Yang_Full_26 LDBB Zhao Yang
User Manual: LDBB
Open the PDF directly: View PDF ![]() .
.
Page Count: 79
PROCESSING OF THE DIBAL ADDUCT OF A PROLINE-DERIVED ESTER TO
GENERATE A SINGLE DIASTEREOMER OF AN ALLYL ALCOHOL FOR USE IN A
NOVEL SYNTHETIC METHOD FOR PYRROLIZIDINES
by
Yang Zhao
BS, Tsinghua University, 2002
Submitted to the Graduate Faculty of
University of Pittsburgh in partial fulfillment
of the requirements for the degree of
Master of Science
University of Pittsburgh
2005
ii
UNIVERSITY OF PITTSBURGH
FACULTY OF ARTS AND SCIENCES
This dissertation was presented
by
Yang Zhao
It was defended on
December 8th, 2005
and approved by
Toby M. Chapman
Scott G. Nelson
Theodore Cohen
Dissertation Director
iii
Advisor: Professor Theodore Cohen
PROCESSING OF THE DIBAL ADDUCT OF A PROLINE-DERIVED ESTER TO
GENERATE A SINGLE DIASTEREOMER OF AN ALLYL ALCOHOL FOR USE IN A
NOVEL SYNTHETIC METHOD FOR PYRROLIZIDINES
Yang Zhao, MS
University of Pittsburgh, 2005
Using the method of intramolecular carbolithiation in which the organolithium is generated
by reductive lithiation of a phenyl thioether, annulations on to pyrrolidine derivatives have been
accomplished to produce virtually enantiomerically and diastereomerically pure pyrrolizidines.
However, the main part of the thesis involves mechanistic and theoretical studies of the highly
diastereoselective process by which a key intermediate, (S,S)-2-pyrrolidinyl vinyl carbinol 6 used,
in the synthesis of a hydroxylated pyrrolizidine, is generated from N-Boc-L-proline methyl ester.
The process involves the treatment of this ester with DIBAL at -78 °C, warming to -20 °C,
cooling to -78 °C, and treatment with vinylmagnesium bromide. It was demonstrated that there
is virtually no stereoselectivity when the vinylmagnesium bromide is added to the corresponding
aldehyde in the presence of di-isobutylaluminum methoxide, the products expected if the
DIBAL-ester adduct decomposes before Grignard addition. Further evidence that an aldehyde is
not involved was obtained when it could not be detected by 1H NMR in the solution after warm-
up.
The theoretical study was designed to test a postulated mechanism in which a mixture of
diastereomeric adducts R1 and R2 of DIBAL and the ester, generated at -78 °C, undergoes
equilibration by reversible ionization of the methoxide ion when warmed and that the isomer R1
iv
greatly predominates at equilibrium. Both diastereomers are believed to involve a seven-
membered ring, afforded by coordination of the Al atom of the adduct with the carbonyl oxygen
atom of the Boc group, fused to the pyrrolidine. Reaction of the diastereomer R1 with
vinylmagnesium bromide via a SNi mechanism would yield the observed diastereomer of the
allylic alcohol.
Calculations do indeed predict that R1 is substantially more stable than its diastereomer R2
providing evidence for the mechanism. As a bonus, it has been discovered that the same high
stereoselectivity can be attained without raising the temperature by adding a catalytic amount of
the Lewis acid ZnCl2 at -78 °C; the Lewis acid probably aids the ionization of the methoxide ion
thus increasing the rate of equilibration, providing an additional piece of evidence for the
mechanism as well as simplifying the experimental procedure.
v
TABLE OF CONTENTS
PREFACE...................................................................................................................................... xi
1. INTRODUCTION .................................................................................................................. 1
2. PROCESSING OF THE DIBAL ADDUCT OF A PROLINE-DERIVED ESTER TO
GENERATE A SINGLE DIASTEREOMER OF AN ALLYL ALCOHOL ................................. 2
2.1 Introduction........................................................................................................................... 2
2.1.1. Previous work on the addition of Grignard reagents to DIBAL adducts of α-
aminoester derivatives to generate β-amino secondary alcohols diastereoselectively ........... 2
2.1.2. Mechanism study of DIBAL reduction followed by addition of organometallic to
generate β-amino secondary alcohols diastereoselectively..................................................... 7
2.2 Results and Discussions..................................................................................................................... 12
2.2.1. Wide application of advanced ester DIBAL reduction/alkylation with
organometallics..................................................................................................................... 12
2.2.2. Mechanism study on advanced DIBAL reduction................................................ 18
2.2.3. Improvement in the advanced ester reduction/alkylation method ........................ 30
2.3 Conclusions......................................................................................................................... 32
2.4 Experimental. ...................................................................................................................................... 34
3. ASYMMETRIC SYNTHESIS METHOD FOR NITROGEN HETEROCYCLES............. 40
3.1 Introduction. ........................................................................................................................................ 40
vi
3.1.1. Background for methods to produce organolithiums by intramolecular
carbolithiation ....................................................................................................................... 40
3.1.2. Lithium oxyanion effect in accelerating and exerting stereocontrol over
intramolecuar carbolithiation reactions................................................................................. 45
3.2 Results and Discussions.................................................................................................................... 48
3.3 Conclusions......................................................................................................................... 53
3.4 Experimental....................................................................................................................... 54
APPENDIX A............................................................................................................................... 59
B3LYP/6-31+G(d) Cartesian coordinates (Å) for optimized stationary points........................ 59
APPENDIX B ............................................................................................................................... 62
Certain O-H distances (Å) between the oxygen atom on the MeO group and the hydrogens (HA
and HB) on the 3 methylene group in H1, H2, M1, M2, R1 and
R2……………………………………………………………………………………….……..62
BIBLIOGRAPHY......................................................................................................................... 63
vii
LIST OF TABLES
Table 2.1 Total Energies of R1 and R2 calculated from different methods…………………….24
Table 2.2 Distances between Al atom and O atoms in R1 and R2 …………………………...25
Table 2.3 The energy and structure data of penta-coordinated structures for R1 or R2 after
optimization………………………………………………………………………………….27
Table 2.4 Free energies of the optimized M, H and R by B3LYP/6-31+G*………………...….30
Table 2.5 Distances between Al atom and O atoms in M, H and R by B3LYP/6-31+G*…...….30
viii
LIST OF FIGURES
Figure 2.1 Several proposed transition states for stereoselective additions to protected amino
aldehydes………………………………………………………………………………… .….9
Figure 2.2 Mechanistic hypotheses by Polt…………………………………………...………....11
Figure 2.3 NMR spectra for diastereomers 6 and 10…………………………………………….15
Figure 2.4 Predicted mechanism for the advanced ester reduction/alkylation of 5……………...22
Figure 2.5 Crystal structure data of tetra-coordinated aluminium compounds………………….26
Figure 2.6 R1 and R2 optimized by B3LYP/6-31+G*………………………………………….26
Figure 2.7 Structures of H (H1 or H2), M (M1 or M2) and R (R1 or R2)………………….….28
Figure 2.8 M (M1 or M2) and H (H1 or H2) optimized by B3LYP/6-31+G*………………….29
Figure 3.1 Radical anion reducing agents………………………………………………………..44
ix
LIST OF SCHEMES
Scheme 1.1 Synthetic route of compound 4…………………………………………….……..….1
Scheme 1.2 Synthetic route of compound 8 or 9…………………………………………….…....1
Scheme 2.1 Taguchi’s reduction/alkylation of N-Boc-L-proline methyl ester 5………….……....3
Scheme 2.2 Modified reduction/alkylation of N-Boc-L-proline methyl ester 5……….…….……3
Scheme 2.3 Advanced ester reduction/alkylation of Boc-(S)- methylalaninate 11 and addition of
vinylmagnisium chloride to Boc-(S)-alaninal 14 by Ibuka, Fujii and Yamamoto………….....5
Scheme 2.4 Advanced ester reduction/alkylation of N-Boc protected amino acid methyl esters
(15 and 18) and addition of vinylmagnisium chloride to N-Boc protected aminoaldehyde (21
and 24) by Angle……………………………………………………………………………...6
Scheme 2.5 DIBAL-reduction/alkylation of Schiff base esters to phenylpropanolamines...……..7
Scheme 2.6 Addition of different Grignard reagents to aldehydes…………………………….…9
Scheme 2.7 Cram chelate model for initial hydride delivery to the ester…………………….…10
Scheme 2.8 Synthesis of N-Boc-L-proline methyl ester 5………………………………………12
Scheme 2.9 Advanced ester reduction/alkylation of N-Boc-L-proline methyl ester 5 by
vinylmagnesiumbromide………………………………………………………………..…...13
Scheme 2.10 Advanced ester reduction of N-Boc-L-proline methyl ester 5 followed by
organometallic addition……………………………………………………………………...16
Scheme 2.11 Stereochemical assignments for amino alcohols………………………………….17
Scheme 2.12 Literature synthesis of 32 from β- hydroxy sulfoxide 41………………………….18
x
Scheme 2.13 Mechanistic study on the advanced ester reduction/alkylation……………………21
Scheme 2.14 Advanced ester reduction/alkylation with Lewis acid catalyzed equilibration....…32
Scheme 3.1 Intramolecular carbolithiation by halogen-lithium exchange………………………41
Scheme 3.2 Mechanism of iodide-lithium exchange……………………………………………42
Scheme 3.3 Bailey’s cyclization of a secondary alkyllithium…………………………..………42
Scheme 3.4 Tin-lithium exchange in intramolecular carbolithiation……………………………43
Scheme 3.5 Selenium-lithium exchange…………………………………………………………43
Scheme 3.6 Mechanism of reductive lithiation…………………………………………..………44
Scheme 3.7 Examples of earlier intramolecular carbolithiations by reductive lithiation…..……45
Scheme 3.8 Intramolecuar carbolithianion reactions with a tertiary organolithium...………...…46
Scheme 3.9 Intramolecuar carbolithianion reactions with oxyanionic groups...………………...46
Scheme 3.10 Intramolecuar carbolithianion reaction with an oxyanionic group exo to the ring..47
Scheme 3.11 Intramolecuar carbolithianion reaction with a homo allylic oxyanionic group…...47
Scheme 3.12 Procedure to synthesize compound 1 through Beak’s method……………………48
Scheme 3.13 Asymmetric deprotonation of N-Boc-pyrrolidine 50…………………………...…49
Scheme 3.14 Asymmetric synthesis for pyrrolizidine 5……………………………..……..……49
Scheme 3.15 Synthesis of 1-(phenylthiomethyl)pyrrolidine through SN2 reaction…………..…50
Scheme 3.16 Intramolecular cabanionic cyclization………………………………………….…51
Scheme 3.17 Asymmetric synthesis for pyrrolizidine 10……………………………………..…51
Scheme 3.18 Unsuccessful methods to obtain Compound 57…………………………………...53
xi
PREFACE
I wish to express sincere gratitude to my advisor Dr. Cohen for his guidance, inspiration, and
encouragement throughout my graduate program. This work could not have been done without
Dr. Cohen expert advice and strong support. I also wish to warmly thank Dr. Cohen for his
understanding and help on my personal issues.
My sincere appreciation extends to my graduate committee members: Dr. Chapman and Dr.
Nelson for critical review of my thesis and their invaluable assistance. I am grateful to Dr.
Jordan for his help during the computation calculation.
My special thanks also go to my labmates, my friends and family who have supported me
throughout my research.
Finally, this work is in memorial of Xueying Shan, my always beloved mom, who passed
away last year in her early fifties. My mom is always the support of my life.
1
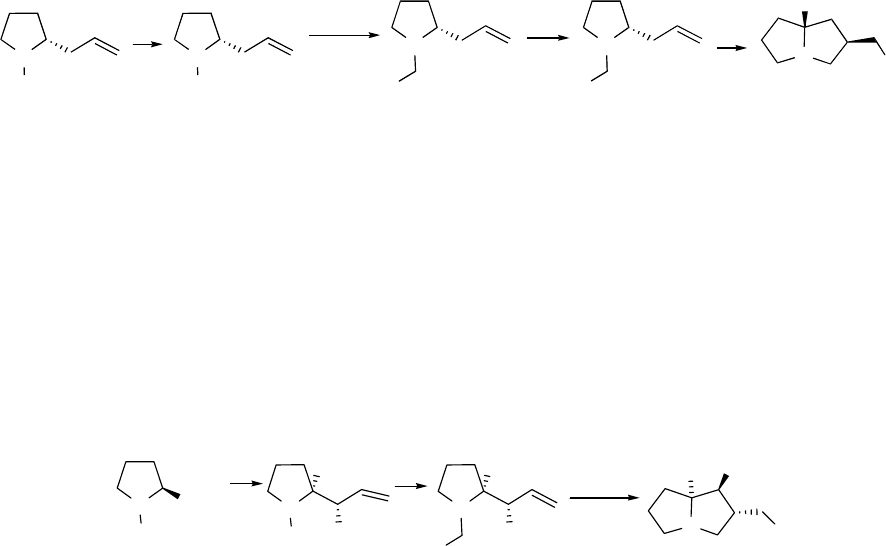
1. INTRODUCTION
The original goal of this research was the development of a new method of preparation of
pyrrolizidines utilizing cyclization by intramolecular carbolithiation whereby the organolithium
is prepared by reductive lithiation of phenyl thioethers by aromatic radical-anions.1 For example
in Scheme 1.1, the known compound 1 could be deprotected and converted to 2 which, upon
reductive lithiation with the aromatic radical-anion lithium 4,4’-di-tert-butylbiphenylide (LDBB),
would yield the organolithium 3 that would be expected to cyclize to 4; the background for such
a reaction scheme is given in Chapter 3.
N
Boc 1
N
H
PhSH
(CH2O)nN
PhS
LDBB
N
Li
N
H
Li
23 4
Scheme 1.1 Synthetic route of compound 4
The more functionalized pyrrolizidine 8 could arise from similar processing of the known
allyl alcohol 6, generated from the protected proline ester 5 by treatment with
diisobutylaluminum hydride (DIBAL) at -78 °C, warming the adduct to -20 °C and adding
vinylmagnesium bromide (see Scheme 1.2).2 A study of this type of stereoselective conversion
of 5 to 6 is discussed in Chapter 2 while the cyclization of 7 is discussed in Chapter 3.
N
Boc OH 6
H
N
Boc
COOMe
5
N
PhS OH
H
7
1. BuLi
2. LDBB N
HOLi
Li
8
Scheme 1.2 Synthetic route for compound 8
2
2. PROCESSING OF THE DIBAL ADDUCT OF A PROLINE-DERIVED ESTER
TO GENERATE A SINGLE DIASTEREOMER OF AN ALLYL ALCOHOL
2.1. Introduction
2.1.1. Previous work on the addition of Grignard reagents to DIBAL adducts of α-
aminoester derivatives to generate β-amino secondary alcohols diastereoselectively
As mentioned in Chapter 1, we required vinyl 2-pyrrolidinyl alcohol 6 for our projected
synthesis of pyrrolizidines. Taguchi2 has reported the addition of vinylmagnesium bromide 9 to
the DIBAL adduct of N-Boc-L-proline methyl ester with some diastereoselectivity. In his
experiment, 5 was treated with DIBAL at -78 oC followed by a warm-up step to -23 oC before
addition of the Grignard reagent at -78 oC as shown in Scheme 2.1. He obtained the β-amino
secondary alcohol 6 (see Scheme 2.1) and its diastereomer in 83% yield and 5:1 diastereomer
ratio as determined by the MTPA method. According to this method, the diastereomers were
converted into the S- and R- 2-methoxy-2-trifluoromethylphenylacetic acid (MTPA) esters,
which had different chemical shifts.2
3
N
Boc
OCH
3
H
O
(S)
N
Boc
H
5
83% 5:1
OH
MgBr
1) DIBAL, CH2Cl2/Et2O, -78 oC, 30 min
2) -23 oC, 1 h
3) , -78 oC to rt
4) sat. NH4Cl solution
6 and 10
N
Boc
H
HOH
(S)
(S)
N
Boc
H
HOH
(S)
(R)
610
9
Scheme 2.1 Taguchi’s reduction/alkylation of N-Boc-L-proline methyl ester 5
In our study, a similar reaction as shown in Scheme 2.2 was performed with a change in
solvent and a minor change in temperature of the warm-up. We obtained one diastereomeric
protected β-amino secondary alcohol 6 in 62% yield. The stereochemical assignment for
secondary alcohol 6 is based on the NMR data of the two known diastereomers.3 The purified
diastereomer ratio of 6 to 10 was found to be greater than 32 to 1 by NMR analysis of the crude
product. This ratio is also consistent with the gas chromatographic (GC) analysis (see
Experimental section) .
N
Boc
OCH
3
H
O
(S)
N
Boc
H
HOH
(S)
(S)
MgBr
1) DIBAL, CH
2
Cl
2
, -78
o
C, 30 min
2) -20
o
C, 1 h
3) , -78
o
to rt
4) sat. NH
4
Cl solution
56
9
Scheme 2.2 Modified reduction/alkylation of N-Boc-L-proline methyl ester 5
This high diastereoselectivity was very attractive. A literature search revealed that Taguchi
was not the first to use the addition of an organometallic to the DIBAL adduct of an amino ester
derivative to generate a β-amino secondary alcohol. The earliest work on this method was
reported by Ibuka, Fujii, Yamamoto and co-workers.4
4
These authors observed a dramatically increased diastereoselectivity (29:2), as comparing to
that 7:3 obtained from the reaction of the Grignard reagent to the corresponding aldehyde as
starting material when t-Boc-protected methyl alaninate was treated sequentially with DIBAL
and vinylmagnesium chloride (Scheme 2.3). In their report, it is notable that they creatively
added a warm-up step from -78 oC to -20 oC before Grignard reagent treatment at -78 oC. This
makes their method different from the previously used DIBAL reduction methods of derivatives
of esters of α-amino acids when addition of DIBAL is directly followed by addition of the
Grignard reagent without any warm-up.5 To simplify the later discussions, we call the method
with a warm-up step after the addition of DIABL and before the addition of Grignard reagent
“the advanced ester reduction/alkylation”.
In their experiment (Scheme 2.3), N-Boc-(S)-methylalaninate 11 was first reduced by
DIBAL at -78 oC and the reaction mixture was then warmed to -20 oC for 30 min. It was then re-
cooled to -78 oC before the addition of vinylmagnesium chloride. This experimental procedure
gave excellent diastereoselectivity. The diastereomer ratio was 29:2 for syn 12 and anti 13 allyl
alcohols in 60% combined yield. This ratio is superior to that obtained from the reaction of Boc-
(S)-alaninal 14 with vinylmagnesium bromide (THF, -70 oC to 0 oC). The latter gave a mixture
(7:3) of syn and anti allyl alcohols in 53% combined yield.
