NUPACK User Guide 3.0
NUPACK-UserGuide-3.0
User Manual: Pdf
Open the PDF directly: View PDF ![]() .
.
Page Count: 20
- Overview
- Executables
- pfunc: calculate the partition function
- pairs: calculate base-pairing observables
- mfe: find the minimum free energy (MFE) secondary structure(s)
- subopt: find all secondary structures within a specified free energy gap of the MFE
- count: count the number of secondary structures in the ensemble
- energy: calculate the free energy of a secondary structure
- prob: calculate the equilibrium probability of a secondary structure
- defect: calculate the ensemble defect
- complexes: calculate the partition functions of all strand complexes up to a specified size
- concentrations: calculate the equilibrium concentration of each complex in a dilute solution
- distributions: calculate the equilibrium population distribution and expected value for a few complexes in a dilute solution
- design: design the sequence of one or more strands intended to adopt a target secondary structure at equilibrium
- References
NUPACK 3.0
User Guide
BRIAN R. WOLFE, JUSTIN S. BOIS, NILES A. PIERCE
California Institute of Technology
January 27, 2015
Contents
1 Overview 2
1.1 Directories ............................................ 2
1.2 Compilation ........................................... 2
1.3 Example job files ........................................ 2
1.4 Notation and terminology .................................... 2
1.5 Secondary structure model ................................... 3
1.6 Conventions ........................................... 3
1.7 License ............................................. 6
2 Executables 7
2.1 pfunc: calculate the partition function ............................ 7
2.2 pairs: calculate base-pairing observables .......................... 7
2.3 mfe: find the minimum free energy (MFE) secondary structure(s) .............. 9
2.4 subopt: find all secondary structures within a specified free energy gap of the MFE .... 9
2.5 count: count the number of secondary structures in the ensemble .............. 9
2.6 energy: calculate the free energy of a secondary structure .................. 9
2.7 prob: calculate the equilibrium probability of a secondary structure ............. 10
2.8 defect: calculate the ensemble defect ............................ 10
2.9 complexes: calculate the partition functions of all strand complexes up to a specified size 10
2.10 concentrations: calculate the equilibrium concentration of each complex in a dilute
solution ............................................. 13
2.11 distributions: calculate the equilibrium population distribution and expected value
for a few complexes in a dilute solution ............................ 16
2.12 design: design the sequence of one or more strands intended to adopt a target secondary
structure at equilibrium ..................................... 17
References 20
1

1 Overview
NUPACK is a growing software suite for the analysis and design of nucleic acid systems. The package cur-
rently enables thermodynamic analysis of dilute solutions of interacting nucleic acid strands, and sequence
design for complexes of nucleic acid strands intended to adopt a target secondary structure at equilibrium.
NUPACK algorithms are formulated in terms of nucleic acid secondary structure. In most cases, pseudo-
knots are excluded from the structural ensemble. Much of this software may be conveniently run through
the NUPACK web server at http://www.nupack.org (Zadeh et al., 2010b).
Please direct questions, concerns, comments, and bug reports to support@nupack.org.
1.1 Directories
After compiling, the root directory nupack contains a Makefile and the following sub-directories:
src
Source code.
bin
Executables.
lib
Static libraries defining functions for linking at compile time.
parameters
Parameter files for RNA and DNA free energy models.
doc
Documentation, including this User Guide and example input and output files.
It is convenient to set the environment variable NUPACKHOME, specifying an absolute path to the root direc-
tory nupack (e.g., NUPACKHOME=/usr/local/nupack or NUPACKHOME=/home/username/nupack).
1.2 Compilation
Core routines are written in C. To compile the code, type make in the root nupack directory. This will
create the executables described in this user guide.
1.3 Example job files
While using this user guide, it may be useful to refer to the examples in the doc/examples directory. It
contains many sample input and output files. The README file in that directory gives detailed information
about its contents.
1.4 Notation and terminology
To facilitate the precise description of certain quantities without including lengthy explanations in the
NUPACK User Guide, notation and technical terms are drawn from (Dirks et al., 2007).
2

1.5 Secondary structure model
The secondary structure of multiple interacting strands is defined by a list of base pairs (Dirks et al., 2007).
Apolymer graph for a secondary structure can be constructed by ordering the strands around a circle,
drawing the backbones in succession from 50to 30around the circumference with a nick between each
strand, and drawing straight lines connecting paired bases. A secondary structure is pseudoknotted if every
strand ordering corresponds to a polymer graph with crossing lines. A secondary structure is connected if
no subset of the strands is free of the others. Algorithms are formulated in terms of ordered complexes,
each corresponding to the structural ensemble, Ω(π), of all connected polymer graphs with no crossing
lines for a particular ordering, π, of a set of strands. The free energy of an unpseudoknotted secondary
structure is calculated using nearest-neighbor empirical parameters for RNA in 1M Na+(Serra & Turner,
1995;Mathews et al., 1999) or for DNA in user-specified Na+and Mg++ concentrations (SantaLucia, 1998;
SantaLucia & Hicks, 2004;Koehler & Peyret, 2005); additional parameters are employed for the analysis of
pseudoknots (single RNA strands only) (Dirks & Pierce, 2003;Dirks & Pierce, 2004). NUPACK calculates
free energies and equilibrium concentrations of ordered complexes as described in (Dirks et al., 2007) (in
particular, see endnote 13 regarding strand association penalties).
1.6 Conventions
The following formatting standards apply to all NUPACK input and output:
•All executables except concentrations,distributions and design take input from an
input file prefix.in, where prefix is a command line argument. If prefix is not specified or
prefix.in is absent or improperly formatted, the user is prompted for input on the screen.
