Bayes Traits V3.Manual
User Manual: Pdf
Open the PDF directly: View PDF ![]() .
.
Page Count: 81

Table of Contents
Disclaimer................................................................................................................................................ 5
New features / models ........................................................................................................................... 6
Introduction ............................................................................................................................................ 7
Methods and Approach .......................................................................................................................... 8
BayesTraits methods ............................................................................................................................... 8
Tree Format ............................................................................................................................................ 9
Data Format ............................................................................................................................................ 9
Branch lengths ...................................................................................................................................... 10
Running BayesTraits .............................................................................................................................. 10
Running BayesTraits with a command file ........................................................................................ 11
Continuous-time Markov models of trait evolution for discrete traits ................................................ 11
The Generalised Least Squares model for continuously varying traits ................................................. 13
Model Testing: Likelihood ratios and Bayes Factors ............................................................................. 13
Stepping stone sampler .................................................................................................................... 14
Harmonic mean ................................................................................................................................. 15
Priors ..................................................................................................................................................... 15
Burn-in and sampling in MCMC analysis ............................................................................................... 16
The parameter proposal mechanism and mixing in MCMC analysis .................................................... 17
Mixing................................................................................................................................................ 17
Monitoring Acceptance Rates ........................................................................................................... 17
Over parameterisation .......................................................................................................................... 18
Parameter restrictions .......................................................................................................................... 18
Reverse Jump MCMC ............................................................................................................................ 18
MultiState ML example ......................................................................................................................... 19
MultiState MCMC example ................................................................................................................... 20
Parameter restriction example ............................................................................................................. 21
Tags ....................................................................................................................................................... 23
Ancestral state reconstruction MultiState / Discrete ........................................................................... 23
Fixing node values / fossilising .............................................................................................................. 25
Discrete ................................................................................................................................................. 26
Discrete independent ........................................................................................................................... 26
Discrete dependent .............................................................................................................................. 28
Reverse Jump MCMC and model reduction ......................................................................................... 29
Covarion model ..................................................................................................................................... 30
Discrete: Covarion ................................................................................................................................. 31
Discrete: Heterogeneous ...................................................................................................................... 32
Local Heterogeneous models of evolution (MultiState and Discrete) ................................................. 32
Continuous: Random Walk (Model A) ML ............................................................................................ 35
Continuous: Random Walk (Model A) MCMC ...................................................................................... 35
Testing trait correlations: continuous ................................................................................................... 35
Continuous: Directional (Model B) MCMC ........................................................................................... 37
Continuous: Regression ........................................................................................................................ 37
Testing trait significance ................................................................................................................... 38
Continuous: Estimating ancestral sates and tip values ......................................................................... 38
Estimating unknown values internal nodes .......................................................................................... 38
Estimating unknown values for tips ...................................................................................................... 39
Independent contrast ........................................................................................................................... 40
Independent contrast: Correlation ....................................................................................................... 41
Independent contrast: regression ........................................................................................................ 42
Tree transformations, kappa, lambda, delta, OU ................................................................................. 43
Kappa ................................................................................................................................................ 43
Delta .................................................................................................................................................. 45
Lambda .............................................................................................................................................. 46
Ornstein Uhlenbeck (OU) .................................................................................................................. 48
Tree transformations table ................................................................................................................... 49
Tree transformations commands ......................................................................................................... 49
Variable rates model ............................................................................................................................. 51
Geo (Geographical) model .................................................................................................................... 52
Samples of trait data ............................................................................................................................. 53
Local transformations, kappa, lambda, delta, OU, nodes and branches .............................................. 54
Reverse Jump local transformations, kappa, lambda, delta, and OU ................................................... 56
Maximum likelihood search options ..................................................................................................... 58
MLTries (Maximum Likelihood Tries) ................................................................................................ 58
MLAlg (Maximum Likelihood Algorithm) .......................................................................................... 58
MLTol (Maximum Likelihood Tolerance) .......................................................................................... 59
MLMaxEval (Maximum Likelihood Maximum evaluations) .............................................................. 59
SetMinMaxRate (Set Minimum Maximum Rate) .............................................................................. 59
Model options table .............................................................................................................................. 60
Output files ........................................................................................................................................... 61
BayesTraits versions .............................................................................................................................. 62
Quad Precision .................................................................................................................................. 62
Threaded ........................................................................................................................................... 62
OpenCL .............................................................................................................................................. 62
OpenCL graphics hardware ........................................................................................................... 62
OpenCL driver ............................................................................................................................... 62
Building BayesTraits from source ......................................................................................................... 63
Common problem / Frequently Asked Questions ................................................................................ 64
1) Problems starting the program ................................................................................................. 64
2) Common tree and data errors .................................................................................................. 64
3) “Memory allocation error in file …” .......................................................................................... 64
4) Chain is not mixing between trees. .......................................................................................... 64
5) Cannot find a valid set of starting parameters. ........................................................................ 64
6) Too many free parameters to estimate ................................................................................... 65
7) Run to run variation .................................................................................................................. 65
Command table ..................................................................................................................................... 66
Command List ....................................................................................................................................... 68
References ............................................................................................................................................ 80
Disclaimer
Software development is a notoriously error prone activity. While efforts are made to make
the programme as accurate as possible, due to the size and complexity of BayesTraits it will contain
bugs, care must be taken when using the software. If you notice any unusual behaviour including
crashes or inconsistent results please contact the authors with the data, trees and commands used.
New features / models
Version 3 of BayesTraits includes a range of heterogeneous models, removing the assumption that
the model of evolution is constant threw the tree, these can be particularly important for large or
diverse trees.
Automatically detect shifts in rates of evolution (Variable Rates model) for MultiState / discrete data,
as well as continuous.
Kappa, lambda, delta and rate scalars can be applied to nodes within a tree
Allow patterns of evolution to vary within a tree for MultiState / discrete data
Improved parallelism
Integration of a fast / high precision likelihood calculation for multi-state and discrete models
Reverse Jump MCMC (RJ-MCMC) methods to detect changes in evolutionary patterns (kappa,
lambda, and delta)
Improved Maximum Likelihood searching
Geographical models
Distributions of trait data instead of single values
More control over priors
Introduction
BayesTraits is a computer package for performing analyses of trait evolution among groups
of species for which a phylogeny or sample of phylogenies is available. It can be applied to the
analysis of traits that adopt a finite number of discrete states, or to the analysis of continuously
varying traits. The methods can be used to take into account uncertainty about the model of
evolution and the underlying phylogeny. Evolutionary hypotheses can be tested including
Finding rates of evolution
Establishing correlations between traits
Calculating ancestral state values
Building regression models
Predicting unknown values
Testing for modes of evolution
Accelerated / decelerated rates of evolution through time
Magnitude of phylogenetic signal
Variable rate of evolution through time and within the tree
Ornstein-Uhlenbeck processes
If trait change is concentrated at speciation events
Test for covarion evolution
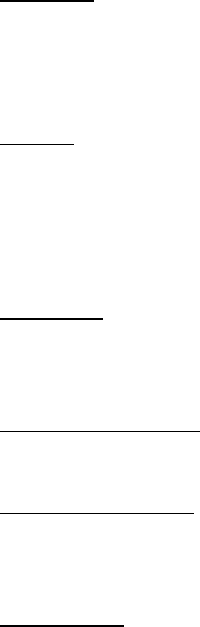
Methods and Approach
BayesTraits uses Markov chain Monte Carlo (MCMC) methods to derive posterior
distributions and maximum likelihood (ML) methods to derive point estimates of, log-likelihoods, the
parameters of statistical models, and the values of traits at ancestral nodes of phylogenies. The user
can select either MCMC or reversible-jump MCMC. In the latter case the Markov chain searches the
posterior distribution of different models of evolution as well as the posterior distributions of the
parameters of these models (see below).
BayesTraits can be used with a single phylogenetic tree in which case only uncertainty about
model parameters is explored, or, it can be applied to suitable samples of trees such that models are
estimated and hypotheses are tested taking phylogenetic uncertainty into account.
BayesTraits is designed to be as flexible as possible, but users must treat this flexibility with
care, as it allows models which may not have a valid interpretation, for example you can create
complex models which cannot be estimated from the given data, such as estimating 100 parameters
for a 30 taxa tree or build a regression model from unsuitable data.
BayesTraits methods
• MultiState is used to reconstruct how traits that adopt a finite number of discrete states
evolve on phylogenetic trees. It is useful for reconstructing ancestral states and for testing models
of trait evolution. It can be applied to traits that adopt two or more discrete states (Pagel, Meade et
al. 2004)
• Discrete is used to analyse correlated evolution between pairs of discrete binary traits. Most
commonly the two binary states refer to the presence or absence of some feature, but could also
include “low” and “high”, or any two distinct features. Its uses might include tests of correlation
among behavioural, morphological, genetic or cultural characters (Pagel 1994, Pagel and Meade
2006)
• Continuous is for the analysis of the evolution of continuously varying traits using a GLS
framework. It can be used to model the evolution of a single trait, to study correlations among pairs
of traits, or to study the regression of one trait on two or more other traits (Pagel 1999).
• Continuous regression is used to build regression models and use these models to
reconstruct unknown values (Organ, Shedlock et al. 2007).
• Independent contrast methods (Felsenstein 1973, Freckleton 2012) provides a very fast
alternative to the GLS methods. They are useful for analysing large trees, but some model
parameters cannot be estimated.
• Variable rates is used to detect variations in the rate of evolution threw the tree, accounting
for changes in rate on a single lineage or for a group of taxa (Venditti, Meade et al. 2011).
This manual is designed to show how to use the programs that implement these models.
Detailed information about the methods can be found in the papers listed at the end (some are
available as pdfs on our website). Syntax and a description of available commands in BayesTraits is
listed below.

Model
Data
MultiState
Multistate
Discrete: Independent
Two binary traits
Discrete: Dependent
Two binary traits
Continuous: Random Walk
Continuous traits
Continuous: Directional
Continuous traits
Continuous: Regression
Two or more continuous traits
Independent Contrasts
Continuous traits
Independent Contrasts: Correlation
Two or more continuous traits
Independent Contrasts: Regression
Two or more continuous traits
Discrete: Covarion
Two binary traits
Discrete: Heterogeneous
Two binary traits
Geographical
Two traits longitude and latitude
Tree Format
BayesTraits requires trees to be in Nexus format, trees can include hard polytomies but must
be correctly rooted and include branch lengths. Taxa names must not be included in the description
of the tree but should be linked to a number in the translate section of the tree file, a number of
example trees are included with the program.
Data Format
Data is read from a plain text file (ASCII), with one line for each species or taxon in the tree.
The names must be spelled exactly as in the trees and must not have any spaces within them but do
not need to be in the same order. Following a species name, leave white space (tab or space) and
enter the first column of data, repeat this for additional columns of data (see below). Data for
MultiState analysis should take values such as “0”, “1”, “2” or “A”, “B”, “C” etc. Data for discrete
must be exactly two columns of binary data and must take the values “0” or “1”. Continuous data
should be integers or floating points. If data are missing it should be represented using “-“, for a trait
in MultiState or Discrete the remaining data for the taxa is used, if data are missing for Continuous
models the taxa are removed from the tree. Example data files for MultiState, Discrete and
Continuous data are included with the program.
Example of MultiState data
Taxon01
A
A
C
Taxon02
B
B
C
Taxon03
A
B
-
Taxon04
C
BC
B
….
TaxonN
C
A
B
Taxon 3 has missing data for the third site. In Discrete and Multistate missing data are
treated as if the trait could take any of the other states, with equal probability. Alternatively you can
indicate uncertainty about a traits value. For example, the second trait for Taxon 4 is uncertain. The
code BC signifies that it can be in states B or C (with equal probability) but not in state A.
Example of Discrete (binary) data
Taxon01
0
0
Taxon02
0
-
Taxon03
1
0
Taxon04
0
1
….
TaxonN
1
1
Example of Continuous data
Taxon01
10
9.0
Taxon02
1.06
-
Taxon03
5.3
2
Taxon04
3
4
….
TaxonN
1
1.1
Branch lengths
Model parameters are dependent on branch lengths, as branch lengths can come in differing
units, years, millions of years, expected number of substitutions, it is recommended that the branch
lengths are scaled to have a mean of 0.1 for MultiState and discrete models, this prevents the rates
becoming small, hard to estimate or search for. The command “ScaleTrees” can be used to scale the
branch lengths, if no parameters are supplied the branches are scaled to have a mean of 0.1,
otherwise the branch lengths are scaled by the supplied factor.
Running BayesTraits
BayesTraits is run from the command prompt (Windows) or terminal (OS X and Linux), it is
not run by double clicking on it. The program, tree file and data file should be placed in the same
directory / folder. Start the command prompt / terminal and change to the directory that the
program, tree and data are in and type.
Windows
BayesTraitsV3.exe TreeFile DataFile
Linux / OSX
./BayesTraitsV3 TreeFile DataFile
Where TreeFile is the name of the tree file and DataFile is the name of the data file.
Running BayesTraits with a command file
If you need to run an analysis multiple times or if it is complex it can be more convenient to
place the commands into a command file, instead of typing them in each time. A command file is a
plane ASCII text file, that contains the commands to run.
An example command file is included with the program, “ArtiodactylMLIn.txt”. The file has
three lines.
1
1
Run
The first line selects MultiState, the second is for ML analysis and the third is to run the
program. To run BayesTraits using the Artiodacty tree, data and input file use the following
command. The command and their order can be found by running the program normally and noting
your inputs.
Windows
BayesTraitsV3.exe Artiodactyl.trees Artiodactyl.txt < ArtiodactylMLIn.txt
Linux / OS X
./BayesTraitsV3 Artiodactyl.trees Artiodactyl.txt < ArtiodactylMLIn.txt
Continuous-time Markov models of trait evolution for discrete traits
MultiState and Discrete fit continuous-time Markov models to discrete character data. This
model allows the trait to change from the state it is in at any given moment to any other state over
infinitesimally small intervals of time. The rate parameters of the model estimate these transition
rates (see (Pagel 1994) for further discussion). The model traverses the tree estimating transition
rates and the likelihood associated with different states at each node.
The table below shows an example of the model of evolution for a trait that can adopt three
states, 0, 1, and 2. The qij are the transition rates among the three states, and these are what the
method estimates on the tree, based on the distribution of states among the species. If these rates
differ statistically from zero, this indicates that they are a significant component of the model. The
main diagonal elements are constrained to be equal to minus the sum of the other elements in the
row.

