Guide
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 21
Lithium ELNES with WIEN2k
October 12, 2018
Contents
1 Tools 2
2 Path 2
3 Setup 3
3.1 LoadStructure .................................. 3
3.2 Initialization.................................... 4
4 Convergence 6
4.1 Cellparameters:.................................. 6
4.2 Setting the Muffin Tin Size . . . . . . . . . . . . . . . . . . . . . . . . . . . 7
4.3 K point and RKMax convergence . . . . . . . . . . . . . . . . . . . . . . . . 8
5 Density of States 9
6 TELNES3 10
6.1 Monopoleeffects ................................. 12
7 Core Hole 13
8 Density Calculations 16
8.1 Xcrysden...................................... 16
8.2 Critic2....................................... 16
9 Common Errors/Issues Encountered in Wien2k 19
9.1 SettingRMT/RKMax .............................. 19
9.2 Ghostbands .................................... 19
9.3 NNinOptimization................................ 20
9.4 NRandNT .................................... 21
9.5 GMax Value less than Gmin . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
9.6 Ordering...................................... 21
1
1 Tools
Here’s a list of software I use to run simulations. I’ve put some installation instructions for
linux as needed.
•Wien2k - If you are reading this guide, you either already have it installed somewhere,
or should figure out how to do that before buying a license. Otherwise, read the
userguide where the installation process is well explained.
•VESTA: http://jp-minerals.org/vesta/en/download.html. Download the .rpm
file, install it with your package manager in the directory it was downloaded to, eg.
“sudo apt-get vesta...rpm”
•Xcrysden: http://www.xcrysden.org/Download.html. Download the appropriate
tar for your operating system, and make sure to select the semishared version. Unzip
it, and just run “$./xcrysden”. Add the executable to your path in .bashrc to be able
to run it anywhere.
•Critic2: https://github.com/aoterodelaroza/critic2. Download the zip from
GitHub, unzip where you want to install it (eg ∼/Programs or something), use your
package manager (dnf/apt-get) to install autoconf, automake, and your favourite flavour
of fortran (if unsure, it is probably gfortran). Then run the 4 commands from the
readme: “autoreconf -i”, “./configure”, “make”, and “(sudo) make install”. You may
or may not need the sudo for the last one.
2 Path
Here is the overall list of things that need to be done to rigorously obtain ELNES spectra
with WIEN2k:
•Load a structure
•Initialize
•Volume convergence/structure optimization
•Set RMT values
•K point/RKmax convergence
•Calculate DOS
•Calculate ELNES
•Include Core Hole, if indicated by DOS, calculate PDOS
•Calculate Screening from density
•Calculate ELNES from partial hole
2
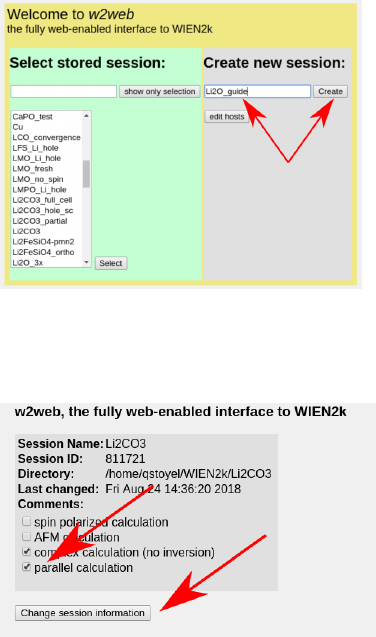
3 Setup
3.1 Load Structure
There a couple things required to start ELNES simulations. Foremost is a crystal struc-
ture. This can be obtained from the literature, XRD, or alternatively Materials Project:
https://materialsproject.org/. Download a cif (the primative cell typically) or enter
the coordinates directly into the wien2k struct gen tool, set the rmt and save the file.
Make a new Wien2k session:
Create/change a working directory, and change the session information for parallel cal-
culation.
You will also need to make a “.machines” file which you can either steal from one of my
directories, look in the user guide and make your own, cut and paste the one from below, or
wait for w2web to automatically generate one at some point after it inevitably crashes on
something. Sample .machines file:
#=======================================
#This i s a v a l i d . machines f i l e
#
granularity :1
1 : l o c a l h o s t #as many o f t h e se l i n e s as you want cpu c or e s running
1 : l o c a l h o s t #i d e a l l y pi ck a mu lt ip le of kp oint number
1 : l o c a l h o s t #so something l i k e 5 c or e s f o r 10 k po in ts .
3

1: localhost
1: localhost
1: localhost
Next, go make a struct file, with struct gen, either by importing the cif, or entering
the positions manually. Use VESTA (drag n’ drop the .struct file) to make sure that the
structure is what you’d expect. In the Li2O case this looks like:
3.2 Initialization
The next step is initialization. This is best done via the command line as it give a little more
flexibility and intuition than in w2web, and will need to be run multiple times. The first
time will be to set up the calculation for volume optimization, which needs smaller muffin
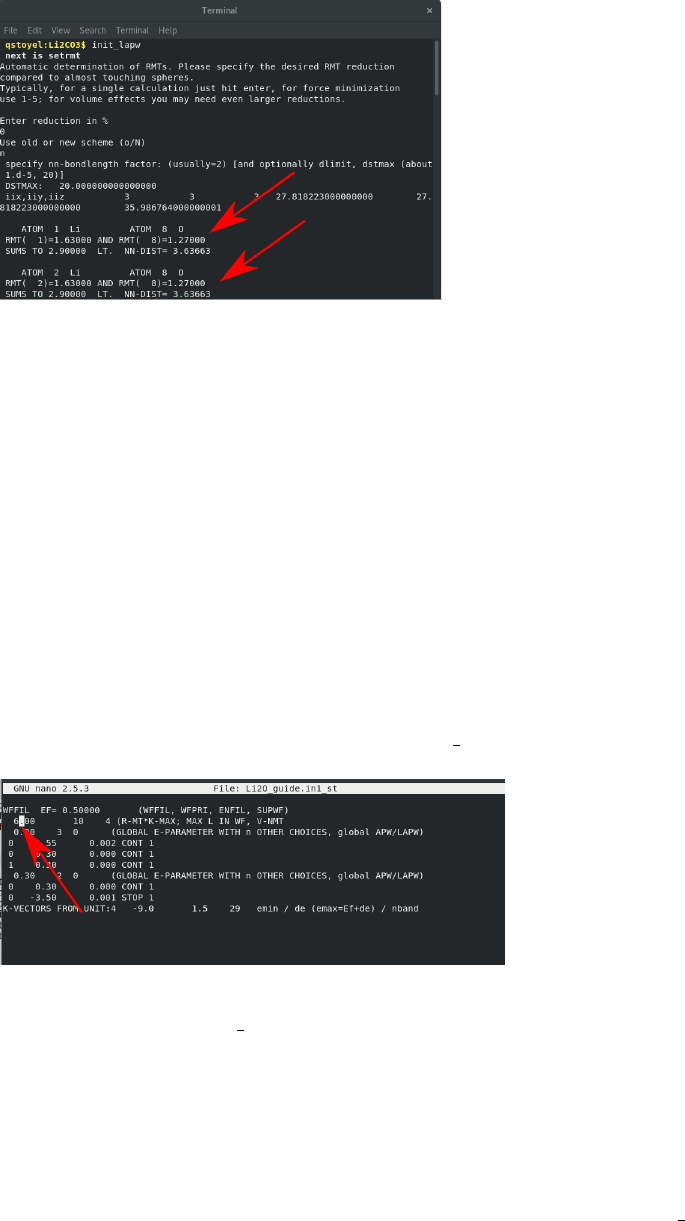
tins. Run init lapw and go through the following steps:
•setrmt. Sets the muffin tin size on the atoms. Reduce the sphere size by 5-10% (I
used 7%), using either old or new scheme and accept.
