Wannier90 User Guide
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 166 [warning: Documents this large are best viewed by clicking the View PDF Link!]
wannier90: User Guide
Version 3.0
28th February 2019
Contents
I Introduction 5
II wannier90.x 11
1 Methodology 13
2 Parameters 17
3 Projections 51
4 Code Overview 59
5wannier90 as a post-processing tool 61
6wannier90 as a library 71
7 Transport Calculations with wannier90 77
8 Files 81
9 Some notes on the interpolation 101
10 Sample Input Files 103
III postw90.x 107
11 Parameters 109
12 Overview of the berry module 141
13 Overview of the gyrotropic module 145
3

Part I
Introduction
5
Introduction
Getting Help
The latest release of wannier90 and documentation can always be found at http://www.wannier.org.
The development version may be cloned/downloaded from the official repository of the wannier90 code
on GitHub (see https://github.com/wannier-developers/wannier90).
There is a wannier90 mailing list for discussing issues in the development, theory, coding and algorithms
pertinent to MLWF. You can register for this mailing list by following the links at http://www.
wannier.org/forum.html. Alternatively, for technical issues about the wannier90 code, check the
official repository of wannier90 on GitHub where you may raise issues or ask questions about about
its functionalities.
Finally, many frequently asked questions are answered in Appendix B. An expanded FAQ session may
be found on the Wiki page of the GitHub repository at https://github.com/wannier-developers/
wannier90/wiki/FAQ.
Citation
We ask that you acknowledge the use of wannier90 in any publications arising from the use of this
code through the following reference
[ref] A. A. Mostofi, J. R. Yates, G. Pizzi, Y.-S. Lee, I. Souza, D. Vanderbilt and N. Marzari,
An updated version of wannier90: A Tool for Obtaining Maximally-Localised Wannier
Functions, Comput. Phys. Commun. 185, 2309 (2014)
http://dx.doi.org/10.1016/j.cpc.2014.05.003
It would also be appropriate to cite the original articles:
Maximally localized generalized Wannier functions for composite energy bands,
N. Marzari and D. Vanderbilt, Phys. Rev. B 56, 12847 (1997)
Maximally localized Wannier functions for entangled energy bands,
I. Souza, N. Marzari and D. Vanderbilt, Phys. Rev. B 65, 035109 (2001)
7

8wannier90: User Guide
Credits
The Wannier90 Developer Group includes Giovanni Pizzi (EPFL, CH), Valerio Vitale (Cambridge,
GB), David Vanderbilt (Rutgers University, US), Nicola Marzari (EPFL, CH), Ivo Souza (Universi-
dad del Pais Vasco, ES), Arash A. Mostofi (Imperial College London, GB), and Jonathan R. Yates
(University of Oxford, GB).
The present release of wannier90 was written by the Wannier90 Developer Group together with Ry-
otaro Arita (Riken and U. Tokyo, JP), Stefan Blügel (FZ Jülich, DE), Frank Freimuth (FZ Jülich, DE),
Guillame Géranton (FZ Jülich, DE), Marco Gibertini (EPFL and University of Geneva, CH), Dominik
Gresch (ETHZ, CH), Charles Johnson (Imperial College London, GB), Takashi Koretsune (Tohoku
University and JST PRESTO, JP), Julen Ibañez-Azpiroz (Universidad del Pais Vasco, ES), Hyungjun
Lee (EPFL, CH), Daniel Marchand (EPFL, CH), Antimo Marrazzo (EPFL, CH), Yuriy Mokrousov (FZ
Jülich, DE), Jamal I. Mustafa (UC Berkeley, USA), Yoshiro Nohara (Tokyo, JP), Yusuke Nomura (U.
Tokyo, JP), Lorenzo Paulatto (Sorbonne Paris, FR), Samuel Poncé (Oxford University, GB), Thomas
Ponweiser (RISC Software GmbH, AT), Florian Thöle (ETHZ, CH), Stepan Tsirkin (Universidad del
Pais Vasco, ES), Małgorzata Wierzbowska (Polish Academy of Science, PL).
Contributors to the code include: Daniel Aberg (w90pov code), Lampros Andrinopoulos (w90vdw
code), Pablo Aguado Puente (gyrotropic routines), Raffaello Bianco (k-slice plotting), Marco Buon-
giorno Nardelli (dosqc v1.0 subroutines upon which transport.f90 is based), Stefano De Gironcoli
(pw2wannier90.x interface to Quantum ESPRESSO), Pablo Garcia Fernandez (matrix elements of the
position operator), Nicholas D. M. Hine (w90vdw code), Young-Su Lee (specialised Gamma point
routines and transport), Antoine Levitt (preconditioning), Graham Lopez (extension of pw2wannier90
to add terms needed for orbital magnetisation), Radu Miron (constrained centres), Nicolas Poilvert
(transport routines), Michel Posternak (original plotting routines), Rei Sakuma (Symmetry-adapted
Wannier functions), Gabriele Sclauzero (disentanglement in spheres in k-space), Matthew Shelley
(transport routines), Christian Stieger (routine to print the U matrices), David Strubbe (various bug-
fixes/improvements), Timo Thonhauser (extension of pw2wannier90 to add terms needed for orbital
magnetisation).
We also acknowledge individuals not already mentioned above who participated in the first Wannier90
community meeting (San Sebastian, 2016) for useful discussions: Daniel Fritsch, Victor Garcia Suarez,
Jan-Philipp Hanke, Ji Hoon Ryoo, Jürg Hutter, Javier Junquera, Liang Liang, Michael Obermeyer,
Gianluca Prandini, Paolo Umari.
wannier90 Version 2.x was written by: Arash A. Mostofi, Giovanni Pizzi, Ivo Souza, Jonathan R.
Yates. wannier90 Version 1.0 was written by: Arash A. Mostofi, Jonathan R. Yates, Young-Su Lee.
wannier90 is based on Fortran 77 codes written for isolated bands by Nicola Marzari and David
Vanderbilt and for entangled bands by Ivo Souza, Nicola Marzari, and David Vanderbilt.
wannier90 c
2007-2019 The Wannier Developer Group and individual contributors
Licence
All the material in this distribution is free software; you can redistribute it and/or modify it under
the terms of the GNU General Public License as published by the Free Software Foundation; either
version 2 of the License, or (at your option) any later version.
This program is distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY;

wannier90: User Guide 9
without even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR
PURPOSE. See the GNU General Public License for more details.
You should have received a copy of the GNU General Public License along with this program; if not,
write to the Free Software Foundation, Inc., 51 Franklin Street, Fifth Floor, Boston, MA 02110-1301,
USA.
Part II
wannier90.x
11

Chapter 1
Methodology
wannier90 computes maximally-localised Wannier functions (MLWF) following the method of Marzari
and Vanderbilt (MV) [1]. For entangled energy bands, the method of Souza, Marzari and Vanderbilt
(SMV) [2] is used. We introduce briefly the methods and key definitions here, but full details can be
found in the original papers and in Ref. [3].
First-principles codes typically solve the electronic structure of periodic materials in terms of Bloch
states, ψnk. These extended states are characterised by a band index nand crystal momentum k.
An alternative representation can be given in terms of spatially localised functions known as Wannier
functions (WF). The WF centred on a lattice site R,wnR(r), is written in terms of the set of Bloch
states as
wnR(r) = V
(2π)3ZBZ "X
m
U(k)
mnψmk(r)#e−ik.Rdk,(1.1)
where Vis the unit cell volume, the integral is over the Brillouin zone (BZ), and U(k)is a unitary
matrix that mixes the Bloch states at each k.U(k)is not uniquely defined and different choices will
lead to WF with varying spatial localisations. We define the spread Ωof the WF as
Ω = X
nhwn0(r)|r2|wn0(r)i − |hwn0(r)|r|wn0(r)i|2.(1.2)
The total spread can be decomposed into a gauge invariant term ΩIplus a term ˜
Ωthat is dependant
on the gauge choice U(k).˜
Ωcan be further divided into terms diagonal and off-diagonal in the WF
basis, ΩDand ΩOD,
Ω=ΩI+˜
Ω=ΩI+ ΩD+ ΩOD (1.3)
where
ΩI=X
n"hwn0(r)|r2|wn0(r)i − X
Rm|hwnR(r)|r|wn0(r)i|2#(1.4)
ΩD=X
nX
R6=0|hwnR(r)|r|wn0(r)i|2(1.5)
ΩOD =X
m6=nX
R|hwmR(r)|r|wn0(r)i|2(1.6)
The MV method minimises the gauge dependent spread ˜
Ωwith respect the set of U(k)to obtain
MLWF.
wannier90 requires two ingredients from an initial electronic structure calculation.
13

14 wannier90: User Guide
1. The overlaps between the cell periodic part of the Bloch states |unki
M(k,b)
mn =humk|unk+bi,(1.7)
where the vectors b, which connect a given k-point with its neighbours, are determined by
wannier90 according to the prescription outlined in Ref. [1].
2. As a starting guess the projection of the Bloch states |ψnkionto trial localised orbitals |gni
A(k)
mn =hψmk|gni,(1.8)
Note that M(k,b),A(k)and U(k)are all small, N×Nmatrices1that are independent of the basis set
used to obtain the original Bloch states.
To date, wannier90 has been used in combination with electronic codes based on plane-waves and
pseudopotentials (norm-conserving and ultrasoft [4]) as well as mixed basis set techniques such as
FLAPW [5].
1.1 Entangled Energy Bands
The above description is sufficient to obtain MLWF for an isolated set of bands, such as the valence
states in an insulator. In order to obtain MLWF for entangled energy bands we use the “disentangle-
ment” procedure introduced in Ref. [2].
We define an energy window (the “outer window”). At a given k-point k,N(k)
win states lie within this
energy window. We obtain a set of NBloch states by performing a unitary transformation amongst
the Bloch states which fall within the energy window at each k-point:
|uopt
nki=X
m∈N(k)
win
Udis(k)
mn |umki(1.9)
where Udis(k)is a rectangular N(k)
win ×Nmatrix2. The set of Udis(k)are obtained by minimising the
gauge invariant spread ΩIwithin the outer energy window. The MV procedure can then be used to
minimise ˜
Ωand hence obtain MLWF for this optimal subspace.
It should be noted that the energy bands of this optimal subspace may not correspond to any of the
original energy bands (due to mixing between states). In order to preserve exactly the properties of a
system in a given energy range (e.g., around the Fermi level) we introduce a second energy window.
States lying within this inner, or “frozen”, energy window are included unchanged in the optimal
subspace.
1Technically, this is true for the case of an isolated group of Nbands from which we obtain NMLWF. When using
the disentanglement procedure of Ref. [2], A(k), for example, is a rectangular matrix. See Section 1.1.
2As Udis(k)is a rectangular matrix this is a unitary operation in the sense that (Udis(k))†Udis(k)=1N.

wannier90: User Guide 15
se
Chapter 2
Parameters
2.1 Usage
wannier90.x can be run in parallel using MPI libraries to reduce the computation time.
For serial execution use: wannier90.x [-pp] [seedname]
•seedname: If a seedname string is given the code will read its input from a file seedname.win.
The default value is wannier. One can also equivalently provide the string seedname.win instead
of seedname.
•-pp: This optional flag tells the code to generate a list of the required overlaps and then exit.
This information is written to the file seedname.nnkp.
For parallel execution use: mpirun -np NUMPROCS wannier90.x [-pp] [seedname]
•NUMPROCS: substitute with the number of processors that you want to use.
Note that the mpirun command and command-line flags may be different in your MPI implementation:
read your MPI manual or ask your computer administrator.
Note also that this requires that the wannier90.x executable has been compiled in its parallel version
(follow the instructions in the file README.install in the main directory of the wannier90 distribution)
and that the MPI libraries and binaries are installed and correctly configured on your machine.
2.2 seedname.win File
The wannier90 input file seedname.win has a flexible free-form structure.
The ordering of the keywords is not significant. Case is ignored (so num_bands is the same as
Num_Bands). Characters after !, or # are treated as comments. Most keywords have a default value
that is used unless the keyword is given in seedname.win. Keywords can be set in any of the following
ways
17


wannier90: User Guide 19
Keyword Type Description
System Parameters
num_wann I Number of WF
num_bands I Number of bands passed to the code
unit_cell_cart P Unit cell vectors in Cartesian coor-
dinates
atoms_cart * P Positions of atoms in Cartesian co-
ordinates
atoms_frac * R Positions of atoms in fractional co-
ordinates with respect to the lattice
vectors
mp_grid I Dimensions of the Monkhorst-Pack
grid of k-points
kpoints R List of k-points in the Monkhorst-
Pack grid
gamma_only L Wavefunctions from underlying ab
initio calculation are manifestly real
spinors L WF are spinors
shell_list I Which shells to use in finite differ-
ence formula
search_shells I The number of shells to search when
determining finite difference formula
skip_B1_tests L Check the condition B1 of Ref. [1]
nnkpts I Explicit list of nearest-neighbour k-
points.
kmesh_tol R The tolerance to control if two
kpoint belong to the same shell
Table 2.1: seedname.win file keywords defining the system. Argument types are represented by, I for
a integer, R for a real number, P for a physical value, L for a logical value and S for a text string.
*atoms_cart and atoms_frac may not both be defined in the same input file.

20 wannier90: User Guide
Keyword Type Description
Job Control
postproc_setup L To output the seedname.nnkp file
exclude_bands I List of bands to exclude from the
calculation
select_projections I List of projections to use in Wan-
nierisation
restart S Restart from checkpoint file
iprint I Output verbosity level
length_unit S System of units to output lengths
wvfn_formatted L Read the wavefunctions from a
(un)formatted file
spin S Which spin channel to read
devel_flag S Flag for development use
timing_level I Determines amount of timing infor-
mation written to output
optimisation I Optimisation level
translate_home_cell L To translate final Wannier centres to
home unit cell when writing xyz file
write_xyz L To write atomic positions and final
centres in xyz file format
write_vdw_data L To write data for futher processing
by w90vdw utility
write_hr_diag L To write the diagonal elements of
the Hamiltonian in the Wannier ba-
sis to seedname.wout (in eV)
Table 2.2: seedname.win file keywords defining job control. Argument types are represented by, I for
a integer, R for a real number, P for a physical value, L for a logical value and S for a text string.
translate_home_cell only relevant if write_xyz is .true.
Keyword Type Description
Plot Parameters
wannier_plot L Plot the WF
wannier_plot_list I List of WF to plot
wannier_plot_supercell I Size of the supercell for plotting the
WF
wannier_plot_format S File format in which to plot the WF
wannier_plot_mode S Mode in which to plot the WF,
molecule or crystal
wannier_plot_radius R Cut-off radius of WF*
wannier_plot_scale R Scaling parameter for cube files
wannier_plot_spinor_mode S Quantity to plot for spinor WF
wannier_plot_spinor_phase L Include the “phase” when plotting
spinor WF

wannier90: User Guide 21
bands_plot L Plot interpolated band structure
kpoint_path P K-point path for the interpolated
band structure
bands_num_points I Number of points along the first sec-
tion of the k-point path
bands_plot_format S File format in which to plot the in-
terpolated bands
bands_plot_project I WF to project the band structure
onto
bands_plot_mode S Slater-Koster type interpolation or
Hamiltonian cut-off
bands_plot_dim I Dimension of the system
fermi_surface_plot L Plot the Fermi surface
fermi_surface_num_points I Number of points in the Fermi sur-
face plot
fermi_energy P The Fermi energy
fermi_energy_min P Lower limit of the Fermi energy
range
fermi_energy_max P Upper limit of the Fermi energy
range
fermi_energy_step R Step for increasing the Fermi energy
in the specified range
fermi_surface_plot_format S File format for the Fermi surface
plot
hr_plot LThis parameter is not used anymore.
Use write_hr instead.
write_hr LWrite the Hamiltonian in the WF
basis
write_rmn LWrite the position operator in the
WF basis
write_bvec LWrite to file the matrix elements of
the bvectors and their weights
write_tb LWrite lattice vectors, Hamiltonian,
and position operator in WF basis
hr_cutoff P Cut-off for the absolute value of the
Hamiltonian
dist_cutoff P Cut-off for the distance between WF
dist_cutoff_mode S Dimension in which the distance be-
tween WF is calculated
translation_centre_frac R Centre of the unit cell to which final
WF are translated
use_ws_distance LImprove interpolation using mini-
mum distance between WFs, see
Chap. 9
ws_distance_tol RAbsolute tolerance for the distance
to equivalent positions.
22 wannier90: User Guide
ws_search_size IMaximum extension in each direc-
tion of the super-cell of the Born-von
Karmann cell to search for points in-
side the Wigner-Seitz cell
write_u_matrices LWrite U(k)and Udis(k)matrices to
files
Table 2.5: seedname.win file keywords controlling the plot-
ting. Argument types are represented by, I for a integer,
R for a real number, P for a physical value, L for a logical
value and S for a text string. * Only applies when wan-
nier_plot_format is cube.
wannier90: User Guide 23
Keyword Type Description
Disentanglement Parameters
dis_win_min P Bottom of the outer energy window
dis_win_max P Top of the outer energy window
dis_froz_min P Bottom of the inner (frozen) energy
window
dis_froz_max P Top of the inner (frozen) energy win-
dow
dis_num_iter I Number of iterations for the minimi-
sation of ΩI
dis_mix_ratio R Mixing ratio during the minimisa-
tion of ΩI
dis_conv_tol R The convergence tolerance for find-
ing ΩI
dis_conv_window I The number of iterations over which
convergence of ΩIis assessed.
dis_spheres_num I Number of spheres in k-space where
disentaglement is performed
dis_spheres_first_wann I Index of the first band to be consid-
ered a Wannier function
dis_spheres R List of centres and radii, for disen-
tanglement only in spheres
Table 2.3: seedname.win file keywords controlling the disentanglement. Argument types are repre-
sented by, I for a integer, R for a real number, P for a physical value, L for a logical value and S for a
text string.

24 wannier90: User Guide
Keyword Type Description
Wannierise Parameters
num_iter I Number of iterations for the minimi-
sation of Ω
num_cg_steps I During the minimisation of Ωthe
number of Conjugate Gradient steps
before resetting to Steepest Descents
conv_window I The number of iterations over which
convergence of Ωis assessed
conv_tol P The convergence tolerance for find-
ing Ω
precond L Use preconditioning
conv_noise_amp R The amplitude of random noise ap-
plied towards end of minimisation
procedure
conv_noise_num I The number of times random noise
is applied
num_dump_cycles I Control frequency of check-pointing
num_print_cycles I Control frequency of printing
write_r2mn L Write matrix elements of r2between
WF to file
guiding_centres L Use guiding centres
num_guide_cycles I Frequency of guiding centres
num_no_guide_iter I The number of iterations after which
guiding centres are used
trial_step * R The trial step length for the
parabolic line search during the min-
imisation of Ω
fixed_step * R The fixed step length to take dur-
ing the minimisation of Ω, instead
of doing a parabolic line search
use_bloch_phases ** L To use phases for initial projections
site_symmetry*** L To construct symmetry-adapted
Wannier functions
symmetrize_eps*** R The convergence tolerance used in
the symmetry-adapted mode
scdm_proj L Whether to use the SCDM-k
method of Ref. [6]
scdm_entanglement I The entanglement method to use in
SCDM-k
scdm_mu P The value of the parameter µin
SCDM-k
scdm_sigma P The value of the parameter σin
SCDM-k
slwf_num I The number of objective WFs for se-
lective localization
slwf_constrain L Whether to constrain the centres of
the objective WFs
slwf_lambda R Value of the Lagrange multiplier for
constraining the objective WFs
slwf_centres P The centres to which the objective
WFs are to be constrained
Table 2.4: seedname.win file keywords controlling the wannierisation. Argument types are represented
by, I for a integer, R for a real number, P for a physical value, L for a logical value and S for a text
string. *fixed_step and trial_step may not both be defined in the same input file. **Cannot be used in
conjunction with disentanglement. ***Cannot be used in conjunction with the inner (frozen) energy window.

wannier90: User Guide 25
Keyword Type Description
Transport Parameters
transport L Calculate quantum conductance and
density of states
transport_mode S Bulk or left-lead_conductor_right-
lead calculation
tran_win_min P Bottom of the energy window for
transport calculation
tran_win_max P Top of the energy window for trans-
port calculation
tran_energy_step R Sampling interval of the energy val-
ues
fermi_energy R The Fermi energy
tran_num_bb I Size of a bulk Hamiltonian
tran_num_ll I Size of a left-lead Hamiltonian
tran_num_rr I Size of a right-lead Hamiltonian
tran_num_cc I Size of a conductor Hamiltonian
tran_num_lc I Number of columns in a left-
lead_conductor Hamiltonian
tran_num_cr I Number of rows in a
conductor_right-lead Hamilto-
nian
tran_num_cell_ll I Number of unit cells in PL of left
lead
tran_num_cell_rr I Number of unit cells in PL of right
lead
tran_num_bandc I Half-bandwidth+1 of a band-
diagonal conductor Hamiltonian
tran_write_ht L Write the Hamiltonian for transport
calculation
tran_read_ht L Read the Hamiltonian for transport
calculation
tran_use_same_lead L Left and right leads are the same
tran_group_threshold R Distance that determines the group-
ing of WFs
hr_cutoff P Cut-off for the absolute value of the
Hamiltonian
dist_cutoff P Cut-off for the distance between WF
dist_cutoff_mode S Dimension in which the distance be-
tween WF is calculated
one_dim_axis S Extended direction for a one-
dimensional system
translation_centre_frac R Centre of the unit cell to which final
WF are translated
Table 2.6: seedname.win file keywords controlling transport. Argument types are represented by, I for
a integer, R for a real number, P for a physical value, L for a logical value and S for a text string.

26 wannier90: User Guide
2.4 System
2.4.1 integer :: num_wann
Number of WF to be found.
No default.
2.4.2 integer :: num_bands
Total number of bands passed to the code in the seedname.mmn file.
Default num_bands=num_wann
2.4.3 Cell Lattice Vectors
The cell lattice vectors should be specified in Cartesian coordinates.
begin unit_cell_cart
[units]
A1xA1yA1z
A2xA2yA2z
A3xA3yA3z
end unit_cell_cart
Here A1xis the x-component of the first lattice vector A1,A2yis the y-component of the second lattice
vector A2, etc.
[units] specifies the units in which the lattice vectors are defined: either Bohr or Ang.
The default value is Ang.
2.4.4 Ionic Positions
The ionic positions may be specified in fractional coordinates relative to the lattice vectors of the unit
cell, or in absolute Cartesian coordinates. Only one of atoms_cart and atoms_frac may be given in
the input file.
Cartesian coordinates
begin atoms_cart
[units]
P RP
xRP
yRP
z
Q RQ
xRQ
yRQ
z
.
.
.
end atoms_cart

wannier90: User Guide 27
The first entry on a line is the atomic symbol. The next three entries are the atom’s position R=
(Rx, Ry, Rz)in Cartesian coordinates. The first line of the block, [units], specifies the units in which
the coordinates are given and can be either bohr or ang. If not present, the default is ang.
Fractional coordinates
begin atoms_frac
P F P
1FP
2FP
3
Q F Q
1FQ
2FQ
3
.
.
.
end atoms_frac
The first entry on a line is the atomic symbol. The next three entries are the atom’s position in
fractional coordinates F=F1A1+F2A2+F3A3relative to the cell lattice vectors Ai,i∈[1,3].
2.4.5 integer, dimension :: mp_grid(3)
Dimensions of the regular (Monkhorst-Pack) k-point mesh. For example, for a 2×2×2grid:
mp_grid : 2 2 2
No default.
2.4.6 K-points
Each line gives the coordinate K=K1B1+K2B2+K3B3of a k-point in relative (crystallographic)
units, i.e., in fractional units with respect to the primitive reciprocal lattice vectors Bi,i∈[1,3]. The
position of each k-point in this list assigns its numbering; the first k-point is k-point 1, the second is
k-point 2, and so on.
begin kpoints
K1
1K1
2K1
3
K2
1K2
2K2
3
.
.
.
end kpoints
There is no default.
Note: There is an utility provided with wannier90, called kmesh.pl, which helps to generate the
explicit list of kpoints required by wannier90. See Sec. A.1.
2.4.7 logical :: gamma_only
If gamma_only=true, then wannier90 uses a branch of algorithms for disentanglement and localisation
that exploit the fact that the Bloch eigenstates obtained from the underlying ab initio calculation are
manifestly real. This can be the case when only the Γ-point is used to sample the Brillouin zone. The
localisation procedure that is used in the Γ-only branch is based on the method of Ref. [7].

28 wannier90: User Guide
The default value is false.
2.4.8 logical :: spinors
If spinors=true, then wannier90 assumes that the WF correspond to singularly occupied spinor states
and num_elec_per_state=1.
The default value is false.
2.4.9 Shells
The MV scheme requires a finite difference expression for ∇kdefined on a uniform Monkhorst-Pack
mesh of k-points. The vectors {b}connect each mesh-point kto its nearest neighbours. Nsh shells of
neighbours are included in the finite-difference formula, with Msvectors in the sth shell. For ∇kto be
correct to linear order, we require that the following equation is satisfied (Eq. B1 of Ref. [1]):
Nsh
X
s
ws
Ms
X
i
bi,s
αbi,s
β=δαβ ,(2.1)
where bi,s,i∈[1, Ms], is the ith vector belonging to the sth shell with associated weight ws, and α
and βrun over the three Cartesian indices.
2.4.10 integer :: shell_list(:)
shell_list is vector listing the shells to include in the finite difference expression. If this keyword is
absent, the shells are chosen automatically.
2.4.11 integer :: search_shells
Specifies the number of shells of neighbours over which to search in attempting to determine an
automatic solution to the B1 condition Eq. 2.1. Larger values than the default may be required in
special cases e.g. for very long thin unit cells.
The default value is 36.
2.4.12 logical :: skip_B1_tests
If set to .true., does not check the B1 condition Eq. 2.1. This should only be used if one knows why
the B1 condition should not be verified. A typical use of this flag is in conjunction with the Z2PACK
code: http://www.physics.rutgers.edu/z2pack/.
The default value is .false..
2.4.13 integer, dimension(:, 5) :: nnkpts
Specifies the nearest-neighbour k-points which are written to the .nnkp file. This can be used to
explicitly specify which overlap matrices should be calculated.

wannier90: User Guide 29
begin nnkpts
1 2 0 0 0
.
.
end nnkpts
Each nearest neighbour k+bis given by a line of 5 integers. The first specifies the k-point number
nkp of k. The second is the k-point number of the neighbour. The final three integers specify the
reciprocal lattice vector which brings the k-point specified by the second integer to k+b.
This format is the same as in the .nnkp file, except that the number of neighbours per k-point is not
specified. However, the number of neighbours still needs to be a multiple of the number of k-points.
This input parameter can be used only if postproc_setup = .true., and is not intended to be used
with a full Wannier90 run. It can be used also if the k-points do not describe a regular mesh.
2.4.14 real(kind=dp) :: kmesh_tol
Two kpoints belong to the same shell if the distance between them is less than kmesh_tol. Units are
Ang.
The default value is 0.000001 Ang.
2.5 Projection
The projections block defines a set of localised functions used to generate an initial guess for the
unitary transformations. The projection block can be specified in conjunction with scdm_proj=true
(see below). This is only used to read the centres of the projections, which in some cases could help
the optimisation if guiding_centres=true is added to the input file. This data will be written in the
seedname.nnkp file to be used by a first-principles code.
begin projections
.
.
end projections
If guiding_centres=true, then the projection centres are used as the guiding centres in the Wan-
nierisation routine.
For details see Section 3.1.
2.6 Job Control
2.6.1 logical :: postproc_setup
If postproc_setup=true, then the wannier code will write seedname.nnkp file and exit. If wannier90
is called with the option -pp, then postproc_setup is set to true, over-riding its value in the
seedname.win file.