Example of the model of evolution for a trait that adopts three states
State
0
1
2
0
--
q01
q02
1
q01
--
q12
2
q20
q21
--
For a trait that adopts four states, the matrix would have twelve entries, for a binary trait
the matrix would have just two.
Discrete tests for correlated evolution in two binary traits by comparing the fit (log-
likelihood) of two of these continuous-time Markov models. One of these is a model in which the
two traits evolve independently on the tree. Each trait is described by a 22 matrix in the same
format as the one above, but in which the trait adopts just two states, “0” and “1”. This creates two
rate coefficients per trait.
The other model, allows the traits to evolve in a correlated fashion such that the rate of
change in one trait depends upon the background state of the other. The dependent model can
adopt four states, one for each combination of the two binary traits (0,0; 0,1; 1,0; 1,1). It is
represented in the program as shown below and the transition rates describe all possible changes in
one state holding the other constant. The main diagonal elements are estimated from the other
values in their row as before. The other diagonal elements are set to zero as the model does not
allow ‘dual’ transitions to occur, the logic being that these are instantaneous transition rates and the
probability of two traits changing at exactly the same instant of time is negligible. Dual transitions
are allowed over longer periods of time, however. See Pagel, 1994 for further discussion of this
model.
State
0,0
0,1
1,0
1,1
0,0
--
q12
q13
--
0,1
q21
--
--
q24
1,0
q31
--
--
q34
1,1
--
q42
q43
--
The values of the transition rate parameters will depend upon the units of measurement
used to estimate the branch lengths in the phylogeny. If the branch lengths are increased by a factor
‘c’ the transition rates will be decreased by the same factor ‘c’. This has implications for modelling
the rate parameters in Markov chains (see Branch lengths).
BayesTraits implements the covarion model for trait evolution (Tuffley and Steel 1998). This
is a variant of the continuous-time Markov model that allows for traits to vary their rate of evolution
within and between branches. It is an elegant model that deserves more attention, although users
may find it of limited value with small trees.
The Generalised Least Squares model for continuously varying traits
Phylogenetically structured continuously varying data is analysed using a generalised least
squares (GLS) approach that assumes a Brownian motion model of evolution (see (Pagel 1997, Pagel
1999)). In the GLS model, non-independence among the species is accounted for by reference to a
matrix of the expected covariances among species. This matrix is derived from the phylogenetic
tree. The model estimates the variance of evolutionary change (the Brownian motion parameter),
sometimes called the ‘rate’ of change, and the ancestral state of traits at the root of the tree (alpha).
It can also estimate the covariance of changes between pairs of traits, and this is how it tests for
correlation.
The GLS approach as implemented in Continuous makes it possible to transform and scale
the phylogeny to test the adequacy of the underlying model of evolution, to assess whether
phylogenetic correction of the data is required, and to test hypotheses about trait evolution itself –
for example, is trait evolution punctuational or gradual, is there evidence for adaptive radiation, is
the rate of evolution constant.
Generalised Least Squares (GLS) and independent contrasts
The generalised least squares (GLS) method requires a number of computationally intensive
calculations, including matrix inversions, Kronecker products and matrix multiplications. The time it
takes to calculate a solution for GLS methods increases rapidly with the size of the tree, making
analysis on large trees computationally intensive. Independent contrast (Felsenstein 1973,
Freckleton 2012) uses a restricted likelihood method, these methods are computationally efficient at
the expense of not estimating some parameters, especially when using MCMC, see individual model
description for information about which parameters are estimated. If speed is an issue, for data sets
with hundreds or thousands of taxa, independent contrast should be favoured.
Model Testing: Likelihood ratios and Bayes Factors
BayesTraits does not test hypotheses for you but prints out the information needed to make
hypothesis tests. These will be discussed in more detail in conjunction with the examples below, but
here we outline the two kinds of tests most often used.
The likelihood ratio (LR) test is often used to compare two maximum likelihoods derived
from nested models (models that can be expressed such that one is a special or general case of the
other). The likelihood ratio statistic is calculated as
LR= 2[log-likelihood(better fitting model) – log-likelihood(worse fitting model)]

The likelihood ratio statistic is asymptotically distributed as a 2 with degrees of freedom
equal to the difference in the number of parameters between the two models. However, in some
circumstances (Pagel 1994, Pagel 1997) the test may follow a 2 with fewer degrees of freedom.
Variants of the LR test include the Akaike Information Criterion and the Bayesian
Information Criterion. We do not describe these tests here. They are discussed in many works on
phylogenetic inference (see for example, (Felsenstein 2004)).
The LR, Akaike and Bayesian Information Criterion tests presume that the likelihood is at or
near its maximum likelihood value. In a MCMC framework tests of likelihood often rely on Bayes
Factors (BF). The logic is similar to the likelihood ratio test, except here we compare the marginal
likelihoods of two models rather than their maximum likelihoods. The marginal likelihood of a
model is the integral of the model likelihoods over all values of the models parameters and over
possible trees, weighted by their priors. In practice this marginal likelihood is difficult to calculate
and must be estimated.
Stepping stone sampler
The stepping stone sampler (Xie, Lewis et al. 2011) estimates the marginal likelihood by
placing a number of ‘stones’ which link the posterior with the prior, the stones are successively
heated, forcing the chain from the posterior towards the prior, this provides an effective estimate of
the marginal likelihood. The “stones” command, is used to set the sampler, the command takes the
number of stones and the number of iterations to run the chain on each stone. An example of
setting the stones sampler is below, the command sets the sampler to use 100 stones and run each
stone for 10,000 iterations.
Stones 100 10000
The sampler runs after the chain has finished and produces a file with the extension
“Stones.txt”, the log marginal likelihood is recorded on the last line of the file. Other information
such as temperature, the stones likelihood and marginal likelihood of each stone is also included but
this is mainly for diagnostic purposes.
The marginal likelihood from the stepping stone sampler are expressed on a natural log
scale, these values can be converted into Log Bayes Factors using the formula below. Raffety in
(Gilks, Richardson et al. 1996) Pages 163–188, provides an interpretation of these values.
Log Bayes Factors = 2(log marginal likelihood complex model – log marginal likelihood simple model)
Log Bayes Factors
Interpretation
<2
Weak evidence
>2
Positive evidence
5-10
Strong evidence
>10
Very strong evidence
The stepping stone estimate of the marginal likelihood is sensitive to a number of factors,
including, priors, length of the chain, number of estimated parameters and run to run variation. Care
should be taken to ensure estimates are accurate and stable, multiple independent run should be
used, the accuracy of the sampler can be increased by using more stones and/or sampling each
stone for longer. These options should be investigated if there is large run to run variation. Model
testing is a controversial topic with Bayesian analysis, and other options such as BIC, AIK, DIC may be
considered.
The stepping stone sampler places the stones connecting the posterior distribution with the
prior according to a beta distribution, the default uses an α = 0.4, β = 1.0, these parameters seem to
work well but other beta parameters can be specified using the stones command. The stones
command can take the number of stones, the number of iterations per stone, α and β. The
command below uses 250 stones each for 5000 iterations drawn from a beta distribution with an α
of 2.2 and β of 5.7
Stones 250 5000 2.2 5.7
Harmonic mean
Previous versions of BayesTraits produced a running harmonic mean to estimate the
marginal likelihood, this has been removed in version 3, as the stepping stone sampler produces a
more stable estimate and is computationally more efficient. For backwards compatibility a web site
that calculates the log harmonic mean from a sample of likelihoods has been created.
www.evolution.reading.ac.uk/HMeanCalc/
The site take a list of log likelihoods and calculates the log harmonic mean from them. An
example of 10,000 log likelihoods can be found in the file “HarmonicMeanLh.txt”, if the likelihoods
are passed into the text box on the web site, the Log Harmonic mean should be -141.448
Priors
When using the MCMC analysis method, the prior distributions of the parameters of the
model of evolution must be chosen. Uniform or uninformative priors should be used if possible as
these assume all values of the parameters are equally likely a priori and are therefore easily justified.
Uniform priors can be used when the signal in the data is strong. But in a comparative study there
will typically only be one or a few data points (unlike the many hundreds or thousands in a typical
gene-sequence alignment) so a stronger prior than a uniform may be required.
Priors are the soft underbelly of Bayesian analyses. The guiding principle is that if the choice
of prior is critical for a result, you must have a good reason for choosing that prior. It is often useful
to run maximum likelihood analyses on your trees to get a sense of the average values of the
parameters. One option if a uniform with a wide interval does not constrain the parameters is to use
a uniform prior with a narrower range of values, and this might be justified either on biological
grounds or perhaps on the ML results. The ML results will not define the range of the prior but can
give an indication of its midpoint.
NOTE: A rule of thumb when choosing a constrained or informed (non-uniform) prior is that if the
posterior distribution of parameter values seems truncated at either the upper or lower end of the
constrained range, then the limits on the prior must be changed.
The program allows uniform, exponential, gamma, Chi-squared, log normal and normal
distributed priors, specified as “uniform”, “exp”, “gamma”, “chi”, “lognormal”, “normal” and
“sgamma”. The uniform prior requires the user to specify a range, the exponential distribution
always has its mode at zero and then slopes down, whereas the gamma can take a variety of uni-
modal shapes or even mimic the exponential. The exponential prior is useful when the general
feeling is that smaller values of parameters are more likely than larger ones. If the parameters are
thought to take an intermediate value, a gamma prior with an intermediate mean can be used. The
scaled gamma (Venditti, Meade et al. 2011), sgamma, is useful for scalars.
Priors are set using the prior command, the Prior command takes a parameter to set the
prior for, a distribution and the parameters of the distribution. For example,
Prior q01 exp 10
is used to set an exponential prior with a mean of 10 for the rate parameter q01
Prior delta uniform 0 100
sets the prior on delta to a uniform 0 – 100
In many cases you will want to use the same prior on all rate parameters, the PriorAll
command can be used to do this. It is identical to the prior command but does not take a parameter.
For example,
PriorAll exp 10
sets all rate priors to an exponential with a mean of 10
Because it can be difficult to arrive at suitable values for the parameters of the prior
distributions, BayesTraits allows the use of a hyper-prior. A hyper-prior is simply a distribution –
usually a uniform -- from which are drawn values to seed the values of the exponential or gamma
priors. We recommend using hyper-priors as they provide an elegant way to reduce some of the
uncertainty and arbitrariness of choosing priors in MCMC studies. For an example of selecting priors
and using a hyper-prior see (Pagel, Meade et al. 2004)
When using the hyper-prior approach you specify the range of values for the uniform
distribution that is used to seed the prior distribution. Thus, for example
HyperPriorAll exp 0 10
seeds the mean of the exponential prior from a uniform on the interval 0 to 10.
HyperPriorAll gamma 0 10 0 10
seeds the mean and variance of the gamma prior from uniform hyper priors both on the
interval 0 to 10. For a full list of commands see Command List.
Burn-in and sampling in MCMC analysis
The burn-in period of a MCMC run is the early part of the run while the chain is reaching
convergence. It is impossible to give hard and fast rules for how many iterations to give to burn-in.
We often find that a minimum of 10,000 and seldom more than 1,000,000 is sufficient for simple
models. With more complex models (Variable rates model, ect) or larger trees often requiring longer
burn-in periods. The length of burn-in is set with the burnin command. During burn-in nothing is
printed.
Because successive iterations of most Markov chains are autocorrelated, there is frequently
nothing to be gained from printing out each line of output. Instead the chain is sampled or thinned
to ensure that successive output values are roughly independent. This is the job of the sample
command. It instructs the program only to print out every nth sample of the chain. Choose this
value such that the autocorrelation among successive points is low (this can be checked in most
statistics programs or Excel). For many comparative datasets, choosing every 1000th or so iterations
is more than adequate to achieve a low autocorrelation.
The chain is run for 1,010,000 iterations by default, this can be changed with the iterations
command, which takes the number of iterations to run for or -1 for an infinite chain, which can be
stopped by holding Ctrl and pressing C.
The parameter proposal mechanism and mixing in MCMC analysis
Mixing
Mixing, the proportion of proposed changes to a chain that is accepted, is key to a successful
MCMC analysis, MCMC proceeds by proposing changes to parameters. If proposed changes to a
parameter are too large the likelihood will change dramatically, and at convergence many of the
proposed changes will have a poor likelihood. This will cause the chain to have a low acceptance rate
and the chain will mix poorly or even become stuck. The other side of the coin is, if small changes are
proposed the likelihood does not change much, leading to a high acceptance rate, but the chain
typically does not explore the parameter space effectively. An ideal acceptance rate is often
between 20-40% when the chain is at convergence.
Parameter values can vary widely between data sets and trees, as the units data and branch
lengths are in can vary orders of magnitude. This makes it very hard to find a universal proposal
mechanism. An automatic tuning method is used in BayesTraits to adapt the proposal mechanism to
achieve an acceptance rate of 35%.
Monitoring Acceptance Rates
BayesTraits produces a schedule file, which is used to monitor how the chain is mixing, the
file contains the schedule, the percentage of operators tried, followed by a header. The header
shows the number of times an operator was tried and the percentage of times it was accepted, if
auto tune is used the rate deviation values, acceptance rate for that parameter, the average
acceptance for that iteration and the running mean acceptance rate is recorded. The schedule file
should be reviewed to make sure the chain is mixing correctly.
Over parameterisation
Due to the statistical nature of the methods it is possible to create over parameterised
models, were too many parameters are estimated from not enough data. Indications of over
parameterisation include, poorly estimated parameters, parameters trading off against each other,
suboptimal likelihoods, and poor convergence / parameter optimisation. Model complexity can be
reduced by combining parameters with the restrict command, or by using reverse jump MCMC and
ensuring the ratio of parameters to data is not high.
Parameter restrictions
For MultiState and discrete models the number of parameters increases roughly as a square
of the number of states, it is important to have sufficient data to estimate them. MultiState and
discrete models allow parameters to be combined, reducing the number of free parameters. The
restrict command (res) is used to restrict parameters, the command takes two or more parameter
names, restricting all supplied parameters to the first, it can also be used to restrict parameters to a
constant.
The following command restricts alpha2 to alpha1
Res alpha1 alpha2
To restrict all parameters, in an independent model, to alpha1 use
Res alpha1 alpha2 beta1 beta2
Or
ResAll alpha1
Parameters can also be restricted to constants, including zero, in the same way
Res alpha1 1.5
Or
Res alpha1 alpha2 1.5
Model testing (see Model Testing: Likelihood ratios and Bayes Factors) can be used to test if
a parameter is statistically justified, when rates are restricted the number of free parameters is
reduced.
Reverse Jump MCMC
For a complex model the number of possible restrictions is large, and may be impossible to
test. A reverse jump MCMC method (Green 1995) was developed to integrate results over model
parameter and model restrictions, for a detailed description see (Pagel and Meade 2006).
The RevJump (RJ) command is used to select reverse jump MCMC, the command takes a
prior and prior parameters. For example, the command below uses reverse jump with an
exponential prior with a mean of 10. The second command uses reverse jump with a hyper
exponential prior where the mean of the exponential is drawn from a uniform 0 - 100
RevJump exp 10
Or
RJHP exp 0 100
For the general case
RevJump Prior Name Prior parameters
Or
RJHP Prior Name Prior parameters range
Where the prior name is “exp”, “gamma”, “uniform”
Parameter restrictions can be used in combination with Reverse Jump, if you would like to
set parameters to specific values or use a specific set of restrictions.
MultiState ML example
Start the program using the “Artiodactyl.trees” tree file and the “Artiodactyl.txt” file. The
following screen should be presented to you
Please select the model of evolution to use.
1) MultiState
Select 1 for the MultiState model
Please select the analysis method to use.
1) Maximum Likelihood.
2) MCMC
Select 1 for Maximum Likelihood analysis.
The default options will be printed, displaying basic information. This should always be
checked to ensure it is what you expect.
Type
run
to start the analysis
The options for the run will be printed followed by a header row.

