Phonon Relaxation Manual
User Manual: Pdf
Open the PDF directly: View PDF ![]() .
.
Page Count: 34
- First-principles calculation of phonon-induced electron-hole relaxation dynamics for finite systems within linear response theory: Manual and Example
First-principles calculation of phonon-induced electron-hole
relaxation dynamics for finite systems within linear response
theory: Manual and Example
Jonathan Trinastic - jptrinastic@gmail.com
Department of Physics and Quantum Theory Project,
University of Florida, Gainesville, Florida, 32611, USA
(Dated: June 21, 2016)
1
CONTENTS
I. Overview 3
II. Theory 3
A. Reduced density matrix and Redfield equations 3
B. Nonadiabatic system-reservoir coupling 4
C. Electronic nonadiabatic coupling within LR-TDDFT 5
D. Franck-Condon-weighted density of states 7
E. Phonon-induced relaxation dynamics 7
F. Further References 8
III. Example Calculation: Porphyrin 8
A. Electronic excitations with linear response theory 9
B. Electronic nonadiabatic coupling 12
1. Ab initio molecular dynamics 12
2. DFT and LR-TDDFT calculations 17
3. Kohn-Sham orbital overlap calculations 18
4. LR-TDDFT electronic coupling 20
C. Franck-Condon-weighted density of states 23
1. Phonons 24
2. Excited state geometry relaxation 25
3. Huang-Rhys factors 26
4. Franck-Condon-weighted density of states 29
D. Phonon-induced relaxation dynamics 31
References 34
2
I. OVERVIEW
This manual reviews the theory and computational methods to calculate phonon-induced
relaxation dynamics of electron-hole pairs in finite systems within linear response the-
ory. The method employs linear-response, time-dependent density functional theory (LR-
TDDFT) to calculate excited states and the reduced density matrix formalism (RDM) to
investigate the nonequilibrium relaxation dynamics of an initially excited electron-hole pair.
Systems with hundreds of atoms can be explored using the steps described below.
Required software includes a C++ compiler, Fortran compiler, Matlab, Gaussian09, and
VASP (optional for use in generating molecular dynamics trajectories). Input for the latter
two DFT programs is only described briefly, and details about their use can be found at
their respective websites.
The manual is organized as follows. Section II provides a brief introduction into the
underlying theory, which is described in more detail in several references included in Section
II F. Section III then provides a step-by-step example of how to use the code, applying
the method to study phonon-induced relaxation of an excitation in the Soret state of the
porphyrin molecule, which has been previously studied experimentally.
II. THEORY
A detailed overview of the theory and computational steps can be found in published
work1. Only primary equations and descriptions of major variables will be described here -
please see references given for more details.
A. Reduced density matrix and Redfield equations
Within the RDM formalism,2dynamics of a system of interest are determined due to
its interaction with a reservoir that contains many more degrees of freedom and cannot be
described in detail. If a density matrix is constructed of the combined system and reservoir,
then the reduced density matrix (σαβ ) can be made by taking the trace of the density matrix
with respect to the reservoir coordinates. The reduced density matrix then only depends
on the system degrees of freedom. For the model described within, electronic ground and
3
excited states described by LR-TDDFT are the collective system interacting with a phonon
reservoir that causes downward energy relaxation from an initial excited state.
If we construct the RDM with the electronic eigenstates of the system, then the Redfield
equations describe both population transfer (diagonal elements) and coherence dephasing
(offdiagonal) between eigenstates αand β:
1) Population relaxation:
dσI
αα(t)
dt =−X
µ
Rαα,µµσI
µµ(t) (1)
Rαα,µµ =δαµ X
κ
kακ −kµα (2)
2) Coherence dephasing:
dσI
αβ(t)
dt =−Rαβ,αβσI
αβ(t) (3)
Rαβ,αβ =X
µ
1
2(kαµ +kβµ) + γ0,(4)
Here, Rαβ,µν is the Redfield relaxation tensor, kαβ is the relaxation rate due to the system-
reservoir interaction, and γ0is a dephasing constant determined from experiment or other
computational methods. Thus, determining energy relaxation rates of excited states requires
the calculation of kαβ, which has been shown to be:2,3
kαβ =2π
~DX
j|hφiΨα|V|φjΨβi|2δ(~ωαβ −~ωij)E,(5)
where Vis the system-reservoir interaction to be defined below, Ψαis an electronic excited or
ground state, φiis a nuclear state, ωαβ and ωij are electronic transition energies and phonon
energies, respectively, and D...E=Pifi=e−βEi/(Pie−βEi), the thermal average over
initial conditions. The sum over final reservoir states preserves conservation of energy. The
principle of detailed balance ensures proper behavior as time goes to infinity and provides the
following relationship between forward and backward transition rates kαβ =e~ωαβ /kBTkβα.
