Isomir Sea Manual V0 1
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 20

Contents
1 Introduction 3
1.1 isomiR-SEA philosophy . . . . . . 3
1.2 Main features . . . . . . . . . . . . 3
2 Program usage 5
2.1 Installation procedures . . . . . . 5
2.1.1 Pre-compiled release . . 5
2.2 Dependencies . . . . . . . . . . . 5
2.3 The miRNAs databases . . . . . . 5
2.4 Program options . . . . . . . . . . 5
2.4.1 Alignment parameters . . 6
2.4.2 Paths and files . . . . . . . 6
2.4.3 DNA/RNA conversion
and filtering . . . . . . . . 6
3 Analysis work-flow 7
3.1 The interaction sites point of view 7
3.2 The isomiRs point of view . . . . 9
3.3 isomiR-SEA Algorithm . . . . . . 10
3.3.1 Parameters setting . . . . 10
3.3.2 miRNAs DB and tags
counts file processing . . 12
3.3.3 isomiR-SEA alignment
procedure . . . . . . . . . 12
3.3.4 isomiR-SEA output files . 15
4 Files formats 16
4.1 Input files . . . . . . . . . . . . . . 16
4.2 Output files . . . . . . . . . . . . . 17
Bibliography 20
2
1 Introduction
1.1 isomiR-SEA philosophy
The main idea behind isomiR-SEA is to provide users with an integrated and user-friendly tool capa-
ble to extract miRNAs and isomiR expression levels from high-throughput sequencing data ensuring
high degrees of accuracy and detail.
The innovation introduced by isomiR-SEA consists in the pragmatic approach implemented to detect
and quantify miRNAs into the analysed samples.
Indeed the tool identifies as reads accounting for miRNAs all the sequences that retain specific miR-
NAs features and that, as a consequence, can interact in some way with mRNA target molecules.
Clearly these interactions strictly depend on the amount and kind of differences detected in the read
with respect to the miRNA from which it derives. Furthermore, specific miRNA positions are nowa-
days considered fundamental to guarantee miRNA:mRNA target interaction, as proven in several re-
searches conducted in the last decade and exploited in most of the miRNAs target prediction algo-
rithms.
In the light of these considerations and given the lack of software tools capable to take into account
these miRNAs features during the reads alignment phase, we have developed isomiR-SEA.
isomiR-SEA is the first tool implemented in order to perform reads alignment on miRNAs databases
by considering the miRNA:mRNA interaction pairing aspects. This novelty allows isomiR-SEA to
reach high accuracy in miRNAs expression levels extraction and at the same time to provide users
with a very detailed description of the detected expression profiles.
In other words, why biologists have to look pictures from 0.5 Mega pixel cameras when the available
knowledge allows them to look at the same images from 10 Mega pixel cameras? Additionally it is
usually simpler reduce the degree of detail of a picture than increase it thus isomiR-SEA has been
implemented in order to provide users with the most complete and detailed information that can be
extracted from data.
This precise representation, made possible by an accurate and fast alignment procedure, includes the
mature miRNAs and miRNA isoforms detected in the sample and their possible mode of interaction
with a target mRNA. The obtained information can be freely managed by users in order to highlight
those aspects considered as more meaningful.
1.2 Main features
isomiR-SEA is a software tool for the accurate and detailed processing of miRNAs sequencing data.
It requests as input a set of reads coming from miRNAs sequencing or a pre-elaborated tags file and
provides as output the tags counts attributed to the different miRNAs, distinguishing among their
isoforms. Moreover, conserved miRNA:mRNA interaction sites are reported in the output files.
High accuracy levels in miRNAs quantification are reached by isomiR-SEA thanks to a seed-based
alignment procedure. This procedure begins with the search and recognition of an exact miRNA-
seed match between read and miRNA and proceed with the extension of the alignment. Furthermore
isomiR-SEA allows for multi-species miRNAs analyses: One or more species contained in miRBase
database can indeed be selected during the parameters selection in order to assign reads to the most
similar miRNA. This last feature can be considered very useful in different situations such as during
the analysis of pathogens and host organisms or when inter-species comparisons have to be per-
formed.
isomiR-SEA advantages can be summarized as follows:
3
• High level of accuracy in miRNAs detection and quantification thanks to a miRNA-specific
alignment procedure;
• High degree of detail and precision in the isomiRs detection and profiling thanks to accurate
reads assignment;
• Ability in detecting, for each read supporting a specific miRNA, all the possible modes of inter-
action with a target mRNA;
• Computational costs contained despite the high degree of detail provided;
• Ability to perform multi-species analyses;
• High degree of flexibility.
