disease in
pregnancy and other management issues. Hematology
2006; 1: 246–252.
2. Robinson S, Bewley S, Hunt BJ, Radia DH, Harrison
CN. e management and outcome of 18 pregnancies
in women with polycythemia vera. Haematologica
2005; 90: 1477–1483.
3. Tupule S, Bewley S, Robinson SE et al. e
management and outcome of 4 pregnancies in women
with idiopathic myelobrosis. British Journal of
Haematology 2008; 142: 480–482.
4. Griesshammer M, Struve S, Harrison CM. Essential
thrombocythemia/polycythemia vera and pregnancy:
theneedforanobservationalstudyinEurope.
Seminars in rombosis and Hemostasis 2006; 32:
422–429.
5. Elliott MA, Teeri A. rombocythaemia and
pregnancy. Best Practice Research Clinical
Haematology 2003; 16: 227–242.
6. Harrison CN. Platelets and thrombosis in
myeloproliferative diseases. Hematology 2005; 1:
409–415.
7. McMullinMF,BarefordD,CampbellPet al.
Guidelines for the diagnosis, investigation and
management of polycythaemia/erythrocytosis.
British Journal of Haematology 2005; 130: 174–
195.
8. Bain BJ. Blood Cells, 3rd edn. Oxford, UK: Blackwell
Science, 1995.
9. Bellucci S, Janvier M, Tobelem G et al.Essential
thrombocythemias. Clinical evolutionary and
biological data. Cancer 1986; 58: 2440–2447.
10. BeardJ,HillmenP,AndersonCCet al.Primary
thrombocythaemia in pregnancy. British Journal of
Haematology 1991; 77: 371–374.
11. LeoneG,DeS,V,D’AddosioA.[Essential
thrombocythemia pregnancy]: Haematologica 1991;
76: 365–367.
12. Pagliaro P, Arrigoni L, Muggiasca ML et al.Primary
thrombocythemia and prgnancy: treatment and
outcome in een cases. American Journal of
Hematology 1996; 53: 6–10.
13. RandiML,RossiC,FabrisF,GirolamiA.Essential
thrombocythemia in young adults: treatment and
outcome of 16 pregnancies. Journal of Internal
Medicine 1999; 246: 517–518.
14. Cincotta R, Higgins JR, Tippett C et al. Managament
of essential thrombocythaemia during pregnancy.
Australia and New Zealand Journal of Obstetrics and
Gynaecology 2000; 40: 33–37.
15. Bangerter M, Guthner C, Beneke H
et al. Pregnancy in essential thrombocythaemia:
treatment and outcome of 17 pregnancies.
European Journal of Haematology 2000; 65: 165–
169.
16. Wright CA, Teeri A. A single institutional experience
with 43 pregnancies in essential thrombocythemia.
European Journal of Haematology 2001; 66: 152–
159.
17. Candoni A, Fanin R, Michelutti T et al. Pregnancy and
abortion in women with essential thrombocythemia.
American Journal of Hematology 2002; 69: 233–234.
18. Niittyvuopio R, Juvonen E, Kaaja R et al. Pregnancy in
essential thrombocythaemia: experience with 40
pregnancies. European Journal of Haematology 2004;
73: 434–436.
19. Crowley JP, Barcohana Y, Sturner WQ. Disseminated
intravascular coagulation following rst trimester
abortion in polycythemia vera. Increased platelet
count apparently contributed to fatal outcome.
Rhode Island Medical Journal 1987; 70: 109–
112.
20. Centrone AL, Freda RN, McGowan L. Polycythemia
rubra vera in pregnancy. Obstetrics and Gynecology
1967; 30: 657–659.
21. Ruch WA, Klein RL. Polycythaemia vera and
pregnancy: report of a case. Obstetrics and Gynecology
1964; 23: 107–111.
22. Subtil D, Deruelle P, Trillot N, Jude B. Preclinical phase
of polycythemia vera in pregnancy. Obstetrics and
Gynecology 2001; 98: 945–947.
23. Hochman A, Stein JA. Polycythemia and pregnancy.
Report of a case. Obstetrics and Gynecology 1961; 18:
230–235.
24. Harris RE, Conrad FG. Polycythemia vera in the
childbearing age. Archives of Internal Medicine 1967;
120: 697–700.
25. Ruggeri M, Tosetto A, Castaman G, Rodeghiero F.
Pulmonary embolism aer pregnancy in a patient with
polycythemia vera. American Journal of Hematology
2001; 67: 216–217.
26. Pata O, Tok CE, Yazici G et al. Polycythemia vera and
pregnancy: a case report with the use of hydroxyurea
in the rst trimester. American Journal of Perinatology
2004; 21: 135–137.
27. Robinson S, Bewley S, Hunt BJ et al. e management
and outcome of 18 pregnancies in women with
polycythemia vera. Haematologica 2005; 90;
1477–1483.
2
8
. TaylorUB,BardeguezAD,IglesiasN,GasconP.
Idiopathic myelobrosis in pregnancy: a case report 241

Section 7. Malignant conditions
and review of the literature. American Journal of
Obstetrics and Gynecology 2004; 167: 38–39.
29. Gotic M, Cvetkovic M, Bozanovic T, Cemerikic V.
[Successful treatment of primary myelobrosis
with thrombocytosis during pregnancy with
alfa-interferon] Srp Arh Celok Lek 2001; 129: 304–308.
30. Tulpule S, Bewley S, Robinson SE, Radia D,
Nelson-Piercy C, Harrison CN. e management and
outcome of 4 pregnancies in women with idiopathic
myelopbrosis. British Journal of Haematology 2008;
142: 480–482.
242

Section 7 Malignant conditions
Chapter
20 Eects of chemoradiotherapy
for hematological malignancy
on fertility and pregnancy
Seonaid Pye and Nina Salooja
Introduction
Advances in treatment for hematological malignancies
over the last two decades have led to marked improve-
ments in survival, and for many patients this trans-
lates to cure. Consideration of the long-term seque-
lae of treatments administered are therefore becoming
increasingly relevant to the overall management strate-
gies of these disorders and, since many of the poten-
tially treatable hematological cancers occur in chil-
dren and young adults, this includes concerns about
future fertility. e chemotherapy agents and radio-
therapy used to treat leukemias and lymphomas can
aect reproductive potential in a variety of ways. In
this chapter we will consider:
rthe general eects of chemoradiotherapy on
female fertility;
rthe incidence of infertility following radiation and
chemotherapy;
rthelikelyoutcomeofpregnancyinapatient
treated for hematological malignancy;
rthe strategies available for the preservation of
fertility in patients who require potentially
sterilizing treatment.
General eects of chemoradiotherapy
on fertility
Normal reproduction requires interplay between the
gonads and the hypothalamic–pituitary–endocrine
axis. In addition, the uterus must be receptive to
implantation and capable of eecting appropriate
growth in pregnancy. Damage to hormone producing
cells in the hypothalamus, pituitary, or gonads can lead
to infertility as well as more direct damage to the germ
cells, reproductive tracts, or sexual organs.
In females the production of germ cells (oocytes)
ceases before birth. ereaer, the number of oocytes
decrease throughout life, either by a mechanism of pre-
programmed cell death (physiological apoptosis) or
else post-menarche, in menstruation. When the num-
ber of oocytes falls below a critical number, ovulation
and ovarian function cease. A female’s fertility poten-
tial is therefore related to the number of oocytes, which
arepresentintheovary.Chemotherapyandradiother-
apy both lead to an additional irreversible reduction in
oocyte numbers by mechanisms which involve activa-
tion of apoptotic pathways.
e chance of retaining ovarian function following
such external insults to germ cell numbers depends on
the starting number of oocytes and this is related to the
age of the patient. us young women who start with
high numbers of oocytes are more likely to recover
menses and fertility following chemoradiotherapy,
although they remain at risk of premature ovarian
failure. Older women with relatively low numbers of
oocytes remaining in the ovary pre-treatment, fre-
quently experience immediate and irreversible cessa-
tion of ovarian function. Clinically, this may be accom-
panied by severe vasomotor symptoms and biochemi-
callyitisassociatedwithlowserumlevelsofestradiol
with signicantly elevated levels of FSH and LH.
The incidence of infertility
following radiation
e clinical eects of radiotherapy on fertility depend
on the dose and radiation eld in addition to patient
age as discussed above. Animal studies have shown
that increasing doses of ovarian radiation lead to loss
of primordial follicles in a dose-dependent manner.
e dose at which 50% of human oocytes are lost
(LD50) has been estimated to be ⬍2Gy.1Considering
243
e Obstetric Hematology Manual, ed. Sue Pavord and Beverley Hunt. Published by Cambridge University Press.
CCambridge University Press 2010.