5
CHOMe
NHBoc
COOMeMe
NHBoc
Me
NHBoc
OH
Me
NHBoc
OH
-70
o
C to 0
o
C
MgBr , THF +
53% 7:3
1) DIBAL, CH
2
Cl
2
, -78
o
C to -20
o
C
MgCl, -78
o
C to 0
o
C
2)
Me
NHBoc
OH
Me
NHBoc
OH
+
60% 29:2
11 12 13
12 13
14
Scheme 2.3 Advanced ester reduction/alkylation of Boc-(S)- methylalaninate 11 and addition of
vinylmagnesium bromide to Boc-(S)-alaninal 14 by Ibuka, Fujii and Yamamoto4
Angle later also achieved high diastereoselectivity when he applied the advanced ester
reduction/alkylation in the synthesis of β-amino secondary alcohols after he did not obtain ideal
selectivity using aldehydes as starting material.6 Illustrated in the upper panel of Scheme 2.4 are
two reactions starting from the aldehydes 15 and 18. The desired amino alcohols 16 and 19 are
the products of a chelation-controlled (cyclic Cram) addition to the aldehyde. The mechanism of
the chelated transition state will be discussed in detail in section 2.1.2 (see (c) in Figure 2.1,
R=CH3). This reaction gave allyl alcohols in 62% yield as a 3:l mixture of syn/anti
diastereomers 16 and 17 when R=CH3 and in 78% yield as a 7:l mixture of syn/anti
diastereomers 19 and 20 when R=CH2Ph. However, when the advanced ester
reduction/alkylation is used, they observed an enhancement in the stereoselectivity during the
transformation of N-Boc-alanine methyl ester to amino alcohol in a one pot reaction upon the
sequential addition of DIBAL and vinylmagnesium chloride. The advanced ester
reduction/alkylation afforded allyl alcohol products in 59% yield as an 8:1 mixture of
diastereomers 22 and 23 when R=CH3 and in 31% yield of alcohol 25 as a single diastereomer
6
when R=CH(CH)3. The excellent selectivity makes this one-pot procedure the method of choice
for selectively preparing amino alcohols.
RH
N
O
BocH
R
NBocH
OH
R
NBocH
OH
EtO
2
, -20
o
C to 0
o
C
MgBr , ZnCl
2
+
62% 3:1 R=CH
3
78% 7:1 R=CH
2
Ph
ROMe
N
O
BocH
R
NBocH
OH
R
NBocH
OH
1) DIBAL, CH
2
Cl
2
, -70
o
C to -20
o
C
MgCl
+
59% 8:1 R=CH
3
31% >15:1 R=CH(CH)
3
, -70
o
C to 0
o
C
2)
15
18 16
19 17
20
21
24 22
25 23
26
Scheme 2.4 Addition of vinylmagnesium chloride to N-Boc protected aminoaldehyde (15
and 18) and advanced ester reduction/alkylation of N-Boc protected amino acid methyl esters (21
and 24) by Angle
In summary, there are several ways starting from α-amino acid derivatives to make α-amino
secondary alcohols diastereoselectively.
i) In one of the methods, where an α-amino acid is utilized as a source of chirality, a
suitably protected amino acid ester is first converted to its corresponding aldehyde. The
optically active protected aminoaldehyde then reacts with various carbon nucleophiles. This
method is straightforward and is of potential synthetic value.4,7-25 However, it usually suffers
from configurational instability (enolization) under a range of reaction conditions, and the
stereoselectivities in these reactions are often not ideal.5,26-34
ii) By virtue of the stability of α-aminoesters to epimerization, D-esters are better starting
materials for syntheses of β-amino secondary alcohols than aldehyde. The method of DIBAL
reduction followed by alkylation of the α-aminoesters to β-amino secondary alcohols is

7
straightforward. The DIBAL reduction/alkylation method by treating some chiral protected α-
amino esters with DIBAL and Grignard reagent sequentially without the warm-up step can give
good selectivity under some circumstances.35-39 As illustrated in Scheme 2.5, Polt40 observed a
high threo- α-amino secondary alcohol yield (73-85%) and excellent “syn” stereoselectivity (8:1
to 11:1, threo or like product preferred) in the experiment when he treated optically pure imine-
protected amino esters with DIBAL or DIBAL/TIBAL(i-Bu3Al), followed by RMgX or RLi.
OR
N
Me
O
Ph2C
1) 1 equiv DIBAL/TIBAL, CH2Cl2, -78 oC
2) 3 equiv PhMgBr, Et2O, -78 oC to 0 oC
3) H3O+
Ph
Me
NH2
OH
Ph
Me
NH2
OH
+
R=Me 7.6:1
R=Et 8.8:1
R=CH2Ph 6.3:1
R=CHPh2 10.7:1
R=tBu 11.0:1
~80%
threo- erythro-
Scheme 2.5 DIBAL-reduction/alkylation of Schiff base esters to phenylpropanolamines
iii) The advanced ester reduction/alkylation involves a sequential treatment of N-Boc-α-
amino esters with DIBAL and, after a warm-up step, Grignard reagents, as demonstrated in
Schemes 2.1 and 2.2 mentioned above. The advanced ester reduction/alkylation method gives
higher selectivity than that from treating the aldehyde with a Grignard reagent or not employing
the warm-up period.
2.1.2. Mechanism study of DIBAL reduction followed by addition of organometallic to
generate β-amino secondary alcohols diastereoselectively
In brief, there have been mainly three methods to synthesize α-amino secondary alcohols
diastereoselectively from α-amino acid derivatives. The first method of reacting the
8
aminoaldehyde with carbon nucleophiles has been extensively studied and its mechanism has
been well established. The mechanism of the second method, the sequential addition of hydride
and C-nucleophile has been studied without much success. The third method, as described above,
which is the advanced ester reduction/alkylation method to synthesize β-amino secondary
alcohols from α-amino esters greatly increases the stereoselectivity. Thus, it is an ideal method
to synthesize optically pure amino alcohols. However, to date, no mechanistic explanation has
been provided for this high stereoselectivity. Thus, in the following section of this Chapter, the
reported mechanistic studies for the first two methods will be summarized.
Many researchers had made efforts to elucidate the mechanism(s) leading to
diastereoselectivity in the method of synthesizing β-amino secondary alcohols from α-
aminoaldehydes through addition of organometallic reagents. As Duhamel demonstrated in his
work with racemic N,N-dialkyl-α-amino aldehydes, a Felkin-Ahn-type transition state, as shown
in Figure 2.1 (a), can explain the erythro products41,42 that are formed; it is believed that there is
steric interference by the bulky benzyl groups with the chelating-ability of the nitrogen lone pair.
When smaller groups are attached to nitrogen (e.g. N,N-dimethyl substitution), chelation is
allowed.43 However, removal of the protection from nitrogen poses a problem here when using
groups such as methyl. Fortunately, with the efforts of many researchers in this field, several
solutions have been provided for this problem. Reetz "tied back" the benzyl groups to favor the
chelated transition state as shown in Figure 2.1 (b). In this transition state, the benzylic
protection could be easily removed in the downstream reactions.21 It has been reported by
several other groups4,13,15 that acyl-protected amines can provide an anionic chelated transition
state as shown in Figure 2.1 (c), when the N-H proton is removed to generate an anionic nitrogen.

9
H
N
R
O
H
M+
Nu-
(b)
H
N
R
O
H
M+
Nu-
(c)
O
O
Bu
(a)
N
R
H
H
O
Ph
Ph
Nu-
t
Figure 2.1 Several proposed transition states for stereoselective additions to protected amino
aldehydes
As demonstrated in Scheme 2.6, a minor modification in the reaction conditions can
dramatically change the course of this reaction. With a small reaction condition change (e.g.
H2C=CHMgC1 vs H2C=CHMgBr), deprotonation can generate a chelating substrate from a
substrate which normally undergoes Felkin-Ahn addition.41-43 Thus, the relative rate of
deprotonation vs addition becomes extremely important in these reactions. With the chelated
transition state mechanism described previously, this phenomenon becomes readily
understandable. H2C=CHMgBr can deprotonate the nitrogen atom more efficiently than
H2C=CHMgC1 and form a better chelated transition state.
RH
N
O
BocH
R
NBocH
OH
R
NBocH
OH
EtO
2
, -20
o
C to 0
o
C
MgBr , ZnCl
2
+
62% 3:1 R=CH
3
78% 7:1 R=CH
2
Ph
RH
N
O
BocH
R
NBocH
OH
R
NBocH
OH
EtO
2
, -20
o
C to 0
o
C
MgCl , ZnCl
2
+
R=CH
3
R=CH
2
Ph
very low yield
Scheme 2.6 Addition of different Grignard reagents to aldehydes
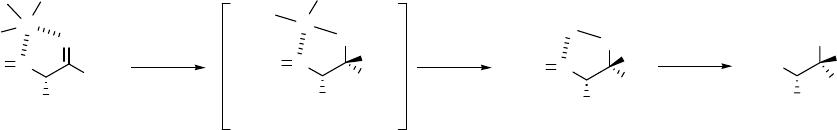
10
Scheme 2.7 and Figure 2.2 are directly adapted from Polt’s paper. The discussion is also
based on his paper in which his explanation didn’t fit his experimental data. We also have many
questions concerning on his explanation.
To date, there is no universally accepted mechanistic interpretation for the results of the
sequential addition of hydride and C-nucleophiles to the protected α-amino ester. Some
researchers believe that the observed threo selectivity is due to an aluminum-chelated N-t-Boc-
amino aldehyde (Cram cyclic transition state) as the intermediate. As shown in Scheme 2.7, Polt
attempted to interpret the stereoselectivity for his reactions in Scheme 2.5 by invoking the cyclic
Cram chelate model, with tri-sec-butylaluminum behaving as a chelation agent, for initial
hydride delivery to the ester, followed by subsequent inversion of configuration in the
displacement of the methoxide ion by the incoming nucleophile.
OMe
N
R
O
Ph2C
Al
OMe
N
R
O
Ph2C
Al
H-Ph-
H
N
R
O
Ph2C
+M
Ph H3O+
H
H2N
R
OH
Ph
H
Scheme 2.7 Cram chelate model for initial hydride delivery to the ester
In his report, Polt postulated that Schiff base esters permit the chelation controlled addition
of hydride (transition state (a) in Figure 2.2) at low temperature. He believes that a portion of the
desired threo products arises from transition state (b) after the methoxide ion has been lost (SN1-
like pathway), although some products may arise from transition state (c) (SN2-like pathway after
delivery of the hydride ion). Currently, it is not possible to either confirm or deny the possibility
that the aluminoxy acetals can exist as tight ion-pairs (d) or (e) (Figure 2.2) based on the
available data. The conversion from an SN2-like to an SN1-like mechanism may not account for
the decrease in diastereoselectivity observed with coordinating solvents. This decrease may be
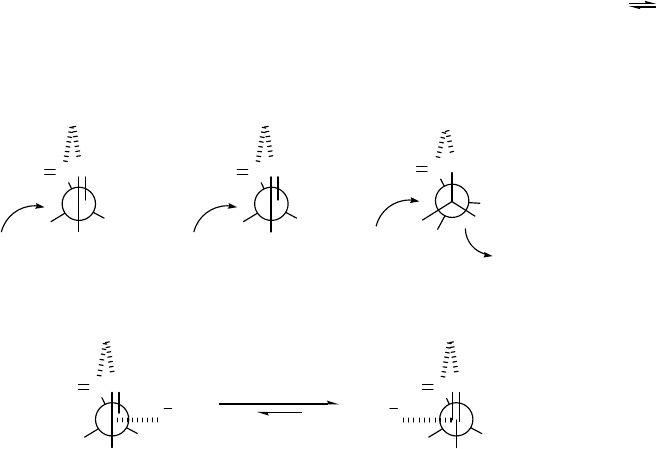
11
due to increased "leakage” between the two structures (d) and (e). If there was an equilibration
between the two ion-pairs, eclipsing interactions between the R group and the R'O group should
favor of structure (e). As presented in Scheme 2.5, this may not be the case; that is to say,
configurational equilibration (inversion) of the aluminoxy acetal via ion-pair rearrangement may
not be a major pathway, at least under the condition of low temperature and absence of THF.
The parallel increase in stereoselectivity with steric bulk of the ester in Scheme 2.5 is in the
opposite direction to what one would expect from the ion-pair rearrangement. With the ion-pair
rearrangement, one would expect decreased selectivity because the equilibrium (d) (e)
should shift to the right as the steric bulk of the R'O group increases.
.
H
N
R
O
OR'
M
+
Ph
2
C
H
-
(a)
H
N
R
O
H
M
+
Ph
2
C
Nu
-
(b)
H
N
R
O
OR'
H
M
+
Ph
2
C
Nu
-
(c)
H
N
R
O
H
M
+
Ph
2
C
OR'
H
N
R
O
H
M
+
Ph
2
C
R'O
(d) (e)
Figure 2.2 Mechanistic hypotheses by Polt
12
2.2. Results and Discussion
2.2.1. Wide application of advanced ester DIBAL reduction/alkylation with
organometallics
As mentioned above, the advanced ester reduction/alkylation method, with a warm-up step,
provides the significant advantage of high stereoselectivity, for the DIBAL reduction and
subsequent Grignard reaction using protected α-amino esters as starting material. To date, there
has been no systematic study on this method and its mechanism has not been elucidated. Thus,
further study on this advanced ester reduction/alkylation method, with a warm-up step, is a
worthwhile project in the development of procedures to make optically pure amino acids with
high diastereoselectivity.
The requisite N-Boc-L-proline methyl ester 5 was readily prepared in good yield by known
methods from L-proline 27 (Scheme 2.8). Briefly, L-proline 27 was first esterified with
methanol via the acid chloride to give the corresponding methyl ester as the hydrochloride. The
salt of the methyl ester 28 was then neutralized, followed by the treatment of the resulting amine
with di-tert-butyl dicarbonate. This method afforded N-Boc-L-proline methyl ester 5 in 85%
yield over two steps after purification by flash chromatography.44 The optical rotation of the
product 5 agreed with the reported values for optically pure N-Boc-L-proline methyl ester 5.45-47
The optical purity establishes that there was no racemization during this synthesis.
N
H
COOH
H
(S)
N
H2
COOMe
H
(S)
N
Boc
OCH3
H
O
(S)
SOCl2, MeOH (Boc)2O, NEt3
CH2Cl2
Cl-
585%
27 28
Scheme 2.8 Synthesis of N-Boc-L-proline methyl ester 5
13
N-Boc-L-proline methyl ester 5 was used as the substrate for the advanced DIBAL ester
reduction. It is very interesting and exciting that only a single distereomer was observed. As
shown in Scheme 2.9, N-Boc-L-proline methyl ester 5 was first reduced by DIBAL at -78 oC and
the reaction mixture was then warmed to -20 oC for 1 h. The reaction mixture was then re-
cooled to -78 oC before the addition of vinylmagnesium bromide. This experimental procedure
gave secondary alcohol 6 in approximately 80% yield. The by-products mainly include i) the
over-reduced primary alcohol 29, which is the common side-product of DIBAL reduction of
esters, and ii) the tertiary alcohol 30 which is generated from reaction of one molecule of the
ester 5 and two molecules of the Grignard reagent.
N
Boc
OCH
3
H
O
(S)
N
Boc
H
HOH
(S)
(S)
MgBr
1) DIBAL, CH
2
Cl
2
, -78
o
C, 30 min
2) -20
o
C, 1 h
3) , -78
o
C to rt
4) sat. NH
4
Cl solution
56
N
Boc
OH
H
N
Boc
H
OH
by-products
29 30
Scheme 2.9 Advanced ester reduction/alkylation of N-Boc-L-proline methyl ester 5 by
vinylmagnesium bromide
The stereochemical assignment for secondary alcohol 6 is based on the NMR data of the
two known diastereomers.3 In fact, it is simple to determine the stereochemistry of the reaction
products by comparison of their NMR spectra. As shown in Figure 2.3, (a) is the NMR spectrum
of the product from the reaction in Scheme 2.9,and (b) represents the NMR spectrum of a
mixture of two diastereomers 6 and 10 which are produced from other reactions that will be
discussed in the later chapter. In (b), there are four peaks at around 4 ppm. Based on the known
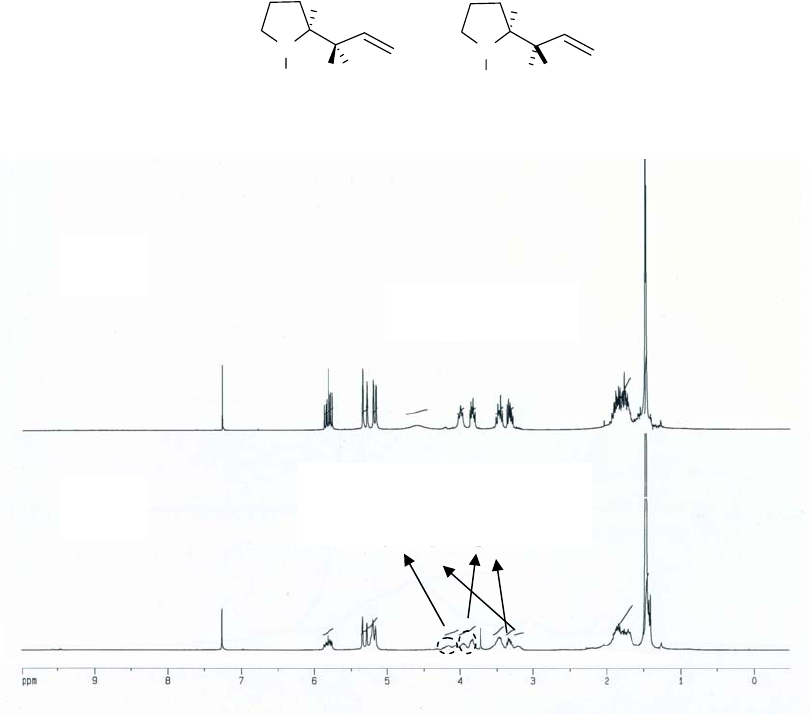
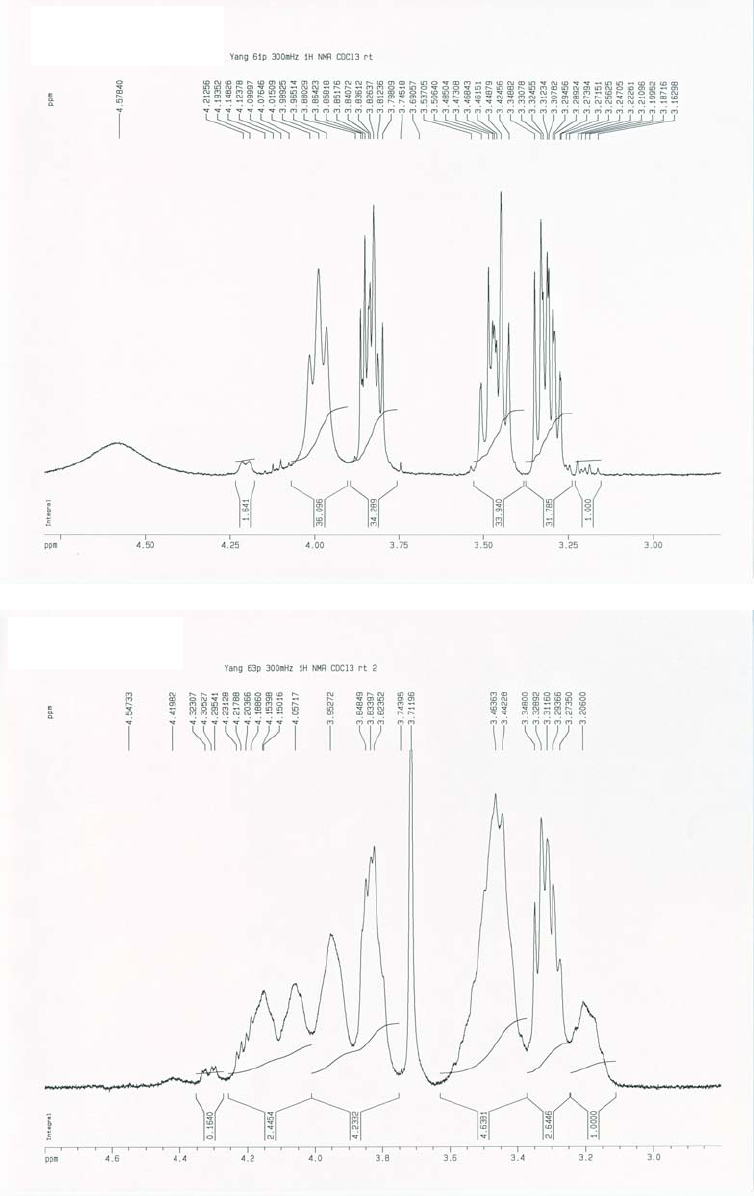
14
NMR data of diastereomers 6 and 10, we can assign the left two peaks, at lower field than 4 ppm,
to 10, and the right two peaks, at higher field than 4 ppm, to 6. Another area in the spectrum that
we can use to distinguish 6 and 10 is that between 3.0-3.5 ppm. In this range of the spectrum,
the two marked peaks belong to 6 and 10, respectively. It is obvious from comparison of the
expanded spectra (c) and (d) in the vicinity of 2.40-4.80 ppm of (a) and (b), respectively, that the
diastereomers 6 and 10 are present in a ratio of at least 32:1.
