Manual V2.1.x
User Manual: Pdf
Open the PDF directly: View PDF ![]() .
.
Page Count: 28

PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
1
PartitionFinder2
Manual
Rob Lanfear, December 2016
Icon © Ainsley Seago. Thanks Ainsley!
Questions, suggestions, problems, bugs? Search on or post in the discussion group at:
http://groups.google.com/group/partitionfinder
!
Step-by-step!tutorial:!http://www.robertlanfear.com/partitionfinder/tutorial/5
FAQs:!http://www.robertlanfear.com/partitionfinder/faq/!
Citations
PartitionFinder2 incorporates many years of hard work from many people, presented in
various different papers. Citations are the only way that we know people are using our
methods, and are all we have to demonstrate to funders that the methods are useful. So
if you use PartitionFinder2 in your published work please cite the appropriate papers
(there will often be more than one). The correct citations are listed in the ‘Citations’
section below, and will also included in the program’s output.
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
2
!
Disclaimer! 35
What!PartitionFinder2!is!for! 35
Operating!systems!(Mac,!Windows!and!Linux!work)! 45
QuickStart!–!simple!use!cases! 55
For$a$small$multilocus$dataset$(e.g.$~10$loci)$ 55
For$a$larger$dataset$(e.g.$~100$loci)$ 55
For$a$really$big$dataset$(e.g.$~1000$loci)$ 65
To$compare$all$possible$models$of$evolution$ 65
Overview! 75
Installing!and!Running!PartitionFinder2!on!a!Mac!or!Linux! 85
1.$Install$Python$and$dependencies$using$Anaconda$or$otherwise$ 85
2.$Install$PartitionFinder2$ 85
3.$Run$PartitionFinder2$ 85
Installing!and!Running!PartitionFinder2!on!Windows!105
1.$Install$Python$and$dependencies$using$Anaconda$or$otherwise$105
2.$Install$PartitionFinder2$105
3.$Run$PartitionFinder2$105
Input!Files!125
Alignment$File$in$phylip$format$125
Configuration$File$135
alignment 135
branchlengths 135
models 145
model_selection 185
[data_blocks] 195
[schemes] 195
search 195
user_tree_topology 215
Output!files!235
best_schemes.txt 235
subsets folder 235
schemes folder 235
Command!line!options!245
--all-states 245
--force-restart 245
--min-subset-size 245
--no-ml-tree 245
--processors N, -p N 245
--quick, -q 245
--raxml 255
--rcluster-max N 255
--rcluster-percent N 255
--save-phylofiles 265
--weights “Wrate, Wbase, Wmodel, Walpha” 265
Citations!275

PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
3
Disclaimer
Copyright (C) 2011-2015 Robert Lanfear, Paul Frandsen, and Brett Calcott
This program is free software: you can redistribute it and/or modify it under the terms
of the GNU General Public License as published by the Free Software Foundation,
either version 3 of the License, or (at your option) any later version. This program is
distributed in the hope that it will be useful, but WITHOUT ANY WARRANTY; without
even the implied warranty of MERCHANTABILITY or FITNESS FOR A PARTICULAR
PURPOSE. See the GNU General Public License for more details. You should have
received a copy of the GNU General Public License along with this program. If not, see
<http://www.gnu.org/licenses/>. PartitionFinder also includes the PhyML program and
the RAxML program, using PartitionFinder implies that you agree with those licences
and conditions as well.
What PartitionFinder2 is for
PartitionFinder2 is a program for selecting best-fit partitioning schemes and models of
evolution for nucleotide, amino acid, and morphology alignments. The user provides
an alignment, and optionally some pre-defined data blocks (e.g. 9 data blocks defining
the 1st, 2nd and 3rd codon positions of 3 protein-coding genes, see Figure 1). The
program then finds the best partitioning scheme for this dataset, at the same time as
selecting best-fit models for each subset of sites/columns. Here are a few things you can
do with the program:
1. Find the best-fit partitioning scheme nucleotide, amino acid, or morphology
datasets
2. Compare any number of user-defined partitioning schemes
3. Find best-fit models of evolution for each subset in any partitioned dataset
(much like you might do with ModelTest or ProtTest).
The idea is that finding best-fit partitioning schemes and models of evolution will
improve any downstream analyses of your data, like estimating phylogenetic trees or
molecular dates. All of those kinds of analyses assume that your model of evolution is
correct, and PartitionFinder2 helps make the model as good as it can be.
PartitionFinder2 can be downloaded from www.robertlanfear.com/partitionfinder. It is
designed to take the hard work out of comparing partitioning schemes, and to help find
a scheme that maximises the fit of the model to your data without including more
parameters than are necessary. PartitionFinder2 implements three information-theoretic
measures for comparing models of molecular evolution and partitioning schemes: the
Akaike Information Criterion (AIC), the corrected Akaike Information Criterion (AICc),
and the Bayesian Information Criterion (BIC). We recommend that you use the AICc,
see below.
At the end of a run, you are given output files that tell you the best partitioning scheme,
along with the best-fit model of evolution for each subset (sometimes called a
‘partition’, but that term is a bit misleading) in that scheme. So you can then move
straight on to your phylogenetic analyses.
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
4
Operating systems (Mac, Windows and Linux work)
Mac OSX, Windows, and Linux are supported.
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
5
QuickStart – simple use cases
In all of these examples, things in quotes and brackets (i.e. “<>”) indicate that you
should use full file paths. E.g. “<PartitionFinder.py>” might be
/Desktop/partitionfinder/PartitionFinder.py
Also, defining data blocks by gene and codon position refers DNA datasets from
protein coding genes. For amino acid datasets, or DNA datasets from non-coding
regions, just define data blocks by gene. For morphological datasets, there is often no
intuitive way to define data blocks. In this case you can try the k-means algorithm.
For a small multilocus dataset (e.g. ~10 loci)
For DNA, use a greedy search with PhyML. For amino acids, use a greedy search with
RAxML. These options give, respectively, the most models for DNA and amino acids.
1. Define data blocks by gene and codon position
2. In the .cfg file, set the following options:
branchlengths = linked;
models = all;
model_selection = aicc;
search=greedy;
3. Run PartitionFinder from the commandline as follows for DNA:
python “<PartitionFinder.py>” “<InputFoldername>”
or as follows for amino acids:
python “<PartitionFinderProtein.py>” “<InputFoldername>” --raxml
For a larger dataset (e.g. ~100 loci)
Use a greedy search with RAxML, for both DNA and amino acids. This will usually be
fairly quick.
1. Define data blocks by gene and codon position
2. Set the .cfg file options as above.
3. Run PartitionFinder using RAxML, as follows for DNA:
python “<PartitionFinder.py>” “<InputFoldername>” --raxml
or as follows for amino acids:
python “<PartitionFinderProtein.py>” “<InputFoldername>” –raxml
If that’s too slow for proteins, it’s probably because there are a lot of models
being compared, so try specifying a smaller list of models in the .cfg file, e.g.:
models = LG, LG+G, LG+I+G, LG+I+G+F, LG4X;
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
6
For a really big dataset (e.g. ~1000 loci)
For datasets of this size, the greedy algorithm is likely to be too slow. So we can use
some faster algorithms that try and guess the best partitioning schemes instead.