4

2 Program usage
2.1 Installation procedures
isomiR-SEA current version is provided only in a pre-compiled format.
2.1.1 Pre-compiled release
In order to make as easy as possible isomiR-SEA installation, we provide the binary package for the
three most common operating systems. To use these pre-build packages, simply download that ad-
equate for your operating system, decompress it and check if binaries are in stored a folder in your
PATH environment variable.
2.2 Dependencies
isomiR-SEA is implemented in C++ as stand alone tool. The installation of third party tools is not
required if the standard input file format is used.
However, the most-common pre-elaboration activities can be executed taking advantege of the fol-
lowing programs:
• The SRA Toolkit to convert .sra files into other formats like .fasta or .fastq. http://www.ncbi.
nlm.nih.gov/books/NBK158900/
•Flexbar - flexible barcode and adapter removal to remove 3’ end read adapter. The adapter
sequence used during the sequencing procedure has to be provided. http://sourceforge.net/
p/flexbar/wiki/Manual/
• The FASTX Toolkit and in particular fastx_collapser program, to generate the tags counts file.
http://hannonlab.cshl.edu/fastx_toolkit/index.html.
2.3 The miRNAs databases
isomiR-SEA current version adopts as miRNAs reference database miRBase http://www.mirbase.
org/. In particular it uses miRBase file that stores mature miRNAs sequences.
To download the complete last miRBase release from unix:
1$ wget −r ftp://mirbase.org/pub/mirbase/CURRENT/
To download only miRBase mature miRNAs file:
1$ wget −nd ftp://mirbase.org/pub/mirbase/CURRENT/mature.fa.gz
To download from browser click the following link:
ftp://mirbase.org/pub/mirbase/CURRENT/mature.fa.gz
2.4 Program options
Run isomiR-SEA from command-line:
5

1$ miR−SEA_1_6 −s hsa −l 16 −b 4 −i DIRECTORY_NAME
2−p DIRECTORY_NAME/test_16_4 −ss 6 −h 11 −m MATURE_DB −t COUNTFILE
2.4.1 Alignment parameters
-s, species_codes STR (hsa)
Specie or species that have to be selected as reference(s) during tags alignment procedure.
-l, min\_tag\_length INT (10)
Minimum alignment length to perform the third ungapped extension.
-ss, seed_size INT (6)
miRNA seed size to be searched into tags sequences.
-sb, begin_seed INT (2)
miRNA seed start position.
-se, end_seed INT (7)
miRNA seed end position.
-b, tag_begin INT (4)
Maximum coordinate in the tag at which miRNA seed has to be searched.
2.4.2 Paths and files
-p, path_out STR (NULL)
Output folder path.
-i, path_in STR (NULL)
Input folder path.
-m, db_file_name STR (NULL)
Reference database file name (formatted in fasta file and converted in .txt). The extension must not be specified.
-t, tag_file_name STR (NULL)
Tags counts file name without .txt extension.
2.4.3 DNA/RNA conversion and filtering
-r, dna_or_rna BOOL (1)
If this parameter is equal to 0 the conversion between DNA to RNA is avoided.
-h, tag_selection_thr INT (11)
Threshold used to discard, during the alignment score check phase and the tag/miRNA
assignment procedure, multi-mapped tags with higher scores.
6

3 Analysis work-flow
The analysis of miRNA-Seq data is nowadays considered a central step in the characterization of bio-
logical samples. In order to perform such an analysis (Figure 3.1) the huge number of reads obtained
as output of sequencing machine runs needs to be reduced. This is performed by grouping all the
identical reads and counting their occurrence: The output file obtained is called tags count file.
State of the art tools such as miRanalyzer and miRExpress detect miRNAs expression levels in the
sample under investigation by aligning each tag on a selected miRNAs database.
A tag is then considered expression of a specific miRNA if it can be attributed to the same miRNA with
a high level of confidence.
However the alignment engines used by these tools are in the most of the cases general purpose align-
ment algorithms that don’t take into account miRNA-specific evidences.
Figure 3.1: Example of miRNA expression levels analysis pipeline.
In order to improve the information that can be derived from miRNA-Seq data, we have designed
isomiRNA Seed Extension Aligner (isomiR-SEA).
Differently from state of the art tools, its whole pipeline has been developed taking into accounts
miRNAs features as seed region, miRNAs families, miRNAs isoforms and possible interaction sites.