N
Boc
H
HOH
(S)
(S)
N
Boc
H
HOH
(S)
(R)
610
6
10 and 6
(a)
(b)
15
Figure 2.3 NMR spectra for diastereomers 6 and 10
(c) 6
(d) 10 and 6
16
Taking advantages of the high selectivity and good yield of the advanced DIBAL reduction
method, we synthesized several secondary amino alcohols by using different organometallic
compounds in this reaction.
The diastereoselectivity of the reactions of N-Boc-L-proline methyl ester 5 with DIBAL and
different organometallics was examined (Scheme 2.10). N-Boc protected β-amino alkanols (6,
10, 31-34) were isolated by quenching the reaction with saturated NH4Cl solution. As shown in
the table of Scheme 2.10, all of the organometallic compounds afforded high threo selectivity.
The two by-products consisted of the erythro isomer and the primary alcohol from over-
reduction. The yields (%) were the combined yields of both diastereomers, and the ratios were
determined from the GC spectra of the crude products.
N
Boc
OCH
3
H
O
(S)
1) DIBAL, CH
2
Cl
2
, -78
o
C, 30 min
2) -20
o
C, 1 h
3) R-M, -78
o
C to rt
4) sat. NH
4
Cl solution
N
Boc
R
H
HOH
(S)
N
Boc
R
H
HOH
(S)
+
5
(S) (R)
6 10 R= vinyl
31 32 R= methyl
33 34 R= ethyl
Entry R M % Yield Ratio
1 CH2 =CH MgBr 80 6:10 > 32:1
2 CH3 Li 49 31:32 = 8:1
3 CH2 CH3 Li 52 33:34 = 9:1
4 CH3 MgBr 57 31:32 = 6:1
5 CH2 CH3 MgBr 67 33:34 = 28:1
6 (CH3) 2 Zn 52 31:32 = 10:1
7 (CH2 CH3) 2 Zn 54 33:34 > 50:1
8 CH3CH2 CH2 ZnCl NA No reaction
Scheme 2.10 Advanced ester reduction of N-Boc-L-proline methyl ester 5 followed by
organometallic addition
17
The NMR spectra of diastereomers 6 and 10 are known.3 The stereochemical assignments
of these diastereomers were further confirmed by the lH-lH NOESY NMR analysis of the
oxazolidone derivatives obtained from a cyclization reaction (as shown in Scheme 2.11,
R=vinyl).3 For diastereomer 32, prepared as in Scheme 2.12, the lH NMR and l3C NMR data
have been reported46 but not the corresponding data for it diastereomer 31. However, the lH
NMR and l3C NMR data of 37 and 38, which are the corresponding cyclized products of 31 and
32, are known.48 With the lH NMR and l3C NMR data of 37 and 38 available, the
stereochemistry of both 31 and 32 could be readily deduced. The configurations of 33 and 34
were assigned based on NMR comparisons with diastereomers 31 and 32, since their structures
are very similar; the isomer with the lower field chemical shift for the methyl protons of the ethyl
group is tentatively assigned as the threo isomer. This assignment also leads to the reasonable
conclusion that the formation of the secondary alcohols 33 and 34 is analogous to those of 6 and
10 and of 31 and 32 by analogous processes.
N
Boc
R
H
HOH
(S)
(S)
N
O
O
H
A
RH
B
(S)
(S)
NaH
THF
N
O
O
H
A
H
B
R
(S)
(R)
N
Boc
R
H
HO H
(S)
(R)
NaH
THF
35 R= vinyl
37 R= methyl
39 R= ethyl
36 R= vinyl
38 R= methyl
40 R= ethyl
Scheme 2.11 Stereochemical assignments for amino alcohols
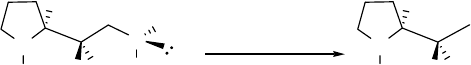
18
N
Boc
H
HO H
(S)
(R) S
O
Tol Rayney nickel
MeOH, rt, 1 h
41
N
Boc
H
HO H
(S)
(R)
32
Scheme 2.12 Literature synthesis of 32 from β- hydroxy sulfoxide 41
In summary, the advanced DIBAL reductions on N-Boc-L-proline methyl ester 5 produced
N-Boc protected β-amino secondary alcohols in very high diastereoselectivity and in good yield
when organolithiums, Grignard reagents and dialkylzincs were used. A monoalkylzinc chloride
does not bestow these benefits since it is apparently not as reactive as dialkylzinc reagents.
2.2.2. Mechanism study on advanced DIBAL reduction
It has been previously reported that there is better selectivity when advanced the ester
reduction/alkylation is used rather than the addition of organometallics to the aldehydes.3,6 In
our current study, we also observed a high selectivity of advanced DIBAL reduction of N-Boc
protected α-amino acid methyl ester 5 followed by the addition of organolithiums, Grignards,
and dialkylzincs. However, the mechanism of this high selectivity is still obscure. In this part of
the Chapter, our attempts to elucidate the mechanism from both experimental and theoretical
perspectives are described.
First, we compared the ester reduction/alkylation method (without a warm-up step) with the
advanced ester reduction/alkylation method (with a warm-up step). In Scheme 2.13, reaction (a)
is the advanced ester reduction/alkylation with the warm-up step. N-Boc-L-proline methyl ester
5 was first mixed with DIBAL at -78 oC and the solution was then warmed to -20 oC for 1 h.
The reaction mixture was then re-cooled to -78 oC before the addition of vinylmagnesium
bromide. This experimental procedure gave a single diastereomer 6. Reaction (b) is the basic
19
DIBAL reduction without the warm-up step. N-Boc-L-proline methyl ester 5 was added to
DIBAL at -78 oC followed by the addition of vinylmagnesium bromide directly at -78 oC. The
product was a 2:1 mixture of diastereomers 6 and 10. This data indicates that the warm-up step,
which is the only difference between reaction (a) and (b), is very likely the key to the high
diastereoselectivity.
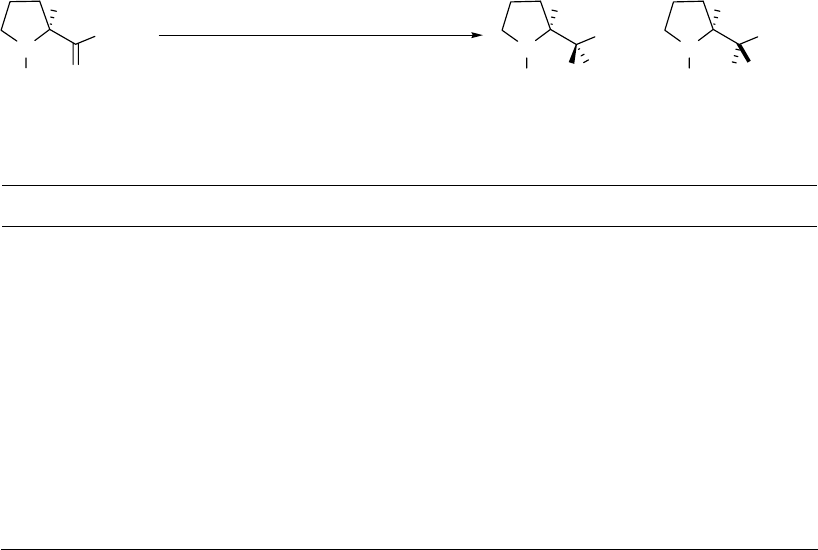
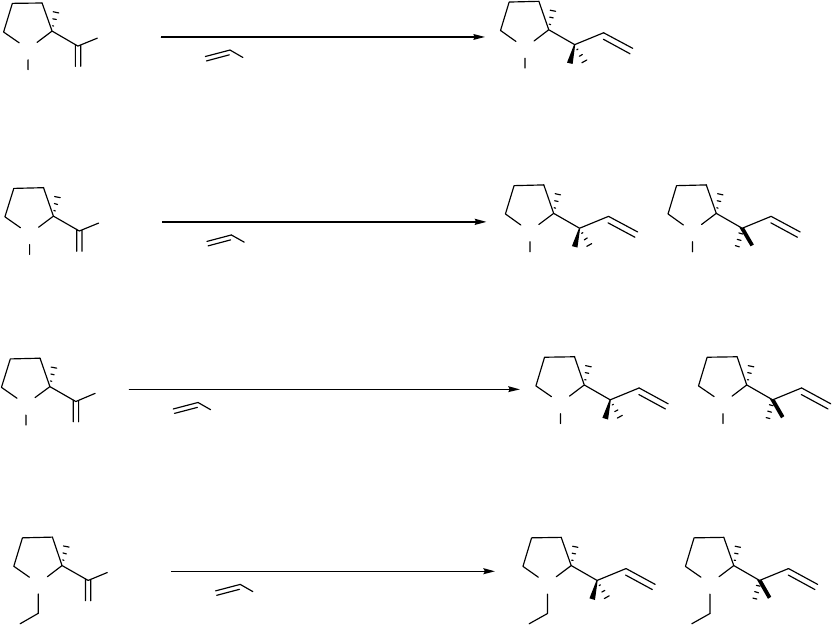
Two possible reaction mechanisms may be envisioned for the advanced ester
reduction/alkylation. The first mechanism is that N-Boc-L-proline methyl ester 5 reacts with
DIBAL to produce aluminoxy-acetals R1 and R2 (Figure 2.4). The higher temperature allows
equilibration of R1 and R2, leading to a very high ratio of R1 to R2. Vinylmagnesium bromide
then replaces the MeO group of R1 with retention of configuration (SNi process) to give 6. The
second possible mechansim is the conversion of the aluminoxy-acetals R1 and R2 to an aldehyde
and (i-Bu)2AlOMe, and the addition of vinylmagnesium bromide to the aldehyde in the presence
of (i-Bu)2AlOMe. We set up a model reaction (as shown in reaction (c) in Scheme 2.13) to
simulate the second predicted mechanism. N-Boc-L-prolinal 42 was mixed with (i-Bu)2AlOMe
at -78 oC and vinylmagnesium bromide was then added to the reaction mixture. This model
reaction gave diastereomers 6 and 10 (2:1), suggesting that the second predicted mechanism
involving an aldehyde intermediate is unlikely.
To experimentally determine that no aldehyde is involved in the mechanism, we tracked the
warm-up step in reaction (a), Scheme 2.13, by NMR. In brief, N-Boc-L-proline methyl ester 5
was mixed with DIBAL in CD2Cl2 at -78 oC in an NMR tube. The NMR tube was sealed before
it was placed in the NMR spectrometer. The NMR tube was warmed from -78 oC to -20 oC
stepwise with increments of 15 oC; generally for each increment it took about 20 minutes to
reach the higher temperature and another 15 minutes for temperature stabilization. At every 15
20
oC increment, a proton NMR spectrum was collected. In all the spectra collected, no aldehyde
peak around 10 ppm was found during the warm-up process from -78 oC to -20 oC. An
extremely small aldehyde peak was observed after the reaction mixture had been maintained at -
20 oC for 1 h. Based on this experimental data, we can be sure that no aldehyde is involved in
the mechanism.
In the first predicted aluminoxy-acetal mechanism, aluminum can coordinate with an
oxygen atom on the Boc group or with the nitrogen of the pyrrolidine ring. To test whether the
Boc group is required, reaction (d) is Scheme 2.13 was designed in which the substrate bore an
N-benzyl group instead of an N-Boc group. N-Benzyl-L-proline methyl ester 43 was first treated
with DIBAL at -78 oC and then the mixture was warmed up to -20 oC and maintained at that
temperature for 1 h. The reaction mixture was then re-cooled to -78 oC before the addition of
vinylmagnesium bromide. This experimental procedure gave two diastereomers 44 and 45 in an
approximate ratio of 1:1. By comparing reactions (a) and (d), we conclude that an oxygen atom
on the Boc group probably plays an important role in the advanced DIBAL reaction with high
diastereoselectivity. This concept is supported by the theoretical calculations that are discussed
below.
21
N
Boc
OCH
3
H
O
(S)
MgBr
1) DIBAL, CH2Cl2, -78 oC, 30 min
2) , -78 oC to rt
3) sat. NH4Cl solution
N
Boc
H
HOH
(S)
(S)
N
Boc
H
HOH
(S)
(R)
+
MgBr
1) (i-Bu)2AlOMe, CH2Cl2, -78 oC, 30 min
2) , -78 oC to rt
3) sat. NH4Cl solution
N
Boc
H
HOH
(S)
(S)
N
Boc
H
HOH
(S)
(R)
+
N
Boc
H
H
O
(S)
N
Boc
OCH
3
H
O
(S)
N
Boc
H
HOH
(S)
(S)
MgBr
1) DIBAL, CH2Cl2, -78 oC, 30 min
2) -20 oC, 1 h
3) , -78 oC to rt
4) sat. NH4Cl solution
56
5610
42 6 10
53% 2:1
86% 2:1
NOCH
3
H
O
(S)
MgBr
1) DIBAL, CH2Cl2, -78 oC, 30 min
2) -20 oC, 1 h
3) , -78 oC to rt
4) sat. NH4Cl solution
Ph
N
H
HOH
(S)
(S)
N
H
HOH
(S)
(R)
+
Ph Ph
43
(a)
(b)
(c)
(d)
44 45 85% 1:1
80%
Scheme 2.13 Mechanistic study on the advanced ester reduction/alkylation
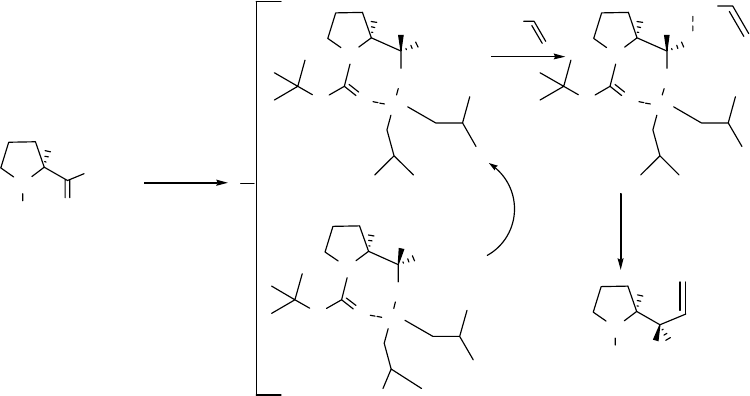
Thus, the first aluminoxy-acetal mechanism is a reasonable one to explain the advanced
DIBAL reduction reaction (a) in Scheme 2.13. As elucidated in detail in Figure 2.4, N-Boc-L-
proline methyl ester 5 reacts with DIBAL and results in aluminoxy-acetal intermediates R1 and
R2. During the warm-up step, R2 epimerizes to R1. When the reaction mixture is re-cooled to
-78 oC, the aluminoxy-acetal R1 undergoes an SNi reaction with vinylmagnesium bromide with
retention of configuration to give 6 as the dominant diastereomer.
22
NH
H
O
O
OAl
OCH3
(S) (S)
NOCH3
H
O
O
OAl
H
(S) (R)
N
Boc
H
HOH
(S)
(S)
DIBAL-H
retention
epimerization
R1
R2
CH2Cl2-CH3OMgBr
NOCH3
H
O
O
OAl
H
(S) (R)
BrMg
BrMg
SNi
N
Boc
OCH3
H
O
(S)
5
6
Figure 2.4 Postulated mechanism for the advanced ester reduction/alkylation of 5
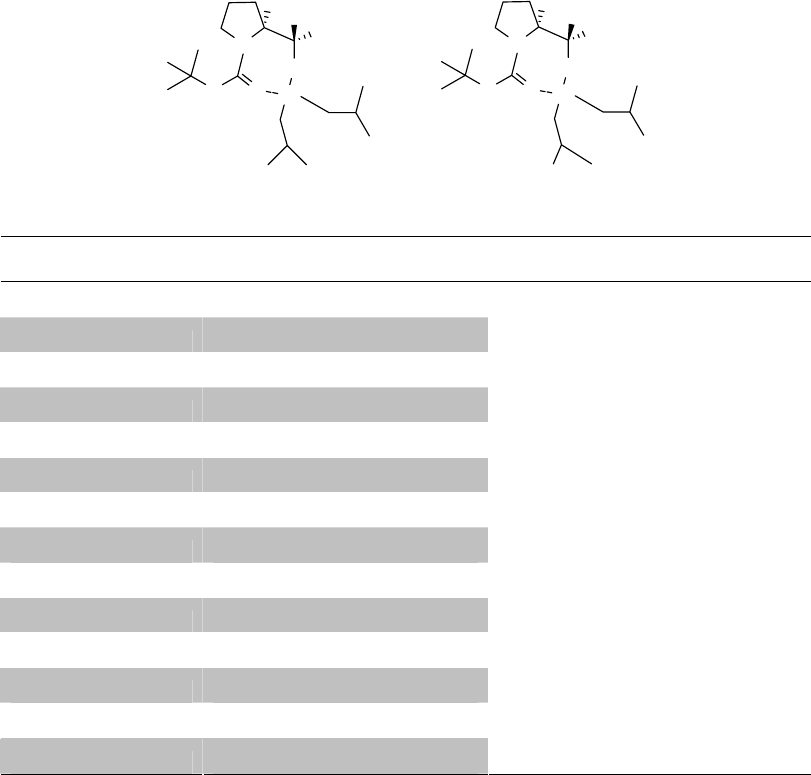
Theoretical calculations have been performed on the structure of the DIBAL adducts. It was
postulated from evidence given above that the Al atom in the tetrahedral intermediates R1 and
R2 is coordinated with the most basic and least crowded of the two oxygen atoms of the Boc
group, the carbonyl oxygen atom. The computations were designed to determine if complexes
R1 and R2 are reasonable structures for the DIBAL adducts and, if so, whether R1 is
substantially more thermodynamically stable than R2.
The theoretical calculations were done using Gaussian 03 software49 with different semi-
empirical and ab-initio methods. By using these methods, optimization of R1 and R2 was
performed and the total energy and the optimized structures of R1 and R2 were studied.
