Bcl2fastq2 Conversion Software V2.19 User Guide 15051736 V2
bcl2fastq2_guide_15051736_v2
bcl2fastq2_guide_15051736_v2
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 30

bcl2fastq2 Conversion v2.19
User Guide
Introduction 3
Install bcl2fastq2 Conversion Software 4
BCL Conversion Input Files 5
Sample Sheet 12
Run BCL Conversion and Demultiplexing 14
BCL Conversion Output Files 18
Troubleshooting 25
Appendix: Installation Requirements 26
Revision History 27
Technical Assistance
Document # 15051736 v02
March 2017
ILLUMINA PROPRIETARY
For Research Use Only. Not for use in diagnostic procedures.
This document and its contents are proprietary to Illumina, Inc. and its affiliates ("Illumina"), and are intended solely for the
contractual use of its customer in connection with the use of the product(s) described herein and for no other purpose. This
document and its contents shall not be used or distributed for any other purpose and/or otherwise communicated, disclosed, or
reproduced in any way whatsoever without the prior written consent of Illumina. Illumina does not convey any license under its
patent, trademark, copyright, or common-law rights nor similar rights of any third parties by this document.
The instructions in this document must be strictly and explicitly followed by qualified and properly trained personnel in order to
ensure the proper and safe use of the product(s) described herein. All of the contents of this document must be fully read and
understood prior to using such product(s).
FAILURE TO COMPLETELY READ AND EXPLICITLY FOLLOW ALL OF THE INSTRUCTIONS CONTAINED HEREIN MAY RESULT
IN DAMAGE TO THE PRODUCT(S), INJURY TO PERSONS, INCLUDING TO USERS OR OTHERS, AND DAMAGE TO OTHER
PROPERTY.
ILLUMINA DOES NOT ASSUME ANY LIABILITY ARISING OUT OF THE IMPROPER USE OF THE PRODUCT(S) DESCRIBED
HEREIN (INCLUDING PARTS THEREOF OR SOFTWARE).
© 2017 Illumina, Inc. All rights reserved.
Illumina, the pumpkin orange color, and the streaming bases design are trademarks of Illumina, Inc. and/or its affiliate(s) in the U.S.
and/or other countries. All other names, logos, and other trademarks are the property of their respective owners.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
2
bcl2fastq2 Conversion Software v2.19 Guide
Introduction
The Illumina sequencing instruments generate per-cycle base call (BCL) files at the end of the sequencing run. A
majority ofanalysisapplications use per-read FASTQ files asinput for analysis. You can use the bcl2fastq2 Conversion
Software v2.19 to convert base call(BCL) files from a sequencing run into FASTQ files.
Use thisguide to installthe bcl2fastq2 Conversion Software and run the BCL conversion and demultiplexing process.
Supported Instruments
The bcl2fastq2 Conversion Software supports the following instruments:
uMiniSeq
uMiSeq
uNextSeq 500, 550
uHiSeq X
uHiSeq 2000, 2500, 3000, 4000
uNovaSeq 5000, 6000
If your Illumina sequencing system runs an earlier software version of Real-Time Analysis (RTA) than v1.18.54 and you
want to convert BCL to FASTQ, install bcl2fastq v1.8.4, and refer to the
bcl2fastq Conversion User Guide Version
v1.8.4 (part # 15038058)
for instructions.
BCL Conversion and Demultiplexing Directory
The bcl2fastq2 Conversion Software performs BCL conversion and demultiplexing in a single step. By default, the
software puts the resulting demultiplexed compressed FASTQ files in <run
folder>/Data/Intensities/BaseCalls.
The software puts reads with undetermined indexes in files that begin with Undetermined_S0_. If unindexed
samples are included in a lane with indexed information, the software exits with an error (missing a barcode).
If the Sample_Project column is specified for a sample in the sample sheet, the FASTQ files for that sample are placed
in <run folder>/Data/Intensities/BaseCalls/<Project>.
Multiple samples can use the same project directory. If the Sample_ID and Sample_Name columns are specified but
do not match, the FASTQ files are placed in an additional sub-directory called <SampleId> with files named using
the Sample_Name value.
BCLto FASTQ Conversion Process
The bcl2fastq2 Conversion Software converts the base calls in the per-cycle BCLfiles to the per-read FASTQformat.
As an option, the software can trim adapters and remove Unique Molecular Identifier (UMI)bases from reads.
Adapter Trimming—The bcl2fastq2 Conversion Software checks whether a read extends past the sample DNA
insert and into the adapter sequence. The software uses an approximate string matching algorithm to identify all or
part ofthe adapter, and treats the insertions and deletions as a single mismatch. If an adapter sequence isdetected,
base calls matchingthe adapter and beyond the match are masked or removed in the FASTQ file.
Unique Molecular Indentifiers (UMIs) Removal—UMIs are random k-mers attached to the genomic DNAbefore
polymerase chain reaction (PCR)amplification. After the UMI isamplified with amplicons, the software can retrieve
these bases and place them into the read name in the FASTQfiles. Also, when the TrimUMI sample sheet setting is
active, the software can remove the bases from the reads.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
3
bcl2fastq2 Conversion Software v2.19 Guide
Demultiplexing—First, the software reorganizes the FASTQ files based on the index sequencing information. For best
practices, avoid choosing indexes that differ by fewer than 3 bases during sample preparation. After generating the
FASTQ files, the software generates the statistics and reports for the demultiplexed FASTQ files. The software also
recalculates the base calling analysis statistics and store the statistics in the InterOp folder. You can view the statistics
with the Sequencing Analysis Viewer (SAV) software from Illumina.
Output Files
uFASTQFiles
uInterOp Files
uConversionStats File
uDemultiplexingStats File
uAdapter Trimming File
uFastqSummary and DemuxSummary
uHTML Reports
uJSONFile
Install bcl2fastq2 Conversion Software
You can download the bcl2fastq2 Conversion Software from the Downloads page on the Illumina website.
For installation requirements, see
Appendix: Installation Requirements
on page 26.
Install from RPMPackage
You need to have access the root system to install.
1 Toinstall the RPM file, use the followingcommand line:
yum install -y <rpm package-name>
The starting point forthe bcl2fastq converter is the binary executable /usr/local/bin/bcl2fastq.
2 Toinstall the RPM package in a user specified location, use the followingcommand line:
rpm --install --prefix <user specified directory>
<rpm package-name>
Install from Source
For installation, the directory locations are specified with the followingenvironment variables:
Variables Description
SOURCE Location of the bcl2fastq2 source code
BUILD Location of the build directory
INSTALL_DIR Location where the executable is installed
For example, the environment variables can be set as:
export TMP=/tmp
export SOURCE=${TMP}/bcl2fastq
export BUILD=${TMP}/bcl2fastq2-v2.19.x-build
export INSTALL_DIR=/usr/local/bcl2fastq2-v2.19.x
The build directory must be different from the source directory.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
4
bcl2fastq2 Conversion Software v2.19 Guide

Follow these steps to install from source:
1 Decompress and extract the source code.
cd ${TMP}
tar -xvzf bcl2fastq2-v2.19.x.tar.gz
This command populates the directory ${TMP}/bcl2fastq.
2 Configure the build using the following commands:
mkdir ${BUILD}
cd ${BUILD}
chmod ugo+x ${SOURCE}/src/configure
chmod ugo+x ${SOURCE}/src/cmake/bootstrap/installCmake.sh
${SOURCE}/src/configure --prefix=${INSTALL_DIR}
The first two commands create a build directory to work from. The next two lines ensure necessary files can be
executed. Executingthe final configure command populates the ${BUILD} directory with that necessary files
needed to build bcl2fastq2 in step 3 . The --prefix parameter provides the absolute path to the installation
directory. Make sure you have write permission to the ${INSTALL_DIR} directory. The ${BUILD} directory will be
created.
