Sanger_paper Sanger Paper
User Manual: sanger_paper
Open the PDF directly: View PDF ![]() .
.
Page Count: 6
- A large genome center’s improvements to the Illumina sequencing system
- Fragmentation
- A-tailing, ligation and size selection
- PCR
- Quantification
- Denaturation
- Amplification quality control
- Conclusion
- ACKNOWLEDGMENTS
- COMPETING INTERESTS STATEMENT
- REFERENCES
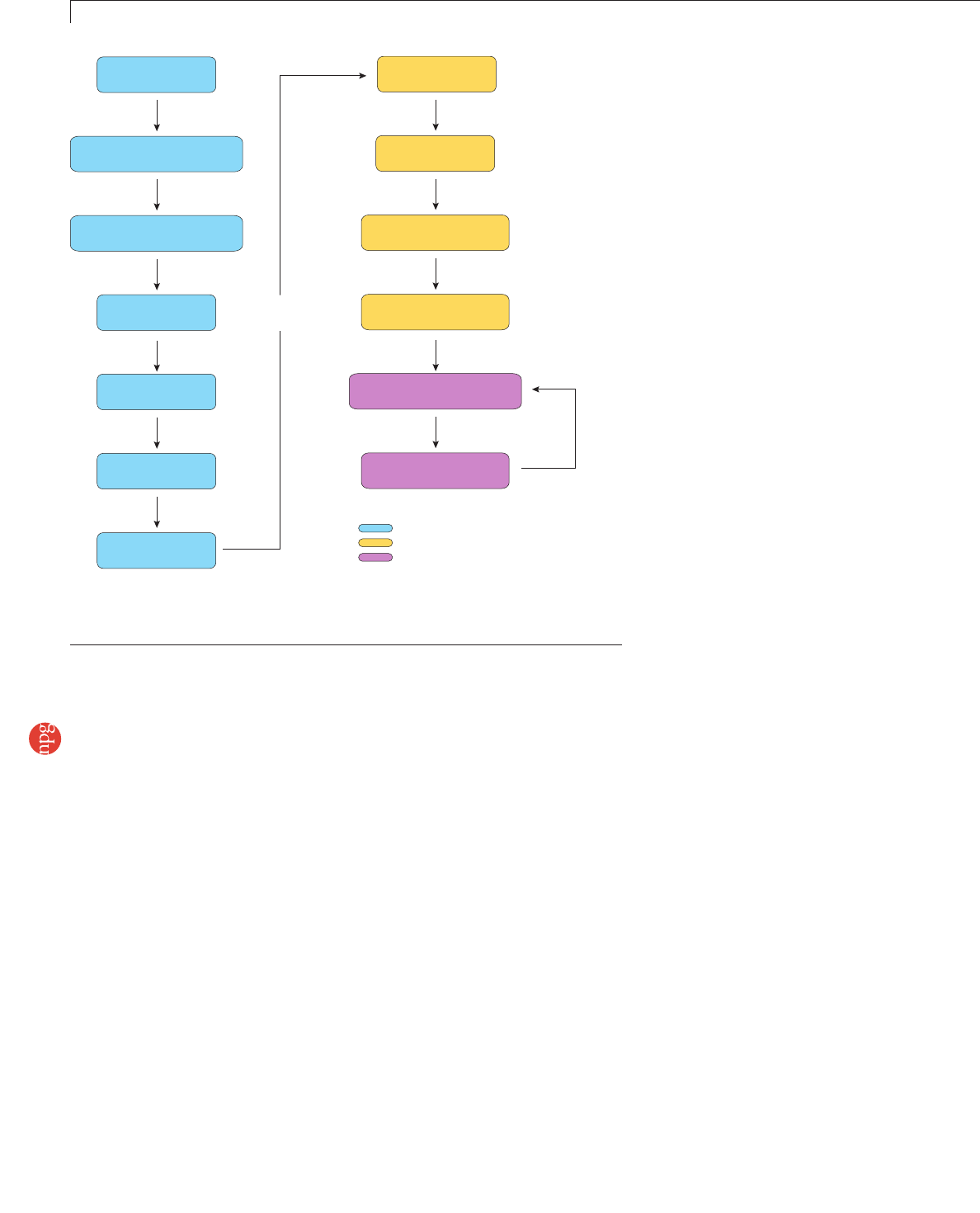
- Figure 1 | Illumina sequencing workflow
- Figure 2 | Sample fragmentation
- Figure 3 | A-tailing, ligation and size selection
- Figure 4 | PCR
- Figure 5 | Quantification
- Figure 6 | Denaturation

The Wellcome Trust Sanger Institute is one of the
world’s largest genome centers, and a substantial
amount of our sequencing is performed with
‘next-generation’ massively parallel sequencing
technologies: in June 2008 the quantity of purity-
filtered sequence data generated by our Genome
Analyzer (Illumina) platforms reached 1 terabase,
and our average weekly Illumina production output
is currently 64 gigabases. Here we describe a set of
improvements we have made to the standard Illumina
protocols to make the library preparation more reliable
in a high-throughput environment, to reduce bias,
tighten insert size distribution and reliably obtain
high yields of data.
Next-generation DNA sequencers, such as the 454-FLX
(Roche), SOLiD (Applied Biosystems) and Genome
Analyzer (Illumina) have transformed the landscape of
genetics through their ability to produce hundreds of
megabases of sequence information in a single run. This
has enabled us to design genome-wide and ultra-deep
sequencing projects that, because of their enormity, would
not otherwise have been possible (for reviews see refs. 1,
2 and for an evaluation of the performance of these three
platforms see ref. 3).
At the Wellcome Trust Sanger Institute we currently
have all three of these sequencing platforms, though the
Genome Analyzer is the platform we have invested most
heavily in: we have 28 machines on site, all capable of
generating paired-end data. The Illumina data analysis
pipeline performs ‘purity filtering’ of these data to elimi-
nate sequence data from clusters that appear to be mixed
as a consequence of their proximity on the flowcell. We
typically generate 4–5 gigabases (Gb) of filtered sequence
data, with an error rate of <0.9% per seven-day, 36-cycle
paired-end run, making us one of the world’s largest and
most productive users of Illumina sequencers.