3.1 The interaction sites point of view
Recent researches by Bartel and colleagues on miRNAs have proven that almost all these short non-
coding molecules are characterized by similar structures [3]. In particular they present a common
standard region between nucleotides 2 and 7 that is fundamental for miRNA target recognition (Fig-
ure 3.2).
Moreover this region is usually conserved among miRNAs belonging to the same family.
Figure 3.2: miRNA seed region.
As stated in Section 1.1 one of isomiR-SEA mile stones consists in the pragmatic approach adopted
to detect miRNAs expression levels. This approach required an extensive literature review to identify
those miRNA features able to guarantee the correct attribution of a tag to a specific miRNA. Among
these the modes of miRNA:mRNA interaction have been considered fundamental.
7

Figure 3.3 reports on the most common miRNA:mRNA interaction modes. Among them 4 interaction
sites have been identified as responsible for these interaction modes:
• Seed site interaction: A perfect seed match is required in all the presented cases with exception
of G and H;
• Offset site interaction: Frequently the pairing of miRNA 8 nucleotide is required for miRNA:mRNA
interaction;
• Supplementary site interaction: In cases F, G and H the pairing of miRNA region between nu-
cleotides 13 and 16 is required for the interaction;
• Central site interaction: It has been recently identified in [4] as the only mode of interaction
able to compensate the lack of a strong pairing between miRNA seed and target mRNA.
Figure 3.3: miRNA:mRNA interaction regions (extracted from [3] and [4]).
Even if miRNA seed region is considered fundamental by all miRNAs target recognition tools, this fea-
ture has not been yet exploited in relation to miRNAs expression levels extraction.
isomiR-SEA algorithm is developed just considering this feature (Figure 3.2) and so miRNA seed iden-
tification is the first activity performed during the alignment procedure.
As a consequence only those tags exactly matching miRNA seed sequences in positions allowed by
miRNA structure (usually nt 1-10) are provided as input for the second alignment step of the pipeline.
8

3.2 The isomiRs point of view
Being miRNA:mRNA interaction sites identified on miRNA structure, even reduced variation in miRNA
sequences (determining isomiRs) can lead to the loss of interaction sites. For example an isomiR
characterized by a single nucleotide polymorphism in one of its interaction site will not be able to
interact and perhaps downregaluate the target genes of its miRNA of origin (as shown in Figure 3.3).
For this reason all the different isoforms that contribute to the miRNAs expression level value are re-
ported as output of isomiR-SEA run. Furthermore isomiR-SEA is able to distinguish among several
isoforms as illustrated in Figure 3.4 and reported in the following:
1. miRNA exact (mirna_exact), the original sequence reported in the database;
2. 5p isoforms (iso_5p), characterized by insertions or deletions in the 5p miRNA side;
3. single nucleotide polymorphism isoforms (iso_snp), presenting a mismatch with respect to
miRNA sequence;
4. multiple nucleotide polymorphism isoforms (iso_multi_snp), presenting more than a mismatch
with respect to miRNA sequence;
5. 3’ isoforms (iso_3p), characterized by insertions or deletions in the 3p miRNA side.
Figure 3.4: isomiR sequences aligned on the original miRNA sequence.
Moreover, since tags can also derive from two or more isoforms, also their combinations have to be
considered. Summarizing tags can be classified as deriving from:
1. mirna_exact
2. iso_5p-iso_snp-iso_3p
3. iso_5p-iso_snp
4. iso_5p-iso_multi_snp-iso_3p
9
5. iso_5p-iso_multi_snp
6. iso_5p-iso_3p
7. iso_5p_only
8. iso_snp-iso_3p
9. iso_snp_only
10. iso_multi_snp-iso_3p
11. iso_multi_snp_only
12. iso_3p_only
The proposed classification allows users to easily discard from the whole number of mapped tags
those attributed to a specific kind of isoforms.
3.3 isomiR-SEA Algorithm
isomiR-SEA analysis pipeline is implemented in C++ and takes advantage of SeqAn library. [?].
Thanks to a series of data analysis algorithms, SeqAn library usage, allows isomiR-SEA to assume a
modular frame-work and to be very fast even when computationally expensive tasks have to be per-
formed. The isomiR-SEA algorithm, shown in Figure 3.5, is basically divided in the following phases:
• The preprocessing step in which parameters are set and input files loaded in memory;
• The alignment procedure in which each miRNA seed is compared with all the tags to check
for similarities in order to discard too divergent sequences with remarkable reduction in time
search space usage. This activity is performed by considering all the rules developed taking into
account biological evidences relative to miRNAs structure;
• The output file generation that allow to distinguish between the so called unique and multi
mapped tags.