Header
Output
Tree No
The tree number, 1-500 for this data
Lh
Maximum likelihood value for the tree
qDG
The transition rate from D to G
qGD
The transition rate from G to D
Root P(D)
The probability the root is in state D
Root P(G)
The probability the root is in state G
For each tree in the sample a line of output will be printed. Once all trees have been
analysed the program will terminate.
MultiState MCMC example
Start the program using the “Artiodactyl.trees” tree and “Artiodactyl.txt” data file, the
commands below are used to select MultiState (1) and MCMC (2). The default options will be
printed. The third line sets all priors to an exponential with a mean of 10, and the fourth line starts
the chain with the run command.
1
2
PriorAll exp 10
Run
A header will be printed
Header
Output
Iteration
Current iteration of the chain
Lh
Current likelihood of the chain
Tree No
Current tree number
qDG
Transition rate from D to G
qGD
Transition rate from G to D
Root P(D)
Probability the root is in state D
Root P(G)
Probability the root is in state G
Followed by some output.
Iteration
Lh
Tree No
qDG
qGD
Root
P(D)
Root
P(G)
11000
-7.93307
187
3.702561
2.5446
0.351023
0.648977
12000
-8.98846
495
3.120973
4.914959
0.475628
0.524372
13000
-8.37416
99
3.799383
4.489798
0.417393
0.582607
14000
-10.2806
95
17.07613
27.54498
0.499972
0.500028
15000
-10.7122
95
6.945588
5.865219
0.48436
0.51564
…
…
…
…
…
…
…
1006000
-8.94481
400
8.97661
8.407563
0.473517
0.526483
1007000
-8.53244
147
0.33644
1.477702
0.012116
0.987884
1008000
-8.03562
338
2.454093
3.334973
0.270869
0.729131
1009000
-8.41139
107
2.217407
4.183507
0.365974
0.634026
1010000
-9.72812
61
7.50541
9.538785
0.484114
0.515886
The output will be saved in a log file, ending “.Log.txt”. Output from the chain is tab
separated and is designed to be used in programs such as Excel and JMP. Run to run output will vary
and is dependent on the random seed used.
Parameter restriction example
The previous example assumed that the transition rates from state D to G (qDG) and from
state G to D (qGD) were different and both were estimated. The parameter estimates from a longer
run are plotted below, the two distributions show considerable overlap.
To test if the rate D changes to G (qDG) is significantly different from the rate G changes to D
(qGD), re-run the analysis restricting qGD to take the same value as qDG. First calculate the marginal
likelihood of a model which estimates qGD and qDG separately using the commands below. The first
two commands select MultiState (1) and MCMC (2), the third commands uses an exponential prior
with a mean of 10 for both rates, the fourth command uses the stepping stone sampler with 100
stones and 1000 iterations per stone to estimate the marginal likelihood, the fifth command starts
the analysis.
1
2
PriorAll exp 10
Stones 100 1000
Run
Once the analysis has finished a file containing the marginal likelihood will be created,
Artiodactyl.txt.Stones.txt, the last line will contain the marginal likelihood, and should be similar to
the line below. There will be run to run variation so the numbers will not be identical, but should be
roughly -8.7
Log marginal likelihood: -8.774642
Rerun the analysis restricting qDG to qGD using the commands below.
1
2
PriorAll exp 10
Stones 100 1000
Restrict qDG qGD
Run
The options are the same, except the 5th command sets qGD equal to qDG, the output
should be very similar but the rate parameters (qDG and qGD) will take the same value each
iteration. Once the run has finished, the stepping stone file’s last line should be similar to the one
below.
Log marginal likelihood: -8.296317
Bayes Factors can be used to test if qDG is significantly different from qGD, in this case the
model where qDG = qGD is the simple model as it has one fewer parameters, the model where qDG
≠ qGD is the complex one.
Log Bayes Factor = 2(log marginal likelihood complex model – log marginal likelihood simple model)
Log BF = 2(-8.774642- -8.296317)
Log BF = -0.95665
The log BF is less than two so the simpler model should be favoured (see (Gilks, Richardson
et al. 1996) or the table in Stepping stone sampler)
Values of the marginal likelihood calculated from the stepping stone sampler will vary
between runs, depending on the random seed, values are only for illustrative purposes. These values
are only used to demonstrate basic model testing.
The same restrict command can be used in Maximum Likelihood analysis.
1
1
PriorAll exp 10
Restrict qDG qGD
Run
Tags
Tags are used to identify nodes within a sample of trees, they can be used to reconstruct
ancestral states, fix nodes to specific values (fossilise), test if a node has a different rate or mode of
evolution and fit different evolutionary models to subsets of the tree. The AddTag (AT shortcut) is
used to create a tag, it takes a unique name to identify the tag and a list of taxa names that define
the node.
For example, the command below creates a tag called TestTag on a node defined by Sheep
Goat, Cow and Buffalo.
AddTag TestTag Sheep Goat Cow Buffalo Pronghorn
Ancestral state reconstruction MultiState / Discrete
Note: The syntax for reconstructing a node has changed since version 2.0. Tags are now used to
identify nodes within a sample of trees.
The AddMRCA and AddNode commands are used to reconstruct ancestral states in
MultiState and discrete models. The syntax for the two commands is similar.
The commands take a name to identify the reconstructed node in the output, and the name
of a tag (see Tags) that defines the node. BayesTrees
(http://www.evolution.reading.ac.uk/BayesTrees.html) is a graphical tree viewer which can be used
to generate the commands by clicking on the appropriate node.
Start BayesTraits with the Artiodactyl tree and data file (Artiodactyl.trees, Artiodactyl.txt),
use the commands below to reconstruct a node.
1
2
AddTag TRecNode Porpoise Dolphin FKWhale Whale
AddNode RecNode TRecNode
Run
The first two commands select the MultiState model and MCMC analysis, the third
command creates a tag called TRecNode defined by four taxa Porpoise, Dolphin, FKWhale and
Whale. The AddNode command is used to reconstruct the tag, it takes a name (RecNode in this
instance), so the node can be identified in the output and the name of the tag to reconstruct.
Two new columns will be added to the output “RecNode P(D)” and “RecNode P(G)”, these
represent the probability of reconstructing a D or a G at RecNode.
BayesTraits uses a sample of trees and some nodes may not be present in all trees, the node
defined by Sheep, Goat, Cow, Buffalo and Pronghorn is only present in 58% of the trees. The
posterior probability of node reconstruction will not be present in some trees, some samples of the
chain will record the ancestral sate as "--" because the node is not present in those trees.
The MRCA command reconstructs the Most Recent Common Ancestor, while a MRCA will be
present in every tree it may not be the same node (see (Pagel, Meade et al. 2004) for more details).
Rerun the analysis using MRCA.
1
2
PriorAll exp 10
Res qDG qGD
AddTag TVarNode Sheep Goat Cow Buffalo Pronghorn
AddNode VarNode TVarNode
Run
Any number of nodes can be reconstructed in a single analysis without affecting each other.
Fixing node values / fossilising
Internal nodes can be set to take a fixed value, if external information is available or to test if
the value of one state is significant. The fossil command takes a name, so the node can be identified
in the output, a tag that defines the node and the state or states to fossilise the node in. Fossilised
nodes are found using the most recent common ancestor method.
The command below fossilises a node defined by sheep, goats, cows, buffalo and pronghorn
to state D.
1
2
AddTag FNode Sheep Goat Cow Buffalo Pronghorn
Fossil Node01 FNode D
Run
Be aware that fossilising nodes will influence the models, by forcing a node to take a specific
value the model parameters will be affected. The fossil command can be used to fossilise in multiple
states, if the data had three states, A, B and C the fossil command
Fossil Name Tag AC
Fossilises the node in states A and C but not B.
Nodes can be fossilised for continuous models (not currently independent contrast) in the
same way.
4
2
AddTag Tag-01 Dorcopsulus_macleayi Dorcopsulus_vanheurni
Fossil Node01 Tag-01 90.95
Run
Fossilising states for discrete models requires a number instead of a state, as there are more
combinations of fossil sates. The table below shows the numbers and their corresponding states. X
denotes the likelihood is left unchanged, - sets the likelihood to zero.

Number
0,0
0,1
1,0
1,1
0
X
-
-
-
1
-
X
-
-
2
-
-
X
-
3
-
-
-
X
10
X
X
-
-
11
X
-
X
-
12
X
-
-
X
13
-
X
X
-
14
-
X
-
X
15
-
-
X
X
20
X
X
X
-
21
X
X
-
X
22
X
-
X
X
23
-
X
X
X
Discrete
Discrete is used to test if two binary traits are correlated, significance is established by
comparing the likelihoods of two models, one which assumes the traits evolve independently, with
one which assumes the traits evolution is correlated. The examples focus on MCMC but maximum
likelihood can also be used. The examples use a sample of 500 primate trees (“Primates.trees”) and
a data set of two binary traits, estrus advertisement and multi-male mating (“Primates.txt”). Two
binary traits have 4 possible states, written as “0,0”, “0,1”, “1,0” and “1,1”.
Discrete independent
The independent model assumes the two traits evolve independently, e.g. the transition
from 0 1 in the first trait is independent of the state of the second trait. The independent model
has 4 rate parameters, alpha1, beta1, alpah2 and beta2.
Parameter
Symbol
Trait
Transitions
alpha1
1
1
0 1
beta1
1
1
1 0
alpha2
2
2
0 1
beta2
2
2
1 0

0,0
0,1
1,0
1,1
0,0
-
2
1
0
0,1
2
-
0
1
1,0
1
0
-
2
1,1
0
1
2
-
Start BayesTraits with the tree file “Primates.trees” and the data file “Primates.txt”. Using
the commands below to select the independent model (2) and MCMC analysis (2). Set all the priors
to an exponential with a mean of 10 and use the stepping stone sampler with 100 stones and 1000
iterations per stone to estimate the marginal likelihood, the run command is used to start the
analysis. The commands are listed below. The mean for the prior was found by analysing the tree
using Maximum Likelihood and studying the resulting parameter rate estimates.
2
2
PriorAll exp 10
Stones 100 1000
Run
The output will be similar to the MultiState analysis, the header will contain.
The stepping stone sampler will produce a file “Primates.txt.Stones.txt”, the marginal
likelihood is recorded in the last line,
Log marginal likelihood: -46.674444
The marginal likelihood will show run to run variation but it should be roughly -46.67.
Header
Output
Iteration
Current iteration of the chain
Lh
Current likelihood of the chain
Tree No
Current tree number
alpha1
The alpha1 transition rate
beta1
The beta1 transition rate
alpha2
The alpha2 transition rate
beta2
The beta2 transition rate
Root – P(0,0)
Probability the root is in state 0,0
Root – P(0,1)
Probability the root is in state 0,1
Root – P(1,0)
Probability the root is in state 1,0
Root – P(1,1)
Probability the root is in state 1,1

Discrete dependent
The dependent model assumes that the traits are correlated and the rate of change in one
trait is dependent on the state of the other. The dependent model has 8 parameters, q12, q13, q21,
q24, q31, q34, q42 and q43. Double transitions from state 0,0 to 1,1 or from 0,1 to 1,0 are set to
zero.
Parameter
Dependent on
Trait
Transitions
q12
Trait 1 = 0
2
0 1
q13
Trait 2 = 0
1
0 1
q21
Trait 1 = 0
2
1 0
q24
Trait 2 = 1
1
0 1
q31
Trait 2 = 0
1
1 0
q34
Trait 1 = 1
2
0 1
q42
Trait 2 = 1
1
1 0
q43
Trait 1 = 1
2
1 0
0,0
0,1
1,0
1,1
0,0
-
q1,2
q1,3
0
0,1
q2,1
-
0
q2,4
1,0
q3,1
0
-
q3,4
1,1
0
q4,2
q4,3
-
Start BayesTraits with the tree file “Primates.trees” and the data file “Primates.txt”, select
the dependent model (3) and MCMC analysis (2). Set all the priors to an exponential with a mean of
10 and use the stepping stone sampler with 100 stones and 1000 iterations per stone to estimate
the marginal likelihood, the final command starts the analysis.
3
2
PriorAll exp 10
Stones 100 1000
Run
The output will be very similar to the independent model except that the dependent
parameters are estimated. The marginal likelihood can be found in “Primates.txt.Stones.txt” and
should be roughly -41.62. To test if the traits are correlated calculate a log Bayes Factor (see Model
Testing: Likelihood ratios and Bayes Factors) between the dependent and independent models, in
this case the dependent model is the complex one as it has more parameters. The calculations for
Log Bayes factors is given below.
Log BF = 2(log marginal likelihood complex model – log marginal likelihood simple model)
Log BF = 2(-41.62- -46.67)
Log BF = 10.1
The Log BF of 10.1 suggests there is evidence for correlated evolution. Marginal likelihoods
vary between runs and it is important to get a stable estimate by using multiple independent runs.
Reverse Jump MCMC and model reduction
Given the size of the data and complexity of the models not all parameters may be
statistically distinguishable. The previous parameter restriction example demonstrated how a model
could be simplified by setting parameters equal to each other and how to test if restrictions were
significant. There are 51 possible restrictions for the independent model and over 21,000 for the
dependent model, which would take a long time to test. Reverse jump MCMC (RJ-MCMC) offers an
alternative by integrating results over the model space, weighting naturally by their probabilities,
allowing the users to select viable models and parameters, see (Pagel and Meade 2006) for more
information.
The reverse jump command takes a prior as a parameter, one prior must be applied to all
parameters, the command below uses an RJ MCMC model with an exponential prior with a mean of
10
RevJump exp 10
RJ MCMC can also be used with a hyper-prior, (RJHP command).
RevJumpHP exp 0 100
Run the primates data and tree, with the dependent model and MCMC analysis, using the
commands below
3
2
RevJump exp 10
Stones 100 1000
Run

The output will contain 4 new columns.
Header
Output
No Of Parameters
Number of parameters
No Of Zero
Number of parameters set to zero
Model string
A model string showing parameter restrictions
Dep / InDep
A flag showing if the model is dependent (D)
or independent (I)
Model strings are used to characterise the models restrictions, the string start with ' and is
followed by numbers indicating which parameters are in which groups or a Z if the parameters have
been restricted to zero. For example the model string for a dependent model will have 8
components one for each parameter, the model string “'1 Z 0 0 0 1 1 Z”, has two parameters and
two rates set to zero. The first group consists of the 1st, 6th and 7th parameters (q12, q34 and q42),
the second group is formed of the 3rd, 4th and 5th parameters (q21, q24 and q31), and the 2nd and 8th
parameter is set to zero. This can be checked against the parameter estimates.
To test if a data set is correlated compare the marginal likelihood of an independent model
using RJ MCMC and a dependent model using RJ MCMC.
Covarion model
BayesTraits implements a basic on / off covarion model as described by (Tuffley and Steel
1998) for MultiState and Discrete models, the model requires one additional parameter, the
switching rate between the on / off states. The model allows the rate of evolution to vary through
the tree. The “CV” command is used to activate the covarion model, two additional columns will be
included in the output, “Covar On to Off” and “Covar Off to On”. The switching rate between the on
and off states will be the same.
Below is an example of how to fit a covarion model, using a large bird phylogeny (Jetz,
Thomas et al. 2012) (http://birdtree.org/) and territory data taken from (Tobias, Sheard et al. 2016),
it is used purely as an example and not to make a biological point. First calculate the likelihood and
parameters using a homogenous model. Start BayesTraits with the tree “Bird.trees” and the data file
“BirdTerritory.txt”
1
2
ScaleTrees 0.001
Stones 100 1000
Run