Sequencing library preparation involves the produc-
tion of a random collection of adapter-modified DNA
fragments, with a specific range of fragment sizes, which
are ready to be sequenced. We have found the standard
Illumina sequencing library preparation protocols (Fig. 1)
to be suboptimal in several respects, and we enhanced our
output by developing and implementing many modifica-
tions and improvements to these protocols, all with the
aim of obtaining the maximum number of high-quality
sequence reads per run from the lowest mass of starting
DNA, in a robust and reproducible way. The modifica-
tions and improvements we describe here can be adopted
en masse as an alternative library preparation pipeline.
However, because some steps are additional rather than
alternative, we tend to select different modifications for
different sequencing projects, depending on the specific
requirements of that project (Supplementary Table 1
online). Here we have attempted to describe each modifi-
cation in the order in which it would fit in to the standard
library preparation pipeline.
Fragmentation
The first stage in a standard genomic DNA library prepa-
ration for the Genome Analyzer is DNA fragmentation
by nebulization (in 30–60% glycerol at 30–35 p.s.i.). This
generates fragments with a typical size range of 0–1200
base pairs (bp) and a peak around 5–600 bp. Nebulization
is a fairly reproducible technique, is sequence-indepen-
dent, and is rapid and inexpensive4. However, the wide
size distribution of generated fragments is uneconomical:
by mass, the 200 ± 20-bp fragments represent only ~10%
of the total DNA after nebulization. Moreover, approxi-
mately half of the DNA vaporizes during nebulization,
meaning that only 5% of the original DNA is used for
subsequent library generation. Even under much more
extreme nebulization conditions (for example, 90 p.s.i.
for 18 min) it is not possible to ‘move’ the fragment-
size peak below around 400 bp, and doing so still does
not improve the yield at 200 bp (ref. 4 and unpublished
observations). Thus we have evaluated alternative meth-
ods of sample fragmentation.
A large genome center’s improvements to
the Illumina sequencing system
Michael A Quail, Iwanka Kozarewa, Frances Smith, Aylwyn Scally, Philip J Stephens,
Richard Durbin, Harold Swerdlow & Daniel J Turner
Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridgeshire, CB10 1SA, UK. Correspondence should be
addressed to D.J.T. (djt@sanger.ac.uk).
PUBLISHED ONLINE 25 NOVEMBER 2008; DOI:10.1038/NMETH.1270
NATURE METHODS | VOL.5 NO.12 | DECEMBER 2008 | 1005
PERSPECTIVE
©2008 Nature Publishing Group http://www.nature.com/naturemethods
tocol: (i) bias in the base composition of
sequences; (ii) high frequency of chimeric
sequences, produced when two template
strands are ligated during the adapter liga-
tion step; and (iii) imperfect distribution of
insert sizes. These have all been overcome by
the use of several protocol modifications,
described here.
Paired-end oligonucleotides. We no longer
use the Illumina single-end adapters or PCR
primers because paired-end oligos generate
sequencing libraries that are compatible with
both single and paired-end flowcells. The
adapters themselves are modified to confer
protection from digestion at the 3′ thymine
(T) overhang. Though we do not have the
details of the modification used by Illumina,
we have obtained comparable results using
our own adapters and PCR primers modi-
fied with a phosphorothioate between the
two bases at the 3′ end (Supplementary
Protocol 2 online).
Gel extraction. During the size selection step
of the standard library prep protocol, a gel
slice is selected and the DNA extracted. We
identified that melting this gel slice by heating
to 50 °C in chaotropic buffer decreased the
representation of A+T-rich sequences, pos-
sibly reflecting a higher affinity of spin col-
umns for double-stranded DNA, as strands
with a high A+T content will be most likely to
become denatured during this step and may not reanneal. To improve
the representation of these A+T-rich sequences, we modified the gel-
extraction protocol, melting agarose gel slices in the supplied buffer
