User's Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 6
User’s Manual
1. Running environment
ProGeo-neo requires a Linux operation system (centos6) with Python (V2.7) , Perl and Java
installed.
2. External reference datasets
In order to run normally, some third-party software such as BWA ,Gatk,and Annovar need extra
databases. Here we provided these files in the reference_files, such as Hg38.fasta. In addition,
during annotating genetic variants, annovar software needs lots of databases including: refGene,
ensGene, cytoBand, avsnp147, dbnsfp30a, MT_ensGeneMrna, refGeneWithVerMrna, etc. of hg
38, putting them into humandb folder for the sake of convenience.
3. Usage
cd ProGeo-neo
Users with root privileges can ignore the following:
chmod 755 soft/bwa/bwa
chmod 755 soft/samtools/samtools
chmod 755 soft/bcftools/bcftools
chmod 755 soft/gatk/gatk
chmod 755 soft/annovar/convert2annovar.pl
chmod 755 soft/annovar/table_annovar.pl
chmod 755 soft/annovar/annotate_variation.pl
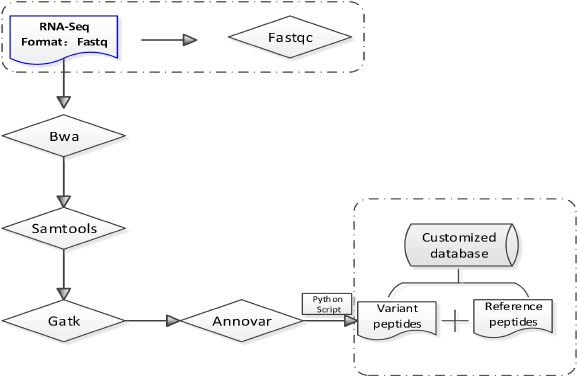
3.1 Construction of customized protein sequence database[1-5]
python get_variant-fasta.py /path/to/RNA-seq1_1.fastq /path/to/RNA-seq1_2.fastq
eg: python get_variant-fasta.py test/rna/rnaseq-sample1_1.fastq test/rna/rnaseq-sample1_2.fastq
Figure1. Construction of customized protein sequence database
Reference method:
In order to generate the customized protein sequence database, protein sequences with
missense mutation sites can be generated by substituting the mutant amino acid in normal protein
sequences and all mutan sequences were appended to the normal protein and cRAP fasta file. Here