The methods used to optimize R1 and R2 include semi-empirical methods, such as AM1
and PM3, and ab-initio methods, such as HF (Hartree-Fork) 3-21G*, DFT (density function
theory) B3LYP/6-31G*, B3LYP/6-31+G*, B3LYP/6-31++G**, B3LYP/Gen and ONIOM
(B3LYP/6-31++G**:B3LYP/6-31G*). A series of "standard" basis sets is stored internally in
Gaussian; these basis sets may be specified by including the appropriate keyword within the
23
route section for the calculation. The Gen keyword allows a user-specified basis set to be used in
the Gaussian calculation. In the ONIOM procedure, the molecular system being studied is
divided into two or three layers which are treated with different model chemistries. The results
are then automatically combined into the final predicted results. Layer assignments are specified
as part of the molecule specification.
Density function theory (DFT) was found to be the most time expensive but the best
calculation method compared to semi-empirical methods (AM1 and PM3) and Hartree-Fock
theory. Many published reports use density function theory for their theoretical calculations,
especially for organic chemistry. It is the method we finally adopted for our theoretical
calculations. As shown in Table 2.1, the results calculated by different methods agree with each
other, and all of them suggest that the total energy of R2 is higher than R1. Using method
B3LYP/6-31+G*, the total energy of R2 is 6.017 kcal/mol higher than R1, a decisive difference.
24
NOCH
3
H
O
O
OAl
H
(S) (R)
R1
NH
H
O
O
OAl
OCH
3
(S) (S)
R2
Total energy (H)
1 H = 627.51 kcal/mol
Energy difference R1-R2
(kcal/mol)
R1_AM1_G -0.41254
R2_AM1_G -0.40691
-3.53511
R1_PM3_G -0.41240
R2_PM3_G -0.40268
-6.09923
R1_HF3-21G*_G -1330.31620
R2_HF3-21G*_G -1330.30153
-9.20351
R1_DFT6-31G*_G -1345.12318
R2_DFT6-31G*_G -1345.11416
-5.659412
R1_DFT6-31+G*_G -1345.15546
R2_DFT6-31+G*_G -1345.14587
-6.01734
R1_ONIOM -1345.20675
R2_ONIOM -1345.19840
-5.24441
R1_Gen -1345.20828
R2_Gen -1345.19864
-6.04725
Table 2.1 Total Energies of R1 and R2 calculated by different methods
In the optimized structures of R1 and R2, the distances between the aluminum atom and the
four oxygen atoms were measured. As shown in Table 2.2, calculations by all of the different
methods indicate that the Al-O bond distance is around 1.80 Å and the distance between the Al
and the carbonyl oxygen atom of the Boc group is around 1.95 Å, except that in the
semiempirical methods it is 2.4-2.5 Å in the case of R1; such a distance indicates coordination
between the Al and this oxygen atom. On the other hand, the distances between Al and the
oxygen atoms on the methoxy group and the butoxy group in Boc are around 3.8-4.0 Å,
25
suggesting that no coordination exists. Therefore, the calculation results tell us that the Al atoms
in R1 and R2 bonds with the O atom on the stereo center and coordinates with the carbonyl O on
the Boc group, to form the aluminoxy-acetal.
NOCH3
H
O
O
OAl
H
(S) (R)
NH
H
O
O
OAl
OCH3
(S) (S)
R1 R2
Al-O(carbonyl on Boc) length
(Å)
Al-O bond
length (Å)
Al-O (OMe)
length (Å)
Al-O (on Boc)
length (Å)
R1_AM1_G 2.41365 1.74494 3.82501 4.46249
R2_AM1_G 1.83017 1.77085 3.76605 3.88320
R1_PM3_G 2.45997 1.76147 3.61861 4.67955
R2_PM3_G 1.86865 1.79708 3.61433 4.05575
R1_HF_G 1.87222 1.74815 3.80520 4.06886
R2_HF_G 1.88299 1.76116 3.95279 3.92682
R1_DFT6-31+G*_G 1.94888 1.78720 3.63919 4.12067
R2_DFT6-31+G*_G 1.95960 1.79390 3.92336 3.99970
R1_ONIOM 1.94649 1.78762 3.78663 4.13982
R2_ONIOM 1.96418 1.79763 3.93527 4.0157
R1_Gen 1.94949 1.78871 3.63171 4.11993
R2_Gen 1.96056 1.7954 3.92392 4.00130
Table 2.2 Distances between Al atom and O atoms in R1 and R2
As presented in Figure 2.5, the distances between the corresponding Al atoms and oxygen
atoms measured from the crystal structures of many tetra-coordinated aluminum compounds are
in excellent agreement with those determined from our theoretical calculations.50 This
agreement gives us confidence in the reliability of our calculation.
26
NOCH
3
H
O
O
OAl
~1.95
~1.80
R1 or R2
CH
3
tbu
tBu
O
Al
Et
Et
O
pTol
O
H
3
C
O
Al
CH
3
CH
3
H
3
C
Al
O
OMe
CH
3
CH
3
BrBr
tbu tBu
O OO
Al Al
1.887
1.748
1.862
1.854
1.969
1.957
1.780
O
Al
O
N
Ph
Ph Ph
Al
1.929
1.859
1.842
tBu
tBu
O
Al
O O
1.737
1.866
Figure 2.5 Crystal structure data of tetra-coordinated aluminium compounds
Figure 2.6 shows the conformations of R1 and R2 after they were optimized by B3LYP/6-
31+G*. Thus, as suggested in our original hypothesis, the main intermediates are probably also
fused 5- and 7-membered rings.
R1 R2
Figure 2.6 R1 and R2 optimized by B3LYP/6-31+G*
It is known that Al can undergo tetra-coordination and penta-coordination as well as hexa-
coordination.50 To test whether there is a higher degree of coordination in R1 or R2 than 4, we
27
attempted to form a penta-coordinated structure (with O of the butoxy group in Boc or of the
methoxy group). Pre-formed penta-coordination structures of R1 or R2 were optimized by AM1
and PM3. Table 2.3 shows the energy and structure data after optimization. In all cases, the
penta-coordination was broken up during the optimization process and the resulting
conformations had energies higher than those associated with tetra-coordination. Based on this
data, we can conclude that penta-coordination does not exist in R1 and R2.
Total energy
(kcal/mol)
Al-
O(cabonyl
on Boc)
length (Å)
Al-O bond
length
(Å)
Al-O
(OMe)
length (Å)
Al-O (on
Boc)
length
(Å)
Energy
difference R1-
R2 (kcal/mol)
R1_AM1_pentaBoc -246.24734 3.80323 1.74407 3.781 2.489
R2_AM1_pentaBoc -256.27232 2.43567 1.75016 3.630 4.289
10.0250
R1_AM1_pentaOMe -258.87453 2.41363 1.74494 3.825 4.462
R2_AM1_pentaOMe -256.24343 2.43567 1.75016 3.630 3.883
-2.6311
R1_PM3_pentaBoc -236.62970 4.22043 1.75830 3.601 2.532
R2_PM3_pentaBoc -246.26372 2.47137 1.77524 3.616 2.556
9.6340
R1_PM3_pentaOMe -251.56474 2.55690 1.75114 2.646 4.835
R2_PM3_pentaOMe -251.99134 2.54561 1.74876 2.639 4.809
0.4266
Table 2.3 The energy and structure data of penta-coordinated structures for R1 or R2 after
optimization
To reveal the structural basis for the energy difference of R1 and R2, calculations were
performed on simplified structures in which the isobutyl groups of R1 and R2 were replaced by
hydrogens, (H1, H2 in Figure 2.7) and by methyl groups (M1, M2 in Figure 2.7) and the
structures were optimized by the same method (B3LYP/6-31+G*) used for R1, R2 optimization.
Figure 2.8 shows the conformations of M and H after optimization by B3LYP/6-31+G*. All
optimized M and H structures are aluminoxy-acetals in accord with the data from R.
28
NOCH
3
H
O
O
HO AlH
2
H
(S) (R)
H1
NH
H
O
O
HO AlH
2
OCH
3
(S) (S)
H2
NOCH
3
H
O
O
OAl
H
(S) (R)
R1
NH
H
O
O
OAl
OCH
3
(S) (S)
R2
NOCH
3
H
O
O
OAl
H
(S) (R)
M1
NH
H
O
O
OAl
OCH
3
(S) (S)
M2
Figure 2.7 Structures of H (H1 or H2), M (M1 or M2) and R (R1 or R2)
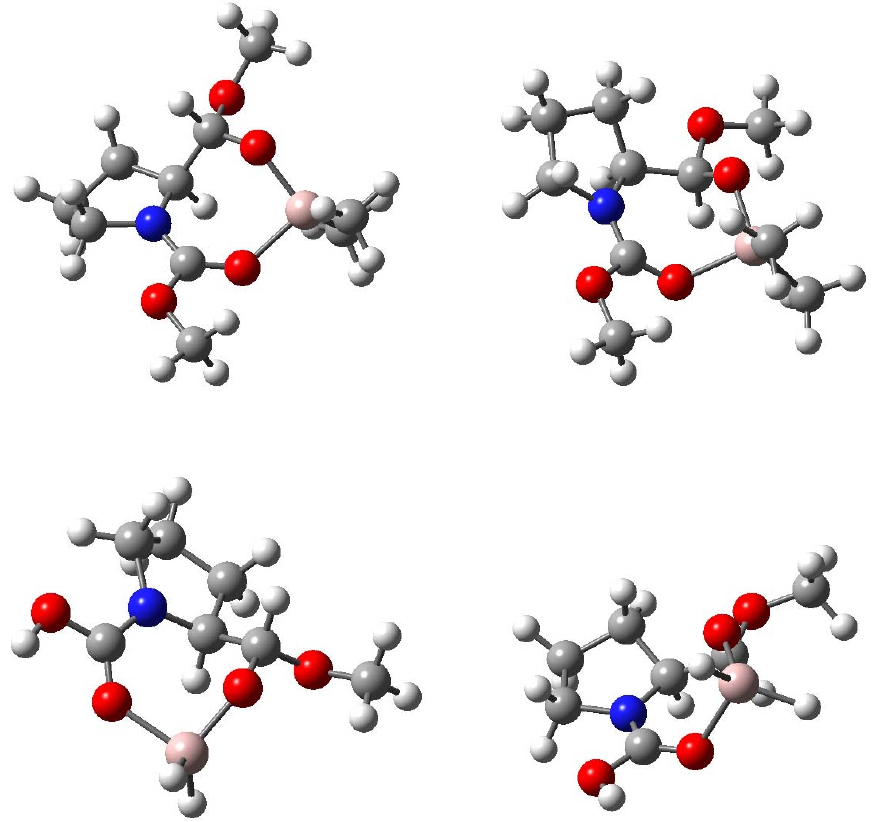
29
M1 M2
H1 H2
Figure 2.8 M (M1 or M2) and H (H1 or H2) optimized by B3LYP/6-31+G*
Tables 2.4 and 2.5 show the free energy and structure data of the optimized M (M1, M2), H
(H1, H2), R1 and R2. The free energy difference between M1 and M2, H1 and H2, and R1 and
R2 are -5.19 kcal/mol, -4.48 kcal/mol and -5.64 kcal/mol respectively. The energy differences
within M (M1 or M2), H (H1 or H2) and R (R1 or R2) are remarkably close to each other and
the nature of the two substituents on Al thus has very little influence on the energy difference
between the two diastereomers. Therefore, the energy difference between the two diastereomers

30
R1 and R2 is probably due to interactions between groups on the 7-membered rings bearing the
Al. There is no obvious crowding between the MeO group and the hydrogens on the 3
methylene group in trans isomer R2. (see Appendix B)
Energy
H
Zero-point
correction
H/particle
Sum of electronic and thermal
free energies H
(T= -20 oC, 253 K)
ΔG(R1-R2)
kcal/mol
R1 -1345.15546 0.55650 -1344.662958
R2 -1345.14587 0.55661 -1344.653965
-5.64320
H1 -873.36520 0.21596 -873.189492
H2 -873.35810 0.21619 -873.181922
-4.47503
M1 -991.32719 0.30140 -991.074073
M2 -991.31959 0.30162 -991.065796
-5.19390
Table 2.4 Free energies of the optimized M, H and R by B3LYP/6-31+G*
Al-O(cabonyl
on Boc) length
(Å)
Al-O
bond
length
(Å)
Al-O
(OMe)
length (Å)
Al-O (on
Boc) length
(Å)
R1 1.94888 1.78720 3.63919 4.12067
R2 1.95960 1.79390 3.92336 3.99970
H1 1.94413 1.78053 3.60887 4.07657
H2 1.94954 1.78851 3.88375 3.84009
M1 1.95294 1.78785 3.63404 4.08154
M2 1.96272 1.79532 3.91362 3.9657
Table 2.5 Distances between Al atom and O atoms in M, H and R by B3LYP/6-31+G*
2.2.3. Improvement in the advanced ester reduction/alkylation method
In this advanced ester reduction/alkylation mechanism, which was first proposed based on
experimental data and later confirmed by theoretical calculations, we found that the existence of
an equilibration between the reaction intermediates is the key to the high reaction
stereoselectivity. Any factor affecting the equilibration may also affect this selectivity. A warm-
31
up step was applied in our previous study and the higher temperature indeed increased the
selectivity. However, a warm-up step is time consuming and requires careful temperature
control. In an attempt to discover a simpler method than the warm-up step, we have utilized
ZnCl2 in the ester reduction/alkylation in the hope that this Lewis acid would help remove the
MeO group and allow the methoxy group to be re-deposited on the other side of the 7-memered
ring, thus facilitating the epimerization.
As described in reaction (a) in Scheme 2.14, DIBAL was added to the mixture of N-Boc-L-
proline methyl ester 5 and 1 equiv ZnCl2, followed by adding vinylmagnesium bromide. The
reaction did indeed produce a single diastereomer 6. Furthermore, this reaction also worked well
with a catalytic amount ZnCl2 and yielded a single diastereomer 6, as shown in reaction (b)
Scheme 2.14. This reaction is of significant mechanistic and practical importance. First, it
significantly simplifies the reaction but still maintains the high selectivity. Second, it strongly
supports the mechanism that we advocate. To ensure that the high selectivity originated from the
effect of ZnCl2 on the equilibration, but not from the ZnCl2 causing chelation between the
oxygen or nitrogen atoms inducing selectively in the DIBAL addition step, reaction (c) in
Scheme 2.14 was performed. After DIBAL was added to N-Boc-L-proline methyl ester 6, 0.1
equiv ZnCl2 was added to the reaction mixture, followed by the addition of vinylmagnesium
bromide. Reaction (c) indeed affords one single diastereomer 6. Therefore, reaction (a), (b) and
(c) provide solid evidence for our suggested mechanism.
32
N
Boc
OCH
3
H
O
(S)
N
Boc
H
HOH
(S)
(S)
MgBr
1) 1 equiv ZnCl
2
solution, CH
2
Cl
2
2) DIBAL, -78
o
C, 30 min
3) , -78
o
C to rt
4) sat. NH
4
Cl solution
N
Boc
OCH
3
H
O
(S)
N
Boc
H
HOH
(S)
(S)
MgBr
1) DIBAL, CH
2
Cl
2
, -78
o
C, 30 min
2) 0.1 equiv ZnCl
2
solution, 30min
3) , -78
o
C to rt
4) sat. NH
4
Cl solution
N
Boc
OCH
3
H
O
(S)
N
Boc
H
HOH
(S)
(S)
MgBr
1) 0.1 equiv ZnCl
2
solution, CH
2
Cl
2
2) DIBAL, -78
o
C, 30 min
3) , -78
o
C to rt
4) sat. NH
4
Cl solution
56
56
56
(a)
(b)
(c)
62%
69%
63%
Scheme 2.14 Advanced ester reduction/alkylation with Lewis acid catalyzed equilibration
2.3. Conclusions
The addition of vinylmagnesium bromide to the DIBAL adduct of N-Boc-L-proline methyl
ester, after a warm-up step, gives a Boc-protected β-amino secondary allylic alcohol with high
diastereoselectivity of greater than 32:1. This method can be expanded to other Grignard
reagents, organolithiums and dialkylzincs with slightly less stereoselectivity.
N-Boc-L-proline methyl ester reacts with DIBAL to produce aluminoxy-acetals R1 and R2.
The higher temperature probably causes equilibration of R1 and R2, leading to a very high ratio
of R1 to R2. The ratio is consistent with the high computed energy differences between R1 and
R2 and favoring the former. Vinylmagnesium bromide then reacts with R1 to cause replacement
of the methoxide ion with the vinyl nucleophile with retention of configuration (SNi process) and
gives the protected β-amino secondary allylic alcohol 6.
33
Adding ZnCl2 before or after the addition of DIBAL, followed by vinylmagnesium bromide,
significantly simplifies the reaction allowing one to avoid the warm-up step, which is time
consuming and requires careful temperature control. The high stereoselectivity is still maintained.
The Lewis acid ZnCl2 is postulated to aid the removal of the MeO group; this group may be re-
deposited on the other side of the 7-memered ring, thus facilitating the epimerization.
34
2.4. Experimental
General Considerations: 1H and 13C NMR spectra were recorded on a Bruker DPX-300
spectrometer operating at 300 MHz for 1H and 75 MHz for 13C. Chemical shift data are reported
in units of δ (ppm) relative to CHCl3 as δ = 7.26 for 1H NMR spectra and CDCl3 as δ = 77.09
for 13C NMR spectra. Multiplicities are given as: s (singlet), d (double), t (triplet), q (quartet), m
(multiplet), and br (broad). Coupling constants, J, are reported in Hz and refer to apparent peak
multiplicities and not true constants. Silica gel 60 (40-60 μm, Sorbent Technologies) was used
for flash column chromatography. Thin-layer chromatography was performed on glass
supported 250-μm silica GF plates (Analtech). Visualization of TLC plates was accomplished
with one or more of the following: 254 nm UV light; 7% phosphomolybdic acid in ethanol; 5%
anisaldehyde in ethanol containing 5% sulfuric acid and a trace amount of acetic acid. The ratios
between diastereomers were determined by 1H NMR spectroscopy and/or GC analysis. GC
analyses were carried out with the Agilent 6850 Series GC System by using the Agilent 19091Z-
413E HP-1 methyl siloxane column, heated from 50 oC to 315 oC with a rate of 10 oC/min, and
were detected by FID. Anhydrous magnesium sulfate was used as the drying reagent. All
reactions were performed under an argon atmosphere and standard precautions against moisture
were taken. A Dry Ice/acetone bath was used to obtain a temperature of -78 oC and -20 oC. An
ice bath was used to obtain 0 oC. Tetrahydrofuran (THF) and diethyl ether were distilled over
sodium benzophenone ketyl. Hexane was distilled over sodium hydride and toluene was distilled
from sodium. All reagents used were purchased from Aldrich.
(S)-1-tert-butyl 2-methylpyrrolidine-1,2-dicarboxylate (5).44 A solution of (0.59 g, 5.1
mmol) of L-proline in 5 mL of methanol was cooled to 0 oC and thionyl chloride (0.40 mL, 5.5
35
mmol) was added dropwise over 20 min. After the solution had been refluxed for 1 h, the
solvent was removed in vacuo to afford a yellow oil which was then dissolved in 6 mL of
CH2Cl2 under argon before triethylamine (1.03 g, 1.4 mmol) and di-tert-butyl dicarbonate (1.33
g, 6.1 mmol) were added at 0 oC. The reaction mixture was stirred at 0 oC for 1 h and then at
room temperature overnight. It was made acidic by adding saturated citric acid solution. The
organic layer was separated and washed with H2O, saturated NaHCO3 and then brine. The
combined organic layer was dried over MgSO4 and concentrated in vacuo to give a yellow oil
that was purified by column chromatography (15% acetone in hexanes) to yield 0.99 g (85%) of
the title product as a yellow oil. 1H NMR (CDCl3) δ 4.27 (dd, J=8, 4 Hz, 0.5 H), 4.17 (dd, J=8, 5
Hz, 0.5 H), 3.68 (s, 3 H), 3.51-3.32 (m, 2 H), 2.18 (m, 1 H), 1.87 (m, 3 H), 1.41 (s, 3.7 H), 1.36
(s, 6.6 H); 13C NMR (CDCl3) δ 173.67, 173.42, 154.33, 153.69, 79.73, 59.00, 58.62, 51.98, 51.82,
46.45, 46.21, 30.78, 29.81, 28.22, 24.24, 23.59; [α]589 = -65.5 (c=0.44, MeOH).