at room temperature (18–22 °C). This reduces G+C bias considerably
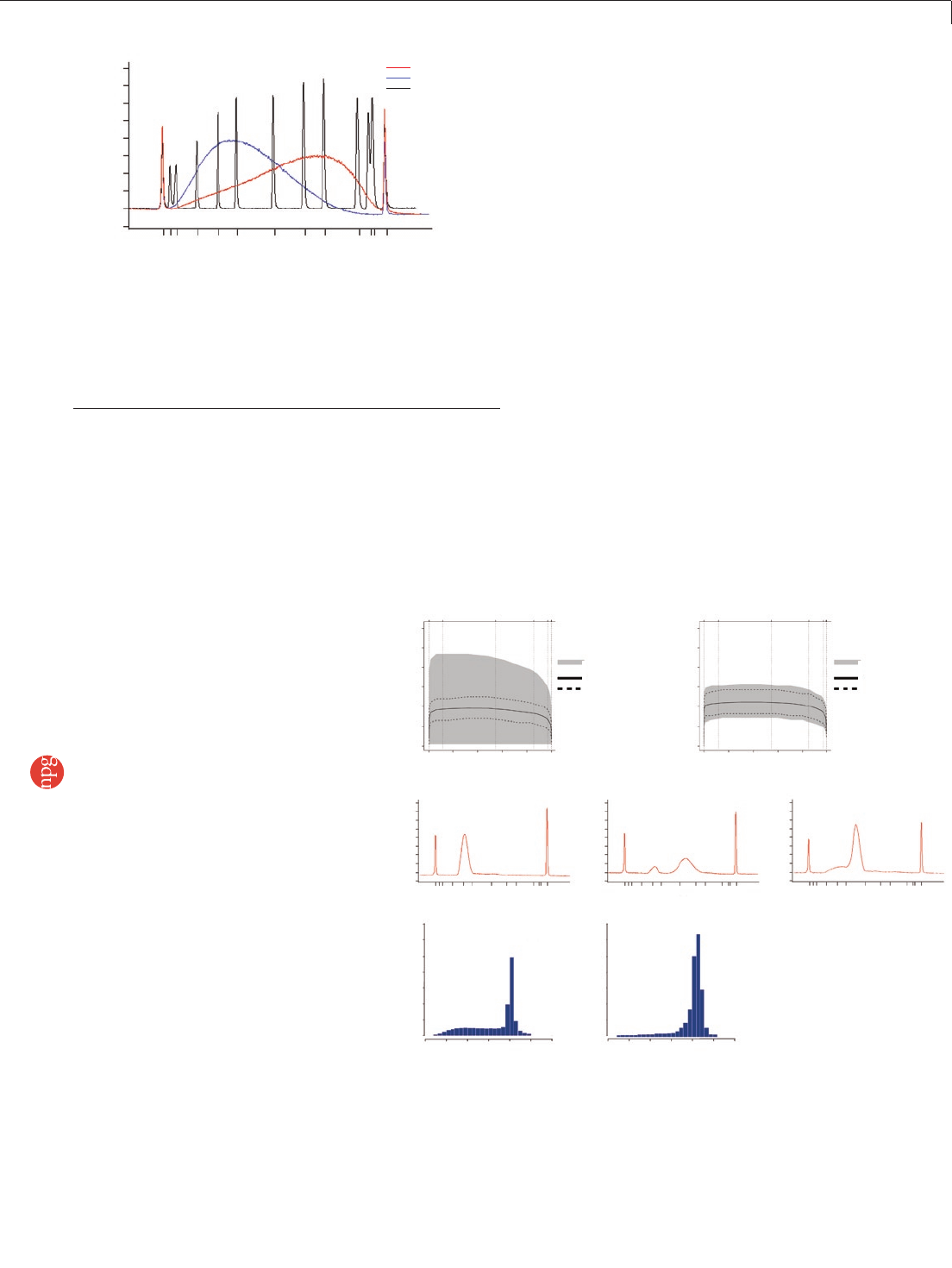
(Supplementary Protocol 3 online and Fig. 3a,b).
Double size selection. Partially complementary adapters, which
essentially consist of the sequences to which the sequence primers
hybridize during the sequencing reaction, are ligated onto the A-tailed
fragments7 via a T overhang (Fig. 1). Their structure ensures that each
template strand receives different sequences at the opposite ends8 and
works in much the same way as a vectorette9.
Inefficient end-repair or A-tailing reactions will result in a lower
concentration of template to which adapters can be successfully ligat-
ed, and so the relative concentration of adapters is increased, which
will promote the formation of adapter dimers. If these dimers are not
removed, they will ultimately be sequenced along with the intended
template, wasting the capacity of the flowcell. Additionally, inefficient
A-tailing will result in a high proportion of blunt-ended template
molecules, which can self-ligate, generating chimeric sequences.
It is likely that the efficiency of the A-tailing step will be improved
by the use of alternative polymerases and higher concentrations of
magnesium ions. Additionally, the efficiency of the ligation step
appears to be improved by the use of ultrapure ligases, such as
those from Enzymatics, which are virtually free of the contaminat-
ing exonuclease activity generally found in standard commercial
Adaptive focused acoustics. We now routinely fragment DNA sam-
ples using adaptive focused acoustics technology in a 24-well format
(AFA; Covaris). In this process, acoustic energy is controllably focused
into the aqueous DNA sample by a dish-shaped transducer, resulting
in cavitation events within the sample. The collapse of bubbles in the
suspension creates multiple, intense, localized jets of water, which dis-
rupt the DNA molecules in a reproducible and predictable way.
After disruption, 200-bp fragments comprise 17% of the total
fractionated DNA by mass, but in contrast to nebulization, very
little DNA is lost during the fragmentation process, generating a
four- to fivefold higher yield of the intended fragment size range
(Supplementary Protocol 1 online and Fig. 2). Additionally, because
of its high-throughput capability, AFA has enabled highly multiplexed
sequencing using indexing tags, where each sample needs to be pro-
cessed separately until after PCR amplification (Fig. 1). Also, because
the size distribution of DNA fragmented by AFA is narrow, for some
applications, such as array enrichment of targeted loci5,6, we are able
to omit the gel electrophoresis–based size selection step altogether
from the library preparation, decreasing the workload and increasing
yields further.
A-tailing, ligation and size selection
After close scrutiny of paired-end reads obtained from the Genome
Analyzer, that is, those in which each cluster was sequenced in both
forward and reverse directions, we discovered a number of artifacts
that could be attributed to the standard library preparation pro-
Genomic DNA PCR-amplified
library
Fragmented DNA
sample (0–1200 bp)
Blunt-ended, 5'-
phosphorylated fragments
3' A-tailed
fragments
Adapter-ligated
fragments
Size-selected
fragment library
PCR-amplified
library
Fragmentation
End repair
A-tailing
Ligation
Size selection
PCR
1
2
3,4
4,5
6,7,8,9,10
Single-stranded
DNA library
Clonal DNA clusters
on flowcell surface
Single-stranded clusters
ready for sequencing
Incorporation of fluorescent
reversible terminator nucleotides
Cleavage of fluorophores
and blocking groups
12
13
Denaturation
Linearization, blocking and hybridization
Flowcell transfer to Genome Analyzer
Imaging
Cycles of cluster amplification
Quantification
11
Library preparation
Cluster growth
Sequencing by synthesis
Figure 1 | Illumina sequencing workflow. Stages in the library preparation. Steps accompanied by
numbers are those for which we suggest alternatives to the standard Illumina protocols. Numbers
correspond to those given in Supplementary Protocols 1–13 online.