•nn. Checks for overlapping muffin tins. Enter “2.0”, close the first file and use the
new NN file if suggested, run nn with 2.0 again, look at how much “wiggle room” you
have on the spheres, by comparing the “sums to” and the “nn-dist” for every atom,
see below:
4
•sgroup. Verifies the space group. Again, accept any changes the program makes, this
first run is all about reducing the cell to the smallest unit cell. Again, the files can be
largely ignored at this point, unless there is an indication of a Bravais lattice change, in
which case, accept the new struct file. If so, nn and sgroup will run again with “nice”
results.
•symmery. Generates all the symmetry operations. Run it and continue (enter “c”)
•lstart. Sets the spin state, selects which XC kernel, and defines cutoff between core and
valence states. Accept default spins (up, the no spin case), unless there is a transition
metal involved. Select GGA PBE as the XC potential, again, unless there is reason to
suspect otherwise. For the purposes of volume optimization, the defualt energy value
(-6.0 Ry) works well.
•kgen Set the RkMax value in case.in1 st from 7 to the desired value:
and then pick a k point number. Both of these values should initially be taken for fast
convergence, in this case I chose RkMax=6.0, and 16 k points (Li2O is an insulator,
for metals 1000 k points is a good starting point).
•Dstart: make sure to pick non spin polarized, unless there is reason to believe other-
wise (is there a transition metal in the sample?)
Assuming there were no warnings in the final run through init lapw, the case can begin
to be converged.
5
4 Convergence
Ideally, every variable should be converged regarding the simulation. Typically this is cell
parameters, k points and Rkmax. The first step is to just make sure the calculation converges,
running it with a small RKmax and few kpoints and making sure it finishes without error.
Once finished altering parameters in init lapw, run $run lapw -p to start a calculation. If
errors occur, search for them in the userguide, or in the final chapter of this guide. Otherwise
proceed with convergence.
4.1 Cell parameters:
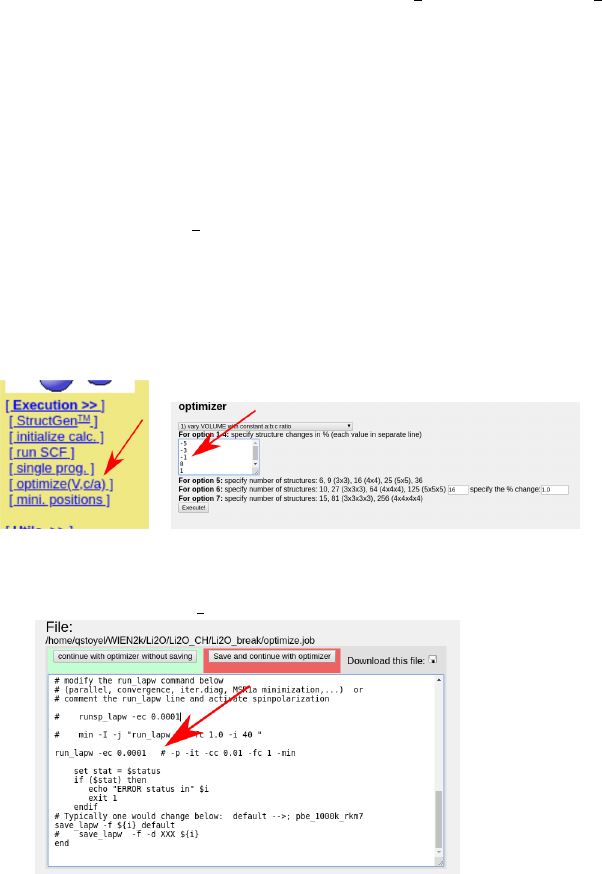
This process works best from W2Web, as described in the tutorials and using the muffin
tins from setrmt reduced by a healthy percentage (∼5-10%) to avoid nn errors (you may
need to rerun init lapw). As a number (5-11+) of calculations are run in this process, a
low number of k points and RKMax values is ideal here. For Li2O, I used 16 kpoints and an
RKmax of 6. In the x “optimize” tab, choose what you want to optimize, the first option
(volume) works well, unless there are suspicions otherwise. Enter a range of values of test
volumes, see picture:
Also make sure to edit “optimize.job” to enable parallization by moving the “#” to after
the “-p” in the run lapw line:
You can then “run optimize.job” from w2web. This job is okay to run in the background,
which means the browser can be closed without the job stopping. When it is done, plot the
“Energy vs Volume,” which should look something like this:
6

The important feature here is that the overall trend of the curve is clear, if too few k
points are chosen, the points will be “noisy.” In the case of Li2O, the used lattice parameter
of 4.6590˚
A results in the minimum energy, so no further changes are needed. If we needed a
structure with a different lattice parameter, search the case directory for all the struct files
and rename the appropriate one to case.struct, eg “Li2O vol 3.0.struct →Li2O.struct”.