(S)-tert-butyl 2-((S)-1-hydroxyallyl)pyrrolidine-1-carboxylate (6) and (S)-tert-butyl 2-
((R)-1-hydroxyallyl)pyrrolidine-1-carboxylate (10)
Procedure (a) :4,6 Ester reduction/alkylation method.
DIBAL (2.62 mL of a 1.0 M solution in hexane, 2.62 mmol) was added to a solution of N-
Boc-proline methyl ester 5 (0.50 g, 2.18 mmol) in CH2Cl2 (10 mL) at -78 oC. The resulting
solution was stirred at -78 oC for 30 min, followed by the addition of vinylmagnesium bromide
(6.54 mL of a 1.0 M solution in THF, 6.54 mmol) dropwise at -78 oC. The solution was then
allowed to slowly warm to room temperature overnight. Saturated aqueous NH4Cl solution (10
mL) was added to quench the reaction. Saturated sodium tartrate solution (10 mL) was added to
resulting gel. The mixture was stirred at room temperature for 30 min. The organic layer was
36
extracted with CH2Cl2 (3 x 15 mL) and the combined organic layer was dried over anhydrous
MgSO4, and concentrated in vacuo to give an inseparable mixture of diastereomers 6 and 10 at
2:1 ratio. Flash chromatography (30% ethyl acetate in hexanes) gave 0.26 g of the title
compound (yield 53%) as a yellow oil. 1H NMR (CDCl3) δ 5.79 (m, 1 H), 5.33-5.16 (m, 2 H),
4.12 (m, 0.67 H), 3.92 (m, 1.33 H), 3.45 (m, 1 H), 3.31 (m, 0.7 H), 3.21 (m, 0.3 H) 1.90-1.68 (m,
4 H), 1.47 (s, 6 H), 1.46 (s, 3H).
Procedure (b):4,6 Advanced ester reduction/alkylation method.
DIBAL (2.62 mL of a 1.0 M solution in hexane, 2.62 mmol) was added to a solution of N-
Boc-L-proline methyl ester 5 (0.50 g, 2.18 mmol) in CH2Cl2 (10 mL) at -78 oC. The resulting
solution was stirred at -78 oC for 30 min, and then at -20 oC for 1 h. The mixture was re-cooled
to -78 oC, followed by the dropwise addition of vinylmagnesium bromide (6.54 mL of a 1.0 M
solution in THF, 6.54 mmol). The solution was then allowed to slowly warm to room
temperature overnight. Saturated aqueous NH4Cl (10mL) solution was added to quench the
reaction. Saturated sodium tartrate solution (10 mL) was added to the resulting gel and the
mixture was stirred at room temperature for 30 min. The organic layer was extracted with
CH2Cl2 (3 x 15 mL) and the combined organic layer was dried over anhydrous MgSO4, and
concentrated in vacuo. Flash chromatography (30% ethyl acetate in hexanes) gave 0.40 g of the
title compound (yield 80%) as a yellow oil. 1H NMR (CDCl3) δ 5.81 (m, 1 H), 5.31 (d, J=17 Hz,
1 H), 5.18 (d, J=10 Hz, 1 H), 4.00 (m, 1 H), 3.85 (m, 1 H), 3.49 (m, 1 H), 3.29 (m, 1 H), 1.90-
1.69 (m, 4 H), 1.48 (s, 9 H); 1H NMR (C6D6) δ 5.77 (m, 1 H), 5.35 (m, 1 H), 5.06 (d, J=10 Hz, 1
H), 4.11 (m, 1 H), 3.82 (m, 1 H), 3.17 (m, 1 H), 3.00 (m, 1 H), 1.47-1.20 (m, 13 H); 13C NMR
(CDCl3) δ 157.85, 138.33, 116.69, 80.43, 77.21, 62.39, 47.32, 29.62, 28.38 (3 C), 23.80; 13C
NMR (C6D6) δ 157.13, 138.96, 115.92, 79.88, 75.91, 62.84, 47.49, 28.46 (3 C), 27.56, 24.05.
37
Procedure (c):4,6 Addition of vinylmagnesium bromide to N-Boc-L-prolinal in the
presence of (i-Bu)2AlOMe.
Dry methanol (0.02 mL, 0.52 mmol) was added to DIBAL (0.52 mL of a 1.0 M solution in
hexane, 0.52 mmol) at -78 oC. The resulting mixture was stirred at -78 oC for 30 min, followed
by the addition of a solution of N-Boc-L-prolinal 42 (0.10 g, 0.44 mmol) in CH2Cl2 (4 mL).
Vinylmagnesium bromide (1.32 mL of a 1.0 M solution in THF, 1.32 mmol) was then added to
the mixture dropwise. After the addition was complete, the reaction mixture was allowed to
warm to room temperature overnight. Saturated aqueous NH4Cl solution was added to quench
the reaction. Saturated sodium tartrate solution (8 mL) was added to the resulting gel and the
mixture was stirred at room temperature for 30 min. The organic layer was extracted with
CH2Cl2 (3 x 10mL) and the combined organic layer was dried over anhydrous MgSO4, and
concentrated in vacuo to give an inseparable mixture of diastereomers 6 and 10 at 2:1 ratio
(0.085 g, 86%) as a yellow oil. 1H NMR (CDCl3) δ 5.79 (m, 1 H), 5.33-5.16 (m, 2 H), 4.12 (m,
0.67 H), 3.92 (m, 1.33 H), 3.45 (m, 1 H), 3.28 (m, 0.6 H), 3.21 (m, 0.3 H) 1.90-1.68 (m, 4 H),
1.47 (s, 6 H), 1.46 (s, 3H).
Procedure (d): With the use of ZnCl2 catalysis of equilibration instead of warming.
DIBAL (0.48 mL of a 1.0 M solution in hexane, 0.48 mmol) was added to a solution of N-
Boc-L-proline methyl ester 5 (0.10 g, 0.44 mmol) and ZnCl2 (0.44 mL of a 1.0 M solution in
ether, 0.44 mmol) in CH2Cl2 (2 mL) at -78 oC. The resulting solution was stirred at -78 oC for 30
min, followed by the addition of vinylmagnesium bromide -78 oC (1.31 mL of a 1.0 M solution
in THF, 1.31 mmol) dropwise. The solution was then allowed to warm to room temperature
overnight. Saturated aqueous NH4Cl solution was added to quench the reaction. Saturated
sodium tartrate solution (3 mL) was added to the resulting gel and the mixture was stirred at
38
room temperature for about 30 min. The aqueous layer was extracted with CH2Cl2 (3 x 10 mL)
and the combined organic layer was dried over anhydrous MgSO4, and concentrated in vacuo to
give crude product as a yellow oil. Flash chromatography (30% ethyl acetate in hexanes) gave
0.057 g of the title compound (yield 62%) as a yellow oil. 1H NMR (CDCl3) δ 5.84 (m, 1 H),
5.30 (d, J= 13 Hz, 1 H), 5.18 (d, J=10 Hz, 1 H), 3.98 (m, 1 H), 3.85 (m, 1 H), 3.48 (m, 1 H), 3.31
(m, 1 H), 2.04-1.72 (m, 4 H), 1.47 (s, 9 H).
Analogous experiment were performed in which only 10% of ZnCl2 was used (i) before
addition of DIBAL and (ii) after the addition of DIBAL and the results were virtually the same.
(S)-tert-butyl 2-((S)-1-hydroxyethyl)pyrrolidine-1-carboxylate (31) and (S)-tert-butyl
2-((R)-1-hydroxyethyl)pyrrolidine-1-carboxylate (32).46 The procedure was the same as for 6
and 10 except that methylmagnesium bromide or methyl lithium or dimethyl zinc was used
instead of vinylmagnesium bromide. It gave 0.54 g (57%) 31 and 32 at 6:1 ratio when
methylmagnesium bromide was used. It gave 0.46 g (49%) 31 and 32 at 8:1 ratio when methyl
lithium was used. It gave 0.49 g (52%) 31 and 32 at 10:1 ratio when dimethyl zinc was used. 1H
NMR (CDCl3) δ 5.19 (br, 1 H), 3.74-3.65 (m, 2 H), 3.47 (m, 1 H), 3.25 (m, 1 H), 2.00 (m, 1 H),
1.89-1.70 (m, 2 H), 1.59 (m, 1 H), 1.47 (s, 9 H), 1.14 (d, J=6 Hz, 3 H).
(1S,7aS)-1-methyl-tetrahydropyrrolo[1,2-c]oxazol-3(1H)-one (38).3 31 (0.089 g, 0.41
mmol) in THF (4 mL) was treat with NaH (0.033 g of 60% wt dispersed in mineral oil, 0.83
mmol) at 0 oC. The suspension was stirred at 0 oC for 5 min, and then at room temperature
overnight. The reaction was quenched with H2O (5 mL). The mixture was extracted with
CH2Cl2 (3 x 5 mL) the combined organic extract was dried over anhydrous MgSO4 and
39
concentrated in vacuo. Flash chromatography (50% ethyl acetate in hexanes) gave 0.040 g of the
title compound (yield 69%) as a yellow oil. 1H NMR (CDCl3) δ 4.0 (m, 1 H), 3.63 (m, 1 H),
3.49 (m, 1 H), 3.15 (m, 1 H), 2.12-1.82 (m, 4 H), 1.48-1.46 (d, J=6.4 Hz, 3 H); 13C NMR (C6D6)
δ 160.97, 75.98, 65.67, 45.83, 30.17, 25.51, 21.06.
(S)-methyl 1-benzylpyrrolidine-2-carboxylate (43). A mixture of L-proline methyl ester
hydrochloride (0.30 g, 1.78 mmol), benzyl bromide (0.30 g, 1.8 mmol) and K2CO3 (0.98 g, 7.1
mmol) in dry CH2Cl2 was stirred at room temperature for 24 h. The reaction mixture was poured
into a mixture of water (5 mL) and ethyl acetate (10 mL). The aqueous layer was washed with
ethyl acetate (3 x 10 mL). The combined organic layer was dried over anhydrous MgSO4 and
concentrated in vacuo. Flash chromatography (15% ethyl acetate in hexanes) gave 0.28 g of the
title compound (yield 69%) as a yellow oil. 1H NMR (CDCl3) δ 7.41-7.35 (m, 5 H), 3.97 (d,
J=13 Hz, 1 H), 3.73 (s, 3 H), 3.66 (d, J=13 Hz, 1 H), 3.33 (m, 1 H), 3.17-3.12 (m, 2 H), 2.28-
1.26 (m, 4 H); 13C NMR (CDCl3) δ 174.46, 138.36, 129.12, 128.10, 127.01, 65.22, 58.60, 53.16,
51.55, 29.32, 22.98.
40
3. ASYMMETRIC SYNTHESIS METHOD FOR NITROGEN HETEROCYCLES
3.1. Introduction
3.1.1. Background for methods to produce organolithiums by intramolecular
carbolithiation
There have been an increasing number of papers about the intramolecular addition of
alkyllithiums to unactivated alkenes as a preparative method for cyclopentylmethyllithiums, their
heterocyclic analogues and, less effectively, the corresponding six-membered rings.51-53
Although recent significant advances has been made by many in this field, the methods to
produce organolithiums by intramolecular carbiolithiation still have considerable limitations.
Previously, a major limitation has been the lack of a general method for preparing
organolithiums. For the most part, the conventional generation methods can only be used for
primary organolithiums or those with special stabilizing features such as adjacent heteroatom
groups that direct lithiations or sp2 character of the carbon atom bearing the lithium. In most
cases, the organolithium was produced by halogen-lithium or tin-lithium exchange or by
heteroatom-directed lithiation.
Halogen-lithium exchange is a method to generate primary alkyllithiums, aryllithiums, and
vinyllithiums, which can undergo intramolecular carbolithiation (Scheme 3.1). All three
reactions54-56 in Scheme 3.1 give cyclized products in good yield. Reaction (a)57,58 has become a
standard method to generate primary alkyllithiums and is widely used in organic synthesis.
41
These organolithiums could be formed at -78 oC. However, the carbolithiation reaction requires
a higher temperature, such as 0 oC or ambient temperature.
(a) Primary alkyllithium cyclization
I
2.2 eq t-BuLi
n-C
5
H
12
/ Et
2
O (3:2)
-78
o
C
Li 23
o
C H
2
O
86% (trans:cis=10.7:1)
Li
(b) Arylithiums cyclization
Br 2.2 eq n-BuLi
THF, -78
o
C
Li Li
23
o
CD
2
O
D
92%
(c) Vinyllithium cyclization
Br
2.2 eq t-BuLi
n-C
5
H
12
/ Et
2
O (9:1)
-78
o
CLi
0
o
C, 2.5 h Li TMSCl TMS
79%
Scheme 3.1 Intramolecular carbolithiation by halogen-lithium exchange
The mechanism of halogen-lithium exchange (Scheme 3.2) involves a step proceeding
through an ate-complex intermediate.59 Two equivalents of t-BuLi are needed for the reaction.
The first equivalent of t-BuLi reacts with the alkyl iodide 46 to form an ate-complex 47, which
decomposes to t-BuI and the alkyllithium 48. The first two reactions are reversible. The second
equivalent of t-BuLi reacts with t-BuI and drives the equilibra to the alkyllithium product side.
A limitation of halogen-lithium exchange is that secondary and tertiary alkyllithiums can not be
formed through halogen-lithium exchange due to the severe Wurtz-type coupling and elimination
reactions of secondary and tertiary halides.

42
(CH
3
)
3
CLi I
+I
(CH
3
)
3
CLi
(CH
3
)
3
ILi
+
LiI
++(CH
3
)
3
CLi
46 47
48
Scheme 3.2 Mechanism of iodide-lithium exchange
However, there is an exception. Bailey’s group reported a cyclization of a secondary
alkyllithium (Scheme 3.3).60 They found that the ratio of the trans and cis products depended on
the order of addition of the reactants t-BuLi and alkyl iodide. If t-BuLi was added to the alkyl
iodide, both product yields and reaction selectivity were relatively poor. Almost pure trans
product was obtained if the alkyl iodide was added to t-BuLi. The authors explained the
differences in product yield and reaction selectivity by two competing mechanisms. When t-
BuLi was added to the alkyl iodide, a single electron transfer mechanism applied. The radical
intermediate affords more cis procuct than trans, which matches the results for similar radical
reactions.61 When the alkyl iodide is added to t-BuLi, an ate-complex is generated and the
secondary alkyllithium is produced. The latter cyclizes to the trans product. The yield is fairly
low because of Wurtz-type coupling.
(CH
3
)
3
CLi I
++
Addition of t-BuLi to alkyl iodide 5.3% 7.9%
trans cis
Addition of alkyl iodide to t-BuLi 44.0% <1%
Scheme 3.3 Bailey’s cyclization of a secondary alkyllithium
43
Tin-lithium exchange has also been used in intramolecular carbolithiations (Scheme 3.4).62-
64 The reaction between organolithiums and stannanes is very fast. Aryllithiums, vinyllithium
and α-heterosubstituted organolithiums can be formed through tin-lithium exchange.
O
RSnBu
3
n-BuLi
-78
o
CO
RLi
-78
o
C to 0
o
C
O
R
54% (cis:trans =11:1)
O
RSnBu
3
n-BuLi
-78
o
C
O
R
87% (cis:trans =10:1)
OMe
O
RLi
OMe
N
SnBu
3
n-BuLi
-78
o
CN
Li
N
H
87% (96% ee)
-78
o
C to 0
o
C
-78
o
C to 0
o
C
Scheme 3.4 Tin-lithium exchange in intramolecular carbolithiation.
Selenium-lithium exchange can also be used but only when the resultant organolithium is
more stable than the starting organolithium. This method is usually used to form stabilized
organolithiums, such as allyllithiums and benzyllithiums, which can not be generated by
halogen-lithium exchange due to the coupling between the newly formed organolithium and the
unreacted halide (Scheme 3.5).65
SeMe
Ph Li
Ph Ph
t-BuLi
THF, -78 oC
30 min
85% (98:2)
Scheme 3.5 Selenium-lithium exchange
44
Recently, an extensive investigation of the use of reductive lithiation of phenyl thioethers
for the production of the alkyl- and vinyllithium substrates for intramolecular carbolithiation
appeared from this laboratory.1
The reductive lithiation of phenylthioethers by lithium naphthalenide (LN), independently
discovered by Cohen66-68 and Screttas69,70 in the late 70’s, has proved to be a general method to
produce organolithiums. While lithium naphthalenide was used in the early work, two
alternative reducing agents, lithium(1-dimethylamino)naphthalenide (LDMAN)71 and lithium
4,4’-di-tert-butylbiphenylide (LDBB),72 were soon introduced (Figure 3.1). All three
radical-anion reducing agents are widely used today.
Li
N
Li
Li
Figure 3.1 Radical anion reducing agents
As shown in Scheme 3.6, the mechanism73 of reductive lithiation of phenylthioethers is
generally believed to involve the reversible transfer of an electron from the reducing agent (LN,
LDMAN, or LDBB) to the substrate. The C-S bond in the radical-anion intermediate is
homolytically cleaved to form a carbon radical. This carbon radical receives electron from
another equivalent of reducing agent and forms carbanion. The formation of the carbon radical
is the rate-determined step and the rate is largely determined by the stability of this radical.
R
1
R
2
SPh
R
3
e
-
R
1
R
2
SPh
R
3
-PhS
-
slow
C
R
1
R
2
R
3
e
-
fast
C :
R
1
R
2
R
3
..
Scheme 3.6 Mechanism of reductive lithiation
Lithium
naphthalenide (LN)
Lithium
1-(dimethylamino)naphthalenide
(LDMAN)
Lithium
4,4’-di-tert-butylbiphenylide
(LDBB)
45
A few examples of intramolecular carbolithiation of nonconjugated alkyllithiums prepared
by reductive lithiation were published prior to or almost concurrently with the recent
publication1a from this laboratory (Scheme 3.7). Broka62,63 reported two examples of reactions
that produce tetrahydrofuran and pyrrolidine with good selectivity and fairly good yield.
Rychnovsky74 reported an example of cyclization of the tertiary carbanion generated by
reductive lithiation of a nitrile substitutent.
OSPh
Li-Naphthalenide
THF, 0
o
C, 1.5h O
52% (trans:cis =7:1)
RR
NSPh
n-hexyl
n-Bu
Li-Naphthalenide
THF, 0
o
C, 1.5h
56% (cis:trans=6:1)
N
n-Bu
n-hexyl
O
CN
LDBB
THF, -78
o
C
10 min
O
Li
O
HLi
O
HCOOMe
1. CO
2
, -78
o
C
2. CH
2
N
2
Scheme 3.7 Examples of earlier intramolecular carbolithiations by reductive lithiation
3.1.2. Lithium oxyanion effect in accelerating and exerting stereocontrol over
intramolecuar carbolithiation reactions
Recent work from our laboratory has greatly advanced cyclizations to 5-membered rings by
intramolecular carbolithiation. Organolithiums have been shown to be easily available by
reductive lithiation of phenyl thioethers by aromatic radical anions. Intramolecular
carbometallations were found to be greatly accelerated in the presence of a suitably placed
oxyanionic group. The oxyanionic group also controls the stereochemistry of the products.