1. Define data blocks by gene and codon position
2. Set the .cfg file options as above, except:
search=rcluster;
3. Run PartitionFinder using RAxML, as follows (for amino acids, change
‘PartitionFinder.py’ to ‘PartitionFinderProtein.py’:
python “<PartitionFinder.py>” “<InputFoldername>” --raxml
This implements the relaxed clustering algorithm described in Lanfear et al
2014 (Selecting optimal partitioning schemes for phylogenomic datasets. BMC
evolutionary biology, 14(1), 82). If the default behaviour is still too slow on
your dataset, reduce rcluster-max from the default of at least 1000 to e.g. 100
like this (see below for an explanation of what this does):
python “<PartitionFinder.py>” “<InputFoldername>” --raxml --rcluster-max 100
The rcluster algorithm gives you a lot of control over the balance between speed
and accuracy with two parameters: --rcluster-max and --rcluster-percent. Read
below for more information on these
To compare all possible models of evolution
PartitionFinder2 implements lots more models of evolution than PF1. Most of the above
examples use models=all which implements all of the models most people are
interested in most of the time. However, there are some models that are not included in
models=all. These are mostly models in which state frequencies (e.g. frequencies of A,
C, T, and G) are estimated with maximum likelihood instead of just by counting
frequencies from the data. These kinds of models are usually just a tiny bit better in
terms of their fit to the data, but they can take a lot longer to optimise because you are
optimising additional parameters (a lot of them in the case of protein datasets). If you
really want a list of models with all possible models (though I wouldn’t recommend it,
see below for more information) use this setting in the .cfg file:
models = allx;
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
7
Overview
Partitioning involves splitting sites in your alignment into sets that have evolved under
similar models. For example, if you have a dataset of 3 protein-coding genes you might
suspect that each of the three genes has been evolving differently – perhaps they come
from different chromosomes or have experienced different evolutionary constraints.
Furthermore, you might think that each codon position within each gene has been
evolving differently – different codon positions tend to evolve at different rates, and
experience different substitution processes thanks to the triplet structure of the genetic
code. Because of this, you might split your data into 9 sets of sites (we call those data
blocks) for this alignment – one for each codon position in each gene. But is this too
many different sets? Perhaps it would be better to join together the 1st and 2nd codon
sites of each gene, so defining 6 sets of sites. Or perhaps it would be better to forget the
divisions between genes and define only 2 sets of sites – 1st and 2nd codon sites versus
3rd codon sites. The trouble is that if you start with 9 possible sets of sites, there are a lot
of different possible partitioning schemes you might consider, 21147 in fact. This
creates a problem – how do we find the best scheme from that many schemes?
Even worse, what if you have a dataset (like one made up of UCE’s, for example) that
doesn’t have convenient features like codon positions? How do you partition that?
PartitionFinder2 (PF2) solves these problems in one of two ways. If you have pre-defined
data blocks, PF2 can quickly and efficiently search for the best scheme from the set of
all possible schemes by trying lots of combinations of data blocks (this is what the
greedy, rcluster, and hcluster algorithms do). In the example above, all you would need
to do is define your 9 possible sets of sites (i.e. the largest number of sets of sites you
think is sensible to define) as data blocks, and PF2 will do the rest. If you do not have
pre-defined data blocks, you can specify a single set of sites and use the k-means
algorithm in PF2 which will attempt to find a good partitioning scheme by splitting your
alignment into sets of sites which evolve in similar ways.
At the end of a run you are told the best partitioning scheme that PF2 could find (in the
best_scheme.txt file) and also which model of molecular evolution you should use for
each subset of sites in that scheme (i.e., you don’t have to use ModelTest or ProtTest or
similar programs on your partitioned dataset, PartitionFinder does all of this model
selection for you at the same time as finding a partitioning scheme). You can then go
straight on to performing your phylogenetic analysis, without any additional model-
testing or comparisons of partitioning schemes.
PF2 is pretty flexible and should be able to accommodate most of the popular kinds of
partitioned analyses people like to do. This includes options for how to treat branch
lengths between subsets, which models of molecular evolution to consider, and many
other things. This manual describes in detail all of these steps.

PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
8
Installing and Running PartitionFinder2 on a Mac or Linux
1. Install Python and dependencies using Anaconda or otherwise
PartitionFinder needs Python 2.7.10 or higher (but not 3.x!) and some additional
libraries to run. By far the simplest way to set this up is to install the Anaconda Python
distribution, which is a simple point-and-click installer which can be downloaded from
here:
http://continuum.io/downloads
Follow the link for the Python 2.7 graphical installer, then open it and follow the
prompts. You need to make sure that you have version 2.3.0 or higher of the Anaconda
Python distribution.
If you don’t want to install with Anaconda, you can install Python 2.7.x however you
like, and then install the following dependencies:
numpy
pandas
pytables
pyparsing
scipy
sklearn
2. Install PartitionFinder2
1. Download the latest version of PartitionFinder2 from
www.robertlanfear.com/partitionfinder
2. Double-click the .zip file, and it will automatically unzip. You will get a folder
called something like ‘PartitionFinder2.0.0’
3. Move it to wherever you want to store PartitionFinder2 (e.g. in /Applications)
3. Run PartitionFinder2
These instructions describe how to run the ‘example/nucleotide’ analysis using
PartitionFinder. This is a DNA alignment with 9 data blocks. To use partitionfinder with
amino acid alignments, just follow these instructions but replace ‘PartitionFinder’ with
‘PartitionFinderProtein’ in step 2, and ‘example/nucleotide’ with ‘example/aminoacid’.
To use partitionfinder with morphology alignments, just follow these instructions but
replace ‘PartitionFinder’ with ‘PartitionFinderMorphology’ in step 2, and
‘example/nucleotide’ with ‘example/morphology’.
1. Open Terminal (on most Macs, this is found in Applications/Utilities)
2. In the Terminal, you need to tell the computer where to find Python,
PartititionFinder2, and your input files. The easiest way to do this is as follows:
a. Type “python“ followed by a space
b. Drag and drop the “PartitionFinder.py” file (which is in the PartitionFinder
folder you just unzipped) onto the command prompt. The path to
‘PartitionFinder.py’ will be added automatically.
c. Type another space
d. Drag and drop the blue ‘example/nucleotide’ folder (in the PartitionFinder
folder) onto the command prompt
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
9
3. Hit Enter/Return to run PartitionFinder2
That’s it!
More generally, you run PartitionFinder2 by typing a command line that looks like this:
python “<PartitionFinder.py>” “<InputFoldername>”
Where <PartitionFinder.py> is the full path to the PartitionFinder.py (or
PartitionFinderProtein.py) file, and <InputFoldername> is the full path to your input
folder, which should contain an alignment and a .cfg file. Note that the input folder can
be anywhere on your computer, it doesn’t have to be in the PartitionFinder folder like
the example file.
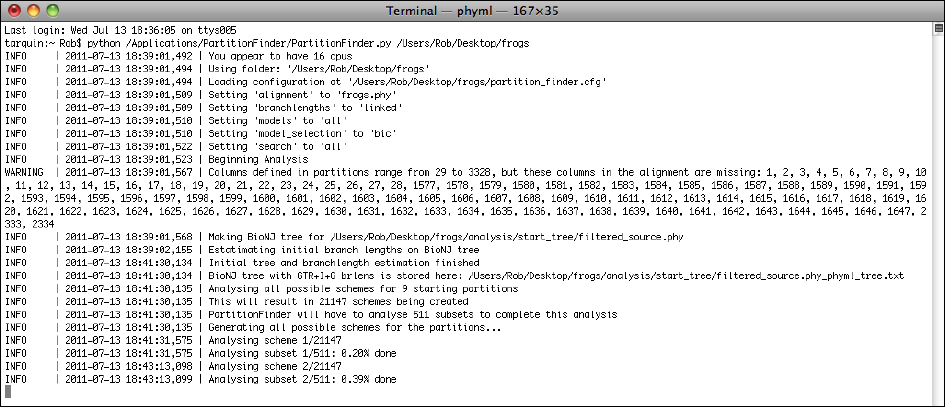
Once PartitionFinder2 is running, it will keep you updated about its progress. If it hits a
problem, it will (hopefully) provide you with a useful error message that will help you
correct that problem. Hopefully, you won’t have too many problems and your terminal
screen will look something like that shown below.

PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
10
Installing and Running PartitionFinder2 on Windows
1. Install Python and dependencies using Anaconda or otherwise
PartitionFinder needs Python 2.7.10 or higher (but not 3.x!) and some additional
libraries to run. By far the simplest way to set this up is to install the Anaconda Python
distribution, which is a simple point-and-click installer which can be downloaded from
here:
http://continuum.io/downloads
Follow the link for the Python 2.7 graphical installer, then open it and follow the
prompts. You need to make sure that you have version 2.3.0 or higher of the Anaconda
Python distribution.
If you don’t want to install with Anaconda, you can install Python 2.7.x however you
like, and then install the following dependencies:
numpy
pandas
pytables
pyparsing
scipy
sklearn
2. Install PartitionFinder2
1. Download the latest version of PartitionFinder2 from
www.robertlanfear.com/partitionfinder
2. Double-click the .zip file, and it will automatically unzip. You will get a folder
called something like ‘PartitionFinder2.0.0’
3. Move it to wherever you want to store PartitionFinder2
3. Run PartitionFinder2
These instructions describe how to run the ‘example/nucleotide’ analysis using
PartitionFinder. This is a DNA alignment. To use partitionfinder with amino acid
alignments, just follow these instructions but replace ‘PartitionFinder’ with
‘PartitionFinderProtein’ in step 2, and ‘example/nucleotide’ with ‘example/aminoacid’.
To use partitionfinder with morphology alignments, just follow these instructions but
replace ‘PartitionFinder’ with ‘PartitionFinderMorphology’ in step 2, and
‘example/nucleotide’ with ‘example/morphology’.
1. Open a command prompt. To do this, click on the Start Menu, then navigate to the
command prompt like this: “All Programs” -> “Accessories” -> “Command Prompt”.
On Windows 7 you can just type “cmd” into the search box area, and you’ll see it.
2. In the command prompt, you need to tell the computer where to find Python,
PartititionFinder2, and your input files. The easiest way to do this is as follows:
a. Type “python“ followed by a space
b. Drag and drop the “PartitionFinder.py” file (which is in the
PartitionFinder folder you just unzipped) onto the command prompt. The
path to ‘PartitionFinder.py’ will be added automatically.
c. Type another space
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
11
d. Drag and drop the blue ‘example/nucleotide’ (in the PartitionFinder
folder) onto the command prompt
3. Hit Enter/Return to run PartitionFinder
That’s it!
More generally, you run PartitionFinder2 by typing a command line that looks like this:
python “<PartitionFinder.py>” “<InputFoldername>”
Where <PartitionFinder.py> is the full path to the PartitionFinder.py (or
PartitionFinderProtein.py) file, and <InputFoldername> is the full path to your input
folder, which should contain an alignment and a .cfg file. Note that the input folder can
be anywhere on your computer, it doesn’t have to be in the PartitionFinder folder like
the example file.
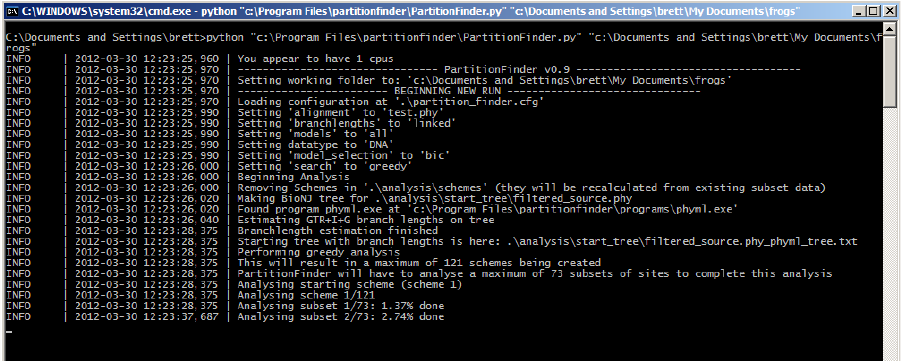
Once PartitionFinder is running, it will keep you updated about its progress. If it hits a
problem, it will (hopefully) provide you with a useful error message that will help you
correct that problem. Hopefully, you won’t have too many problems and your terminal
screen will look something like that shown below.

PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
12
Input Files
PartitionFinder2 needs two input files, a Phylip alignment and a configuration file. The
best way to get a feel for how this works is to have a look in the examples we’ve
provided in the ‘example’ folder. There is also an online tutorial at
www.robertlanfear.com/partitionfinder/tutorial. You can copy and paste these folders
onto your desktop (or anywhere) and try running them by following the instructions
above. Playing around with the options in the .cfg files give you a good idea of what’s
possible.
In the rest of this section, we describe in detail exactly what the two input files should
look like, and what they do.
Alignment File in phylip format
The phylip format: Your alignment needs to be in phylip format. We use the same
version of phylip format that PhyML uses, which is described in detail here
http://www.atgc-montpellier.fr/phyml/usersguide.php?type=phylip. In brief, this format
should contain a line at the top with the number of sequences, followed by the number
of sites in the alignment. After that, there should be one sequence on each line, where a
sequence contains a name, followed by some whitespace (either spaces or tabs) and the
sequence. Names can be up to 100 characters long. There should be nothing else on
the line other than the name and the sequence – watch out if you use MacClade, which
adds some extra things to the end of each line.
Converting other formats to phylip: If you have an alignment in some other format and
want to convert it into phylip format, the best (free!) tool to use is Geneious. Other
alignment editors tend to cut the names short in phylip files (the original definition had
a 10 character limit on names), but Geneious doesn’t. If you don't have Geneious, it's
free and you can download it from http://www.geneious.com/. Once you have
Geneious, follow these steps to convert your alignment file to phylip:
1. Open up your alignment file in Geneious, and highlight it
2. Go the 'File' menu and click 'Export', then 'Selected documents...'
3. Scroll down the list of options and choose 'Phylip (*.phy)', and click 'OK'.
4. Now a box of options will come up, choose 'Export full length'.
5. Save the phylip alignment file in the same folder as your .cfg file for
PartitionFinder.
One more thing: Often you’ll have sites in your alignment that you don’t intend to use
in your final analysis, or perhaps you have an alignment of mixed data types like DNA,
protein, and morphological data. In PartitionFinder2 this is OK. You don’t need to
make separate alignments of each datatype. You can just ignore the sites you’re not
interested in by setting the ‘[data_blocks]’ option to only focus on the data you want
PartitionFinder2 to analyse, more instructions below.
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
13
Configuration File
PartitionFinder2 gets most of its information on the analysis you want to do from a
configuration file. This file should always be called “partition_finder.cfg”. The easiest
thing to do is to base your own .cfg on one the examples provided in the “example”
folder. An exhaustive list of everything in that file follows. Note that all lines in the .cfg
file except comments and lines with square brackets have to end with semi-colons.