3.3.1 Parameters setting
Twelve parameters allow users to select the preferred configuration during isomiR-SEA run:
• specie_codes: Allows to select multiple species as reference on which tags will be aligned. De-
fault: Human (has);
• min_tag_length: Specifies the minimum length of alignment after the second ungapped exten-
sion that allows to further proceed into the alignment procedure. Default: 10;
• seed_size: Indicates the size of the seed to be searched into tags. Default: 6;
• begin_seed: Fixes the location of the first miRNA seed nucleotide. Default: 2;
• end_seed: Locates the last miRNA seed nucleotide. Default: 7;
• tag_begin: Specifies the maximum location of the tag on which the seed has to be searched.
Default: 4 (in order to allow iso 5p predictions);
• path_out: Specifies the location of the output files;
• path_in: Sets the location of the input file;
10
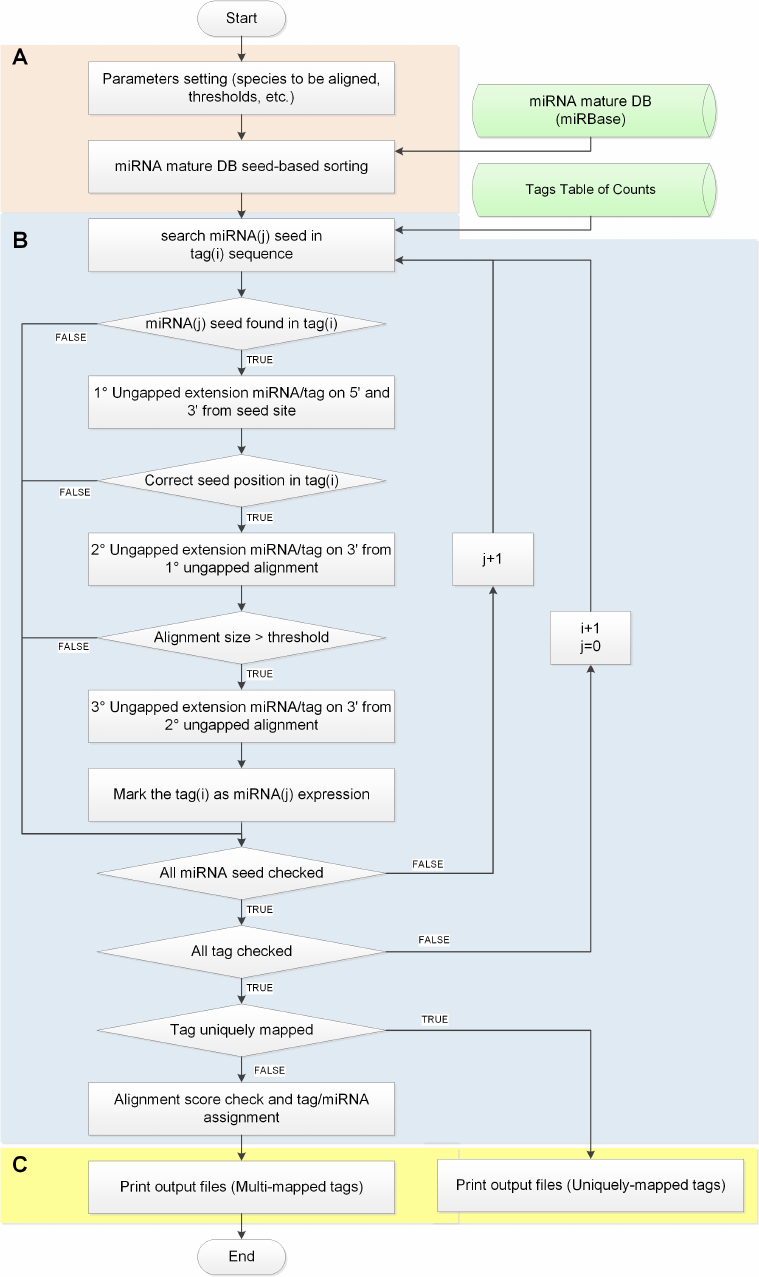
Figure 3.5: isomiR-SEA flowchart.
11
• db_file_name: Specifies the name of the reference database that has to be used (without .txt
extension). The current isomiR-SEA release requires a .txt file;
• tag_file_name: Specifies the input file tags name (without .txt extension). The current isomiR-
SEA release requires a .txt file;
• dna_or_rna: Boolean flag used to avoid conversion from DNA to RNA nucleotides when input
RNA reads are provided. Default: 1;
• tag_selection_thr: Specifies the maximum alignment score for multi-mapped tags reporting in
the output file. Default: 11.