46
Shown in Scheme 3.81 is the first example of a tertiary carbanionic cyclization. It should be
noted that this cyclization was performed at a far lower temperature than that at which such
cyclizations usually occur. Owing to the unique properties of sulfur, rapid construction of the
substrates from the thioacetal of acetone is possible.
SPh
SPh
1. LDBB, -78
o
C
2. CuBr·Me
2
S
3. Br
SPh
LDBB
-45
o
C, 2 h
SC
6
H
4
OMe
94%89%
(MeOC
6
H
4
S)
2
Li
Scheme 3.8 Intramolecuar carbolithianion reactions with a tertiary organolithium
Reductive lithiation using appropriately placed allylic or homoallylic alcohol groups on the
alkene has two major advantages. First, the allylic or homoallylic oxyanionic groups on the
alkene have a powerful accelerating effect on the intramolecular carbometalation. If we compare
the reaction in Scheme 3.8 that takes 2 h at -45 °C with reaction (c) in Scheme 3.9 that takes 1 h
at -78 °C,1 we find that an allylic lithium oxyanionic group greatly accelerates the cyclization of
an unconjugated alkyllithium and/or allows lower temperatures to be used. In the presence of the
allylic lithium oxyanionic group, even the cyclizations of primary alkyllithiums occur in THF at
-78 °C.1 Second, using this technology the cyclization product contains the useful alcohol
functionality in addition to the lithiomethyl group.
RSPh
O
R' R'
Li ROH
R'
SPhR'
1. BuLi
2. LDBB, -78
o
C, time
3. (PhS)
2
ROH
R'
R'
SPh
(a) time=12 h, R=R'=H 73%
(b) time=12 h, R=Me, R'=H 81%
(c) time=1 h, R=H, R'=Me 86%
(d) time=1 h, R=R'=Me 80%
Scheme 3.9 Intramolecuar carbolithianion reactions with oxyanionic groups
47
In Scheme 3.9, the most unexpected phenomenon is that the single diastereomers isolated in
all four cases have the oxygen function and the function derived from the CH2Li group on the
opposite side of the cyclopentane ring. The directing effect of the lithium oxyanionic group is
complete. It is also in the opposite sense to that in the reaction of intramolecular allylmetallic
carbometalations metallo-ene cyclization.1
Although the allylic lithium oxyanionic group shown in Scheme 3.10,1 is in a different
position as comparing to that in Scheme 3.9, it was found to be equally effective at promoting
cyclization. In Scheme 3.9, the allylic lithium oxyanionic group is positioned such that it is a
ring substituent in the cyclized organolithium, whereas in Scheme 3.10 the alcohol function is
positioned exo to the ring.
HO 1. BuLi/TMEDA, hexanes, -78
o
C
2. 25
o
C, 12 h
3. PhS(CH
2
)
3
Br, THF, 25
o
C, 12 h
HO
SPh
1. BuLi, -78
o
C
2. LDBB, -78
o
C
3. -78
o
C, 12 h then H
2
O
53% 71%
OH
Scheme 3.10 Intramolecuar carbolithianion reaction with an oxyanionic group exo to the ring
A homo allylic lithium oxyanion placed exo to the forming ring shown in Scheme 3.111 is
even more effective in accelerating the cyclization than the allylic lithium oxyanionic group
shown in Scheme 3.10. The stereochemistry of the product 49 is still trans. On the other hand,
if the homo allylic lithium oxyanion was a substituent on the forming ring, an apparent
retardation of cyclization was observed.
1. LDE, HMPA, THF, -78 oC~0 oC
2. PhS(CH2)3Br
3. LiAlH4SPh
1. BuLi, -78 oC
2. LDBB, -78 oC
3. -78 oC, 1 h then (PhS)2
HOH2C
SPh
64% 64%
COOH CH2OH
49
Scheme 3.11 Intramolecuar carbolithianion reaction with a homo allylic oxyanionic group

48
The versatility of intramolecular carbolithiation for cyclizations is greatly increased by
taking advantage of the combined powers of reductive lithiation of phenyl thioethers in substrate
preparation and of the accelerating and remarkable directing effect of allylic and homoallylic
lithium oxyanionic groups.
3.2. Results and Discussions
Based on the previous work described above, we developed procedures for the asymmetric
synthesis of fused functionalized pyrrolidines, namely pyrrolizidines such as 52 and 55. The
pyrrolizidine skeleton is found in a large class of alkaloids.80 A previous worker from this
laboratory, Yixiong Lei, had done preliminary work on the cyclization to compound 52.
As shown in Scheme 3.12, compound 1 was obtained by asymmetrically deprotonation of
N-Boc-pyrrolidine 50 by s-BuLi in the presence (-)-sparteine. This general asymmetric
deprotonation method was developed by Beak’s group.81,82
N
Boc
1. s-BuLi/(-)-sparteine
2. allyl iodide
63%
N
Boc
50 1
Scheme 3.12 Procedure to synthesize compound 1 through Beak’s method
As seen in Scheme 3.13,81 (-)-sparteine plays a crucial role in asymmetric deprotonation.
When (-)-sparteine is present in the reaction, high enatioselectivity is observed.
49
N
Boc
1. s-BuLi/(-)-sparteine
2. E
+
N
Boc
E
N
N
H
H
(-)-Sparteine
76% ee. 96% E= Si(CH
3
)
3
75% ee. 90% E= (C
6
H
5
)
2
COH
68% ee. 88% E= CO
2
H
88% ee. 94% E= CH
3
70% ee. 94% E= Sn(C
4
H
9
)
3
60% ee. 59% E= CH
2
OH
12% ee. 91% E= (CH
3
)
2
COH
50
Scheme 3.13 Asymmetric deprotonation of N-Boc-pyrrolidine 50
A more recent publication reported a high yield method of preparation of compound 1
(Scheme 3.14).83 We adopted this method in our study and achieved a much better yield with
acceptable enantioselectivity. Beak deprotonation of N-Boc-pyrrolidine 50 in diethyl ether
followed by treatment with CuCN.2LiCl gave lithium alkylcyanocuprate. The reaction of N-
Boc-2-pyrrolidinylcuprates with allyl bromide gave compound 1 in high yield with acceptable
enantioselectivity.
N
Boc
1. s-BuLi, Et
2
O, -78
o
C
(-)-sparteine
2. CuCN·2LiCl, THF
N
Boc
CuCN·Li
50
Allyl bromide N
Boc
195%
ee. 89:11
TFA/CH
2
Cl
2
(1:1, excess)
0
o
C, 1 h
96%
N
H
51
N
PhS
1. LDBB
2. H
2
O
N
H
52 2
PhSH
(CH
2
O)
n
100℃
81%
Scheme 3.14 Asymmetric synthesis of pyrrolizidine 52
When compound 1 is treated with TFA in dichloromethane (1:1 volume ratio) for 1 h, it
undergoes a de-protection reaction and results in compound 51 in almost quantitative yield,84
50
fortunately requiring no further purification since compound 51 is volatile. In toluene,
compound 51 reacts with thiophenol and formaldehyde85,86 at 100 oC to produce compound 2.
In this study, different approaches for the preparation of compound 2 were tested. One is
the successful method that we have described above. The other potential approach to prepare
compound 2 could be the SN2 reaction by treating compound 51 with chloromethyl phenyl
sulfide.87,88 To test the feasibility of this approach, several conditions, for the model reaction
shown in Scheme 3.15, were investigated. However, none of them succeeded. One possible
explanation for the failure is that chloromethyl phenyl sulfide might instantly react with the
moisture in air to give PhSOH when the cap of the container is opened. Another explanation is
that the chloromethyl phenyl sulfide that we purchased and used in our reaction was not
completely dry. In the NMR spectra of the products obtained from reactions in Scheme 3.15, we
did observe PhSOH as the main product.
N
H
PhSCl+
N
PhS
THF
Additive
Additives:
(a) K
2
CO
3
(c) Bu
4
NI
(b) NaOH (d) K
2
CO
3
, LiI
Scheme 3.15 Synthesis of 1-(phenylthiomethyl)pyrrolidine through SN2 reaction
Compound 2 undergoes a reductive lithiation reaction with LDBB to form a primary
organolithium. This primary organolithium in turn appears undergo an intramolecular
cyclization to produce compound 52. (Scheme 3.16) Compound 52 could not be readily purified
but a picrate salt was characterized and the 1H and 13C spectra were consistent with the salt of the
pyrrolizidine 52. The work will require subsequent refinement.
51
N
PhS
2
LDBB
NLi
H
N
H
52
H
2
O
1. -78
o
C, 2 h
2. -35
o
C, 12 h
N
H
Li
Scheme 3.16 Intramolecular cabanionic cyclization
Allyl alcohol 6 was also utilized in preliminary experiments for a cyclization by similar
processing. Compound 6 was synthesized as mentioned previously by asymmetrically reducing
the ester with DIBAL and ZnCl2 followed by addition of vinyl magnesium bromide (Scheme
3.17).
N
Boc
1. DIBAL, ZnCl
2
-78
o
C, 30 min
2.
62%
PhSH
(CH
2
O)n
82%
1. BuLi
2. LDBB, -78
o
C
3. HCl
N
H
MgBr N
Boc OH
OH
56
55
COOMe
HTFA/CH
2
Cl
2
(1:1, excess)
0
o
C, 1h
98%
N
HOH
53
H
54
N
PhS OH
H
Scheme 3.17 Asymmetric synthesis of pyrrolizidinol 55
Similar to compound (R)-tert-butyl 2-allylpyrrolidine-1-carboxylate 51, compound 53 was
made by treating compound 6 with TFA in dichloromethane (1:1 volume ratio) for 1 h.
Fortunately, no rearrangement of the acid-sensitive allyl alcohol function occurs during the N-
deprotection.
52
Compound 53 reacts with thiophenol and formaldehyde at 100 oC in toluene to afford
compound 54 which upon alcohol deprotonation and reductive lithiation undergoes an allylic
oxyanionic accelerated reductive lithiation and cyclization.
As above, 55 could not be purified to the point of characterization. Flash chromatography
was tried without success. Compound 55 did not elute from a silican column pre-washed with
triethyl amine, and it could not be separated efficiently with a basic aluminium oxide column.
However, the 1H NMR of the crude product 55 has a methyl doublet that was consistent with the
pyrrolizidinol 55. This work will require subsequent refinement.
As shown in Scheme 3.18, in order to obtain compound 57, the 2 diastereomers of 6 but
possessing the enantiomeric configuration, several efforts have been made without success.
These efforts include: (a) Repeating the reported procedure of using s-BuLi to asymmetrically
remove the proton on the pyrrolidine ring followed by the addition of DMF to generate aldehyde
56.82 However, when vinyl magnesium bromide is used to react with 56, although the yield of
compound 57 is good. But the ratio of diastereoisomers is nearly 1:1. (b) treating compound 56
with a vinyl zinc chloride in hopes of a better diastereoselectivity.6,15 The yield was good and
the selectivity was improved as a major and a minor diastereoisomer were obtained. However,
the selectivity is still not as good as the DIBAL reduction method starting from N-Boc-L-proline
methyl ester. (c) Treating the lithium compound with CeCl390 followed by acrolein. The desired
products were not produced.
53
N
Boc
1. s-BuLi/(-)-sparteine
2. DMF
60%
N
Boc
CHO
MgBr
82%
N
Boc OH
50 56 57
N
Boc
CHO
MgBr
N
Boc OH
56 57
ZnCl
2
N
Boc
s-BuLi/(-)-sparteine
N
Boc
Li
50
O
CeCl
3
N
Boc OH
57
(a)
(b)
(c)
Scheme 3.18 Unsuccessful methods to obtain Compound 57
3.3. Conclusions
Previous study in our lab has demonstrated the potential for very greatly extending the
versatility of the cyclization method (1) by generating the organolithiums by reductive lithiation
of phenyl thioethers with aromatic radical-anions such as lithium 1(-
dimethylamino)naphthalenide (LDMAN) and 4,4'-di-tert-butylbiphenylide (LDBB) and (2) by
using allylic or homoallylic alcohol groups on the receiving alkene. In this current study, we
developed a new method for asymmetric synthesis of fused functionalized pyrrolidines, namely
pyrrolizidines. With this new method, we largely expanded the scope of reductive lithiation in
organolithium synthesis. The allylic or homoallylic oxyanionic group on the alkene greatly
accelerates the reaction and leads in most cases to completely stereoselective cyclization at
-78 oC. Furthermore, because the pyrrolizidine skeleton is found in a large class of biological
alkaloids, these pyrrolizidines may lead to alkaloids with high stereoselectivity.
54
3.4. Experimental
General Considerations: see p. 34.
Lithium 4,4'-di-t-butylbiphenylide (LDBB).1 To a flame-dried three-neck round-bottom
flask, equipped with a glass-coated stirring bar, argon inlet and rubber septum was added 4,4'-di-
tert-butylbiphenyl (DBB) (4.00 g, 15.0 mmol). Lithium ribbon was prepared by scraping the
dark oxide coating off of the surface while it was immersed in mineral oil. The shiny metal was
dipped in hexanes in order to remove the oil and then weighed (104 mg, 15.0 mmol) in a tared
beaker containing mineral oil. The metal was sliced into small pieces while it was still immersed
in mineral oil. The lithium pieces were dipped again in hexanes prior to addition to the flask.
THF (40 mL) was added to the DBB/lithium mixture via syringe. The reaction mixture was
stirred at room temperature for about 5 min until a dark-blue color appeared on the lithium
surface and it was then cooled to 0 °C and stirred for 5 h. The resulting dark-blue solution of
LDBB was ready for use in reductive lithiation.
(R)-tert-butyl 2-allylpyrrolidine-1-carboxylate (1).
Procedure (a).81 To (-)-sparteine (23.0 mL, 100 mmol) and N-Boc-pyrrolidine (8.56 g, 50
mmol) in Et2O (135 mL) at -78 °C was added s-BuLi (76.9 mL of 1.4 M solution in cyclohexane,
100 mmol). The reaction mixture was stirred for 5 h at -78 °C and then allyl iodide (11.4 mL,
125 mmol) was added. The mixture was allowed to slowly warm to room temperature overnight.
Workup consisted of addition of water (80 mL), extraction of the aqueous layer with Et2O (3 x
80 mL), extraction of the combined Et2O extracts with 5% phosphoric acid (H3PO4) (80 mL),
55
drying over anhydrous magnesium sulfate (MgSO4), filtration, and concentration in vacuo. The
crude product was purified by flash chromatography (10% ethyl acetate in hexanes) to yield 6.64
g (63%) of the title product as a yellow oil. 1H NMR (CDCl3) δ 5.73 (m, 1 H), 5.08-5.00 (m, 2
H), 3.80 (m, 1 H), 3.32-3.30 (m, 2 H), 2.47 (m, 1 H), 2.11 (m, 1 H), 1.89-1.66 (m, 4 H), 1.46 (s,
9 H); 13C NMR (CDCl3) δ 154.5, 135.21, 116.90, 78.92, 56.69, 46.41, 38.59, 29.68, 28.41 (3 C),
23.17.
Procedure (b).83 N-Boc pyrrolidine (0.87 g, 5.0 mmol) was dissolved in freshly distilled
Et2O (15 mL) along with (-)-sparteine (1.76 g, 7.5 mmol). The reaction mixture was cooled to -
78 °C and s-BuLi (5.77 mL of a 1.3 M solution in cyclohexane/hexanes, 7.5 mmol) was added
dropwise by syringe. The resulting solution was stirred at -78 °C for 1 h. Then a solution
containing CuCN (0.67 g, 7.5 mmol) and LiCl (0.64 g, 15 mmol) in THF (20 mL) was added
dropwise by syringe. The mixture was allowed to stir at -78 °C for 30 min before the addition of
allyl bromide (0.91 g, 7.5 mmol). The reaction mixture was allowed to warm quickly to -50 °C
and then slowly to room temperature overnight. The reaction mixture was diluted with Et2O
(100 mL) and the reaction was quenched with 5% phosphoric acid (H3PO4) (125 mL). The
layers were separated and the organic layer was dried (MgSO4) and concentrated in vacuo to
give an yellow oil that was purified by column chromatography (10% ethyl acetate in hexanes)
to yield 1.0 g (95%) of the title product as a yellow oil. 1H NMR (CDCl3) δ 5.74 (m, 1 H), 5.06-
5.00 (m, 2 H), 3.75 (m, 1 H), 3.36-3.28 (m, 2 H), 2.47 (m, 1 H), 2.09 (m, 1 H), 1.94-1.69 (m, 4
H), 1.46 (s, 9 H).
(R)-2-allyl-1-((phenylthio)methyl)pyrrolidine (2).63 A solution of the 51 (0.75 g, 6.7
mmol) in 7 mL of toluene was treated with PhSH (0.74 g, 6.7 mmol), paraformaldehyde (0.20 g,
56
6.7 mmol), and a few crystals of 4,4'-methylenebis(2,6-di-tert-butylphenol). The mixture was
stirred overnight at 100 °C. The volatiles were removed under reduced pressure, leaving the
crude product (1.3 g, 81%). 1H NMR (CDCl3) δ 7.49 (m, 1 H), 7.30-7.13 (m, 4 H), 5.66 (m, 1 H),
5.03-4.91 (m, 2 H), 4.71 (d, J=13 Hz, 1 H), 4.64 (d, J=13 Hz, 1 H), 2.95-2.78 (m, 3 H), 2.24-2.14
(m, 2 H), 1.94-1.81 (m, 2 H), 1.69 (m, 1 H), 1.50 (m, 1 H).
(R)-2-allylpyrrolidine (51).84 A solution of 1 (2.03 g, 9.63 mmol) in CH2Cl2 (8 mL) was
treated with trifluoroacetic acid (8 mL) at 0 °C and the mixture was stirred for 1 h at the same
temperature. After dilution with CH2Cl2 (60 mL), the pH of the solution was adjusted to 7–8
using saturated aqueous NaHCO3 solution. The crude product was extracted with CH2Cl2 (10 x
15 mL) and dried with K2CO3 and concentrated in vacuo to give 51 (1.02 g, 96%) as a yellow oil.
The product was used for the next step without further purification. 1H NMR (CDCl3) δ 5.75 (m,
1 H), 5.21-5.11 (m, 2 H), 3.50 (m, 1 H), 3.29-3.20 (m, 2 H), 2.54-2.40 (m, 2 H), 2.12-1.95 (m, 3
H), 1.71 (m, 1 H); 13C NMR (CDCl3) δ 132.47, 118.99, 59.39, 44.70, 36.19, 29.87, 23.42.