30 wannier90: User Guide
The default value is false.
2.6.2 integer :: iprint
This indicates the level of verbosity of the output from 0 (“low”), the bare minimum, to 3 (“high”),
which corresponds to full debugging output.
The default value is 1.
2.6.3 integer :: optimisation
This indicates the level of optimisation used in the code. This is a trade between speed and memory. A
positive number indicates fastest execution time at the cost of more memory. Zero or negative numbers
indicates a smaller memory footprint - at increased execution time.
At the moment the only values that have an effect are optimisation<=0 (low memory) and optimisation>0
(fast)
The default value is 3.
2.6.4 character(len=20) :: length_unit
The length unit to be used for writing quantities in the output file seedname.wout.
The valid options for this parameter are:
–Ang (default)
–Bohr
2.6.5 character(len=50) :: devel_flag
Not a regular keyword. Its purpose is to allow a developer to pass a string into the code to be used
inside a new routine as it is developed.
No default.
2.6.6 integer :: exclude_bands(:)
A k-point independent list of states to excluded from the calculation of the overlap matrices; for example
to select only valence states, or ignore semi-core states. This keyword is passed to the first-principles
code via the seedname.nnkp file. For example, to exclude bands 2, 6, 7, 8 and 12:
exclude_bands : 2, 6-8, 12
2.6.7 integer :: select_projections(:)
A list of projections to be included in the wannierisation procedure. In the case that num_proj is
greater than num_wann, this keyword allows a subset of the projections in the projection matrices to

wannier90: User Guide 31
be used. For example, to select the projections given by the indices 2, 6, 7, 8 and 12:
select_projections : 2, 6-8, 12
2.6.8 character(len=20) :: restart
If restart is present the code will attempt to restart the calculation from the seedname.chk file.
The value of the parameter determines the position of the restart
The valid options for this parameter are:
–default. Restart from the point at which the check file seedname.chk was written
–wannierise. Restart from the beginning of the wannierise routine
–plot. Go directly to the plotting phase
–transport. Go directly to the transport routines
2.6.9 character(len=20) :: wvfn_formatted
If wvfn_formatted=true, then the wavefunctions will be read from disk as formatted (ie ASCII) files;
otherwise they will be read as unformatted files. Unformatted is generally preferable as the files will
take less disk space and I/O is significantly faster. However such files will not be transferable between
all machine architectures and formatted files should be used if transferability is required (i.e., for test
cases).
The default value of this parameter is false.
2.6.10 character(len=20) :: spin
For bands from a spin polarised calculation spin determines which set of bands to read in, either up
or down.
The default value of this parameter is up.
2.6.11 integer :: timing_level
Determines the amount of timing information regarding the calculation that will be written to the
output file. A value of 1 produces the least information.
The default value is 1.
2.6.12 logical :: translate_home_cell
Determines whether to translate the final Wannier centres to the home unit cell at the end of the
calculation. Mainly useful for molecular systems in which the molecule resides entirely within the
home unit cell and user wants to write an xyz file (write_xyz=.true.) for the WF centres to compare
with the structure.

32 wannier90: User Guide
The default value is false.
2.6.13 logical :: write_xyz
Determines whether to write the atomic positions and final Wannier centres to an xyz file, seedname_centres.xyz,
for subsequent visualisation.
The default value is false.
2.6.14 logical :: write_vdw_data
Determines whether to write seedname.vdw for subsequent post-processing by the w90vdw utility (in
the utility/w90vdw/ directory of the distribution) for calculating van der Waals energies. Brillouin
zone sampling must be at the Gamma-point only.
The default value is false.
2.7 Disentanglement
These keywords control the disentanglement routine of Ref. [2], i.e., the iterative minimisation of ΩI.
This routine will be activated if num_wann <num_bands.
2.7.1 real(kind=dp) :: dis_win_min
The lower bound of the outer energy window for the disentanglement procedure. Units are eV.
The default is the lowest eigenvalue in the system.
2.7.2 real(kind=dp) :: dis_win_max
The upper bound of the outer energy window for the disentanglement procedure. Units are eV.
The default is the highest eigenvalue in the given states (i.e., all states are included in the disentan-
glement procedure).
2.7.3 real(kind=dp) :: dis_froz_min
The lower bound of the inner energy window for the disentanglement procedure. Units are eV.
If dis_froz_max is given, then the default for dis_froz_min is dis_win_min.
2.7.4 real(kind=dp) :: dis_froz_max
The upper bound of the inner (frozen) energy window for the disentanglement procedure. If dis_froz_max
is not specified, then there are no frozen states. Units are eV.
No default.

wannier90: User Guide 33
2.7.5 integer :: dis_num_iter
In the disentanglement procedure, the number of iterations used to extract the most connected sub-
space.
The default value is 200.
2.7.6 real(kind=dp) :: dis_mix_ratio
In the disentanglement procedure, the mixing parameter to use for convergence (see pages 4-5 of
Ref. [2]). A value of 0.5 is a ‘safe’ choice. Using 1.0 (i.e., no mixing) often gives faster convergence,
but may cause the minimisation of ΩIto be unstable in some cases.
Restriction: 0.0<dis_mix_ratio ≤1.0
The default value is 0.5
2.7.7 real(kind=dp) :: dis_conv_tol
In the disentanglement procedure, the minimisation of ΩIis said to be converged if the fractional
change in the gauge-invariant spread between successive iterations is less than dis_conv_tol for
dis_conv_window iterations. Units are Å2.
The default value is 1.0E-10
2.7.8 integer :: dis_conv_window
In the disentanglement procedure, the minimisation is said to be converged if the fractional change in
the spread between successive iterations is less than dis_conv_tol for dis_conv_window iterations.
The default value of this parameter is 3.
2.7.9 integer :: dis_spheres_num
Number of spheres in reciprocal space where the k-dependent disentanglement is performed. No dis-
entanglement is performed for those k-points that are not included in any of the spheres.
The default is 0, which means disentangle at every k-point in the full BZ (the standard mode in
Wannier90).
2.7.10 integer :: dis_spheres_first_wann
Index of the first band that has to be considered as a Wannier function. Used only if dis_spheres_num
is greater than zero. At k-points where disentanglement is not performed the bands from dis_spheres_first_wann
to dis_spheres_first_wann+num_wann are used to wannierise. The bands excluded using exclude_bands
should not be counted.
The default is 1, the band at the lowest energy.

34 wannier90: User Guide
2.7.11 dis_spheres
Each line gives the coordinate K=K1B1+K2B2+K3B3of a k-point representing the center of one of
the spheres used for k-dependent disentanglement. The same crystallographic units as for kpoints are
used here. Each k-point coordinate Kimust the followed by the respectice sphere radius riin inverse
angstrom (on the same line).
The number of lines must be equal to dis_spheres_num.
begin dis_spheres
K1
1K1
2K1
3r1
K2
1K2
2K2
3r2
.
.
.
end dis_spheres
There is no default.
2.8 Wannierise
Iterative minimisation of e
Ω, the non-gauge-invariant part of the spread functional.
2.8.1 integer :: num_iter
Total number of iterations in the minimisation procedure. Set num_iter=0 if you wish to generate
projected WFs rather than maximally-localized WFs (see Example 8 in the Tutorial).
The default value is 100
2.8.2 integer :: num_cg_steps
Number of conjugate gradient steps to take before resetting to steepest descents.
The default value is 5
2.8.3 integer :: conv_window
If conv_window>1, then the minimisation is said to be converged if the change in Ωover conv_window
successive iterations is less than conv_tol. Otherwise, the minimisation proceeds for num_iter itera-
tions (default).
The default value is -1
2.8.4 real(kind=dp) :: conv_tol
If conv_window >1, then this is the convergence tolerance on Ω, otherwise not used. Units are Å2.
The default value is 1.0E-10

wannier90: User Guide 35
2.8.5 logical :: precond
Whether or not to use preconditioning to speed up the minimization of the spreads. This is based on
the same idea as the classical Tetter-Payne-Allan preconditionning for DFT and dampens the high-
frequency oscillations of the gradient due to contributions from large real lattice vectors. It is useful
when the optimization is slow, especially on fine grids. When optimisation<3, this uses a slower
algorithm to save memory.
The default value is false.
2.8.6 real(kind=dp) :: conv_noise_amp
If conv_noise_amp>0, once convergence (as defined above) is achieved, some random noise fis added
to the search direction, and the minimisation is continued until convergence is achieved once more. If
the same value of Ωas before is arrived at, then the calculation is considered to be converged. If not,
then random noise is added again and the procedure repeated up to a maximum of conv_noise_num
times. conv_noise_amp is the amplitude of the random noise fthat is added to the search direction:
0<|f|<conv_noise_amp. This functionality requires conv_window >1. If conv_window is not
specified, it is set to the value 5 by default.
If conv_noise_amp ≤0, then no noise is added (default).
The default value is -1.0
2.8.7 integer :: conv_noise_num
If conv_noise_amp >0, then this is the number of times in the minimisation that random noise is
added.
The default value is 3
2.8.8 integer :: num_dump_cycles
Write sufficient information to do a restart every num_dump_cycles iterations.
The default is 100
2.8.9 integer :: num_print_cycles
Write data to the master output file seedname.wout every num_print_cycles iterations.
The default is 1
2.8.10 logical :: write_r2mn
If write_r2mn =true, then the matrix elements hm|r2|ni(where mand nrefer to WF) are written
to file seedname.r2mn at the end of the Wannierisation procedure.
The default value of this parameter is false.

36 wannier90: User Guide
2.8.11 logical :: guiding_centres
Use guiding centres during the minimisation, in order to avoid local minima.
wannier90 uses a logarithm definition of the spread functional. As we are taking the log of a complex
argument there is a possibility that the algorithm might make inconsistent choices for the branch cut.
This manifests itself as complex WF with a large spread. By using guiding centres the code will attempt
to make a consistent choice of branch cut. Experience shows that with guiding_centres set to true
this problem is avoided and doing so does not cause any problems. For this reason we recommend
setting guiding_centres to true where possible (it is only not possible if an explicit projection block
is not defined).
The default value is false.
2.8.12 integer :: num_guide_cycles
If guiding_centres is set to true, then the guiding centres are used only every num_guide_cycles.
The default value is 1.
2.8.13 integer :: num_no_guide_iter
If guiding_centres is set to true, then the guiding centres are used only after num_no_guide_iter
minimisation iterations have been completed.
The default value is 0.
2.8.14 real(kind=dp) :: trial_step
The value of the trial step for the parabolic fit in the line search minimisation used in the minimisation of
the spread function. Cannot be used in conjunction with fixed_step (see below). If the minimisation
procedure doesn’t converge, try decreasing the value of trial_step to give a more accurate line search.
The default value is 2.0
2.8.15 real(kind=dp) :: fixed_step
If this is given a value in the input file, then a fixed step of length fixed_step (instead of a parabolic
line search) is used at each iteration of the spread function minimisation. Cannot be used in conjunction
with trial_step. This can be useful in cases in which minimisation with a line search fails to converge.
There is no default value.
2.8.16 logical :: use_bloch_phases
Determines whether to use the Bloch functions as the initial guess for the projections. Can only be
used if disentanglement = false.
The default value is false.

wannier90: User Guide 37
2.8.17 logical :: site_symmetry
Construct symmetry-adapted Wannier functions. For the detail of the theoretical background, see
Ref. [8]. Cannot be used in conjunction with the inner (frozen) energy window.
The default value is false.
2.8.18 real(kind=dp) :: symmetrize_eps
Convergence threshold to check whether the symmetry condition (Eq. (19) in Ref. [8]) on the unitary
matrix U(k)is satisfied or not. See also Eq. (29) in Ref. [8]. Used when site_symmetry = .true.
The default value is 1.0E-3.
2.8.19 logical :: scdm_proj
If scdm_proj=true then the A(k)
mn matrices are generated with the SCDM-k method of Ref. [6]. In this
case, one also needs to specify the scdm_entanglement keyword. One then needs to run wannier90.x
-pp seedname to generate the seedname.nnkp file, to be used by a first-principle code (at the moment
only interface available is with the QuantumEspresso code).
The default value is false.
2.8.20 integer :: scdm_entanglement
Select the functional form for the occupation number matrix f(nk)for the SCDM-k method. Only
three integer values are allowed:
isolated:f(nk)is the identity matrix Ink
erfc: The occupation number matrix is given by:
f(nk) = 1
2ERFC nk−µ
σ
gaussian: The occupation number matrix is given by
f(nk) = EXP −(nk−µ)2
σ2
The default value is isolated.
2.8.21 real(kind=dp) :: scdm_mu
The value of the µparameter in the formulas above. It is strictly required only when scdm_entanglement=erfc
or gaussian. It defines the characteristic energy for the occupation numbers matrix, in units of
eV. If scdm_entanglement=erfc, it gives the mean value of the complementary error function. If
scdm_entanglement=gaussian, it gives the mean value of the gaussian instead.
The default value is 0 eV.

38 wannier90: User Guide
2.8.22 real(kind=dp) :: scdm_sigma
The value of the σparameter in the formulas for the occupation numbers matrix. It is strictly required
only when scdm_entanglement=erfc or gaussian. It defines the spread of the occupation numbers
matrix around µ, and as such it must be a positive real number. It must be given in units of eV.
The default value is 1.0 eV.
2.8.23 integer :: slwf_num
The number of objective Wannier functions for selective localisation in the selectively localised Wan-
nier function (SLWF) method of Ref. [9]. These functions are obtained by minimising the spread
functional only with respect to the degrees of freedom of a subset of slwf_num ≤num_wann functions.
At convergence, the objective WFs will have a minimum cumulative spread, whereas the remaining
num_wann −slwf_num functions are left unoptimised. The initial guesses for the objective WFs are
given by the first slwf_num orbitals in the projections block.
The default is num_wann.
2.8.24 logical :: slwf_constrain
If slwf_constrain=true, then the centres of the objective Wannier functions are constrained to either
the centres of the first slwf_num orbitals in the projections block or to new positions specified in the
slwf_centres block (see Sec. 2.8.26). In this case, a modified spread functional, Ωc, with the addition
of a constraint term, as described in Ref. [9].
The default is false
2.8.25 real(kind=dp) :: slwf_lambda
The value of the Lagrange multiplier λfor the constraint term in term added to modify the spread
functional: λPJ0
n=1 (rn−r0n)2, where J0is slwf_num, and rnand r0nare the centre and target centre,
respectively, for the nth objective WF.
The default is 0.0.
2.8.26 Constraints on centres
If slwf_constrain=true, then by default the centres to which the slwf_num objective Wannier function
centres are constrained are given by the first slwf_num rows of the projections block.
Optionally, the slwf_centres block may be used to define alternative target centres for some or all of
the slwf_num objective Wannier functions.
The block below shows an example of how to set the constraints:
begin centre_constraints
2 0.0 0.0 0.0
4 0.25 0.0 0.0
end centre_constraints

wannier90: User Guide 39
•The first line sets the constraint for the centre of objective WF number 2 (as defined by the order
of WFs in the projections block) to (0.0,0.0,0.0) in fractional co-ordinates.
•The second line sets the constraint for the centre of objective WF number 4 (as defined by the
order of WFs in the projections block) to (0.25,0.0,0.0) in fractional co-ordinates.
•The target centres of all other objective Wannier functions remain as the centres given in the
corresponding rows of the projections block.
2.9 Post-Processing
Capabilities:
–Plot the WF
–Plot the interpolated band structure
–Plot the Fermi surface
–Output the Hamiltonian in the WF basis
–Transport calculation (quantum conductance and density of states)
2.9.1 logical :: wannier_plot
If wannier_plot =true, then the code will write out the Wannier functions in a format specified by
wannier_plot_format
The default value of this parameter is false.
2.9.2 integer :: wannier_plot_list(:)
A list of WF to plot. The WF numbered as per the seedname.wout file after the minimisation of the
spread.
The default behaviour is to plot all WF. For example, to plot WF 4, 5, 6 and 10:
wannier_plot_list : 4-6, 10
2.9.3 integer :: wannier_plot_supercell
The code generates the WFs on a grid corresponding to a ‘super-unit-cell’. If wannier_plot_supercell
is provided as a single integer, then the size of the super-unit-cell is wannier_plot_supercell times
the size of the unit cell along all three linear dimensions (the ‘home’ unit cell is kept approxi-
mately in the middle); otherwise, if three integers are provided, the size of the super-unit-cell is
wannier_plot_supercell(i) times the size of the unit cell along the i−th linear dimension.
The default value is 2.

40 wannier90: User Guide
2.9.4 character(len=20) :: wannier_plot_format
WF can be plotted in either XCrySDen (xsf) format or Gaussian cube format. The valid options for
this parameter are:
–xcrysden (default)
–cube
If wannier_plot_format=xsf: the code outputs the WF on the entire super-unit-cell specified by
wannier_plot_supercell.
If wannier_plot_format=cube: the code outputs the WF on a grid that is smaller than the super-
unit-cell specified by wannier_plot_supercell. This grid is determined by wannier_plot_mode,
wannier_plot_radius and wannier_plot_scale, described in detail below.
The code is able to output Gaussian cube files for systems with non-orthogonal lattice vectors. Many vi-
sualisation programs (including XCrySDen), however, are only able to handle cube files for systems with
orthogonal lattice vectors. One visualisation program that is capable of dealing with non-orthogonal
lattice vectors is VESTA (http://jp-minerals.org/vesta/en/).1
2.9.5 character(len=20) :: wannier_plot_mode
Choose the mode in which to plot the WF, either as a molecule or as a crystal.
The valid options for this parameter are:
–crystal (default)
–molecule
If wannier_plot_format=cube:
•if wannier_plot_mode = molecule, then wherever the WF centre sits in the supercell, the origin
of the cube is shifted (for the purpose of plotting only, ie, nothing is done to the U matrices etc)
to coincide with the centre of mass of the atomic positions specified by the user in the *.win
input file. These atomic positions are also written to the cube file, so when it is visualised, the
WF appears superimposed on the molecular structure.
•if wannier_plot_mode = crystal, then the WF is not shifted, but instead the code searches
for atoms that are within a radius of wannier_plot_scale ×wannier_plot_radius of the WF
centre and writes the coordinates of these atoms to the cube file. In this way, when the cube file
is visualised, the WF appears superimposed on the nearest atoms to the WF centre.
•crystal mode can be used for molecules, and molecule mode can be used for crystals.
1It’s worth noting that another visualisation program, VMD (http://www.ks.uiuc.edu/Research/vmd), is able to
deal with certain special cases of non-orthogonal lattice vectors; see http://www.ks.uiuc.edu/Research/vmd/plugins/
molfile/cubeplugin.html for details.

wannier90: User Guide 41
2.9.6 real(kind=dp) :: wannier_plot_radius
If wannier_plot_format=cube, then wannier_plot_radius is the radius of the sphere that must fit
inside the parallelepiped in which the WF is plotted. wannier_plot_radius must be greater than 0.
Units are Å.
The default value is 3.5.
2.9.7 real(kind=dp) :: wannier_plot_scale
If wannier_plot_format=cube and wannier_plot_mode=crystal, then the code searches for atoms
that are within a radius of wannier_plot_scale ×wannier_plot_radius of the WF centre and writes
the coordinates of these atoms to the cube file. In this way, when the cube file is visualised, the WF
appears superimposed on the nearest atoms to the WF centre. wannier_plot_scale must be greater
than 0. This parameter is dimensionless.
The default value is 1.0.
2.9.8 character(len=20) :: wannier_plot_spinor_mode
If spinors =true then this parameter controls the quantity to plot. For a spinor WF with components
[φ, ψ]the quatity plotted is
–total (default). p[|φ|2+|ψ|2
–up.|φ| × sign(Re{φ})if wannier_plot_spinor_mode =true, otherwise |φ|
–down.|ψ| × sign(Re{ψ})if wannier_plot_spinor_mode =true, otherwise |ψ|
Note: making a visual representation of a spinor WF is not as straightforward as for a scalar WF.
While a scalar WF is typically a real valued function, a spinor WF is a complex, two component spinor.
wannier90 is able to plot several different quantities derived from a spinor WF which should give you
a good idea of the nature of the WF.
2.9.9 logical :: wannier_plot_spinor_phase
If wannier_plot_spinor_phase =true phase information will be taken into account when plotting a
spinor WF.
2.9.10 logical :: bands_plot
If bands_plot =true, then the code will calculate the band structure, through Wannier interpolation,
along the path in k-space defined by bands_kpath using bands_num_points along the first section of
the path and write out an output file in a format specified by bands_plot_format.
The default value is false.

42 wannier90: User Guide
2.9.11 kpoint_path
Defines the path in k-space along which to calculate the bandstructure. Each line gives the start and
end point (with labels) for a section of the path. Values are in fractional coordinates with respect to
the primitive reciprocal lattice vectors.
begin kpoint_path
G0.0 0.0 0.0L0.0 0.0 1.0
L0.0 0.0 1.0N0.0 1.0 1.0
.
.
.
end kpoint_path
There is no default
2.9.12 integer :: bands_num_points
If bands_plot =true, then the number of points along the first section of the bandstructure plot
given by kpoint_path. Other sections will have the same density of k-points.
The default value for bands_num_points is 100.
2.9.13 character(len=20) :: bands_plot_format
Format in which to plot the interpolated band structure. The valid options for this parameter are:
–gnuplot (default)
–xmgrace
Note: it is possible to request output in both formats eg bands_format =gnuplot xmgrace
2.9.14 integer :: bands_plot_project(:)
If present wannier90 will compute the contribution of this set of WF to the states at each point of the
interpolated band structure. The WF are numbered according to the seedname.wout file. The result is
written in the seedname_band.dat file, and a corresponding gnuplot script to seedname_band_proj.dat
.
For example, to project on to WFs 2, 6, 7, 8 and 12:
bands_plot_project : 2, 6-8, 12
2.9.15 character(len=20) :: bands_plot_mode
To interpolate the band structure along the k-point path, either use the Slater-Koster interpolation
scheme or truncate the Hamiltonian matrix in the WF basis. Truncation criteria are provided by
hr_cutoff and dist_cutoff.
The valid options for this parameter are:

wannier90: User Guide 43
–s-k (default)
–cut
2.9.16 integer :: bands_plot_dim
Dimension of the system. If bands_plot_dim <3 and bands_plot_mode =cut, lattice vector R=
N1A1+N2A2+N3A3, where Ni= 0 if Aiis parallel to any of the confined directions specified by
one_dim_axis, are exclusively used in the band structure interpolation.
The valid options for this parameter are:
–3 (default)
–2
–1
2.9.17 logical :: fermi_surface_plot
If fermi_surface_plot =true, then the code will calculate, through Wannier interpolation, the
eigenvalues on a regular grid with fermi_surface_num_points in each direction. The code will write
a file in bxsf format which can be read by XCrySDen in order to plot the Fermi surface.
The default value is false.
2.9.18 integer :: fermi_surface_num_points
If fermi_surface_plot =true, then the number of divisions in the regular k-point grid used to
calculate the Fermi surface.
The default value for fermi_surface_num_points is 50.
2.9.19 real(kind=dp) :: fermi_energy
The Fermi energy in eV. This parameter is written into the bxsf file. If fermi_energy is specified,
fermi_energy_min,fermi_energy_max, and fermi_energy_step should not be specified, and vice-
versa.
The default value is 0.0
2.9.20 real(kind=dp) :: fermi_energy_min
Instead of specifyfing a single Fermi energy, it is possible to scan the Fermi level over a range of values,
and recompute certain quantities for each εF.2This is the minimum value in the range (in eV).
There is no default value.
2Scanning the Fermi level is currently supported only by the postw90 module berry, for berry_task=ahc,morb. For
all other functionalities that require a knowledge of εF, use fermi_energy instead.

44 wannier90: User Guide
2.9.21 real(kind=dp) :: fermi_energy_max
The maximum value in the range of Fermi energies. Units are eV.
The default value is fermi_energy_min+1.0.
2.9.22 real(kind=dp) :: fermi_energy_step
Difference between consecutive values of the Fermi energy when scanning from fermi_energy_min to
fermi_energy_max. Units are eV.
The default value is 0.01.
2.9.23 character(len=20) :: fermi_surface_plot_format
Format in which to plot the Fermi surface. The valid options for this parameter are:
–xcrysden (default)
2.9.24 logical :: write_hr
If write_hr =true, then the Hamiltonian matrix in the WF basis will be written to a file seedname_hr.dat.
The default value is false.
2.9.25 logical :: write_rmn
If write_rmn =true, then the position operator in the WF basis will be written to a file seedname_r.dat.
The default value is false.
2.9.26 logical :: write_bvec
If write_bvec =true, then the the matrix elements of bvector and their weights will be written to a
file seedname.bvec.
The default value is false.
2.9.27 logical :: write_tb
If write_tb =true, then the lattice vectors, together with the Hamiltonian and position-operator
matrices in the WF basis, will be written to a file seedname_tb.dat, in units of Angstrom and eV.
The default value is false.

wannier90: User Guide 45
2.9.28 logical :: transport
If transport =true, then the code will calculate quantum conductance and density of states of a
one-dimensional system. The results will be written to files seedname_qc.dat and seedname_dos.dat,
respectively. Since both quantities are a function of energy, they will be evaluated from tran_win_min
to tran_win_max with an interval of tran_energy_step.
The default value of this parameter is false.
2.9.29 character(len=20) :: transport_mode
If transport_mode =bulk, quantum conductance and density of states are calculated for a perfectly-
periodic one-dimensional system. In this case, the transport part can either use the Hamiltonian
matrix in the WF basis generated by wannier90 or a Hamiltonian matrix provided by the external file
seedname_htB.dat.
If transport_mode =lcr, quantum conductance and density of states are calculated for a system
where semi-infinite, left and right leads are connected through a central conductor region. In this
case, the transport part will work independently from the disentanglement and wannierise procedure.
Details of the method is described in Ref. [10].
If tran_read_ht =true then the Hamiltonian matrices must be provided by the five external files:
seedname_htL.dat, seedname_htLC.dat, seedname_htC.dat, seedname_htCR.dat, seedname_htR.dat.
If tran_read_ht =false then the Hamiltonian matrices are found automatically provided the super-
cell adheres to conditions outlined in Section 7.3.
The valid options for this parameter are:
–bulk (default)
–lcr
2.9.30 real(kind=dp) :: tran_win_min
The lower bound of the energy window for the transport calculation. Units are eV.
The default value is -3.0.
2.9.31 real(kind=dp) :: tran_win_max
The upper bound of the energy window for the transport calculation. Units are eV.
The default value is 3.0.
2.9.32 real(kind=dp) :: tran_energy_step
Sampling interval of the energy values from tran_win_min to tran_win_max. Units are eV.
The default value is 0.01.

46 wannier90: User Guide
2.9.33 real(kind=dp) :: fermi_energy
The Fermi energy in eV. The energy axis of the quantum conductance and density of states data will
be shifted rigidly by this amount.
The default value is 0.0
2.9.34 integer :: tran_num_bb
Size of a bulk Hamiltonian matrix. This number is equal to the number of WFs in one principal layer.
A one-dimensional system can be viewed as an array of principal layers which are defined in a way
that localized basis functions inside a certain principal layer only interact with those in the nearest
neighbor principal layer. In wannier90 a principal layer will be an integer multiple of a unit cell, and
the size is determined by hr_cutoff and/or dist_cutoff. The criterion is rather arbitrary when WFs
are adopted as a localized basis set, and it is up to a user’s choice.
The default value is 0.
2.9.35 integer :: tran_num_ll
Size of a left-lead Hamiltonian matrix. If transport_mode =lcr and tran_read_ht =false then
tran_num_ll is the number of Wannier functions in a principal layer.
The default value is 0.
2.9.36 integer :: tran_num_rr
Size of a right-lead Hamiltonian matrix.
The default value is 0.
2.9.37 integer :: tran_num_cc
Size of a conductor Hamiltonian matrix.
The default value is 0.
2.9.38 integer :: tran_num_lc
Number of columns in a left-lead_conductor Hamiltonian matrix. Number of rows must be equal to
tran_num_ll.
The default value is 0.

wannier90: User Guide 47
2.9.39 integer :: tran_num_cr
Number of rows in a conductor_right-lead Hamiltonian matrix. Number of columns must be equal to
tran_num_rr.
The default value is 0.
2.9.40 integer :: tran_num_cell_ll
Number of unit cells in one principal layer of left lead. Used if transport_mode =lcr and tran_read_ht =
false.
The default value is 0.
2.9.41 integer :: tran_num_cell_rr
Number of unit cells in one principal layer of right lead. Not used at present.
The default value is 0.
2.9.42 integer :: tran_num_bandc
Half-bandwidth+1 of a band-diagonal conductor Hamiltonian matrix.
The Hamiltonian matrix of a central conductor part, which is read from seedname_htC.dat, will
be diagonally dominant when tran_num_cc is very large. tran_num_bandc is used to construct a
compact matrix which contains the non-zero band-diagonal part of a full conductor Hamiltonian matrix.
Setting this parameter is only meaningful when tran_num_bandc is greater than tran_num_lc and
tran_num_cr.
The default value is 0.
2.9.43 logical :: tran_write_ht
If tran_write_ht =true, then the Hamiltonian matrix formatted for the transport calculation will
be written to a file seedname_htB.dat.
The default value is false.
2.9.44 logical :: tran_read_ht
If tran_write_ht =true, then the Hamiltonian matrix formatted for the transport calculation will
be read from a set of files described in the parameter transport_mode. Set tran_write_ht =false
to perform automated lcr calculations (see Section 7.3).
The default value is false.