In the configuration file, white spaces, blank lines, and lines beginning with a “#”
(comments) don’t matter. You can add or remove these as you wish. All the other lines
do matter, and they must all stay in the file in the order they are in below. There is one
exception – the user_tree_topology option (see below).
The basic configuration file looks like this:
# ALIGNMENT FILE #
alignment = test.phy;
# BRANCHLENGTHS #
branchlengths = linked;
# MODELS OF EVOLUTION #
models = all;
model_selection = aicc;
# DATA BLOCKS #
[data_blocks]
Gene1_pos1 = 1-789\3;
Gene1_pos2 = 2-789\3;
Gene1_pos3 = 3-789\3;
# SCHEMES #
[schemes]
search = greedy;
# user schemes (see manual)
The options in the file are described below. Where an option has a limited set of
possible commands, they are listed on the same line as the option, separated by vertical
bars like this “|”.
alignment
The name of your sequence alignment. This file should be in the same folder as the .cfg
file, and must be in the correct phylip format (see above).
branchlengths: linked | unlinked
This determines how branch lengths of will be estimated during your analysis. How you
set this will depend to some extent on which program you intend to use for you final
phylogenetic analysis. Almost all phylogeny programs support linked branch lengths,
but only some support unlinked branch lenghts (e.g. MrBayes, BEAST, and RaxML).
branchlengths = linked; only one underlying set of branch lengths is estimated.
Each subset has its own scaling parameter (i.e. its own subset-specific rate). This allows
subsets to evolve at different rates, but doesn’t change the length of any one branch
relative to any other. The total number of branch length parameters here is quite small.
If there are N species in your dataset, then there are 2N-3 branch lengths in your tree,
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
14
and each subset after the first one adds an extra scaling parameter. For instance, if you
had a scheme with 10 subsets and a dataset with 50 species, you would have 106
branch length parameters.
branchlengths = unlinked; each subset has its own independent set of branch
lengths. In this case, branch lengths are estimated independently for each subset, so
each subset has it’s own set of 2N-3 branch length parameters. With this setting, the
number of branch length parameters can be quite large (2NS – 3S, where S is the
number of species in your alignment). So, a scheme with 10 subsets and a dataset with
50 species would have 970 branch length parameters.
models all | allx | beast | mrbayes | gamma | gammai | <list>
models
Most people will just want to set models to ‘all’ (an excpetion is for morphology
alignments, see below). Below is a very long description of what all of the models are,
and what the other options do. This is written mostly for those who are confused about,
or new to, models of molecular evolution in phylogenetics. If you just want to know
what the options do, skip to the description of the options themselves.
The ‘models’ option sets which models of evolution to consider during model selection.
The models that are available for any particular analysis will depend on two things: your
data type (i.e. nucleotides, amino acids, or morphology) and the phylogeny program
you are using (i.e. PhyML, which is the default, or RAxML which you can specify using
the --raxml command line option). There are many models you can specify, and
although lists are provided below, perhaps the best way to understand what is available
is to look at the models.csv file, which is located in the /partfinder folder. This lists all
available models, as well as details of the models themselves, and whether each model
is implemented in PhyML or RAxML.
Whatever you set for ‘models’, PF2 will print out the list of models it will use at the start
of the analysis. You should check this and make sure it is what you want. For every
subset of sites that PF2 looks at, it will fit every single model in your list of models and
pick the best one according to your chosen information theoretic score (AIC, AICc, or
BIC). Your results therefore tell you not only the best partitioning scheme, but also
which model of evolution is most appropriate for each subset in that scheme. This
means that you don’t need to do any further model selection after PartitionFinder2 is
done.
A very short primer on models of molecular evolution in phylogenetics
Over the years I’ve had a lot of questions about models in phylogenetics. So here’s a
primer if you’re interested. There are two important things to understand: the differences
between models and how the models are named.
The difference between all of the models is in which parameters are fixed a-priori vs
estimated from the data (we call these free parameters), and how the free parameters are
estimated. In general, all models (both amino acid and nucleotide models) have three
components: the frequencies of nucleotides or amino acids (e.g. the proportion of A, C,
T, and G in your data), the relative rates at which the different nucleotides or amino are
replaced by each other (e.g. the rate at which A replaces G in your data), and the
distribution of rates of evolution among sites in your dataset. There are many possible
models of evolution for nucleotide and amino acid alignments. All of the models
implemented in PF (plus a few that are not possible) are listed in the models.csv file,
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
15
which is in the partitionfinder folder, under /partfinder/models.csv. As you read the next
paragraph, it might help to open up this file and take a look at the models to familiarise
yourself with what’s possible.
Frequencies of nucleotides or amino acids can be fixed in advance, estimated from the
data by simply counting the proportions observed in your alignment, or estimated using
maximum likelihood. In the models.csv file, this is described in the base_frequencies
column. There are four possible types of base frequency: ‘equal’ is when all frequencies
are set to equal (e.g. proportions of 0.25 for A, C, T, and G in the JC model) and is only
implemented for nucleotide models in PF2; ‘model’ is when frequencies are determined
a-priori from some other dataset, which is common for amino acid models (e.g. the JTT
model); ‘empirical’ is when the frequencies are determined by just counting the
proportions of nucleotides or amino acids in your data; ‘ML’ is when the frequencies are
determined using maximum likelihood. The last two options tend to give very similar
answers (ML is guaranteed to be better, but the difference is usually so marginal that it
makes no difference to the phylogeny) but ‘empirical’ is much faster, and so most
people never use ML base frequencies. One more important thing – thanks to historical
naming conventions, the way that these models are named differs for nucleotide and
amino acid models. For amino acid models, it’s easy, we’ll use the LG model as an
example. If you specify ‘LG’, then amino acid frequencies will be estimated from the
model (the LG model comes with some best-guess amino acid frequencies estimated
from a large collection of datasets). If you specify ‘LG+F’ then the amino acid
frequencies are estimated empirically from your data. If you specify ‘LG+X’ then the
amino acid frequencies are estimated with maximum likelihood from your data. For
nucleotide models, it’s different. Take a look at rows 2-22 of the models.csv file. First,
no nucleotide models have a-priori base frequencies included in the model (i.e. the
‘model’ option), so your only options are to have them all equal, estimate them
empirically, or estimate them with maximum likelihood. Historically, the first two of
these options were given completely different model names, rather than using the ‘+F’
notation that we use for amino acid models. Let’s take the JC model as an example. If
you specify ‘JC’, that assumes all base frequencies are equal. But there is no JC+F
model, it does exist, but it’s just called the F81 model. This is the reason why there are
no ‘+F’ models in the list of possible models for DNA sequence alignments. Rows 2-22
spell this out: each model with ‘equal’ base frequencies is paired with a model with
empirical base frequencies. For example, the model in row 1 (JC) matches the model in
row 8 (F81), row 2 (K80) matches row 9 (HKY), and so on. To complete the picture, we
can specify that we want our base frequencies for nucleotide models estimated with ML
(few people use these models though). To do that, we just specify the name of the
empirical model with ‘+X’, e.g. ‘F81+X’. Note that most of these kinds of nucleotide
models are only implemented in PhyML, not RAxML.