B. Nonadiabatic system-reservoir coupling
To model electron-phonon interactions, we follow previous literature that treats the nu-
clear kinetic energy operator as a perturbation to the adiabatically determined electronic
4

excited states: hφiΨα|V|φjΨβi=Vαβ,ij =hφiΨα|Pk−~2
2Mk∇2
k|φjΨβi, where Mkis the nu-
clear mass and kruns over the Knuclear degrees of freedom, |φii=|φ1
ii|φ2
ii...|φK
ii.2If we
only include terms to first order in the nuclear gradient operator, previous work has shown
that the system-reservoir coupling can be rewritten to good approximation as4
Vαβ,ij ≈X
k−i~
MkhΨα|∇k|ΨβihˆpiY
khφk
i|φk
ji,(6)
where hˆpiis the expectation value of the momentum operator and obeys classical equa-
tions of motion according to Ehrenfest’s theorem. If we plug this expression for Vαβ into
Equation 5, then we express the relaxation rates as
kαβ =2π
~D|Vαβ|2EF(ωαβ),(7)
where Vαβ =Pk−i~
MkhΨα|∇k|Ψβihˆpiis the nonadiabatic electronic coupling and F(ωαβ) =
Pi,j fiQk|hφk
i|φk
ji|2δ(~ωαβ −~ωij) is the Franck-Condon-weighted density of states (FC-
DOS). Vαβ describes the coupling of states αand βdue to atomic motion and F(ωαβ)
describes the density of phonon states available to couple to the transition.
Equation 7 is the primary equation we will refer to during the example description in
Section III. The calculation requires three major quantities: 1) the ground and excited
electronic states used to construct the reduced density matrix, which we calculate within
LR-TDDFT; 2) the nonadiabatic electronic coupling (Vαβ), which we calculate using ab
initio molecular dynamics; and 3) the Franck-Condon-weighted density of states, which we
calculate within the harmonic approximation using the minimum-energy configuration of
each excited state and the ground state normal modes.
C. Electronic nonadiabatic coupling within LR-TDDFT
We follow theory developed by Tavernelli et al.5,6 to calculate Vαβ within LR-TDDFT.
The theory is exact for first-order nonadiabatic coupling between ground and excted states
and is a good approximation for coupling between excited states (exact within the Tamm-
Dancoff approximation). Tavernelli showed that the nonadiabatic electronic coupling can
be expressed as follows, replacing the nuclear gradient operator with a time derivative and
using finite differences in the last expression:
Vαβ =−i~hΨα|∂
∂t|Ψβi≈− i~
2∆t[hΨα(t)|Ψβ(t+ ∆t)i−hΨα(t+ ∆t)|Ψβ(t)i],(8)
5

where ∆tis the time step. The next step is to define the wave function used to solve
Equation 8. Within the Casida ansatz,7an excited state within LR-TDDFT can be written
as
|Ψαi=X
ma
(Xma
α+Yma
α)ˆa†
aˆam|Ψ0i=X
ma
(Xma
α+Yma
α)|Ψm−>ai,(9)
where mand aindex occupied and unoccupied Kohn-Sham orbitals, respectively, ˆa†
aand
ˆamare creation and annihilation operators, Xma
αand Yma
αare the normalized coefficients
corresponding to an excitation from the mth to ath orbital and de-excitation from the
ath to mth orbital, respectively, |Ψ0iis the ground state Slater determinant, and |Ψm−>ai
is a singly excited Slater determinant. Plugging this wave function definition into Equa-
tion 8, the wave function overlaps can be expressed by singly excited Slater determi-
nant overlaps: hΨα(t)|Ψβ(t+ ∆t)i=Pma Pnb(Xma
α(t) + Yma
α(t))(Xnb
β(t+ ∆t) + Ynb
β(t+
∆t))hΨm−>a(t)|Ψn−>b(t+ ∆t)i. These Slater determinant overlaps can be further decom-
posed into single-particle Kohn-Sham orbital overlaps using Slater-Condon rules:
hΨα(t)|Ψβ(t+ ∆t)i=X
mab
(Xma
α(t) + Yma
α(t))†(Xmb
β(t+ ∆t) + Ymb
β(t+ ∆t))vab
−X
amn
(Xma
α(t) + Yma
α(t))†(Xna
β(t+ ∆t) + Yna
β(t+ ∆t))vmn,
(10)
where vab =hψa|ψbi, and a,bare unoccupied and m,nare occupied Kohn-Sham orbitals,
respectively.
Thus, Equation 10 lays out all the required variables to calculate Vαβ. To do so, we
perform an ab initio molecular dynamics simulation at 300K (or any relevant temperature)
to generate a set of configurations - the time step of the simulation is ∆t. Changes in
the LR-TDDFT wave functions between time steps represent coupling of the electronic
structure to phonon modes in the system. Thus, calculating Vαβ at adjacent time steps
and averaging across them will provide the thermally averaged, nonadiabatic electronic
coupling we require. This requires knowledge of the LR-TDDFT coefficients and Kohn-
sham orbital wave functions at each step, which come from a self-consistent calculation for
each configuration at each of the two time steps, and are entered into Equation 10. Results
from this equation enter into Equation 8 to find Vαβ for a given pair of adjacent time steps.
The thermally averaged electronic nonadiabatic coupling that enters into Equation (7) is
then calculated by averaging the value of Vαβ across all time steps of the AIMD trajectory
consisting of Ntime steps: D|Vαβ|2E=1
NPN
i=1 |Vαβ|2.
6

D. Franck-Condon-weighted density of states
The final term required to calculate relaxation rates is F(ωαβ), describing the density of
phonon states that couple to a given α−βtransition. Assuming a quantum harmonic oscil-
lator phonon bath and that the ground state normal modes are an accurate approximation
for all excited state modes, F(ωαβ) describes the density of phonon states coupled to a given
nonadiabatic transition:2
F(ωαβ) = 1
2π~Z∞
−∞
dteiωαβ teG(t),(11)
where G(t) = PkSk
αβ[(e−iωkt−1)(n(ωk) + 1) + (eiωkt−1)n(ωk)]. Sk
αβ is the Huang-Rhys
factor, a dimensionless quantity representing the coupling strength of the kth normal mode
to the transition, and n(ωk) is the Bose-Einstein distribution for a phonon with energy ~ωk
at a given temperature. The function F(ω) peaks at energies corresponding to modes with
nonzero Sαβ.