48 wannier90: User Guide
2.9.45 logical :: tran_use_same_lead
If tran_use_same_lead =true, then the left and the right leads are the same. In this case, seedname_htR.dat
is not required.
The default value is true.
2.9.46 real(kind=dp) :: tran_group_threshold
Used to group and sort Wannier functions according to the positions of their centres. Wannier functions
in a group are within tran_group_threshold from one another in x,y and zdirections. Units are Å
The default is 0.15
2.9.47 real(kind=dp) :: translation_centre_frac(3)
Centre of the unit cell to which the final Wannier centres are translated. Numbers are in fractional
coordinates with respect to the lattice vectors.
The default value is (0.0,0.0,0.0).
2.9.48 logical :: use_ws_distance
Improves the interpolation of the k-space Hamiltonian, by applying a translation to each WF by a
basis vector of the super-lattice that minimises the distance between their centres. The translation
is dependent on both WF and on the unit cell vector to which they belong, i.e., translate function
Wj(r−R)inside the Wigner-Seitz cell centred on WF Wi(r).
For a longer explanation, see Chapter 9.
If false the code puts all the WF in the home cell, only possible choice until wannier90 v2.0.1.
The default value is true (default changed since v.3.0). Introduced in v2.1.
2.9.49 real(kind=dp) :: ws_distance_tol
Tolerance when determining whether two values kdijR+˜
Rnmlkand kdijR+˜
Rn0m0l0k(as defined in
chapter 9) for the shortest distance between two Wannier functions are equivalent. If the difference in
distance (in Angstrom) is less than ws_distance_tol, they are taken to be equivalent.
The default value is 10−5.
2.9.50 :: ws_search_size
Maximum absolute value for the integers n, m, l that identify the super-lattice vectors ˜
Rnml (see
chapter 9) when searching for points inside the Wigner-Seitz cell. If ws_search_size is provided as
a single integer, then the number of repetitions of the Born-von Karman cell is the same along all
three linear dimensions; otherwise, if three integers are provided, the number of repetitions along the
i−th linear dimension is ws_search_size(i). The variable is used both in hamiltonian.F90 and in

wannier90: User Guide 49
ws_distance.F90. In the latter case, its value is incremented by one in order to account for WFs
whose centre wanders away from the original reference unit cell.
The default value is generally sufficient, but might need to be increased in case of elongated cells.
The default value is 2.
2.9.51 logical :: write_u_matrices
Write the U(k)and Udis(k)matrices obtained at the end of wannierization to files seedname_u.mat
and seedname_u_dis.mat, respectively.
The default value is false.
2.9.52 real(kind=dp) :: hr_cutoff
The absolute value of the smallest matrix element of the Hamiltonian in the WF basis. If hmn(R)>
hr_cutoff, then the matrix element hmn(R)is retained and used in the band structure interpola-
tion (when bands_plot_mode =cut) or in the transport calculation. Otherwise it is deemed to be
insignificant and is discarded. Units are eV.
The default value is 0.0.
2.9.53 real(kind=dp) :: dist_cutoff
The largest distance between two WFs for which the Hamiltonian matrix element is retained and used
in the band interpolation (when bands_plot_mode =cut) or in the transport calculation. Units are
Å.
The default value is 1000.0.
2.9.54 character(len=20) :: dist_cutoff_mode
Dimension in which the distance between two WFs is calculated. The vector connecting two WFs may
be projected to a line (one_dim) or a plane (two_dim). The size of the projected vector is calculated,
and dist_cutoff is applied. When one_dim or two_dim is used, one_dim_axis must be given to
specify extended or confined direction.
The valid options for this parameter are:
–three_dim (default)
–two_dim
–one_dim
2.9.55 character(len=20) :: one_dim_axis
Extended direction for a one-dimensional system or confined direction for a two-dimensional system.
This direction must be parallel to one of the Cartesian axes.

50 wannier90: User Guide
The valid options for this parameter are:
–x
–y
–z
No default.
Chapter 3
Projections
3.1 Specification of projections in seedname.win
Here we describe the projection functions used to construct the initial guess A(k)
mn for the unitary
transformations.
Each projection is associated with a site and an angular momentum state defining the projection
function. Optionally, one may define, for each projection, the spatial orientation, the radial part, the
diffusivity, and the volume over which real-space overlaps Amn are calculated.
The code is able to
1. project onto s,p,d and f angular momentum states, plus the hybrids sp, sp2, sp3, sp3d, sp3d2.
2. control the radial part of the projection functions to allow higher angular momentum states, e.g.,
both 3s and 4s in silicon.
The atomic orbitals of the hydrogen atom provide a good basis to use for constructing the projec-
tion functions: analytical mathematical forms exist in terms of the good quantum numbers n,l
and m; hybrid orbitals (sp, sp2, sp3, sp3d etc.) can be constructed by simple linear combination
|φi=Pnlm Cnlm|nlmifor some coefficients Cnlm.
The angular functions that use as a basis for the projections are not the canonical spherical harmonics
Ylm of the hydrogenic Schrödinger equation but rather the real (in the sense of non-imaginary) states
Θlmr, obtained by a unitary transformation. For example, the canonical eigenstates associated with
l= 1,m={−1,0,1}are not the real px, pyand pzthat we want. See Section 3.4 for our mathematical
conventions regarding projection orbitals for different n,land mr.
We use the following format to specify projections in <seedname>.win:
Begin Projections
[units]
site:ang_mtm:zaxis:xaxis:radial:zona
.
.
.
End Projections
Notes:
51

52 wannier90: User Guide
units:
Optional. Either Ang or Bohr to specify whether the projection centres specified in this block (if given
in Cartesian co-ordinates) are in units of Angstrom or Bohr, respectively. The default value is Ang.
site:
C,Al, etc. applies to all atoms of that type
f=0,0.50,0 – centre on (0.0,0.5,0.0) in fractional coordinates (crystallographic units) relative to the
direct lattice vectors
c=0.0,0.805,0.0 – centre on (0.0,0.805,0.0) in Cartesian coordinates in units specified by the optional
string units in the first line of the projections block (see above).
ang_mtm:
Angular momentum states may be specified by land mr, or by the appropriate character string. See
Tables 3.1 and 3.2. Examples:
l=2,mr=1 or dz2 – a single projection with l= 2,mr= 1 (i.e., dz2)
l=2,mr=1,4 or dz2,dx2-y2 – two functions: dz2and dxz
l=-3 or sp3 – four sp3hybrids
Specific hybrid orbitals may be specified as follows:
l=-3,mr=1,3 or sp3-1,sp3-3 – two specific sp3hybrids
Multiple states may be specified by separating with ‘;’, e.g.,
sp3;l=0 or l=-3;l=0 – four sp3hybrids and one s orbital
zaxis (optional):
z=1,1,1 – set the z-axis to be in the (1,1,1) direction. Default is z=0,0,1
xaxis (optional):
x=1,1,1 – set the x-axis to be in the (1,1,1) direction. Default is x=1,0,0
radial (optional):
r=2 – use a radial function with one node (ie second highest pseudostate with that angular momentum).
Default is r=1. Radial functions associated with different values of rshould be orthogonal to each other.
zona (optional):
zona=2.0 – the value of Z
afor the radial part of the atomic orbital (controls the diffusivity of the radial
function). Units always in reciprocal Angstrom. Default is zona=1.0.
Examples
1. CuO, s,p and d on all Cu; sp3hybrids on O:
Cu:l=0;l=1;l=2
O:l=-3 or O:sp3
2. A single projection onto a pzorbital orientated in the (1,1,1) direction:
c=0,0,0:l=1,mr=1:z=1,1,1 or c=0,0,0:pz:z=1,1,1
3. Project onto s, p and d (with no radial nodes), and s and p (with one radial node) in silicon:
Si:l=0;l=1;l=2
Si:l=0;l=1:r=2

wannier90: User Guide 53
3.2 Spinor Projections
When spinors=.true. it is possible to select a set of localised functions to project onto ‘up’ states
and a set to project onto ‘down’ states where, for complete flexibility, it is also possible to set the local
spin quantisation axis.
Note, however, that this feature requires a recent version of the interface between the ab-initio code
and Wannier90 (i.e., written after the release of the 2.0 version, in October 2013) supporting spinor
projections.
Begin Projections
[units]
site:ang_mtm:zaxis:xaxis:radial:zona(spin)[quant_dir]
.
.
.
End Projections
spin (optional):
Choose projection onto ‘up’ or ‘down’ states
u– project onto ‘up’ states.
d– project onto ‘down’ states.
Default is u,d
quant_dir (optional):
1,0,0 – set the spin quantisation axis to be in the (1,0,0) direction. Default is 0,0,1
Examples
•18 projections on an iron site
Fe:sp3d2;dxy;dxx;dyz
•same as above
Fe:sp3d2;dxy;dxx;dyz(u,d)
•same as above
Fe:sp3d2;dxy;dxz;dyz(u,d)[0,0,1]
•same as above but quantisation axis is now x
Fe:sp3d2;dxy;dxz;dyz(u,d)[1,0,0]
•now only 9 projections onto up states
Fe:sp3d2;dxy;dxz;dyz(u)
•9 projections onto up-states and 3 on down
Fe:sp3d2;dxy;dxz;dyz(u)
Fe:dxy;dxz;dyz(d)
•projections onto alternate spin states for two lattice sites (Cr1, Cr2)
Cr1:d(u)
Cr2:d(d)

54 wannier90: User Guide
3.3 Short-Cuts
3.3.1 Random projections
It is possible to specify the projections, for example, as follows:
Begin Projections
random
C:sp3
End Projections
in which case wannier90 uses four sp3orbitals centred on each C atom and then chooses the appropriate
number of randomly-centred s-type Gaussian functions for the remaining projection functions. If the
block only consists of the string random and no specific projection centres are given, then all of the
projection centres are chosen randomly.
3.3.2 Bloch phases
Setting use_bloch_phases = true in the input file absolves the user of the need to specify explicit
projections. In this case, the Bloch wave-functions are used as the projection orbitals, namely A(k)
mn =
hψmk|ψnki=δmn.
3.4 Orbital Definitions
The angular functions Θlmr(θ, ϕ)associated with particular values of land mrare given in Tables 3.1
and 3.2.
The radial functions Rr(r)associated with different values of rshould be orthogonal. One choice would
be to take the set of solutions to the radial part of the hydrogenic Schrödinger equation for l= 0, i.e.,
the radial parts of the 1s, 2s, 3s. . . orbitals, which are given in Table 3.3.
wannier90: User Guide 55
l mrName Θlmr(θ, ϕ)
0 1 s1
√4π
1 1 pz q3
4πcos θ
1 2 px q3
4πsin θcos ϕ
1 3 py q3
4πsin θsin ϕ
2 1 dz2 q5
16π(3 cos2θ−1)
2 2 dxz q15
4πsin θcos θcos ϕ
2 3 dyz q15
4πsin θcos θsin ϕ
2 4 dx2-y2 q15
16πsin2θcos 2ϕ
2 5 dxy q15
16πsin2θsin 2ϕ
3 1 fz3 √7
4√π(5 cos3θ−3 cos θ)
3 2 fxz2 √21
4√2π(5 cos2θ−1) sin θcos ϕ
3 3 fyz2 √21
4√2π(5 cos2θ−1) sin θsin ϕ
3 4 fz(x2-y2) √105
4√πsin2θcos θcos 2ϕ
3 5 fxyz √105
4√πsin2θcos θsin 2ϕ
3 6 fx(x2-3y2) √35
4√2πsin3θ(cos2ϕ−3 sin2ϕ) cos ϕ
3 7 fy(3x2-y2) √35
4√2πsin3θ(3 cos2ϕ−sin2ϕ) sin ϕ
Table 3.1: Angular functions Θlmr(θ, ϕ)associated with particular values of land mrfor l≥0.
56 wannier90: User Guide
l mrName Θlmr(θ, ϕ)
−1 1 sp-1 1
√2s+1
√2px
−1 2 sp-2 1
√2s−1
√2px
−2 1 sp2-1 1
√3s−1
√6px +1
√2py
−2 2 sp2-2 1
√3s−1
√6px −1
√2py
−2 3 sp2-3 1
√3s+2
√6px
−3 1 sp3-1 1
2(s+px +py +pz)
−3 2 sp3-2 1
2(s+px −py −pz)
−3 3 sp3-3 1
2(s−px +py −pz)
−3 4 sp3-4 1
2(s−px −py +pz)
−4 1 sp3d-1 1
√3s−1
√6px +1
√2py
−4 2 sp3d-2 1
√3s−1
√6px −1
√2py
−4 3 sp3d-3 1
√3s+2
√6px
−4 4 sp3d-4 1
√2pz +1
√2dz2
−4 5 sp3d-5 −1
√2pz +1
√2dz2
−5 1 sp3d2-1 1
√6s−1
√2px −1
√12 dz2 +1
2dx2-y2
−5 2 sp3d2-2 1
√6s+1
√2px −1
√12 dz2 +1
2dx2-y2
−5 3 sp3d2-3 1
√6s−1
√2py −1
√12 dz2 −1
2dx2-y2
−5 4 sp3d2-4 1
√6s+1
√2py −1
√12 dz2 −1
2dx2-y2
−5 5 sp3d2-5 1
√6s−1
√2pz +1
√3dz2
−5 6 sp3d2-6 1
√6s+1
√2pz +1
√3dz2
Table 3.2: Angular functions Θlmr(θ, ϕ)associated with particular values of land mrfor l < 0, in
terms of the orbitals defined in Table 3.1.
wannier90: User Guide 57
r Rr(r)
12α3/2exp(−αr)
21
2√2α3/2(2 −αr) exp(−αr/2)
3q4
27 α3/2(1 −2αr/3+2α2r2/27) exp(−αr/3)
Table 3.3: One possible choice for the radial functions Rr(r)associated with different values of r:
the set of solutions to the radial part of the hydrogenic Schrödinger equation for l= 0, i.e., the radial
parts of the 1s, 2s, 3s. . . orbitals, where α=Z/a =zona.
3.5 Projections via the SCDM-k method
For many systems, such as aperiodic systems, crystals with defects, or novel materials with complex
band structure, it may be extremely hard to identify a-priori a good initial guess for the projection
functions used to generate the A(k)
mn matrices. In these cases, one can use a different approach, known
as the SCDM-kmethod[6], based on a QR factorization with column pivoting (QRCP) of the density
matrix from the self-consistent field calculation, which allows one to avoid the tedious step of specifying
a projection block altogether, hence to avoid . This method is robust in generating well localised
function with the correct spatial orientations and in general in finding the global minimum of the
spread functional Ω. Any electronic-structure code should in principle be able to implement the
SCDM-kmethod within their interface with Wannier90, however at the moment (develop branch
on the GitHub repository November 2017) only the QuantumEspresso package has this capability
implemented in the pw2wannier90 interface program. The SCDM-kcan operate in two modes:
1. In isolation, i.e., without performing a subsequent Wannier90 optimisation (not recommended).
This can be achieved by setting num_iter=0 and dis_num_iter=0 in the <seedname>.win input
file. The rationale behind this is that in general the projection functions obtained with the
SCDM-kare already well localised with the correct spatial orientations. However, the spreads of
the resulting functions are usually larger than the MLWFs ones.
2. In combination with the Souza-Marzari-Vanderbilt (recommended option). In this case, the
SCDM-kis only used to generate the initial A(k)
mn matrices as a replacement scheme for the
projection block.
The following keywords need to be specified in the input file <seedname>.win:scdm_proj scdm_entanglement
scdm_mu scdm_sigma
Chapter 4
Code Overview
wannier90 can operate in two modes:
1. Post-processing mode: read in the overlaps and projections from file as computed inside a first-
principles code. We expect this to be the most common route to using wannier90, and is
described in Ch. 5;
2. Library mode: as a set of library routines to be called from within a first-principles code that
passes the overlaps and projections to the wannier90 library routines and in return gets the
unitary transformation corresponding to MLWF. This route should be used if the MLWF are
needed within the first-principles code, for example in post-LDA methods such as LDA+U or
SIC, and is described in Ch. 6.
59
60 wannier90: User Guide
Wannier_libWannier_prog
Wannerise PlotOverlap Disentangle Transport
Hamiltonian
Kmesh
Constants
Parameters
io
Utility
Figure 4.1: Schematic overview of the module structure of wannier90. Modules may only use data
and subroutines from lower modules.
Chapter 5
wannier90 as a post-processing tool
This is a description of how to use wannier90 as a post-processing tool.
The code must be run twice. On the first pass either the logical keyword postproc_setup must be set
to .true. in the input file seedname.win or the code must be run with the command line option -pp.
Running the code then generates the file seedname.nnkp which provides the information required to
construct the M(k,b)
mn overlaps (Ref. [1], Eq. (25)) and A(k)
mn (Ref. [1], Eq. (62); Ref. [2], Eq. (22)).
Once the overlaps and projection have been computed and written to files seedname.mmn and seedname.amn,
respectively, set postproc_setup to .false. and run the code. Output is written to the file seedname.wout.
5.1 seedname.nnkp file
OUTPUT, if postproc_setup =.true.
The file seedname.nnkp provides the information needed to determine the required overlap elements
M(k,b)
mn and projections A(k)
mn. It is written automatically when the code is invoked with the -pp
command-line option (or when postproc_setup=.true. in seedname.win. There should be no need
for the user to edit this file.
Much of the information in seedname.nnkp is arranged in blocks delimited by the strings begin block_name
. . . end block_name, as described below.
5.1.1 Keywords
The first line of the file is a user comment, e.g., the date and time:
File written on 12Feb2006 at 15:13:12
The only logical keyword is calc_only_A, eg,
calc_only_A : F
5.1.2 Real_lattice block
begin real_lattice
61

62 wannier90: User Guide
2.250000 0.000000 0.000000
0.000000 2.250000 0.000000
0.000000 0.000000 2.250000
end real_lattice
The real lattice vectors in units of Angstrom.
5.1.3 Recip_lattice block
begin recip_lattice
2.792527 0.000000 0.000000
0.000000 2.792527 0.000000
0.000000 0.000000 2.792527
end recip_lattice
The reciprocal lattice vectors in units of inverse Angstrom.
5.1.4 Kpoints block
begin kpoints
8
0.00000 0.00000 0.00000
0.00000 0.50000 0.00000
.
.
.
0.50000 0.50000 0.50000
end kpoints
The first line in the block is the total number of k-points num_kpts. The subsequent num_kpts lines
specify the k-points in crystallographic co-ordinates relative to the reciprocal lattice vectors.
5.1.5 Projections block
begin projections
n_proj
centre l mr r
z-axis x-axis zona
centre l mr r
z-axis x-axis zona
.
.
end projections
Notes:
n_proj: integer; the number of projection centres, equal to the number of MLWF num_wann.

wannier90: User Guide 63
centre: three real numbers; projection function centre in crystallographic co-ordinates relative to the
direct lattice vectors.
l mr r: three integers; land mrspecify the angular part Θlmr(θ, ϕ), and rspecifies the radial part
Rr(r)of the projection function (see Tables 3.1, 3.2 and 3.3).
z-axis: three real numbers; default is 0.0 0.0 1.0; defines the axis from which the polar angle θin
spherical polar coordinates is measured.
x-axis: three real numbers; must be orthogonal to z-axis; default is 1.0 0.0 0.0 or a vector per-
pendicular to z-axis if z-axis is given; defines the axis from with the azimuthal angle ϕin spherical
polar coordinates is measured.
zona: real number; the value of Z
aassociated with the radial part of the atomic orbital. Units are in
reciprocal Angstrom.
5.1.6 spinor_projections block
begin spinor_projections
n_proj
centre l mr r
z-axis x-axis zona
spin spn_quant
centre l mr r
z-axis x-axis zona
spin spn_quant
.
.
end spinor_projections
Notes: Only one of projections and spinor_projections should be defined. Variables are the same as
the projections block with the addition of spin and spn_quant.
spin: integer. ‘1’ or ‘-1’ to denote projection onto up or down states.
spn_quant: three real numbers. Defines the spin quantisation axis in Cartesian coordinates.
5.1.7 nnkpts block
begin nnkpts
10
1 2 0 0 0
.
.
end nnkpts
First line: nntot, the number of nearest neighbours belonging to each k-point of the Monkhorst-Pack
mesh
Subsequent lines: nntot×num_kpts lines, ie, nntot lines of data for each k-point of the mesh.

64 wannier90: User Guide
Each line of consists of 5 integers. The first is the k-point number nkp. The second to the fifth specify
it’s nearest neighbours k+b: the second integer points to the k-point that is the periodic image of the
k+bthat we want; the last three integers give the G-vector, in reciprocal lattice units, that brings
the k-point specified by the second integer (which is in the first BZ) to the actual k+bthat we need.
5.1.8 exclude_bands block
begin exclude_bands
8
1
2
.
.
end exclude_bands
To exclude bands (independent of k-point) from the calculation of the overlap and projection matrices,
for example to ignore shallow-core states. The first line is the number of states to exclude, the following
lines give the states for be excluded.
5.1.9 An example of projections
As a concrete example: one wishes to have a set of four sp3projection orbitals on, say, a carbon atom at
(0.5,0.5,0.5) in fractional co-ordinates relative to the direct lattice vectors. In this case seedname.win
will contain the following lines:
begin projections
C:l=-1
end projections
and seedname.nnkp, generated on the first pass of wannier90 (with postproc_setup=T), will contain:
begin projections
4
0.50000 0.50000 0.50000 -1 1 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
0.50000 0.50000 0.50000 -1 2 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
0.50000 0.50000 0.50000 -1 3 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
0.50000 0.50000 0.50000 -1 4 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
end projections
where the first line tells us that in total four projections are specified, and the subsquent lines provide
the projection centre, the angular and radial parts of the orbital (see Section 3.4 for definitions), the
zand xaxes, and the diffusivity and cut-off radius for the projection orbital.
pwscf, or any other ab initio electronic structure code, then reads seedname.nnkp file, calculates the
projections and writes them to seedname.amn.

wannier90: User Guide 65
5.2 seedname.mmn file
INPUT.
The file seedname.mmn contains the overlaps M(k,b)
mn .
First line: a user comment, e.g., the date and time
Second line: 3 integers: num_bands,num_kpts,nntot
Then: num_kpts ×nntot blocks of data:
First line of each block: 5 integers. The first specifies the k(i.e., gives the ordinal corresponding to
its position in the list of k-points in seedname.win). The 2nd to 5th integers specify k+b. The
2nd integer, in particular, points to the k-point on the list that is a periodic image of k+b, and in
particular is the image that is actually mentioned in the list. The last three integers specify the G
vector, in reciprocal lattice units, that brings the k-point specified by the second integer, and that thus
lives inside the first BZ zone, to the actual k+bthat we need.
Subsequent num_bands ×num_bands lines of each block: two real numbers per line. These are the real
and imaginary parts, respectively, of the actual scalar product M(k,b)
mn for m, n ∈[1,num_bands]. The
order of these elements is such that the first index mis fastest.
5.3 seedname.amn file
INPUT.
The file seedname.amn contains the projection A(k)
mn.
First line: a user comment, e.g., the date and time
Second line: 3 integers: num_bands,num_kpts,num_wann
Subsequently num_bands ×num_wann ×num_kpts lines: 3 integers and 2 real numbers on each line.
The first two integers are the band indices mand n. The third integer specifies the kby giving the
ordinal corresponding to its position in the list of k-points in seedname.win. The real numbers are the
real and imaginary parts, respectively, of the actual A(k)
mn.
5.4 seedname.dmn file
INPUT.
The file seedname.dmn contains the data needed to construct symmetry-adapted Wannier functions [8].
Required if site_symmetry = .true.
First line: a user comment, e.g., the date and time
Second line: 4 integers: num_bands,nsymmetry,nkptirr,num_kpts.
nsymmetry: the number of symmetry operations
nkptirr: the number of irreducible k-points
Blank line

66 wannier90: User Guide
num_kpts integers: Mapping between full k- and irreducible k-points. Each k-point is related to some
k-point in the irreducible BZ. The information of this mapping is written. Each entry corresponds to a
k-point in the full BZ, in the order in which they appear in the k-point list in seedname.win file. The
(integer) value of each entry is the k-point index in the IBZ to which the k-point maps. The number
of unique values is equal to the number of k-points in the IBZ. The data is written 10 values per line.
Blank line
nkptirr integers: List of irreducible k-points. Each entry corresponds to a k-point of the IBZ. The
(integer) value of each entry is the k-point index corresponding to the k-point list in seedname.win
file. The values should be between 1 and num_kpts. The data is written 10 values per line.
Blank line
nkptirr blocks of nsymmetry integer data (each block separated by a blank line): List of k-points
obtained by acting the symmetry operations on the irreducible k-points. The data is written 10 values
per line.
Blank line
nsymmetry ×nkptirr blocks of data:
The information of Dmatrix in Eq. (15) of Ref. [8]. Each block contains num_wann ×num_wann
lines and is separated by a blank line. The data are stored in d_matrix_wann(m,n,isym,ikirr) with
m,n∈[1,num_wann],isym ∈[1,nsymmetry], and ikirr ∈[1,nkptirr]. The order of the elements is
such that left indices run faster than right indices (m: fastest, ikirr: slowest).
Blank line
nsymmetry ×nkptirr blocks of data:
The information of ˜
dmatrix in Eq. (17) of Ref. [8]. Each block contains num_bands ×num_bands
lines and is separated by a blank line. The data are stored in d_matrix_band(m,n,isym,ikirr) with
m,n∈[1,num_bands],isym ∈[1,nsymmetry], and ikirr ∈[1,nkptirr]. The order of the elements is
such that left indices run faster than right indices (m: fastest, ikirr: slowest).
5.5 seedname.eig file
INPUT.
Required if any of disentanglement,plot_bands,plot_fermi_surface or write_hr are .true.
The file seedname.eig contains the Kohn-Sham eigenvalues εnk(in eV) at each point in the Monkhorst-
Pack mesh.
Each line consist of two integers and a real number. The first integer is the band index, the second
integer gives the ordinal corresponding to the k-point in the list of k-points in seedname.win, and the
real number is the eigenvalue.
E.g.,
1 1 -6.43858831271328
2 1 19.3977795287297
3 1 19.3977795287297
4 1 19.3977795287298

wannier90: User Guide 67
5.6 Interface with pwscf
Interfaces between wannier90 and many ab-initio codes such as pwscf,abinit (http://www.abinit.
org), siesta (http://www.icmab.es/siesta/), fleur,VASP and Wien2k (http://www.wien2k.
at) are available. Here we describe the seamless interface between wannier90 and pwscf, a plane-wave
DFT code that comes as part of the Quantum ESPRESSO package (see http://www.quantum-espresso.
org). You will need to download and compile pwscf (i.e., the pw.x code) and the post-processing inter-
face pw2wannier90.x. Please refer to the documentation that comes with the Quantum ESPRESSO
distribution for instructions.
1. Run ‘scf’/‘nscf’ calculation(s) with pw
2. Run wannier90 with postproc_setup =.true. to generate seedname.nnkp
3. Run pw2wannier90. First it reads an input file, e.g., seedname.pw2wan, which defines prefix
and outdir for the underlying ‘scf’ calculation, as well as the name of the file seedname.nnkp, and
does a consistency check between the direct and reciprocal lattice vectors read from seedname.nnkp
and those defined in the files specified by prefix.pw2wannier90 generates seedname.mmn,
seedname.amn and seedname.eig.seedname.dmn and seedname.sym files are additionally cre-
ated when write_dmn = .true. (see below).
4. Run wannier90 with postproc_setup =.false. to disentangle bands (if required), localise
MLWF, and use MLWF for plotting, bandstructures, Fermi surfaces etc.
Examples of how the interface with pwscf works are given in the wannier90 Tutorial.
5.6.1 seedname.pw2wan
A number of keywords may be specified in the pw2wannier90 input file:
•outdir – Location to write output files. Default is ‘./’
•prefix – Prefix for the pwscf calculation. Default is ‘ ’
•seedname – Seedname for the wannier90 calculation. Default is ‘wannier’
•spin_component – Spin component. Takes values ‘up’,‘down’ or ‘none’ (default).
•wan_mode – Either ‘standalone’ (default) or ‘library’
•write_unk – Set to .true. to write the periodic part of the Bloch functions for plotting in
wannier90. Default is .false.
•reduce_unk – Set to .true. to reduce file-size (and resolution) of Bloch functions by a factor of
8. Default is .false. (only relevant if write_unk=.true.)1
•wvfn_formatted – Set to .true. to write formatted wavefunctions. Default is .false. (only
relevant if write_unk=.true.)
1Note that there is a small bug with this feature in v3.2 (and subsequent patches) of quantum-espresso. Please use
a later version (if available) or the CVS version of pw2wannier90.f90, which has been fixed.

