ACR 3231 WC500054888
User Manual: ACR 3231
Open the PDF directly: View PDF ![]() .
.
Page Count: 55
- 1. BACKGROUND INFORMATION ON THE PROCEDURE
- 2 SCIENTIFIC DISCUSSION
- 2.1 Introduction
- 2.2 Quality aspects
- 2.3 Non-clinical aspects
- Introduction
- Non-clinical toxicity studies were conducted in compliance with the good laboratory practice (GLP) regulations. Safety pharmacology studies, including cardiovascular safety studies in the cynomolgus monkey, conducted during preclinical development were not done under formal GLP requirements. In addition, a further series of non-GLP investigations was also conducted as part of regulatory GLP-studies in order to address specific safety aspects of tocilizumab.
- Pharmacology
- Pharmacokinetics
- Toxicology
- Ecotoxicity/environmental risk assessment
- Discussion on the non-clinical aspects
- 2.4 Clinical aspects
- 2.5 Pharmacovigilance
- 2.6 Overall conclusions, risk/benefit assessment and recommendation
- Quality
- In general, the different aspects of the chemical, pharmaceutical and biological documentation comply with existing guidelines. The fermentation and purification of the drug substance are adequately described, controlled and validated. The drug substance is well characterised with regard to its physicochemical and biological characteristics, using state-of the-art methods, and appropriate specifications are set. The manufacturing process of the drug product has been satisfactorily described and validated. The quality of the drug product is controlled by adequate test methods and specifications. The viral safety and the safety concerning other adventitious agents including TSE have been sufficiently assured. Except for a number of quality points, which will be addressed as part of post-approval follow-up measures, the overall Quality of RoActemra is considered acceptable.
- Non-clinical pharmacology and toxicology
- Efficacy
- Safety
- Risk-benefit assessment
- Recommendation
European Medicines Agency
Evaluation of Medicines for Human Use
7 Westferry Circus, Canary Wharf, London, E14 4HB, UK
Tel. (44-20) 74 18 84 00 Fax (44-20) 74 18 86 13
E-mail: mail@emea.europa.eu http://www.emea.europa.eu
© European Medicines Agency, 2009. Reproduction is authorised provided the source is acknowledged.
Doc.Ref.: EMEA/26276/2009
ASSESSMENT REPORT
FOR
RoActemra
International Nonproprietary Name: tocilizumab
Procedure No. EMEA/H/C/000955
Assessment Report as adopted by the CHMP with
all information of a commercially confidential nature deleted.
TABLE OF CONTENTS
1. ................................................. 5 BACKGROUND INFORMATION ON THE PROCEDURE
1.1 ........................................................................................................ 5 Submission of the dossier
1.2 .......................................................................... 5 Steps taken for the assessment of the product
2 ....................................................................................................... 6 SCIENTIFIC DISCUSSION
2.1 .............................................................................................................................. 6 Introduction
2.2 ......................................................................................................................... 7 Quality aspects
2.3 ............................................................................................................... 13 Non-clinical aspects
2.4 ...................................................................................................................... 18 Clinical aspects
2.5 ................................................................................................................. 46 Pharmacovigilance
2.6 ...................................... 51 Overall conclusions, risk/benefit assessment and recommendation
2/55
List of Abbreviations
ACR American College of Rheumatology
ALT Alanine aminotransferase
ANC Absolute neutrophil count
AST Aspartate aminotransferase
AUC Area under the serum concentration-time curve
BMI Body Mass Index
CBR Cytokine binding region
CDR Complementarity determining region
CHO Chinese hamster ovary
CI Confidence Interval
CIA Collagen-induced arthritis
CL Clearance
Cmax Maximum Concentration
CMH Cochran-Mantel Haenszel
Cmin Concentration at the End of the Dosing Interval (Trough Concentration)
CRP C-reactive protein
Ctrough trough concentrations
CV Coefficient of variation
CYP Cytochrome
DAS Disease Activity Score
DMARD Disease modifying anti-rheumatic drug
DTH delayed-type hypersensitivity reaction
EIA Enzyme immunoassay
ELISA Enzyme-Linked Immunosorbent Assay
ESR Erythrocyte sedimentation rate
EULAR European League Against Rheumatism
FACIT Functional Assessment of Chronic Illness Therapy
FcRN Neonatal Fc receptor
GCP Good Clinical Practice
GD Gestation day
GI Gastrointestinal
gp130 Glycoprotein 130 (signaling complex)
h Hour
HAHA Human Anti-Human Antibodies
HAQ-DI Health Assessment Questionnaire – Disability Index
HCP host cell protein
HDL High density lipoprotein
IEC ion exchange chromatography
IgG Immunoglobulin G
IL-1β Interleukin-1 beta
IL-2 Interleukin-2
3/55
IL-6 Interleukin-6
IL-6R Interleukin-6 receptor
IPC In-process control
ITT Intent-to-Treat
IV Intravenous
KD Equilibrium dissociation constant
MCB master cell bank
mIL-6R Membrane bound interleukin-6 receptor
MR16-1 Mouse specific interleukin-6 receptor antibody
MRA Myeloma receptor antibody
MTX Methotrexate
NOAEL Non observed adverse effect level
NSAID Non-steroidal anti-inflammatory drug
PD Pharmacodynamic(s)
pJIA Polyarticular juvenile idiopathic arthritis
PK Pharmacokinetic(s)
PP Per Protocol
RA Rheumatoid arthritis
RF Rheumatoid factor
SD Standard deviation
SF-36 Short form health survey
sIL-6R Soluble interleukin-6 receptor
SJC Swollen joint count
sJIA Systemic Juvenile Idiopathic Arthritis
T½ (α) Initial half-life
T½ (β) Terminal half-life
TB Tuberculosis
TCZ Tocilizumab
TFF tangential flow filtration
TJC Tender joint count
TNF Tumor necrosis factor
ULN Upper Limit of Normal
VAS Visual analogue scale
Vss Volume of Distribution at Steady-State
4/55
1. BACKGROUND INFORMATION ON THE PROCEDURE
1.1 Submission of the dossier
The applicant Roche Registration Ltd. submitted on 29 November 2007 an application for Marketing
Authorisation to the European Medicines Agency (EMEA) for RoActemra, through the centralised
procedure falling within the Article 3(1) and point 1.
The legal basis for this application refers to Article 8.3 of Directive 2001/83/EC, as amended
The applicant applied for the following indication:
Treatment of moderate to severe active rheumatoid arthritis in adult patients who:
have not been previously treated with traditional disease modifying anti-rheumatic drugs
(DMARDs) or
have an inadequate response to one or more traditional DMARDs or
have an inadequate response or are intolerant to a tumour necrosis factor antagonist.
Tocilizumab Roche can be given as monotherapy or in combination with methotrexate (MTX) and / or
other traditional DMARDs.
Scientific Advice:
The applicant received Scientific Advice from the CHMP on 24 May 2007 and 20 September 2007. The
Scientific Advice pertained to quality and clinical aspects of the dossier.
Licensing status:
At the time of submission of the application, tocilizumab was licensed in Japan for the treatment of
Castleman’s disease.
The Rapporteur and Co-Rapporteur appointed by the CHMP were:
Rapporteur: Christian Schneider Co-Rapporteur: János Borvendég
1.2 Steps taken for the assessment of the product
The application was received by the EMEA on 29 November 2007.
The procedure started on 26 December 2007.
The Rapporteur's first Assessment Report was circulated to all CHMP members on 19 March 2008.
The Co-Rapporteur's first Assessment Report was circulated to all CHMP members on 14 March
2008.
During the meeting on 21-24 April 2008, the CHMP agreed on the consolidated List of Questions to
be sent to the applicant. The final consolidated List of Questions was sent to the applicant on 25
April 2008.
The applicant submitted the responses to the CHMP consolidated List of Questions on 25 July
2008.
An inspection of the following site Chugai Pharma Manufacturing Co., Ltd. was carried out
between 23 June and 3 July 2008.
The Rapporteurs circulated the Joint Assessment Report on the applicant’s responses to the List of
Questions to all CHMP members on 9 September 2008.
During the CHMP meeting on 22-25 September 2008, the CHMP agreed on a list of outstanding
issues to be addressed in writing by the applicant.
The applicant submitted the responses to the CHMP list of outstanding issues on 20 October 2008.
5/55

The Rapporteurs circulated the Joint Assessment Report on the applicant’s responses to the list of
outstanding issues to all CHMP members on 4 November 2008.
The Rapporteurs circulated the updated Joint Assessment Report on the applicant’s responses to the
list of outstanding issues to all CHMP members on 16 November 2008.
During the meeting on 17-20 November 2008, the CHMP, in the light of the overall data submitted
and the scientific discussion within the Committee, issued a positive opinion for granting a
Marketing Authorisation to RoActemra on 20 November 2008. The applicant provided the letter of
undertaking on the follow-up measures to be fulfilled post-authorisation on 20 November 2008.
2 SCIENTIFIC DISCUSSION
2.1 Introduction
Rheumatoid arthritis (RA) is a chronic, potentially debilitating disease that is currently treated by
conventional and biotechnologically produced immunosuppressive agents. Although a several potent
drugs are available there is still a considerable therapeutic failure rate observed, necessitating new
treatment strategies.
The rationale for Development of tocilizumab in RA has been supported by extensive studies
demonstrating that IL-6 is a pleiotropic pro-inflammatory multi-functional cytokine produced by a variety
of cell types including various types of lymphocyte, fibroblasts, synoviocytes, endothelial cells, neurons,
adrenal glands, mast cells, keratinocytes, Langerhans cells, astrocytes and colonic epithelial cells.
Elevated levels of IL-6 have been implicated in the disease pathology of several inflammatory and
autoimmune disorders including RA. Instrumental in RA pathophysiology, IL-6 has been shown to be
involved in processes such as T-cell activation, differentiation of B cells into immunoglobulin-secreting
plasma cells, maturation of megakaryocytes leading to platelet production and is now well recognized to
stimulate the production of acute phase proteins by hepatocytes. IL-6 also induces the synthesis of the iron
regulatory peptide hepcidin during inflammation.
Tocilizumab (RO4877533, TCZ), also referred to as myeloma receptor antibody (MRA), is a recombinant
humanized anti-human monoclonal antibody of the immunoglobulin G1 (IgG1) sub-class directed against
the soluble and membrane-bound interleukin 6 receptor (IL-6R). In vivo, tocilizumab has been shown to
prevent onset of bone and cartilage destruction in a collagen-induced arthritis model in cynomolgus
monkeys.
The clinical development of tocilizumab for the treatment of RA includes two dose-finding Phase II
studies and five well-controlled Phase III studies investigating the use of tocilizumab, administered either
as monotherapy or in combination with methotrexate (MTX) and/or other commonly prescribed disease
modifying anti-rheumatic drugs (DMARDs), to adults with moderate to severe, active RA. Long-term
safety information and data supporting the durability of efficacy are derived from two long-term
open-label uncontrolled observation studies into which patients who completed the 24-week pivotal Phase
III studies were eligible for enrolment.
The initially proposed therapeutic indication is as follows:
“for the treatment of moderate to severe active rheumatoid arthritis in adult patients who:
have not been previously treated with traditional disease modifying anti-rheumatic drugs
(DMARDs) or
have an inadequate response to one or more traditional DMARDs or
have an inadequate response or are intolerant to a tumour necrosis factor antagonist.
6/55
Tocilizumab Roche can be given as monotherapy or in combination with methotrexate (MTX) and / or
other traditional DMARDs.”
After review of the dossier, the indication has been revised and approved by the CHMP as follows:
“RoActemra, in combination with methotrexate (MTX) is indicated for the treatment of moderate to
severe active rheumatoid arthritis (RA) in adult patients who have either responded inadequately to, or
who were intolerant to, previous therapy with one or more disease modifying anti-rheumatic drugs
(DMARDs) or tumour necrosis factor (TNF) antagonists. In these patients, RoActemra can be given as
monotherapy in case of intolerance to MTX or where continued treatment with MTX is inappropriate.”
RoActemra is supplied as a sterile liquid concentrate for solution for intravenous (iv) infusion available at
a concentration of 20 mg/mL. Three presentations of tocilizumab are intended for registration. These are
80 mg, 200 mg and 400 mg and have been selected to provide flexible combinations over the likely body
weight range of patients
2.2 Quality aspects
Introduction
Tocilizumab, the active ingredient of RoActemra, is a humanised monoclonal antibody directed against
the human interleukin-6 receptor (IL-6R) and is produced in a Chinese Hamster Ovary (CHO) cell line by
recombinant DNA technology.
The fermentation process is a serum-free process. The cell culture filtrate is purified by a series of
chromatography steps, ultra-diafiltration steps and viral inactivation and filtration steps.
The drug product manufacture consists of the formulation of the drug substance with excipients, sterile
filtration, aseptic filling into vials, stoppering and capping.
RoActemra is presented as a concentrate for solution for infusion (20 mg/ml) in single-use vials.
Active Substance
Nomenclature
INN Name: tocilizumab
Compendial Name: not applicable
USAN/JAN: tocilizumab
Laboratory Code Name: RO4877533
CAS Registry Number: 375823-41-9
Other Names: - recombinant humanised anti-human IL-6R monoclonal antibody
- MRA
Description of the active substance
Tocilizumab is a recombinant humanised monoclonal antibody of IgG1κ subclass composed of two heavy
chains and two lights chains, with 12 intra-chain and 4 inter-chain disulfide bonds.
The N-linked glycostructures present in tocilizumab include complex-type oligosaccharide structures (see
Section “Characterisation”).
The total molecular weight of the glycoprotein is approximately 149 kDa.
7/55

Manufacture
The drug substance manufacturing, release testing and stability testing for commercial batches is
performed by Chugai Pharma Manufacturing Co. Ltd, Utsunomiya Plant, 16-3 Kiyohara Kogyodanchi,
Utsunomiya-city, Tochigi, 321-3231, Japan.
Development genetics
CHO DXB11 cells, derived from a CHO K1 strain, were transfected with an expression vector containing
the genes encoding tocilizumab heavy and light chains as well as the DHFR gene.
Transfected CHO DXB11 cells with the DHFR+ phenotype were selected and then cultured in stepwise-
increasing concentrations of MTX. Cells were selected for resistance to MTX. By this process, an
integrated copy of the DHFR sequence and the flanking regions (i.e. the tocilizumab-encoding sequence)
were co-amplified. From the cells obtained, CHO V4 cells were cloned for use as seed cells for
tocilizumab production.
Cell bank system
A two-tiered cell banking system of Master Cell Bank (MCB) and Working Cell Bank (WCB) has been
developed and maintained in accordance to cGMP and ICH guidelines.
The CHO V4 seed cells were adapted to growth in suspension culture in a serum-free medium, leading to
the establishment of the original MCB (MCB-M1) and the original WCB (WCB-M1).
MCB-M1 was modified during development to generate a new MCB (MCB-M2971) and WCB (WCB-
M2971). Finally, the current WCB (WCB-M2033) was established from MCB-M2971 by replacement of
animal-derived raw materials, with the exception of some bovine milk and salmon-derived additives.
Procedures followed for the preparation of MCB and WCB have been appropriately described. An
extensive range of tests has been performed for their characterisation, in accordance with ICH guidelines,
including identity, viability, stability, presence of adventitious agents.
Fermentation process
A vial of WCB-M2033 is thawed and cells are expanded in a series of spinner flasks in a selective serum-
free growth medium to generate the cell inoculum. A series of bioreactors with increasing volumes is then
used to expand the cell mass to generate sufficient cells for the inoculation of a production bioreactor.
Following the production phase, the bioreactor content is harvested using tangential flow filtration (TFF)
in order to remove cells from the cell culture medium. The resulting cell culture filtrate is then further
purified (see below).
Cell culture conditions and in-process controls (IPC) have been sufficiently described and are considered
appropriate.
Purification process
The purification process starting from the cell culture filtrate comprises the following steps, successively:
- Protein A chromatography;
- Viral inactivation step;
- Anion exchange chromatography;
- Mixed-mode ion exchange chromatography;
- Ultra-diafiltration (UF-DF);
8/55