•All sequences are listed 50to 30. The bases in an ordered complex are indexed starting with 1 at the
50-most base of the first strand and ending at the 30-most base of the last strand. For example, if an
ordered complex has three strands of length 15, 20, and 13, respectively, the fifth base of the third
strand has index 40.
•Valid bases are A,C,G,T, and U. For RNA calculations, Tis automatically converted to U, and vice
versa for DNA calculations.
•Secondary structures may be specified in dot-parens-plus notation (each unpaired base is represented
by a dot, each base pair by matching parentheses, and each nick between strands by a plus). For
example, ((...)) specifies that bases 1 and 2 are paired to bases 7 and 6, respectively, while bases
3, 4, and 5 are unpaired. ((+...)) specifies that bases 1 and 2 of strand 1 are paired to bases 5
and 4 of strand 2. Four types of “parentheses” are accepted: (),[],{}, and <>. Within a specified
structure, each type of parentheses must satisfy a nesting property but different types need not be
nested, allowing specification of pseudoknotted structures (though highly nested pseudoknots may
not be specifiable with only four types of parentheses).
•Secondary structures may also be specified in pair list format, where each line consists of two
whitespace-separated integers [i j], i < j, specifying that base iis paired to base j. Any secondary
structure, including highly-nested pseudoknots, may be specified in this way.
•Comment lines begin with a %symbol.
•In input files, comment lines may be interspersed with input data. However, blank lines are not
permitted in input files.
The following physical considerations are universal throughout NUPACK:
3
•Except where noted, all energy units are kcal/mol and all concentration units are molar.
•The zero free energy reference state for all calculations is a system where all relevant strands are
present with no base pairs.
•The base pairs considered in the calculations include Watson-Crick (A·U/Tand G·C) and wobble
(G·U/T) pairs.
•Except where noted, results appropriately reflect distinguishability corrections that arise in the multi-
stranded setting (Dirks et al., 2007).
The following option flags are recognized by multiple NUPACK executables:
-material parameters
The parameter files defining the nucleic acid material are specified via the argument parameters
which represents either a filename prefix or a shorthand identifier for an included parameter set. If the
filename does not contain a relative or absolute path, then the program will look for the files first in
the current directory, and then in the directory $NUPACKHOME/parameters. Available filename
prefixes currently include:
•rna1995 (default; shorthand: rna)
Parameter files *.dG and *.dH for RNA allowing calculations at different temperatures (Serra
& Turner, 1995); includes pseudoknot parameters from (Dirks & Pierce, 2003).
•dna1998 (shorthand: dna)
Parameter files *.dG and *.dH for DNA allowing calculations at different temperatures (San-
taLucia, 1998); there are no pseudoknot parameters.
•rna1999
Parameter file *.dG for RNA for calculations at 37 ◦C (Mathews et al., 1999); includes pseu-
doknot parameters from (Dirks & Pierce, 2003).
DNA/RNA hybrids are not allowed.
-sodium concentration
The Na+concentration of the solution in units of molar (default is 1.0) is specified by concentration.
This flag is only valid when the -material dna is also selected because no RNA salt correction
parameters are available. Otherwise, [Na+] = 1.0 M by default.
-magnesium concentration
The Mg2+ concentration of the solution in units of molar (default is 0.0) is specified by concentration.
This flag is only valid when the -material dna is also selected. Otherwise, no magnesium is
present by default.
-dangles treatment
The way in which dangle energies are incorporated is specified by treatment, which may have the
following values:
none: No dangle energies are incorporated.
some: (default) A dangle energy is incorporated for each unpaired base flanking a duplex (a base flank-
ing two duplexes contributes only the minimum of the two possible dangle energies).
all: A dangle energy is incorporated for each base flanking a duplex regardless of whether it is paired.
4

-T temperature
Temperature specified in ◦C (default is 37).
-multi
Specify a calculation involving complexes of multiple interacting strands.
-pseudo
Augment the structural ensemble of ordered complex Ω(π)with the class of pseudoknots defined
in (Dirks & Pierce, 2003). This option is currently only available for single-stranded RNA calcula-
tions. An error message is returned if -pseudo is specified in combination with either -multi or
-material dna.
5
1.7 License
NUPACK Software License Agreement
Copyright c
2015. California Institute of Technology. All rights reserved.
Use and redistribution in source form and/or binary form, with or without modification, are permitted for
non-commercial academic purposes only, provided that the following conditions are met:
1. Redistributions in source form must retain the above copyright notice, this list of conditions and the
following disclaimer.
2. Redistributions in binary form must reproduce the above copyright notice, this list of conditions and
the following disclaimer in the documentation provided with the distribution.
3. Web applications that use the software in source form or binary form must reproduce the above copy-
right notice, this list of conditions and the following disclaimer in online documentation provided with
the web application.
4. Neither the name of the copyright holder nor the names of its contributors may be used to endorse or
promote derivative works without specific prior written permission.
Disclaimer
This software is provided by the copyright holders and contributors “as is” and any express or implied war-
ranties, including, but not limited to, the implied warranties of merchantability and fitness for a particular
purpose are disclaimed. In no event shall the copyright holder or contributors be liable for any direct, in-
direct, incidental, special, exemplary, or consequential damages (including, but not limited to, procurement
of substitute goods or services; loss of use, data, or profits; or business interruption) however caused and
on any theory of liability, whether in contract, strict liability, or tort (including negligence or otherwise)
arising in any way out of the use of this software, even if advised of the possibility of such damage.