Section 7. Malignant conditions
Table 20.1 Radiation sites relevant to reproductive potential
Site of irradiation
Tissues relevant to
fertility potential
Irradiation to the cranium, for
example:
(a) total body irradiation
(b) craniospinal irradiation
(c) direct cranial irradiation
Hypothalamic–pituitary
axis
Irradiation to the abdomen or pelvis,
for example:
(a) total body irradiation
(b) total lymphoid irradiation
(c) craniospinal irradiation
(d) direct irradiation to the pelvis or
abdomen
Ovaries
Uterus
Reproductive tract
the radiation elds, treatment impinging on either the
cranium or reproductive tract can lead to impairment
of fertility (Table 20.1).
Direct cranial irradiation
Irradiation of the hypothalamic–pituitary axis leads to
a classical pattern of hormone loss, with growth hor-
mone being the most sensitive and rst to be aected
followed by the gonadotrophins. In patients with
intracranial disease associated with acute leukemia,
direct cranial radiation may be administered. Doses
in the range 18–24 Gy may result in isolated growth
hormone (GH) deciency. In addition, subtle disturb-
ances in the menstrual cycle may occur, although their
relevance to future reproductive potential is unclear.
Delayed puberty has been described in young girls who
receivedosesabove24Gy.
Patients with pituitary and primary brain tumors
can be given higher doses of radiation exceeding 50 Gy.
Such doses can damage pituitary function directly,
but features of hypopituitarism in patients who have
received doses less than 50 Gy are more likely to be sec-
ondary to hypothalamic dysfunction.
Direct abdomino-pelvic radiation
As discussed above, radiation to the ovaries leads to
damage which is dose related. Abdomino-pelvic irra-
diation also damages the uterus with adverse eects
documented on the endometrium, myometrium, and
vasculature. In a study which investigated length and
blood ow of the uterus in ten women aged 15–
31 following 20–30 Gy abdominal radiotherapy, there
were signicant reductions in uterine length and blood
ow compared with women whose treatment had not
included abdominal radiation.2
Radiation induced damage might also be expected
to impair implantation and/or growth and develop-
ment of a fetus and there is data to support this. In a
study where 38 patients were given 20–26.5 Gy abdom-
inal radiotherapy in childhood, there were four docu-
mented conceptions but no live births, all miscarrying
in the second trimester.3
Total body irradiation
is aects all of the radiation sites relevant to fertility
potential. It is usually given together with chemother-
apy, as conditioning prior to stem cell transplanta-
tion(SCT) and serves two separate functions:
rsuppression of the host immune system to allow
donor engrament;
reradication of hemopoiesis in the host bone
marrow.
Doses of 8–15 Gy are administered either as a sin-
gle dose or in fractions. At these doses, the eects on
the hypothalamic–pituitary axis are usually minimal,
but both ovarian function and uterine function are
compromised. e incidence of ovarian failure is high.
In a single center study, which included 144 women
who had received total body irradiation (TBI) as con-
ditioning for SCT, all became amenorrheic immedi-
ately post-transplant and only 9 of the 144 recovered
menses at a median of 4 years following treatment.4All
who regained ovarian function were aged less than 25
atthetimeofSCT.
Uterine function following TBI has been less exten-
sively studied, but in a study which included 12 women
who had received TBI in childhood, there was a reduc-
tion in uterine volume to 40% of adult size despite the
use of sex steroid replacement therapy.5ese patients
had received either unfractionated TBI at a midline
dose of 8.5–10 Gy (n=4) or a total midline dose of
10.9–11.7 Gy in three fractions (n=8). ese data
suggest that the adverse eects of radiation may be
more marked if given pre-pubertally, before optimum
growth of the uterus has been achieved.
The incidence of infertility following
chemotherapy
e likelihood of infertility following chemotherapy
depends on
rthe drug(s) administered;
rthe doses to which the patient is exposed;
244
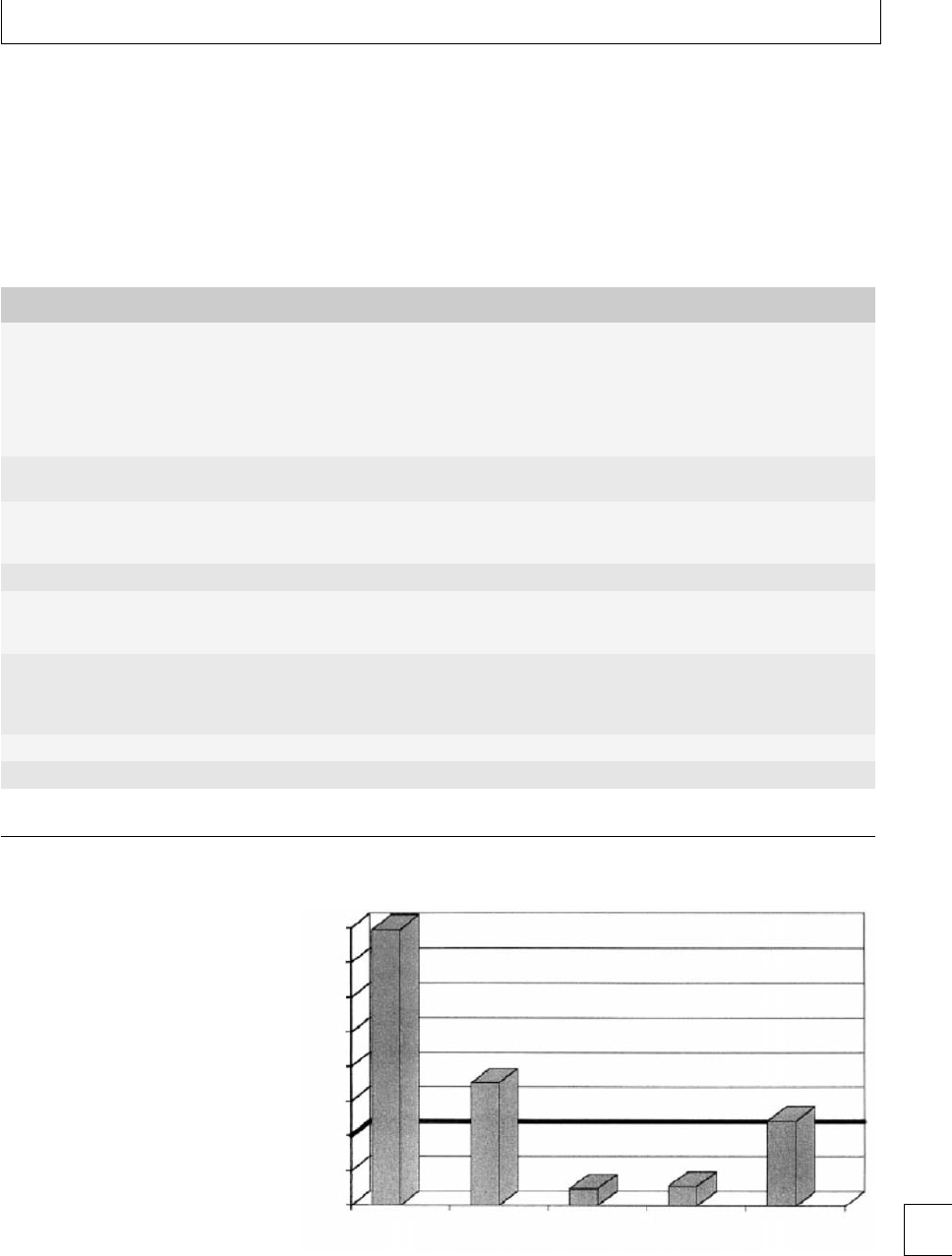
Chapter 20. Effects of chemoradiotherapy
rthe underlying disease;
rpatient age.
Chemotherapy agents can be divided into classes
based upon their mechanism of action (Table 20.2).
Meirow6provides an elegant analysis of sterilizing
eects of dierent classes of chemotherapeutic agents
(Fig. 20.1). Data on 168 patients treated with com-
bination chemotherapy were evaluated and the odds
ratio for ovarian failure calculated for exposed versus
Table 20.2 Classes of chemotherapeutic agents and their action
Drug class/subclasses Examples Mechanism of action
Alkylating agents
Nitrogen mustards
Nitrosureas
Alkyl sulfonates
Triazines
Ethylenimines
Platinum drugs∗
Cyclophosphamide, chlorambucil, melphalan
BCNU (carmustine), Lomustine
Busulfan
Dacarbazine
Thiotepa, altretamine
Cisplatin, carboplatin
DNA damage
Antimetabolites Methotrexate, 5-fluorouracil, 6-mercaptopurine,
gemcitabine, cytarabine (Ara-C), fludarabine
Interfere with nucleic acid or nucleotide synthesis
Antibiotics
Anthracyclines Daunorubicin, doxorubicin, epirubicin, idarubicin Various mechanisms, e.g. interference with
enzymes involved in DNA synthesis
Other Bleomycin, mitomycin-C. mitoxantrone∗∗
Topoisomerase Inhibitors
Topoisomerase I
Topoisomerase II
Topotecan
Etoposide (VP-16), Mitoxantrone∗∗
Mitotic inhibitors
Taxanes
Vinca alkaloids
Epothilones
Paclitaxel, docetaxel
Vinblastine, vincristine
Ixabepilone (ixempra)
Tyrosine kinase inhibitors Imatinib, dasatinib
Miscellaneous Bortezomib (Velcade), L-asparaginase
∗platinum drugs are grouped here with alkylating agents because they have a similar mechanism of action.
∗∗ similar to doxorubicin but also acts as topoisomerase II inhibitor,
3
2.5
2
1.5
1
0.5
0
Alkylating
agents
Cis-platinum
Odds ratio adjusted for age
Antibiotics Antimetabolites Plant alkaloids
4
3.5
Fig. 20.1 In 168 cancer patients treated
by combination chemotherapy, the
overall ovarian failure rate was 34%,
representing an odds ratio of 1.0.
Medications were in five drug categories
(alkylating agents, platinum derivatives,
antibiotics, anti-metabolites, and plant
alkaloids) and analysis was performed on
these groups. The fraction contributed by
each of the chemotherapeutic classes was
analyzed by the odds ratio of exposed
versus non-exposed patients. The results
were adjusted for age. (
Figure reproduced
with permission from Preservation of
Fertility, Tulandi and Gosden 2004, page
31.)
245

Section 7. Malignant conditions
Table 20.3 Relationship between dose of cyclophosphamide and age
Age
Number of
patients
Number developing
amenorrhea
Average cumulative dose
at onset of amenorrhea
⬎40 13 13 5.2 g (range 1.4–8.4 g)
30–40 5 4 (2 subsequently resumed menses) 9.3 g (7–11 g)
20–30∗5 3 20.4 g (14–24.5 g)
From Ref. 7.
∗these patients received other treatment modalities in addition to CY.
unexposed patients. Results were then adjusted for age
by logistic regression analysis. ese data show that
alkyating agents and platinum derivatives are associ-
ated with the highest risks of ovarian failure with odds
ratios of 3.98 and 1.7, respectively.
For many individual drugs, however, the true
age-related, dose-related incidence of infertility is
unknown because there are insucient longitudinal
data using them as single agents. A notable exception
to this is cyclophosphamide for which there are exten-
sivepublisheddata.isisbecausethedrugisuse-
ful in the treatment of a variety of diseases that aect
women of child-bearing age: these include breast can-
cer, autoimmune disorders such as SLE, and hemato-
logical malignancies. Cyclophosphamide also plays an
important part in conditioning treatment given prior
to allogeneic stem cell transplantation where it can be
used as:
ra high dose single agent (for example, in patients
transplanted for severe aplastic anemia, SAA);
rin combination with busulfan (for example,
pediatricandadulttransplantationforleukemia);
rtogether with total body irradiation (for example,
in adults transplanted for leukemia).
e relationship between ovarian failure and
age in women administered cyclophosphamide was
clearlydemonstratedbyastudyinwhichpre-
menopausal women with breast cancer were treated
with cyclophosphamide (CY) at a dose of 100 mg/day.7
e data are illustrated in Table 20.3 and show that a
total cumulative dose in excess of 11 g will lead to ces-
sation of menstruation in most women over the age of
30 but not younger women.
Although the cumulative dose administered is
important as illustrated above, data from transplant
centers where cyclophosphamide is administered in
a single high dose as pre-transplant conditioning
suggest that this may be a particularly gonadotoxic
approach. Follow-up of 43 women with SAA who
received CY in doses of 200 mg/kg as pre-transplant
conditioning demonstrated acute cessation of men-
struation in all 27 of these patients who were less than
26 years of age at the time of transplant subsequently
recovered ovarian function in comparison with only 5
of the 16 women aged ⬎26.4
More limited data are available on other
chemotherapeutic drugs used as single agents.
An association of busulfan with ovarian failure has
been noted as far back as the 1950s with cumulative
doses of 150–400 mg associated with acute amenor-
rhea. More recent data from transplant patients in
which high doses of busulfan (BU) are incorporated
into pre-transplant conditioning regimens further
highlights the gonado-toxicity of this agent. In a large
European multicenter evaluating pregnancy following
SCT, the combination of BUCY as pre-transplant
conditioning appeared more gonado-toxic than
CY/TBI as there were no pregnancies in patients
with malignant disease who had received BUCY in
standard doses for pre-transplant conditioning.8
ere are fewer protocols involving use of chloram-
bucil as a single agent in young females. In a small
study of 10 pre-pubertal girls exposed to cumulative
doses of chlorambucil ranging from 9–28 mg/kg for
autoimmune disease, all had normal pubertal devel-
opment including normal age at onset of menarche.9
Larger cumulative doses of 535–750 mg/m2adminis-
tered to women with breast cancer, however, are asso-
ciated with ovarian failure.10
Data on drug combinations will now be discussed
in the context of the underlying disease.
Acute leukemias
Conventional treatments for acute myeloid leukemia
(AML) and acute lymphocytic leukemia (ALL) are
generally less gonado-toxic than those used to treat
lymphomas. Typical induction regimens for AML
246