To calculate Sk
αβ, we relax the atomic positions for all excited states and project the
change in atomic configuration between two states αand βonto the normal modes of the
system. The Huang-Rhys factor is then calculated as
Sk
αβ = (µkωkd2
kαβ )/~,(12)
where µkis the modal reduced mass and dkαβ is the configurational difference between
excitations αand βprojected onto the kth mode eigenvector. These values of Sk
αβ enter
Equation 11 along with phonon modes (ωk) calculated within the harmonic approximation.
E. Phonon-induced relaxation dynamics
Once we have determined the LR-TDDFT excited sates, Vαβ, and F(ωαβ), we can examine
phonon-induced relaxation dynamics. We consider a system bathed in light with frequency
ωat t < 0. After the light is switched off at t= 0, a nonequilibrium carrier distribution
will relax due to interactions with phonons as described by Equations (1) and (7). The
population change in σαβ is then defined as ∆η(E, t) = Pασαα(t)δ(Eα−E)−ηeq, where
Eαis the energy of the αth excitation and ηeq is the equilibrium population.
7
F. Further References
Reduced density matrix and Redfield equations:
May, V and Kuhn, O. Charge and energy transfer dynamics in molecular systems. John
Wiley and Sons, 2008.
Nonadiabatic system-reservoir coupling:
Prezhdo, O and Rossky, P. Evaluation of quantum transition rates from quantum-classical
moleculary dynamics simulations. J. Chem. Phys., 107(15), 5863-5878, 1997.
Electronic nonadiabatic coupling within LR-TDDFT:
Tavernelli, I et al. Nonadiabatic coupling vectors for excited states within time-dependent
density functional theory in the Tamm-Dancoff approximation and beyond. J. Chem. Phys.,
133(19), 194104, 2010.
Franck-Condon-weighted density of states:
May, V and Kuhn, O. Charge and energy transfer dynamics in molecular systems. John
Wiley and Sons, 2008.
III. EXAMPLE CALCULATION: PORPHYRIN
Phonon-induced relaxation has been well studied in the porphyrin molecule, which
demonstrates a strong absorption peak known as the Soret band between 350-400 nm
depending the solvent (see Figure 1).8Several experimental studies8–10 have measured re-
laxation times from the Soret band to lower states ranging from 50-200 fs. The availablility
of experimental data combined with the small size of the molecule makes this a nice system
to demonstrate the steps for the present method with minimal computational resources.
All calculations and post-processing codes are developed to work with Gaussian09, version
D.01, except for the ab initio molecular dynamics simulations, which can be done using either
Gaussian09 or VASP. This manual will focus on describing the codes specific to calculating
relaxation rates; see the Gaussian09 and VASP websites for detailed information about
input. All files for submitting jobs are tailored to NERSC’s Edison system - changes will
need to be made as appropriate for the particular high-performance computing server being
used.
8

FIG. 1. Porphyrin absorption spectrum8as a function of wavelength in the following solvents: 1)
benzene/cyclohexane 1:10, 2) benzene/cyclohexane 1:10 added with CF3COOH, and 3) concen-
trated HClO4. The Soret band is the large peak between 350-400 nm and the lowest excited state
occurs between 500-600 nm.
A. Electronic excitations with linear response theory
The first step requires the calculation of excited states within LR-TDDFT and confir-
mation that the results generally match experimental absorption spectra. This is a crucial
check because a general match with experimental data is a good indication that the excited
state wave functions are well described with the basis set and functional being used. If this
is not the case, then the electronic nonadiabatic couplings (Vαβ) may be inaccurate and
subsequent relaxation rates will not be correct.
•After untarring Phonon Induced Relaxation LRTDDFT.tar attached with this man-
ual, move into Porphyrin Example/Electronic Excitations.
•Within this folder, porphyrin.xyz provides the atomic coordinates for porphyrin in
XYZ format. Look at the molecule using any visualization software (e.g., VESTA) to
understand its general structure as shown in Figure 2.
•Next, open porph lrtddft.com, which is the input file for Gaussian09 that will cal-
culate the first six excitations (TD command). It is assumed that the geometry has
already been optimized to the minimum-energy configuration. Several lines are of
particular importance:
9

FIG. 2. Atomic structure of porphyrin. Brown spheres are carbon atoms, small pink spheres are
hydrogen atoms, and gray spheres are nitrogen atoms.
–IOP(9/40=2) forces Gaussian to output the LR-TDDFT coefficients that are
greater than 0.01 for each excitation, where IOP(9/40=X) corresponds to print-
ing coefficients greater than 10−X. This value should be at least 2 in order to
accurately calculate Vαβ.
–b3lyp/6-31g* denotes the functional and Gaussian basis set used in the calcu-
lation. Both the B3LYP functional and 6-31g* basis set reproduce excitations in
porphyrin well, but this should be tested for each new system.
–NOSYMMETRY prevents Gaussian from rotating the molecule during any calculation.
This command should be included in all calculations as both Vαβ and
F(ωαβ) require comparisons between configurations with the same orientation.
–SCRF initiates the Polarizable Continuum Model to represent the benzene solvent
used in most experiments studying porphyrin, which is important to capture the
correct excitation energies.