Relative rates of substitution (this is usually called the rate matrix) can be fixed in
advance, or estimated from the data. For DNA models, there are really just 6 types of
relative rate matrix. JC and F81 have all rates equal (no free parameters), other models
set certain parameters to be equal (1-4 free parameters, depending on the model), and at
the other end of the scale the SYM and GTR models allow all six reversible rate
parameters to differ (5 free parameters, since one is set arbitrarily to 1.0). In the amino
acid models, almost all models use pre-estimated rate matrices. The matrices tend not to
be estimated from the data, because most datasets do not contain enough information to
estimate so many parameters (189 free parameters in an amino acid replacement
matrix). These matrices have names like WAG, LG, JTT, etc. There is one exception –
one can estimate a GTR model for amino acid data – this has a LOT of parameters (189

PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
16
free parameters), but for very big datastets it might be appropriate and so might be worth
including in your list of possible models (note that only this model is only supported
when you are using the ‘--raxml‘ commandline option (see below).
The distribution of rates across sites is modelled in one of five ways. The first four work
the same for most nucleotide and amino acid models, so I’ll just use the HKY model as
an example. First, one can assume that all sites evolve at the same rate (not a very good
assumption in most cases), in which case the model would be just ‘HKY’. Second, we
could assume that some sites never change, and so model a proportion of invariant
sites. In this case, we estimate one free parameter (the proportion), and the model would
be ‘HKY+I’. Third, we could assume that the sites evolve according to some distribution
of rates, which we can model with a gamma distribution (we use this because it can
take lots of different shapes). In this case, we estimate 1 free parameter, which
determines the shape of the distribution, and the model would be ‘HKY+G’. Fourth and
last, we could combine the proportion of invariant sites and the gamma distribution, in
this case we estimate two free parameters and the model would be ‘HKY+I+G’. Finally,
there is a new class of rate distribution models which are often called ‘free rate’ models.
In these models, one specifies some number of categories of rate, and instead of
modelling them as coming from a distribution (like the gamma distribution) one
estimates the rate of each category. These models have been implemented for
nucleotides and amino acids, but because of various technical limitations the only
model like this that’s implemented in PartitionFinder is the LG4X model (which works
only with the the ‘--raxml‘ commandline option, see below). The LG4X model has four
rate categories, and it also has four separate sets of amino acid frequencies. It’s a very
neat model, and often fits the data better than other amino acid models. If you use it,
you should read and reference this paper:
http://mbe.oxfordjournals.org/content/29/10/2921.
Models of morphological evolution in PartitionFinder2.
PartitionFinder2 implements four different models of evolution that can be used on
morphological data: BINARY+G, BINARY+G+A, MULTISTATE+G, MULTISTATE+G+A.
All of these models require the --raxml commandline option, and more details on their
implementation is available in the RAxML manual. The BINARY models are for binary
data, and the MULTISTATE models are MK models for multistate data. You can only
analyse your data under a single morphological model at a time, since the choice of
binary vs. multistate, and whether or not you need an ascertainment bias for your data
(the +A option) are ones that need to be made ahead of time based on the properties of
your data. The AIC/AICc/BIC are not appropriate methods for choosing between these
models for a given dataset.
models = all; Chooses the largest sensible set of models possible, depending on the
analysis being conducted (i.e. whether you are using DNA or amino acid alignments,
and whether you are using PhyML or RAxML). Note that this list does NOT include
models in which the base or amino acid frequencies are estimated with maximum
likelihood, because these models are very rarely used and, in practice, take a lot longer
to estimate for extremely marginal gains in performance.
If you are analysing DNA sequences with the default options in PF2, then models = all
will compare 56 models of nucleotide evolution for each subset. These 56 models
comprise the 14 most commonly used models of molecular evolution (JC, K80, TrNef,
K81, TVMef, TIMef, SYM, F81, HKY, TrN, K81uf, TVM, TIM, and GTR), each of which

PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
17
comes in four flavours: on its own, with invariant sites (+I), with gamma distributed rates
across sites (+G), or with both gamma distributed rates and invariant sites (+I+G).
If you are analysing DNA sequences with the --raxml commandline option, then models
= all will compare the 3 models of nucleotide evolution available in RAxML: GTR,
GTR+G, GTR+I+G.
If you are analysing amino acid sequences with the default options in PF2, then models
= all will compare 112 models of evolution, comprising 14 amino acid rate matrices
(LG, WAG, MTREV, DAYHOFF, DCMUT, JTT, VT, BLOSUM62, CPREV, RTREV,
MTMAM, MTART, HIVB, HIVW), each of which comes in 8 flavours: with or without
empirical amino acid frequencies (+F), combined with the four types of distributions of
rates across sites described above (i.e. all rates equal, +G, +I, and +I+G). E.g. the 8
flavours of the LG model are: LG, LG+F, LG+G, LG+G+F, LG+I, LG+I+F, LG+I+G,
LG+I+G+F.
If you are analysing amino acid sequences with the --raxml commandline option, then
models = all will compare 128 models of evolution. Most of these work as above –
there are 21 models (LG, WAG, MTREV, DAYHOFF, DCMUT, JTT, VT, BLOSUM62,
CPREV, RTREV, MTMAM, MTART, HIVB, HIVW, MTZOA, PMB, JTTDCMUT, FLU,
STMTREV, DUMMY, DUMMY2), each of which comes in 6 flavours: with or without
empirical amino acid frequencies (+F), combined with 3 types of rate distribution across
sites (all rates equal, +G, and +I+G). Note that RAxML does not estimate models with
just +I, and that’s why there are 6 not 8 flavours of each model when using the --raxml
option. The final two models are the LG4X model (described above), and the LG4M+G
model. There is only one flavor of each of these models implemented in RAxML, if
you’re interested to know why, there is a great description in the RAxML manual and
the paper that describes the original models
http://mbe.oxfordjournals.org/content/29/10/2921.
models = allx; this is a list of models that includes every single model listed in the
‘models = all’ description above, but also includes models in which base or amino acid
frequencies are estimated using maximum likelihood (+X) rather than empirically (+F)
where possible.
For DNA sequences: with the default options this list comprises 84 models (all the 56
models from the ‘models = all;’ list, plus the following base models in with +X, +I+X,
+G+X and +I+G+X: F81, HKY, TrN, K81uf, TVM, TIM, and GTR); with the --raxml
commandline option it comprises 6 models (GTR, GTR+X, GTR+G, GTR+G+X,
GTR+I+G, GTR+I+G+X).
For amino acid sequences: with the default options this list comprises the same 112
models as ‘models = all;’ because PhyML does not allow for amino acid frequencies to
be calculated with maximum likelihood (i.e. +X models are not implemented in PhyML,
so are not possible to estimate with the default options of PF2); with the --raxml
commandline option it comprises 195 models. These 195 models include all the
models from the ‘models = all;’ list, plus variants of the 21 rate matrices in RAxML
combined with amino acid frequencies estimated from the data (e.g. LG+X, LG+G+X,
LG+I+G+X) as well as all 6 variants of the protein GTR model, in which the rate matrix
is itself estimated from the data. You should be careful before you use the GTR model,
since it can take a very long time to run and is only likely to provide good information
theoretic scores on exceptionally large datasets.
