Gulp4.4 Manual
gulp4.4_manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 173 [warning: Documents this large are best viewed by clicking the View PDF Link!]
General Utility Lattice Program
Version 4.4
Julian D. Gale
Nanochemistry Research Institute, Curtin Institute for Computation,
Department of Chemistry,
Curtin University,
P.O. Box U1987, Perth, WA 6845,
Australia
email: gulpcode@curtin.edu.au
1
Chapter 1
Introduction & background
The General Utility Lattice Program (GULP) is designed to perform a variety of
tasks based on force field methods. The original code was written to facilitate the
fitting of interatomic potentials to both energy surfaces and empirical data. How-
ever, it has expanded now to be a general purpose code for the modelling of con-
densed phase problems. While version 1.0 focussed on solids, clusters and embed-
ded defects, the latest version is also capable of handling surfaces, interfaces, and
polymers.
As with any large computer program (and GULP currently runs to about 460,000
lines) there is always the possibility of bugs. While every attempt is made to ensure
that there aren’t any and to trap incorrect input there can be no guarantee that a user
won’t find some way of breaking the program. So it is important to be vigilant and
to think about your answers - remember GIGO! Immature optimising compilers can
also be a common source of grief. As with most programs, the author accepts no
liability for any errors but will attempt to correct any that are reported.
As from GULP3.4, the program has migrated to Fortran 90 and should compile
with any standard f90 compiler. From version 4.0 the code included several ma-
jor additions, the most significant of which are the addition of the ReaxFF reactive
force field and also the facility to use continuum dielectric solvation in multiple
dimensions through the COSMIC algorithm, derived as the name would suggest
from COSMO. A further major improvement is the addition of a stochastic thermo-
stat/barostat with a new integrator in molecular dynamics. This is far more robust
than the old schemes used and less sensitive to the choice of parameters. Finally,
the ability to calculate PDF data has been added courtesy of Beth Cope and Mar-
tin Dove (University of Cambridge at the time; now Queen Mary, University of
London). In version 4.2 the calculation of thermal conductivity has been added
based on the Allen-Feldman approach for diffusons. This was achieved through a
collaboration with Jason Larkin (Carnegie Mellon, Pittsburgh).
2
1.1 References for GULP
The following papers describe various aspects of GULP:
Basic algorithms and symmetry adaption:
•J.D. Gale, J.C.S. Faraday Transactions, 93, 629-637 (1997)
Detailed background theory:
•J.D. Gale and A.L. Rohl, Molecular Simulation, 29, 291-341 (2003)
Free energy minimisation:
•J.D. Gale, J. Phys. Chem. B, 102, 5423 (1998)
Review of status and functionality:
•J.D. Gale, Z. Krist., 220, 552-554 (2005)
Solvent models in GULP:
•J.D. Gale and A.L. Rohl, Molecular Simulation, 33, 1237-1246 (2007)
ReaxFF in GULP:
•J.D. Gale, P. Raiteri and A.C.T. van Duin, PCCP, 13, 16666-16679 (2011)
Pair Distribution Functions in GULP:
•E.R. Cope and M.T. Dove, J. Appl. Cryst., 40, 589-594 (2007)
Thermal conductivity in GULP:
•J.M. Larkin and A.J.H. McGaughey, Phys. Rev. B, 89, 144303 (2014)
1.2 Overview of program
The following is intended to act as a brief summary of the capabilities of GULP to
enable you to decide whether your required task can be performed without having
to read the whole manual. Alternatively it may suggest some new possibilities for
calculations!
3
System types
•0-D (clusters and embedded defects)
•1-D (polymers, screw dislocations)
•2-D (slabs, surfaces, grain boundaries, interfaces)
•3-D (bulk materials)
Energy minimisation
•constant pressure / constant volume / unit cell only / isotropic / orthorhombic
•thermal/optical calculations
•application of external isotropic or anisotropic pressure
•user specification of degrees of freedom for relaxation
•relaxation of spherical region about a given ion or point
•symmetry constrained relaxation
•unconstrained relaxation
•constraints for fractional coordinates and cell strains
•Newton/Raphson, conjugate gradients or Rational Function optimisers
•BFGS or DFP updating of hessian
•limited memory variant of BFGS for large systems
•search for minima by genetic algorithms with simulated annealing
•free energy minimisation with analytic first derivatives
•choice of regular or domain decomposition algorithms for first derivatives
•minimisation of gradient/force norm instead of energy
•uniaxial stress
Transition states
•location of n-th order stationary points
•mode following
4
Crystal properties
•elastic constants
•bulk modulus (Reuss/Voigt/Hill conventions)
•shear modulus (Reuss/Voigt/Hill conventions)
•Young’s modulus
•Poisson ratios
•compressibility
•piezoelectric stress and strain constants
•static dielectric constants
•high frequency dielectric constants
•frequency dependent dielectric constants
•static refractive indices
•high frequency refractive indices
•NMR chemical shifts
•stress tensor
•phonon frequencies
•phonon densities of states (total and projected)
•phonon dispersion curves
•S/P-wave velocities
•Born effective charges
•zero point vibrational energies
•heat capacity (constant volume)
•entropy (constant volume)
•Helmholtz free energy
•pair distribution function (PDF) calculation
•thermal conductivity (Allen-Feldman model)
•Raman susceptibility tensors
5
Molecular properties
•centre of mass
•moment of inertia tensor
•rotational partition function
•translational partition function
•rotational/translational free energy
Defect calculations
•vacancies, interstitials and impurities can be treated
•explicit relaxation of region 1
•implicit relaxation energy for region 2
•energy minimisation and transition state calculations are possible
•defect frequencies can be calculated (assuming no coupling with 2a)
Surface calculations
•calculation of surface and attachment energies
•multiple regions allowed with control over rigid or unconstrained movement
•can be used to simulate grain boundaries and interfaces
•calculation of phonons allowed for region 1
•continuum solvation of surfaces
Fitting
•empirical fitting to structures, energies and most crystal properties
•fit to multiple structures simultaneously
•simultaneous relaxation of shell coordinates during fitting
•fit to structures by either minimising gradients or displacements
•variation of potential parameters, charges and core/shell charge splits
•constraints available for fitted parameters
6
•simplex or BFGS minimisation algorithms for fitting
•generate initial parameter sets by the genetic algorithm for subsequent refine-
ment
•fit to quantum mechanically derived energy hypersurfaces
•bond lengths, bond angles, volume and reaction energies now available as
observables
Structure analysis
•calculate bond lengths/distances
•calculate bond angles
•calculate torsion angles
•calculate improper torsion angles
•calculate out of plane distances
•calculation of the density and cell volume
•electrostatic site potentials
•electric field gradients
Structure manipulation
•convert centred cell to primitive form
•creation of supercells
Electronegativity equalisation method
•use EEM to calculate charges for systems containing H, C, N, O, F, Al, Si, P
•use QEq to calculate charges for any element
•new modified scheme for hydrogen within QEq that has correct forces
•Gasteiger charge calculation based on bond increments
•calculation of charges from ReaxFF
7
Generation of files for other programs
•GDIS (.gin/.res)
•THBREL/THBPHON/CASCADE (.thb)
•MARVIN (.mvn)
•Insight (.xtl file)
•Insight (.arc/.car files)
•G-Vis (.xr)
•Cerius2 (.arc/.xtl/.cssr)
•Materials Studio
•COSMO solvent accessible surface for Materials Studio
•SIESTA (.fdf)
•Molden (.xyz)
•QMPOT (.frc)
•General (.cif/.xml)
•DLV (.str)
•lammps_pot (Table of potentials for use in LAMMPS)
Output files for post-processing
•Pressure tensor (.pre)
•Oscillator strengths
•Pair distribution function (.pdf)
•Charges and bond orders (.qbo)
•MD trajectory file (.trg)
8
Interatomic potentials available
•Buckingham
•Four-range Buckingham
•Lennard-Jones (with input as A and B)
•Lennard-Jones (with input in εand σformat)
•Lennard-Jones (with ESFF combination rules)
•Morse potential (with or without Coulomb subtract)
•Harmonic (with or without Coulomb subtract)
•General potential (Del Re) with energy and gradient shifts
•Spline
•Spring (core-shell)
•Spring with cosh functional form
•Coulomb subtract
•Coulomb with erfc
•Coulomb with short range taper
•Inverse Gaussian
•Damped dispersion (Tang-Toennies)
•Rydberg potential
•Covalent exponential form
•Breathing shell harmonic
•Breathing shell exponential
•Coulomb with complementary error function
•Coulomb with short range taper
•Covalent-exponential
•Fermi-Dirac form
9
•Three body potentials - harmonic with or without exponential decay
•Exponential three-body potential
•Urey-Bradley three-body potential
•Stillinger-Weber two- and three-body potentials
•Stillinger-Weber with charge softening
•Axilrod-Teller potential
•Four-body torsional potential
•Ryckaert-Bellemans cosine expansion for torsional potential
•Out of plane distance potential
•Tsuneyuki Coulomb correction potential
•Squared harmonic
•Mei-Davenport twobody
•Glue potential
•Short range potentials based on (complementary) error functions
•Embedded atom method for metals (Sutton-Chen potentials and others)
•Modified Embedded Atom Method of Baskes and co-workers
•Two-body potentials can be intra- or inter-molecular, or both
•Two-body potentials can be tapered to zero using cosine, polynomial or Voter
forms
•Universal force field functional forms for three-, four-body cases and out of
plane
•Universal force field combination rules available
•Angle-angle cross out of plane potential
•Lennard-Jones potential between atoms and a plane
•Radial force between all atoms and a point in space
•Sixbody out of plane - out of plane cross potentials
10
•Grimme C6 damped dispersion
•Environmentally Dependent Interatomic Potential (EDIP)
•ReaxFF
•Central Force Model (CFM) potentials
•Baskes twobody potential
•Ziegler-Biersack-Littmark (ZBL) potential
•Torsion potentials can be specified as improper
•Externally defined potentials via OpenKIM
Coulomb summations
•Ewald sum for 3-D
•Parry sum for 2-D
•Saunders et al sum for 1-D
•Cell multipole method for 0-D
•Wolf et al sum for 0-,1-,2-, & 3-D
Molecular dynamics
•Shell model (dipolar and breathing) molecular dynamics
•Finite mass or adiabatic algorithms
•Forward extrapolation of shells added for adiabatic algorithms
•NVE or NVT (Fixed cell) or NPT (Variable cell shape)
•Stochastic integrator with thermostat/barostat
•isotropic or orthorhombic cell constraints
•Atom-based potential energy decomposition and forces added to trajectory
file
11
Monte Carlo
•Rigid molecules allowed for
•Displacement or rotation of species
•NVT or Grand Canonical ensembles allowed
1.3 Introduction
The simulation of ionic materials has a long history going back over most of the
last century. It began with lattice energy calculations based on experimental crys-
tal structures through the use of Madelung’s constant [1]. This was then expanded
through the inclusion of short-range repulsive interactions, as found in the work
of Born-Lande and Born-Mayer [2], in order that the crystal structure be a mini-
mum with respect to isotropic expansion or compression. For many simple ionic
materials a reasonable estimate of the lattice energy may even be obtained without
knowledge of the structure, as demonstrated by the work of Kapustinskii [3]. Over
the last few decades atomistic simulation, in which we are only concerned with
atoms, rather than electrons and sub-atomic particles, has developed significantly
with the widespread use of computers. Correspondingly the field has evolved from
one that was initially concerned with reproducing experimental numbers, to one
where predictions are being made, and insight is being offered.
The widespread use of atomistic simulation for solid state materials clearly re-
sulted from the availability of computer programs for the task, just as much as the
advent of the hardware to run the calculations. In the early days of solid state
forcefield simulation for ionic materials much of the work in the UK was centred
around the Atomic Energy Authority at Harwell. Consequently a number of com-
puter codes arose from this work, such as HADES [4], MIDAS [5], PLUTO [6],
METAPOCS and CASCADE [7]. Eventually these migrated into the academic
domain, leading to the THB suite of codes, including THBREL, THBFIT and
THBPHON from Leslie. Further development of these programs led to the PARA-
POCS code from Parker and co-workers [8], for free energy minimisation of solids
using numerical derivatives, and the DMAREL code from the group of Price for
the simulation of molecular crystals through the use of distributed multipoles [9].
There were also several other prominent codes developed contemporaneously to the
above family, in particular the WMIN code of Busing [10], the PCK series of pro-
grams from Williams [11], and the UNISOFT program of Eckold et al [12]. While
the codes mentioned above focus specifically on static lattice and quasiharmonic
approaches to simulation, it should not be forgotten that there was a much larger,
parallel, development of forcefield software for performing molecular dynamics
simulation leading to programs such as GROMOS [13], AMBER [14], CHARMM
[15], and DL_POLY [16, 17], to name but a few.
12

This article focuses on the General Utility Lattice Program which began devel-
opment in the early 90’s, and is therefore subsequent to much of the aforemention
software, but implements many of the same ideas. However, there are increasingly
many new, and unique, developments as well. The key philosophy was to try to
bring together many of the facilities required for solid state simulation, with partic-
ular emphasis on static lattice/lattice dynamical methods, in a single package, and
to try to make it as easy to use as possible. Of course, this is an aim and the degree
of success depends on the perspective of the end user! It is important to also men-
tion here the programs METADISE from Parker and co-workers[18], and SHELL
from the group of Allan [19], which are also contemporary simulation codes sharing
some of the same ideas.
In this work, the specific aim is to document the very latest version of GULP,
4.0, which includes many new features over previous versions. Firstly, we detail
the background theory to the underlying methods, some of which is not readily
available in the literature. Secondly, we present a brief review of the utilisation of
the code to date, in order to highlight the scope of its previous application. Finally,
we present some results illustrating the new capabilities of the latest version.
1.4 Methods
The starting point for the majority simulation techniques is the calculation of the
energy, and so will it be for this article. Most methods are based around the ini-
tial determination of the internal energy, with subsequent treatment of the nuclear
degrees of freedom in order to determine the appropriate free energy to the ensem-
ble of interest. In principle, the internal energy of a solid is a manybody quantity
that explicitly depends upon the positions and momenta of all electrons and nuclei.
However, this is an intractable problem to solve at any level of theory, and thus ap-
proximations must be made to simplify the situation. To tackle this we assume that
the effect of the electrons will largely be subsumed into an effective atom, and that
the energy can be decomposed into an expansion in terms of interactions between
different subsets of the total number of atoms, N:
U=
N
∑
i=1
Ui+1
2
N
∑
i=1
N
∑
j=1
Ui j +1
6
N
∑
i=1
N
∑
j=1
N
∑
k=1
Ui jk +....
where the first term represents the self energies of the atoms, the second the pairwise
interaction, etc. This decomposition is exact if performed to a high enough order.
However, we know that the contribution from higher order terms becomes progres-
sively smaller for most systems, and so we choose to neglect the terms beyond
a certain point and introduce a degree of parameterisation of the remaining terms
to compensate. Justification for this is forthcoming from quantum mechanics. It
is well known that the Hartree-Fock method is a reasonable first approximation for
13

the description of many systems, albeit with a systematic quantitative error for most
observables. Here the highest term included is a four-centre integral, which indi-
cates that including up to four-body terms should be reasonable approach, which is
indeed found to be the case for most organic systems, for example. Furthermore, it
is intuitively obvious that the further apart two atoms are, the weaker their interac-
tion will be. Thus the introduction of distance cut-offs is a natural way to simplify
the computational task.
The form of the explicit interaction between atoms is usually chosen based on
physical insights as to the nature of the forces between the particles. For instance, if
considering a covalent diatomic molecule the natural representation of the potential
energy surface would be a Morse potential since this is harmonic at the minimum
and leads to dissociation at large bond lengths, in accord with spectroscopic ob-
servation. In the following sections we will review some of the common types
of potential that are widely used, as well as some novel approaches which point
towards the future of forcefield methods.
1.4.1 Coulomb interaction
When considering ionic materials, the Coulomb interaction is by far the dominant
term and can represent, typically, up to 90% of the total energy. Despite having the
simplist form, just being given by Coulomb’s law;
UCoulomb
i j =qiqj
4πε0ri j
it is in fact the most complicated to evaluate for periodic systems (subsequently
atomic units will be employed and the factor of 4πε0will be omitted). This is be-
cause the Coulomb energy is given by a conditionally convergent series, i.e. the
Coulomb energy is ill-defined for an infinite 3-D material unless certain additional
conditions are specified. The reason for this can be readily understood - the inter-
action between ions decays as the inverse power of r, but the number of interacting
ions increases with the surface area of a sphere, which is given by 4πr2. Hence, the
energy density of interaction increases with distance, rather than decaying. One so-
lution to the problem, proposed by Evjen [20], is to sum over charge-neutral groups
of atoms. However, by far the most widely employed approach is the method of
Ewald [21] for three-dimensional materials. Here the conditions of charge neu-
trality and zero dipole moment are imposed to yield a convergent series with a
well-defined limit. To accelerate the evaluation, the Coulomb term is subjected to
a Laplace transformation and then separated into two components, one of which
is rapidly convergent in real space, and a second which decays quickly in recipro-
cal space. Conceptually, this approach can be viewed as adding and subtracting a
Gaussian charge distribution centred about each ion [22]. The resulting expressions
14

for real and reciprocal space, as well as the self-energy of the ion, are given below:
Ureal =1
2
N
∑
i=1
N
∑
j=1
qiqj
ri j
er f cη1
2ri j
Urecip =1
2
N
∑
i=1
N
∑
j=1∑
G
4π
VqiqjexpiG.ri jexp−G2
4η
G2
Usel f =−
N
∑
i=1
q2
iη
π1
2
Uelectrostatic =Ureal +Urecip +Usel f
Here qis the charge on an ion, Gis a reciprocal lattice vector (where the special
case G=0 is excluded), Vis the volume of the unit cell, and ηis a parameter that
controls the division of work between real and reciprocal space. It should also be
noted that although the reciprocal space term is written as a two-body interaction
over pairs of atoms, it can be rewritten as a single sum over ions for more effi-
cient evaluation. The above still leaves open the choice of cut-off radii for real and
reciprocal space. One approach to defining these in a consistent fashion is to min-
imise the total number of terms to be evaluated in both series for a given specified
accuracy, A[23]. This leads to the following expressions:
ηopt =Nwπ3
V21
3
rmax =−ln(A)
η1
2
Gmax =2η1
2(−ln(A))1
2
Note that the above expressions contain one difference from the original derivation,
in that a weight parameter, w, has been included that represents the relative com-
putational expense of calculating a term in real and reciprocal space. Tuning of
this parameter can lead to significant benefits for large systems. There have been
several modifications proposed for the basic Ewald summation that accelerate its
evaluation for large systems, most notably the particle-mesh [24], and fast multi-
pole methods [25, 26]. Furthermore, there are competitive approaches that operate
purely in real space for large unit cells and that scale linearly with increasing size,
such as the hierarchical fast multipole methods, though care must be taken to ob-
tain the same limiting result by imposing the zero dipole requirement. This latter
approach can also be applied to accelerating the calculation of the Coulomb energy
of finite clusters.
15

In principle, it is possible to calculate the Coulomb energy of a system with a
net dipole, µ, as well. The nature of the correction to the Ewald energy can be
determined as a function of the shape of the crystal, and the formula below can also
be employed [27]:
Udipole =2π
3Vµ2
However, the complication of using the above correction is that it depends on the
macroscopic dipole of the crystal, and even on any compensating electric field due
to the environment surrounding the particle. Hence the dipole moment is usually
ill-defined, since it depends on the surfaces, as well as the bulk material. Even if
we neglect surface effects, the definition of the dipole moment is ambiguous since
the operator is not invariant under translation of atomic images by a lattice vector.
Consequently, we will take the Ewald result as being definitive.
Similarly, it is possible to relax the charge neutrality constraint, and to perform
calculations on charged supercells [28], provided care is taken when constructing
thermodynamic cycles. This is often used when probing defect energetics as an
alternative to the Mott-Littleton method. Here the net charge, Q(Q=∑qi)is neu-
tralised by a uniform background charge, leading to an energy correction of:
Ubackground =−π
2VηQ2
It should be noted that this correction ensures that a consistent Ewald sum energy
is computed regardless of the value of eta. However, to correct the energy to the
limit of no net interaction between the charged cells (and therefore no cell stress)
requires the addition of a positive Madelung correction. For the special case of a
simple cubic lattice, the correction is given by:
Umadelung =2.837297
2aεQ2
Here the value 2.837297 is the Madelung constant, ais the cell length and εis the
dielectric constant. The general situation is more complex and therefore only the
simple cubic case correction can be automatically added at present.
So far we have only considered how to handle infinite 3-D solids, but the same
issues exist for lower dimensionalities. Again for a 2-D slab, the Coulomb sum
is only conditionally convergent and so an analogous approach to the Ewald sum
is usually taken, originally devised by Parry [29, 30]. Here the slab, or surface,
is taken so as to be oriented with the surface vectors in the xy plane and with the
surface normal lying parallel to z. The energy contributions in real and reciprocal
space are given by:
Ureal =1
2
N
∑
i=1
N
∑
j=1
qiqj
ri j
er f cη1
2ri j
16

Urecip
1=1
2
N
∑
i=1
N
∑
j=1∑
G
π
A
qiqjexpiG.ri j
|G|"exp|G|zi jer f c |G|
2η1
2
+η1
2zi j!+
exp−|G|zi jer f c |G|
2η1
2−η1
2zi j!#
Urecip
2=−1
2
N
∑
i=1
N
∑
j=1
2πqiqj
A
zi j.er f η1
2zi j+
exp−ηz2
i j
(πη)1
2
Usel f =−
N
∑
i=1
q2
iη
π1
2
Note, there are now two terms in reciprocal space involving the 2-D reciprocal
lattice vector, G, and here Ais the surface area of the repeat unit, while zi j is the
component of the distance between two ions parallel to the surface normal. Again
it is possible to relax the dipolar and charge neutrality constraints within the repeat
directions. However, the approach to correcting the energy is far more uncertain,
and it is necessary to make approximations [31]. As per the 3-D case, the optimum
value of the convergence parameter can also be determined [32]:
ηopt =πw
A
Recently, another approach has been proposed for the calculation of the 2-D Coulomb
sum, which is reported to be faster than the Parry method for all cases, and espe-
cially beneficial as the number of atoms increases [33].
Lowering the dimensionality further, we arrive at 1-D periodic systems which
represents a single polymer strand, for example. At this dimensionality the Coulomb
sum becomes absolutely convergent, though at a very slow rate when performed di-
rectly in real space. Summing over charge neutral units accelerates the process,
though convergence is still somewhat tardy. While there have been several pro-
posed approaches to accelerating the Coulomb sum, we find that the method pro-
posed by Saunders et al [34], in which a neutralising background charge is applied,
is effective. Here there are three contributions to the energy given by:
Ureal
1=1
2
+M
∑
m=−M
N
∑
i=1
N
∑
j=1
qiqj
ri j +ma
Ureal
2=−1
2
N
∑
i=1
N
∑
j=1
qiqj
alnq(u+x)2+y2+z2+u+x+
lnq(u−x)2+y2+z2+u−x−2lna
17
Ureal
3=1
2
N
∑
i=1
N
∑
j=1
qiqjξM,ri j+ξM,−ri j
where the first term is summed over all images of iand jin the unit cells from −M
to M,ais the 1-D repeat parameter in the xdirection, and the remaining variables
and functions are defined as below:
u=aM+1
2
ξM,ri j=−
M
∑
i=1
Eia2i−1W2i−1u+x,y2+z2
Wn(u+x,α) = ∂
∂un(u+x)2+α−1
2
Although the required extent of the summations, given by M, to achieve a given
precision is not known a priori, the method can be implemented in an iterative
fashion so that the degree of convergence is tested as the number of lattice repeats
is increased.
One alternative approach to the above methods for performing the Coulomb sum
in any dimensionality is that due to Wolf et al [35]. Their approach involves a purely
real space summation that is asymptotic to the Ewald limit given a suitable choice
of convergence parameters. It is based around the concept of ensuring that the sum
of the charges of all ions within a spherical cut-off region is equal to zero and that
the potential goes smoothly to zero at that cut-off. This is achieved by placing an
image of every ion that a given atom interacts with at the cut-off boundary, but in
the diametrically opposite direction. While this approach can be applied directly to
the Coulomb potential, convergence is slow. Better results are obtained by using
a damped form of the potential, which is chosen to be equivalent to the real space
component of the Ewald sum, with subtraction of the associated self-energy of the
corresponding Gaussian. In this form the expression for the energy is:
UWol f =1
2∑
i
∑
j
qiqj er f cαri j
ri j −lim
ri j→rcut (er f c αri j
ri j )!−
∑
i
q2
ier f c(αrcut )
2rcut
+α
π1
2
where αis the convergence parameter, closely related to the factor ηin the Ewald
sum, and rcut is the cut-off radius. There is a trade-off to be made in the choice
of parameters within this sum. The smaller the value of α, the closer the con-
verged value will be to the Ewald limit. However, the cut-off radius required to
achieve convergence is also increased. This summation method has been imple-
mented within GULP for 1-D, 2-D and 3-D calculations, though by default the
18

term due to the limit of the distance approaching the cut-off is omitted from the
derivatives in order to keep them analytically correct at the expense of the loss of
smoothing.
Before leaving the topic of how Coulomb interactions are evaluated, it is impor-
tant to note the special case of molecular mechanics forcefields. Here the Coulomb
interaction, and usually the dispersion one too, is subtracted for interactions which
are between neighbours (i.e. bonded or 1-2) and next nearest neighbours (i.e.
which have a common bonded atom, 1-3) according to the connectivity. This is
done so that the parameters in the two- and three-body potentials can be directly
equated with experimentally observable quantities, such as force constants from
spectroscopy. Furthermore, long-range interactions for atoms that are 1-4 con-
nected are often scaled, usually by 1
2.
So far we have discussed the methods for evaluating Coulomb sums. However,
we have yet to comment on how the atomic charges are determined. In most simu-
lations, the charges on ions are fixed (i.e. independent of geometry) for simplicity
and their magnitude is determined parametrically along with other forcefield pa-
rameters, or extracted from quantum mechanical information. In the later situation
there is the question as to what is the appropriate charge to take since it depends
on how the density matrix is partitioned. Although Mulliken analysis [36] is com-
monly used as a standard, this is not necessarily the optimum charge definition for
use in a forcefield. Arguably a better choice would be to employ the Born effective
charges [37] that describe the response of the ions to an electric field, about which
more will be said later. If the charges are purely regarded as parameters then the use
of formal charges is convenient as it removes a degree of freedom and maximises
the transferability of the forcefield, especially to charged defects.
An alternative to using fixed charges is to allow the charges to be determined as
a function of the geometry. It is well documented that this environment dependance
of the charge is very important in some cases. For example, the binding energy of
water in ice is much greater than that in the water dimer. This arises due to the
increased ionicity of the O-H bond in the solid state and cannot be described cor-
rectly by a simple two-body model. In order to implement a variable charge scheme,
a simple Hamiltonian is needed that is practical for forcefield simulations. Conse-
quently most approaches to geometry-dependent charges have been based around
the concept of electronegativity equalisation [38]. Here the energy of an atom is
expanded with respect to the charge, q, where the first derivative of the energy with
respect to charge is the electronegativity, χ, and the second is the hardness, µ:
Ui=U0
i+χ0
i(qi−q0) + 1
2µ0
i(qi−q0)2+1
2qiVi
The final term in the above expression is the interaction with the Coulomb potential
due to other atoms within the system. In this equation the term q0is the charge
about which electronegativity parameters are assumed to be quadratic. Usually this
19

is value is 0 (i.e. the parameters are determined from the ionisation potential and
first electron affinity of the neutral atom). However, an exception will be noted
below.
By solving the coupled set of equations for all atoms simultaneously, this leads
to a set of charges that balance the chemical potential of the system. There are two
variants of this method in general use. In the first, typified by the work of Mortier
and co-workers [39], the Coulomb interaction, J, is described by a simple 1
rform.
The alternative, as used by Rappe and Goddard in their QEq method [40], is to
use a damped Coulomb potential that allows for the fact that at short distances the
interaction arises from the overlap of electron density, rather than from just simple
point ions. Hence, in QEq the potential is calculated based on the interaction of s
orbitals with appropriate exponents. A further variant tries to encapsulate the short
range damping of the Coulomb interaction in a mathematically more efficient form
[41];
Ji j =1
r3
i j +γ−3
i j 1
3
where the pairwise terms γi j are typically determined according to combination
rules in order to minimise the number of free parameters:
γi j =√γiγj
In the case of hydrogen, the variation of charge is so extreme between the hy-
dride and proton limits that it was necessary to make the electronegativity a func-
tion of the charge itself in the original QEq scheme. As a result, the solution for
the charges now requires an iterative self-consistent process. Here we propose a
modified formulation from that of Rappe and Goddard that drastically simplifies
the calculation of analytic derivatives:
UH=U0
H+χ0
HqH+1
2J0
HH 1+2qH
3ζ0
Hq2
H+1
2qHVH
The reason for the simplification is based on the Hellmann-Feynman theorem and
can be understood as follows. If we consider the Cartesian first derivatives of the
variable charge energy we arrive at:
dU
dα=∂U
∂ α q
+∂U
∂qα∂q
∂ α
where the first term represents the conventional fixed charge derivative, and the sec-
ond term is the contribution from the variation in charge as a function of structure.
However, if the charges at each geometry are chosen so as to minimise the total
energy of the system, then the first derivative of the internal energy with respect to
20

charge is zero and so the correction disappears. Consequently it only becomes nec-
essary to evaluate the first derivatives of the charges with respect to position when
calculating the second derivative matrix.
A variant of the QEq method has been proposed, known as EQeq [42]. This
method is essentially the same as the QEq method, except that the charge about
which the quadratic expansion of the energy as a function of charge is made (i.e.
q0) is no longer zero for all elements.
An alternative parameterisation of electronegativity equalisation has been de-
veloped by Marc Henry known as PACHA [43]. The parameters for this method
are also included in GULP. Through the PACHA formalism it is now possible to
estimate NMR chemical shifts for groups of species based on the PACHA variable
charges [44].
1.4.2 Dispersion interactions
After the Coulomb energy, the most long-ranged of the energy contributions is usu-
ally the dispersion term. From quantum theory we know that the form of the inter-
action is a series of terms in increasing inverse powers of the interatomic distance,
where even powers are usually the most significant:
Udispersion
i j =−C6
r6
i j −C8
r8
i j −C10
r10
i j −.....
The first term represents the instanteous dipole - instanteous dipole interaction en-
ergy, and the subsequent terms correspond to interactions between higher order
fluctuating moments. Often in simulations only the first term, C6
r6, is considered as
the dominant contribution. Again, the dispersion term can cause difficulties due
to the slow convergence with respect to a radial cut-off. Although the series is
absolutely convergent, unlike the Coulomb sum, the fact that all contributions are
attractive implies that there is no cancellation between shells of atoms. The problem
can be remedied by using an Ewald-style summation [45] to accelerate convergence
for materials where the dispersion term is large:
Urecip
C6=−1
2
N
∑
i=1
N
∑
j=1
Ci j π3
2
12V!∑
G
expiG.ri jG3"π1
2er f c G
2η1
2!+
4η3
2
G3−2η1
2
G!exp−G2
4η#
Ureal
C6=−1
2
N
∑
i=1
N
∑
j=1
Ci j
r6 1+ηr2
i j +η2r4
i j
2!exp−ηr2
i j
21

Usel f
C6=1
2
N
∑
i=1
N
∑
j=1−Ci j
3V(πη)3
2+
N
∑
i=1
Ciiη3
6
There can also be problems with the dispersion energy at short-range since it
tends to negative infinity faster than some expressions for the repulsion between
atoms, thus leading to collapse of the system. This can be resolved by recognising
that the above expansion for the dispersion energy is only valid for non-overlapping
systems, and that at short-range the contribution decays to zero as it becomes part
of the intrinsic correlation energy of the atom. To allow for this, Tang and Toennies
[46] proposed that the dispersion energy is exponentially damped as the distance
tends to zero according to the function:
f2nri j=1−(2n
∑
k=0bri jk
k!)exp−bri j
1.4.3 Two-body short-range interactions
Contributions to the energy must be included that represent the interaction between
atoms when they are bonded, or ions when they are in the immediate coordination
shells. For the ionic case, a repulsive potential is usually adequate, with the most
common choices being either a positive term which varies inversely with distance,
or an exponential form. These lead to the Lennard-Jones and Buckingham poten-
tials, respectively, when combined with the attractive C6term:
UBuckingham
i j =Aexp−ri j
ρ−C6
r6
i j
ULennard−Jones
i j =Cm
rm
i j −C6
r6
i j
The Buckingham potential is easier to justify from a theoretical perspective
since the repulsion between overlapping electron densities, due to the Pauli princi-
ple, which take an exponential form at reasonable distances. However, the Lennard-
Jones potential, where the exponent is typically 9-12, is more robust since the re-
pulsion increases faster with decreasing distance than the attractive dispersion term.
For covalently bonded atoms, it is often preferable to Coulomb subtract the
interaction and to describe it with either a harmonic or Morse potential. In doing
so, the result is a potential where the parameters have physical significance. For
instance, in the case of the Morse potential the parameters become the dissociation
energy of the diatomic species, the equilibrium bond length and a third term, which
coupled with the dissociation energy, is related to the vibrational frequency for the
stretching mode.
This does not represent an exhaustive list of the forms used to describe short-
range interactions, but most other forms are closely related to the above functional
22

Table 1.1: Common two-body interatomic potentials currently available within
GULP. Here rrepresents the interatomic distance of which the potential is a func-
tion. All other values are parameters of the potentials.
Potential name Functional form
Buckingham Aexp−r
ρ−C
r6
Lennard-Jones A
rm−B
rn
Lennard-Jones (ε,σ)ε n
m−nσ
rm−m
m−nσ
rn
Lennard-Jones (ε,σ)zero εhn
m−nm
nm
m−nσ
rm−m
m−nm
nn
m−nσ
rni
Lennard-Jones buffered A
(r+r0)m−B
(r+r0)n
Morse Deh(1−exp(−a(r−r0)))2−1i
Harmonic 1
2k2(r−r0)2+1
6k3(r−r0)3+1
24k4(r−r0)4
Coulomb-subtract -qiqj
r
Stillinger-Weber 2-body Aexpρ
r−rcuto f f B
r4−1
Polynomial c0+c1r+c2r2+c3r3+c4r4+c5r5
Spring 1
2k2r2+1
24k4r4
Breathing shell harmonic 1
2k(r−r0)2
Squared harmonic 1
4kr2−r2
02
forms, with the exception of the use of a spline function, which consists of a tab-
ulation of function values versus distance. A full list of the two-body potential
functional forms presently available is given in Tables 1.1, 1.2 and 1.3.
1.4.4 Polarisability
The Coulomb interaction introduced previously is just the first term of an expansion
involving moments of the charge density of an atom which includes the monopole,
dipole, quadrupole, etc. Unlike the monopole term, it is generally unreasonable to
assume that the dipole moment of an atom is fixed, since both the magnitude and
direction readily alter within the crystalline environment according to the polaris-
ability of the species. There are two approaches to modelling the polarisability that
have been widely used, which we will now introduce.
The first, and most intuitive model is to use a point ion dipolar polarisability, α,
which, in the presence of an electric field, Vf, will give rise to a dipole moment, µ,
and energy of interaction as given below:
µ=αVf
Upolarisation =−1
2αV2
f
23

Table 1.2: Less common two-body interatomic potentials currently available within
GULP. Here rrepresents the interatomic distance of which the potential is a func-
tion, and qdenotes the atomic charge of the species. All other values are parameters
of the potentials.
Potential name Functional form
General/Del Re Aexp−r
ρ
rm−C
rn
Four-range Buckingham Aexp−r
ρ,a0+a1r+a2r2+a3r3+a4r4+a5r5
b0+b1r+b2r2+b3r3,−C
r6
Inverse Gaussian −Aexp−b(r−r0)2
Tang-Toennes −C6
r6f6(r)−C8
r8f8(r)−C10
r10 f10 (r)
Qtaper qiqj
rf(r) +C(1−f(r)) where f(r)is a taper function
Polynomial-harmonic (c0+c1r+c2r2+c3r3+c4r4+c5r5)(r−r0)2
Qerfc qiqj
rer f cr
ρ
Covalent exponential −Dexp−a(r−r0)2
2r
Rydberg −A1+Br
r0−1exp−Br
r0−1
Fermi-Dirac A
(1+exp(B(r−r0)))
Cosh-spring k2d2coshr
d−1
Tsuneyuki - form 1 (Q1Q2−q1q2)g(r)
r
where g(r) = (1+ζr)exp(−2ζr)
Tsuneyuki - form 2 (Q1Q2−q1q2)g(r)
r
where g(r) = 1+11ζr
8+3(ζr)2
4+(ζr)3
6exp(−2ζr)
Q over r2 QiQj
r2
Force constant d2E
da.db = (KL−KT)a.b
r2+δabKT
where a,bare Cartesian coordinates
Erf-Erfc Aer f (r
α)er f c(r
β)
r
Repulsive Erfc Aer f c(r
β)
r
Erf Aer f (r
β)
r
24

Table 1.3: More of the less common two-body interatomic potentials currently
available within GULP. Here rrepresents the interatomic distance of which the
potential is a function. All other values are parameters of the potentials.
Potential name Functional form
Short-range glue a14(r−d)4+a13(r−d)3+a12(r−d)2+a11(r−d) + a10 if r<d
a26(r−d)6+a25(r−d)5+a24(r−d)4+a23(r−d)3+
a22(r−d)2+a21(r−d) + a20 if d<r<rmax
Mei-Davenport −φ0(1+δ(r
r0−1))exp(−γ(r
r0−1))
Baskes 2-body −2
Z(1+a+d∗a3)exp(−a)−Ax lnx
where x=ρre f (r)
ρ0
and a=α
r0(r−r0)
VBO 2-body Nexp −γ
1−q(r
δ)!
where N=2exp γ
1−q(r0
δ)!
Exp powers AexpB0+B1r+B2r2+B3r3
Grimme C6 −C6fdamp(r)
r6
where fdamp (r) = 1
1+exp−dr
r0−1
CFM_harmonic 1
2k(r−r0)2(1−t(r))
where t(r) = 1
21+tanhr−R
w
CFM_Gaussian kexp−ζ(r−r0)2t(r)
where t(r) = 1
21+tanhr−R
w
CFM_power A
rnt(r)
where t(r) = 1
21+tanhr−R
w
CFM_Fermi k
1+exp(ζ(r−r0))t(r)
where t(r) = 1
21+tanhr−R
w
GCoulomb −A
(r3+γ)1
3
25