we only provide mutant protein sequences (Var-proSeq.fasta) based on RNASeq data, users can
add other reference protein sequences as needed.
3.2 Precision HLA typing from next-generation sequencing data[6]
3.2.1 Install all required software and libraries
1. Include samtools, razers3, hdf5 and cbc in your PATH environment variable. Add HDF5's lib
directory to your LD_LIBRARY_PATH.
2. Installation of samtools
cd soft/samtools
./configure --prefix= /path/to/soft/
make &&make install
3. Installation of cbc
cd soft/Cbc-2.9.9
BuildTools/get.dependencies.sh
./configure
make && make install
4.export HDF5_DIR=/path/to/hdf5-1.8.15
5. pip install numpy
pip install pyomo
pip install pysam
pip install matplotlib
pip install tables
pip install pandas
pip install future
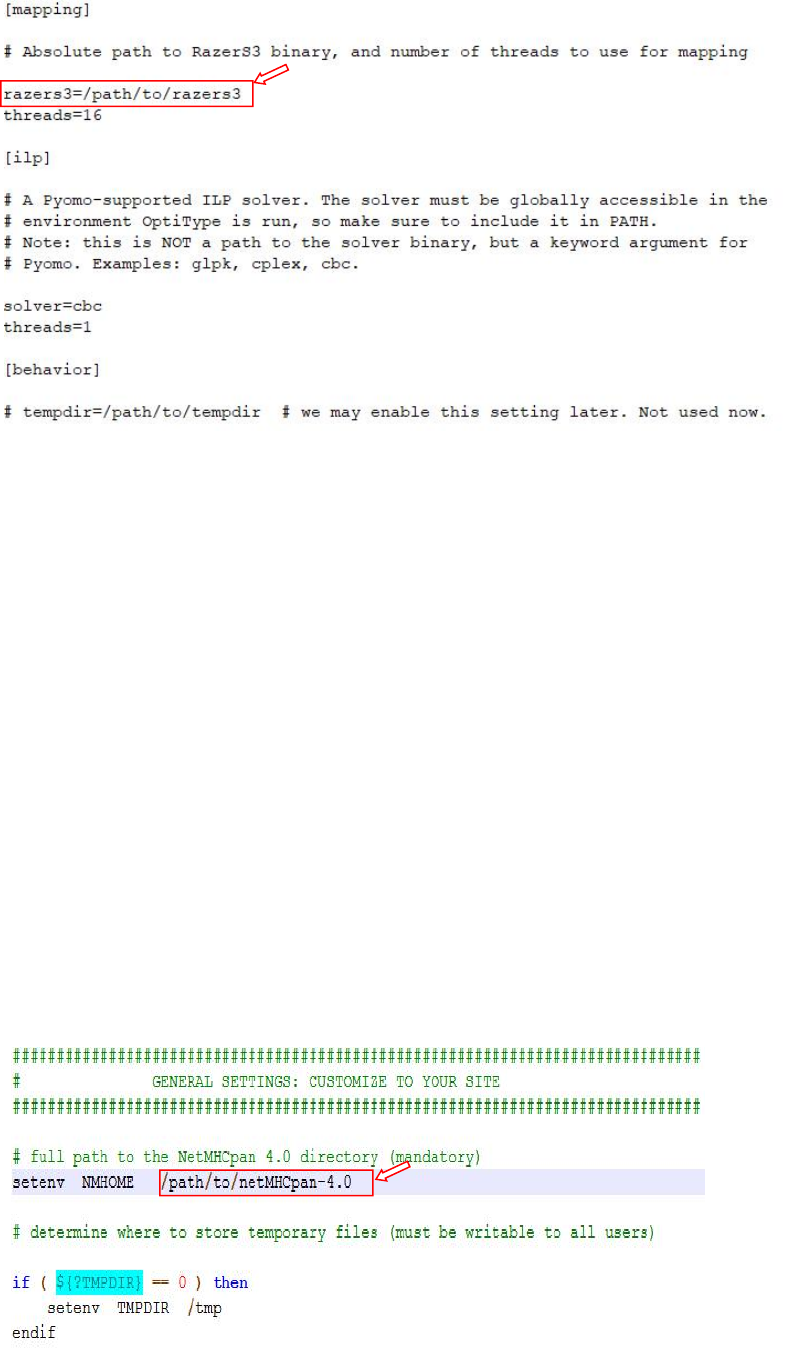
6. Create a configuration file following config.ini
In the 'OptiType' directory edit the script config.ini'
3.2.2 Predicting HLA typing from next-generation sequencing data
cd soft/OptiType
python OptiTypePipeline.py -i /path/to/RnaSeq_1.fastq /path/to/RnaSeq_2.fastq --rna -v -o
rna-hla_output
eg: python OptiTypePipeline.py -i ./test/rna/CRC_81_N_1_fished.fastq ./test/rna/
CRC_81_N_2_fished.fastq --rna -v -o ./test/rna/
3.3 Prediction and Filtration of Neontigens[2,7-10]
3.3.1 Install all required software
1. Installation of NetMHCpan-4.0
cd soft/NetMHCpan-4.0
In the 'netMHCpan-4.0' directory edit the script 'netMHCpan' [7]:
At the top of the file locate the part labelled "GENERAL SETTINGS: CUSTOMIZE TO
YOUR SITE”, set the 'NMHOME' variable to the full path to the 'netMHCpan-4.0' directory on
your system.