6

2 Executables
2.1 pfunc: calculate the partition function
Command: pfunc [-T temperature] [-multi] [-pseudo] [-material parameters]
[-dangles treatment] prefix
Description: Computes the partition function, Q, for an ordered complex over the set Ω(π).
Input: For single-stranded calculations, the input file contains the strand sequence specified on a single line.
If -multi is specified, the input file must contain the following entries on separate lines:
•The number of distinct strand species, |Ψ0|.
•The sequences for each distinct strand species, each on a separate line.
•Lintegers from the range 1 to |Ψ0|representing the distinct circular permutation π∈Πof the L
strands in the ordered complex.
Note that strand species defined on different lines are treated as distinct even if they have the same sequence.
Example 1
Partition function for a single RNA strand at 37◦C including pseudoknots.
Input file contents:
GGGCUGUUUUUCUCGCUGACUUUCAGCCCCAAACAAAAAAUGUCAGCA
Command: pfunc -pseudo
$NUPACKHOME/doc/examples/jcc04 telomerase/jcc04 telomerase
Example 2
Partition function for an ordered complex of four DNA strands at 23◦C, two of which are indistinguishable.
Input file contents:
3
AGTCTAGGATTCGGCGTGGGTTAA
TTAACCCACGCCGAATCCTAGACTCAAAGTAGTCTAGGATTCGGCGTG
AGTCTAGGATTCGGCGTGGGTTAACACGCCGAATCCTAGACTACTTTG
1223
Command: pfunc -T 23 -multi -material dna
$NUPACKHOME/doc/examples/pnas04 hcr/pnas04 hcr basic
Output:
Following header comments, the free energy of the ordered complex (given by ∆G=−kT log Q) is written
to the screen. The value of the partition function is written immediately below.
2.2 pairs: calculate base-pairing observables
Command: pairs [-T temperature] [-multi] [-pseudo] [-material parameters]
[-dangles treatment] [-cutoff cutoffvalue] prefix
7
Description: Computes pair probabilities p(in·jm;π)for the ordered complex corresponding to the spec-
ified circular permutation π∈Π. When -multi is selected, also computes the expected number of base
pairs E(i{A}·j{B};π).
Additional option:
-cutoff cutoffvalue
Only probabilities and expected values at or above cutoffvalue (default is 0.001) are saved in the
output file(s).
Input: Same as for the executable pfunc.
Output: The output is written to the files:
•prefix.ppairs
Contains the probability of each type of base pair in the ordered complex. The relevant quantities are
p(in·jm;π), the probability that base iof strand nis paired to base jof strand min the ordered
complex corresponding to distinct circular permutation π. All strands in the ordered complex are
considered to be distinct; there are no distinguishability corrections. For example, the two strands
labeled 2 in Example 2 are considered distinct. One might think of them as strand 2aand 2b, and
a given base of strand 2amay have different pair probabilities than the corresponding one in strand
2b. The total number of bases in the complex is N=PL
l=1 Nl, so indexing bases from 1 to N, the
pair probabilities can be stored in a symmetric N×Nmatrix. Augmentation by an N+ 1st column
containing the probability that each base is unpaired causes the rows to sum to unity.
By default, the file is formatted as follows. Following header comments, the first entry is the integer
N. The remaining entries come in triplets of the form [i j p], where 1≤i≤Nand 1≤j≤N+ 1
are base numbers and pis the probability of the corresponding pair. Values corresponding to j=N+1
represent the probability that base iis unpaired. If -pseudo is selected, each row is augmented by
two additional columns. The first is the probability that bases iand jform a nested pair and the second
is the probability that bases iand jform a non-nested pair. In the case of j=N+ 1, these additional
columns store the probability that bases iand jdo not form a nested pair and the probability that they
do not form a non-nested pair, respectively.
•prefix.epairs
Generated when -multi is selected. Similar to prefix.ppairs except strands of the same
species are considered to be indistinguishable. The relevant quantities are E(i{A}·j{B};π), the
expected number of base iof strand species Athat are paired to base jof strand species Bin the
ordered complex corresponding to distinct circular permutation π. The number of distinct bases in the
complex is Ndistinct ≡Pk∈Ψ0Nk, representing the total number of bases in all |Ψ0|strand species.
Numbering the distinct bases from 1 to Ndistinct, the distinct base pairs may be represented as a sym-
metric Ndistinct ×Ndistinct matrix; by augmenting the matrix with an extra column that contains the
expected number of base iof strand species Athat are unpaired, each row sums to the number of base
iof strand species Ain the complex. Note that this numbering system is used even if some sequences
listed in the input file are absent from the specified ordered complex.
The file is formatted as follows. Following header comments, the first entry is the integer Ndistinct, and
the remaining entries come in triplets of the form [i j E], analogously to the .ppairs file, except
Eis the expected number of the corresponding pair. Information is stored only for bases included in
the specified ordered complex.
8

2.3 mfe: find the minimum free energy (MFE) secondary structure(s)
Command: mfe [-T temperature] [-multi] [-pseudo] [-material parameters]
[-dangles treatment] [-degenerate] prefix
Description: Compute and store the minimum free energy and MFE secondary structure(s) in Ω(π). If
the -degenerate flag is selected, all secondary structures that share the same minimum free energy are
stored; otherwise only one MFE structure is stored.
Input: Same format as for the executable pfunc.
Output: Output is written to the file prefix.mfe. After header comments, each entry describes one
of the possibly many degenerate MFE structures. The entries are separated by comment lines (repeated %
signs). The first line in each entry is the number of bases in the ordered complex. The second line is the
minimum free energy. The third line is the dot/parentheses depiction of the MFE structure. Subsequent lines
contain the MFE structure in pair list notation.