68 wannier90: User Guide
•write_amn – Set to .false. if A(k)
mn not required. Default is .true.
•write_mmn – Set to .false. if M(k,b)
mn not required. Default is .true.
•write_spn – Set to .true. to write out the matrix elements of Sbetween Bloch states (non-
collinear spin calculation only). Default is .false.
•spn_formatted – Set to .true. to write spn data as a formatted file. Default is .false. (only
relevant if write_spn=.true.)
•write_uHu – Set to .true. to write out the matrix elements
hunk+b1|Hk|umk+b2i.
Default is .false.
•uHu_formatted – Set to .true. to write uHu data as a formatted file. Default is .false. (only
relevant if write_uHu=.true.)
•write_uIu – Set to .true. to write out the matrix elements of
hunk+b1|umk+b2i.
Default is .false.
•uIu_formatted – Set to .true. to write uIu data as a formatted file. Default is .false. (only
relevant if write_uIu=.true.)
•write_unkg – Set to .true. to write the first few Fourier components of the periodic parts of
the Bloch functions.
•write_dmn – Set to .true. to construct symmetry-adapted Wannier functions. Default is
.false.
•read_sym – Set to .true. to customize symmetry operations to be used in symmetry-adapted
mode. When read_sym = .true., an additional input seedname.sym is required. Default is
.false. (only relevant if write_dmn=.true.).
For examples of use, refer to the wannier90 Tutorial.
5.6.2 seedname.sym
If read_sym = .true., then this additional input file is required for pw2wannier90.x
if read_sym = .false., then this file is written by pw2wannier90.x (only for reference – it is not used
in subsequent calculations)
The file seedname.sym contains the information of symmetry operations used to create symmetry-
adapted Wannier functions. If read_sym = .false. (default), pw2wannier90.x uses the full symme-
try recognized by pw.x. If read_sym = .true., you can specify symmetry operations to be used in
symmetry-adapted mode.
First line: an integer: nsymmetry (number of symmetry operations)
Second line: blank
Then: nsymmetry blocks of data. Each block (separated by a blank line) consists of four lines. The
order of the data in each block is as follows:

wannier90: User Guide 69
R(1,1) R(2,1) R(3,1)
R(1,2) R(2,2) R(3,2)
R(1,3) R(2,3) R(3,3)
t(1) t(2) t(3)
Here, Ris the rotational part of symmetry operations (3×3matrix), and tis the fractional translation
in the unit of “alat” (refer the definition of “alat” to the manual of pwscf). Both data are given in
Cartesian coordinates. The symmetry operations act on a point ras rR−t.
Chapter 6
wannier90 as a library
This is a description of the interface between any external program and the wannier code. There
are two subroutines: wannier_setup and wannier_run. Calling wannier_setup will return informa-
tion required to construct the M(k,b)
mn overlaps (Ref. [1], Eq. (25)) and A(k)
mn =hψmk|gniprojections
(Ref. [1], Eq. (62); Ref. [2], Eq. (22)). Once the overlaps and projection have been computed, calling
wannier_run activates the minimisation and plotting routines in wannier90.
IMPORTANT NOTE: the library mode ONLY works in serial. Please call it from a serial code, or
if compiled in parallel, make sure to run it from a single MPI process.
You can find a minimal example of how the library mode can be used among the tests, in the file
test-suite/library-mode-test/test_library.F90 in the Wannier90 git repository.
6.1 Subroutines
6.1.1 wannier_setup
wannier_setup(seed_name,mp_grid,num_kpts,real_lattice,recip_lattice,
kpt_latt,num_bands_tot,num_atoms,atom_symbols,atoms_cart,
gamma_only,spinors,nntot,nnlist,nncell,num_bands,num_wann,proj_site,
proj_l,proj_m,proj_radial,proj_z,proj_x,proj_zona,
exclude_bands,proj_s,proj_s_qaxis)
•character(len=*), intent(in) :: seed_name
The seedname of the current calculation.
•integer, dimension(3), intent(in) :: mp_grid
The dimensions of the Monkhorst-Pack k-point grid.
•integer, intent(in) :: num_kpts
The number of k-points on the Monkhorst-Pack grid.
•real(kind=dp), dimension(3,3), intent(in) :: real_lattice
The lattice vectors in Cartesian co-ordinates in units of Angstrom.
•real(kind=dp), dimension(3,3), intent(in) :: recip_lattice
The reciprocal lattice vectors in Cartesian co-ordinates in units of reciprocal Angstrom.
71

72 wannier90: User Guide
•real(kind=dp), dimension(3,num_kpts), intent(in) :: kpt_latt
The positions of the k-points in fractional co-ordinates relative to the reciprocal lattice vectors.
•integer, intent(in) :: num_bands_tot
The total number of bands in the first-principles calculation (note: including semi-core states).
•integer, intent(in) :: num_atoms
The total number of atoms in the system.
•character(len=20), dimension(num_atoms), intent(in) :: atom_symbols
The elemental symbols of the atoms.
•real(kind=dp), dimension(3,num_atoms), intent(in) :: atoms_cart
The positions of the atoms in Cartesian co-ordinates in Angstrom.
•logical, intent(in) :: gamma_only
Set to .true. if the underlying electronic structure calculation has been performed with only
Γ-point sampling and, hence, if the Bloch eigenstates that are used to construct A(k)
mn and M(k,b)
mn
are real.
•logical, intent(in) :: spinors
Set to .true. if underlying electronic structure calculation has been performed with spinor
wavefunctions.
•integer, intent(out) :: nntot
The total number of nearest neighbours for each k-point.
•integer, dimension(num_kpts,num_nnmax), intent(out) :: nnlist
The list of nearest neighbours for each k-point.
•integer,dimension(3,num_kpts,num_nnmax), intent(out) :: nncell
The vector, in fractional reciprocal lattice co-ordinates, that brings the nnth nearest neighbour
of k-point nkp to its periodic image that is needed for computing the overlap M(k,b)
mn .
•integer, intent(out) :: num_bands
The number of bands in the first-principles calculation used to form the overlap matricies (note:
excluding eg. semi-core states).
•integer, intent(out) :: num_wann
The number of MLWF to be extracted.
•real(kind=dp), dimension(3,num_bands_tot), intent(out) :: proj_site
Projection function centre in crystallographic co-ordinates relative to the direct lattice vectors.
•integer, dimension(num_bands_tot), intent(out) :: proj_l
lspecifies the angular part Θlmr(θ, ϕ)of the projection function (see Tables 3.1, 3.2 and 3.3).
•integer, dimension(num_bands_tot), intent(out) :: proj_m
mrspecifies the angular part Θlmr(θ, ϕ), of the projection function (see Tables 3.1, 3.2 and 3.3).
•integer, dimension(num_bands_tot), intent(out) :: proj_radial
rspecifies the radial part Rr(r)of the projection function (see Tables 3.1, 3.2 and 3.3).
•real(kind=dp), dimension(3,num_bands_tot), intent(out) :: proj_z
Defines the axis from which the polar angle θin spherical polar coordinates is measured. Default
is 0.0 0.0 1.0.

wannier90: User Guide 73
•real(kind=dp), dimension(3,num_bands_tot), intent(out) :: proj_x
Must be orthogonal to z-axis; default is 1.0 0.0 0.0 or a vector perpendicular to proj_z if
proj_z is given; defines the axis from with the azimuthal angle ϕin spherical polar coordinates
is measured.
•real(kind=dp), dimension(num_bands_tot), intent(out) :: proj_zona
The value of Z
aassociated with the radial part of the atomic orbital. Units are in reciprocal
Angstrom.
•integer, dimension(num_bands_tot), intent(out) :: exclude_bands
Kpoints independant list of bands to exclude from the calculation of the MLWF (e.g., semi-core
states).
•integer, dimension(num_bands_tot), optional,intent(out) :: proj_s
’1’ or ’-1’ to denote projection onto up or down spin states
•real(kind=dp), dimension(3,num_bands_tot), intent(out) :: proj_s_qaxisx
Defines the spin quantisation axis in Cartesian coordinates.
Conditions:
?num_kpts =mp_grid(1) ×mp_grid(2) ×mp_grid(3).
?num_nnmax = 12
This subroutine returns the information required to determine the required overlap elements M(k,b)
mn
and projections A(k)
mn, i.e., M_matrix and A_matrix, described in Section 6.1.2.
For the avoidance of doubt, real_lattice(1,2) is the y−component of the first lattice vector A1, etc.
The list of nearest neighbours of a particular k-point nkp is given by nnlist(nkp,1:nntot).
Additionally, the parameter shell_list may be specified in the wannier90 input file.
6.1.2 wannier_run
wannier_run(seed_name,mp_grid,num_kpts,real_lattice,recip_lattice,
kpt_latt,num_bands,num_wann,nntot,num_atoms,atom_symbols,
atoms_cart,gamma_only,M_matrix_orig,A_matrix,eigenvalues,
U_matrix,U_matrix_opt,lwindow,wann_centres,wann_spreads,
spread)
•character(len=*), intent(in) :: seed_name
The seedname of the current calculation.
•integer, dimension(3), intent(in) :: mp_grid
The dimensions of the Monkhorst-Pack k-point grid.
•integer, intent(in) :: num_kpts
The number of k-points on the Monkhorst-Pack grid.
•real(kind=dp), dimension(3,3), intent(in) :: real_lattice
The lattice vectors in Cartesian co-ordinates in units of Angstrom.

74 wannier90: User Guide
•real(kind=dp), dimension(3,3), intent(in) :: recip_lattice
The reciprical lattice vectors in Cartesian co-ordinates in units of inverse Angstrom.
•real(kind=dp), dimension(3,num_kpts), intent(in) :: kpt_latt
The positions of the k-points in fractional co-ordinates relative to the reciprocal lattice vectors.
•integer, intent(in) :: num_bands
The total number of bands to be processed.
•integer, intent(in) :: num_wann
The number of MLWF to be extracted.
•integer, intent(in) :: nntot
The number of nearest neighbours for each k-point.
•integer, intent(in) :: num_atoms
The total number of atoms in the system.
•character(len=20), dimension(num_atoms), intent(in) :: atom_symbols
The elemental symbols of the atoms.
•real(kind=dp), dimension(3,num_atoms), intent(in) :: atoms_cart
The positions of the atoms in Cartesian co-ordinates in Angstrom.
•logical, intent(in) :: gamma_only
Set to .true. if the underlying electronic structure calculation has been performed with only
Γ-point sampling and, hence, if the Bloch eigenstates that are used to construct A(k)
mn and M(k,b)
mn
are real.
•complex(kind=dp), dimension(num_bands,num_bands,nntot,num_kpts),
intent(in) :: M_matrix
The matrices of overlaps between neighbouring periodic parts of the Bloch eigenstates at each
k-point, M((k,b))
mn (Ref. [1], Eq. (25)).
•complex(kind=dp), dimension(num_bands,num_wann,num_kpts),
intent(in) :: A_matrix
The matrices describing the projection of num_wann trial orbitals on num_bands Bloch states at
each k-point, A(k)
mn (Ref. [1], Eq. (62); Ref. [2], Eq. (22)).
•real(kind=dp), dimension(num_bands,num_kpts), intent(in) :: eigenvalues
The eigenvalues εnkcorresponding to the eigenstates, in eV.
•complex(kind=dp), dimension(num_wann,num_wann,num_kpts),
intent(out) :: U_matrix
The unitary matrices at each k-point (Ref. [1], Eq. (59))
•complex(kind=dp), dimension(num_bands,num_wann,num_kpts),
optional, intent(out) :: U_matrix_opt
The unitary matrices that describe the optimal sub-space at each k-point (see Ref. [2], Sec-
tion IIIa). The array is packed (see below)
•logical, dimension(num_bands,num_kpts), optional, intent(out) :: lwindow
The element lwindow(nband,nkpt) is .true. if the band nband lies within the outer energy
window at kpoint nkpt.

wannier90: User Guide 75
•real(kind=dp), dimension(3,num_wann), optional, intent(out) :: wann_centres
The centres of the MLWF in Cartesian co-ordinates in Angstrom.
•real(kind=dp), dimension(num_wann), optional, intent(out) :: wann_spreads
The spread of each MLWF in Å2.
•real(kind=dp), dimension(3), optional, intent(out) :: spread
The values of Ω,ΩIand ˜
Ω(Ref. [1], Eq. (13)).
Conditions:
?num_wann ≤num_bands
?num_kpts =mp_grid(1) ×mp_grid(2) ×mp_grid(3).
If num_bands =num_wann then U_matrix_opt is the identity matrix and lwindow=.true.
For the avoidance of doubt, real_lattice(1,2) is the y−component of the first lattice vector A1, etc.
M_matrix(m,n,nn,nkp) =humk|unk+bi
A_matrix(m,n,nkp) =hψmk|gni
eigenvalues(n,nkp) =εnk
where
k=kpt_latt(1:3,nkp)
k+b=kpt_latt(1:3,nnlist(nkp,nn)) +nncell(1:3,nkp,nn)
and {|gni} are a set of initial trial orbitals. These are typically atom or bond-centred Gaussians that
are modulated by appropriate spherical harmonics.
Additional parameters should be specified in the wannier90 input file.
Chapter 7
Transport Calculations with wannier90
By setting transport =TRUE,wannier90 will calculate the quantum conductance and density of states
of a one-dimensional system. The results will be written to files seedname_qc.dat and seedname_dos.dat,
respectively.
The system for which transport properties are calculated is determined by the keyword transport_mode.
7.1 transport_mode = bulk
Quantum conductance and density of states are calculated for a perfectly periodic one-dimensional
conductor. If tran_read_ht =FALSE the transport properties are calculated using the Hamiltonian in
the Wannier function basis of the system found by wannier90. Setting tran_read_ht =TRUE allows
the user to provide an external Hamiltonian matrix file seedname_htB.dat, from which the properties
are found. See Section 2.9 for more details of the keywords required for such calculations.
7.2 transport_mode = lcr
Quantum conductance and density of states are calculated for a system where semi-infinite, left and
right leads are connected through a central conductor region. This is known as the lcr system. Details
of the method is described in Ref. [10].
In wannier90 two options exist for performing such calculations:
•If tran_read_ht =TRUE the external Hamiltonian files seedname_htL.dat, seedname_htLC.dat,
seedname_htC.dat, seedname_htCR.dat, seedname_htR.dat are read and used to compute the
transport properties.
•If tran_read_ht =FALSE, then the transport calculation is performed automatically using the
Wannier functions as a basis and the 2c2 geometry described in Section 7.3.
77
78 wannier90: User Guide
7.3 Automated lcr Transport Calculations: The 2c2 Geometry
Calculations using the 2c2 geometry provide a method to calculate the transport properties of an lcr
system from a single wannier90 calculation. The Hamiltonian matrices which the five external files
provide in the tran_read_ht =TRUE case are instead built from the Wannier function basis directly.
As such, strict rules apply to the system geometry, which is shown in Figure 7.1. These rules are as
follows:
•Left and right leads must be identical and periodic.
•Supercell must contain two principal layers (PLs) of lead on the left, a central conductor region
and two principal layers of lead on the right.
•The conductor region must contain enough lead such that the disorder does not affect the principal
layers of lead either side.
•A single k-point (Gamma) must be used.
PL3 PL4PL1 ConductorPL2
h
H
LC
L
00
hCR
10
L
H , HR
01
C
H
PL1
Figure 7.1: Schematic illustration of the supercell required for 2c2 lcr calculations, showing where
each of the Hamiltonian matrices are derived from. Four principal layers (PLs) are required plus the
conductor region.
In order to build the Hamiltonians, Wannier functions are first sorted according to position and then
type if a number of Wannier functions exist with a similar centre (eg. d-orbital type Wannier functions
centred on a Cu atom). Next, consistent parities of Wannier function are enforced. To distingiush
between different types of Wannier function and assertain relative parities, a signature of each Wannier
function is computed. The signature is formed of 20 integrals which have different spatial dependence.
They are given by:
I=1
VZV
g(r)w(r)dr(7.1)
where Vis the volume of the cell, w(r)is the Wannier function and g(r)are the set of functions:
g(r) = n1,sin 2π(x−xc)
Lx,sin 2π(y−yc)
Ly,sin 2π(z−zc)
Lz,sin 2π(x−xc)
Lxsin 2π(y−yc)
Ly,
sin 2π(x−xc)
Lxsin 2π(z−zc)
Lz, ...o(7.2)
upto third order in powers of sines. Here, the supercell has dimension (Lx, Ly, Lz)and the Wannier
function has centre rc= (xc, yc, zc). Each of these integrals may be written as linear combinations of
the following sums:

wannier90: User Guide 79
Sn(G) = eiG.rcX
m
Umn ˜u∗
mΓ(G)(7.3)
where nand mare the Wannier function and band indexes, Gis a G-vector, Umn is the unitary matrix
that transforms from the Bloch reopresentation of the system to the maximally-localised Wannier
function basis and ˜u∗
mΓ(G)are the conjugates of the Fourier transforms of the periodic parts of the
Bloch states at the Γ-point. The complete set of ˜umk(G)are often outputted by plane-wave DFT
codes. However, to calculate the 20 signature integrals, only 32 specific ˜umk(G)are required. These
are found in an additional file (seedname.unkg) that should be provided by the interface between the
DFT code and wannier90 . A detailed description of this file may be found in Section 8.31.
Additionally, the following keywords are also required in the input file:
•tran_num_ll : The number of Wannier functions in a principal layer.
•tran_num_cell_ll : The number of unit cells in one principal layer of lead
A further parameter related to these calculations is tran_group_threshold.
Examples of how 2c2 calculations are preformed can be found in the wannier90 Tutorial.
Chapter 8
Files
8.1 seedname.win
INPUT. The master input file; contains the specification of the system and any parameters for the run.
For a description of input parameters, see Chapter 2; for examples, see Section 10.1 and the wannier90
Tutorial.
8.1.1 Units
The following are the dimensional quantities that are specified in the master input file:
•Direct lattice vectors
•Positions (of atomic or projection) centres in real space
•Energy windows
•Positions of k-points in reciprocal space
•Convergence thresholds for the minimisation of Ω
•zona (see Section 3.1)
•wannier_plot_cube: cut-off radius for plotting WF in Gaussian cube format
Notes:
•The units (either ang (default) or bohr) in which the lattice vectors, atomic positions or projection
centres are given can be set in the first line of the blocks unit_cell_cart,atoms_cart and
projections, respectively, in seedname.win.
•Energy is always in eV.
•Convergence thresholds are always in Å2
•Positions of k-points are always in crystallographic coordinates relative to the reciprocal lattice
vectors.
81

82 wannier90: User Guide
•zona is always in reciprocal Angstrom (Å−1)
•The keyword length_unit may be set to ang (default) or bohr, in order to set the units in which
the quantities in the output file seedname.wout are written.
•wannier_plot_radius is in Angstrom
The reciprocal lattice vectors {B1,B2,B3}are defined in terms of the direct lattice vectors {A1,A2,A3}
by the equation
B1=2π
ΩA2×A3etc., (8.1)
where the cell volume is V=A1·(A2×A3).
8.2 seedname.mmn
INPUT. Written by the underlying electronic structure code. See Chapter 5 for details.
8.3 seedname.amn
INPUT. Written by the underlying electronic structure code. See Chapter 5 for details.
8.4 seedname.dmn
INPUT. Read if site_symmetry = .true. (symmetry-adapted mode). Written by the underlying
electronic structure code. See Chapter 5 for details.
8.5 seedname.eig
INPUT. Written by the underlying electronic structure code. See Chapter 5 for details.
8.6 seedname.nnkp
OUTPUT. Written by wannier90 when postproc_setup=.TRUE. (or, alternatively, when wannier90
is run with the -pp command-line option). See Chapter 5 for details.
8.7 seedname.wout
OUTPUT. The master output file. Here we give a description of the main features of the output. The
verbosity of the output is controlled by the input parameter iprint. The higher the value, the more
detail is given in the output file. The default value is 1, which prints minimal information.

wannier90: User Guide 83
8.7.1 Header
The header provides some basic information about wannier90, the authors, the code version and
release, and the execution time of the current run. The header looks like the following different (the
string might slightly change across different versions):
+---------------------------------------------------+
| |
| WANNIER90 |
| |
+---------------------------------------------------+
| |
| Welcome to the Maximally-Localized |
| Generalized Wannier Functions code |
| http://www.wannier.org |
| |
| Wannier90 Developer Group: |
| Giovanni Pizzi (EPFL) |
| Valerio Vitale (Cambridge) |
| David Vanderbilt (Rutgers University) |
| Nicola Marzari (EPFL) |
| Ivo Souza (Universidad del Pais Vasco) |
| Arash A. Mostofi (Imperial College London) |
| Jonathan R. Yates (University of Oxford) |
| |
| For the full list of Wannier90 3.x authors, |
| please check the code documentation and the |
| README on the GitHub page of the code |
| |
| |
| Please cite |
.
.
| |
+---------------------------------------------------+
| Execution started on 18Dec2018 at 18:39:42 |
+---------------------------------------------------+
8.7.2 System information
This part of the output file presents information that wannier90 has read or inferred from the master
input file seedname.win. This includes real and reciprocal lattice vectors, atomic positions, k-points,
parameters for job control, disentanglement, localisation and plotting.
------
SYSTEM

84 wannier90: User Guide
------
Lattice Vectors (Ang)
a_1 3.938486 0.000000 0.000000
a_2 0.000000 3.938486 0.000000
a_3 0.000000 0.000000 3.938486
Unit Cell Volume: 61.09251 (Ang^3)
Reciprocal-Space Vectors (Ang^-1)
b_1 1.595330 0.000000 0.000000
b_2 0.000000 1.595330 0.000000
b_3 0.000000 0.000000 1.595330
*----------------------------------------------------------------------------*
| Site Fractional Coordinate Cartesian Coordinate (Ang) |
+----------------------------------------------------------------------------+
| Ba 1 0.00000 0.00000 0.00000 | 0.00000 0.00000 0.00000 |
| Ti 1 0.50000 0.50000 0.50000 | 1.96924 1.96924 1.96924 |
.
.
*----------------------------------------------------------------------------*
------------
K-POINT GRID
------------
Grid size = 4 x 4 x 4 Total points = 64
*---------------------------------- MAIN ------------------------------------*
| Number of Wannier Functions : 9 |
| Number of input Bloch states : 9 |
| Output verbosity (1=low, 5=high) : 1 |
| Length Unit : Ang |
| Post-processing setup (write *.nnkp) : F |
.
.
*----------------------------------------------------------------------------*
8.7.3 Nearest-neighbour k-points
This part of the output files provides information on the b-vectors and weights chosen to satisfy the
condition of Eq. 2.1.
*---------------------------------- K-MESH ----------------------------------*
+----------------------------------------------------------------------------+
| Distance to Nearest-Neighbour Shells |
| ------------------------------------ |
| Shell Distance (Ang^-1) Multiplicity |

wannier90: User Guide 85
| ----- ----------------- ------------ |
| 1 0.398833 6 |
| 2 0.564034 12 |
.
.
+----------------------------------------------------------------------------+
| The b-vectors are chosen automatically |
| The following shells are used: 1 |
+----------------------------------------------------------------------------+
| Shell # Nearest-Neighbours |
| ----- -------------------- |
| 1 6 |
+----------------------------------------------------------------------------+
| Completeness relation is fully satisfied [Eq. (B1), PRB 56, 12847 (1997)] |
+----------------------------------------------------------------------------+
8.7.4 Disentanglement
Then (if required) comes the part where ΩIis minimised to disentangle the optimally-connected sub-
space of states for the localisation procedure in the next step.
First, a summary of the energy windows that are being used is given:
*------------------------------- DISENTANGLE --------------------------------*
+----------------------------------------------------------------------------+
| Energy Windows |
| --------------- |
| Outer: 2.81739 to 38.00000 (eV) |
| Inner: 2.81739 to 13.00000 (eV) |
+----------------------------------------------------------------------------+
Then, each step of the iterative minimisation of ΩIis reported.
Extraction of optimally-connected subspace
------------------------------------------
+---------------------------------------------------------------------+<-- DIS
| Iter Omega_I(i-1) Omega_I(i) Delta (frac.) Time |<-- DIS
+---------------------------------------------------------------------+<-- DIS
1 3.82493590 3.66268867 4.430E-02 0.36 <-- DIS
2 3.66268867 3.66268867 6.911E-15 0.37 <-- DIS
.
.
<<< Delta < 1.000E-10 over 3 iterations >>>
<<< Disentanglement convergence criteria satisfied >>>
Final Omega_I 3.66268867 (Ang^2)
+----------------------------------------------------------------------------+

86 wannier90: User Guide
The first column gives the iteration number. For a description of the minimisation procedure and
expressions for Ω(i)
I, see the original paper [2]. The procedure is considered to be converged when
the fractional difference between Ω(i)
Iand Ω(i−1)
Iis less than dis_conv_tol over dis_conv_window
iterations. The final column gives a running account of the wall time (in seconds) so far. Note that
at the end of each line of output, there are the characters “<– DIS”. This enables fast searching of the
output using, for example, the Unix command grep:
my_shell> grep DIS wannier.wout | less
8.7.5 Wannierisation
The next part of the output file provides information on the minimisation of e
Ω. At each iteration, the
centre and spread of each WF is reported.
*------------------------------- WANNIERISE ---------------------------------*
+--------------------------------------------------------------------+<-- CONV
| Iter Delta Spread RMS Gradient Spread (Ang^2) Time |<-- CONV
+--------------------------------------------------------------------+<-- CONV
------------------------------------------------------------------------------
Initial State
WF centre and spread 1 ( 0.000000, 1.969243, 1.969243 ) 1.52435832
WF centre and spread 2 ( 0.000000, 1.969243, 1.969243 ) 1.16120620
.
.
0 0.126E+02 0.0000000000 12.6297685260 0.29 <-- CONV
O_D= 0.0000000 O_OD= 0.1491718 O_TOT= 12.6297685 <-- SPRD
------------------------------------------------------------------------------
Cycle: 1
WF centre and spread 1 ( 0.000000, 1.969243, 1.969243 ) 1.52414024
WF centre and spread 2 ( 0.000000, 1.969243, 1.969243 ) 1.16059775
.
.
Sum of centres and spreads ( 11.815458, 11.815458, 11.815458 ) 12.62663472
1 -0.313E-02 0.0697660962 12.6266347170 0.34 <-- CONV
O_D= 0.0000000 O_OD= 0.1460380 O_TOT= 12.6266347 <-- SPRD
Delta: O_D= -0.4530841E-18 O_OD= -0.3133809E-02 O_TOT= -0.3133809E-02 <-- DLTA
------------------------------------------------------------------------------
Cycle: 2
WF centre and spread 1 ( 0.000000, 1.969243, 1.969243 ) 1.52414866
WF centre and spread 2 ( 0.000000, 1.969243, 1.969243 ) 1.16052405
.
.
Sum of centres and spreads ( 11.815458, 11.815458, 11.815458 ) 12.62646411
2 -0.171E-03 0.0188848262 12.6264641055 0.38 <-- CONV
O_D= 0.0000000 O_OD= 0.1458674 O_TOT= 12.6264641 <-- SPRD
Delta: O_D= -0.2847260E-18 O_OD= -0.1706115E-03 O_TOT= -0.1706115E-03 <-- DLTA

wannier90: User Guide 87
------------------------------------------------------------------------------
.
.
------------------------------------------------------------------------------
Final State
WF centre and spread 1 ( 0.000000, 1.969243, 1.969243 ) 1.52416618
WF centre and spread 2 ( 0.000000, 1.969243, 1.969243 ) 1.16048545
.
.
Sum of centres and spreads ( 11.815458, 11.815458, 11.815458 ) 12.62645344
Spreads (Ang^2) Omega I = 12.480596753
================ Omega D = 0.000000000
Omega OD = 0.145856689
Final Spread (Ang^2) Omega Total = 12.626453441
------------------------------------------------------------------------------
It looks quite complicated, but things look more simple if one uses grep:
my_shell> grep CONV wannier.wout
gives
+--------------------------------------------------------------------+<-- CONV
| Iter Delta Spread RMS Gradient Spread (Ang^2) Time |<-- CONV
+--------------------------------------------------------------------+<-- CONV
0 0.126E+02 0.0000000000 12.6297685260 0.29 <-- CONV
1 -0.313E-02 0.0697660962 12.6266347170 0.34 <-- CONV
.
.
50 0.000E+00 0.0000000694 12.6264534413 2.14 <-- CONV
The first column is the iteration number, the second is the change in Ωfrom the previous iteration, the
third is the root-mean-squared gradient of Ωwith respect to variations in the unitary matrices U(k),
and the last is the time taken (in seconds). Depending on the input parameters used, the procedure
either runs for num_iter iterations, or a convergence criterion is applied on Ω. See Section 2.8 for
details.
Similarly, the command
my_shell> grep SPRD wannier.wout
gives
O_D= 0.0000000 O_OD= 0.1491718 O_TOT= 12.6297685 <-- SPRD
O_D= 0.0000000 O_OD= 0.1460380 O_TOT= 12.6266347 <-- SPRD
.
.
O_D= 0.0000000 O_OD= 0.1458567 O_TOT= 12.6264534 <-- SPRD
which, for each iteration, reports the value of the diagonal and off-diagonal parts of the non-gauge-
invariant spread, as well as the total spread, respectively. Recall from Section 1 that Ω=ΩI+ΩD+ΩOD.