–TD performs the LR-TDDFT calculation, where the included subcommands in-
struct the program to calculate 6 singlet states.
•After submitting and completing the calculation, porph lrtddft.out will contain
information about the excited state energies and LR-TDDFT coefficients. Within the
10

TABLE I. Excitation energies and oscillator strengths for first six excited states in porphyrin.
Excited State Energy (eV) Energy (nm) Oscillator Strength (f)
1 2.28 544 0.0003
2 2.44 509 0.0000
3 3.22 385 0.8905
4 3.33 372 1.2500
5 3.42 362 0.0000
6 3.62 342 0.0000
file, look for the keyword Excited State to find energies listed for the first six states
as listed in Table I. The Soret band corresponds to excited states 3 and 4, which show
strong absorption between 350-400 nm as seen in experiment. This example will look
at phonon-induced relaxation rates of an electron-hole pair initially excited to these
states.
•Below each excited state, a list of Kohn-Sham electron-hole transitions are listed
that make up each LR-TDDFT excitation along with their LR-TDDFT coefficient
or weight. For example, under Excited State 1, ’64 -> 83 -0.01058’ indicates
that the transition from the 64th to 83rd KS orbital has a coefficient of -0.01058*√2
(Gaussian normalizes the coefficients such that their sum of squares is 1 only if you
multiply them by √2).
Having confirmed that the choice of functional and basis set provides an accurate descrip-
tion of excited states, the next step will be the calculation of the electronic nonadiabatic
coupling (Vαβ) between these six states using AIMD and post-processing codes. The energies
of the excitations will also be required as input for calculating F(ωαβ) in Section III C.
11

B. Electronic nonadiabatic coupling
1. Ab initio molecular dynamics
Calculating Vαβ first requires an AIMD trajectory that provides the input configurations
for subsequent LR-TDDFT and post-processing calculations. This can be done in two ways:
1) using Gaussian09 and the same functional used to calculate electronic structure or 2)
using VASP and a more computationally efficient functional. The former is technically
more accurate and consistent, however non-hybrid, GGA functionals often provide accurate
strutural information and describe phonon modes well. This makes them advantageous for
calculating AIMD trajectories that can be computationally burdensome for large systems.
Each option is outlined below:
GAUSSIAN AIMD
Performing molecular dynamics in Gaussian09 requires two sequential steps: 1) an initial
density matrix propagation method (ADMP) in which the system is connected to a velocity-
rescaling thermostat and nuclear velocities are obtained after reaching thermal equilibration,
and 2) a second NVE run using Born-Oppenheimer dynamics (BOMD) with the ADMP
velocities included as input. The configurations from the BOMD trajectory are then used
to calculate Vαβ.
•Navigate to Porphyrin Example/Electronic NAC/AIMD/Gaussian09. In this folder,
porph admp.com contains all the input commands required to run the initial ADMP
dynamics to obtain nuclear velocities:
–The input configuration should be the optimized geometry of the system.
–ADMP turns on the density matrix propagation method.
∗Maxpoints is the maximum number of time steps in the simulation.
∗Stepsize indicates the time step size Nin units of N∗0.0001 femtoseconds
(fs). Thus, 2500 is a 0.25 fs time step - a small enough time step must be used
to find good temperature and energy behavior during the NVE run. Thus,
the run will last 4000*0.25 = 1000 fs - increase Maxpoints if the system
requires longer time to reach equilibrium.
12

∗NKE indicates the initial nuclear kinetic energy in units of microHartree. The
relevant formula is NKE = (Natoms −1)(3/2)kT , where Natoms is the
number of atoms, k= 3.1668 microHartree/K, and Tis temperature in
Kelvin. Thus, a 38-atom system (porphyrin) at 300K corresponds to NKE =
52727.
–The IOps are described in the input file and provide controls over the velocity
rescaling.
•Submit the job in this directory to perform the AIMD calculation, which will take at
least several hours to complete.
•After the AIMD calculation complete, open the porph admp.out file. This will contain
information, such as energetics and nuclear velocities, about each time step of the
trajectory (search for the Summary information keyword). Move to the last time
step and copy the geometry and nuclear velocities into the input file for the BOMD
NVE run (example found in the BOMD folder).
•In the BOMD folder, open porph bomd.com to understand the input for the final NVE
run:
–BOMD initiates the BOMD NVE AIMD trajectory. Change Maxpoints to a smaller
value, but large enough to have sufficient configurations to converge Vαβ (at least
150 fs).
∗Readmvwel tells the program to read in the nuclear velocities that will be
included below.
∗Sample=Microcanonical will perform the dynamics using the NVE ensem-
ble.
∗Units=Bohr tells the program to look for coordinates and nuclear velocities
in units of bohr (make sure you do this!).
–Below the 0 1, copy in the configuration from the last ADMP time step in units
of bohr. Below the 0, copy in the nuclear velocities matrix from the ADMP
output file. There are already numbers in these regions to show formatting, but
these do not correspond to positions and velocities in equilibrium.
13

–Submit the job. Once finished, plot the temperature and energy as a function
of time step to make sure the system was thermally equilibrated. If the energy
is not roughly constant compared to the potential energy, or the temperature
oscillations are too alrge, try running the ADMP calculation for a longer time to
achieve equilibration, then rerun the BOMD calculation.