(R)-2-methyl-hexahydro-1H-pyrrolizine (52).1 Freshly prepared LDBB (16.9 mmol in 45
mL of THF) at -78 °C was cannulated to a flask containing compound 2 (0.79 g, 3.39 mmol) in
22 mL of THF at -78 °C. The reaction mixture was stirred at -78 °C for 2 h and then at -35 °C
overnight. The reaction was quenched with 2 M HCl solution (20 mL) at -78 °C. After the
reaction mixture had been further stirred for 10 minutes, the aqueous layer was washed with
ether (3 x 30 mL). The resulting aqueous layer was neutralized with 2 M NaOH solution and
extracted with CH2Cl2 (3 x 20 mL). The combined organic layer was dried over anhydrous
K2CO3 and concentrated in vacuo to give a yellow oil (0.36 g). The yellow oil was crystallized
57
with picric acid in ethanol to form a yellow solid. NMR data of the picrate salt of compound 52:
1H NMR (CDCl3) δ 8.92 (s, 2 H), 2.68-2.12 (m, 5 H), 1.97-1.55 (m, 7 H), 1.14 (d, J= 6Hz, 3 H);
13C NMR (CDCl3) δ 160.61, 152.48, 141.22 (2 C), 125.95 (2 C), 68.10, 61.19, 54.30, 39.42,
34.76, 29.93, 24.23, 15.21.
(S)-1-((S)-pyrrolidin-2-yl)prop-2-en-1-ol (53). A solution of 6 (0.12 g, 0.53 mmol) in
CH2Cl2 (1 mL) was treated with trifluoroacetic acid (1 mL) at 0 °C and the mixture was stirred
for 1 h at the same temperature. After dilution with CH2Cl2 (5 mL), the mixture was washed
with 3.5mL 2 M NaOH solution. The crude product was extracted with ether (10 x 5 mL) and
the extract was dried with anhydrous K2CO3 and concentrated in vacuo. The product was used
for the next step without further purification (0.066 g, 98%). 1H NMR (CDCl3) δ 5.80 (ddd, J=6,
10, 17 Hz, 1 H), 5.27 (m, 1 H), 5.12 (m, 1 H), 3.25-2.86 (m, 4 H), 1.78-1.66 (m, 4 H).
(S)-1-((S)-1-((phenylthio)methyl)pyrrolidin-2-yl)prop-2-en-1-ol (54). A solution of the
53 (0.25 g, 2 mmol) in 3 mL of toluene was treated with PhSH (0.22 g, 2 mmol),
paraformaldehyde (0.060 g, 2 mmol), and a few crystals of 4,4'-methylenebis(2,6-di-tert-
butylphenol). The mixture was stirred overnight at 100 °C. The volatiles were removed under
reduced pressure, leaving the crude product (0.41 g, 82%). 1H NMR (CDCl3) δ 7.52-7.16 (m, 5
H), 5.72 (m, 1 H), 5.08-4.94 (m, 2 H), 4.67 (m, 1 H), 3.83 (d, J=12 Hz, 1H), 3.76 (d, J=12 Hz,
1H), 3.18-2.49 (m, 3 H), 2.00-1.65 (m, 4 H).
(S)-2-methyl-hexahydro-1H-pyrrolizin-1-ol (55). To a stirred solution of compound 54
(0.41 g, 1.64 mmol) in 6 mL of THF at -78 °C under argon, was added n-BuLi (1.1 mL of 1.6 M
58
solution in hexanes, 1.73 mmol) via syringe. The reaction mixture was stirred at -78 °C for 1
h. Freshly prepared LDBB (4.93 mmol in 14 mL THF) at -78 °C was cannulated to the reaction
flask. The reaction mixture was stirred at -78 °C overnight. The reaction was quenched with 2
M HCl solution (10 mL) at -78 °C. After the reaction mixture had been further stirred for 10
minutes, the aqueous layer was washed with ether (3 x 15 mL). The resulting aqueous layer was
neutralized with 2 M NaOH solution and extracted with CH2Cl2 (3 x 10 mL). The combined
organic layer was dried over anhydrous K2CO3 and concentrated in vacuo to give a yellow oil
(0.18 g, 79%). 1H NMR (CDCl3) δ 3.37 (m, 1 H), 2.02-1.68 (m, 5 H), 1.44-1.43 (d, J=8 Hz, 3 H),
1.29-0.83 (m, 5 H).
59
APPENDIX A
B3LYP/6-31+G(d) Cartesian coordinates (Å) for optimized stationary points.
R1 R2
C 1.840579 -0.539625 3.377844 C -0.769278 0.361858 3.566714
C 3.203111 0.143561 3.234873 C -2.286279 0.377021 3.846094
C 2.192671 -0.024383 1.004856 C -1.954499 -0.857134 1.816142
N 1.188887 -0.236552 2.086229 N -0.643172 -0.272516 2.228872
C -0.099739 0.115805 1.97054 C 0.531078 -0.29175 1.587729
O -0.630868 0.515133 0.900854 O 0.702349 -0.604276 0.374635
O -0.780484 0.037774 3.114103 O 1.546389 0.061917 2.38007
C -2.227857 0.377511 3.24676 C 2.97562 0.097669 1.953912
C -2.450696 1.852169 2.902655 C 3.416583 -1.299359 1.50996
C -3.068282 -0.56916 2.388499 C 3.183095 1.15841 0.872857
C -2.471054 0.123021 4.736424 C 3.677096 0.502674 3.253026
C 3.545306 -0.067968 1.751682 C -2.93876 0.10894 2.480266
H 1.236725 -0.164577 4.203219 H -0.203769 -0.202961 4.31482
H 1.94424 -1.627051 3.489281 H -0.342602 1.367865 3.516984
H 3.952474 -0.279697 3.911453 H -2.60172 1.325148 4.292479
H 3.105352 1.211916 3.464793 H -2.548411 -0.420897 4.550854
H 2.01604 0.959468 0.555859 H -2.02865 -1.839232 2.307967
C 2.098445 -1.085344 -0.116228 C -2.174445 -1.096345 0.305169
H -1.78764 2.490989 3.497115 H 3.20238 -2.036734 2.291849
H -2.280062 2.051501 1.843497 H 2.921719 -1.605837 0.586837
H -3.48507 2.123131 3.143727 H 4.498908 -1.294322 1.337456
H -2.919625 -0.397207 1.321852 H 2.704178 0.884195 -0.067327
H -2.826492 -1.613148 2.616643 H 2.788775 2.126878 1.198389
H -4.128585 -0.410534 2.616843 H 4.257466 1.274196 0.68902
H -2.259758 -0.920385 4.993105 H 3.324028 1.479818 3.599154
H -1.837158 0.769358 5.352893 H 3.493145 -0.233727 4.042372
H -3.517789 0.333231 4.980565 H 4.757377 0.566117 3.084409
H 4.239714 0.6758 1.354811 H -3.005636 1.020985 1.876541
H 4.009066 -1.051031 1.606123 H -3.936329 -0.326712 2.556997
H 2.324665 -2.081736 0.314614 O -3.546708 -1.462879 0.233695
C -2.121275 -0.700909 -1.51746 A -0.239287 0.086844 -1.198803
O 3.167531 -0.733162 -0.992498 O -1.874642 -0.002609 -0.4669
C 3.367463 -1.658863 -2.052076 C 0.109264 -1.322147 -2.568964
O 0.882277 -1.145595 -0.738682 C 0.273231 2.005992 -1.402842
A -0.374919 0.100325 -0.986088 C 1.531268 -1.908345 -2.733294
C 0.163869 1.87664 -1.72871 C 2.535309 -0.851387 -3.222755
60
C -2.286973 -1.270084 -2.947744 C 1.546107 -3.122138 -3.680824
H 3.582722 -2.665492 -1.65938 C -0.807497 2.936196 -2.008396
H 2.491134 -1.711499 -2.70758 C -0.366978 4.411443 -1.989118
H 4.230633 -1.301552 -2.619444 C -1.188381 2.518705 -3.438745
C -1.42386 -2.525137 -3.160241 H -0.310508 2.542639 -4.099729
C -3.759088 -1.580525 -3.275114 H -1.599683 1.501681 -3.470493
C 0.011339 2.138229 -3.245193 H -1.944136 3.192021 -3.864251
C 0.908379 1.207822 -4.078248 H -0.218493 -0.934436 -3.550122
C 0.296302 3.607548 -3.607153 H -0.58393 -2.153526 -2.353589
H -0.362395 4.286825 -3.05099 H 1.193816 2.105664 -2.00398
H 1.333337 3.8734 -3.357588 H 0.537855 2.398219 -0.405659
H 0.150198 3.799096 -4.679577 H 1.875577 -2.262763 -1.748048
H -2.884403 0.084004 -1.365944 H 2.598891 0.002204 -2.536723
H -2.383885 -1.499199 -0.800861 H 2.234917 -0.458884 -4.204195
H -0.43517 2.630336 -1.18617 H 3.544042 -1.272758 -3.328117
H 1.210478 2.091282 -1.447146 H 0.869902 -3.909804 -3.325051
H -1.950679 -0.508999 -3.669841 H 1.214215 -2.830962 -4.687217
H -1.742991 -3.32948 -2.482356 H 2.551595 -3.557009 -3.772083
H -0.364829 -2.33189 -2.95438 H -1.713367 2.853247 -1.388935
H -1.511601 -2.901446 -4.188288 H 0.532997 4.555485 -2.603748
H -4.156368 -2.338287 -2.584814 H -0.127543 4.740125 -0.969482
H -4.384457 -0.683656 -3.178497 H -1.149964 5.075537 -2.381491
H -3.876623 -1.964837 -4.298143 C -3.955902 -1.876353 -1.063189
H -1.033257 1.932476 -3.529927 H -1.58507 -1.982848 -0.004022
H 0.670276 0.15078 -3.905199 H -3.362503 -2.737447 -1.408632
H 1.965487 1.353726 -3.816999 H -3.861191 -1.059946 -1.787882
H 0.798535 1.40096 -5.153744 H -5.004241 -2.173486 -0.977457
H1 H2
C -1.368589 -0.534744 -2.171096 C -1.181075 0.205127 2.391143
C -2.504619 0.398099 -1.741768 C -2.549511 0.3465 1.704703
C -0.785993 0.43602 0.013489 C -1.038728 -0.501457 0.037835
N -0.268931 -0.132013 -1.266298 N -0.280019 -0.28059 1.308479
C 1.001398 -0.050751 -1.642404 C 1.023333 -0.387456 1.510854
O 1.954923 0.413533 -0.964861 O 1.916315 -0.636363 0.64996
O 1.256059 -0.488636 -2.890476 O 1.396154 -0.210597 2.793852
C -2.311027 0.514639 -0.220411 C -2.21124 0.469817 0.21212
H -1.063342 -0.42183 -3.211511 H -1.183144 -0.500566 3.226814
H -1.618062 -1.58835 -1.992659 H -0.798891 1.163178 2.756853
H -3.487747 0.004337 -2.017785 H -3.104012 1.206017 2.093149
H -2.383126 1.374082 -2.227474 H -3.157711 -0.548513 1.882778
H -0.34508 1.429359 0.153967 H -1.411868 -1.53508 0.064612
C -0.409388 -0.432318 1.23726 C -0.232005 -0.328573 -1.274319
H -2.72762 1.432633 0.199415 H -1.876899 1.482238 -0.040999
H -2.798734 -0.32414 0.290322 H -3.043054 0.20958 -0.444213
H -0.901236 -1.421392 1.142952 O -1.235105 -0.282038 -2.274567
O -1.010277 0.260338 2.324689 Al 2.291867 0.530421 -0.86605
C -0.888168 -0.412567 3.572044 O 0.568169 0.792296 -1.264852
O 0.936271 -0.633084 1.389209 C -0.713592 -0.27281 -3.599063
61
Al 2.3215 0.393301 0.944287 H 0.365628 -1.24509 -1.450078
H -1.351933 -1.410585 3.526695 H -0.08372 -1.15726 -3.779991
H 0.161749 -0.519306 3.866073 H -0.126346 0.633023 -3.785495
H -1.417661 0.198839 4.306751 H -1.575539 -0.299388 -4.270078
H 2.236124 1.933294 1.353057 H 2.36979 -0.239659 2.811418
H 3.686715 -0.409218 1.078254 H 3.081757 -0.423444 -1.866392
H 2.215475 -0.394026 -3.030883 H 2.962524 1.830356 -0.233865
M1 M2
C 2.345521 -0.417838 1.728592 C 2.611153 0.229449 -1.293882
C 3.350977 0.482732 1.004305 C 3.50211 0.504394 -0.070218
C 1.310329 0.368198 -0.356915 C 1.467513 -0.461453 0.767473
N 1.077618 -0.101549 1.037858 N 1.35172 -0.309552 -0.714523
C -0.095595 0.026808 1.66082 C 0.287668 -0.532636 -1.48168
O -1.158599 0.441239 1.133516 O -0.876482 -0.820736 -1.085657
O -0.072501 -0.298866 2.960928 O 0.549601 -0.429927 -2.793955
C -1.326672 -0.214157 3.67384 C -0.570787 -0.560245 -3.69737
C 2.849009 0.475988 -0.449183 C 2.514807 0.605767 1.101813
H 2.254892 -0.218652 2.79639 H 3.047361 -0.488451 -1.994922
H 2.582032 -1.481057 1.592088 H 2.376717 1.142775 -1.849778
H 4.378448 0.118432 1.103375 H 4.102483 1.409016 -0.207373
H 3.309673 1.494363 1.426805 H 4.194671 -0.330841 0.09002
H 0.829345 1.345143 -0.478187 H 1.890379 -1.459037 0.956877
C 0.713651 -0.591425 -1.41408 C 0.14725 -0.360203 1.572337
H 3.150357 1.360101 -1.015189 H 2.027082 1.586773 1.13216
H 3.241682 -0.397656 -0.98317 H 2.975524 0.42177 2.073688
H 1.249987 -1.560756 -1.360409 O 0.592984 -0.256785 2.917635
C -3.551065 -0.947532 -0.288754 Al -1.969367 0.370991 0.026849
O 1.059308 0.033598 -2.649 O -0.640292 0.698158 1.188607
C 0.697056 -0.725059 -3.795054 C -3.436158 -0.792178 0.664417
O -0.624175 -0.826654 -1.27452 C -2.363714 1.964837 -1.082262
Al -1.9578 0.173383 -0.628162 H -4.11627 -0.24806 1.33609
C -2.182289 2.000168 -1.361317 H -3.083425 -1.675971 1.214698
H 1.187534 -1.711537 -3.785256 H -3.076889 1.750384 -1.891916
H -0.387901 -0.86716 -3.852285 H -1.464904 2.404278 -1.540083
H 1.042286 -0.159538 -4.664272 C -0.463834 -0.305057 3.86756
H -4.295022 -0.434434 0.338508 H -0.396338 -1.321066 1.482661
H -3.296094 -1.89409 0.209386 H -1.031381 -1.244609 3.778537
H -2.845931 2.611477 -0.731417 H -1.147517 0.542048 3.74212
H -1.236962 2.551503 -1.46597 H 0.003816 -0.259152 4.85431
H -2.639699 1.97146 -2.361523 H -4.049411 -1.161209 -0.170938
H -4.061954 -1.212651 -1.226225 H -2.817505 2.759782 -0.472027
H -2.069678 -0.86791 3.212604 H -1.033408 -1.543192 -3.587476
H -1.091651 -0.547354 4.684601 H -0.137173 -0.446255 -4.690844
H -1.691065 0.815348 3.680324 H -1.306608 0.223262 -3.505349
62
APPENDIX B
Certain O-H distances (Å) between the oxygen atom on the MeO group and the hydrogens
(HA and HB) on the 3 methylene group in H1, H2, M1, M2, R1 and R2.
NOCH3
H
O
O
O
RAl
RR
H
NH
H
O
O
O
R
Al
RR
OCH3
HAHBHB
HA
H1 H2 R=H
M1 M2 R=methyl
R1 R2 R=butyl
O- HA distance
Å
O- HB distance
Å
H1 2.771 2.970
H2 2.918 2.610
M1 2.335 3.210
M2 2.940 2.610
R1 2.750 2.940
R2 3.027 2.610
63
BIBLIOGRAPHY
1. (a) Deng, K.; Bensari, A.; Cohen, T. "Cyclization by intramolecular carbolithiation of alkyl-
and vinyllithiums prepared by reductive lithiation: Surprising stereochemistry in the lithium
oxyanion accelerated cyclization" J. Am. Chem. Soc. 2002, 124, 12106-12107. (b) Deng, K.;
Bensari-Bouguerra, A.; Whetstone, J.; Cohen, T. "Cyclization by Intramolecular Carbolithiation
of Alkyl- and Vinyllithiums Prepared by Reductive Lithiation of Phenyl Thioethers. High
Stereoselectivity in the Cyclization Accelerated by an Allylic Lithium Oxyanion" J. Org. Chem.
2006, 71, 2360-2372.
2. Ito, H.; Ikeuchi, Y.; Taguchi, T.; Hanzawa, Y.; Shiro, M. "Zirconium-Mediated, Highly
Diastereoselective Ring Contraction of Vinylmorpholine Derivatives from α-Amino Acids: An
Application to the Synthesis of (-)-Macronecine" J. Am. Chem. Soc. 1994, 116, 5469-5470.
3. Ibuka, T.; Taga, T.; Habashita, H.; Nakai, K.; Tamamura, H.; Fujii, N.; Chounan, Y.; Nemoto,
H.; Yamamoto, Y. "Syn-SN2' pathway in the reaction of certain g-(mesyloxy)-α,β-enoates with
RCu(CN)MgX.BF3 reagents. Importance of MgX and bulky R group upon the
diastereoselectivity" J. Org. Chem. 1993, 58, 1207-1214.
4. Ibuka, T.; Habashita, H.; Otaka, A.; Fujii, N.; Oguchi, Y.; Uyehara, T.; Yamamoto, Y. "A
highly stereoselective synthesis of (E)-alkene dipeptide isosteres via organocyanocopper-Lewis
acid mediation reaction" J. Org. Chem. 1991, 56, 4370-4382.
5. Newman, H. "Stereoselective synthesis of D-erythro-sphingosine" J. Chem. Soc. 1973, 95,
4098-4099.
6. Angle, S. R.; Breitenbucher, J. G.; Arnaiz, D. O. "An efficient stereoselective synthesis of D-
4,5-pipecolic esters" J. Org. Chem. 1992, 57, 5947-5955.
7. Andrew, R. G.; Conrow, R. E.; Elliott, J. D.; Johnson, W. S.; Ramezani, S. "Asymmetric
synthesis via acetal templates. 14. Preparation of enantiomerically pure (3S,4S)- and (3S,4R)-
statine derivatives" Tetrahedron Lett. 1987, 28, 6535-6538.
8. Claremon, D. A.; Lumma, P. K.; Phillips, B. T. "Organolithium addition to aldehyde
dimethylhydrazones: a highly diastereocontrolled synthesis of threo 2-amino alcohols, and
(1R,2R)-(-)-norpseudoephedrine" J. Am. Chem. Soc. 1986, 108, 8265-8266.
9. Danishefsky, S.; Kobayashi, S.; Kerwin, J. F., Jr. "Cram rule selectivity in the Lewis acid
catalyzed cyclocondensation of chiral aldehydes. A convenient route to chiral systems of
biological interest" J. Org. Chem. 1982, 47, 1981-1983.
10. Davidsen, S. K.; Chu-Moyer, M. Y. "Synthesis of stereochemically defined y[CH(alkyl)NH]
pseudopeptides" J. Org. Chem. 1989, 54, 5558-5567.
11. De Frutos, M. P.; Dolores Fernandez, M.; Fernandez-Alvarez, E.; Bernabe, M. ''Synthesis of
asymmetric (E)-α-(2-phenylcyclopropyl)glycines" Tetrahedron Lett. 1991, 32, 541-542.