This approach has the advantage that it is readily extended to higher order polar-
isabilities, such as quadrupolar, etc [47]. It has been applied both in the area of
molecular crystals, though often fixed moments are sufficient here [48], and, more
recently, to ionic materials by Wilson, Madden and co-workers [49]. The only
disadvantage of this approach is that the polarisability is independent of the envi-
ronment, which implies that it is undamped at extreme electric fields and can lead
to a polarisation catastrophy. It is well documented that the polarisability of the
oxide ion is very sensitive to its location, since in the gas phase the second electron
is unbound and only associates in the solid state due to the Madelung potential [50].
A further complication is that the scheme must involve a self-consistency cycle if
the induced multipoles on one atomic centre are allowed to interact with those on
another, though in some approaches this is neglected for simplicity.
The second approach to the inclusion of dipolar polarisability is via the shell
model first introduced by Dick and Overhauser [51]. Here a simple mechanical
model is used, whereby an ion is divided into a core, which represents the nucleus
and inner electrons of the ion and therefore has all of the mass associated with
it, and a shell, which mimics the valence electrons. Although it is convenient to
think in terms of this physical picture, it should not be taken too literally as in
some situations the shell can carry a positive charge, particularly for metal cations.
The core and shell are Coulombically screened from each other, but coupled by a
harmonic spring of force constant kcs. If the shell charge is qs, then the polarisability
of the ion in vacu is given by;
α=q2
s
kcs
By convention, the short-range forces are specified to act on the shell, while the
Coulomb potential acts on both. Hence, the short-range forces act to damp the po-
larisability by effectively increasing the spring constant, and thus the polarisability
is now environment dependent. The shell model has been widely adopted within the
ionic materials community, particularly within the UK. Although the same issue ex-
ists as for point ion polarisabilities, namely that self-consistency has to be achieved
for the interaction of the dipoles due to the positions of the shells, the problem is
transformed into a coordinate optimisation one. This can be solved concurrently
with the optimisation of the atomic core positions. The main disadvantage of this
approach is that is not naturally extensible to higher order moments, though some
attempts have been made, such as the spherical and elliptical breathing shell models.
Furthermore, when performing molecular dynamics special treatment of the shells
must be made by either using an adiabatic approach, in which the shells are opti-
mised at every timestep, or by using a technique analoguous to the Car-Parrinello
method [52], in which a fictious mass is assigned to the shell [53].
As a final note on the topic of polarisability, it is impossible to distinguish from
a phenomenological point of view between on site ion polarisation and charge trans-
fer between ions. This may explain why the combination of formal charges with the
26

shell model has been so successful for modelling materials that are quite covalent,
such as silica polymorphs. Provided the crystal symmetry is low enough, the shell
model could be viewed as representing charge transfer/covalency.
1.4.5 Radial interactions
There is a refinement to the conventional point particle shell model, which is the so-
called breathing shell model, that introduces non-central ion forces [54]. Here the
ion is assigned a finite radius, R0, and then all the short-range repulsion potentials
act upon the radius of the ion, rather than the nuclear position. A radial constraining
potential is then added which represents the self-energy of the ion. Two functional
forms are most commonly used:
UBSM−Harmonic
i=1
2KBSM (Ri−R0)2
UBSM−Exponential
i=K0
BSM (exp(ρ(Ri−R0)) + exp(−ρ(Ri−R0)))
This model has two important consequences. Firstly, it allows the change of radius
between two different coordination environments to be modelled - for example,
octahedral versus tetrahedral. This represents an alternative to using different re-
pulsive parameters in the Buckingham potential by scaling the Aterm according to
exp(−ρtet /ρoct )to correct for this effect. Secondly, the coupling of the repulsive
interactions via a common shell radius creates a many-body effect that is able to
describe the Cauchy violation (C12 6=C44)for rock salt structured materials.
1.4.6 Three-body interactions
There are two physical interpretations for the introduction of three-body terms, de-
pending on whether you take a covalent or ionic perspective. Within the former
view, as adopted by molecular mechanics, the three-body potential represents the
repulsion between bond pairs, or even occasionally lone pairs. Hence, the form
chosen is usually a harmonic one that penalises deviation from the expected angle
for the coordination environment, such 120ofor a trigonal planar carbon atom:
Ui jk =1
2k2(θ−θ0)2
At the other end of the spectrum, ionic materials possess three-body forces due to
the three-centre dispersion contribution, particularly between the more polarisable
anions. This is typically modelled by the Axilrod-Teller potential [55]:
Ui jk =k1+3cosθi jkcosθjkicosθki j
r3
i jr3
jkr3
ik
27
As with two-body potentials, there are many variations on the above themes, such
as coupling the three-body potential to the interatomic distances, but the physical
reasoning is often the same. A full tabulation of the three-body potentials is given
in Table 1.4.
1.4.7 Four-body interactions
Specific four-body interactions are usually only included in molecular mechanics
forcefields where they act to describe torsional angles. Hence the functional form
usually involves the cosine of the torsional angle with factors that reflect the equi-
librium torsional angle, φ0, and the periodicity with respect to rotation about the
central bond. The most widely used form is therefore:
Ui jkl =k4(1+mcos(nφ−φ0))
The other form of torsional potential more occasionally found is one that employs
a harmonic potential to describe the out of plane bending mode of a central atom
that has a planar coordination geometry. This is of utility when describing aromatic
systems, and has also been used in the modelling of the carbonate anion. An al-
ternative to this potential is to use so-called improper torsions, where the planar
geometry is maintained by specifying a torsional potential between atoms that are
not bonded. The option of specifying a sub-option of improper has been added to
some torsional potentials to allow them to be handled using the bonding topology.
A full list of the available functional forms of four-body potential is given in Table
1.5.
1.4.8 Six-body interactions
Although for many molecular systems four-body interactions are the highest order
needed to describe the properties, there are some cases where six-body interactions
are important. The many case is for two sp2or resonant atoms that are bonded
together, where the six-body interaction aims to keep the both groups of atoms in
the same plane and to prevent rotation about the resonant or double bond.
At present there is only one potential type for this kind of interaction, which is
the cross out of plane potential that couples two out of plane interactions for groups
of atoms that share a common bond and the energy takes the form of the product of
the out of plane distances times a constant.
1.4.9 Many-body interactions
Some important interactions for particular systems cannot be described within the
above forcefield framework. Below we describe a few selected higher order inter-
action potentials that are of significance and that are becoming more widely used,
despite the greater computational cost.
28

Table 1.4: Three-body interatomic potentials currently available in GULP. For po-
tentials with a unique pivot atom, this atom is taken to be atom 1 and θis the angle
between the vectors r12 and r13. All terms other than θ,θ123,θ231,θ312,r12,r13,
r23 are parameters of the potential.
Potential name Functional form
Three (harmonic) 1
2k2(θ−θ0)2+1
6k3(θ−θ0)3+1
24k4(θ−θ0)4
Three (exponential-harmonic) 1
2k2(θ−θ0)2exp−r12
ρ12 exp−r13
ρ13
Three (exponential) kexp−r12
ρ12 exp−r13
ρ13 exp−r23
ρ23
Axilrod-Teller k(1+3cos(θ123)cos(θ231)cos(θ312))
r3
12r3
13r3
23
Stillinger-Weber 3-body kexpρ12
r12−rcuto f f
12
+ρ13
r13−rcuto f f
13 (cos(θ)−cos(θ0))2
Bcross kr12 −r0
12r13 −r0
13
Urey-Bradley 1
2k2r23 −r0
232
Vessal k2((θ0−π)2−(θ−π)2)2
8(θ0−π)2exp−r12
ρ12 exp−r13
ρ13
Cosine-harmonic 1
2k2(cos(θ)−cos(θ0))2
Murrell-Mottram kexp(−ρQ1)fMM (Q1,Q2,Q3)
fMM =c0+c1Q1+c2Q2
1+c3Q2
2+Q2
3+c4Q3
1+c5Q1Q2
2+Q2
3+
(c6+c10Q1)Q3
3−3Q3Q2
2+c7Q4
1+c8Q2
1Q2
2+Q2
3+c9Q2
2+Q2
32
Q1=R1+R2+R3
√3,Q2=R2−R3
√2,Q3=2R1−R2−R3
√6
R1=r12−r0
12
r0
12 ,R2=r13−r0
13
r0
13 ,R3=r23−r0
23
r0
23
BAcross k12 r12 −r0
12+k13 r13 −r0
13(θ−θ0)
Linear-three-body k(1±cos(nθ))
Bcoscross k(1+bcosm(nθ))r12 −r0
12r13 −r0
13
Hydrogen-bond (A
rm−B
rn)(cosθ)pif θ>900, else 0
Equatorial 2K
n2(1−cos(nθ)) + 2Kexp(−β(r13 −r0))
UFF3 K(C0+C1cosθ+C2cos2θ)
BAcoscross k12 r12 −r0
12+k13 r13 −r0
13(cos(θ)−cos(θ0))
3Coulomb −sQ2Q3
r23
exp2 Kexp(−ρ1(r12 −r0))exp(−ρ13 (r13 −r0))
g3coulomb −A
(r3+γ)1
3
29

Table 1.5: Four-body interatomic potentials currently available in GULP. Here the
potential acts on the sequence of atoms 1-2-3-4 (except for the out of plane poten-
tial), where the torsion angle φlies between the plane containing atoms 1-2-3 and
atoms 2-3-4, θi jk is the angle between the vectors rji and rjk, and drepresents the
distance of atom 1 out of the plane of atoms 2-3-4. Note, the standard torsional
and ESFF forms can also be multiplied by exponential decays or taper functions to
create smooth potentials.
Potential name Functional form
Torsional k4(1±cos(nφ−φ0))
Ryckaert-Bellemanns ∑5
n=0cncosnφ
Out of plane k2d2+k4d4
ESFF torsion k1sin2θ123 sin2θ234 +k2sinnθ123 sinnθ234 cos(nφ)
Torsional harmonic 1
2k2(φ−φ0)2
Inversion k(1−cosφ)
Inversion squared 1
2(k
sin2(k0))(cosφ−cosk0)2
UFF4 1
2k(1−cosnφcosnφ0)
Angle-angle cross k213/4(θ213 −θ0,213)(θ214 −θ0,214) + k312/4(θ312 −θ0,312)(θ314 −θ0,314)+
k412/3(θ412 −θ0,412)(θ314 −θ0,314)
UFF out of plane k(c0+c1cosφ+c2cos2φ)
Torsion-angle cross kcosφ(θ−θo)(θ0−θ0
o)
1.4.9.1 The Embedded Atom Method
The Embedded Atom Model (EAM) is an approach that has been successful in the
description of metallic systems. Its foundations lie within density functional theory,
and is based on the tenet that the energy is a function of the electron density. To
simplify things, the EAM considers that the electron density is a superposition of
the atomic densities, and that instead of integrating the density across all space it is
sufficient just to express the energy as a function of the density at the nucleus of an
atom, summed over all particles:
UEAM =−
N
∑
i=1
f(ρi)
The above equations encapsulate the idea that the interaction between any given
pair of atoms is dependent on the number of other atoms within the coordination
sphere. Within this generic scheme there are a number of variations, based around
different functionals of the density (Table 1.6) and different representations of how
the density varies with distance (Table 1.7).
In the original work of Sutton and Chen [57], which developed and extended the
ideas of Finnis and Sinclair [58], a square root was used as the density functional,
while the density itself was represented as an inverse power of the interatomic dis-
tance. The densities from the paper of Finnis and Sinclair, both the quadratic and
30

Table 1.6: Density functionals available within the Embedded Atom Method.
Functional Functional form
Power law f(ρ) = Aρ1
n
Banerjea and Smith [56] f(ρ) = c01−1
nlnρ
ρ0ρ
ρ01
n+c1ρ
ρ0
Numerical splined data from data file
Johnson f(ρ) = F0(1−ln(x))x+F1y
where x= ( ρ
ρ0)α
βand y= ( ρ
ρ0)
γ
β
Glue f(ρ) = c14(ρ−ρ1)4+c13(ρ−ρ1)3+c12(ρ−ρ1)2+c11(ρ−ρ1) + c10 if ρ<ρ1
f(ρ) = c24(ρ−ρ2)4+c23(ρ−ρ2)3+c22(ρ−ρ2)2+c21(ρ−ρ2) + c20 if ρ1<ρ<ρ2
f(ρ) = c33(ρ−ρ2)3+c32(ρ−ρ2)2+c31(ρ−ρ2) + c30 if ρ>ρ2
Foiles f(ρ) = F0ρ2+F1ρ+F2ρ5
3
F3+ρ
Mei-Davenport −Ech1−α
βln(ρ)iρα
β+
∑3
m=1φ0sm
2exp(−γ(√m−1))h1+δ(√m−1)−δ√mlnρ
βiρ
γ√m
β
Baskes AE0xln(x)
where x=ρ
ρ0
VBO f(ρ) = Aρrn
Spline f(ρ) = A(ρ−ρ0)3+B(ρ−ρ0)2+C(ρ−ρ0) + D
combined quadratic-cubic form, can both also be used. One of the beauties of the
EAM is that, in principle, once the metal is parameterised it can be studied in other
environments, such as alloys, without further modification. On the downside, the
prediction of the relative stability of phases can be sensitive to the cut-off radius
chosen, though if care is taken this problem can be surmounted [59].
1.4.9.2 The Modified Embedded Atom Method
In the Embedded Atom Method it is assumed that the density of an atom is spheri-
cally symmetric. However, for some atoms, such as transition metals, this may not
be such a good approximation. In the Modified Embedded Atom Method (MEAM)
of Baskes [60] the density becomes a sum of terms including higher order contri-
butions:
ρ0=∑
i
ρ0
i
ρ12=∑
α ∑
i
ρ1
i
ri
α
ri!2
ρ22=∑
α,β ∑
i
ρ2
i
ri
α
ri
ri
β
ri!2
−1
3 ∑
i
ρ2
i!2
31

Table 1.7: Functional forms for the distance dependence of the atomic density avail-
able within the Embedded Atom Method.
Density Functional form
Power law ρi j =cr−n
i j
Fractional power ρi j =cr−rn
i j
Exponential ρi j =crn
i j exp−dri j −r0
Gaussian ρi j =crn
i j exp−dri j −r02
Cubic ρi j =cri j −r03for ri j <r0
Quadratic ρi j =cri j −r02for ri j <r0
Quartic ρi j =cri j −r04for ri j <r0
Voter-Chen cr6
i j exp−βri j+29exp−2βri j
EVoter cr6
i j exp−βri j+29exp−2βri jexp(−1
rmax−ri j )
Glue ρi j =b13(r−r1)3+b12(r−r1)2+b11(r−r1) + b10 if r<r1
ρi j =b23(r−r2)3+b22(r−r2)2+b21(r−r2) + b20 if r1<r<r2
ρi j =b33(r−rm)3+b32(r−rm)2+b31(r−rm) + b30 if r2<r<rm; else 0
Mei-Davenport ∑5
l=0cl
12(r
r0)l
Baskes ρi j =Aexp−βri j−r0
r0
VBO ρi j =cNσexp −γ
1−qri j
δ!
where N=exp γ
1−qr0
δ!
32

ρ32=∑
α,β,γ ∑
i
ρ3
i
ri
α
ri
ri
β
ri
ri
γ
ri!2
The total density is then the combination of these terms weighted by a separate
coefficient, tmfor each order, m:
ρ=
3
∑
m=0
tmρm
ρ02
The radial forms of the density and the embedding expressions closely resemble
those found for the EAM scheme and so they are not repeated here.
In the MEAM approach the interaction between a pair of atoms is weighted by a
screening function that naturally truncates the terms at long-range in the solid. Un-
like the more common use of a taper function, the screening function for MEAM
is a many-body term that depends on the neighbours of the pair of atoms. The idea
is that any neighbouring atom that comes between a pair of atoms will shield the
interaction. Because of the somewhat complex nature of this many-body screen-
ing function, which is then coupled with the many-body embedding potential, at
present only analytic first derivatives are available for MEAM when the screening
function is employed. In the absence of this term, analytic second derivatives are
also available. Both the MEAM-1NN (first nearest neighbour) and MEAM-2NN
(second nearest neighbour) schemes can be used in GULP.
1.4.9.3 Bond Order Potentials
Related in many ways to the embedded atom method, but with a more sophisticated
formalism, are so-called bond order potentials. It was recognised by Abell [61] that
the local binding energy could be expressed as follows:
UBO =
N
∑
i=2
i−1
∑
j=1hUrepulsive ri j−Bi jUattractive ri ji
where Bi j is the bond order between the atoms iand j. The bond order is dependent
on the local environment of both atoms and thereby converts an apparent two-body
interaction into a many-body one. Several different formulations have been pro-
posed, most notably by Tersoff [62, 63], and also more recently by Pettifor and
co-workers [64], where the latter use a more extensive analysis of the contributions
to the bond order and appeal to first principles methods to extract the parameters.
One particular model has had an enormous impact in recent years due to its appli-
cability to carbon polymorphs and hydrocarbon systems, that due to Brenner and
co-workers [65]. Unsurprisingly, it has been extensively applied to fullerenes, nan-
otubes and diamond, as systems of topical interest. One of the other reasons for the
popularity of the model is the fact that Brenner makes his code freely available. An
33

independent implementation of the Brenner model has been made within GULP,
since the capabilities of the program require that analytic derivatives to at least sec-
ond order, and preferably third, are present, which is not the case for the existing
code. To date, there exist three published variants of the Brenner potentials, but we
have implemented just the latest of these models since it superceeds the previous
two [66].
The terms in the expression for the energy in the Brenner model are expressed
as follows:
Urepulsive (r) = A f c(r)1+Q
rexp(−αr)
Uattractive (r) = fc(r)
3
∑
n=1
Bnexp(−βnr)
where A,Q,α,B1−3, and β1−3are parameterised constants that depend on the
atomic species, C or H, involved and fc(r)is a cosine tapering function to ensure
that the potential goes smoothly to zero of the form:
1r<rmin
fc(r) = 1
21+cos(r−rmin)
(rmax−rmin)π rmin <r<rmax
0r>rmax
The bond order term itself is composed of several terms;
Bi j =1
2bσ−π
i j +bσ−π
ji +1
2ΠRC
i j +bDH
i j
Note that the above expression for Bi j differs from the one given in the defining
manuscript due to the factor of a half for the second term, but is required to obtain
results that agree with those quoted. The first two terms in the above equation
represent the influence of local bond lengths and angles about the atoms i and j,
respectively, while the third term is a correction for radical character, and the fourth
one for the influence of dihedral angles. Both of the last two terms are related to the
degree of conjugation present. Full defining equations for these terms, along with
the parameters, can be found in the original reference and the subsequent errata.
In the above many-body contributions to the bond order, bicubic and tricubic
splines are used to interpolate parameter values. For the distributed Brenner po-
tential code, the spline coefficients are precomputed and supplied as data files. In
the present implementation the splines are performed internally on the fly. This has
two advantages in that it both avoids possible transcription errors, as well as loss
of precision through I/O, and allows for the possibility of parameter fitting to be
readily implemented.
Because of the short-ranged nature of the Brenner potential we have imple-
mented two different algorithms for the evaluation of the interactions. The first
involves a conventional search over all atoms to find neighbours with a non-zero
34
interaction. The second uses a spatial decomposition of the system into cubes of
side length equal to the maximum range of the potential. Consequently only atoms
within neighbouring cubes can possibly interact. This leads to a linear scaling al-
gorithm that is far more efficient for large systems. A comparison is presented in
the results section.
While the Brenner model does have many strengths, such as its ability to de-
scribe bond dissociation, there are also a few limitations. Perhaps the most sig-
nificant is the difficulty in describing long-range forces. For instance, there is no
bonding between the sheets for graphite. There have been a number of remedies
proposed, including adding on two-body potentials to describe these effects, either
only between different molecules, or with a tapering that removes the interaction at
short-range so as not to invalidate the parameterisation. However, there are limita-
tions to these approaches, though a more sophisticated expression for removing the
contribution of long-range forces where the existing interactions due to the Brenner
potential are significant shows promise [67]. As yet, this is still to be implemented.
1.4.9.4 ReaxFF
The ReaxFF force field is a reactive bond order potential model that explicitly in-
cludes long-range interactions and electrostatic effects. Developed by Adri van
Duin and co-workers, this is model is far more sophisticated (and therefore com-
plex) than the other bond order potentials currently available in GULP. The bond
order can contain separate contributions for σ,πand π−πbonding. From this
bond order, several energy contributions are calculated:
•Bond energy
•Bond penalty energy
•Lone pair energy
•Overcoordination energy
•Undercoordination energy
•Three-body valence energy
•Three-body penalty energy
•Torsional energy
•Conjugation energy
•Hydrogen bonding
35

On top of these terms, there are energy contributions from van der Waals inter-
actions that are damped at short-range where the bonding terms are important. In
addition, a charge equilibration calculation is performed to determine geometry de-
pendent charges, and thereby electrostatic and self energy contributions. It should
be noted that the electrostatics in ReaxFF use a screened Coulomb expression:
UCoulomb =1
2
N
∑
i=1
N
∑
j=1
qiqj
r3
i j +1
γi j 31
3
fri j
In the interests of speed, the Coulomb and van der Waals interactions are truncated
at 10 Å by use of taper function, f, and so no Ewald or Parry summation is nec-
essary. This is an implicit part of ReaxFF and so any errors associated with this
truncation are subsumed into the parameterisation. When computing the charges
there are two approaches available in GULP. The default method is to solve the
matrix equations directly. Alternatively an iterative approach can be used which
employs sparse matrix storage of the Coulomb interactions. For medium to large
systems the iterative method will be far more efficient and is recommended. When
running in parallel then the iterative approach must be used. It is worth noting that
the domain decomposition in GULP is particularly effective for ReaxFF because
of the relatively short cut-off. Using the spatial keyword in combination with
setting a domain size of 2-2.5 Å can often lead to maximum efficiency.
For full details of the ReaxFF formalism we refer the reader to the original
paper by van Duin et al [68]. There are a few important points to note though relat-
ing to the implementation in GULP. Firstly, the formalism for ReaxFF has evolved
since the original paper and some of the expressions for the bond order terms have
changed. Therefore older parameter sets from the literature (approximately pre-
2006) may not give exactly the right results. Secondly, at present there are only
analytic first derivatives implemented for this force field. However, second deriva-
tive properties can be computed using finite difference methods with the caveat that
there will be some numerical error.
Because there are a large number of parameters for a ReaxFF force field then
the easiest way to run calculations is by using a library file. A number are provided
already with GULP and more can be generated from the format of Adri van Duin’s
program (ffield/fort.4) using a utility that is provided. When testing converted pa-
rameter files it is important to note that the cut-offs for bond order terms (which are
in the control file of Adri’s program) can make a significant difference and so it is
important to set these values to be the same. Furthermore, there is some additional
tapering of terms added in GULP for smoothness that might lead to small changes
in the values. The verbose keyword can be useful for checking as this causes GULP
to print the energy decomposition and all the bond orders that arise from ReaxFF.
36

1.4.9.5 EDIP
Another form of bond-order potential that is supported is the Environment-Dependent
Interatomic Potential (EDIP). This was originally proposed by Bazant et al [69] and
parameterised for silicon, including defective structures [70]. It consists of two- and
three-body energy terms that depend on interatomic distances and the coordination
number, which in turn is determined by a sum over neighbouring atoms. The origi-
nal form of the potential was extended to carbon by Marks [71], which involved the
addition of πcontributions to the coordination number and a modification of the
three-body term. Both forms of EDIP are supported by GULP and library files are
provided containing the parameters.
1.4.10 One-body interactions
Going to the other extreme of complexity from many-body interactions, we have the
simplist possible atomistic model, namely the Einstein model [72]. This approach
deviates from those that have gone before in that there is no interaction between
particles and all atoms are simply tethered to their lattice site by harmonic springs:
UEinstein =1
2
N
∑
i=1
kixi−x0
i2+yi−y0
i2+zi−z0
i2
This model acts as a reference state since all interactions are purely harmonic and
consequently the phonon density of states, and also the structure, are independent
of temperature. This implies that all the quantities that can be derived from the
vibrational partition function can be analytically determined for any temperature
without approximation, unlike other potential models. Even a structure that con-
sistents of atoms coupled together by a series of harmonic potentials has implied
anharmonicity that arises from the derivatives of the transformation matrix from
the bond-oriented pairwise frame of reference to the Cartesian one.
Given that the results of the Einstein model can be derived without recourse to a
structural description, there is no need to employ an atomistic simulation program
in order to calculate the required quantities. However, it can be useful to combine
the Einstein model with a conventional, more accurate, representation of the in-
teractions for use in thermodynamic integration [73]. Since the free energy of the
Einstein crystal is known under any conditions, it is possible to extract free ener-
gies from molecular dynamics via a series of perturbative runs in which the Einstein
model is introduced to an anharmonic potential in a series of steps, as a function
of a switching parameter, λ, in order to obtain the difference relative to the known
value.
It is worth highlighting the differences between the Einstein model and all others
within the program. Because of the lack of interatomic interactions, there can be
no optimisation of the structure and no strain related properties calculated. For
37
the same reason, there is no phonon dispersion across the Brillouin zone. Finally,
because the particles are tethered to lattice sites there is no translational invariance,
and consequently all vibrational frequencies are positive and non-zero (assuming
all force constants are specified to be likewise).
1.4.11 External forces
All of the above interactions are the result of interatomic forces. However, it is
sometimes useful to include external forces on a system. One of the most common
scenarios is to applied an external electric field. While the application of an electric
field is allowed in any arbitrary direction, it is usually only sensible to apply this in a
non-periodic direction. Only attempt to apply an electric field in a periodic direction
if you are an expert and really know what you are doing. You have been warned!
For example, in a slab calculation then a field can be applied normal to the surface
leading to a polarisation of the atoms based on their charges. There are several
important points to note when using an electric field. Firstly, when optimising the
system it is important to fix at least one atom other the force will cause the system
to translate indefinitely since translational invariance is broken. Secondly, because
a field will generally not conform to the space group of a material, the turning off of
symmetry is enforced when a field is applied. Finally, there is no absolute energy of
interaction with an electric field and so the energy is computed relative to the initial
configuration, α0:
Uf ield =
N
∑
i=1∑
α
qiVααi−αi
0
A consequence of this relative definition of the energy is that there will be a discon-
tinuity in the absolute energy, but not the forces, on restarting.
In the same way that an external force can be applied by an electric field, it is
also possible to specify an external force on specific atoms without reference to their
charge. This can be static or time-dependent if performing molecular dynamics. For
the time-dependent case, the force is of the oscillatory form;
F=Acos(2π(Bt +C))
where trepresents the time.
1.4.12 Potential truncation
As previously mentioned, all potentials must have a finite range in order to be cal-
culable. However, there are a range of conditions for a potential to act between
two species, as well as various methods for handling the truncation of potentials.
Hence, it is appropriate to describe a few of these issues here.
38
1.4.12.1 Radial truncation
The natural way to truncate a potential is through the use of a spherical cut-off
radius. It is also possible to specify a minimum radius from which the potential
should act too. Hence, it is possible to create multiple distance ranges within a
potential with different forcefield parameters, as well as to overlay different poten-
tials. Of course, where there is a cut-off boundary, there will be a discontinuity in
the energy for most interaction terms, unless the potential smoothly tends to zero
at that distance by design. This can lead to problems during energy minimisation,
since the point of lowest energy may not be a point of zero force anymore, and in
other simulation techniques, such as molecular dynamics, where energy conserva-
tion will be affected by discontinuities. Other than increasing the cut-off radius,
there are several approaches to minimising these difficulties as described briefly in
the subsequent subsections.
1.4.12.2 Cut and shift
To avoid a discontinuity in the energy at the potential cut-off, a constant shift can
be added to the potential so that the energy at the cut-off boundary becomes zero:
Ushi f ted
i j ri j=Ui j ri j−Ui j (rcut )
where rcut is the cut-off radius. However, the gradient is still discontinuous at the
cut-off distance, so the procedure can be extended by adding a linear term in the
distance such that the gradient is also zero at this point. While in principle this
method can be applied to make any order of derivative go exactly to zero at the
boundary by construction, the increasing powers of distance lead to the correction
terms modifying the variation of the potential with distance more strongly as the
order rises. This characteristic, that the potential is modified away from the point
of cut-off, makes this method of smoothing less desirable than some.
1.4.12.3 Tapering
In this approach, the potential is multiplied by a smooth taper function that goes
to zero, both for its value and its derivatives typically up to second order. This
is usually applied over a short range from an inner radius, rtaper, to the cut-off
distance:
Ui j ri jri j <rtaper
Ui j ri jftaper ri jrtaper <ri j <rcut
0ri j >rcut
Hence, within rtaper the potential is unchanged. There are numerous possible
functional forms that satisfy the required criteria for a taper function. Perhaps the
two most commonly used are a fifth order polynomial or a cosine function with a
39
half a wavelength equal to rcut −rtaper. Both of these are available within GULP as
part of the overall two-body potential cut-off.
1.4.12.4 Molecular mechanics
In the simulation of molecular systems, it is often preferable to use a molecular
mechanics forcefield. This implies that certain cut-offs are determined by connec-
tivity, rather than by distance alone. For example, particular potentials may only act
between those atoms that are bonded, such as a harmonic force constant, whereas
others may specifically only act between those that aren’t covalently linked. Fun-
damental to this is the notion of a bond between atoms. Within GULP these can
either be determined automatically by comparing distances between pairs of atoms
with the sum of the covalent radii, multiplied by a suitable tolerance factor, or alter-
natively the connectivity can be user specified. From this information, it is possible
for the program to determine the number of molecules and, in the case of periodic
systems, their dimensionality.
A number of options are possible that control how both the potentials and the
Coulomb terms act, as described below:
•Bonded: A potential may act only between atoms that are bonded (1-2).
•Exclude 1-2: The case of bonded atoms is specifically omitted from the al-
lowed interactions.
•Exclude 1-3: Interactions between bonded atoms and those with a common
bonded atom are excluded.
•Only 1-4: Potentials only act between atoms that are three bonds apart. This
can be useful in the description of torsional interactions.
•1-4 and greater: Potentials only act between atoms that are three bonds apart
or further. This can be useful in the description of torsional interactions.
•Intramolecular: Only interactions within a given molecule are permitted for
the potential.
•Intermolecular: Only interactions between atoms of different molecules are
allowed.
•Bond order: An interaction may be limited to bonds of a specific order. Valid
bond orders are; single, double, triple, quadruple, resonant, amide, custom,
half, quarter and third. Note, that the automatic bond calculation assigns all
bonds to be single bonds. Hence, to use this facility it is necessary to use the
connect option to set the bond order between particular atoms.
40
•Molecular Coulomb subtraction: All electrostatic interactions between atoms
within the same molecule are excluded. This implies that the charges on the
atoms within a molecule purely serve to describe its interaction with other
molecules.
•Molecular mechanics Coulomb treatment: This implies that Coulomb terms
are excluded for all 1-2 and 1-3 interactions. In addition, it is sometimes
desirable to remove, or scale down, 1-4 electrostatic interactions too. The
benefit of this is that the parameters of the intramolecular potentials then have
a direct correspondance with the equilibrium bond lengths, bond angles and
localised vibrational frequencies. It should be noted that two-body potentials
can also be scaled for the 1-4 interaction, if so desired.
1.4.13 Partial occupancy
Many materials have complex structures which include disorder of one form or an-
other. This can typically consist of partial occupancy where there are more symme-
try degenerate sites than there are ions to occupy them, or where there are mixtures
of ions that share the same structural position. In principle, the only way to accu-
rately model such systems is to construct a supercell of the material containing a
composition consistent with the required stoichiometry and then to search through
configuration space for the most stable local minima, assuming the system is in
thermodynamic equilibrium (which may not always be the case). This includes al-
lowing for the contribution from the configurational entropy to the relative stability.
While this approach has been taken for several situations, most notably some of the
disordered polymorphs of alumina [74, 75], it is demanding because of the shear
number of possibilities which increases with a factorial dependance.
There is an approximate approach to the handling of disorder, which is to intro-
duce a mean-field approach. Here all atoms are assigned an occupancy factor, oi,
where 0 ≤oi≤1, and all interactions are then scaled by the product of the relevant
occupancies:
Um−f
i j =oiojUi j
Um−f
i jk =oiojokUi jk
and so on for high order terms. This approach can be utilised in any atomistic
simulation program by scaling all the relevant potential parameters according to
the above rules. However, for complex forcefields this can be tedious, and it also
precludes fitting to multiple structures where the occupancies of ions of the same
species are different. Hence, the inclusion of partial occupancies has been auto-
mated in GULP so that only the site occupancies have to be specified and every-
thing else is handled by the program. This includes the adding of constraints so
that atoms that share the same site move together as a single particle, as well as
41

checking that the sum of the occupancies at any given site do not exceed one. It
should be noted that the handling of partial occupancies requires particular care in
determining phonons where the matrix elements for all coupled species must be
condensed in order to obtain the correct number of modes.
The use of a mean field model is clearly an approximation. For structures, it can
often work quite well, since crystallography returns an average anyway. However,
for other quantities, such as thermodynamic data it has limitations. For example,
the excess enthalpy of mixing of two phases is typically overestimated since the
stabilisation that arises from local structural distortions to accommodate particular
species is omitted [76].
1.4.14 Structural optimisation
Having defined the internal energy of a system, the first task to be performed is to
find the minimum energy structure for the present material. To be more precise,
this will typically be a local minimum on the global potential energy surface that
the starting coordinates lie closest to. Trying to locate the global energy minimum
is a far more challenging task and one that has no guarantee of success, except for
the simplist possible cases. There are several approaches to searching for global
minima, including simulated annealing [77], via either the Monte Carlo or molec-
ular dynamics methods, and genetic algorithms [78, 79]. Here we will focus the
quest for a local minimum.
At any given point in configuration space, the internal energy may be expanded
as a Taylor series:
U(x+δx) = U(x) + ∂U
∂xδx+1
2!
∂2U
∂x2(δx)2+...
where the first derivatives can be collectively written as the gradient vector, g, and
the second derivative matrix is referred to as the Hessian matrix, H. This expan-
sion is usually truncated at either first or second order, since close to the minimum
energy configuration we know that the system will behave harmonically.
If the expansion is truncated at first order then the minimisation just involves
calculating the energy and first derivatives, where the latter are used to determine
the direction of movement and a line search is used to determine the magnitude of
the step length. In the method of steepest descents this process is then repeated
until convergence. However, this is known to be an inefficient strategy since all the
previous information gained about the energy hypersurface is ignored. Hence it is
far more efficient to use the so-called conjugate gradients algorithm [80] where sub-
sequent steps are made orthogonal to the previous search vectors. For a quadratic
energy surface, this will converge to the minimum in a number of steps equal to the
number of variables, N.
42

If we expand the energy to second order, and thereby use a Newton-Raphson
procedure, then the displacement vector, ∆x, from the current position to the mini-
mum is given by the expression;
∆x=−H−1g
which is exact for a harmonic energy surface (i.e. if we know the inverse Hessian
matrix and gradient vector at any given point then we can go to the minimum in one
step). For a realistic energy surface, and starting away from the region close to the
minimum, then the above expression becomes increasingly approximate. Further-
more, there is the danger that if the energy surface is close to some other stationary
point, such as a transition state, then simply applying this formula iteratively may
lead to a maximum, rather than the minimum. Consequently, the expression is
modifed to be
∆x=−αH−1g
where αis a scalar quantity which is determined by performing a line search along
the search direction to find the one-dimensional minimum and the procedure be-
comes iterative again, as per conjugate gradients.
By far, the most expensive step of the Newton-Raphson method, particularly
once the size of the system increases, is the inversion of the Hessian. Furthermore,
the Hessian may only vary slowly from one step to the next. It is therefore wasteful
and undesirable to invert this matrix at every step of the optimisation. This can be
avoided through the use of updating formulae that use the change in the gradient and
variables between cycles to modify the inverse Hessian such that it approaches the
exact matrix. Two of the most widely employed updating schemes are those due to
Davidon-Fletcher-Powell (DFP) [81] and Broyden-Fletcher-Goldfarb-Shanno [82],
which are given below:
HDFP
i+1=HDFP
i+∆x⊗∆x
∆x.∆g−HDFP
i.∆g⊗HDFP
i.∆g
∆g.HDFP
i.∆g
HBFGS
i+1=HBFGS
i+∆x⊗∆x
∆x.∆g−HBFGS
i.∆g⊗HBFGS
i.∆g
∆g.HBFGS
i.∆g+
h∆g.HBFGS
i.∆giv⊗v
v=∆x
∆x.∆g−HBFGS
i.∆g
∆g.HBFGS
i∆g
The BFGS algorithm is generally recognised as the more efficient one and so is
the default optimiser. However, if performing a transition state search then DFP
is preferred due to the different tendances of the updates with regard to targetting
a positive definite character for the Hessian. The practical approach taken in a
43
BFGS optimisation is to initialise the Hessian by performing an exact inversion
of the second derivatives and then subsequently updating for a number of cycles.
Occassionally it is necessary to calculate the exact inverse Hessian again. This is
triggered by one of a number of possible situations:
•The maximum number of cycles for updating is exceeded
•The angle between the gradient vector and the search vector exceeds a given
threshold
•The energy has dropped by more than a certain threshold in one cycle, so the
curvature is likely to have altered
•The energy cannot be lowered by line minimisation along the current search
vector
There is one variation upon the BFGS strategy above, in which, rather than calcu-
lating the exact second derivative matrix and inverting it, the Hessian is initialised
approximately and then updated such that it tends to the true Hessian. Two possible
starting approximations to the Hessian are either to use a unit matrix or to perform a
finite difference estimate of the on-diagonal elements only in order to achieve better
preconditioning. By taking this approach the cost is similar to conjugate gradients,
though with a higher memory requirement, but the convergence is typically twice as
fast since more information from previous energy/gradient evaluations is retained.
The choice of appropriate algorithm for minimisation depends on the size of the
system and number of variables. For small systems, the Newton-Raphson meth-
ods that use second derivatives will be far more efficient. As the size of system
increases, the computational cost of building and inverting the Hessian matrix will
ultimately grow as N3while the memory requirements also increase as N(N+1)/2
for lower-half triangular storage. Hence, ultimately it will become necessary to use
first the unit Hessian approach and then ultimately the conjugate gradients method
as Ncontinues to increase. This balance may also be influenced by the use of
symmetry, where possible, as will be discussed later. A further issue is that for
minimisation where the initial structure lies outside the harmonic region, Newton-
Raphson methods may not be particularly advantageous, whereas they will be as
convergence is approached. Hence GULP includes the facility to change the min-
imiser from, for example, conjugate gradients, to BFGS when a criterion is met,
such as the gradient norm falling below a specified threshold.
Aside from the minimisation algorithm itself, something should be said about
the criteria for convergence of an optimisation. Typically some or all of the follow-
ing can be checked for convergence:
•the gradient norm (root mean square gradient per variable)
44
•the maximum individual gradient component
•the norm of the estimated displacement vector
•the change in the function (energy, enthalpy or free energy) between succes-
sive cycles
•the change in the variables between successive cycles
Normally all of these quantities are well converged (and often excessively so - i.e.
well beyond chemical accuracy) with Newton-Raphson optimisation for a force-
field. Occassionally convergence is not achieved, which is nearly always indicative
of a significant discontinuity in the energy surface, such as that caused by inter-
atomic potential cut-offs. Ideally all potentials should be tapered so that their value,
first and second derivatives go smoothly to zero at the cut-off radius. In practice,
most repulsive potentials are negligibly small by a typical cut-off distance of 10
Angstroms and so it is usually the attractive potentials that mimic dispersion that
are responsible. The other common cause are discontinuities in bonding - i.e. an
atom is displaced beyond a bond cut-off during minimisation and so the whole
nature of the interaction potential changes. While this can be overcome by main-
taining a fixed connectivity list, the real issue is that the forcefield is inadequate for
the system being modelled.
All of the above discussion has related to the search for a local minimum where
the first derivatives are zero and the second derivative matrix is positive definite.
However, there are many instances in which it is necessary to locate other stationary
points on the potential energy surface, and in particular transition states, such as for
ion diffusion. There are a number of algorithms available for locating transition
states, of which the most widely used class in solid state optimisations tend to be
constrained minimisations. Here the reaction path is discretised in some way and
the system minimised so that the variable changes are orthogonal to the direction
of the pathway. Arguably the most successful and widely used approach is the
Nudged Elastic Band method [83]. However, the disadvantage of these constrained
techniques is that some assumption usually has to be made about the nature of the
mechanism before the calculations are performed. In contrast, there are algorithms
which use Newton-Raphson techniques in order to locate stationary points with an
arbitrary number of negative eigenvalues for the Hessian (where a transition state is
the special case of a single negative eigenvalue). In the case of GULP, the method
implemented is so-called Rational Functional Optimisation (RFO) [84].
In the RFO method, the inverse Hessian matrix is diagonalised to obtain the
eigenvalues and eigenvectors. From the eigenvalues it is possible to examine whether
the matrix has the required characteristics for the stationary point being sought. If
the number of negative eigenvalues is incorrect, then the spectrum is level-shifted to
correct this and the search direction constructed appropriately. By default, the Hes-
sian modes with the smallest eigenvalues are followed towards the corresponding
45
stationary point. However, eigenvector following can also be performed in which
different modes from the spectrum are selected. Hence the RFO optimiser will,
in principle, locate various possible transition states starting from a given position.
There are a few provisos though, most notably that there must be a non-zero pro-
jection of the gradient vector in the search direction leading to the stationary point.
Furthermore, the step size must be quite small in order to ensure convergence to a
transition state. Since the line search can no longer be used, the optimisation relies
on a fixed proportion of the search vector to achieve convergence. In the implemen-
tation in GULP, this step magnitude is correlated to the gradient norm, so that as the
calculation approaches convergence, and therefore enters the harmonic region, the
proportion of the search vector used approaches 100%. Hessian updating can also
be applied while optimising towards a transition state. However, because the rate of
convergence depends more critically on the inverse Hessian being accurate, more
frequent exact calculations are usually required. RFO can also be used to accelerate
optimisations to minima once the harmonic region is approached, since it is able
to better handle soft modes. In extreme cases, the number of cycles is reduced by
an order of magnitude by use of RFO towards the end of an optimisation, as well
confirming that the Hessian has the required structure. A final note of caution is
that this method does not guarantee that a local minimum has been achieved, even
if the Hessian is confirmed as having all positive eigenvalues. This is because the
Hessian only spans the space of the allowed optimisation variables and there may
be directions of negative curvature with respect to constrained degrees of freedom.
Hence, a Γ-point phonon calculation should always be used as a final validation.
Often it is desirable to apply external forces to a system when performing a
minimisation. The most common example is the application of an isotropic external
pressure, such as used in the determination of the equation of state for a material.
This situation can be straightforwardly handled by making the objective function
for minimisation the enthalpy instead of the internal energy:
H=U+pV
More complex is the case when an external force is to be applied to individual
atoms. This arises when the program is being coupled to a separate quantum
mechanical calculation as part of a QM/MM method (i.e. a quantum mechan-
ics/molecular mechanics coupled calculation). Here the forces on the forcefield
atoms arise from the quantum mechanical region, which is not explicitly consid-
ered in the present calculation. Adding the negative of the external forces to the
internal derivatives is simple to implement. However, for minimisation the inter-
nal energy is no longer a well-behaved objective function (i.e. the minimum of
the internal energy does not correspond to a configuration of zero gradients). One
solution to this would be to minimise the gradient norm instead, but this requires
either one order higher of energy derivatives or that conjugate gradients are used to
minimise, which is less efficient.
46