- Nanofiltration;
- Final filling and storage
Each step of the purification process has been adequately described, including description of the different
buffers used, column regeneration and storage conditions of both columns and product after each step.
Suitable IPC controls are in place, with acceptable limits.
Reprocessing is not performed in the manufacturing process of the drug substance.
Manufacturing process development and process validation
The manufacturing process for tocilizumab drug substance has evolved over time in four main stages: “1st
generation” (G1) process to “4th generation” (G4) process corresponding to the commercial process.
During process development, the cell culture media used for tocilizumab fermentation process changed
several times to reduce the use of components derived from animal sources.
The G4 process was developed to increase product yield. This included manufacturing site transfer with
scale up in fermentation and purification, optimisation of the cell culture media and fermentation
parameters.
Materials obtained from the G4 process were used in phase III clinical studies.
Manufacturing process development data were considered satisfactory. For filiation assessment, extensive
structural, physicochemical and biological analyses of materials manufactured pre- and post-change were
conducted prior to the implementation of each new manufacturing generation.
The tocilizumab manufacturing process was validated using data from commercial scale and scale-down
models with respect to consistency and robustness of process performance and quality attributes,
according to approved validation protocols. It was demonstrated that the G4 process consistently
maintains process parameters within specified ranges and meets acceptance criteria for performance
indicators. Overall, process validation was considered satisfactory.
Characterisation
A) Elucidation of structure and other characteristics:
A comprehensive set of analytical methods was applied to characterise the structure of tocilizumab drug
substance derived from the G4 process. Characterisation was performed with respect to the covalent as
well as the higher order structure of the tocilizumab molecule and also included the assessment of the
heterogeneity with respect to glycosylation and charge-based isoforms.
A1) Physicochemical characterisation:
The complete amino acid sequence of tocilizumab was confirmed and the primary, secondary and tertiary
structure were analysed.
It was confirmed that the disulfide linkages in tocilizumab drug substance reflect the disulfide structure
known for IgG1 molecules.
Monosaccharide composition was analysed and the types and amounts of monosaccharides identified
(N-acetylglucosamine, fucose, mannose and galactose) reflect what is expected for IgG1 molecules.
Analysis of the oligosaccharide composition has shown that the major glycostructures are constituted by
core-fucosylated biantennary complex-type oligosaccharide structures differing in the degree of terminal
galactosylation, i.e. containing two (G(2)), one (G(1)-1, G(1)-2) or no (G(0)) galactose residues. Besides
the major glycostructures, afucosylated (G(0)-F, G(1)-1-F, G(1)-2-F, G(2)-F) and high mannose type
9/55
oligosaccharides (M5) are present in tocilizumab. Sialylated oligosaccharides and other high mannose
type structures (for example M6, M7) are present at even lower levels.
Ion exchange chromatography (IEC) revealed the presence of several isoforms. Structural characterisation
of these isoforms demonstrated that differences between the isoforms are largely due to C- and N-terminal
heterogeneity of the heavy chain and incomplete cleavage of the signal sequence from the N-terminus of
the light chain.
Investigation of charged-based isoforms was performed.
The structural integrity of the tocilizumab molecule was tested.
Size exclusion chromatography (SEC) was performed to analyse the size distribution of tocilizumab
molecule. The two peaks detected in the chromatograms correspond to the monomer and dimer of
tocilizumab molecule.
A2) Biological characterisation:
In the cell-based bioassay, the cell growth-inhibiting activity by tocilizumab was evaluated by addition of
tocilizumab and IL-6 to the cells such that they compete for the IL-6R on the cell.
The binding activities of tocilizumab to human soluble IL-6R were also assessed.
In vitro data confirmed that tocilizumab has essentially no or minimal complement dependent cytotoxicity
(CDC) activity and no significant antibody-dependent cellular cytotoxicity (ADCC) activity.
B) Impurities:
Product-related substances correspond to isoform peaks observed by IEC as well as the dimer and the
degradation peaks observed by SEC of tocilizumab drug substance.
Potential process-related impurities include:
- Cell substrate derived impurities: host cell proteins (HCP) and DNA;
- Cell culture derived impurities;
- Downstream-derived impurities such as leached Protein A;
- Other impurities including endotoxin, bioburden;
Specifications
The drug substance release specifications have been suitably justified and are supported by consistent data
from multiple lots. The specifications contain tests for pharmacopoeial methods as well as specific
methods to ensure sufficient safety and quality with respect to identity, purity, quantity, potency.
Stability
The design of the stability program, including the testing intervals and temperature storage conditions, are
in accordance with current ICH guidelines. The tests chosen are a subset of tests from the release
specifications selected for stability-indicating properties.
The stability data provided were within the specifications and support a shelf life of 24 months at -50°C
for the drug substance.
10/55
Medicinal Product
Pharmaceutical development
RoActemra is presented as a concentrate for solution for infusion in a single-use Type I glass vial. The
concentrate is to be diluted in 0.9% sodium chloride prior to administration.
Each vial contains 80 mg, 200 mg or 400 mg of tocilizumab formulated with sucrose, polysorbate 80,
disodium phosphate dodecahydrate and sodium dihydrogen phosphate dihydrate and water for injections.
These excipients are commonly used in formulating protein pharmaceuticals. Buffer, polysorbate 80 and
sucrose are optimised to prevent protein aggregation that may occur in the vial on storage.
The main changes to the formulation occurred during early clinical development and consisted of the
removal of D-mannitol followed by the change of sodium chloride for sucrose.
Adventitious agents
Tocilizumab is produced in a serum-free culture medium without use of human- or animal-derived
components; only fish, milk-derived and salmon-derived raw materials are added during the fermentation
of tocilizumab. This minimises a possible contamination with adventitious agents.
Compliance with the Note for Guidance on “Minimising the Risk of Transmitting Animal Spongiform
Encephalopathy Agents via Human and Veterinary Medicinal Products” (EMEA/410/01 rev 02) has been
sufficiently demonstrated.
Extensive screening for viruses was performed. The tests did not reveal the presence of any viral
contaminant in the cells used for production of tocilizumab, with the exception of intracellular A-type and
C-type retroviral particles. Such particles are well known to be present in CHO cells. This is acceptable
since there is sufficient capacity within the tocilizumab manufacturing process for reduction of this type of
viral particles.
The purification process of tocilizumab includes several steps for inactivation/removal of enveloped
viruses. Viral safety has been sufficiently demonstrated.
Manufacture of the product
The drug product is manufactured at Chugai Pharma Manufacturing Co. Ltd, Utsunomiya Plant, 16-3
Kiyohara Kogyodanchi, Utsunomiya-city, Tochigi, 321-3231, Japan.
The secondary packaging of the drug product is carried out at F. Hoffmann-La Roche Ltd, Wurmisweg,
CH-4070 Kaiseraugs, Switzerland.
Quality control testing and EU batch release of the drug product is performed at Roche Pharma AG, Emil-
Barell-Strasse 1, 79639 Grenzach-Wyhlen, Germany.
Frozen tocilizumab drug substance is thawed and then stored until use (validated maximum storage period
of 7 days).
The drug product manufacturing process consists of formulation steps followed by an initial filtration of
the formulated bulk solution (optional re-processing step), sterile filtration of the formulated bulk solution,
aseptic filling into vials, stoppering and capping steps.
There are no intermediates isolated during the manufacture of the drug product.
The media fill and process validation results, lot-to-lot consistency data and critical process controls have
shown that the sterile filtration and aseptic filling steps are robust and well controlled and that the drug
product can be consistently manufactured.
11/55
Specifications
Appropriate specifications have been developed. The specifications contain tests for pharmacopoeial
methods as well as specific methods.
Stability of the drug product
Real-time and accelerated stability studies were initiated in accordance with ICH guidelines and per
protocol to monitor the time-temperature stability of cGMP lots of drug product. On the basis of the data
provided, the approvable shelf life for the drug product is 30 months at 2-8°C.
Discussion on chemical, pharmaceutical and biological aspects
The different aspects of the chemical, pharmaceutical and biological documentation comply with existing
guidelines.
From a Quality point of view, no Major Objection was raised during the evaluation procedure. However,
the applicant was asked to clarify several aspects. These points have been solved.
The generation of the original cell line, from the expression construct to the selection process was well
described. Cell banks have been established and adequately characterised.
The drug substance manufacturing process is well described. In-process controls (IPC) have been defined
and the proposed action limits are acceptable.
Process validation data is satisfactory. In the scale-down models for the chromatography steps, the column
diameters were linearly reduced while the bed heights and linear flow rates were maintained. The
applicant confirmed the suitability of these models for viral clearance validation.
Manufacturing process development data is satisfactory. For filiation assessment, extensive structural,
physicochemical and biological analyses of materials manufactured pre- and post-change were conducted
prior to the implementation of each new manufacturing process generation (G1, G2, G3 and G4,
successively).
The drug substance has been well characterised. Sources of heterogeneity have been assessed in detail
using a wide variety of state-of-the-art techniques. The test methods chosen are considered adequate.
Overall, impurities have been adequately identified and characterised.
The pharmaceutical development of the drug product was considered satisfactory.
The drug product manufacturing process, together with IPCs, have been adequately described; critical
steps have been defined and the proposed limits are acceptable. Process validation was considered
acceptable.
The description and validation of analytical procedures for the drug substance and drug product were
generally satisfactory except for certain methods for which additional information was requested. These
issues have been solved.
The applicant has revised the specifications; the acceptance criteria for several tests have been tightened as
requested and the available data re-analysed accordingly. In addition, the suitability of the current tests to
detect the major variants has been demonstrated.
12/55
On the basis of the stability data provided, the proposed shelf life of 24 months at ≤-50°C for the drug
substance and 30 months at 2-8°C for the drug product are considered acceptable.
Viral safety and safety concerning other adventitious agents including TSE are sufficiently assured.
Chugai Pharma Manufacturing Co. Ltd, which is the manufacturing site for the drug substance and drug
product, was inspected by the German inspectorate in July-August 2008. Several major deficiencies were
identified. The applicant provided a corrective action plan that is acceptable. It can now be considered that
this site is operated in accordance to current EU Good Manufacturing Practices (GMP).
2.3 Non-clinical aspects
Introduction
Non-clinical toxicity studies were conducted in compliance with the good laboratory practice (GLP)
regulations. Safety pharmacology studies, including cardiovascular safety studies in the cynomolgus
monkey, conducted during preclinical development were not done under formal GLP requirements. In
addition, a further series of non-GLP investigations was also conducted as part of regulatory GLP-studies
in order to address specific safety aspects of tocilizumab.
Pharmacology
Primary pharmacodynamics
Tocilizumab is a recombinant humanized anti-human interleukin-6 receptor (IL-6R) monoclonal antibody
of the immunoglobulin IgG1 subclass. IL-6 is a multi-functional cytokine, produced by a variety of cell
types and involved in T-cell activation, induction of acute phase proteins and stimulation of
haematopoiesis.
IL-6 has been implicated in the pathogenesis of various diseases, including inflammatory diseases, RA,
inflammatory bowel disease, osteoporosis and neoplasia. IL-6 exerts its biological activities through its
receptors, membrane-bound IL-6 receptor (mIL-6R) and soluble IL-6 receptor (sIL-6R). Due to restricted
expression of the cognate mIL-6R, signalling via the membrane-bound pathway is confined to only a
small population of cell types, which includes neutrophils, monocytes, T-lymphocytes, B-lymphocytes,
hepatocytes, osteoblasts and keratinocytes. However, the number of cell types expressing mIL-6R does
not reflect the full spectrum of cell types that can respond to IL-6. IL-6 can activate cells that do not
express the IL-6R through a process known as trans-signalling. In this process, IL-6 first binds to free sIL-
6R, then the IL-6/sIL-6R complex binds to the common signal-transducing molecule [glycoprotein 130
(signalling complex) gp130, which is expressed on the surface of most cells. This process enables cells
that do not possess the cognate mIL-6R to respond to IL-6 signalling.
The preclinical pharmacology programme showed that tocilizumab specifically binds to the IL-6 binding
site of both sIL-6R and mIL-6R with similar affinity. Therefore, tocilizumab is able to block IL-6 from
binding to both receptors and thereby blocks the activity of IL-6. In vitro studies demonstrated that
tocilizumab can inhibit IL-6 binding to and displace already bound IL-6 from sIL-6R and that tocilizumab
has a strong anti- IL-6 effect. Tocilizumab is specific to the IL-6R with no binding to other receptors
associated with gp130 or to receptors for other cytokines. Preclinical studies showed specificity of
tocilizumab to the IL-6R with no direct cross-reactive inhibitory effect on TNF-α, IL-1β, IL-15 or IL-2 in
vitro.
The cynomolgus monkey was chosen as the pharmacologically relevant species because tocilizumab
cross-reacts with monkey IL-6R under in vitro and in vivo conditions. In a cynomolgus monkey model of
collagen-induced arthritis (CIA), tocilizumab was shown to prevent both the local joint and the systemic
inflammatory disease manifestations.
13/55