1006 | VOL.5 NO.12 | DECEMBER 2008 | NATURE METHODS
PERSPECTIVE
©2008 Nature Publishing Group http://www.nature.com/naturemethods
Paired-end size selection. Using standard protocols, we found single-
end library preparations, using single- or paired-end adapters, to be
considerably more robust at the step of size selection by electropho-
resis than their paired-end counterparts. With single-end preps, we
excise a band of 50 bp or larger, which generally yields more than
enough DNA to give a high yield of PCR products. However, for the
paired-end protocol, to generate as narrow an insert size range as
possible, a scalpel is inserted into the gel at the desired position, the
blade is washed with Tris buffer, and this buffer acts as the template
for the PCR amplification. We found this practice to yield enough
DNA to give successful amplification only in approximately 30–40%
of attempts. To overcome this, we now excise a 2-mm-wide gel slice
containing DNA of the desired size and extract that following the
Illumina protocol, though with no heating during the melting step,
as discussed above (Supplementary Protocol 3). In our hands this
typically yields 10–20 times more DNA than the standard protocol,
has an almost 100% success rate and generates an acceptably narrow
size distribution of paired-end reads (Fig. 3f,g).
PCR
As well as increasing robustness, extracting more DNA from gel slices
enables the DNA to be quantified more accurately before PCR ampli-
fication. The PCR step introduces into the adapter-ligated template
molecules the oligonucleotide sequences required for hybridization
to the flowcell surface.
preparations of ligases. Using this enzyme we have achieved a
20–30% increase in yield of successfully ligated fragments (as deter-
mined by quantitative PCR (qPCR); Supplementary Protocol 4
online), presumably because the reduced exonuclease activity
regenerates fewer blunt-ended fragments after A-tailing.
However, although the steps described
above may reduce the formation of blunt-
ended ligation concatamers, enzymatic
reactions are rarely 100% efficient, and thus
a small proportion of template strands will
still be chimeric. In many applications, a low
frequency of chimeric sequences will pres-
ent no problem and can simply be removed
informatically. In other applications, such as
detection of infrequent de novo recombinant
molecules, chimeric sequences will generate
false positives, so their frequency needs to be
reduced to minimize the amount of subse-
quent confirmatory work.
Chimeric templates will be longer than the
singletons, which provides a way of prevent-
ing them from contaminating the DNA frac-
tion: for applications in which a low frequency
of chimeric templates is required, we perform
an additional size selection after shearing but
before ligation (Supplementary Protocol 5
online). This results in a narrow size range
being available for ligation. Any blunt-ended
concatemers are appreciably longer than the
singletons, and we remove these in the post-
ligation size-selection step. This additional
size selection reduces the incidence of chime-
ras to 0.02%, compared to up to 5% with the
standard library preparation protocol, and we
have found this step to have the added benefit
of reducing the shoulder of small insert sizes,
giving a tighter insert-size distribution of the
desired fraction (Fig. 3c–e), which leads to
clusters with more uniform diameter.
140
120
100
80
60
40
20
0
–20
Relative fluorescence units
15 100 150 300 400 500 700 1,500
Size (bp)
Nebulization
AFA
Ladder
140
120
100
80
60
40
20
0
–20
Relative fluorescence units
15 100150 300 400 500 700 1,500
Size (bp)
60
50
40
30
20
10
0
Mapped depth (bin size, 500 bp)
0 20 40 60 80 100
Percentile of unique sequence
ordered by G+C content
10 30 40 50 60
G+C content (%)
Distribution of reads with
indicated G+C content
Mean read depth
S.d.
60
50
40
30
20
10
0
Mapped depth (bin size, 500 bp)
0 20 40 60 80 100
Percentile of unique sequence
ordered by G+C content
10 30 40 50 60
G+C content (%)
Distribution of reads with
indicated G+C content
Mean read depth
S.d.
140
120
100
80
60
40
20
0
–20
Relative fluorescence units
15 100150 300 400 500 700 1,500
Size (bp)
140
120
100
80
60
40
20
0
–20
Relative fluorescence units
15 100150 300 400 500 700 1,500
Size (bp)
3.5
3.0
2.5
2.0
1.5
1.0
0.5
0.0
Number of reads (millions)
0 100 200 300 400 500 600
Size (bp)
3.5
3.0
2.5
2.0
1.5
1.0
0.5
0.0
Number of reads (millions)
0 100 200 300 400 500 600
Size (bp)
ab
cde
fg
Figure 2 | Sample fragmentation. Comparison of fragmentation by
nebulization with AFA technology. We fragmented 4.5 µg of human genomic
DNA by nebulization or AFA, purified the samples using a spin column,
eluted the DNA in 30 µl of 10 mM Tris pH 8.5, and ran 1 µl of each eluate on
an Agilent Bioanalyzer 2100 DNA 100 chip. For a 200-bp (±20 bp) library,
the yield produced by AFA was four- to fivefold greater than that produced
by nebulization.
Figure 3 | A-tailing, ligation and size selection. (a,b) G+C plots before (a) and after (b) optimization
of gel extraction. The figures show the total area in which reads with a particular G+C content are
distributed, with the mean and s.d. The greater width of the shaded area in plot a indicates a wider
dispersion of coverage for all values of G+C content for which sequences were obtained. (c–e) Agilent
Bioanalyzer 2100 traces for three libraries, a 60-bp insert library with optimized PCR (c), the same
60-bp library with excess DNA in PCR (d) and a 200bp insert library, showing shoulder of small
fragments (e). (f,g) Insert size distribution from sequenced human DNA using the standard (f) and
modified (g) paired-end library preparation protocols.
NATURE METHODS | VOL.5 NO.12 | DECEMBER 2008 | 1007
PERSPECTIVE
©2008 Nature Publishing Group http://www.nature.com/naturemethods
PCR yield. By the use of alternative high-fidelity polymerases in
a more optimized reaction, we have found it possible to increase
the yield of the enrichment PCR five- to tenfold (Supplementary
Protocol 7 online and Fig. 4a), which allows fewer cycles of amplifi-
cation to be performed.