Chapter 20. Effects of chemoradiotherapy
involve drugs such as cytarabine, daunorubicin, and
etoposide followed by consolidation treatment, which
may incorporate amsacrine or mitoxantrone. e inci-
dence of persistent gonadal damage following treat-
ment with anthracycline-based regimens during child-
hood or adulthood has been reported as ⬍10%. AML
survivors had a 6% incidence of acute ovarian failure
in the Childhood Cancer Survivors Study published in
2006.11
Acute lymphoblastic leukemia is the common-
est childhood cancer, although it also aects adults.
In addition to induction and consolidation phases,
treatment of ALL also incorporates a maintenance
phase and CNS-directed therapy. e latter is generally
administered as intrathecal chemotherapy with cranial
or craniospinal irradiation reserved for those at high
risk (5%–20%) of CNS relapse. e drugs commonly
used in the treatment of ALL are glucocorticoids, vin-
cristine, anthracycline, and asparaginase. High dose
methotrexate may be administered to those with high
risk disease and a tyrosine kinase inhibitor, imatinib, is
used for patients who have Philadelphia positive ALL.
e incidence of persistent gonadal damage in females
following treatment of childhood ALL with standard
UKALL protocols is less than 20%. ose who received
craniospinal irradiation or cyclophosphamide as part
of their treatment are at greatest risk. Data from the
Childhood Cancer Survivors Study demonstrated an
acute ovarian failure rate in ALL survivors of 14%.11
Chronic leukemias
Chronic lymphocytic leukemia is predominantly a dis-
order of the elderly and so will not be discussed further.
Chronic myeloid leukemia (CML) is typically a disor-
der of middle-aged adults, but a signicant number of
cases occur in women of child-bearing age (15–49). In
the past, the mainstay of treatment for chronic myeloid
leukemia (CML) has been treatment with hydroxycar-
bamide (formerly known as hydroxyurea) followed by
stem cell transplantation. In the last 10 years, however,
there has been considerable success in managing CML
with tyrosine kinase inhibitors such as imatinib: the
rst example of a molecularly targeted therapy. Ima-
tinib was rst administered to patients with CML in
1998 and has now been used to treat more than 60 000
patients worldwide. It is given orally and is generally
well tolerated. To date, there are limited data avail-
able on the eects of imatinib on fertility. Reproduct-
ive studies in animals have shown imatinib is terato-
Table 20.4 Treatment regimens for Hodgkin’s disease and
likelihood of gonadal failure
Risk of
gonadal
failure
Combination chemotherapy
regimens for Hodgkin’s
lymphoma
High risk (⬎80%) MVPP
MOPP
ChlVPP/EVA
COPP
Intermediate risk BEAM (BCNU, etoposide, cytarabine
and melphalan)
VEBEP
Alternating ABVD/MOPP or COPP
CHOP
Low risk (⬍20%) VAPEC-B
BEACOPP
VEEP
ABVD
genic in rats, so patients are advised to avoid pregnancy
while taking it. Nonetheless, 180 pregnancies have
been reported in patients who were taking imatinib.12
Dosage data were not available in every case, but many
were receiving standard doses of 300–400 mg. Recent
data, however, suggest that higher doses of imatinib
may be associated with premature ovarian failure.13
Lymphomas
Hodgkin’s and non-Hodgkin’s lymphoma (HL and
NHL) together account for approximately 10% of
pediatric cancers (4% HL, 6% NHL). With mod-
ern treatments for HL in excess of 90% of children
and adolescents can expect to be cured. Alkylating
agents have played a major role in many of the com-
bination chemotherapy protocols proposed for the
treatment of HL and many of these have therefore
been associated with infertility (Table 20.4). In the
1970s, treatment regimens containing nitrogen mus-
tard such as MOPP (nitrogen mustard, vincristine,
procarbazine, and prednisolone) and MVPP (nitrogen
mustard, vinblastine, procarbazine, and prednisolone)
were used and were associated with oligo- or amenor-
rhea in approximately 20%–40% of women. In the mid
1970s, however, it was discovered that a new regimen
combining doxorubicin, bleomycin, vinblastine, and
dacarbazine (ABVD) was as ecacious a treatment
as MOPP or MVPP, but lacked the gonado-toxicity.
ABVD has become the modern gold standard of treat-
ment and the risk of sterilization in women under the
ageof25isalmostzero.
14 Women treated with inverted
Y-irradiation, in addition to alkylating agents, have 247

Section 7. Malignant conditions
been shown to have a signicantly higher risk of pre-
mature menopause, however.
Non-Hodgkin’s lymphomas (NHL) can be broadly
subdivided into low grade and high grade lymphomas.
Low dose oral chemotherapy such as chlorambucil is
appropriate for many patients with low grade disease
and it has been discussed above. e gold standard
treatment for high grade NHL is a combination of
the following drugs: cyclophosphamide (750 mg/m2),
doxorubicin, vincristine, and prednisolone (CHOP).
Pre-menopausal women treated using this regimen
are likely to develop amenorrhea during chemother-
apy, but the majority (95%) will resume menstruation
shortly aer completion of treatment. e risk of per-
manent ovarian failure is highest in those aged 40 or
moreatthetimeoftreatment.Evenwhenhigherdoses
of cyclophosphamide are used as in “mega-CHOP,”
in which 2–3 g/m2cyclophosphamide are adminis-
tered, it appears that the risk of persistent ovarian fail-
ure may be low with 92% (12/13) of women regain-
ing ovarian function in one study.15 Women in this
latter study who were aged ⬍40 years were oered
GnRH analogs in parallel with their chemotherapy in
an attempt to preserve fertility. A second study which
combined data on various chemotherapy regimens for
NHL in premenopausal women found a higher inci-
denceofovarianfailureof44%,
16 but the patient
characteristics and details of chemotherapy regimens
used were not reported and this is likely to under-
lie the discrepancy. Childhood NHL is treated with
similar protocols to childhood ALL and the risks of
infertility and delayed puberty are low. In a recent
prospective study of survivors of childhood NHL or
ALL, all 40 females treated with chemotherapy alone
or chemotherapy plus cranial irradiation underwent
spontaneous menarche. Whether these patients will
subsequently undergo premature ovarian failure is not
known, however.17
The likely outcome of pregnancy in
a patient treated for hematological
malignancy
ere are several reasons why pregnancy outcome
might be adversely aected by prior treatment with
chemoradiotherapy. Irradiation to the uterus may
aect implantation, potentially predisposing to
miscarriage or intra-uterine growth retardation.
Chemotherapy agents such as cyclophosphamide
can cause gene mutations, chromosomal breaks, and
rearrangements raising the possibility of an increased
risk of congenital malformations.
Although the focus of this chapter is on the
relationship between chemoradiotherapy and fertility
potential, eects on other maternal organs may cause
complications for pregnancy and delivery. Cardiac and
pulmonary toxicities, for example, are well described
following some regimens. Patients at risk of such com-
plications should have a cardiorespiratory review early
in pregnancy, including an echocardiogram and pul-
monary function tests and may require assessment by
an anesthetist prior to delivery. Similarly, patients with
renalimpairmentwillrequireexpertreviewandmoni-
toring throughout pregnancy.
ere are some data to support concerns of an
adverse pregnancy outcome resulting from pelvic irra-
diation. A retrospective multicenter study identied
139 pregnancies in 111 female patients who had
received SCT.8Of these 111 women, 39 had received
autologous stem cells and 74 had received allogeneic
stem cells. Of the latter group, 21 had been condi-
tioned with TBI-containing regimens. In this study, the
majority of pregnancies were uncomplicated; however,
20% of female allogra recipients had pre-term sin-
gleton deliveries (normal incidence approximately 6%)
and 23% had low birth weight singleton babies (nor-
mal incidence approximately 6%). ese complica-
tions were conned to women who had received total
body irradiation. e incidence of Cesarean section
was signicantly higher amongst allograed women at
42% compared with approximately 16% in the normal
population, but the incidence of congenital anomalies
amongst ospring was not increased.
A further report from the Childhood Cancer Sur-
vivor Study looked at pregnancy outcome for 4029
pregnancies in 1915 women previously treated for can-
cers in childhood. e pregnancy outcome of the sib-
ling closest in age to the patient was used for control
data. eir results showed that women treated with
pelvic irradiation tended to have smaller babies than
the controls and delivered earlier, at an average of 37.23
weeks vs. 38.47 weeks for controls. Use of daunoru-
bicin or doxorubicin was also linked adversely to birth
weight but there was no clear dose–response relation-
ship.18
Despite the theoretical concern of congenital
abnormalities arising in ospring of survivors of can-
cer treatment, available data do not demonstrate a sub-
stantial increase in risk in patients where conventional
248

Chapter 20. Effects of chemoradiotherapy
chemotherapyhasbeenusedpriortoconception.It
may be, however, that existing studies do not have suf-
cient power to detect a small dierence.
Eects of chemotherapy
during pregnancy
e risks of teratogenicity are signicant if chemother-
apy is administered during pregnancy, particularly
in the rst trimester. Management of hematological
malignancy in pregnancy will therefore depend on the
stage of pregnancy when a diagnosis is made and the
balance of risk between delaying treatment and terato-
genicity. In patients who require curative chemother-
apy in the rst trimester, then therapeutic termination
should be discussed. is would include patients with
acute leukemia and also some patients with aggressive
or extensive/bulky lymphomas. In some cases it may
be possible to delay treatment to later in pregnancy
or even until aer delivery. In patients with CML, for
example, leukapheresis can be used to temporarily
control the white cell and platelet counts. ere
have been reports of certain chemotherapy agents
being administered in the second and third trimester
without complication, for example, adriamycin,
CHOP, and rituximab; however, data is limited.
Some highly teratogenic drugs, such as methotrexate
and dacarbazine, should be avoided at all stages of
pregnancy.
Recent data also highlight concerns about the
potential teratogenic eects of imatinib. As discussed
above, this tyrosine kinase inhibitor is used in the man-
agement of both CML and Philadelphia positive ALL.
In rats imatinib leads to exencephaly, encephaloceles,
and deformities of the skull bones. Female rats admin-
istered doses ⬎45 mg/kg (which equates to approxi-
mately half the maximum human dose of 800 mg/day,
based on body surface area) experienced signicant
post-implantation loss with increased fetal resorption,
stillbirths, non-viable pups, and early pup mortality. In
astudywhichincludeddataonpregnancyoutcomes
for 125 women exposed to imatinib at conception and
during part or all of the rst trimester there were 12 o-
spring identied with abnormalities, 3 of which were
terminated electively. Of the ospring with identi-
able anomalies, 3 had strikingly similar complex mal-
formations, which were unlikely to have occurred by
chance.12 Imatinib does not appear to damage chro-
mosomes, but it is capable of interacting with sev-
eral target proteins of relevance to embryonic devel-
opment, such as cKIT and PDGFR.
Strategies for fertility preservation
Some women who require treatment for cancer have
more than one therapeutic option. In such cases,
women who hope to commence a family aer treat-
ment may be able to avoid potentially sterilizing treat-
ment.Inwomenwhorequirepelvicirradiation,itmay
be possible to laparoscopically transpose the ovaries
outside the eld of radiation, leaving the ovarian blood
supply intact. is is not always successful, however;
not only can the ovaries migrate back into the eld of
radiation, but complications can occur as a result of the
procedure such as chronic pain or formation of ovar-
ian cysts. Alternatively, modied eld radiation can
sometimes be planned to omit/reduce radiation to the
ovaries. Scatter radiation can nonetheless contribute to
ovarian failure and follow-up remains important.
In women whose treatment is highly likely to result
in infertility, cryopreservation of embryos prior to
treatment oers the best hope for parenthood post-
chemoradiotherapy. is technique requires ovarian
stimulation over a 2-week period, following which
mature oocytes in their second metaphase are col-
lected. ese oocytes are fertilized in vitro before freez-
ing. is option is not open to all patients with cancer,
however. Ovarian stimulation takes 2 weeks, but it has
to be timed in relation to the menstrual cycle. Treat-
ment delays can therefore extend to 6 weeks and this is
prohibitive for many patients with cancer. Additional
complexity arises if the patient lacks a male partner to
provide sperm. e option of donor sperm can be con-
sidered, but this requires careful counseling in relation
to future implications (see below).
In healthy women who attempt pregnancy with
transfer of thawed embryos, the pregnancy rate is
20%–30%. ere are several reasons why the out-
come of articial reproductive techniques (ART) may
be lower in women with cancer, however. Firstly,
women with cancer do not always respond well to
stimulation regimens and the quality and number of
oocytes may be lower than expected. Secondly, many
patients will have compromised endometrial function
in addition to ovarian failure as a result of their treat-
ment. is could potentially impede implantation or
fetal growth and development. ere are some data
to support this from a European multicenter study
which included 9 women who conceived using ART 249