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
18
models = mrbayes; models = beast; tells PartitionFinder to use only the
nucleotide models available in MrBayes3.2, or BEAST2 respectively. This can be useful
if you intend to use one of these programs for your phylogenetic analysis, as it restricts
the models that are compared to only those that are implemented in the particular
programs. This is not the most appropriate thing to do for a Bayesian analysis though, so
be careful (see above).
models = gamma; models = gammai; tells PartitionFinder to use only a subset of
models from the ‘models = all;’ list. ‘gamma’ uses only those models from ‘models =
all;’ that have ‘+G’. ‘gammai’ uses only those models from models = all that have
‘+I+G’ but not ‘+G’ for their rate distribution.
models = <list>; This can be any list of models appropriate for the data type. If
you are not sure which models are possible, you can either study the models.csv file (in
the /partfinder folder) or just try out a list. If you include a model that won’t work, PF2
will tell you which models didn’t work an error message before your analysis gets
underway. Each model in the list should be separated by a comma. For example, if I
was only interested in a few nucleotide models in PartitionFinder, I might do this:
models = JC, JC+G, HKY, HKY+G, GTR, GTR+G;
Or, for protein models in PartitionFinderProtein I might do this:
models = LG, LG+G, LG+G+F, WAG, WAG+G, WAG+G+F;
Note that in this list you can specify either nucleotide models, or amino acid models,
but not a mixture of both. If you have a mixed dataset (i.e. some data blocks are amino
acid, some are nucleotides), you have to run PartitionFinder on the nucleotide data,
then PartitionFinderProtein on the amino acid data.
A complete list of all models implemented in PF2 is provided in the models.csv file. This
list also includes notes on some of the models, including many models that look like
they could be implemented but are not for various reasons.
model_selection: AIC | AICc | BIC
Sets which metric to use for model selection. It also defines the metric for comparing
partitioning schemes if you use search=greedy (see below).
The AIC, AICc, and BIC are similar in spirit – they all reward models that fit the data
better, but penalise models that have more parameters. The idea is include parameters
that help the model fit the data more than some specified amount, but to avoid
including too many parameters (overparameterisation). The BIC penalises extra
parameters the most, followed by the AICc, and then the AIC. Which model_selection
approach you use will depend on your preference. There are lots of papers comparing
the merits of the different metrics, and my current favourite is the AICc. In general, you
should never use the AIC since the AICc is always preferable. However, it’s included in
PartitionFinder mostly for historical reasons.

PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
19
[data_blocks]
On the lines following this statement you define the starting subsets for your analysis
(we call these data blocks). Each data block has a name, followed by an “=” and then a
description. The description is built up as in most Nexus formats, and tells
PartitionFinder which sites of your original alignment correspond to each data block.
Based on our research (http://mbe.oxfordjournals.org/content/32/6/1611) we
recommend that you use the data blocks to give PartitionFinder as much biological
information about your sequences as you possibly can. The best way to understand this
it to look at a couple of examples.
Imagine a DNA sequence alignment with 1000bp of protein-coding DNA, followed by
1000bp of intron DNA. Your data block definitions might look like this:
Gene1_codon1 = 1-1000\3; !
Gene1_codon2 = 2-1000\3; "
Gene1_codon3 = 3-1000\3; #
intron = 1001-2000; $
!-# are typical of how you might separate out codon positions for a protein coding
gene. The numbers either side of the dash define the first and last sites in the data block,
and the number after the backslash defines the spacing of the sites. Every third site will
define a codon position, as long as your alignment stays in the same reading frame
throughout that gene.
$ shows the single block of sites for the intron.
Note that data blocks cannot be overlapping. That is, each site in the original alignment
can only be included in a single data block.
To help with cutting and pasting from Nexus files (like those used by MrBayes) you can
leave “charset” at the beginning of each line. So, the following would be treated exactly
the same as the example above:
charset Gene1_codon1 = 1-1000\3;
charset Gene1_codon2 = 2-1000\3;
charset Gene1_codon3 = 3-1000\3;
charset intron = 1001-2000;
[schemes]
On the lines following this statement, you define how you want to look for good
partitioning schemes, and any user schemes you want to define. You only need to
define user schemes if you choose search=user.
search: all | greedy | rcluster | rclusterf | hcluster |
kmeans | user
This option defines which partitioning schemes PartitionFinder will analyse, and how
thorough the search will be. In general ‘all’ is only practical for analyses that start with
12 or fewer data blocks defined (see below). A rough guide is to use ‘all’ for very small
datasets, ‘greedy’ for datasets of ~10 loci, and ‘rcluster’ for datasets of 100’s of loci. We
do not recommend you use ‘hcluster’, but rather that if ‘rcluster’ is too slow, you make it

PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
20
quicker using the ‘--rcluster-max’ commandline argument (see below). ‘kmeans’ has
been disabled for all but morphological data, for which it remains experimental (see
search = kmeans below). We suggest that you prefer biologically-motivated
partitioning schemes (like genes and codon positions) where possible.
search = all; Tells PartitionFinder to analyse all possible partitioning schemes.
That is, every scheme that includes all of your data blocks in any combination at all.
Whether you can analyse all schemes will depend on how much time you have, and on
what is computationally possible. If you have any more than 12 data blocks to start
with you should not choose ‘all’. This is because the number of possible schemes can
be extremely large. For instance, with 13 data blocks there are almost 28 million
possible schemes, and for 16 data blocks the number of possible schemes is over 10
billion. It’s just not possible to analyse that many schemes exhaustively. For 12 data
blocks the number of possible schemes is about 4 million, so it might be possible to
analyse all schemes if you have time to wait, and a fast computer with lots of
processors.
search = greedy; Tells PartitionFinder to use a greedy algorithm to search for a
good partitioning scheme. This is a lot quicker than using search=all, and will often give
you the same answer. However, it is not 100% guaranteed to give you the best
partitioning scheme. If you use this algorithm, please cite the 2012 PartitionFinder paper
(see citations, below, or here: http://mbe.oxfordjournals.org/content/29/6/1695) in
addition to the PF2 paper.
search = rcluster; Tells PartitionFinder to use a relaxed hierarchical clustering
algorithm to search for a good partitioning scheme. This option only works with the
--raxml commandline option (see above). It works by measuring the similarity of
different subsets, then looking at schemes that combine the most similar subsets. It
usually performs worse than the greedy search option, and always performs better than
the hcluster option. You can control this algorithm using the ‘--rcluster-max’, ‘—rcluster-
percent’ and ‘--weights‘ command line options (see below). The rcluster algorithm is a
very efficient way to search, and can be used even on large phylogenomic datasets with
1000s of loci. It’s designed for use with datasets that are too large to analyse with the
greedy algorithm. If you use this algorithm, please cite the 2014 paper in which it is
described (see Citations, or here: http://www.biomedcentral.com/1471-2148/14/82).
search = rclusterf; Tells PartitionFinder to use a variant of the relaxed
hierarchical clustering algorithm described above. This option only works with the
--raxml commandline option (see above). It works very similarly to the rcluster
algorithm, but instead of putting together the best pair of subsets found at each step (as
in the rcluster algorithm) it puts together the top 50% of subsets found at each step. As a
result, the algorithm can complete in many fewer steps than the rcluster algorithm. This
can be particularly helpful in situations where you are examining some models of
molecular evolution that take a lot longer than others to optimise (e.g. the LG4X models
for protein evolution), and/or where you have many available processors and a very
large dataset. In these cases, the rcluster algorithm often spends much of its time in each
step (>90%) waiting for a single analysis to complete on a single processor, which is a
huge waste of available resources. The rclusterf algorithm avoids this by having fewer
steps. However, it is not guaranteed to be faster on all datasets. The best thing to do is
try the rcluster algorithm, and if you notice that it spends a long time waiting for a small
number of analyses at the end of each step, switch to using the rclusterf algorithm.
Control this algorithm as you would the rcluster algorithm.

PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
21
search = hcluster; Not recommended for empirical analyses. Tells
PartitionFinder to use a strict hierarchical clustering algorithm to search for a good
partitioning scheme. This option only works with the --raxml commandline option. This
algorithm often performs a great deal worse than the rcluster algorithm. In general, I do
not recommend using this algorithm under any circumstances. It is better to use the
‘rcluster’ algorithm with ‘--rcluster-max’ set to some very low number (e.g. 10, see
below) instead. The hcluster algorithm is almost the same as using the rcluster algorithm
with ‘--rcluster-max’ set to 1. You can control this algorithm using the ‘--weights‘
command line options (see below). The algorithm remains in PartitionFinder purely
because it makes our research that proved it was not worth using replicable. If you use
this algorithm, please cite the 2014 paper in which it is described (see Citations, or here:
http://www.biomedcentral.com/1471-2148/14/82)
search = kmeans; This method has been discontinued for all but morphological
data, and we caution that it remains experimental for morphological data. There is
increasing evidence that the kmeans algorithm can lead to poor inferences, so we have
discontinued its use for most data types. You should instead use other approaches (e.g.
partitioning by locus and codon position). If you have any questions, please get in touch
on the google group. More information on the empirical issues can be found in this
paper: http://www.sciencedirect.com/science/article/pii/S1055790316302780.
We have kept the method available for morphological data, but warn users that the
method is: experimental, untested on morphological data (either empirical or
simulated), and may give incorrect topologies and branch lengths (see link to paper
above). We are working on improved methods (that build on the original method here:
http://www.biomedcentral.com/1471-2148/15/13), but these are likely to take some
time to finalise.
search = user; Use this option to compare partitioning schemes that you define by
hand. User-defined schemes are listed, one-per-line, on the lines following
“search=user”. A scheme is defined by a name, followed by an “=” and then a
definition. To define a scheme, simply use parentheses to join together data blocks that
you would like to combine. Within parentheses, each data block is separated by a
comma. Between parentheses, there is no comma. All user schemes must contain all of
the data blocks defined in [data_blocks].
Here’s an example. If I’m working on my one protein-coding gene plus intron alignment
above, I might want to try the following schemes: (i) all data blocks analysed together;
(ii) intron analysed separately from protein coding gene; (iii) intron separate, 1st and 2nd
codon positions analysed separately from 3rd codon positions; (iv) all data blocks
analysed separately. I could do this as follows, with one scheme on each line:
together = (Gene1_codon1, Gene1_codon2, Gene1_codon3, intron);
intron_123 = (Gene1_codon1, Gene1_codon2, Gene1_codon3) (intron);
intron_12_3 = (Gene1_codon1, Gene1_codon2) (Gene1_codon3) (intron);
separate = (Gene1_codon1) (Gene1_codon2) (Gene1_codon3) (intron);
user_tree_topology
This is an additional option which can be added into the .cfg file after the ‘alignment’
line. It’s used if you’d like to supply PartitionFinder with a fixed topology, rather than
relying on the neighbour joining topology that the program estimates by default. This
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
22
might be useful if you know ahead of time what the true tree is, for instance when
doing simulations. To use the option, just add in an extra line to the .cfg file like this:
# ALIGNMENT FILE #
alignment = test.phy;
user_tree_topology = tree.phy;
Where “tree.phy” is the name (not the path) of the file containing a newick formatted
tree topology (with or without branch lengths). The file name can be anything – it
doesn’t have to be ‘tree.phy’. The tree file must be in the same folder as the alignment
and the .cfg file. When you use this option, the topology you supply in the tree file will
be fixed throughout the analysis. Branch lengths will be re-estimated using a GTR+I+G
model on the whole dataset, as in a standard analysis.
If you don’t want to use this option, you can just leave out the user_tree_topology line
from the .cfg file.
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
23
Output files
All of the output is contained in a folder called “analysis” which appears in the same file
as your alignment. There is a lot of output, but in general you are likely to be interested
in three things, maybe this order:
best_schemes.txt++
has information on the best partitioning scheme found, and the settings used to find it.
This includes a detailed description of the scheme, as well as the model of molecular
evolution that was selected for each subset in the scheme. It also contains a description
of the each scheme in RAxML and Nexus formats.
subsets+folder+
is a folder which contains the results of the model selection on each subset of sites that
was analysed. These are .txt files, in which each model you included in your analysis is
listed, in order of increasing AICc score (i.e. best model is at the top). The default is to
save model selection results of only those subsets that made it into the best partitioning
scheme. If you want to save the model selection results from all subsets of sites that
were analysed, then you can use the --save-phylofiles commandline option (but beware,
this results in writing a lot of files – see info on the option, below). The subsets folder
also contains a database (data.db, an hdf5 file) of information on the subsets of sites that
were analysed, so that PartitionFinder can re-run analyses without re-calculating lots of
results.
schemes+folder+
is a folder which contains detailed information on the schemes that were analysed
during the analyses, each in a separate .txt file that is very like the best_scheme.txt file.
For the greedy and clustering algorithms, this folder contains only the starting scheme
and the best scheme that was found at each step of the algorithm. For the kmeans
algorithm, it will just contain the start_scheme.txt and final_scheme.txt files, because we
cannot save schemes along the way during the kmeans algorithm (read the paper to find
out why). This folder will also contain a .csv file that summarises all of the schemes. If
you analyse really huge datasets, you may want to turn off writing these files using the
command line option -q (see below).
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
24
Command line options
There are a number of additional commands you can pass to PartitionFinder from the
commandline. These can be used to fine-tune your analyses.
--all-states
Only affects the k-means algorithm. Specifically, this limits the k-means algorithm to
only produce subsets that contain all possible states. We allow ambiguous DNA states
that code for <4 states, and we count these as representing all possible resolutions of
the ambiguity. We do not count ‘?’, ‘N’, or ‘-‘ states as representative of anything. This
option is designed to try and ensure that the partitioning schemes produced by the k-
means algorithm are easy to analyse in downstream software like RAxML.
--force-restart
This will delete all previous workings (by deleting the ‘analysis’ folder) before restarting
a run. The default is not to do this so PartitionFinder can use results that it has already
calculated.
--min-subset-size
Default: 100
Only affects the k-means algorithm. This option limits the k-means algorithm to
produce subsets that are at least as big as min-subset-size. The default is 100 sites, so
by default the k-means algorithm will never produce a subset of less than 100 columns.
--no-ml-tree
PartitionFinder 2’s default is to estimate a maximum-likelihood tree from your data as a
starting tree for the analysis. This should help avoid any biases that can come from
using a sub-optimal starting tree in an analysis. However, this only works if every
column in your alignment is assigned to a data block, which is almost always the case.
If for some reason you cannot do this, you should remove the un-assigned sites from
your alignment before doing your analysis. If that’s not possible, then the –no-ml-tree
option is there to help you. If you add this to the commandline, then PF2 will estimate
a Neighbour Joining starting tree (if you are using PhyML) or a Maximum Parsimony
starting tree (if you are using RaxML).
--processors N, -p N
Default – use all available processors.
N is the number of processors you want PartitionFinder to use. This controls the
number independent PhyML or RAxML runs that PartitionFinder will run at any one
time. The default is for PartitionFinder to use all of the available processors (look for
this message at the start of the run, to see how many it found: “You appear to have N
cpus”). However, if you don’t want it to use all the processors, control with this option.
E.g. –p 5 would tell PartitionFinder to use up to 5 processors at once.