3 Build and installthe package using the followingcommands:
cd ${BUILD}
make
make install
Depending on the ${INSTALL_DIR} directory, you may need root privilege.
BCL Conversion Input Files
After sequencing, the instruments generate a BaseCalls directory, which containsthe base calls files (BCL), for
demultiplexing.
For demultiplexing, the bcl2fastq2 Conversion Software requiresthe followinginput files:
Instrument Input Files
MiSeq and HiSeq 2000/2500 • BCL Files (*.bcl.gz)
• STATS Files
• FILTERFiles
• Position Files
• RunInfo Files
• Config Files
• Sample Sheet Files (optional)
MiniSeq and NextSeq 500/550 • BCL Files (*bcl.bgzf)
• BCI Files
• FILTERFiles
• Position Files
• RunInfo Files
• Sample Sheet Files (optional)
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
5
bcl2fastq2 Conversion Software v2.19 Guide
Instrument Input Files
HiSeq X and HiSeq 3000/4000 • BCL Files (*.bcl.gz)
• FILTERFiles
• Position Files
• RunInfo Files
• Sample Sheet Files (optional)
NovaSeq • CBCL Files (*.cbcl)
• FILTERFiles (*.filter)
• Position Files (s.locs)
• Runinfo FIles (Runinfo.xml)
• Samples Sheet Files (SampleSheet.csv, optional)
BCL Conversion Input Files Diagram
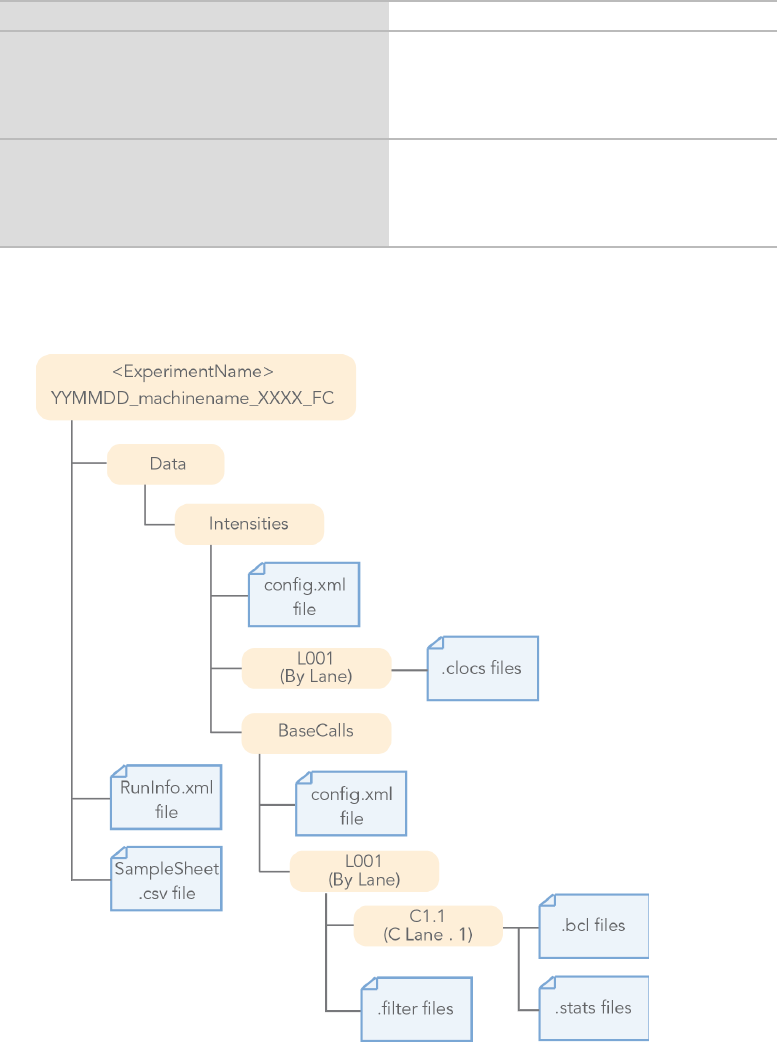
Figure 1 BCL Conversion Input Files from the MiSeq or HiSeq 2000/2500 System
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
6
bcl2fastq2 Conversion Software v2.19 Guide
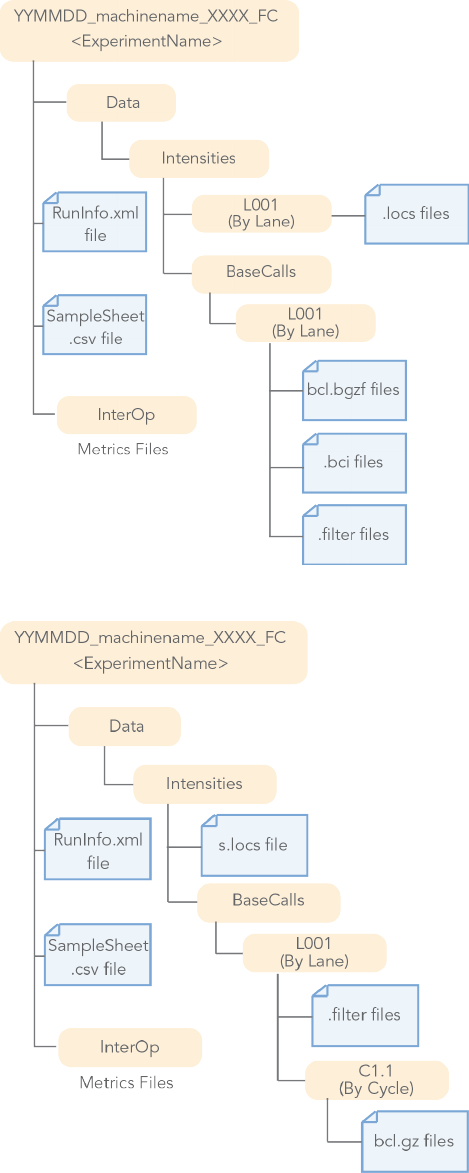
Figure 2 BCL Conversion Input Files from the MiniSeq or NextSeq System
Figure 3 BCL Conversion Input Files from the HiSeq X System
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
7
bcl2fastq2 Conversion Software v2.19 Guide
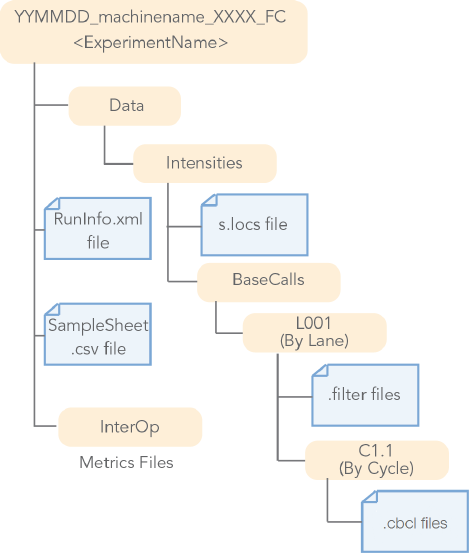
Figure 4 BCL Conversion Input Files from the NovaSeq System
Folder and File Naming
The top-level run folder name is generated using 3 fields to identify the <ExperimentName>, separated by
underscores.
The software generates the top-level run folder using 3 fields separated by underscores to identify the
<ExperimentName>.
Example:
YYMMDD_machinename_NNNN
For best practices, do not deviate from the run folder naming convention because doing so can cause the software to
stop.
uThe first field is a six-digit number (YYMMDD)specifying the date ofthe run.
uThe second field specifies the name of the sequencing machine. The field can consist of any combination of upper
or lower case letters, digits, or hyphens, but it
cannot
contain any other characters or underscore.
uThe third field isa four-digit number that specifies the experiment ID on that instrument. Each instrument supplies
a series of consecutively numbered experiment IDsfrom the on-board sample trackingdatabase or a LIMS.