Section 7. Malignant conditions
following TBI.8Among the pregnancies to these
women, the incidence of preterm delivery and low
birthweightospringwashigh,andmedianbirth
weights were lower than expected for gestational age.
Although there are a number of case reports of success-
ful pregnancies using cryopreserved embryos follow-
ing systemic cancer therapy, it is dicult to quote accu-
rate success rates for these patients and some will elect
to use a surrogate if available to carry their embryos.
Experimental approaches
to restoring fertility
Freezing unfertilized oocytes
Although this is a promising option for women requir-
ing sterilizing treatment who lack a partner to pro-
vide sperm, live birth rates following this procedure
are currently low at about 2%.19 isistoolowto
justify routine use of this technique in clinical prac-
tice. Low pregnancy rates partly reect the suscep-
tibility of the mature unfertilized oocyte to thermal
and osmotic injury, which exceeds that of the pre-
implantation embryo. Furthermore, it is probable that
poorer quality embryos are generated following oocyte
cryopreservation. Vitrication, which involves ultra-
rapid cooling, has improved post-thaw oocyte survival
and pregnancy rates in small studies, but further infor-
mation is required on the eciency and safety of this
technique. Nonetheless, in recent years there have been
reports of successful pregnancies in women treated
forcancerusingthistechnique.Yangandcolleagues
20
report a patient with Hodgkin’s disease who had frozen
oocytes thawed and fertilized by ICSI aer cryopreser-
vation for 6 years. During the time that the oocytes
were frozen, the patient had multiple relapses and was
treated with combination chemotherapy (ABVD fol-
lowed by COPP followed by udarabine) and total
body irradiation (200 cGy) in the context of a non-
myeloablative SCT. Nine embryos were obtained from
her frozen oocytes and all of these were implanted over
the course of three separate cycles. A surrogate was
used because of concerns of radiation damage to the
patient’s uterus, and a successful pregnancy occurred
aer the nal cycle of implantation.
Freezing ovarian tissue
is is currently the only option open to pre-pubertal
patients or to those women whose disease will not tol-
erate a signicant delay in treatment. Cortical frag-
ments containing primordial follicles with immature
oocytes can be obtained by laparoscopy and frozen.
Ideally, ovarian tissue should be obtained before the
patient has been exposed to chemotherapy, but this
is not always possible and is not an absolute require-
ment.Attemptstorestoreovarianfunctionandfer-
tility have involved reimplanting the ovarian tissue,
either orthotopically adjacent to the ovary or het-
erotopically, for example, into the anterior abdomi-
nal wall. A major concern with this technique is the
possibility of reintroducing cancer cells, and a thor-
ough histological assessment of the tissue should be
made before re-transplantation. In 2005 Meirow et al.
described a successful pregnancy in a patient with non-
Hodgkin’s lymphoma using cryopreserved ovarian tis-
sue in conjunction with IVF and additional pregnan-
cies have been reported since.21 However, it is likely
that ovarian tissue transplanted in this way will have
a limited lifespan and transplantation of ovarian tissue
should probably be reserved for assisting the restora-
tion of fertility rather than for restoring hormone pro-
duction.
e use of assisted reproductive techniques in can-
cer patients raises a range of ethical concerns, includ-
ing several issues relating to consent. Consent takes
place when two or more people agree upon a course
of action and it implies that agreement occurred:
rwithout coercion;
rbasedontheprovisionofinformation;and
rthat the participants have the ability to
understand the facts and implications of the
action (“competence”).
In the UK, consent for long-term cryopreservation of
gametesisgovernedbytheHumanFertilization&
Embryology Authority (HFEA) and they have con-
structed guidance and consent forms, which are avail-
able at www.hfea.gov.uk. e consent of both partners
is required when embryos are cryopreserved and also
when the embryos are replaced. If either partner with-
draws consent, the embryos cannot be used. In the
UK, young people aged 16–18 can consent to treat-
ment under the Family Law Reform Act 1969 (“com-
petent minors”). e position in younger patients was
established in the case of Gillick v West Norfolk Area
Health Authority (1985). As a result of this case, chil-
dren who are of sucient understanding and cap-
able of expressing their own wishes (Gillick compe-
tent) can also make informed decisions. Under HFEA
250

Chapter 20. Effects of chemoradiotherapy
regulations, parents or guardians cannot give consent
on behalf of a child for the storage or use of gametes.
Immature germ cells obtained from gonadal tissue of
pre-pubertal children do not come under this remit,
however. e tissue can therefore be recovered with
parental consent if it is considered to be in the best
interest of the child.
Conclusions
Treatment of hematology cancers is constantly evolv-
ing to produce improved survival data and incorpo-
rate better tolerated agents. As a result of this, an
increasing number of young patients diagnosed with
hematological malignancies can now hope to lead rel-
atively normal adult lives and for many this includes
the expectation of parenthood. Management of possi-
ble infertility should start before cancer treatment is
administered and, ideally, should include a full dis-
cussion of: (1) treatment options and the likelihood
of infertility associated with each option; (2) strategies
for preserving fertility if the chance of sterilization as
aresultoftreatmentishigh.Fulldataarenotalways
available, however, particularly where new drugs are
used or when experimental methods for preserving
fertility are considered. Long-term follow-up studies
of patients treated for cancer remains a central prior-
ity to provide the core information required for such
pre-treatment counseling.
251

Section 7. Malignant conditions
References
1. Wallace WHB, omson AB, Kelsey TW. e
radiosensitivity of the human oocyte. Human
Reproduction. 2003; 18: 117–121.
2. Critchley HO, Wallace WH, Shalet SM et al.
Abdominal irradiation in childhood: the potential for
pregnancy. British Journal of Obstetrics and
Gynaecology. 1992; 97: 804–810.
3. Wallace WH, Shalet SM, Crowne EC et al. Ovarian
failure following abdominal irradiation in childhood:
naturalhistoryandprognosis.Clinical Oncology. 1989;
1: 75–79.
4. Sanders JE, Buckner CD, Amos D et al. Ovarian
function following marrow transplantation for aplastic
anemia or leukemia. Journal of Clinical Oncology.
1988; 6: 813–818.
5. Holm K, Nysom K, Brocks V et al. Ultrasound B-mode
changes in the uterus and ovaries and Doppler changes
in the uterus aer total body irradiation and allogeneic
bone marrow transplantation. Bone Marrow
Transplantation. 1999; 23: 259–263.
6. Meirow D. Reproduction post-chemotherapy in young
cancer patients. Molecular Cell Endocrinology. 2000;
169: 123–131.
7. Koyama H, Wada T, Nishizawa Y et al.
Cyclophosphamide-induced ovarian failure and its
therapeutic signicance in patients with breast cancer.
Cancer 1977; 39: 1403–1409.
8. Salooja N, Szydlo RM, Socie G et al. Pregnancy
outcomes aer peripheral blood or bone marrow
transplantation: a retrospective study. e Lancet 2001;
358: 271–276.
9. Callis L, Nieto J, Vila A, Rende J. Chlorambucil
treatment in minimal lesion nephrotic syndrome: a
reappraisal of its gonadal toxicity. Journal of Pediatrics
1980; 97: 653–656.
10. Freckman HA, Fry HL, Mendez FL et al.
Chlorambucil–prednisolone therapy for disseminated
breast carcinoma. Journal of the American Medical
Society 1964; 189: 23–26.
11. Chemaitilly W, Mertens AC, Mitby P et al. Acute
ovarian failure in the Childhood Cancer Survivor
Study. Journal of Clinical Endocrinology and
Metabolism 2006; 91: 1723–1728.
12. Pye S, Cortes J, Ault P et al. e eects of imatinib on
pregnancy outcome. Blood 2008; 111: 5505–5508.
13. Christopoulos C, Dimakopoulou V, Rotas E.
Primary ovarian insuciency associated with imatinib
therapy. New England Journal of Medicine 2008; 358:
1079–1080.
14. Meirow D, Dor J. Epidemiology and infertility in
cancer patients. In Preservation of Fertility, Tulandi T
and Gosden RG, ed. Taylor & Francis. 2004.
15. DannEJ,EpelbaumR,AviviIet al. Fertility and
ovarian function are preserved in women treated with
an intensied regimen of cyclophosphamide,
adriamycin, vincristine and prednisone (Mega-CHOP)
for non-Hodgkin lymphoma. Human Reproduction
2005;20; 2247–2249.
16. Meirow D. Reproduction post-chemotherapy in young
cancer patients. Molecular Cell Endocrinology 2000;
169:123–131.
17. Steens M, Beauloye V, Brichard B et al. Endocrine
and metabolic disorders in young adult survivors of
childhood acute lymphoblastic leukemia (ALL) or
non-Hodgkin lymphoma (NHL). Clinical
Endocrinology 2008; 69: 819–827.
18. GreenDM,WhittonJA,StovallMet al. Pregnancy
outcomes in female survivors of childhood cancer: a
report from the Childhood Cancer Survivor Study.
American Journal of Obstetrics and Gynecology 2002;
187: 1070–1080.
19. Oktay K, Cil AP, Bang H et al. Eciency of oocyte
cryopreservation: a meta-analysis. Fertility and
Sterility 2006; 86: 70–80.
20. Yang D, Brown SE, Nguyen K et al. Live birth aer the
transfer of human embryos developed from
cryopreserved oocytes harvested before cancer
treatment. Fertility and Sterility 2007; 87: 1469.
e1–4.
21. Meirow D, Baum M, Yaron R et al. Ovarian tissue
cryopreservation in hematologic malignancy: ten
years’ experience. Leukemia and Lymphoma 2007; 48:
1569–1576.
252

Index
Note: page numbers in italics refer to gures and tables
abdomino-pelvic radiotherapy 244
ABO blood group
incompatibility 86
red cell products 166
abortion, recurrent spontaneous
antiphospholipid syndrome
133
see also miscarriage
ABVD drug regimen 247–248
acidosis, prevention in obstetric
hemorrhage 160, 159–160
activated partial thromboplastin time
(APTT) 5
disseminated intravascular
coagulation 212
factor XI deciency 189–190
activated protein C (APC)
resistance in pregnancy 7
sensitivity ratio test
acute chest syndrome
blood transfusion 36
sickle cell disease 36
prevention 37
acute fatty liver of pregnancy 214,
223–224
clinical signs 223
diagnosis 223
pathogenesis 223–224
acute lymphocytic leukemia
(ALL) 246–247
acute myeloid leukemia
(AML) 246–247
ADAMTS 13 219–221, 222
adjunctive ante-natal treatments 83
adult respiratory distress syndrome
(ARDS), catastrophic
antiphospholipid
syndrome 134
alpha gene deletion 39
alpha thalassemia 38
carrier 38
distribution 29
amniocentesis 78–79
disadvantages 79
hemophilia 197–198
mutation detection 198
miscarriage risk 197–198
amniotic uid embolism, cell
salvage 161
anemia
Fanconi’s 47
isoimmunized pregnancy 78–79
megaloblastic 23
microangiopathic hemolytic 214
postpartum 21
pregnancy 57
see also autoimmune hemolytic
anemia (AIHA); iron
deciency
anesthesia
cesarean section
epidural catheter 122–123
full anticoagulation 125–126
epidural 122, 122–123
factor XI deciency 191
labor
full anticoagulation 124
heparin use 122
obstetric hemorrhage
management 158–164
single shot subarachnoid 122,
122–123
spinal block 122
timing 123
see also general anesthesia; regional
anesthesia
analgesia
hemophilia 188
labor 123
full anticoagulation
124–125
von Willebrand disease 181
angiogenic factors, pre-eclampsia
205
antepartum hemorrhage 151
diagnosis 153
management 153
anti-2glycoprotein I antibodies 131,
135
procoagulant eects 132
anticardiolipin antibodies (aCL) 131,
135
anticoagulation
cardiopulmonary bypass 126
delivery 120–126
full anticoagulation 123–126
life-saving treatment 126
management during
pregnancy 110–112
prosthetic heart valves 109
cesarean section 116
labor induction 116
management 111,114
maternal/fetal outcomes 112
maternal/fetal risk 114
monitoring 115, 115
anti-D immunoglobulin 50,76
administration 86–87
feto-maternal hemorrhage
prevention 76
immune/idiopathic
thrombocytopenic purpura
treatment 50, 51–52
miscarriage 76
prophylaxis
refusal 77
routine ante-natal 76–77
recombinant monoclonal
antibodies 86–87
mutated 87
safety 86–87
anti-D isoimmunization
measurement 78
antibrinolytics, obstetric
hemorrhage 167–168
antihypertensive drugs,
pre-eclampsia 211
prevention 209 253