--quick, -q
This option will stop PartitionFinder from writing out unnecessary summaries of
partitioning schemes during the analysis. Most people will not need to use this option,
but if you are running really big analyses, particularly with the greedy algorithm, it can
marginally speed things up.
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
25
--raxml
This tells PartitionFinder and PartitionFinderProtein to use RAxML rather than PhyML
(the default). You might want to do this because RAxML is faster than PhyML, or
because it implements the models you are interested in (NB, RAxML implements fewer
nucleotide models, but many more amino acid models, than PhyML). Because of the
nature of RAxML, we can’t guarantee that the RAxML executables we have provided in
the ‘programs’ folder will work on all Windows and Mac machines. So if you use this
option and RAxML doesn’t work, you’ll need to download and compile RAxML
yourself, on your own computer.
--rcluster-max N
Default: --rcluster-max the larger of 1000 and 10 times the number of data blocks.
See below for a description. If you want this option to be infinite, set it to -1
--rcluster-percent N
Default: --rcluster-percent 10
rcluster-max and rcluster-percent control the thoroughness of the relaxed clustering
algorithm together. Setting either of them higher will tend to make the search more
thorough and slower. Setting them lower will tend to make the search quicker but less
thorough. The rcluster algorithm works by finding the rcluster-max most similar pairs of
data blocks, OR the top rcluster-percent of similar datablocks, whichever is smaller. It
then calculates the information score (e.g. AICc) of all of these data blocks and keeps
the best one. Setting --rcluster-max to 1000 and --rcluster-percent to 10 (i.e. the default
values) is usually sufficient to ensure that PF2 will estimate a robust partitioning
scheme, even on very large datasets in which there may be millions of possible pairs of
data blocks. Please note that it is better to use rcluster with --rcluster-max set to a very
small number (e.g. 10) than to use the hcluster algorithm. The hcluster algorithm is in
PartitionFinder more to make sure that old analyses can be replicated than for use in
empirical research.
Why do we have two parameters to control the rcluster algorithm? What's important is
that the smaller value of --rcluster-max and --rcluster-percent changes as the algorithm
progresses. Let's imagine that you have rcluster-max at 1000 and rcluster-percent at 10,
and that you have a large dataset with a lot of data blocks, for which the optimal
partitioning scheme happens to have a small number of subsets. In the early stages of
the algorithm, there are A LOT of potential combinations of subsets, so the algorithm
will consider the most similar 1000 subsets (because 10% of A LOT will be bigger than
1000, so the rcluster-max cutoff will be working; and remember that similarity is
defined by measuring attributes of subsets like their rate of evolution). But as we get
towards the end of the algorithm, we will get to a partitioning scheme with just a few
subsets, because we've merged most of the subsets. Specifically, let's imagine that that
the algorithm is currently working on a partitioning scheme with 100 subsets. If there
are 100 subsets then there are 100 choose 2 possible combinations, which is 4950.
Now, rcluster-max is 1000, but rcluster-percent is 10, and 10 percent of 4950 is 495.
So in this case the rcluster algorithm will consider just the most similar 495 subsets. It
will not consider the top 1000 subsets, because 495 (determined by rcluster-percent =
10) is smaller than 1000 (determined by rcluster-max = 1000).
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
26
Why is this sensible? There are two answers to this. One is empirical: I've tested this
algorithm on a huge range of empirical datasets, and the default settings seem to
provide the best balance between speed and accuracy across those datasets. The other
answer is theoretical: when you have rcluster-percent set to 100, you sometimes spend
a long time at the end of the algorithm analysing really big subsets because at the end
of the algorithm you have joined together a lot of the initial data blocks into larger
subsets. And here's the important bit – all this searching seems to almost never matter.
The improvements in AICc scores we get at the end of the algorithm are very small, and
on top of that it seems to be much easier to guess which are the best subsets to merge
as the subsets get bigger, probably because we can more accurately guess which subset
combinations will lead to improvements in the AICc score when the subsets are bigger
and have more information. So it's much better to analyse fewer subsets at the end of
the algorithm - we may lose a tiny bit of the improvement in AICc score (my tests show
that we often lose nothing in terms of AICc score though!), but we gain an awful lot in
terms of speed. So, because the whole point of the rcluster algorithm is that it's less
thorough but a lot quicker than the greedy algorithm I think setting the default for
rcluster-percent to 10 is sensible. Of course, you can set these parameters however you
like.
--save-phylofiles
This option will make PartitionFinder write a lot of extra files to disk. In general, you
will not need to use this. Specifically, it will write the input and output of every single
phylogenetic analysis into the /analysis/phylofiles folder (beware, this can be many
millions of files, and can take up a lot of disk space). It will also write the results of
model selection on every single subset encountered, rather than just the subets in the
best scheme that was estimated. This option can be particularly useful if PartitionFinder
(or the programs it uses, like RAxML and PhyML) can’t analyse your dataset. It can help
track down bugs in the programs, or errors in your input files.
--weights “Wrate, Wbase, Wmodel, Walpha”
Default: --weights “1, 0, 0, 0”
A list of weights to use in the clustering algorithms (NB, this only works in combination
with the --raxml option and either the hcluster or rcluster search options). This list
allows you to assign different weights to the overall rate for a subset, the base/amino
acid frequencies, the model parameters, and the alpha parameter (which describes
gamma distributed rates across sites). This will affect how subsets are clustered
together. For instance:
--weights '1, 1, 1, 0.1'
would weight the subset rate, base frequencies, and the model parameters equally, but
the alpha parameter as 10x less important. You can play around with these parameters
to try and find the best scheme that you can.
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
27
Citations
Depending on your analysis, you may need to cite up to three papers. One for
PartitionFinder2, one for the algorithm you use (if you use the rcluster, hcluster, or
kmeans options), and one for either PhyML or RAxML.
PartitionFinder2
If you any of this program in any published work please cite:
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., Calcott, B. (2016)
PartitionFinder 2: new methods for selecting partitioned models of evolution for
molecular and morphological phylogenetic analyses. Molecular biology and evolution.
DOI: dx.doi.org/10.1093/molbev/msw260
Using search = ‘rcluster’ or search = ‘hcluster’
These algorithms are described in the following paper, if you use them please cite:
Lanfear, R., Calcott, B., Kainer, D., Mayer, C., & Stamatakis, A. (2014). Selecting
optimal partitioning schemes for phylogenomic datasets. BMC evolutionary
biology, 14(1), 82.
Using search = ‘kmeans’
This algorithm is described in the following paper, if you use it please cite:
Frandsen, P. B., Calcott, B., Mayer, C., & Lanfear, R. (2015). Automatic selection of
partitioning schemes for phylogenetic analyses using iterative k-means clustering of site
rates. BMC Evolutionary Biology, 15(1), 13.
PhyML
If you use PF2 without the --raxml command line option, PF2 relies heavily on PhyML
version 3.0, so please cite:
New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing
the Performance of PhyML 3.0. Guindon S., Dufayard J.F., Lefort V., Anisimova M.,
Hordijk W., Gascuel O. Systematic Biology, 59(3):307-21, 2010.
Using the --raxml command line option
If you use the --raxml commandline option, PF2 uses RAxML v8.0 for calculations. If
you use it, please cite:
A. Stamatakis, RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with
thousands of taxa and mixed models, Bioinformatics 22, 2688–2690 (2006)
PartitionFinder2 = new methods for selecting partitioned models of evolution for phylogenetic analyses.
28