The approach implemented within GULP to handle the application of external
forces is to define a new additional term in the internal energy, which is the work
due to the external force:
Uexternal =−Zf inal
initial
Fexternal .α
where αrepresents the vector of atomic coordinates and Fexternal is the vector of
external forces. With this correction to the internal energy, the minimum coincides
with force balance and so the full range of standard minimisers are applicable. How-
ever, the final energy is an arbitrary quantity unless taken with respect to a standard
reference set of atomic positions.
The framework of structural optimisation in GULP is designed to be flexible and
to allow the user as much freedom as possible to control the degrees of freedom.
Intrinsically, there are four main classes of variables possible within the forcefield
models available, namely, atomic coordinates, the unit cell periodicity (for poly-
mers, surfaces, and bulk materials), shell coordinates, and breathing shell radii.
Each variable can be controlled by optionally specifying a binary flag that indicates
whether it is to be varied or held fixed. Alternatively there are keywords that select
default settings of the flags, such that those quantities not constrained by symmetry
are allowed to vary. In particular, the defaults automatically handle the fact that one
atom must be fixed in order to remove the three translational degrees of freedom.
For bulk systems, the first atom is usually the one chosen to be fixed, since the
choice is arbitrary. In the case of slab calculations, the atom nearest the centre of
the slab is fixed since this is the one least likely to move by a substantial amount.
When there is a centre of symmetry in the space group, then there is no need to fix
any atoms since the origin is already immoveable. The default optimisation options
also automatically construct the constraints necessary to preserve the space group
symmetry as the atoms are displaced.
When minimising periodic systems there are a couple of choices that have to be
made. The first concerns the representation of the unit cell. This can be achieved
through either the unit cell parameters, a,b,c,α,β,γ, or via the Cartesian cell
vectors as a (3x3) matrix. Correspondly, when optimising the unit cell this can
be done with respect to the cell parameters directly, the Cartesian cell vectors or
by using a strain tensor. Here we chose to use the Voigt strain tensor to scale the
Cartesian lattice vectors:
1+ε11
2ε61
2ε5
1
2ε61+ε21
2ε4
1
2ε51
2ε41+ε3
Through using the six Voigt strains as the variables, we have the same advan-
tage as working with the unit cell parameters in that cell rotation is eliminated by
construction. However, as will be demonstrated later, the derivatives with respect
47
to strain are closely related to the Cartesian internal derivatives and therefore read-
ily calculated at almost no extra cost. We should note that when strain derivatives
are taken, they are done so with respect to infinitesimal strain about the current
configuration, rather than with respect to a single constant reference cell. For 2-D
systems, the strain tensor is correspondingly reduced to a (2x2) matrix and for poly-
mers there is a single scalar strain value. At this point we should also comment on
the absolute spatial orientation of systems, since this a matter of convention, rather
than an absolute choice. For 3-D systems, GULP orients the acell vector along the
x-axis, the bcell vector in the xy-plane, and then the orientation of the ccell vector
is fixed, except for an issue of chirality, which is resolved by selecting the orienta-
tion that has a positive dot product with the z-axis. For 2-D systems, the same two
conventions are followed for the surface vectors, which leaves the surface normal
parallel to the z-axis. Finally, for 1-D systems, the repeat direction is taken to lie
along the x-axis.
1.4.15 Genetic algorithms
The approach to minimisation of a structure has already been discussed when con-
sidering the case of a local stationary point. Now we turn to consider the quest for a
global minimum. In order to do this, a technique is required that allows barriers to
be overcome in some way. While hyperdynamic [85]and basin-filling approaches
are becoming increasingly useful, the more traditional approach has been to employ
methods that incorporate random changes in the structure with a tendancy to min-
imise a so-called cost function. Here the cost function is a quantity that represents a
measure of the quality of the structure. In the most obvious case, this might be just
the total energy of the system. However, because stochastic processes require many
moves, it is often convenient to use a more computationally efficient cost function.
For example, one form that has previously been successfully used for oxides is a
combination of bond-valence sum rules with a short-ranged Coulomb penalty that
prevents ions of like charge approaching each other [86].
There are two main approaches to the global optimisation of structures via a
random walk - the Monte Carlo method [87] and Genetic Algorithms (GAs) [78,
86]. Although both are in principle equally capable of performing the task, it could
be argued that genetic algorithms are more naturally suited to coarse grained global
optimisation, while the Monte Carlo technique is also able to yield an integration
within the phase space of a statistical ensemble. Here we will concentrate on the
use of GAs.
In the genetic algorithm, each optimisation variable is discretised within a spec-
ified range. This is particularly well suited to fractional coordinates where there are
natural bounds of 0 and 1. Then all that need be specified is the number of allowed
intervals within the range. If a coarse grid of points is specified then configura-
tion space will be rapidly searched, but the solution will be very approximate and
48
the global minimum may not be resolvable with the level of resolution available.
Conversely, if a fine grid is utilised then the search is capable of locating a more
accurate estimate of the global minimum. However, because of the large number of
possible configurations, the optimal state may never be visited. Clearly a hierarchi-
cal approach may be ideal, where a coarse discretisation is initially used and then
refined as the process progresses. The state of each configuration is represented by
a binary number which is a concatination of the binary representations of the grid
point numbers for each individual variable.
The fundamental idea behind genetic algorithms is to perform a process that
mimics natural selection through survival of the fittest. Here the concept of fitness
equates to the value of the chosen cost function. Each cycle of the GA begins with
an even number of so-called parents. In the first step these are typically randomly
chosen initial configurations. These parents then are randomly paired up in order to
breed to produce two children in order to maintain the size of the population. This is
the tournament phase, in which a probability is set for the parent with the lower cost
function to go through to the next generation and to form the children. A random
number is chosen and compared to this probability in order to make the decision.
Choosing this probability threshold is a balance between wanting to ensure that the
better configurations survive against trying to maintain diversity in the population
during the initial stages. More sophisticated tournament procedures have also been
proposed where an exponential weighting is used in favour of the fitter candidates.
Usually the number of children is set equal to the number of parents in order to
maintain a stable population size.
Apart from the tournament phase, there are two other processes that can occur,
which are mutation and crossover. In the mutation phase, a number of binary digits
are changed from 0 to 1 or vice versa. Again the chance of this occuring is de-
termined by comparison of a random number to the mutation probability. During
crossover, two configurations are selected, the binary strings split at a random point,
and the two parts switched between configurations. Both mutation and crossover
represent genetic defects that are designed to introduce randomness with the aim of
exploring configuration space.
The above three genetic algorithm steps are applied repeatedly with the aim
of gradually reducing the cost functions of the population down and focussing in
on the global minimum. When the procedure is stopped after the specified num-
ber of steps, then hopefully the configuration with the lowest cost function will be
the global minimum, though this can never been known for certain for complex
systems. If the cost function used was an approximation to the total energy that
is ultimately desired to be the criterion for selection, then a selection of the best
configurations can be subsequently minimised according to the total energy and
hopefully the global minimum will lie amongst this set, even though it is not the
structure with the absolutely lowest cost function.
There are several possible refinements to the genetic algorithm procedure, which,
49

somewhat appropriately, is itself still evolving. However, the above basic formu-
lation is capable of correctly predicting the atomic coordinates of binary, and even
some ternary oxides, given the unit cell. Indeed, the method had an early success
through assisting in the solution of the previously unknown structure of Li3RuO4
[79]. More details of the background to, and application of, the genetic algorithm
for solid state structural optimisation can be found elsewhere [86].
1.4.16 Calculation of bulk properties
Having determined the optimised structure for a material it is then possible to cal-
culate a wide range of physical properties based on the curvature of the energy
surface about the minimum. These include both mechanical properties, such as the
bulk modulus and elastic constants, as well as dielectric properties. The expres-
sions used for the calculation of the individual properties that may be determined
routinely from the second derivative matrix are detailed below.
1.4.16.1 Elastic constants
The elastic constants represent the second derivatives of the energy density with
respect to strain:
Ci j =1
V∂2U
∂ εi∂ ε j
thereby describing the mechanical hardness of the material with respect to deforma-
tion. Since there are 6 possible strains within the notation scheme we employ, the
elastic constant tensor is a 6 x 6 symmetric matrix. The 21 potentially independent
matrix elements are usually reduced considerably by symmetry [88]. For example,
for a cubic material the only unique elements are C11,C12, and C44. The calculation
of elastic constants is potentially very useful, since the full tensor has only been
measured experimentally for a very small percentage of all known solids. This is
primarily because the practical determination typically requires single crystals with
a size of a few micrometres at least.
In calculating the second derivatives of the energy with respect to strain it is
important to allow for the response of all internal degrees of freedom of the crystal
to the perturbation. If we introduce the following notation for the second derivative
matrices:
Dεε =∂2U
∂ ε∂ ε internal
Dεi=∂2U
∂ ε∂ αiε
Di j =∂2U
∂ αi∂ β jε
50

then the full expression for the elastic constant tensor can be written as:
C=1
VDεε −DεiD−1
i j Djε
The elastic compliances, S, can be readily calculated from the above expression by
inverting the matrix (i.e. S=C−1). The above expressions are valid for adiabatic
conditions and at zero pressure. The adiabatic elastic constants can be extended to
the case of finite external pressure according to the corrections:
Cαβ γζ =1
V ∂2U
∂ εαβ ∂ εγζ !+p
22δαβ δγζ −δαζ δβγ −δαγ δβ ζ
Here the strains are expressed directly in terms of the Cartesian components, as
opposed to in the Voigt notation, so as to make the corrections more transparent.
Under isothermal conditions the expressions for the elastic constant tensor are
analogus, except for the fact that the differentials are with respect to the Helmholtz
free energy, rather than the internal energy. Because the evaluation of the isothermal
elastic constants requires the fourth derivative of the internal energy with respect to
internal coordinates and strains, this is not presently implemented within GULP
analytically. However, finite differences may be used in combination with free
energy minimisation in order to determine this tensor.
1.4.16.2 Bulk and shear moduli
Like the elastic constant tensor, the bulk (K) and shear (G) moduli contain infor-
mation regarding the hardness of a material with respect to various types of de-
formation. Experimentally a bulk modulus is much more facile to determine than
the elastic constant tensor. If the structure of a material is studied as a function of
applied isotropic pressure, then a plot of pressure versus volume can be fitted to an
equation of state where the bulk modulus is one of the curve parameters. Typically
a third or fourth order Birch-Murgnahan equation of state is utilised. Alternatively,
the bulk and shear moduli are also clearly related to the elements of the elastic con-
stant. However, there is no unique definition of this transformation. Here we give
three different definitions due to Reuss, Voigt and Hill [88]. Below are the equa-
tions for the Reuss and Voigt definitions, while the Hill values are defined as the
average of the other two:
KVoigt =1
9(C11 +C22 +C33 +2(C12 +C13 +C23))
KReuss = (S11 +S22 +S33 +2(S12 +S13 +S23))−1
GVoigt =1
15 (C11 +C22 +C33 +3(C44 +C55 +C66)−C12 −C13 −C23)
GReuss =15
4(S11 +S22 +S33 −S12 −S13 −S23) + 3(S44 +S55 +S66)
51

1.4.16.3 Young’s moduli
When a uniaxial tension is applied to a material then the lengthening of the material
is measured according to the strain. The ratio of stress to strain defines the value of
the Young’s modulus for that axis:
Yα=σαα
εαα
Since a material will always increase in length under tension, the value of this quan-
tity should always be positive. The Young’s moduli in each of the Cartesian direc-
tions can be calculated from the elastic compliances:
Yx=S−1
11
Yy=S−1
22
Yz=S−1
33
1.4.16.4 Poisson’s ratio
Complementary to Young’s modulus is the Poisson ratio, which measures the change
in a material at right angles to the uniaxial stress. Formally it is defined as the ratio
of lateral to longitudinal strain under a uniform, uniaxial stress. The expression
used to calculate this property, assuming an isotropic medium, is given below [88]:
σα(β) = −Sααβ βYβ
Because most materials naturally shrink orthogonal to an applied tension this leads
to positive values for Poisson’s ratio, with a theoretical maximum of 0.5. Typi-
cal values for many materials lie in the range 0.2-0.3, though negative values are
also known. For an isotropic material this quantity can also be related to the bulk
modulus:
K=1
3
Y
(1−2σ)
1.4.16.5 Acoustic velocities
The acoustic velocities are key quantities in the interpretation of seismic data. The
polycrystalline averages of these acoustic velocities in a solid can be derived from
the bulk and shear moduli of the material, as well as the density, ρ. There are two
values, that for a transverse wave, Vs, and that for a longitudinal wave, Vp, which
are given by:
Vs=sG
ρ
52

Vp=s4G+3K
3ρ
As to be expected, there is some degree of variability in the calculated values ac-
cording to the definition of the bulk and shear moduli employed.
1.4.16.6 Static and high frequency dielectric constants
The dielectric properties of a material are of crucial importance in many contexts,
including those beyond the strictly bulk properties. For example, the response of
a solid to a charged defect depends on the inverse of the dielectric constant. The
actual value of the dielectric constant varies according to the frequency of the elec-
tromagnetic field applied. Commonly two extreme values are quoted, namely the
static and high frequency dielectric constants. In the static limit all degrees of free-
dom of the crystal, both nuclear and electronic, are able to respond to the electric
field and therefore to provide screening. At the high frequency limit the oscillation
is greater than the maximum vibrational frequency of the material and therefore
only the electrons are able to respond to the perturbation fast enough. Here we
describe how to calculate these extremal values, while the case of intermediate fre-
quencies will be presented later.
The static dielectric constant (3 x 3) tensor can be determined from the Cartesian
second derivative matrix of all particles, Dαβ , and the vector, q, containing the
charges of all particles:
ε0
αβ =δαβ +4π
VqD−1
αβ q
The expression for the high frequency dielectric constant is identical to that for the
static equivalent, except that the second derivative matrix, Dαβ , now only includes
the Cartesian components for any shells present within the model. If a core only
model is being used then the high frequency dielectric tensor is just a unit matrix.
Hence information regarding the high frequency dielectric constants is particularly
useful in determining the parameters of a shell model due to the relatively direct
correlation.
Because the dielectric constant tensor depends on the inverse second derivative
matrix, it has many of the characteristics of the Hessian matrix and is therefore
quite a sensitive indicator of whether a potential model is sensible. Extreme values,
particularly negative ones, instantly point to the fact that the potential model is
inadequate or that the system wishes to undergo a symmetry change.
1.4.16.7 Refractive indices
The refractive indices of a material, n, are simply related to the dielectric constant
by:
n=√ε
53

For orthorhombic, tetragonal or cubic unit cells, in the standard orientation, this
represents a trivial mapping since the dielectric constant tensor is a diagonal matrix
and so the square roots can be taken directly. For other crystal systems it is neces-
sary to diagonalise the tensor first and then find the refractive indices in this new
axis eigensystem.
1.4.16.8 Raman susceptibility tensors
The calculation of Raman intensities uses the Raman susceptibilities. Here the key
quantities are related to the derivatives of the high frequency dielectric constant
tensor with respect to the atomic displacements through the quantity χ:
χ=ε∞−δ
4π
If the keyword is specified to compute the Raman properties then the derivatives of
χwith respect to the Cartesian coordinates of each atom in the system are output in
units of inverse Angstroms. Because this requires the use of analytical third deriva-
tives this functionality will only work for a subset of force field models where the
third derivatives are available, which includes two-, three- and four-body potentials.
Note that because this is the derivative of the high frequency dielectric constant then
this is only useful if you are working with a shell model, otherwise the answer will
be zero. Strictly speaking the derivatives output are components of the Raman sus-
ceptibility tensor and they must be projected onto the eigenvectors of the phonons
and scaled by a volume factor to get the true susceptibility tensor. If the Raman
susceptibilities are computed then these will be used to determine the intensities of
the vibrations, if this keyword is specified. The Raman intensity depends on the
direction of the incoming and outgoing wave, which can be set using an option.
1.4.16.9 Piezoelectric constants
The piezoelectric constants are key quantities in many technological applications,
since they govern the correlation between strain in a material and applied elec-
tric field for non-centrosymmetric materials (centrosymmetric materials necessarily
have zero for all piezoelectric constants). There are several different types of piezo-
electric constant too, depending on whether it is the polarisation being induced by
a given strain that it is being considered or the stress/strain induced by an applied
electric field. In GULP, both the piezoelectric stress constants, d, and the piezoelec-
tric strain constants, e, are calculated:
dαi=∂P
α
∂ σi
eαi=∂P
α
∂ εi
54

The two sets are related by a transformation involving either the elastic constant, or
the elastic compliance tensor, depending on the direction. The piezoelectric strain
constants are calculated from the Cartesian second derivative matrices according to:
eαi=−
N−1
∑
k=1"4π
Vqk∂2U
∂ α∂ β −1∂2U
∂ β ∂ εi#
The above piezoelectric strain constants can be readily transformed into piezoelec-
tric stress constants by multiplication by the elastic compliance tensor.
1.4.16.10 Electrostatic potential, electric field and electric field gradients
The electrostatic site potential is a measure of the Coulomb interaction per unit
charge experienced by an ion at a given position in space. This can be formally
defined by;
Vi=
N
∑
j=1
qj
ri j
where the summation explicitly excludes the case where i=j. Clearly this is closely
defined to the definition of the electrostatic energy, the two simply being related by;
Uelectrostatic =1
2
N
∑
i=1
qiVi
Similar considerations also apply with regard to periodic systems (i.e. an appropri-
ate charge summation technique must be utilised with a net charge and dipole of
zero).
The electrostatic potential is a useful quantity for qualitative predictions as to
certain properties of a material. For instance, if a structure contains several distinct
oxygen sites one might expect that there would be a correlation between site poten-
tial and basicity. However, the limitations of the point ion, or dipolar shell, model
must be kept in mind. Certainly the absolute site potential will be totally dependent
on the choice of charges in the potential model, and therefore will be arbitrary, so
at best only trends should be considered.
Based on the calculation of the site potential, the electric field, and the electric
field gradient may also be determined as the first and second derivatives, respec-
tively, of this quantity, while again excluding the case where i=j;
∂Vi
∂ α =
N
∑
j=1−qj
r3
i j
αi j
∂2Vi
∂ α∂ β =
N
∑
j=13αi jβi j −r2
i jδαβ
r5
i j
55

The electric field gradient tensor is perhaps the most useful quantity since it may
be transformed into the so-called asymmetry parameter, η, which can be important
in the interpretation of solid state NMR data [89]. If we represent the tensorial
components of the electric field gradient as Vαβ , then first the (3x3) matrix can be
diagonalised to yield the three principal axis components, Vxx,Vyy, and Vzz. If the
components are labelled V1,V2, and V3, such that |V3|>|V2|>|V1|, then the asymmetry
parameter is defined as;
η=|V2|−|V1|
|V3|
which ensures that the values lie in the range 0-1. A high symmetry site, such as
one of Ohpoint group where all components are equal, would therefore have an
asymmetry of zero.
1.4.16.11 Born effective charges
The quantification of the charges associated with atoms/ions is one of the most
problematic tasks in theoretical chemistry. Given that the atomic charge is not a
direct quantum mechanical observable, it is therefore an artificial quantity, but one
that is nonetheless useful in the development of understanding from quantitative
results. Not surprisingly there are many different definitions of charge, the most
famous of which is that due to Mulliken [36], where overlap density is apportioned
equally to both nuclear centres. More recently, there has been considerable interest
in Bader analysis [90] as a means of partitioning electron density. Both of these
aforementioned schemes are based on the analysis of the density matrix, or the
density itself. However, there is another family of charge definitions which define
the atomic charges in terms of the response of the dipole moment of the system
with respect to perturbations. Since the dipole is a genuine observable, such charge
definitions are particularly attractive for forcefields as they describe the charges that
are consistent with the response of the material to atomic displacements.
The most widely used definition in this second category is the Born effective
charge:
qBorn
i=∂ µα
∂ βi
Here the charge of an atom is now a symmetric (3x3) tensorial quantity since it
describes the derivative of the three Cartesian components of the dipole moment
with respect to the three Cartesian atomic displacements. While the definition of
the dipole moment is complicated by the choice of the particular images of each
ion, this complication doesn’t affect the derivatives of the dipole moment within
the present model. Differentiation of the dipole moment leads to the following
expression for the Born effective charge;
qBorn
i=qcore
iδαβ −Dcore−shellD−1
shell−shell qshelli
56

where the first term on the right-hand side is the core charge of the ion, and the sec-
ond term is the corresponding component of the product of the core-shell second
derivative matrix with the inverse of the shell-shell second derivative matrix, scaled
by the vector of shell charges, qshell . Physically, the second term corresponds to the
response of all the shells present to the atomic displacement of atom i, or in other
words, the electronic contribution. Hence for a rigid ion model, the Born effective
charge tensor is equal to a diagonal matrix with all the diagonal elements equal
to the core charge. Increasingly the Born effective charges are being determined
from ab initio calculations, thus creating a new avenue for determining shell model
parameters beyond the fitting of dielectric constants. Beyond this, the Born effec-
tive charges are also especially important in the determination of Γ-point phonon
frequencies, as will be discussed in the next section.
1.4.16.12 Phonons
Atoms must constantly be in motion as a consequence of the Heisenberg uncertainty
principle and this is achieved through vibrations. At low temperatures the vibrations
correspond to simple harmonic motion about the position of minimum energy, while
as the temperature increases they become increasingly anharmonic.
For a molecule, there will be 3N−6 vibrational modes (or 3N−5 for a linear
system). The remaining modes consistent of three translations and either 2 or 3
rotations depending on whether the molecule is linear or not. It should be noted
that for molecules the rotations can sometimes contaminate the vibrations of the
system. In GULP there is the option to explicitly remove rotation and translation
by using an Eckart transformation [91].
In the case of an infinitely perfect 3D solid, there will be a corresponding infinite
number of phonons. These phonons are described by calculating their values at
points in reciprocal space, usually within the first Brillouin zone. Hence we will
obtain 3Nphonons per k-point. The lowest three modes represent the so-called
acoustic branch which tend to values of zero at the centre of the Brillouin zone
(k=0,0,0), known as the Γ-point. At this point, the acoustic modes correspond to
the pure translation of the crystal lattice, and thus they are modes of zero frequency.
A plot of the vibrational frequencies versus kgives rise to phonon dispersion curves.
To calculate the vibrations or phonons of a system, the starting point is the
force constant matrix, given by the second derivatives with respect to the atoms
in Cartesian space. In the case of a solid, the terms must be multiplied by the
corresponding phase factor, exp(ik.r). Thus the force constant matrix, F, between
two atoms iand jis given by;
Fiαjβ(k) = ∑
R∂2U
∂ α∂ β expik ri j +R
The summation over Rrepresents the sum over lattice vectors within the cut-off
radius. This matrix of force constants is then converted to the dynamical matrix, D,
57

by multiplying by the inverse square root masses of the ions:
Diαjβ(k) = 1
mimj1
2
Fiαjβ(k)
The origin of the three acoustic phonons at the Γ-point for a solid, or in the
regular vibrational spectrum for a molecule, arises from the sum rules that govern
the energy derivatives. Firstly, the sum of all first derivatives, or alternatively forces,
must be equal to zero in the absence of an external force:
N
∑
i=1∂U
∂ αi=0
Secondly, by further differentiating the above expression, it can be shown that the
on-diagonal elements of the force constant matrix are equal to the negative sum of
the off-diagonal elements:
∂2U
∂ αi∂ βi=−∑0N
j=1∂2U
∂ αi∂ β j
where the summation now excludes the case when i=j. It should be noted that if
a phonon calculation is performed for a surface using a two-region strategy, as will
be discussed later, then there will no longer be three modes of zero frequency at
the zone-centre. This is because the influence of region 2 acts as an external force,
which breaks the translational invariance.
In forming the dynamical matrix from the force constant matrix there are a
number of issues relating to particular forcefields and system types. The most com-
mon issue relates to the use of the shell model, in which the shells have zero mass.
Since the number of vibrational modes is strictly given by three times the num-
ber of atoms, which in this case also corresponds the number of cores, then the
shell co-ordinates cannot appear directly in the dynamical matrix. Instead the shell
contributions are factored into the force constants of the cores according to the ex-
pression:
Ftotal
cc =Fcc −FcsF−1
ss Fsc
where Fcc,Fss, and Fsc are the core-core, shell-shell and shell-core force constant
matrices, respectively, and Fcs is just the transpose of Fsc. In the case of the breath-
ing shell model, the shell index is extended to run over the shell radius, as well as
its Cartesian co-ordinates.
At the Γ-point there is an extra complication in the calculation of the phonons.
In materials where the atoms possess a charge, the degeneracy of the Transverse
Optical (TO) and Longitudinal (LO) Optical modes is broken due to the electric
field that is generated during vibration. This effect is not allowed for in the usual
analytic evaluation of the dynamical matrix. Furthermore, the precise splitting that
58

occurs also depends on the direction of approach to the Γ-point in reciprocal space,
kΓ(i.e. the LO and TO modes are likely to be discontinuous at this point in phonon
dispersion plots). If the Born effective charges are known then this non-analytic
term [92] can be corrected for by adding a correction to the dynamical matrix [37]:
Dna
iαjβ=4π
Vmimj1
2kΓ.qborn
iαkΓ.qborn
jβ
(kΓε∞kΓ)αβ
where terms have the meanings as previously defined. As to be expected, the in-
fluence of the charges on the splitting is mediated in inverse proportion to the high
frequency dielectric constant tensor.
Two scenarios arise with regard to the calculation of the Γ-point phonons. If the
value for a specific direction of approach is required, such as in the case of a phonon
dispersion curve, then the value of kΓmay be explicitly specified. The direction
of approach is automatically set equal to the direction of the phonon dispersion
curve when the Γ-point is part of one in GULP. Alternatively, if the intention is to
compute the infra-red spectrum as a polycrystalline average, then the phonons at
this point should be a spherical average over all possible orientations. To allow for
this last possibility, there is the possibility within GULP to perform a numerical
integration by sampling the phonon modes as a function of θand φin spherical
polar coordinates and then averaging the resulting frequencies.
1.4.16.13 Vibrational partition function
Statistical mechanics allows the connection to be made between microscopic quan-
tum energy levels and macroscopic thermodynamic observables [93]. Pivotal to
this process is the partition function, Ztot , for the system. In order to determine
this quantity we make the assumption that all forms of energy are independent and
therefore that the total energy is just the sum of contributions from translation, ro-
tation, vibration and the electronic state. In the case of a solid, we can neglect
the translational and rotational components, and furthermore since we are consid-
ering forcefield methods the electronic energy is not directly calculated. Hence,
the problem reduces down to one of determining the vibration energy levels, and
subsequently the corresponding partition function.
The vibrational energy levels for a harmonic oscillator for the mth mode are
simply given by;
Uvib
m(n,k) = n+1
2hω(m,k)
if the frequency is specified in Hz. By summing over all energy levels it is possible
59

to show that the vibrational partition function is equal to;
Zvib =∑
m
∑
k
exp−hω
2kBT
1−exp−hω
kBT
Here, and subsequently, the explicit dependance of the frequency on mode and
k-point has been dropped for brevity. Substituting this expression into the various
statistical mechanical expressions fors thermodynamic quantities, we obtain the fol-
lowing relationships:
Uvib =∑
m
∑
k
wk
1
2hω+hω
exphω
kBT−1
Avib =∑
m
∑
k
wk1
2hω+kBTln1−exp−hω
kBT
Cvib
v=∑
m
∑
k
wkkBhω
kBT2exp−hω
kBT
exp−hω
kBT−12
Note that the first term in the internal energy is just the zero point vibrational energy.
In the above equations a sum over points in the Brillouin zone is included and
the term wkrepresents the weight of that particular point, such that the sum of all
weights is equal to one. Formally speaking, this sum should be an integration over
the phonon density of states. In practice, a discrete sum of points is typically used
to numerically integrate the quantities.
There are a number of different choices possible for the set of points in the
Brillouin zone for integration of the thermodynamic properties, just as there are for
integration of the band structure in an electronic calculation. For high symmetry
materials there are occasionally special point(s), such as those due to Baldereschi
[94], and Chadi and Cohen [95]. However, the most appropriate in general is usually
the Monkhorst-Pack scheme [96]. This involves an evenly spaced mesh of k-points,
given by three so-called shrinking factors, one along each axis of the Brillouin zone.
This still leaves the issue of the translational position of the mesh relative to the
origin. The optimal choice is to maximise the distance of all points from the origin
along each axis by using an offset of half a mesh spacing from there to the first
mesh point. The benefits of this choice are that it increases the rate of convergence
as a function of increasing shrinking factor, as well as avoiding the issue of the
non-analytic correction to the LO/TO splitting at the Γ-point.
If the space group symmetry has been specified for the material, then it is pos-
sible to determine the Patterson group, which is the equivalent of the space group
in reciprocal space. Note that all systems have at least inversion symmetry (also
60

known as time-reversal symmetry) in the Brillouin zone - a fact that is always used
to accelerate the Ewald sum. By using the Patterson group, it is only necessary to
sum over k-points that lie within the asymmetric wedge of the Brillouin zone, with
appropriate weighting of points situated on the boundary [97]. This can lead to
substantial computational savings for systems with small unit cells (and therefore
high shrinking factors) and high symmetry.
When calculating thermodynamic quantities it is important to ensure conver-
gence to the desired precision with respect to the shrinking factors. Because the
mesh is in reciprocal space, the shrinking factor required for a given precision is
inversely proportional to the unit cell length along that axis.
1.4.16.14 Frequency-dependent dielectric constants and reflectivity
We have already seen that the limiting dielectric properties in the static and high-
frequency limits can be readily calculated for a system. However, the dielectric
constant may also be calculated as an explicit function of the frequency of applied
field, ωf. In order to determine this, first it is necessary to calculate the oscillator
strength, Ω, for each vibrational mode based on the Born effective charges and the
eigenvector, e, for that mode:
Ωαβ =
N
∑
i=1
qBorn
iαγ eiγ
m
1
2
i
N
∑
i=1
qBorn
iβγ eiγ
m
1
2
i
The dielectric constant at the applied field is then given by:
εαβ ωf=εαβ (∞) + 4π
V
modes
∑
m=1
Ωm
αβ
ω2
m−ω2
f
From the form of the above relationship, it can be readily seen that there is a sin-
gularity in the dielectric constant when the applied frequency exactly matches that
of one of the vibrational modes of the system. The limiting behaviour of the above
expression is such that the results are equivalent to the previously calculated values
for the dielectric constant as the frequency of the applied field tends to both zero
and infinity.
One further property can be readily extracted from the above data, which is the
bulk reflectivity. This measures the ratio of the reflected to absorbed power at a
given frequency. While the frequency-dependent dielectric constant is a tensor, the
reflectivity, R, has a value for each specific direction of incidence, kin, and reflection,
kout , typically specified as vectors in reciprocal space:
Rωf=
qεωf−1
qεωf+1
2
61

εωf=kinεωfkout
1.4.16.15 Thermal Conductivity
Heat transport in materials is an important property that is quantified through the
thermal conductivity. Because this depends on phonon-phonon scattering processes,
the usual methods to calculate this property typically involve molecular dynamics
simulations. However, a number of approximate schemes have been proposed that
work within the framework of lattice dynamics. The method implemented in GULP
is that of Allen and Feldman [98] and describes the contribution to thermal conduc-
tivity of so-called diffusons. Here the phonons are calculated for a supercell and by
broadening the phonon density of states using a Lorentzian the thermal conductiv-
ity, κ, can be computed according to the following expression:
κ=1
V
modes
∑
i=1
Ci(T)Di
Here Vis the cell volume, Ci(T)is the heat capacity of mode iat temperature T,
and Di, is the mode diffusivity. The full equations for the method are given in the
original reference [98]. The key points to note is that the method, as implemented
in GULP, uses the phonons at the Γpoint only and therefore the size of the super-
cell needs to checked for convergence. Furthermore, the final value will depend
on the broadening factor used for the density of states, and to a lesser extent the
drop tolerance for the Lorentzian broadening. Since the Allen-Feldman approach
in finite super cells only captures the higher frequency contributions to the thermal
conductivity, the correct approach is to only compute the contribution above a cut-
off frequency that is given by the omega_af option. The propagating contribution
due to the acoustic branch below this cut-off frequency can then be computed more
accurately by a number of approximate formulae that describe the quadratic nature
of the density of states as it tends to zero frequency.
1.4.16.16 Pair Distribution Functions
Another use of the phonon information is in the calculation of the Pair Distribution
Function (PDF). This has been used (under various names) for many years to pro-
vide an understanding of both structure and dynamics on the atomic scale. It was
initially developed for liquids [99], and has continued to be useful with amorphous
materials [100]. As early as the 1960’s, workers were making use of the dynamic
contributions to the PDF [101]. More recently it has become an important tool for
use with crystalline materials [102].
The PDF, and related correlations functions (see Pair Distribution Functions in
Further Background section), are found experimentally through a Fourier transform
of the observed total scattering on neutron or X-ray diffraction experiments, such
62
as those performed on the GEM diffractometer [103] at the ISIS pulsed neutron
source. Total scattering experiments involve collecting the complete diffraction
pattern; diffuse scattering as well as Bragg peaks. A Fourier transform of the exper-
imental data gives the relative probability of finding a pair of atoms of type mand n
separated by a distance between rand r+δr. The atomic positions that contribute
to the Bragg peaks in the elastic scattering give the peak positions in the PDF, the
crystal arrangement (number of neighbours) gives the area under each peak, while
the dynamic properties of the material are responsible for the width of the peak.
Working within harmonic lattice dynamics, the PDF can be modelled using Gaus-
sians. Recently, Chung and Thorpe [104] proposed a method for calculating these
Gaussian peak widths from phonon calculations, thus providing the phonon-based
model of PDFs implemented here.
Taking advantage of crystal symmetry, every atom within the primitive unit cell
is used to generate atomic pairs up to a given maximum radius. Phonon informa-
tion for every k-point within a sufficiently dense Monkhorst-Pack grid is used to
calculate the contribution to the width of the PDF peak from that pair. These are
summed and suitably averaged to give the total ρPDF(r)function, which is con-
verted into each of the total correlation functions described in Further Background
section. The contributions from all pairs of each type are also used to output the
partial PDFs. Other useful data and statistics are included in the standard out-
put. Standard neutron scattering lengths are supplied with the program, available in
the eledata Library file, but can be overwritten using bbar and siginc with the
element input option.
There are two main applications for this new modelling approach. First, to assist
in the design of experiments, giving a theoretical model of experimental outcome.
Second, to ‘experiment’ on the model, for example changing cation distribution, or
investigating different phonon contributions.
Citation: Please cite the primary reference [105] in any publications produced
using the PDF module, together with the citation for the main GULP program [106].
1.4.17 Calculation of surface properties
The properties of the surfaces of materials are every bit as important as the bulk
properties, since they control the interaction between the substance and the external
environment. At the most obvious level, the very shape of the particles or crys-
tallites formed is determined by the properties of the surface relative to the bulk,
while catalysis and reactions of the material also predominantly occur at the sur-
face. From the earlier discussion of electrostatics, it is apparent that the surface
structure is a key factor in determining the bulk polarisation and net dipole of the
material, which has consequences even for the bulk region.
Due to the interest in surface phenomena, the history of modelling surfaces us-
ing atomistic simulation is also a long one spanning a number of different computer
63
codes. Of the recent era, the dominant computer code for surface simulation of ionic
materials has been MARVIN [107], which has been developed alongside GULP for
many years. However, to ensure the maximum integration of functionality between
bulk and surface simulations, both in terms of forcefield models, as well as accessi-
ble properties, it was decided to incorporate much of the functionality of MARVIN
into GULP to produce a single code capable of simulating systems with any type of
boundary condition from 0-D, through to 3-D. Here we describe the methodology
employed for surface calculations.
There are two phases to any surface calculation, namely the creation of the sur-
face from the bulk material and the subsequent calculation of its optimised struc-
ture and properties. Each surface is specified by at least two pieces of information.
Firstly, there are the Miller indices (hkl)of the plane that defines the orientation of
the bulk cleavage. Secondly, there is the so-called shift - i.e. the displacement of
the plane relative to the unit cell origin. For simple cases, such as the (001)surface
of a rock salt structure there is only one unique choice of shift. However, for more
complex cases there may be several shifts for a given plane that lead to distinct sur-
faces. When cleaving surfaces there are also other important considerations to take
into account, in particular the type of surface. As illustrated in Figure 1.1, there
are three basic types of surface. In type 1, the atomic structure consists of charge
neutral sheets of ions parallel to the surface plane and thus all shifts are guaranteed
to yield a surface with no dipole moment in the direction of the surface normal. For
type 2 surfaces, there are combinations of layers of cations and anions that possess
zero net dipole in the appropriate direction. Hence for well chosen values of the
shift a non-dipolar surface can be obtained. Finally, for type 3, all cleavage planes
result in a dipolar surface, which is therefore likely to be less stable. While dipolar
surfaces can exist, this normally leads to twinning of crystals or strong solvation
in order to anul the dipole. Alternatively, the surfaces can reconstruct in order to
remove the dipole. This typically involves the creation of cation or anion vacancies
at the surface or chemical modification. When an ion is conceptually removed, in
practice it is moved to the bottom of the surface slab in a calculation in order to
maintain charge neutrality [108]. Real examples of the three types of surface are
presented for polymorphs of calcium carbonate in Figure 1.2.
From the above, it can be seen that often the creation of the surface is a signif-
icant task in its own right and can be a complex process. In previous programs for
surface calculations, the structure manipulation has usually been performed via the
input deck of the code. Clearly, when reconstructions are involved this can become
rather unwieldy. As a result, a different strategy has been adopted for use with
GULP. All construction of the surface and structural manipulation is performed in-
dependently from the main forcefield engine by graphical means. This is achieved
through an interface to the freely available program GDIS developed by Dr Sean
Fleming (http://gdis.seul.org/). This interface allows surfaces to be specified by
their Miller indices, valid shifts to be searched for, and the geometries then to be
64

Figure 1.1: The three types of surface as categorised by Tasker (a) type I, where
each layer consists of coplanar anions and cations, and all planar cuts lead to a
nonpolar surface; (b) type IIa, where the anions and cations comprising the layers
are not coplanar, but which allows for some surface cuts to split the layers in such
a way as to produce no dipole; (c) type IIb, which is as per type IIa, except that
some ions must be moved from the surface to the bottom of region 2 in order to
achieve zero dipole; and (d) type III, where there are alternating layers of cations
and anions, and all possible planar cuts result in a surface with a dipole moment.
(a) (b)
(c) (d)
65