For best practices, we recommend that you create unique names for the experiment or sample IDs for each
instrument to avoid naming conflicts.
For example, a run folder named 150108_instrument1_3147 indicates that the experiment ID is 3147; the run is on
instrument 1, and the date is on January 8, 2015 (YYMMDD). The date and instrument name specify a unique run
folder for any number of instruments.
Also, you can view the flow cellnumber in the run folder name.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
8
bcl2fastq2 Conversion Software v2.19 Guide
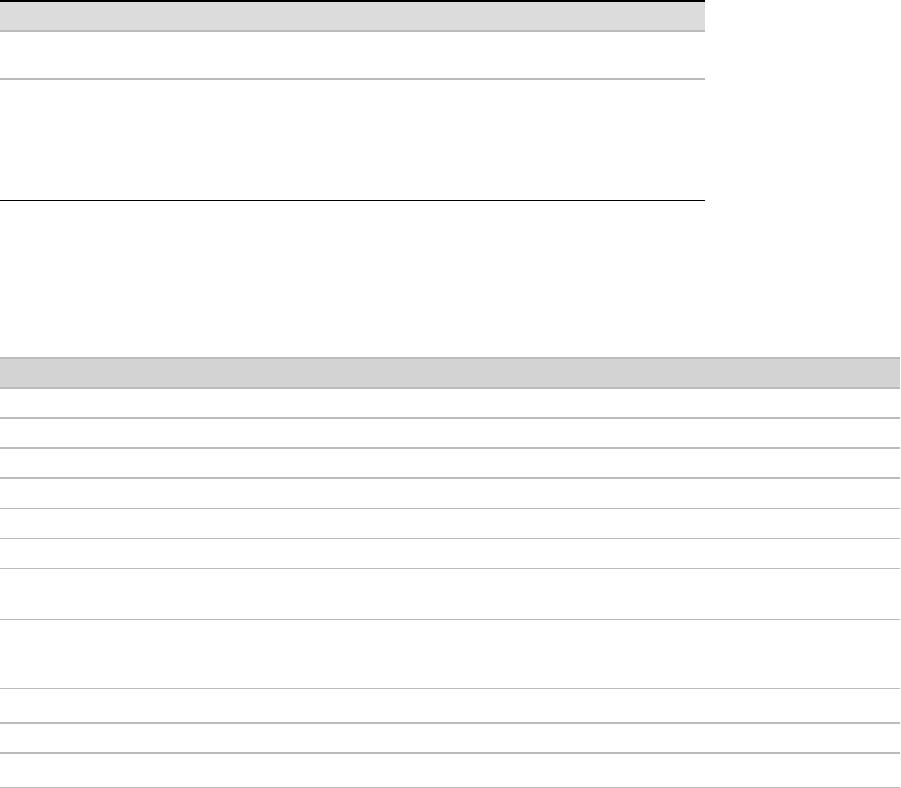
Example:
YYMMDD_machinename_NNNN_FCYYY
When you publish the data to a public database, we recommend that you use a prefix for each instrument with the
identity of the sequencing center.
BCL Files
The BCL files are compressed with the gzip (*.gz) or the blocked GNU zip (*.bgzf) format.
The BaseCalls directory containsthe BCL files. The NextSeq and MiniSeq files are located in the followingdirectory:
Data/Intensities/BaseCalls/L<lane>
You can locate the HiSeq and MiSeq files in the following directory:
Data/Intensities/BaseCalls/L<lane>/C<Cycle>.1
Bytes Description Data type
Bytes 0–3 Number N of cluster Unsigned 32 bits
integer
Bytes 4–(N+3)
N—Cluster
index
Bits 0–1 are the bases, [A, C, G, T] for [0, 1, 2, 3]:
bits 2–7 are shifted by 2 bits and contain the quality
score.
All bits with 0 in a byte is reserved for no call.
Unsigned 8 bits
integer
Table 1 BCL File Format
CBCL Files
The BCL data are aggregated and written out in the CBCL format when aggregation is on (the current aggregation
scheme is per lane/surface). The CBCL file format is as follows:
Table 2 CBCL File Format
CBCLFile Header
Bytes/Field Description Data Type
Bytes 0 - 1 Version number, current version is 1 unsigned 16 bits little endian integer
Bytes 2 - 5 Header size unsigned 32 bits little endian integer
Byte 6 Number of bits per basecall unsigned
Byte 7 Number of bits per q-score unsigned
q-val mapping info
Bytes 0-3 Number of bins (B), zero indicates no
mapping
B pairs of 4 byte values (if B > 0) {from,to}, {from,to}, {from,to} …
from: quality score bin
to: quality score
Number of tile records unsigned 32bits little endian integer
gzip virtual file offsets, one record per tile
Bytes 0-3: tile number
Bytes 4-7 Number of clusters that were written
into the current block (required due to
bit-packed q-scores)
unsigned 32 bit integer
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
9
bcl2fastq2 Conversion Software v2.19 Guide
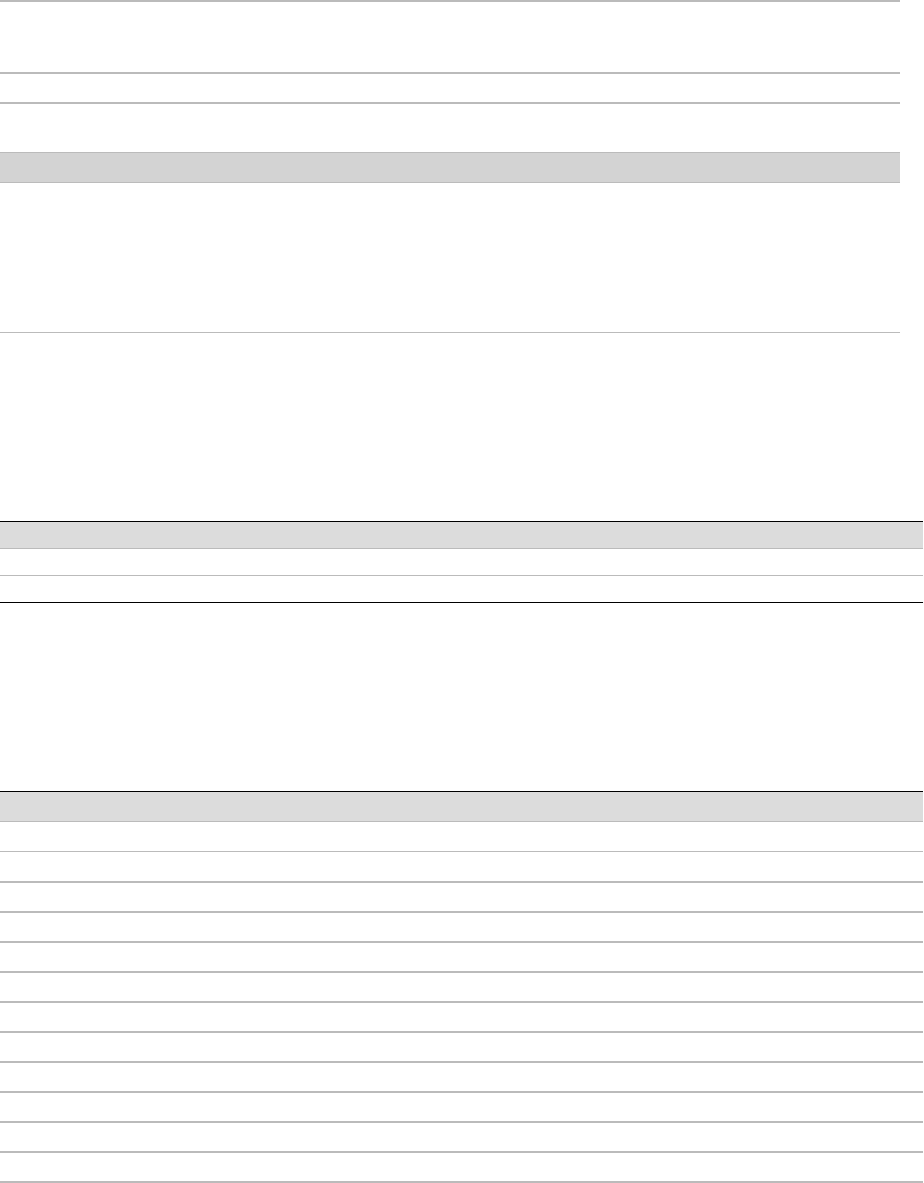
Bytes 8-11 Uncompressed block size of the tile
data (useful for sanity check when
excluding non-PF clusters)
unsigned 32 bit integer
Bytes 12-15 Compressed block size of the tile data unsigned 32 bit integer
non-PF clusters excluded flag 1: non-PF clusters are excluded
0: non-PF clusters are included
CBCL File Content
N blocks of gzip files, where N is the number of tiles. Each block consists of C number of basecall, quality score pairs where C
is the number of clusters for the given tile.