2.4 subopt: find all secondary structures within a specified free energy gap of the MFE
Command: subopt [-T temperature] [-multi] [-pseudo] [-material parameters]
[-dangles treatment] prefix
Description: Similar to mfe except that all secondary structures in Ω(π)with free energies within the spec-
ified (non-negative) free energy gap of the MFE are calculated and stored. This can be very slow and the
output very large if the specified gap is too large. The output is sorted by increasing free energy.
Input: Same format as for the executable pfunc, plus one additional row containing the energy gap.
Output: Output is written to the file prefix.subopt with the same format as for the executable mfe.
2.5 count: count the number of secondary structures in the ensemble
Command: count [-multi] [-pseudo] prefix
Description: Similar to pfunc but sets all energy parameters to zero, thereby giving a count of the number
of secondary structures in Ω(π). Note that this ensemble over-counts rotationally symmetric structures.
Input: Same format as for the executable pfunc.
Output: The number of secondary structures in Ω(π)is written to the screen, preceded by header comments.
2.6 energy: calculate the free energy of a secondary structure
Command: energy [-T temperature] [-pseudo] [-multi] [-material parameters]
[-dangles treatment] prefix
Description: Calculate the free energy of a given sequence and secondary structure.
9

Input: Same format as for executable pfunc, plus one additional row specifying the secondary structure
in dot/parentheses notation. Alternatively, the structure may be represented in pair list notation.
Output: The free energy of the structure is written to the screen, preceded by header comments.
2.7 prob: calculate the equilibrium probability of a secondary structure
Command: prob [-T temperature] [-pseudo] [-multi] [-material parameters]
[-dangles treatment] prefix
Description: Calculates the equilibrium probability of a given secondary structure.
Input: Same format as for the executable energy.
Output: The equilibrium probability of the given structure is written to the screen, preceded by header
comments.
2.8 defect: calculate the ensemble defect
Command: defect [-T temperature] [-pseudo] [-multi] [-material parameters]
[-dangles treatment] [-mfe] prefix
Description: Calculate the ensemble defect, n(φ, s), for a sequence φ, and secondary structure s, defined
as the average number of incorrectly paired nucleotides at equilibrium evaluated over the ensemble of the
ordered complex (Dirks et al., 2004;Zadeh et al., 2010a).
Additional option:
-mfe
Calculate the MFE defect (Zadeh et al., 2010a) instead of the ensemble defect.
Input: Same format as for the executable energy.
Output: Following header comments, the ensemble defect, n(φ, s), and the normalized ensemble defect,
n(φ, s)/|s|, are written to the screen. If the -mfe flag is selected, the MFE defect ( µ(φ, s)) and the
normalized MFE defect (µ(φ, s)/|s|) are written to the screen.
2.9 complexes: calculate the partition functions of all strand complexes up to a specified size
Command: complexes [-T temperature] [-material parameters] [-ordered]
[-pairs] [-mfe] [-degenerate] [-dangles treatment] [-timeonly] [-quiet]
prefix
Description: First calculates the identities of all distinct circular permutations π∈Πof strands for all
possible (unpseudoknotted) complexes up to a user-defined size Lmax and then calculates their respective
partition functions Q(π)(Dirks et al., 2007). The partition function for a complex, Q, is obtained by sum-
ming over the partition functions of its constituent ordered complexes, Q(π)for all π∈Π. Significant
additional functionality can be specified via command line flags. The output of complexes can be used as
the input to the executables concentrations and distributions.
10

Additional options:
-ordered
Also store properties for ordered complexes, each corresponding to one distinct circular permutation
π∈Π, in addition to the summed properties for complexes.
-pairs
Calculate base-pairing observables as for the pairs executable.
-cutoff cutoffvalue
Only probabilities and expected values at or above cutoffvalue (default is 0.001) are saved in the
output file(s) generated when the -pairs flag is selected.
-mfe
Calculate all minimum free energy structures for each ordered complex as for the mfe executable.
Must be used in conjunction with the -ordered flag. The -degenerate flag is only applicable in
conjunction with the -mfe flag.
-timeonly
After generating all distinct circular permutations, estimate the time it would take to compute all of
the partition functions. The partition function calculations are not performed, the time estimate is
written to the screen, and no output files are generated.
-quiet
Suppress output to the screen.
Input: The input file must contain the following entries on separate lines:
•The number of distinct strand species (|Ψ0|).
•Sequence for each distinct strand species (each on a separate line).
•Maximum complex size (Lmax).
In addition to considering all complexes up to a specified maximum number of strands Lmax, the optional
file prefix.list can be used to manually specify ordered complexes with more than Lmax strands. Each
ordered complex of size L > Lmax is specified on a separate line by:
•A list of Lintegers from the range 1 to |Ψ0|representing the distinct circular permutation π∈Πof
the Lstrands in the ordered complex.
11

Example 3
For a system containing three DNA strand species at 23◦C, calculate partition functions, pair probabilities,
and MFE structures for all ordered complexes of up to four strands, and for additional larger complexes
specified in a .list file.