The commands select MultiState (1), MCMC (2), the command “ScaleTrees 0.001” is used to
rescale the branch lengths to prevent the rates from becoming very small and the stones command
is used to estimate the marginal likelihood using 100 stones and 1000 iterations per stone, the final
command starts the analysis. This should produce a marginal likelihood of roughly -1942
Then run the data with a covarion model, the commands are the same except the covarion
command turns on the model.
1
2
Covarion
ScaleTrees 0.001
Stones 100 1000
Run
This should produce a marginal likelihood of roughly -1781, giving a Bayes Factor of over
300, suggesting it is highly significant.
Discrete: Covarion
The discrete covarion model removes the assumption that a trait is either correlated or not
threw out the tree. It simultaneously fits both an independent and dependent model and uses a
covarion method to switch between them. The model requires 14 parameters, 4 for the
independent model, 8 for the dependent model and 2 that switch between the dependent and
independent models. As this model has a large number of parameters the use of reverse jump is
recommended, in combination with a large tree. If the discrete covarion model is significant this is
not necessarily evidence for correlated evolution, the dependent model may be acting as a second
independent model identifying changes in rates or patterns of evolution, this can be tested for by
comparing models that restrict the dependent parameters to independent ones. The discrete
covarion model supports RJ MCMC, to reduce the large number of parameters required.
Header
Output
Iteration
Current iteration of the chain
Lh
Current likelihood of the chain
Tree No
Current tree number
alpha1
The independent alpha1 transition rate
beta1
The independent beta1 transition rate
alpha2
The independent alpha2 transition rate
beta2
The independent beta2 transition rate
q12
The dependent q12 transition rate
q13
The dependent q13 transition rate
q21
The dependent q21 transition rate
q24
The dependent q24 transition rate
q31
The dependent q31 transition rate

q34
The dependent q34 transition rate
q42
The dependent q42 transition rate
q43
The dependent q43 transition rate
qDI
The switching rate between the dependent and independent models
qID
The switching rate between the independent and dependent models
Root - I P(0,0)
Probability the root is in independent state 0,0
Root - I P(0,1)
Probability the root is in independent state 0,1
Root - I P(1,0)
Probability the root is in independent state 1,0
Root - I P(1,1)
Probability the root is in independent state 1,1
Root - D P(0,0)
Probability the root is in dependent state 0,0
Root - D P(0,1)
Probability the root is in dependent state 0,1
Root - D P(1,0)
Probability the root is in dependent state 1,0
Root - D P(1,1)
Probability the root is in dependent state 1,1
Discrete: Heterogeneous
The discrete heterogeneous model is an experimental model which fits both an independent
and a dependent model on a branch by branch bases, each branch is assigned one of the two
models. It can only be used on a single tree with MCMC. The output contains a table “Hetro Model
Key” which links each node in the tree with a number, each iteration of the chain will contain a map
string which indicates which model is assigned to which branch. If the map has a 0 the independent
model is applied to the branch, if the map has a 1 the dependent model is applied. In the same way
a significant result for the discrete covarion model does not necessarily signal correlated evolution
but could be two patterns or rates of evolution, restricting the dependent model to the independent
one can distinguish between these cases. While the model may improve the likelihood the models
assigned to each branch can be highly variable. The discrete covarion model supports RJ MCMC, to
reduce the large number of parameters required.
Local Heterogeneous models of evolution (MultiState and Discrete)
The assumption of a homogenous model of evolution may not be suitable for large
phylogenies, as evolutionary processes may not be constant over long time periods or across a
diverse range of taxa. BayesTraits allows different models of evolution to be fitted to different parts
of the tree. Fitting a different model of evolution allows changes in both rates and patterns of
evolution to be modelled, local transformations can be used to model changes in rates. Fitting a
different model of evolution estimates a separate transition matrix for a subset of the tree, this can
be estimated using MCMC or ML. The command “AddPattern” is used to estimate a separate pattern
of evolution on a node, it takes a name for the pattern and a tag to identify the node on the trees
(see Tags for creating tags).
Below is an example of how to fit a heterogeneous model, using a large bird phylogeny (Jetz,
Thomas et al. 2012) (http://birdtree.org/) and territory data taken from (Tobias, Sheard et al. 2016),
it is used purely as an example and not to make a biological point. First calculate the likelihood and
parameters using a homogenous model, run BayesTraits with the tree file Bird.trees and data file
BirdTerritory.txt, and the following commands

1
2
ScaleTrees 0.001
Stones 100 1000
Run
The commands select MultiState (1), MCMC (2), use “ScaleTrees 0.001” to scale the branch
lengths to prevent the rates from becoming small and start the analysis. The stepping stone sampler
is used with 100 stones and 1000 iterations per stone to estimate the marginal likelihood, the
analysis is started with the run command. This should produce a marginal likelihood of roughly -1942
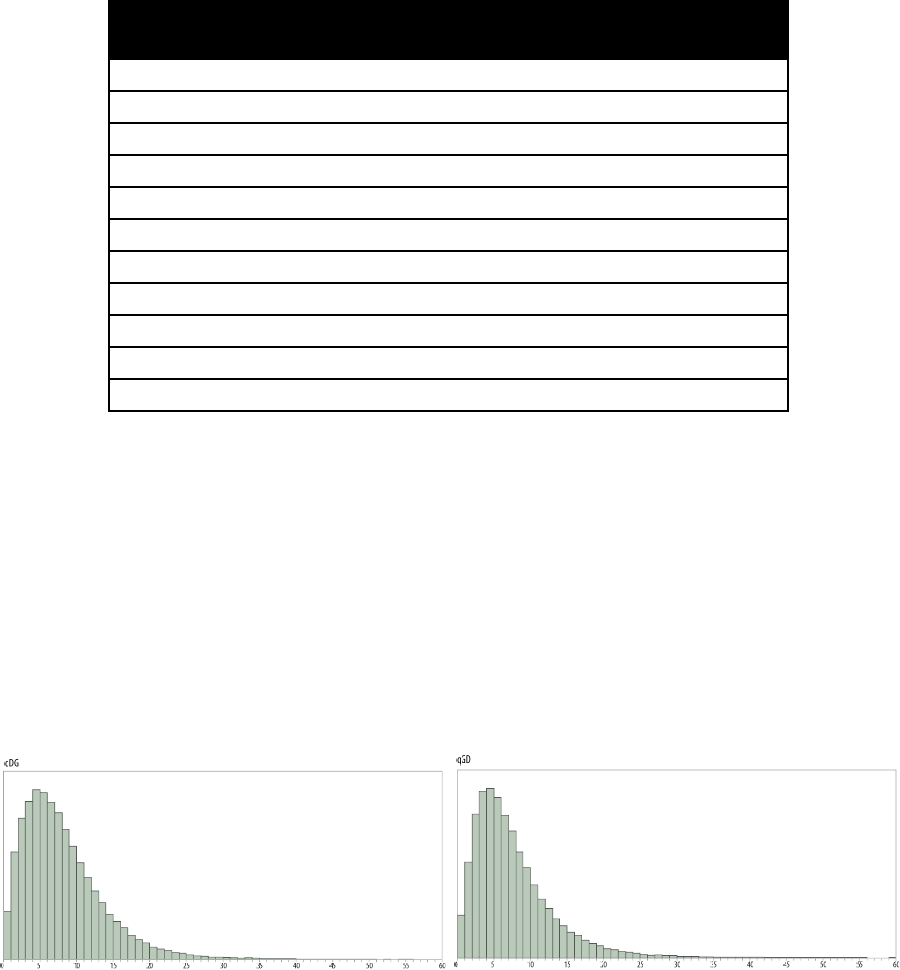
(in BirdTerritory.txt.Stones.txt) and rate parameters (q01 and q10) similar to the ones below.
The commands to run a heterogeneous model, where a different evolutionary model is
fitted to the Passeriformes can be found in the command file “BirdHetCom.txt”, as the tag to define
the Passeriformes has over 3500 taxa and is too large to include.
The commands are
1
2
ScaleTrees 0.001
AddTag PasseriformesTag Strigops_habroptila Nestor_notabilis …
AddPattern Passeriformes PasseriformesTag
Stones 100 1000
Run
The run BayesTraits with the trees, data and command file (see Running BayesTraits with a
command file).
0246810 12 14 16 18 20 22 24 0246810 12 14 16 18 20 22 24
q01 q10
The first three commands select MultiState model, MCMC and scale the tree by a factor of
0.001, the fourth command creates a tag called “PasseriformesTag” defined by the supplied list of
taxa, the fifth command estimates a different pattern of evolution on the node defined by the
PasseriformesTag, the pattern is called “Passeriformes”, the stepping stone sampler is used to
estimate the marginal likelihood. This should produce a marginal likelihood of roughly, -1912.4,
giving a log Bayes Factor of 59.2, suggesting very strong evidence for a heterogeneous pattern of
evolution within the birds. This is born out in the differences in the rate parameters, the parameters
estimated in the Passeriformes are different from the rest of the tree. Interestingly fitting a separate
pattern of evolution alters the estimated ancestral state, from a probability of 0.804 for a
reconstruction of 1 at the root to 0.995.
Different patterns of evolution can be fitted using maximum likelihood or MCMC, they can
be used with MultiState or Independent or Dependent discrete models. Parameters with in a pattern
behave the same as other transition rates, they can be fixed to values, set equal to each other or
modelled using RJ MCMC.
0246 8 10 12 14 16 18 20 22 24 26 28 30
0246 8 10 12 14 16 18 20 22 24 26 28 30
0246 8 10 12 14 16 18 20 22 24 26 28 30
0246 8 10 12 14 16 18 20 22 24 26 28 30

Continuous: Random Walk (Model A) ML
Start BayesTraits with the tree file “Mammal.trees” and data file “MammalBody.txt”, the
tree file is a sample of 50 mammal trees and the data is there corresponding body size, the trees and
data are for illustrative purposes and are not accurate or a good sample. The commands below run a
basic maximum likelihood Brownian motion analysis, 4 selects “Continuous: Random Walk (Model
A)”, 2 selects maximum likelihood and the final command starts the chain.
4
1
Run
Basic information will be printed followed by a header
Header
Output
Tree No
The tree number, 1-50 for this data
Lh
Maximum likelihood value for the tree
Alpha 1
The phylogenetically corrected mean of the data, also the estimated root value
Sigma^2 1
The phylogenetically corrected variance of the data
Continuous: Random Walk (Model A) MCMC
Start BayesTraits with the tree file “Mammal.trees” and data file “MammalBody.txt”, select
“Continuous: Random Walk (Model A)” (4) and MCMC (2), start the analysis with run
4
2
Run
Basic information will be printed followed by a header
Header
Output
Iteration
Current iteration of the chain
Lh
Current likelihood of the chain
Tree No
Current tree number
Alpha Trait 1
The phylogenetically corrected mean of the data, also the estimated root value
Sigma^2 1
The phylogenetically corrected variance of the data
Testing trait correlations: continuous
To test if two traits are correlated, the results from two analysis are compared, one in which
a correlation is assumed (the default) and one where the correlation is set to zero. Run an analysis
using the tree file “Mammal.trees” and a data file “MammalBrainBody.txt”, containing brain and
body size data. The commands select “Continuous: Random Walk (Model A)” (4) and MCMC analysis
(2), the stones command is used to estimate the marginal likelihood using 100 stones and 1000
iterations per stone. The final command starts the analysis.

4
2
Stones 100 1000
Run
Basic information will be printed followed by a header
Header
Output
Iteration
Current iteration of the chain
Lh
Current likelihood of the chain
Tree No
Current tree number
Alpha 1
The phylogenetically corrected mean of the first trait
Alpha 2
The phylogenetically corrected mean of the second trait
Sigma^2 1
The phylogenetically corrected variance of the first trait
Sigma^2 2
The phylogenetically corrected variance of the second trait
R Trait 1 2
R correlation between trait 1 and trait 2
The marginal likelihood is recorded in the last line of the “MammalBrainBody.txt.Stones.txt”
file, it should be roughly -80.93
Rerun the analysis forcing the correlation to be zero using the TestCorrel (TC) command.
4
2
TestCorrel
Stones 100 1000
Run
The output should be similar except the “R Trait 1 2” value will be 0, the marginal likelihood
should be roughly -140.64. The significance of the correlation can be tested by computing a Bayes
Factor between the two runs, the model that estimates the correlation will be the complex one.
If the analysis allowing a correlation produced a marginal likelihood of -80.93 and the
analysis with the correlation fixed to zero gave a marginal likelihood of -146.64, this would lead to a
log Bayes Factor of 119.42, suggesting they are highly correlated.
Log BF = 2(log marginal likelihood complex model – log marginal likelihood simple model)
Log BF = 2(-80.93 - -140.64)
Log BF = 119.42
Continuous: Directional (Model B) MCMC
The directional model can be used to test if there is a directional change in a traits evolution,
by testing if a trait is correlated with the root to tip distance of the taxa. Model B cannot be used
with ultrametric trees as there is no root to tip variation between taxa. A fictional data set
“MammalModelB.txt” can be used to test if there is a significant directional trend by performing a
model test between Model A and Model B. If Model B is significant there is signal for a directional
trend in the data. To test if model B is significant, first run an analysis with the tree file
Mammal.trees and the data file MammalModelB.txt, the following commands select model A and
MCMC, the stones command is used to estimate the marginal likelihood using 100 stones and 1000
iterations per stone and the final command starts the analysis.
4
2
Stones 100 1000
Run
The marginal likelihood, found in the last line of MammalModelB.txt.Stones.txt should be
roughly 60.5 log units.
The commands below rerun the analysis using Model B, where a directional trend is
estimated in the data.
5
2
Stones 100 1000
Run
The marginal likelihood should be roughly 67.28, giving a Log Bayes Factor of roughly 13.56,
suggesting significance.
Log BF = 2(log marginal likelihood complex model – log marginal likelihood simple model)
Log BF = 2(67.28- 60.5)
Log BF = 13.56
Continuous: Regression
The continuous regression model is used to perform regression analysis, test trait
significance and predict unknown values. The regression model takes two or more traits, the first
trait is assumed to be the dependent variable. MammalBrainBodyGt.txt is a data set of mammal
brain, body and gestation time. Run BayesTraits with the “Mammal.trees” tree file and
“MammalBrainBodyGt.txt” data. The commands below select the regression model (6) and MCMC
(2) and the last line runs the analysis.