Alternatively, cut and paste the lattice parameters from this file into the w2web structgen
tool.
4.2 Setting the Muffin Tin Size
This is the first step of the process that is specific to calculating the Lithium ELNES. In
order to have the ability to insert core holes, the lithium 1S state needs to be treated as a
core state. This requires playing with the muffin tin radius and the energy cutoff value in
lstart. Rerun init lapw with no reduction in muffin tin size, and during lstart, use a cuutoff
energy of -3.5 Ry (The Li 1 S state has an energy of -3.8 Ry). Investigate the case.outputst
file (it should pop up) for the following lines for the lithium atom and look at the 1S states:
E−up (Ry) E−dn (Ry) Occupancy q/ sp her e core−state
1S −3.801968 −3.785309 1 . 00 1. 0 0 0.9 90 4 T
1S −3.801968 −3.785309 1 . 00 1. 0 0 0.9 90 4 T
2S −0.236711 −0.003297 1 . 00 0. 0 0 0.0 59 5 F
2S −0.236711 −0.003297 1 . 00 0. 0 0 0.0 59 5 F
These indicate the core states (T/F), their energy levels (in this case ∼-3.8eV) and how
much the electrons in these states are contained in the muffin tins (0.9904). As this is less
than 1, it means that some 1S lithium electron is leaking out of the muffin tins, which is
why there should now be all kinds of warnings popping up. So go ahead and “ctrl-c” out of
init lapw.
To fix the leakage problem, the Lithium muffin tins need to be bigger. Ideally, they should
be just big enough to hold all of the 1S electrons, without making them too different from the
other muffin tins, as the larger this difference, the harder things get to calculate/converge.
In this case, try Li=2.0, O=1.8 and try init lapw again making sure to discard the suggested
7

muffin tins from setrmt. If that still didn’t work (lstart still has leakage errors), keep going
until it does. If the RMT values are too different (eg if RMTLi ≈1.5RMTO) they will cause
errors (ghostbands) further along. The solution then is to try and minimize the leakage (and
ignore the warnings) and acknowledge that it might be unavoidably causing artifacts. Once
the spheres are set in the struct file, rerun init lapw.
4.3 K point and RKMax convergence
To converge these parameters, again start with very low values and then increase them,
checking the total energy to determine when they are converged. Generally k points are
easier to converge, so start with them and then move on to RKMax. The procedure for
converging both of these values is:
•set/increase kpoints or RKMax, either by re-running “x kgen” or editing case.in1 and
case.in1 st.
•Run the scf cycle using “run lapw -p -NI”, the NI flag means it will continue from
where the previous calculation left off which can save time. The final converged choice
should still be run from scratch though (rerun init lapw, rm *broyd* before running
run lapw)
•Check the energy in case.scf. To do this, find it in “scf files” on w2web and use ctrl+f
in your browser to search the file for “:ene” , which should appear in a line that looks
like:
:ENE : ∗∗∗∗∗∗∗∗∗∗ TOTAL ENERGY IN Ry = −180.82588608
There will be one of these lines for each scf cycle, so find the last one in the document
and note the energy.
•loop through the first 3 steps until the energy no longer changes significantly when you
increase the kpoints/RKMax. A table is useful here to track these effects eg:
kpoints RKMax Energy
16 6.0 -180.82588608
32 6.0 -180.85055185
64 6.0 -180.85166466
32 7.0 -180.85723055
32 8.0 -180.85860820
32* 8.0* -180.85977640
Depending on the case, the energy will only converge so far. For the above table, I would
choose 32 k points and RKMax of 8.0. there were only minimal changes (<0.001Ry) to the
energy beyond those. The starred case indicate the calculation being run from scratch.
8
5 Density of States
In order to determine whether or not a core hole is necessary, the first step is to analyze the
DOS. These are best calculated in w2web, clicking the buttons.
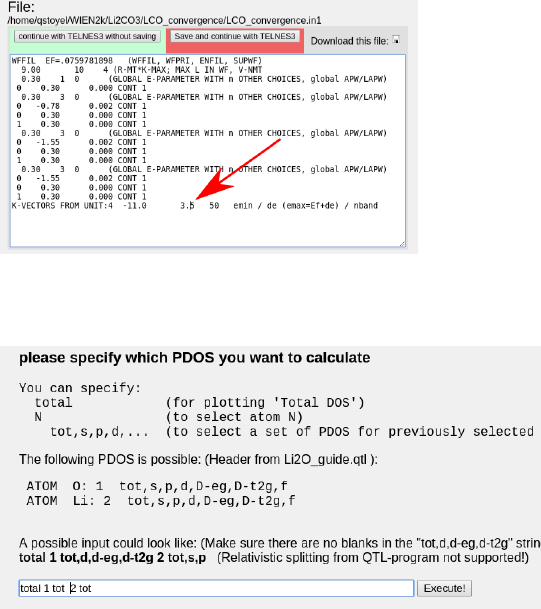
To start, run the optional steps and increase the upper energy limit in case.in1 from 1.5
to ∼2-3.5, see picture. This value defines how many higher energy states are included in
Ry (1 Ry ≈13.6 eV, 1.5Ry ≈20eV). Therefore, to obtain the correct DOS and ELNES for
features more than 20eV from the onset, this should be increased to match.