Secondary pharmacodynamics
Tocilizumab does not bind to IL-6R of rats, mice and rabbits. Tests in non-responder species are of very
limited value for target-specific molecules, such as therapeutic antibodies. Studies did not show any
evidence for off-target effects of tocilizumab.
Safety pharmacology programme
The in vivo immunomodulatory consequences of inhibition of IL-6R signalling were investigated in
studies in mice using the mouse specific IL-6R antibody MR16-1. The studies demonstrated that IL-6
inhibition per se does not affect the primary antibody response to a T-cell dependent antigen. The delayed-
type hypersensitivity reaction (DTH) was only reduced when IL-6R signalling was blocked during the
induction phase but had no effect at later phases. The data suggests an overall effect of inhibition of IL-6R
signalling on T-cell priming rather than on T-cell differentiation, IL-6R inhibition did not affect the
development of T-cell memory and T-helper cell activity.
The cardiovascular safety of tocilizumab has been investigated in a series of preclinical in vivo studies in
cynomolgus monkeys. Tocilizumab showed no effect on the cardiac electrophysiological performance,
cardiac tissue integrity or systemic pro-thrombotic activities IV at doses up to 50 mg/kg.
Pharmacodynamic drug interactions
Pharmacodynamic drug interaction studies were not conducted with tocilizumab.
Pharmacokinetics
In a single dose intravenous study in rats values of ca. 200 h for terminal half-life, ca. 0.6 ml/h/kg for total
CL and ca. 160 ml/kg for the distribution volume (Vss) were calculated after doses of 0.5, 5 and 50 mg/kg,
respectively. Dose proportional increase in AUC was observed.
From single dose intravenous studies in male cynomolgus monkeys values of 0.5 and 0.2 ml/h/kg for total
CL and of ca. 60 ml/kg for the distribution volume (Vss) were calculated after the 5 and 50 mg/kg dose,
respectively. A clear biphasic decline was observed. The comparability of the results for the 5 mg/kg dose
obtained in a study with female monkeys suggests that there are no pharmacokinetic differences due to
gender in monkeys. An apparent non-linear pharmacokinetic behaviour in the monkeys was observed
(dose dependent values for CL and over-proportional increase in AUC) which was more pronounced
between the lower doses (0.5 and 5 mg/kg) than between the 5 and 50 mg/dose groups. This resembles the
findings in humans.
The small values for the volume of distribution found in both species of about 1.5 to 2 times the plasma
volume indicate a low tissue penetration of tocilizumab comparable to other IgG molecules. From one
study in monkeys using the s.c. route of administration a bioavailability of 72% was calculated after a
dose of 5 mg/kg.
A multiple-dose study in cynomolgus monkeys indicated that pharmacokinetics of tocilizumab did not
change upon repeated administration over 8 weeks. Neutralizing anti-tocilizumab antibodies could be
detected in plasma of cynomolgus monkeys after single and repeated dosing which were possibly
responsible for an apparently accelerated decline in plasma concentration of tocilizumab at later time
points in the single dose study.
A distribution study in cynomolgus monkeys revealed tissue/plasma ratios of tocilizumab > 1 for some
tissues at day 28 p.i.. In most cases the ratios increased from day 7 to day 28 suggesting slow equilibration
between plasma and tissues. A 10fold higher concentration in the synovial fluid than in plasma was
observed at day 28. Binding to blood cells was rather low, ca. 20% of total tocilizumab in blood
14/55
circulation seems to be bound to/in blood cells. The extent of binding to plasma proteins, especially to the
soluble receptor sIL-6R, has not been determined in animal plasma.
Consistent with its nature as IgG, tocilizumab seems to be able to cross the placental barrier. In a segment
II-embryo-foetal development study in cynomolgus monkeys foetal plasma concentrations of tocilizumab
were 39% and 60% of the maternal plasma concentration after application of 10 and 50 mg/kg/day, resp.
There is agreement that catabolism pathways similar to the known pathways for IgG molecules can be
assumed for tocilizumab. Regarding the Neonatal Fc receptor (FcRn)-mediated pathway a critical value is
the affinity of tocilizumab to this receptor. The binding of tocilizumab to the human neonatal Fc-receptor
(FcRn) was studied in vitro by surface plasmon resonance analysis. Based on the results of this study, it is
concluded that tocilizumab shows an affinity to human FcRn similar to that of other therapeutic IgGs
(trastuzumab and rituximab). Thus, these data provide evidence that tocilizumab undergoes the same
FcRn-mediated clearance and transcytosis processes that have been described for other IgGs. From in
vitro binding studies at Fc-receptors on PBMC, it is assumed that binding of tocilizumab to the FcγI
receptor occurs in the expected nanomolar range for an IgG.
Chromatographic measurements allowing for the detection of metabolites in plasma indicate that
tocilizumab is present in plasma mainly as unchanged tocilizumab. An additional peak (5%) was found in
monkey plasma which was attributed to a tocilizumab-2xsIL6R-complex. The meaning of this observation
is unclear unless circulating levels of s-IL6R and of the possible complex are measured in parallel and are
compared in monkeys/humans/different disease states.
Renal excretion of intact tocilizumab (measured by Enzyme Immunoassay (EIA)) was negligible in both
rats and monkeys. Radioactivity was primarily recovered from urine as small molecular weight entities
(small peptides, amino acids and/or free 125I) in both rats and monkeys, faecal recovery was very low.
These results are consistent with the known elimination behaviour of IgG which undergo little if any renal
or biliary excretion but mainly catabolism by proteolysis in lysosomes. The resulting small peptides and
amino acids subsequently may be excreted or added to the endogenous amino acid pool.
An in vitro study on hepatozytes indicates that the down-regulation of cytochrome (CYP) isoenzymes by
IL-6 (mainly CYP3A4) is mediated via the IL-6 receptor, since tocilizumab is shown to inhibit this IL-6
effect on hepatozytes in vitro. However, the relevance is not clear since this IL-6 down-regulating effect
occurred at very high concentrations only. But it cannot be excluded that tocilizumab might indirectly
influence the expression level of CYP enzymes in RA patients by inhibiting the down-regulating effect of
IL-6. Co-administered drugs metabolised by this pathway could then be metabolised faster in the presence
of tocilizumab.
Comparison of the pharmacokinetics of tocilizumab with that of an intentionally altered tocilizumab
variant, in which the terminal galactose of the glycosylation had been enzymatically removed (G0-
enriched tocilizumab) following single intravenous administration to rats revealed very similar
noncompartmental pharmacokinetic parameters indicating that changes in galactosylation did not change
tocilizumab pharmacokinetics in rats. Together with results from in vitro binding studies with lectins it
can be assumed that tocilizumab has no relevant affinity for galactose-recognizing glycoprotein receptors
and that such glycoprotein receptors are unlikely to contribute to the in vivo clearance of tocilizumab.
Toxicology
The cynomolgus monkey was chosen as the relevant responder species and a comprehensive toxicology
program was conducted with single- and multiple-dose studies up to duration of 6 months to characterize
the overall safety of tocilizumab. Additionally supportive data from diverse studies conducted with the
rodent analogue MR16-1 also were included in the assessment of the data.
15/55
The cardiovascular safety of tocilizumab has been investigated in a series of rigorously designed
preclinical in vivo studies in cynomolgus monkeys. These results indicate that tocilizumab does not
adversely affect cardiac integrity or electrophysiology; neither was an alteration of blood pressure
observed in any of the preclinical studies.
Toxicity studies have shown tocilizumab to be well tolerated in cynomolgus monkeys, both as single
intravenous (IV) doses up to 100 mg/kg and when given in multiple IV doses up to 50 mg/kg/day for 4
weeks or at IV doses up to 100 mg/kg/week for 6 months. No major abnormal findings were observed in
either the clinical pathology investigations or in the histopathological evaluation of tissues. The systemic
steady state exposure to tocilizumab in these monkey studies was 8-to10-fold above the maximum human
exposure comparing trough levels in the animals with the maximum level measured in clinical trials.
Changes in haematological parameters were observed, e.g. decrease in red blood cell count or increased
lymphocyte count.
A signal toward reduction of neutrophils was observed in the 2-week toxicity study with a clear
pronounced manifestation in the 4-week daily treatment cynomolgus study with no manifestation in the
bone marrow. The absence of bone marrow myeloid hyper or hypoplasia in the presence of reduced
absolute neutrophil counts (ANCs) along with the lack of neutrophil morphological abnormalities strongly
suggests that neither peripheral sequestration nor incomplete granulopoiesis is the underlying mechanism
of the reduced circulating neutrophils.
As expected from differences between human and cynomolgus monkey heavy- and light chain
immunoglobulin sequences, tocilizumab is immunogenic in the monkey. The observed anti-tocilizumab
response showed a clear inverse dose relationship, an effect which is frequently observed with molecules
of this type. Neutralizing antibodies were either greatly diminished or not detected in animals treated with
high dose tocilizumab. Thus, the observed immunogenicity of tocilizumab in cynomolgus monkey studies
did not compromise the results of these studies and their relevance for risk extrapolation to humans.
The studies demonstrated that inhibition of IL-6 normalizes the inflammation-driven osteoclastic bone
destruction and safety studies conducted with tocilizumab demonstrated that a morphologically and
functionally normal bone homeostasis is maintained under continuous chronic IL-6 inhibition with
tocilizumab.
Reproduction Toxicity
A reproductive teratology study was performed to address potential effects on embryo-foetal development,
and supplementary data from IL-6 knock out (k.o.) and IL-6 transgenic mouse models were evaluated for
functional and developmental risk assessments. The effect of tocilizumab on embryonic development has
been evaluated in an embryofoetal toxicity study in cynomolgus monkeys in doses up to 50 mg/kg/day.
The absence of any teratogenic/dysmorphogenic potential of tocilizumab concurs with the normal
phenotype reported for IL-6 deficient mice strains. Neither an abnormal phenotype nor any effect on
reproductive performance as consequence of IL-6 depletion has been reported. Nonclinical data suggest
that IL-6 is not regarded as a growth factor critical for the development of the musculo-skeletal system
and growth and development of other organ systems and is obviously also not critical for the
immunological control of the maternal/foetal interface.
A higher incidence of abortion/embryo-foetal death was observed in the cynomolgus teratology study in
the 50 mg/kg/day high-dose group with systemic exposure of factor >100 above the human targeted
efficacious plasma concentration. Therefore the NOAEL for this study is 10 mg/kg/day due to the possibly
treatment-related abortion observed as statistically significant at a dose level of 50 mg/kg/day. But already
in the 10 mg/kg/day group there were more abortion compared to the control and the 2 mg/kg/day group.
The abortions observed in the study are unlikely to be a consequence of direct embryo-toxicity induced by
tocilizumab. The transport of immunoglobulin G (IgG) across the transplacental barrier is dependent on
the stage of gestation. The IgG transfer is low or absent in the first trimester, increases at around gestation
16/55
day (GD) 60 and is most effective in the last trimester of gestation. However, foetal exposure to
tocilizumab at the time when the abortions occurred is considered to be nearly absent. This suggests an
underlying maternal rather than a direct foetotoxic effect. The findings cannot be explained by the
tocilizumab mechanism of action. Interleukin-6 (IL-6) does not play a critical regulatory role in the
control of pregnancy. In female IL-6 deficient mice, no abnormality of implantation was observed. This is
in contrast to other factors of the IL-6 cytokine family that have a central regulatory function in the early
physiological processes of implantation. Both in-vitro and in-vivo studies consistently demonstrated that
IL-6 does not have a recognized role in the physiology of pregnancy. IL-6 deficient mice represent the
most sensitive experimental model to assess such functionality. In IL-6 deficient mice, the reproductive
performance investigated did not show a clinically relevant difference in female reproductive parameters
such as litter size and neonatal loss compared to the wild-type background strains of mice
Nonclinical data do not suggest an effect on fertility under treatment with tocilizumab as effects on
endocrine active organs or on organs of the reproductive system were not seen in a chronic primate
toxicity study. There is also no preclinical evidence, that IL-6 signalling is involved in processes of
reproduction, and, accordingly, the reproductive performance is not affected in IL-6 deficient mice.
Neither monkeys exposed to tocilizumab over more than 6 months, nor IL-6 k.o. mice showed
morphological alterations to the primary or secondary tissues of their immune system nor in any other
organ or tissues, demonstrating that IL-6 does not play a critical role in organ or tissue development at any
stage.
Other toxicity studies
In clinical trials, mild and moderate elevations of hepatic transaminases have been observed with
tocilizumab treatment, but without progression to serious hepatic injury. Increased frequency of these
elevations was observed when hepatotoxic drugs were used in combination with tocilizumab or added to
tocilizumab monotherapy. Tocilizumab did not affect liver enzymes in cynomolgus monkey studies in
which IL-6 was elevated due to CIA induced inflammation. IL-6 is, however, known to have antiapoptotic
and growth-promoting effects in hepatocytes. In vitro studies conducted with tocilizumab on human
hepatoma cell lines have however not been able to clarify this clinical finding. Clinical Precautions and
safety monitoring in patients and long-term-extension studies are in development to address this issue.
Furthermore serious hepatic events are an endpoint to be followed in pharmacovigilance and in registries;
such events will be fully queried using a Guided Questionnaire in clinical studies and in response to
spontaneous adverse event reports. Guidance will be provided in the product labelling under Section 2
“Posology and Administration” and Section 4.4. “Special warnings and precautions for use, regarding the
monitoring of liver enzymes”.
Ecotoxicity/environmental risk assessment
Tocilizumab is a monoclonal antibody, a protein and as such formally exempted from environmental risk
assessment according to the EMEA guideline 4447/00 (2006), which assumes no significant
environmental risks arising from proteins. Based on standard acute ecotoxicity tests with algae, daphnia
and fish and on lack of evidence for bacterial toxicity, the ecotoxic potential of tocilizumab is considered
to be low. The excipients are degradable too and show no significant ecotoxic potential.
Discussion on the non-clinical aspects
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity and genotoxicity.
The embryo-foetal toxicity study conducted in cynomolgus monkeys showed no evidence of a teratogenic
potential of tocilizumab. A slight increase of abortion/embryo-foetal death was observed with high
systemic exposure in the highest dose group (exposure > 100 above the expected human efficacious
concentration). The relevance of this finding for human pregnancy cannot be excluded.
17/55
2.4 Clinical aspects
Introduction
GCP
The Clinical trials were performed in accordance with GCP as claimed by the applicant.
The applicant has provided a statement to the effect that clinical trials conducted outside the community
were carried out in accordance with the ethical standards of Directive 2001/20/EC.
Pharmacokinetics
The assay for measurement of tocilizumab in serum (ELISA) measures “free tocilizumab” only, i.e.
tocilizumab with one or two free IL-6R binding sites. It is assumed that the measurement of tocilizumab
reflects the total amount of free tocilizumab plus an uncertain fraction of tocilizumab which has been
bound to sIL-6R. Furthermore, it is assumed that antibody-bound tocilizumab would not be measured in
the assay as well. Therefore, when interpreting PK data it has to be kept in mind that the concentrations
refer mainly to free drug. However, the majority of tocilizumab present in human is expected to be free,
i.e. not complexed to sIL-6R, as its molar concentration largely exceeds that of sIL-6R.
The main PK results were based on the population PK analysis from 4 Phase III studies in 1793 RA
patients with supportive data from non-compartmental analyses of clinical pharmacology studies.
Non-compartmental PK analysis from single and multiple dose studies showed similar PK characteristics
in RA patients compared to healthy volunteers. The PK of tocilizumab was characterized by non-linear
kinetics over the dose range tested. CL was concentration-dependent. There was no deviation from a dose
proportional increase for Maximum Concentration (Cmax) and a more than dose-proportional increase in
AUCinf and Concentration at the End of the Dosing Interval (Trough Concentration) (Cmin). The over-
proportional increase in AUCinf with increasing dose seemed more pronounced between the 2 and 10
mg/kg doses than between the higher doses (10, 20 and 28 mg/kg).RA patients showed mean values of
0,26 ml/h/kg for CL, ca. 160 h for t1/2 and ca. 60 ml/kg for Vss after 10 mg/kg. These values were
comparable to healthy volunteers in this dose range. The PK parameters of tocilizumab did not change
with time.
The observed concentration-dependency of the tocilizumab CL could be best described by a population
PK model rather than by non-compartmental analysis. A two-compartment disposition model with parallel
first-order (linear CL) and Michaelis-Menten elimination (nonlinear or concentration-dependent CL)
kinetics was obtained which could explain convincingly the dose proportionality of Cmax, the over-
proportional increase in AUC and Cmin and the apparent increase of half-life with dose observed in single
and multiple dose ascending studies:
The total CL of tocilizumab is concentration-dependent and is the sum of linear CL and nonlinear CL. The
portion of the nonlinear CL is reflecting a zero-order process which is saturated at very low concentrations
(estimated KM: 2.7µg/ml). This is believed to reflect the target-mediated CL via binding to IL-6R. At
higher tocilizumab concentrations, total CL is mainly determined by linear CL which was calculated to be
12.5 mL/h.
Non-linear CL plays a major role at low tocilizumab concentrations. The average contribution of nonlinear
CL to the total CL was less for 8 mg/kg than for 4 mg/kg tocilizumab every 4 weeks.
Due to the concentration-dependent total CL a calculated terminal half-life must appear in a
“concentration-dependent” manner as well. Therefore, it should not be derived using non-compartmental
analysis. Nevertheless, the mean apparent t1/2 of tocilizumab following a single dose of 10 mg/kg was
18/55
estimated by non-compartmental analysis to be about 8 days. Based on parameter estimates from the
population PK model, the effective t1/2 at steady-state ranged from 8 to 14 days for 8 mg/kg dosing every
4 weeks.
At high concentrations, when nonlinear CL becomes negligible, the apparent t1/2 can be calculated using
the formula for a two-compartmental model with the linear CL term only. Applying this formula to
tocilizumab results in an apparent t1/2 of 21 days. This estimate of apparent t1/2 is comparable to the t1/2
of human IgG antibodies (ie, around 23 days).
A major conclusion is that due to the concentration-dependent PK, the total CL and/or apparent half-life
estimated is only valid for a given dose and dosing interval and should not be translated to other dosing
regimens.
As with other immunoglobulins, tocilizumab is not excreted via the renal or biliary route. It can be
assumed that tocilizumab is predominantly eliminated via catabolism. Therefore, the total plasma CLt
reflects both degradation (in plasma) and distribution to tissues/endothelial cells where also degradation
takes place.
After multiple dosing with 8 mg/kg every 4 weeks, the predicted systemic accumulation ratio is small for
Cmax and AUC (1.06 and 1.22, respectively), but higher for Cmin (2.35). For the 8 mg/kg dose, steady-
state is achieved following the first administration, after 8 weeks and after 20 weeks for Cmax, AUC and
Cmin, respectively. The predicted coefficient of variation (CV) for AUC at steady-state for 8 mg/kg
tocilizumab every 4 weeks was 44%.
After administration of the 8 mg/kg dose in a 4-week interval, a mean (± SD) Cmin at steady-state
(Ctrough) of 9.7 µg/ml is estimated showing a high variability (CV) of about 100%. This mean Cmin
value is close to the KM value (2.7 μg/mL) estimated for the nonlinear CL component, with Cmin being
about 3.6-fold higher than KM. Thus, the Ctrough values are in a concentration range where small
changes in serum tocilizumab concentrations result in a large change in nonlinear CL. Furthermore, the
expected Ctrough value is 65 fold higher than the calculated value for KD of tocilizumab for the binding
at the IL-6R (KD about 1 nM = 0.15 µg/ml), thus indicating complete receptor occupancy even at the end
of each 8 mg/kg dosing interval.
Estimated volume of distribution during steady state (Vss) was small (6.4 L). This corresponds to 1-2
times the plasma volume which is in accordance with values obtained for other monoclonal antibodies.
Protein binding in-vitro and ex-vivo or binding to blood cells has not been determined in human plasma.
Results with monkey plasma indicate that during steady state ca. 20% of total tocilizumab concentration in
plasma might be bound at/in blood cells.
Consistent with its nature as IgG, tocilizumab seems to be able to cross the placental barrier. In a segment
II-embryo-foetal development study in cynomolgus monkeys, foetal plasma concentrations of tocilizumab
were 39% and 60% of the maternal plasma concentration after application of 10 and 50 mg/kg/day,
respectively. Data in humans are not available.
It is unknown whether tocilizumab is excreted in human breast milk. From its IgG nature it has to be
expected.
Special populations
From the evaluation of the influence of different co-variates on the PK of tocilizumab (population PK) it
can be concluded that no dose adjustments are necessary for age, gender or race as these did not affect the
PK of tocilizumab. Body size had an effect on CL: With a flat dose, an increase in BW would result in a
19/55
decrease in the secondary PK parameters AUC and Cmin. With a BW-adjusted dosing regimen the effect
of BW on CL is accounted for. However, the BW-adjusted dosing results in higher exposures with higher
BWs (>100 kg) and lower exposure in patients with low body weight < 60 kg (predicted AUC increase
almost two-fold from lowest to highest body weight). This did not affect efficacy or safety parameters in a
clinically relevant manner. Nevertheless, it is recommended not to exceed a dose of 1200 mg as this was
the maximum dose in clinical trials. In order to minimise the risk of insufficient therapeutic response in
patients with a body weight <60 kg, a recommendation for a dose capping at 480 mg was introduced.
Although no formal renal impairment studies were conducted, both the results from one study with renal
impaired RA patients and from the population PK analysis do not indicate an influence of renal
impairment on the main PK variables of tocilizumab. Thus, at least mild renal impairment does not change
the PK of tocilizumab. For patients with moderate to severe renal impairment (CLCr < 30 ml/min) no data
are available.
Due to the lack of specific metabolism in the liver it is not expected that PK of tocilizumab is altered in
hepatic impaired patients. Therefore the influence has not been studied.
Other covariates with a statistically significant influence on the primary PK parameters were High density
lipoprotein (HDL)-cholesterol, RF, total protein and albumin, however, these covariates did not change
the PK of tocilizumab in a clinically relevant manner.
Pharmacokinetic interaction studies
The influence on the PK of tocilizumab by other drugs has been addressed in studies for methotrexate
(MTX) only. It is known that MTX can decrease the CL of antibodies. The PK data from two studies
indicate that MTX appears to have no/little influence on the PK of tocilizumab. However, it cannot be
excluded that a small interference is causative for the slight but not significant increase in all four Cmin
values during the four dose intervals in the 8 mg/kg groups with co-medication of MTX.
The influence of tocilizumab on the PK of other drugs was investigated in one study for the drugs
dextromethorphan and omeprazol. A noticeable influence could only be detected for omeprazol.
The bioavailability of omeprazol (10 mg oral dose) was decreased by about 20-30% after intravenous
administration of 8 mg/kg tocilizumab. Since omeprazole is a substrate (and inhibitor) of CYP2C19 this
can be explained with an inhibition of the down-regulating effect of IL-6 by tocilizumab.
However, the finding that the CL of dextromethorphan (a CYP3A4 and CYP2D6 substrate) was slightly
decreased while the CL of its metabolite dextrorphan (CYP3A4 substrate) was increased (total CL
unchanged) is not consistent with the suggestion of a general inhibition of tocilizumab on the down-
regulating effect of IL-6 which appeared to affect almost all CYP isoforms in vitro. However, this IL-6
effect occurred only at very high concentrations in vitro. Thus, the relevance is not clear.
For all other clinically relevant RA medications (NSAIDs, steroids etc.), no data about PK interactions are
available except the results from the population PK analysis in which none of the co-medication in all
clinical trials was identified as a co-variate influencing the PK of tocilizumab. Since it cannot be excluded
that tocilizumab can potentially increase the CL of all co-administered drugs which are metabolized by
CYP450 enzymes in the liver, also the CL of several NSAIDs and steroids might be affected. The possible
interaction is most important for CYP450 substrates with a narrow therapeutic index (eg, warfarin,
cyclosporin). This has been addressed accordingly in the SPC.
There is an ongoing study (WP18663) in 24 RA patients to investigate the PK of MTX and the CYP3A4
substrate simvastatin in combination with a single i.v. dose of 10 mg/kg tocilizumab. Since MTX and
simvastatin are important co-medications in RA patients the results of this study are not without
significance. This issue is addressed in the Risk Management Plan (RMP).
20/55
Pharmacodynamics
Mechanism of action
Tocilizumab specifically binds to the IL-6 binding site of both sIL-6R and mIL-6R receptors with similar
affinity in the nanomolar range. Data from binding studies to modified hIL-6R variants suggested that the
binding region of tocilizumab was within in the cytokine-binding region (CBR) to which IL-6 binds.
Therefore, tocilizumab is able to block IL-6 from binding to both receptors and thereby blocks the activity
of IL-6.
Tocilizumab inhibits both the IL-6 classical and trans-signalling pathways by binding to
mIL-6R and sIL-6R. The binding of tocilizumab to the receptor prevents receptor binding to IL-6. The
tocilizumab/receptor complex cannot be bioactive since it is unable to effect the dimerization of the gp130
molecule. In the absence of this dimerization, the IL-6 signal is completely blocked.
Primary and Secondary pharmacology
In healthy volunteers, an increase in IL-6 levels was observed following administration of single
tocilizumab doses of 1 and 2 mg/kg. Baseline values of IL-6 were all well below 10 pg/ml as it is expected
for healthy volunteers. A clear but variable increase was observed for the 1 and 2 mg/kg doses with peak
levels of 10-60 pg/ml occurring between 24 and 72 h post-dose.
In RA patients, administration of single tocilizumab doses resulted in a marked increase in IL-6 after 8
mg/kg and 10 mg/kg. In one multiple dose study peak values of up to 1800 pg/ml have been observed
after the 8 mg/kg dose during the first days after administration.
During the 4 pivotal Phase III studies the only sampling time points were pre-infusion and 2 weeks after
infusion. For both 4 and 8 mg/kg doses, mean IL-6 levels peaked at week 2. Since this was the only
assessed time point these are no true peak values, from the former studies peak values are expected earlier
during the first days after infusion. Due to the sparse sampling also the postulated tendency for a decrease
in peak levels over time cannot be followed.
The observed increase in IL-6 serum concentrations following infusion of tocilizumab is believed to
reflect the binding of tocilizumab to the soluble and membrane-bound IL-6 receptors. Following
tocilizumab dosing, tocilizumab binds to the IL-6 receptors and blocks the receptor-mediated CL of IL-6,
leading to an increase in serum concentration of IL-6. Across the studies, lower mean trough IL-6 levels
were observed for the 4 mg/kg dose compared with the 8 mg/kg dose. Overall, IL-6 trough levels tended
to decrease over time. This is believed to reflect the declining production of Il-6 as the inflammatory
process is controlled.
A marked and dose-dependent increase in sIL-6R was observed in healthy volunteers and RA patients at
single doses ≥ 0.5 mg/kg tocilizumab. With increasing doses, both peak levels (up to 7-fold increase of
baseline levels) and time to reach these peak levels increased (about 3 to 4 weeks after the 8 mg/kg dose).
Following a single dose of 10 mg/kg to RA patients, peak sIL-6R levels were achieved at 4 weeks post-
administration with values returning to baseline approximately 8 weeks post-administration. At multiple
doses of 8 mg/kg every 4 weeks, high and sustained sIL-6R levels, with up to a 14-fold increase from
baseline, were observed with only a slight fluctuation within the dosing interval.
The increase in soluble IL-6 receptor levels with tocilizumab exposure is believed to be a consequence of
the binding of tocilizumab to those receptors. It is hypothesized that the accumulation of sIL-6R in serum
with increasing tocilizumab exposure reflects the slow CL of the TCZ/sIL-6R complex.
21/55
Using a gel filtration method it has been shown in healthy volunteers after a 2 mg/kg dose, that more than
99% of sIL-6R was bound to tocilizumab. Since the sIL-6R assay measures only an unknown amount of
these complexes, i.e. it is underestimating the total sIL-6R levels. Since the clinical relevance for the
increase in sIL-6R levels is not clear this discrepancy might not be important. The population PK analysis
revealed that sIL-6R did not affect the pharmacokinetics of tocilizumab.
A feedback rise of IL-6 and an up-regulation of IL-6 receptors (and hence sIL-6R) due to the receptor
blockade by tocilizumab is conceivable. However, the observed new increase after each dose is not fully
consistent with the idea of complete receptor occupancy by tocilizumab throughout the whole interval.
During the first-in-man study MRA001JP a decrease in the complement titres and IL-2 was observed. The
observed decreased IL-2 reactivity was questionable due to the inappropriateness of the IL-2 reactivity
assay method. IL-2 reactivity has not been measured in subsequent clinical trials. Reduction in
complement levels is believed to be consecutive to the inhibition by tocilizumab of IL-6 stimulation of
hepatocyte acute phase protein synthesis. Further measurements of the time course of mean CH50, C3 and
C4 in patients with RA in several studies in Japan showed that mean concentrations decreased to values
around the lower limit of normal and were maintained thereafter. The changes in complement were not
clinically significant and were not associated with symptoms suggesting extensive immune complex
formation.
After a single dose of 5 and 10 mg/kg tocilizumab in RA patients mean C-reactive protein (CRP) levels
decreased markedly between week 1 and week 3 to 4, respectively. For the 5 mg/kg dose, mean CRP
levels decreased by about 80%, from 1.7 ± 1.2 mg/dL at day 1 to 0.3 ± 0.1 mg/dL at week 1. By week 6,
mean CRP levels had returned to baseline.
In multiple dose studies, a dose-dependent decrease in CRP levels was observed in RA patients with
tocilizumab doses ≥ 4 mg/kg. For 8 mg/kg every 4 weeks, CRP levels were markedly suppressed as early
as week 2 and sustained around the normal range during the entire dose interval (4 weeks) compared to
MTX (no effect on CRP). Only slight fluctuations in CRP were observed with this dose. For multiple
doses of 4 mg/kg every 4 weeks, fluctuations in CRP levels were greater than with 8 mg/kg. CRP levels at
week 2 post-dose were low and similar to levels for 8 mg/kg. However, for the 4 mg/kg dose, CRP
transiently increased at trough which is in clear contrast to what was observed for the 8 mg/kg dose.
Therefore, a sustained decrease in CRP throughout treatment duration was achieved for 8 mg/kg every 4
weeks dose only, supporting this dosing regimen for the treatment of RA.
Immunogenicity was monitored in all clinical trials with tocilizumab, the sampling time points during the
four pivotal Phase III studies were every 2 weeks up to 24 weeks. A total of 18 out of 1793 RA patients
(1%) were positive for anti-tocilizumab antibodies (HAHAs) in the confirmation assay. In addition, the
immunogenicity of tocilizumab was assessed as part of the population PK analysis, which investigated the
effect of anti-tocilizumab HAHAs on the PK of tocilizumab. HAHAs were not identified as a covariate
influencing the PK of tocilizumab.
However, neither the screening/confirmation assay nor the inhibition ELISA are capable to measure
already built complexes of tocilizumab-HAHA. Thus, the HAHA-assays measure free antibodies only.
Since it must be assumed that most of the HAHAs will be bound to tocilizumab (especially in the presence
of high tocilizumab concentrations), the measured HAHAs are probably only a small amount of the total
HAHAs built. Secondly, due to the small free portion of the antibodies they might not be detected at all in
the studies. Thus, the true incidence of patients who built HAHAs and the quantity HAHAs measured in
these patients might be underestimated. The issue of further monitoring immunogenicity is addressed in
the RMP.
The exposure-efficacy relationship showed an exposure-dependent decrease in DAS28 (clinical endpoint).
Exposure safety relationship showed an exposure-dependent decrease in neutrophil count. PK/PD
modelling of both relationships revealed EC50 values for tocilizumab of 3.7 µg/ml (for decrease in
22/55