2
6
Run
The header will contain.
Header
Output
Iteration
Current iteration of the chain
Lh
Current likelihood of the chain
Tree No
Current tree number
Alpha
Intercept
Beta Trait 2
Regression coefficient for trait 2
Beta Trait 3
Regression coefficient for trait 3
Var
Brownian motion variance
R^2
R2 - Coefficient of determination
SSE
Sum of squared error
SST
Total sum of squared
s.e. Alpha
Standard error Alpha
s.e. Beta-2
Standard error Beta-2
s.e. Beta-3
Standard error Beta-3
Testing trait significance
There are a number of ways to test if a trait is significant in the regression model, the first is
to compare marginal likelihood (MCMC) or likelihood ratios (ML) from runs with and without the
trait. The second is the ratio of the time the regression coefficient crosses the zero point, if a
regression coefficient is well supported it will not switch from positive to negative, or vice versa.
Continuous: Estimating ancestral sates and tip values
Continuous models can be used to estimate unknown values on the tree, either internal
nodes or tips. Estimating unknown values is a two-step process, first a distribution of models is
estimated from available data, secondly the models are used to estimate unknown values. The two-
step process prevents estimated data from affecting the model parameters. Estimating unknown
values can be used with model A, model B and the regression model but only using MCMC.
The “SaveModels” command is used to save models to a specified file, the “LoadModels”
command is used to load the models into BayesTraits. The same model parameters, including tree
transformations, has to be specified when creating a model file and when estimating unknown
values, only very basic error checking is implemented.
Estimating unknown values internal nodes
Start BayesTraits with the “Mammal.trees” file and “MammalBody.txt” data. Select model A
(4) and MCMC analysis (2). Save the models and run the analysis with the commands below, the
models will be saved into a file called “MamBodyModels.bin”

4
2
SaveModels MamBodyModels.bin
Run
Once the program has finished a file called “MamBodyModels.bin” will be created.
To estimate data, start BayesTraits with the same tree and data files and use the commands
below to select model A (4) and MCMC (2), the third line loads the models from
MamBodyModels.bin, the fourth and fifth lines define two tags called Tag01 and Tag02 (see Tags)
and the six and seventh lines reconstruct both tags labelling them Node-01 and Node-02 in the
output.
4
2
LoadModels MamBodyModels.bin
AddTag Tag01 Whale Hippo Llama Ruminant Pig
AddTag Tag02 Mouse Rat Hystricid Caviomorph
AddMRCA Node-01 Tag01
AddMRCA Node-02 Tag02
Run
The output header will contain
Header
Output
Iteration
Current iteration of the chain
Lh
Current likelihood of the chain
Tree No
Current tree number
Model No
Current model number from the model file
Alpha Trait 1
Phylogenetic mean
Sigma^2 1
Brownian motion variance
Est Node-02 - 1
Estimated values for Node-02 trait 1
Est Node-01 - 1
Estimated values for Node-01 trait 1
Estimating unknown values for tips
Data for taxa, as well as internal nodes can be estimated. A data file
“MammalBrainBodyNoTapir.txt” has been created with the data for tapir removed and replaced by
‘-‘. Start BayesTraits with the “Mammal.trees” tree file and “MammalBrainBodyNoTapir.txt” data
file. The commands below select the regression model (6) and MCMC analysis (2), save the models
to a file (MamRegModels.bin) and run the analysis.
6
2
SaveModels MamRegModels.bin
Run
A data file “MammalBrainBodyPredTapir.txt” which contains the tapir body size but with the
brain size set to “?”. The use of a question mark in the data is used to indicate the value should be
estimated. Run an analysis using the “Mammal.trees” tree file and
“MammalBrainBodyPredTapir.txt” data file. Select the regression model (6) and MCMC analysis (2).
Use the commands below to load the models and run the analysis
6
2
LoadModels MamRegModels.bin
Run
The output will contain a column labelled “Est Tapir – Dep”, with the predicted tapir brain
size, the predicted brain size should be roughly 2.1 ± 0.15, the actual brain size is 2.2.
Independent contrast
BayesTraits implements a range of independent contrast models (Felsenstein 1973,
Freckleton 2012), independent contrast offers a significantly faster alternative to Generalised Least
Squares (GLS) methods for large data sets. Independent contrast models can be run using MCMC or
ML. Start BayesTraits with the tree file “Mammal.trees” and data file “MammalBrainBody.txt”. The
commands below run the independent contrast model (7) using MCMC (2)
7
2
Run
The independent contrast model assumes sites are independent, e.g the covariance is set to zero.
The output will contain

Header
Output
Iteration
Current iteration of the chain
Lh
Current likelihood of the chain
Tree No
Current tree number
Alpha 1
Phylogenetic mean of the first trait
Alpha 2
Phylogenetic mean of the second trait
Sigma^2 1
Brownian motion variance for the first trait
Sigma^2 2
Brownian motion variance for the second trait
Independent contrast: Correlation
The independent contrasts correlation model allows correlations to be tested between two
traits using the independent contrast framework.
Start BayesTraits with the tree file “Mammal.trees” and data file “MammalBrainBody.txt”.
The commands below are used to select Independent Contrast: Correlation (8) and MCMC (2), the
stones command is used to estimate the marginal likelihood using 100 stones and 1000 iterations
per stone and the final command starts the analysis.
8
2
Stones 100 1000
Run
The output will contain
Header
Output
Iteration
Current iteration of the chain
Lh
Current likelihood of the chain
Tree No
Current tree number
Alpha 1
Phylogenetic mean of the first trait
Alpha 2
Phylogenetic mean of the second trait
Sigma^2 1
*Brownian motion variance for the first trait
Sigma^2 2
*Brownian motion variance for the second trait
R Trait 1 2
*The covariance between the first and second trait
*Due to the independent contrast framework the variance and covariance parameters are
set to their maximum likelihood values even when using MCMC.
The log marginal likelihood, found in the last line of MammalBrainBody.txt.Stones.txt,
should be roughly -79.3 log units.

To test a correlation between two traits re run the analysis using the “TestCorrel” (TC)
command to set the covariance to zero and calculate a Log Bayes Factor. Start BayesTraits with the
tree file “Mammal.trees” and data file “MammalBrainBody.txt”. The commands below are used to
select Independent Contrast: Correlation (8) and MCMC (2), the TestCorrel command sets the co-
variance to zero, the stones command is used to estimate the marginal likelihood using 100 stones
and 1000 iterations per stone and the final command starts the analysis.
8
2
TestCorrel
Stones 100 1000
Run
The log marginal likelihood, found in the last line of MammalBrainBody.txt.Stones.txt,
should be roughly -140.6 log units. The significance can be assessed by calculating Log Bayes Factors,
in this case the complex model assumes the correlation and the simple model sets it to zero.
Log BF = 2(log marginal likelihood complex model – log marginal likelihood simple model)
Log BF = 2(-79.3 - -140.6)
Log BF = 122.6
Independent contrast: regression
The independent contrast models supports simple and multiple regression, using ML and
MCMC. The first trait is assumed to be the dependent variable, subsequent traits are assumed to be
the independent variables. Run an analysis using the tree file “Mammal.trees” and the data file
“MammalBrainBodyGt.txt”. Select “Independent Contrast: Regression”, 9 and MCMC 2, and run the
analysis. The log file will contain
Header
Output
Iteration
Current iteration of the chain
Lh
Current likelihood of the chain
Tree No
Current tree number
Alpha
*Analytical value of Intercept
Beta 1
Regression coefficient for trait 2
Beta 2
Regression coefficient for trait 3
* Due to the independent contrast framework the maximum likelihood intercept value is
used, calculated from the regression coefficient, even when using MCMC, it is not an estimated
value.
Tree transformations, kappa, lambda, delta, OU
BayesTraits supports a number of tree transformations including, kappa (κ), lambda (λ),
delta (δ) and Ornstein Uhlenbeck (OU) for both continuous and independent contrast models. These
scaling parameters allow tests of the tempo, mode, and phylogenetic associations of trait evolution.
Kappa, lambda, and delta take the value 1.0 by default, OU takes the value 0. These default values
assume that the phylogeny and its branch lengths accurately describe a constant-variance random
walk model. However, if trait evolution has not followed the topology or the branch lengths, these
values will depart from 1.0. When they do, incorporating them into the analysis of the data (e.g.,
when estimating the correlation between two traits) significantly improves the fit of the data to the
model.
Below is a subset of the primates tree, it will be used to demonstrate tree transforms.
Kappa
The kappa parameter differentially stretches or compresses individual phylogenetic branch
lengths and can be used to test for a punctuational versus gradual mode of trait evolution. Kappa >
1.0 stretches longer branches more than shorter ones, indicating that longer branches contribute
more to trait evolution (as if the rate of evolution accelerates within a long branch). Kappa < 1.0
compresses longer branches more than shorter ones. In the extreme of Kappa = 0.0, trait evolution
is independent of the length of the branch, all branch lengths are set to 1. Kappa = 0.0 is consistent
with a punctuational mode of evolution. Below are examples of the tree transform by a) kappa = 2 b)
kappa = 0.5 and c) kappa = 0
a) Kappa = 2
b) Kappa = 0.5

c) Kappa = 0
Delta
The parameter delta scales overall path lengths in the phylogeny - the distance from the
root to the tips, as well as the shared path lengths. It can detect whether the rate of trait evolution
has accelerated or slowed over time as one moves from the root to the tips, and can find evidence
for adaptive radiations. If the estimate of Delta < 1.0, this says that shorter paths (earlier evolution in
the phylogeny) contribute disproportionately to trait evolution - this is the signature of an adaptive
radiation: rapid early evolution followed by slower rates of change among closely related species.
Delta > 1.0 indicates that longer paths contribute more to trait evolution. This is the signature of
accelerating evolution as time progresses. Seen this way, delta is a parameter that detects
differential rates of evolution over time and re-scales the phylogeny to a basis in which the rate of
evolution is constant. Below are examples of the tree transform by a) delta = 0.5 and b) delta = 2

a) Delta 0.5
b) Delta 2
Lambda
The parameter lambda reveals whether the phylogeny correctly predicts the patterns of
covariance among species on a given trait. This important parameter in effect indicates whether one
of the key assumptions underlying the use of comparative methods - that species are not
independent - is true for a given phylogeny and trait. If a trait is in fact evolving among species as if
they were independent, this parameter will take the value 0 and indicate that phylogenetic
correction can be dispensed with. A lambda value of 0 corresponds to the tree being represented as
a star or big-bang phylogeny. If traits are evolving as expected given the tree topology and the
random walk model, lambda takes the value of 1.0. Values of lambda = 1.0 are consistent with the
constant-variance model (sometimes called Brownian motion) being a correct representation of the
data. Intermediate values of lambda arise when the tree topology over-estimates the covariance
among species. Below are examples of the tree transform by a) lambda = 0.75 and b) lambda = 0
a) Lambda = 0.7
b) Lambda = 0

The value of lambda can differ for different traits on the same phylogeny. If the goal is to
estimate the correlation between two traits then lambda should be estimated while simultaneously
estimating the correlation. If, on the other hand, the goal is to characterise traits individually, a
separate lambda can be estimated for each.
Ornstein Uhlenbeck (OU)
The Ornstein Uhlenbeck (OU) transform has traditionally been associated with stabilising
selection, where the OU parameter measures the strength of a return to a theoretical optimum
(Hansen 1997) this may be due to other factors and care should be taken when using and
interpreting OU results. OU parameter values of zero correspond to the default branch lengths,
parameter values >0 are evidence of an OU process. The OU model should only be used with
ultrametric trees for independent contrast models, the correction for non-ultrametric trees (Slater
2014) is implemented for GLM. Below are examples of the tree transform by a) OU = 0.05 and b) OU
= 1, the OU process can produce trees which are very similar to delta transformed trees but the
interpretation is very different. High values of OU can also produce trees which are similar to lambda
trees but again the interpretation is very different.
a) OU = 0.05

b) OU = 1
Tree transformations table
Parameter
Action
0
<1
1
>1
lambda
Assess contribution
of phylogeny
Star phylogeny
(species
independent)
phylogenetic
history has
minimal effect
default
phylogeny
not defined
kappa
Scale branch
lengths in tree
punctuational
evolution
stasis in longer
branches
default
gradualism
longer
branches
more change
delta
Scale total path
(root to tip) in tree
not defined
temporally
early change
important
(adaptive
radiation)
default
gradualism
temporally
later change
(species
specific
adaptation)
OU
default
>0
evidence of an
OU process
Four scaling parameters and their interpretation when applied to trait evolution on a
phylogeny
Tree transformations commands
All four parameters can be estimated using ML or MCMC, the syntax for the four parameters
is the same, either the scaling parameter on its own to toggle its estimation (they are not estimated
by default) or the scaling parameter followed by a number to fix the parameter to a given value.
The first command estimates lambda, the second fixes it to 0.5
Lambda
Lambda 0.5
The first command estimates kappa, the second fixes it to 0.5
Kappa
Kappa 0.5
The first command estimates delta, the second fixes it to 0.5
Delta
Delta 0.5
The first command estimates OU, the second fixes it to 0.5
OU
OU 0.5
Model testing, Bayes Factors (MCMC) or Likelihood ratios (ML), can be used to determine if
a transform is significant or if a value of a transform is significant, e.g. is lambda significantly
different from 0.