Then boost the k points value by an order of magnitude to pull out the finer features in
the DOS, and run x lapw -p and x lpaw2 -qtl -p. Then select the total DOS of each atom
in case.int, using the w2web interface:
Run x tetra and plot the DOS with dosplot, which should give you something like this:
9
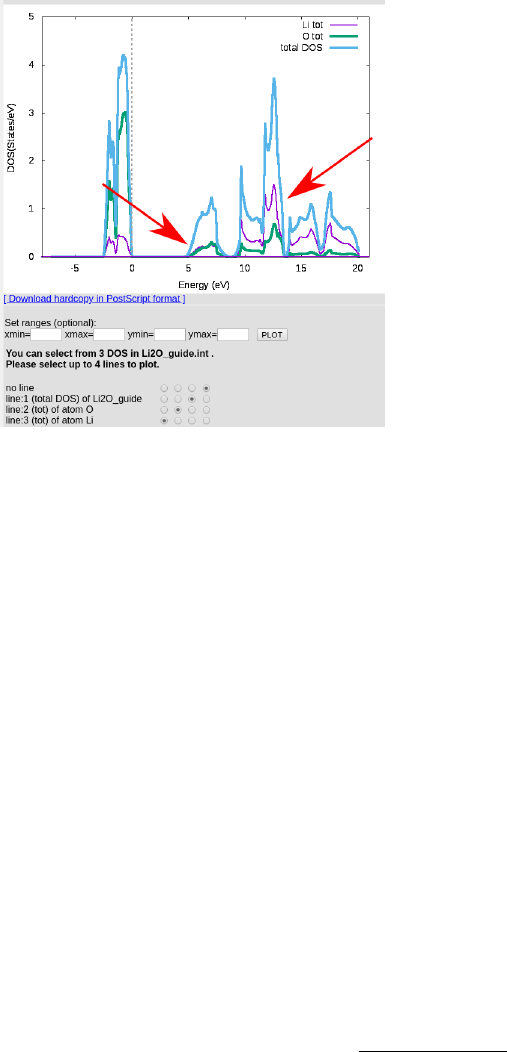
What we are looking for here is whether the conduction band has a large component
of unoccupied lithium states, which it does in this case, indicated by the arrows. If the
unoccupied states were purely oxygen in nature, no core hole effect would be expected, as
there would be no states to alter effecting the ELNES. For more insight on this behavior,
the reader is referred to Mauchamp, Jaouen and Schattschneider (2009) “Core-hole effect in
the one-particle approximation revisited from density functional theory.”
As there is indeed a large component of lithium states in the low conduction band, a core
hole is necessary. Before that, we calculate an initial ELNES for comparison later.
6 TELNES3
Once the calculation is converged, ELNES can be calculated. There is a good deal of useful
information and a complete description of the input file in the wien2k userguide, which
should solve most issues. Again, there is a large list of parameters that must be set for in
order to obtain reasonable results. It is also worth converging Kpoints and RKMax against
the spectra as well.
The majority of the important parameters need to be set in the case.innes file, which is
easiest through w2web. Choose the right atom (in this case Li1) for the edge, and the right
atomic numbers:
Edge n l
K 1 0
L1 2 0
L23 2 1
M45 3 2
Next, set the edge onset, edge values can be found at http://www.kayelaby.npl.co.
uk/atomic_and_nuclear_physics/4_2/4_2_1.html as well as at a number of other loca-
tions. Set the beam energy to it’s correct value, same goes for the collection and convergence
10
angles, although TELNES is relatively robust to these: eg 5mrad produces very similar re-
sults to 1 mrad.
Set the energy grid to a large range of values, eg -20-50eV so you can see all of the
features that might appear. The defaults for the remaining values should be fine.
In addition to the case.innes parameters, increase the number of kpoints, to at least dou-
ble, or 10×, so that there is less doubt about this being converged, use x kgen for this. This
should already have been done for calculating the DOS.
Increase the upper energy limit in case.in1 from 1.5 to ∼2-3.5, see picture. This value
defines how many higher energy states are included in Ry (1 Ry ≈13.6 eV, 1.5Ry ≈20eV).
Therefore, to obtain the correct ELNES for features more than 20eV from the onset, this
should be increased to match.
x lapw1 -p, x qtl -telnes -p, and x telnes3 can then all be run in succession. These
are relatively slow commands, so to avoid waiting on them, run all three in sequence from
the command line: $x lapw1 -p && x qtl -telnes -p && x telnes3.
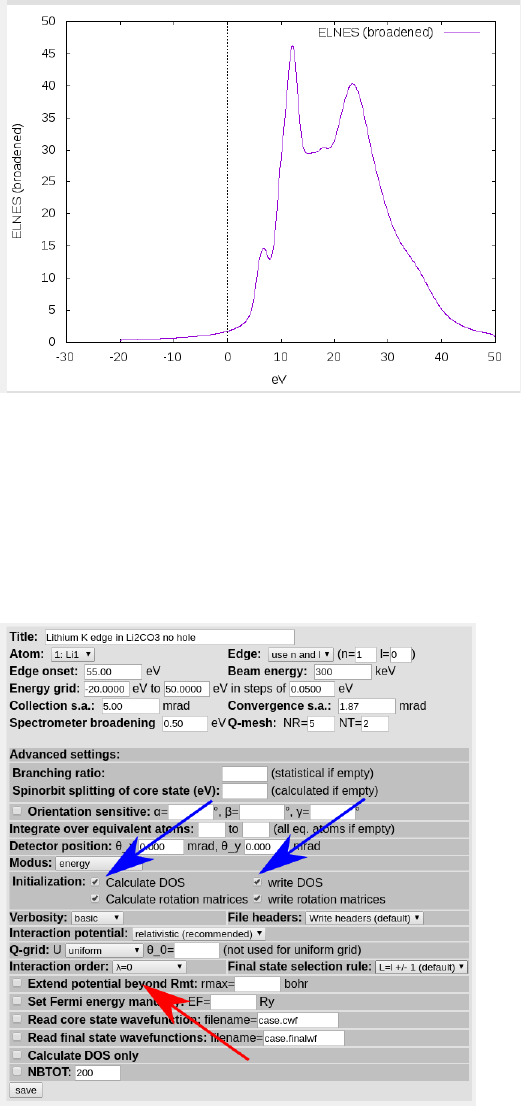
Once telnes3 is finished, edit case.inb and play with the spectrometer broadening on
the last line, before clicking “x broadening” to generate the final spectra. To experiment
with different broadening values, just repeat these steps. Sometimes w2web gets stuck and
stops re-broadening the spectra, in which case, just delete the case.broadspec file and run
“x broadening” again. Plot the spectrum in a new tab (ctrl+click on “plot”) for easy com-
parison to new spectra without having to save it. Rerun all the TELNES steps, but with
a higher/lower number of kpoints to confirm that that is not effecting the spectra. At this
point you can also uncheck the “calculate and write DOS” checkboxes in case.innes, which
allow you to rerun telnes without needing to rerun lapw1 each time (unless you change
the k points/case.in1 file). If everything went well, the ELNES should look something like
(broadening = 0.5):
11
6.1 Monopole effects
Because the lithium 1S state is quite delocalized, it is susceptible to monopole effects. These
cause artifacts in the spectra resulting from non orthogonal states at the muffin tin bound-
ary. Fortunately, it is easy to check for them. Return to the case.innes file and change the
interaction order to 0.