DAS28) and 7.4 µg/ml (for neutrophil loss rate), respectively. Since both the dose-response curves are
close together it becomes clear that a positive response will necessarily be accompanied by a certain
neutrophil loss rate. Comparing these EC50 values with the predicted values of about 10 µg/ml for
Ctrough and about 180 µg/ml for Cmax of tocilizumab during a 8 mg/kg dosing interval, a reduction in
Cmax (reduction in infusion rate) might be considerable in order to reduce the risk of very high neutrophil
loss rate.
Clinical efficacy
Tocilizumab (TCZ, MRA) is intended for the treatment of moderate to severe active RA. The applicant
initially applied for a broad indication covering all clinical situations from first line therapy in previously
untreated patients to patients that have failed on one or more anti-TNF medications.
In the pivotal trials TCZ has been studied at two doses, the applied dose of 8 mg/kg every 4 weeks
corresponds to the higher of the two studied doses. A combination therapy with MTX, as is common in
the setting of RA treatment with biologics, is left at the discretion of the physician.
The clinical development involved five pivotal phase III studies and two long-term extension studies.
There are four supportive studies, two phase II studies supporting dosing recommendations and two phase
III studies that are central to licensing in Japan.
Table 4; Overview of main studies
Study ID Design Diagnosis
Incl. criteria Study Posology Subjs
by arm
entered/
compl.
Duration Primary
Endpoint
WA17822 Three-armed,
randomised,
double-blind,
placebo-
controlled,
parallel group,
multicentre
Moderate to
severe active
RA who had
an
inadequate
response to
MTX
Placebo +
MTX
TCZ 4 mg/kg
+ MTX
TCZ 8 mg/kg
+ MTX
204
214
205
24 weeks ACR 20
response rate
WA
17823
Three-armed,
randomised,
double-blind,
placebo-
controlled,
parallel group,
multicentre
Moderate to
severe active
RA who had
an
inadequate
response to
MTX
Placebo +
MTX
TCZ 4 mg/kg
+ MTX
TCZ 8 mg/kg
+ MTX
394
401
401
2 years
(ongoing)
ACR 20
response rate
at week 24
Change from
baseline in
modified
Sharp score
and change in
physical
function at 1
and 2 years
WA18063 Two-armed,
double blind,
randomised,
placebo-
controlled
Moderate to
severe active
RA who had
an
inadequate
response to
DMARDs
Placebo +
DMARD
TCZ 8 mg/kg
+ DMARD
415
805
24 weeks ACR 20
response rate
23/55

WA17824 Two armed,
double blind,
double
dummy,
randomised,
placebo-
controlled
Active RA;
MTX naïve
or MTX
discontinued,
but not due
to lack of
efficacy or
AE
MTX 7.5-20
mg/week
TCZ 8 mg/kg
q4weeks
284
288
24 weeks ACR 20
response rate
at week 24
WA18062 Three-armed,
double blind,
randomised,
placebo-
controlled
Moderate to
severe active
RA who had
an
inadequate
response to
anti-TNF
agent(s)
Placebo +
MTX
TCZ 4 mg/kg
+ MTX
TCZ 8 mg/kg
+ MTX
160
164
174
24 weeks ACR 20
response rate
at week 24
WA18695 Open label
extension
study, single
arm
Patients
completing
17822
TCZ 8 mg/kg
q4weeks +
MTX
537 Long term
safety/efficacy
WA18696 Open label
extension
study, single
arm
Patients
completing
WA17824,
WA18062,
WA18063,
WP18663
TCZ 8 mg/kg
q4weeks alone
or plus
MTX/DMARD
1902 Long term
safety/efficacy
The applicant has designed a comprehensive clinical program that took several relevant patient
populations into account, i.e. MTX/DMARD failure patients, anti-TNF failure patients and MTX naïve
patients. Comparator for MTX/DMARD failure (study WA17822, WA17823, WA18063) and anti-TNF
failure populations (study WA18062) was placebo on the background of stable doses of standard therapy.
Study WA17824 compared tocilizumab to MTX in a non-inferiority study.
Endpoints in the clinical trials were very similar in all trials and involved response evaluation according to
ACR, DAS28 and patient reported outcomes and were fully compliant with the current “Points to
Consider on the Clinical Investigation of Medicinal Products other than NSAIDs in Rheumatoid
Arthritis”.
Dose response studies
LRO301 was a 20-week Phase II, double-blind, parallel-group, placebo-controlled, randomized, seven-
arm, dose-finding study conducted in Europe, with TCZ given alone or in combination with MTX. This
was the primary study used to support the doses investigated in the pivotal trials.
The trial consisted of a 4-week run-in phase with weekly MTX (10 mg) and folic acid, after which
patients entered a 20-week active treatment phase (of four study infusions) and were allocated to one of
seven treatment groups: TCZ 2 mg/kg plus MTX placebo, TCZ 4 mg/kg plus MTX placebo, TCZ 8 mg/kg
plus MTX placebo, TCZ 2 mg/kg plus MTX, TCZ 4 mg/kg plus MTX, TCZ 8 mg/kg plus MTX or
placebo infusion plus MTX. Patients took 5 mg folic acid weekly for the duration of the study. Patients
were assigned to one of the seven treatment groups using the minimisation technique stratifying for centre
and MTX dose level.
24/55

Tocilizumab at a dose of 2 mg/kg in the monotherapy setting was clearly not superior to MTX
monotherapy and proved inferior in some aspects. Treatment with doses higher than 2 mg/kg was superior
to MTX monotherapy. The 8 mg/kg dose appeared to be the most effective dose across monotherapy as
well as combination therapy with MTX. However, there was no clear evidence showing improved efficacy
with combination therapy.
The following figure depicts ACR response rate at week 16 (LOCF, FAS population).
Figure 4
In the monotherapy setting, a linear dose-response relationship could be observed, this was less
pronounced in combination therapy with MTX. Doses higher than 8 mg/kg were not tested, this is
considered acceptable given the dose-response relationship that was observed.
MRA009JP was a randomized, double-blind, placebo-controlled, parallel-group study that evaluated two
different dose regimens of TCZ in patients who were not receiving concomitant DMARD. A total of 163
patients from 29 sites in Japan received at least one dose of study medication. The primary endpoint was
the proportion of ACR20 responders at week 12 (or the last observation prior to week 12). There was a
significantly higher proportion of ACR20 responders in the TCZ groups compared with the placebo group
(p <0.0001). Additionally, ACR50 and ACR70 response rates were higher in the TCZ groups than the
placebo group (see Table 5 below).
Table 5
25/55
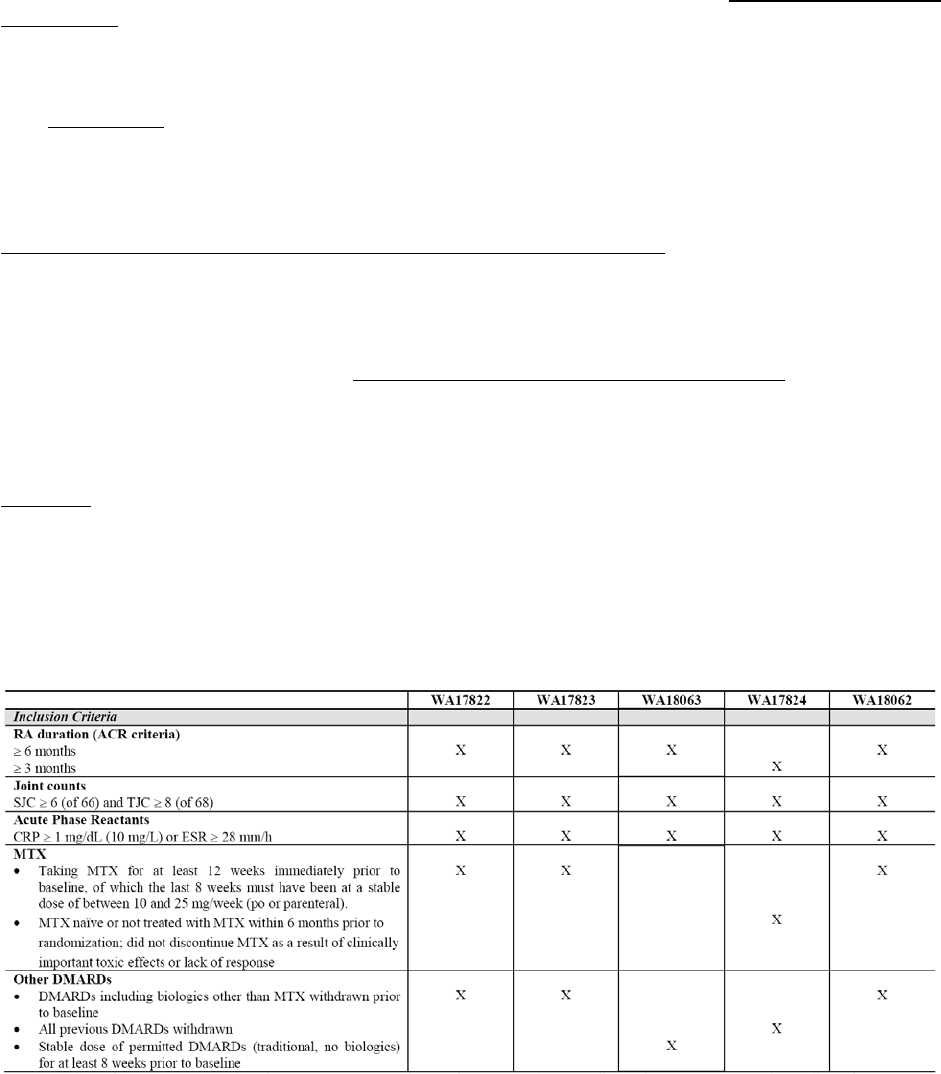
Main studies
WA17822: A randomized, double-blind, parallel group study of the safety and reduction of signs and
symptoms during treatment with TCZ vs placebo, in combination with methotrexate (MTX), in patients
with moderate to severe active rheumatoid arthritis (RA).
WA17823: A randomized, double-blind, parallel group study of the safety and prevention of structural
joint damage during treatment with TCZ versus placebo, in combination with methotrexate (MTX), in
patients with moderate to severe active rheumatoid arthritis (RA).
WA17824: A randomized, double-blind, double-dummy, parallel group study of the safety and efficacy of
TCZ monotherapy, versus methotrexate (MTX) monotherapy, in patients with active rheumatoid arthritis.
WA18062: A randomized, double-blind, placebo-controlled, parallel group study of the safety and
reduction of signs and symptoms during treatment with TCZ versus placebo, in combination with
methotrexate (MTX) in patients with moderate to severe active rheumatoid arthritis (RA) and an
inadequate response to previous anti-tumour necrosis factor (TNF) therapy.
WA18063: A randomized, double-blind, placebo-controlled, parallel group study of the safety and
reduction of signs and symptoms during treatment with TCZ versus placebo, in combination with
traditional disease modifying antirheumatic drug (DMARD) therapy in patients with moderate to severe
active rheumatoid arthritis (RA) and an inadequate response to current DMARD therapy.
WA 18695/18696: Long-term extension study of safety during treatment with TCZ (TCZ) in patients
completing treatment in TCZ core studies
METHODS
Study Participants
A total of 4211 adult RA patients from 725 centres worldwide were enrolled in the pivotal Phase III trials.
The key patient selection criteria for the pivotal Phase III studies are shown in Table 6 below:
Table 6
26/55
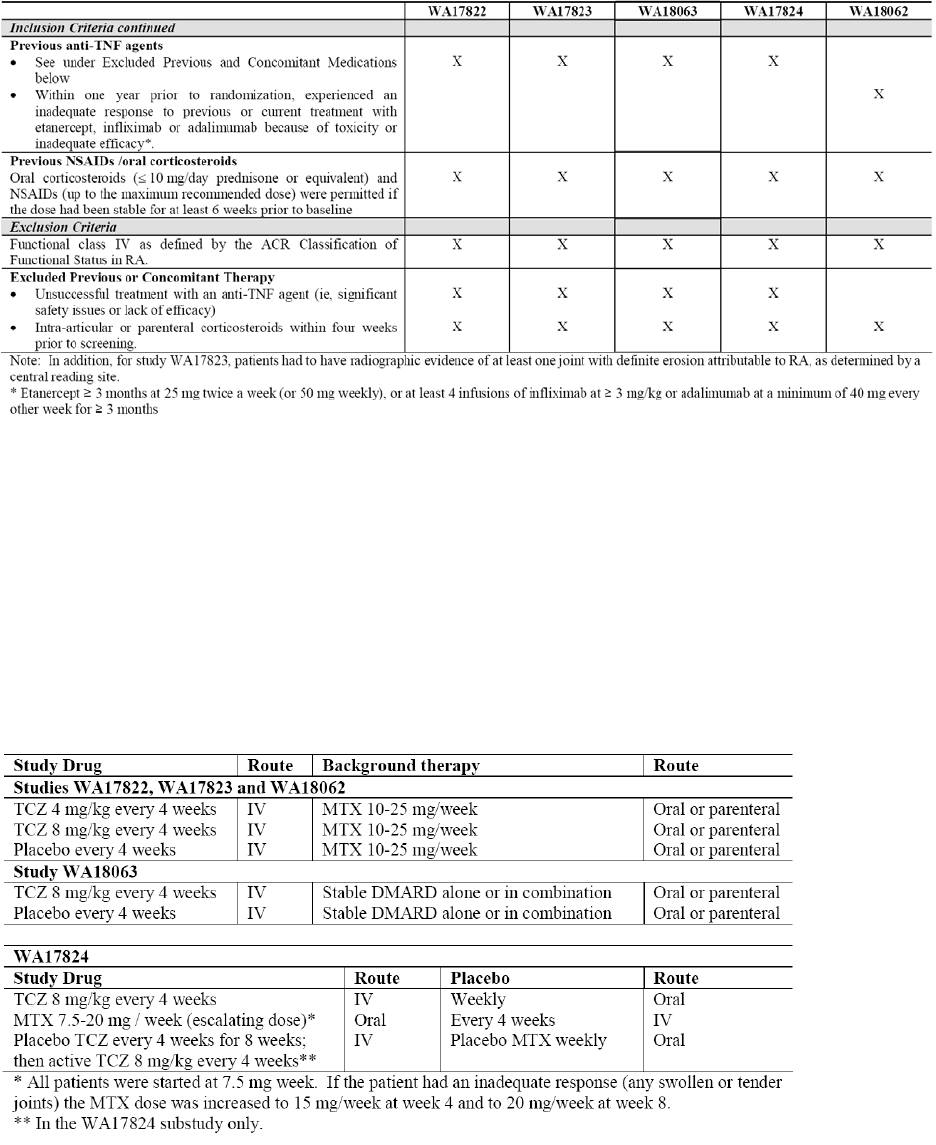
For all studies, the treatment groups within each protocol were well balanced with respect to demographic
and baseline RA characteristics. The study populations were representative of the heterogeneity of
characteristics among RA patients with moderate to severe, active disease in terms of the range of disease
duration and prior treatments as well as comorbidities and concomitant treatments such as corticosteroids
and NSAIDs.
Treatments
In studies WA17822, WA17823, WA18062 and WA18063, patients received TCZ or placebo infusion in
combination with MTX or other background DMARD therapy. WA17824 was a monotherapy study.
Table 7
In study WA17824, patients were randomly assigned (in a 1:1 ratio) to either TCZ 8 mg/kg or MTX. In
the WA17824 substudy, patients were randomly assigned (in 1:1:1 ratio) to either TCZ 8 mg/kg or MTX
(as in the main study), or to receive placebo MTX weekly plus iv placebo every 4 weeks for 8 weeks (2
infusions), followed by TCZ 8 mg/kg as an iv infusion every 4 weeks for the remaining 4 months of the
study. Patients continued to receive placebo MTX capsules to maintain the blind (TCZ: placebo). In this
study, one 5 mg (2.5 mg capsules) reduction in medication (MTX or its placebo) was permitted for
patients who, in the opinion of the treating physician, experienced dose-limiting MTX-related side effects.
27/55