Index
antiphospholipid antibodies
(aPL) 131
acquired abnormalities 141–142
2glycoprotein I-dependent 132
laboratory evaluation 135
management of pregnancy
with 136–137
pre-eclampsia 208
systemic lupus erythematosus 225
venous thrombosis 133
antiphospholipid syndrome 131–139
etiology 131–132
anti-2glycoprotein I
antibodies 131, 135
anticardiolipin antibodies 131, 135
arterial thrombosis 133
aspirin 137
2glycoprotein I
lupus anticoagulant 135
procoagulant eects 131–132
catastrophic 134
classication criteria 133
clinical features
CNS 134
heart valve defects 134
thrombocytopenia 134
thrombosis 132–134
CNS eects 134
heart valve defects 134
heparin replacement of
warfarin 137
lupus anticoagulant 135
management 135–139
dilemmas 138–139
pharmacological 138
plan 136
pregnancy morbidity 137–139
pre-pregnancy 136
women with previous venous
events 137
miscarriage 133–134, 138, 144
neonates 139
obstetric complications
early 133
late 134
morbidity 132
pathophysiology 131–132
placental dysfunction 134
pre-eclampsia 134, 208
preterm delivery 134
prevalence 131
primary 131
recurrent spontaneous
abortion 133
secondary 131
thrombocytopenia 134, 138
thrombosis 132–134
management 136
ultrasonography 137–138
warfarin replacement with
heparin 137
antiplatelet agents
delivery 120–121
pre-eclampsia prevention 209–210
see also aspirin
antithrombin
pre-eclampsia 210
pregnancy levels
antithrombin concentrates,
pre-eclampsia prevention 210
antithrombotics
pre-eclampsia prevention 209–210
pregnancy loss 146–147
thrombophilia 146–147
aprotinin, obstetric hemorrhage 167
arterial thrombosis, antiphospholipid
syndrome 133
articial reproductive technologies
(ART) 249–250
ethics 250–251
ascites, fetal 74
aspirin
antiphospholipid syndrome 136,
137
delivery 120–121
labor 120–121
myeloproliferative disorders 236,
238
pre-eclampsia prevention 209–210
prosthetic heart valves 113,
114–115
thrombotic thrombocytopenic
purpura 222
venous thromboembolism
treatment 102
von Willebrand disease 181
autoantibodies
autoimmune hemolytic
anemia 57–58
maternal autoimmune
cytopenias 45
autoimmune cytopenias, maternal 58
autoantibodies 45
see also named conditions and
diseases
autoimmune hemolytic anemia
(AIHA) 45, 56–58
autoantibodies 57–58
blood transfusion 57–58
cold type 56, 58
diagnosis 57
epidemiology 56–57
examination 57
fetus 58
hemolysis treatment 58
laboratory studies 57
management 57–58
neonates 58
pathogenesis 56–57
thromboprophylaxis 58
warm type 56, 58
autoimmune neutropenia (AIN) 45,
54–56
diagnosis 54–55
granulocyte colony-stimulating
factor 55–56
history taking 55
incidence 54
laboratory assessment 55
management 55–56
pathogenesis 54
postpartum period 56
pregnancy 56
sepsis 55
azathioprine, immune/idiopathic
thrombocytopenic purpura 52
Bcells 4
balloon tamponade 156
Bart’s hydrops 38–39
Bernard–Soulier syndrome 183–184
neonates 184
beta thalassemia carrier 39
beta thalassemia intermedia 39
beta thalassemia major 28, 39
screening 28–30
2glycoprotein I 131
lupus anticoagulant 135
procoagulant eects 131–132
bilirubin 73, 78
serum levels 84
blood component therapy, obstetric
hemorrhage 160–161, 167
blood group systems 74–75
blood patch, epidural 125
blood transfusion
acute chest syndrome 36
autoimmune hemolytic
anemia 57–58
complications 84
fetal 79–80
historical landmarks 79
hematocrit 80
hemolytic disease of the
newborn 79–80
indicators 84
254

Index
iron deciency 19
obstetric hemorrhage 160–161
sickle cell disease 36
prophylaxis 37–38
thalassemia 38, 39
pregnancy 41
ultrasound-guided direct
intravascular transfusion 79
B-Lynch brace suture 156
bone, thalassemia
complications 40–41
bone marrow
folate deciency 23
immune/idiopathic
thrombocytopenic
purpura 46, 49
breastfeeding
glucose-6-phosphate dehydrogenase
deciency 43
heparin use
low molecular weight heparin 122
busulfan, infertility incidence 246
cesarean section
anesthesia
epidural catheter 122–123
full anticoagulation 125–126
single shot
subarachnoid 122–123
anticoagulant management with
prosthetic heart valves 116
bleeding risk with LMW
heparin 125
factor XI deciency 190
heparin use 122–123
immune/idiopathic
thrombocytopenic purpura
53
life-saving treatment 126
postpartum hemorrhage 173
sickle cell disease 37
thromboprophylaxis 122–123
carboprost, obstetric hemorrhage 159
cardiac problems
pre-eclampsia 207
thalassemia 41–42
cardiopulmonary bypass 126
cardiovascular disease,
pre-eclampsia 207
cauda equina syndrome 125, 125–126
cell salvage 162
machine 163
obstetric hemorrhage 161
principles 161–162
central nervous system (CNS),
antiphospholipid
syndrome 134
central venous catheterization,
obstetric hemorrhage 158
central venous pressure, monitoring in
obstetric hemorrhage 158
cephalhematoma,
hemophilia 187–188
chemoradiotherapy 243–251
fertility
eects 243
preservation 249–250
teratogenicity risk 248–249
teratogens 249
toxicity 248
chemotherapy 243–251
agents 245
infertility incidence 244–246
ovarian failure rate 245
childhood cancer treatment,
pregnancy outcome 248
children, consent 250–251
chlorambucil, infertility incidence 246
CHOP drug regimen 248
chorionic villus sampling,
hemophilia 197
chronic lymphocytic leukemia
(CLL) 247
chronic myelogenous leukemia
(CML) 229, 247
circle of Willis 81
clopidogrel 120–121
clot lysis time 10
coagulation factor(s)
activity assays 196
hormonal inuences 191
mutations 141–142
pregnancy 5, 6
loss 143
replacement in von Willebrand
disease 179–180
coagulation factor deciencies
management
ante-natal 191
neonatal 192
pre-pregnancy 191
miscarriage 192
neonates 192
rare 191–192
thrombosis risk 192
treatment 191–192
see also coagulopathy, inherited
coagulation screen, obstetric
hemorrhage 169
coagulopathy
inherited 186–192
factor XI deciency 189–191
genetic diagnosis 198
hemophilia 186–189
rare 191–192
prevention in obstetric
hemorrhage 160, 159–160
colloid uids, obstetric
hemorrhage 159
competence 250–251
competitive heme oxygenase
inhibitors 87
compression stockings see venous
compression stockings
(TEDS)
computed tomography pulmonary
angiography (CTPA)
pulmonary thromboembolism
radiation exposure
congenital heart defects, scanning 115
consent 250–251
constitutional thrombocytopenia 47
contraception, sickle cell disease 30
cord traction, controlled 152–153
corticosteroids 50
antiphospholipid
syndrome 135–136
autoimmune hemolytic anemia 58
autoimmune neutropenia 55
fetal and neonatal alloimmune
thrombocytopenia maternal
treatment 68
HELLP syndrome 213–214
immune/idiopathic
thrombocytopenic purpura
treatment 50, 51–52
thrombotic thrombocytopenic
purpura 221–222
counseling
genetic for hemophilia 194–197
pre-natal in immune/idiopathic
thrombocytopenic purpura 54
pre-pregnancy
antiphospholipid syndrome 136
coagulation factor
deciencies 191
cranial irradiation, direct 244
Creutzfeldt–Jakob disease, variant
(vCJD) 182 255

Index
crystalloid uids, obstetric
hemorrhage 159
cyclophosphamide 248
dose and age relationship 246
infertility incidence 246
cytopenia see autoimmune cytopenias,
maternal
cytoreductive therapy,
myeloproliferative
disorders 236–238
postpartum 238–239
danaparoid 103
D-dimers, pregnancy 8–10
testing in venous
thromboembolism
deep vein thrombosis
diagnosis
epidemiology
management
post-thrombotic syndrome
deferasirox 41
deferiprone 41
delivery
anticoagulation 120–126
full 123–126
antiplatelet agents 120–121
aspirin 120–121
factor XI deciency 190, 190
hemophilia 188, 187–188
hemorrhagic complications 124
heparin
low molecular weight 121–123
timing 123
immune/idiopathic
thrombocytopenic purpura
maternal considerations 52–53
mode 53
neonatal considerations 53
myeloproliferative disorders 238
pre-eclampsia 210–211
induced 210
prosthetic heart valves 115–116
sickle cell disease
management 36–37
thalassemia 41
thromboprophylaxis 124
unfractionated heparin
use 123–124
venous compression stockings
124
venous thromboembolism
management
von Willebrand disease
management 180–181
delta OD450 79, 78–79
desamino-8-D-arginine vasopressin
(DDAVP)
hemophilia 187, 188
obstetric hemorrhage 169
platelet function inherited
disorders 183
von Willebrand disease 179, 182
contraindications 180
desferrioxamine 41
diabetes mellitus type 1,
thalassemia 41
disseminated intravascular
coagulation (DIC) 225
activated partial thromboplastin
time 212
catastrophic antiphospholipid
syndrome 134
hypertensive thrombocytopenia 47
management 212
obstetric hemorrhage 169
placental abruption 214
pre-eclampsia 212
prothrombin time 212
dura mater puncture 125
dysbrinogenemia 192
thrombosis risk 192
eclampsia 47, 203
elliptocytosis 42
embryo
cryopreservation 249–250
loss in thrombophilia 144, 143–144
embryopathy, warfarin 103, 112,
113
emergencies, obstetric, time to
death 152
endocrine conditions, thalassemia 41
endoglin 205, 208–209
endothelial cell activation,
pre-eclampsia 205
endothelial dysfunction,
pre-eclampsia 205–206
Entonox
epidural procedures
anesthesia for cesarean
section 122–123
dura mater puncture 125
vertebral canal hematoma risk 122
ergometrine, obstetric
hemorrhage 159
erythroblastosis fetalis 84
erythrocytosis 235
erythropoiesis
iron deciency 14–15
megaloblastic 23
erythropoietin
recombinant 84–85
supplementation in iron
deciency 18–19
essential thrombocythemia (ET) see
primary thrombocythemia
(PT)
ethics, articial reproductive
technologies 250–251
evacuation of retained products of
conception (ERPC) 155–156
Evan’s syndrome 45
factor V Leiden
fetal 144
pregnancy loss 141–142, 143, 143
factor VIIa, recombinant
activated 183, 190, 192
factor VIII
ante-natal management 179
assays 177, 196
deciency 186
hormonal inuences in
pregnancy 177–178, 186, 187
mutations 196
postpartum levels 181
prophylaxis 186
replacement 179–180
thrombotic thrombocytopenic
purpura 219–220
von Willebrand disease 177
von Willebrand factor ratio 196
see also hemophilia
factor IX
assays 196
deciency 186
mutations 196
prophylaxis 186
see also hemophilia
factor XI concentrate 190
factor XI deciency 189–191
cesarean section 190
clinical features 189
complications 189
delivery 190, 190
incidence 189
labor 190, 190
management
ante-natal 190, 189–190
256