Telnes now only calculates the monopole component, look in case.outputelnes to confirm
this. Compare the monopole contribution to the full spectra, it should be much smaller
(∼100×). If it is not, set the interaction order to 1 to enforce dipole selection and use that
for all future spectra.
12
7 Core Hole
To introduce a core hole, start an entirely new case, and copy only the struct and the .ma-
chines files. Because of the periodic boundary conditions, it is often necessary to use a
supercell to avoid core holes interacting with themselves in neighbouring cells. This depends
largely on the size of the original unit cell, for a single atom case (metallic lithium) a 3×3×3
cell is required, for large calcium phosphates, no cell is needed.
Li2O is a small sized cell, so a 2×2×2 supercell is a good starting point, which would
need to be compared to a 3×3×3 supercell for full rigorousness.
To generate the supercell, run x supercell in the new directory and use the copied struct
file, with no shift.
With all core holes, it is essential to remove the symmetry in the unit cell, so there is
only a single hole per cell:
To achieve this, the cell must be reduced to a P1 space group. This can be done in
VESTA. Import the struct file and “remove symmetry”, under “Edit →Edit Data →Unit
Cell.” The spacegroup should switch to P-1, then select “apply”
To save these changes select “File →Export data” and save the structure as a .cif.
13
Running Cif2struct name of cif.cif should then be able to generate a valid struct file,
which should be renamed as case.struct. For some reason, I am only able to do this with my
local install of wien2k/cif2struct, the remote version complains about “ Space group name
nor symmetry operations are not given!” I have attached the final struct file that I used to
the github directory. It might be necessary to install wien2k after all...
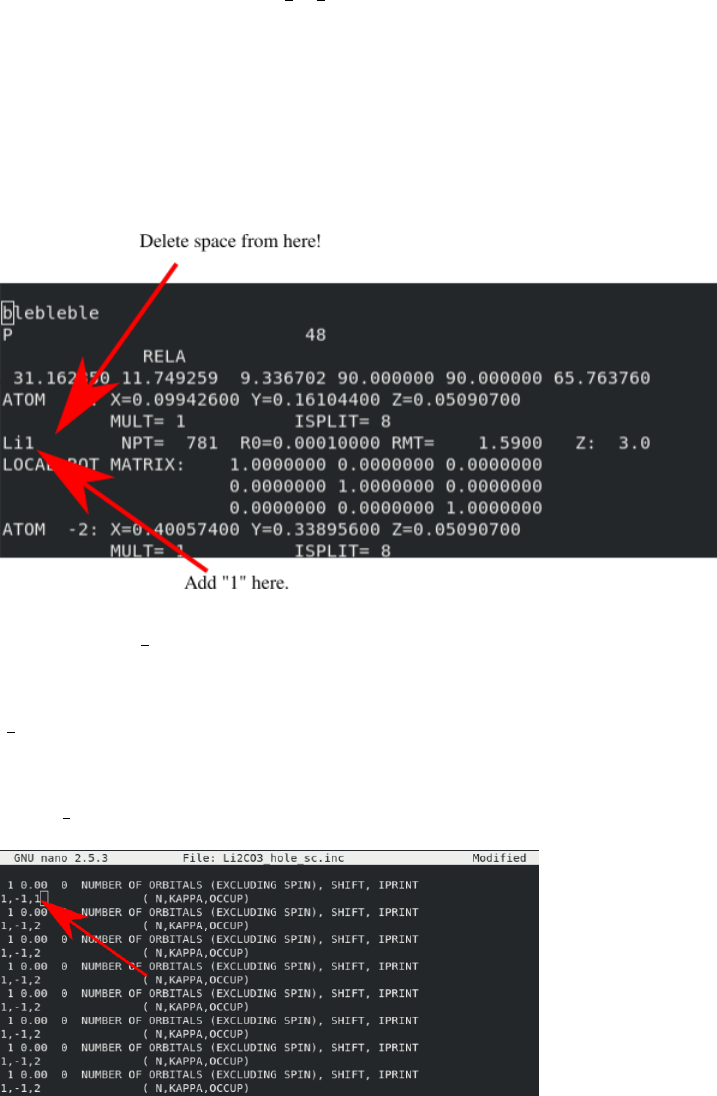
In the new struct file, add a “1” to the end of a lithium atom (AND DELETE A SPACE).
The file should look like this:
Now run init lapw and accept all of the defaults proposed by nn, which may require
cycling through nn a couple times. sgroup should not have any suggestions here, if it does,
something has gone wrong and it is trying to restore all of the symmetry. At the end of
init lapw, make sure to double check that the struct file is still what it should be in VESTA.