PCR cleanup. Surplus PCR primers may interfere with quantification
and will compete with the amplicon for hybridization to the flowcell
surface. Consequently, it is necessary to remove surplus oligos after
amplification. We have found that solid-phase reversible immobiliza-
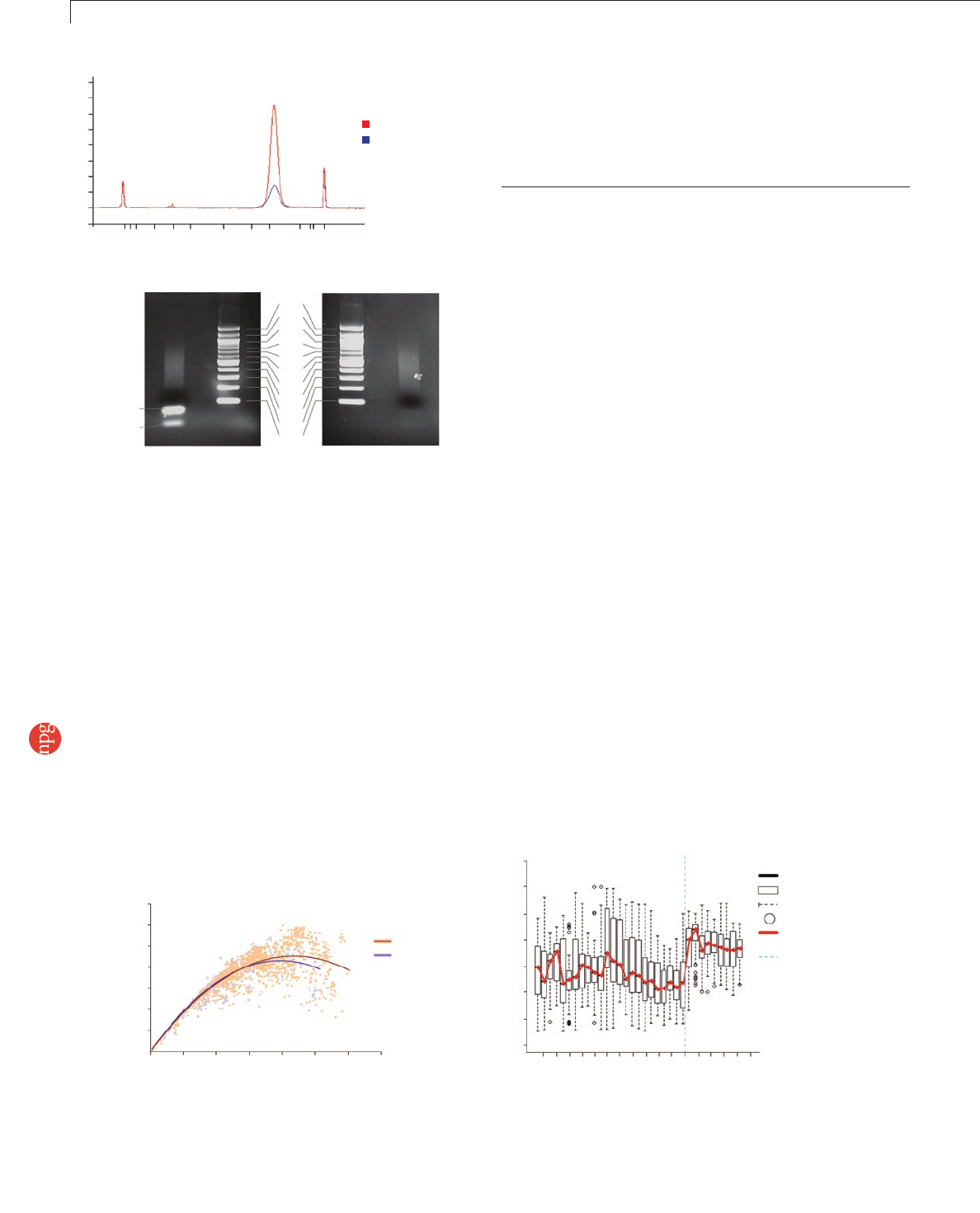
tion (SPRI) technology10 can be used to remove a higher proportion
of primers and adapter dimers than spin columns, while producing
a comparable yield of amplicon DNA, and allows elution in a wider
variety of buffers (Supplementary Protocol 8 online and Fig. 4b).
Sequencing without PCR. We have found that it is unnecessary to
retain the PCR step to enrich for properly ligated fragments, so long
as only those fragments with an adapter at either end can be quanti-
fied, as only they will yield clusters that can be sequenced. This can
be done by quantitative PCR, discussed below. Thus we can elimi-
nate the PCR step entirely, simply by ligating on appropriate adapters
after A-tailing (Supplementary Protocol 9 online). For this purpose
we use high-performance liquid chromatography–purified, partially
noncomplementary oligos with a phosphorothioate linkage between
the two bases at the 3′ end of one strand. From a starting amount
of 5 µg of DNA and fractionation by AFA, we can obtain sufficient
paired-end DNA for > 400 lanes of high-density clusters, or 100 lanes
if nebulization is used to fragment the DNA. The obvious benefits
of this are that PCR duplicates are absent: the observed duplication
Template quantity. By using optimized quantities of template in the
PCR, we can ensure a clean library, free of adapter-dimer or single-
stranded DNA. We routinely analyze our sequencing libraries after
PCR by microfluidic capillary electrophoresis and have noticed that
the quality of the library obtained decreases with increasing concen-
tration of template DNA: too much template DNA often results in
the accumulation of an apparently higher-molecular-weight peak
(Fig. 3d), which may represent a single-stranded template product
that accumulates as primers become depleted. Conversely, the lower
the mass of DNA used in the PCR, the fewer the number of template
strands for the same fragment size, and the greater the incidence of
PCR product duplicates in the resulting sequences: we have observed
libraries from which as many as 60% of sequences were PCR product
duplicates. Thus it is essential to choose the appropriate set of condi-
tions for each PCR (Supplementary Protocol 6 online).