The usage of this parameters will be further detailed in the course of the guide when encountered.
3.3.2 miRNAs DB and tags counts file processing
In this phase both the database containing reference miRNA sequences and the input tags counts
file are elaborated. miRNAs reference sequences can be downloaded from several pubblic repository
such as TargetScan database [5], HMDD [6], PolymiRTS [7], miRNEST [8], CoGemiR [9] and the widely
adopted miRBase [10].
The format currently accepted by isomiR-SEA for the reference file is that proper of miRBase mature
miRNAs. In Section 4.1 more details about this file format will be provided.
The miRBase v21 set includes 35828 mature miRNAs from 223 different species among animals, bac-
teria and plants.
In the miRNA mature DB seed-based sorting phase, seeds are extracted from miRNAs sequences. All
the miRNAs belonging to the same family and as a consequence sharing the same seed are then clas-
sified depending on the species and grouped together. The seed based groups are stored in a conve-
nient data structure in order to be easily accessible during the alignment procedure Figure 3.5.
Regarding the inputs, isomiR-SEA accepts the standard tags counts file format characterized by
the presence of the tag sequence followed by a number representing the occurrence of identical reads
supporting that tag (Section 4.1). This file is loaded in memory.
Even if the conversion from fastq reads to tags counts file is out of the scope of this manual, for com-
pleteness we will briefly report on it in the following lines. Firstly the adapter sequences introduced
in the library preparation process are removed from the reads thanks to Flexbar tool [10]. Then the
clipped reads are collapsed by means of an ad hoc script to obtain the so called table of counts file
which stores all the different clipped read sequences detected in the sample (tags), with their occur-
rence (tag_count).
3.3.3 isomiR-SEA alignment procedure
Each miRNA seed is searched into the different tags contained in the input file. If the seed is found
in the tag, the alignment between the miRNA and the tag is extended without gaps in both 5’ and 3’
directions (ungapped extension). Similarly the tag is aligned on all the miRNAs that share the identi-
fied seed.
For each alignment, the position on the tag where the first match has been encountered is evaluated
to check if compatible with the standard miRNA seed position. Tags characterized by a seed located
in an incompatible position are discarded as shown in Figure 3.6.G.
All the tags with a correctly located miRNA seed are instead extended in 3’ direction allowing for the
presence of a second mismatch.
A threshold, called min_tag_length, is imposed in this phase in order to evaluate the distance of this
second mismatch from to the first one: Tags not satisfying the threshold value, since having two adja-
cent mismathces, will be not considered in the next alignment steps as depict in Figure 3.6.H. Instead,
alignments with two mismatches enough far away from each other, are once a time extended without
12

gaps until the third mismatch which marks the end of the alignment procedure.
isomiR-SEA alignment policy is further detailed in eight different cases reported in Figure 3.6 in
relation to hsa-miR-15a-5P miRNA. In all these examples miRNA sequence is shown as first line, the
tag in the third line and the alignment representation in the middle. The numbered line in each sub-
figure is instead used to identify the algorithm step in which sub-sequences have been evaluated:
1. The numbered 1 red line represents the miRNA seed sequence searching activity in the tag;
2. The numbered 2 blue line reports on the seed extension step in both 5’ and 3’ directions until a
first encountered mismatch;
3. The numbered 3 green line describes the alignment extension in 3’ direction until a second
found mismatch;
4. The numbered 4 violet line depicts the last extension step executed if a minimum alignment
length is reached in the previous phases, until a last allowed mismatch.
isomiR-SEA parameters have been set for the previous tests as: Sizemi n =11 (minimum alignment
length for last extension) and Begin_Tagmi n <4(start tag position alignment on miRNA seed). The
imposed parameters values led to the discarding of those alignments reported in Figure 3.6 with light
red background and to consider those characterized by the light green one. Figure 3.6.A reports on
atag alignment characterized by a mismatch in position 10. This mismatch lead the algorithm to
continue the alignment procedure in its second phase since no additional mismatches are detected
between miRNA and tag sequence.
As depicted in Figure 3.6.B the mismatch encountered in the tag at position 14 leads isomiR-SEA to
assume the same behaviour highlighted in Figure 3.6.A.
In Figure 3.6.C the alignment procedure comprises all the four phases of the algorithm, indeed the
two mismatches found in the tag sequence are located in positions compatible with the imposed pa-
rameters.
