Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 6

HaploHammer Manual
Tools for manipulating re-sequencing data.
David C. Tack, Ángel Ferrero-Serrano, Phil C. Bevilacqua, Sarah M. Assmann
Dependencies Link
╔Python 2.7.X ╔https://www.python.org/
╠vcftools ╠https://vcftools.github.io/man_latest.html
╚BioPython ╚http://biopython.org/
Recommended Link
╔R ╔https://www.r-project.org/
╠R Package (??) ╠(??)
╚R Package (??) ╚(??)
Introduction
HaploHammer is a suite of Python scripts designed to quickly apply a large number of <.vcf>
files back to their respective genome, yielding all inferred changes to whole transcripts or particular
transcript features. These features may then be sorted into a whole haplotype matrix, cataloging the full
range of haplotypes present among all surveyed <.vcf>s, along with an accompanying ‘master’ <.fasta>
file containing an entry for every haplotype referenced by the matrix. Thus, it effectively aims to
summarize the variation among re-sequenced lines and organize that information into a useful and
halotype aware format.
Installation and Support
Download the entire package from Github and place in a directory of your choosing, or you
may use git directly to clone the repository from Github with the command:
git clone https://github.com/StructureFold2/HaploHammer
Next, set the path to the folder containing the scripts, such that they may be called from any
directory. This may be done by adding the following to your shell profile:
PATH=$PATH:/home/path/to/new/folder/HaploHammer
File permissions to the installation folder must now be set. Typically this will entail running the
following command on the HaploHammer folder which will grant all users read and execute
permissions and all permissions to the owner:
chmod -R 755 HaploHammer/
This manual makes the best attempt possible to detail the present HaploHammer tool set and
demonstrate its use. However, it is nearly impossible to plan for all contingencies. While the
software is provided ‘as is’, we are eager to improve and further streamline our analysis pipeline. If
you encounter a bug or require support, or have an idea for a feature that would improve your
overall HaploHammer experience, please include HaploHammer in the subject line when writing to
David Tack (kujiratan@gmail.com).

File Type Contains Used with Module:
╔FASTA ╔Nucleotide Sequences ╔
╠VCF ╠Variant Calls ╠
╠GFF/GTF ╠Genome Annotation ╠
╚CSV ╚Any data, delimited by comas ╚
Scripts Letter Function
╔ vcf_manager.py ╔A ╔Batch prepares <.vcf>s with vcftools
╠haplo_hammer.py ╠B ╠Applies <.vcf>s to a reference <.fasta>
╠batch_translate.py ╠C ╠Translates <.fasta>s to peptide sequence
╠allele_matrix.py ╠D ╠Summarizes <.fasta>s to a matrix and a <.fasta>
╠matrix_expand.py ╠E ╠Converts a haplotype matrix to a GWAS style matrix
╠allele_extract.py ╠F ╠Creates a <.fasta> of all alleles of a given sequence
╚annotation_parser.py ╚G ╚Classes for dealing with GFF3/GTF
Input Data
This tool matches variation from vcf files back to the reference genome they were generated
against, thus you must have the same version of the reference genome that was used to generate the vcf
files, i.e. what the variants were called from. There is no known limit on the number of vcfs that can be
incorporated into a single haplotype matrix. It is possible to use an updated GFF3/GTF file as long as it
still references the same build of the genome, thus giving more or updated gene models without
perturbing the genomic coordinates. The pipeline was built around the IRRI 3000 genomes project and
the 1001 Arabidopsis genomes project, where each re-sequenced line is both diploid and contained
within it’s own vcf file; support for vcf files detailing the changes from multiple lines may be added in
the future.
Workflow
1 Prepare <.vcf> files
Filtering out non-variant lines and variant types allows for a streamlined <vcf> to be applied in
future steps, making the regeneration of line specific <fasta> files much more efficient. This
step will batch run vcftools with a Python script on a directory of vcf files, unzip them if
needed, apply filters, and write filtered vcfs with their respective filtering log into a new
directory while automatically naming them. Intermediate files can be automatically deleted after
a run.
The important option here is the -method option of the script which has the options ‘fixed’
(default) and ‘poly’ which correspond to the vcftools options of ‘--non-ref-af 1’ and ‘--non-ref-
ac-any’. In fixed mode, only fixed variants in the re-sequenced line are retained, while poly
(polymorphic) mode retains cases where there is a a single non-fixed (heterozygous) variant in
the re-sequenced line, or two new variants.
Since vcf files, especially uncompressed, can run very large, and re-sequencing projects can
have thousands of files, it is highly recommended to let the script drive the uncompressing,
filtering, and then subsequently delete the intermediate unfiltered vcf. This results in a ton of
saved space, as the compressed raw vcf and the filtered new vcf should be comparatively very

