User Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 10
Genetic type 1 Error Calculator (GEC)
(Version 0.2)
User Manual
Miao-Xin Li
Department of Psychiatry and State Key Laboratory for Cognitive and Brain Sciences;
the Centre for Reproduction, Development and Growth; and Genome Research Centre,
the University of Hong Kong, Pokfulam, Hong Kong
Contents
1. Introduction ................................................................................................................ 3
2. Installation............................................................................................................... 4
2.1 Installation of Java Runtime Environment (JRE) ................................................ 4
2.2 Installation of GEC .............................................................................................. 4
3. Input files ................................................................................................................... 4
3.1 Input files used to obtain LD information of SNPs ............................................. 4
3.2. Input files for multiple testing............................................................................. 5
4. Examples .................................................................................................................... 6
4.1 Estimate effective number of independent test ............................................... 6
4.2 Do multiple-testing in a set of p-values ............................................................... 8
1. Introduction
The Genetic Type I error calculator (GEC) is a Java-based application developed to
address multiple-testing issue with dependent Single-nucleotide polymorphisms
(SNPs). The core part is a new measure of effective number of independent tests Me
[Hum Genet. 2012 May;131(5):747-56.], which is more roust that available methods
[Figure 1]. Based on this new measure, several popular multiple-testing methods
including Bonferroni, Holm, Simes correction was improved to evaluate significance
level of SNP p-values in genome-wide association studies. A standalone version of
this tool was provided to process large datasets on users’ local computers and an
on-line version (GEC,
http://statgenpro.psychiatry.hku.hk/gec/estimateB.php?function=Bonferroni) was
made for users to quick handle a SMALL dataset conveniently.
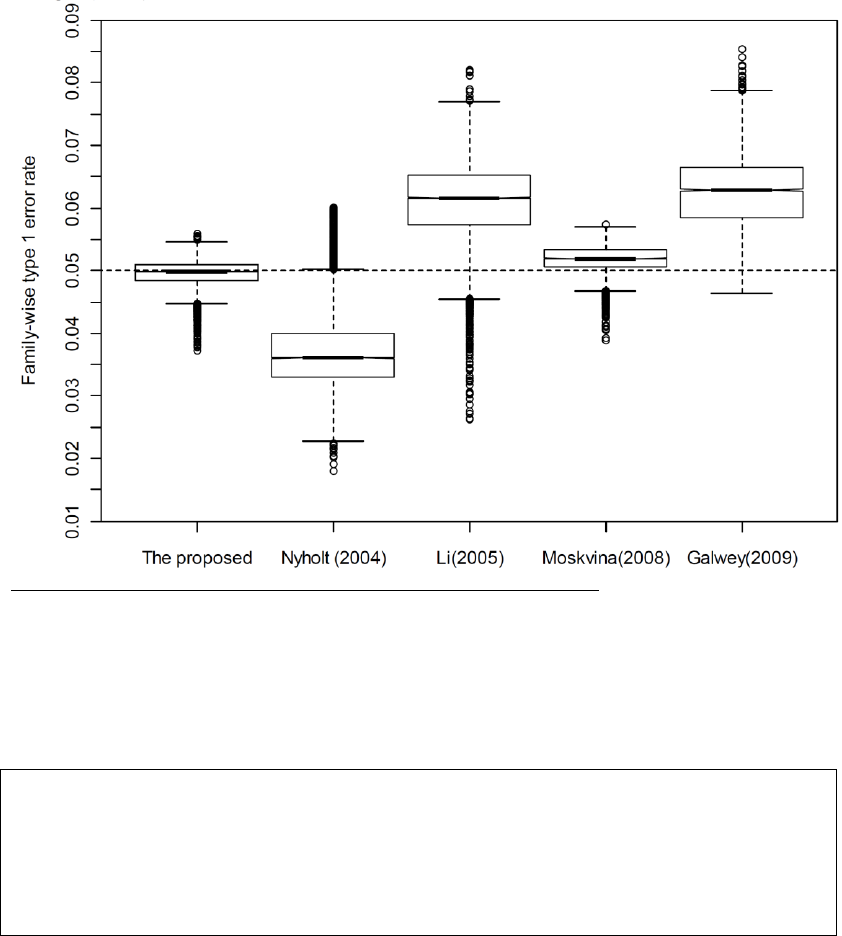
Figure 1: Box plot of MVN-derived FWERs for 5 different methods.