Index
intrapartum 190, 190
neonatal 191
postpartum 191
prepregnancy 189
neonates 191
peripartum bleeding risk 189
regional anesthesia 191
tranexamic acid 191
vaginal delivery 190
factor XIII
deciency and miscarriage risk 192
maternal 192
Family Law Reform Act
(1969) 250–251
Fanconi’s anemia 47
fentanyl analgesia in labor 125
ferric carboxymaltose 18
ferritin
iron deciency 14
pregnancy levels 14
ferrous salts, iron supplementation 17
fertility
articial reproductive technologies
aer
chemoradiotherapy 249–250
chemoradiotherapy eects 243
preservation with
chemoradiotherapy 249–250
restoration 250–251
thalassemia 39–40
fetal and neonatal alloimmune
thrombocytopenia
(FNAIT) 63–71
ante-natal management 70, 66–70,
71
optimal approach 71
ante-natal screening 65
clinical diagnosis 64
clinical signicance 64
epidemiology 63
history in previous pregnancies 65
incidence 63–64
information for mother 66
intracranial hemorrhage 64, 65
intravenous immunoglobulin 66,
83
laboratory diagnosis 64
laboratory testing 65
management 65–71
maternal treatment 68–71
complications 68
non-responders 70
neonates 64
platelets 63, 65–66, 68
post-natal management 65
red cell antigens 86
severity prediction 65
ultrasound-guided fetal blood
sampling 66, 67, 67–68
fetal blood sampling (FBS),
ultrasound-guided 66, 67,
67–68
feto-maternal hemorrhage 73, 76
risk 74,76
size quantication 76
fetus
abdomino-pelvic irradiation 244
autoimmune hemolytic anemia
58
blood transfusion 79–80
genes contributing to
pre-eclampsia 207–208
myeloproliferative disorder
monitoring 238
pericardial eusion 74
platelet transfusion
serial 66–67
in utero 66
thrombophilia 144, 144
warfarin eects 103, 112
dosage in pregnancy 113
see also teratogens
brin 10
brin degradation products (FDP) 8,
10
brinogen 167, 192
brinolysis 8–10
markers in pregnancy 7
uid resuscitation
obstetric hemorrhage 159
warmer 160
folate deciency 21–24
blood count 23
blood lm 23
bone marrow 23
clinical signs/symptoms 25
diagnosis 23
epidemiology 21–23
hematinic assays 23
management 23–24
pathogenesis 23
prevention 24
prophylaxis 23
treatment 24
folic acid
autoimmune hemolytic anemia
treatment 58
food supplementation 24
preconceptual 23, 24
thrombophilia 146
requirements 24
fondaparinux 103
free fetal DNA (DNA) 80–81
hemophilia diagnosis 197, 198
fresh frozen plasma (FFP) 167
coagulation factor
deciencies 191–192
factor XI deciency 190
Gelfoam embolization 172, 173, 173
generalanesthesia
cesarean section 125
obstetric hemorrhage 162
genetic counseling,
hemophilia 194–197
genetic diagnosis
hemophilia
carriers 196–197
rst trimester 197–198
pre-natal 197–198
inherited coagulopathy 198
gestational hypertension 47, 203
gestational thrombocytopenia 46–47,
219
Gillick competence 250–251
Glanzmann’s thromabasthenia 183
glucose-6-phosphate dehydrogenase
deciency 42–43
ante-natal management 43
breastfeeding 43
neonatal management 43
glycoprotein IIb/IIIa deciency 183
graduated compression stockings see
venous compression stockings
(TEDS)
granulocyte colony-stimulating factor
(GCSF) 55–56
growth hormone (GH) deciency 244
hematinic deciencies 13–26
see also folate deciency; iron
deciency; vitamin B12
deciency
hematocrit
blood transfusion 80
myeloproliferative
disorders 237–238
pregnancy 3
hematological malignancy
childhood 248
pregnancy outcomes 248–249
see also leukemia; lymphomas 257

Index
hematological variables in
pregnancy 237
hemoglobin Bart’s hydrops 38
hemoglobin H disease 38
hemoglobin levels
ante-natal management 22
iron deciency 15
peripheral decrease 15
pregnancy 3
hemoglobin S 30
polymerization 30
hemoglobinopathies 28–43
ante-natal screening 28–30
neonatal screening 28–30
red cell membrane disorders 42–43
structural 28
unbalanced globin chain
production 28
see also named conditions and
diseases
hemolysis
autoimmune hemolytic anemia 56
causes 56, 57
immune-mediated 78
laboratory studies 57
sickle cell disease 30
treatment 58
see also HELLP (hemolysis, elevated
liver enzymes and low
platelets) syndrome
hemolytic disease of the
newborn 73–74
blood transfusion 79–80
indicators 84
intravenous immunoglobulin 84
light therapy 84
management 83–86
severe disease 83
routine ante-natal anti-D
prophylaxis 77
see also red cell alloimmunization
hemolytic uremic syndrome (HUS)
HELLP dierential diagnosis 214
liver involvement 224–225
pre-eclampsia dierential
diagnosis 214
pregnancy 47
hemophilia 186–189, 194
amniocentesis 197–198
mutation detection 198
analgesia 188
carrier status prediction 195, 195,
195
carriers 194–195
genetic detection 196
genetic diagnosis
limitations/hazards 196–197
laboratory detection 196
cephalhematoma 187–188
chorionic villus sampling 197
clinical features 186
complications in
pregnancy 186–187
DDAVP 187, 188
delivery 188, 187–188
fetal sexing 197
rst trimester genetic
diagnosis 197–198
genetic counseling 194–197
heritability 194–195
incidence 186
intracranial hemorrhage 186–189
labor 188, 187–188
management
ante-natal 187, 187
intrapartum 188, 187–188
neonatal 188–189
postpartum 188
prepregnancy 187
maternal bleeding 186
mutations 196–197
neonatal risk 186–187
neonates 188–189
pedigree analysis 195, 195
pre-implantation diagnosis
198
pre-natal diagnosis 197–198
future techniques 198
severity 187
somatic mosaicism 196–197
sporadic 195, 195
tranexamic acid 188
transmission potential 195, 196
vitamin K 188
hemorrhage, obstetric 151–156
access 158
anesthetic management 158–164
general 162
regional 162
antepartum 151
management 153
antibrinolytics 167–168
blood/blood component
therapy 160–161
cell salvage 161
principles 161–162
classication 160
coagulation screen 169
communication 158
denition 166
diagnosis 153
disseminated intravascular
coagulation 169
documentation 163–164
drills 164
early warning scoring system 159
epidemiology 151
factor XI deciency 189
brinogen 167
uid resuscitation 159
fresh frozen plasma 167
full blood count 169
general anesthesia 162
hemophilia 186
hemostatic replacement
therapy 166–167
hypothermia, acidosis and
coagulopathy ‘lethal triad’
prevention 160, 159–160
interventional radiology
elective management 172–173
emergency 172
emergency postpartum 172,
171–172
prophylactic
management 172–173
investigations 162
management 153–154, 169
anesthetic 158–164
hemostatic 166–169
practical points 169
monitoring 158, 169
mortality 158
oxytocics 158–159
pharmacological agents 167–169
placental abruption 214
platelet transfusion 166–167
post-hemorrhage care 163, 169
prevention 151–153, 155
protocols 164
radiological management
171–174
recognition 155
red cell products 166
regional anesthesia 162
time to death in emergencies 152
treatment 154
von Willebrand disease 178
see also postpartum hemorrhage;
primary postpartum
hemorrhage (PPH)
hemostasis, inherited
disorders 176–184
hemostatic markers, pregnancy 9
hemostatic replacement
therapy 166–167
blood/blood products 160–161, 167
desamino-8-D-arginine
vasopressin 169
brinogen 167
fresh frozen plasma 167
platelet transfusion 166–167
258

Index
prothrombinase
complexes 168–169
recombinant factor VIIa 168
red cell products 166
see also blood transfusion
headache, post-dural puncture 125
heart valve defects in antiphospholipid
syndrome 134
heart valves, prosthetic 109–117
ante-natal care 115
anticoagulation 109
aspirin 113, 114–115
cesarean section 116
heparin 109, 110–112,
113–116
labor induction 116
management 111,114
maternal/fetal outcomes 112
maternal/fetal risk 114
monitoring 115, 115
warfarin 112, 115–116
aspirin 113, 114–115
biprosthetic 109–110
congenital heart defect
scanning 115
delivery management 115–116
infective endocarditis
prevention 116
labor management 115–116
management 111
replacement indications 109
thromboembolism
prevention 110
risk 110
valve thrombosis management
116
valve types 110, 109–110
warfarin anticoagulation 112
replacement with
heparin 115–116
HELLP (hemolysis, elevated liver
enzymes and low platelets)
syndrome 47, 224
acute fatty liver of pregnancy
dierential diagnosis 214
antiphospholipid syndrome 134
clinical presentation 213
complications 213
corticosteroids 213–214
denition 212
diagnosis 213
dierential diagnosis 213, 214
epidemiology 212–213
hypertension 213
management 213–214
pathophysiology 213
platelet transfusion 214
pre-eclampsia 212–214
thrombocytopenia 218
HemocueTm 163
heparin
antiphospholipid syndrome 136,
137
breastfeeding
cesarean section 122–123
delivery 121–123
timing 123
labor 121–122
anesthesia 122
analgesia 123
induction 121
recommencing 121–122
low molecular weight 102–103
ante-natal management 103,
104–105
antiphospholipid syndrome
137
bleeding risk at cesarean
section 125
breastfeeding 122
cesarean section 122–123
delivery 121–123
labor 121–122
labor analgesia 123
myeloproliferative disorders 236,
238
post-natal management 105
postpartum treatment
pregnancy loss 147
prosthetic heart valves 109,
110–112, 113–116
thrombophilia 147
thromboprophylaxis 101
thrombotic thrombocytopenic
purpura 222
treatment monitoring
with warfarin 103
warfarin replacement 115–116,
123, 137
myeloproliferative disorders 236,
238
pre-eclampsia prevention 210
prosthetic heart valves in pregnant
women 109, 110–112,
113–115
warfarin replacement 115–116
side-eects 102–103
unfractionated 102
delivery management
prosthetic heart valves 109,
110–112, 113, 114–116
vaginal delivery 123–124
warfarin replacement 115–116
venous thromboembolism
treatment 102–103
ante-natal management 104–105
delivery
maintenance
monitoring
post-natal management 105
postpartum
heparin-induced
thrombocytopenia 102, 103
hepatitis C, thalassemia 41
hereditary persistence of fetal
hemoglobin (HPFH) 29
hereditary spherocytosis 42
Hodgkin’s lymphoma 247–248
treatment 247
homocysteine
folate deciency 23
pregnancy 11
Human Fertilisation and Embryology
Authority (HEFA) 250–251
human platelet alloantigens
(HPAs) 63
immunization against 64
hydrops fetalis 73, 83–84
hydroxycarbamide 37
hyperemesis gravidarum 222–223
hyperhomocystinemia treatment
24
thrombophilia 146
hypertension
chronic 203
gestational 47, 203
HELLP syndrome 213
sickle cell disease 34
thrombocytopenia 47
hypertension in pregnancy (HIP) 47
hyperuricemia, pre-eclampsia
marker 208
hypervolemia, physiological in
pregnancy 14
hypogonadotrophic hypogonadism,
thalassemia 39–40
hypothalamic–pituitary axis,
radiotherapy 244
hypothermia, acidosis and
coagulopathy ‘lethal triad’
prevention 160, 159–160
hysterectomy, primary postpartum
hemorrhage 156
imatinib 247, 249
immune response, maternal 83 259