•After obtaining the BOMD trajectory, the last step is to extract each configuration
from the porph bomd.out output file into separate files that will be used as input for
the next step. To do this, copy porph bomd.out into the Porphyrin Example/Electronic NAC/NAC
folder. In addition, from Codes/Electronic NAC/Config Generator, copy Config
Generator 3Atoms G09.f90 to the same folder.
•Rename the BOMD output file as MD G09 and look at the input cfg gen file to un-
derstand its contents (the code discusses its details). Compile and run this code with
MD G09 and input cfg gen in the same directory, and a posX file should be created
for each timestep of the trajectory. Each file should list atom labels and correspond-
ing coordinates in the Gaussian09 input format (an example can be found in the
sample output folder). These files will be used as input for performing DFT and
LR-TDDFT calculations at each step in the next section.
VASP AIMD
Generating the AIMD trajectory using VASP requires three steps: 1) optimizing the
geometry in VASP using the new, more computationally efficient functional; 2) annealing
the structure to room temperature and reaching equilibrium within the NVT ensemble; and
3) obtaining a final NVE trajectory that will be used as input for calculating Vαβ in the
next section. This manual assumes that the new geometry optimization has already been
performed and begins with the annealing steps using the NVT ensemble. Note that this
method uses velocity rescaling, but VASP also has a thermostat that may be more accurate
and can be used in a similar manner.
•Move to Porphyrin Example/Electronic NAC/AIMD/VASP. Several files should be
found that will be used to obtain the AIMD trajectory:
–POTCAR contains the pseudopotential for each atom in the system. For this ex-
ample, the PBE-GGA pseudopotential is uised.
14

–KPOINTS only includes Gamma point since the porphyrin molecule is a finite
system.
–POSCAR initial is the molecule in the minimum-energy configuration using the
PBE functional. Make sure the system is optimized using the PBE functional -
don’t just use the same geometry from the Gaussian09 calculation!
–INCAR files contain the necessary control inputs to tell VASP what type of molec-
ular dynamics simulations to perform, using a 0.25 fs time step for 1200 time
steps.
•Copy POSCAR initial to POSCAR, which VASP looks for as the input configuration file,
and copy INCAR 0 300 NVT to INCAR. This first simulation will anneal the structure
from 0K to 300K using velocity rescaling. Submit the job.
•Once the job has completed, check the vasp.out file to make sure the system has
heated up to roughly 300K as expected. If so, then copy the CONTCAR file, which
contains the coordinates and nuclear velocities from the end of the run, as the new
INCAR file. Copy INCAR 300 300 NVT as INCAR - this run will now equilibrate the
system at 300K for another 1200 time steps. Submit the job.
•Once that job is done, again check the vasp.out file and confirm that the system is
staying roughly around 300K - if not, run another simulation within the NVT ensemble
until equilibrium is reached.
•Finally, again copy CONTCAR to POSCAR file and copy INCAR NVE as INCAR. Submit this
job, which will run an NVE ensemble with initial coordinates and nuclear velocities
supplied from the end of the NVT simulation.
•Once this job is complete, plot the temperature stored in vasp.out as well as the
total energy and potential energy as a function of time (see example in Figure 3).
Make sure temperature is oscillating reasonably (about 15-20% above and below the
average) and that the total energy is conserved relative to the potential energy. If any
of these are not true, rerun the NVT ensemble for a longer period of time to get closer
to equilibrium, and then rerun the NVE simulation. In the sample output folder,
example OUTCAR and vasp.out files can be found from the final NVE simulation. The
15
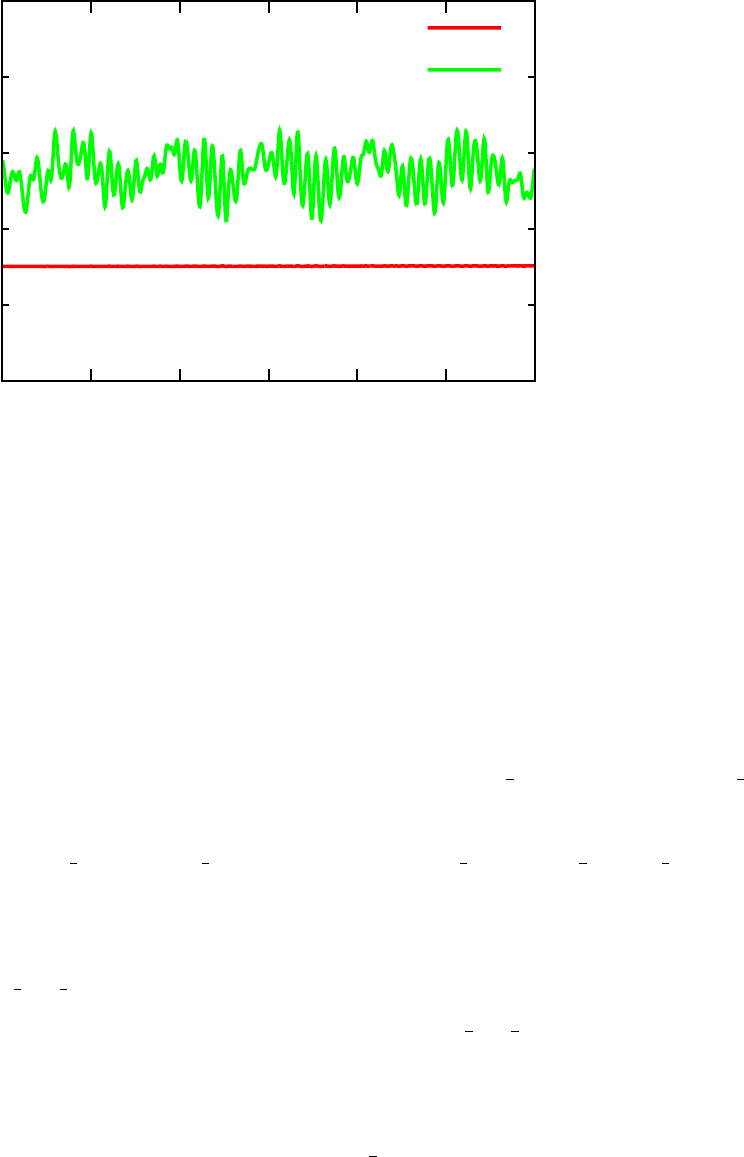
-265
-264
-263
-262
-261
-260
0 50 100 150 200 250
Energy (eV))
Time (fs)
Total Energy
Potential Energy
FIG. 3. Total energy and potenteial energy as a function of time over the course of AIMD trajectory
using VASP.