The dose could not be increased at any time after the dose had been reduced nor could it be reduced to less
than 4 capsules / week.
Adjustments to study medication made for insufficient therapeutic response were permitted in all studies
(escape therapy).
Objectives
The primary objective of study WA18063 was to assess the efficacy of TCZ vs. placebo in patients with
moderate to severe active RA, with regard to reduction in signs and symptoms over 6 months of treatment
in combination with background DMARD therapy. This study was conducted in patients with an
inadequate clinical response to current DMARD therapy.
The primary objective of study WA17824 was to assess the efficacy of TCZ monotherapy vs. MTX in
patients who had not been treated with MTX within 6 months prior to randomization and who had not
discontinued previous MTX treatment as a result of clinically important toxic effects or lack of response
(as determined by the investigator). This study included a 3-arm randomized, double-blind, double-
dummy, parallel-group substudy with a placebo arm (8 weeks of placebo treatment followed by 16 weeks
of TCZ 8 mg/kg) as an internal control for efficacy.
With the exception of WA17823, all studies had a 24-week treatment period and the primary endpoint was
the proportion of ACR20 responders at week 24. Study WA17823 is an ongoing study with two planned
interim analyses, primary endpoints are evaluated at 6, 12 and 24 months. The 6-month primary endpoint
was the proportion of ACR20 responders at week 24. The 12 and 24 month primary endpoints are the
change from baseline in modified Sharp total radiographic score and change in physical function as
measured by the area under the curve for the change from baseline in the Health Assessment
Questionnaire - Disability Index (HAQ-DI). After year 2, patients can enter an optional open-label
extended treatment period of up to 3 years.
WA18695 and WA18696 are Phase III, open-label, international, multicentre studies, the primary
objective of which is to assess the long-term safety of TCZ 8 mg/kg as monotherapy or in combination
with background DMARD therapy(ies) with regard to adverse events and laboratory result abnormalities.
Secondary objectives include assessment of continuing clinical benefit using the same measures as in the
core studies.
Outcomes/endpoints
The primary endpoint in all pivotal studies was based on response criteria defined by the ACR at week 24.
The ACR20 response rate (the proportion of patients with an ACR20 response) at week 24 was the
primary efficacy parameter in the pivotal Phase III studies. An ACR20 response is defined as at least 20%
improvement compared with baseline in both tender joint count (TJC) and swollen joint count (SJC), as
well as in 3 out of 5 of the additional parameters shown in Table 7 below.
Secondary efficacy endpoints were based on additional ACR response criteria and the European League
Against Rheumatism (EULAR) measures of disease activity, which include defined changes to a disease
activity score (DAS) and patient-reported outcomes. ACR and DAS28 are both based on a core set of
outcome measures combined to quantify disease activity (continuous variables), together with definitions
of improvement (response variables). The core set of parameters is shown below:
28/55
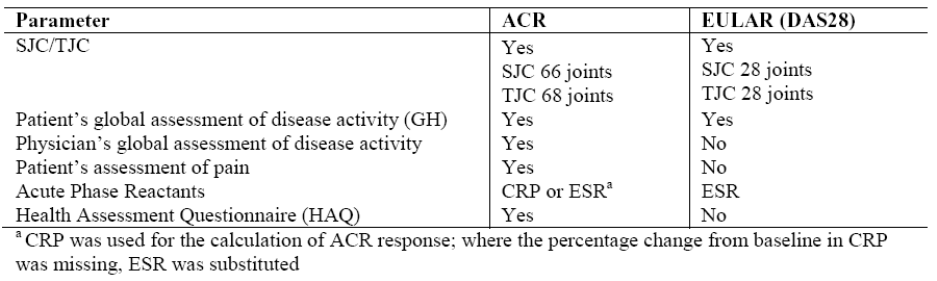
Table 8
In addition to the patient-reported outcomes included in the ACR and EULAR criteria (eg, HAQ-DI,
patient’s global assessment of disease activity, patient’s assessment of pain), other secondary outcomes
relating to degree of fatigue and general mental and physical health were assessed for each patient.
The degree of anaemia (haemoglobin) and rheumatoid factor (RF) assessment was also included as a
secondary endpoint.
Sample size
The sample size was selected based on data from a Phase II dose-finding study (LRO301) in which
ACR20 response rates of 60% to 70% were observed in the TCZ 4 mg/kg + MTX and TCZ 8 mg/kg +
MTX treatment arms, and a response rate of 40% was observed in the placebo + MTX arm. The response
rates in the present study were expected on clinical grounds to be lower than these rates, in view of the
relatively treatment refractory patient population being studied. Allowing for 15% of patients in each
treatment arm being classified as non-responders because of missing data or early withdrawal and using an
alpha level of 0.03 to accommodate for multiplicity, a selected sample size of 150 enrolled patients per
treatment group (450 overall) was sufficient to provide 80% power to detect a difference between the
proportion of patients achieving ACR20 scores in the TCZ 8 mg/kg + MTX and placebo + MTX treatment
arms, assuming that the proportion of patients achieving an ACR20 response would be 50% in the TCZ 8
mg/kg + MTX group and 30% in the placebo + MTX group.
Randomisation
Randomization was administered centrally via an interactive voice response system (IVRS) and was
stratified by ‘site’ using a randomization list provided by Sponsor. A patient’s eligibility was evaluated by
the investigator to ensure that the inclusion and exclusion criteria were met and that the patient was
eligible for participation in the study. Eligible patients were then randomized and assigned a unique
randomization number. Medication numbers were assigned by the IVRS prior to dosing at each dosing
visit depending on the patient’s weight and allocated treatment arm in order to ensure that the correct
dosage was provided.
Blinding (masking)
In case of blinding, the study was blinded with the sponsor, investigators, and patients unaware of the
treatment assignment of each patient. A patient’s treatment assignment was only to be unblinded in cases
where knowledge of the identity of the test medication was essential for further patient management.
Patients whose treatment assignments were unblinded did not receive any further study treatment.
29/55
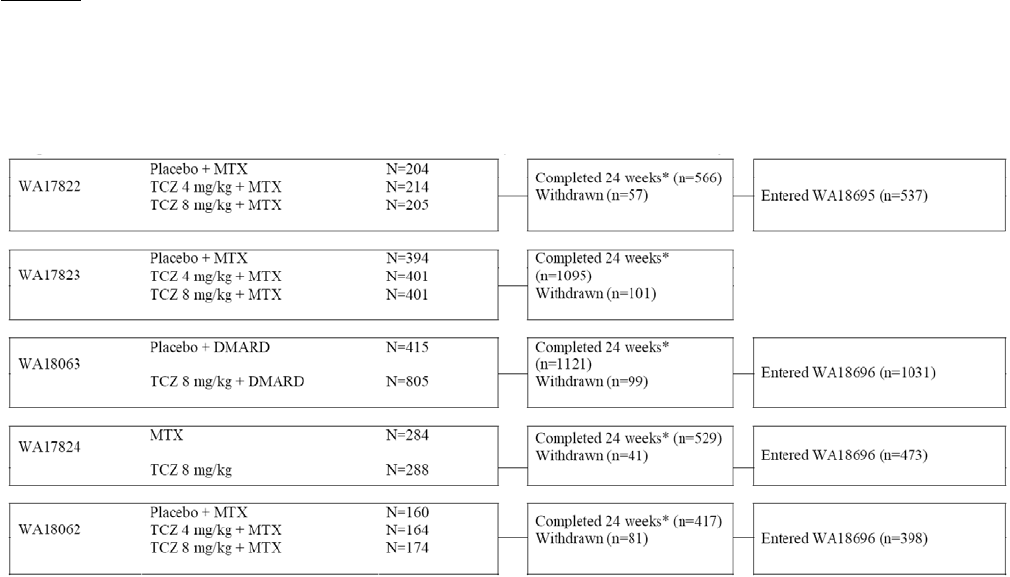
Statistical methods
Statistical methods were appropriate. The approaches taken were appropriately conservative. The intent-
to-treat (ITT) population was defined as the primary analysis population for all trials other than
WA17824. This study was a non-inferiority trial and the PP population was the primary analysis
population, as is standard in studies of this type. Categorical data were analyzed using the Cochran-Mantel
Haenszel (CMH) chi-square test, adjusted for site, which is a standard test used for analyses of this type.
In addition, the data were analyzed using logistic regression, allowing for the odds ratio to be presented
and adjusted for site as well as an assessment of other covariates in the model. The logistic regression
analyses provided confirmation of the results using CMH. Appropriate measures for handling of missing
data were employed
RESULTS
Participant flow
The most important data (Nr. of patients randomized, completed/withdrawn, distribution of patients per
arms, etc) of pivotal studies are shown below:
Of the 4211 patients randomized into the TCZ Phase III programme, 4098 received study medication:
1170 (28%) received placebo + DMARD, 774 (18%) received TCZ 4 mg/kg + MTX, 1582 (38%)
received TCZ 8 mg/kg + DMARD, 284 patients (7%) received MTX monotherapy, 288 (7%) patients
received TCZ 8 mg/kg monotherapy and 101 patients (2%) received 8 weeks of placebo followed by 16
weeks of TCZ 8 mg/kg (placebo / TCZ 8 mg/kg). Table 8 below presents the number of patients in each
treatment group, by study.
The disposition of patients in study WA18695/8696 is shown below:
30/55

Table 9
Recruitment
The earliest pivotal study started in 2004, the latest ones in 2005. One was completed in November 2006;
all the others in 2007. The extension study started in August 2005 and the cut off date of these ongoing
studies was on 20 April 2007.
Conduct of the study
Minor amendments were made in some studies, either not affecting outcomes or statistical analysis or in
order to make studies uniform.
Baseline data
For all studies, the treatment groups within each protocol were balanced with respect to demographic and
baseline RA characteristics. The study populations were representative of the heterogeneity of
characteristics among RA patients with moderate to severe, active disease in terms of the range of disease
duration and prior treatments as well as comorbidities and concomitant treatments such as corticosteroids
and NSAIDs. Unfortunately the number of DMARD naive patients were only relatively small (129
patients in the MTX group and 115 patients in the TCZ group). (Study WA17824).
Numbers analysed
ITT population is shown in the Table below:
31/55

Table 10
When ITT and per-protocol (PP) analysis were compared, no statistically significant differences were
observed
Outcomes and estimation
A summary of results of the main studies is given below:
WA17822:
ACR20 (%)
Placebo + MTX (n=204): 26.5%
TCZ 4 mg/kg + MTX (n=213): 47.9%
TCZ 8 mg/kg + MTX (n=205): 58.5%
For both TCZ + MTX groups, there was a highly statistically significant difference from the placebo +
MTX group in the proportion of ACR20 responders at week 24 (p < 0.0001 for both groups). Similar
results were obtained for the ITT robustness and PP population analyses of the primary efficacy
parameter. Logistic regression analysis showed that the odds of achieving an ACR20 response at week 24
were 3 times higher for patients receiving TCZ 4 mg/kg + MTX and 6 times higher for patients receiving
TCZ 8 mg/kg + MTX than for patients receiving placebo + MTX. ACR20 response rates were higher in
the TCZ + MTX groups compared with the placebo + MTX group at all time points from the first
scheduled assessment at week 2, with the highest rates being consistently observed in the TCZ 8 mg/kg +
MTX group. Secondary endpoint analyses (not shown) supported the primary efficacy findings.
WA17823:
ACR20 (%)
Placebo + MTX (n=393): 27%
TCZ 4 mg/kg + MTX (n=399): 51%
TCZ 8 mg/kg + MTX (n=398): 56%
32/55

For the primary endpoint, ACR20 response at week 24 (=interim analysis), significantly more patients in
both TCZ + MTX arms compared with the placebo + MTX arm achieved a response (p < 0.0001 for both
comparisons). Analysis of all secondary endpoints related to disease activity demonstrated significantly
better efficacy with TCZ 8 mg/kg + MTX compared with placebo + MTX (data not shown). This included
improvements from baseline in all ACR core set parameters (TJC, SJC, pain VAS, CRP, physician global
VAS, patient global VAS, Erythrocyte sedimentation rate (ESR), HAQ-DI) as well as the higher clinical
hurdle secondary endpoints such as ACR50, ACR70, DAS28 remission and EULAR good’ response.
Treatment with TCZ 8 mg/kg + MTX was associated with a significant increase in mean haemoglobin
levels compared with the placebo + MTX arm, with the most profound effects being observed in anaemic
patients. More patients in the placebo + MTX arm compared with the TCZ + MTX arms either withdrew
as a result of lack of efficacy or switched to escape therapy due to failure to achieve a > 20% improvement
in SJC and TJC.
Evaluation of the inhibition of progression of structural damage was a pre-defined endpoint in study
WA17823. The mean change of the total Sharp-Genant Score at week 52 was significantly lower for
patients treated with TCZ + MTX (0.34 and 0.29 for the 4 mg/kg and 8 mg/kg groups, respectively) than
for patients who received placebo + MTX (1.13, p < 0.0001 for both comparisons).
WA17824:
The primary endpoint was the proportion of patients with an ACR20 response at Week 24. The proportion
of ACR20 responders at Week 24 was 52.1% in the MTX group and 70.6% in the TCZ group, with a
weighted difference of 0.21 (95% CI 0.13 to 0.29). The lower limit of the CI was 0.13. Since the lower
limit is greater than –0.12 (pre-defined non-inferiority limit), treatment with TCZ was considered non-
inferior to treatment with MTX. As TCZ was shown to be at least non inferior to MTX, further testing for
superiority to MTX was conducted. For the purposes of this assessment the ITT population was used. The
weighted difference in ACR20 response at Week 24 was 0.19 (95% CI 0.11 to 0.27). Since the lower limit
of the 95% CI of the treatment difference was greater than 0, treatment with TCZ 8 mg/kg was
demonstrated to be superior to treatment with MTX. This result was highly statistically significant (p <
0.0001). To support the conclusions from the primary analysis a comparison was made between all
patients treated with TCZ and the placebo treated patients enrolled into the placebo controlled substudy.
The ITT population was used for this assessment. As patients in this study received placebo only for the
first 8 weeks, this analysis compared proportions of patients achieving an ACR20 response at Week 8.
The proportion of ACR20 responders at Week 8 was 13.1% in the placebo/TCZ group and 55.6% in the
TCZ group. The weighted difference in ACR20 response at 8 weeks was 0.43 (95% CI 0.34 to 0.52).
Since the lower limit of the 95% CI for the weighted difference was greater than 0, TCZ 8 mg/kg is
considered to be superior to treatment with placebo at Week 8. In general, all secondary endpoints tested
were positive and supported the improved efficacy of TCZ compared with MTX observed with the
primary efficacy endpoint. Logistic regression analyses showed the odds of achieving an ACR20 response
at Week 24 were approximately 3 times higher in the TCZ group. In addition to the differences observed
at Week 24, onset of response occurred earlier in the TCZ group with differences between the two
treatment groups apparent as early as Week 2 (ie, first scheduled assessment) for ACR20 response. By
Week 2, an increase in mean haemoglobin value of 7 g/l was observed in the TCZ group compared with a
mean decrease of 3 g/l in the MTX group.
WA18062:
The proportion of patients achieving ACR20, 50, or 70 responses at Week 24 was consistently higher in
TCZ 8 mg/kg + MTX group than the TCZ 4 mg/kg + MTX group or the placebo + MTX group (ACR20:
50% vs 30% and 10%, ACR50: 29% vs 17% and 4%, ACR70: 12% vs 5% and 1%, respectively). In
addition, irrespective of the most recently failed anti-TNF medication and the number of previously failed
anti-TNF medications, TCZ 8 mg/kg + MTX was shown to be beneficial in this difficult-to-treat
33/55