Index
immune/idiopathic thrombocytopenic
purpura (ITP) 45, 54, 218
bone marrow 46, 49
clinical examination 48
delivery planning
maternal considerations 52–53
mode 53
neonatal considerations 53
diagnosis 46–48
epidemiology 46
evaluation of suspected disease 49
history 48
laboratory assessment 48–49
labor management 53–54
life-threatening bleeding 51–52
management 54
monitoring 50
neonates 53–54
pathogenesis 46
platelets 46, 48–49, 50
postpartum care 53–54
pregnancy monitoring 50
pre-natal counseling 54
refractory case management 52
splenectomy 52
thrombocytopenia 46–49
treatment 51, 50–51
immunoglobulin G (IgG)
maternal 73–74
maternal autoimmune
cytopenias 45
implantation, abdomino-pelvic
irradiation 244
infections
sickle cell disease 34
thrombocytopenia 47–48
transfusion transmitted in von
Willebrand disease 182
infective endocarditis, prevention with
prosthetic heart valves 116
infertility incidence
chemotherapy 244–246
radiotherapy 243–244
inammatory bowel disease,
antiphospholipid
syndrome 131
internal iliac artery embolization
172
internal iliac vessel ligation 156
interventional radiology
indications for use 172
obstetric hemorrhage 171–174
postpartum hemorrhage
elective management 172–173
emergency 172, 171–172
intracranial hemorrhage
fetal and neonatal alloimmune
thrombocytopenia 64, 65
hemophilia 186–189
immune/idiopathic
thrombocytopenic purpura
delivery 53
intrahepatic cholestasis of
pregnancy 222–223
intrauterine growth restriction
(IUGR)
pre-eclampsia 203, 207
sickle cell disease 34
uterine artery abnormal
ow 218–219
intravascular transfusion,
ultrasound-guided direct
79
intravenous immunoglobulin
(IVIG) 50
autoimmune hemolytic anemia
58
autoimmune neutropenia 55
fetal and neonatal alloimmune
thrombocytopenia
treatment 66, 68, 83
hemolytic disease of the
newborn 84
immune/idiopathic
thrombocytopenic purpura
treatment 50, 51–52
Rhesus D antigen isoimmunization
prevention 83
serum plasmapheresis
combination 83
iron
absorption 13, 17
dietary 16–17
homeostasis 13–14
intramuscular 18
liquid form 17
overload in thalassemia 39, 39–40
parenteral 17–18
placental regulation of transfer to
fetus 14
requirements in pregnancy 13, 14
supplementation 16–18, 20
preventive 19–20
tablet form 17
iron chelation, thalassemia 38, 39–40,
41
iron deciency 13–21
blood transfusion 19
clinical signs/symptoms 15, 15
diagnosis 14
eects 16
epidemiology 13
erythropoiesis 14–15
erythropoietin 18–19
hemoglobin levels 15
iron supplementation 16–18
laboratory investigations 16
management 16–19
maternal 14
pathogenesis 13–14
postpartum 21
prevention 19–20
screening 20–21
storage decrease 14
treatment 19
iron dextran 18
iron sucrose 18
Janus kinase 2 (JAK2) protein 233
mutations 233, 235
jaundice, ABO incompatibility 86
Kell antigen 85–86
kernicterus, risk 84
labetalol, pre-eclampsia treatment 211
labor
anesthesia
full anticoagulation 124
heparin use 122
analgesia 123
full anticoagulation 124–125
aspirin 120–121
factor XI deciency 190, 190
hemophilia 188, 187–188
heparin
analgesia 123
induction 121
recommencing 121–122
immune/idiopathic
thrombocytopenic
purpura 53–54
induction
heparin use 121
with prosthetic heart valves 116
low molecular weight
heparin 121–122
prosthetic heart valves 115–116
sickle cell disease
management 36–37
third stage 124
active management 152–153
von Willebrand disease
management 180–181
warfarin 124
leukocyte lters 163
260

Index
leukemia
acute 246–247
chronic 229, 247
Libman–Sacks endocarditis 134
light therapy, hemolytic disease of the
newborn 84
liver, thalassemia complications 41
liver disease in pregnancy 222–223
acute fatty liver of pregnancy 214,
223–224
intrahepatic cholestasis of
pregnancy 223
see also hemolytic uremic syndrome
(HUS); HELLP (hemolysis,
elevated liver enzymes and low
platelets) syndrome;
pre-eclampsia
lupus anticoagulant 135
lymphocytes, pregnancy 4
lymphomas 247–248
magnesium sulphate, pre-eclampsia
treatment 211
May Hegglin anomaly 47
mean corpuscular hemoglobin
concentration (MCHC), iron
deciency 15
mean corpuscular volume (MCV)
iron deciency 15
pregnancy 3
mechanical valves see heart valves,
prosthetic
megakaryocytes, immune/idiopathic
thrombocytopenic purpura 49
megakaryopoiesis, immune/idiopathic
thrombocytopenic purpura
46
megaloblastic anemia 23
megaloblastic erythropoiesis, folate
deciency 23
metamyelocytes 4
methyldopa, pre-eclampsia
treatment 211
microangiopathic hemolytic
anemias 214
microangiopathies,
pregnancy-related 219
middle cerebral artery (MCA) 81
peak systolic ow velocity 81, 81,
82,83
mirror syndrome 39
miscarriage
amniocentesis risk 197–198
anti-D use 76
antiphospholipid syndrome
133–134, 138, 144
coagulation factor deciencies 192
denition 141
dysbrinogenemia 192
factor XIII deciency 192
sickle cell disease 34
see also pregnancy loss
misoprostol, obstetric
hemorrhage 159
monoclonal antibody-specic
immobilization of platelet
antigens (MAIPA) assay 64
monocytes 4
myelocytes 4
myelobrosis (PMF) 229, 232
case study 240
diagnosis 235
myeloproliferative disorders 229–240
aspirin 236, 238
case studies 239–240
clinical conditions 229–233
cytoreductive therapy 236–238
postpartum 238–239
delivery 238
diagnosis 233–235
epidemiology 229–233
familial 239
fetal monitoring 238
hematocrit 237–238
heparin 236, 238
high risk criteria 237
management 237–238
plan 235
postpartum 238–239
monitoring 238
myelobrosis 229, 232
case study 240
diagnosis 235
pathogenesis 233
PCV 235–238
postpartum 238–239
Philadelphia-negative 229
placental infarction 233
platelet count 235–238
postpartum 238–239
polycythemia vera 229, 231
case study 239–240
diagnosis 234–235
treatment 236
postpartum
thromboprophylaxis 238
preconceptual meeting 237
primary thrombocythemia 229,
234
case study 239, 240
incidence 230
polycythemia vera 236
thrombosis 233
risk management 238
treatment 235–237
venesection 237
venous compression stockings 236,
238
MYH-9 disorders 47
myocardial infarction (MI), cardiac
compromise 126
natural anticoagulants, pregnancy 6,
7,6–7
neonates
antiphospholipid syndrome 139
autoimmune hemolytic anemia 58
Bernard–Soulier syndrome 184
coagulation factor deciencies 192
factor XI deciency 191
fetal and neonatal alloimmune
thrombocytopenia 64
Glanzmann’s thromabasthenia 183
hemophilia 186–187, 188–189
immune/idiopathic
thrombocytopenic
purpura 53–54
von Willebrand disease 182, 183
neural tube defects
folic acid supplementation 24
preconceptual folic acid 23
neutropenia 54
severity 55
see also autoimmune neutropenia
(AIN)
NHS sickle and thalassemia
screening 28–30
nifedipine, pre-eclampsia
treatment 211
nitrogen mustard drug regimens 247
non-Hodgkin’s lymphoma 247–248
obesity, maternal and venous
thromboembolism 100–101,
120
obstetric shock, non-hemorrhagic
oocytes
freezing of unfertilized 250
production 243 261

Index
operative delivery
sickle cell disease 37
see also cesarean section
opioid analgesia in labor 124–125
oral anticoagulants (OACs) 110–112
labor/delivery
management 115–116
osteopenia, thalassemia
complications 40–41
osteoporosis
heparin-induced 102–103
thalassemia complications 40–41
ovarian function, chemoradiotherapy
eects 243
ovaries
abdomino-pelvic irradiation 244
failure rate with chemotherapy 245
laparoscopic transposition 249
tissue freezing 250
total body irradiation 244
oxytocic drugs, obstetric
hemorrhage 158–159
oxytocin 152–153, 158
packed cell volume (PCV),
myeloproliferative
disorders 235–238
postpartum 238–239
partial D alleles 85
partner testing, thalassemia 39–40
parvovirus B19 182
peak systolic ow velocity (PSV),
middle cerebral artery 81, 81,
82,83
pedigree analysis, hemophilia carrier
prediction 195, 195
pelvic irradiation
fertility preservation 249
pregnancy outcome 248
pericardial eusion, fetal 74
physiological changes during
pregnancy 222
placenta
circulation in pregnancy loss 141
dysfunction in antiphospholipid
syndrome 134
high-risk pregnancy 218–219
infarction in myeloproliferative
disorders 233
manual removal 155
pathology in pregnancy loss 142
placenta accreta 172–173
placental abruption 214
placentation, normal pregnancy 142
plasma exchange, thrombotic
thrombocytopenic
purpura 221, 222
plasma products, von Willebrand
disease 182
complications 182
plasmin inhibitors 9–10
plasminogen activator inhibitors
(PAI) 8–10
platelet function inherited
disorders 183–184
platelet transfusion
HELLP syndrome 214
obstetric hemorrhage 166–167
serial fetal 66–67, 67
in utero 67
platelets
fetal and neonatal alloimmune
thrombocytopenia 63, 65–66,
68
function assays 177
gestational
thrombocytopenia 46–47
immune/idiopathic
thrombocytopenic
purpura 46, 48–49, 50
myeloproliferative
disorders 235–238
postpartum 238–239
pregnancy 5
thrombocytopenia in
pre-eclampsia 211–212
pneumatic compression boots, venous
thromboembolism 101–102
polycythemia vera (PV) 229, 231
case study 239–240
diagnosis 234–235
treatment 236
porphyrin ring 15
post-dural puncture headache 125
postpartum hemorrhage
elective management 172–173
emergency
cesarean section 173
interventional
radiology 171–172
management protocol 172
interventional radiology 171–173
prophylactic management 172–173
see also primary postpartum
hemorrhage (PPH)
postpartum period
autoimmune neutropenia 56
fetal and neonatal alloimmune
thrombocytopenia
management 66
immune/idiopathic
thrombocytopenic
purpura 53–54
myeloproliferative
disorders 238–239
sickle cell disease management 37
venous thromboembolism
management
post-thrombotic syndrome
pre-eclampsia 47, 203–215
angiogenic factors 205
antihypertensive drugs 211
antiphospholipid antibodies 208
antiphospholipid syndrome 134
antithrombin 210
cardiovascular risk 207
with chronic hypertension 203
complications 204
constitutional factors 207
delivery 210–211
induced 210
diagnosis 203–204
dierential diagnosis 214
disseminated intravascular
coagulation 212
early onset 207
endoglin 205, 208–209
endothelial cell activation 205
endothelial dysfunction 205–206
endothelial markers 208
epidemiology 203
fetal genes 207–208
hematological
complications 211–214
HELLP syndrome 212–214
intrauterine growth restriction 203,
207
late onset 207
liver involvement 224
management 209–211
guidelines 211
microparticles 206
pathogenesis 206, 205–206, 208
placental markers 208
prediction 208–209
prevention 209–210
recurrence 203
regional anesthesia 210
risk factors 204
severe 204
sickle cell disease 34
signs/symptoms 204
thalassemia 39
262