For each method, the nominal FWER was set to be 0.05. The bottom and top of each box mark the
25th and 75th percentile, respectively, and the band in the box denotes the 50th percentile. The
lines above and below each box denote the upper and lower 1.5 interquartile range (IQR).
This result indicated the our new measure (The proposed) is more robust (See detailed description
about the analysis in our paper)
When may you need GEC to help you?
I want to know
- the independent number of tests or redundancy degree in a set of dependent
SNPs.
- the p-value thresholds to declare significant SNPs for association in a set of
dependent p-values.
2. Installation
2.1 Installation of Java Runtime Environment (JRE)
The JRE is required to run GEC on any operating systems (OS). It can be downloaded
from http://java.sun.com/javase/downloads/index.jsp for free.
2.2 Installation of GEC
GEC has not had an installation wizard by far. After downloaded from our website and
decompressed, it can be launched through a command, java -jar –Xms256m –Xmx1300m
"./GEC.jar” <arguments >, in a command prompt window provided by OS. In the command,
-Xms<size> and -Xmx<size> set the initial and maximum Java heap sizes for GEC
respectively. A larger maximum heap size can speed up the process of analysis. A higher
setting like –Xmx1300m is suggested dealing with large number of SNPs, say more than
2,000,000. The number, however, should be less than the size of physical memory.
3. Input files
3.1 Input files used to obtain LD information of SNPs
GEC now is able to recognize dataset containing LD information of SNPs in 4
different formats. Use can choose any one of appropriate formats.
3.1.1 HapMap LD dataset format.
Uses can download the LD data from the HapMap website
(http://hapmap.ncbi.nlm.nih.gov/downloads/ld_data/latest/) as input of GEC without
any modification. Notably, it is not necessary to uncompress the downloaded file form
HapMap because GEC is able to recognize the “.gz” compressed format.
3.1.2 Plink Binary format
GEC can directly read genotypes in the binary format generated by
Plink(http://pngu.mgh.harvard.edu/~purcell/plink/data.shtml#bed), which compressed
format and can be stored and processed more efficiently. GEC will calculate the
genotypic correlation to approximate the LD degree between SNPs. The Plink binary
file set always includes three linked files *.fam, *.bim and *.bed, which should be put
in the same folder.

3.1.3 Linkage format
Genotypes in the linkage format are also a valid input of GEC. The linkage file
set include two linked files: Pedigree file and Map file. A detailed description
about this format can be referred to
http://pngu.mgh.harvard.edu/~purcell/plink/data.shtml#ped and
http://pngu.mgh.harvard.edu/~purcell/plink/data.shtml#map. The
following is a simple example. GEC is also able to recognize the “.gz”
compressed format of these files.
Linkage Pedigree Format (Example):
1
100
0
0
1
0
c g
t g
t g
c a
…
2
101
0
0
2
2
c c
t t
t t
c c
…
3
307
0
0
2
2
c g
t g
t g
c a
…
4
502
0
0
1
1
0 0
0 0
t t
a a
…
5
501
0
0
1
1
c c
t t
0 0
c c
…
6
306
0
0
1
1
g g
g g
g g
a a
…
The first five columns indicate the Pedigree ID, Individual ID, Father ID, Mother ID,
and Gender respectively. The sixth column is for phenotype and the remaining
columns are for genotypes. Genotypes must be denoted by the standard single
nucleotide symbols (a, t, g and c), which are case-insensitive. Missing genotypes are
indicated by “0 0”.
Hint: In the current version, GEC assume all subjects are independent when calculating the genotypic
correlation between SNPs. So please do not input sample with genetically related individuals.
Linkage Map File Format (Example):
1
rs2980300
0
775852
1
rs10907175
0
1120590
1
rs2887286
0
1145994
1
rs307378
0
1258710
1
rs7540231
0
1495898
The map file describes the SNP information with genotypes in the pedigree file. Four
attributes are required, chromosome, rsID, genetic map, and physical position on the
human genome assembly. The columns of chromosome, rsID and physical position
are required.
3.1.4 Haplotype data of 1000 Genome project in VCF files
This is a simple and easy format of phased haplotype data provided by us
http://statgenpro.psychiatry.hku.hk/limx/kgg/phasedgty.html . You can download the
ancestry matched VCF data to account for LD in your own dataset.
3.2. Input files for multiple testing
GEC can read p-values and report significant SNPs. The output of many genetic

association tools can be input to GEC without any modification. Users need specify
the column order of chromosome, SNP ID, the physical position of SNP and p-values.