Each basecall, quality score pair has the following format (assuming 2 bits are used for the basecalls):
Bits 0-1: Basecalls (respectively [A, C, G, T] for [00, 01, 10, 11])
Bits 2 and up: Quality score (unsigned Q bit little endian integer where Q is the number of bits per q-score).
For a two bit quality score, this is two clusters per byte where the bottom 4 bits are the first cluster and the higher 4 bits are the
second cluster.
BCI Files
The BCI (*.bci) files contain one record per tile for the sequencing run in binary format. You can locate these files from
the following directory:
<run directory>/Data/Intensities/BaseCalls/L<lane>
Bytes Description
Bytes 0–3 Tile number
Bytes 4–7 Number of clusters in the tile
Table 3 BCI File Format
STATS Files
The STATSfile (*.stats) is a binary file that containsbase calling statistics. You can locate these files from the following
directory:
Data/Intensities/BaseCalls/L00<lane>/C<cycle>.1
Start Description Data Type
Byte 0 Cycle number integer
Byte 4 Average Cycle Intensity double
Byte 12 Average intensity for A over all clusters with intensity for A double
Byte 20 Average intensity for C over all clusters with intensity for C double
Byte 28 Average intensity for G over all clusters with intensity for G double
Byte 36 Average intensity for T over all clusters with intensity for T double
Byte 44 Average intensity for A over clusters with base call A double
Byte 52 Average intensity for C over clusters with base call C double
Byte 60 Average intensity for G over clusters with base call G double
Byte 68 Average intensity for T over clusters with base call T double
Byte 76 Number of clusters with base call A integer
Byte 80 Number of clusters with base call C integer
Byte 84 Number of clusters with base call G integer
Table 4 Stats File Format
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
10
bcl2fastq2 Conversion Software v2.19 Guide
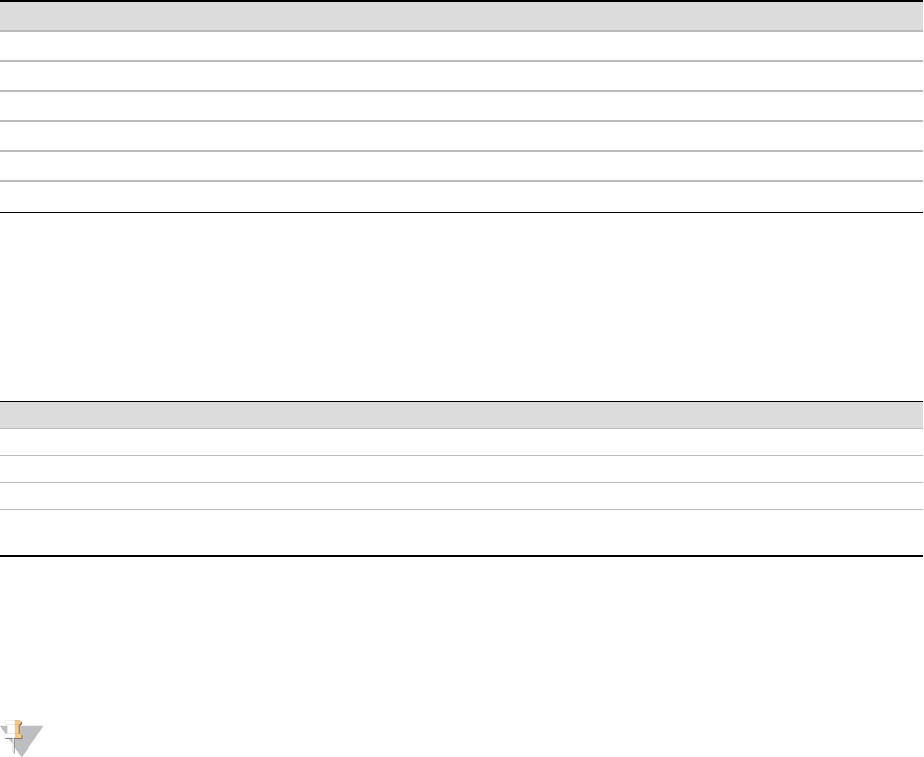
Start Description Data Type
Byte 88 Number of clusters with base call T integer
Byte 92 Number of clusters with base call X integer
Byte 96 Number of clusters with intensity for A integer
Byte 100 Number of clusters with intensity for C integer
Byte 104 Number of clusters with intensity for G integer
Byte 108 Number of clusters with intensity for T integer
FILTER Files
The FILTER file (*.filter) is a binary file that containsthe filter results. You can locate these files from the following
directory:
Data/Intensities/BaseCalls/L<lane>
Bytes Description
Bytes 0–3 Zero value (for backwards compatibility)
Bytes 4–7 Filter format version number
Bytes 8–11 Number of clusters
Bytes 12–(N+11)
N—cluster number
Unsigned 8 bits integer
Bit 0 is pass or failed filter
Table 5 Filter File Format
CONTROL Files
CONTROL files (*.control) are deprecated as ofbcl2fastq v2.19and are no longer used by the software.
CONFIG Files
NOTE
The CONFIG filesare only created on RTA1 systems (MiSeq and HiSeq 2500). They are not produced or
expected on newer platforms.
The CONFIG (*config.xml) file records information specific to the generation of the subfolders. The file contains a tag-
value list that describes the cycle-image folders used to generate each folder of intensity and sequence files. You can
locate the file from the followingdirectory:
<run directory>/Data/Intensities/
The other CONFIG (*config.xml) file is in the BaseCalls directory, which containsthe meta-information on the base
caller runs. You can locate the file from the followingdirectory:
<run directory>/Data/Intensities/BaseCalls/
Position Files
The BCL to FASTQ converter can use different types of position files.
The LOCS(*.locs) file is a binary file that containsthe cluster positions. Additionally, the *.clocs files are compressed
versions of LOCS files.
The *_pos.txt files are text-based files with 2 columns and a number of rows equal to the number of clusters. The first
column is the X-coordinate and the second column is the Y-coordinate. Each line has a <cr><lf> at the end.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
11
bcl2fastq2 Conversion Software v2.19 Guide
You can locate these files in the following directory:
Data/Intensities/L<lane>
RunInfo File
The RunInfo.xml file is located at the top-level run folder <run directory>. The file contains information on the run,
flow cell, and instrument IDs, date and read structure. Also, the file provides the number of reads, the number of
cycles per read, and the index reads.
Sample Sheet
The sample sheet (*SampleSheet.csv) file provides information on the relationship between samples and indexes
during library creation. The sample sheet is optional and the default location is the top-level run folder. You can use the
--sample-sheet command line option to specify any CSV file in any location. When a sample sheet isnot provided, all
reads are assigned to the default sample Undetermined_S0, which includes one file per lane per read.
Settings Section
The bcl2fastq2 Conversion Software uses the Settings section of the SampleSheet to specify adapter trimming, UMI,
and index-fastq options..