Figure 1.2: The examples of the three types of surface illustrated using polymorphs
of calcium carbonate; (a) type I (b) type II and (c) type III. The type I and III
surfaces illustrated are from the crystal structure of calcite, which due to its high
symmetry has no type II surfaces present. The type II example is from aragonite.
(a) Type I (b) Type II (c) Type III
manipulated, if necessary. Once the desired surface structure has been generated,
then the necessary GULP calculation can be performed.
1.4.17.1 Surface energy
The thermodynamic penalty for cleaving a surface from a bulk material is measured
according to the surface energy. Given a bulk energy of Ubulk and an energy for the
same system with a surface created of Usur f ace, then the surface energy, ∆USE , is
defined as an intensive quantity according to:
∆USE =Usur f ace −Ubulk
A
where Ais the resulting surface area. By definition, for any stable material the sur-
face energy will be endothermic. A calculated negative surface energy implies that
a material should dissociate, i.e. the crystal should disperse into the surrounding
medium.
There are two practical approaches that are widely used to determine the surface
energy by computational means. In the first, a two-dimensional slab of material is
created from the bulk, thus creating two surfaces overall. This method has the ad-
vantage that it can be used within programs that only allow for three-dimensional
boundary conditions through the introduction of a sufficently large vacuum gap
between the images, such that the surfaces don’t interact across the vacuum. In
addition, it becomes necessary to assess whether the slab is also thick enough since
the properties must converge to those of the bulk at the centre of the slab. In the
second method, a single surface is created by employing a two region strategy, as
66

shown in Figure 1.3. Here the solid is divided into region 1, which contains the sur-
face and all layers of atoms below it that exhibit a significant atomic relaxation, and
region 2, which contains the rest of the bulk material where it is assumed that no
displacements from the three-dimensional crystal structure are induced. In practice,
only the atoms of region 2 that have an interaction with region 1 need be explicitly
considered, and so the depth of region 2 is controlled by the cut-offs of the force-
field and the Parry sum used for the electrostatics. This second method is the most
efficient and precise for atomistic techniques. However, it is considerably harder
to extend to quantum mechanical methods since the electronic perturbations may
extend further into the bulk and embedding, typically via Green’s functions, is re-
quired to determine the influence of the electronic structure of region 2 on region
1. Through the GDIS interface, it is possible to automatically estimate the required
region 1 and 2 sizes needed to converge the surface energy (with a default toler-
ance of 0.001J/m2) based on the unrelaxed surface energies. However, if there
are strong relaxations of the surface, it may be necessary to further verify that the
relaxed surface energy is sufficiently converged.
The total energy for a surface calculation comprising two regions can be written
in terms of the interaction energies within, and between, the different regions:
Utot =U11 +U12 +U22
The energy of region 2 with itself, U22, is not particularly meaningful, since the re-
gion 2 is just a partial representation of the effectively infinite bulk material below,
and any given particle within this region will not experience the full set of interac-
tions that it should. However, this term is just an additive constant that is unaltered
on energy minimisation, or any other displacement of region 1. Consequently it
can be ignored in calculations. In the two region model, the energy required to
determine the surface energy is given by:
Usur f ace =U11 +1
2U12
Note that while the above energy only includes half of the region 1-region 2 in-
teraction energy, the objective quantity for energy minimisation is the total energy,
which includes the full value of U12. This is because the energy change of region 2
must be allowed for when optimising.
1.4.17.2 Attachment energy
The surface energy, described above, provides a measure of the thermodynamic
stability of a cleavage plane. However, there is a widely used alternative criterion
which is the attachment energy. This is the energy associated with the addition of a
stoichiometric layer of material on to the surface cut:
Uattachment =Un+1
tot −Un
tot −U1
tot
67
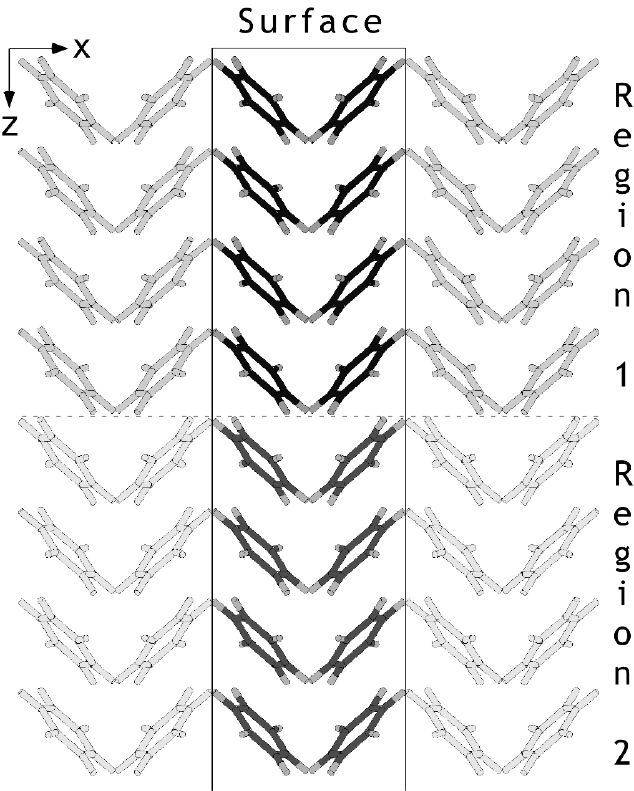
Figure 1.3: The two region surface simulation cell viewed at right angles to the
surface normal, where solid vertical lines denote the boundaries between two-
dimensional periodic images of the cell and the dash line indicates the boundary
between region 1 and the frozen region 2.
68
where Un
tot represents the total internal energy of a surface model consisting of n
growth layers, and U1
tot is the energy of the growth layer alone. For any stable
material, this implies that the attachment energy must be an exothermic quantity.
In practice, the calculation of the attachment energy is obtained from the energy of
interaction of the growth layer at the surface with the rest of the underlying material.
This benefits from the fact that the attachment energy can be obtained from a single
calculation, just as is the case for the surface energy, rather than by performing the
actual addition of a layer as part of a multistage process.
Although the attachment energy is also a strictly thermodynamic quantity, it is
often regarded as representing the kinetics of crystal growth because of the concep-
tual link between the ease of the addition of a growth layer and the rate at which a
surface is added to. Consequently, those faces where the attachment energy is most
exothermic will tend to grow most rapidly.
1.4.17.3 Morphology
The morphology of a crystal is the macroscopic shape that it adopts. Because this
can be readily observed for nearly all materials, either under an electron micro-
scope or, in the case of many naturally occuring minerals, by visual inspection with
the naked eye, it should provide a ready means to test the validity of a simulation
model. Of course, the reality is rather more complex, since the morphology is sen-
sitive to the presence of impurities, the nature of the solvent used as the growth
medium, and many other factors relating to the sample preparation. Consequently
there are materials where many different morphologies can be observed for the same
compound. A classic example, is that of calcite (CaCO3), where there are both sev-
eral different polymorphs of the bulk material and several hundred different known
morphologies. Many of these variations result from biomineralisation by different
species. Despite this, for many pure inorganic materials morphological prediction
using atomistic techniques is surprisingly successful.
Crystal morphologies can be calculated based on either the surface energy or
attachment energy, which are typically taken to represent growth under conditions
of thermodynamic and kinetic control, respectively. In order to do this, it is first
necessary to determine the objective quantity for all significant faces. Given that
dipolar faces are usually unstable, the number of likely cleavage planes for most
materials is actually considerably smaller than initially might be conceived of based
on permutations of the Miller indices. Furthermore, where there is space group
symmetry present for the bulk, many surface planes are equivalent, thus reducing
the number of unique faces. Finally, only those faces with the largest interplanar
spacings are likely to appear in the morphology [109]. The actual morphology is
generated as a three-dimensional Wulff plot [110]. Here the ratio of the surface
normal distances of all planes from the centre of the polyhedron are determined
according to the either the surface or attachment energy. The final shape of the
69
polyhedron is then determined by the intersection of the cleavage planes. Unstable
surfaces lie outside the polyhedron and never intersect. Morphological plots can
also be produced through the GDIS interface to GULP.
In the equilibrium morphology approach, the contribution of a given plane to
the total surface area is inversely proportional to its surface energy [111]. For the
growth morphology methodology [112], the surface area contribution is again in-
versely proportional, but now to the negative of the attachment energy. This is
because surfaces with a highly exothermic attachment energy will rapidly grow out
of the morphology to leave the slow growing bounding faces.
1.4.17.4 Surface phonons
The calculation of the phonons of a 2-D slab is exactly analogous to that for the
three dimensional case, except that the Brillouin zone is also now two dimensional
leading to dispersion only in the xy-plane (taking zto be the direction of the surface
normal).
Turning to consider the standard two region surface model, there are some im-
portant issues to consider. Because region 2 is quasi-infinite, it is only possible to
determine the phonons for the particles in region 1. Therefore, the dynamical ma-
trix is constructed based on region 1 alone, but with contributions to the on-diagonal
matrix blocks from the potential experienced due to a rigid, non-vibrating region 2.
Consequently it is assumed that the vibrations of the two regions are completely
decoupled. Since this is an approximation, the frequencies near the interface of
the regions will be slightly in error, particularly in the low frequency regime where
coupling is generally strongest. However, the surface modes, which are usually the
ones of primary interest, will be less affected. As always, it is essential to monitor
the convergence of all quantities with respect to increasing the region 1 depth. Fi-
nally, because region 1 is vibrating under the influence of an external potential, the
first three frequencies at the Γ-point will no longer be zero, though they typically
will be small.
1.4.18 Continuum solvation of surfaces
In the above description of surface properties the surface is always assumed to be
in contact with vacuum. However, the properties of surfaces in contact with solvent
are equally as important. While the interface between a solid and liquid is most
accurately studied using molecular dynamics simulations, there are some situations
where more approximate methods are valuable. For example, to predict the solvent
dependent morphology of a crystal only requires the ratio of the surface energies
to be correct and not the absolute magnitude. To perform molecular dynamics to
extract the relative free energies of solvation for a large collection of surface is very
challenging and time consuming. Here the use of a more approximate approach is
70
valuable.
In GULP the option to employ a continuum solvation model has been added.
Here the solvent accessible surface is described using a grid of points and the re-
sponse of the solvent due to its dielectric properties calculated at this surface in
response to the electrostatic potential of the solid. Full details of the method im-
plemented in GULP can be found in the literature [113]. In brief, the method uses
the COnductor-like Screening MOdel (COSMO) of Klamt and Schuurmann [114],
but with a few modifications. The solvent accessible surface (SAS) is smoothed by
use of tapering functions, and octahedral triangulation is used to generate the points
in order to maximise symmetry. Furthermore, the frame of a molecule (if applied
in 0-D) is rotated according to the eigenvectors of the moment of inertia tensor
in order to ensure rotational invariance. The final change is more significant; be-
cause the induced charge on the solvent accessible surface is unconstrained the total
charge of a surface and solvent could be non-zero (and non-integer). When work-
ing within periodic boundary conditions it is essential to maintain charge-neutrality
such that a convergent electrostatic energy can be obtained. To do this GULP uses
the COSMO method under the constrain of Integer Charge (COSMIC) to ensure
that the electrostatics remain valid.
The current implement of the COSMIC continuum solvation model can be ap-
plied to surfaces, polymers, solids and molecules. Both analytic first and second
derivatives are available, though phonons can only be evaluated at the gamma point
at present.
1.4.19 Free energy minimisation
One of the most important issues in solid state modelling is the variation of ma-
terials properties with the applied conditions. While isotropic external pressure is
trivial to incorporate, as has already been shown, inclusion of the effect of temper-
ature is more complex. This process is exacerbated by the fact that the majority of
potentials have been derived through the use of some empirical (i.e. experimental)
data which has been measured under a given set of conditions. Hence the force-
field itself can often already contain an implicit temperature, or worse, if multiple
pieces of experimental data have been employed that were measured at different
conditions, it can be a convolution of several temperatures. One solution to this is
to extract forcefields from quantum mechanical methods so that everything is ob-
tained explicitly at absolute zero. For now, we will assume that the forcefield has
been derived so as to be free of implicit temperature effects, by whatever means.
There are several distinct approaches to the inclusion of temperature into simu-
lations. Which one is most appropriate depends on the particular temperature and
nature of the system. In the low temperature regime, the atoms of the crystal struc-
ture just execute vibrations about their lattice sites, which in the limit of absolute
zero will be purely harmonic. This situation is best described through the use of
71
lattice dynamics, which is the quasi-static approach to treating a vibrating lattice.
As the temperature increases, the motions will become increasingly anharmonic.
In principle, this can be handled, to a point, through the use of anharmonic correc-
tions to the harmonic lattice dynamics, in order to allow for weak phonon-phonon
coupling. However, when the temperature becomes sufficiently high that diffusion
can occur it is necessary to switch to an alternative approach. Typically one of two
approaches can then be employed. If detailed information is required concerning
the atomic motions and related properties, then the method of choice is molecular
dynamics [115]. This propagates Newton’s equations of motion through time using
a finite difference formalism. Hence it retains time correlation functions and the
trajectories of all atoms. Its strength is that anharmonic effects are fully included.
However, since it regards nuclei as classical particles, it is invalid at low temper-
atures due to the neglect of the quantisation of vibration and zero point motion,
unless the Path-Integral formalism is employed. It can be seen therefore that lattice
dynamics and molecular dynamics are largely complementary in their regions of
applicability. Although GULP also includes the capability to perform molecular
dynamics, this topic will not be discussed here since this area, in regard to other
codes, has been discussed elsewhere [16], and the more unique, static, features
will be focussed on here. Likewise, the effect of temperature can also be explored
through Monte Carlo simulation [87], if the focus is integration over phase space,
with no regard to timescale or kinetic factors, but this topic will not be discussed
further because it is not one of the more novel features of GULP.
Concentrating now on the use of lattice dynamics for examining the temper-
ature dependance of crystal properties, the dominant effect is the change in the
crystal structure. When heated, most materials undergo thermal expansion which
correspondingly leads to softening of many of the mechanical properties. There are
a few celebrated examples of materials that actually contract on heating, through
the rotation of quasi-rigid polyhedra, which is technologically very important in the
quest for zero thermal expansion composites.
It has already been shown that it is possible to calculate the Helmholtz free en-
ergy for a given structure as a function of temperature through the determination of
the vibrational partition function from the phonon density of states. Hence, a nat-
ural approach to determine the dependance of structure on temperature is through
free energy minimisation. Here the key foundation is the quasi-harmonic approxi-
mation, which assumes that the vibrational frequencies can be determined as if the
atoms are vibrating purely harmonically while the cell parameters are adjusted to
minimise the free energy. Previous studies have indicated that this is a reasonable
approximation until a temperature of approximately half the melting point has been
reached.
The major barrier to free energy minimisation is that we have already seen that
efficient optimisation requires at least the first derivatives of the quantity with re-
spect to the structural variables. Hence a number of approaches evolved for tackling
72

this problem. LeSar et al [116] developed a method whereby the atoms where rep-
resented as Gaussian distributions, whose width represented the vibrational motion.
The Gaussian exponent was then regarded as a variational parameter that minimised
the free energy. Hence the free energy could be obtained without direct recourse
to a phonon density of states, making derivatives straight forward. While this ap-
proach is easy to apply to metals, the extension to a more complex ionic systems
is more demanding. A method that approximated the phonon density of states was
introduced by Sutton [117], which involved taking moments of the dynamical ma-
trix, as pioneered by Montroll [118] several decades earlier, and in the spirit of tight
binding theory. This approach has the advantage that the derivatives are taken of
the dynamical matrix elements, rather than of phonon frequencies, which are the
product of a matrix diagonalisation.
In the field of ionic materials, Parker and Price [8] pioneered the use of free
energy minimisation through the use of numerical derivatives. Here central finite
differences were taken with respect to the cell strains, with the internal degrees of
freedom being formally optimised at every point. This approach had the advantage
over the other methods that no approximation was being introduced beyond the
quasiharmonic one. However, because this requires (2N+1)optimisations and
phonon evaluations per energy/gradient evaluation with respect to the free energy,
the minimisation was restricted to the strains alone in order to reduce the number
of variables, N, to a maximum of six.
More recently, Kantorovich [119] has derived expressions for the analytical
derivatives of the free energy, which were implemented contemporaneously in the
program SHELL of Allan and co-workers [120], and in GULP [121]. If we consider
the differential with respect to strain, though the expressions would be identical for
any degree of freedom, then the first derivative of the free energy is given by:
∂A
∂ ε =∂Ustatic
∂ ε +∑
k
∑
m
h
2ω
1
2+1
exphω
kBT−1
∂ ω2
∂ ε
where the expression is written in terms of the derivatives of the square of the
frequencies, since these values represent the eigenvalues of the dynamical matrix.
Their derivatives can be calculated through the application of perturbation theory
and expressed as the derivatives of the dynamical matrix projected onto the corre-
sponding eigenvectors:
∂ ω (k,m)2
∂ ε !=e∗
m(k)∂D(k)
∂ ε em(k)
In order to determine the derivatives of the phonon frequencies, we therefore require
the phased third derivatives of the energy with respect to either three Cartesian
coordinates, or two Cartesian coordinates and one strain. for internal and external
variables, respectively.
73

For the most efficient optimisers, based on Newton-Raphson techniques, we
strictly need the second derivatives with respect to the free energy, which corre-
sponds to the fourth derivatives of the internal energy. However, this is considerably
more expensive and complex, so the Hessian matrix is usually approximated by the
conventional internal energy Hessian by assuming that the free energy contribution
to the curvature is small. Furthermore, the use of updating formulae will ultimately
correct for the discrepancy given sufficient optimisation steps.
With the advent of analytical derivatives, it is now possible to consider two types
of free energy minimisation. The first has been christened the Zero Static Internal
Stress Approximation (ZSISA) by Allan and co-workers [122], which resembles the
approach taken with numerical first derivatives (i.e. the unit cell is minimised with
respect to the free energy, while maintaining the internal degrees of freedom at a
minimum with respect to the internal energy). When working within this formalism
there is an additional contribution to the strain derivatives, which corrects for the
fact that internal degrees of freedom are not at a minimum with respect to the free
energy:
∂A
∂ ε ZSISA
=∂A
∂ ε −∂2A
∂ ε∂ α ∂2A
∂ α∂ β −1∂A
∂ β
Here the approximation is again made that the second derivative matrices can be
approximated by neglecting the free energy contribution; an approximation that
should be enhanced by the cancelation that results from taking a ratio. The second
type of optimisation can be called full free energy minimisation (FFEM), in which
the internal degrees of freedom are also minimised with respect to the free energy.
This is potentially appealing since sometimes it is the details of the internal changes
that might be of interest, for example, the nature of an adsorption complex within a
microporous material.
Results for silicate materials show a number of interesting features regarding
the merits of both approaches. Firstly, in the case of α-quartz where accurate ex-
perimental data is available for comparison, it appears that the ZSISA approach
underestimates the thermal expansion, while the FFEM method is more accurate,
though of course this is subject to the limitations of the specific potential model cho-
sen [121]. However, for all silicates tried so far the full minimisation approach goes
catasrophically wrong at about ambient conditions. This is illustrative of a general
problem with this approach which tends to drive systems to instability. This can
be readily understood, since the free energy is minimised by creating phonons that
tend to zero frequency and hence the structure is motivated to create soft modes.
This behaviour does not tend to happen in the ZSISA approach where only the cell
strains are directly coupled to the free energy and thus the relaxations tend to lead
only to a scaling of modes, rather than more individual changes. Hence the use of
ZSISA is far more robust and generally recommended for most purposes.
74

1.4.20 Monte Carlo
An alternative approach to including temperature into results, if one is not inter-
ested in the time correlation properties, just the ensemble averages, is Monte Carlo
simulation. Here the distribution of positions according to a Boltzmann distribu-
tion is determined numerically for a specified temperature and conditions. Under
the Metropolis importance sampling scheme, if a randomly chosen move for the
system leads to a lowering in the energy then the move is accepted and the system
evolves to the new state. If the energy increases, then the move is accepted with a
probability given by:
P=exp−∆U
kBT
where ∆Uis the energy change associated with the move. There are several types
of move available for a system as listed below:
•Translation - this allows atoms or molecules to move in x, y or z, according
to the flags set
•Rotation - for rigid molecules, this allows the molecule to sample the rota-
tional distribution
•Creation/destruction - in the Grand Canonical ensemble, molecules can be
inserted or removed to a reservoir of molecules whose chemical potential is
specified
Note that the Monte Carlo feature is relatively new and should be considered as a
beta release available to those who wish to test.
1.4.21 Defect calculations
While the simulation of bulk material properties is important, just as crucial is the
study of both intrinsic and extrinsic defects. Many of the key applications of solid
state systems, such as catalysis, electronic and ionic conductivity, ion exchange and
waste immobilisation, critically depend on the utlisation of the characteristics of de-
fect centres. Consequently, from the early days of the field of atomistic simulation
defect studied have been one of the most vigorously pursued topics.
There are two widely used approaches for performing defect calculations on
solids; the supercell and the cluster methods, with or without embedding in the
latter case. Both approaches have their merits and demerits. Putting computational
implementation factors aside, the use of embedded clusters is ideal for the infinitely
dilute limit, while the supercell method is more appropriate for high concentrations
of defects where there exists significant defect-defect interaction. In practice, the
nature of the computational method often biases the method of choice. However,
75

with atomistic techniques both approaches are feasible. Since the supercell method
is simply the extension of a bulk calculation, we will focus here on the embedded
cluster approach. In this particular context, the technique is generally referred to
as the Mott-Littleton method [123], after the authors of the pioneering work in the
field, though the implementational details differ a little from their original work.
The basis of the Mott-Littleton method is the so-called two region strategy. Here
a point called the defect centre is defined, which typically lies at a point concentric
with the initial defect site, or, where there is more than one defect, at the mid-
point of the ensemble of point defects. The crystal around the defect centre is
divided into two spherical regions, with the inner sphere being labelled region 1,
and the outer spherical shell of ions being region 2a. Atoms outside of these spheres
belong to region 2b, which then extends to infinity. The dimensions of these regions
are typically specified either by their radii or the number of ions contained within
them. The ions in region 1 are assumed to be strongly perturbed by the defect
and therefore are relaxed explicitly with respect to their Cartesian coordinates. In
contrast, the ions in region 2 are assumed to be weakly perturbed and therefore
their displacements, with the associated energy of relaxation, can be approximated
in some way. Clearly, as the region 1 radius is increased these approximations will
become increasingly valid, and thus an important stage of a defect calculation is
to ensure that the defect energy is sufficiently converged with respect to the region
radii. In some cases the convergence of the absolute defect energy can be slow, in
which case it may be sufficent to monitor the convergence of relative defect energies
instead. As a guideline, the radius of region 1 and the difference of the radii of
regions 1 and 2 should be both be greater than the short-range potential cut-off to
achieve convergence, though for charged defects this may not be adequate.
Within the Mott-Littleton scheme, we can express the total energy of the two
region system as the sum of contributions from the energies within the regions and
between them:
Utot (x,ξ) = U11 (x) +U12 (x,ξ) +U22 (ξ)
where U11 (x)represents the energy of region 1 as a function of the Cartesian coor-
dinates, x,U22 (ξ)represents the energy of region 2 as a function of the Cartesian
displacements, ξ, and U12 (x,ξ)is the energy of interaction between the two re-
gions. At this stage we do not distinguish between regions 2a and 2b. If the forces
acting on region 2 are small, then we can assume that the response of the atoms in
this region will be purely harmonic. Hence, the energy of region 2 can be written
as:
U22 (ξ) = 1
2ξTH22ξ
where H22 is the Hessian matrix for region 2. If we now apply the condition that the
displacements in region 2 will be the equilibrium values this yields the following
76

condition: ∂Utot (x,ξ)
∂ ξ x
=∂U12 (x,ξ)
∂ ξ x
+H22ξ=0
Combining this equation, and the previous one, it is possible to eliminate the energy
of region 2 from the total energy without direct recourse to the Hessian matrix
(which would be of infinite dimension):
Utot (x,ξ) = U11 (x) +U12 (x,ξ)−1
2∂U12 (x,ξ)
∂ ξ x
ξ
Thus the problem of calculating the energy of region 1 in the potential of region 2
has been reduced to evaluating the energy of region 1 and its interaction with region
2, without having to evaluate the self energy of region 2. Furthermore, in order to
lead to partial cancellation of terms, the quantity calculated is the defect energy -
i.e. the difference between the energy of the perfect region 1 and 2, Up
tot , and the
defective case, Ud
tot , rather than the individual contributions:
Ude f ect (x,ξ) = Ud
tot (x,ξ)−Up
tot (x,ξ)
It should be noted that it is assumed that the energy of any species removed or
added during the formation of the defect has an energy of zero at infinite separation.
If this is not the case, then the defect energy must be corrected a posteriori for
this. However, such corrections have no influence on the outcome of the defect
calculation itself.
There is one important consequence of the above formulation of the total de-
fect energy, in that it becomes necessary to find the optimised energy with respect
to the Cartesian coordinates of region 1 by force balance, rather than by energy
minimisation. The point at which the forces tend to zero is usually only slightly
different from the minimum in the internal energy, depending on the degree of per-
turbation of region 2. Hence, during an optimisation of the defect energy GULP
initially minimises the energy, as per a conventional Newton-Raphson procedure.
Once the gradient norm falls below a specified tolerance, and region 1 lies within
the harmonic region, then the harmonic displacements according to the Hessian and
gradients are applied without the use of a line search until the forces drop below the
specified tolerance.
A further important point relates to the state of the bulk crystal when performing
defect calculations. Because of the use of displacements in region 2, it is crucial
that the bulk structure is at an energy minimium with respect to the internal co-
ordinates before performing a defect calculation. Hence the bulk crystal must be
relaxed to equilibrium at least at constant volume, if not at constant pressure. Fur-
thermore, it is also important that there are no imaginary phonon modes within the
Brillouin zone otherwise the displacements in region 2 may correspond to unsta-
ble harmonic equilibrium. The presence of such modes is the most common cause
77

of unphysical results, for instance obtaining negative defect energies for intrinsic
defects. Because the presence of defects lowers the symmetry, a Mott-Littleton
calculation may encounter instabilities not apparent for the bulk material.
Now it is necessary to consider the treatment of region 2 in more detail, and
in particular the difference between regions 2a and 2b. In region 2a, the forces on
the individual ions due to short-range interatomic potentials and the Coulomb term
are explicitly calculated and the local displacement evaluated. While the forces on
region 2a are technically due to all ions in region 1, a commonly used approximation
is to evaluate the forces due to defect species only. This is the approach taken in the
default calculation method for compatability with earlier results.
In contrast to the above situation for region 2a, for region 2b the energy of
relaxation must be determined implicitly since this region extends to infinity. It is
assumed that in region 2b the only force acting is that due to the Coulomb potential
in order to simplify the problem. By choosing the radius of region 2a to always
be greater than that of region 1 by the short-range potential cut-off this can always
be arranged to be valid. Even having retained only the Coulomb interaction in
the perturbation of region 2b, this still leaves many interactions to consider. To
simplify the problem, the electrostatic potential due to defects in region 1 can be
represented by the multipole moments of the deformation situated at the defect
centre. If region 2a is sufficiently large, then the only significant term will be the
monopole moment of the defect (i.e. the net charge). Hence, the energy of region
2b is evaluated as the induced relaxation energy due to the net charge of the defect
situated at the specified defect centre. While this is a significant approximation, it
usually works well in practice and again becomes valid with increasing region sizes.
In the original Mott-Littleton method, the interaction with region 2b was described
by a continuum approximation. However, in subsequent implementations a sum of
the induced polarisation energy is performed over the atomic sites. For the general
case of an anisotropic dielectric constant tensor [124], the energy of region 2b is
calculated as:
U2b=−1
2Q2 ∑
i∈2b
∑
αβ
Mαβ
irα
cirβ
ci
r6
ci !
where Qis the net charge of the defect, rci is the distance of the ith atom from the
defect centre, and Mαβ
iis a 3 x 3 matrix for each atomic site, in the Cartesian frame,
that represents the on-site polarisability. The quantitative definition of the matrix
elements Mαβ
iis [4]:
Mαβ
i=∑
γD−1αγ qiε−1γβ
where D−1
irepresents the on-diagonal block of a modified inverse second derivative
matrix and εis the static dielectric constant tensor. Note that the inverse of the
second derivative matrix is singular and therefore a further approximation must
78

be made. Physically, this problem corresponds to the division of the polarisation
between the sublattices being arbitrary. The usual solution taken is to assume that
the polarisation is divided equally between the cation and anion sublattices, with
the second derivative matrix being modified correspondingly. In the special case of
a cubic crystal, the expression for the region 2b energy can be simplified to:
U2b=−1
2Q2 ∑
i∈2b
mi
r4
ci !
where miis the average of the diagonal elements of the matrix M(for the cubic case
these will all be equal, but if the isotropic formula were to be applied in a non-cubic
case this would not be so).
Because the expression for the region 2b energy is not particularly short-ranged,
it is appropriate to evaluate it using lattice summation techniques analogous to those
applied to the monopole-monopole term. Hence the term is summed to convergence
based on a perfect infinite lattice and then the contributions from ions in regions 1
and 2a are subtracted off. The distance dependent factor within the expression for
the energy can be rewritten as:
rαrβ
r6=1
8
∂21
r4
∂rα∂rβ
+δαβ
41
r4
Hence, by evaluating the equivalent of the Ewald summation for the inverse fourth
power of distance, and its second derivatives with respect to Cartesian displace-
ments, it is possible to achieve a rapidly convergent expression for the energy of
region 2b.
Having described how defect energies are calculated in theory, we mention a
few practical points. There are three types of defect that can be specified within
GULP:
•Vacancy
•Interstitial
•Impurity
The last one of these three is clearly the combination of a vacancy and an intersti-
tial at the same atomic position in the structure. The location of a vacancy or an
impurity can be specified by reference to either a spatial position or an atom po-
sition by referencing the site. An interstitial, by its very nature, must be specified
by coordinates. When an atom is designated to be vacant, then both the core and
shell will be removed automatically since it would not be sensible to leave one or
79
the other present in the system. It is also possible to specify that a whole molecule
be removed from the system, when the connectivity has been defined.
Most types of calculation, that are logically applicable, can also be applied to
defect runs, such as optimisation to a minimum or a transition state. In the case of
vibrations, the frequencies are calculated for a dynamical matrix based on region
1. In must be noted that this is a large approximation, since any modes that are
coupled between regions 1 and 2 will not be properly described. Only localised
modes relating to atoms near the centre of region 1 will be correctly predicted.
Consequently such calculations of vibrations must be interpreted cautiously.
Finally, a degree of point group symmetry may be utilised during defect calcu-
lations. An automatic search for common symmetry elements is performed about
the defect centre, including mirror planes and C2axes that are aligned with, or be-
tween, the Cartesian axes. Such symmetry is used to reduce the number of region
1 and region 2 atoms stored, thereby reducing the number of degrees of freedom
for optimisation, as well as taking advantage of symmetry adapted algorithms to
accelerate the calculation of the energy and its derivatives.
1.4.22 Derivation of interatomic potentials
One of the major challenges facing anyone wishing to use forcefield methods is to
determine the functional form and parameters required to calculate the energy and
properties. In the field of organic chemistry and biomolecules, there are a number
of well-established forcefields, such as MM3 [125], and those associated with the
programs AMBER [14], and CHARMM [15], which are, in principle, capable of
handling most elements typically found in these systems (C, H, O, N, S, P, etc)
in their common hybridisation states. These are constructed around the molecular
mechanics approach where the forcefield is connectivity driven and interactions are
described in terms of two-, three-, and four-body bonding terms, plus long-range
Coulomb and non-bonded interactions. While there have been several attempts to
generate general forcefields that cover the entire periodic table, such as UFF [126],
ESFF [127], etc, none have been completely successful. Because of the enormity
of the amount of data required when spanning the whole range of elements, it is
impractical to determine such a universal forcefield by empirical fitting. Instead
general rules must be used to predict parameters based on extrapolations and intrin-
sic properties of the element that are known - for instance the electronegativity. Not
surprisingly, this leads to limitations in the quality of the results. For most inor-
ganic systems it is usually necessary to derive a forcefield for the specific material,
or family of materials, of interest.
There are two means by which a forcefield can generally be derived, if we ex-
clude rule based extrapolations. Firstly, it is possible to obtain parameters by fitting
to a potential energy surface obtained from a non-empirical theoretical method.
This would typically consist of results from ab initio calculations, ideally on the
80
periodic solid [128], or perhaps on a gas phase cluster, or even better, both of the
aforementioned sources. The potential energy surface can be fitted either as a se-
quence of geometries with their corresponding energies, or derivatives of the energy
could also be included to maximise the amount of information from each higher
level calculation. Many of the early forcefields for ionic materials were determined
using electron gas methods [129], in which the energies of interaction between pairs
of ions were determined by a density functional calculation using the overlapping
ionic electron densities, where the anion is confined in an appropriate potential well.
Secondly, parameters can be obtained by empirical fitting in which the normal pro-
cess of using a forcefield to determine the properties of a material is inverted. This
approach depends on the availability of a range of experimental data. Knowledge
of the crystal structure is a definite prerequisite for this method, but is insufficient
alone since information is required as to the curvature of the energy surface about
the minimum. This later data may come from quantities such as elastic constants,
bulk moduli, piezoelectric constants, dielectric constants, or phonon frequencies.
In order to perform a fit, first it is necessary to define a quantity that measures
the quality of the results, known as the sum of squares;
F=
Nobs
∑
i=1
wifobs
i−fcalc
i2
where Nobs is the number of observables, fobs
iand fcalc
iare the fitted and calculated
values of the observable, respectively, and wiis the weighting factor for the given
observable. Because of the weighting factor, there are an infinite number of possible
solutions all of which are equally valid. Hence, one of the most important skills
is knowing how to choose appropriate and sensible weighting factors. There are
several criteria that can be used for guidance though. Firstly, if fitting experimental
data, the weighting factor should be inversely proportional to the uncertainty in the
measured value. Obviously trusted, precise values should be given more priority
than data where there are large error bars. Secondly, the weight factor should be
inversely proportional to the magnitude of the observable squared. This ensures
that all values are fitted on an equal footing, regardless of units. For example,
fitted vibrational frequencies in wavenumbers are typically two to three orders of
magnitude larger than structural variables.
The fitting process itself involves minimising the above function F. To do this,
the default approach is similar to that used in optimisation. For many terms, the
evaluation of the derivatives of the sum of squares with respect to the variables is
complex, and in some of the fitting algorithms that will be discussed subsequently
it is even impossible. As a result, numerical derivatives are employed during fitting
since it greatly simplifies the process. Because of this, the default optimiser is to
use a BFGS update of an initial on-diagonal only Hessian, obtained by finite dif-
ferences, in a Newton-Raphson process. However, for particularly difficult cases,
81
where correlation between variables is strong, there is the option to use a full Hes-
sian matrix, again obtained by finite differences.
Now we turn to specifically consider the fitting methodology for the case of
empirical data. Traditionally, the experimental structure is fitted by varying the po-
tential parameters so as to minimise the forces at this configuration, and this is the
default strategy. Other observables, such as elastic constants etc, are then calcu-
lated at the experimental structure too. When working with the shell model, either
dipole or breathing shell, there is an additional complication though in that while
the cores are fully specified since they are equated with the nuclei of the ions, the
positions/radii of the shells are undefined. One approach is to fit with the shells po-
sitioned to be concentric with the cores. However, this is unphysical since it implies
that there is no ionic polarisation, which defeats the object of including the model
in the first place. A second approach might be to place the shell according to spec-
ified ion dipoles, but this information is almost impossible to come by. Only data
about the total polarisation of the unit cell is typically available and a unique atomic
decomposition is not possible. In order to handle this issue, the simultaneous re-
laxation of shells approach was introduced into GULP [130]. Here the shells are
allowed to move during fitting. Formally, the most correct approach is to allow the
shells to be energy minimised at every evaluation of the fitting function. However,
a simpler approach has been implemented in which the shell forces are added as ob-
servables and the shell positions become fitting variables. In this way, the shells are
minimised directly within the fitting procedure. In the absence of any observables
other than the structure, this is exactly equivalent to minimising the shells at every
step of fitting. When observables are present there will be small differences, but
these are usually an acceptable price to pay for the greater ease of implementation.
There is actually a more fundamental flaw with the approach of fitting forces at
the experimental geometry as a method of fitting. Often we judge the quality of a fit
by the percentage error in the structural variables rather than the forces. Although
lowering the forces during a fit generally improves the optimised structure with
respect to experiment, this is not guaranteed to be the case (and indeed we have
found examples where it is not). This can be understood by making a harmonic
estimate of the atomic displacements, x, based on the forces, f:
x=H−1f
It can be readily seen that the magnitude of the displacements also depends on the
inverse of the Hessian matrix. Thus if the forces improve, but the description of
the curvature about the minimum deteriorates, then the errors can potentially in-
crease. If curvature information is included in the fit, then this can tend to reduce
this problem. However, there is a further difficulty here. Formally speaking, the ex-
pressions for the elastic constants and other properties are defined about a position
of zero stress and zero internal derivative. Therefore, calculating the properties at
the experimental structure when the forces are non-zero leads to errors also. The
82
solution to both of these dilemas is to use the so-called relax fitting methodology
[130] in which the structure is fully optimised at every evaluation of the fitting func-
tion and the properties calculated at the optimised configuration. Obviously this is
a far more expensive procedure, but does yield the most reliable results. Also there
is the requirement that the initial potential set is reasonable enough to actually give
a valid minimisation of the structure.
Having obtained an apparently successful fit, it is important to assess the qual-
ity of the results, since there are plenty of pitfalls and so convergence shouldn’t be
taken to represent a good quality solution. Firstly, the potential model should be
tested for all reasonable properties, not just those used in the fit. It could easily be
the case that a forcefield reproduces a high symmetry structure and, say, a single
curvature observable, such as the bulk modulus. However, examination of the full
elastic constant tensor, dielectric properties and phonons might reveal that the sys-
tem is unstable with respect to a lowering of symmetry which wouldn’t show up in
the fit. Secondly, the forcefield could be transferred to a different material, not used
in the original fit, to test whether the results are sensible. Finally, it is important to
look at the potential parameters and assess whether the numbers are physically sen-
sible. For instance, it is not uncommon for dispersion terms to become extremely
large if allowed to fit unconstrained. While this might have improved the sum of
squares for one particular system, it means all hope of transferability has been lost.
Similarly, there can often be problems with fitting Aand ρof a Buckingham poten-
tial concurrently due to the strong correlation coefficent between the parameters.
The focus above has been primarily on empirical derivation of interatomic po-
tentials. However, with the increasing ability to perform periodic ab initio calcula-
tions on solids an attractive alternative is to derive forcefields that attempt to repro-
duce the results of such methods. There are several reasons to take this approach.
Firstly, by fitting the outcomes of a single Hamiltonian it is possible to guarantee
that the training set is fully consistent (i.e. there are no differences in temperature,
pressure, sample quality, or variable uncertainities in the observables). Secondly,
data can be obtained for materials were no experimental information exists or at ge-
ometries that are significantly perturbed from the equilibrium one. Thirdly, the data
from quantum mechanical methods is free of statistical mechanical effects, such
as thermal vibrations and zero point motion. Hence, if the aim is to perform free
energy minimisation then the interatomic potentials will represent a proper starting
point for the inclusion of these quantities.
Fitting of quantum mechanical data can be performed in one of two ways, either
by proceeding in the same fashion as for empirical derivation, or by use of an energy
hypersurface. In the latter case, this is achieved by specifying a series of structures
with their corresponding energies, and optionally first derivatives. Typically the
structures would include the equilibrium configuration and as many distinct dis-
tortions about this point in order to probe as many different interatomic distances
between atoms as possible. Perhaps the most difficult decision is how to weight
83
the configurations. Unless the forcefield is able to reproduce the ab initio data
very accurately, then it is usually desirable to weight the fit in favour of configura-
tions nearer the equilibrium structure. One approach that has been taken is to use a
Boltzmann factor weighting based on the energy difference to the minimum energy
configuration, with an appropriate choice of temperature to the task ultimately to be
performed [131]. However, there are many other possible choices too. A further is-
sue concerns the fitting of quantum mechanical energies. Unless these values have
been converted to a binding or lattice energy with reference to the dissociated state
of the species within the system then it is inappropriate to fit the absolute values
of the energies. Consequently, the easiest solution is to include an additive energy
shift parameter in the fit, such that only relative energies are actually fitted.
Finally, the option exists within GULP to perform fitting using genetic algo-
rithms as well as via least squares techniques. This may be potentially useful in
cases where a complex system is being fitted when there is no reasonable starting
approximation to the forcefield available or where there may be multiple local min-
ima in the parameter space. However, to date we have yet to encounter a situation
where this approach has proved beneficial over the more conventional methodology.
This emphasizes that there is no substitute for making physically sensible choices
for the functional form of the forcefield and the initial parameters.
1.4.23 Calculation of derivatives
In order to be able to optimise structures efficiently and to calculate many proper-
ties requires the availability of derivatives. While finite difference methods can be
used to determine these, this is both inefficient and inaccurate for forcefields, since
numerical errors can cause problems, especially when potentials do not go exactly
to zero at the cut-off distance. Consequently all derivatives are determined ana-
lytically in GULP. All functional forms for the energy have up to analytic second
derivatives available, while two-, three- and four-body interactions include third
derivatives. In addition, analytic third derivatives can be calculated for the embed-
ded atom method, but currently not for any other many-body potential functions.
Because the determination of derivatives is central to many quantities, a brief
description of the approach to their calculation is given here, while fuller details can
be found in both the original Harwell Report [132], and the subsequent publication
of many of these details[133]. Ultimately two types of derivative are required -
those with respect to atomic degrees of freedom and those with respect to the unit
cell. In GULP, all atomic coordinate derivatives are calculated in the Cartesian
frame of reference and then transformed to the fractional coordinate derivatives,
when appropriate to the periodicity. For the unit cell, strain derivatives are taken as
previously described.
Both the Cartesian and strain derivatives can be related to a set of pivotal quanti-
ties, which are the first derivatives with respect to the interatomic distances, divided
84