Adapter dimers
PCR primers
1,400
1,000
1,000
900
800
700
600
500
400
300
200
Column
cleanup Ladder
Band
size (bp) Ladder
SPRI
cleanup
280
240
200
160
120
80
40
0
15 100 150 300 400 500 700 1,500
Size (bp)
Optimal PCR conditions
Standard PCR conditions
a
b
Figure 4 | PCR. (a) An ~200-bp fragment library was prepared, and 10 ng
was amplified for 18 cycles using standard Illumina PCR conditions or
optimized PCR conditions. (b) A comparison of methods of PCR amplicon
purification. We prepared a paired-end library with phiX DNA using conditions
that would promote the formation of adapter dimers and unextended PCR
primers. After PCR, we divided the library into two: half was purified following
the standard Illumina protocol, through a QiaQuick PCR cleanup column (left),
whereas the other was purified using SPRI technology (right).
35,000
30,000
25,000
20,000
15,000
10,000
5,000
0
Number of clusers after purity filtering
0 10,000 20,000 30,000 40,000 50,000 60,000 70,000
Total number of clusters detected
500-bp fragments
200-bp fragments
a
70,000
50,000
30,000
10,000
0
Unfiltered cluster number per tile
75 150 250 350 450 550 650 750 850
Run number
Median cluster number
Upper and lower quartiles
1.5 interquartile range
Outlier
Fluctuation in median cluste
r
number from bin to bin
qPCR assay introduced
b
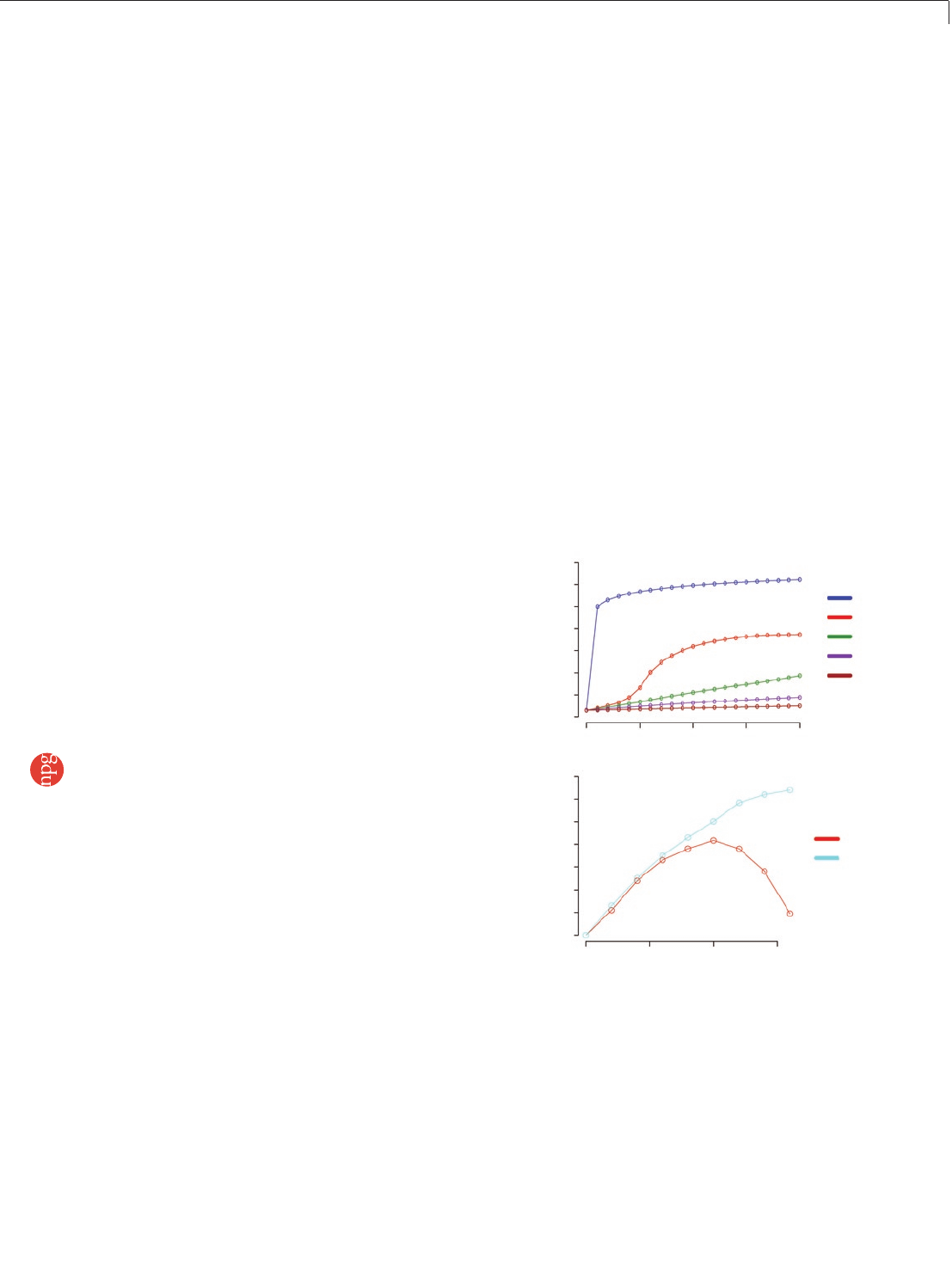
Figure 5 | Quantification. (a) Cluster throughput as a function of total clusters for 200- and 500-bp inserts. The 500-bp inserts underwent fewer cycles of
cluster amplification (28, compared to 35 for the 200-bp libraries), resulting in smaller clusters, and so a cluster density of 40–44k per tile (GA1) will produce
the maximum yield from either insert size. (b) Standardization of cluster density with qPCR quantification. Runs were grouped into 25-run bins, and a boxplot
was generated. After some initial problems with degradation of standards, cluster number has leveled out at ~35,000–40,000 per tile.