12. DeCamp, A. E.; Kawaguchi, A. T.; Volante, R. P.; Shinkai, I. "Stereocontrolled addition of
propionate homoenolate equivalents to chiral α-amino aldehydes" Tetrahedron Lett. 1991, 32,
1867-1870.
64
13. Denis, J. N.; Correa, A.; Greene, A. E. "Direct, highly efficient synthesis from (S)-(+)-
phenylglycine of the taxol and taxotere side chains" J. Org. Chem. 1991, 56, 6939-6942.
14. Fujita, M.; Hiyama, T. "Highly diastereocontrolled reduction of ketones by means of
hydrosilanes. Practical synthesis of optically active 1,2-diols and 2-amino alcohols of threo or
erythro configuration" J. Am. Chem. Soc. 1984, 106, 4629-4630.
15. Hanson, G. J.; Lindberg, T. "Synthesis of new dipeptide analogs containing novel ketovinyl
and hydroxyethylidene isosteres via Grignard addition to chiral α-amino aldehydes" J. Org.
Chem. 1985, 50, 5399-5401.
16. Jouin, P.; Castro, B.; Nisato, D. "Stereospecific synthesis of N-protected statine and its
analogs via chiral tetramic acid" J. Chem. Soc. Perk. T. 1 1987, 1177-1182.
17. Kano, S.; Yokomatsu, T.; Shibuya, S. "Stereocontrolled convergent synthesis of
hydroxyethylene dipeptide isosteres by the reaction of α-amino aldehyde with alkoxytitanium
homoenolates" Tetrahedron Lett. 1991, 32, 233-236.
18. Kiyooka, S.; Nakano, M.; Shiota, F.; Fujiyama, R. "Dramatic changes in diastereoselectivity
with the quantity of titanium tetrachloride used in Lewis acid mediated reactions of allylsilane
with α-amino aldehydes" J. Org. Chem. 1989, 54, 5409-5411.
19. Kogen, H.; Nishi, T. "Diastereoselective epoxidation of cis-4-amino allylic alcohol with m-
chloroperbenzoic acid. An efficient synthesis of statine" J. Chem. Soc. Chem. Comm. 1987, 311-
312.
20. Mikami, K.; Kaneko, M.; Loh, T. P.; Terada, M.; Nakai, T. "Aminoaldehyde ene reaction:
stereoselective route to β-amino alcohols" Tetrahedron Lett. 1990, 31, 3909-3912.
21. Reetz, M. T.; Drewes, M. W.; Schmitz, A. "Stereoselective synthesis of β-amino alcohols
from optically active α-amino acids" Angew. Chem. 1987, 99, 1186-1188.
22. Rich, D. H.; Sun, E. T.; Boparai, A. S. "Synthesis of (3S,4S)-4-amino-3-hydroxy-6-
methylheptanoic acid derivatives. Analysis of diastereomeric purity" J. Org. Chem. 1978, 43,
3624-3626.
23. Stork, G.; Nakamura, E. "A simplified total synthesis of cytochalasins via an intramolecular
Diels-Alder reaction" J. Am. Chem. Soc. 1983, 105, 5510-5512.
24. Trost, B. M.; Ohmori, M.; Boyd, S. A.; Okawara, H.; Brickner, S. J. "Palladium-catalyzed
synthesis of macrocycles. A total synthesis of (-)-aspochalasin B'' J. Am. Chem. Soc. 1989, 111,
8281-8284.
25. Woo, P. W. K. "Chiral synthesis of statine" Tetrahedron Lett. 1985, 26, 2973-2976.
26. Boutin, R. H.; Rapoport, H. "α-Amino acid derivatives as chiral educts for asymmetric
products. Synthesis of sphingosine from α'-amino-α,β-ynones" J. Org. Chem. 1986, 51, 5320-
5327.
27. Fehrentz, J. A.; Castro, B. "An efficient synthesis of optically active α-(t-
butoxycarbonylamino)-aldehydes from α-amino acids" Synthesis 1983, 676-678.
28. Garner, P.; Park, J. M.; Malecki, E. "A stereodivergent synthesis of D-erythro-sphingosine
and D-threo-sphingosine from L-serine" J. Org. Chem. 1988, 53, 4395-4398.
29. Herold, P. "Synthesis of D-erythro- and D-threo-sphingosine derivatives from L-serine" Helv.
Chim. Acta. 1988, 71, 354-362.
30. Kozikowski, A. P.; Wu, J. P. "Synthesis of fluorine-containing isosteres of sphingosine as
inactivators of protein kinase C" Tetrahedron Lett. 1990, 31, 4309-4312.
31. Mori, K.; Funaki, Y. "Synthesis of sphingosine relatives. III. Synthesis of (4E,8E,2S,3R,2'R)-
N-2'-hydroxyhexadecanoyl-1-O-β-D-glucopyranosyl-9-methyl-4,8-sphingadienine, the fruiting-
65
inducing cerebroside in a basidiomycete Schizophyllum commune" Tetrahedron 1985, 41, 2379-
2386.
32. Nimkar, S.; Menaldino, D.; Merrill, A. H.; Liotta, D. "A stereoselective synthesis of
sphingosine, a protein kinase C inhibitor" Tetrahedron Lett. 1988, 29, 3037-3040.
33. Rittle, K. E.; Homnick, C. F.; Ponticello, G. S.; Evans, B. E. "A synthesis of statine utilizing
an oxidative route to chiral α-amino aldehydes" J. Org. Chem. 1982, 47, 3016-3018.
34. Tkaczuk, P.; Thornton, E. R. "Useful syntheses of erythro- and threo-N-oleoyl-D-
sphingosines (ceramides) and galactosylceramides (cerebrosides) from L-serine" J. Org. Chem.
1981, 46, 4393-4398.
35. Bolis, G.; Fung, A. K. L.; Greer, J.; Kleinert, H. D.; Marcotte, P. A.; Perun, T. J.; Plattner, J.
J.; Stein, H. H. "Renin inhibitors. Dipeptide analogs of angiotensinogen incorporating transition-
state, nonpeptidic replacements at the scissile bond" J. Med. Chem. 1987, 30, 1729-1737.
36. Comins, D. L.; Herrick, J. J. "A one-step conversion of esters to secondary alcohols with
lithium borohydride (LiBH4)/alkylmagnesium halide (RMgX)" Tetrahedron Lett. 1984, 25,
1321-1324.
37. Jackson, W. R.; Jacobs, H. A.; Matthews, B. R.; Jayatilake, G. S.; Watson, K. G.
"Stereoselective syntheses of ephedrine and related 2-amino alcohols of high optical purity from
protected cyanohydrins" Tetrahedron Lett. 1990, 31, 1447-1450.
38. Kano, S.; Yuasa, Y.; Yokomatsu, T.; Shibuya, S. "Aluminum-mediated one-pot conversion
of α-amino acid esters to threo 2-amino alcohols with high diastereoselectivity" Chem. Pharm.
Bull. 1989, 37, 2867-2869.
39. Meyers, A. I.; Comins, D. L. "N-Methylamino pyridyl amides (MAPA). II. An efficient
acylating agent for various nucleophiles and sequential addition to give unsymmetrical tert-
alcohols" Tetrahedron Lett. 1978, 5179-5182.
40. Polt, R.; Peterson, M. A.; DeYoung, L. "Aluminoxy acetals from α-amino esters: chirality
transfer via sequential addition of hydride and C-nucleophiles. 2-Amino alcohols and
sphingosines" J. Org. Chem. 1992, 57, 5469-5480.
41. Duhamel, P.; Duhamel, L.; Gralak, J. "Addition to carbonyls of α-(N,N-
dialkylamino)aldehydes and ketones. Asymmetric induction and effect of nitrogen substituents"
Tetrahedron Lett. 1972, 2329-2332.
42. Nichols, M. A.; McPhail, A. T.; Arnett, E. M. "Chelation of 2-substituted 1-lithoxides:
structural and energetic factors of relevance to synthetic organic chemistry" J. Am. Chem. Soc.
1991, 113, 6222-6233.
43. Reetz, M. T. "New synthetic methods. (44). Chelate- or nonchelate control in addition
reactions of chiral α- and β-alkoxycarbonyl compounds" Angew. Chem. 1984, 96, 542-555.
44. Harris, B. D.; Bhat, K. L.; Joullie, M. M. "Synthetic studies of detoxin complex. II.
Syntheses of detoxin B1 and B3" Heterocycles 1986, 24, 1045-1060.
45. Price, M. D.; Kurth, M. J.; Schore, N. E. ''Comparison of Solid-Phase and Solution-Phase
Chiral Auxiliaries in the Alkylation/Iodolactonization Sequence to g-Butyrolactones" J. Org.
Chem. 2002, 67, 7769-7773.
46. Yuste, F.; Ortiz, B.; Carrasco, A.; Peralta, M.; Quintero, L.; Sanchez-Obregon, R.; Walls, F.;
Garcia Ruano, J. L. "Synthesis of enantiomerically pure 2-amino alcohols from amino acids
mediated by sulfoxides" Tetrahedron: Asymmetry 2000, 11, 3079-3090.
47. Sibi, M. P.; Christensen, J. W. "Enantiospecific Synthesis of (-)-Slaframine and Related
Hydroxylated Indolizidines. Utilization of a Nucleophilic Alaninol Synthon Derived from
Serine" J. Org. Chem. 1999, 64, 6434-6442.
66
48. Parker, K. A.; O'Fee, R. "Halonium-initiated cyclizations of allylic urethanes: stereo- and
regioselectivity in functionalizing the olefinic bond" J. Am. Chem. Soc. 1983, 105, 654-655.
49. Frisch, M. J. T., G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.;
Montgomery, J. A., Jr.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.;
Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.;
Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima,
T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.;
Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.;
Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.;
Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.;
Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul,
A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.;
Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara,
A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; Pople,
J. A. Gaussian 03, Revision C.01. In Gaussian, Inc., Wallingford CT, 2004.
50. Bodor, A.; Banyai, I.; Toth, I. "H-1- and C-13-NMR as tools to study aluminium
coordination chemistry - aqueous Al(III)-citrate complexes" Coordin. Chem. Rev. 2002, 228,
175-186.
51. Bailey, W. F.; Ovaska, T. V. In Advances in Detailed Reaction Mechanisms; Coxon, J. M.,
Ed.; JAI Press: Greenwich, CT.; Vol. 3, 1994; p 251-273.
52. Mealy, M. J.; Bailey, W. F. "Synthesis of heterocycles by cyclization of unsaturated
organolithiums: a review" J. Organomet. Chem. 2002, 646, 59-67.
53. Clayden, J. Organolithiums: Selectivity for Synthesis. Pergamon Press: New York, 2002; p
293-335.
54. Bailey, W. F.; Khanolkar, A. D.; Gavaskar, K.; Ovaska, T. V.; Rossi, K.; Thiel, Y.; Wiberg,
K. B. "Stereoselectivity of cyclization of substituted 5-hexen-1-yllithiums: regiospecific and
highly stereoselective insertion of an unactivated alkene into a carbon-lithium bond" J. Am.
Chem. Soc. 1991, 113, 5720-5727.
55. Ross, G. A.; Koppang, M. D.; Bartak, D. E.; Woolsey, N. F, "Cyclization of 2-(3-
butenyl)bromobenzene via the aryllithium derivative" J. Am. Chem. Soc. 1985, 107, 6742-6743.
56. Bailey, W. F.; Mealy, M. J. "Asymmetric Cyclization of Achiral Olefinic Organolithiums
Controlled by a Stereogenic Lithium: Intramolecular Carbolithiation in the Presence of (-)-
Sparteine" J. Am. Chem. Soc. 2000, 122, 6787-6788.
57. Bailey, W. F.; Patricia, J. J.; DelGobbo, V. C.; Jarret, R. M.; Okarma, P. J. "Cyclization of 5-
hexenyllithium to (cyclopentylmethyl)lithium" J. Org. Chem. 1985, 50, 1999-2000.
58. Negishi, E.; Swanson, D. R.; Rousset, C. J. "Clean and convenient procedure for converting
primary alkyl iodides and a,w-diiodoalkanes into the corresponding alkyllithium derivatives by
treatment with tert-butyllithium" J. Org. Chem. 1990, 55, (19), 5406-5409.
59. Bailey, W. F.; Patricia, J. J. "The mechanism of the lithium-halogen interchange reaction: a
review of the literature" J. Organomet. Chem. 1988, 352, 1-46.
60. Bailey, W. F.; Nurmi, T. T.; Patricia, J. J.; Wang, W. "Preparation and regiospecific
cyclization of alkenyllithiums" J. Am. Chem. Soc. 1987, 109, 2442-2448.
61. Curran, D. P. P., N.A.; Giese, B. Stereochemistry of Radical Reactions. VCH: Weinhein,
1996; p Chapter 2.
62. Broka, C. A.; Lee, W. J.; Shen, T. "Anionic cyclizations of α-alkoxy lithiums. New,
stereoselective routes to substituted tetrahydrofurans" J. Org. Chem. 1988, 53, 1336-1338.
67
63. Broka, C. A.; Shen, T. "Reductive lithiation mediated anionic cyclizations and [2,3]-
sigmatropic rearrangements" J. Am. Chem. Soc. 1989, 111, 2981-2984.
64. Coldham, I.; Hufton, R.; Snowden, D. J. "Anionic Cyclizations of α-Aminoorganolithiums.
Determination of the Stereoselectivity at the Carbanion Center and the Synthesis of (+)-
Pseudoheliotridane" J. Am. Chem. Soc. 1996, 118, 5322-5323.
65. Krief, A.; Barbeaux, P. "Original syntheses of arylcycloalkanes" J. Chem. Soc. Chem. Comm.
1987, 1214-1216.
66. Cohen, T.; Bhupathy, M. "Organoalkali compounds by radical anion induced reductive
metalation of phenyl thioethers" Acc. Chem. Res. 1989, 22, 152-161.
67. Cohen, T.; Daniewski, W. M. "A convenient route to cyclopropanone dithioketals. The
thiophenoxide anion as a nucleofugal leaving group" Tetrahedron Lett. 1978, 2991-2994.
68. Cohen, T.; Weisenfeld, R. B.; Gapinski, R. E. "One-step synthesis of complex
cyclopropanone phenylthioketals by the reaction of sulfur-stabilized anions with ketene
bis(phenylthio)acetal" J. Org. Chem. 1979, 44, 4744-4746.
69. Screttas, C. G.; Micha-Screttas, M. "Hydrolithiation of α-olefins by a regiospecific two-step
process. Transformation of alkyl phenyl sulfides to alkyllithium reagents" J. Org. Chem. 1978,
43, 1064-71.
70. Screttas, C. G.; Micha-Screttas, M. "Markownikoff two-step hydrolithiation of α-olefins.
Transformation of secondary and tertiary alkyl phenyl sulfides to the relevant alkyllithium
reagents" J. Org. Chem. 1979, 44, 713-719.
71. Cohen, T.; Matz, J. R. "Reductive lithiation of some thioketals using lithium 1-
(dimethylamino)naphthalenide" Synthetic Commun. 1980, 10, 311-317.
72. Freeman, P. K.; Hutchinson, L. L. "Alkyllithium reagents from alkyl halides and lithium
radical anions" J. Org. Chem. 1980, 45, 1924-1930.
73. Severin, M. G.; Arevalo, M. C.; Farnia, G.; Vianello, E. "Homogeneous and heterogeneous
electron transfer to benzylphenylsulfide" J. Phys. Chem. 1987, 91, 466-472.
74. Rychnovsky, S. D.; Marumoto, S.; Jaber, J. J. "Role of 2-Oxonia Cope Rearrangements in
Prins Cyclization Reactions" Org. Lett. 2001, 3, 3815-3818.
75. Yus, M.; Ortiz, R.; Huerta, F. F. "DTBB-catalyzed lithiation of 6-chloro-1-hexene and
related systems: synthetically useful temperature-dependent behavior" Tetrahedron Lett. 2002,
43, 2957-2960.
76. Chen, F.; Mudryk, B.; Cohen, T. "Tandem reductive lithiations-carbanionic cyclizations
yielding sulfur stabilized cyclopropyl- and cyclobutylcarbinyllithiums" Tetrahedron 1999, 55,
3291-3304.
77. Bakuzis, P.; Bakuzis, M. L. F.; Fortes, C. C.; Santos, R. "The sulfide group as an aldehyde
precursor" J. Org. Chem. 1976, 41, 2769-2770.
78. Lipshutz, B. H.; Sharma, S.; Dimock, S. H.; Behling, J. R. "Preparation of C-4 alkylated
dideoxyribosides: potential precursors to a novel series of nucleosides" Synthesis 1992, 191-5.
79. Aurell, M. J.; Gil, S.; Mestres, R.; Parra, M.; Parra, L. "Alkylation of lithium dienediolates of
butenoic acids. Regioselectivity effects of structure and leaving group of the alkylating agent"
Tetrahedron 1998, 54, 4357-4366.
80. Dictionary of alkaloids. Chapman & Hall / CRC: London; New York, 1989.
81. Beak, P.; Kerrick, S. T.; Wu, S.; Chu, J. "Complex induced proximity effects:
enantioselective syntheses based on asymmetric deprotonations of N-Boc-pyrrolidines" J. Am.
Chem. Soc. 1994, 116, 3231-3239.
68
82. Beak, P.; Lee, W. K. "α-Lithioamine synthetic equivalents: syntheses of diastereoisomers
from Boc derivatives of cyclic amines" J. Org. Chem. 1993, 58, 1109-1117.
83. Dieter, R. K.; Oba, G.; Chandupatla Kishan, R.; Topping Chris, M.; Lu, K.; Watson Rhett, T.
"Reactivity and enantioselectivity in the reactions of scalemic stereogenic alpha-(N-
carbamoyl)alkylcuprates" J. Org. Chem. 2004, 69, 3076-3086.
84. Park, S. H.; Kang, H. J.; Ko, S.; Park, S.; Chang, S. "A short and concise synthetic route to (-
)-coniceine" Tetrahedron: Asymmetry 2001, 12, 2621-2624.
85. Coldham, I.; Holman, S.; Lang-Anderson, M. M. S. "Preparation of β-amino alcohols by
carbon-carbon bond formation using substituted lithiomethylpyrrolidines" J. Chem. Soc. Perk. T.
1 1997, 1481-1485.
86. Grillot, G. F.; Felton, H. R.; Garrett, B. R.; Greenberg, H.; Green, R.; Clementi, R.;
Moskowitz, M. "The condensation of thiophenols with secondary amines and formaldehyde" J.
Am. Chem. Soc. 1954, 76, 3969-3971.
87. Aahman, J.; Somfai, P. "An improved procedure for the N-alkylation of aziridines" Synthetic
Commun. 1994, 24, 1121-1127.
88. Shiozaki, M.; Hiraoka, T. "Stereo-controlled synthesis of intermediates of (±)-thienamycin"
Tetrahedron Lett. 1980, 21, 4473-4776.
89. Li, Y.; Marks, T. J. "Organolanthanide-Catalyzed Intra- and Intermolecular Tandem C-N and
C-C Bond-Forming Processes of Aminodialkenes, Aminodialkynes, Aminoalkeneynes, and
Aminoalkynes. New Regiospecific Approaches to Pyrrolizidine, Indolizidine, Pyrrole, and
Pyrazine Skeletons" J. Am. Chem. Soc. 1998, 120, 1757-1771.
90. Dimitrov, V.; Kostova, K.; Genov, M. "Anhydrous cerium(III) chloride - effect of the drying
process on activity and efficiency" Tetrahedron Lett. 1996, 37, 6787-6790.