88 wannier90: User Guide
Wannierisation with selective localization and constrained centres
For full details of the selectively localised Wannier function (SLWF) method, the reader is referred to
Ref. [9]. When using the SLWF method, only a few things change in the output file and in general the
same principles described above will apply. In particular, when minimising the spread with respect to
the degrees of freedom of only a subset of functions, it is not possible to cast the total spread functional
Ωas a sum of a gauge-invariant part and a gauge-dependent part. Instead, one has Ω0= ΩIOD + ΩD,
where
Ω0=
J0<J
X
n=1 hr2in−r2
n
and
ΩIOD =
J0<J
X
n=1 "hr2
ni − X
R|hRn|r|nRi|2#.
The total number of Wannier functions is J, whereas J0is the number functions to be selectively
localized (so-called objective WFs). The information on the number of functions which are going to be
selectively localized (Number of Objective Wannier Functions) is given in the MAIN section of the
output file:
*---------------------------------- MAIN ------------------------------------*
| Number of Wannier Functions : 4 |
| Number of Objective Wannier Functions : 1 |
| Number of input Bloch states : 4 |
Whether or not the selective localization procedure has been switched on is reported in the WANNIERISE
section as
| Perform selective localization : T |
The next part of the output file provides information on the minimisation of the modified spread
functional:
*------------------------------- WANNIERISE ---------------------------------*
+--------------------------------------------------------------------+<-- CONV
| Iter Delta Spread RMS Gradient Spread (Ang^2) Time |<-- CONV
+--------------------------------------------------------------------+<-- CONV
------------------------------------------------------------------------------
Initial State
WF centre and spread 1 ( -0.857524, 0.857524, 0.857524 ) 1.80463310
WF centre and spread 2 ( 0.857524, -0.857524, 0.857524 ) 1.80463311
WF centre and spread 3 ( 0.857524, 0.857524, -0.857524 ) 1.80463311
WF centre and spread 4 ( -0.857524, -0.857524, -0.857524 ) 1.80463311
Sum of centres and spreads ( -0.000000, -0.000000, 0.000000 ) 7.21853243
0 -0.317E+01 0.0000000000 -3.1653368719 0.00 <-- CONV
O_D= 0.0000000 O_IOD= -3.1653369 O_TOT= -3.1653369 <-- SPRD
------------------------------------------------------------------------------

wannier90: User Guide 89
Cycle: 1
WF centre and spread 1 ( -0.853260, 0.853260, 0.853260 ) 1.70201498
WF centre and spread 2 ( 0.857352, -0.857352, 0.862454 ) 1.84658331
WF centre and spread 3 ( 0.857352, 0.862454, -0.857352 ) 1.84658331
WF centre and spread 4 ( -0.862454, -0.857352, -0.857352 ) 1.84658331
Sum of centres and spreads ( -0.001010, 0.001010, 0.001010 ) 7.24176492
1 -0.884E-01 0.2093698260 -3.2536918930 0.00 <-- CONV
O_IOD= -3.2536919 O_D= 0.0000000 O_TOT= -3.2536919 <-- SPRD
Delta: O_IOD= -0.1245020E+00 O_D= 0.0000000E+00 O_TOT= -0.8835502E-01 <-- DLTA
------------------------------------------------------------------------------
.
.
------------------------------------------------------------------------------
Final State
WF centre and spread 1 ( -0.890189, 0.890189, 0.890189 ) 1.42375495
WF centre and spread 2 ( 0.895973, -0.895973, 0.917426 ) 2.14313664
WF centre and spread 3 ( 0.895973, 0.917426, -0.895973 ) 2.14313664
WF centre and spread 4 ( -0.917426, -0.895973, -0.895973 ) 2.14313664
Sum of centres and spreads ( -0.015669, 0.015669, 0.015669 ) 7.85316486
Spreads (Ang^2) Omega IOD = 1.423371553
================ Omega D = 0.000383395
Omega Rest = 9.276919811
Final Spread (Ang^2) Omega Total = 1.423754947
------------------------------------------------------------------------------
When comparing the output from an SLWF calculation with a standard wannierisation (see Sec. 8.7.5),
the only differences are in the definition of the spread functional. Hence, during the minimization O_OD
is replaced by O_IOD and O_TOT now reflects the fact that the new total spread functional is Ω0. The
part on the final state has one more item of information: the value of the difference between the global
spread functional and the new spread functional given by Omega Rest
ΩR=
J−J0
X
n=1 hr2in−r2
n
If adding centre-constraints to the SLWFs, you will find the information about the centres of the
original projections and the desired centres in the SYSTEM section
*----------------------------------------------------------------------------*
| Wannier# Original Centres Constrained centres |
+----------------------------------------------------------------------------+
| 1 0.25000 0.25000 0.25000 | 0.00000 0.00000 0.00000 |
*----------------------------------------------------------------------------*
As before one can check that the selective localization with constraints is being used by looking at the
WANNIERISE section:

90 wannier90: User Guide
| Perform selective localization : T |
| Use constrains in selective localization : T |
| Value of the Lagrange multiplier : 0.100E+01 |
*----------------------------------------------------------------------------*
which also gives the selected value for the Lagrange multiplier. The output file for the minimisation
section is modified as follows: both O_IOD and O_TOT now take into account the factors coming from
the new term in the functional due to the constraints, which are implemented by adding the following
penalty functional to the spread functional,
λc
J0
X
n=1
(rn−r0n)2,
where r0nis the desired centre for the nth Wannier function, see Ref. [9] for details. The layout of the
output file at each iteration is unchanged.
1 -0.884E-01 0.2093698260 -3.2536918930 0.00 <-- CONV
As regarding the final state, the only addition is the information on the value of the penalty functional
associated with the constraints (Penalty func), which should be zero if the final centres of the Wannier
functions are at the target centres:
Final State
WF centre and spread 1 ( -1.412902, 1.412902, 1.412902 ) 1.63408756
WF centre and spread 2 ( 1.239678, -1.239678, 1.074012 ) 2.74801593
WF centre and spread 3 ( 1.239678, 1.074012, -1.239678 ) 2.74801592
WF centre and spread 4 ( -1.074012, -1.239678, -1.239678 ) 2.74801592
Sum of centres and spreads ( -0.007559, 0.007559, 0.007559 ) 9.87813534
Spreads (Ang^2) Omega IOD_C = -4.261222001
================ Omega D = 0.000000000
Omega Rest = 5.616913337
Penalty func = 0.000000000
Final Spread (Ang^2) Omega Total_C = -4.261222001
------------------------------------------------------------------------------
8.7.6 Plotting
After WF have been localised, wannier90 enters its plotting routines (if required). For example, if you
have specified an interpolated bandstucture:
*---------------------------------------------------------------------------*
| PLOTTING |
*---------------------------------------------------------------------------*
Calculating interpolated band-structure

wannier90: User Guide 91
8.7.7 Summary timings
At the very end of the run, a summary of the time taken for various parts of the calculation is given.
The level of detail is controlled by the timing_level input parameter (set to 1 by default).
*===========================================================================*
| TIMING INFORMATION |
*===========================================================================*
| Tag Ncalls Time (s)|
|---------------------------------------------------------------------------|
|kmesh: get : 1 0.212|
|overlap: read : 1 0.060|
|wann: main : 1 1.860|
|plot: main : 1 0.168|
*---------------------------------------------------------------------------*
All done: wannier90 exiting
8.8 seedname.chk
INPUT/OUTPUT. Information required to restart the calculation or enter the plotting phase. If we
have used disentanglement this file also contains the rectangular matrices Udis(k).
8.9 seedname.r2mn
OUTPUT. Written if write_r2mn =true. The matrix elements hm|r2|ni(where mand nrefer to
MLWF)
8.10 seedname_band.dat
OUTPUT. Written if bands_plot=.TRUE.; The raw data for the interpolated band structure.
8.11 seedname_band.gnu
OUTPUT. Written if bands_plot=.TRUE. and bands_plot_format=gnuplot; A gnuplot script to plot
the interpolated band structure.
8.12 seedname_band.agr
OUTPUT. Written if bands_plot=.TRUE. and bands_plot_format=xmgrace; A grace file to plot the
interpolated band structure.

92 wannier90: User Guide
8.13 seedname_band.kpt
OUTPUT. Written if bands_plot=.TRUE.; The k-points used for the interpolated band structure, in
units of the reciprocal lattice vectors. This file can be used to generate a comparison band structure
from a first-principles code.
8.14 seedname.bxsf
OUTPUT. Written if fermi_surface_plot=.TRUE.; A Fermi surface plot file suitable for plotting with
XCrySDen.
8.15 seedname_w.xsf
OUTPUT. Written if wannier_plot=.TRUE. and wannier_plot_format=xcrysden. Contains the wth
WF in real space in a format suitable for plotting with XCrySDen or VMD, for example.
8.16 seedname_w.cube
OUTPUT. Written if wannier_plot=.TRUE. and wannier_plot_format=cube. Contains the wth WF
in real space in Gaussian cube format, suitable for plotting in XCrySDen, VMD, gopenmol etc.
8.17 UNKp.s
INPUT. Read if wannier_plot=.TRUE. and used to plot the MLWF. Read if transport_mode=lcr
and tran_read_ht=.FALSE. for use in automated lcr transport calculations.
The periodic part of the Bloch states represented on a regular real space grid, indexed by k-point p
(from 1 to num_kpts) and spin s(‘1’ for ‘up’, ‘2’ for ‘down’).
The name of the wavefunction file is assumed to have the form:
write(wfnname,200) p,spin
200 format (’UNK’,i5.5,’.’,i1)
The first line of each file should contain 5 integers: the number of grid points in each direction (ngx,
ngy and ngz), the k-point number ik and the total number of bands num_band in the file. The full file
will be read by wannier90 as:
read(file_unit) ngx,ngy,ngz,ik,nbnd
do loop_b=1,num_bands
read(file_unit) (r_wvfn(nx,loop_b),nx=1,ngx*ngy*ngz)
end do
If spinors=true then s=‘NC’, and the name of the wavefunction file is assumed to have the form:

wannier90: User Guide 93
write(wfnname,200) p
200 format (’UNK’,i5.5,’.NC’)
and the file will be read by wannier90 as:
read(file_unit) ngx,ngy,ngz,ik,nbnd
do loop_b=1,num_bands
read(file_unit) (r_wvfn_nc(nx,loop_b,1),nx=1,ngx*ngy*ngz) ! up-spinor
read(file_unit) (r_wvfn_nc(nx,loop_b,2),nx=1,ngx*ngy*ngz) ! down-spinor
end do
All UNK files can be in formatted or unformatted style, this is controlled by the logical keyword
wvfn_formatted.
8.18 seedname_centres.xyz
OUTPUT. Written if write_xyz=.TRUE.; xyz format atomic structure file suitable for viewing with
your favourite visualiser (jmol,gopenmol,vmd, etc.).
8.19 seedname_hr.dat
OUTPUT. Written if write_hr=.TRUE.. The first line gives the date and time at which the file was
created. The second line states the number of Wannier functions num_wann. The third line gives the
number of Wigner-Seitz grid-points nrpts. The next block of nrpts integers gives the degeneracy of
each Wigner-Seitz grid point, with 15 entries per line. Finally, the remaining num_wann2×nrpts lines
each contain, respectively, the components of the vector Rin terms of the lattice vectors {Ai}, the
indices mand n, and the real and imaginary parts of the Hamiltonian matrix element H(R)
mn in the WF
basis, e.g.,
Created on 24May2007 at 23:32:09
20
17
412141121461112
1 2
0 0 -2 1 1 -0.001013 0.000000
0 0 -2 2 1 0.000270 0.000000
0 0 -2 3 1 -0.000055 0.000000
0 0 -2 4 1 0.000093 0.000000
0 0 -2 5 1 -0.000055 0.000000
.
.
.

94 wannier90: User Guide
8.20 seedname_r.dat
OUTPUT. Written if write_rmn =true. The matrix elements hm0|r|nRi(where nRrefers to MLWF
nin unit cell R). The first line gives the date and time at which the file was created. The second
line states the number of Wannier functions num_wann. The third line states the number of Rvectors
nrpts. Similar to the case of the Hamiltonian matrix above, the remaining num_wann2×nrpts lines
each contain, respectively, the components of the vector Rin terms of the lattice vectors {Ai}, the
indices mand n, and the real and imaginary parts of the position matrix element in the WF basis.
8.21 seedname.bvec
OUTPUT. Written if write_bvec =true. This file contains the matrix elements of bvector and their
weights. The first line gives the date and time at which the file was created. The second line states
the number of k-points and the total number of neighbours for each k-point nntot. Then all the other
lines contain the b-vector (x,y,z) coordinate and weigths for each k-points and each of its neighbours.
8.22 seedname_wsvec.dat
OUTPUT. Written if write_hr =true or write_rmn =true or write_tb =true. The first line gives
the date and time at which the file was created and the value of use_ws_distance. For each pair of
Wannier functions (identified by the components of the vector Rseparating their unit cells and their
indices) it gives: (i) the number of lattice vectors of the periodic supercell Tthat bring the Wannier
function in Rback in the Wigner-Seitz cell centred on the other Wannier function and (ii) the set
of superlattice vectors Tto make this transformation. These superlattice vectors Tshould be added
to the Rvector to obtain the correct centre of the Wannier function that underlies a given matrix
element (e.g. the Hamiltonian matrix elements in seedname_hr.dat) in order to correctly interpolate
in reciprocal space.
## written on 20Sep2016 at 18:12:37 with use_ws_distance=.true.
00011
1
000
00012
1
000
00013
1
000
00014
1
000
00015
1
000
00016
2

wannier90: User Guide 95
0 -1 -1
1 -1 -1
.
.
.
8.23 seedname_qc.dat
OUTPUT. Written if transport =.TRUE.. The first line gives the date and time at which the file was
created. In the subsequent lines, the energy value in units of eV is written in the left column, and the
quantum conductance in units of 2e2
h(e2
hfor a spin-polarized system) is written in the right column.
## written on 14Dec2007 at 11:30:17
-3.000000 8.999999
-2.990000 8.999999
-2.980000 8.999999
-2.970000 8.999999
.
.
.
8.24 seedname_dos.dat
OUTPUT. Written if transport =.TRUE.. The first line gives the date and time at which the file
was created. In the subsequent lines, the energy value in units of eV is written in the left column, and
the density of states in an arbitrary unit is written in the right column.
## written on 14Dec2007 at 11:30:17
-3.000000 6.801199
-2.990000 6.717692
-2.980000 6.640828
-2.970000 6.569910
.
.
.
8.25 seedname_htB.dat
INPUT/OUTPUT. Read if transport_mode =bulk and tran_read_ht =.TRUE.. Written if tran_write_ht =
.TRUE.. The first line gives the date and time at which the file was created. The second line gives
tran_num_bb. The subsequent lines contain tran_num_bb×tran_num_bb Hmn matrix, where the in-
dices mand nspan all tran_num_bb WFs located at 0th principal layer. Then tran_num_bb is recorded
again in the new line followed by Hmn, where mth WF is at 0th principal layer and nth at 1st principal
layer. The Hmn matrix is written in such a way that mis the fastest varying index.

96 wannier90: User Guide
written on 14Dec2007 at 11:30:17
150
-1.737841 -2.941054 0.052673 -0.032926 0.010738 -0.009515
0.011737 -0.016325 0.051863 -0.170897 -2.170467 0.202254
.
.
.
-0.057064 -0.571967 -0.691431 0.015155 -0.007859 0.000474
-0.000107 -0.001141 -0.002126 0.019188 -0.686423 -10.379876
150
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
.
.
.
0.000000 0.000000 0.000000 0.000000 0.000000 -0.001576
0.000255 -0.000143 -0.001264 0.002278 0.000000 0.000000
8.26 seedname_htL.dat
INPUT. Read if transport_mode =lcr and tran_read_ht =.TRUE.. The file must be written in
the same way as in seedname_htB.dat. The first line can be any comment you want. The second line
gives tran_num_ll.tran_num_ll in seedname_htL.dat must be equal to that in seedname.win. The
code will stop otherwise.
Created by a WANNIER user
105
0.316879 0.000000 -2.762434 0.048956 0.000000 -0.016639
0.000000 0.000000 0.000000 0.000000 0.000000 -2.809405
.
.
.
0.000000 0.078188 0.000000 0.000000 -2.086453 -0.001535
0.007878 -0.545485 -10.525435
105
0.000000 0.000000 0.000315 -0.000294 0.000000 0.000085
0.000000 0.000000 0.000000 0.000000 0.000000 0.000021
.
.
.
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000
8.27 seedname_htR.dat
INPUT. Read if transport_mode =lcr and tran_read_ht =.TRUE. and tran_use_same_lead =
.FALSE.. The file must be written in the same way as in seedname_htL.dat.tran_num_rr in

wannier90: User Guide 97
seedname_htR.dat must be equal to that in seedname.win.
8.28 seedname_htC.dat
INPUT. Read if transport_mode =lcr and tran_read_ht =.TRUE.. The first line can be any com-
ment you want. The second line gives tran_num_cc. The subsequent lines contain tran_num_cc×tran_num_cc
Hmn matrix, where the indices mand nspan all tran_num_cc WFs inside the central conductor region.
tran_num_cc in seedname_htC.dat must be equal to that in seedname.win.
Created by a WANNIER user
99
-10.499455 -0.541232 0.007684 -0.001624 -2.067078 -0.412188
0.003217 0.076965 0.000522 -0.000414 0.000419 -2.122184
.
.
.
-0.003438 0.078545 0.024426 0.757343 -2.004899 -0.001632
0.007807 -0.542983 -10.516896
8.29 seedname_htLC.dat
INPUT. Read if transport_mode =lcr and tran_read_ht =.TRUE.. The first line can be any
comment you want. The second line gives tran_num_ll and tran_num_lc in the given order. The
subsequent lines contain tran_num_ll×tran_num_lc Hmn matrix. The index mspans tran_num_ll
WFs in the surface principal layer of semi-infinite left lead which is in contact with the conductor
region. The index nspans tran_num_lc WFs in the conductor region which have a non-negligible
interaction with the WFs in the semi-infinite left lead. Note that tran_num_lc can be different from
tran_num_cc.
Created by a WANNIER user
105 99
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
.
.
.
-0.000003 0.000009 0.000290 0.000001 -0.000007 -0.000008
0.000053 -0.000077 -0.000069
8.30 seedname_htCR.dat
INPUT. Read if transport_mode =lcr and tran_read_ht =.TRUE.. The first line can be any
comment you want. The second line gives tran_num_cr and tran_num_rr in the given order. The
subsequent lines contain tran_num_cr×tran_num_rr Hmn matrix. The index mspans tran_num_cr
WFs in the conductor region which have a non-negligible interaction with the WFs in the semi-infinite

98 wannier90: User Guide
right lead. The index nspans tran_num_rr WFs in the surface principal layer of semi-infinite right
lead which is in contact with the conductor region. Note that tran_num_cr can be different from
tran_num_cc.
Created by a WANNIER user
99 105
-0.000180 0.000023 0.000133 -0.000001 0.000194 0.000008
-0.000879 -0.000028 0.000672 -0.000257 -0.000102 -0.000029
.
.
.
0.000000 0.000000 0.000000 0.000000 0.000000 0.000000
0.000000 0.000000 0.000000
8.31 seedname.unkg
INPUT. Read if transport_mode =lcr and tran_read_ht =.FALSE.. The first line is the number
of G-vectors at which the ˜umk(G)are subsequently printed. This number should always be 32 since
32 specific ˜umkare required. The following lines contain the following in this order: The band index
m, a counter on the number of G-vectors, the integer co-efficient of the G-vector components a, b, c
(where G=ab1+bb2+cb3), then the real and imaginary parts of the corresponding ˜umk(G)at the
Γ-point. We note that the ordering in which the G-vectors and ˜umk(G)are printed is not important,
but the specific G-vectors are critical. The following example displays for a single band, the complete
set of ˜umk(G)that are required. Note the G-vectors (a, b, c) needed.
32
1 1 0 0 0 0.4023306 0.0000000
1 2 0 0 1 -0.0000325 0.0000000
1 3 0 1 0 -0.3043665 0.0000000
1 4 1 0 0 -0.3043665 0.0000000
1 5 2 0 0 0.1447143 0.0000000
1 6 1 -1 0 0.2345179 0.0000000
1 7 1 1 0 0.2345179 0.0000000
1 8 1 0 -1 0.0000246 0.0000000
1 9 1 0 1 0.0000246 0.0000000
1 10 0 2 0 0.1447143 0.0000000
1 11 0 1 -1 0.0000246 0.0000000
1 12 0 1 1 0.0000246 0.0000000
1 13 0 0 2 0.0000338 0.0000000
1 14 3 0 0 -0.0482918 0.0000000
1 15 2 -1 0 -0.1152414 0.0000000
1 16 2 1 0 -0.1152414 0.0000000
1 17 2 0 -1 -0.0000117 0.0000000
1 18 2 0 1 -0.0000117 0.0000000
1 19 1 -2 0 -0.1152414 0.0000000
1 20 1 2 0 -0.1152414 0.0000000
1 21 1 -1 -1 -0.0000190 0.0000000
1 22 1 -1 1 -0.0000190 0.0000000

wannier90: User Guide 99
1 23 1 1 -1 -0.0000190 0.0000000
1 24 1 1 1 -0.0000190 0.0000000
1 25 1 0 -2 -0.0000257 0.0000000
1 26 1 0 2 -0.0000257 0.0000000
1 27 0 3 0 -0.0482918 0.0000000
1 28 0 2 -1 -0.0000117 0.0000000
1 29 0 2 1 -0.0000117 0.0000000
1 30 0 1 -2 -0.0000257 0.0000000
1 31 0 1 2 -0.0000257 0.0000000
1 32 0 0 3 0.0000187 0.0000000
2 1 0 0 0 -0.0000461 0.0000000
.
.
.
8.32 seedname_u.mat
OUTPUT. Written if write_u_matrices =.TRUE.. The first line gives the date and time at which the
file was created. The second line states the number of kpoints num_kpts and the number of wannier
functions num_wann twice. The third line is empty. Then there are num_kpts blocks of data, each
of which starts with a line containing the kpoint (in fractional coordinates of the reciprocal lattice
vectors) followed by num_wann * num_wann lines containing the matrix elements (real and imaginary
parts) of U(k). The matrix elements are in column-major order (ie, cycling over rows first and then
columns). There is an empty line between each block of data.
written on 15Sep2016 at 16:33:46
64 8 8
0.0000000000 +0.0000000000 +0.0000000000
0.4468355787 +0.1394579978
-0.0966033667 +0.4003934902
-0.0007748974 +0.0011788678
-0.0041177339 +0.0093821027
.
.
.
0.1250000000 0.0000000000 +0.0000000000
0.4694005589 +0.0364941808
+0.2287801742 -0.1135511138
-0.4776782452 -0.0511719121
+0.0142081014 +0.0006203139
.
.
.

100 wannier90: User Guide
8.33 seedname_u_dis.mat
OUTPUT. Written if write_u_matrices =.TRUE. and disentanglement is enabled. The first line
gives the date and time at which the file was created. The second line states the number of kpoints
num_kpts, the number of wannier functions num_bands and the number of num_bands. The third line
is empty. Then there are num_kpts blocks of data, each of which starts with a line containing the
kpoint (in fractional coordinates of the reciprocal lattice vectors) followed by num_wann * num_bands
lines containing the matrix elements (real and imaginary parts) of Udis(k). The matrix elements are
in column-major order (ie, cycling over rows first and then columns). There is an empty line between
each block of data.
written on 15Sep2016 at 16:33:46
64 8 16
0.0000000000 +0.0000000000 +0.0000000000
1.0000000000 +0.0000000000
+0.0000000000 +0.0000000000
+0.0000000000 +0.0000000000
+0.0000000000 +0.0000000000
.
.
.
0.1250000000 0.0000000000 +0.0000000000
1.0000000000 +0.0000000000
+0.0000000000 +0.0000000000
+0.0000000000 +0.0000000000
+0.0000000000 +0.0000000000
.
.
.
Chapter 9
Some notes on the interpolation
In wannier90 v.2.1, a new flag use_ws_distance has been introduced (and it is set to .true. by
default since version v3.0). Setting it to .false. reproduces the “standard” behavior of wannier90 in
v.2.0.1 and earlier, while setting it to .true. changes the interpolation method as described below. In
general, this allows a smoother interpolation, helps reducing (a bit) the number of k−points required
for interpolation, and reproduces the band structure of large supercells sampled at Γonly (setting
it to .false. produces instead flat bands, which might instead be the intended behaviour for small
molecules carefully placed at the centre of the cell).
The core idea rests on the fact that the Wannier functions wnR(r)that we build from N×M×L
k−points are actually periodic over a supercell of size N×M×L, but when you use them to interpolate
you want them to be zero outside this supercell. In 1D it is pretty obvious want we mean here, but in
3D what you really want that they are zero outside the Wigner–Seitz cell of the N×M×Lsuperlattice.
The best way to impose this condition is to check that every real-space distance that enters in the
R→kFourier transform is the shortest possible among all the N×M×L−periodic equivalent copies.
If the distances were between unit cells, this would be trivial, but the distances are between Wannier
functions which are not centred on R= 0. Hence, when you want to consider the matrix element of a
generic operator O(i.e., the Hamiltonian) hwi0(r)|O|wjR(r)iyou must take in account that the centre
τiof wi0(r)may be very far away from 0and the centre τjof wjR(r)may be very far away from R.
There are many way to find the shortest possible distance between wi0(r)and wjR(r−R), the one
used here is to consider the distance dijR=τi−(τj+R)and all its superlattice periodic equivalents
dijR+˜
Rnml, with ˜
Rnml = (Nna1+Mma2+Lla3)and n, l, m =−L, −L+ 1, ...0, ..., L −1, L, with L
controlled by the parameter ws_search_size.
Then,
1. if dijR+˜
Rnml is inside the N×M×Lsuper-WS cell, then it is the shortest, take it and quit
2. if it is outside the WS, then it is not the shortest, throw it away
3. if it is on the border/corner of the WS then it is the shortest, but there are other choices of
(n, m, l)which are equivalent, find all of them
In all distance comparisons, a small but finite tolerance is considered, which can be controlled with
the parameter ws_distance_tol.
101

102 wannier90: User Guide
Because of how the Fourier transform is defined in the wannier90 code (not the only possible choice)
it is only R+˜
Rnml that enters the exponential, but you still have to consider the distance among
the actual centres of the Wannier functions. Using the centres of the unit-cell to which the Wannier
functions belong is not enough (but is easier, and saves you one index).
Point 3 is not stricly necessary, but using it helps enforcing the symmetry of the system in the resulting
band structure. You will get some small but evident symmetry breaking in the band plots if you just
pick one of the equivalent ˜
Rvectors.
Note that in some cases, all this procedure does absolutely nothing, for instance if all the Wannier
function centres are very close to 0 (e.g., a molecule carefully placed in the periodic cell).
In some other cases, the effect may exist but be imperceptible. E.g., if you use a very fine grid of
k−points, even if you don’t centre each functions perfectly, the periodic copies will still be so far away
that the change in centre applied with use_ws_distance does not matter.
When instead you use few k−points, activating the use_ws_distance may help a lot in avoiding
spurious oscillations of the band structure even when the Wannier functions are well converged.
Chapter 10
Sample Input Files
10.1 Master input file: seedname.win
num_wann : 4
mp_grid : 4 4 4
num_iter : 100
postproc_setup : true
begin unit_cell_cart
ang
-1.61 0.00 1.61
0.00 1.61 1.61
-1.61 1.61 0.00
end unit_cell_cart
begin atoms_frac
C -0.125 -0.125 -0.125
C 0.125 0.125 0.125
end atoms_frac
bands_plot : true
bands_num_points : 100
bands_plot_format : gnuplot
begin kpoint_path
L 0.50000 0.50000 0.50000 G 0.00000 0.00000 0.00000
G 0.00000 0.00000 0.00000 X 0.50000 0.00000 0.50000
X 0.50000 0.00000 0.50000 K 0.62500 0.25000 0.62500
end kpoint_path
begin projections
C:l=0,l=1
end projections
begin kpoints
103