1008 | VOL.5 NO.12 | DECEMBER 2008 | NATURE METHODS
PERSPECTIVE
©2008 Nature Publishing Group http://www.nature.com/naturemethods
library to 10 pM, based on the concentration as measured using
an Agilent Bioanalyzer 2100. With this assay, we take the Agilent-
derived concentration values to be arbitrary, allowing us to dilute
unknowns to a concentration that lies within the 1–100 pM range,
but Agilent values also provide a useful double-check. We have found
that cluster density can be predicted reliably in this way (Fig. 5b).
We have found that the ability to quantify DNA in the picomolar
concentration range also opens up the opportunity for sequencing
much lower DNA concentrations than those permitted by the stan-
dard protocol, such as unamplified array eluates from a sequence-
capture experiment5,6.
Denaturation
Being single-stranded, array eluates require no particular steps to
denature the DNA12 before sequencing. However, for low concen-
trations (<1 nM) of double-stranded DNA it is more problematic:
denaturation by heating has the potential both to damage the DNA
and to introduce anti-(G+C) bias13.
Modified hybridization buffers. For all denaturation we pre-
fer the use of 0.1 M NaOH to heating, though for subnanomolar
libraries this requires an alternative hybridization buffer to be used
(Supplementary Protocol 12 online). We have found the addi-
rate in mapped gorilla DNA sequences prepared without PCR was
approximately 0.5%. This rate of duplication is caused by noise in the
cluster detection and sequence analysis software.
Direct sequencing of short amplicons. To avoid unnecessary PCR
amplification steps, which would potentially exacerbate biases, we can
perform extremely deep sequencing of short amplicons using locus-
specific primers that possess tails that can hybridize to the oligos teth-
ered to the flowcell surface. The tailless forward and reverse oligos
are then used as primers in the sequencing steps (Supplementary
Protocol 10 online).
Quantification
At close to neutral pH, the concentration of DNA going onto the
flowcell governs the number of clusters produced. Thus, for differ-
ent fragment sizes undergoing a given number of cycles of cluster
amplification, there is an optimal concentration range of DNA that
will yield clusters in the optimal density range, enabling the maxi-
mum amount of data to be obtained. For fragments with a mean
insert size of 500 bp or lower, we aim for 40,000–44,000 clusters per
imaged area (tile) on the Genome Analyzer model 1, giving an aver-
age of 20,000–25,000 filtered clusters per tile, equating to 2.0–2.4
Gb per single end run (150–170,000 clusters per tile for the Genome
Analyzer model 2; Fig. 5a).
Overestimation of DNA concentration results in too few clus-
ters, which may make the flowcell uneconomical to sequence.
Underestimation results in too high a cluster density, which can
greatly reduce the amount of data obtained, owing to cluster overlap.
Quantification of DNA before sequencing is thus one of the key fac-
tors in the process.
Electrophoresis. We found the accuracy of spectrophotometry to be
inadequate for quantification: cluster density based on this method
tended to be inconsistent, but typically five- to tenfold lower than
anticipated, presumably because spectrophotometry analysis mea-
sures not only the intended amplicon but also adapter dimers and
unextended primers, with no way of distinguishing between them,
and also struggles to measure low DNA concentrations accurately. By
quantifying libraries electrophoretically, with an Agilent Bioanalyzer,
we have been able to achieve a much more consistent cluster den-
sity. Additionally, because electrophoresis can be used to distinguish
between DNA species on the basis of size, it provides a way to check
the quality of the library preparation. However, for a small proportion
of libraries, we obtained far higher cluster densities, and consequently
far less useful data, than anticipated. We assume that this is a result
of single-stranded DNA generated in the PCR that cannot be easily
quantified when mixed with double-stranded DNA.
Quantitative PCR. This led us to develop a qPCR quantification
assay (for discussion see ref. 11) because such an approach should
be capable of detecting and quantifying all amplifiable molecules.
We designed amplification primers and a dual-labeled probe to
target the Illumina paired-end adapter sequences (Supplementary
Protocol 11 online). We quantify unknown libraries against stan-
dard libraries that have been sequenced previously, and for which
we know the accurate cluster number, and how this relates to the
Agilent concentration of that library. Because amplification in the
qPCR with these primers is not perfectly efficient, we use 3 dilutions
of standard libraries (100, 10 and 1 pM), and dilute the unknown
14
13
12
11
10
9
8
7
pH
0 5 10 15 20
Volume 0.1 M NaOH added (µl)
30,000
20,000
10,000
0
Cluster/tile
0 5 10 15
Volume denatured template added (µl)
Water
5× SSC
5× SSC + 2 mM Tr is
5× SSC + 10 mM Tr is
5× SSC + 50 mM Tr is
5× SSC
5× SSC + 5 mM Tr is
a
b
Figure 6 | Denaturation. (a) pH titration of hybridization buffers. Following
denaturation, the concentration of NaOH in DNA templates is 0.1 M
NaOH. Adding more than 8 µl of this denatured template to the 1 ml of
Hybridization Buffer (5× SSC, 0.1% Tween-20), before loading DNA onto the
flowcell, increases the pH to above 10. This prevents efficient hybridization,
and thus the cluster density falls. The addition of Tris-HCl pH 7.3 to the
supplied bottles of Hybridization Buffer dramatically increases buffering
capacity, making template hybridization more robust. (b) The addition
of 5 mM Tris-HCl pH 7.3 to Illumina Hybridization Buffer allows a greater
volume of denatured template to be added before high pH prevents effective
annealing of templates to the oligos on the flowcell surface. This increases
the robustness of cluster generation by counteracting pipetting errors in the
denaturation step.
NATURE METHODS | VOL.5 NO.12 | DECEMBER 2008 | 1009
PERSPECTIVE
©2008 Nature Publishing Group http://www.nature.com/naturemethods

software and a faster run time, both of which we are currently testing,
a conservative prediction is that by the end of 2008, our output will
reach 6–10 terabases of high-quality sequence per year, equivalent to
180 human genomes at 15-fold coverage, or approximately 200,000
bases per second.