Variable rates model
The variable rates model allows the rate of change to vary threw time and identifies areas of
the tree where the rate of evolution differs significantly, for an in-depth description see (Venditti,
Meade et al. 2011). The variable rates model can be used with MultiState, Discrete or any of the
independent contrast models. The variable rates model uses RJ MCMC to identify areas of the tree in
which the rate of evolution varies significantly. The model works with a single tree and requires
MCMC analysis. A tree “Marsupials.trees” of roughly 250 marsupials and their body sizes
“Marsupials.txt” is included. Start BayesTraits with the tree and data file, the commands below
select the independent contrast model (7) and MCMC (2), the VarRates command selects the
variable rates option, both burn-in and the number of iterations are increased as reverse jump
models can be harder to converge and have more parameters than the standard Brownian motion
models. Because variable rates models are more complex chains may fail to converge, it is important
to check convergence using multiple chains.
7
2
VarRates
Burnin 10000000
Iterations 110000000
Run
The log file will contain
Header
Output
Iteration
Current iteration of the chain
Lh
Current likelihood of the chain
Tree No
Current tree number
Alpha 1
Phylogenetic mean of the first trait
Sigma^2 1
Brownian motion variance for the first trait
No RJ Local Branch
Number of branch lengths scaled using variable rates
No RJ Local Node
Number of nodes scaled using variable rates
Two additional data files are created, “Marsupials.txt.Output.trees” contains the trees
scaled by the rate of change, areas which are stretched have an increased rate of change, areas
which are shrunken have a decreased rate. The file “Marsupials.txt.VarRates.txt” contains a detailed
description of the changes, the format of the file is, line 1, number of taxa, followed by a unique
taxa ID and taxa name. The second part is the number of internal nodes, followed by a list of internal
nodes consisting of a unique node ID, branch length (-1 for root), number of taxa which define the
node and the list of taxa ID. The third section details the results of the chain, the columns are
Header
Output
It
Iteration of the chain
Lh
Likelihood of the chain
Lh + Prior
Likelihood + prior
No Pram
Number of rate changes on the tree
Alpha
Estimated phylogenetic mean
Sigma^2
Brownian motion
Alpha Scale
Scale of the prior (unchanging, for diagnostics only)
For each change of rate of the tree (No Param) there is
Header
Output
Node ID
The node ID the change is on
Scale
The value of the scale parameter
Crate It
The iteration the change was created on
Scaler type
The type of the scaler, this can be node,
branch, kappa, lambda or delta
Bayes Factors calculated from marginal likelihoods can be used to test if there is rate
variability by comparing a model with and without variable rates.
A web site that processes the output from a variable rates model is available at
http://www.evolution.reading.ac.uk/VarRatesWebPP/
it can be used to determine how often each node is scaled and by how much.
Geo (Geographical) model
Geographical coordinates (longitude and latitude) cannot be modelled using Brownian
motion due to the spherical nature of the earth. The geo model maps longitude and latitude onto a 3
dimensional Cartesian coordinates system which can be modelled using Brownian motion. The
model requires two sites, longitude and latitude, it can only be used with a single tree, as all
ancestral sates must be simultaneously estimated, the geo model can only be used with MCMC, no
ML option is available.
A phylogenetic tree of 85 Northeast Bantu languages and their geographical coordinates
(longitude and latitude) can be found in NortheastBantu.trees and NortheastBantu.txt. Run
BayesTraits with the tree file and data, the commands below select the Geo model, number 13,
MCMC will automatically be selected, start the model using the run command. The log file will
contain standard MCMC information, iteration, Lh ect and Alpha which will be fixed to two and scale
parameter of roughly 71. The scale parameter is the variance of the normal distribution, with a mean
of 0, that expected changes are drawn from.
13
Run
The Geo model records the estimated ancestral states in the “.AnsStates.txt” file. The file
assigns a node number to each internal node defined by a list of taxa.
For example
Node-00012 G35_Luguru G36_Kami
Defines Node 12 by two taxa G35_Luguru, G36_Kami.
The second part of the file contains the iteration number, likelihood, parameters and
estimated longitude and latitude for each internal node.
Samples of trait data
Traditionally comparative methods assume trait values are known without error but traits
often show within species variation. BayesTraits allows samples of data to be used, results are
integrated over the sample. There are two types of samples, linked where multiple traits with in a
taxa are taken from the same source, for example if you had brain and body measurements from a
sample and could identify what brains measurements came from what bodies, unlinked data is
where the traits for a taxa are not from the same source.
Samples of data are placed in a text file, the format of the data file is, taxa name, followed
by Linked or UnLinked, then the sample of data separated by comers (,) without spaces or tabs.
Spaces are used if there is more than one trait. The example below defines a sample of data for two
taxa, Taxa1 and Taxa2, the data set has two traits. Taxa1 is linked and the data will be paired, both
the first and second trait must have the same number of data points, and are sampled together.
Taxa2 is unlinked, a sample is not available for the first trait a single value is used, four values are
available for the second trait.
Taxa1 Linked 5.5,6.4,4.9 8.3,9.2,7.7
Taxa2 Unlinked 8 5.5,9.1,6.6,8.3
An example of sample data can be seen in MammalBrainBodySampleData.txt, it has samples
of data for Hippo and Whale, Hippo has 100 linked samples of brain and body size, Whale has 30
unlinked samples of brain and 10 samples of body size. Start BayesTraits with the supplied tree and
data file Mammal.trees and MammalBrainBody.txt, and use the following commands.
7
2
DistData MammalBrainBodySampleData.txt
Run
7 selects Independent Contrast and 2 for MCMC, the command “DistData
MammalBrainBodySampleData.txt” is used to read in the sample of data, start the chain with “run”.
The output will have four new columns, Hippo-1 and Hippo-2 that draws from the sample for the
first and second site for Hippo and the same for Whale.
Local transformations, kappa, lambda, delta, OU, nodes and branches
Commonly transformations are applied to the whole tree, assuming a single evolutionary
process, but as trees become large this assumption may not hold. BayesTraits allows local
transforms to be applied to nodes within trees as well as to the root. Local transforms include,
kappa, lambda, delta, OU (see Tree transformations, kappa, lambda, delta, OU), as well as node and
branch scalars. A node transform scales all branches in a node by a value, a branch transform scales
a single branch. A significant scalar, on a branch suggests a change in the trait value which is
inherited by all members of the clade, for example there is a significant branch scaler on the nodes
containing the bats within the mammal tree, as there was a decrease in body size along that branch.
A significant scalar, greater than one, on a node suggests an increase in trait variance for that group.
While the OU model can be fitted to nodes of the tree the implementation does not include the
correction for non-ultrametric trees (Slater 2014).
The syntax to create a local transform (shortcut LT) is
LocalTransform Name TagList Type Value
Where “Name” is an identifier associated with the transform, allowing it to be identified in
the output. Tag List is a list of one or more tags to apply the transform to, BayesTraits can apply the
same transform to one or more nodes on the tree, for example if an ecological factor is associated
with a change in the rate of a traits evolution. “Type” is the transform to apply, this can be Node,
Branch, Kappa, Delta, Lambda or OU. “Value” is optional if you what to set the transform parameter
to a fixed value, if none is supplied the value is estimated using either ML or MCMC.
An example of using local transforms applied to the Marsupials tree and body size
(Marsupials.trees and Marsupials.txt) is given below, this is for illustrative purposes and should not
be viewed as the correct model. First run a model assuming a homogenous evolutionary process
using the commands below, selecting Independent Contrasts and MCMC, the stones command is
used to estimate the marginal likelihood (see Model Testing: Likelihood ratios and Bayes Factors)
using 100 stones and 1000 iterations per stone, the final command starts the analysis.
7
2
Stones 100 1000
Run
The estimated marginal log likelihood, found in the last line of the Marsupials.txt.Stones.txt
file, should be roughly -155.5.
Secondly run a model where a local node transform is applied to the clade that includes
Petrogale_brachyotis Petrogale_burbidgei Petrogale_concinna, using the commands below.
7
2
AddTag Tag01 Petrogale_brachyotis Petrogale_burbidgei Petrogale_concinna
LocalTransform TransNode Tag01 Node
Stones 100 10000
Run
The first two command select Independent Contrasts and MCMC, the third command
creates a tag called Tag01 that is defined by three taxa, Petrogale_brachyotis Petrogale_burbidgei
Petrogale_concinna. The fourth command applies a local node transform to the defined tag, the
stones command is used to estimate the marginal likelihood. The default prior on the node scalar
called VRNode is a scaled gamma (see (Venditti, Meade et al. 2011)) with an alpha of 1.1 and a beta
of 1, this can be changed (see Priors). Once the chain has run the Marsupials.txt.Stones.txt file
should contain an estimate of the marginal log likelihood, roughly -148
Log BF = 2(log marginal likelihood complex model - log marginal likelihood simple model)
Log BF = 2(-148 - -155.5)
Log BF = 15
The Log Bayes factor suggests “very strong evidence” for a rate shift on node.
The estimated rate scaler is recorded in the output as “TransNode – Node” column of the log
file, below is a plot of the frequency histogram. This shows that the scale is different from 1, the
default, this is borne out by the significant Bayes factor.
Any number of local transforms can be simultaneously applied to a tree, the transforms can
be mixed, allowing different parts of the tree to be modelled by different processes. Transforms can
be nested within each other, when transforms are nested the nodes deeper in the tree (the nodes
defined by the most taxa) are applied first. This gives the flexibility to create complex models but
they may be hard to interpret or not have a biological foundation. The resulting scaled trees are
logged in an output file ending “.Output.trees” this can be used to visualise the transforms and can
be used for further analysis.
Reverse Jump local transformations, kappa, lambda, delta, and OU
The method for fitting local transformations, described above, requires the number and
location of the transform to be known in advance. This is not often the case, the reverse jump local
transform method can be used to estimate the number and location of transforms with in a tree.
The method is similar to the Variable rates model (see Variable rates model), the variable rates
method estimates the number and location of branch and node scalars, reverse jump local
transform allow the number and location of other evolutionary processes to be modelled, such as
kappa, lambda, and delta. The RJLocalTransform command turns on the use of reverse jump local
transforms, it takes a transform type, kappa, lambda, delta or OU.
For example
RJLocalTransform Delta
Sets reverse jump local transform using delta, more than one reverse jump transform can be
used at the same time but the interpretation can become complex. By default a node must comprise
of at least 10 taxa to be transformed, A minimum number of taxa are used to prevent small nodes
from being transformed, as the transform may fit the data better but may not be interpretable as
the evolutionary process. This limit can be changed with the SetMinTransTaxaNo command.
50
100
150
200
250
0510 15 20 25 30 35 40 45 50 55 60 65
An example of using reverse jump local transform is given below, using the Marsupials tree
and data set (Marsupials.trees and Marsupials.txt). First run an analysis to establish the marginal
likelihood for the Brownian motion model, using the commands below.
7
2
Burnin 1000000
Iterations 10000000
Stones 250 10000
Run
This should produce a marginal likelihood of roughly -155.5. The number of stones, burn-in
and iterations have been increased as reverse jump models can be harder to fit. Run a second
analysis using reverse jump local transform delta, using the commands below.
7
2
Burnin 1000000
Iterations 10000000
RJLocalTransform Delta
Stones 250 10000
Run
This should produce a marginal likelihood of roughly -119, a variable rates log file
“Marsupials.txt.VarRates.txt” will be created identifying each node and recording which nodes have
had delta transforms applied and their value, the number of deltas will also be recorded in the log
file. The transformed tree is stored in the nexus tree file “Marsupials.txt.Output.trees”. While the RJ
delta marginal likelihood is significantly higher than the Brownian motion, this may be due to RJ
delta acting as a proxy for variable rates, to test if RJ delta is significant on top of variable rates run
an analysis with both variable rates and RJ delta using the commands below.
7
2
VarRates
Burnin 1000000
Iterations 10000000
RJLocalTransform Delta

Stones 250 10000
Run
This should produce a marginal likelihood of roughly -110, suggesting the RJ delta is
significant on top of the variable rates model.
Model
Approximate marginal likelihood
Brownian motion
-155.5
Reverse jump Delta
-119.8
Variable rates
-119.3
Reverse jump Delta + Variable rates
-110.4
Maximum likelihood search options
Under maximum likelihood, estimated parameters which don’t have an analytical solution
have to be searched for, this can be computationally hard if there are a large number of parameters,
they trade off with each other or if the search space is complex to traverse. BayesTraits uses the
NLopt library (Johnson) (http://ab-initio.mit.edu/nlopt). There are a number of options which can be
set to help parameter optimisation. It is important to run the analysis multiple times to ensure that
the results are stable, if each run produces different results the options below can be used to modify
the search algorithm. Limiting the search space using Set Minimum Maximum Rate (SetMinMaxRate)
and increasing the number of maximum likelihood tries (MLTries) can be effective.
MLTries (Maximum Likelihood Tries)
The MLTries command sets the number of times the maximum likelihood algorithm is called,
10 is the default, only the model with the best likelihood is retained. Increasing this number of tries
will produce more stable results at the cost of computational time.
MLAlg (Maximum Likelihood Algorithm)
The MLAlg command is used to set the optimisation algorithm. It takes the name of the
algorithm to use, the default is BOBYQA. The different algorithms have different run times and
behaviours which can vary with data sets and models. For details of the algorithms and references
see (http://ab-initio.mit.edu/wiki/index.php/NLopt_Algorithms)

Name
Reference
BOBYQA
M. J. D. Powell, "The BOBYQA algorithm for bound constrained optimization
without derivatives," Department of Applied Mathematics and Theoretical
Physics, Cambridge England, technical report NA2009/06 (2009).
NEWUOA
M. J. D. Powell, "The NEWUOA software for unconstrained optimization without
derivatives," Proc. 40th Workshop on Large Scale Nonlinear Optimization (Erice,
Italy, 2004).
NELDERMEAD
J. A. Nelder and R. Mead, "A simplex method for function minimization," The
Computer Journal 7, p. 308-313 (1965).
PRAXIS
Richard Brent, Algorithms for Minimization without Derivatives (Prentice-Hall,
1972). (Reprinted by Dover, 2002.)
COBYLA
M. J. D. Powell, "A direct search optimization method that models the objective
and constraint functions by linear interpolation," in Advances in Optimization and
Numerical Analysis, eds. S. Gomez and J.-P. Hennart (Kluwer Academic:
Dordrecht, 1994), p. 51-67.
SBPLX
T. Rowan, "Functional Stability Analysis of Numerical Algorithms", Ph.D. thesis,
Department of Computer Sciences, University of Texas at Austin, 1990.
AUGLAG
Andrew R. Conn, Nicholas I. M. Gould, and Philippe L. Toint, "A globally
convergent augmented Lagrangian algorithm for optimization with general
constraints and simple bounds," SIAM J. Numer. Anal. vol. 28, no. 2, p. 545-572
(1991).
MLTol (Maximum Likelihood Tolerance)
The MLTol command sets the algorithm stopping tolerance, how the stopping tolerance is
used varies between algorithms but it’s commonly used to stop the search. If the likelihood does not
improve by more than a specified tolerance for a given number of tries the algorithm will terminate.
Setting the tolerance lower increases accuracy at the expense of run time, the default is 0.000001
MLMaxEval (Maximum Likelihood Maximum evaluations)
The MLMaxEval command set the maximum number of times the likelihood function is
evaluated, if the number of evaluation exceeds this limit the algorithm terminates, this can stop the
algorithm getting stuck. The default is 20000, using -1 removes any limit.
SetMinMaxRate (Set Minimum Maximum Rate)
The SetMinMaxRate command sets the minimum and maximum boundaries for the
transition rates used by the Multistate and Discrete models. The transition rates are dependent on
the supplied branch lengths (see Branch lengths), if the branch lengths are in millions of years the
transition rates will be very small compared to branch lengths that are in expected number of
substitutions. The default is 1.0e-32 to 100, designed for branch lengths in expected number of
substitutions. If the run to run results are highly variable reducing the search space range can
increase accuracy, if the parameters are bumping up against the range increasing the limit may find
a solution with a better likelihood.
Model options table
1 local transform does not have the same interpretation for MultiState / Discrete data
2 OU transforms assumes the tree is ultrametric (Slater 2014)
MultiState
Discrete: Independent
Discrete: Dependant
Continuous: Random Walk
Continuous: Directional
Continuous: Regression
Independent Contrast
Independent Contrast: Correlation
Independent Contrast: Regression
Discrete: Covarion
Discrete: Heterogeneous
Geo
Fix (Fossilise) internal nodes
✔
✔
✔
✔
✔
✔
-
-
-
✔
✔
-
Reconstruct Ancestral states
✔
✔
✔
✔
✔
✔
-
-
-
✔
✔
✔
Multiple Patters
✔
✔
✔
-
-
-
-
-
-
-
-
-
Covarion
✔
✔
✔
-
-
-
-
-
-
-
-
-
Variable Rates
✔
✔
✔
-
-
-
✔
✔
✔
✔
✔
-
Reverse jump model reduction
✔
✔
✔
-
-
-
-
-
-
✔
✔
-
Gamma Rate Heterogeneity
✔
-
-
-
-
-
-
-
-
-
-
-
Kappa
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
✔
-
Lambda
-
-
-
✔
✔
✔
✔
✔
✔
-
-
-
Delta
-
-
-
✔
✔
✔
✔
✔
✔
-
-
-
OU
-
-
-
✔
✔
✔
✔2
✔2
✔2
-
-
-
Local Kappa
✔
✔
✔
-
-
-
✔
✔
✔
✔
✔
-
Local Lambda
✔1
✔1
✔1
-
-
-
✔
✔
✔
✔1
✔1
-
Local Delta
✔1
✔1
✔1
-
-
-
✔
✔
✔
✔1
✔1
-
Local OU
✔1
✔1
✔1
-
-
-
✔2
✔2
✔2
✔1
✔1
-
Local Node
✔
✔
✔
-
-
-
✔
✔
✔
✔
✔
-
Local Branch
✔
✔
✔
-
-
-
✔
✔
✔
✔
✔
-
Distribution of tip data
-
-
-
✔
✔
✔
✔
✔
✔
✔
✔
✔