small, allowing for the preservation of the original and the use of the smaller effective copy. It
also has the benefit of keeping everything named and organized properly. Run the script in the
directory containing your vcfs. Running the example below in a directory would sequentially
uncompress each .gz file, filter the vcf with vcftools and write this to biouser/variation, then
remove the intermediate file.
vcf_manager.py -in_ext gz -outdir /home/biouser/variation -rm_vcf
Ⓐ vcf_manager.py
Options
-h, --help show this help message and exit
-single SINGLE Operate on this single file, rather than the directory
-quality QUALITY [default = 30] vcftools ‘--minQ’
-in_ext {gz, vcf} Operate on files with this extension
-method {FIXED,POLY} [default = FIXED] Variants to keep
-outdir OUTDIR [default = filtered_vcf] Out Directory
-rm_vcf remove unfiltered vcf after filtering
-zip_out compress new vcf after filtering
When filtering the vcfs, we highly recommended only using fixed differences to generate your
haplotypes at this time.
2 Create haplotype-specific <fasta> files
This step will apply the filtered <vcf> files to the reference genome (fasta), using the reference
annotation as a guide (GFF3/GTF). Line specific variants are applied in whichever scope the
user prefers; by selecting the 5’UTR, only changes specific to the 5’UTR are applied and the
resultant output fasta files would only contain those regions. In cases of polymorphic SNP
variants , the mixed base code is inserted into the new sequence (i.e. A or G would be an R). In
polymorphic cases of Indels, the shortest is chosen. This behavior may be more configurable in
a future release.
Similar to the previous script, haplo_hammer.py is run in the directory containing all of the
filtered <.vcf> files. It requires the corresponding genome fasta and one annotation file
(GFF3/GTF). Likewise, the user specifies an out directory, where new fasta files incorporating
the variants from the vcf files will be written to. New fasta file names will include the scope of
the features used when generating them.
Ⓑ haplo_hammer.py
Positional arguments
genome Genome <.fasta>
annotation Annotation <.gff/.gtf/.gff3>
{RNA,CDS,FP,TP} Target Features
Options

-h, --help show this help message and exit
-method {FIXED,POLY} [default = FIXED] VCF Filtering Used
-outdir OUTDIR [default = out_fasta] Out Directory
-single SINGLE [<.vcf>] Run on single <.vcf>, rather than directory
-chr_names CHR_NAMES [<.txt>] Convert <.vcf> chromosomes
3 Translate CDS fasta files (Optional)
If your questions center around changes in peptide sequence, you may wish to effectively
ignore synonymous variants in the CDS region by translating the sequence. This module will
translate all of the fasta files in a directory of CDS fasta files into their peptide equivalents. In
order for this to work (for now), these files must be generated off only fixed changes.
Ⓒ batch_translate.py
Options
-h, --help show this help message and exit
-no_truncate Do not truncate peptide at premature stop codons
-single SINGLE [default = current directory] Operate on this single file
-in_ext IN_EXT [default = fa] <.fasta> file extension
-suffix SUFFIX [default = peptide] Suffix for out files
4 Assemble Matrix and Unified Fasta
After applying all the vcf changes (and possibly translating) your sequences of interest, the next
step is to consolidate all of this information into a more usable format, i.e. the actual goal of this
pipleline. The allele_matrix script will operate on every fasta in a directory and generate a
matrix where rows are the re-sequenced lines and columns are the sequences. For each
sequence, every haplotype found will be assigned a code (A-ZZZ, 18,278 max haplotype limit
currently) internally, and the matrix will be populated with these codes. For example, if you had
26 haplotypes of a sequence, that sequence could be any letter from A-Z; to sort these lettter
codes by the frequency of the allele, use the -sort option. Every haplotype of every sequence is
logged to a universal fasta file, where the letter code is suffixed to the sequence name, therefore
all sequence haplotypes are contained in one file, and the matrix shows which lines have which
haplotype.
Ⓓ allele_matrix.py
Options
-h, --help show this help message and exit
-sort Alleles are named by frequency
-fasta FASTA Specify output <.fasta> name
-matrix MATRIX Specify output <.csv> name