by the distance:
∂U
∂ α =1
r
∂U
∂rα
∂U
∂ ε =1
r
∂U
∂rαβ
where, in the expression for the strain derivative, the components αand βare the
appropriate Cartesian directions for the given strain (e.g. ε4implies α=yand
β=z). It is implicit that all quantities are subscripted with i j to indicate that the
terms refer to a specific pairwise interaction. Let us introduce the shorthand:
U1=1
r
∂U
∂r
U2=1
r
∂
∂r1
r
∂U
∂r
U3=1
r
∂
∂r1
r
∂
∂r1
r
∂U
∂r
The second derivatives can then be obtained by differentiating the above expres-
sions once more and written as:
∂2U
∂ α∂ β =U2αβ +U1δαβ
∂2U
∂ α∂ ε =U2αβ γ +U1βδαγ +γδαβ
∂2U
∂ εαβ ∂ εγζ
=U2αβ γζ +U1δαβ δγζ +1
4U1δαγ β ζ +δαζ βγ +δβ γ αζ +δβ ζ αγ
Likewise, the third derivatives can be obtained by further differentiation:
∂3U
∂ α∂ β ∂ γ =U3αβ γ +U2αδβ γ +β δαγ +γδαβ
∂3U
∂ α∂ β ∂ εγζ
=U3αβ γζ +U2αγδβ ζ +ζ δβ γ +βγδαζ +ζ δαγ +γζ δαβ +U1δαγ δβ ζ +δαζ δβ γ
Note that for free energy minimisation, which is were the third derivatives are re-
quired, only the derivatives with respect to two Cartesian coordinates and one strain,
or three Cartesian coordinates are needed.
In both three-body and four-body terms there exist derivatives with respect to
either a trignometric function of an angle, or the angle itself. These derivatives can
be converted into the above forms through the use of the cosine rule:
cos(θ) = r2
12 +r2
13 −r2
23
2r12r13
85
where θis the angle at the pivot atom 1, lying between the vectors r12 and r13.
Derviatives with respect to angles are therefore obtained through the expression:
∂ θ
∂r=−1
sin(θ)
∂cos(θ)
∂r
Care must be taken in handling the limit as θtends to either 0oor 180o.
1.4.24 Crystal symmetry
An important topic, particularly in the context of optimisation, is crystal symmetry.
For periodic crystals, there is the option to specify the solid via the asymmetric unit
atoms, the space group number/symbol, and the origin setting. It should be noted
that the use of space group symbols is preferable since it distinguishes between dif-
ferent settings of the same space group. Apart from the building of the full unit cell
from the asymmetric unit, the symmetry can be utilised to greatly accelerate the
structural optimisation through several different means. The first benefit of sym-
metry is that it lowers the number of independent geometric variables, since many
atomic coordinates are related via a roto-translation operation. Furthermore, the
existence of special positions implies that some coordinates are not allowed to vary,
typically for sites such as 1
2,1
2,1
2, and that in other cases coordinates are not inde-
pendent and must be related by constraints. Even larger reductions in scale can be
achieved by using unit cell centring to reduce the centred-cell down to the primitive
representation, thereby reducing the number of atoms by a factor or between 2 and
4. All of these factors reduce the number of variables, and since the rate of con-
vergence is usually correlated to the number of independent variables, depending
on the algorithm, this can lead to fewer optimisation steps being required. Further-
more, the amount of memory to store the Hessian matrix is reduced, as well as there
being a large improvement in the cost of inverting this matrix.
The second gain from the use of symmetry is that it is possible to use new algo-
rithms to calculate the energy and its derivatives that involve fewer computations,
provided the symmetry is high enough [134]. Considering a two-body potential
model, instead of looping over all possible pairs of atoms in order to compute the
energy, when symmetry is present it is sufficient to only calculate the interactions
between the atoms of the asymmetric unit and all other atoms. For a system of
Nfatoms in the full unit cell and Naatoms in the asymmetric unit, then the execu-
tion times for the algorithms, Tfand Ta, respectively, will be given by:
Tf∝1
2NfNf+1
Ta∝NaNf
It can therefore be seen that provided the number of atoms in the asymmetric unit
is roughly less than half the number in the full unit cell then the symmetry adapted
86
algorithm will be faster. The extension of symmetry to other types of potential
is also straightforward. For example, for a three-body angular potential it is only
necessary to calculate the terms that arise when an asymmetric unit atom lies within
a valid triad of species.
While the symmetry-adapted evaluation of the energy is trivial, more care is
required for the derivatives since these are vector/tensorial properties. For the first
derivatives with respect to atomic positions, it is sufficient to again only evaluate
the derivatives with respect to the asymmetric unit atoms and then to scale these
terms by the number of symmetry-equivalent atoms in the full cell. Conversely,
the first derivatives with respect to cell strain are more complex, since although the
interaction of the asymmetric unit with all other atoms spans all possible unique
derivatives, the orientation of the terms now matters. In short, the strain derivatives
will violate the crystal symmetry if the terms are evaluated for the asymmetric unit
interacting with the rest of the crystal alone. However, given that the crystal sym-
metry is specified and that the sum of the terms is correct, when again scaled by
the site multiplicities, it is possible to obtain the correct first strain derivatives by
appropriate averaging.
Turning now to the second derivatives, the symmetry-adapted generation of the
Hessian matrix is more complex. Considering the process by which the Hessian is
generated in the absence of symmetry, there are three steps. Firstly, the Cartesian
derivatives that are initially generated have to be transformed into fractional space
by multiplying each 3 x 3 block by the unit cell vector matrix and its transpose.
This generates the matrix Df f , where the subscript fsignifies a dimension equal to
number of atomic coordinates in the full unit cell. Secondly, Df f is reduced to the
smaller matrix Daa, where the subscript anow signifies that the dimension is equal
to the number of atomic coordinates in the asymmetric unit. This is achieved using
the transformation matrix, Tf a, and its transpose:
Daa =Ta f Df f Tf a
The transformation matrix is sparse and contains 3 x 3 blocks between each asym-
metric unit atom and its symmetry equivalent images, which are just the rotation
matrices, R, that created those images. The third, and final, step is to reduce the
matrix Daa according to any constraints that are present between coordinates. It
is possible to combine the second and third steps into one by pre-multiplying the
transformation matrix by the constraint matrix.
All the symmetry unique information concerning the second derivatives is con-
tained within the columns between the asymmetric unit and the full cell. Hence,
it is more efficient just to calculate the sub-matrix Df a instead. This can then by
transformed to the required matrix according to:
Daa =Sa f Df a
This not only reduces the computational effort in calculating the second derivatives,
87

but also reduces the memory required and lowers the number of matrix multiplica-
tions required to form the Hessian. The 3 x 3 blocks of the required transformation
matrix are given by:
Sa f =1
Na
eqv
Na
eqv
∑
n=1
R−1
nRf0
where Na
eqv is the number of symmetry equivalent atoms to the asymmetric unit
atom a, and f0is the atom to which the atom fmaps under the transformation
R−1
n. Again the second derivatives with respect to strain-strain and strain-internal
are more complex since they are initially generated such that symmetry is violated.
However, resymmetrisation by averaging symmetry related matrix elements solves
this problem.
The use of crystal symmetry in reciprocal space is even more straightforward
than in real space. Because the Ewald sum can be written to be a sum over one-
centred terms in reciprocal space, instead of a pairwise expression, the only change
necessary is to restrict the sum to the asymmetric unit with appropriate weighting
for site multiplicity. The same strategy is used in the symmetrisation of other one-
centre terms in real space, such as the Einstein model. Crystal symmetry is also
exploited in the calculation of charges via the electronegativity equalisation method,
which is thereby accelerated, especially through the reduction of the size of the
matrix to be inverted.
1.4.25 Code details
The original version of GULP [134] was written in Fortran 77 since the more recent
standards had yet to be released. This implied that memory was statically allocated
via a series of parameters. Subsequently, non-standard extensions were introduced
to allow the second derivative arrays to be dynamically declared, since they repre-
sented the dominant use of memory. For the current version Fortran 90 has now
been adopted leading to full use of dynamic memory.
The program has been compiled and tested for most Unix-style operating sys-
tems, including Linux and Apple-Macintosh OS X (under which it is developed),
using most Fortran 90 compilers (g95, gfortran, ifort, etc). While compilation un-
der MS-DOS is in principle possible, this operating system is not supported since it
is the only operating system that cannot be automatically catered for within a single
standard Makefile.
The code has also been parallelised for the evaluation of the energy and first
derivatives using MPI, based upon a replicated data paradigm. When performing
calculations on sufficiently large systems that require the use of parallel computers,
then the most appropriate types of calculation are usually either conjugate gradient
optimisation or molecular dynamics. Hence, the absence of second derivatives is
less critical. However, a distributed data algorithm for the second derivatives us-
88
ing Scalapack for matrix diagonalisation/inversion would be feasible and may be
implemented in the future. Because GULP is currently targetted primarily at crys-
talline systems, where unit cells are typically small, the distribution of parallel work
does not use a spatial decomposition. Instead the Brode-Ahlrichs algorithm [135]
is used for the pairwise loops in real space in order to try to ensure load balancing
over the processors. A similar approach is used for the four-body potentials based
on the first two atoms of the sequence of four. In the case of three-body potentials,
the work is divided by a straight distribution of pivot atoms over the nodes. Paral-
lelisation in reciprocal space is achieved by an equal division of reciprocal lattice
vectors over nodes. Given that the number of operations per k-vector is equal, this
should guarantee load balancing as long as the number of reciprocal lattice vectors
is large compared with the number of processors (which is almost always the case).
89

Chapter 2
Results
In this section we present a few illustrations of the application of the new function-
ality within GULP, including validation studies to compare with previous imple-
mentations.
2.0.26 Mechanical properties
The range of mechanical, and related, properties computed by GULP has been sig-
nificantly extended for the present version. Since no article on simulations of ionic
materials would be complete without a mention of the ubiquitous and evergreen
perennial MgO, we choose to take this well-studied system as an example.
Magnesium oxide adopted the cubic rock salt structure and possesses the well-
known characteristic of exhibiting a Cauchy violation in the elastic constants (i.e.
C12 6=C44). No simple two-body forcefield is capable of reproducing this many
body effect. Consequently, it is necessary to use a breathing shell model to describe
this material accurately. While there have been previous breathing shell models
for MgO [136], we choose to fit a new set of potentials here that reproduce the
structure, elastic constants, and high and low frequency dielectric constants under
ambient conditions. The resulting potential model is described in Table 2.1, while
the calculated properties are given in Table 2.2.
The calculated properties for magnesium oxide can be seen to be in excellent
agreement with experiment under ambient conditions, with the exception of the
Poisson ratio. Of course, this agreement is a consequence of fitting a model with
the correct essential physics to a subset of the experimental data. The disagreement
in the Poisson ratios is because the values are calculated using different expressions.
If the Poisson ratio is evaluated based on the sound velocities according to:
σ=Vp
Vs2−2
2Vp
Vs2−1
90

Table 2.1: Breathing shell model for magnesium oxide. Here “shel” denotes a
potential that acts on the central position of the shell, while “bshel” denotes an
interaction that acts on the radius of the shell which was fixed at 1.2Å. The charge
on Mg is +2, while the core and shell charges for Oare +0.8 and −2.8, respectively.
Species 1 Species 2 A(eV) ρ(Å) C6(eVÅ6)kcs(eVÅ−2)kBSM(eVÅ−2)
Mg core O bshel 28.7374 0.3092 0.0 - -
O shel O shel 0.0 0.3 54.038 - -
O core O shel - - - 46.1524 -
O shel O bshel - - - - 351.439
Table 2.2: Calculated and experimental properties for magnesium oxide under am-
bient conditions. All experimental elastic properties are taken from reference [137].
Note, for the calculated bulk (K)and shear (G)moduli the Hill value is taken,
though the variation between definitions is small.
Properties Experiment Calculated
a(Å) 4.212 4.2123
C11 (GPa)297.0 297.1
C12 (GPa)95.2 95.1
C44 (GPa)155.7 155.7
ε09.86 9.89
ε∞2.96 2.94
K(GPa)162.5 162.4
G(GPa)130.4 130.9
Vs(km/s)6.06 6.05
Vp(km/s)9.68 9.70
σ0.18 0.24
91
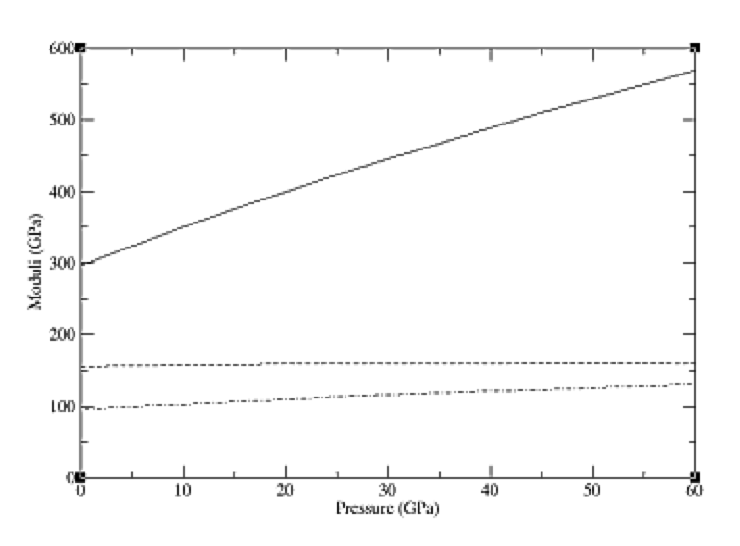
Figure 2.1: The variation of the elastic constants of magnesium oxide with applied
pressure as determined by breathing shell model calculation. C11,C12, and C44 are
presented by a solid, dashed and dot-dashed line, respectively.
then our calculated value becomes 0.182 in good agreement with the determination
of Zha et al [137].
To provide a test of the model, it is possible to calculate the variation of the elas-
tic properties of magnesium oxide as a function of applied pressure. The variation
of the elastic constants up to 60 GPa is shown in Figure 2.1.
When compared to the experimental results of Zha et al, the calculated trend in
the value of C11 is in good agreement in that it consistently increases under pressure
and approximately doubles in magnitude by the time that 60 GPa is reached. Unfor-
tunately, the trends for the other elastic constants are not so good, since the curve for
C12 flattens with increasing pressure, rather than becoming steeper, and the curve
for C44 passes through a maximum which is not observed in the experimental data
from the aforementioned group. However, the calculated trends do match the ex-
trapolated behaviour based upon the results of ultrasonic measurements [138].
2.0.27 Born effective charges
The Born effective charges represent a useful means of characterising the response
of a potential model to perturbations, particularly those that create an electric field.
92

Table 2.3: Born effective charges (in a.u.) calculated according to the shell model
of Sanders et al [139] and from first principles techniques [140]. Values are shown
for the asymmetric unit atoms with the approximate positions of the Si atom at
(0.46,0,0) and the O atom at (0.41,0.27,0.11) in space group 154, with the origin set
to (0,0,1/3).
Si O
3.122 0.0 0.0 -1.406 0.368 0.252
Shell model 0.0 3.530 0.292 0.364 -1.920 -0.517
0.0 -0.171 3.422 0.176 -0.568 -1.711
3.016 0.0 0.0 -1.326 0.429 0.222
LDA 0.0 3.633 0.282 0.480 -1.999 -0.718
0.0 -0.324 3.453 0.298 -0.679 -1.726
Increasingly values are becoming available from ab initio techniques for solids
through the application of linear response methods. Hence, this quantity can pro-
vide a useful comparison between the results of formal-charge shell model calcula-
tions and more accurate first principles methods.
One of the first materials for which the Born effective charges were determined
using planewave techniques is α-quartz. In Table 2.3 the values obtained from the
shell model of Sanders et al [139] are compared with those yielded by a planewave
calculation using the Local Density Approximation and norm-conserving pseu-
dopotentials [140].
The comparison of the Born effective charge tensors demonstrates that the oxy-
gen shell model is surprisingly successful at reproducing the quantum mechanical
data, especially in comparison to rigid ion models, which would yield a diagonal
matrix with all components equivalent. Furthermore, the polarisability of the shell
leads to the ions behaving as partially charged species with realistic magnitudes.
Consequently, this explains why the seemingly unreasonable use of a formal charge
for Si4+actually works extremely well in practice. Similar observations have been
previously made for perovskite materials [141].
2.0.28 Frequency-dependent optical properties
Ever since the early days of atomistic simulation within the shell model context it
has been routine to calculate the static and high frequency dielectric constant ten-
sors. Indeed this data has often been used during the fitting process as well. How-
ever, the values obtained from experiment will always be measured at a particular
frequency and this will never exactly correspond to the limiting values determined
by the direct means of calculation. As described in the background theory section,
it is possible to evaluate the dielectric properties and refractive indices as a function
of radiation frequency for the gamma point.
93
Here we present results for the frequency variation of the dielectric constant
tensor of quartz, shown in Figure 2.2, as calculated using the previously mentioned
shell model potential of Sanders et al [139]. Note that the limiting values are the
same as the ones obtained without reference to the phonon frequencies.
In accord with experiment, the dielectric constant decreases slowly with the fre-
quency of measurement until the highest phonon mode of quartz - the Si-O stretch
- is approached. At frequencies below this the curve contains considerable vari-
ation caused by the discontinuities when a lattice phonon mode is reached. For
simplicity, the curve shown is for the dielectric constant in the ab plane only. The
corresponding curve for the principal tensor component parallel to the 001 direc-
tion is almost identical, except that the limiting values are slightly different and the
positions of the discontinuities due to coincidence with phonons are displaced to
higher frequency.
While the qualitative agreement with experimental data is good, there is a quan-
titative discrepancy in the dielectric constant variation. This is a result of the im-
perfection of the original fit to the dielectric data for quartz, though there are also
issues concerning the variation with temperature since the calculations are formally
performed at absolute zero, while the experiment data was measured at 293 K. How-
ever, the use of empirical fitting implies that the interatomic potentials do partially
account for the temperature difference already. At the lowest measured frequency,
the calculated values are an almost perfect match due to the faster rate of decrease
of the dielectric constant in the experiment data.
2.0.29 Surface calculations
Before applying GULP to surfaces problems that were previously not possible, it
must first be validated. Firstly, we focus on comparing our results to MARVIN,
starting with the simple inorganic salt corundum. In the original MARVIN paper
[107], the twelve faces with the lowest interplanar spacings were identified and their
surfaces relaxed using several different potential models. The calculations utilis-
ing the QM5 model were repeated using both MARVIN with the BFGS minimiser
and the same surfaces generated using GDIS and minimised in GULP using its
BFGS minimiser. The unrelaxed surface and attachment energies were compared
and both sets of calculations agreed to better than 0.0001Jm−2for the surface en-
ergies and within 0.0001eV mol−1for the attachment energies. This indicates that
the 2-D Ewald sum incorporated into GULP is correct. Next the relaxed surface
and attachment energies were compared. Except for the (310) face, all the GULP
calculations returned the same relaxed surface energy as the corresponding MAR-
VIN run to within 0.0001Jm−2. The relaxed attachment energies agreed to within
0.003eV mol−1. Excluding the (310) face, the MARVIN calculations took 138 s on a
1133MHz Intel Pentium III CPU Linux system, whilst the GULP calculations took
just 108 s. This difference in timing is primarily due to the faster energy calculation
94
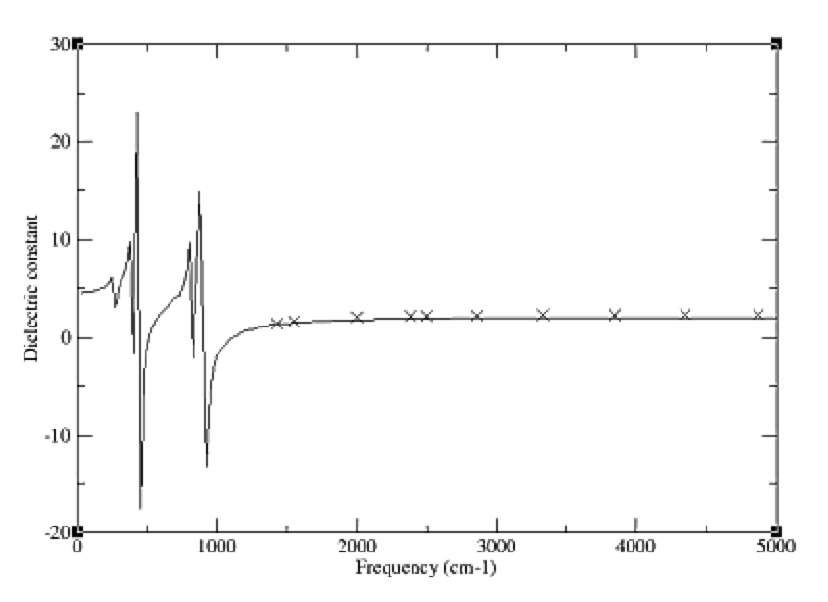
Figure 2.2: The on-diagonal component of the dielectric constant tensor for α-
quartz in the ab plane as a function of frequency of measurement. The solid line
represents the calculated shell model values, while the crosses represent values es-
timated from experimental measurements of the refractive index as a function of
frequency.
95
time in GULP since the MARVIN minimiser is based on the GULP algorithm and
consequently the number of cycles to minimise a face in GULP and MARVIN only
differed by at most one cycle, except for the (21-1) where GULP took 31 cycles ver-
sus the 24 for MARVIN. The (310) face is interesting as the relaxed surface energy
calculated by GULP is the same as that reported in the original paper [107] and it
is the minimised surface energy from the present calculation with MARVIN that is
different. Although not stated in the MARVIN paper, the minimisations were done
using a combination of minimisers; conjugate gradients until the gradient norm is
unity, followed by a BFGS minimisation. If the MARVIN calculations are repeated
with this combination, all surface energies between MARVIN and GULP agree to
within 0.0001Jm−2. In conclusion then, we can say that for this simple inorganic
system, GULP and MARVIN produce essentially the same results.
As an example of the application of the new GULP surface capabilities, they
have been recently utilised to search for surface reconstructions of the (10-14)
cleavage plane of calcite [142]. The previous modelling studies have not found
any evidence of surface reconstructions, despite the LEED results of Stipp [143].
A very efficient and assumption free way of finding reconstructions is to determine
the surface phonon dispersion across the Brillouin zone, where the presence of any
imaginary phonons will indicate that a reconstruction wants to occur. This calcula-
tion was performed on the cleavage plane of calcite, using a new calcite potential
model that we have recently developed. An imaginary mode was found to be present
at (1/2, 0) in reciprocal space, which indicates that the system is unstable within the
(1x1) surface cell and that the reconstruction requires the formation of a (2x1) su-
percell. On creation of the surface supercell, the system was perturbed along the
eigenvector of the imaginary mode and reminimised using the rational function op-
timisation technique to ensure that the final Hessian matrix had the correct character
for an energy minimum. Finally, the optimised (2x1) cell was examined to ensure
that there were no imaginary phonon modes anywhere within the Brillouin zone.
The calculations were repeated using other calcite models in the literature, which
were found not to possess any imaginary modes.
In the reconstructed surface, as shown in Figure 2.3, every second row of sur-
face carbonate anions are found to rotate. The rotation moves the O atom of each
carbonate such that it is 13ocloser to the vertical. This is accompanied by signifi-
cant change in the carbonate geometry with an increase of the O-C-O angle of 3o,
where the two oxygen atoms are those pointing out of the surface and a compensat-
ing decrease in the other two O-C-O angles. Finally, we note that the reconstruction
is confined to the top layer of the surface. In order to confirm the correct nature of
the surface reconstruction, we have calculated the LEED pattern that would result,
which is found to be an excellent match to the experimental pattern measured by
Stipp under low pressure conditions.
96
Figure 2.3: Comparison of the geometry for (a) a doubled unit cell of the (1x1)
structure for the (10-14) surface of calcite and (b) the optimised (2x1) reconstruc-
tion of the same surface. The reconstruction results in every second vertical row,
labelled B, adopting a different configuration to the unreconstructed rows, labelled
A.
(a)
(b)
97
Table 2.4: Calculated properties of diamond based on the model of Brenner et al
[66]. Note that the calculated values for the bond length and energy are not quoted
in the original reference. However, since the experimental values were part of the
fitted data, we take these values to be equal to the observed ones.
Property Experiment Original value GULP value
Bond length(Å) 1.54 1.54 1.5441
Bond energy (eV) 3.68 3.68 3.684
C11(GPa) 10.8 10.7 10.75
C12(GPa) 1.3 1.0 1.26
C44(GPa) 5.8 6.8 7.21
2.0.30 Bond-order potentials
Given that new implementation of the Brenner model has been introduced into
GULP we present here some results for diamond, Table 2.4, as previously studied
in the original paper [66], in order to validate the code. For the second derivative
properties, there is also the difference that the values obtained here are fully analytic
whereas the published values are obtained via finite differences. This may explain
the small discrepancies in the elastic constants.
As stated in the background section, two algorithms have been implemented for
the evaluation of the Brenner potential based on either pairwise atom looping or a
spatial decomposition in order to determine the neighbour list. The computational
cost of the two approaches for increasing supercells of diamond are illustrated in
Figure 2.4. The linear scaling behaviour of the spatial decomposition can clearly
be seen, as can the fact that this algorithm is effectively superior in performance for
systems containing beyond a 100 atoms. Obviously this trade off point is dependent
on the density of the system, though there are few cases for hydrocarbons where the
density is greater than that of diamond. For systems much below a 100 atoms the
cost of evaluating the Brenner potential is so negligible in comparison to other tasks,
such as a matrix inversion for property calculation, that the choice of algorithm is
irrelevant.
98
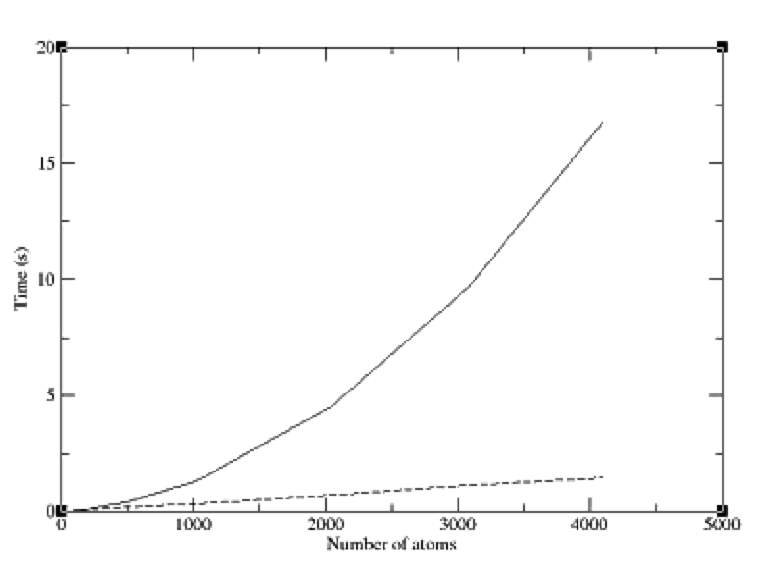
Figure 2.4: A comparison of the computer times required for a single energy-force
evaluation using the Brenner model according to whether the algorithm used in-
volves a pairwise sum (solid line) or a spatial decomposition (dashed line) to eval-
uate the neighbour list. Timings given are for a Mac PowerBook G4 laptop with a
clock speed of 700 MHz.
99
Chapter 3
Further background
In this section some of the theory behind GULP is explained and references are
supplied for those who require a more detailed description of the methods involved.
3.0.30.1 Cut-offs and molecules
All short-ranged two-, three- and four-bodied potentials have finite cut-offs in real
space which must be set by the user in some way. Unless the cut-off chosen is so
large that convergence is genuinely achieved then it effectively becomes a param-
eter of the potential. Hence when publishing new potentials it is good practice to
publish the cut-offs. Similarly, if you are trying to reproduce the results of previ-
ously published potentials make sure you use the same cut-offs.
The main effect of finite cut-offs is to introduce discontinuities into the energy
surface as atoms move across the boundary. Generally speaking, the energy min-
imisation procedure in GULP is not too sensitive to these because of the use of
analytical second derivatives. However, if working with only first derivatives or
particularly short cut-offs this can be the reason for a minimisation failing to satisfy
the required convergence criteria.
An important difference between GULP and some other programs is that it is
perfectly allowable for potentials to overlap, i.e. two or more potentials can act be-
tween the same species at the same distance. Hence, there are no resulting restric-
tions for the cut-offs and complex potential functions can be built by combining
several potentials together. Conversely, it is important not to duplicate potentials
when not intended.
For some types of potential the cut-offs may correspond to chemical criteria
such as bond lengths or they may only need to act between molecules or conversely
only within them. In such cases it is best not to use distance cut-offs to achieve
the correct effect, but instead to use the molecule handling facilities within GULP.
There are three keywords which when specified activate the molecule facility within
the program - molecule, molq and molmec. If any of these words are present
100
Table 3.1: Common functional forms for two-body interatomic potentials incorpo-
rated into GULP (where rrepresents the distance between two atoms iand j). For
full documentation see help.txt.
Potential Name Formula Units for input
Buckingham Aexp(−r/ρ)−Cr−6Ain eV, ρin Å, Cin eVÅ6
Lennard-Jones†Ar−m−Br−nAin eVÅm,Bin eVÅn
or
ε(c1(σ
r)m−c2(σ
r)n)εin eV, σin Å
c1= (n/(m−n))(m/n)(m/(m−n))
c2= (m/(m−n))(m/n)(n/(m−n))
Harmonic?1
2k2(r−r0)2+1
6k3(r−r0)3+1
12k4(r−r0)4k2in eVÅ−2,r0in Å
k3in eVÅ−3,k4in eVÅ−4
Morse D{[1−exp(−a(r−r0))]2−1}Din eV, ain Å−2,r0in Å
Spring (core-shell) 1
2k2r2+1
24k4r4k2in eVÅ−2,k4in eVÅ−4
Cosh-spring k2r2(cosh(r
/d)−1)k2in eVÅ−2
General Aexp(−r/ρ)r−m−Cr−nAin eVÅm,ρin Å,
Cin eVÅn
Stillinger-Weber Aexp(ρ/(r−rmax))(Br−4−1)Ain eV, ρin Å, Bin Å4
(sw2)
†combination rules permitted
?k3,k4are optional
101

Table 3.2: Common functional forms for three-body potentials incorporated into
GULP (where rrepresents the distance between two atoms iand j,θi jk represents
the angle between the two interatomic vectors i-jand j-k). For full documentation
of all the functional forms available see help.txt.
Potential Name Formula Units for input
Stillinger-Weber Kexp(ρ/(r12 −rmax) + ρ/(r13 −rmax))(cos(θ213)−cos(θ0))2K in eV,
(sw3) ρin Å
Three-body †1
2k2(θ−θ0)2+1
6k3(θ−θ0)3+1
12k4(θ−θ0)4θ0in ◦
k2in eVrad−2
k3in eVrad−3
k4in eVrad−4
Three-body?1
2k2(θ213 −θ0)2exp(−r12/ρ)exp(−r13/ρ)k2in eVrad−2
θ0in ◦,ρin Å
Axilrod-Teller K(1+3cosθ213 cosθ123 cosθ132)/(r12r13r23)3Kin eVÅ9
Exponential Aexp(−r12/ρ)exp(−r13/ρ)exp(−r23/rho)Ain eV, rin Å
Urey-Bradley 1
2k(r23 −r0)2kin eVÅ−2
r0in Å
Cosine-harmonic 1
2k2(cosθ−cosθ0)2k2in eV
Linear3 (lin3) k(1±cos(nθ)) kin eV
Hydrogen-bond (A
rm−B
rn)cospθA in eVÅm, B in eVÅn
UFF3 K(C0+C1cosθ+C2cos2θ)Kin eV
Equatorial 2K
n2(1−cos(nθ)) + 2Kexp(−β(r13 −r0)) Kin eV, βin Å−1
Bond-angle cross k1ri j −r0
i j+k2rjk −r0
jk(θ−θ0)k1&k2in eV Å−1deg−1
(bacross)
Bond cos cross k1ri j −r0
i jrjk −r0
jk(1+bcos(nθ)2)k1in eV Å−2
Bond cross k1ri j −r0
i jrjk −r0
jk k1in eVÅ−2
†harmonic, option three
?harmonic + exponential, option three expo
102
Table 3.3: Common functional forms for four-body potentials incorporated into
GULP (where φi jkl is the torsional angle between the planes i jk and jkl). For full
documentation see the file help.txt.
Potential Name Formula Units for input
Torsion k(1+cos(nφ−φ0)) kin eV, φ0in ◦
ESFF torsion k1sin2
1sin2
2±k2sinn
1sinn
2cos(nφ)kin eV
Ryckaert-Bellemans ∑kn(cosφ)nknin eV
Tortaper/torexp As per torsion multiplied by taper or exponential decay
Torharm 1
2k(φ−φ0)2kin eV rad−2,φ0in rad
UFF4 k
2(1−cos(nφ0)cos(nφ)) kin eV
Torangle kcos(φ)(θ123 −θ0
123)(θ234 −θ0
234)kin eV rad−2
Torcosangle kcos(φ)(cos(θ123)−cos(θ0
123))(cos(θ234)−cos(θ0
234)) kin eV
Out of plane k2d2+k4d4k2in eVÅ−2,k4in eVÅ−4
Inversion k(1−cos(φ)) kin eV
Cross angle (xangleangle) k213/4(θ213 −θ0
213)(θ214 −θ0
214)+ k213/4,k312/4,k412/3in eV rad−2
k312/4(θ312 −θ0
312)(θ314 −θ0
314)+
k412/3(θ412 −θ0
412)(θ413 −θ0
413)
Cross angle cosine (xcosangleangle) k213/4(cos(θ213)−cos(θ0
213))(cos(θ214)−cos(θ0
214))+ k213/4,k312/4,k412/3in eV
k312/4(cos(θ312)−cos(θ0
312))(cos(θ314)−cos(θ0
314))+
k412/3(cos(θ412)−cos(θ0
412))(cos(θ413)−cos(θ0
413))
UFF out of plane k(c0+c1cos(φ) + c2cos(2φ)) kin eV
then a search will be performed to locate any molecules within the structures input.
This is done by searching for bonds based on the sum of the covalent radii plus a
percentage tolerance factor. For most common compounds the default covalent radii
will be sufficient to locate all the bonds - if this is not the case then it is possible for
the user to either increase the tolerance factor or to adjust the covalent radii using
the covalent option from the element group of commands.
An alternative scenario is that atoms become bonded which shouldn’t be. For
example, metal atoms often can become bonded in ionic compounds because the
covalent radii is no longer relevant for a positively charged ion. These bonds can be
removed either by manually setting the radii of the element to zero or by using the
nobond option to exclude the formation of certain bond types. Whether the correct
molecules have been located or not can be seen from the molecule print out in the
output file. The three molecule-based keywords mentioned above differ in what
they imply for the treatment of intramolecular electrostatics:
molecule ⇒exclude all Coulomb interactions within the molecule
molq ⇒retain all Coulomb interactions within the molecule
molmec ⇒exclude all Coulomb interactions between atom which are
bonded (1-2) or two bonds away (1-3)
The specification of molmec does not automatically imply that all potentials will
be treated in a molecular mechanics fashion, only the electrostatic terms. Provid-
ing one of the above three terms is present then optional words may be added to a
103

potential specification line which control aspects of the potential cut-offs. Below
is a list of the words that are available and whether it is necessary to still give any
cut-offs on the potential parameter line:
Option Effect Cut-offs?
intra only act within a molecule yes
inter only act between molecules yes
bond only act between bonded atoms no
x12 do not act between bonded atoms yes
x13 do not act between 1-2 and 1-3 atoms yes
o14 only act between 1-4 atoms yes
g14 act between 1-4 atoms and beyond yes
single only act where atoms are singly bonded no
resonant only act where atoms are resonantly bonded no
double only act where atoms are doubly bonded no
triple only act where atoms are triply bonded no
quadruple only act where atoms are quadruply bonded no
amide only act where atoms are part of an amide group no
custom only act where atoms are joined by a custom bond no
half only act where atoms are half bonded no
quarter only act where atoms are a quarter bonded no
third only act where atoms are one third bonded no
In addition to atoms being bonded, GULP now offers the ability to specify a bond
type for each bond. This can be used so a potential only acts between two species
when they have a particular bond order. At present the bond order has to be specified
via the connect option (see below).
Although with some options it is necessary to still specify a cut-off for gener-
ality, the value may not be important any more. For example, if an O-H potential
for water is specified as being intramolecular then as long as the maximum dis-
tance cut-off is greater than about 1.0 Å then it doesn’t matter particularly what it
is. Similarly for a potential which is given as being x12 then it doesn’t matter if the
minimum cut-off distance is zero - the potential won’t act between bonded atoms.
By default, GULP dynamically calculates the molecular connectivity during a
calculation. The reason for this is that it ensures that the restart file will yield the
same answer as the point in the calculation where it left off. However, sometimes
difficulties occur because a bond becomes too long and the molecule breaks into
two. When this happens GULP will stop with an error message as this often in-
dicates that the potential model is not working well for the system under study. If
the user wants to proceed regardless then there is a keyword fix which tells the
program to fix the connectivity as that at the starting geometry and not to update it.
This means that the program will never stop with this error, but it does mean that
a restart may not give the same answer as the initial run if atoms have moved too
104

far. There is now also the option for the user to specify the connectivity explicitly
in the input deck using the connect option. Note that when using symmetry the
connect option must be specified based on the full cell atom numbers and for all
images at present.
In the case of ionic materials where the user would like to try to remove some
of the numerical problems associated with cut-offs then there are some other op-
tions. The normal way of doing this is with a cut-and-shifted potential. In this
approach the potential is forced to go to zero at the cut-off by adding a constant
to the energy. This makes the energy continuous, but the gradient still has a dis-
continuity. Again this can be resolved by adding a second term which shifts the
gradient to be zero at the cut-off. In GULP this takes the form of a linear term in
the distance which, provided the cut-off isn’t very short, will have minimal effect in
the region of the potential minimum. These corrections are activated using the po-
tential options energy or gradient after the potential type, but are only currently
applicable to certain two-body potentials where it is appropriate. It should be noted
that some potential functions go to zero by construction at the cut-off, for example
the Stillinger-Weber two- and three-body potentials.
3.0.30.2 Combination rules
When using Lennard-Jones potentials it is common to use combination rules to
determine the interaction parameters between two species. This means that the
parameters for the interaction are determined from one-centre only parameters by
some form of averaging. The main advantage of this approach is that it reduces the
number of parameters to be determined and aids transferability of potentials. Con-
versely, the resulting potentials may not be as accurate for any one given system.
There are two types of combination rule used, depending on whether the potential
is being used in the ε/σor A/B format (see Table 2 for details). If the potentials are
being used in the A/B form then the average is taken using a geometric mean:
Ai j =pAiAj
Bi j =pBiBj
However, if the ε/σform is being employed then a more complex relationship is
needed:
εi j =2(εiεj)1
2(σ3
iσ3
j)
(σ6
i+σ6
j)
σi j = σ6
i+σ6
j
2!1
6
Within GULP it is possible to specify the parameters by species, rather than by
pairs of species, using the atomab or epsilon options. If the word combine is
105
added to the specification of a lennard-type potential then the parameters can be
omitted from the input and they will be generated using the appropriate combination
rules. In turn this makes it possible to fit potentials based on combination rules
without having to do this via a series of constraints.
3.0.30.3 Mean field theory
One of the biggest problems that can face someone attempting to simulate complex
materials is the fact that often they can be partly disordered or involve partial occu-
pancies of sites. One approach to treating such systems is to generate a supercell
so that lots of permutations can be examined. However, the number of possibili-
ties is usually too large to examine each one individually to locate the most stable
configuration. Furthermore, this process may alter the symmetry of crystal. Fitting
potentials to such structures also becomes rather difficult.
An alternative approach to handling partial occupancies is to use mean field
theory. The effect of this is that each site experiences a potential which is the
mean of all possible configurations on the disordered positions. In doing so we are
assuming that all possible configurations are equally as likely, i.e. the less stable
configurations are equally as likely as the more stable ones. This may apply to
materials were there is little energetic difference between configurations or to ones
which were formed under kinetic rather than thermodynamic control and haven’t
had the chance to achieve a Boltzmann distribution. It must be decided for any
given material whether it is therefore appropriate to use this approach.
The practical upshot of the mean field method is that all interactions just become
scaled by the site occupancies of both atoms. This has been implemented in GULP
such that the user can specify the site occupancy in addition to the coordinates (see
the later section on the input for further details) and the program will automatically
handle most aspects of the mean field approach. This includes ensuring the total
occupancy on a site does not exceed unity and where two different ions share a site
with partial occupancy they are constrained to move as a single ion in optimisations.
One important word of warning - it is important that the user thinks through
interactions carefully when using the partial occupancy feature to ensure that ev-
erything is handled properly. The biggest danger comes in systems where there are
two partially occupied sites very close to each other such that in the real system
their occupancy would be mutually exclusive. When this happens it is often nec-
essary to specifically exclude potentials between these atoms to obtain the correct
behaviour.
3.0.30.4 Algorithms for energy and derivative evaluations
GULP actually contains several different algorithms for calculating the energy and
its first and second derivatives. By default the program will try to choose the most
efficient for any given system, excluding possibilities such as the cell multipole
106
method which would actually lead to slight changes in the answer. Normally the
user will need to know nothing about what algorithm is being used, so this section
is really for the curious.
Usually real-space interactions are calculated in a lower-half triangular fashion
to avoid double counting of interactions which would give rise to loops of the form
shown below:
do i= 2, numat
do j= 1, i-1
[Calculate interaction between iand j]
enddo
enddo
If there is the possibility of self-terms or interactions between periodic replica-
tions of the same atom then the i=jterm would not be excluded, though it may
be more efficient to handle this case in a separate loop. For solids where there is
significant space group symmetry then a different algorithm may be more efficient:
do i= 1, nasym
do j= 1, numat
[Calculate interaction between iand j]
enddo
enddo
where nasym is the number of species in the asymmetric unit and numat is the
number of atoms in the full unit cell. Both the symmetry adapted and standard
algorithms are present in GULP with selection being made based on the amount
of symmetry in the crystal. The use of symmetry can result in up to an order of
magnitude speed-up in favourable cases and therefore is well worth using. More
details concerning the use of symmetry, in particular with respect to the calculation
of derivatives, can be found elsewhere [11].
The second algorithmic aspect to mention applies to the situation when a con-
stant volume optimisation is being performed and some atoms are held fixed. Typ-
ical cases where this occurs are in an optical calculation, in which only shells are
relaxed, or where a molecule is docked within a rigid microporous material. In
this case the energy of interaction between certain atoms is a constant term and the
forces on them are ignored. When this happens these atoms are excluded or frozen
out of the energy calculation after the first point to save computational expense.
107
3.0.31 Phonons
3.0.31.1 Phonon density of states
We may also be interested in the phonon density of states for a solid as the num-
ber of frequencies versus frequency value becomes a continuous function when
integrated across the Brillouin zone. While full analytical integration across the
Brillouin zone is not readily carried out, this integral can be approximated by a
numerical integration. We can imagine calculating the phonons at a grid of points
across the Brillouin zone and summing the values at each point multiplied by the
appropriate weight (which for a simple regular grid is just the inverse of the number
of grid points). As the grid spacing goes to zero the result of this summation tends
to towards the true result.
For performing these integrations GULP uses a standard scheme developed by
Monkhorst and Pack [16] for choosing the grid points. This is based around three
so-called shrinking factors, n1,n2and n3- one for each reciprocal lattice vector.
These specify the number of uniformly spaced grid points along each direction.
The only remaining choice is the offset of the grid relative to the origin. This is
chosen so as to maximise the distance of the grid from any special points, such as
the gamma point as this gives more rapid convergence.
In many cases it is not necessary to utilise large numbers of points to achieve
reasonable accuracy in the integration of properties, such as phonons, across the
Brillouin zone. For high symmetry systems several schemes have been devised to
reduce the number of points to a minimum by utilising special points in kspace.
However, because GULP is designed to be general the Monkhorst-Pack scheme is
used. The user can input special points instead, if known for the system of interest.
Often it is not necessary to integrate across the full Brillouin zone due to the
presence of symmetry. By using the Patterson group (the space group of the recip-
rocal lattice) GULP reduces the integration region to that of the asymmetric wedge
which may only be 1/48-th of the size of the full volume [17].
When producing plots of the phonon density of states the critical factor, apart
from the resolution of the integration grid, is the ‘box’ size. The continuous density
of states curve has to be approximated by a series of finite regions of frequency or
boxes. Each phonon mode at each point in k space is assigned to the box whose
frequency region it falls into. The smaller the box size the better the resolution of
the plot. However, more points will be needed to maintain a smooth variation of
number density.
3.0.31.2 Infra-red phonon intensities
In order to make comparison between theoretically calculated phonon spectra and
experiment it is important to know something about the intensity of the vibrational
modes. Of course the intensity depends on the technique being used to determine
108