temperature oscillates slightly more than desired, but the results work well enough for
this manual.
•With the NVE trajectroy in hand, copy the OUTCAR file to Porphyrin Example/Electronic NAC/NAC,
where it will be used to generate the input configurations for the next section. Also,
from Codes/Electronic NAC/Config Generator, copy Config Generator 3Atoms VASP.f90
to the same folder.
•Look at the input cfg gen file to understand its contents (the code discusses more
details). Compile and run the code with OUTCAR and input cfg gen in the same
directory, and a posX file should be created for the X time steps of the trajectory.
Each file should list atom labels and corresponding coordinates in the Gaussian09
input format (an example can be found in the sample output folder). These files will
be used as input for performing DFT and LR-TDDFT calculations at each step in the
next section.
16

2. DFT and LR-TDDFT calculations
Using the posX files obtained in the previous section, DFT and LR-TDDFT calculations
will be completed to obtain Kohn-Sham orbitals and LR-TDDFT coefficients at each time
step. These orbitals and coefficients will be used as input into a post-processing code that
calculates Vαβ according to Equation 10.
•Copy the contents of Codes/Electronic NAC/DFT TDDFT Calc to the NAC folder where
the posX files were created. These files include several short bash scripts and header
and footer files that will be used to create a folder and input files for each time step.
•Open cycle dft script to understand its contents. This will be the first script to
run, as it will create a folder for each time step X, labeled scfX, in which the DFT
and LR-TDDFT calculations are performed. Inside each folder, a Gaussian09 input
file (gauss dft.com) used to perform a standard DFT calculation will be created by
combining the head dft,posX, and foot files. The foot file lists the name of the
.wfx file that will contain the orbital information for computing overlaps later in the
section. Check the head dft file to make sure all entries make sense - all commands
should be identical to those used for the linear response calculation in Section III A,
except for two important changes:
–The TD command has been removed since only a ground state calculation is being
performed.
–IOP(99/18=100) has been added, which instructs Gaussian09 to calculate 100
empty orbitals. This number must be large enough to include all empty Kohn-
Sham orbitals necessary to describe electron-hole transitions constituting each of
the LR-TDDFT excited states as computed in Section III A. Look through all the
excited states from the linear response calculation and make sure that enough
empty orbitals are included, otherwise future steps will not work correctly. One
hundred orbitals should be sufficient for porphyrin, but this must be tested on a
case by case basis.
•Run the cycle dft script from the command line in the NAC folder, which will create
10 scf folders with a different configuration and Gaussian09 input file in each. It will
also submit the job in each folder.
17

•While the DFT calculations are running, the next step will be to submit jobs to cal-
culate the LR-TDDFT excitations for each time step. These calculations will provide
the LR-TDDFT coefficients required to solve Equation 10.
•Open cycle lrtddft script. This bash script is very similar to cycle dft script,
however now a tddft subfolder will be created and head lrtddft will be combined
with posX to create a Gaussian09 input file to calculate LR-TDDFT excitations for
each configuration. Again, the head lrtddft should contain identical commands as
those used in the earlier linear response calculations. POP=None has been included to
remove some orbital population analysis and save computational time.
•Submit cycle lrtddft script and 10 jobs should be submitted, one in each scfX/tddft
folder.
It is important to note that more than 10 configurations will be necessary to converge
Vαβ, however this small number will be enough to understand the basic steps. Convergence
with respect to number of time steps must be checked rigorously for each system studied.
Roughly 500-600 steps should be necessary in the porphyrin case.
3. Kohn-Sham orbital overlap calculations
Now that Kohn-Sham orbitals have been found for every time step, the overlaps (vab) can
be calculated that appear in Equation 10.
•Copy the contents of Codes/Electronic NAC/KS Orbital Overlap to the NAC folder
where all the scfX folders have been created. These files consist of a parallelized
or serialized version of the code to calculate Kohn-Sham overlaps between two wave
functions (ks ovlap gauss mpi.cpp and ks ovlap serial.cpp), the input file to run
this code (input ovlap), and a bash script to run this overlap code in each scfX folder
(cycle ovlap).