Setting Description
Adapter or TrimAdapter The adapter sequence to be trimmed. If an AdapterRead2 is provided, this sequence is
only used to trim Read 1. To trim two or more adapters, separate the sequences by a plus
sign (+). The plus sign between the adapters signifies that these are independent
adapters and that they need to be assessed for trimming independently for each read.
AdapterRead2 or
TrimAdapterRead2
The adapter sequence to be trimmed in Read 2. If not provided, the same sequence
specified in Adapter is used. To trim two or more adapters, separate the sequences by a
plus sign (+). The plus sign between the adapters signifies that these are independent
adapters and that they need to be assessed for trimming independently for each read.
MaskAdapter The adapter sequence to be masked rather than trimmed. If MaskAdapterRead2 is
provided, this sequence is only used to mask Read 1.
MaskAdapterRead2 The adapter sequence to be masked in Read 2. If not provided, the same sequence
specified in MaskAdapter is used.
FindAdaptersWithIndels 1 (default) or 0. If 1 (true), an approximate string matching algorithm is used to identify the
adapter, treating insertions and deletions as a single mismatch (Myers 1999, J.ACM). If 0
(false), a sliding window algorithm is used, in which insertions and deletions of bases
inside the adapter sequence is not tolerated.
Table 6 Adapter Specifications
Setting Description
Read1EndWithCycle The last cycle to use for Read 1.
Read2EndWithCycle The last cycle to use for Read 2.
Read1StartFromCycle The first cycle to use for Read 1.
Read2StartFromCycle The first cycle to use for Read 2.
Read1UMILength The length of the UMIused for Read 1.
Read2UMILength The length of the UMIused for Read 2.
Table 7 Cycle and Tile Specifications
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
12
bcl2fastq2 Conversion Software v2.19 Guide
Setting Description
Read1UMIStartFromCycle The first cycle to use for UMI in Read 1.
The cycle index is absolute and not affected by Read1StartFromCycle. The software
supports UMIs only at the beginning or end of reads. This sample sheet setting must be
used in conjunction with the Read1UMILength sample sheet setting or it will be ignored.
Read2UMIStartFromCycle The first cycle to use for UMI in Read 2.
The cycle index is absolute and not affected by Read2StartFromCycle. The software
currently supports UMIs only at the beginning or end of reads. This sample sheet setting
must be used in conjunction with the Read2UMILength sample sheet setting or it will be
ignored.
TrimUMI 0 (default) or 1 (true). When TrimUMI setting is set to 1, the software trims the UMI bases
from Read 1 and Read 2.
ExcludeTiles Tiles to exclude. Separate tiles using a plus sign [+], or specified as a range with a hyphen
[-]. For example,ExcludeTiles,1101+2201+1301-1306 meansskip tiles 1101, 2201,
and 1301 through 1306.
ExcludeTilesLaneX Tiles to exclude for Lane X. For example, ExcludeTilesLane6,1101–1108 means skip
tiles 1101 through 1108 for lane 6 only.
Setting Description
CreateFastqForIndexReads 0 (default) or 1. If 1 (true), generate FASTQ files for index reads. Normally, these FASTQ
files are not needed, because demultiplexing is carried out automatically based on the
sample sheet. Also, the index sequence is already placed in the sequence identifiers in
the FASTQ files.
Generating FASTQ files is based on the following:
• The index read masks are specified from the --use-bases-mask option.
• The RunInfo.xml file when the --use-bases-mask option is not used.
ReverseComplement 0 (default) or 1. If 1 (true), all reads are reverse complemented as they are written to
FASTQ files. This step is necessary in certain unusual cases (eg processing of mate-pair
data using BWA, which expects paired-end data).
Table 8 FASTQ Specifications
DataSection
The bcl2fastq2 Conversion Software uses the information in the columns of the Data section.
Column Description
Lane When specified, the software generates FASTQ files for only the samples with the
specified lane number.
Sample_ID The sample ID. Do not use "all" or "unknown" as the sample ID. If either of these is used as
the name, the sample will be omitted from the report.
Sample_Name The sample name. Note: Do not use "all" or "undetermined" as the sample name. If either
of these is used as the name, the sample will be omitted from the report.
Sample_Project The sample project name. The software creates a directory with the specified sample
project name and stores the FASTQ files there. You can use multiple samples in the same
project. Note: Do not use "all" or "default" as the sample project name. If either of these is
used as the name, the sample will be omitted from the report.
index The index sequence.
index2 The index sequence for index 2.
If the Sample_ID and Sample_Name columns do not match, the FASTQ files are placed in an additional sub-directory
called <SampleId>.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
13
bcl2fastq2 Conversion Software v2.19 Guide

You can use alphanumeric characters, hyphens [-], and underscores [_] for the Sample_Project, Sample_ID,
andSample_Name. Sample_ID, Sample_Name, and Sample_Project field entriesin the sample sheet cannot
contain illegal characters that are not allowed by some file systems. Examples ofcommon characters that are not
allowed are the space character and the following: ?()[]/\=+<>:;"',*^| &.
Sample Sheet Demultiplexing Scenarios
The Illumina Experiment Manager performs the following for sample sheet BCL conversion and demultiplexing:
uAll reads are placed in the Undetermined_S0 FASTQ files when there is no sample sheet.
uAll reads are placed in the Undetermined_S0 FASTQ files when there is a sample sheet but no data section.
uAll reads are placed in the sample FASTQ file asdefined in the sample sheet when there is a sample sheet and one
sample with no indexes.
uWhen there isa sample sheet and the samples have indexes, the software performs the following:
uReads without a matchingindex are placed in the default Undetermined_S0 FASTQ files.
uReads with a valid index are placed in the sample FASTQ file as defined in the sample sheet.
For each sample, there isone file per lane per read number when reads exist for that sample, lane, and read number.
NOTE
Whenthe Lane column ofthe sample sheet Data section ispopulated, only those lanesare converted. When
the Lane column isnot used, all lanesare converted.
Create a Sample Sheet with IEM
The Illumina Experiment Manager (IEM) software helps you create and edit sample sheets for Illumina sequencers and
analysis software. You can use IEM to create sample sheets for any Illumina sequencer.
You can download IEM at support.illumina.com/sequencing/sequencing_software/experiment_
manager/downloads.html.
View the Illumina Experiment Manager User Guide for creating a sample sheet.
Run BCL Conversion and Demultiplexing
Use the following command to run the bcl2fastq2 Conversion Software :
nohup /usr/local/bin/bcl2fastq [options]
An example of a command with options:
nohup /usr/local/bin/bcl2fastq --runfolder-dir <RunFolder>
--output-dir <BaseCalls>
This command produces a set of FASTQ files in the BaseCalls directory. Reads with an unresolved or erroneous index
are placed in the Undetermined_S0 FASTQ files. By default, --runfolder-dir is the current directory and --
output-dir is the Data/Intensities/BaseCalls sub-directory ofthe run folder.
BCL2FASTQ Options
The main command line options are the --runfolder-dir and --output-dir. For command line options that
have a corresponding sample sheet setting, the value passed on the command line overwrites the value found in the
sample sheet.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
14
bcl2fastq2 Conversion Software v2.19 Guide
Option Description
-R, --runfolder-dir Path to run folder directory
Default: ./
-o, --output-dir Path to demultiplexed output
Default: <runfolder-
dir>/Data/Intensities/BaseCalls/
Table 9 Main Options
You can use the followingadvanced options for non-default settings or for customized settings.
Option Description
-i, --input-dir Path to input directory
Default: <runfolder-dir>/Data/Intensities/BaseCalls/
--sample-sheet Path to sample sheet, so you can specify the location and name of
the sample sheet, if different from default.
Default: <runfolder-dir>/SampleSheet.csv
Table 10 Directory Options
The following directory options and thread control options provide more control of the conversion process, but are not
needed for standard usage.