When the struct file is ready, initialize use the converged parameters (RMT, RKMax)
from the single cell case. (divide the number of k points appropriately, in this case by 8)
After init lapw, insert a core hole into the cell by editing case.inc and case.inm:
In case.inc, a hole is inserted by changing the occupancy of an orbital, the trick in this
case is picking out one of the isolated lithium atoms (with mult=1) which depends on where
you added the 1 to the struct file. Each set of lines corresponds to the core states of each atom
defined in the struct file. This is why it is important to be able to treat the lithium states as
core states, otherwise it would not be possible to insert a hole. This is also why it is impor-
tant to break the symmetry in the unit cell as if not, this would insert holes into multiple
14
atoms. In the case.inm file, the excited electrons need to be added to the background charge:
The value is negative as this operation adds charge, not electron number. Once the
background charge is added, execute run lapw -p and calculate the ELNES. This might
take some time, so you may want to run it remotely using tmux so you can leave it overnight
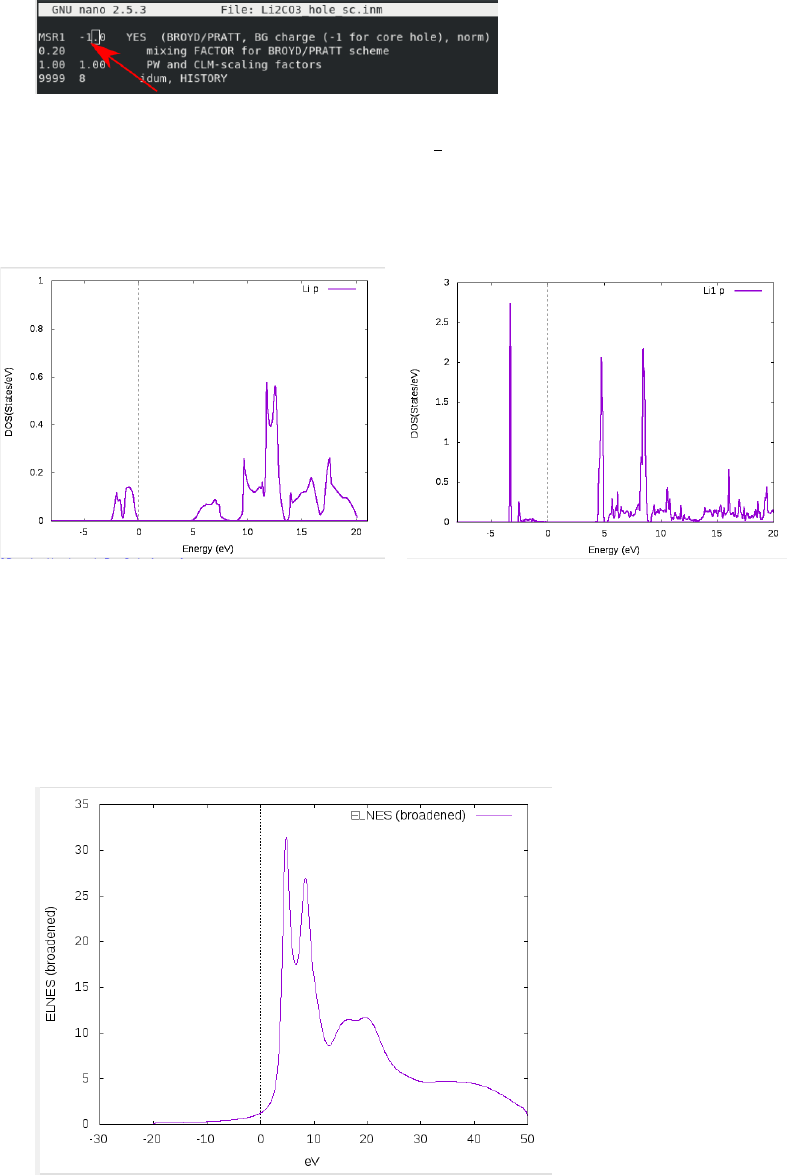
if need be. Once done, calculate the DOS of the Lithium p states and compare to the no
hole case:
Figure 1: Li P states from no-hole case (left) and hole case (right). Note the dramatic peak
growth at 4eV and 7 eV in the hole case, indicating excitonic effects.
The dramatic differences in the unoccupied band when a core hole is introduced, combined
with the significant fraction of total unoccupied states represented by the lithium band,
indicates core hole effects in the ELNES. Calculating the ELNES, should look something
like this (broadening = 0.9), if it all went well:
Having determined the need to include a core hole in the calculations, the next step is
15
determining if the hole is shielded, which can be done using density calculations.
8 Density Calculations
Density calculations can be done using either xcrysden (qualitative) or Critic2 (quantitative).
8.1 Xcrysden
Xcyrsden density rendering is well explained in the online wien2k tutorials to which the
reader is referred to. Depending on your particular setup, density rendering with xcrysden
may need to be performed locally, which will also require install wien2k locally. For Li2O,
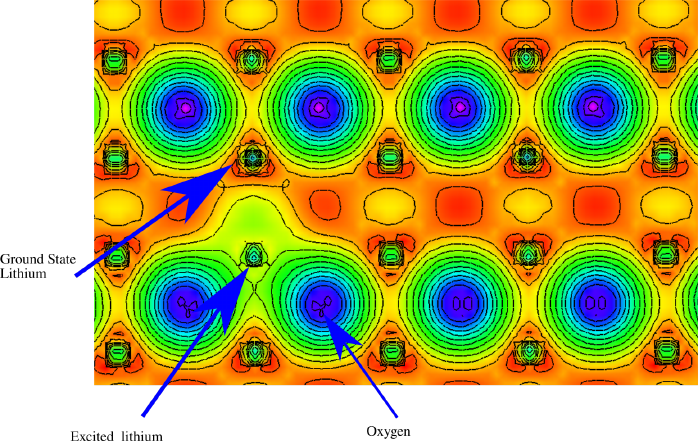
the density looks something like:
Qualitative investigation of plot shows the excited lithium atom (atom with a hole)
distorting the oxygen electron clouds. The atomic basin appears smaller, defined by the ring
of yellow surrounding the excited atom, although the entire region around the excited atom
has experienced a net gain in electron count (more green than red). These features suggest
that there is a non negligible response from the material to the core hole. The regularity
of the ground state lithium’s appear to indicate that the distance between excited lithium
atom is sufficient to isolate the core holes. In this case this distance is more than 9˚
A which
is considered suitable for this purpose.
8.2 Critic2
While Xcrysden is ideal for qualitative analysis, Critic2 is better suited to quantifying elec-
tron density features. The documentation for critic, while very complete, is in a giant txt
file. The code takes three inputs: the case.struct file, the case.clmsum file, and an input
parameters file “more.cri”. A sample .cri file is below:
c r y s t a l . / Li2O . s t r u c t
16
load . / Li2O . clmsum . / Li2O . s t r u c t
auto
i n t e g r a l s ga u le g 50 50 cp 2 v erb os e
s p h e r e i n t e g r a l s gau le g 50 50 cp 2 R0 1d−1 REND 2 . 0
The first line inputs the crystal structure, and the second line tells the code where to find
the clmsum/struct files in the second line.
The third line determines the location of all the critical points.
The 4th line calculates the electron population inside the atomic basin surrounding crit-
ical point 1. “Gauleg” is an integration option, look in the critic documentation for more
options. The two 50’s are sampling parameters, increase/decrease these for more/less accu-
rate calculations.
The 5th line calculates the electron population in spheres surrounding critical point 2.