Output files
BayesTraits produces several output files, there purpose, format and file extensions are
listed below.
Extension
File
.Log.txt
Log file contains the model options and output
.Schedule.txt
Schedule from the MCMC chain containing information about mixing
.AncStates.txt
Estimated ancestral sates from the Geographical model
.VarRates.txt
Transforms applied to the tree from the variable rates and local transform
models
.Stones.txt
Stepping stone sampler file, the last line will contain the estimated log
marginal likelihood, additional information about the stones is recorded.
.Output.trees
The output trees scaled by variables rates or other transforms, saved in nexus
format, can be turned on using the SaveTrees command

62
BayesTraits versions
Quad Precision
Quad precision is no longer required as the likelihood is scaled internally to prevent
underflows, this is significantly faster.
Threaded
The threaded version of BayesTraits allows the likelihood calculation, the computationally
intensive part of the program, to be executed over multiple cores, this can speed up the runtime of
the program. The speed increase is data set and model dependent, and for small trees or simple
models the parallel version can run slower than the serial version if the overhead associated with the
threads outstrips the calculation gain. The command “cores” can be used to set how many cores the
program uses.
Note: The threaded version of BayesTraits requires additional library’s to be installed, for
windows the dll file “libiomp5md.dll” must be installed or be in the same directory as the
executable, it is supplied with the program. For OS X “libgomp” and “libgcc_s” must be installed they
are available as part of the homebrew gcc install (https://brew.sh)
OpenCL
OpenCL allows computationally intensive operations to be performed on specialised
hardware, such as graphics cards, which can give large speed increases, especially for big trees,
typically thousands of taxa. To use OpenCL you need three things, graphics hardware that supports
OpenCL, an OpenCL driver installed and the OpenCL version of BayesTraits.
OpenCL graphics hardware
The Khronos group, who oversee the standard, keep a list of OpenCL compatible hardware
(http://www.khronos.org/conformance/adopters/conformant-products/). Typically, any AMD and
NVIDA graphics cards purchased in the last couple of year should support OpenCL but please check.
Each month hundreds of graphics cards are released with different processors, memory and
interfaces. The web site CompuBench (www.compubench.com) provides a comprehensive
comparison of OpenCL performance for different graphics cards.
It is possible to get over a 40 fold speed increase compared to Version 1, the performance
increase is heavily dependent on a number of factors, including tree and data size, model type and
complexity, the underlying hardware both graphics cards and system, and the drivers and operating
system used. As with the OpenMP version of BayesTraits it is possible for the OpenCL version to run
slower.
OpenCL driver
An OpenCL driver is software which provides a level of abstraction between the hardware
and software, allowing a program written using OpenCL to run on a large number of different
hardware platforms. The OpenCL driver is supplied by the hardware manufacture, normally AMD or
NVIDIA, on some systems it comes with the graphics driver and does not need to be installed
separately. If the OpenCL version of BayesTraits fails to start or reports and error before the run
starts, suitable graphics hardware or OpenCL driver may be missing.

63
Building BayesTraits from source
The source code for BayesTraits is available on the web site and is released under GNU
General Public License V3. The basic build of BayesTraits requires three libraries, NLOpt a nonlinear
optimiser, the GNU Scientific Library (GSL) and Linear Algebra Package (LAPACK) or equivalent (MKL,
ScaLAPACK). The program supports OpenMP and OpenCL if available, the code has been built using
Visual studios (windows), gcc (Linux) and clang (OSX).
The command below will build a basic Linux version,
gcc *.c -O3 -lm -lgsl -lgslcblas -lnlopt -llapack
the command will vary depending on the name of the libraries installed
to build the threaded version,
gcc *.c -O3 -lm -lgsl -lgslcblas -lnlopt -llapack -DOPENMP_THR -fopenmp
a threaded version of lapack library (such as MKL) should be used.
The command below will build a basic OS X version,
clang *.c -O3 -lm -lgsl -lgslcblas -lnlopt -framework Accelerate
clang does not support OpenMP and cannot build the threaded version, the homebrew
(http://brew.sh/) package manager can be used to install gcc.
64
Common problem / Frequently Asked Questions
1) Problems starting the program
Q) Double clicking on the program does not work.
A) BayesTraits is run from the command line and not by double clicking on it. See
Running BayesTraits section.
2) Common tree and data errors
A) Tree must be in nexus format with a valid translate block. Use the example files as a
template.
B) Tree descriptions must have numbers and not taxa name in them.
C) Trees must be rooted.
D) Trees and data must be encoded using ASCII format not Unicode
E) The error “Could not load data for taxa X”, this error is caused by a taxa being specified
in the tree file but not in the data file. Check spelling and taxa numbers
F) The error “Tree file does not have a valid nexus tag.” Is because a nexus tag is not found
in the tree file. Possible causes are specifying the data file before the tree file.
3) “Memory allocation error in file …”
Error message “Memory allocation error in file …”, is a catch all memory allocation error, the
two main causes of memory allocation errors are running out of memory, this can be due to
too many trees in the tree file or a complex memory intensive model. Try running the
program with a smaller number of trees and simpler models. The second cause of the crash
is due to programming errors, if you believe this is the case, please send along the tree file,
data file and commands used.
4) Chain is not mixing between trees.
This problem can be caused when one tree’s likelihood is significantly better than other
trees in the sample. Trees are sampled in proportion to their likelihood, if one is much better
than the rest it will be sample much often. This can be a particular problem with a large
number of trees when the topology is poorly supported, a chance combination creates a
much better likelihood preventing the chain from mixing. To test if this is the problem run
the sample using ML, this will determine if the tree which the chain gets stuck on has the
best likelihood. Two options are available, the first is to remove the tree from the sample if
you believe it is anomalous for some reason. The second is to use the Equal Tree command
to force the chain to spend an equal amount of time on each tree. The equal tree command
will produce a separate posterior sample of rates, parameters and ancestral states per tree,
instead of a single set integrated over the sample of trees.
5) Cannot find a valid set of starting parameters.
A valid set of parameters to start the chain cannot be found. The model may be invalid, the
combination of restrictions, priors or data may produce an invalid model, for example
65
setting all the transition rates to zero. Try using simpler models, restricting the number of
model to a single transition rate and slowly building to more complex models.
6) Too many free parameters to estimate
The number of free parameters is limited to 25 or fewer for maximum likelihood. Likelihood
methods allow parameters to be estimated from data, comparative methods data can be
limited, typically consisting of one or more sites for a number of taxa. This limits the number
of parameters which can be accurately estimated from the data. Accurately estimating 25 or
more parameters would require a vast amount of data. So the number of free parameters
under ML is limited, if you believe your data can support more parameters please contact
the authors for this limitation to be removed.
7) Run to run variation
It is important to run the same analysis a number of times, to check for convergence when
using MCMC and to ensure that optimal parameter values have been found when using ML.
Large differences between runs is often a warning sign, commonly this is because of to many
parameters for the data to support or the parameters create a deceptive likelihood surface,
for MCMC this can prevent the chain from converging and / or mixing, for ML different runs
may produce different results. Multiple runs can help identify if this is a problem, for ML
analysis increasing the number of maximum likelihood tries (MLT) per tree can help, the
default of 10 gives a balance between speed and accuracy, increasing this number can help if
there is large run to run variation but also increases the run time. See Maximum likelihood
search options for more information and options about searching.
66
Command table
The table below lists the commands and gives an overview of them, detailed information is
available in the Command List section below.
Command
Function
#
A comment, used for annotating input files
AddErr
Adds values to terminal branch lengths to correct for sampling errors
AddMRCA
Reconstruct a node using the most recent common ancestor method
AddNode
Reconstruct a specific node on a tree if present.
AddPattern
Apply a different pattern of evolution to a subset of the tree
AddTag
Create a tag defining a node
AlphaZero
Set the alpha parameter to zero for continuous models
BurnIn
Set the number of iterations to burn-in the MCMC chain
CapRJRates
Cap the maximum number of estimated rates in an RJ MCMC analysis
Cores
Set the number of cores to use in the threaded version of the program
CoVarion
Use a covarion model for MultiState and discrete
CustomSchedule
Sets the MCMC schedule, selecting the frequency of each operator
Delta
Estimate or fixes delta to a specific value
DistData
Use a sample of data instead of single values
EqualTrees
Run the chain on each tree an equal number of iterations
EvenRoot
Split the branch length equally between the in group and outgroup
Exit
Quit the program
Fossil
Fix an internal node at a specific value
Gamma
Use gamma rate heterogeneity for MultiState models with multiple sites
Help
Print a list of options
HyperPrior
Set a hyper prior on a parameter
HyperPriorAll
Set the same hyper prior on all rate parameters
Info
Print the current options
Iterations
Set the number of iterations the chain will run for
Kappa
Estimate or fix kappa to a specific value
Lambda
Estimate or fix lambda to a specific value
LoadModels
Load models from a file
LocalTransform
Transform a node on the tree by scaling a node/branch or using kappa,
lambda, delta or OU
LogFile
Set the name of the log file
MakeUM
Convert the tree to ultrametric using a simple transform
MLAlg
Specify the maximum likelihood algorithm
MLMaxEval
Specify the number of times the maximum likelihood algorithm can
evaluate the likelihood
MLTol
Set the maximum likelihood tolerance
MLTries
Set the number of times to call the maximum likelihood optimiser
NoLh
Turn off the likelihood calculation for MCMC, for testing only
NormaliseQMatrix
Normalise the Q matrix, allowing rates to be compared between data sets
OU
Estimate or fix Ornstein–Uhlenbeck to a specific value
Pis
Set the type of base frequencies to use, none, uniform or empirical
Prior
Set the prior on an estimated parameter
PriorAll
Set all the priors on available parameters
PriorCats
Set the number of categories to discretise the prior distribution into, for
MultiState and discrete rate parameters, default 100

67
Restrict
Restrict rate parameters, set them equal to each other or a constant
RestrictAll
Restrict all rate parameters to a given parameter or constant
RevJump
Use reverse jump to integrate results over possible models
RevJumpHP
Use reverse jump, with a hyper prior, to integrate results over possible
models
RJLocalTransform
Use reverse jump to apply multiple local transforms (Kappa, Lambda, Delta)
Run
Start the analysis
Sample
Set the sample frequency of the MCMC chain, default 1000
SaveInitialTrees
Save the initial sample of trees
SaveModels
Save the model parameters to a file
SaveTrees
Save the posterior sample of trees, with transforms ect applied
ScaleTrees
Scale the tree by a given value or set the sample to have a mean branch
length of 0.1
Schedule
Toggle the recording of the schedule in an MCMC chain, default is on
Seed
Set the random seed
SetMinMaxRate
Set the minimum and maximum transition rates, ML only
SetMinTransTaxaNo
Set the minimum size node to apply RJ local transform to, default 10
Stones
Use a stepping stone sampler to estimate the marginal likelihood
Symmetrical
Make the transition matrix symmetrical for MultiState models
TestCorrel
Set the correlation between continuous traits to zero
Unrestrict
Remove a restriction
VarRates
Use the variable rates model
68
Command List
Command: #
Purpose: Add a comment.
Shortcut: //
Parameters: None
Example: # This is a comment.
Command: AddErr
Purpose: Add a specified amount of branch length to terminal nodes to account for
measurement / sampling error
Shortcut: ER
Parameters: A text file containing a list of taxa names and amount of branch length to add for
each taxa
Example: AddErr TaxaErrorFile.txt
Command: AddMRCA
Purpose: To reconstruct an internal node using the most recent common ancestor approach
Shortcut: MRCA
Parameters: A node name and the name of a tag that defines the node.
Example: AddMRCA Node1 Tag1
MRCA Node1 Tag1
Command: AddNode
Purpose: To reconstruct an internal node
Shortcut: AddN
Parameters: A node name and the name of a tag that defines the node.
Example: AddNode Node1 Tag1
AddN Node1 Tag1
Command: AddPattern
Purpose: Estimate a different model of evolution on a subset of the tree
Shortcut: AP
Parameters: A name to identify the pattern and a tag that defines the MRCA
Example: AddPattern Pattern1 Tag1
AP Pattern1 Tag1
Command: AddTag
Purpose: Create a tag that defines a most recent common ancestor across a sample of trees
Shortcut: AT
Parameters: A name to identify the tag and a list of taxa names that define it
Example: AddTag Tag1 Taxa1 Taxa2 Taxa3
AP Primates Macaca Gorilla Pan Hylobates
69
Command: AlphaZero
Purpose: Sets the intercept (alpha) to zero in a regression model
Shortcut: AZ
Parameters: None
Example: AlphaZero
Command: BurnIn
Purpose: To set the number of iterations to burn the MCMC chain in for, use -1 for an infinite
chain.
Shortcut: BI
Parameters: An integer
Example: BurnIn 50000
BI -1
Command: CapRJRates
Purpose: Cap the maximum number of reverse jump rates to use
Shortcut: Cap
Parameters: An integer, >0
Example: CapRJRates 2
Cap 1
Command: Cores
Purpose: Set the number of cores to use, results will be model / data set dependent, requires
a threaded version of the program.
Shortcut: cor
Parameters: An integer, >1
Example: cores 2
cor 4
Command: CoVarion
Purpose: Turn on/off the convarion model
Shortcut: CV
Parameters: None
Example: CoVarion
CV
Command: CustomSchedule
Purpose: Set a custom schedule for the MCMC chain, the schedule determines how often
each component of the model is perturbed, for example how often delta is changed
relative to alpha. It can be used to turn off changes to a parameter. Setting the
schedule does not have any error checking and should be used with care, for
example scheduling a variable rates operator when the variable rates option has not
been set will cause the program to crash.