Option Description
--intensities-dir Path to intensities directory
If intensities directory is specified, then the input directory must
also be specified.
Default: <input-dir>/../
--interop-dir Path to demultiplexing statistics directory
Default: <runfolder-dir>/InterOp/
--stats-dir Path to human-readable demultiplexing statistics directory
Default: <output-dir>/Stats/
--reports-dir Path to reporting directory
Default: <output-dir>/Reports/
Table 11 Additional Directory Options
For processing, ifyour computingplatform supports threading, the software managesthe threads by the following
defaults:
u4 threads for reading the data
u4 threads for writing the data
u20% for demultiplexing data
u100% for processing demultiplexed data
The file i/o threads spend most oftheir time sleeping, and so take little processing time. The processing of
demultiplexed data is allocated 1 thread per CPU to make sure that there are no idle CPUs, resulting in more threads
than CPUs by default. You can use the following options to provide control on threading. If, for example, you share your
computing resources with colleagues and wish to limit your usage, these options are useful.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
15
bcl2fastq2 Conversion Software v2.19 Guide

Option Description
-r, --loading-threads Number of threads used for loading BCL data.
Default depends on architecture.
-p,
--processing-threads
Number of threads used for processing demultiplexed data.
Default depends on architecture.
-w,
--writing-threads
Number of threads used for writing FASTQ data. This number should not be set higher
than number of samples.
Default depends on architecture.
Table 12 Processing Options
If you want to use these options to assign multiple threads, consider the following:
uThe most CPU demandingstage is the processingstep (-p option). Assign this step the most threads.
uReadingand writing stages are lightweight and do not need many threads. Thisconsideration is especially
important for a local hard drive where too many threads mean too many parallel read write actions giving
suboptimal performance.
uUse one thread per CPU core plus a little more to supply CPU with work. This method prevents CPUsbeingidle
due to a thread being blocked while waiting for another thread.
uThe number of threads depends on the data. If you specify more writing threads than samples, the extra threads
do no work but can cost time due to context switching.
Option Description
--adapter-stringency The minimum match rate that would trigger the masking or trimming process. This value is
calculated as MatchCount / (MatchCount + MismatchCount) and ranges from 0 to 1, but
it is not recommended to use any value <0.5, as this value would introduce too many
false positives. The default value for this parameter is 0.9, meaning that only reads with
>90% sequence identity with the adapter are trimmed.
Default: 0.9
--barcode-mismatches Number of allowed mismatches per index
Multiple entries, comma delimited allowed. Each entry is applied to the corresponding
index; last entry applies to all remaining indexes.
Default: 1. Accepted values: 0, 1 or 2.
--create-fastq-for-index-
reads
Create FASTQ files also for Index Reads.
Generating FASTQ files is based on the following:
• The index read masks are specified from the --use-bases-mask option.
• The RunInfo.xml file when the --use-bases-mask option is not used.
--ignore-missing-bcls Missing or corrupt BCL files are ignored. Assumes 'N'/'#' for missing calls
--ignore-missing-filter Missing or corrupt filter files are ignored. Assumes Passing Filter for all clusters in tiles
where filter files are missing.
--ignore-missing-positions Missing or corrupt positions files are ignored. If corresponding position files are missing,
bcl2fastq writes unique coordinate positions in FASTQ header.
--minimum-trimmed-read-
length
Minimum read length after adapter trimming. bcl2fastq trims the adapter from the read
down to the value of this parameter. If there is more adapter match below this value, then
those bases are masked, not trimmed (replaced by N rather than removed).
Default: 35
Table 13 Behavioral Options
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
16
bcl2fastq2 Conversion Software v2.19 Guide

Option Description
--mask-short-adapter-reads This option applies when a read is shorter than the length specified by --minimum-
trimmed-read-length (note that the read does not specifically have to be trimmed for
this option to trigger, it need only fall below the —minimum-trimmed-read-length for
any reason). These parameters specify the following behavior:
If the number of bases left after adapter trimming is less than --minimum-trimmed-
read-length, force the read length to be equal to --minimum-trimmed-read-length
by masking adapter bases (replace with Ns) that fall below this length.
If the number of ACGT bases left after this process falls below --mask-short-adapter-
reads, mask all bases, resulting in a read with --minimum-trimmed-read-length
number of Ns. In addition, if a read is shorter than--mask-short-adapter-reads for any
reason, it will be masked with Ns. Because it applies when a read is shorter than the value
of --minimum-trimmed-read-length, it should be set to a value that is less than or
equal to this parameter. If it is set to a greater value, it will automatically default to the
same value as--minimum-trimmed-read-length.
Default: 22
--tiles The --tiles argument takes a regular expression to select for processing only a subset
of the tiles available in the flow cell. Multiple selections can be made by separating the
regular expressions with commas. Examples:
To select all the tiles ending with 5 in all lanes:
--tiles [0–9][0–9][0–9]5
To select tile 2 in lane 1 and all the tiles in the other lanes:
--tiles s_1_0002,s_[2-8]
--use-bases-mask The --use-bases-mask string specifies how to use each cycle.
An nmeans ignore the cycle.
AY(or y) means use the cycle.
An Imeans use the cycle for the Index Read.
A number means that the previous character is repeated that many times.
An asterisk [*] means that the previous character is repeated until the end of this read or
index (length according to the RunInfo.xml).
The read masks are separated with commas: ,
The format for dual indexing is as follows: --use-bases-mask Y*,I*,I*,Y* or
variations thereof as specified.
You can also specify the --use-bases-mask multiple times for separate lanes, like this
way:
--use-bases-mask 1:y*,i*,i*,y* --use-bases-mask y*,n*,n*,y*
Where the 1: means: Use this setting for lane 1. In this case, the second --use-bases-
mask parameter is used for all other lanes.
If this option is not specified, the mask is determined from the 'RunInfo.xml file in the run
directory. If it cannot do this determination, supply the --use-bases-mask.
When the --use-bases-mask option is specified, the number of index cycles and the
length of index in the sample sheet should match.
--with-failed-reads Include all clusters in the output, even clusters that are non-PF. These clusters would
have been excluded by default.
Note: This option cannot be applied to CBCLdata.
On RTA 2 systems, clusters that fail filter are no longer read after cycle 25. On systems
other than MiSeq and HiSeq 2500, you will get 25 bases, then all Ns.
--write-fastq-reverse-
complement
Generate FASTQ files containing reverse complements of actual data.
--no-bgzf-compression Turn off BGZF compression, and use GZIP for FASTQ files. BGZF compression allows
downstream applications to decompress in parallel. This parameter is available in case a
consumer of FASTQ data cannot handle all standard GZIP formats.
--fastq-compression-level Zlib compression level (1–9) used for FASTQ files.
Default: 4
--no-lane-splitting Do not split FASTQ files by lane.
--find-adapters-with-
sliding-window
Find adapters with simple sliding window algorithm. Insertions and deletions of bases
inside the adapter sequence are not handled.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
17
bcl2fastq2 Conversion Software v2.19 Guide
NOTE
Donot use the --no-lane-splitting option if you want to upload the resulting FASTQ files toBaseSpace.
The FASTQ files generated from the --no-lane-splitting option are not compatible with the
BaseSpace file uploader. Files generated without this option (the default setting) are compatible for upload to
BaseSpace.
NOTE
FASTQ filescontainingfailed reads cannot be uploaded to BaseSpace.
Option Description
-h,
--help
Produce help message and exit
-v,
--version
Print program version information
-l,
--min-log-level
Minimum log level
Recognized values: NONE, FATAL, ERROR, WARNING, INFO, DEBUG, TRACE
Default: INFO
Table 14 General Options
BCL Conversion Output Files
The bcl2fastq2 Conversion Software provides the followingoutput files: output directory has the following
characteristics:
uFASTQFiles
uInterOp Files
uConversionStats File
uDemultiplexingStats File
uAdapterTrimming File
uFastqSummary and DemuxSummary
uHTML Reports
uJSONFile
FASTQ Files
The bcl2fastq2 Conversion Software converts *.bcl, *.bcl.gz, *.bcl.bgzf, and .cbcl files into FASTQ files, which can be
used as input for secondary analysis. When there is no sample sheet, the software generates a Undetermined_S0
FASTQ file for each lane and read number combination.