The alignment in Figure 3.6.D is instead ended in the second phase of the alignment after the first
encountered mismatch located in the last admissible nucleotide.
In Figure 3.6.E the tag alignment is ended before the third gap at position 20.
The mismatch detected in the tag at position 1 allows isomiR-SEA to proceed in the alignment as de-
picts in Figure 3.6.F when the detailed parameters are imposed. This mismatch will influence how-
ever the overall alignment size with consequences on the calculated alignment score.
According to Begin_Tagmi n <4, the tag in Figure 3.6.G is discarded after the second algorithm step
because tag alignment start position is beyond the third nucleotide; this condition, that is a high
number of nucleotides flanking seed 5’ position on the tag, could indeed account for a different kind
of RNA structure such as a pre-miRNA as discussed in Bartel et al. [?].
In Figure 3.6.H the tag is discarded because a second mismatch can be detected in position 12 not
satisfying the imposed Sizemi n threshold. Once a time this choice depends on experimental observa-
tions: Bartel et al. [?] shown indeed that sequences having two not distant mismatches, close also to
the seed sequence in the most of the cases do not derive from miRNAs.
At the end of the alignment procedure, an alignment score is assigned to each alignment. This score
is very useful when the same tag is found to be mapped on more than a miRNA or if the user is inter-
ested in performing some kind of filtering to isolate the most significant aligned tags (characterized
by a higher degree of similarity to the miRNA).
The alignment score (al i g nscore ), based on several alignment parameters, is calculated as reported
in Equation 3.1.
al i g nscore =miRN Asi ze −(ali g nsi ze −#g aps)
mi RN Asi ze
∗100 (3.1)
where miRN Asi ze is the length of the miRNA sequence, #g aps represents the number of gaps in the
13

Figure 3.6: isomiR-SEA alignment cases.
alignment (maximum 2) and ali g nsi ze the miRNA-tag alignment length (remember that an align-
ment is stopped when the third gap is found). This parameter allows to evaluate the alignment qual-
14
ity: Lower scores indicate better alignments.
Moreover in order to consider also the tag length during the selection of the alignments output of
isomiR-SEA run, a second parameter is computed as shown in Equation 3.2:
mi RN At agd i f f =mi R N Asi ze −t ag si ze (3.2)
Obviously positive values of this parameter account for miRNA sequences longer than tags and vice
versa.
These two parameters are adopted in the last phase of the algorithm in order to choose the best align-
ment for tags that have been mapped on more than a miRNA.
Moreover in future users will be left free to introduce the proposed scoring scheme also for the uniquely
mapped tags, in order to give them the possibility to require a minimum alignment quality to report
tag assignments.
3.3.4 isomiR-SEA output files
All the tags found to be aligned on the miRNAs sequences are finally saved in two files according to
the kind of alignment detected. In particular if a tag is aligned only on a miRNA, it is saved into a
unique tags file (see Section 4.2), otherwise if it is mapped on multiple miRNAs it is saved into an
ambiguous tags file.
Moreover the multiple mapped tags are scanned and assigned to the miRNA for which they present
a lower alignment ali g nscore and a smaller mi RN At agdi f f . After this selection the obtained tags
are organized into a new file having the same format of that used for storing the unique mapped tags
but reporting also, for each tag, the number of miRNAs and miRNAs families on which the same tag
has been mapped and the alignment score difference between the score calculated for that tag on the
chosen miRNA and that on the second scored one.
Furthermore two miRNAs expression files are also provided to the user: One relative to the unique
mapped tags and one for those ambiguously aligned. These files contain useful information such as
the number of different tags attributed to the miRNA, the tags counts of the detected isomiRs and
interaction sites (details in Section 4.2).
15

4 Files formats
4.1 Input files
Two input files are required in order to run isomiR-SEA tool: A file containing the mature miRNAs
reference sequences and the tags counts file. The mature miRNAs file that has to be provided is in the
standard fasta format even if, to be compatible with isomiR-SEA execution, its extension has to be .txt
(this limitation will be removed in the next versions).