“gualeg 50 50” mean the exact same as on the previous line, R0 is the starting radius, 1d-1
sets the radius step size, REND is the final radius (in bohr).
The program is run using $critic2 <more.cri >case.cro in the command line in
the case directory. The final file (case.cro) is just where the output is stored and can be
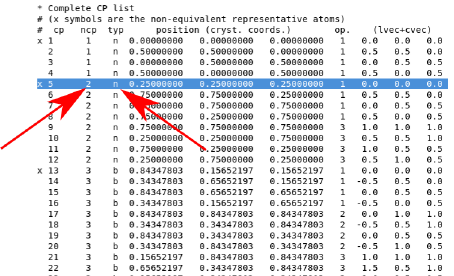
called anything. To determine which critical points are needed for the integrals, look at the
complete cp list in the output file:
There are a number of similar lists which can be used to identify which atom is of interest.
The critical point should be a nuclear critical point (typ = n), and as a double check, the
coordinates should match up to those in the struct file. The name should also match, in this
case, the ncp 1 corresponds to “Li1.”
The electron populations are located towards the bottom of the document. The first case
looks like this:
17
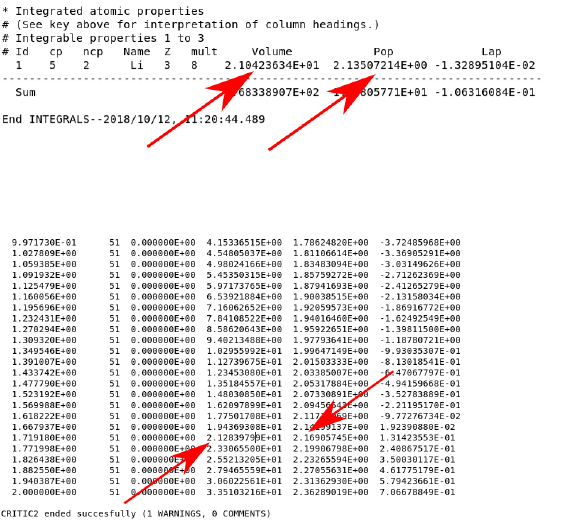
The two values indicated by the arrows are the volume of the basin and how many elec-
trons are inside. The second population is calculated directly below this in a table:
By matching to the volume value, relatively comparable values should be attainable (2.14
vs 2.17), in the no hole case. The reason for performing two types of population calculation
is due to the fact that the atomic basins can become ill defined when a core hole is inserted.
This is revealed when the volume of the basin is dramatically larger (>20%) than the no
hole case. In these situations, a comparison of the sphere integrals at the same volume (set
by the basin volume in the no hole case) is more appropriate.
In the case of Li2O, the no hole case had a population of 2.135 and the no hole case had a
population of 1.283 (and a smaller basin volume). This indicates that the core hole is slightly
screened and has an effective strength of 2.135-1.283 = 0.852. To implement this effect, a
third, final supercell calculation is required, with a reduced hole inserted. case.inc would
need an occupancy of 1.15 and case.inm would need a background charge of -0.85. This can
be rapidly set up by copying the struct file from the full hole calculation and initializing
from there.
For this partial hole calculation, the only relevant property is the ELNES calculation as
the DOS are somewhat unphysical. This ELNES should represent the best agreement with
experiment, if all has gone well and the calculations are converged with all aspects. If all
goes well, it should like this:
18
9 Common Errors/Issues Encountered in Wien2k
9.1 Setting RMT/RKMax
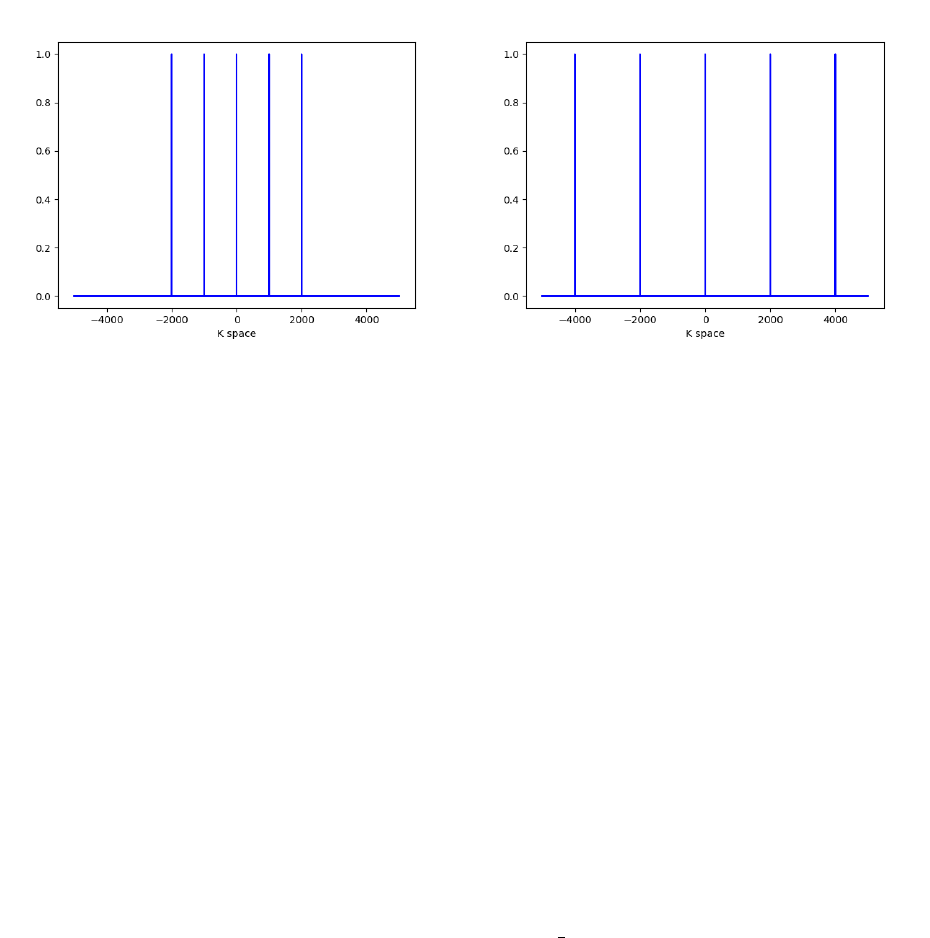
The RKMax value is a little obscure and takes some getting used to. It is defined as the
Muffin Tin Radii ×Maximum K point vector. The maximum k point corresponds to the
highest frequency plane wave used in the calculation. RKMax defines a cutoff for sampling
k space, based on the size of each atom. A visualization of this effect is depicted below:
Two case, both with 5 kpoints and different RKMax’s. The plot on the left depicts a smaller
RKMax relative to the one on the right.