104 wannier90: User Guide
0.00 0.00 0.00
0.00 0.00 0.25
0.00 0.50 0.50
.
.
.
0.75 0.75 0.50
0.75 0.75 0.75
end kpoints
10.2 seedname.nnkp
Running wannier90 on the above input file would generate the following nnkp file:
File written on 9Feb2006 at 15:13: 9
calc_only_A : F
begin real_lattice
-1.612340 0.000000 1.612340
0.000000 1.612340 1.612340
-1.612340 1.612340 0.000000
end real_lattice
begin recip_lattice
-1.951300 -1.951300 1.951300
1.951300 1.951300 1.951300
-1.951300 1.951300 -1.951300
end recip_lattice
begin kpoints
64
0.00000 0.00000 0.00000
0.00000 0.25000 0.00000
0.00000 0.50000 0.00000
0.00000 0.75000 0.00000
0.25000 0.00000 0.00000
.
.
.
0.50000 0.75000 0.75000
0.75000 0.00000 0.75000
0.75000 0.25000 0.75000
0.75000 0.50000 0.75000
0.75000 0.75000 0.75000
end kpoints

wannier90: User Guide 105
begin projections
8
-0.12500 -0.12500 -0.12500 0 1 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
-0.12500 -0.12500 -0.12500 1 1 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
-0.12500 -0.12500 -0.12500 1 2 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
-0.12500 -0.12500 -0.12500 1 3 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
0.12500 0.12500 0.12500 0 1 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
0.12500 0.12500 0.12500 1 1 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
0.12500 0.12500 0.12500 1 2 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
0.12500 0.12500 0.12500 1 3 1
0.000 0.000 1.000 1.000 0.000 0.000 2.00
end projections
begin nnkpts
8
1 2 0 0 0
1 4 0 -1 0
1 5 0 0 0
1 13 -1 0 0
1 17 0 0 0
1 22 0 0 0
1 49 0 0 -1
1 64 -1 -1 -1
2 1 0 0 0
2 3 0 0 0
2 6 0 0 0
2 14 -1 0 0
2 18 0 0 0
2 23 0 0 0
2 50 0 0 -1
2 61 -1 0 -1
.
.
.
64 1 1 1 1
64 16 0 0 1
64 43 0 0 0
64 48 0 0 0
64 52 1 0 0
64 60 0 0 0
64 61 0 1 0
64 63 0 0 0

106 wannier90: User Guide
end nnkpts
begin exclude_bands
4
1
2
3
4
end exclude_bands
Part III
postw90.x
107
Chapter 11
Parameters
11.1 Introduction
The wannier90.x code described in Part II calculates the maximally-localized Wannier functions.
The postw90.x executable contains instead a series of modules that take the Wannier functions cal-
culated by wannier90.x and use them to calculate different properties. This executable is parallel
(by means of MPI libraries), so it can be run on multiple CPUs. The information on the calculated
Wannier functions is read from the checkpoint seedname.chk file. Note that this is written in an un-
formatted machine-dependent format. If you need to use this file on a different machine, or you want
to use a version of postw90.x compiled with a different compiler, refer to Sec. A.2 in the Appendices
for a description of how to export/import this file.
11.2 Usage
postw90.x can be run in parallel using MPI libraries to reduce the computation time.
For serial execution use: postw90.x [seedname]
•seedname: If a seedname string is given the code will read its input from a file seedname.win.
The default value is wannier. One can also equivalently provide the string seedname.win instead
of seedname.
For parallel execution use: mpirun -np NUMPROCS postw90.x [seedname]
•NUMPROCS: substitute with the number of processors that you want to use.
Note that the mpirun command and command-line flags may be different in your MPI implementation:
read your MPI manual or ask your computer administrator.
Note also that this requires that the postw90.x executable has been compiled in its parallel version
(follow the instructions in the file README.install in the main directory of the wannier90 distribution)
and that the MPI libraries and binaries are installed and correctly configured on your machine.
109

110 wannier90: User Guide
11.3 seedname.win File
The postw90.x uses the same seedname.win input file of wannier90.x. The input keywords of
postw90.x must thus be added to this file, using the same syntax described in Sec. 2.2.
Note that wannier90.x checks if the syntax of the input file is correct, but then ignores the value of
the flags that refer only to modules of postw90.x, so one can safely run wannier90.x on a file that
contains also postw90.x flags.
Similarly, postw90.x ignores flags that refer only to wannier90.x (as number of iterations, restart
flags, . . . ). However, some parts of the input file must be there, as for instance the number of Wannier
functions, etc.
The easiest thing to do is therefore to simply add the postw90 input keywords to the seedname.win
file that was used to obtain the Wannier functions.
11.4 List of available modules
The currently available modules in postw90.x are:
•dos: Calculation of the density of states (DOS), projected density of states (PDOS), net spin
etc.
•kpath: Calculation of k-space quantities such as energy bands and Berry curvature along a
piecewise linear path in the BZ (see examples 17 and 18 of the tutorial).
•kslice: Calculation of k-space quantities on a planar slice of the BZ (see examples 17 and 18 of
the tutorial).
•berry: Calculation of properties related to the BZ integral of the Berry curvature and Berry
connection, including anomalous Hall conductivity, orbital magnetisation, optical conductivity
and nonlinear shift current (see Chap. 12 and examples 18, 19 and 25 of the tutorial).
•gyrotropic: Calculation of gyrotropic properties, including natural and current0induced optical
rotation, and the current-induced magnetization (see Chap. 13 and examples of the tutorial).
•BoltzWann: Calculation of electronic transport properties for bulk materials using the semiclas-
sical Boltzmann transport equation (see Chap. 14 and example 16 of the tutorial).
•geninterp (Generic Band Interpolation): Calculation band energies (and band derivatives) on a
generic list of kpoints (see Chap. 15).
11.5 Keyword List
On the next pages the list of available postw90 input keywords is reported. In particular, Table 11.1
reports keywords that affect the generic behavior of all modules of postw90. Often, these are “global”
variables that can be overridden by module-specific keywords (as for instance the kmesh flag). The
subsequent tables describe the input parameters for each specific module.
A description of the behaviour of the global flags is described Sec. 11.6; the description of the flags
specific to the modules can be found in the following sections.
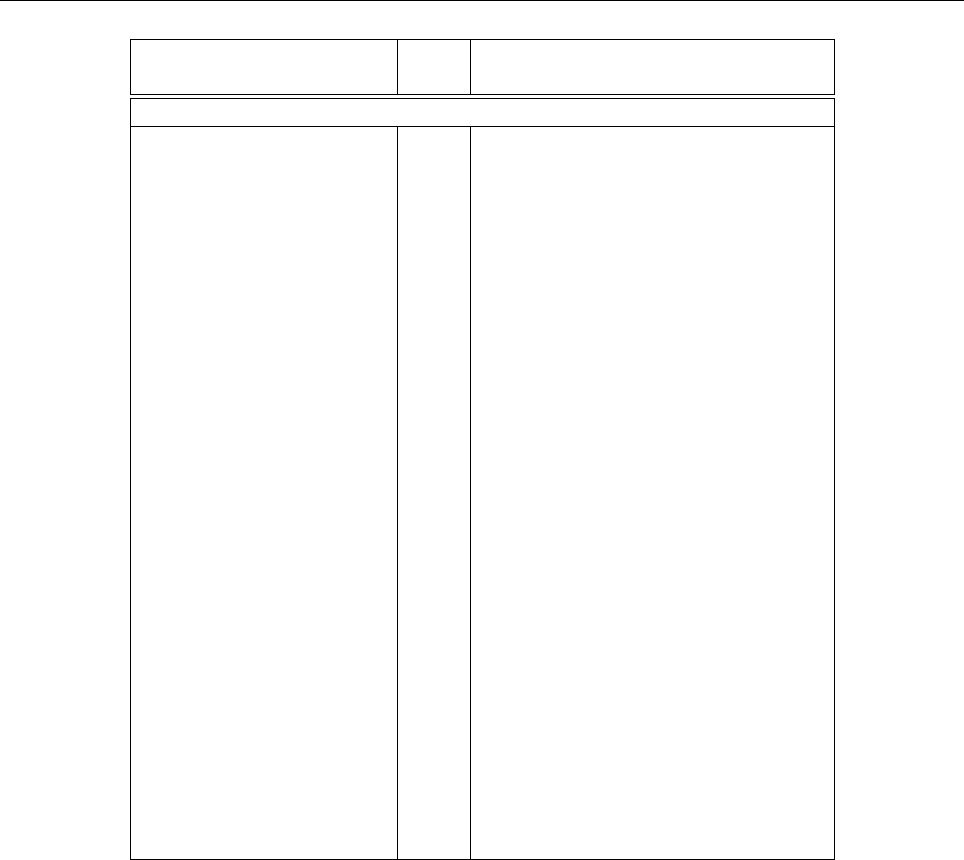
wannier90: User Guide 111
Keyword Type Description
Global Parameters of postw90
kmesh I Dimensions of the uniform interpo-
lation k-mesh (one or three integers)
kmesh_spacing R Minimum spacing between kpoints
in Å−1
adpt_smr L Use adaptive smearing
adpt_smr_fac R Adaptive smearing prefactor
adpt_smr_max P Maximum allowed value for the
adaptive energy smearing (eV)
smr_type S Analytical form used for the broad-
ened delta function
smr_fixed_en_width P Energy smearing (if non-adaptive)
num_elec_per_state I Number of electrons per state
scissors_shift P Scissors shift applied to the conduc-
tion bands (eV) (deprecated)
num_valence_bands I Number of valence bands
spin_decomp L Decompose various properties into
up-spin, down-spin, and possibly
spin-flip parts
spin_axis_polar P Polar angle of the spin quantization
axis (deg)
spin_axis_azimuth P Azimuthal angle of the spin quanti-
zation axis (deg)
spin_moment∗L Determines whether to evaluate the
spin magnetic moment per cell
uHu_formatted L Read a formatted seedname.uHu file
spn_formatted L Read a formatted seedname.spn file
berry_curv_unit S Unit of Berry curvature
Table 11.1: seedname.win file keywords controlling the general behaviour of the modules in postw90.
Argument types are represented by, I for a integer, R for a real number, P for a physical value, L for
a logical value and S for a text string.
The keyword spin_moment does not affect the behavior of the modules in postw90, and does not really
belong to any of them. It is listed here for lack of a better place.
112 wannier90: User Guide
Keyword Type Description
dos Parameters
dos L Calculate the density of states and
related properties
dos_task S List of properties to compute
dos_energy_min P Lower limit of the energy range for
computing the DOS (eV)
dos_energy_max P Upper limit of the energy range for
computing the DOS (eV)
dos_energy_step R Step for increasing the energy in the
specified range (eV)
dos_project I List of WFs onto which the DOS is
projected
[dos_]kmesh I Dimensions of the uniform interpo-
lation k-mesh (one or three integers)
[dos_]kmesh_spacing R Minimum spacing between kpoints
in Å−1
[dos_]adpt_smr L Use adaptive smearing for the DOS
[dos_]adpt_smr_fac R Adaptive smearing prefactor
[dos_]adpt_smr_max P Maximum allowed value for the
adaptive energy smearing (eV)
[dos_]smr_fixed_en_width P Energy smearing (if non-adaptive)
for the DOS (eV)
[dos_]smr_type S Analytical form used for the broad-
ened delta function when computing
the DOS.
Table 11.2: seedname.win file keywords controlling the dos module. Argument types are represented
by, I for a integer, R for a real number, P for a physical value, L for a logical value and S for a text
string.
Keyword Type Description
kpath Parameters
kpath L Calculate properties along a piece-
wise linear path in the BZ
kpath_task L List of properties to evaluate
kpath_num_points I Number of points in the first kpath
segment
kpath_bands_colour S Property used to colour the energy
bands along the path
Table 11.3: seedname.win file keywords controlling the kpath module. Argument types are represented
by, I for a integer, R for a real number, P for a physical value, L for a logical value and S for a text
string.

wannier90: User Guide 113
Keyword Type Description
kslice Parameters
kslice L Calculate properties on a slice in the
BZ
kslice_task S List of properties to evaluate
kslice_corner R Position of the corner of the slice
kslice_b1 R First vector defining the slice
kslice_b2 R Second vector defining the slice
kslice_2dkmesh I Dimensions of the uniform interpo-
lation k-mesh on the slice (one or
two integers)
kslice_fermi_level PThis parameter is not used anymore.
Use fermi_energy instead.
kslice_fermi_lines_colour S Property used to colour the Fermi
lines
Table 11.4: seedname.win file keywords controlling the kslice module. Argument types are repre-
sented by, I for a integer, R for a real number, P for a physical value, L for a logical value and S for a
text string.
114 wannier90: User Guide
Keyword Type Description
berry Parameters
berry L Calculate Berry-type quantities
berry_task L List of properties to compute
[berry_]kmesh I Dimensions of the uniform interpo-
lation k-mesh (one or three integers)
[berry_]kmesh_spacing R Minimum spacing between kpoints
in Å−1
berry_curv_adpt_kmesh I Linear dimension of the adaptively
refined k-mesh used to compute the
anomalous Hall conductivity
berry_curv_adpt_kmesh_thresh P Threshold magnitude of the Berry
curvature for adaptive refinement
kubo_freq_min P Lower limit of the frequency range
for optical spectra, JDOS and shift
current (eV)
kubo_freq_max P Upper limit of the frequency range
for optical spectra, JDOS and shift
current (eV)
kubo_freq_step R Step for increasing the optical fre-
quency in the specified range
kubo_eigval_max P Maximum energy eigenvalue in-
cluded when evaluating the Kubo-
Greenwood conductivity, JDOS and
shift current
[kubo_]adpt_smr L Use adaptive energy smearing for
the optical conductivity, JDOS and
shift current
[kubo_]adpt_smr_fac R Adaptive smearing prefactor
[kubo_]adpt_smr_max P Maximum allowed value for the
adaptive energy smearing (eV)
[kubo_]smr_type S Analytical form used for the broad-
ened delta function when computing
the optical conductivity, JDOS and
shift current
[kubo_]smr_fixed_en_width P Energy smearing (if non-adaptive)
for the optical conductivity, JDOS
and shift current (eV)
sc_eta R Energy broadening of energy differ-
ences in the sum over virtual states
when computing shift current
sc_phase_conv I Convention for phase factor of Bloch
states when computing shift current
sc_w_thr R Frequency threshold for speeding
up delta function integration when
computing shift current
Table 11.5: seedname.win file keywords controlling the berry module. Argument types are represented
by, I for a integer, R for a real number, P for a physical value, L for a logical value and S for a text
string.

wannier90: User Guide 115
Keyword Type Description
berry Parameters
gyrotropic L Calculate gyrotropic quantities
gyrotropic_task L List of properties to compute
[gyrotropic_]kmesh I Dimensions of the uniform interpo-
lation k-mesh (one or three integers)
[gyrotropic_]kmesh_spacing R Minimum spacing between kpoints
in Å−1
gyrotropic_freq_min P Lower limit of the frequency range
for optical rotation (eV)
gyrotropic_freq_max P Upper limit of the frequency range
for optical rotation (eV)
gyrotropic_freq_step P Step for increasing the optical fre-
quency in the specified range
gyrotropic_eigval_max P Maximum energy eigenvalue in-
cluded when evaluating the inter-
band natural optical activity
gyrotropic_degen_thresh P threshold to exclude degenerate
bands from the calculation
[gyrotropic_]smr_type S Analytical form used for the broad-
ened delta function
[gyrotropic_]smr_fixed_en_width P Energy smearing (eV)
[gyrotropic_]band_list I list of bands used in the calculation
gyrotropic_box_center R The center and three basis vectors,
defining the box for integration (in
reduced coordinates, three real
numbers for each vector)
gyrotropic_box_b1 R
gyrotropic_box_b2 R
gyrotropic_box_b3 R
Table 11.6: seedname.win file keywords controlling the gyrotropic module. Argument types are
represented by, I for a integer, R for a real number, P for a physical value, L for a logical value and S
for a text string.
116 wannier90: User Guide
Keyword Type Description
BoltzWann Parameters
boltzwann L Calculate Boltzmann transport co-
efficients
[boltz_]kmesh I Dimensions of the uniform interpo-
lation k-mesh (one or three integers)
[boltz_]kmesh_spacing R Minimum spacing between kpoints
in Å−1
boltz_2d_dir S Non-periodic direction (for 2D sys-
tems only)
boltz_relax_time P Relaxation time in fs
boltz_mu_min P Minimum value of the chemical po-
tential µin eV
boltz_mu_max P Maximum value of the chemical po-
tential µin eV
boltz_mu_step R Step for µin eV
boltz_temp_min P Minimum value of the tempera-
ture Tin Kelvin
boltz_temp_max P Maximum value of the tempera-
ture Tin Kelvin
boltz_temp_step R Step for Tin Kelvin
boltz_tdf_energy_step R Energy step for the TDF (eV)
boltz_tdf_smr_fixed_en_width P Energy smearing for the TDF (eV)
boltz_tdf_smr_type S Smearing type for the TDF
boltz_calc_also_dos L Calculate also DOS while calculat-
ing the TDF
boltz_dos_energy_min P Minimum value of the energy for the
DOS in eV
boltz_dos_energy_max P Maximum value of the energy for the
DOS in eV
boltz_dos_energy_step R Step for the DOS in eV
[boltz_dos_]smr_type S Smearing type for the DOS
[boltz_dos_]adpt_smr L Use adaptive smearing for the DOS
[boltz_dos_]adpt_smr_fac R Adaptive smearing prefactor
[boltz_dos_]adpt_smr_max P Maximum allowed value for the
adaptive energy smearing (eV)
[boltz_dos_smr_]fixed_en_width P Energy smearing (if non-adaptive)
for the DOS (eV)
boltz_bandshift L Rigid bandshift of the conduction
bands
boltz_bandshift_firstband I Index of the first band to shift
boltz_bandshift_energyshift P Energy shift of the conduction bands
(eV)
Table 11.7: seedname.win file keywords controlling the BoltzWann module (calculation of the Boltz-
mann transport coefficients in the Wannier basis). Argument types are represented by, I for a integer,
R for a real number, P for a physical value, L for a logical value and S for a text string.

wannier90: User Guide 117
Keyword Type Description
geninterp Parameters
geninterp L Calculate bands for given set of k
points
geninterp_alsofirstder L Calculate also first derivatives
geninterp_single_file L Write a single file or one for each
process
Table 11.8: seedname.win file keywords controlling the Generic Band Interpolation (geninterp) mod-
ule. Argument types are represented by, I for a integer, R for a real number, P for a physical value, L
for a logical value and S for a text string.

118 wannier90: User Guide
11.6 Global variables
11.6.1 integer :: kmesh(:)
Dimensions of the interpolation grid used in postw90.x.
Not to be confused with the mp_grid input flag, which instead specifies the Monkhorst–Pack grid used
in the ab-initio calculation!
If three integers l m n are given, the reciprocal-space cell subtended by the three primitive translations
is sampled on a uniform l×m×ngrid (including Γ). If only one integer mis given, an m×m×m
grid is used.
If you use a module which needs a k-mesh, either kmesh_spacing or kmesh must be defined.
11.6.2 real(kind=dp) :: kmesh_spacing
An alternative way of specifying the interpolation grid. This flag defines the minimum distance for
neighboring kpoints along each of the three directions in kspace.
The units are Å−1.
If you use a module which needs a k-mesh, either kmesh_spacing or kmesh must be defined.
11.6.3 logical :: adpt_smr
Determines whether to use an adaptive scheme for broadening the DOS and similar quantities defined
on the energy axis. If true, the values for the smearing widths are controlled by the flag adpt_smr_fac.
The default value is true.
11.6.4 real(kind=dp) :: adpt_smr_fac
The width ηnkof the broadened delta function used to determine the contribution to the spectral
property (DOS, ...) from band nat point kis calculated as
ηnk=α|∇kεnk|∆k,
where εnkis the energy eigenvalue and the dimensionless factor αis given by adpt_smr_fac.∆kis
taken to be the largest of the mesh spacings along the three reciprocal lattice vectors b1,b2, and b3.
If the calculated value of ηnkexceeds adpt_smr_max, the latter value is used.
The default value is √2.
11.6.5 real(kind=dp) :: adpt_smr_max
See description given immediately above.
The units are eV. The default value is 1.0.

wannier90: User Guide 119
11.6.6 character(len=120) :: smr_type
Defines the analytical form used for the broadened delta function in the computation of the DOS and
similar quantities defined on the energy axis.
•gauss: Gaussian smearing
•m-pN: derivative of the N-th order Methfessel-Paxton function (N≥0). Example: m-p2 for the
second-order Methfessel-Paxton function. If only m-p is provided, the first-order function is used,
i.e., it is equivalent to m-p1.
•m-v or cold: derivative of the Marzari–Vanderbilt cold-smearing function
•f-d: derivative of the Fermi-Dirac distribution function
The default value is gauss.
11.6.7 logical :: smr_fixed_en_width
Energy width for the smearing function for the DOS. Used only if adpt_smr is false.
The units are eV. The default value is 0 eV. Note that if the width is smaller than twice the energy
step (e.g. dos_energy_step for the dos module), the DOS will be unsmeared (thus the default is to
have an unsmeared properties when adpt_smr is set to false.).
11.6.8 integer :: num_elec_per_state
Number of electrons per state. It can only take the values one or two.
The default value is 1 if spinors=true, 2 otherwise.
11.6.9 real(kind=dp) :: scissors_shift
Scissors shift applied to the conduction bands.
Note! This variable is deprecated and will be removed in future versions of the code. This applies the
scissors shift only to the Hamiltonian, but also other matrices might need to be updated if a scissors
shift is applied. If you are using BoltzWann, consider using boltz_bandshift instead.
The units are eV. The default value is 0 eV (i.e., no scissors shift applied).
11.6.10 integer :: num_valence_bands
Number of valence bands of the system. Used in different modules and for the scissors shift.
No default value.

120 wannier90: User Guide
11.6.11 logical :: spin_decomp
If true, extra columns are added to some output files (such as seedname-dos.dat for the dos module,
and analogously for the berry and BoltzWann modules).
For the dos and BoltzWann modules, two further columns are generated, which contain the decomposi-
tion of the required property (e.g., total or orbital-projected DOS) of a spinor calculation into up-spin
and down-spin parts (relative to the quantization axis defined by the input variables spin_axis_polar
and spin_axis_azimuth). For the berry module with berry_task = kubo, three extra columns are
added to seedname-jdos.dat, containing the decomposition of the JDOS into up →up, down →
down, and spin-flip transitions. In the same way, six extra columns are added to the data files
seedname-kubo*.dat where the complex optical conductivity is stored.
The file seedname.spn must be present at input. Furthermore, if this variable is set to true it requires
num_elec_per_state = 1.
The default value is false.
11.6.12 real(kind=dp) :: spin_axis_polar
Polar angle of the spin quantization axis.
The units are degrees. The default value is 0.
11.6.13 real(kind=dp) :: spin_axis_azimuth
Azimuthal angle of the spin quantization axis.
The units are degrees. The default value is 0.
11.6.14 logical :: spin_moment
Determines whether to evaluate the spin moment.
The default value is false.
11.6.15 logical :: uHu_formatted
If uHu_formatted=true, then the uHu matrix elements will be read from disk as formatted (ie ASCII)
files; otherwise they will be read as unformatted files.
The default value of this parameter is false.
11.6.16 logical :: spn_formatted
If spn_formatted=true, then the spin matrix elements will be read from disk as formatted (ie ASCII)
files; otherwise they will be read as unformatted files. Unformatted is generally preferable as the files
will take less disk space and I/O is significantly faster. However such files will not be transferable

wannier90: User Guide 121
between all machine architectures and formatted files should be used if transferability is required (i.e.,
for test cases).
The default value is false.
11.6.17 character(len=20) :: berry_curv_unit
Unit in which the Berry curvature is specified at input (in berry_curv_adpt_kmesh_thresh) or written
to file (when kpath_task=curv or kslice_task=curv).
•ang2: Angstrom2
•bohr2: Bohr2(atomic units)
The default value is ang2.
11.6.18 real(kind=dp) :: sc_eta
The width ηused to broaden energy differences in denominators of the form
1
εnk−εmk→Re 1
εnk−εmk+iη .
The above is needed in shift-current calculations in order to avoid numerical problems caused by
near-degeneracies in the sum over virtual states.
The units are eV. The default value is 0.4.
11.6.19 integer :: sc_phase_conv
Convention for the expansion of the Bloch states in shift-current calculations. It can only take the
values one or two. We follow the convention of Ref. [11]:
•1: Include Wannier centre τn=hwn0|r|wn0iin the phase factor (so-called tight-binding conven-
tion):
|unki=X
R
e−ik(r−R−τn)|wnRi
•2: Do not include Wannier centre in the phase factor (usual Wannier90 convention):
|unki=X
R
e−ik(r−R)|wnRi
The convention does not affect the full shift-current matrix element, but it does affect the weights of
the internal components that compose it (see Ref. [12]).
The default value is 1.

122 wannier90: User Guide
11.6.20 real(kind=dp) :: sc_w_thr
Parameter αtfor speeding up the frequency integration in shift-current calculations. It settles the
frequency threshold ωt=αtηnk(a factor times the broadening) beyond which the delta functions are
taken as zero.
The default value is 5.0.

wannier90: User Guide 123
11.7 DOS
Note that the behavior of the dos module is also influenced by the value of some global flags (listed in
Table 11.1), as spin_decomp,spin_axis_polar,spin_axis_azimuth,scissors_shift, etc. Some of
the global flags can be possibly overridden by local flags of the DOS module, listed below, which have
the same name of the global flag but are prefixed by dos_.
11.7.1 logical :: dos
Determines whether to enter the DOS routines.
The default value is false.
11.7.2 character(len=20) :: dos_task
The quantity to compute when dos=true
The valid options for this parameter are:
–dos_plot Density of states. An output data file seedname-dos.dat is created, containing the
energy values in eV in the first column, and the total DOS per unit cell and unit energy range
(in eV−1) in the second. Two additional columns are present if spin_decomp=true
The default value is dos_plot.
11.7.3 real(kind=dp) :: dos_energy_min
Lower limit of the energy range for computing the DOS. Units are eV.
The default value is the minimum value of the energy eigenvalues stored in seedname.eig, minus
0.6667.
11.7.4 real(kind=dp) :: dos_energy_max
Upper limit of the energy range for computing the DOS. Units are eV.
If an inner energy window was specified, the default value is the upper bound of the innter energy win-
dow, plus 0.6667. Otherwise it is the maximum value of the energy eigenvalues stored in seedname.eig,
plus 0.6667.
11.7.5 real(kind=dp) :: dos_energy_step
Energy step for the grid of energies used to plot the dos. Units are eV.
The default value is 0.01 eV.

124 wannier90: User Guide
11.7.6 integer :: dos_project(:)
If present postw90 computes, instead of the total DOS, the partial DOS projected onto the WFs listed.
The WFs are numbered according to the file seedname.wout.
For example, to project onto WFs 2, 6, 7, 8, and 12:
dos_project : 2, 6-8, 12
The DOS projected onto a set Sof orbitals is calculated as
ρS(E) = 1
NkX
kX
nhψ(H)
nk|ˆ
Pk(S)|ψ(H)
nkiδ(εnk−E)(11.1)
ˆ
Pk(S) = X
m∈S |ψ(W)
nkihψ(W)
nk|,(11.2)
where Nkis the number of mesh points used to sample the BZ, and the superscript (H) and (W) refer
to Hamiltonian gauge and Wannier gauge [13].
11.7.7 integer :: dos_kmesh(:)
Overrides the kmesh global variable (see Sec. 11.6).
11.7.8 real(kind=dp) :: dos_kmesh_spacing
Overrides the kmesh_spacing global variable (see Sec. 11.6).
11.7.9 logical :: dos_adpt_smr
Overrides the adpt_smr global variable (see Sec. 11.6).
11.7.10 real(kind=dp) :: dos_adpt_smr_fac
Overrides the adpt_smr_fac global variable (see Sec. 11.6).
11.7.11 real(kind=dp) :: dos_adpt_smr_max
Overrides the adpt_smr_max global variable (see Sec. 11.6).
11.7.12 logical :: dos_smr_fixed_en_width
Overrides the smr_fixed_en_width global variable (see Sec. 11.6).
Note that if the width is smaller than twice the energy step dos_energy_step, the DOS will be
unsmeared (thus the default is to have an unsmeared DOS).