•This manual assumes the use of the parallelized code, but the same instructions follow
for the serial version. Compile ks ovlap gauss mpi.cpp using a C++ compiler and
rename the executable ovlap code. This code requires three inputs:
18

–input ovlap should have two lines: the first line gives the minimum and max-
imum Kohn-Sham band index that will be included in the overlap matrix. So,
40 120 means that overlaps between Kohn-Sham orbitals indexed from 40 to 120
in the SCF calculations will be calculated. Again, make sure this encompasses
all the electron-hole transitions making up each LR-TDDFT excitation of the
system of interest. The second line is the time step from the molecular dynam-
ics trajectory. This is included because the code calculates the single-particle
coupling between Kohn-Sham states, even though the current method applies to
LR-TDDFT state coupling.
–The two wave functions used to calculate the overlap must be in the same folder
and named wfx1 and wfx2.
•Check that input ovlap includes the correct quantities (it should for the porphyrin
example - the 40-120 band range should be plenty). Next, open cycle ovlap and
understand its contents. Running this script from the command line will copy the
compiled code and input file into each scfX folder, then copy the current time step’s
wave function file as wfx1 and the adjacent time step’s wave function file as wfx2 into
the same directory. For this reason, the bash script should only cycle up to N−1
folders, where Nis the total number of time steps, since the adjacent wave function
file that is required will not exist for the final time step.
•Make sure that the job file is suited for your server, and then run cycle ovlap from
the command line. Nine jobs should now be submitted, one for each of the first nine
time steps, and each should only take a few minutes. The default number of processors
to be used is four, but this can be changed.
The ks ovlap gauss mpi.cpp file contains information about the different outputs (all
written out with a output prefix), but the primary output required for the next step is
output ovlap. This file will contain the overlap matrix (vab) for the included bands as
denoted in the input ovlap file. Diagonal elements should be close to but less than one, as
the nuclear motion creates nonzero offdiagonal overlaps. This file will be used as input for
the next step that calculates Vαβ between adjacent time steps.
19

4. LR-TDDFT electronic coupling
Now that Kohn-Sham orbital overlaps and LR-TDDFT excitation coefficients have been
calculated for each time step, every quantity in Equation 10 is known and Vαβ can be found.
This step will use a post-processing code to calculate Vαβ using the output ovlap and
gauss lrtddft.out files created in the previous sections.
•Copy the contents of Codes/Electronic NAC/LRTDDFT Coupling into the NAC folder,
the directory that should contain all the scf folders. These files consist of a post-
processing code (lrtddft elec coupling.cpp) to calculate Vαβ between adjacent time
steps and the input file for the code to run properly (input nac).
•First, open lrtddft elec coupling.cpp and take time to understand the contents of
the code. The code provides a description of each input and output. This code will
take as input:
–the output ovlap file calculated in the previous section containing the vab matrix.
–input nac, which must list fifteen parameters for the code to run correctly:
∗dft dir name: prefix of all folders containing DFT and overlap calculations
from previous steps.
∗td dir name: name of folder within dft dir name folders that contain Gaus-
sian LR-TDDFT calculation.
∗overlap file name: Name of output file within dft dir name folder for each
time step containing the K-S overlap matrix.
∗tddft file name: name of output file in td dir name directory containing
LR-TDDFT output information.
∗nsteps: total number of MD time steps for which DFT, overlap, and LR-
TDDFT calculations have been completed (number distinct dft dir name
directories you have).
∗gaus homo: HOMO orbital index
∗gaus emin: lowest orbital index, must match lowest index in output ovlap
file.
20
∗gaus emax: highest orbital index, must match highest index in output ovlap
file.
∗num exc: number of LR-TDDFT excited states in input exc gaussian files.
All files must have the same number of excitations and all must be included
in coupling calculation.
∗time step: time step of MD trajectory in femtoseconds.
∗dft ordering: (’yes’ or ’no’) will enforce same ordering of K-S states
across all time-steps by calculating K-S overlaps and switching the order of
states in the overlap matrix to ensure the largest value is along the diagonal.
This ensures that the same K-S states are being used to calculate the coupling
even if their ordering changes throughout the MD trajectory.
∗td ordering: (’yes’ or ’no’) will enforce same ordering of LR-TDDFT
states across all time-steps by calculating LR-TDDFT excitation overlaps
and switching the order of states based on this overlap matrix such that the
largest values are along the diagonal. This ensures that the same LR-TDDFT
states are being used to calculate the coupling even if their ordering changes
throughout the MD trajectory.
∗same sign: (’yes’ or ’no’) program will enforce the sign of all LR-TDDFT
coefficients to not change between time steps. This is used in rare cases when
all LR-TDDFT coefficients flip signs between adjacent time steps, which leads
to unphysically large coupling values.
∗use thresh: (’yes’ or ’no’) program will output all LR-TDDFT excita-
tion coefficients that change by more than the thresh value provided below.
This can be helpful in determining whether nearly degenerate states are flip-
ping positions between time steps, which leads to unphysically large coupling
values.
∗thresh: value of the threshold change in LR-TDDFT excitation coefficients
between time steps above which it is reported in the output threshold file.
•Compile lrtddft elec coupling.cpp using a C++ compiler and run the code. In
this case, we will only calculate the nonadiabatic coupling matrix using 10 time steps.
More will be necessary in general to converge the matrix elements.
21
•As the code runs, it will calculate the thermally averaged, squared nonadiabatic cou-
pling matrix by going into each dft dir name directory, using the DFT, LR-TDDFT,
and overlap matrix information to calculate the coupling between adjacent time steps,
and output information in a coupling folder created within dft dir name. It will
do this for every dft dir name, and then calculate the thermally averaged nonadia-
batic coupling matrix by averaging the square of each matrix output in each coupling
folder.