population in all three of these parameters. Similar results were obtained in the PP population analysis and
in several sensitivity analyses investigating different methods of imputation for missing data. Logistic
regression analysis indicated that the odds of achieving an ACR20 response at Week 24 were 9 times
higher for patients receiving TCZ 8 mg/kg + MTX and 4 times higher for patients receiving TCZ 4 mg/kg
+ MTX than for patients receiving placebo + MTX. Importantly, a statistically significant and clinically
meaningful benefit over placebo + MTX was observed in the TCZ 8 mg/kg + MTX group in the higher
clinical disease hurdles such as ACR50 and ACR70 response, EULAR ‘good’ response, and DAS28
remissions rates. In addition to the demonstrated benefits at Week 24, the onset of response occurred early
in the TCZ 8 mg/kg + MTX group, with differences from the placebo + MTX group becoming apparent as
early as the first scheduled assessment (Week 2). Finally, the benefits of TCZ treatment were reflected in
the large numbers of placebo + MTX patients who either withdrew from the study due to insufficient
therapeutic response (11%) or entered the escape phase at Week 16 (41%) compared with early
withdrawal and escape rates of 2% and 11% for patients receiving TCZ 8 mg/kg + MTX.
WA18063:
The proportion of ACR20 responders at week 24 was 61% (488/803 patients) in the TCZ 8 mg/kg +
DMARDs group and 25% (101/413 patients) in the placebo + DMARDs group. For the TCZ 8 mg/kg +
DMARDs group, there was a highly statistically significant difference from the placebo + DMARDs
group in the proportion of ACR20 responders at week 24 (p < 0.0001). Similar results were obtained for
the ITT robustness and PP population analyses of the primary efficacy parameter. Logistic regression
analysis showed the odds of achieving an ACR20 response at week 24 were 6 times higher for patients
receiving TCZ 8 mg/kg + DMARDs than for patients receiving placebo + DMARDs. ACR 20 response
rates were higher in the TCZ 8 mg/kg + DMARDs group compared with the placebo + DMARDs group at
all time points from the first scheduled assessment at week 2.
Secondary endpoint analyses supported the primary efficacy findings. At week 24, statistically significant
differences from the placebo + DMARDs group were achieved for the TCZ 8 mg/kg + DMARDs group
for all secondary endpoints related to disease activity. In addition to the differences observed at week 24,
onset of response occurred early in the TCZ 8 mg/kg + DMARDs group and differences between the TCZ
8 mg/kg + DMARDs group and the placebo + DMARDs group were apparent by week 2 (ie, the first
scheduled assessment). A highly statistically significant increase in haemoglobin was also observed in the
TCZ compared with the placebo + DMARDs group. An increase of 6 g/L in mean haemoglobin values in
the TCZ 8 mg/kg + DMARDs group was observed early at the first scheduled assessment for haemoglobin
at week 2 and mean haemoglobin increase at week 24 was 9.753 g/L in TCZ + 8 mg/kg + DMARDs
group vs a mean decrease of 1.280 g/L in placebo + DMARDs group. For the primary and secondary
efficacy endpoints at week 24 and throughout the 24-week treatment period, the greatest responses were
consistently observed in the TCZ 8 mg/kg + DMARDs group.
WA18695/18696:
At the time of the data cut, the median treatment duration was 41 weeks in the WA17824 group, 52 weeks
in the WA18062 group, and 61 weeks in the pooled group. Response rates to therapy with 8 mg/kg TCZ
(+DMARD) were generally maintained or continued to improve with duration of treatment. For all
efficacy endpoints, the highest response rates were consistently achieved and maintained in the WA17824
group, followed by the pooled group, and then the WA18062 group. However, the pattern of response was
similar across all groups, ie, increasing response rate after the first 24 weeks of treatment as well as
durability of responses. The very low number of withdrawals due to insufficient therapeutic response
(2.4%) supports the observed improvement in efficacy during long-term TCZ treatment. Reported
categorical endpoints (ACR20, ACR50, ACR70, ACR90, and EULAR response) were affected by
imputation of missing data, as response was generally assessed every 12 weeks, and the study was still
ongoing at the data cut. The mean ACRn response increased over time in all 3 study groups and at week
72 reached 48.2 in the WA17824 group, 25.5 in the WA18062 group, and 44.9 in the pooled group. Mean
ACR core set parameters and the mean DAS28 showed further improvement after the first 24 weeks of
34/55
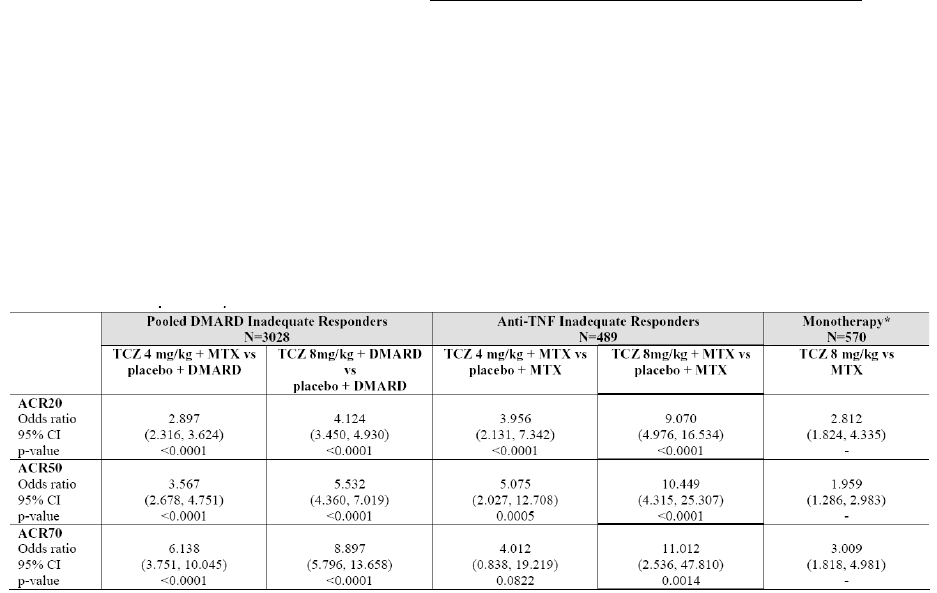
treatment in all 3 groups. The number of patients who reduced or stopped corticosteroid/DMARD use due
to sustained efficacy was 31 and 15 patients, respectively. An improvement in mean haemoglobin level
Improvements in RA disease activity were accompanied by an improvement in the patients’ quality of life.
Results from the SF-36 patient questionnaire demonstrated that both the mean physical and mental
component summary scores improved from baseline by more than the minimal clinically important
difference. At the week 48 visit, change from baseline in all 8 domains included in the SF-36 score
exceeded the minimal clinically important difference in all 3 study groups. The mean Functional
Assessment of Chronic Illness Therapy (FACIT)-fatigue score increased from baseline and then remained
stable for the duration of the studies. A substantial proportion of patients were able to achieve the highest
degrees of efficacy indicative of clinical remission or low disease activity.
Ancillary analyses
Analysis performed across trials (pooled analyses and meta-analysis)
The results from the individual studies provide evidence of the efficacy of TCZ in patients with moderate
to severe active RA. However, to provide an estimate of the treatment effect of TCZ in the DMARD
inadequate responder patient population and to investigate the effect of TCZ where there are likely to be
small differences between the treatment groups, studies WA17822, WA17823 and WA18063 were
pooled.
These studies have been considered appropriate to pool based on study design, demographic and baseline
characteristics and homogeneity of treatment effect. With this pooling, the results of pivotal studies can be
grouped into:
a) DMARD inadequate responders
b) anti-TNF-inadequate responders
c) TCZ monotherapy
The comparison of results of the primary efficacy criteria is shown below:
Table 11
35/55
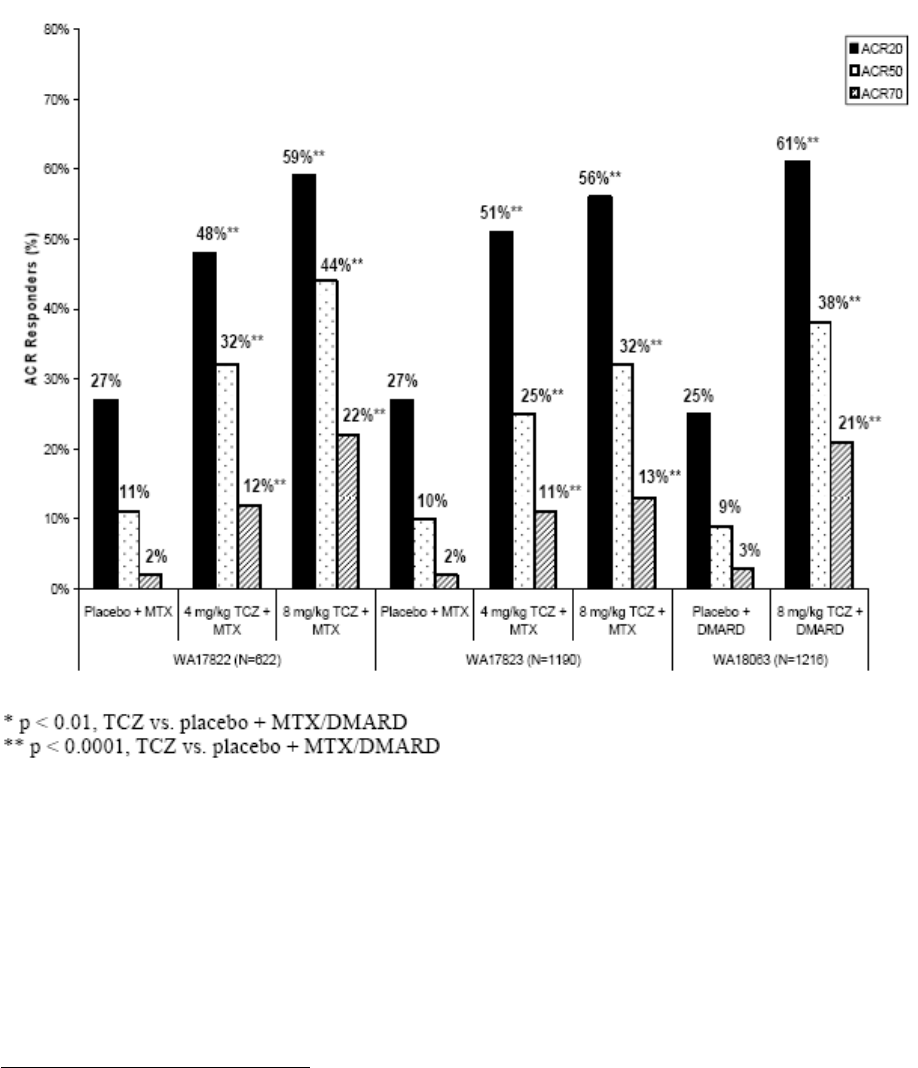
Figure 5: Proportion of ACR20, ACR50 and ACR70 responders at week 24 for studies WA17822
(inadequate MTX responders), WA17823 (inadequate MTX responders) and WA18063 (inadequate
DMARD responders)
Clinical studies in special populations
Of the various groups (children, elderly, gender, race and weight/BMI), only body weight (BW) has an
impact upon PK of TCZ. There was a slight decrease in clinical efficacy among patients with body weight
over 100 kg. However, a logistic regression model failed to show any significant differences between
treatment subgroups.
Supportive studies
Joint Damage Study MRA012JP: At the time of this submission, there are no radiographic data from the
pivotal studies. However, data showing the effects of TCZ on inhibiting structural joint damage, bone
densitometry and markers of bone metabolism are available from a supportive study, MRA012JP, in
Japanese patients from the Chugai development program.
This was a multi-centre, parallel group, open-label study conducted at 26 centres in Japan in RA patients
who had an inadequate response to current DMARD or immunosuppressive therapy. Patients were
randomized to remain on existing DMARD or immunosuppressant treatment or TCZ 8 mg/kg iv every 4
36/55

weeks for 52 weeks. In the control group, the concomitant use of drugs that slow bone or joint destruction
(eg, infliximab, etanercept, leflunomide) was prohibited. No restrictions were placed on switching
DMARDs or the doses of DMARDs and immunosuppressants for treating RA from the first observations
onwards. The primary endpoint was the change in erosion score at week 52, according to the modified
Total Sharp Score methodology of radiograph reading. Radiographs were scored by two blinded
independent readers using the van der Heijde modified Sharp method. The time course of absolute change
in lumbar and femoral bone mineral density (BMD) measured by dual energy X-ray absorptiometry
(DEXA) at weeks 28 and 52 was assessed as a secondary endpoint. The treatment groups were generally
well-balanced with respect to baseline demographic and disease characteristics. The mean duration of RA
was 2.4 years in the control group and 2.2 years in the TCZ group. Mean baseline DAS28 scores were 6.4
in the control group and 6.5 in the TCZ group. Mean baseline Total Sharp Scores were 30.6 and 28.3 in
the control and TCZ groups, respectively, and the rate of annual progression was 12.3/year in the control
group and 14.1/year in the TCZ group. At week 52, the TCZ group had statistically significantly less
radiographic change in Total Sharp Score, Erosion Score and Joint Space Narrowing compared with the
control group. Additionally, there were significant differences between the treatment groups in changes in
serum osteocalcin (marker of bone formation), urinary deoxypyridinoline (marker of bone resorption) and
lumbar and femoral BMD. TCZ treatment prevented bone loss with a significant increase in serum
osteocalcin levels, significant decrease in deoxypyridinoline levels and a smaller decrease in lumbar and
femoral BMD compared with conventional DMARD treatment.
These results provide evidence to support the hypothesis that inhibition of IL-6 by TCZ may have
beneficial effects in slowing bone and joint destruction.
Table 12 Change in Radiographic Scores (Full Analysis Set)
Study MRA213JP was a Phase III, two-arm, parallel-group, double-blind, multicentre study comparing
TCZ 8 mg/kg monotherapy every 4 weeks with MTX 8 mg weekly in RA patients with an inadequate
response to MTX. The study was designed to assess safety and signs and symptoms of RA after 24 weeks
of TCZ therapy.
37/55

38/55
Clinical safety
The pivotal clinical safety data supporting this application derives from five adequate and well-controlled,
double blind, international, Phase III studies in adult patients with RA: WA17822, WA17823, WA17824,
WA18062 and WA18063.
These 5 studies are referred to as the core studies and were conducted by Roche. The core studies are
completed, with the exception of the ongoing 2-year study WA17823, for which only pre-planned
24-week interim data are included in this submission. Long term safety information from 2439 RA
patients treated with open-label TCZ 8 mg/kg was also provided. The data are derived from 2 open-label
ongoing extension studies (cut off date April 20th, 2007). The duration of the extension studies will be 5
years.
Additional information is available from studies conducted by the applicant/sponsor’s co-development
partner, Chugai Pharmaceutical in healthy volunteers, PK studies in RA patients, RA patients who had
inadequate response to DMARDs or MTX and 6 studies in Castleman’s Disease (121 pts.), 1 in Crohn’s
(24 pts), 3 in multiple myeloma (37 pts.) and 1 in systemic lupus erythematosus (14 pts).
Patient exposure
The trials represent a broad range of both of RA patients and other auto-immune conditions. The RA
patients include those with early disease (<2 years), MTX-naïve patients, partial responders to standard
non-biologic DMARDs, and those who had failed treatment with anti-tumour necrosis factor (TNF)
agents.
The core safety data set encompasses TCZ when used alone or in combination with methotrexate (MTX)
or other non-biological disease-modifying anti-rheumatic drugs (DMARDs) for reducing signs and
symptoms in adult patients with moderate to severe active RA. For the proposed posology (8 mg/kg every
4 weeks) and indications data have been collected in 1870 patients for 6 months.
Long-term safety analyses were based on all patients who completed the 24-week controlled studies and
received TCZ in the open-label extension studies. A total of 2439 patients completed the core studies and
entered the extension studies providing, 2188/2439 patients (89.7%) had been receiving TCZ treatment for
at least 24 weeks, 1507 patients (61.8%) had been receiving TCZ treatment for at least 48 weeks and 574
patients (24%) for at least 18 months. The mean and median extent of exposure to TCZ treatment from
the first dose was 1.08 years.
Taken together, this represents an acceptable safety database, both in number of patients and exposure
time with TCZ. Upon request the applicant provided a tabular overview showing 6, 12, 18, month data for
the number of patients belonging to the proposed target population at the proposed dose (8 mg/kg) and
proposed SPC indications, i.e. stratified by prior therapy and concomitant treatment, thereby giving exact
numbers and duration for each proposed indication.

Table 13: Exposure to Tocilizumab 8 mg/kg Every Four Weeks by Patient Population
Moderate to Severe RA Monotherapy Combination therapy
MTX DMARD
6M 12M 18M 6M 12M 18M 6M 12M 18M
Not previously treated with DMARDs1
No. of Patients 132 94 52 118 48 14
Patient years exposure 161.4 137.6 87.8 110.4 59.8 21.9
Inadequate response to DMARDs
No. of Patients2 182 124 66 177 96 34
Patient years exposure 214.6 180.8 112.5 172.1 118.2 50.3
Non Escape3
No. of Patients - - - 1295 880 646 453 382 270
Patient years exposure 1672.8 1452.3 1180.9 661.8 613.1 481.5
Escape4
No. of Patients - - - 135 95 80 - - -
Patient years exposure 180.3 160.6 142.2
Inadequate response to anti-TNF
Non escape patients5
No. of Patients - - - 277 215 125 - - -
Patient years exposure 356.5 313.1 207.9
Escape patients6
No. of Patients - - - 101 76 41 - - -
Patient years exposure 124.1 105.9 65.7
1. WA17824: patients were permitted to add DMARDs in the extension. Exposure on monotherapy is summarised separately from combination therapy
2. Patients from WA17824 who were not DMARD-naïve at baseline;
3. WA17822, WA17823 (6 month data only) and WA18063: patients who did not receive escape therapy during the studies. WA18063: a wide range of DMARDs
were permitted as background therapy whereas other studies have MTX-only background medication Thus, exposure data from WA18063 separates MTX only
(shown in MTX combination therapy columns) and other background therapies excluding MTX alone (shown in DMARD combination columns). Patients taking
2 background DMARDs including MTX are included in the combination column. Patients who switched DMARD treatments are counted in the original active
treatment group actually received, i.e. if they were on MTX alone in the core study and increased to 2 DMARDs in the extension study, they are included in the
MTX-only group.
4. Patients from WA17822 and WA17823 who received escape therapy In WA18063, no’s on escape not included as escape was increase in DMARDs, not switch to
TCZ.
5. Patients from WA18062 who did not take escape therapy
6. Patients from WA18062 who received escape therapy
39/55

Additional 4 month safety data update (4MSU)
A 4 month update of the safety information was provided from the open label extension studies
WA18695 and WA18696, (cut-off date of October 1, 2007).
An update was also provided on deaths and SAEs occurring from September 1, 2007 to January 31,
2008, in the following:
The ongoing Japanese development programme (including non-RA indications);
Spontaneous reports from the treatment of multicentric Castleman’s Disease;
Compassionate use of TCZ in children with systemic onset inflammatory juvenile arthritis;
The ongoing study WA17823; and
The ongoing studies WA18695 and WA18696 (beyond October 1, 2007).
Data was also included regarding the safety and efficacy of TCZ 8 mg/kg monotherapy from patients
in study WA17824 who achieved a ≥ 50% decrease in the number of active swollen and tender joints
(assessed from baseline) and opted to continue blinded treatment in the so-called “transition phase”.
Further information was submitted from the immunogenicity testing program including data from
patients during the first 6 months of WA17823 and long-term follow-up of patients in WA18695 and
WA18696.
By October 1, 2007, an additional 123 patients had entered study WA18696, providing a total of 2562
patients who received at least one dose of TCZ in the long-term studies. Of the additional 123 patients
who were included in the 4MSU population, 93 were from the “transition phase” of WA17824.
The data was compared to the existing long-term data from the studies WA18695 and WA18696 in
2439 patients and AEs of special interest (infections, infusion reactions, cardio-vascular AEs,
neoplasms, gastrointestinal (GI) ulcerations and perforations, skin AEs, blood and lymphatic AEs,
hepatic AEs, autoimmune events and demyelinating disorders, and fractures) were focused on.
The 4 MSU population showed a ~ 3-5% increase in severe AEs, related AEs, serious AEs and AEs
leading to dose modification compared to the initial application. This may have to do with the
relatively short time period addressed in this report (30th April 2007 – 1st Oct. 2007) or may indicate
a trend. Any tendencies of increases would have to be captured by the RMP. In general the types of
AEs observed in the 4MSU population were similar to those reported previously. The new events (3x
cutaneous lesions of discoid lupus, and 1x arteritis, hypertensive encephalopathy, ischemic colitis,
intestinal obstruction, hepatic fibrosis, pulmonary hypertension, glioblastoma and B cell lymphoma)
should be kept in mind for the evaluations of the PSURs.
In the long-term studies the development of HAHAs (2.3%) neutralizing antibodies (<1%) was low.
The increased exposure to TCZ did not appear to result in an increase in the proportion of patients who
developed anti-TCZ HAHA and had associated allergic events. However, it remains uncertain as to
how the methods and assay for determining HAHAs affected the outcome (see also RMP)
Adverse events
Overall, adverse effects associated with the mechanism of IL-6R inhibition were observed in all TCZ
treatment groups. The safety profile in the 5 core studies was fairly consistent (also between the 4 and
8 mg/kg dosing regimens) and the following main safety signals emerged:
Infections
IL6 is a pleiotropic 24 kDa cytokine influencing antigen-specific immune responses and inflammatory
reactions. It is one of the major physiological mediators of acute phase reaction. As pathogenic
bacterial lipoproteins and lipopolysaccharides stimulate the production of IL-6 and sIL-6R, which then
act on multiple cell types to activate the ‘innate’ immune response, tocilizumab would be expected to
have an immunosuppressive effect on the development of effective immunity to bacterial and viral
infections.
In the studies, the TCZ-combination therapy treated patients had higher rates of infection compared to
the placebo patients, more patients with 2 or more infections, more severe infections and more serious
infections (placebo: 1.5% vs TCZ 2.1%). Within the category of infections, respiratory infections, skin
40/55

infections and GI were predominant. Systemic infections were rare (3 events of sepsis). The nature of
the serious infections in 2644 TCZ-treated patients in the controlled trials were pneumonia (0.5%),
cellulitis (0.3%), and herpes zoster (0.2%), sepsis (0.1%), gastroenteritis (0.1%). Opportunistic
infections were isolated cases (Pneumocystis carinii, Mycobacterium avium intracellulare), and in the
ongoing Japanese studies intracellular or opportunistic infections included 2 cases of tuberculosis
(TB), one case of bronchopulmonary aspergillosis and one case of candida osteomyelitis.
A higher incidence of infections was observed in patients who were previously exposed to anti-TNF
medications in all treatment groups in study WA18062 compared with patients in the other studies.
Cellulitis was questionably observed more frequently in the TCZ compared to the control groups and
the applicant had been requested to clarify this issue. Although there is an overlap of the 95% CI
between the placebo and bapineuzumab groups for cellulitis, it cannot thus necessarily be attributed to
TCZ, the sponsor has nevertheless included cellulitis in the list of adverse reactions in the SPC, in
recognition of the increased risk of infections in patients treated with TCZ.
As IL-6 is known to differentiate B cells into antibody-producing plasma cells the concern was raised
that by blocking IL-6, one may expect a decrease also in B cell functions and hence IgG levels and
antigen-specific antibodies (such as anti-pneumococcal-antibody titres/anti-tetanus titres) The sponsor
committed to continue monitoring the levels of immunoglobulins in the ongoing open label extension
studies and to conducting an additional vaccination study as a part of ongoing open-label long-term
extension study.
The timely detection of serious infection may be difficult due to the suppression of the acute phase
reaction (CRP, neutrophils). This aspect has been included in the SPC.
Gastrointestinal disorders
In the TCZ preclinical programme, no signals of GI toxicity were observed. However, in healthy
volunteers, events of diarrhoea, mouth ulceration, vomiting, abdominal pain, gingival pain, oral pain
and flatulence were reported. Although the wide use of corticosteroids and NSAIDs in patients with
RA contributes to these side-effects, gastric disorders (mainly gastritis and upper GI ulcers) were
observed more frequently with TCZ combination treatment compared with the control. In one study
(17824) related GI AEs in the monotherapy TCZ 8 mg/kg group(~20%) were the same as the MTX
monotherapy group, but practically double that of the combination therapy TCZ + MTX (9%) and
quadruple that of MTX + placebo in another study (17823; 5%). The discrepancies between the studies
were thought to be due to the different focusing of the patient in the Informed Consent Form. In 5
cases the increased inflammation led to perforations and in 2 cases to death. The rate of total GI
perforations is 0.184 /100 pt-years which is slightly higher than that reported in the literature for RA
patients. Gastritis, mouth ulceration, diverticulitis and its complications (peritonitis, perforation,
fistulae and abscess) are listed as adverse reactions in the SPC.
Infusion reactions
Infusion reactions occurred mainly during the first or second infusion. In the monotherapy groups, 6%
in the TCZ 8 mg/kg group and 2% in the MTX group had infusion reactions. In the combination
therapy groups ~5% in the TCZ groups and 3% in the placebo groups had infusion reactions. In the
long term safety population, these defined events were reported by 6.5% of patients. Infusion reactions
comprised mostly hypertension, rash and pruritus. Overall, 12 patients experienced medically
significant hypersensitivity reactions (six anaphylactic reactions).
Skin disorders
There was no evidence of skin toxicity with TCZ from preclinical data. Single events of rash were
reported in studies in healthy volunteers. Therefore the clinical data with increased skin and
subcutaneous tissue disorders raise a new concern. More skin disorders (rash, dermatitis and pruritus,
skin ulcers) occurred in the TCZ groups. No clear connection could be seen in the patients who had
higher eosinophils. Skin related AEs occurred independently of dose level (for skin cancer, see next
paragraph on malignancies).
41/55