the frequency as different methods have different selection rules. While Raman in-
tensities are not readily calculable from most potential models, due normally to the
absence of polarisabilities higher than dipolar ones, approximate values for infra-
red spectra can be determined [18]:
IIR ∝(∑
all species
qd)2
where qis the charge on each species and dis the Cartesian displacement asso-
ciated with the normalised eigenvector.
3.0.31.3 Pair Distribution Functions
The calculation of Pair Distribution Functions is in accordance with the theory of
Chung and Thorpe [104]. It makes use of phonon information calculated within
GULP, for an optimised structure, using a Monkhorst-Pack grid (as described in
section 3.0.28.1).
Chung and Thorpe [104] state that the probability of finding a pair of atoms i
and j, with position riand rjrespectively, at position ris given by
ρi j(r) = δ(r−(rj−ri))(3.1)
where <··· >is the statistical average implying both configurational and thermal
averages. Summing over all such pairs gives the density function ρ(r), which is
averaged by using each atom in turn as the starting point. Working with a crystal
lattice, the complexity of such calculations is reduced because only atoms in the first
unit cell are used as starting points. Moreover, GULP reduces the crystal symmetry
to a primitive cell, minimizing the required number of calculations. (It is, of course,
still possible to specific a conventional cell and use these k points instead.)
Consider a lattice of unit cells each containing natoms. Denote the position of
atom iin the original unit cell as ri0and similarly atom jin the `th unit cell as rj`.
Define the pair separation vector between two atoms i0(in the original unit cell)
and j`(in the `th unit cell) as ri0j`=rj`−ri0.
The density function (with units of 1/volume) is the weighted sum over all pairs
between atom i0and atom jin all unit cells, averaged over the number of atoms
in the unit cell, n. The spherical average is taken, dividing by 4πr2, to remove
orientational dependence.
ρ(r) = 1
4πr2n∑
` ∑
i0
0
∑
j
wi jρi0j`(r)!(3.2)
where the prime indicates i06=j0(i.e. ri0j`6=0). The guassian peak (with units of
1/length), is given in equation 3.8. The weighting is dependent on the fraction of
109
atoms of type i,ci, and coherent bound scattering length, ¯
bi, and is expressed as
wi j =¯
bi¯
bj
∑n
k=1ck¯
bk2(3.3)
As suggested by equation 3.1, if the atoms were completely stationary, the density
function would be a series of delta functions located at the interatomic spacings. To
account for thermal motion, Chung and Thorpe [144] demonstrated that, within the
harmonic approximation, the Debye-Waller theorem can be used to justify the use
of a series of weighted Gaussian peaks ρi j(r), centred at ri j with width σi j. Taking
ˆri j to be the unit vector between atoms iand j, and ui j =uj−ujwhere uiis the
displacement of atom i, then the width is given by
σi j =Dui j ·ˆri j2E1
2(3.4)
This can be expressed in terms of phonon modes as
σ2
i0j`=¯
h
2N∑
k,ν
2n[ω(ν,k)] + 1
ω(ν,k)|ri0j`|2|uj`(ν,k)−ui0(ν,k)·ri0j`|2(3.5)
where the displacements ui`are as given in equation 3.6. It should be noted that
this corrects a typographical error by Reichard et al [145] equation 3, where the
numerator is multiplied by a factor of √mirather than divided by it.
ui`=esigi(ν,k)exp[ik·ri`]
√mi
(3.6)
Nis the number of k-points, νis the mode index, n[ω(ν,k)] is the Bose occupation
number
n=1
exp(¯
hω
kT )−1(3.7)
ω(ν,k)is the frequency from the eigenvalues of the dynamical matrix, and esigi(ν,k)
is the eigenvector for atom i(see section 3.0.32). The mass of atom iis mi. When
this is implemented within GULP, a Monkhorst-Pack grid [96] of a specified density
is used to generate an even distribution of k-points.
In summary, the Gaussian peak (with units of 1/length) for each pair is calcu-
lated from the width
ρi0j`(r) = 1
q2πσ2
i0j`
exp"|ri0j`|−r
2σ2
i0j`#(3.8)
These are summed and averaged as in equation 3.2to give the Chung & Thorpe
(1999) density function (with units of 1/volume). The partial density function for
atomic pair i j is the contribution from ρi0jl(r)for that pair: the sum of all partials
is the total density function
110

3.0.32 Eigenvectors
Within the literature, there are two commonly used settings for calculating the dy-
namical matrix depending on whether the atomic position in the unit cell is included
in the eigenvector, or taken out as a phase factor. The default setting in GULP is
the same as that used by Lovesey [146], where the eigenvector of atom jat a given
k-point is phased by the position of the atom in the unit cell (rj0). Lovesey [146]
writes this as σ, but we have used esig here to avoid confusion with peak widths.
This is the eigenvector used by Chung and Thorpe[144] and here. Lovesey [146]
also describes an alternative setting, written as e, used in several books e.g. Willis &
Pryor [147] and Dove[148]. The two settings are related by the phase factor shown
(from eq. 4.28 in Lovesey [146])
esig j(ν,k) = ej(ν,k)·expik·rj0(3.9)
3.0.33 Commonly Used Correlation Functions
A number of different formalisms for PDFs exist in the literature. Keen [149] per-
formed an extensive survey of these and we follow his recommendations. The three
main real space correlation functions, G(r),D(r)and T(r)are variously used de-
pending on the purpose. T(r), which scales as rat large r, is often used for peak
fitting, and for analyzing structural detail at low r(e.g. in amorphous systems).
D(r)is similar to T(r), but has a term subtracted that scales with r, making it the
correlation function of choice for studying mid to high r structural detail. G(r)is
also used as it makes the low r peaks prominent. A comparison of the different
forms is given by Dove [150].
Keen [149] writes the density function of Chung and Thorpe [104] as ρPDF (r).
It is the same as that used in the PDFFIT program [151] as well as by several current
workers in this field, e.g. [152] [153]. This real space correlation function tends to
ρoat high rand is zero below the minimum interpair spacing.
The PDFFIT program also outputs a radial distribution function [144], also
known as a pair distribution function [104], with units of 1/area. It is written as
GPDF (r)by Keen [149], and defined as
GPDF (r) = 4πrρPDF (r)−ρo(3.10)
where ρo=n
Vunitcell , the average number density (units of 1/volume). While ρPDF (r)
oscillates around the number density, GPDF (r)oscillates around zero. This can be
converted to a Keen [149] total radial distribution function,G(r), which has units
of area. At values of rless than the minimum interpair spacing, this function tends
to −(∑n
i=1cibi)2, and to ∞at high r.
G(r) = GPDF (r)(∑n
i=1cibi)2
4πrρo
(3.11)
111

G(r)is often expressed in units of Barns (1×10−28 m2=1×10−8Å2), but in the
GULP output we use Å2to be consistent with the other correlation functions.
The differential correlation function,D(r), and total correlation function,T(r),
are used as part of the ATLAS suit of programs [154], as used at the ISIS pulsed
spallation neutron source, and defined as
D(r) = 4πrρoG(r)(3.12)
=GPDF (r) n
∑
i=1
cibi!2
(3.13)
T(r) = D(r) + 4πrρo n
∑
i=1
cibi!2
(3.14)
=GPDF (r) + 4πrρo n
∑
i=1
cibi!2
(3.15)
D(r)and T(r)have units of 1/length.
In addition to the total pair distributions functions written to the .pdfs file, a .pdfs
file is produced for every pair-type (root_pairnumber_atom1_atom2.pdfs) contain-
ing a selection of partial pair distribution functions. First, the weighted ρij(r)func-
tion: the sum of all of these is the total pair distribution function of Chung and
Thorpe. Secondly, a weighted version of gij(r), which again sums directory to
give G(r). Finally, an unweighted (true) gij(r), which corresponds to the gij(r)of
Keen[149]. This output can be supressed using the nopartial keyword.
3.0.33.1 Thermodynamic quantities from phonons
There are a range of quantities that can be readily calculated from the phonon den-
sity of states. The accuracy with which they are determined though clearly depends
on the kpoints or shrinking factors selected for the Brillouin zone integration. For
systems with large unit cells a small number of kpoints, perhaps even the Γ-point
alone, will be sufficient. However, for those systems with small to medium unit
cells it is important to examine how converged the properties calculated are with
respect to the grid size.
If a phonon calculation is performed then GULP will automatically print out
the relevant thermodynamical quantities. This output depends partly on whether a
temperature has been specified for the given structure. If the calculation is set for
zero Kelvin then only the zero point energy is output:
ZPE =∑
k−points
wk∑
all modes
1
2hν
where wkis the weight associated with the given kpoint. In principal, the zero
point energy should be added to the lattice energy when determining the relative
112

stability of two different structures. However, because the derivatives of the zero
point energy are non-trivial it is normally neglected in an energy minimisation.
For temperatures above absolute zero we can calculate the vibrational partition
function, which in turn can be readily used to calculate three further properties:
Vibrational partition function:
Zvib =∑
k−points
wk∑
all modes 1−exp−hν
kT −1
Vibrational entropy:
Svib =RlnZvib +RT ∂lnZvib
∂T
Helmholtz free energy:
A=U−T Svib
where
U=Ulattice energy +Uvibrational energy
Heat capacity at constant volume:
Cv=RT 2∂lnZvib
∂T+T∂2lnZvib
∂T2
3.0.34 Free energies
Although the most common methods for studying the properties of materials as a
function of temperature are molecular dynamics and Monte Carlo simulations, there
is an alternative based on static methods within the quasi-harmonic approximation.
This is to directly minimise the free energy of the system at a given temperature,
where the free energy is calculated from the lattice energy combined with contribu-
tions from the phonons including the entropy and zero point energy.
The advantages of working with free energy minimisation are that MD simula-
tions are quite expensive due to the need to reduce the uncertainty by sampling large
amounts of phase space. Molecular dynamics and free energy minimisation are in
fact complementary techniques. The latter approach breaks down at high tempera-
tures as anharmonic effects become important - typically it works at temperatures
up to half the melting point as a rough guide. Conversely, molecular dynamics is
not strictly valid at low temperatures because the zero point motions and quantum
nature of the vibrational levels is ignored.
Although in principle it is possible to analytically fully minimise the free energy
of a solid, in practice this is extremely difficult as it requires the fourth derivatives
of the energy with respect to the Cartesian coordinates. Hence, a number of ap-
proximations are normally made - the main one being that the principal effect of
113

temperature is to expand or contract the unit cell and the effect on internal degrees
of freedom is less important.
When changing unit cell parameters we are concerned with the Gibbs free en-
ergy as this is appropriate to a constant pressure calculation. This quantity is related
to the Helmholtz free energy, whose relationship to the vibrational entropy has al-
ready been given previously, by the expression;
G=A+PV
with
P=P
ext −P
int
where Pis the pressure. The pressure has two components - any external applied
pressure plus the internal phonon pressure coming from the vibrations. The phonon
pressure is given by:
P
int =−∂A
∂V
In order to calculate the Gibbs free energy it is therefore necessary to calculate
the derivative of the Helmholtz free energy with respect to the unit cell volume.
This can be done numerically by finite differences. Central differencing is more
expensive than using forward differences. However, it is generally necessary to
determine the phonon pressure with sufficient accuracy. In turn each calculation of
the Helmholtz free energy requires a constant volume minimisation for the given
set of unit cell parameters, followed by a phonon calculation.
Once the Gibbs free energy has been calculated then the next stage of a free
energy minimisation is to isotropically expand or contract the unit cell until the ex-
ternal pressure balances the internal pressure. Having done this then the derivatives
of the Gibbs free energy can be evaluated numerically by finite differences and the
unit cell optimised with respect to this quantity.
Because of the three levels of optimisation plus phonon calculations involved,
free energy minimisations are rather expensive and shouldn’t be undertaken lightly!
Due to the numerical nature of several of the derivatives it may be necessary for the
user to adjust the finite differencing interval for a calculation to work optimally.
Also the calculations are very sensitive to the quality of the underlying energy sur-
face. Potentials with short cutoffs, leading to discontinuities, and soft modes can
cause difficulties for the method, so always check your model well before starting.
Free energy minimisation can be used in conjunction with fitting to allow a
series of structures at different temperatures to be fitted with inclusion of the thermal
effects, though again this is an expensive procedure. It is important to note that a
free energy minimisation at 0 K is not the same as an ordinary static calculation.
This is due to the presence of the zero point energy in the former method.
114
3.0.35 Defects
3.0.35.1 The Mott-Littleton method
The calculation of defect energies is more difficult and approximate than the calcu-
lation of bulk properties. In theory, a defect can cause very long range perturbations,
particularly if it is not charge-neutral. Consequently the user must always check the
convergence of the approximations made.
The simplification in the modelling of defects is to divide the crystal that sur-
rounds the defect into three spherical regions known as regions 1, 2a and 2b [19-
21]. In region 1 all interactions are treated exactly at an atomistic level and the
ions are explicitly allowed to relax in response to the defect. Except in the case of
very short-ranged defects it is not generally possible to achieve the desired degree
of convergence by increasing region 1 before running out of computer resources.
Consequently, in region 2a some allowance is made for the relaxation of ions but in
a way that is more economical.
In region 2a the ions are assumed to be situated in an harmonic well and they
subsequently respond to the force of the defect accordingly [22]. This approxima-
tion is only thus valid for small perturbations and also requires that the bulk lattice
has been optimised prior to the defect calculation. For region 2a individual ion dis-
placements are still considered, whereas for region 2b only the implicit polarisation
of sub-lattices, rather than specific ions, is considered.
If the vector xrepresents the positions of ions in region 1, while ζrepresents
the displacements of ions in region 2a, then the total energy of the system may be
written as:
E=E1(x) + E12(x,ζ) + E2(ζ)
where E1and E2are the energies of regions 1 and 2 respectively, and E12 is the
energy of interaction between them. We now assume that the energy of region 2 is
a quadratic function of the displacements:
E2(ζ) = 1
2ζTWζ
We also know that we wish to obtain the displacements in region 2 for which
the energy is a minimum:
∂E
∂ ζ =0=∂E12(x,ζ)
∂ ζ +Wζ
This expression can be used to eliminate E2from the total energy, leaving it
purely in terms of E1and E12:
E=E1(x) + E12(x,ζ)−1
2
∂E12(x,ζ)
∂ ζ ζ
115

The displacements in region 2 are formally a function of xfor region 1 which
makes the minimisation of the total energy with respect to both the positions of
region 1 and the displacements of region 2 potentially complicated. This problem
can be avoided by using force balance in region 1 as the criteria for convergence
(i.e. all forces on ions in region 1 must be zero), rather than purely minimising the
energy. The two approaches are equivalent provided that region 2 is at equilibrium
also. This will be achieved provided that the displacements in region 2 are small
enough that they are genuinely quadratic.
In terms of the minimisation procedure employed for defect calculations the
force balance process leads to a slightly different approach to the bulk optimisation.
Initially the same Newton-Raphson procedure with BFGS hessian updating and
line searches is employed to avoid convergence to stationary points which are not
minima. After at least one cycle of the above and when the gradient norm falls
below a certain threshold the minimiser abandons the line search procedure and
aims purely to reduce the gradients to zero, regardless of the energy. In practice
positive changes in the energy near convergence are only ever small.
The defect energy is now the difference in the total energies for the defective
and perfect lattice, Edand Eprespectively, with corrections due to the energy of
any interstitial or vacancy species at infinite separation from the lattice, E∞:
Edefect =Ed−Ep+E∞
Two final aspects must be dealt with in order to obtain the final working equa-
tions for the defect energy. Firstly, due to the slow convergence of electrostatic
terms in real space alone we cannot evaluate the region 1 - region 2 energy directly.
Instead we must calculate the energy of region 1 interacting with the perfect lattice
to infinity and then explicitly subtract and add back the terms due to ions which
are no longer on their perfect lattice sites. Secondly, because the displacements in
region 2 depend on the force acting on a given ion, which in turn is a function of
other region 2 ions, there is in fact a linear dependency of the energy on ζ. By suit-
able manipulation of the energy terms this may be removed to leave the following
expression for the defect energy:
Edefect =E11(dd)−E11(d p) + E1∞(d p)−E1∞(pp)
+E12a(dd)−E12a(d p) + E12a(pp)−E12a(pd)
−∑∂E12a(dd)
∂r−∂E12a(pd)
∂r
where the general symbol Ei j(kl)denotes the energy of interaction summed
over all ions in region iinteracting with ions in region jwhere iand jcan be 1,
2a or ∞(signifying a sum over 1, 2a and 2b out to infinity). The letters kand l
indicate whether the energy is for the perfect or defective coordinates in regions i
and jrespectively, depending on whether they are por d.
116

3.0.35.2 Displacements in region 2a
Expanding the energy as a Taylor series and truncating at second order gives the
Newton-Raphson estimate of the vector from the current ion position to the energy
minimum position in terms of the force, g, acting on the ion:
ζ=−W−1g
Hence if we know the local second derivative matrix and the force acting on
the ion we can calculate its displacement. There are a number of possible ways of
calculating the force acting on the ions in region 2a. The most common approach
is to use the electrostatic force due to only the defect species - i.e. the force due
to any interstitial species based on their current positions, less the force due to any
vacancies at the position of the original vacant site. In this way region 2a responds
to the change in the multipole moments of the defect species in region 1, but not the
influence of other forces. Hence for this approximation to strictly hold the distance
between any defects and the boundary of region 2a should be greater than the short-
range cutoff.
3.0.35.3 Region 2b energy
Region 2b is assumed to be sufficiently far from the defects that the ions only re-
spond by polarising according to the electrostatic field resulting from the total defect
charge placed at the centre of region 1. This can be written for cubic systems as
follows:
E2b=−1
2Q2∑
i6=1,2a
qimi
R4
i
Because this expression is just dependent on the distance and a couple of lattice
site related parameters the region 2b energy can be evaluated using a method analo-
gous to the Ewald sum and then subtracting off the contribution from ions in regions
1 and 2a. An alternative more general, but still not completely general, expression is
the following where the lattice site dependent property is now an anisotropic tensor,
rather than a scalar [23]:
E2b=−1
2Q2∑
i6=1,2a
∑
αβ
qiMαβ
iRα
iRβ
i
R6
i
This can again be calculated by partial reciprocal space transformation based on the
second derivatives of the R−4lattice sum.
117
3.0.36 Fitting
3.0.36.1 Fundamentals of fitting
Before any production runs can be performed with an interatomic potential program
it is necessary to obtain the potential parameters. If you are lucky there may be good
parameters for your system of interest already published in the literature so you can
just type them in and get going straight away. Unfortunately most people are not
so lucky! The fitting facility within GULP [24] allows you to derive interatomic
potentials in either of two possible ways. Firstly, you can determine the parameters
by fitting to data from some higher quality calculation, such as an ab initio one,
normally by attempting to reproduce an energy hypersurface. Secondly, you could
attempt to derive empirical potentials by trying to reproduce experimental data.
Regardless of which method of fitting you are using the key quantity is the ’sum
of squares’ which measures how good your fit is. Ideally this should be zero at the
end of a fit - in practice this will only happen for trivial cases where the potentials
can be guaranteed to completely reproduce the data (for example fitting a Morse
potential to a bond length, dissociation energy and frequency for a diatomic should
always work perfectly). The sum of squares, F, is defined as follows:
F=∑
all observables
w(fcalc −fobs)2
where fcalc and fobs are the calculated and observed quantities and wis a weighting
factor. There is no such thing as a unique fit as there are an infinite number of pos-
sible fits depending on the choice of the weighting factors. The choice of weighting
factor for each observable depends on several factors such as the relative magnitude
of the quantities and the reliability of the data (for instance a crystal structure will
generally be more reliable than an elastic constant measurement).
The aim of a fit is to minimise the sum of squares by varying the potential
parameters. There are several standard techniques for solving least squares prob-
lems. By default GULP uses a Newton-Raphson functional minimisation approach
to solving the problem, rather than the more conventional methods. This is because
it avoids storing the co-variance matrix. The downside is that near-redundant vari-
ables are not eliminated. Currently the minimisation of the sum of squares is per-
formed using numerical first derivatives. The reason for using numerical derivatives
is because many of the properties, particularly those derived from second deriva-
tives, are rather difficult to implement analytical derivatives for. Consequently the
value of the gradient norm output during fitting should only be taken as a rough
guide to convergence. In cases where the numerical gradients prove noisy then
the use of the simplex algorithm for fitting can be useful as this requires only the
function.
The choice of which potential parameters to fit belongs to the user and is con-
trolled by a series of flags on the potential input line (0 ⇒fix, 1 ⇒vary). There are
118
also options contained within the variables sub-section for allowing more general
parameters to fit, such as charge distributions. Note that when fitting charges at
least two charges must be varied to have any effect as the program eliminates one
variable due the charge neutrality constraint. There is also the option to vary the
charge split between a core and shell while maintaining a constant overall charge
on the ion. The user may also impose their own constraints on fitting variables
through the constrain fit option.
It is generally recommended that a small number of parameters are fitted ini-
tially and the number gradually increased in subsequent restarts. Often if all pa-
rameters are allowed to vary from the start unphysical parameters may result. Dis-
persion terms of Buckingham or Lennard-Jones potentials are particularly prone to
poor behaviour during fitting, as they tend to go to zero or become exceedingly
large. It is generally recommended that such terms are set equal to a physically
sensible value (based on quantum mechanical estimates or polarisability-based for-
mulae) and held fixed until everything else is refined.
A final check that the program looks for is that the total number of variables
being fitted is less than the total number of observables!
3.0.36.2 Fitting energy surfaces
To fit an energy surface it is basically necessary to input all the structures and the en-
ergies that correspond to them. To do this it is just a matter of putting one structure
after another in the input file (within the limit of the maximum number of structures
for which the program is dimensioned). It is possible to fit the gradients acting on
the atoms as well the energy of each structure, though often just the energies are
fitted. If the latter is the case, then the easiest way to turn off the fitting of the
gradients is to specify noflag as a keyword to prevent the program for looking for
gradient flags in the absence of a keyword to specify them.
Perhaps the only unique feature of fitting an energy surface is the need to in-
clude an energy shift in some cases. This is a single additive energy term which is
the same for all structures and just moves the energy scale up and down. The justi-
fication for this is that often it is impossible to calculate the energy that corresponds
exactly to the interatomic potential one from a quantum mechanical calculation
[25]. Most commonly this arises where the potential model has partial charges in
which case there is an unknown term in the lattice energy due to ionisation poten-
tials and electron affinities for fractions of an electron.
To simplify the specification of this shift value in the input, if you give the shift
option after the first structure then this value will apply to all subsequent structures
until a different value is input. Similarly, its magnitude can be altered by using the
variables sub-section to specify the shift as a variable and this will apply to all
structures. It is generally recommended that the shift is fitted first and allowed to fit
through out the procedure.
119
3.0.36.3 Empirical fitting
An alternative to fitting quantum mechanical data to derive an interatomic potential
is to actually fit experimental data. In this case the procedure serves two purposes.
Firstly, the degree to which all the data can be reproduced may serve as some guide
as to the physical correctness of the model used. Secondly, it provides a means of
extrapolation of experimental data for one system to a different one where the data
may not be known, or alternatively to unknown properties of the same material.
Any of the properties that can be calculated for the bulk solid or gas phase
molecule can also be used in reverse to fit a potential to. Obviously the essential
ingredient in the fit is the experimental structure, without which you won’t get very
far! The conventional way to fit the structure is by requiring that the forces on the
atoms are zero. This is clearly not a perfect strategy as it could be satisfied by a
transition state rather than a minimum, though in practice it is rare, except when
symmetry constraints are imposed.
Normally a good fit requires some second derivative information as well as the
structure. For very high symmetry systems, such as rock salt, the structural data
alone is completely inadequate. If we imagine a potential as being a binomial ex-
pansion about the experimental geometry, then unless the first and second deriva-
tives are reasonably well reproduced by our model then the range of applicability
will be almost zero. Typical sources of second derivative information are elastic,
dielectric and piezoelectric (where applicable) constants. Also vibrational frequen-
cies contain far more information than any of the above. However, the fitting of
frequencies is not straightforward. To fit the frequency magnitudes is certainly pos-
sible, however, you have no guarantee that the correct mode has been fitted to the
correct eigenvalue. Hence, frequency fitting only tends to be useful from empirical
data for special cases, such as O-H stretching modes which are well separated from
other modes and for diatomics where there is no problem in assignment!
One other case where frequency fitting can be useful is at the lower end of the
spectrum. For an isolated molecule or a solid at its Γpoint the first three modes
should have zero frequency as they are just translations. In some cases there may
be imaginary modes due the potentials not correctly reproducing the true symmetry.
Hence by fitting the first three modes to be zero it is possible to encourage the
potentials to yield the correct symmetry.
3.0.36.4 Simultaneous fitting
There is one main difficulty in the conventional scheme for fitting in which the
forces on the atoms are minimised by variation of the potential parameters which
arises when using a shell model. Normally we don’t know what the shell coor-
dinates are at the outset unless the ions are sited at centres of symmetry. In the
past people have tried fitting with the shells placed on top of the cores. However,
this means that the potentials are tuned to minimise the polarisation in the system
120
and leads to the shell model having only a small beneficial effect. It also doubles
the number of observables connected with gradients, but only introduces a small
number of extra variables thus making it harder to get a good fit.
The solution to this problem is allow the shell positions to evolve in some way
during the fit. There are two possibilities - either we can minimise the shell posi-
tions at every point during the fit or we can added the shell coordinates as fitting
parameters. In the case where only structural data is being fitted the two meth-
ods are equivalent except in the way that they evolve towards the answer. When
other properties are included the second approach is not strictly correct, though the
difference is usually small.
After experimenting with several test cases it was found that the second scheme
in which the shell coordinates become fitted variables was far more stable in con-
vergence and more efficient. Hence this is the scheme that has been adopted and
is referred to as ’simultaneous’ fitting due to the concurrent fitting of shell posi-
tions. Whenever working with shell models it is recommended that the keyword
simultaneous is added during conventional fitting - it can improve the sum of
squares by several orders of magnitude! Not only does this scheme apply to the
coordinates of shells, but also to the radii of breathing shells as well.
3.0.36.5 Relax fitting
It has been observed that sometimes in conventional fitting getting an improved sum
of squares doesn’t always get you what is considered to be a better quality fit. This
is because people often use different criteria to make their judgement to the ones
input into the fitting process. In particular they look at the difference between the
optimised structural parameters and those from experiment, rather than looking at
the forces. The reason why the forces can be lower, but lead to a worse structure is
because in a harmonic approximation the displacements in the structure are given
by the gradient vector multiplied by the inverse hessian. Hence, if the gradients get
smaller but the inverse hessian gets much larger then the situation may get worse.
The solution to this problem is to fit according to the criteria by which the struc-
tures are judged - this is what relax fitting does. This means that at every point in
the fit the structure is optimised and the displacements of the structural parameters
calculated instead of the gradients. In this approach the shell model is naturally
handled correctly and so there is no need for simultaneous fitting. The downside is
that it is much more expensive in computer time than conventional fitting. Also you
can only start a relax fit once you have a reasonable set of potential parameters - i.e.
one which will give you a valid minimisation. Hence a conventional fit is often a
prerequisite for a relax fit.
There is a further benefit to using relax fitting. In a conventional fit the prop-
erties are calculated at the experimental structure normally with non-zero gradients
which is not strictly correct. In a relax fit the properties are calculated for the opti-
121
mised structure where they are valid.
3.0.37 Genetic algorithms
Conventional minimisation techniques based upon methods such as Newton-Raphson
are excellent ways of locating local minima. However, they are of limited use in
finding global minima. For example, if we know the chemical composition of a
compound and its unit cell, but don’t know the structure then we would want to
locate the most stable arrangement for placing the atoms within the unit cell. To
search systematically for a reasonable set of atomic coordinates may take a very
long time by hand. Genetic algorithms [26] are a method by which we can search
for global minima rather than local minima, though there can never be an guarantee
of finding a global minimum. In many respects it resembles Monte Carlo methods
for minima searching, though is regarded by some as being more efficient.
The concept behind the method, as the name might suggest, is to carry out a
’natural selection’ procedure within the program in the same way that nature does
this in real life. We start off with an even numbered sample of randomly chosen
configurations. This is our trial set which is allowed to evolve according to a num-
ber of principles described below. Before we can do this we need to consider how
to represent our data for each configuration. To do this we encode each number as
a binary string by dividing the range between the maximum and minimum possi-
ble values (for example 1 and 0 for fractional coordinates) into a series of intervals
where the number of such intervals is an integer power of 2. Given this data repre-
sentation the system now evolves according to the following steps:
(a) Reproduction (tournament) - pairs of configurations are chosen at random and
the parameters which measure the relative quality of the two are compared (this is
the energy for genetic optimisation or the sum of squares for genetic fitting). The
best configuration goes forward to the next iteration, except that there is a small
probability, which can be set, for the weaker configuration to win the tournament.
This process is repeated as many times as there are configurations so that the total
number remains constant.
(b) Crossover - a random point is chosen at which to split two binary strings, after
which the two segments are swapped over.
(c) Mutation - a random binary digit is switched to simulate genetic mutations.
This can help to search for alternative local minima.
The default output from a genetic algorithm run is a given number of the final
configurations, where the ones with the best fitness criteria are selected. However,
unless the run smoothly progresses to the region of a single minimum it may be
122
more interesting to look at a sample of the best configurations from the entire run.
This can be done with GULP using the best option.
Genetic algorithms can only locate minima to within the resolution allowed by
the discretisation used in the binary representation. Also they are very slow to
converge within the region of a minimum. Hence, the genetic algorithm should be
used to coarsely locate the regions associated with minima on the global surface,
after which conventional Newton-Raphson methods will most efficiently pin-point
the precise minimum in each case.
123
3.1 Getting started
3.1.1 Setting environment variables
In previous versions of GULP it was necessary to specify where library files and
the documentation could be found by editing the source code. This has now been
replaced by the use of two environment variables, GULP_LIB and GULP_DOC,
respectively. Under tcsh these can be set as follows:
setenv GULP_DOC /Users/myname/gulp4.2/Docs
setenv GULP_LIB /Users/myname/gulp4.2/Libraries
or by using appropriate commands under other Unix shells.
3.1.2 Running GULP
Under UNIX:
To run GULP on a machine with the UNIX operating system simply type:
<directory>gulp < inputfile
where <directory> is the path name for the location of gulp on your machine, or
if the executable is in your current directory or lies in your path then this may be
omitted. In this case the output will come to your terminal. If you wish to save it to
an output file then type
<directory>gulp < inputfile > outputfile
You may like to try using one of the example input files (called exampleN, where N
is a number) to see what happens! Input may also be typed directly into the program
line by line if no input file is specified. Having finished typing all the required input
just type ‘start’ to commence the run.
3.1.3 Getting on-line help
To obtain on-line help on GULP type
<directory>gulp <CR>
help <CR>
A list of all the possible help topics can then be accessed by typing topics or
alternatively just type the particular keyword or option that you require help on.
124

Only sufficient characters to specify a unique topic are required. To finish with help
type stop if you wish to exit the program or quit if you want to return to interactive
use.
If the help command fails to work it means that the path for the location of the
file help.txt (which is an ordinary ASCII text file containing all the help informa-
tion) has not been set at compile time and that the file is not in the present directory
either.
An alternative way of accessing help is to generate an HTML file using the
gulp2html utility (courtesy of Dr. Jörg-R. Hill) which produces a file help.html
which can then be inspected with a suitable browser, such as netscape.
3.1.4 Example input files
With the program you should have received a number of sample input files which
illustrate how GULP works for a number of particular run types. They also serve as
a test to ensure that the program works correctly on your machine type. Please note
that the interatomic potentials should not be taken as correct for general use - some
are made up for the purposes of demonstration only! Below is a brief description
of what each example file is doing.
Table 3.4: List of examples provided
example1 optimises the structure of alumina to constant pressure and then calculates the properties
at the final point
example2 simultaneous fit of a shell model potential to the structure of α-quartz, followed by an
optimisation with the fitted potentials - the general potential is used with energy and
gradient shifts for the Si-O instead of the usual Buckingham potential
example3 an electronegativity equalisation calculation is used to derive partial charges for quartz
and are then used to calculate the electrostatic potential and electric field gradients at
each site - bond lengths are also calculated
example4 simultaneous fit of a shell model potential to La2O3 using an Ewald-style sum to evaluate
the C6 terms, followed by an optimisation with the production of a table comparing the
initial and final structures at the end
example5 calculation of a phonon dispersion curve for MgO from 0,0,0 to 1/2,1/2,1/2 - note that
normally the structure should be optimised first and that although a phonon density of
states curve is produced this may not be accurate due to restricted sampling of k space.
example6 calculation of the defect energy for replacing a Mg2+ ion in MgO by a Li+ ion to create
a negatively charged defect
example7a location of the transition state for a magnesium cation migrating to a vacant cation site in
MgO in a defect calculation
example7b this shows an alternative way of obtaining the same result as in 7a by starting the magne-
sium in a special position and using the resulting symmetry constraints to allow a ordinary
minimisation to the saddle point
125

example8 a molecular defect calculation in which a sulphate anion is removed from BaSO4 - note
that the use of the mole keyword to Coulomb subtract within the sulphate anion.
example9 an example of how to use a breathing shell model for MgO - including fitting the model,
optimising the structure and calculating the properties
example10 optimisation of urea showing how to handle intermolecular potentials
example11 an example of how to map out the potential energy surface for the migration of a sodium
cation parallel to the c axis through a crystal of quartz with an aluminium defect using the
translate option
example12 optimisation of two structures within the same input file - also illustrates the use of the
name option
example13 shows how to use a library to access potentials for an optimisation of corundum
example14 relaxed fit to structure and properties
example15 simple NVE molecular dynamics
example16 example of constant pressure shell model MD
example17 Sutton-Chen calculation for bulk Ni
example18 example of shell model MD in NVT ensemble
example19 shell model MD run for a zeolite with finite mass
example20 shell model MD run for a zeolite with adiabatic algorithm
example21 charged defect optimisation in a supercell
example22 energy surface fit for a molecular crystal
example23 evaluation of the cost function for a particular structure
example24 example of structure prediction for polymorphs of TiO2
example25 free energy minimisation of quartz within the ZSISA approximation
example26 Using the "ditto" option to run the same structure at 3 pressures in the same file
example27 Interface calculation in which a rigid block of MgO is optimised over the 001 surface of
MgO
example28 Tersoff model calculation on bulk silicon followed by a vacancy defect calculation
example29 Streitz-Mintmire model calculation on bulk alumina
example30 REBO model calculation for a diamond surface
example31 Constant pressure optimisation of a corundum slab with a 2-D phonon calculation
example32 1-D calculation on MgO using manual specification of flags and the switch minimiser
option
example33 Grand Canonical Monte Carlo for a single species being introducted into a box
example34 Grand Canonical Monte Carlo for a rigid molecule being introducted into a box
example35 Voter-Chen EAM for Ag
example36 Frequency dependent property calculation for quartz
example37 SiH4 molecule using the Smith and Dyson REBO 1 model
example38 Electric field applied to a slab from example31
example39 Glue potential for Au with print out of EAM atomic densities and energy contributions
example40 Simple example of 3coulomb potential for a water molecule for validation
example41 Force calculation for a urea molecule in the presence of a continuum solvent model with
qsas keyword
126