Input file contents:
3
AGTCTAGGATTCGGCGTGGGTTAA
TTAACCCACGCCGAATCCTAGACTCAAAGTAGTCTAGGATTCGGCGTG
AGTCTAGGATTCGGCGTGGGTTAACACGCCGAATCCTAGACTACTTTG
4
List file contents:
12233
12323
23232
122233
Command: complexes -T 23 -material dna -ordered -pairs -mfe
$NUPACKHOME/doc/examples/pnas04 hcr/pnas04 hcr
Output: Unless the -quiet flag is selected, complexes reports progress to the screen. By default there
is one output file:
•prefix.cx
Contains the composition and free energy of each complex. The first column is an integer complex
identifier, and the next |Ψ0|+ 1 columns are L1L2. . . L|Ψ0|∆G.
Depending on the command line options, the following output files may also be written:
•prefix.ocx
Generated if -ordered is selected. Contains the composition and free energy of each ordered com-
plex. The first and second columns are integer complex and ordered complex identifiers, respectively,
and the remaining |Ψ0|+ 1 columns are L1L2. . . L|Ψ0|∆Gfor the ordered complex.
•prefix.ocx-key
Generated if -ordered is selected. Contains the distinct circular permutation of strands for each
ordered complex. The first and second columns are integer complex and ordered complex identifiers,
respectively, and the remaining Lcolumns are integers from the range 1 to |Ψ0|. Note that the value
of Lmay be different for each complex.
•prefix.cx-epairs
Generated if -pairs is selected. Contains the base-pairing expectation values for each type of
distinct base pair in each complex. The relevant quantities are E(i{A}·j{B}), the expected number
of base iof strand species Athat are paired to base jof strand species Bin the complex. The file
contains a list of entries each with the same format as the .epairs file generated by the executable
pairs. Each entry is separated by comment lines (repeated %symbols). Additionally, each entry
begins with a comment line containing the complex identifier id, expressed as “% complexid”.
•prefix.ocx-epairs
Generated if -pairs and -ordered are selected. Similar to .cx-epairs but for ordered com-
plexes. The relevant quantities are E(i{A}·j{B};π), the expected number of base iof strand species
12
Athat are paired to base jof strand species Bin the ordered complex corresponding to distinct cir-
cular permutation π. The entries are separated by comment lines (repeated %symbols), and each
entry begins with a comment line containing the complex identifier id and order identifier iorder,
expressed as “% complexid-orderiorder”.
•prefix.ocx-ppairs
Generated if -pairs and -ordered are selected. Similar to .ocx-epairs except that all strands
in the ordered complex are assumed to be distinct. The data in each entry are the same as those in the
.ppairs file produced by the executable pairs.
•prefix.ocx-mfe
Generated if -mfe and -ordered are selected. Contains the minimum free energy and MFE struc-
ture(s) for each ordered complex. Each entry is formatted the same as the output for the mfe ex-
ecutable. The entries are separated by comment lines (repeated %symbols), and each entry begins
with a comment line containing the complex identifier id and order identifier iorder, expressed
as “% complexid-orderiorder”. If the -degenerate flag is selected, the degenerate MFE
structures for a given entry are separated by a comment line of repeated %symbols.
2.10 concentrations: calculate the equilibrium concentration of each complex in a dilute solu-
tion
Command: concentrations [-ordered] [-pairs] [-sort method] [-quiet] prefix
Description: Given user-defined concentrations for each strand species, calculates the equilibrium concen-
tration of each complex species or base pair in a large dilute solution, typical of experimental conditions in
a test tube (Dirks et al., 2007). Partition function information is read in from output files generated with the
executable complexes.
Additional options:
-ordered
Performs the calculation on ordered complexes rather than complexes. The input is read from the
prefix.ocx (output from the executable complexes).
-pairs
Compute base-pairing information for the entire solution using results from prefix.cx-epairs
or, if -ordered is selected, prefix.ocx-epairs, as output by the executable complexes.
-cutoff cutoffvalue
Only ensemble pair fractions at or above cutoffvalue (default is 0.001) are saved in the output file
prefix.fpairs generated when the -pairs flag is selected. Note that cutoffvalue should
not be less than that used with complexes to generate the input files.
-sort method
The argument method is one of the following integers:
0: Output is listed in the same order as in the input file.
1: (default) Output is sorted by the concentration of each complex (default) or ordered complex (if
-ordered is selected).
13
2: Output is sorted first by the concentration of each complex and then, if -ordered is selected,
by the concentration of each constituent ordered complex.
3: Output is sorted first by the integer complex identifier and then, if -ordered is selected, by
the ordered complex identifier.
4: Output is sorted first by the number of strands in each complex, then by the integers L1L2. . . L|Ψ0|
defining the number of each strand type in a given complex (with L1having the highest prece-
dence, followed by L2, and so on), and finally, if -ordered is selected, by the integer ordered
complex identifier.
-quiet
Suppress output to the screen.
Input: By default, input is read from the file prefix.cx. Alternatively, specifying -ordered causes in-
put to be read from prefix.ocx. These files are formatted as for the output of the executable complexes.
The temperature at which the calculation is done is read from a line in the comments of the .cx or .ocx
input file that reads “% T = temperature”, where temperature is the temperature in ◦C. This line
is automatically included in all output files of the executable complexes.
The input file prefix.con specifies the total molar concentration of each of |Ψ0|strand species on a
separate line. The concentration may be in scientific notation (e.g., 1e-6 for a strand species at µM con-
centration).
Output: Unless -quiet is selected, the following information is written to the screen:
•The error in conservation of mass for each strand species in molar.
•The free energy of the entire solution in kcal/L.
•The wall clock time for the calculation.
The output is written to the files:
•prefix.eq
The content is the same as the input file (except resorted, depending on the -sort option) with
an extra column containing the concentration of the species in molar inserted after the free energy
column.