Malignancies
Malignancies are increased in patients with RA and additionally with some treatments commonly used
in RA (MTX and DMARDs). The risk appears to be particularly higher for lymphoproliferative
malignancies, such as non-Hodgkin’s lymphoma and multiple myeloma, in RA patients compared
with the general population.
IL-6 is recognized as a potent growth factor in the pathogenesis of some forms of cancers; in non-
clinical TCZ pharmacology studies anti-proliferative effects were observed. However, IL-6 also is
claimed to have a role as a therapeutic anti-tumour agent. Thus, it remains to be seen what long-term
effects TCZ will have in tumorigenesis.
Overall the malignancies did not seem to be increased compared to the placebo groups. For solid
cancers it was slightly lower than the MTX group. Non-melanoma skin cancers in the TCZ groups
were 0.4/100pt-y – 0.71/100 pt-y (placebo 0.6/100 pt-y). In the overall clinical trials (including long-
term data) the rate of malignancies is 1.27/100 pt-y, this may indicate an increase. The 4/5 thyroid
neoplasms were shown to be benign in character (one patient was lost to follow-up).
The SPC text on malignancies refers to the pre-clinical data and now includes a section on the limited
clinical data to date. Long-term safety evaluations are ongoing.
Cardio/vascular disorders
In the monotherapy groups, 2 patients (0.7%) in the TCZ 8 mg/kg group had coronary events and none
were reported in the MTX group, in the combination therapy TCZ and placebo group rates were
similar (0.6% and 0.5%). In the long-term safety population, 14 patients had serious coronary ischemic
events. The rate of myocardial infarction in patients treated with TCZ during the Phase III studies was
0.35 per 100 patient years of exposure. The observed rate of myocardial infarction with TCZ is within
the rates reported in literature for myocardial infarction in patients with RA (0.4 – 0.7/100 pt-years).
Premature cardiovascular disease is a feature of extra-articular RA, leading to an increased risk for
myocardial infarctions, heart failure and cerebrovascular disease. In the studies, vascular disorders
(mainly hypertension) occurred with up to double the frequency in the TCZ group vs the control
groups. In addition¸ lipid parameters are increased in all TCZ groups, in some studies there is also an
increase in atherogenic indices. Viewed long-term, the RA population may thus be at an increased risk
for ischemic events (in addition to the underlying risk). As the 6 month data is relatively short, it is
momentarily difficult to come to a clear conclusion with regard to ischemic events. Post-authorisation
the applicant will be entering the treated patients into registries where data on this aspect will be
obtainable. The sponsor commits to track ischaemic events in registry studies and will submit annual
reports to the EMEA. The role of TCZ as a possible trigger to either a pre-existing cardiac/
cerebrovascular history or to a medication known to increase the risk of vascular events is difficult to
assess. In the updated Risk Management Plan the sponsor has initiated a dedicated study to assess the
effect of TCZ on surrogate markers of atherosclerosis.
Nervous system and psychiatric disorders
Nervous system disorders were experienced more frequently in the TCZ groups compared to the
control groups. The most common events were headache and dizziness. The rate of cerebrovascular
accident events in patients treated with TCZ during the Phase III studies was 0.26 per 100 patient
years of exposure. This is slightly lower than rates reported in the literature for the RA adult
population (~0.5 /100 pt-years).
Two events suggestive of central demyelination reported in the original submission were enrolled in
clinical trials WA17823 and WA18696 (extension of WA18062). After completion of the original
application dossier, the sponsor received a report of a case of leukoencephalopathy (patient 114002) in
open-label, long-term extension study MRA215JP in RA in Japan. The extensive work-up of the case
showed that a drug related encephalopathy could not be ruled out by exclusion. However, the PCR
testing for the JC virus in the spinal fluid was negative on three occasions, thereby making the
possibility of PML very unlikely.
42/55

Further investigation revealed three neurological events of interest: cranial neuropathy, abnormal
nuclear magnetic resonance imaging and peripheral demyelination. The applicant has modified the
RMP and the SPC to incorporate this potential adverse event
Although the monotherapy TCZ group shows slightly higher percentages of patients with insomnia,
anxiety and depression, the overall TCZ group does not show an increase compared to the placebo
DMARD group, thus an inclusion in the SPC is not necessarily warranted.
Eye disorders
Eye disorders were reported more frequently in the TCZ groups; the difference being mainly due to
conjunctivitis (2.75 in the all TCZ group vs. 1.77 in DMARD+ placebo/r 100 patient-years).
However, the confidence intervals for all TCZ groups overlapped with the placebo + DMARD group,
showing that there were no significant differences between TCZ and control. However, recognizing
the increased risk of infections in patients treated with TCZ, conjunctivitis has been included in the list
of adverse reactions terms in the SPC.
Swelling / edema
The diverse nature of oedema makes it difficult to assess. Although the TCZ 8 mg/kg + DMARD
group was higher compared to placebo + DMARD (5.84 vs. 3.75, respectively), the overlapping
confidence intervals do not allow to draw any precise conclusion.
The 6-month data for angioedema revealed 17/2644 (0.64%) cases in the TCZ group and 3/1170
(0.25%) in the placebo group. In the TCZ 8mg/kg groups (SPC recommended dose), there are 16/1870
(0.85%) and in the all-exposure TCZ population there are 33/3778 (0.87%) cases of angioedema,
indicating that there may be a trend. The sponsor has proposed to include the evaluation of risk of
angioedema as part of the evaluation of serious hypersensitivity reactions. This will encompass
reporting of angioedema events by means of guided questionnaires (see RMP; post-marketing reports)
and detailed evaluation of angioedema events in the ongoing clinical trials
Other biological agents and risks
Clinical experience both in RA and a number of other autoimmune diseases has been gathered with
other biological medicinal agents that block TNF activity. The main safety concerns include serious
infections (TB), infusion-related reactions, antibody development, worsening of cardiac disorders,
neurological disorders, blood disorders, and possible increased risk of malignancies. These safety
concerns remain after a patient has been switched to TCZ combination therapy.
Bone function
IL-6 stimulates osteoclast activity and bone resorption. By inhibiting this activity through exposure to
TCZ (preclinical data) normal bone function may possibly be maintained and thus not deemed to be a
risk factor. Nevertheless, fractures were addressed separately in the studies. In addition, approximately
50% of patients with fractures were taking oral steroids at baseline and approximately 60% of them
had a history of osteopenia/osteoporosis and the majority of those patients with a history were on
bisphosphonates.
Excipient:sucrose
Sucrose has been thought to cause or enhance the development of acute renal failure (ARF) when
administered in larger amounts e.g. in some sucrose-containing intravenous immunoglobulins (IVIG)
esp. in the presence of pre-existing renal insufficiency, creatinine > 1.5 mg/dl and concomitant
nephrotoxic drugs. The content of sucrose in the TCZ formulation is 50 mg/ml (or 2.5 mg sucrose/1
mg TCZ). Therefore administration of TCZ 8 mg/kg every 4 weeks corresponds to a sucrose uptake of
0.02 g/kg every 4 weeks. For sucrose-containing IVIGs the sucrose load would be considerably
higher, namely ~0.35 – 1.67 g/kg. Nevertheless it is not known precisely in patients with pre-existing
renal conditions what the minimal amount of sucrose is that might trigger further damage. In older
preclinical literature a single 0.05 mg sucrose infusion was sufficient to cause renal damage with
tubular swelling in healthy dogs. One 71 year-old patient died (in study WA18063) of
cardiopulmonary arrest secondary to end stage renal failure after having elevated renal function tests
throughout the study (9 TCZ infusions). He was diagnosed with an obstructive uropathy with
43/55

secondary renal insufficiency. The death was considered by the investigator to be unrelated to study
drug. However, the SPC should reflect that additional caution should be exercised in patients with pre-
existing renal conditions
Serious adverse event/deaths/other significant events
The general pattern of serious adverse events was similar in nature to the adverse events listed above.
The most common serious adverse events in all treatment groups were infections. Serious infections
were reported with a higher frequency in all TCZ 8 mg/kg groups compared with their respective
controls, and compared with the TCZ 4 mg/kg + MTX arm. Few infections led to withdrawal (< 1%),
and similar proportions of patients withdrew because of infections in each TCZ treatment group. Most
patients with serious infection temporarily interrupted or reduced dosing.
By the April 20th, 2007 cut-off date, a total of 26 deaths were reported in all studies conducted in the
RA indication, which enrolled 6315 patients. Twenty-one of these deaths were reported in the Roche
clinical trial programme, which had 4098 patients in the safety population In the 6 month controlled
clinical studies, the rate of deaths was similar in TCZ treated patients compared with control groups
(0.41 per 100 patient years exposure in the pooled TCZ groups vs 0.8 per 100 patient years exposure
in the placebo + DMARD group and 0.75 per 100 patient years exposure in the MTX group. The rate
of deaths did not increase in long term extension studies (0.42 per 100 patient years exposure).
In the core 24-week studies 5 deaths occurred in patients receiving treatment with the TCZ 8 mg/kg
dose, two of these (2/1582 [0.13%]) occurred in patients enrolled in trials comparing the 8 mg/kg and
4 mg/kg dose regimens and three occurred in trials evaluating the 8 mg/kg dose alone. Causes of death
in the TCZ patients were myocardial ischemia, cardiopulmonary arrest, stroke, postoperative infection,
GI haemorrhage. Five deaths occurred in the control groups during the 6-month double-blind period
due to pneumonia, Wegener’s granulomatosis, coronary artery thrombosis and intestinal obstruction in
the placebo + DMARD group and due to lung cancer in the MTX group.
An additional 11 patients died during treatment in the long-term safety analysis, 5 of them due to
infections. In the other patients the causes of death were myocardial infarction (2 patients), cancer,
suicide, progressive idiopathic neuropathy (one patient each), and in one patient the cause of death is
unknown.
Two additional patient deaths occurred following the cut-off date for reporting data in the clinical
safety summary (1x acute myocardial infarction 8 months after completion of the core study
(WA17822) while awaiting transition to the long-term extension study; 1x cardiopulmonary arrest
during the long-term extension study)
Laboratory findings
Neutrophils
Decreases in neutrophil counts without associated decreases in other haematology laboratory
parameters have been reported in animal studies. This was seen in the clinical studies in healthy
volunteers and in patients with RA and was felt to constitute a pharmacodynamic effect.
From the submitted data, decreases in neutrophil counts were recorded more frequently in the TCZ
groups mainly with shifts in CTC grade from normal to -1. These were observed after the first dose of
treatment and were sustained throughout the dosing interval. There was little to no clear temporal
association between low neutrophil counts and the occurrence of infections. In patients with
infections, approximately one-third of infection AEs were associated with an increase of neutrophils,
one-third with a decrease, and approximately one-third did not change. The pattern of these changes
was similar in both treatment groups. Severe decreases in neutrophil count to < 0.5 x 109/L were
infrequent (8 patients in the combination therapy studies and 4 patients in the long-term safety
analysis) and led to discontinuation of TCZ after which the neutrophil counts returned to pre-treatment
values. Neutropenia as an AE was not reported in the placebo or MTX groups, but in up to 1.4% in the
44/55

TCZ groups. There also seems to be an increased risk of neutropenia in patients who have previously
been treated with a TNF antagonist. This has been added to the SPC.
Platelets
A decrease in the formerly slightly elevated platelet count to within the normal range was observed in
all the TCZ groups with only isolated incidences of thrombocytopenia (as an AE it occurred in 0.1-
0.3% in the 6 month data). The SPC has been revised to include wording relating to monitoring of
platelets, as well as reference to the occurrence of rare cases of grade 3-4 thrombocytopenia.
Hepatic transaminase elevations
As there was no evidence of increases of hepatic transaminases in the pre-clinical toxicology studies
or in the healthy volunteers, the clinical data raise a new concern. In addition, in vitro studies
conducted with TCZ on human hepatoma cell lines have however not been able to clarify this clinical
finding.
The applicant suggested that these effects in humans are related to the combined effects of
concomitant medications (MTX, leflunomide, NSAIDs), as well as concomitant conditions (obesity,
diabetes, metabolic syndrome) and thus may influence liver function in patients with RA. However,
the exact cause is unclear and not fully elucidated.
In the studies elevations in liver function tests were more frequent in the TCZ groups compared to
placebo groups, but slightly less frequent in the monotherapy compared to MTX. The applicant
stresses that these elevations were single events; however, this could not be extracted from the data
which showed prevailing percentages of patients with increases through Week 24. In general these
elevations were managed by simple or no intervention. ALT + AST elevations were not associated
with simultaneous elevations of bilirubin. However, bilirubin alone was also more frequently
increased in TCZ patients compared to placebo patients. Patients were required to discontinue PP
when elevations reached > 5 ULN. In some cases there was evidence of steatosis. Only one biopsy
was performed showing a fatty liver.
The aspect of increases of hepatic enzymes is addressed in the SPC, as are the necessary dose
adjustments in case of laboratory abnormalities.
Lipid parameters
IL6 has also been implicated in regulating adipose mass. However, no effect of TCZ on lipid
parameters was seen healthy volunteers or in animal studies.
Patients with chronic inflammatory conditions, such as active RA, have lower lipid levels in
comparison with the general population and an increase in lipid parameters has been associated with
other biological agents such as anti-TNFs. An increase in lipids by TCZ may thus reflect the
pharmacodynamic effect of TCZ on suppression of inflammation in patients with RA. However, such
increases could pose a long-term risk for the development of atherosclerosis and subsequently of
cardiovascular/cerebrovascular incidents.
Elevations in lipid parameters and atherogenic indices were more frequent in patients on TCZ
compared to control groups. These elevations occurred early after initiation of treatment and stabilized
on continuation. They were associated with decreases in CRP and other acute phase proteins. There
did not seem to be a direct association with the occurrence of cardiovascular events, however, 6 month
data would not really suffice to assess this risk. In addition, under TCZ treatment hypertension and in
some cases cardiac events were increased. Thus, the long-term effect of increased lipids and
atherogenic indices in this chronically ill patient population remains to be seen. Elevations in total
cholesterol generally responded to treatment with lipid-lowering agents. This aspect is addressed in the
SPC.
Immunogenicity HAHAs)
Tocilizumab was produced by selecting a mouse anti-human IL-6R monoclonal antibody with the
most potent inhibitory activity in an in vivo nude mouse myeloma cell xenograft system.
45/55