example42 Optimisation of the (001) surface of urea in the presence of a continuum solvent model
example43 Example of PDF calculation for MgO - phonons < wmax
example44 Example of PDF calculation for MgO - full phonon range
example45 Example of PDF calculation for MgO - phonons > wmin only
example46 Example of PDF calculation for (Ca,Sr)TiO3
example47 Example of Eckart keyword for removing rotation/translation from molecular vibrations
example48 Fitting of vibration modes using specific eigenvectors for a water molecule
example49 UFF input file with manual specification of bonds for meta-xylene
example50 Example of Dreiding input
example51 Example of a thermal conductivity calculation for Allen-Feldman diffusons in amorphous
silicon
example52 Example of a thermal conductivity calculation that uses estimated B and v_s from prop-
erties
example53 Calculation of Raman intensities for the vibrations of alpha-quartz
example54 Example of ReaxFF for an ethene molecule
example55 Example of a MEAM-2NN calculation for FCC Cu metal
3.2 Guide to input
3.2.1 Format of input files
On the whole it is only necessary to use up to the first four letters of any word,
unless this fails to specify a unique word, and the input is not case sensitive as all
characters are converted to lower case on being read in.
The first line of the input is the only special line and is referred to as the key-
word line. Keywords should all be given on this line. These consist of control words
which require no further parameters and generally specify the tasks to be performed
by the program. For example a typical keyword line would look like:
optimise conp properties phonon
or in abbreviated form:
opti conp prop phon
This combination of words tells GULP to do a constant pressure optimisation
and then to calculate the lattice properties and phonons at the optimised geometry.
The order of words within the keyword line is not significant.
All subsequent lines can be given in any order unless that line relates to a pre-
vious piece of input. Such lines contain ‘options’ which generally also require the
specification of further information. This information can normally follow on the
127
same line or on the subsequent line. For example the pressure to be applied to a
structure could be input as either
pressure 10.0 or pressure
10.0
In many cases the units may also be specified if you don’t wish to use the default:
pressure 1000 kbar
Any lines beginning with a ‘#’ and anything that follows a ‘#’ part way through
a line is treated as a comment and as such is ignored by the program.
When performing runs with multiple structures any structure dependent options
are assumed to apply to the last structure given, or the first structure if no struc-
ture has yet been specified. Some options should be specified as sub sections of
a particular option. For example, elastic, sdlc, hfdlc, piezo, energy and
gradients all are sub sections of the observables command and should appear
as follows:
observables
elastic 2
1 1 54.2
3 3 49.8
hfdlc
1 1 2.9
end
Provided there is no ambiguity, GULP will accept these options even if observables
is omitted, however, it makes the input more readable if the section heading is in-
cluded.
GULP reads only the first 80 characters on a line in an input file. Should an
input line be two long to fit within this limit then the line can be continued on a
second or further lines by adding the continuation character ‘&’ to the end of the
line.
3.2.2 Atom names
Many parts of the input to GULP require the specification of atom names, be it when
giving their coordinates or when specifying potential parameters. The convention
adopted in GULP is that an atom should be referred to by its element symbol,
optionally followed by a number to distinguish different occurrences of the same
element. Numbers between 1 and 999 are valid numbers. Hence examples of valid
128
atom specifiers are Si,Si12, O3 and H387. Something like Si4+ would not be a
valid symbol. The reason for using the element symbol is because several calcu-
lations use elemental properties such as the mass or covalent radii in dynamical or
molecular runs, respectively.
Sometimes it is desirable to label all the atoms in the structure with numbers
to identify them, but with the same interatomic potential acting on them. To avoid
having to input the potential multiple times for each symbol there is a convention
within GULP which it is important to know. Any reference to just an atomic symbol
applies to all occurrences of that element, whereas any reference to an atom type
with a number only applies to that specific species. For example a Buckingham
potential specified as follows:
buck
Si core O core 1283.0 0.299 10.66 0.0 12.0
would apply to all Si atoms, regardless of whether they are called Si or Si1 etc.
However, the following potential:
buck
Si1 core O core 1283.0 0.299 10.66 0.0 12.0
would only act on Si1. It is important to remember this as people have labelled
one atom Si and the Si1 in the past and put potentials for both which resulted in
twice the potential acting on Si1 as there should have been. If the potential had
been specified as just acting on Si then the correct answer would be obtained as it
would act on both atoms once.
In addition to the atom label there is optionally a species type specifier which
should be one of the following:
core - represents the main part of an atom including all its mass
shel - represents the mass-less component in a shell model
bcor - a core, but with a spherical breathing radius
bshe - a shell, but with a spherical breathing radius
If not given, then core is the default type. Note that the bcor and bshe types
only need to be used in the structure specification. There after they can be treated
as an ordinary core or shell in the potential specification and the program will select
whether the potential should act on the radius or the centre of the species.
129
3.2.3 Input of structures
The structure for a three-dimensional solid requires the input of three main sets of
information - the unit cell, the fractional coordinates and types of the atoms, and
finally the space group symmetry. Taking these in order, the unit cell can be input
either as the cell parameters:
cell
4.212 4.212 4.212 90.0 90.0 90.0
or as the cell vectors:
vectors
4.212 0.000 0.000
0.000 4.212 0.000
0.000 0.000 4.212
Normally it is easiest to use the cell parameter form and this is recommended.
The main reason why you might chose to use the cell vectors is because you want to
calculate the properties in a non-standard reference frame (given that quantities such
as the elastic constants depend on the unit cell orientation relative to the Cartesian
frame). If the cell parameters are input, then the acell vector is aligned along the
xaxis, the bcell vector in the xy plane and the ccell vector in the general xyz
direction.
If the keywords conp or conv are not specified, then the program will expect
6 flags to indicate how the unit cell is to be optimised. Here a flag of 1 implies
that a degree of freedom should be allowed to vary and 0 will imply that it should
be kept fixed. Because GULP works with the strain tensor, these flags refer to the
strain components in the order xx, yy, zz, yz, xz and xy, respectively. Examples of
how to input the flags for the case given above when only the on-diagonal strain
components are to be varied are given below:
cell
4.212 4.212 4.212 90.0 90.0 90.0 1 1 1 0 0 0
or as the cell vectors:
vectors
4.212 0.000 0.000
0.000 4.212 0.000
0.000 0.000 4.212
111000
130
When GULP transposes a system between the primitive and centred unit cells
the orientation of the atoms is preserved so that any properties calculated will be
the same regardless of the cell used. It is recommend that the cell parameters be
used for input where possible as this ensures that symmetry can be used to acceler-
ate optimisations. Turning now to the internal coordinates of the atoms, these can
again be given either in fractional or Cartesian form, though the former is the more
natural for a periodic system. Each line of input must contain at least the atom label
followed by the coordinates, in which ever units. For example for the case of MgO:
fractional
Mg core 0.0 0.0 0.0
O core 0.5 0.5 0.5
Note that if the space group symmetry is to be given then it is only necessary to
specify the atoms of the asymmetric unit. Furthermore in any cases where a frac-
tional coordinate is a recurring decimal, such as 1/3, then it is necessary to specify
this value to six decimal places to be sure of it being recognised correctly as a
special position. If we were to include a shell model for oxygen then the input of
coordinates would now look like the following:
fractional
Mg core 0.0 0.0 0.0
O core 0.5 0.5 0.5
O shel 0.5 0.5 0.5
There is no need to specify the number of atoms to be input or to terminate the
section as this is automatically done when the program finds something which is
not an element symbol or a special character at the start of a line.
If a shell model has been specified for a given atom in the species section (see
later), but the shells are omitted from the input for the structure then GULP will
attempt to add them automatically. When this happens, the shell be placed at the
same location as the core by default.
In addition to the coordinates, there are a number of optional parameters which
can follow the zcoordinate on the line. These are, in order, the charge, the site oc-
cupancy (which defaults to 1.0), the ion radius for a breathing shell model (which
defaults to 0.0) and 3 flags to identify geometric variables (1 ⇒vary, 0 ⇒fix). Note
that the flags will only be read if there is no keyword to specify the geometric vari-
ables (e.g. conp or conv). Hence in full the input for MgO could look as follows:
fractional
Mg core 0.0 0.0 0.0 2.00000 1.0 0.0 0 0 0
131
O core 0.5 0.5 0.5 0.86902 1.0 0.0 0 0 0
O shel 0.5 0.5 0.5 -2.86902 1.0 0.0 0 0 0
In the case of MgO all the flags can be set to 0 as there are no geometric variables
within the unit cell by symmetry.
If we wanted to run a breathing shell calculation for MgO then the input might
look like the following for a constant pressure run:
fractional
Mg core 0.0 0.0 0.0 2.00000 1.0 0.0
O core 0.5 0.5 0.5 0.86902 1.0 0.0
O bshe 0.5 0.5 0.5 -2.86902 1.0 1.2
or for a mean field calculation of the energy of a 40/60 MgO/CaO material:
fractional
Mg core 0.0 0.0 0.0 2.00000 0.4 0.0
Ca core 0.0 0.0 0.0 2.00000 0.6 0.0
O core 0.5 0.5 0.5 0.86902 1.0 0.0
O shel 0.5 0.5 0.5 -2.86902 1.0 0.0
The space group symmetry can be specified either through the space group num-
ber or through the standard Hermann-Mauguin symbol. Again for MgO, either of
the following would be valid:
space
225
or space
F M 3 M
In general it is better to use the symbol rather than the number as the structure
may be in a non-standard setting. The help file contains a full list of the standard
symbols for each space group to illustrate how the symbol should be written in the
input, though further non-standard settings will be accepted. The space option is
not compulsory in the input of a structure and if it is absent then GULP will assume
that the structure is in P 1 (i.e. no symmetry).
Related to the space option is the origin option which allows non-standard
origins to be handled. The input for this option can take the form of a single integer
(1 or 2) if you want to select one of the standard alternative origin settings. Alter-
natively if three floating point numbers are input then they are taken to be an origin
shift in fractional coordinates, or if three integer numbers are input then they are
divided by 24 to obtain the shift.
The structural input for a molecular system is just the Cartesian coordinates.
Currently the use of point group symmetry is unavailable for isolated systems so
132
there is no equivalent command to space for molecules. There is unlikely to be
much benefit from the addition of point group symmetry as most molecular calcu-
lations are much faster than their solid state analogues.
Multiple structures can be included in the same file by placing one after another,
including mixtures of solid and molecular compounds. A useful option for keeping
track of different structures is the name option. This must precede the structure and
allows the user to give a one word name to the compound which will then be used
as a label in the output file. Using this the structural input for a file containing both
corundum and quartz might look as follows:
name corundum
cell
4.7602 4.7602 12.9933 90.0 90.0 120.0
frac
Al core 0.00000 0.0 0.35216
O core 0.30624 0.0 0.00000
space
167
name quartz
cell
4.91485 4.91485 5.40629 90.0 90.0 120.0
frac
Si core 0.4682 0.0000 0.333333
O core 0.4131 0.2661 0.213100
space
152
In the situation where you want to run the same structure at several different condi-
tions then the ditto option can be particularly useful to avoid repeating the struc-
ture. For example, to run corundum at a pressure of 1 GPa and 2 GPa you could use
the following input for the structures:
name corundum
cell
4.7602 4.7602 12.9933 90.0 90.0 120.0
frac
Al core 0.00000 0.0 0.35216
O core 0.30624 0.0 0.00000
space
167
pressure 1 GPa
ditto structure
133
pressure 2 GPa
3.2.4 Species / libraries
In the input for the coordinates there was the option to input the species charge for
each individual atom in the asymmetric unit or even the full cell. Normally this
is unnecessary as all atoms of the same type have the same charge. In this latter
case the charges can be assigned by the species option. So for a zeolite structure,
for example, where there may be lots of different Si and O sites we could assign
charges as follows:
species
Si core 4.00000
O core 0.86902
O shel -2.86902
The species command can also serve another purpose which is to assign poten-
tial library symbols to each atom type. Quite often we may simulate a whole series
of materials with a standard set of potentials. Rather than typing them in every time
we can call a library. GULP at present comes with two libraries - one for zeolite and
aluminophosphate type systems [3,28,29] and one for metal oxides from the work
of Bush et al [30]. All we need to do to call these potentials is to assign the poten-
tial types to the types in the library files. In the case of bush.lib, there is no need
to do anything as the symbols are just the metal element symbols. For the zeolitic
materials there is more than one kind of some atom types and so an assignment is
needed. Using this our input would look like:
species
Si core Si
O core O_O2-
O shel O_O2-
library catlow.lib
3.2.5 Input of potentials
The various types of potentials available in GULP have been tabulated earlier and
detailed descriptions of the input format for each one can be found in the on-line
help. This section will therefore just contain some general pointers as to how to
input potentials.
134
Let us take the example of a Buckingham potential which acts between magne-
sium cores and oxygen shells with the parameters A=1280.0eV, ρ=0.300Å, C=4.5
eVÅ6and acts over the range of 0 to 12 Å. The input for this would look as follows
for an optimisation run:
buck
Mg core O shel 1280.0 0.3 4.5 0.0 12.0
If we want to perform a fitting run then it is also necessary to specify the flags
which indicate which parameters are to be variables (1) and which ones are not (0).
There is one flag for each potential parameter and the order of the flags matches
that of the parameters. Hence a fit in which we want to vary A only would look as
follows:
buck
Mg core O shel 1280.0 0.3 4.5 0.0 12.0 1 0 0
It would not matter if we had put the flags on the end of the line in the input
for an optimisation run - they would have just been ignored. For most potential
types some parameters are optional and can be omitted, normally when they are
zero. There is always a hierarchy to the order of omission of values. For example,
for most two-body potentials if one number is missing then this is assumed to be
the minimum cut-off radius and this value is zero (as it quite commonly is). For a
Buckingham potential, if a second number is omitted then this is assumed to be the
C term which again is often zero. If you are going to omit values it is important to
remove the flags when not needed from the input as this may confuse matters. If in
doubt give all values!
The number of input parameters can also vary according to any options speci-
fied after the potential type. For example, if we wanted the above potential to only
act between atoms which are bonded then the input would be:
buck bond
Mg core O shel 1280.0 0.3 4.5
No potential cut-offs are needed as these are set by the fact that the atoms
must be bonded. Similarly for a Lennard-Jones potential when given as lennard
combine then no potential parameters will appear on the input line as these are
determined by combination rules.
More than one potential can be specified for each occurrence of the potential
type. Hence the following would be perfectly valid:
buck
135
Mg core O shel 1280.0 0.3 4.5 0.0 12.0
Ca core O shel 1420.0 0.3 6.3 0.0 10.0
If the input for one potential is too long to fit on one line then it may be continued
on to the next line by using the continuation character ‘&’ at the end of the line.
For two-body potentials there is no ambiguity about the order of the atoms as
both are equivalent. For some three-body potentials and all four-body potentials
it is important to be aware of the convention regarding the order of input. For a
three-body potential which has a unique pivot atom, typically at which the angle is
measured, then this pivot atom must be given first and then the two terminal atoms
in any order. Hence the O-Si-O angle bending term widely used for zeolites is input
as:
three
Si core O shel O shel 2.09 109.5 1.9 1.9 3.6
In the case of four-body terms there is no unique pivot and so the atoms are
input in the order which they are connected. A piece of good advice is that three-
and four-body terms are often most readily dealt with using connectivity based cut-
offs as part of the molecule set of options.
3.2.5.1 Input of PDF settings
Appropriate keywords for normal GULP operation should be used. Importantly,
the potential model needs to be properly optimised using opti. To run the PDF
module, the keyword PDF should be added. This automatically enables the phonon
and eigenvector keywords, calculating the necessary phonon information. It also
activates the keyword makeEigenArrays (which stores all phonon data in internal
arrays), and rephase which rephases the (complex) eigenvectors so that the com-
ponent with the largest magnitude is all real for all eigenvectors and also checks
the normalisation. This rephasing is a rotation in the complex plane that does
not change the results of the PDF calculations performed using the eigenvectors,
but helps with visualisation. To ensure that the entire Monkhorst-Pack grid of k-
points is used (not the symmetry reduced one), noksymmetry is also enabled. It
is often useful to use the nokpoints keyword to prevent the output of all the k-
points used (a typical PDF calculation may require 8000 k-points before conver-
gence is acheived). Similarly, suppression of phonon output is useful: the keyword
nophonon is used.
In addition to calculating the PDF from the full phonon data, it is possible to
restrict the frequencies (ω) used. A cut-off of ωmin/ωmax can be set in the op-
tions (see below). When the keyword PDFcut or PDFbelow is used, all frequencies
above/below the cut-off frequency are excluded. (There is no need to enter the PDF
136
keyword separately as it is assumed). Alternatively, all frequencies above/below the
cut-off frequency can be set to the cut-off frequency by the addition of PDFkeep.
3.2.5.2 Options
The configuration should be set up in the normal way. Importantly, in addition to
specifying the structure and potentials, a Monkhorst-Pack grid of k-points must be
generated using the shrink option (as described in section 3.0.28.1). (Convergence
of phonon properties with number of k-points is essential before results can be
considered accurate. If the width of all pairs of type xi−yj, as given in the .wid file
are divergent increase the density of the grid.)
As with other output files, the GULP option output can be used to specify the
filename:
output pdf <filename>
It is important to specify an output file for all configurations, otherwise the root
<pdf >or <pdf_config_# >will be used. Two filestypes are produced
.wid file Lists every atomic pair and the width (use of keyword nowidth supresses
this)
.pdfs file Outputs Pair Distribution Functions up to rmax with the number of bins
specified at input
Within the literature, there are a number of different correlation functions; the
module expresses the results in several of these: rho(r),GPDF(r),G(r),D(r), and
T(r). More details are available in the ‘Further Background’ section of this manual.
Where phonon selection has been used, ωmax/ωmin is also stated in the output files,
using the frequency units specified (see below), together with the temperature and
number of k-points.
All other input options for PDF calculations are entered in lower case within a
pdf input block.
The pdf input block is opened with the word pdf and closed with the word end.
i.e.
pdf
<PDF options>
end
When working with multiple configurations, it is possible to open the pdf block with
"pdf all": it then operates on all configurations, ensuring that comparable PDFs are
produced with the same cut-offs, maximum radius and binning. Alternatively, a
seperate pdf block can be given for each configuration.
For PDFs, the maximum radius (in Å) should be specified (default = 5 Å), and
the number of bins used for the output (default = 100). These can be set, within the
pdf input block, using rmax and rbins respectively, or the default values used.
137
pdf
rmax [real]
rbins [int]
end
When using the frequency cut-off facility, either wmin or wmax (as appropriate)
must be set, again in the pdf input block. While the default units for frequency
input/output in the PDF module is THz, it can (optionally) be changed as shown
using the units options: (cm or wav can be used for wavenumbers)
pdf
rmax [real]
rbins [int]
units freq [rad/Thz/cm/wav/meV]
wmin [real]
or
wmax [real]
end
In neutron scattering, advantage is often taken of the different scattering lengths
of different isotopes. The library file only contains element information. However,
this can be overwritten using the element option input block with bbar or siginc
(mass, ionic radius, etc, can also be changed in this way, see command help for
more). The units are Å and Å squared. NB. The Sears table, used to produce the
eledata Library file, uses Fm and Barns (=1×10−28m).
3.2.6 Defects
In this section we shall cover the basic input required to perform a Mott-Littleton
calculation for an isolated defect in an otherwise perfect solid, as activated by the
presence of the keyword defect. The main run type that we will be concerned
with for defects is optimisation as we normally wish to obtain the defect energy and
structure. We may also wish to locate transition states for the migration of defects
- this follows the same approach as an optimisation, but with the keyword trans
rather than opti. There is currently no facility to fit to defect quantities.
The first issue to consider is the bulk calculation that must precede a defect
calculation. For a correct calculation the bulk structure must be optimised at least to
constant volume otherwise negative defect energies may result from the removal of
bulk forces, rather than defect related ones. If you intend to perform several defect
calculations and the bulk unit cell is a reasonable size it is sensible to optimise
the bulk as a first job and then use the restart file for the defect calculations to
avoid wasted effort. There are also other reasons for optimising the bulk separately.
Firstly, if you want to perform a transition state run then the trans keyword will try
138
to be applied to the bulk as well as the defect with strange results (this can avoid by
using the bulk_noopt keyword). Secondly, when creating defects it is important
to know where the bulk atoms are so that they can be placed correctly.
At the end of the bulk calculation a property evaluation must be performed as
the second derivatives and dielectric constants are needed for the response tensors
of region 2. This is automatically invoked and there is no need to add the keyword
property. Again for some materials the calculation of the properties can be ex-
pensive. Hence, if multiple defect calculations are to be performed then this step
can be minimised by adding the keyword save to the first run (which will write
out a temporary file fort.44 which contains the quantities needed for future runs in
a binary form to save space) and the keyword restore to subsequent runs (which
will cause them to read in the information from fort.44 rather than re-calculating
it).
Having dealt with the preliminaries, we are now ready to consider how to input
the details of the defect calculation. Remember, the following commands should
appear after the structure to which they refer. Firstly, we need to determine the
defect centre around which the regions are based. This is given using the option
centre (center will also work for the benefit of those of you who are American-
minded!). The defect centre is normally placed at the same position as the defect,
in the case of a single defect site, or at the middle of a series of defects so as to
maximise the distance between any defect and the region 1 boundary. Symmetry
is not explicitly input by the user for a defect calculation. However, the program
will automatically try to search for any simple symmetry elements. These can then
be used to accelerate the calculation. In order to do this it requires that the defect
centre is chosen so as to maximise the point group symmetry about itself. This is
worth keeping in mind when choosing the location of the defect centre. A gen-
eral feature of the centre option, and those which specify the positions of defect
species, is that there are a number of alternative methods for specifying the location:
(a) Atom symbol : this is the label for a species within the unit cell. It is best to use
a unique specifier (by changing the type number of one atom if necessary) - if there
is ambiguity the program will normally chose the first occurrence of the symbol.
centre Mg2 core
This will place the defect centre at the final bulk position of the Mg2 core.
(b) Atom number : here the position is given by the number of the atom in the
asymmetric unit as input.
centre 3
139
This will place the defect centre at the final bulk position of the third atom in the
asymmetric unit.
(c) Fractional coordinates : here the position is explicitly given in fractional units
- the frac option in the following command is optional as it is the default:
centre frac 0.25 0.25 0.25
(d) Cartesian coordinates: here the position is explicitly given in Cartesian coor-
dinates, the origin of the unit cell being at 0,0,0:
centre cart 1.5 2.4 0.8
(e) Molecule number: this places the defect centre at the middle of the molecule
whose number is given (which corresponds to that in the output):
centre mol 2
Having located the defect centre the next thing we need to do is to specify the region
1 and region 2a radii. This is done with the size command:
size 4.0 10.0
would result in a region 1 radius of 4.0 Å and a region 2a radius of 10.0 Å. It is im-
portant to check how sensitive the defect energy is to these values and to increase
them until satisfactory convergence is achieved. One way of doing this while min-
imising computational expense is by using the restart file. If we wanted to restart a
defect calculation run with the above radii, but with region 1 increased to 6.0 Å and
region 2a to 12.0 Å then we would just need to edit the restart file to contain the new
values, plus the old region 1 size (needed for correct restarting) at the end of the line:
size 6.0 12.0 4.0
We now have specified the regions - next we need to create some defects. There
are three options for this:
vacancy - removes an ion from the structure to infinity
interstitial - inserts an ion into the structure from infinity
impurity - replaces one ion with a different one
The last one, an impurity, is obviously just a short-cut combination of the other two.
The actual input for each option for specifying the ion(s) involved follows that for
centre in that the atom label, atom number, fractional or Cartesian coordinates can
be used. The molecule number can also be used with the vacancy option, in which
140
case it removes all the atoms of the molecule from the structure (see example8).
Note that when molecules are removed and inserted it is important to correct the
defect energy for the molecule at infinity, if this is not zero, as this is not done
automatically.
An important part of the interstitial and impurity commands is to specify the
type of species to be inserted. For example, the impurity command to replace O2
with Swould be:
impurity S O2
The key thing to note is that the inserting species is always specified first. To
make life easy shells are handled automatically in most cases. So if both O2 and S
were specified as shell model atoms then the above command would remove both
the O2 core and shell, and also insert the S core and shell (initially at the same
position). It becomes important when introducing species with a type different to
any of the bulk species that all the necessary properties of the inserting ion are given
in the species option otherwise the atom will be assumed to have no charge and
no shell.
There is one further option when introducing an interstitial to make life easier.
For example, imagine we want to protonate an oxygen (typically in a zeolite frame-
work) to generate a hydroxyl group at O2. This can be readily achieved using the
bond option:
interstitial H bond O2
this will place the H into the structure at the sum of the covalent radii from O2. To
determine the direction for the bond the program maximises the angles to any other
atoms to which O2 has bonds.
Putting all these keywords together, we shall illustrate how a lithium impurity
could be created in magnesium oxide to generate a negatively charged defect. For
the purposes of this example we will work with the full unit cell structure as given
by the following:
opti conp defect
cell
4.212 4.212 4.212 90.0 90.0 90.0
frac
Mg core 0.0 0.0 0.0
Mg core 0.0 0.5 0.5
Mg1 core 0.5 0.0 0.5
Mg core 0.5 0.5 0.0
O core 0.5 0.5 0.5
141
O core 0.5 0.0 0.0
O core 0.0 0.5 0.0
O core 0.0 0.0 0.5
species
Mg core 2.0
O core -2.0
Li core 1.0
Based on the above basic input (+ interatomic potentials) all the following
would be valid ways of creating the defect:
(a)
centre Mg1
size 6.0 12.0
vacancy Mg1
interstitial Li 0.5 0.0 0.5
(b)
centre Mg1
size 6.0 12.0
impurity Li Mg1
(c)
centre 0.5 0.0 0.5
size 6 12
impurity Li 0.5 0.0 0.5
(d)
centre 3
size 6 12
impurity Li cart 2.106 0.0 2.106
There are more permutations than this, but hopefully it gives you the general idea.
3.2.7 Restarting jobs
Because on the whole GULP doesn’t require very much CPU time per step (the ex-
ceptions normally being second derivative calculations on large systems) it doesn’t
maintain a binary dumpfile with all the details for a restart from exactly where it
left off. Instead there is the option to dump out a restart file which is just a copy
of the original input (slightly rearranged!) but with any coordinates or potential
parameters updated as the run progresses. The frequency with which this is written
142

out can be controlled by the user. If the following is specified:
dump every 4 gulp.res
then a restart file will be written after every 4 cycles. If the ‘4’ is omitted then it
defaults to being one - i.e. a dumpfile is written after every cycle. As the cost of
writing out this file is normally small compared to the cost of a cycle this is the
usual choice. If the every option is omitted then the restart file is written just at the
end of the run.
3.2.8 Memory management
From version 3.4 of GULP the code was written in Fortran90 and therefore pretty
much all memory is dynamically allocated. Hence there should rarely be any need
to alter the code to increase a parameter. The only main parameter in the code is the
number of elements, which is set to 107 by default. In the event that you wish to
study elements further down the periodic table than this, you need to edit this value,
add the appropriate element data and recompile.
Even though the code is fully dynamic, you can still exhaust the memory of
your computer under certain circumstances. Remember that any runs that require
the analytic second derivatives, such as phonons or Newton-Raphson optimisation,
will cause a memory requirement of roughly 5
2(3N+6)2double precision words.
Hence if you run out of memory or the machine starts swapping you should first
of all try to reduce the memory demands by using an optimisation algorithm that
doesn’t require full second derivatives, such as unit-Hessian based BFGS, limited
memory BFGS or conjugate gradients.
143
3.2.9 Summary of keywords
The following is a concise summary of all the valid keywords available in GULP -
for more detail consult the on-line help.
Table 3.5: Valid keywords in GULP
angle calculate valid three body angles
anneal perform simulated annealing
average output average bond lengths
bond calculate valid bond lengths based on covalent radii
breathe only calculate gradients for breathing shell radii
broaden_dos apply Lorentzian broadening to density of states data
bulk_noopt fix bulk structure prior to a defect calculation
c6 calculate C6 terms using lattice sum method
cartesian output Cartesian coordinates for initial structure
cellonly only calculate gradients for and optimise cell parameters
cmm calculate cluster electrostatics using cell multipole method
compare produce a table comparing the initial and final geometries
conjugate use conjugate gradients
conp perform constant pressure calculation - cell to vary
conv perform constant volume calculation - hold cell fixed
coreinfo output atomic information (for cores not shells) used in phonon calculations
cosmo use the COSMO continuum solvation model
cosmic use the COSMIC continuum solvation model
cost perform cost function calculation
dcharge output the first derivatives of the atomic charges
defect perform a defect calculation after bulk calculation
dfp use Davidon-Fletcher-Powell update rather than BFGS
dipole add the dipole correction energy for the unit cell
distance calculate interatomic distances
eem calculate charges using electronegativity equalisation
efg calculate the electric field gradients
eigenvectors write out eigenvectors for phonons/frequencies
fit perform a fitting run
fix_molecule fix connectivity for molecules at start and do not update
free_energy perform a free energy instead of internal energy calc
frequency calculate defect frequencies
full write out structure as full rather than primitive cell
gear use the Gear fifth order algorithm for molecular dynamics
genetic perform a genetic algorithm run
global after global optimisation, dump out restart file before opt
gradients perform a single point calculation of energy and gradients
144
hessian output hessian matrix
hexagonal write out structure as hexagonal rather than rhombohedral
hideshells suppress output of shells
include_imaginary include imaginary frequencies in phonon DOS/dispersion output
intensity calculate IR intensities for phonon/vibrational modes
isotropic allow cell parameters to vary isotropically
linmin output details of line minimisation
lower_sym try to lower the symmetry using imaginary modes
madelung include the Madelung correction for a charged simple cubic system
makeeigenarrays store all eigenvectors and frequencies after calculation
md perform a molecular dynamics run
minimum_image use the minimum image convention during MD
molecule locate molecules and coulomb subtract within them
molmec locate molecules and coulomb subtract 1-2 and 1-3
molq locate molecules for intramolecular potentials only
montecarlo perform a Monte Carlo simulation
noaddshells do not automatically add missing shells
noanisotropic use isotropic formula for region 2b energy
nobreathe freeze breathing shell radii during optimisation
nod2sym do not use symmetry for second derivatives
nodpsym do not use symmetry for defect potential calculation
noelectrostatics turns off Ewald sum even when charges are present
noenergy do not calculate the energy - useful for debugging datasets
noexclude do not use atom freezing algorithm during optimisation
nodensity do not output density of state plot after phonon calculation
nodsymmetry do not use symmetry during a defect calculation
nofirst_point skip first point in a translate run
noflags no variable flags are present and all variables to be excluded
nofrequency do not print out frequencies at each k point
nokpoints do not print out k point list
noksymmetry do not use Patterson symmetry in Brillouin zone
nolist_md do not use list method for 3- and 4-body terms in MD
nomcediff controls algorithm for Monte Carlo energy evaluation
nomolecularinternalkeremove initial intramolecular kinetic energy in MD
nononanal exclude non-analytic correction at gamma point
nopartial suppress output of partial PDFs
noreal exclude all real space two-body interactions
norecip exclude all reciprocal space two-body interactions
norepulsive do not truncate repulsive terms based on Ewald accuracy
norxQ turn off charge calculation in ReaxFF
nosasinitevery do not initialise solvent accessible surface at each step
nosderv do not use symmetry for gradient calculations
145

nosymmetry turn off symmetry once unit cell has been generated
nowidth suppress output of peak widths for PDF calculations
nozeropt exclude zero point energy from free energy calculation
numdiag estimate on-diagonal hessian elements numerically
numerical forces use of numerical second derivatives for properties
operators print out listing of symmetry operators used
optimise minimise the energy with respect to geometrical variables
outcon output constraints in restart file
pacha compute variable charges using the PACHA formalism of Marc Henry
PDF calculate the peak widths for each atomic pair and the Pair Distributions
PDFcut as per PDF but cut-off all phonon contributions greater than ωmax
PDFbelow as per PDF but cut-off all phonon contributions below ωmin
PDFkeep as with PDFcut and PDFbelow but set all ω>ωmax to ωmax (or ω<ωmin to
ωmin)
phonon calculate the lattice phonon modes and cluster frequencies
positive force hessian to remain positive on the diagonal
pot calculate the electrostatic potential at the atomic sites
potgrid calculate the electrostatic potential over a grid
predict perform structure prediction calculation
property calculate the bulk lattice properties
pureq use pure Coulomb potential in COSMO/COSMIC molecular case
qeq calculate charges using the QEq scheme of Rappe and Goddard
qiter solve for charges using iterative algorithms rather than matrix diagonalisation
qok run with a non-charge neutral unit cell
qsas causes charges and details of solvent accessible surface to be output
raman causes calculation and output of Raman susceptibility tensors
regi_before output region 1 coordinates before defect calculation
relax use relax fitting
rephase change the eigenvector phases so largest magnitude component is all real for
all eigenvectors and renormalize to unity
restore read in defect calculation restart info and skip property calcn
rfo use the rational function optimisation method
save write out defect calculation restart information
shell only calculate gradients for and optimise shell positions
site_energy output energy per atomic site
simultaneous simultaneously optimise shells while fitting
simplex use simplex algorithm for fitting
single perform a single point calculation of the energy
sopt output shell iterations during molecular dynamics
static_first perform a static optimisation before a free energy one
spatial use domain decomposition algorithm where possible
146

thermalconductivity calculate the Allen-Feldman thermal conductivity for diffusons in an amor-
phous solid
torsion calculate valid four body torsion angles
transition_state optimise using RFO to first order transition state
unit use a unit matrix as the initial hessian for optimisation
zero_potential set average potential across lattice sites to zero
zsisa use the ZSISA approximation in a free energy minimisation
3.2.9.1 Groups of keywords by use
(a) Control of calculation type
angle, bond, cosmo, cosmic, cost, defect, distance, eem, efg, fit,
free_energy, gasteiger, genetic, gradients, md, montecarlo, noautobond,
noenergy, optimise, pacha, pot, predict, preserve_Q, property, phonon,
qeq, qbond, raman, single, sm, static_first, thermalconductivity,
torsion, transition_state
(b) Geometric variable specification
breathe, bulk_noopt, cellonly, conp, conv, isotropic, orthorhombic,
nobreathe, noflags, shell, unfix
(c) Algorithm
c6, dipole, fbfgs, fix_molecule, full, hill, kfull, marvinSE, madelung,
minimum_image, molecule, molmec, molq, newda, noanisotropic_2b, nod2sym,
nodsymmetry, noelectrostatics, noexclude, nofcentral, nofirst_point,
noksymmetry, nolist_md, nomcediff, nonanal, noquicksearch, noreal,
norecip, norepulsive, nosasinitevery, nosderv, nozeropt, numerical,
qiter, qok, spatial, sopt, storevectors, nomolecularinternalke, voigt,
zsisa
(d) Optimisation method
conjugate, dfp, lbfgs, numdiag, positive, rfo, unit
(e) Output control
average, broaden_dos, cartesian, compare, conserved, dcharge, dynamical_matrix,
eigenvectors, global, hessian, hexagonal, include_imaginary, intensity,
linmin, meanke, nodensity_out, nodpsym, nofirst_point, nofrequency,
nokpoints, operators, outcon, prt_eam, prt_two, prt_regi_before, qsas,
restore, save, site_energy, terse
(f) Structure control
full, hexagonal, lower_symmetry, nosymmetry
147

(f) PDF control
PDF, PDFcut, PDFbelow, PDFkeep, coreinfo, nowidth, nopartial
(g) Miscellaneous
nomodcoord, oldunits, zero_potential
3.2.9.2 Summary of options
The following is a concise summary of all the valid options available in GULP - for
more detail consult the on-line help.
Table 3.6: Valid options in GULP
14_scale specifies the 1-4 scaling factor for molecular mechanics
accuracy specifies the accuracy of the Ewald summation
atomab specifies the one-centre A and B terms for combination rules
axilrod-teller specifies an Axilrod-Teller three-body potential
bacoscross specifies a bond-angle cosine cross term three-body potential
bacross specifies a bond-angle cross term three-body potential
bagcross specifies a bond-bond-angle general cross term three-body potential
balcross specifies a bond-bond-angle linear cross term three-body potential
bbar used within element input block to overwrite default scattering length
best output best N configurations from genetic algorithm run
bcross specifies a bond-bond cross term three-body potential
both all subsequent potentials are both inter- and intra-molecular
box specify box size/number for dispersion and DOS plots
brenner specifies that the Brenner REBO energy be calculated
broaden_dos alters the default DOS broadening factor
bsm specifies radial parameters for a breathing shell model
buck4 specifies a four range Buckingham potential
buckingham specifies a Buckingham potential
bulk_modulus gives the experimental bulk modulus for fitting (cubic only)
cartesian input crystal structure using Cartesian coordinates
cell input unit cell as a, b, c, alpha, beta, gamma
centre specifies location of defect centre
charge vary specified atomic charges during fitting
cmm selects cell multipole method for Coulomb terms and order
configurations controls the number of configurations in genetic algorithm
constrain input constraints between variables (fitting and optimisation)
contents specifies the unit cell contents for structure prediction
cosmoframe manually specifies frame of reference for a molecular COSMO/COSMIC run
148

cosmoshape selects between octahedral or dodecahedral triangulation in
COSMO/COSMIC
cost set the parameters for the cost function
coulomb specifies a coulomb subtraction potential
covalent specifies the covalent radii for an element
covexp specifies the covalent-exponential potential
crossover specifies the crossover probability in the genetic algorithm
cutd cutoff for distance calculation
cutp overall interatomic potential cutoff
cuts core-shell cutoff distance
cv specifies the constant volume heat capacity for fitting
cwolf controls the Wolf summation used in the COSMO/COSMIC methods
damped_dispersion C6 and C8 potentials with short range damping
deflist input defect species list for restart
delay_field delay start of time-dependent field in MD
delay_force delay start of time-dependent force in MD
delf maximum energy change before hessian is recalculated
delta specifies the differencing interval for numerical gradients
discrete specifies the discretisation interval for the genetic algorithm
dispersion produces phonon dispersion curves
ditto copy information from previous configuration to current one
dump write out a dumpfile for restarts
eam_density specifies the form and parameters for the Embedded Atom density
eam_functional specifies the functional form of the Embedded Atom Method
edip_accuracy specifies truncation of exponential terms in EDIP
edip_coordination specifies coordination number parameters in EDIP
edip_threebody specifies angular parameters in EDIP
edip_twobody specifies pairwise parameters in EDIP
edip_zmax specifies coordination number truncation parameter in EDIP
elastic specifies elastic constant values for fitting
electroneg input new parameters for electronegativity equalisation
element opens the element parameter options section
end_field stop time for time-dependent field in MD
end_force stop time for time-dependent force in MD
energy specifies the lattice energy for fitting
ensemble selects either the NVE or NVT ensemble for MD
entropy specifies the entropy for fitting
epsilon/sigma specifies the one-centre ε/σfor combination rules
equilibriation length of equilibriation period in a molecular dynamics run
erferfc specifies the parameters of an erf x erfc potential
erfpot specifies the parameters of an erf potential
ewaldrealreadius specifies target real space cut-off for Ewald sum
149
exponential specifies an exponential three-body potential
factor temperature reduction factor for simulated annealing
fangle specifies a bond angle as a fitting observable
fbond specifies a bond length as a fitting observable
field apply an electric field in a non-periodic direction
finite use finite differences to evaluate numerical derivatives
forceconstant specifies a force constant model
fractional input crystal structure using fractional coordinates
ftol specifies the function tolerance for optimisations
gamma_angular controls the spherical averaging for non-analytic corrections
gamma_direction specifies the direction of approach to gamma for non-analytic corrections
gastiter sets the maximum number of iterations for the Gasteiger method
gasttol sets the charge tolerance for the Gasteiger method
gcmcexistingmoleculesallows existing GCMC molecules to be specified
gcmcmolecule specifies molecules for GCMC
gcmcspecies specifies species for GCMC
gdcrit controls change from energy to force balance in defect calculation
general specifies a general interatomic potential
genetic general option for genetic algorithm sub-options
gexp specifies expotential weights for genetic algorithms
gradients specifies gradients that are to be used in fitting
grid specifies the grid for genetic algorithms
gtol specifies the gradient tolerance for optimisations
harmonic specifies an harmonic potential
hfdlc specifies high frequency dielectric constants for fitting
hfrefractive specifies the high frequency refractive index for fitting
ignore tells the program to ignore input until "erongi" is found
impurity replace one ion by another for a defect calculation
index_k specifies the maximum range of vectors in reciprocal space
intconserved specifies the integral of the conserved quantity - restart option
integrator specifies MD integrator algorithm to use
intermolecular all subsequent potentials are intermolecular only
interstitial insert an interstitial for a defect calculation
intramolecular all subsequent potentials are intramolecular only
ionic specifies the ionic radii for an element
iterations specifies that the number of cycles of shell optimisation in MD
keyword allows the input of keywords on any line
kpoints specify explicit k points for phonon calculation
lennard specifies a Lennard-Jones potential
library specifies a file containing a library of interatomic potentials
lin3 specifies parameters for the ESFF linear three-body potential
line maximum number of points in a line minimisation
150
lorentzian_tolerance sets the drop tolerance for the Lorentzian broadening
lowest_mode sets the lowest and highest modes to be included in the free energy
manybody specifies that a manybody potential should act between two atoms
marvin input commands to be passed through to a Marvin run
mass specifies the atomic mass for an element
maxcyc specifies the maximum number of cycles
maximum upper bound for genetic algorithm parameters
maxone limits size of second derivative arrays for dynamic memory
mcchemicalpotential controls the chemical potential during Grand Canonical Monte Carlo
mccreate controls the creation probability during Monte Carlo
mcdestroy controls the destruction probability during Monte Carlo
mclowest specifies the lowest energy found during Monte Carlo - restart option
mcmaxdisplacement controls the maximum displacement during Monte Carlo
mcmaxrotation controls the maximum rotation during Monte Carlo
mcmaxstrain controls the maximum strain to be trialled during Monte Carlo
mcmeans specifies running means for Monte Carlo
mcmove controls the translation probability during Monte Carlo
mcoutfreq controls the output frequency during Monte Carlo
mcrotate specifies rotation probability for Monte Carlo
mcsample controls the sampling frequency during Monte Carlo
mcstep specifies step number for Monte Carlo restart
mcstrain specifies strain probability for Monte Carlo
mcswap controls the atom swap probability during Monte Carlo
mctrial specifies the number of trial events during Monte Carlo
mcvolume specifies the volume used in a Grand Canonical Monte Carlo
mdarchive specifies the name for an MD archive file
mdmaxtemp specifies the maximum temperature allowed for an MD run
mdmaxvolume specifies the maximum volume allowed for an MD run
meam_density specifies the form and parameters for the Modified Embedded Atom Method
density
meam_functional specifies the functional form of the Modified Embedded Atom Method
meam_rhotype specifies the functional form of the density for MEAM
meam_screening specifies the Cmin Cmax parameters for the MEAM screening function
mei-davenport specifies the parameters of the Mei-Davenport potential
minimum lower bound for genetic algorithm parameters
mode2a allows the user to chose how region 2a is treated
morse specifies a Morse potential
move_2a_to_1 after a defect calc region 2a ions are moved to region 1
murrell-mottram species parameters for the Murrell-Mottram 3-body potential
mutation specifies the mutation probability in the genetic algorithm
name give a one-word name to a structure
nor specifies the NMR parameters for a species in a PACHA calculation
151
nobond excludes bond formation between species in molecule run
observables opens the observables option section
odirection controls in/out directions for frequency dependent properties
omega controls calculation of frequency dependent properties
omega_damping damping factor in frequency dependent properties
omega_af minimum frequency for Allen-Feldman thermal conductivity
outofplane out of plane distance four-body potential
origin gives the origin setting number or explicit origin
output creates dumpfiles for input to other programs
parallel controls parallel algorithm being used
pdf start of input block used for PDF options
piezo specifies the piezoelectric constants for fitting
plane_lj specifies a Lennard-Jones potential between an atom and a plane
pointsperatom controls number of points on solvent accessible surface
poisson_ratio specifies a Poisson ratio for fitting
polynomial specifies a polynomial potential
potential inputs electrostatic potential at a point for fitting
potgrid specifies a grid of points at which to calculate the potential
pressure specifies the applied external pressure
production controls the length of the production time for MD run
project_dos generate projected densities of states
qeqiter specifies maximum number of iterations for QEq charges
qeqradius sets the radius at which two-centre integrals switch to 1/r
qeqtol tolerance for convergance of QEq charges for H
qerfc specifies that an erfc screened Coulomb terms should be used
qincrement specifies the bond charge increment for the Gasteiger method
qiterations specifies the convergence parameters for an iterative charge solve
qmmm controls the QM/MM embedding scheme used in the energy calculation
qoverr2 specifies the parameters of a charge over distance squared potential
qtaper tapers Coulomb term at short range to a constant value
radial_force specifies a force acting between all atoms and a point in space
random specifies the number of random number calls made - used in restarting
rangeforsmooth controls the smoothing of the solvent accessible surface
rbins sets number of bins to be used in PDF output (pdf block)
rcspatial controls the region size for domain decomposition
rdirection specifies the in and out directions of waves for Raman intensities
reaction specifies a reaction energy as a fitting observable
reaxfftol specifies tolerances for ReaxFF
reaxff_chi specifies element specific electronegativity for ReaxFF
reaxff_mu specifies element specific hardness for ReaxFF
reaxff_gamma specifies element specific Coulomb screening parameter for ReaxFF
reaxff0_bond specifies non-element specific bond parameters for ReaxFF
152
reaxff0_over specifies non-element specific over-coordination parameters for ReaxFF
reaxff0_valence specifies non-element specific valence parameters for ReaxFF
reaxff0_penalty specifies non-element specific penalty parameters for ReaxFF
reaxff0_torsion specifies non-element specific torsion parameters for ReaxFF
reaxff0_vdw specifies non-element specific VDW parameters for ReaxFF
reaxff0_lonepair specifies non-element specific lonepair parameters for ReaxFF
reaxff1_radii specifies element specific radii for ReaxFF
reaxff1_valence specifies element specific valence information for ReaxFF
reaxff1_over specifies element specific over-coordination parameters for ReaxFF
reaxff1_under specifies element specific under-coordination parameters for ReaxFF
reaxff1_lonepair specifies element specific lonepair parameters for ReaxFF
reaxff1_angle specifies element specific three-body angle parameters for ReaxFF
reaxff1_morse specifies element specific two-body Morse parameters for ReaxFF
reaxff2_bo specifies pair-wise bond order parameters for ReaxFF
reaxff2_bond specifies pair-wise bond energy parameters for ReaxFF
reaxff2_over specifies pair-wise over-coordination parameters for ReaxFF
reaxff2_morse specifies pair-wise Morse parameters for ReaxFF
reaxff2_pen specifies pair-wise penalty parameters for ReaxFF
reaxff3_angle specifies angular valence energy parameters for ReaxFF
reaxff3_pen specifies angular penalty energy parameters for ReaxFF
reaxff3_conjugation specifies angular conjugation parameters for ReaxFF
reaxff3_hbond specifies hydrogen-bonding parameters for ReaxFF
reaxff4_torsion specifies torsional energy parameters for ReaxFF
region_1 explicit specification of region ions for defect calculation
reldef maps defect region 1 atoms to perfect ones for restarting
reperfc specifies the parameters of a repulsive erfc potential
rmax sets maximum radius for a PDF calculation (pdf block)
rspeed controls the balance between real and reciprocal space
rtol specifies the bond length tolerance for molecule generation
rydberg specifies the parameters for a Rydberg potential
ryckaert specifies a Ryckaert-Bellemans four-body potential
sample controls sampling frequency during an MD run
sasexclude excludes atoms from being part of the solvent accessible surface
sasparticles controls handling of shell charge in COSMO/COSMIC run
scale specifies scaling factor for vectors and Cartesian coordinates
scmaxsearch sets maximum search range for many body potentials in FEM
sdlc specifies static dielectric constants for fitting
seed specifies seed for random number generator
segmentsperatom controls the number of segments on the solvent accessible surface
shear_modulus specifies the shear modulus for fitting (cubic case only)
shellmass specifies the proportion of atoms mass for the shell in MD
shift adds an offset to the energy for fitting energy hypersurfaces
153
shrink specify shrinking factors for Brillouin zone integration
siginc used within element input block to overwrite default incoherent cross section
size specifies the sizes of regions 1 and 2a for a defect calculation
solventepsilon specifies the dielectric constant in a COSMO/COSMIC run
solventradius sets the solvent radius in a COSMO/COSMIC run
solventrmax sets the cut-off between use of segments vs points in COSMO/COSMIC
spacegroup gives either the space group number or symbol
species specifies the charges for all atomic species
spline specifies spline potential and splining data
split vary specified core-shell charge split during fitting
spring specifies core-shell spring constant
srefractive specifies the static refractive index for fitting
srglue inputs parameters for the short-range part of the glue model
start tells the program to ignore the remaining input and to begin
stepmx controls the maximum step during a minimisation
stop tells the program to stop executing immediately
stress specifies stress values as observables for fitting
strain_derivative specifies strain derivative values as observables for fitting
supercell creates a supercell
sw2 specifies a Stillinger-Weber two-body potential
sw3 specifies a Stillinger-Weber three-body potential
switch_minim changes the minimiser according to a given criteria
symbol changes element symbol from those in eledata
symmetry_number specifies the symmetry number for the rotational partition function
tau_barostat controls the relaxation time for the stochastic barostat
tau_thermostat controls the relaxation time for the stochastic thermostat
td_field applies a time-dependent electric field during MD
temperature specifies temperature for thermodynamic properties and MD
terse suppresses output during run
tether specifies atoms to be fixed during MD instead of flags
three specifies a three-body potential
time places a limit on the run time for the job
timestep controls the timestep in a molecular dynamics run
title inputs title lines for a job
torsion specifies a four-body potential
tournament defines the tournament probability for the genetic algorithm
tpxo species two point crossover in genetic algorithms
translate scans a potential energy surface by translating atoms
tscale controls the temperature scaling in a molecular dynamics run
ttol specifies minimum temperature for simulated annealing
uff_bond allows the user to change the bond orders used in UFF for each bond type
uff1 specifies the species-wise parameters for UFF combination rules
154