4. Examples
4.1 Estimate effective number of independent test
1) By genotype data in conventional linkage format
I. on whole genome:
java -Xmx1g -jar gec.jar --effect-number \
--linkage-file D:/tmp/test \
--genome --out test1
II. in some regions:
java -Xmx1g -jar gec.jar --effect-number \
--linkage-file D:/tmp/test \
--regions ‘chr2:0-233434,chr4:2323-54564554’ \
--coordinate-version hg19>hg18 \
--out test1
Hint:--coordinate-version specifies the reference genome
version of physical positions (coordinates) in the linkage map
file and –-regions respectively.
III. in some chromosomes:
java -Xmx1g -jar gec.jar --effect-number \
--linkage-file D:/tmp/test \
--regions ‘chr2,chr4’ \
--out test1
2) By genotype data in plink binary format
I. on whole genome:
java -Xmx1g -jar gec.jar --effect-number \
--plink-binary D:/tmp/test \
--genome --out test1
II. in some regions:
java -Xmx1g -jar gec.jar --effect-number \
--plink-binary D:/tmp/test \
--regions ‘chr2:0-233434,chr4:2323-54564554’ \
--coordinate-version hg19>hg18 \
--out test1
Hint:--coordinate-version specifies the reference genome
version of physical positions (coordinates) in the plink map file
and –-regions respectively.
III. in some chromosomes:
java -Xmx1g -jar gec.jar --effect-number \
--plink-binary D:/tmp/test \
--regions ‘chr2,chr4’ \
--out test1
3) By genotypes from 1000 Genomes Project VCF format
I. on whole genome:
java -Xmx1g -jar gec.jar --effect-number \

--vcf-file D:/tmp/CEU/
1kg.phase1.v3.shapeit2.eur.hg19.chr_CHROM_.vcf.gz \
--genome \
--out test1
Hint: 1. We have provided a set of compiled 1KG 1000 Genomes data,
http://statgenpro.psychiatry.hku.hk/limx/kgg/phasedgty.html.
2. _CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
II. in some regions:
java -Xmx1g -jar gec.jar --effect-number \
--vcf-file D:/tmp/CEU/
1kg.phase1.v3.shapeit2.eur.hg19.chr_CHROM_.vcf.gz \
--regions ‘chr2:0-233434,chr4:2323-54564554’ \
--coordinate-version hg19>hg18 \
--out test1
Hint: _CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
--coordinate-version specifies the reference genome version of
physical positions (coordinates) in the vcf file and –-regions
respectively.
III. in some chromosomes:
java -Xmx1g -jar gec.jar --effect-number \
--vcf-file D:/tmp/CEU/
1kg.phase1.v3.shapeit2.eur.hg19.chr_CHROM_.vcf.gz \
--regions ‘chr2,chr4’ \
--out test1
Hint: _CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
4) By HapMap LD data
I. on whole genome:
java -Xmx1g -jar gec.jar --effect-number \
--ld-file D:/tmp/CEU/ld_chr_CHROM__CEU.txt.gz \
--genome \
--out test1
Hint: _CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
II. in some regions:
java -Xmx1g -jar gec.jar --effect-number \
--ld-file D:/tmp/CEU/ld_chr_CHROM__CEU.txt.gz \
--regions ‘chr2:0-233434,chr4:2323-54564554’ \
--coordinate-version hg19>hg18 \
--out test1
Hint: _CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
--coordinate-version specifies the reference genome version of
physical positions (coordinates) in the ld file and –-regions
respectively.
IV. in some chromosomes:
java -Xmx1g -jar gec.jar --effect-number \
--ld-file D:/tmp/CEU/ld_chr_CHROM__CEU.txt.gz \
--regions ‘chr2,chr4’ \
--out test1
Hint: _CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
5) By Haploype data with the format the same as MACH
I. on whole genome:
java -Xmx1g -jar gec.jar --effect-number \
--haplotype-file D:/tmp/CEU/EUR.chr_CHROM_.hap
--haplotype-map D:/tmp/CEU/EUR.chr_CHROM_.map \
--genome \
--out test1
Hint: _CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
II. in some regions:
java -Xmx1g -jar gec.jar --effect-number \
--haplotype-file D:/tmp/CEU/EUR.chr_CHROM_.hap \
--haplotype-map D:/tmp/CEU/EUR.chr_CHROM_.map \
--regions ‘chr2:0-233434,chr4:2323-54564554’ \
--coordinate-version hg19>hg18 \
--out test1
Note: _CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
--coordinate-version specifies the reference genome version of
physical positions (coordinates) in the haplotype map file and
–-regions respectively.