Humanization was performed by grafting the Complementarity determining region (CDR) of the
mouse anti-human IL-6R monoclonal antibody onto a human IgG1 antibody framework, followed by
transfection of both light and heavy chain genes into CHO cells to produce a humanized antibody.
A total of 2876 patients have been tested for anti-tocilizumab antibodies in the controlled clinical
trials. Forty-six patients (1.6%) developed positive anti-tocilizumab antibodies, of whom 5 had an
associated medically significant hypersensitivity reaction leading to withdrawal. In 30 patients (1.1%)
who developed neutralizing antibodies, no apparent correlation to clinical response was observed.
Safety in special populations
Elderly: The only difference, i.e. higher frequency of serious infections in older patients does not seem
to be related directly to the TCZ treatment and may rather due to the disease- (RA) and age-related co-
morbidities.
Safety related to drug-drug interactions and other interactions
CYP-450
IL-6 has been shown to cause depression of major P-450 (CYP)-associated drug metabolism in
humans during inflammation, infection and possibly in other disease indications including cancer.
Thus, the possibility cannot be ruled out that treatment with tocilizumab may promote the metabolism
of CYP-metabolized drugs. This is reflected in the SPC.
Discontinuation due to adverse events
The withdrawal of patients due to safety reasons occurred infrequently and was more prominent in the
combination therapy (TCZ 8 mg/kg + DMARD) group. The most common types of AEs leading to
discontinuation were abnormalities of liver function tests and infections. There was no indication that
any particular types of events within any of the MedDRA SOCs led to an increase in withdrawals.
Post marketing experience
No post-marketing experience has been available at time of Marketing Authorisation.
2.5 Pharmacovigilance
Detailed description of the Pharmacovigilance system
The CHMP considered that the Pharmacovigilance system as described by the applicant fulfils the
legislative requirements.
Risk Management Plan
The MAA submitted a risk management plan, which included a risk minimisation plan.
46/55
Table 14: Summary of the risk management plan
Safety Concern Proposed Pharmacovigilance
Activities (Routine and
Additional)
Proposed Risk Minimisation
Activities (Routine and Additional)
Identified risks
Serious infections Routine pharmacovigilance
Special CRF for events of
special interest:
implemented in clinical
trials as of Q4 2007/Guided
Questionnaire (post-
marketing data)
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
Epidemiology data:
o US claims database
o EU registries (BSRBR,
ARTIS, RABBIT)
o US registry (NDB)
Routine risk minimization by means
of labelling
SmPC Section 4.3.
Contraindications
SmPC Section 4.4. Special
warnings and precautions for use/
Infections
SmPC Section 4.8. Undesirable
effects
Patient Information Leaflet,
Sections 2 and 4.
Additional risk minimization:
Educational programme
Patient Alert Card
Complications of
diverticulitis
Routine pharmacovigilance
Guided Questionnaire
(post-marketing data)
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
Epidemiology data:
US claims database
EU registries (BSRBR,
ARTIS, RABBIT)
US registry (NDB)
Routine risk minimization by means
of labelling:
SmPC Section 4.4. Special
warnings and precautions for
use
SmPC Section 4.8
Undesirable effects
Patient Information Leaflet,
Sections 2 and 4.
Additional risk minimization:
Educational programme
Patient Alert Card
Serious hypersensitivity
reactions
Routine pharmacovigilance
Guided Questionnaire
(post-marketing data)
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
Epidemiology data:
US claims database
EU registries (BSRBR,
ARTIS, RABBIT)
US registry (NDB)
Routine risk minimization by means
of labelling:
SmPC Section 4.4. Special
warnings and precautions for use
SmPC Section 4.8 Undesirable
effects
Patient Information Leaflet,
Sections 2 and 4.
Additional risk minimization:
Educational programme
Potential risks
Neutropenia Study to address
mechanism of neutrophil
reduction
Routine risk minimization by means
of labelling:
SmPC section 4.2. Posology and
47/55
Routine pharmacovigilance
Guided Questionnaire for
events of special interest
will collect neutrophil data
in cases of serious infection
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
method of administration
SmPC section 4.4. Special
warnings and precautions for use
SmPC Section 4.8 Undesirable
effects/Laboratory evaluations
Patient Information Leaflet,
Section 4.
See also Serious Infections above
Thrombocytopenia Routine pharmacovigilance
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
Routine risk minimization by means
of labelling:
SmPC section 4.2. Posology and
method of administration
SmPC section 4.4. Special
warnings and precautions for use
SmPC Section 4.8 Undesirable
effects/Laboratory evaluations
Elevated hepatic
transaminases
Routine pharmacovigilance
Guided Questionnaire
(post-marketing data) to
collect information on
serious hepatic events
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
Nature and frequency of
hepatic events representing
potential clinical
manifestations of increased
transaminase levels will be
monitored in the registry
studies:
o US claims database
o EU registries
(BSRBR, ARTIS,
RABBIT)
o US registry (NDB)
Routine risk minimization by means
of labelling:
SmPC section 4.2. Posology and
method of administration
SmPC section 4.4. Special
warnings and precautions for use
SmPC section 4.8. Undesirable
effects/Laboratory evaluations
Patient Information Leaflet,
Sections 2 and 4.
Immunogenicity Routine pharmacovigilance
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
Routine risk minimization by means
of labelling:
SmPC section 4.8. Undesirable
effects/Immunogenicity
Elevated lipids Study WA19923 evaluating
the effects of IL-6 receptor
blockade with tocilizumab
(TCZ) on lipids, arterial
stiffness, and markers of
atherogenic risk in patients
with moderate to severe
active RA
Routine risk minimization by means
of labelling:
SmPC section 4.2. Posology and
method of administration
SmPC section 4.4. Special
warnings and precautions for use
SmPC section 4.8. Undesirable
effects/Laboratory evaluations
48/55

Routine pharmacovigilance
Ongoing clinical trial
programme
Guided Questionnaires on
myocardial infarction/acute
coronary syndrome, stroke.
Regular review by Roche
Pharmacoepidemiology
Board
Rate of clinical events
potentially related to
atherogenesis (e.g. angina,
myocardial infarction,
cerebrovascular accident) as
a potential clinical
manifestation of increased
lipid levels will be
monitored in the registry
studies. The nature and rate
of such events will be
monitored and evaluated on
the basis of reports to the:
o Sponsor’s
pharmacovigilance
database
o US claims database
o EU registries (BSRBR,
ARTIS, RABBIT)
o US registry (NDB)
Patient Information Leaflet,
Sections 2 and 4.
Malignancies Routine pharmacovigilance
Guided Questionnaire
(post-marketing data)
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
Epidemiology data:
US claims database
EU registries (BSRBR,
ARTIS, RABBIT)
US registry (NDB)
Routine risk minimization by means
of labelling:
SmPC section 4.4. Special
warnings and precautions for use
SmPC section 4.8. Undesirable
effects
49/55

Demyelinating disorders Routine pharmacovigilance
Guided Questionnaire (post-
marketing data)
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
Epidemiology data:
US claims database
EU registries (BSRBR,
ARTIS, RABBIT)
US registry (NDB)
Routine risk minimization by means of
labelling:
SmPC section 4.4. Special
warnings and precautions for use
CYP450 enzyme
normalization
Routine pharmacovigilance
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
Routine risk minimization by means of
labelling:
SmPC section 4.5. Interaction with
other medicinal products and other
forms of interaction
Missing information
Elderly patients Routine pharmacovigilance
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
Epidemiology data:
US claims database
EU registries (BSRBR,
ARTIS, RABBIT)
US registry (NDB)
Routine risk minimization by means
of labelling
SmPC section 4.2. Special
populations/ Elderly Patients
Paediatric patients Routine pharmacovigilance
Regular review by Roche
Pharmacoepidemiology
Board
Off-label use managed
under compassionate use
programme
Additional studies on efficacy
and safety in paediatric patients:
Study WA18221 (SJIA)
Study WA19977(PJIA)
Routine risk minimization by means
of labelling:
SmPC Section 4.2. Special
Populations/ Paediatric Patients
Patient Information Leaflet,
Section 2.
Effects during pregnancy Routine pharmacovigilance
Ongoing clinical trial
programme
Regular review by Roche
Pharmacoepidemiology
Board
Registry studies with OTIS
Queries to ENTIS
Pregnancy data from
BSRBR and RABBIT
Routine risk minimization by means
of labelling:
SmPC section 4.6. Pregnancy and
lactation/Pregnancy
Patient Information Leaflet,
Section 2.
50/55
Hepatic impairment Routine pharmacovigilance
Regular review by Roche
Pharmacoepidemiology
Board
Routine risk minimization by means
of labelling:
SmPC section 4.2. Special
populations/Hepatic Impairment
SmPC section 5.2.
Pharmacokinetic properties/
Pharmacokinetics in Special
Populations
Patient Information Leaflet,
Section 2.
Renal impairment Routine pharmacovigilance
Regular review by Roche
Pharmacoepidemiology
Board
Routine risk minimization by means
of labelling
SmPC section 4.2. Special
populations/Renal Impairment
SmPC section 5.2.
Pharmacokinetic properties/
Pharmacokinetics in Special
Populations/Renal Impairment
Patient Information Leaflet,
Section 2.
Combination with
biologics
Routine pharmacovigilance
Regular review by Roche
Pharmacoepidemiology
Board
Epidemiology data:
US claims database
EU registries (BSRBR,
ARTIS, RABBIT)
US registry (NDB)
Routine risk minimization by means
of labelling:
SmPC section 4.4. Special
warnings and precautions for use
Patient Information Leaflet,
Section 2.
Vaccinations Routine pharmacovigilance
Regular review by Roche
Pharmacoepidemiology
Board
Substudy to be performed
Routine risk minimization by means
of labelling:
SmPC section 4.4. Special
warnings and precautions for use
/Vaccinations
Patient Information Leaflet,
Section 2.
The CHMP, having considered the data submitted in the MA application is of the opinion that the
following risk minimisation activities are necessary for the safe and effective use of the medicinal
product: see as detailed in section 2.3 of this CHMP Assessment Report.
2.6 Overall conclusions, risk/benefit assessment and recommendation
Quality
In general, the different aspects of the chemical, pharmaceutical and biological documentation comply
with existing guidelines. The fermentation and purification of the drug substance are adequately
described, controlled and validated. The drug substance is well characterised with regard to its
physicochemical and biological characteristics, using state-of the-art methods, and appropriate
specifications are set. The manufacturing process of the drug product has been satisfactorily described
and validated. The quality of the drug product is controlled by adequate test methods and
specifications. The viral safety and the safety concerning other adventitious agents including TSE have
been sufficiently assured. Except for a number of quality points, which will be addressed as part of
post-approval follow-up measures, the overall Quality of RoActemra is considered acceptable.
51/55
Non-clinical pharmacology and toxicology
Non-clinical data reveal no special hazard for humans based on conventional studies of safety
pharmacology, repeated dose toxicity and genotoxicity.
A study in animals has shown an increased risk of spontaneous abortion/embryo-foetal death at a high
dose. The potential risk for humans is unknown. Adequate labelling is provided in section 4.6 and 5.3
of SPC.
Efficacy
Efficacy has been shown in patients with moderate to severe RA that have an inadequate response to
one or more DMARDs including MTX, as well as patients that have an inadequate response or are
intolerant to anti-TNF medications. However, although superiority to MTX has been shown in patients
that were off MTX at the time of inclusion in the study the supplied data are not considered sufficient
to justify a “first line” indication. This is related to the fact that MTX first line therapy is generally
considered effective for the treatment of signs and symptoms as well as inhibition of progression of
structural damage, while there are no data for tocilizumab showing inhibition of progression of
structural damage in a “first line” setting.
For the justification of a “second line” indication after failure of DMARD and/or anti-TNF the
applicant has provided a meta-analysis of efficacy and safety that compares anti-TNF medications to
tocilizumab in patients that had failed on conventional DMARD. This analysis shows that ACR20
appears to be comparable, and ACR50 and ACR70 appear to be more likely with tocilizumab than
anti-TNF medications when using a 24 week endpoint. This data is considered supportive for the
“second line” indication.
Evaluation of the inhibition of progression of structural damage was a pre-defined endpoint in study
WA17823. The two co-primary 12 month endpoints included prevention of progression of joint
damage and improvement in physical function of TCZ therapy in combination with MTX versus MTX
alone (with confirmation at 24 months). The study met its two primary endpoints at week 52.
The doses of 4 mg/kg and 8 mg/kg were tested in the majority of studies and both show comparable
efficacy. However, patients dosed with 8 mg/kg had consistently higher response rates in the pivotal
trials. These clinical efficacy data are supported by pharmacodynamic investigations relating to
changes in CRP and haemoglobin giving further evidence for choosing the 8 mg/kg dose. From the
performed population PK no dose adjustments appear necessary for age, gender, race or renal
impairment. As no hepatic metabolism was noted no impact of hepatic impairment on PK is
anticipated, although not formally studied.
The design of the studies and the statistical analysis are considered adequate to determine safety and
efficacy in a population with moderate to severe RA. Inclusion and exclusion criteria of the pivotal
studies, the design of the studies as well as the obtained baseline characteristics are considered
adequate to obtain external validity.
Safety
The main risks with tocilizumab therapy are infections, GI disorders, infusion reactions, skin
disorders, neutropenia, elevation in hepatic enzymes and lipid parameters. Certain AE cannot be
reliably evaluated due to the relative scarcity of data, this relates to e.g. cardiovascular disorders and
malignancies.
Serious infections were more common under the study drug (placebo: 1.5% vs TCZ 2.1%). Within the
category of infections, respiratory infections, skin infections and GI were predominant. Systemic
infections were rare (3 events of sepsis) and only isolated cases of opportunistic infections were
observed. A higher infection rate was observed in patients that had previously received anti-TNF
medication.
52/55
Gastric disorders (mainly gastritis and upper GI ulcers) were observed more frequently with TCZ
combination treatment compared with the control. In 5 cases, the increased inflammation led to
perforations and in 2 cases to death. The rate of total GI perforations is 0.184 /100 pt-years which is
slightly higher than that reported in the literature for RA patients.
Infusion reactions occurred mainly during the first or second infusion. In the monotherapy groups, 6%
in the TCZ 8 mg/kg group and 2% in the MTX group had infusion reactions. In the combination
therapy groups ~5% in the TCZ groups and 3% in the placebo groups had infusion reactions. In the
long term safety population, these defined events were reported by 6.5% of patients. Infusion reactions
comprised mostly hypertension, rash and pruritus. Overall, 12 patients experienced medically
significant hypersensitivity reactions (six anaphylactic reactions). Although angioedema was seen
more frequently TCZ population 0.87%) than in the placebo groups (0.25%) this was not statistically
significant, but warrants further focused investigation in the patients’ registries post-authorisation.
Malignancies are increased in patients with RA and additionally with some treatments commonly used
in RA (MTX and DMARDs). In the overall clinical trials (including long-term data) the rate of
malignancies is 1.27/100 pt-y, it is currently unclear whether this could present an increase and the
SPC has been worded accordingly. The 4/5 thyroid neoplasms were shown to be benign in character
(one patient was lost to follow-up).
Premature cardiovascular disease is a feature of extra-articular RA, leading to an increased risk for
myocardial infarctions, heart failure and cerebrovascular disease. In the studies vascular disorders
(mainly hypertension) occurred with up to double the frequency in the TCZ group vs the control
groups. In addition¸ lipid parameters are increased in all TCZ groups, in some studies there is also an
increase in atherogenic indices. Viewed long-term the RA population may thus be at an increased risk
for ischemic events (in addition to the underlying risk). As the 6 month data is relatively short, it is
momentarily difficult to come to a clear conclusion with regard to ischemic events or the possibility of
TCZ triggering such events.
Decreases in neutrophil counts were recorded more frequently in the TCZ groups mainly with shifts in
CTC grade from normal to -1. These were observed after the first dose of treatment and were sustained
throughout the dosing interval. There was little to no clear temporal association between low
neutrophil counts and the occurrence of infections. Severe decreases in neutrophil count to < 0.5 x
109/L were infrequent (8 patients in the combination therapy studies and 4 patients in the long-term
safety analysis) and led to discontinuation of TCZ after which the neutrophil counts returned to pre-
treatment values. Neutropenia as an AE was not reported in the placebo or MTX groups, but in up to
1.4% in the TCZ groups. There also seems to be an increased risk of neutropenia in patients who have
previously been treated with a TNF antagonist.
To date the potential for central demyelination with TCZ is unknown. Initially 2 cases were described
as optic neuritis and chronic brain ischemia most likely secondary to polycythemia. After completion
of the original filing dossier, there was one report of drug-induced leukoencephalopathy (possibly
drug-related), one cranial neuropathy, one abnormal nuclear magnetic resonance imaging and one
peripheral demyelination.
In the studies elevations in liver function tests were more frequent in the TCZ groups compared to
placebo groups but slightly less frequent in the monotherapy compared to MTX. In general these
elevations were managed by simple or no intervention. ALT + AST elevations were not associated
with simultaneous elevations of bilirubin.
Having considered the safety concerns in the risk management plan, the CHMP considered that the
proposed activities described in section 3.5 adequately addressed these.
User consultation
The user testing report should be further developed to be in line with the stated FUM.
53/55

Risk-benefit assessment
Benefits:
The pivotal clinical studies have demonstrated a relevant benefit for patients with moderate to severe
active RA that have failed on MTX or other DMARDs or anti-TNF drugs. Although two doses were
tested in the clinical trials the higher dose of 8 mg/kg shows consistently better efficacy. This benefit
is demonstrated by ACR20, ACR50 and ACR70 responses that are clinically relevant, reproducible
and show consistent effects across all clinical studies. Consistent results were also demonstrated for
the secondary endpoints, e.g. individual ACR components, DAS28 and patient reported outcomes. The
design, the quality of conduct and the analysis of the clinical studies are considered sufficient to
demonstrate this benefit.
The benefit has been demonstrated on the basis of randomised, double-blinded, controlled 24-weeks
trials, maintenance of effect was demonstrated by open label extension trials that allow conclusions for
up to one year. Although superior results to MTX have also been shown in a clinical trial including
patients that were off MTX therapy at baseline and had shorter disease duration, a situation resembling
a first line setting, the current efficacy results are not considered sufficient to grant a marketing
authorisation in this population.
The applicant has provided evidence derived from a meta-analysis of efficacy [and safety] comparing
anti-TNF medications to tocilizumab. Although the strength of evidence derived from this meta-
analysis is considered inferior to results of a clinical trial, the conclusion that tocilizumab and anti-
TNF medications appear to have comparable efficacy is acknowledged.
One year radiological data of joint destruction from a dedicated phase III study confirm earlier results
in the Japanese population, namely that tocilizumab is effective at inhibiting the progression of
structural damage. Unfortunately a direct comparison of effects to other second line therapies is not
possible at present because of different scoring systems used.
Tocilizumab has been administered in combination with MTX in the pivotal studies except WA17824
thus the preferable therapy is combination therapy.
Risks:
As identified risks for patients treated with TCZ, Roche reported serious infections, serious
hypersensitivity reactions and GI perforation as complication of diverticulitis. Potential risks for
further evaluation were neutropenia, thrombocytopenia, liver enzyme elevation and their implications,
elevated lipid levels and their implication, especially with regard to cardiovascular and
cerebrovascular events, and immunogenicity. For all identified and potential risks, risk minimisation
activities have been described. Further to the identified and potential risk, risk identified for other
biological DMARDs such as malignancies, demyelinating disorders have been addressed in the RMP.
Regarding the effect of TCZ on IL-6 and the known suppression of CYP450 by IL-6, an influence on
concentration of drugs metabolized by CYP450 is anticipated.
Missing information for relevant populations as elderly, paediatric patients, pregnant women, patients
with impaired hepatic and also renal function as well as the combination with other biologics and
vaccination have been addressed, including the description of planned risk minimisation activities.
Beside the reported missing information of the combination with other biological DMARDs there
seems to be a tendency of increased frequency of severe neutropenia, when 7 of 8 cases of severe
neutropenia occurred in patients with previous treatment with anti-TNF drugs, but only a limited
number of patients treated with TCZ received previous anti-TNF treatment. Therefore concomitant
treatment with other biological DMARDs is not recommended and a corresponding warning statement
has been included in the SPC. Additionally recommendations for monitoring blood cell count during
treatment and also recommendations regarding further treatment with tocilizumab when neutrophil
count will decrease had been included in the SPC.
54/55

55/55
Regarding the occurrence of anti-TCZ antibodies and the limitation of the assay, which only measures
not bound antibodies, further investigations are necessary. Because of the limitation of the assay there
will be an underestimation of the incidence of patients experiencing anti-TCZ antibodies. Especially
those patients who experience hypersensitivity reactions or infusion reactions should be tested for anti-
TCZ antibodies. The risk for developing anti-TCZ antibodies at re-administration, when TCZ
treatment had been interrupted should be investigated. For further evaluation of anti-TCZ antibodies in
relation to hypersensitivity reactions and infusion reactions the Applicant had included corresponding
questions in the guided questionnaire (see Pharmacovigilance).
From the safety database all the adverse reactions reported in clinical trials have been included in the
Summary of Product Characteristics.
Balance:
The overall benefit-risk ratio for Roactemra is considered positive.
A risk management plan was submitted. The CHMP, having considered the data submitted, was of the
opinion that:
pharmacovigilance activities in addition to the use of routine pharmacovigilance were needed
to investigate further some of the safety concerns.
the following additional risk minimisation activities were required: see as detailed in section
2.3
Recommendation
Based on the CHMP review of data on quality, safety and efficacy, the CHMP considered by
consensus that the risk-benefit balance of RoActemra in the following indication:
RoActemra, in combination with methotrexate (MTX) is indicated for the treatment of moderate to
severe active rheumatoid arthritis (RA) in adult patients who have either responded inadequately to, or
who were intolerant to, previous therapy with one or more disease modifying anti-rheumatic drugs
(DMARDs) or tumour necrosis factor (TNF) antagonists. In these patients, RoActemra can be given as
monotherapy in case of intolerance to MTX or where continued treatment with MTX is inappropriate
was favourable and therefore recommended the granting of the marketing authorisation.