uff3 specifies a three-body potential of UFF form
uff4 specifies a four-body potential of UFF form
uffoop specifies an out of plane potential of UFF form
unfreeze sets optimisation flags to 1 within a spherical region
unique sets cost function difference for structures to be unique
units sets the units for quantities (currently only for frequencies in PDF)
update sets the maximum number of hessian updates
urey-bradley specifies a Urey-Bradley three-body potential
vacancy creates a vacancy for a defect calculation
variables opens the variables option section
vectors input lattice vectors to define unit cell
volume specifies the cell volume as a fitting observable for a 3-D system
weight changes the weights of observables in fitting
wmax sets maximum phonon frequency for PDF calculation (pdf block)
wmin sets minimum phonon frequency for PDF calculation (pdf block)
write controls the frequency of writing for the MD dumpfile
xangleangle specifies a angle-angle cross potential
xtol controls the parameter tolerance in optimisation
youngs_modulus specifies a Youngs modulus for fitting
zbl specifies a Ziegler-Biersack-Littmark potential
3.3 Guide to output
3.3.1 Main output
Hopefully, if you understand what has gone into the input for the calculation the
output will be largely self-explanatory! Hence this section will give only a few
brief pointers as to the nature of the output. Many pieces of the output are specific
to particular options. The following is a guide to the order which things will appear
in the output file, which in turn mirrors the order in which runtypes are executed:
Banner gives the version number of the program
Keywords the program echoes the keywords it has found in the input with the
exception of some debugging keywords which only affect the more ver-
bose pieces of output
Title if input by the user
155

Structural output for each configuration (structure) in turn the program will echo the
structural information that was input and any derived quantities in the
following order:
•Formula for compound (excluding shells)
•Number of species in the asymmetric unit
•Total number of species in the primitive cell
•Dimensionality of system
•Charge if not zero
•Solvation model parameters (if applicable)
•For 3-D systems only
–Symmetry information
–Cell vectors for primitive cell
–Cell parameters for primitive and full cell
–Cell volume
•For non 3-D systems only
–Dipole moment in non-periodic directions
•Electric field (if applicable)
•Shrinking factors for reciprocal space
•Temperature
•Pressure or pressure tensor
•Coordinates (fractional for 3-D / Cartesian for 0-D), including the
site occupancy and charge. Where applicable, coordinates which
are free to vary are indicated by an asterisk following them
•Molecule listing (if requested)
•Geometry analysis output (if requested)
Species output contains all species/element specific data
Electrostatic accuracy
parameter
156

Time limit for run
Interatomic potentials
Fitting output fitting involves all structures and precedes all other calculation types so
that they can use the optimised parameters
Translate output as translate performs the runtype for each point along the specified path
it precedes the run type output
Runtype output the output appears for each configuration in the following order subject
to the runtype having been requested by the user:
•Electronegativity equalisation
•Optimisation / energy / gradient calculation - for non-primitive
unit cells values are given for the primitive cell unless specified
otherwise
•Property calculation
•Phonon calculations
•Electrostatic potential and derivatives
•Molecular dynamics
•Defect calculation
Timing information
File output information
3.3.2 Files for graphical display
Both phonon dispersion and phonon density of states calculations produce infor-
mation which is suitable for graphic display. Although there is a crude picture gen-
erated in the GULP output it is rather limited by the text nature of the output file.
The command output phonon can be used to dump the data generated by GULP
to two files with the extensions ‘.dens’ and ‘.disp’ which can then be exported into
a graph plotting program (after suitable modification) to produce proper plots.
3.3.3 Input files for other programs
The output option allows the user to generate input files for a number of other
programs, both for displaying crystal structures and for other types of run. The file
types available are summarised below:
157

Marvin (.mvn) for a surface calculation
THBREL (.thb) for a calculation using the program THBREL
xtl (.xtl) for input to the Insight graphical interface from Accerlys Inc. Only
applicable to solids.
xr (.xr) for input to the G-Vis interface from Oxford Materials (modified CSSR
file format)
arc (.arc/.car) for input to the Materials Studio graphical interface from Accelrys Inc.
Available for bulk, cluster and defect calculations (each type produces
a separate file with the type appended to the name). This file can be
generated to contain multiple structures for visualisation as a movie.
cssr (.cssr) for input into the Cerius2interface from Accelrys Inc and other pro-
grams. Available for bulk calculations
xyz (.xyz) for input into a range of other molecule codes, such as Molden.
fdf (.fdf) contains the structural information in a form suitable for SIESTA
cif (.cif) writes out a simple cif file for input into many other crystallographic
codes
drv (.drv) writes a file with the derivatives - useful for linking to QM/MM codes
frc (.frc) writes a file with the force constants - useful for linking to QM/MM
codes
str (.str) writes out a file in the CRYSTAL98 format for reading by DLV
history (.his) writes out a file in DLPOLY history file format
Note that when generating input files for other programs there is no guarantee of
compatibility due to differences in features or because of changes in format.
3.3.4 Temporary files
Some use is made by GULP of binary scratch files for certain run types. Most are
transient files which are removed before the end of a run. The normal reason for
their existence is to economise on memory by allowing large arrays to be overlaid.
The following is a list of the Fortran channels that may be used and what they are
used for (a ‘D’ in brackets indicates that the file should be automatically deleted
before successful completion of a job):
31/32 restart files for a molecular dynamics run
44 restart file for a defect calculation to avoid bulk property calcn
51 storage of frequencies for passing between routines (D)
59 projection of phonons needed for project_dos option (D)
158
3.4 Guide to installation
3.4.1 Environment variables
In previous versions of GULP it was necessary to edit the file local.F (now lo-
cal.F90) to set the location of the library files and help text. This is no longer
necessary since the location is now specified by environment variables:
GULP_LIB - specifies the directory path for the libraries
GULP_DOC - specifies the directory path for the documentation for GULP
If you are working in tcsh/csh then you can set these by putting the following lines
in your .cshrc:
setenv GULP_LIB /Users/julian/progs/gulp3.4/Libraries
setenv GULP_DOC /Users/julian/progs/gulp3.4/Docs
3.4.2 Setting compilation options
All of the settings required for compilation of GULP are contained in the file "get-
machine". Once this is correctly set then all that is needed to compile is to type
"make".
You may need to edit the file getmachine to set the options according to your
particular type of f90 compiler. After this all you need to do on most Unix machines
is type "make".
The file getmachine has separate settings for different machine types based on
the result of the command "uname -s". The following is a guide to the environment
variables that can be set in this file:
RUNF90 = name of Fortran compiler
RUNCC = name of C compiler (only needed if compiling with certain options)
FFLAGS = Fortran compile flags unrelated to optimisation - must include "-I.."
CFLAGS = C compiler flags
LDFLAGS = flags for use at link time
LIBS = Any libraries that need to be linked against (not usually needed)
OPT = Default optimisation flags for Fortran (e.g. -O2)
OPT1 = Optimisation flags for Fortran routines that need a lower level of optimisa-
tion (e.g. -O1)
OPT2 = Optimisation flags for Fortran routines that need a higher level of optimi-
159
sation
BAGGER = Flags for debugging code (e.g. -g -Wall -fbounds_check)
LAPACK = This is the source of lapack routines. By default it is "lapack.o" but
could be an optimised library
BLAS = This is the source of blas routines. By default it is "blas.o" but could be an
optimised library
DEFS = Sets flags that enable optional pieces of code during compilation
PLUMEDDEFS = Sets flags that enable optional code for the package PLUMED
(if applicable)
3.4.3 Compiling for parallel execution
If you wish to run the program in parallel using MPI then you will need to alter
the file "getmachine" accordingly. The usual changes would be to add to DEFS the
"-DMPI" option and in some cases change the compiler name (for example to the
wrappers mpif77/mpif90) or include the MPI libraries in the link stage (i.e. add
"-lmpi" to LIBS).
3.4.4 Trapping of Control C
An optional feature during GULP compilation is to enable trapping of Control C
so that the program exits either from optimisation or fitting cycles. This allows the
user to cleanly exit an iterative process that is going nowhere. A second execution
of Control C will then stop the program as normal. To enable this feature you need
to add the -DCTRLC flag to DEFS in getmachine, and also uncomment the 2 refer-
ences to csignal in the Makefile.
3.4.5 Building with OpenKIM
There is now support for energy and forces to be provided to GULP by an external
routine using the OpenKIM standard. To build GULP for use with OpenKIM you
need to modify getmachine in several places:
a) Add -DKIM to DEFS
b) Ensure that the location of the OpenKIM application is defined using KIM_DIR
as an environment variable.
c) Define the variable "KFLAGS=-I’$KIM_DIR/KIM_API’"
d) Define the variable "KLIBS=-L’$KIM_DIR/KIM_API’ -lkim -lstdc++"
160
3.4.6 Building with PLUMED
The PLUMED package for metadynamics can be used within GULP. To do this a
C compiler is required that will cross compile with Fortran (e.g. gfortran) and the -
DPLUMED flag must be part of DEFS. In addition, you should set "PLUMEDDEFS=-
DDL_POLY" for the benefit of PLUMED. The PLUMED code should be placed in
a sub-directory "Plumed" of Src in the GULP distribution.
161
Bibliography
[1] E. Madelung. Das elektrische feld in systemen von regelmaessig angeord-
neten punktladungen. Phys. Z., 19:524–532, 1918.
[2] M. Born and J.E. Mayer. Zur gittertheorie der ionenkristalle. Z. Physik,
75:1–18, 1932.
[3] A.F. Kapustinskii. Lattice energy of ionic crystals. Quart. Rev. Chem. Soc.,
10:283–294, 1956.
[4] M.J. Norgett. A user’s guide to hades. Technical Report AERE-R7015,
AERE Harwell Laboratory, 1972.
[5] J.H. Harding. A guide to midas. Technical Report AERE-R13127, AERE
Harwell Laboratory, 1988.
[6] J.H. Harding. A guide to the harwell pluto program. Technical Report
AERE-R10546, AERE Harwell Laboratory, 1982.
[7] M. Leslie. Cascade. Technical Report DLSCITM36T, Daresbury Laboratory,
1984.
[8] S.C. Parker and G.D. Price. Computer modelling of phase transitions in
minerals. Adv. Solid State Chem., 1:295–327, 1989.
[9] D.J. Willock, S.L. Price, M. Leslie, and C.R.A. Catlow. The relaxation of
molecular crystal structures using a distributed multipole electrostatic model.
J. Comp. Chem., 16:628–647, 1995.
[10] W.R. Busing. Wmin, a computer program to model molecules and crystals
in terms of potential energy functions. Technical Report ORNL-5747, Oak
Ridge National Laboratory, 1981.
[11] D.E. Williams. Molecular packing analysis. Acta Cryst. A, 28:629–635,
1972.
[12] G. Eckold, M. Stein-Arsic, and H.J. Weber. Unisoft - a program package for
lattice dynamical calculations. J. Appl. Cryst., 20:134–139, 1987.
162
[13] W.F. van Gunsteren and H.J.C. Berendsen. Groningen molecular simulation
(gromos) library manual. Technical report, Biomos, Groningen, 1987.
[14] D.A. Pearlman, D.A. Case, J.W. Caldwell, W.S. Ross, T.E. Cheatham III,
S. DeBolt, D. Ferguson, G. Seibel, and P. Kollman. Amber, a package of
computer programs for applying molecular mechanics, normal mode analy-
sis, molecular dynamics and free energy calculations to simulate the struc-
tural and energetic properties of molecules. Comput. Phys. Commun., 91:1–
41, 1995.
[15] B.R. Brooks, R.E. Bruccoleri, B.D. Olfson, D.J. States, S. Swaminathan, and
M. Karplus. Charmm: A program for macromolecular energy, minimization,
and dynamics calculations. J. Comp. Chem., 4:187–217, 1983.
[16] W. Smith and T.R. Forester. Dl_poly_2.0: A general purpose parallel molec-
ular dynamics simulation package. J. Mol. Graphics, 14:136–141, 1996.
[17] W. Smith, C.W. Yong, and P.M. Rodger. Dl_poly: application to molecular
simulation. Mol. Simul., 28:385–471, 2002.
[18] G. Watson, E.T. Kelsey, N.H. de Leeuw, D.J. Harris, and S.C. Parker. Atom-
istic simulation of dislocations, surfaces and interfaces in mgo. J. Chem.
Soc., Faraday Trans., 92:433–438, 1996.
[19] M.B. Taylor, G.D. Barrera, N.L. Allan, T.H.K. Barron, and W.C. Mackrodt.
Shell: A code for lattice dynamics and structure optimisation of ionic crys-
tals. Comp. Phys. Commun., 109:135–143, 1998.
[20] H.M. Evjen. On the stability of certain heteropolar crystals. Phys. Rev.,
39:675–687, 1932.
[21] P.P. Ewald. Die berechnung optischer und elektrostatisher gitterpotentiale.
Ann. Phys., 64:253–287, 1921.
[22] M.P. Tosi. Cohesion of ionic solids in the born model. Solid State Phys.,
16:1–120, 1964.
[23] R.A. Jackson and C.R.A. Catlow. Computer simulation studies of zeolite
structure. Mol. Simul., 1:207–224, 1988.
[24] U. Essmann, L. Perera, M.L. Berkowitz, T. Darden, H. Lee, and L.G. Peder-
sen. A smooth particle mesh ewald method. J. Chem. Phys., 103:8577–8595,
1995.
[25] L. Greengard and V. Rokhlin. A fast algorithm for p@article simulations. J.
Comput. Phys., 73:325–348, 1987.
163
[26] H.G. Petersen, D. Soelvason, J.W. Perram, and Smith E.R. A very fast mul-
tipole method. J. Chem. Phys., 101:8870–8876, 1994.
[27] S.W. de Leeuw, J.W. Perram, and E.R. Smith. Simulation of electrostatic sys-
tems in periodic boundary conditions. i. lattice sums and dielectric constants.
Proc. R. Soc. London, Ser. A, 373:27–56, 1980.
[28] M. Leslie and M.J. Gillan. The energy and elastic dipole tensor of defects
in ionic-crystals calculated by the supercell method. J. Phys. C, Solid State
Phys., 18:973–982, 1985.
[29] D.E. Parry. The electrostatic potential in the surface region of an ionic crys-
tal. Surf. Sci., 49:433–440, 1975.
[30] D.E. Parry. Errata: The electrostatic potential in the surface region of an
ionic crystal. Surf. Sci., 54:195–195, 1976.
[31] E. Wasserman, J.R. Rustad, and A.R. Felmy. Molecular modeling of the
surface charging of hematite i. the calculation of proton affinities and acidites
on a surface. Surf. Sci., 424:19–27, 1999.
[32] J.H. Harding. A guide to midas. Technical Report AERE R-13127, AERE
Harwell Laboratory, 1988.
[33] A. Arnold and C. Holm. A novel method for calculating electrostatic in-
teractions in 2d periodic slab geometries. Chem. Phys. Lett., 354:324–330,
2002.
[34] V.R. Saunders, C. Freyia-Fava, R. Dovesi, and C. Roetti. On the electrostatic
potential in linear periodic polymers. Comp. Phys. Commun., 84:156–172,
1994.
[35] D. Wolf, P. Keblinski, S.R. Philpot, and J. Eggebrecht. Exact method for
the simulation of coulombic systems by spherically truncated, pairwise r−1
summation. J. Chem. Phys., 110:8254–8282, 1999.
[36] R.S. Mulliken. Electronic population analysis on lcao-mo molecular wave
functions. i. J. Chem. Phys., 23:1833–1840, 1955.
[37] S. Baroni, S. de Gironcoli, A. Dal Corso, and P. Giannozzi. Phonons and
related crystal properties from density-functional perturbation theory. Rev.
Mod. Phys., 73:515–562, 2001.
[38] R.T. Sanderson. An interpretation of bond lengths and a classification of
bonds. Science, 114:670–672, 1951.
164
[39] K.A. van Genechten, W.J. Mortier, and P. Geerlings. Intrinsic framework
electronegativity: A novel concept in solid state chemistry. J. Chem. Phys.,
86:5063–5071, 1987.
[40] A.K. Rappe and W.A. Goddard III. Charge equilibration for molecular dy-
namics simulations. J. Phys. Chem., 95:3358–3363, 1991.
[41] S.L. Njo, J.F. Fan, and B. van de Graaf. Extending and simplifying the
electronegativity equalization method. J. Mol. Catal. A, 134:79–88, 1998.
[42] C.E. Wilmer, K. Chul Kim, and R.Q. Snurr. An extended charge equilibration
method. J. Phys. Chem. Lett., 3:2506–2511, 2012.
[43] M. Henry. Nonempirical quantification of molecular interactions in
supramolecular assemblies. Chem. Phys. Chem., 3:561–569, 2002.
[44] H. Senouci, B. Millet, C. Volkringer, C. Huguenard, F. Taulelle, and
M. Henry. Full spectroscopic characterization of an hydrolytically stable
and colored ti(iv)-precursor in solution. Comptes Rendus Chemie, 13:69–96,
2010.
[45] D.E. Williams. Accelerated convergence treatment of r−nlattice sums. Cryst.
Rev., 2:3–25, 1989.
[46] K.T. Tang and J.P. Toennies. An improved simple-model for the van der
waals potential based on universal damping functions for the dispersion co-
efficients. J. Chem. Phys., 80:3726–3741, 1984.
[47] J. Applequist. A multipole interaction theory of electric polarization of
atomic and molecular assemblies. J. Chem. Phys., 83:809–826, 1985.
[48] T. Beyer, G.M. Day, and S.L. Price. The prediction, morphology, and me-
chanical properties of the polymorphs of paracetamol. J. Am. Chem. Soc.,
123:5086–5094, 2001.
[49] P.A. Madden and M. Wilson. ’covalent’ effects in ’ionic’ systems. Chem.
Soc. Rev., pages 339–350, 1996.
[50] J.H. Harding and N.C. Pyper. The meaning of the oxygen 2nd-electron affin-
ity and oxide potential models. Phil. Mag. Lett., 71:113–121, 1995.
[51] B.G. Dick and A.W. Overhauser. Theory of the dielectric constants of alkali
halide crystals. Phys. Rev., 112:90–103, 1958.
[52] R. Car and M. Parrinello. Unified approach for molecular-dynamics and
density-functional theory. Phys. Rev. Lett., 55:2471–2474, 1985.
165
[53] P.J.D. Lindan and M.J. Gillan. Shell-model molecular-dynamics simulation
of superionic conduction in ca f2.J. Phys. Condens. Matter, 5:1019–1030,
1993.
[54] U. Schroeder. A new model for lattice dynamics (breathing shell model).
Solid State Commun., 4:347–349, 1966.
[55] P.M. Axilrod and E. Teller. Interaction of the van der waals type between
three atoms. J. Chem. Phys., 11:299–300, 1943.
[56] A. Banerjea and J.R. Smith. Origins of the universal binding-energy relation.
Phys. Rev. B, 37:6632–6645, 1988.
[57] A.P. Sutton and J. Chen. Long-range Finnis-Sinclair potentials. Phil. Mag.
Lett., 61:139–146, 1990.
[58] M.W. Finnis and J.E. Sinclair. A simple empirical n-body potential for tran-
sition metals. Phil. Mag. A, 50:45–55, 1984.
[59] J. Cai and Y.Y. Ye. Simple analytical embedded-atom-potential model in-
cluding a long-range force for fcc metals and their alloys. Phys. Rev. B,
54:8398–8410, 1996.
[60] M.I. Baskes. Modified embedded-atom potentials for cubic materials and
impurities. Phys. Rev. B, 46:2727–2742, 1992.
[61] G.C. Abell. Empirical chemical pseudopotential theory of molecular and
metallic bonding. Phys. Rev. B, 31:6184–6196, 1985.
[62] J. Tersoff. New empirical-model for the structural-properties of silicon. Phys.
Rev. Lett., 56:632–635, 1986.
[63] J. Tersoff. New empirical-approach for the structure and energy of covalent
systems. Phys. Rev. B, 37:6991–7000, 1988.
[64] D.G. Pettifor, I.I. Oleinik, D. Nguyen-Manh, and V. Vitek. Bond-order po-
tentials: bridging the electronic to atomistic modelling hierarchies. Comp.
Mater. Sci., 23:33–37, 2002.
[65] D.W. Brenner. Empirical potential for hydrocarbons for use in simulating the
chemical vapor deposition of diamond films. Phys. Rev. B, 42:9458–9471,
1990.
[66] D.W. Brenner, O.A. Shenderova, J.A. Harrison, S.J. Stuart, B. Ni, and S.B.
Sinnott. A second-generation reactive empirical bond order (rebo) potential
energy expression for hydrocarbons. J. Phys.: Condens. Matter, 14:783–802,
2002.
166
[67] J. Che, T. Cagin, and W.A. Goddard III. Generalized extended empirical
bond-order dependent force fields including nonbond interactions. Theor.
Chem. Acc., 102:346–354, 1999.
[68] A.C.T. van Duin, S. Dasgupta, F. Lorant, and W.A. Goddard III. Reaxff: A
reactive force field for hydrocarbons. J. Phys. Chem. A,, 105:9396–9409,
2001.
[69] M.Z. Bazant, E. Kaxiras, and J.F. Justo. Phys. Rev. B, 56:8542–8552, 1997.
[70] J.F. Justo, M.Z. Bazant, E. Kaxiras, V.V. Bulatov, and S. Yip. Interatomic
potential for silicon defects and disordered phases. Phys. Rev. B, 58:2539–
2550, 1998.
[71] N.A. Marks. Generalizing the environment-dependent interaction potential
for carbon. Phys. Rev. B, 63:035401, 2000.
[72] A. Einstein. Die plancksche theorie der strahlung und die theorie der spezi-
fischen waerme. Ann. Phys., 22:180–190, 1907.
[73] D. Frenkel and A.J.C. Ladd. New monte carlo method to compute the free en-
ergy of arbitrary solids. application to the fcc and hcp phases of hard spheres.
J. Chem. Phys., 81:3188–3193, 1981.
[74] G. Paglia, A.L. Rohl, C.E. Buckley, and J.D. Gale. A computational inves-
tigation of the structure of κ-alumina using interatomic potentials. J. Mater.
Chem., 11:3310–3316, 2001.
[75] G.W. Watson and D.J. Willock. The enumeration of structures for γ-alumina
based on a defective spinel structure. Chem. Commun., pages 1076–1077,
2001.
[76] N.L. Allan, G.D. Barrera, M.Y. Lavrentiev, I.T. Todorov, and J.A. Purton.
Ab initio calculation of phase diagrams of ceramics and minerals. J. Mater.
Chem., 11:63–68, 2001.
[77] P.J.M. van Laarhoven and E.H.L. Aarts. Simulated annealing: Theory and
applications. Reidel, 1987.
[78] J.H. Holland. Adaption in natural and artificial systems. The University of
Michigan Press, 1975.
[79] T.S. Bush, C.R.A. Catlow, and P.D. Battle. Evolutionary programming tech-
niques for predicting inorganic crystal-structures. J. Mater. Chem., 5:1269–
1272, 1995.
167
[80] W.H. Press, S.A. Teukolsky, W.T. Vetterling, and B.P. Flannery. Numerical
recipes in Fortran: The art of scientific computing. Cambridge University
Press, 1986.
[81] R. Fletcher and M.J.D. Powell. A rapidly convergent descent method for
minimization. Comput. J., 6:163–168, 1964.
[82] D.F. Shanno. Conditioning of quasi-newton methods for function minimiza-
tion. Math. Comput., 24:647–656, 1970.
[83] G. Mills, H. Jonsson, and G.K. Schenter. Reversible work transition-state
theory - application to dissociative adsorption of hydrogen. Surf. Sci.,
324:305–337, 1995.
[84] A. Banerjee, N. Adams, J. Simons, and R. Shepard. Search for stationary
points on surfaces. J. Phys. Chem., 89:52–57, 1985.
[85] A.F. Voter, F. Montalenti, and T.C. Germann. Extending the time scale in
atomistic simulation of materials. Ann. Rev. Materials Research, 32:321–
346, 2002.
[86] S.M. Woodley, P.D. Battle, J.D. Gale, and C.R.A. Catlow. The prediction of
inorganic crystal structures using a genetic algorithm and energy minimisa-
tion. Phys. Chem. Chem. Phys., 1:2535–2542, 1999.
[87] D. Frenkel and B. Smit. Understanding molecular simulation: from algo-
rithms to applications. Academic Press, 2001.
[88] J.F. Nye. Physical properties of crystals. Oxford University Press, 1957.
[89] J.W. Akitt. NMR and chemistry. Chapman and Hall, 1983.
[90] R.F.W. Bader, T.T. Nguyendang, and Y. Tal. A topological theory of
molecular-structure. Rep. Prog. Phys., 44:893–948, 1981.
[91] C. Eckart. Some studies concerning rotating axes and polyatomic molecules.
Phys. Rev., 47:552–558, 1935.
[92] W. Cochran and R.A. Cowley. Dielectric constants and lattice vibrations. J.
Phys. Chem. Solids, 23:447–450, 1962.
[93] J.M. Seddon and J.D. Gale. Thermodynamics and statistical mechanics.
Royal Society of Chemistry, 2002.
[94] A. Baldereschi. Mean-value point in the brillouin zone. Phys. Rev. B,
7:5212–5215, 1973.
168
[95] D.J. Chadi and M.L. Cohen. Special points in the brillouin zone. Phys. Rev.
B, 8:5747–5753, 1973.
[96] H.J. Monkhorst and J.D. Pack. Special points for brillouin-zone integrations.
Phys. Rev. B, 13:5188–5192, 1976.
[97] R. Ramirez and M.C. Boehm. The use of symmetry in reciprocal space inte-
grations - asymmetric units and weighting factors for numerical-integration
procedures in any crystal symmetry. Int. J. Quantum Chem., 34:571–594,
1988.
[98] P.B. Allen and J.L. Feldman. Phys. Rev. B, 48:12581–12588, 1993.
[99] F. Zernike and J.A. Prins. Die beugung von roentgenstrahlen in fluessigkeiten
als effekt der molekuelanordnung. Z. Physik A Hadrons and Nuclei, 41:184–
194, 1927.
[100] B.E. Warren. Calculation of powder-pattern intensity distributions. J. Appl.
Crystallogr., 11:695–698, 1978.
[101] R. Kaplow, B. L. Averbach, and S. L. Strong. Pair correlations in solid lead
near the melting temperature. J. Phys. Chem. Solids, 25(11):1195–1204,
1964.
[102] T. Egami, J.D. Jorgensen, B.H. Toby, and M.A. Subramanian. Observa-
tion of a local structural-change at tc for tl2ba2cacu2o8 by pulsed neutron-
diffraction. Phys. Rev. Lett., 64:2414–2417, 1990.
[103] W. G. Williams, R. M. Ibberson, P. Day, and J. E. Enderby. GEM - General
Materials Diffractometer at ISIS. Physica B, 241:234–236, 1997.
[104] J.S. Chung and M.F. Thorpe. Local atomic structure of semiconductor alloys
using pair distribution functions. ii. Phys. Rev. B, 59:4807–4812, 1999.
[105] E.R. Cope and M.T. Dove. Pair distribution functions calculated from inter-
atomic potential models using the general utility lattice program. J. Appl.
Crystallogr., 40:589–594, 2007.
[106] J.D. Gale and A.L. Rohl. The General Utility Lattice Program (GULP). Mol.
Simul., 29:291–341, 2003.
[107] D.H. Gay and A.L. Rohl. Marvin: A new computer code for studying sur-
faces and interfaces and its application to calculating the crystal morpholo-
gies of corundum and zircon. J. Chem. Soc., Faraday Trans., 91:926–936,
1995.
169
[108] P.W. Tasker. The stability of ionic crystal surfaces. J. Phys. C, 12:4977–4984,
1979.
[109] J.D.H. Donnay and D. Harker. A new law of crystal morphology extending
the law of bravais. Am. Miner., 22:446–467, 1937.
[110] G. Wulff. Zur frage der geschwindigkeitb des wachsthums und der auflosung
der krystallflachen. Z. Kryst., 34:449–530, 1901.
[111] J.W. Gibbs. Collected works. New York, Longman, 1928.
[112] P. Hartman and P. Bennema. The attachment energy as a habit controlling
factor 1. theoretical considerations. J. Crystal Growth, 49:145–156, 1980.
[113] J.D. Gale and A.L. Rohl. An efficient technique for the prediction of solvent-
dependent morphology: the cosmic method. Mol. Simul., 33:1237–1246,
2007.
[114] A. Klamt and G. Schueuermann. J. Chem. Soc., Perkin Trans. 2, pages 799–
805, 1993.
[115] M.P. Allen and D.J. Tildesley. Computer simulation of liquids. Oxford Uni-
versity Press, 1987.
[116] R. LeSar, R. Najafabati, and D.J. Srolovitz. Thermodynamics of solid and
liquid embedded-atom-method metals - a variational study. J. Chem. Phys.,
94:5090–5097, 1991.
[117] A.P. Sutton. Direct free-energy minimization methods - application to grain-
boundaries. Phil. Trans. R. Soc. London A, 341:233–245, 1992.
[118] E.W. Montroll. Frequency spectrum of crystalline solids. J. Chem. Phys.,
10:218–229, 1942.
[119] L.N. Kantorovich. Thermoelastic properties of perfect crystals with non-
primitive lattices. 1. general-theory. Phys. Rev. B, 51:3520–3534, 1995.
[120] M.B. Taylor, G.D. Barrera, N.L. Allan, and T.H.K. Barron. Free-energy
derivatives and structure optimization with quasiharmonic lattice dynamics.
Phys. Rev. B, 56:14380–14390, 1997.
[121] J.D. Gale. Analytical free energy minimization of silica polymorphs. J. Phys.
Chem. B, 102:5423–5431, 1998.
[122] N.L. Allan, T.H.K. Barron, and J.A.O. Bruno. The zero static internal stress
approximation in lattice dynamics, and the calculation of isotope effects on
molar volumes. J. Chem. Phys., 105:8300–8303, 1996.
170
[123] N.F. Mott and M.J. Littleton. Conduction in polar crystals. i. electrolytic
conduction in solid salts. Trans. Faraday Soc., 34:485–499, 1938.
[124] C.R.A. Catlow, R. James, W.C. Mackrodt, and R.F. Stewart. Defect energet-
ics in α−al2o3and rutile tio2.Phys. Rev. B, 25:1006–1026, 1982.
[125] N.L. Allinger, X.F. Zhou, and J. Bergsma. Molecular mechanics parameters.
J. Mol. Struct. (Theochem), 118:69–83, 1994.
[126] A.K. Rappe, C.J. Casewit, K.S. Colwell, W.A. Goddard III, and W.M. Skiff.
Uff, a full periodic table force field for molecular mechanics and molecular
dynamics simulations. J. Am. Chem. Soc., 114:10024–10035, 1992.
[127] S. Barlow, A.L. Rohl, S.G. Shi, C.M. Freeman, and D. O’Hare. Molecular
mechanics study of oligomeric models for poly(ferro cenylsilanes) using the
extensible systematic forcefield (esff). J. Am. Chem. Soc., 118:7578–7592,
1996.
[128] J.D. Gale, C.R.A. Catlow, and W.C. Mackrodt. Periodic ab initio determina-
tion of interatomic potentials for alumina. Modelling Simul. Mater. Sci. Eng.,
1:73–81, 1992.
[129] R.G. Gordon and Y.S. Kim. Theory for the forces between closed-shell atoms
and molecules. J. Chem. Phys., 56:3122–3133, 1972.
[130] J.D. Gale. Empirical potential derivation for ionic materials. Phil. Mag. B,
73:3–19, 1996.
[131] T.J. Grey, J.D. Gale, and D. Nicholson. Parameterization of a potential func-
tion for the ca2+−ne and ca2+−n2interactions using high-level ab initio
data. Mol. Phys., 98:1565–1573, 2000.
[132] C.R.A. Catlow and M.J. Norgett. Lattice structure and stability of ionic ma-
terials. Technical Report AERE-M2936, AERE Harwell Laboratory, 1976.
[133] C.R.A. Catlow and W.C. Mackrodt. Theory of simulation methods for lattice
and defect energy calculations in crystals. Lecture Notes in Physics, 166:3–
20, 1982.
[134] J.D. Gale. Gulp: A computer program for the symmetry-adapted simulation
of solids. J. Chem. Soc., Faraday Trans., 1:629–637, 1997.
[135] S. Brode and R. Ahlrichs. An optimized md program for the vector computer
cyber-205. Comp. Phys. Commun., 42:51–57, 1986.
171
[136] C.R.A. Catlow, I.D. Faux, and M.J. Norgett. Shell and breathing shell model
calculations for defect formation energies and volumes in magnesium oxide.
J. Phys. C: Solid State Phys., 9:419–429, 1976.
[137] C.-S. Zha, H.-K. Mao, and R.J. Hemley. Elasticity of mgo and a primary
pressure scale to 55 gpa. Proc. Natl. Acad. Sci., 97:13494–13499, 2000.
[138] I. Jackson and H. Niesler. In High pressure research in geophysics. Center
for Academic Publishing, Tokyo, 1982.
[139] M.J. Sanders, M. Leslie, and C.R.A. Catlow. Interatomic potentials for sio2.
J. Chem. Soc. Chem. Commun., pages 1271–1273, 1984.
[140] X. Gonze, D.C. Allan, and M.P. Teter. Dielectric tensor, effective charges,
and phonons in α-quartz by variational density-functional perturbation the-
ory. Phys. Rev. Lett., 68:3603–3606, 1992.
[141] Ph. Ghosez, J.-P. Michenaud, and X. Gonze. Dynamical atomic charges: The
case of abo3compounds. Phys. Rev. B, 58:6224–6240, 1998.
[142] A.L. Rohl, K. Wright, and J.D. Gale. Evidence from surface phonons for the
(2x1) reconstruction of the (10-14) surface of calcite from computer simula-
tion. Am. Miner., page in press, 2003.
[143] S.L.S. Stipp. Toward a conceptual model of the calcite surface: Hydra-
tion, hydrolysis, and surface potential. Geochimica Cosmochimica Acta,
63:3121–3131, 1999.
[144] J.S. Chung and M.F. Thorpe. Local atomic structure of semiconductor alloys
using pair distribution functions. Phys. Rev. B, 55:1545–1553, 1997.
[145] W. Reichardt and L. Pintschovius. Influence of phonons on the pair dis-
tribution function deduced from neutron powder diffraction. Phys. Rev. B,
63:174302, 2001.
[146] S.W. Lovesey. Theory of Neutron Scattering from Condensed Matter. Volume
1: Nuclear Scattering. Oxford University Press, 1984.
[147] B.T.M. Willis and A.W. Pryor. Thermal Vibrations in Crystallography. Cam-
bridge University Press, Cambridge, 1975.
[148] M.T. Dove. Introduction to Lattice Dynamics. Cambridge University Press,
Cambridge, 1993.
[149] D.A. Keen. A comparison of various commonly used correlation functions
for describing total scattering. J. Appl. Crystallogr., 34:172–177, 2001.
172
[150] M.G. Tucker, M.T. Dove, and D.A. Keen. Neutron total scattering method:
simultaneous determination of long-range and short-range order in disor-
dered materials. Eur. J. Mineral., 14:331–348, 2002.
[151] T. Proffen and S.J.L. Billinge. PDFFIT, a program for full profile structural
refinement of the atomic pair distribution function. J. Appl. Crystallogr.,
32:572–575, 1999.
[152] S.J.L. Billinge and T. Egami. Short-range atomic-structure of
Nd(2−x)CexCuO(4−y)determined by real-space refinement of neutron-
powder-diffraction data. Phys. Rev. B, 47:14386–14406, 1993.
[153] S.J.L. Billinge, T. Egami, T. Proffen, and D. Louca. Structural analysis of
complex materials using the atomic pair distribution function - a practical
guide. Z. Krist., 218:132–143, 2003.
[154] W.S. Howells, A.K. Soper, and A.C. Hannon. ATLAS - Analysis of Time-of-
Flight Diffraction Data from Liquid and Amorphous Samples, 1989. 1989.
173