Increasing RKMax allows for higher frequency (more precise) plane waves to be used in
the basis set. These high frequency terms are however more computationally costly (CPU
requirements scale as RKMax3) and less essential for describing large features. This is why
the RKMax includes the Muffin Tin radius: large atoms only require lower frequency plane
waves. Each atom has an effective RKMax which is relative to the ratio of its radius and
that of the smallest atom. Choosing very different muffin tin radii for different elements
leads to issues as it becomes more difficult to align the surface features of each atom.
Increasing RKMax arbitrarily does not solve the problem either, as: 1. “approximate
numerical linear dependency” occurs at large RKMax values and 2. Calculations become
prohibitively expensive. RKMax should at most be between 9-10.
9.2 Ghostbands
Ghostbands are an inevitable consequence of investigating lithium with Wien2k. They man-
ifest as an LAPW2 qtl error during the scf cycles, typically during the first cycle, but
sometimes later as well. They are recorded the lapw2( n).error files as:
’ l2main ’ −QTL−B.GT. 1 5 . , Ghostbands , check s c f f i l e s
Ghostbands are arise in a number of situations, each of which requires a different solution.
Some of these are described below:
19
•Muffin Tins badly chosen: These appear when the muffin tins are too different, or
if you ignore everything that setrmt does. Solution: Diagnose this by running the
calculation with the setrmt values (might need to remove the core hole) or make the
muffin tins more reasonable (might need to allow core leakage).
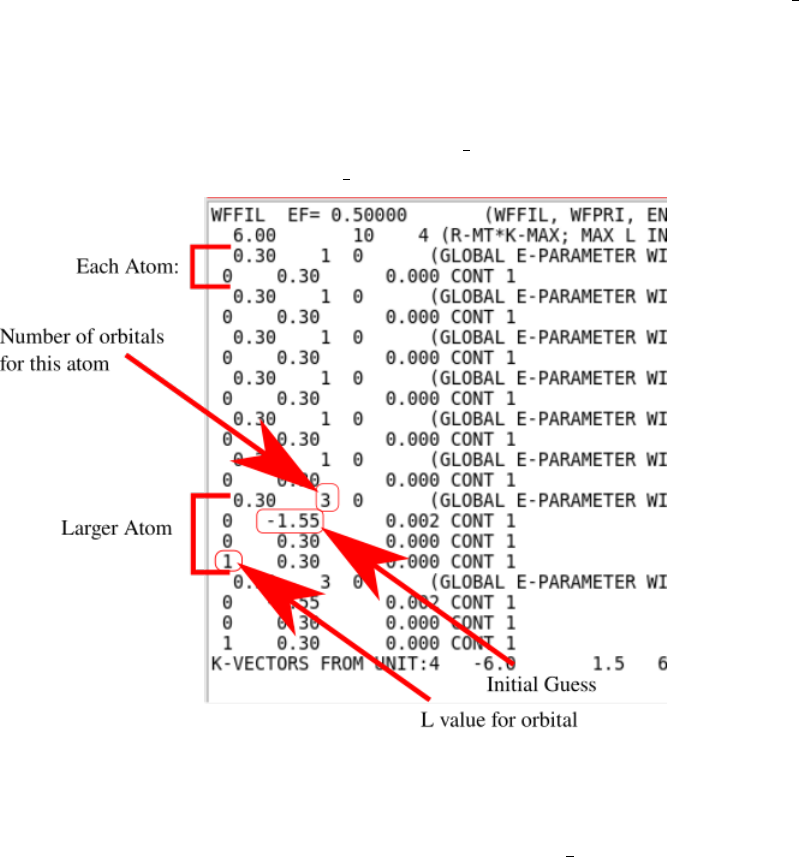
•Local Orbitals need better initial guess: Sometimes the starting points for local
orbitals are not close enough to the converged values resulting in divergence. Solu-
tions: Figure out which orbitals are causing the issue by looking in case.scf2( n). The
last lines of these files should have a line like this:
:WARN : QTL−B v al ue eq . 9 9 .9 5 i n Band o f energy −0.12674 ATOM=
2 L= 0
This message also tells us to look in case.in1( st) and that the problem is with atom 2
and the L=0 orbital. In case.in1( st), every independent atom is listed, eg:
To handle ghostbands, either delete the relevant local orbital line (lines that match
the atom and orbital number with energy guesses that are not 0.30) and reduce the
number of orbitals for that atom correspondingly. A more refined option is to adjust
the initial guesses to another value, and rerunning run lapw.
9.3 NN in Optimization
Crashes the first scf cycle almost immediately, due to overlapping muffin tins resulting from
a decreased cell size. Solution: decrease all muffin tin sizes before running “x optimize”.
20

9.4 NR and NT
Before considering a spectra as final, it is important to verify that it is converged with respect
to the Q mesh as well. The Q mesh defines which changes in momentum are collectable by
the detector. More information on exactly how the mesh is determined can be found in the
Telnes section of the userguide. To test convergence, increase these values from NR=5, and
NT=2 to larger values, (eg. 7,3), rerun telnes( qtl and telnes3) and make sure that the fine
structure does not change.
9.5 GMax Value less than Gmin
Occurs in dstart, fix is to bump up the Gmax value in case.in2 from 12.00 to 14.00 or 16.00.
9.6 Ordering
If a calculation run apparently unchanged, a possible reason is that all the edits were over-
written by init lapw. The order of a calculation should be:
•Make edits to case.struct file, ie. Muffin Tin Radius
•run init lapw
•Edit .in files. eg. setting RKMax, dealing with ghostbands, inserting core hole, in-
creasing k points...
•run lapw
•Increase Kpoints, run case specific calculations, and/or loop back to and run forwards
from there (ie increasing k points/RKMax does not require rerunning init lapw, but
changing muffin tin radii does).
21