Concerning the tags counts file (Figure 4.1) we have implemented an ad hoc script that, given the
fastq file containing the input reads, removes the 3’adapter sequences using the Flexbar tool [10],
then collapse all the identical clipped reads into a unique tag and counts the occurrence of reads
supporting it. The tag file reports on each line in the first column the tag sequence and in the second
column the occurrence of the reads supporting it. The extension of this file should be set as .txt
Figure 4.1: Tags counts file format
16
4.2 Output files
Nine output files are generated by isomiR-SEA run. The names of these files are formatted as out_result_DBNAME_SPECIALIZATION.txt
with the exception of the first two files that store the mature miRNAs sequences grouped and sorted
on the basis of miRNA seed and species. These files are:
• mature_21_db_group.txt
• mature_21_db_group_sorted.txt
• out_result_mature_21_ambigue.txt
• out_result_mature_21_ambigue_ambigue_selected.txt
• out_result_mature_21_tag_ambigue.txt
• out_result_mature_21_tag_ambigue_selected.txt
• out_result_mature_21_tag_unique.txt
• out_result_mature_21_unique.txt
• out_result_mature_21_unique_ambigue_selected.txt
In the following it will be shown the meaning of each field included into these files.
1. mature_21_db_group.txt is a file printed as output of the first phase of the algorithm when the
miRNAs mature database is loaded into memory. It is the representation of the memory struc-
ture used during the alignment procedure and can be stored and then used in all the alignment
performed thanks to isomiR-SEA that adopt the same miRNAs reference sequences. In the first
line of this file are reported all the species codes detected into the input miRNAs database, the
following lines are instead organized as follows:
• The first column reports on the seed sequence;
• The second column indicates the species code of the following miRNAs;
• The third column reports on the first miRNA sequence characterized by that specific seed;
• The fourth column reports on some information related to the first detected miRNA;
• The other columns reports on all the other miRNAs characterized by the same seed and
species.
2. mature_21_db_group_sorted.txt has the same format of the previous one, but it is alphabeti-
cally sorted considering the first field, the miRNA seed sequence.
3. out_result_mature_21_tag_unique.txt file stores for each tag the unique miRNA on which it has
been mapped. This file contains those tags that have been assigned to a unique miRNA with a
series of information extracted during the alignment phase and reported in the following:
• tag_index: An integer value that stores the unique tag index assigned to each tag during
the loading phase (it is equal to the tag row position in the input tags counts file);
• tag_sequence: The original tag sequence;
• tag_quality: In this isomiR-SEA version it stores an “e’" value. In the next releases of
the tool however it will dedicated to store the average tag quality computed for each nu-
cleotide of the tag;
17
• #count_tags: The number of identical reads supporting the tag;
• mirna_id: An identification number for the miRNA (this number is assigned during the
mature miRNAs database loading phase);
• mirna_name: A series of informations relative to the miRNA that has been assigned to the
tag. These data have been recovered from the miRNAs database;
• mirna_seq: The miRNA sequence for the miRNA that has been assigned to the tag;
• seed_index: The identification number of the seed characterizing the miRNA assigned to
the tag (attributed during the loading phase);
• begin_ungapped_mirna: The miRNA nucleotide position where the tag alignment begin;
• begin_ungapped_tag: The tag nucleotide position where the alignment with the selected
miRNA begin;
• size_ungapped: The number of nucleotides aligned during the first ungapped extension.
This number is relative to perfect matches between the two sequences;
• size_ungapped_1: The number of aligned nucleotides during the second ungapped ex-
tension. This number is relative to perfect matches and a unique mismatches between
the two sequences;
• size_ungapped_2: The number of aligned nucleotides during the third ungapped exten-
sion;
• align_score: The alignment score computed with Equation 3.1;
• mir_tag_size_diff: The score calculated as reported in Equation 3.2;
• mirna_exact, iso_5p, iso_snp, iso_multi_snp, iso_3p, offset_site, suppl_compens_site, cen-
tral_site: The flags recovered during the alignment phase that provide informations about
the isoforms identified and the interaction sites conserved during miRNA-tag alignment.
4. out_result_mature_21_tag_ambigue.txt file stores all the tags that have been mapped on more
than a miRNA. It is formatted as the previous unique file with a little difference. Indeed isoform
and interaction site flags are not reported, and each line stores all the miRNAs and alignment
information previously described in relation to out_result_mature_21_tag_unique.txt. All the
miRNAs associated with a tag are so reported in the same tag line, separated by the pipe (|)
character.
5. out_result_mature_21_tag_ambigue_selected.txt reports on all the tags initially evaluated as
ambiguous that, after the analysis of the mapping scores, have been assigned to a specific
miRNA.
This file shares the same format of out_result_mature_21_tag_unique.txt with the addition of
three useful parameters that are:
a) mir2-mir1_align_score: This value is the score difference between the second higher score,
proper of the second best aligned miRNA, and the score of the best aligned one;
b) num_mir_family: It represents the number of different miRNAs families, on which the tag
has been mapped;
c) num_of_mapped_miRNAs: It is the number of miRNAs that has been attributed to the
same tag.