Index
thrombocytopenia 211–212, 218
thrombophilia 208
trophoblasts 207–208
two stages model 206, 205–206
uterine artery abnormal
ow 218–219
pregnancy
hematological variables 237
physiological changes 222
pregnancy loss
antithrombotics 146–147
clotting factors 143
epidemiology 141
factor V Leiden 141–142
incidence 143
management 145–147
dilemmas 147
evidence requirement 147
pathogenesis 141–142
placental circulation 141
placental pathology 142
prevalence 142
recurrent 141
thrombophilia 141–147
diagnosis 145
heritable 142–144
very early 143–144
see also miscarriage
preterm delivery
antiphospholipid syndrome 134
sickle cell disease 34
primary postpartum hemorrhage
(PPH) 151
diagnosis 153
management 153–156
surgical 155–156
volume maintenance 154
pathogenesis 153
prevention 151–153
risk factors 152
primary thrombocythemia (PT) 229
case study 239, 240
diagnosis 234
incidence 230
polycythemia vera 236
prostaglandin E1analogue 154
obstetric hemorrhage 159
protamine sulphate 124, 126
protein C
deciency 141–142
pregnancy 6–7
protein S
deciency 141–142
pregnancy loss 143
pregnancy 6–7
prothrombin complex, coagulation
factor deciencies 191
prothrombin fragments 8
prothrombin G20210A mutation 143
fetal 144
pregnancy loss 143
prothrombin time (PT)
disseminated intravascular
coagulation 212
pregnancy 5
prothrombinase complexes 168–169
protoporphyrin 15
pulmonary thromboembolism
cardiac compromise 126
diagnosis
life-saving treatment 126
management
massive life-threatening
thrombolytic therapy
radiology see interventional radiology
radiotherapy
abdomino-pelvic 244
direct cranial irradiation 244
hypothalamic–pituitary axis 244
infertility incidence 243–244
pelvic irradiation, pregnancy
outcome 248
sites 244
total body irradiation 244
recombinant factor VIIa, obstetric
hemorrhage 168
red cell(s)
fetal leakage into maternal
circulation 76
folate levels 23
pregnancy 3, 4
red cell alloimmunization 73–87
cell salvage 161
genotype 74–75
intervention timing 78
management
recent advances 80–83
traditional 77–80
pathogenesis 73–74
phenotype 74–75
risk 74
red cell disorders, inherited 28–43
red cell membrane disorders 42–43
red cell products, hemostatic
replacement therapy 166
regional anesthesia
cesarean section 125–126
factor XI deciency 191
obstetric hemorrhage 162
pre-eclampsia 210
remifentanil 125
RHCE gene 75
RHD gene 75
DNA sequences 85
RHD pseudogene 85
RHD/CE hybrids 85
Rhesus D antigen 74–75
isoimmunization prevention 75,
75–76
IVIG use 83
negativity 77–78
positivity 77–78
red cell products 166
status determination 78, 85
non-invasive fetal testing 80–81
variants 85
see also anti-D immunoglobulin
Rhesus D disease
blood transfusion 79–80
intervention timing 78
rheumatic fever 109, 116
prevalence 110
rheumatic heart disease 109, 116
rituximab
autoimmune hemolytic anemia 58
immune/idiopathic
thrombocytopenic purpura
52
routine ante-natal anti-D prophylaxis
(RAADP) 76–77
dosing schedule 77
scalp edema 74
sedation, maternal for fetal blood
transfusion 80
sepsis, autoimmune neutropenia 55
shock
hemorrhagic classication 160
obstetric non-hemorrhagic
sickle cell crisis 34
management in pregnancy 35–36
sickle cell disease
acute chest syndrome 36
prevention 37
blood transfusion 36
prophylaxis 37–38
booking time management 34–35
compound heterozygous 30
contraception 30 263

Index
sickle cell disease (cont.)
delivery management 36–37
fetal complications 30–34
hemolysis 30
homozygous (HbSS) 30
hydroxycarbamide 37
hypertension 34
infections 34
intrauterine growth retardation 34
labor management 36–37
management 34–36, 38
booking time 34–35
delivery/labor 36–37
postpartum 37
preconception 34
maternal complications 30–34
maternal mortality 33
miscarriage 34
morbidity/mortality 28
operative delivery 37
pathogenesis 30
peri-natal mortality 33
postpartum management 37
preconception management 34
pre-eclampsia 34
pregnancy management 35–36
prematurity 34
prophylactic blood
transfusion 37–38
screening 28–30
thrombosis risk 34
vaso-occlusion 30
sickling disorders 30–34
spinal block 122
timing 123
splenectomy, immune/idiopathic
thrombocytopenic purpura
52
stillbirth, dysbrinogenemia 192
streptokinase, pulmonary
thromboembolism
systemic lupus erythematosus (SLE)
antiphospholipid antibodies 225
antiphospholipid syndrome 131
exacerbation 225
systemic thromboembolism,
prevention with prosthetic
heart valves 110
Tcells 4
TEDS see venous compression
stockings (TEDS)
teratogens
chemoradiotherapy 248–249
hydroxycarbamide 37
thalassemia 38–42
blood transfusion 38, 39
pregnancy 41
cardiac problems 41–42
delivery 41
fertility 39–40
hepatitis C risk 41
hypogonadotrophic
hypogonadism 39–40
iron chelation 38, 39–40
during pregnancy 41
iron overload 39, 39–40
management 39–42
medical problems in
pregnancy 40–41
partner testing 39–40
pathogenesis 38–39
preconception management 40
pre-eclampsia 39
pregnancy management 40
pregnancy risks 40
risks to baby 40
thrombin 189
thrombin activatable brinolysis
inhibitor (TAFI) 189
thrombin time (TT) 5
thrombin–antithrombin II (TAT)
complexes 8
thrombocytopenia
antiphospholipid syndrome 134,
138
causes 47
constitutional 47
drug-induced 47–48
gestational 46–47, 219
heparin-induced 102, 103
hypertension 47
immune/idiopathic
thrombocytopenic purpura 46
pregnancy 46–49
infections 47–48
moderate to severe 218
pre-eclampsia 211–212
see also fetal and neonatal
alloimmune thrombocytopenia
(FNAIT)
thromboelastograph analyzer 8
thromboelastograph trace 9
thromboelastography 7–8
thromboembolism
prevention with prosthetic heart
valves 110
risk with prosthetic heart valves 110
see also deep vein thrombosis;
pulmonary thromboembolism;
venous thromboembolism
thrombolytic therapy, pulmonary
thromboembolism
thrombophilia
antithrombotics 146–147
embryo loss 144, 143–144
fetal 144, 144
heparin use 147
heritable
incidence 143
pre-eclampsia 208
pregnancy loss 142–144
prevalence 142
management 145–147
dilemmas 147
evidence requirement 147
pathogenesis 141–142
placental pathology 142
pre-eclampsia 208
pregnancy loss 145
diagnosis 141–147
very early 143–144
prevalence rate
screening tests 145
testing 145
venous thromboembolism
risk 100
thromboprophylaxis 99–106, 120
autoimmune hemolytic anemia 58
cesarean section 122–123
delivery 124
myeloproliferative disorders 238
venous thromboembolism
101
recommendations 103–104
thrombosis
antiphospholipid
syndrome 132–134
coagulation factor deciencies
192
dysbrinogenemia 192
myeloproliferative disorders 233
risk management 238
prosthetic heart valves 116
sickle cell disease 34
von Willebrand disease 182
see also deep vein thrombosis;
pulmonary thromboembolism;
venous thromboembolism
thrombotic endocarditis,
non-bacterial 134
thrombotic microangiopathies 218
thrombotic storm, catastrophic
antiphospholipid
syndrome 134
thrombotic thrombocytopenic
purpura (TTP) 218–225
264

Index
acquired 220, 222
previous idiopathic 220–221
acute presentation in
pregnancy 220
ADAMTS 13 219–221, 222
aspirin 222
clinical signs 219
complications 221
congenital 220, 222
diagnosis 219
epidemiology 219
factor VIII 219–220
HELLP dierential diagnosis 214
low molecular weight heparin 222
pathology 219
plasma exchange 221, 222
pre-eclampsia dierential
diagnosis 214
pregnancy 47
presentation during pregnancy 221
previous acquired
idiopathic 220–221
relapse risk 220–221, 222
treatment 221–222
von Willebrand factor 219–220
thyroid dysfunction, thalassemia 41
ticlopidine 120–121
tin-mesoporphyrin 87
tissue plasminogen activator
(t-PA) 8–10
total body irradiation 244
tranexamic acid
coagulation factor deciencies 192
factor XI deciency 191
hemophilia 188
immune/idiopathic
thrombocytopenic purpura 52
obstetric hemorrhage 167–168
platelet function inherited
disorders 183
von Willebrand disease 181–182
transferrin, iron deciency 14
transferrin receptors 14–15
trophoblasts, pre-eclampsia 207–208
Tuohy needle 125
tyrosine kinase inhibitors 247
ultrasonography
antiphospholipid
syndrome 137–138
see also uterine artery, Doppler
screening
ultrasound-guided direct intravascular
transfusion 79
ultrasound-guided fetal blood
sampling 66, 67, 67–68
ursodeoxycholic acid 223
uterine artery
abnormal ow 218–219
Doppler screening 208, 218–219
myeloproliferative disorders 238
uterine artery embolization 156
complications 174
elective management 172–173
emergency postpartum 171–172
obstetric hemorrhage 171
uterine compression, bimanual 155
uterus
abdomino-pelvic irradiation 244
total body irradiation 244
vascularendothelialgrowthfactor
(VEGF) inhibitors 205
venesection, myeloproliferative
disorders 237
venous compression stockings
(TEDS)
delivery 124
myeloproliferative disorders 236,
238
venous thromboembolism 101–102
venous thromboembolism 91–99
antiphospholipid syndrome 133
aspirin treatment 102
D-dimer testing
delivery management
diagnosis
radiation exposure
epidemiology
heparin treatment 102–103
ante-natal management 104–105
delivery
maintenance
monitoring
post-natal management 105
postpartum
management 101–105
ante-natal 104, 104–105
non-pharmacological 101–102
pharmacological 102–104
post-natal 105, 105
thromboprophylaxis 101
maternal obesity 100–101, 120
pathogenesis 99–101
pneumatic compression
boots 101–102
post-natal management 105, 105
risk
assessment 120
with autoimmune hemolytic
anemia 58
with thrombophilia
risk factors 100, 99–100, 100,
101
ante-natal 104
signs/symptoms
thrombophilia 100
testing
thromboprophylaxis 101
ante-natal 104
management strategy 101
post-natal management 105
recommendations 103–104
treatment
initial
maintenance
venous compression
stockings 101–102
Virchow’s triad 99
warfarin treatment 103
vertebral canal hematoma risk 122
Virchow’s triad, venous
thromboembolism 99
vitamin B see folic acid
vitamin B12 deciency 23, 25–26
folic acid supplementation
contraindication 24
homocysteine levels 23
treatment 24
vitamin K
coagulation factor deciencies
192
hemophilia 188
warfarin
in labor 124
in pregnancy 112–113
vitamin K antagonists
von Willebrand disease
176–182
analgesia 181
aspirin 181
classication 177
clinical features 176
coagulation factor replacement
179–180
complications
factor replacement 182
pregnancy 178
DDAVP 179, 182
contraindications 180
factor VIII 177
hormonal inuences in
pregnancy 177–178
inhibitor formation 182
laboratory evaluation 177 265

Index
von Willebrand disease (cont.)
management 178
ante-natal 179, 179
intrapartum 181, 180–181
neonates 182, 183
postpartum 181, 181
pre-pregnancy 178
maternal bleeding 178
neonates 182, 183
plasma products 182
complications 182
pregnancy outcomes 178
prevalence 176–177
subtypes 177
thrombosis 182
tranexamic acid 181–182
transfusion transmitted
infections 182
von Willebrand factor (vWF)
activity 176
alloantibody formation 182
ante-natal management 179
antigen assays 177
deciency 176
factor VIII ratio 196
gene mutations 176–177
hormonal inuences in
pregnancy 177–178
multimers 177
postpartum levels 181
replacement 179–180
thrombotic thrombocytopenic
purpura 219–220
V/Q scan
pulmonary thromboembolism
radiation exposure
warfarin
antiphospholipid syndrome 137
fetal eects 103, 112
labor 124
pregnancy outcome 112–113
dosage 113
prosthetic heart valves 112
replacement with
heparin 115–116
replacement with heparin
antiphospholipid syndrome 137
for delivery 123
prosthetic heart valves 115–116
thromboprophylaxis 101
venous thromboembolism
treatment 103
warfarin embryopathy 103, 112
dosage 113
weak D variants 85
white blood cells 4
266