126 wannier90: User Guide
11.8 kpath
11.8.1 logical :: kpath
Determines whether to enter the kpath routines.
The default value is false.
11.8.2 character(len=20) :: kpath_task
The quantities to plot when kpath=true
The valid options for this parameter are:
–bands Energy bands, in eV. The following files are created:
·seedname-bands.dat (data file)
·seedname-bands.gnu (gnuplot script)
·seedname-bands.py (python script)
·seedname-path.kpt (list of k-points along the path, written in the pwscf format)
–curv Minus the Berry curvature given by Eq. (12.18) of Ch. 12, in units of berry_curv_unit.
The following files are created:
·seedname-curv.dat (data file)
·seedname-curv_{x,y,z}.gnu (gnuplot scripts)
·seedname-curv_{x,y,z}.py (python scripts)
–morb The integrand of the k-space orbital magnetization formula [Eq. (12.20) of Ch. 12] in eV·Å2.
Four output files are created:
·seedname-morb.dat (data file)
·seedname-morb_{x,y,z}.gnu (gnuplot scripts)
·seedname-morb_{x,y,z}.py (python scripts)
–Any combination of the above. The following combinations are of special interest
kpath_task = bands+curv
kpath_task = bands+morb
They generate the following files:
·seedname-bands.dat (data file)
·seedname-{curv,morb}.dat (data file)
·seedname-bands+{curv,morb}_{x,y,z}.py (python scripts)
Two-panel figures are produced, with the energy bands within ±0.65 eV of the Fermi level in the
top panel, and the Berry curvature (or k-space orbital magnetization) in the bottom panel.
The default value is bands.

wannier90: User Guide 127
11.8.3 integer :: kpath_num_points
If kpath =true, then the number of points along the first section of the bandstructure plot given by
kpoint_path. Other sections will have the same density of k-points.
The default value is 100.
11.8.4 character(len=20) :: kpath_bands_colour
When kpath_task=bands, colour code the energy bands according to the specified quantity.
The valid options for this parameter are:
–spin Spin projection (in units of ¯h/2) along the quantization axis defined by the variables
spin_axis_polar and spin_axis_azimuth, for a spinor calculation
–none no colour coding
The default value is none.

128 wannier90: User Guide
11.9 kslice
11.9.1 logical :: kslice
Determines whether to enter the kslice routines.
The default value is false.
11.9.2 character(len=20) :: kslice_task
The quantity to plot when kslice=true
The valid options for this parameter are:
–fermi_lines Lines of intersection between constant-energy surfaces and the slice. The energy
level is specified by the keyword fermi_energy. Output files:
·seedname-kslice-fermi-spn.dat (data file when kslice_fermi_lines_colour = spin)
·seedname-bnd_n.dat (gnuplot data files when kslice_fermi_lines_colour = none)
·seedname-kslice-coord.dat (python data files when kslice_fermi_lines_colour = none)
·seedname-kslice-bands.dat (python data file when kslice_fermi_lines_colour = none)
·seedname-kslice-fermi_lines.gnu (gnuplot script)
·seedname-kslice-fermi_lines.py (python script)
–curv[+fermi_lines] Heatmap of the Berry curvature of the occupied states [together with the
constant-energy contours]. The unit of Berry curvature is berry_curv_unit.
Output files:
·seedname-kslice-coord.dat (data files)
·seedname-kslice-curv.dat (data file)
·[seedname-kslice-bands.dat] (data file)
·seedname-kslice-curv_{x,y,z}[+fermi_lines].py (python scripts)
–morb[+fermi_lines] Heatmap of the k-space orbital magnetization in eV·Å2[together with the
constant-energy contours]. Output files:
·seedname-kslice-coord.dat (data files)
·seedname-kslice-morb.dat (data file)
·[seedname-kslice-bands.dat] (data file)
·seedname-kslice-morb_{x,y,z}[+fermi_lines].py (python scripts)
The default value is fermi_lines.
Note: When kslice_fermi_lines_colour = none the gnuplot scripts draw the k-slices with a square
shape, even when kslice_b1 and kslice_b2 below are not at right angles, or do not have equal lengths.
(The python scripts draw the slices with the correct parallelogram shape.)

wannier90: User Guide 129
11.9.3 real(kind=dp) :: kslice_corner(3)
Reduced coordinates of the lower-left corner of the slice in k-space.
The default value is (0.0,0.0,0.0)
11.9.4 real(kind=dp) :: kslice_b1(3)
Reduced coordinates of the first reciprocal-space vector defining the slice.
The default value is (1.0,0.0,0.0).
11.9.5 real(kind=dp) :: kslice_b2(3)
Reduced coordinates of the second reciprocal-space vector defining the slice.
The default value is (0.0,1.0,0.0).
11.9.6 integer :: kslice_2dkmesh(2)
Dimensions of the k-point grid covering the slice. If two integers m n are given, the slice is sampled
on a uniform m×ngrid. If only one integer mis given, an m×mgrid is used.
The default value for kslice_kmesh is 50.
11.9.7 character(len=20) :: kslice_fermi_lines_colour
When kslice_task=fermi_lines (but not when combined with curv or morb), colour code the Fermi
lines according to the specified quantity.
The valid options for this parameter are:
–spin Spin projection (in units of ¯h/2) along the quantization axis defined by the variables
spin_axis_polar and spin_axis_azimuth, for a spinor calculation
–none no colour coding
The default value is none.

130 wannier90: User Guide
11.10 berry
11.10.1 logical :: berry
Determines whether to enter the berry routines.
The default value is false.
11.10.2 character(len=120) :: berry_task
The quantity to compute when berry=true
The valid options for this parameter are:
–kubo Complex optical conductivity and joint density of states. Output files:
·seedname-kubo-S_{xx,yy,zz,xy,xz,yz}.dat (data files). First column: optical frequency
¯hω in eV. Second and third columns: real and imaginary parts of the symmetric conductivity
σS
αβ(¯hω) = σS
βα(¯hω)in S/cm. Six additional columns are present if spin_decomp = true.
·seedname-kubo-A_{yz,zx,xy}.dat (data files). First column: optical frequency ¯hω in eV.
Second and third columns: real and imaginary parts of the antisymmetric conductivity
σA
αβ(¯hω) = −σA
βα(¯hω)in S/cm. Six additional columns are present if spin_decomp = true.
·seedname-jdos.dat (data file). First column: energy difference ¯hω in eV between conduc-
tion (c) and valence (v) states with the same crystal momentum k. Second column: joint
density of states ρcv(¯hω)(number of states per unit cell per unit energy range, in eV−1).
Three additional columns are present if spin_decomp = true.
–ahc Anomalous Hall conductivity, in S/cm. The three independent components σx=σyz,σy=
σzx, and σz=σxy are computed. Output files:
·seedname-ahc-fermiscan.dat (data file) . The first column contains the Fermi level εFin
eV, and the following three column the values of σx,y,z(εF). This file is written if a range of
Fermi energies is specified via fermi_energy_min and fermi_energy_max. If a single Fermi
energy is given, the AHC is printed in seedname.wpout only.
–morb Orbital magnetisation, in bohr magnetons per cell.
Output files:
·seedname-morb-fermiscan.dat (data file). The first column contains the Fermi level εFin
eV, and the following three column the values of Morb
x,y,z(εF). This file is written if a range of
Fermi energies is specified via fermi_energy_min and fermi_energy_max. If a single Fermi
energy is given, Morb is printed in seedname.wpout only.
There is no default value.
11.10.3 integer :: berry_kmesh(:)
Overrides the kmesh global variable (see Sec. 11.6).

wannier90: User Guide 131
11.10.4 real(kind=dp) :: berry_kmesh_spacing
Overrides the kmesh_spacing global variable (see Sec. 11.6).
11.10.5 integer :: berry_curv_adpt_kmesh
If a positive integer nis given and berry_task=ahc, an n×n×nmesh is placed around points on
the uniform mesh (defined by either berry_kmesh or berry_kmesh_spacing) where the magnitude of
the k-space Berry curvature exceeds the threshold value specified in berry_curv_adpt_kmesh_thresh.
This can be used to densify the BZ integration mesh around spikes in the Berry curvature.
The default value is 1.
11.10.6 real(kind=dp) :: berry_curv_adpt_kmesh_thresh
Magnitude of the Berry curvature (in units of berry_curv_unit) that triggers adaptive mesh refinement
when berry_task=ahc.
The default value is 100.0.
11.10.7 real(kind=dp) :: kubo_freq_min
Lower limit of the frequency range for computing the optical conductivity and JDOS. Units are eV.
The default value 0.0.
11.10.8 real(kind=dp) :: kubo_freq_max
Upper limit of the frequency range for computing the optical conductivity and JDOS. Units are eV.
If an inner energy window was specified, the default value is dis_froz_max-fermi_energy+0.6667.
Otherwise it is the difference between the maximum and the minimum energy eigenvalue stored in
seedname.eig, plus 0.6667.
11.10.9 real(kind=dp) :: kubo_freq_step
Difference between consecutive values of the optical frequency between kubo_freq_min and kubo_freq_max.
Units are eV.
The default value is 0.01.
11.10.10 real(kind=dp) :: kubo_eigval_max
Maximum energy eigenvalue of the eigenstates to be included in the evaluation of the optical conduc-
tivity and JDOS. Units are eV.

132 wannier90: User Guide
If an inner energy window was specified, the default value is the upper bound of the inner energy
window plus 0.6667. Otherwise it is the maximum energy eigenvalue stored in seedname.eig plus
0.6667.
11.10.11 logical :: kubo_adpt_smr
Overrides the adpt_smr global variable (see Sec. 11.6).
11.10.12 real(kind=dp) :: kubo_adpt_smr_fac
Overrides the adpt_smr_fac global variable (see Sec. 11.6).
11.10.13 real(kind=dp) :: kubo_adpt_smr_max
Overrides the adpt_smr_max global variable (see Sec. 11.6).
11.10.14 logical :: kubo_smr_fixed_en_width
Overrides the smr_fixed_en_width global variable (see Sec. 11.6).
11.10.15 character(len=120) :: kubo_smr_type
Overrides the smr_type global variable (see Sec. 11.6).

wannier90: User Guide 133
11.11 Gyrotropic
11.11.1 logical :: gyrotropic
Determines whether to enter the gyrotropic routines.
The default value is false.
11.11.2 character(len=120) :: gyrotropic_task
The quantity to compute when gyrotropic=true
May contain one or more of the following valid options (note that each option starts with a ’-’):
•-D0 The Berry-curvature dipole tensor Eq. (13.1) (dimensionless)
Output file: seedname-gyrotropic-D.dat ( see Sec. 11.11.3 for file format description)
•-Dw The finite-frequency Berry-curvature dipole tensor Eq. (13.2) (dimensionless)
Output file: seedname-gyrotropic-tildeD.dat ( see Sec. 11.11.3 for file format description)
•-C The ohmic conductivity tensor Eq. (13.4) (Ampere/cm)
Output file: seedname-gyrotropic-C.dat ( see Sec. 11.11.3 for file format description)
•-K The orbital contribution to the kME tensor Eq. (13.5) (Ampere)
Output file: seedname-gyrotropic-K_orb.dat ( see Sec. 11.11.3 for file format description)
◦-spin : if this task is present, compute also the spin contribution.
Output file: seedname-gyrotropic-K_spin.dat
•-NOA The orbital contribution to the NOA Eq. (13.5) (Å)
Output file: seedname-gyrotropic-NOA_orb.dat ( see Sec. 11.11.3 for file format description)
◦-spin : if this task is present, compute also the spin contribution.
Output file: seedname-gyrotropic-NOA_spin.dat
•-dos the density of states Output file: seedname-gyrotropic-DOS.dat. First column - energy
(eV), second column - DOS (1/(eV ×3))
There is no default value.
11.11.3 output data format
The calculated tensors are written as functions of Fermi level EF(first column) and frequency ω(second
column). If the tensor does not denend on ω, the second column is filled by zeros. Data is grouped in
blocks of the same ωseparated by two blank lines. In case of natural optical activity the columns 3 to
11 contain the independent components of γabc (antisymmetric in ab): yzx,zxy ,xyz,yzy,yzz,zxz,
xyy,yzz and zxx. For tensors Cab,Dab,e
Dab,Kab the symmetric and antisymmetric components are
writted. Thus, the columns 3 to 11 are marked as xx,yy,zz,xy,xz,yz,x,y,z, wich correspond ,e.g.,
for Dab to Dxx,Dyy,Dzz,(Dxy +Dyx)/2,(Dxz +Dzx)/2,(Dyz +Dzy)/2,(Dyz −Dzy)/2,(Dzx −Dxz)/2,
(Dxy −Dyx)/2

134 wannier90: User Guide
11.11.4 integer :: gyrotropic_kmesh(:)
Overrides the kmesh global variable (see Sec. 11.6).
11.11.5 real(kind=dp) :: gyrotropic_kmesh_spacing
Overrides the kmesh_spacing global variable (see Sec. 11.6).
11.11.6 real(kind=dp) :: gyrotropic_freq_min
Lower limit of the frequency range for computing the optical activity.
Units are eV. The default value 0.0.
11.11.7 real(kind=dp) :: gyrotropic_freq_max
Upper limit of the frequency range for computing the optical activity. Units are eV.
If an inner energy window was specified, the default value is dis_froz_max-fermi_energy+0.6667.
Otherwise it is the difference between the maximum and the minimum energy eigenvalue stored in
seedname.eig, plus 0.6667.
11.11.8 real(kind=dp) :: gyrotropic_freq_step
Difference between consecutive values of the optical frequency between gyrotropic_freq_min and
gyrotropic_freq_max.
Units are eV. The default value is 0.01.
11.11.9 real(kind=dp) :: gyrotropic_eigval_max
Maximum energy eigenvalue of the eigenstates to be included in the evaluation of the Natural optical
activity. Units are eV.
If an inner energy window was specified, the default value is the upper bound of the inner energy
window plus 0.6667. Otherwise it is the maximum energy eigenvalue stored in seedname.eig plus
0.6667.
11.11.10 logical :: gyrotropic_smr_fixed_en_width
Overrides the smr_fixed_en_width global variable (see Sec. 11.6).
11.11.11 character(len=120) :: gyrotropic_smr_type
Overrides the smr_type global variable (see Sec. 11.6).

wannier90: User Guide 135
11.11.12 character(len=120) :: gyrotropic_degen_thresh
The threshould to eliminate degenerate bands from the calculation in order to avoid divergences.
Units are eV. The dfault value is 0.
11.11.13 character(len=120) :: gyrotropic_box_center
- three real numbers. Optionally the integration may be restricted to a parallelogram, centered at
gyrotropic_box_center and defined by vectors gyrotropic_box_b{1,2,3}
In reduced coordinates. Default value is 0.5 0.5 0.5
11.11.14 character(len=120) :: gyrotropic_box_b1
- three real numbers. In reduced coordinates. Default value is 1.0 0.0 0.0
11.11.15 character(len=120) :: gyrotropic_box_b2
- three real numbers. In reduced coordinates. Default value is 0.0 1.0 0.0
11.11.16 character(len=120) :: gyrotropic_box_b3
- three real numbers. In reduced coordinates. Default value is 0.0 0.0 1.0

136 wannier90: User Guide
11.12 BoltzWann
11.12.1 logical :: boltzwann
Determines whether to enter the BoltzWann routines.
The default value is false.
11.12.2 integer :: boltz_kmesh(:)
It determines the interpolation kmesh used to calculate the TDF (from which the transport coefficient
are calculated). If boltz_calc_also_dos is true, the same kmesh is used also for the DOS. Overrides
the kmesh global variable (see Sec. 11.6).
11.12.3 real(kind=dp) :: boltz_kmesh_spacing
Overrides the kmesh_spacing global variable (see Sec. 11.6).
11.12.4 character(len=4) :: boltz_2d_dir
For two-dimensional systems, the direction along which the system is non-periodic. It can assume the
following values: xfor a 2D system on the yz plane, yfor a 2D system on the xz plane, zfor a 2D
system on the xy plane, or no for a 3D system with periodicity along all threee directions.
This value is used when calculating the Seebeck coefficient, where the electrical conductivity tensor
needs to be inverted. If the value is different from zero, only the relevant 2×2sub-block of the electrical
conductivity is inverted.
The default value is no.
11.12.5 real(kind=dp) :: boltz_relax_time
The relaxation time to be used for the calculation of the TDF and the transport coefficients.
The units are fs. The default value is 10 fs.
11.12.6 real(kind=dp) :: boltz_mu_min
Minimum value for the chemical potential µfor which we want to calculate the transport coefficients.
The units are eV. No default value.
11.12.7 real(kind=dp) :: boltz_mu_max
Maximum value for the chemical potential µfor which we want to calculate the transport coefficients.
The units are eV. No default value.

wannier90: User Guide 137
11.12.8 real(kind=dp) :: boltz_mu_step
Energy step for the grid of chemical potentials µfor which we want to calculate the transport coeffi-
cients.
The units are eV. No default value.
11.12.9 real(kind=dp) :: boltz_temp_min
Minimum value for the temperature Tfor which we want to calculate the transport coefficients.
The units are K. No default value.
11.12.10 real(kind=dp) :: boltz_temp_max
Maximum value for the temperature Tfor which we want to calculate the transport coefficients.
The units are K. No default value.
11.12.11 real(kind=dp) :: boltz_temp_step
Energy step for the grid of temperatures Tfor which we want to calculate the transport coefficients.
The units are K. No default value.
11.12.12 real(kind=dp) :: boltz_tdf_energy_step
Energy step for the grid of energies for the TDF.
The units are eV. The default value is 0.001 eV.
11.12.13 character(len=120) :: boltz_tdf_smr_type
The type of smearing function to be used for the TDF. The available strings are the same of the global
smr_type input flag.
The default value is the one given via the smr_type input flag (if defined).
11.12.14 real(kind=dp) :: boltz_tdf_smr_fixed_en_width
Energy width for the smearing function. Note that for the TDF, a standard (non-adaptive) smearing
scheme is used.
The units are eV. The default value is 0 eV. Note that if the width is smaller than twice the energy
step boltz_tdf_energy_step, the TDF will be unsmeared (thus the default is to have an unsmeared
TDF).

138 wannier90: User Guide
11.12.15 logical :: boltz_calc_also_dos
Whether to calculate also the DOS while calculating the TDF.
If one needs also the DOS, it is faster to calculate the DOS using this flag instead of using independently
the routines of the dos module, since in this way the interpolation on the kpoints will be performed
only once.
The default value is false.
11.12.16 real(kind=dp) :: boltz_dos_energy_min
The minimum value for the energy grid for the calculation of the DOS.
The units are eV. The default value is minval(eigval)-0.6667, where minval(eigval) i s the mini-
mum eigenvalue returned by the ab-initio code on the ab-initio qme sh.
11.12.17 real(kind=dp) :: boltz_dos_energy_max
The maximum value for the energy grid for the calculation of the DOS.
The units are eV. The default value is maxval(eigval)+0.6667, where maxval(eigval) i s the maxi-
mum eigenvalue returned by the ab-initio code on the ab-initio qme sh.
11.12.18 real(kind=dp) :: boltz_dos_energy_step
Energy step for the grid of energies for the DOS.
The units are eV. The default value is 0.001 eV.
11.12.19 character(len=120) :: boltz_dos_smr_type
Overrides the smr_type global variable (see Sec. 11.6).
11.12.20 logical :: boltz_dos_adpt_smr
Overrides the adpt_smr global variable (see Sec. 11.6).
11.12.21 real(kind=dp) :: boltz_dos_adpt_smr_fac
Overrides the adpt_smr_fac global variable (see Sec. 11.6).
11.12.22 real(kind=dp) :: boltz_dos_adpt_smr_max
Overrides the adpt_smr_max global variable (see Sec. 11.6).

wannier90: User Guide 139
11.12.23 logical :: boltz_dos_smr_fixed_en_width
Overrides the smr_fixed_en_width global variable (see Sec. 11.6).
11.12.24 logical :: boltz_bandshift
Shift all conduction bands by a given amount (defined by boltz_bandshift_energyshift).
Note: this flag slightly differs from the global scissors_shift flag: with boltz_bandshift, an exact
rigid shift is applied after interpolation; scissors_shift applies instead the shift before interpolation.
As a consequence, results may slightly differ (and this is why we provide both possibilities). Note
also that with scissors_shift you have to provide the number of valence bands num_valence_bands,
while with boltz_bandshift you should provide the first band to shift boltz_bandshift_firstband
=num_valence_bands+1.
The default value is false.
11.12.25 integer :: boltz_bandshift_firstband
Index of the first conduction band to shift.
That means that all bands with index i≥boltz_bandshift_firstband will be shifted by boltz_bandshift_energyshift,
if boltz_bandshift is true.
The units are eV. No default value; if boltz_bandshift is true, this flag must be provided.
11.12.26 real(kind=dp) :: boltz_bandshift_energyshift
Energy shift of the conduction bands.
The units are eV. No default value; if boltz_bandshift is true, this flag must be provided.
11.13 Generic Band Interpolation
11.13.1 logical :: geninterp
Determines whether to enter the Generic Band Interpolation routines.
The default value is false.
11.13.2 logical :: geninterp_alsofirstder
Whether to calculate also the first derivatives of the bands at the given kpoints.
The default value is false.

140 wannier90: User Guide
11.13.3 logical :: geninterp_single_file
Whether to write a single seedname_geninterp.dat file (all I/O is done by the root node); or instead
multiple files (one for each node) with names seedname_geninterp_NNNNN.dat, where NNNNN is the
node number. See also the discussion in Sec. 15.1.2 on how to use this flag.
The default value is true.

Chapter 12
Overview of the berry module
The berry module of postw90 is called by setting berry = true and choosing one or more of the
available options for berry_task. The routines in the berry module which compute the k-space
Berry curvature and orbital magnetization are also called when kpath = true and kpath_task =
{curv,morb}, or when kslice = true and kslice_task = {curv,morb}.
12.1 Background: Berry connection and curvature
The Berry connection is defined in terms of the cell-periodic Bloch states |unki=e−ik·r|ψnkias
An(k) = hunk|i∇k|unki,(12.1)
and the Berry curvature is the curl of the connection,
Ωn(k) = ∇k×An(k) = −Imh∇kunk|×|∇kunki.(12.2)
These two quantities play a central role in the description of several electronic properties of crystals [14].
In the following we will work with a matrix generalization of the Berry connection,
Anm(k) = hunk|i∇k|umki=A∗
mn(k),(12.3)
and write the curvature as an antisymmetric tensor,
Ωn,αβ(k) = αβγ Ωn,γ(k) = −2Imh∇kαunk|∇kβunki.(12.4)
12.2 berry_task=kubo: optical conductivity and joint density of states
The Kubo-Greenwood formula for the optical conductivity of a crystal in the independent-particle
approximation reads
σαβ(¯hω) = ie2¯h
NkΩcX
kX
n,m
fmk−fnk
εmk−εnk
hψnk|vα|ψmkihψmk|vβ|ψnki
εmk−εnk−(¯hω +iη).(12.5)
Indices α, β denote Cartesian directions, Ωcis the cell volume, Nkis the number of k-points used for
sampling the Brillouin zone, and fnk=f(εnk)is the Fermi-Dirac distribution function. ¯hω is the
optical frequency, and η > 0is an adjustable smearing parameter with units of energy.
141
142 wannier90: User Guide
The off-diagonal velocity matrix elements can be expressed in terms of the connection matrix [15],
hψnk|v|ψmki=−i
¯h(εmk−εnk)Anm(k) (m6=n).(12.6)
The conductivity becomes
σαβ(¯hω) = 1
NkX
k
σk,αβ(¯hω)(12.7)
σk,αβ(¯hω) = ie2
¯hΩcX
n,m
(fmk−fnk)εmk−εnk
εmk−εnk−(¯hω +iη)Anm,α(k)Amn,β(k).(12.8)
Let us decompose it into Hermitian (dissipative) and anti-Hermitean (reactive) parts. Note that
δ(ε) = 1
πIm 1
ε−iη ,(12.9)
where δdenotes a “broadended” delta-function. Using this identity we find for the Hermitean part
σH
k,αβ(¯hω) = −πe2
¯hΩcX
n,m
(fmk−fnk)(εmk−εnk)Anm,α(k)Amn,β (k)δ(εmk−εnk−¯hω).(12.10)
Improved numerical accuracy can be achieved by replacing the Lorentzian (12.9) with a Gaussian, or
other shapes. The analytical form of δ(ε)is controlled by the keyword [kubo_]smr_type.
The anti-Hermitean part of Eq. (12.8) is given by
σAH
k,αβ(¯hω) = ie2
¯hΩcX
n,m
(fmk−fnk)Re εmk−εnk
εmk−εnk−(¯hω +iη)Anm,α(k)Amn,β(k).(12.11)
Finally the joint density of states is
ρcv(¯hω) = 1
NkX
kX
n,m
fnk(1 −fmk)δ(εmk−εnk−¯hω).(12.12)
Equations (12.9–12.12) contain the parameter η, whose value can be chosen using the keyword
[kubo_]smr_fixed_en_width. Better results can often be achieved by adjusting the value of ηfor each
pair of states, i.e., η→ηnmk. This is done as follows (see description of the keyword adpt_smr_fac)
ηnmk=α|∇k(εmk−εnk)|∆k. (12.13)
The energy eigenvalues εnk, band velocities ∇kεnk, and off-diagonal Berry connection Anm(k)entering
the previous four equations are evaluated over a k-point grid by Wannier interpolation, as described
in Refs. [13, 16]. After averaging over the Brillouin zone, the Hermitean and anti-Hermitean parts of
the conductivity are assembled into the symmetric and antisymmetric tensors
σS
αβ = ReσH
αβ +iImσAH
αβ (12.14)
σA
αβ = ReσAH
αβ +iImσH
αβ,(12.15)
whose independent components are written as a function of frequency onto nine separate files.

wannier90: User Guide 143
12.3 berry_task=ahc: anomalous Hall conductivity
The antisymmetric tensor σA
αβ is odd under time reversal, and therefore vanishes in non-magnetic
systems, while in ferromagnets with spin-orbit coupling it is generally nonzero. The imaginary part
ImσH
αβ describes magnetic circular dichroism, and vanishes as ω→0. The real part ReσAH
αβ describes
the anomalous Hall conductivity (AHC), and remains finite in the static limit.
The intrinsic dc AHC is obtained by setting η= 0 and ω= 0 in Eq. (12.11). The contribution from
point kin the Brillouin zone is
σAH
k,αβ(0) = 2e2
¯hΩcX
n,m
fnk(1 −fmk)Imh∇kαunk|umkihumk|∇kβunki,(12.16)
where we replaced fnk−fmkwith fnk(1 −fmk)−fmk(1 −fnk).
This expression is not the most convenient for ab initio calculations, as the sums run over the complete
set of occupied and empty states. In practice the sum over empty states can be truncated, but
a relatively large number should be retained to obtain accurate results. Using the resolution of the
identity 1 = Pm|umkihumk|and noting that the term Pn,m fnkfmk(. . .)vanishes identically, we arrive
at the celebrated formula for the intrinsic AHC in terms of the Berry curvature,
σAH
αβ (0) = e2
¯h
1
NkΩcX
k
(−1)Ωαβ(k),(12.17)
Ωαβ(k) = X
n
fnkΩn,αβ(k).(12.18)
Note that only occupied states enter this expression. Once we have a set of Wannier functions spanning
the valence bands (together with a few low-lying conduction bands, typically) Eq. (12.17) can be
evaluated by Wannier interpolation as described in Refs. [13, 17], with no truncation involved.
12.4 berry_task=morb: orbital magnetization
The ground-state orbital magnetization of a crystal is given by [14, 18]
Morb =e
¯h
1
NkΩcX
k
Morb(k),(12.19)
Morb(k) = X
n
1
2fnkIm h∇kunk| × (Hk+εnk−2εF)|∇kunki,(12.20)
where εFis the Fermi energy. The Wannier-interpolation calculation is described in Ref. [17]. Note
that the definition of Morb(k)used here differs by a factor of −1/2from the one in Eq. (97) and Fig. 2
of that work.
12.5 Needed matrix elements
All the quantities entering the formulas for the optical conductivity and AHC can be calculated by
Wannier interpolation once the Hamiltonian and position matrix elements h0n|H|Rmiand h0n|r|Rmi
are known [13, 16]. Those matrix elements are readily available at the end of a standard MLWF

144 wannier90: User Guide
calculation with wannier90. In particular, h0n|r|Rmican be calculated by Fourier transforming the
overlap matrices in Eq. (1.7),
hunk|umk+bi.
Further Wannier matrix elements are needed for the orbital magnetization [17]. In order to calculate
them using Fourier transforms, one more piece of information must be taken from the k-space ab-initio
calculation, namely, the matrices
hunk+b1|Hk|umk+b2i
over the ab-initio k-point mesh [17]. These are evaluated by pw2wannier90, the interface routine
between pwscf and wannier90, by adding to the input file seedname.pw2wan the line
write_uHu =.true.