70
Shortcut: CSched
Parameters: An iteration number to start the custom schedule on followed by 26 frequencies
corresponding to the following operators
Position
Operator
1
Rates
2
Covarion
3
Kappa
4
Delta
5
Lambda
6
RJ split / merge
7
Hyper prior
8
Estimated data
9
Solo tree move
10
Local transform Add / Remove scalar
11
Local Transform Move
12
Local Transform Change Scale
13
Local Transform Hyper Prior
14
Change Hetero
15
Tree Move
16
OU
17
Gamma
18
RJ Dummy Code Add / Remove
19
RJ Dummy Move Node
20
RJ Dummy Change Beta
21
Estimate ancestral sates
22
Change Local Rates
23
Data distribution
24
Time Slice - Time
25
Time Slice - Scale
26
Global Rate
Example: CustomSchedule 1000000 0 0 0 1 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
CustomSchedule 500000 0.5 0 0 0.1 0 0 0 0 0.4 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
Command: Delta
Purpose: Estimate delta or set it to a fixed value
Shortcut: DL
Parameters: None to estimate delta, or a number to fix it to a value
Example: Delta
Delta 0.5
71
Command: DistData
Purpose: Use a sample of data for taxa instead of a single point.
Shortcut: DD
Parameters: A file containing the sample of data for each taxa, see Samples of trait data for file
format.
Example: DistData SampleDataFile.txt
Command: EqualTrees
Purpose: Force the chain to spend an equal amount of time on each tree in the sample. This
results in a separate posterior distribution per tree.
Shortcut: EQT
Parameters: Number of iterations to burn each tree in for.
Example: EqualTrees 20000
Command: EvenRoot
Purpose: Set midpoint rooting for the sample.
Shortcut: ER
Parameters: None
Example: EvenRoot
Command: Exit
Purpose: Exit BayesTraits without running the analysis
Shortcut: Quit
Parameters: None
Example: Exit
Command: Fossil
Purpose: Fix an internal node to a specific value
Shortcut: FO
Parameters: A name, the name of the tag the defines the node and value to fix the node to, see
Fixing node values / fossilising section above for more information.
Example: Fossil AnsNode Tag1 AB
Fossil AnsNode Tag1 1
Fossil Base Tag1 0.3234
Command: Gamma
Purpose: Estimate or fix gamma rate heterogeneity
Shortcut: GA
Parameters: The number of gamma categories and an optional value to fix the parameter
Example: Gamma 4
Gamma 4 0.5
Command: Help
Purpose: Print a list of commands, not all are valid / working
Shortcut: he
72
Parameters: none
Example: Help
Command: HyperPrior
Purpose: Set a hyper prior on a parameter
Shortcut: HP
Parameters: A parameter name, a distribution name, and a range to draw each parameter from
Example: HyperPrior q01 exp 0 100
HP q10 gamma 0 100 0 100
Command: HyperPriorAll
Purpose: Set all priors on rate parameters, to a common hyper prior
Shortcut: HPAll
Parameters: A distribution name and range to draw each parameter from
Example: HyperPriorAll Beta 0 100 0 50
HPAll Exp 0 200
Command: Info
Purpose: Print current options
Shortcut: In
Parameters: None
Example: Info
Command: Iterations
Purpose: Set the number of iterations to run the chain for
Shortcut: IT
Parameters: The number of iterations to run the chain for, or -1 for an infinite chain, use Ctrl+C to
terminate the run.
Example: Iterations 1000000
It -1
Command: Kappa
Purpose: Set the kappa scaling parameter
Shortcut: KA
Parameters: None to estimate kappa, or a number to fix it to a specific value
Example: Kappa
KA 0.1
Command: Lambda
Purpose: Set the lambda scaling parameter
Shortcut: LA
Parameters: None to estimate lambda, or a number to fix it to a specific value
Example: Lambda
LA 0.8
73
Command: LoadModels
Purpose: To load models from a model file, see SaveModels
Shortcut: LM
Parameters: A model file name
Example: LoadModels ModelFile.bin
Command: LocalTransform
Purpose: Apply a local transform to an internal node, this could be a node, branch scalar,
kappa, lambda, delta or OU, See Local transformations, kappa, lambda, delta, OU,
nodes and branches.
Shortcut: LT
Parameters: The name of the transform, one or more tags to apply the transform to, the type of
transform and an optional value to fix the transform to, if emitted the transform
value is estimated.
Example: LocalTransform Trans1 Tag1 Node
LocalTransform Trans1 Tag1 Tag2 Tag3 Branch
LocalTransform Trans1 Tag1 Delta
LocalTransform Trans1 Tag1 Kappa 3.2
Command: LogFile
Purpose: Set the name of the log file and other output files, the default is the name of the
data file.
Shortcut: LF
Parameters: The name of the log file
Example: LogFile Run01
LF ModelALambda
Command: MakeUM
Purpose: Set the branch length to ultrametric using a simple transform
Shortcut: MUM
Parameters: None
Example: MakeUM
MUM
74
Command: MLAlg
Purpose: Specify the maximum likelihood algorithm to use, see MLAlg (Maximum Likelihood
Algorithm) for more information.
Shortcut: MLA
Parameters: Name of the algorithm, valid options are BOBYQA, NEWUOA, NELDERMEAD, MLAlg,
COBYLA, SBPLX and AUGLAG
Example: MLAlg MLAlg
MLA SBPLX
Command: MLMaxEval
Purpose: Set the number of times the maximum likelihood algorithm can evaluate the
likelihood, this can be used to prevent the optimiser getting stuck on local peaks.
The default is 20000, use -1 to remove this termination criteria. See MLMaxEval
(Maximum Likelihood Maximum evaluations) for more information
Shortcut: MLME
Parameters: Number of times to evaluate the likelihood.
Example: MLMaxEval 100000
MLME -1
Command: MLTol
Purpose: Set the maximum likelihood tolerance, the criteria to stop the optimiser, the default
is 0.000001
Shortcut: MLTO
Parameters: The maximum likelihood tolerance.
Example: MLTol 0.001
MLTO 0.1
Command: MLTries
Purpose: Set the number of times to find the maximum likelihood values, higher values are
more consistent but take longer to run
Shortcut: MLT
Parameters: Number of maximum likelihood tries, default 10. See MLTries (Maximum Likelihood
Tries) for more information.
Example: MLTries 35
MLT 100
Command: NormaliseQMatrix
Purpose: Normalise the Q matrix, allowing rates to be compared between data sets. A global
rate is estimated, the rate parameters become relative to each other instead of
absolute values.
Shortcut: NQM
Parameters: None
Example: NormaliseQMatrix
75
NQM
Command: OU
Purpose: Set the Ornstein–Uhlenbeck scaling parameter
Shortcut: OU
Parameters: None to estimate OU, or a number to fix it to a specific value
Example: OU
OU 3.5
Command: Pis
Purpose: Set the state frequencies
Shortcut: Pi
Parameters: Set the state frequencies to empirical values (emp), uniform (uni) or not used
(none). Only valid for MultiState models
Example: Pis emp
Pis uni
Pis none
Command: Prior
Purpose: Set the prior for a parameter
Shortcut: pr
Parameters: A parameter, a distribution type and parameters, distributions include, gamma,
uniform, chi, exp, sgamma, lognormal, normal
Example: Prior alpha1 exp 10
Prior q01 gamma 10 5
Prior q34 Uniform 0 1
Prior OU exp 1
Command: PriorAll
Purpose: Set the prior for all rate parameters
Shortcut: PA
Parameters: A distribution type and parameters, see prior command
Example: PriorAll Exp 10
PriorAll Beta 1 7
Command: PriorCats
Purpose: Specify the number of categories to divide the prior into, default 100, for discrete /
MultiState models.
Shortcut: PCat
Parameters: An integer > 1,
Example: PriorCats 200
PCat 50
76
Command: Restrict
Purpose: Restrict a rate parameter or parameters to another parameter or a fixed value.
Shortcut: Res
Parameters: A list of parameters to restrict, a parameter or fixed value to restrict to.
Example: Restrict alpha1 beta1
Restrict alpha1 beta1 alpha2 beta2
Restrict beta1 beta2 1.5
Command: RestrictAll
Purpose: Restrict all rate parameters to a parameter or fixed value
Shortcut: ResAll
Parameters: A parameter or fixed value
Example: RestrictAll alpha1
ResAll 0.75
Command: RevJump
Purpose: Set a reverse jump analysis to estimate the discrete or MultiState model of evolution
Shortcut: RJ
Parameters: A prior and prior parameter
Example: RevJump exp 10
RevJump Gamma 4 20
RJ Beta 5.0 2.5
Command: RevJumpHP
Purpose: Set a reverse jump analysis to estimate the discrete or MultiState model of evolution
with a hyper prior
Shortcut: RJHP
Parameters: A hyper prior
Example: RevJumpHP exp 0 100
RevJumpHP gamma 0 100 0 50
Command: RJLocalTransform
Purpose: Use reverse jump to apply multiple local transforms (Kappa, Lambda, Delta)
Shortcut: RJLT
Parameters: Transform type, kappa, lambda or delta
Example: RJLocalTransform delta
RJLT lambda
Command: Run
Purpose: Run the analysis
Shortcut: RU
Parameters: None
Example: Run
77
Command: Sample
Purpose: Set the sample frequency
Shortcut: SA
Parameters: An integer > 0
Example: Sample 1000
Sample 250
Command: SaveInitialTrees
Purpose: Save the initial sample of trees, excluding deleted taxa and including internal
estimated nodes
Shortcut: SIT
Parameters: A file name to save the trees to
Example: SaveInitialTrees InitTree.trees
SIT InitTree.trees
Command: SaveModels
Purpose: Save the models to a file
Shortcut: SM
Parameters: A file name to save the models to.
Example: SaveModels ModelFile.bin
SM ModelFile.bin
Command: SaveTrees
Purpose: Save the sample of trees during analysis, including transforms
Shortcut: ST
Parameters: A filename to save the trees to
Example: SaveTrees STrees.trees
Command: ScaleTrees
Purpose: Scale the trees by a given value or set the sample to have a mean branch length of
0.1
Shortcut: SCT
Parameters: None to set the tree to have a mean branch length of 0.1 or a scaler
Example: ScaleTrees
ScaleTrees 0.01
SCT 10
Command: Schedule
Purpose: Toggle the recording of the schedule in an MCMC chain, default is on
Shortcut: SH
Parameters: None
Example: Schedule
SH
78
Command: Seed
Purpose: Set the random seed
Shortcut: Se
Parameters: An integer, > 0, to seed the random number generator from
Example: Seed 39362
Se 483
Command: SetMinMaxRate
Purpose: Set the minimum and maximum transition rates, ML only. Rates cannot be smaller
than 0.00000001, to stop numeric errors. This can help focus maximum likelihood
searching.
Shortcut: SMMR
Parameters: The minimum and maximum transition rates
Example: SetMinMaxRate 0 1
SMMR 5 10
Command: SetMinTransTaxaNo
Purpose: Set the minimum size node to apply RJ local transforms to, default 10
Shortcut: SMTTN
Parameters: The minimum node size to apply an RJ local transform to
Example: SetMinTransTaxaNo 20
SMTTN 5
Command: Stones
Purpose: Initialise the stepping stone sampler
Shortcut: ST
Parameters: The number of stones to use and the number of iterations to use each stone for.
The alpha and beta parameters which specify the beta distribution the stones are
drawn from can also be supplied.
Example: Stones 100 10000
Stone 100 25000 0.6 8.0
Command: Symmetrical
Purpose: Make restrictions to create a symmetrical matrix
Shortcut: SYM
Parameters: None
Example: Symmetrical
SYM
Command: TaxaInfo
Purpose: Show taxa names and numbers
Shortcut: TI
Parameters: None
79
Example: TaxaInfo
Command: TestCorrel
Purpose: Set the correlation between traits to zero, used for model testing.
Shortcut: TC
Parameters: None
Example: TestCorrel
Command: UnRestrict
Purpose: Remove a parameter restriction
Shortcut: UNRes
Parameters: A parameter to un restrict
Example: UnRestrict q01
Command: UnRestrictAll
Purpose: Remove all restrictions
Shortcut: UnResAll
Parameters: None
Example: None
Command: VarRates
Purpose: Use the variable rates model
Shortcut: VR
Parameters: None
Example: VarRates
80
References
Felsenstein, J. (1973). "Maximum-likelihood estimation of evolutionary trees from continuous
characters." American journal of human genetics 25(5): 471.
Felsenstein, J. (2004). Inferring phylogenies, Sinauer Associates Sunderland.
Freckleton, R. P. (2012). "Fast likelihood calculations for comparative analyses." Methods in Ecology
and Evolution 3(5): 940-947.
Gilks, W. R., et al. (1996). Introducing markov chain monte carlo. Markov chain Monte Carlo in
practice, Springer.
Green, P. J. (1995). "Reversible jump Markov chain Monte Carlo computation and Bayesian model
determination." Biometrika 82(4): 711-732.
Hansen, T. F. (1997). "Stabilizing selection and the comparative analysis of adaptation." Evolution:
1341-1351.
Jetz, W., et al. (2012). "The global diversity of birds in space and time." Nature 491(7424): 444-448.
Johnson, S. G. "The NLopt nonlinear-optimization package." from http://ab-initio.mit.edu/nlopt.
Organ, C. L., et al. (2007). "Origin of avian genome size and structure in non-avian dinosaurs." Nature
446(7132): 180-184.
Pagel, M. (1994). "Detecting correlated evolution on phylogenies: a general method for the
comparative analysis of discrete characters." Proceedings of the Royal Society of London. Series B:
Biological Sciences 255(1342): 37-45.
Pagel, M. (1997). "Inferring evolutionary processes from phylogenies." Zoologica Scripta 26(4): 331-
348.
Pagel, M. (1999). "Inferring the historical patterns of biological evolution." Nature 401(6756): 877-
884.
Pagel, M. and A. Meade (2006). "Bayesian analysis of correlated evolution of discrete characters by
reversible‐jump Markov chain Monte Carlo." The American Naturalist 167(6): 808-825.
Pagel, M., et al. (2004). "Bayesian estimation of ancestral character states on phylogenies."
Systematic biology 53(5): 673-684.

81
Slater, G. J. (2014). "Correction to ‘Phylogenetic evidence for a shift in the mode of mammalian body
size evolution at the Cretaceous–Palaeogene boundary’, and a note on fitting macroevolutionary
models to comparative paleontological data sets." Methods in Ecology and Evolution 5(7): 714-718.
Tobias, J. A., et al. (2016). "Territoriality, social bonds, and the evolution of communal signaling in
birds." Frontiers in Ecology and Evolution 4: 74.
Tuffley, C. and M. Steel (1998). "Modeling the covarion hypothesis of nucleotide substitution."
Mathematical biosciences 147(1): 63-91.
Venditti, C., et al. (2011). "Multiple routes to mammalian diversity." Nature 479(7373): 393-396.
Xie, W., et al. (2011). "Improving marginal likelihood estimation for Bayesian phylogenetic model
selection." Systematic biology 60(2): 150-160.