FASTQ File Names
FASTQ files are named with the sample name and the sample number. The sample number is a numeric assignment
based on the order that the sample is listed for the run. For example:
Data\Intensities\BaseCalls\samplename_S1_L001_R1_001.fastq.gz
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
18
bcl2fastq2 Conversion Software v2.19 Guide

usamplename—The sample name listed forthe sample. If a sample name isnot provided, the file name includes
the sample ID.
uS1—The sample number based on the order that samples are listed for the run startingwith 1. In this example, S1
indicates that thissample is the first sample listed for the run.
NOTE
Reads that cannot be assigned toany sample are written toa FASTQ file forsample number 0, and
excluded from downstream analysis.
uL001—The lane number.
uR1—The read. In this example, R1 means Read 1. For a paired-end run, a file from Read 2 includes R2 in the file
name. When generated, the Index Reads are I1 or I2.
u001—The last segment is always 001.
FASTQ files are compressed in the GNU zip format, as indicated by *.gz in the file name. FASTQ files can be
uncompressed using tools such as gzip (command-line) or 7-zip (GUI).
FASTQ File Format
FASTQ file is a text-based file format that contains base calls and quality values per read. Each record contains 4 lines:
uThe identifier
uThe sequence
uA plus sign (+)
uThe quality scores in a +33 offset ASCII encoded format
The identifier is formatted as:
@Instrument:RunID:FlowCellID:Lane:Tile:X:Y:UMI ReadNum:FilterFlag:0:IndexSequence or SampleNumber
Example:
@SIM:1:FCX:1:2106:15337:1063:GATCTGTACGTC 1:N:0:ATCACG
GATCTGTACGTCTCTGCNTCACCTCCACCGTGCAACTCATCACGCAGCTCATGCCCTTCGGCTGCCTCCTGGACTA
+
CCCCCGGGGGGGGGGGG#:CFFGFGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGEGGFGGG
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
19
bcl2fastq2 Conversion Software v2.19 Guide
Identifiers Description
@Each sequence identifier line starts with @.
instrument The instrument ID.
run number The run number on the instrument.
flowcell ID The flowcell ID.
lane The lane number.
tile The tile number.
x_pos The X coordinate of the cluster.
y_pos The Y coordinate of the cluster.
UMI [Optional] The Unique Molecular Identifiers (UMIs) are restricted to A/T/G/C/N. The
UMIsequences for Read 1 and Read 2 are separated by a plus sign (+) when the UMIs are
specified in the sample sheet.
read Read 1—Single read.
Read 2—Paired-end read.
is filtered Y—The read is filtered (only showing when --with-failed-reads option is applied).
N—The read is not filtered.
control number 0—No control bits are turned on.
Even number—Control bits are turned on.
index sequence or sample
number
The Index reads are restricted to A/T/G/C/N. When an indexed sample sheet is used, the
index sequence is written to the end of the read identifier. If an unindexed sample sheet is
used (single sample per lane), the sample number is written to the read identifier.
Table 15 Identifiers Table
FASTQ Compression
FASTQ files are compressed in the GNU zip format, as indicated by *.gz in the file name. FASTQ files can be
uncompressed using tools such as gzip (command-line) or 7-zip (GUI).
The BGZF variant facilitates parallel decompression of the FASTQ files by downstream applications. If a downstream
application cannot handle the BGZF variant, it can be turned off with the --no-bgzf-compression command
line.
Quality Scores
A quality score, or Q-score, is a prediction of the probability of an incorrect base call. A higher Q-score implies that a
base call is more reliable.
Based on the Phred scale, the Q-score serves as a compact way to communicate small error probabilities. Given a
base call, X, the probability that X isnot true, P(~X), results in a quality score, Q(X), according to the relationship:
Q(X) = -10 log10(P(~X))
where P(~X) is the estimated error probability.
The followingtable shows the relationship between the quality score and error probability.
Quality Score Q(X) Error Probability P(~X)
Q40 0.0001 (1 in 10,000)
Q30 0.001 (1 in 1,000)
Q20 0.01 (1 in 100)
Q10 0.1 (1 in 10)
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
20
bcl2fastq2 Conversion Software v2.19 Guide
For more information on the Phred quality score, see en.wikipedia.org/wiki/Phred_quality_score.
During the sequencing run, base call quality scoresare calculated after cycle 25 and results are recorded in base call
(*.bcl) files, which contain the base call and quality score per cycle.
Quality Scores Encoding
In FASTQ files, quality scoresare encoded into a compact form, which uses only 1byte per quality value. In this
encoding, the quality score is represented as the character with an ASCII code equal to its value + 33. The following
table demonstrates the relationship between the encoding character, its ASCII code, and the quality score
represented.
NOTE
WhenQ-score binningis in use, the subset ofQ-scoresapplied by the binsisdisplayed.
Symbol ASCII Code Q-Score
! 33 0
" 34 1
# 35 2
$ 36 3
% 37 4
& 38 5
' 39 6
( 40 7
) 41 8
* 42 9
+ 43 10
, 44 11
- 45 12
. 46 13
/ 47 14
0 48 15
1 49 16
2 50 17
3 51 18
4 52 19
5 53 20
Symbol ASCII Code Q-Score
6 54 21
7 55 22
8 56 23
9 57 24
: 58 25
; 59 26
< 60 27
= 61 28
> 62 29
? 63 30
@ 64 31
A 65 32
B 66 33
C 67 34
D 68 35
E 69 36
F 70 37
G 71 38
H 72 39
I 73 40
Table 16 ASCII Characters Encoding Q-scores 0–40
InterOp Files
You can locate the InterOp files in the directory: <run directory>/InterOp. The directory contains binary files
used by the Sequencing Analysis Viewer (SAV) software to summarize various analysis metrics, such as cluster
density, intensities, quality scores, and overall run quality.
The index metrics are stored in the IndexMetricsOut.bin file generated by bcl2fastq2, which has the following binary
format:
Byte 0: file version (1)
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
21
bcl2fastq2 Conversion Software v2.19 Guide
Bytes (variable length): record:
u2 bytes: lane number (unint16)
u2 bytes: tile number (unint16)
u2 bytes: read number (unint16)
u2 bytes: number of bytes Y for index name (unint16)
uY bytes: index name string (string in UTF8Encoding)
u4 bytes: # clusters identified as index (uint32)
u2 bytes: number of bytes V for sample name (unint16)
uV bytes: sample name string (string in UTF8Encoding)
u2 bytes: number of bytes W for sample project (unint16)
uW bytes: sample project string (string in UTF8Encoding)
ConversionStats File
You can locate the ConversionStats.xmlfile in the directory: <output directory>/Stats/, or in the directory
specified by the --stats-dir option.
The file contains the following information per tile:
uRaw Cluster Count
uRead number
uYieldQ30
uYield
uQualityScore Sum
The file contains the following information per lane:
uLane Number
DemultiplexingStats File
You can locate the DemultiplexingStats.xmlfile in the directory: <output directory>/Stats/, or in the
directory specified by the --stats-dir option. The file contains the following information per lane, barcode, and
sample, project.
Also, the file containsthe followinginformation for flow cell:
uBarcode Count
uPerfectBarcode Count
uOneMismatchBarcode Count
AdapterTrimming File
The AdapterTrimmingfile is a text-based file format that containsa statistic summary ofadapter trimming forthe
FASTQ file. You can locate the file in the <output directory>/Stats/ orin the directory specified by the --
stats-dir option.