•prefix.fpairs
Generated if -pairs is selected. Reports the fraction of each distinct base that is paired to each
of the other distinct bases in solution. The relevant quantity is fA(iA·jB), the expected fraction of
strands of species Afor which base iis paired to base jof strand species B. The number of distinct
bases in the dilute solution is Ndistinct ≡P|Ψ0|
k=1 Nk, representing the total number of bases in all |Ψ0|
strand species. Numbering the distinct bases from 1 to Ndistinct, the quantity fA(iA·jB)may be
stored as an (asymmetric) Ndistinct ×Ndistinct matrix; by augmenting the matrix with an extra column
that contains the expected fraction of base iof strand species Athat are unpaired, each row sums to
unity.
The file is formatted as follows. Following header comments, the first entry is the integer Ndistinct.
The remaining entries come in triplets of the form [i j f], where 1≤i≤Ndistinct and 1≤j≤
Ndistinct + 1 are base numbers and fis the corresponding fraction from the augmented matrix.
14
Example 4
A cautionary tale. NUPACK calculates free energies and equilibrium concentrations of ordered complexes
as described in (Dirks et al., 2007) (in particular, if you plan to calculate equilibrium concentrations by hand,
see endnote 13 regarding strand association penalties). For example, the following holds at equilibrium for
a dilute solution containing strands A and B that can interact to form ordered complex AB:
xAB
xAxB
= exp −∆GAB −∆GA−∆GB
kT ,
=[AB]/ρH2O
([A]/ρH2O) ([B]/ρH2O),
=[AB] ρH2O
[A][B] ,
where for each ordered complex, i,xiis the mole fraction,[i]is the concentration (e.g. in units of mol/L),
∆Giis the free energy as reported by NUPACK, and ρH2O(≈55.14 mol/L at 37◦C) is the concentration of
water.
Consider duplex formation for two RNA strands, A = GCGCG and B = CGCGC, present at concentrations of
[A]0and [B]0, respectively, in 1 M Na+at 37◦C. The free energies given by NUPACK are
∆GA= 0.00 kcal/mol,∆GB= 0.00 kcal/mol,∆GAB =−9.62 kcal/mol.
If only these three ordered complexes are considered, the concentration of AB is determined by finding the
appropriate root of
[AB] ρH2O
([A]0−[AB]) ([B]0−[AB]) = exp −∆GAB −∆GA−∆GB
kT .
For [A]0= [B]0= 1 µM, we get
[A] = [B] = 0.91µM,[AB] = 0.09µM.
A common mistake is to forget to include the ρH2Oin the calculation. Doing so would give the erroneous
result of [AB] = 0.67 µM. It is important to remember that
exp −∆GAB −∆GA−∆GB
kT 6=[AB]
[A] [B] !
Finally, note that we have artificially stipulated that only three ordered complexes are allowed. However, the
sequences of A and B are such that they may form homodimers. If we consider this possibility, we are left
with a system of coupled nonlinear algebraic equations that are difficult to solve. NUPACK performs such
calculations, and the resulting concentrations are
[A] = 0.686 µM,[B] = 0.925 µM,[AB] = 0.069 µM,[AA] = 0.123 µM,[BB] = 0.003 µM,
significantly different from what we calculated neglecting the other ordered complexes. Therefore, one must
exercise caution when applying ordered complex free energies to determination of equilibrium concentra-
tions. It is best to directly use the concentrations executable or the NUPACK web server for these
calculations.
15

2.11 distributions: calculate the equilibrium population distribution and expected value for a
few complexes in a dilute solution
Command: distributions [-ordered] [-maxstates big] [-writestates]
[-sort method] [-quiet] prefix
Description: The executable distributions calculates the partition function Qbox for a box contain-
ing a small number of strands, given user-defined populations for each strand species (Dirks et al., 2007).
This is used to calculate the expected value and probability distribution of the population of each species of
complex (or ordered complex). Partition function information is read from output files generated with the
executable complexes.
Additional options:
-ordered
Performs the calculation on ordered complexes rather than complexes. The input is read from the
prefix.ocx (output from the executable complexes).
-maxstates big
The maximum number of states of the box to be enumerated (|Λ|, default is 1e7). A segmentation
fault will occur if the stack size on your machine is exceeded.
-writestates
Write a (typically large) output file describing properties for all population states of the system.
-sort method
The argument method is one of the following integers:
1: (default) Output is sorted by the expected value of the population of each complex or ordered
complex (if -ordered is selected).
2: Output is sorted first by the expected value of the population of each complex and then, if
-ordered is selected, by the expected value of the population of each constituent ordered
complex.
3: Output is sorted first by the integer complex identifier and then, if -ordered is selected, by
the ordered complex identifier.
4: Output is sorted first by the number of strands in each complex, then by the integers L1L2. . . L|Ψ0|
defining the number of each strand type in a given complex (with L1having the highest prece-
dence, followed by L2, and so on), and finally, if -ordered is selected, by the integer ordered
complex identifier.
-quiet
Suppress output to the screen.
Input: Same as for the executable concentrations, except the file prefix.con file is replaced by
prefix.count, which contains the integer population of each of |Ψ0|strand species on a separate line.
The last line of the file contains the volume of the box in liters. This may be entered in scientific notation
(e.g., 1.4e-18).
Output: Unless the -quiet flag is selected, the following information is written to the screen:
•The number of states of the box.
16
•The free energy of the entire box in units of kT and in units of kcal.
•The wall clock time for the calculation.