6. out_result_mature_21_unique.txt reports, for each miRNA, the overall number of assigned tags
and some other information concerning isoforms and interaction sites. Only tags uniquely
assigned to a miRNA are here considered.
Reported features are:
18
a) mirna_index: The identification number of the miRNA;
b) #different_tags: The number of tags with different sequences mapped on this miRNA;
c) #count_tags: The overall number of reads supporting the miRNA;
d) norm_expressed_all: A field not used in the current version of isomiR-SEA that will be
considered in future versions of the tool;
e) norm_expressed_mapped: A field not used in the current version of isomiR-SEA that will
be considered in future versions of the tool;
f) #mirna_exact_tags: The number of reads having an identical sequence with respect to the
miRNA;
g) #iso_5p_tags: The number of reads having some mutations on the 5’ side. A hierarchi-
cal aggregation method has been adopted for this and the following parameters: If a tag
presents a mutation on both 5’ and on 3’ that tag is counted only for iso_5p. This choice
allows to avoid overestimation in the assignment of tags to more then an isoform bin.
However from the tag files the complete isoform spectrum can be retrieved;
h) #iso_snp_tags: The number of reads characterized by SNP. As for the previous field the
assignment of tags to this category is priority with respect to iso_3p;
i) #iso_multi_snp_tags: The number of reads having more than a SNP in the alignment;
j) #iso_3p_tags: The number of reads having only mutations on the 3’side;
k) #offset_site_tags: The number of reads exactly mapped at the 8 miRNA nucleotide;
l) #suppl_compens_site_tags: The number of reads exactly mapped on miRNA positions 2-
16;
m) seed_index: The miRNA seed identidfication number;
n) mirna_seq: The miRNA sequence;
o) mirna_info: The miRNA information retrieved from miRNAs reference database;
7. out_result_mature_21_ambigue.txt file contains the same information for the re-evaluated tags
initially marked as multi-mapped.
The last two files, out_result_mature_21_ambigue_ambigue_selected.txt and
out_result_mature_21_unique_ambigue_selected.txt, store informations about the discarded
multi-mapped alignments.
19
Bibliography
[1] Peter Wilson, The Memoir Class for Configurable Typesetting – User Guide, 2010.
[2] Vincent Zoonekynd. On-line list of different chapter styles for L
A
T
EX. Available at http://zoonek.
free.fr/LaTeX/LaTeX_samples_chapter/0.html.
[3] Bartel, D.P. (2009) MicroRNAs: Target Recognition and Regulatory Functions. Cell,136, 215–233
[4] Shin, C., Nam, J. W., Farh, K. K. H., Chiang, H. R., Shkumatava, A. and Bartel, D. P. (2010) Expanding
the microRNA targeting code: functional sites with centered pairing. Molecular cell,38, 789–802
[5] Conserved Seed Pairing, Often Flanked by Adenosines, Indicates that Thousands of Human Genes
are MicroRNA Targets Benjamin P Lewis, Christopher B Burge, David P Bartel. Cell, 120:15-20
(2005).
[6] Lu M, Zhang Q, Deng M, Miao J, Guo Y, et al. (2008) An Analysis of Human Mi-
croRNA and Disease Associations. PLoS ONE 3(10): e3420. doi:10.1371/journal.pone.0003420.
http://www.microrna.org/microrna/
[7] Bhattacharya A, Ziebarth JD, Cui Y. PolymiRTS Database 3.0: linking polymorphisms in microR-
NAs and their target sites with human diseases and biological pathways. Nucleic Acids Res. 2014;
42(D1):D86-D91.
[8] Szczes’niak, Michal Wojciech et al. miRNEST Database: An Integrative Approach in microRNA
Search and Annotation. Nucleic Acids Research 40.Database issue (2012): D198-D204. PMC. Web.
11 Dec. 2014.
[9] CoGemiR: a comparative genomics microRNA database Vincenza Maselli, Diego Di Bernardo and
Sandro Banfi BMC Genomics 2008, 9:457 (6 October 2008) http://www.biomedcentral.com/1471-
2164/9/457
[10] Matthias Dodt, Johannes T. Roehr, Rina Ahmed, Christoph Dieterich: Flexbar-flexible barcode
and adapter processing for next-generation sequencing platforms. MDPI Biology 2012, 1(3):895-
905.
20