2. Installation of mono
cd soft/mono-5.18.0.225
./configure --prxfix=path/to/soft
make && make install
3. Include netMHCpan-4.0, kallisto and blast in your PATH environment variable.
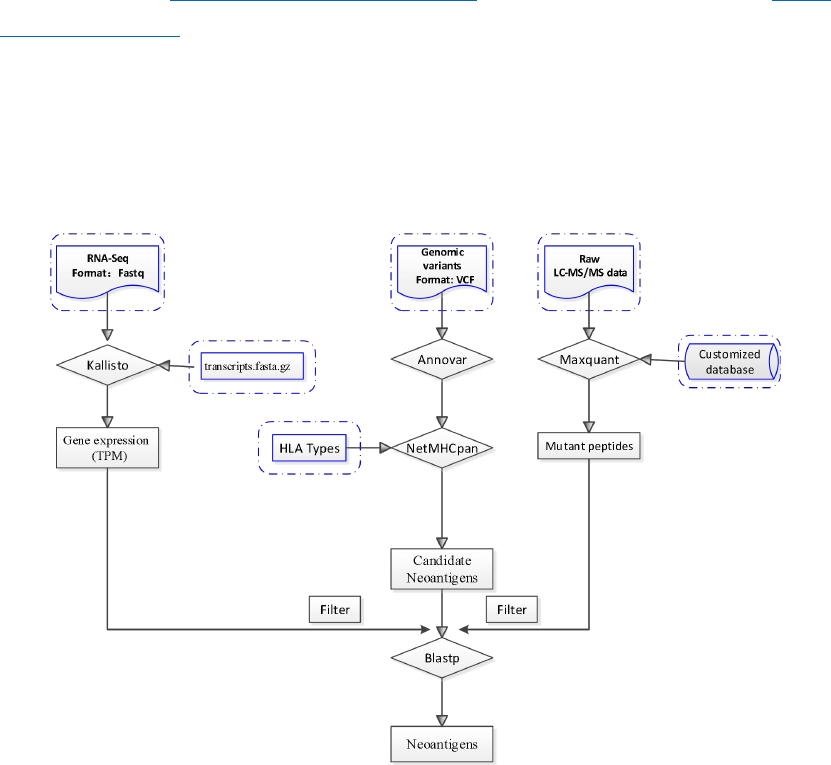
3.3.2 Prediction and Filtration of Neontigens
BLASTDB=~/soft/Balachandran/blast_db
python neoantigen_prediction_filtration.py /path/to/WES.vcf HLA_typing
/path/to/transcripts.fasta.gz /path/to/RnaSeq1_1.fastq /path/to/RnaSeq1_2.fastq /path/to/raw
/path/to/.fasta
note: ' /path/to/raw’, ‘/path/to/.fasta’ need the full path
The transcripts.fasta file supplied can be either in plaintext or gzipped format. Prebuilt indices
constructed from Ensembl reference transcriptomes can be download from the kallisto
transcriptome indices site [9].
eg: python NetMHCpan_Maxquant_lable-free.py test/WGS_20180423.vcf HLA-A03:01
soft/kallisto/test/transcripts.fasta.gz test/rna/rnaseq-sample1_1.fastq test/rna/rnaseq-sample1_2.fastq
/export3/home/user/pipline/test/ms /export3/home/user/pipline/refseq+varseq.fasta
Figure2. Prediction and Filtration of Neontigens

Table 1 summarizes the needed software and download links
Software
Download address
BWA-0.7.17[1]
http://bio-bwa.sourceforge.net/
Samtools-1.9[2]
https://github.com/samtools
Bcftools[3]
https://github.com/samtools/bcftools
GATK4.0.10.1[4]
https://software.broadinstitute.org/gatk/download/
Annovar[5]
http://annovar.openbioinformatics.org/en/latest/user-guide/download/
Optitype [6]
https://github.com/FRED-2/OptiType
NetMHCpan-4.0[7]
http://www.cbs.dtu.dk/services/NetMHCpan/
Maxquant [8]
http://www.coxdocs.org/doku.php?id=maxquant:start
Kallisto [9]
https://github.com/pachterlab/kallisto
Blast [10]
https://blast.ncbi.nlm.nih.gov/Blast.cgi
Reference:
[1] Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform[M].
2009.
[2] Li H , Handsaker B, Wysoker A , et al. The Sequence Alignment/Map format and SAMtools[J].
Bioinformatics, 2009, 25(16):2078-2079.
[3] Li H. A statistical framework for SNP calling, mutation discovery, association mapping and
population genetical parameter estimation from sequencing data. Bioinformatics.
2011;27(21):2987–93.
[4] Ga V D A , Carneiro M , Hartl C, et al. From FastQ data to high confidence variant calls: the
Genome Analysis Toolkit best practices pipeline.[J]. Current Protocols in Bioinformatics, 2013,
43(1110):11.10.1.
[5] Wang K , Li M , Hakonarson H . ANNOVAR: functional annotation of genetic variants from
high-throughput sequencing data[J]. Nucleic Acids Research, 2010, 38(16):e164-e164.
[6] Szolek A , Schubert B , Mohr C , et al. OptiType: precision HLA typing from next-generation
sequencing data[J]. Bioinformatics, 2014, 30(23):3310-3316.
[7] Jurtz V, Paul S, Andreatta M, et al. NetMHCpan-4.0: Improved Peptide-MHC Class I
Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data[J]. Journal of
Immunology, 2017, 199(9):3360.
[8] Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized
p.p.b.-range mass accuracies and proteome-wide protein quantification[J]. Nature Biotechnology,
2008, 26(12):1367.
[9] Bray N L, Pimentel H, Melsted, Páll, et al. Near-optimal probabilistic RNA-seq
quantification.[J]. Nature Biotechnology, 2016, 34(5):525.
[10] Lobo. Basic Local Alignment Search Tool (BLAST)[J]. Journal of Molecular Biology, 2012,
215(3):403-410.