The output is written to the files:
•prefix.dist
The content is the same as the input file (with rows sorted according to -sort) with extra columns af-
ter the free energy column. The first extra column (for complex or ordered complex j) is the expected
value of the population hmji. Subsequent columns are [pj(0) pj(1) . . . pj(max(m0))]. These
represent the probability that complex (or ordered complex) jhas population 0, 1, . . . ,max(m0), at
equilibrium.
•prefix.states
Generated when -writestates is selected. Each row corresponds to a population vector, m, for
the box. The first column is the probability that the population vector occurs at equilibrium. By de-
fault, the remaining entries come in pairs: an integer complex identifier and a nonzero population.
If -ordered is selected, the remaining entries come in triples: integer complex and ordered com-
plex identifiers and then a nonzero population. This pattern continues for all complexes (or ordered
complexes) with non-zero populations.
2.12 design: design the sequence of one or more strands intended to adopt a target secondary
structure at equilibrium
Command: design [-init initmode] [-loadinit] [-outputinit]
[-loadseed] [-outputseed] [-fstop fstopvalue] [-prevent file]
[-mleafopt mleafoptvalue] [-mreopt mreoptvalue] [-pairs]
[-cutoff cutoffvalue] prefix
Description: The executable design designs the sequence of one or more interacting nucleic acid strands
intended to adopt a target secondary structure at equilibrium (Zadeh et al., 2010a). Sequence design is for-
mulated as an optimization problem with the goal of reducing the ensemble defect below a user-specified
stop condition. To reduce the computational cost of accepting or rejecting mutations to a random initial se-
quence, candidate mutations are evaluated on the leaf nodes of a tree-decomposition of the target structure.
During leaf optimization, defect-weighted mutation sampling is used to select each candidate mutation posi-
tion with probability proportional to its contribution to the ensemble defect of the leaf. As subsequences are
merged moving up the tree, emergent structural defects resulting from crosstalk between sibling sequences
are eliminated via reoptimization within the defective subtree starting from new random subsequences.
Additional options:
-init initmode
The argument initmode selects the sequence initialization method from one of the following (using
a sequence that satisfies the base-pairing requirements of the target secondary structure with Watson-
Crick pairs):
AU: Initial sequences are randomly selected from Aand T/U bases only.
CG: Initial sequences are randomly selected Cand Gbases only.
RND: (default) Initial sequences are randomly selected from A,C,G,T/U.
17
SSM: Initial sequences are generated using sequence symmetry minimization (Seeman, 1982;Dirks
et al., 2004).
-loadinit
Initialize the sequence from the file prefix.init.
-outputinit
Output the initial sequence to prefix.init.
-loadseed
Initialize the random number generator with the seed specified in prefix.seed. This can be used
to duplicate design execution.
-outputseed
Output the random number generator’s seed to prefix.seed.
-fstop fstopvalue
Set the stop condition for the design algorithm to fstopvalue (default is 0.01). The design algo-
rithm seeks to achieve n(φ, s)≤fstopvalue |s|.
-prevent preventfile
The file preventfile contains patterns to be prevented from appearing in the sequence design.
-mleafopt mleafoptvalue
Leaf optimization is restarted from new initial conditions up to leafoptvalue times (default 3)
before terminating unsuccessfully (Zadeh et al., 2010a).
-mreopt mreoptvalue
The elimination of emergent defects in a parent node by defect-weighted child sampling and reopti-
mization is attempted up to mreoptvalue times (default is 10) (Zadeh et al., 2010a).
-pairs
Save the pair probabilities in a .ppairs file.
-cutoff cutoffvalue
Only probabilities at or above cutoffvalue are saved in the .ppairs file (default is 0.001).
Input: The target structure and sequence constraints are read from prefix.fold. The first line of the
file is the target structure in dot-parens-plus notation. The second line of the file contains the sequence con-
straints (if any) specified using the standard nucleic acid codes. If no sequence constraints are specified for
a given base, it is assumed to be unconstrained.
Optional inputs are specified in the following files:
prefix.init: Used when -loadinit is specified. The first line in the file is the initial sequence.
prefix.seed: Used when -loadseed is specified. The first line is an integer random seed for the
design algorithm (unique seeds are in the range [0,232 −1]).
preventfile: Specifies patterns to be prevented from appearing in the designed sequences. The file
must contain one pattern per line using standard nucleic acid codes. No design will be produced if the
sequence constraints cannot be satisfied. Sample prevented patterns: AAAA, CCCC, GGGG, UUUU,
KKKKKK, MMMMMM, RRRRRR, SSSSSS, WWWWWW, YYYYYY.