The improved workflow and high yield should maintain the
Genome Analyzer as our next-generation sequencing platform of
choice for the immediate future. How long this remains true depends
upon the performance of existing rival technologies: Roche’s 454,
ABI’s SOLiD, Helicos’ ‘True Single Molecule Sequencing’ and Dover
Systems’ Polonator, and those that are on the horizon, such as nano-
pore technologies, for example Oxford Nanopore Technologies, the
Harvard Nanopore Group, and Pacific Biosciences’ Single Molecule
Real Time technology, which promise to bring us closer to the eagerly
anticipated $1,000 genome.
Note: Supplementary information is available on the Nature Methods website.
ACKNOWLEDGMENTS
We thank all the staff at Illumina for their support, particularly T. Ost, M. Gibbs, J.
Smith, N. Gormley, V. Smith and K. Hall. We also thank C. Brown, A. Brown,
R. Pettett, T. Skelly, N. Whiteford, L. Mamanova, E. Sheridan and E. Huckle for helpful
discussions and assistance.
COMPETING INTERESTS STATEMENT
The authors declare competing financial interests: details accompany the full-text
HTML version of the paper at http://www.nature.com/naturemethods/.
Published online at http://www.nature.com/naturemethods/
Reprints and permissions information is available online at http://npg.
nature.com/reprintsandpermissions/
1. Bentley, D.R. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 16, 545–552
(2006).
2. Mardis, E.R. The impact of next-generation sequencing technology on genetics.
Trends Genet. 24, 133–141 (2008).
3. Smith, D.R. et al. Rapid whole-genome mutational profiling using next-
generation sequencing technologies. Genome Res. 18, 1638–1642 (2008).
4. Surzycki, S. DNA sequencing. in Basic Techniques in Molecular Biology 377–380
(Springer-Verlag, Berlin, 2000).
5. Albert, T.J. et al. Direct selection of human genomic loci by microarray
hybridization. Nat. Methods 4, 903–905 (2007).
6. Hodges, E. et al. Genome-wide in situ exon capture for selective resequencing.
Nat. Genet. 39, 1522–1527 (2007).
7. Sambrook, J., Fritsch, E. & Maniatis, T. Molecular Cloning: A Laboratory Manual,
(Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1989).
8. Smith, D. & Malek, J. Asymmetrical adapters and uses thereof. US patent
0172839 (2007).
9. Riley, J. et al. A novel, rapid method for the isolation of terminal sequences
from yeast artificial chromosome (YAC) clones. Nucleic Acids Res. 18, 2887–2890
(1990).
10. Hawkins, T.L., O’Connor-Morin, T., Roy, A. & Santillan, C. DNA purification and
isolation using a solid-phase. Nucleic Acids Res. 22, 4543–4544 (1994).
11. Meyer, M. et al. From micrograms to picograms: quantitative PCR reduces the
material demands of high-throughput sequencing. Nucleic Acids Res. 36, e5
(2008).
12. Thomas, R. The denaturation of DNA. Gene 135, 77–79 (1993).
13. Mandel, M. & Marmur, J. Use of ultraviolet absorbance-temperature profile for
determining the guanine plus cytosine content of DNA. Methods Enzymol. 12,
195–206 (1968).
tion of Tris to the standard Illumina Hybridization Buffer to be
beneficial to the robustness of the initial hybridization of DNA
to the flowcell for all libraries, because it can counteract pipetting
errors during the denaturation stage that would otherwise raise
the pH to a level that would prevent efficient hybridization (Fig. 6).
Additionally, diluting the supplied 2 M NaOH and adding a greater
volume to the 20 µl denaturation reaction helps to reduce fluctua-
tion in cluster number owing to pipetting errors (Supplementary
Protocol 12).
Amplification quality control
After cluster amplification, DNA on the flowcell is double-stranded,
which allows clusters to be stained by an intercalating dye and to be
detected using a fluorescence microscope (Supplementary Protocol
13 online). This is a valuable quality control step, that we use for all
flowcells before linearization and blocking to confirm that the clus-
ter density is appropriate. We generally do not sequence data from
flowcells that have too high or too low a cluster density beyond the
amplification stage.
Conclusion
The Genome Analyzer is a powerful sequencing technology, yet still
relatively new, and consequently it has not yet reached its full sequenc-
ing potential. Here we have described modifications that allow for
more efficient library preparation and enable a stable workflow in a
production environment.
At the Sanger Institute, in addition to a sequencing research and
development team we have several teams who are responsible for
keeping the production instruments running. A library-making
group processes samples, and generates, quality-controls and quan-
tifies libraries. A production group, working in shifts, prepares and
quality-controls flowcells by SybrGreen staining, prepares reagents for
sequencing and manages washing, priming and loading the instru-
ments seven days per week. Informatics teams are responsible for
facilitating sample tracking, for handling the sequence data and for
performing pipeline analyses. All steps in the process are recorded
using custom-written lab-tracking and run-tracking database soft-
ware. All Genome Analyzers are networked, and the generated image
data are continually uploaded to a large compute and disk-storage
cluster for image and base-calling analysis, alignment and assembly,
and other informatics tasks. We keep images for about 1 month on a
disk server, but we store the run quality control and other run details
in a database and deposit short-read sequences for permanent stor-
age in a large repository. A team of project managers coordinate and
oversee individual sequencing projects.
We have recently upgraded all of our Genome Analyzers to the
model 2. The wider flowcells used by upgraded machines offer a 40%
greater imaging area, with the potential for increased read lengths
(>70 bases) of a higher quality (below 1% error in a phiX control lane
for 1–50 bases). Combined with improvements to the image analysis
1010 | VOL.5 NO.12 | DECEMBER 2008 | NATURE METHODS
PERSPECTIVE
©2008 Nature Publishing Group http://www.nature.com/naturemethods