III. in some chromosomes:
java -Xmx1g -jar gec.jar --effect-number \
--haplotype-file D:/tmp/CEU/EUR.chr_CHROM_.hap \
--haplotype-map D:/tmp/CEU/EUR.chr_CHROM_.map \
--regions ‘chr2,chr4’ \
--out test1
Hint: _CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
4.2 Do multiple-testing in a set of p-values
6) By genotype data in conventional linkage format
java -Xmx1g -jar gec.jar --effect-number [or --multiple-test] --error
0.05 \
--linkage-file D:/tmp/test \
--pvalue-file c:/tmp/test-results.txt \
--chrom-column 1 \
--marker-column 2 \
--marker-position-column 3 \
--coordinate-version hg19-hg18 \
--pvalue-column 7 \
--out test1
Hint:
--multiple-test allows a modified multiple testing by Holm and Simes
procedures. However, it will be very slow for large number of SNPs.
--error is to specify the global type I error rate.
--coordinate-version specifies the reference genome version of
physical positions (coordinates) in the linkage map file and p
value file respectively.
7) By genotype data in plink binary format
java -Xmx1g -jar gec.jar --effect-number [or --multiple-test] --error
0.05 \
--plink-binary D:/tmp/test \
--pvalue-file c:/tmp/test-results.txt \
--chrom-column 1 \
--marker-column 2 \

--marker-position-column 3 \
--coordinate-version hg19-hg18 \
--pvalue-column 7 \
--out test1
Hint:
--multiple-test allows a modified multiple testing by Holm and Simes
procedures. However, it will be very slow for large number of SNPs.
--error is to specify the global type I error rate.
--coordinate-version specifies the reference genome version of
physical positions (coordinates) in the plink map file and p value
file respectively.
8) By genotypes from 1000 Genomes Project VCF format
java -Xmx1g -jar gec.jar --effect-number [or --multiple-test] --error
0.05 \
--vcf-file D:/tmp/CEU/
1kg.phase1.v3.shapeit2.eur.hg19.chr_CHROM_.vcf.gz \
--pvalue-file c:/tmp/test-results.txt \
--chrom-column 1 \
--marker-column 2 \
--marker-position-column 3 \
--pvalue-column 7 \
--coordinate-version hg19-hg18 \
--out test1
Hint: We have provided a set of compiled 1KG 1000 Genomes data,
http://statgenpro.psychiatry.hku.hk/limx/kgg/phasedgty.html.
_CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
--multiple-test allows a modified multiple testing by Holm and Simes
procedures. However, it will be very slow for large number of SNPs.
--error is to specify the global type I error rate.
--coordinate-version specifies the reference genome version of
physical positions (coordinates) in the vcf file and p value file
respectively. If the versions of coordinates are identical, you
can put the same version ID, say, hg19-hg19.
9) By HapMap LD data
java -Xmx1g -jar gec.jar --effect-number [or --multiple-test] --error
0.05 \
--ld-file D:/tmp/CEU/ld_chr_CHROM__CEU.txt.gz \(Assume the
reference genome version is hg18)
--pvalue-file c:/tmp/test-results.txt \
--chrom-column 1 \
--marker-column 2 \
--marker-position-column 3 \
--pvalue-column 7 \
--coordinate-version hg19-hg18 \
--out test1
Hint: _CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
--multiple-test allows a modified multiple testing by Holm and Simes
procedures. However, it will be very slow for large number of SNPs.
--error is to specify the global type I error rate.
--coordinate-version specifies the reference genome version of
physical positions (coordinates) in the ld file and p value file
respectively.
10) By Haploype data with the format the same as MACH
java -Xmx1g -jar gec.jar --effect-number [or --multiple-test] --error
0.05 \
--haplotype-file D:/tmp/CEU/EUR.chr_CHROM_.hap \
--haplotype-map D:/tmp/CEU/EUR.chr_CHROM_.map \

--pvalue-file c:/tmp/test-results.txt \
--chrom-column 1 \
--marker-column 2 \
--marker-position-column 3 \
--pvalue-column 7 \
--coordinate-version hg19-hg18 \
--out test1
Hint: _CHROM_ is a variable standing for the chromosome name
(1,2,…X,Y).
--multiple-test allows a modified multiple testing by Holm and Simes
procedures. However, it will be very slow for large number of SNPs.
--error is to specify the global type I error rate.
--coordinate-version specifies the reference genome version of
physical positions (coordinates) in the haplotype map file and
p value file respectively.