18
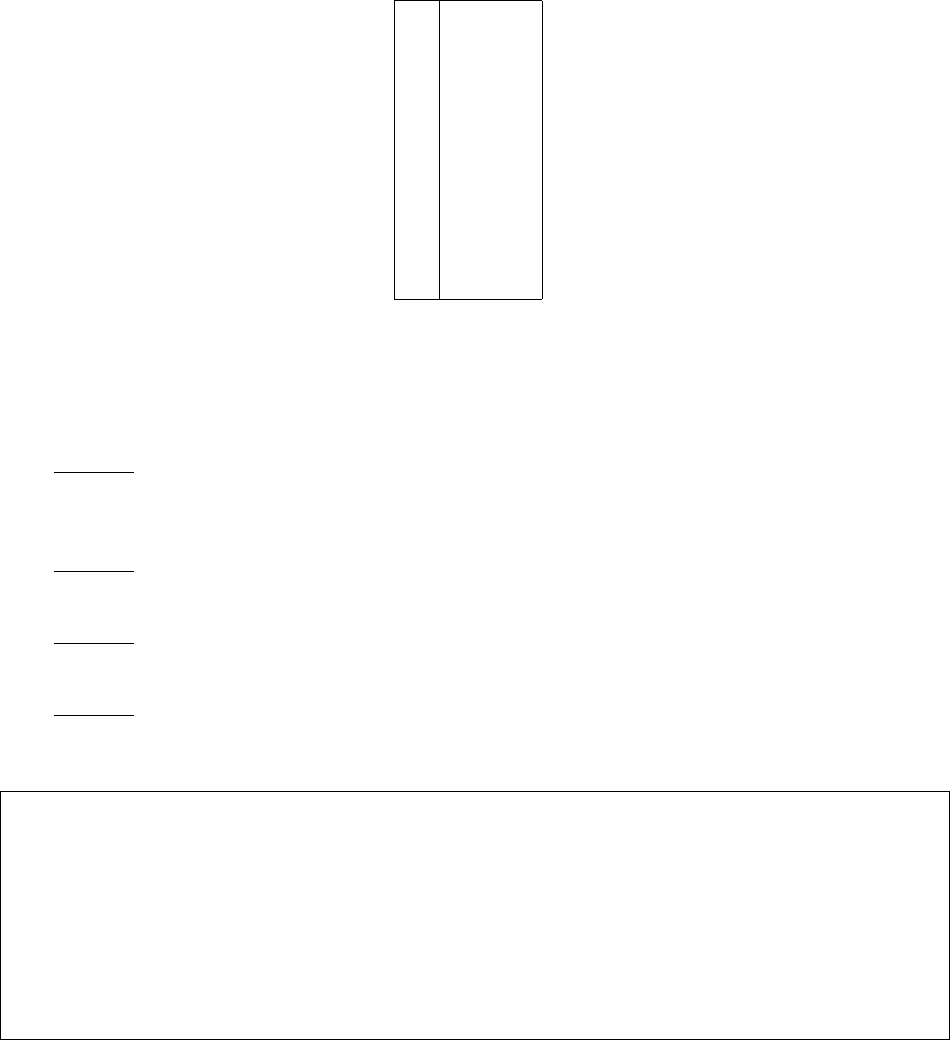
NA,C,G,U
RA,G
YC,U
MA,C
KG,U
SC,G
WA,U
VA,C,G
HA,C,U
BC,G,U
DA,G,U
Table 1. Standard nucleic acid codes for specifying sequence constraints and pattern prevention.
Output: Output is written to the files:
•prefix.summary
The header of this file includes comments about thermodynamic and design parameters used in the
design process. The first line below the header contains the strand sequences separated by +symbols.
•prefix.init
Generated if -outputinit is specified. It contains the initial sequence on the first line of the file.
•prefix.seed
Generated if -outputseed is specified. It contains the random seed on the first line of the file.
•prefix.ppairs
Generated if -pairs is specified. This file specifies the base pairing probabilities for the ordered
complex in the same format as the file generated by the executable pairs.
Example 5
Design of a short DNA duplex with a GC toehold.
Input file contents:
....((((((((((+))))))))))
SSSSNNNNNNNNNN
Command: design -material dna -pairs example
$NUPACKHOME/doc/examples/design/duplex
19
References
Dirks, R.M., & Pierce, N.A. 2003. A partition function algorithm for nucleic acid secondary structure
including pseudoknots. J Comput Chem,24, 1664–1677. (pdf)
Dirks, R.M., & Pierce, N.A. 2004. An algorithm for computing nucleic acid base-pairing probabilities
including pseudoknots. J Comput Chem,25, 1295–1304. (pdf)
Dirks, R.M., Lin, M., Winfree, E., & Pierce, N.A. 2004. Paradigms for computational nucleic acid design.
Nucleic Acids Res,32(4), 1392–1403. (pdf)
Dirks, R.M., Bois, J.S., Schaeffer, J.M., Winfree, E., & Pierce, N.A. 2007. Thermodynamic analysis of
interacting nucleic acid strands. SIAM Rev,49(1), 65–88. (pdf)
Koehler, R.T., & Peyret, N. 2005. Thermodynamic properties of DNA sequences: characteristic values for
the human genome. Bioinformatics,21(16), 3333–3339.
Mathews, D.H., Sabina, J., Zuker, M., & Turner, D.H. 1999. Expanded sequence dependence of thermody-
namic parameters improves prediction of RNA secondary structure. J Mol Biol,288, 911–940.
SantaLucia, Jr., J. 1998. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor
thermodynamics. Proc Natl Acad Sci USA,95(4), 1460–1465.
SantaLucia, Jr., J., & Hicks, D. 2004. The thermodynamics of DNA structural motifs. Annu Rev Bioph Biom
Struct,33, 415–440.
Seeman, N.C. 1982. Nucleic acid junctions and lattices. J Theor Biol,99, 237–247.
Serra, M.J., & Turner, D.H. 1995. Predicting thermodynamic properties of RNA. Methods Enzymol,259,
242–261.
Zadeh, J.N., Wolfe, B.R., & Pierce, N.A. Nucleic acid sequence design via efficient ensemble defect opti-
mization. J Comput Chem. Published online 17 August, 2010, DOI 10.1002/jcc.21633. (pdf)
Zadeh, J.N., Steenberg, C.D., Bois, J.S., Wolfe, B.R., Khan, A.R., Pierce, M.B., Dirks, R.M., & Pierce,
N.A. NUPACK: analysis and design of nucleic acid systems. J Comput Chem. Published online 19
July, 2010, DOI 10.1002/jcc.21596. (pdf)
20