Chapter 13
Overview of the gyrotropic module
The gyrotropic module of postw90 is called by setting gyrotropic = true and choosing one or more
of the available options for gyrotropic_task. The module computes the quantities, studied in [19],
where more details may be found.
13.1 berry_task=-d0: the Berry curvature dipole
The traceless dimensionless tensor
Dab =Z[dk]X
n
∂En
∂ka
Ωb
n−∂f0
∂E E=En
,(13.1)
13.2 berry_task=-dw: the finite-frequency generalization of the Berry
curvature dipole
e
Dab(ω) = Z[dk]X
n
∂En
∂kae
Ωb
n(ω)−∂f0
∂E E=En
,(13.2)
where e
Ωkn(ω)is a finite-frequency generalization of the Berry curvature:
e
Ωkn(ω) = −X
m
ω2
kmn
ω2
kmn −ω2Im (Aknm ×Akmn)(13.3)
Contrary to the Berry curvature, the divergence of ˜
Ωkn(ω)is generally nonzero. As a result, e
D(ω)can
have a nonzero trace at finite frequencies, ˜
Dk6=−2˜
D⊥in Te.
13.3 berry_task=-C: the ohmic conductivity
In the constant relaxation-time approximation the ohmic conductivity is expressed as σab = (2πeτ /¯h)Cab,
with
Cab =e
hZ[dk]X
n
∂En
∂ka
∂En
∂kb−∂f0
∂E E=En
(13.4)
a positive quantity with units of surface current density (A/cm).
145

146 wannier90: User Guide
13.4 berry_task=-K: the kinetic magnetoelectric effect (kME)
A microscopic theory of the intrinsic kME effect in bulk crystals was recently developed [20, 21].
The response is described by
Kab =Z[dk]X
n
∂En
∂ka
mb
n−∂f0
∂E E=En
,(13.5)
which has the same form as Eq. (13.1), but with the Berry curvature replaced by the intrinsic magnetic
moment mknof the Bloch electrons, which has the spin and orbital components given by [14]
mspin
kn=−1
2gsµBhψkn|σ|ψkni(13.6)
morb
kn=e
2¯hImh∂kukn| × (Hk−Ekn)|∂kukni,(13.7)
where gs≈2and we chose e > 0.
13.5 berry_task=-dos: the density of states
The density of states is calculated with the same width and type of smearing, as the other properties
of the gyrotropic module
13.6 berry_task=-noa: the interband contributionto the natural opti-
cal activity
Natural optical rotatory power is given by [22]
ρ0(ω) = ω2
2c2Re γxyz(ω).(13.8)
for light propagating ling the main symmetry axis of a crystal z. Here γxyz (ω)is an anti-symmetric
(in xy) tensor with units of length, which has both inter- and intraband contributions.
Following Ref. [23] for the interband contribution we writewe write, with ∂c≡∂/∂kc,
Re γinter
abc (ω) = e2
ε0¯h2Z[dk]
o,e
X
n,l h1
ω2
ln −ω2Re Ab
lnBac
nl −Aa
lnBbc
nl
−3ω2
ln −ω2
(ω2
ln −ω2)2∂c(El+En)Im Aa
nlAb
lni.(13.9)
The summations over nand lspan the occupied (o) and empty (e) bands respectively, ωln = (El−
En)/¯h, and Aln(k)is given by (12.3) Finally, the matrix Bac
nl has both orbital and spin contributions
given by
Bac (orb)
nl =hun|(∂aH)|∂culi−h∂cun|(∂aH)|uli(13.10)
and
Bac (spin)
nl =−i¯h2
me
abchun|σb|uli.(13.11)

wannier90: User Guide 147
The spin matrix elements contribute less than 0.5% of the total ρinter
0of Te. Expanding H=
Pm|umiEmhum|we obtain for the orbital matrix elements
Bac (orb)
nl =−i∂a(En+El)Ac
nl X
mn(En−Em)Aa
nmAc
ml −(El−Em)Ac
nmAa
mlo.(13.12)
This reduces the calculation of B(orb) to the evaluation of band gradients and off-diagonal elements
of the Berry connection matrix. Both operations can be carried out efficiently in a Wannier-function
basis following Ref. [16].
13.7 berry_task=-spin: compute also the spin component of NOA and
KME
Unless this task is specified, only the orbital contributions are calcuated in NOA and KME, thus
contributions from Eqs. (13.6) and (13.11) are omitted.
Chapter 14
Electronic transport calculations with the
BoltzWann module
By setting boltzwann =TRUE,postw90 will call the BoltzWann routines to calculate some transport
coefficients using the Boltzmann transport equation in the relaxation time approximation.
In particular, the transport coefficients that are calculated are: the electrical conductivity σ, the
Seebeck coefficient Sand the coefficient K(defined below; it is the main ingredient of the thermal
conductivity).
The list of parameters of the BoltzWann module are summarized in Table 11.7. An example of a
Boltzmann transport calculation can be found in the wannier90 Tutorial.
Note: By default, the code assumes to be working with a 3D bulk material, with periodicity along
all three spatial directions. If you are interested in studying 2D systems, set the correct value for the
boltz_2d_dir variable (see Sec. 11.12.4 for the documentation). This is important for the evaluation
of the Seebeck coefficient.
Please cite the following paper [24] when publishing results obtained using the BoltzWann module:
G. Pizzi, D. Volja, B. Kozinsky, M. Fornari, and N. Marzari,
BoltzWann: A code for the evaluation of thermoelectric and electronic transport properties
with a maximally-localized Wannier functions basis,
Comp. Phys. Comm. 185, 422 (2014), DOI:10.1016/j.cpc.2013.09.015.
14.1 Theory
The theory of the electronic transport using the Boltzmann transport equations can be found for
instance in Refs. [25–27]. Here we briefly summarize only the main results.
The current density Jand the heat current (or energy flux density) JQcan be written, respectively, as
J=σ(E−S∇T)(14.1)
JQ=TσSE −K∇T, (14.2)
where the electrical conductivity σ, the Seebeck coefficient Sand Kare 3×3tensors, in general.
149

150 wannier90: User Guide
Note: the thermal conductivity κ(actually, the electronic part of the thermal conductivity), which
is defined as the heat current per unit of temperature gradient in open-circuit experiments (i.e., with
J= 0) is not precisely K, but κ=K−SσST(see for instance Eq. (7.89) of Ref. [25] or Eq. (XI-57b)
of Ref. [26]). The thermal conductivity κcan be then calculated from the σ,Sand Ktensors output
by the code.
These quantities depend on the value of the chemical potential µand on the temperature T, and can
be calculated as follows:
[σ]ij(µ, T ) = e2Z+∞
−∞
dε −∂f (ε, µ, T )
∂ε Σij (ε),(14.3)
[σS]ij(µ, T ) = e
TZ+∞
−∞
dε −∂f (ε, µ, T )
∂ε (ε−µ)Σij (ε),(14.4)
[K]ij(µ, T ) = 1
TZ+∞
−∞
dε −∂f (ε, µ, T )
∂ε (ε−µ)2Σij (ε),(14.5)
where [σS]denotes the product of the two tensors σand S,f(ε, µ, T )is the usual Fermi–Dirac
distribution function
f(ε, µ, T ) = 1
e(ε−µ)/KBT+ 1
and Σij(ε)is the Transport Distribution Function (TDF) tensor, defined as
Σij(ε) = 1
VX
n,k
vi(n, k)vj(n, k)τ(n, k)δ(ε−En,k).
In the above formula, the sum is over all bands nand all states k(including spin, even if the spin
index is not explicitly written here). En,kis the energy of the n−th band at k,vi(n, k)is the i−th
component of the band velocity at (n, k),δis the Dirac’s delta function, V=NkΩcis the total volume
of the system (Nkand Ωcbeing the number of k-points used to sample the Brillouin zone and the unit
cell volume, respectively), and finally τis the relaxation time. In the relaxation-time approximation
adopted here, τis assumed as a constant, i.e., it is independent of nand kand its value (in fs) is read
from the input variable boltz_relax_time.
14.2 Files
14.2.1 seedname_boltzdos.dat
OUTPUT. Written by postw90 if boltz_calc_also_dos is true. Note that even if there are other
general routines in postw90 which specifically calculate the DOS, it may be convenient to use the
routines in BoltzWann setting boltz_calc_also_dos = true if one must also calculate the transport
coefficients. In this way, the (time-demanding) band interpolation on the kmesh is performed only
once, resulting in a much shorter execution time.
The first lines are comments (starting with # characters) which describe the content of the file. Then,
there is a line for each energy εon the grid, containing a number of columns. The first column is the
energy ε. The following is the DOS at the given energy ε. The DOS can either be calculated using the
adaptive smearing scheme1if boltz_dos_adpt_smr is true, or using a “standard” fixed smearing, whose
1Note that in BoltzWann the adaptive (energy) smearing scheme also implements a simple adaptive k−mesh scheme:
if at any given kpoint one of the band gradients is zero, then that kpoint is replaced by 8 neighboring kpoints. Thus,
the final results for the DOS may be slightly different with respect to that given by the dos module.

wannier90: User Guide 151
type and value are defined by boltz_dos_smr_type and boltz_dos_smr_fixed_en_width, respectively.
If spin decomposition is required (input flag spin_decomp), further columns are printed, with the spin-
up projection of the DOS, followed by spin-down projection.
14.2.2 seedname_tdf.dat
OUTPUT. This file contains the Transport Distribution Function (TDF) tensor Σon a grid of energies.
The first lines are comments (starting with # characters) which describe the content of the file. Then,
there is a line for each energy εon the grid, containing a number of columns. The first is the energy
ε, the followings are the components if Σ(ε)in the following order: Σxx,Σxy,Σyy,Σxz,Σyz,Σzz . If
spin decomposition is required (input flag spin_decomp), 12 further columns are provided, with the 6
components of Σfor the spin up, followed by those for the spin down.
The energy εis in eV, while Σis in 1
¯h2·eV ·fs
Å.
14.2.3 seedname_elcond.dat
OUTPUT. This file contains the electrical conductivity tensor σon the grid of Tand µpoints.
The first lines are comments (starting with # characters) which describe the content of the file. Then,
there is a line for each (µ, T )pair, containing 8 columns, which are respectively: µ,T,σxx,σxy,σyy,
σxz,σyz,σzz. (The tensor is symmetric).
The chemical potential is in eV, the temperature is in K, and the components of the electrical conduc-
tivity tensor ar in SI units, i.e. in 1/Ω/m.
14.2.4 seedname_sigmas.dat
OUTPUT. This file contains the tensor σS, i.e. the product of the electrical conductivity tensor and
of the Seebeck coefficient as defined by Eq. (14.4), on the grid of Tand µpoints.
The first lines are comments (starting with # characters) which describe the content of the file. Then,
there is a line for each (µ, T )pair, containing 8 columns, which are respectively: µ,T,(σS)xx,(σS)xy,
(σS)yy ,(σS)xz,(σS)yz,(σS)zz. (The tensor is symmetric).
The chemical potential is in eV, the temperature is in K, and the components of the tensor ar in SI
units, i.e. in A/m/K.
14.2.5 seedname_seebeck.dat
OUTPUT. This file contains the Seebeck tensor Son the grid of Tand µpoints.
Note that in the code the Seebeck coefficient is defined as zero when the determinant of the electrical
conductivity σis zero. If there is at least one (µ, T )pair for which det σ= 0, a warning is issued on
the output file.
The first lines are comments (starting with # characters) which describe the content of the file. Then,
there is a line for each (µ, T )pair, containing 11 columns, which are respectively: µ,T,Sxx,Sxy,Sxz,
Syx,Syy,Syz,Szx,Szy,Szz.

152 wannier90: User Guide
NOTE: therefore, the format of the columns of this file is different from the other three files (elcond,
sigmas and kappa)!
The chemical potential is in eV, the temperature is in K, and the components of the Seebeck tensor ar
in SI units, i.e. in V/K.
14.2.6 seedname_kappa.dat
OUTPUT. This file contains the tensor Kdefined in Sec. 14.1 on the grid of Tand µpoints.
The first lines are comments (starting with # characters) which describe the content of the file. Then,
there is a line for each (µ, T )pair, containing 8 columns, which are respectively: µ,T,Kxx,Kxy,Kyy,
Kxz,Kyz,Kzz. (The tensor is symmetric).
The chemical potential is in eV, the temperature is in K, and the components of the Ktensor are the
SI units for the thermal conductivity, i.e. in W/m/K.

Chapter 15
Generic Band interpolation
By setting geninterp =TRUE,postw90 will calculate the band energies (and possibly the band deriva-
tives, if also geninterp_alsofirstder is set to TRUE) on a generic list of kpoints provided by the
user.
The list of parameters of the Generic Band Interpolation module are summarized in Table 11.8.
The list of input kpoints for which the band have to be calculated is read from the file named
seedname_geninterp.kpt. The format of this file is described below.
15.1 Files
15.1.1 seedname_geninterp.kpt
INPUT. Read by postw90 if geninterp is true.
The first line is a comment (its maximum allowed length is 500 characters).
The second line must contain crystal (or frac) if the k-point coordinates are given in crystallographic
units, i.e., in fractional units with respect to the primitive reciprocal lattice vectors. Otherwise, it must
contain cart (or abs) if instead the k−point coordinates are given in absolute coordinates (in units of
1/Å) along the kx,kyand kzaxes.
Note on units: In the case of absolute coordinates, if alat is the lattice constant expressed in angstrom,
and you want to represent for instance the point X=2π
alat [0.5,0,0], then you have to input for its x
coordinate kx= 0.5∗2∗π/alat. As a practical example, if alat = 4Å, then kx= 0.78539816339745 in
absolute coordinates in units of 1/Å.
The third line must contain the number nof following kpoints.
The following nlines must contain the list of kpoints in the format
kpointidx k1 k2 k3
where kpointidx is an integer identifying the given kpoint, and k1,k2 and k3 are the three coordinates
of the kpoints in the chosen units.
153

154 wannier90: User Guide
15.1.2 seedname_geninterp.dat or seedname_geninterp_NNNNN.dat
OUTPUT. This file/these files contain the interpolated band energies (and also the band velocities if
the input flag geninterp_alsofirstder is true).
If the flag geninterp_single_file is true, then a single file seedname_geninterp.dat is written by
the code at the end of the calculation. If instead one sets geninterp_single_file to false, each pro-
cess writes its own output file, named seedname_geninterp_00000.dat,seedname_geninterp_00001.dat,
. . .
This flag is useful when one wants to parallelize the calculation on many nodes, and it should be used
especially for systems with a small number of Wannier functions, when one wants to compute the
bands on a large number of kpoints (if the flag geninterp_single_file is true, instead, all the I/O
is made by the root node, which is a significant bottleneck).
Important! The files are not deleted before the start of a calculation, but only the relevant files are
overwritten. Therefore, if one first performs a calculation and then a second one with a smaller number
of processors, care is needed to avoid to mix the results of the older calculations with those of the new
one. In case of doubt, either check the date stamp in the first line of the seedname_geninterp_*.dat
files, or simply delete the seedname_geninterp_*.dat files before starting the new calculation.
To join the files, on can simply use the following command:
cat seedname_geninterp_*.dat > seedname_geninterp.dat
or, if one wants to remove the comment lines:
rm seedname_geninterp.dat
for i in seedname_geninterp_*.dat ; do grep -v \# "$i" >> \
seedname_geninterp.dat ; done
The first few lines of each files are comments (starting with #), containing a datestamp, the comment
line as it is read from the input file, and a header. The following lines contain the band energies (and
derivatives) for each band and kpoint (the energy index runs faster than the k-point index). For
each of these lines, the first four columns contain the k-point index as provided in the input, and the
kcoordinates (always in absolute coordinates, in units of 1/Å). The fifth column contains the band
energy.
If geninterp_alsofirstder is true, three further columns are printed, containing the three first
derivatives of the bands along the kx,kyand kzdirections.
The kpoint coordinates are in units of 1/Å, the band energy is in eV.
Part IV
Appendices
155
Appendix A
Utilities
The wannier90 code is shipped with a few utility programs that may be useful in some occasions. In
this chapter, we describe their use.
A.1 kmesh.pl
The wannier90 code requires the definition of a full Monkhorst–Pack grid of kpoints. In the input
file the size of this mesh is given by means of the mp_grid variable. E.g., setting
mp_grid = 4 4 4
tells wannier90 that we want to use a 4×4×4kgrid.
One has then to specify (inside the kpoints block in the the seedname.win file) the list of kpoints of
the grid. Here, the kmesh.pl Perl script becomes useful, being able to generate the required list.
The script can be be found in the utility directory of the wannier90 distribution. To use it, simply
type:
./kmesh.pl nx ny nz
where nx,ny and nz define the size of the Monkhorst–Pack grid that we want to use (for instance, in
the above example of the 4×4×4kgrid, nx=ny=nz=4).
This produces on output the list of kpoints in Quantum Espresso format, where (apart from a header)
the first three columns of each line are the kcoordinates, and the fourth column is the weight of each
kpoint. This list can be used to create the input file for the ab-initio nscf calculation.
If one wants instead to generate the list of the kcoordinates without the weight (in order to copy and
paste the output inside the seedname.win file), one simply has to provide a fourth argument on the
command line. For instance, for a 4×4×4kgrid, use
./kmesh.pl 4 4 4 wannier
and then copy the output inside the in the kpoints block in the seedname.win file.
157

158 wannier90: User Guide
We suggest to always use this utility to generate the kgrids. This allows to provide the kpoint
coordinates with the accuracy required by wannier90, and moreover it makes sure that the kgrid used
in the ab-initio code and in wannier90 are the same.
A.2 w90chk2chk.x
During the calculation of the Wannier functions, wannier90 produces a .chk file that contains some
information to restart the calculation.
This file is also required by the postw90 code. In particular, the postw90 code requires at least the
.chk file, the .win input file, and (almost always) the .eig file. Specific modules may require further
files: see the documentation of each module.
However, the .chk file is written in a machine-dependent format. If one wants to run wannier90 on a
machine, and then continue the calculation with postw90 on a different machine (or with postw90 com-
piled with a different compiler), the file has to be converted first in a machine-independent “formatted”
format on the first machine, and then converted back on the second machine.
To this aim, use the w90chk2chk.x executable. Note that this executable is not compiled by default:
you can obtain it by executing
make w90chk2chk
in the main wannier90 directory.
A typical use is the following:
1. Calculate the Wannier functions with wannier90
2. At the end of the calculation you will find a seedname.chk file. Run (in the folder with this file)
the command
w90chk2chk.x -export seedname
or equivalently
w90chk2chk.x -u2f seedname
(replacing seedname with the seedname of your calculation).
This command reads the seedname.chk file and creates a formatted file seedname.chk.fmt that
is safe to be transferred between different machines.
3. Copy the seedname.chk.fmt file (together with the seedname.win and seedname.eig files) on
the machine on which you want to run postw90.
4. On this second machine (after having compiled w90chk2chk.x) run
w90chk2chk.x -import seedname
or equivalently

wannier90: User Guide 159
w90chk2chk.x -f2u seedname
This command reads the seedname.chk.fmt file and creates an unformatted file seedname.chk
ready to be used by postw90.
5. Run the postw90 code.
A.3 PL_assessment
The function of this utility is to assess the length of a principal layer (in the context of a Landauer-
Buttiker quantum conductance calculation) of a periodic system using a calculation on a single unit
cell with a dense k-point mesh.
The utility requires the real-space Hamiltonian in the MLWF basis, seedname_hr.dat.
The seedname_hr.dat file should be copied to a directory containing executable for the utility. Within
that directory, run:
\$> ./PL_assess.x nk1 nk2 nk3 num_wann
where:
nk1 is the number of k-points in x-direction nk2 is the number of k-points in y-direction nk3 is the
number of k-points in z-direction num_wann is the number of wannier functions of your system
e.g.,
\$> ./PL_assess.x 1 1 20 16
Note that the current implementation only allows for a single k-point in the direction transverse to the
transport direction.
When prompted, enter the seedname.
The programme will return an output file seedname_pl.dat, containing four columns
1. Unit cell number, R
2. Average ’on-site’ matrix element between MLWFs in the home unit cell, and the unit cell R
lattice vectors away
3. Standard devaition of the quantity in (2)
4. Maximum absolute value in (2)
A.4 w90vdw
This utility provides an implementation of a method for calculating van der Waals energies based on
the idea of density decomposition via MLWFs.

160 wannier90: User Guide
For theoretical details, please see the following publication and references therein:
Lampros Andrinopoulos, Nicholas D. M. Hine and Arash A. Mostofi, “Calculating dispersion interac-
tions using maximally localized Wannier functions”, J. Chem. Phys. 135, 154105 (2011).
For further details of this program, please see the documentation in utility/w90vdw/doc/ and the
related examples in utility/w90vdw/examples/.
A.5 w90pov
An utility to create Pov files (to render the Wannier functions using the PovRay utility) is provided
inside utility/w90pov.
Please refer to the documentation inside utility/w90pov/doc for more information.
A.6 k_mapper.py
The wannier90 code requires the definition of a full Monkhorst–Pack grid of k-vectors, which can be
obtained by means of the kmesh.pl utility. In order to perform a GW calculation with the Yambo
code, you need to perform a nscf calculation on a grid in the irreducible BZ. Moreover, you may need a
finer grid to converge the GW calculation than what you need to interpolate the band structure. The
k_mapper.py tools helps in finding the k-vectors indeces of a full grid needed for interpolation into the
reduced grid needed for the GW calculation with Yambo.
Within the /utility folder type:
./k_mapper.py nx ny nz "path_of_the_QE_nscf_output_file_given_to_Yambo"
A.7 gw2wannier90.py
This utility allows to sort in energy the input data of wannier90 (e.g. overlap matrices and energy
eigenvalues). gw2wannier90.py allows to use wannier90 at the G0W0level, where quasi-particle cor-
rections can change the energy ordering of eigenvalues (Some wannier90 modules require states to be
ordered in energy).
Within the work directory type: ./gw2wannier90.py seedname options
NB: Binary files are supported, though not reccommended.
Appendix B
Frequently Asked Questions
B.1 General Questions
B.1.1 What is wannier90?
wannier90 is a computer package, written in Fortran90, for obtaining maximally-localised Wannier
functions, using them to calculate bandstructures, Fermi surfaces, dielectric properties, sparse Hamil-
tonians and many things besides.
B.1.2 Where can I get wannier90?
The most recent release of wannier90 is always available on our website http://www.wannier.org.
B.1.3 Where can I get the most recent information about wannier90?
The latest news about wannier90 can be followed on our website http://www.wannier.org.
B.1.4 Is wannier90 free?
Yes! wannier90 is available for use free-of-charge under the GNU General Public Licence. See the file
LICENSE that comes with the wannier90 distribution or the GNU hopepage at http://www.gnu.org.
B.2 Getting Help
B.2.1 Is there a Tutorial available for wannier90?
Yes! The examples directory of the wannier90 distribution contains input files for a number of tutorial
calculations. The doc directory contains the accompanying tutorial handout.
161

162 wannier90: User Guide
B.2.2 Where do I get support for wannier90?
There are a number of options:
1. The wannier90 User Guide, available in the doc directory of the distribution, and from the
webpage (http://www.wannier.org/user_guide.html)
2. The wannier90 webpage for the most recent announcements (http://www.wannier.org)
3. The wannier90 mailing list (see http://www.wannier.org/forum.html)
B.2.3 Is there a mailing list for wannier90?
Yes! You need to register: go to http://www.wannier.org/forum.html and follow the instructions.
B.3 Providing Help: Finding and Reporting Bugs
B.3.1 I think I found a bug. How do I report it?
•Check and double-check. Make sure it’s a bug.
•Check that it is a bug in wannier90 and not a bug in the software interfaced to wannier90.
•Check that you’re using the latest version of wannier90.
•Send us an email. Make sure to describe the problem and to attach all input and output files
relating to the problem that you have found.
B.3.2 I have got an idea! How do I report a wish?
We’re always happy to listen to suggestions. Email your idea to the wannier90 developers.
B.3.3 I want to help! How can I contribute to wannier90?
Great! There’s always plenty of functionality to add. Email us to let us know about the functionality
you’d like to contribute.
B.3.4 I like wannier90! Should I donate anything to its authors?
Our Swiss bank account number is... just kidding! There is no need to donate anything, please just
cite our paper in any publications that arise from your use of wannier90:
[ref] A. A. Mostofi, J. R. Yates, G. Pizzi, Y.-S. Lee, I. Souza, D. Vanderbilt and N. Marzari,
An updated version of wannier90: A Tool for Obtaining Maximally-Localised Wannier
Functions, Comput. Phys. Commun. 185, 2309 (2014)
http://dx.doi.org/10.1016/j.cpc.2014.05.003

wannier90: User Guide 163
B.4 Installation
B.4.1 How do I install wannier90?
Follow the instructions in the file README.install in the main directory of the wannier90 distribution.
B.4.2 Are there wannier90 binaries available?
Not at present.
B.4.3 Is there anything else I need?
Yes. wannier90 works on top of an electronic structure calculation.
At the time of writing there are public, fully functioning, interfaces between wannier90 and pwscf,
abinit (http://www.abinit.org), siesta (http://www.icmab.es/siesta/), VASP (https://www.
vasp.at), Wien2k (http://www.wien2k.at), fleur (http://www.fleur.de), OpenMX (http://
www.openmx-square.org/), GPAW (https://wiki.fysik.dtu.dk/gpaw/).
To use wannier90 in combination with pwscf code (a plane-wave, pseudopotential, density-functional
theory code, which is part of the quantum-espresso package) you will need to download pwscf from
the webpage http://www.quantum-espresso.org. Then compile pwscf and the wannier90 interface
program pw2wannier90. For instructions, please refer to the documentation that comes with the
quantum-espresso distribution.
For examples of how to use pwscf and wannier90 in conjunction with each other, see the wannier90
Tutorial.
Bibliography
[1] N. Marzari and D. Vanderbilt, Phys. Rev. B 56, 12847 (1997).
[2] I. Souza, N. Marzari, and D. Vanderbilt, Phys. Rev. B 65, 035109 (2001).
[3] A. A. Mostofi, J. R. Yates, Y.-S. Lee, I. Souza, D. Vanderbilt, and N. Marzari, Comput. Phys.
Commun. 178, 685 (2008).
[4] D. Vanderbilt, Phys. Rev. B 41, 7892 (1990).
[5] M. Posternak, A. Baldereschi, S. Massidda, and N. Marzari, Phys. Rev. B 65, 184422 (2002).
[6] A. Damle and L. Lin, ArXiv e-prints (2017), 1703.06958 .
[7] F. Gygi, J. L. Fattebert, and E. Schwegler, Comput. Phys. Commun. 155, 1 (2003).
[8] R. Sakuma, Phys. Rev. B 87, 235109 (2013).
[9] R. Wang, E. A. Lazar, H. Park, A. J. Millis, and C. A. Marianetti, Physical Review B 90 (2014),
10.1103/PhysRevB.90.165125.
[10] M. B. Nardelli, Phys. Rev. B 60, 7828 (1999).
[11] T. Yusufaly, D. Vanderbilt, and S. Coh, “Tight-Binding Formalism in the Context of the PythTB
Package,” http://physics.rutgers.edu/pythtb/formalism.html.
[12] J. Ibañez-Azpiroz, S. S. Tsirkin, and I. Souza, ArXiv e-prints (2018), arXiv:1804.04030 .
[13] X. Wang, J. R. Yates, I. Souza, and D. Vanderbilt, Phys. Rev. B 74, 195118 (2006).
[14] D. Xiao, M.-C. Chang, and Q. Niu, Rev. Mod. Phys. 82, 1959 (2010).
[15] E. I. Blount, Solid State Physics 13, 305 (1962).
[16] J. R. Yates, X. Wang, D. Vanderbilt, and I. Souza, Phys. Rev. B 75, 195121 (2007).
[17] M. G. Lopez, D. Vanderbilt, T. Thonhauser, and I. Souza, Phys. Rev. B 85, 014435 (2012).
[18] D. Ceresoli, T. Thonhauser, D. Vanderbilt, and R. Resta, Phys. Rev. B 74, 024408 (2006).
[19] S. S. Tsirkin, P. Aguado Puente, and I. Souza, ArXiv e-prints (2017), arXiv:1710.03204 [cond-
mat.mtrl-sci] .
[20] T. Yoda, T. Yokoyama, and S. Murakami, Sci. Rep. 5, 12024 (2015).
[21] S. Zhong, J. E. Moore, and I. Souza, Phys. Rev. Lett. 116, 077201 (2016).
[22] E. Ivchenko and G. Pikus, Sov. Phys. Solid State 16, 1261 (1975).
165

166 wannier90: User Guide
[23] A. Malashevich and I. Souza, Phys. Rev. B 82, 245118 (2010).
[24] G. Pizzi, D. Volja, B. Kozinsky, M. Fornari, and N. Marzari, Comput. Phys. Commun. 185, 422
(2014).
[25] J. Ziman, Principles of the Theory of Solids, 2nd ed. (Cambridge University Press, 1972).
[26] G. Grosso and G. P. Parravicini, Solid State Physics (Academic Press, 2000).
[27] G. D. Mahan, in Intern. Tables for Crystallography, Vol. D (2006) Chap. 1.8, p. 7828.