The file contains the following information:
uLane
uRead
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
22
bcl2fastq2 Conversion Software v2.19 Guide

uProject
uSample ID
uSample Name
uSample Number
uTrimmedBases
uPercentageOfBased (being trimmed)
Also, the file containsthe fraction of reads with untrimmed bases for each sample, lane, and read number.
FastqSummaryF1L#
The FastqSummaryF1L#.txt file (the #indicates the lane number) containsthe number of raw and passed filter reads
foreach sample number and tile. You can locate the file in the <output directory>/Stats/ or in the directory
specified by the --stats-dir option.
DemuxSummaryF1L#
The DemuxSummaryF1L#.txt (the #indicates the lane number) file isonly created ifthe sample sheet contains at least
one sample and the sample barcode isprovided. This file contains the percentage ofeach tile that each sample
makes up. The file also containsa list of the 1,000 most common unknown barcode sequences, and the total number
ofreads with each barcode seen (Note: to improve speed, the total for each barcode is estimated using a sampling
algorithm, and is approximate).
You can locate the file in the <output directory>/Stats/ or in the directory specified by the --stats-dir
option.
HTML Report
The HTML reports are generated from data in the DemultiplexingStats.xmland ConversionStats.xmlfiles. You can
locate the reportsin the directory: <output directory>/Reports/html/, or in the directory specified by the -
-reports-dir option.
The Flowcell Summary contains the following information:
uClusters (Raw)
uClusters (PF)
uYield (MBases)
NOTE
For HiSeq X, HiSeq 4000, and HiSeq 3000, the number of raw clustersis actuallythe number ofwellson the flow
cellthat could potentially be seeded. The value is the same in allcases.
The Lane Summary provides the following information for each project, sample, and index sequence specified in the
sample sheet:
uLane #
uClusters (Raw)
u% of the Lane
u% Perfect Barcode
u% One Mismatch
uClusters (Filtered)
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
23
bcl2fastq2 Conversion Software v2.19 Guide
uYield
u% PF Clusters
u%Q30 Bases
uMean Quality Score
The Top Unknown Barcodes table in the HTML report provides the count and sequence for the 10 most common
unmapped bar codes in each lane.
JSON File
The Java Script Object Notification (JSON)file containsthe *.json file extension. The format forthe JSON file makes it
easier to parse the output data. The data in the JSONfile are a combination of allthe followingfiles:
uInterOP
uConversionStats
uDemultiplexingStats
uAdapter Trimming
uFastqSummary and DemuxSummary
uHTMLReport
u
uThe format ofthe JSONfile is similar to the followingexample:
{
Flowcell: string //matches Flowcell from RunInfo.xml
RunNumber: int, //matches Run Number from RunInfo.xml
RunId: string, //matches Run Id from RunInfo.xml
ReadInfosForLanes: [ //details per-lane read information
{
LaneNumber: int,
ReadInfos: [
Number: int, //indicates read 1 or read 2 (possible values: 1 and 2)
NumCycles: int, //indicates number of cycles for this read
IsIndexedRead, bool // indicates whether or not this read is an
index read
]
}
],
ConversionResults:[ //details the conversion/demultiplexing results
{
LaneNumber: int,
TotalClustersRaw: int, //number of raw clusters in this lane (null
for HiSeq X)
TotalClustersPf: int //number of clusters passing filter in this
lane
Yield: int, //total yield in this lane
DemuxResults: [ //do not include undetermined reads in this array
{
SampleId: string,
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
24
bcl2fastq2 Conversion Software v2.19 Guide
SampleName: string,
IndexMetrics: [ //empty array if no indices were used for
demultiplexing this sample
{
IndexSequence: string, //if there are two indices, then
concatenate with '+' character (e.g.
"ATCGTCG+TGATCTA")
MismatchCounts: {
0: int, //count of perfectly matching barcodes
1: int //count of barcodes with one mismatch
}
}
],
NumberReads: int, //number of read pairs identified as
index/index-pair
Yield: int, //number of bases after trimming
ReadMetrics: [
{
ReadNumber: int,
Yield: int,
YieldQ30: int,
QualityScoreSum: int,
TrimmedBases: int
}
]
}
]
}
],
UnknownBarcodes: [ //details all the unknown barcodes for a given lane and
number of times it was encountered
{
Lane: int,
Barcodes: {
string: int //example: "ATGAAGAT": 5888
}
}
]
}
Troubleshooting
uIf the bcl2fastq2 Conversion Software fails to complete a run, it could be missing an input file or have a corrupt file.
View the log file formissing or corrupt files. The exact wording of the file status reported varies dependingon the
nature ofthe file corruption. If the problem is the BCL file, launch the --ignore-missing-bcls option. See
BCLAdvanced Options.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
25
bcl2fastq2 Conversion Software v2.19 Guide

uIf there is a high percentage of reads assigned as undetermined, view the Top Unknown Barcodes table in the
HTMLreport on the index sequence.
uIf the bcl2fastq2 Conversion Software has problems processingSmall RNAsamples, use the --minimum-
trim-read-length 20 and --mask-short-adapter-reads 20 command line instead of the default
settings.
Appendix: Installation Requirements
The bcl2fastq2 Conversion Software requires the following components:
Component Requirements
Network Infrastructure 1 Gigabit minimum.
Server Infrastructure Single multiprocessor or multicore computer running Linux.
Analysis Computer Run software on the Linux operating systems only.
Memory 32 GB RAM.
Software We recommend the either the CentOS 6 or the RedHat Enterprise Linux 6 platform.
NOTE
Other Linuxdistributions maywork if the dependenciesare met, but
are not officially supported for installation.
The following software is required:
• zlib
• librt
• libpthread
The followingsoftware are required to build the bcl2fastq2 Conversion
Software :
• gcc 4.8.2 or later (with support for C++11)
• boost 1.54
• CMake 2.8.9
• zlib
• librt
• libpthread
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
26
bcl2fastq2 Conversion Software v2.19 Guide

Revision History
Part# Revision Date Description of Change
15051736 02 March 2017 • Updated to support bcl2fastq2 v2.19.
• Added NovaSeq file structure information.
• Added CBCL file format section.
• Revised BCL2FASTQ options.
15051736 01 April 2016
• Updated to support bcl2fastq2 v2.18.
• Reformatted the User Guide to Illumina style standards.
• Added JSON file and input files list for MiniSeq.
• Revised BCL2FASTQ options and sample sheet settings.
15051736 G July 2015 Updated to software requirements, gcc version.
15051736 F June 2015 Updated to support bcl2fastq2 v2.17.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
27
bcl2fastq2 Conversion Software v2.19 Guide
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
28
bcl2fastq2 Conversion Software v2.19 Guide
Technical Assistance
For technical assistance, contact Illumina Technical Support.
Website: www.illumina.com
Email: techsupport@illumina.com
Illumina Customer Support Telephone Numbers
North America Germany Singapore
1.800.809.4566 0800.180.8994 1.800.579.2745
Australia Hong Kong Spain
1.800.775.688 800960230 900.812168
Austria Ireland Sweden
0800.296575 1.800.812949 020790181
Belgium Italy Switzerland
0800.81102 800.874909 0800.563118
China Japan Taiwan
400.635.9898 0800.111.5011 00806651752
Denmark Netherlands United Kingdom
80882346 0800.0223859 0800.917.0041
Finland New Zealand Other countries
0800.918363 0800.451.650 +44.1799.534000
France Norway
0800.911850 800.16836
Safety data sheets (SDSs)—Available on the Illumina website at support.illumina.com/sds.html.
Product documentation—Available for download in PDF from the Illumina website. Go to support.illumina.com,
select a product, then select Documentation & Literature.
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
bcl2fastq2 Conversion Software v2.19 Guide
Document # 15051736 v02
For Research Use Only. Not for use in diagnostic procedures.
2
bcl2fastq2 Conversion Software v2.19 Guide