5 Expand Matrix (Optional)
The matrix generated in step 4 may not suit a user’s particular mode of analysis. The matrix can
be ‘expanded’ to more reference a typical ‘GWAS’ type analysis. Every column in the existing
matrix is turned into a number of columns for each haplotype above 5% frequency, and each
letter is turned into a 1 or 0 to indicate the presence or absence of a particular haplotype in a
particular line. For example, if there are three haplotypes (A, B, C) of a particular sequence, that
column becomes three columns; if a line had an A before, it is 1 in the A variant column and 0
in the B and C variant columns.
Ⓔ matrix_expand.py
Positional arguments
csv Haplotype Matrix file to reformat
Options
-h, --help show this help message and exit
-freq FREQ [default = 0.05] alelle frequency threshold for retention
-name NAME Change the name of the outfile, overrides default
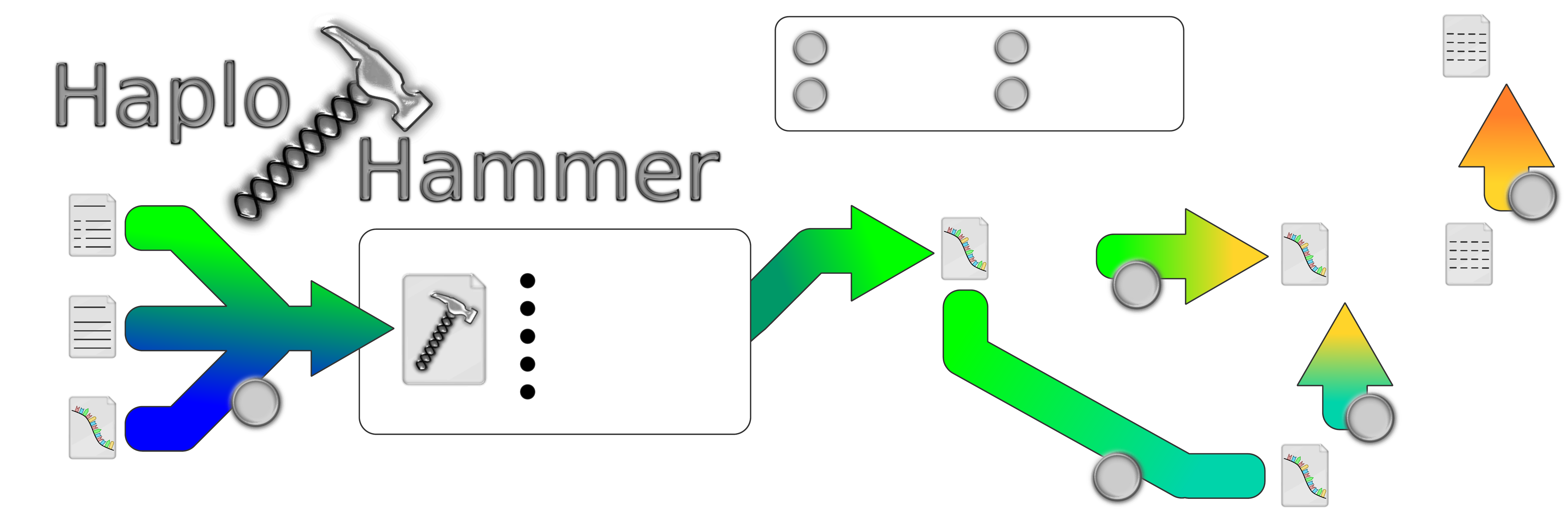
Genome
(.fasta)
Annotation
(.gtf/.gff)
Variation
(.vcf)
1
1
NSelect Features of Interest:
Whole Transcript
Untranslated Regions
Coding Sequences
Introns
Non-Coding RNAs
Haplotypes
(.fasta)
Haplotype
Matrix
(.csv)
Translated
Haplotypes
(.fasta)
B
D
C
D
GWAS
Matrix
(.csv)
E
haplo_hammer.py
B
C
batch_translate.py
D
allele_matrix.py
E
matrix_expand.py
N
N
All
Haplotypes
(.fasta)
11
1