•During the calculation, make sure that the correct number of Excited States are
printed to the screen for each step - in this case, six should be listed twice in a row,
one for each time step. This indicates that the code has correctly read in all excitations
to be included in the coupling matrix.
•Once the calculations have finished, the most important output will be final NAC matrix total,
which contains the squared, averaged nonadiabatic coupling matrix (Vαβ ) to be used
as input in the final step. There is also a NAC timeseries total file, which plots
in rows the cumulative average of the nonadiabatic coupling value for each matrix
element to check for convergence with respect to time steps.
•Next, move into a coupling folder to understand the various outputs specific to one
time step:
–output elec details and output hole details: For each excitation combina-
tion, these files list the electron and hole components (first and second terms in
Equation10, respectively) contributing to Vαβ. These files are helpful to under-
stand which Kohn-Sham orbitals are the dominant contributors to the coupling
values.
–output exc gs details: provides the same information as the other comp files
but applies to matrix elements between the ground and excited states.
–output NAC GS X: files contain the final Vαβ matrix including the ground state for
X=total,X=elec, and X=hole. By default in the cycling script, output nac GS total
is copied to the lrtddft coupling folder to be used to calculate the thermal
average.
22
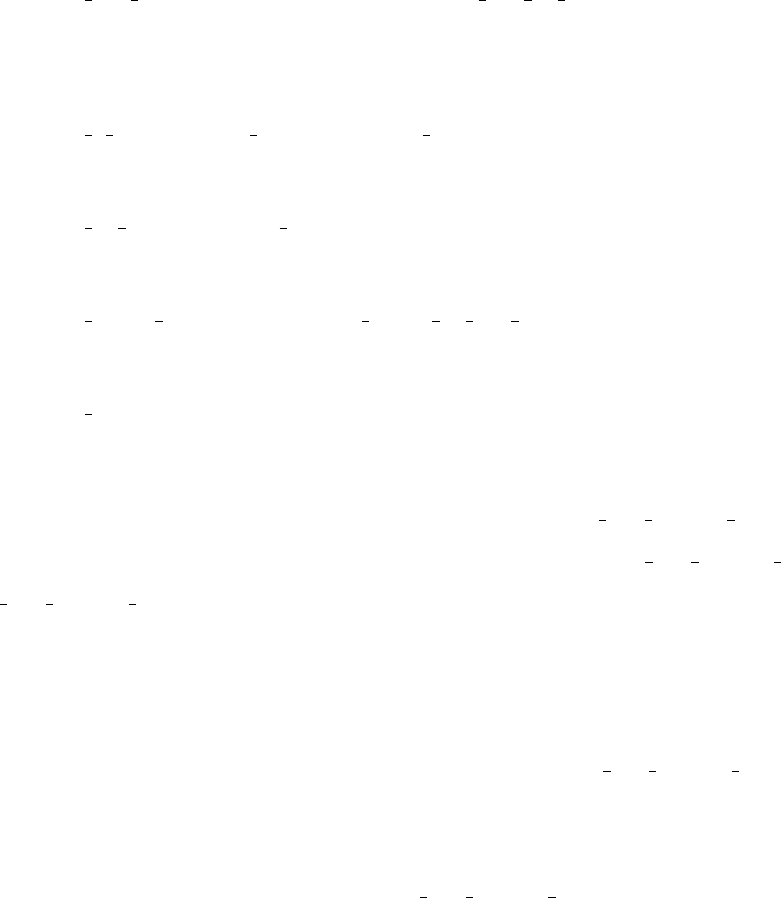
–output NAC X: include same matrix as output nac GS X but does not include the
ground state. Use this matrix if only concerned about excited state dynamics
and not nonradiative recombination to ground state.
–output X flip: If dft ordering or td ordering are turned on, these output
files will list which states have been flipped at this time step (X = KS or TD).
–output TD overlap: If td ordering are turned on, will output the LR-TDDFT
excitation overlap matrix before and after ordering of states.
–output coeff exc1/2 and output coeff de exc 1/2 contain matrices of all LR-
TDDFT excitation coefficients for the first (1) or second (2) time step.
–output threshold lists each excitation combination that includes a change in
LR-TDDFT excitation coefficients greater than the thresh value given as input.
As mentioned above, the squared thermal average of Vαβ is in output NAC matrix total, as
this matrix has been calculated using both terms in Equation 10. output NAC matrix elec
or output NAC matrix hole contain the coupling matrix calculated only using the electronic
or hole component.
With only nine time steps included, Vαβ in the primary output file will not be converged.
After running the same steps above for 400 or more time steps, plot the row corresponding
to the coupling between the 2nd and 3rd excited states from final NAC matrix total to
check convergence. Something similar to Figure 4 should be found, with V32 ≈35 meV and
convergence reached after about 100 fs, or 400 time steps.
Congratulations - the output matrix in output NAC matrix total in this step will en-
ter directly into the phonon-induced relaxation dynamics calculated in Section III D! Now
only the Franck-Condon-weighted density of states are required before we can determine
relaxation rates!
C. Franck-Condon-weighted density of states
The second major component of the relaxation rates is F(ωαβ ), the Franck-Condon-
weighted density of states (FC-DOS, Section II D). To calculate this, three major steps are
required: 1) phonon calculation within the harmonic approximation; 2) Huang-Rhys factors
determined using the phonon modes and minimum-energy geometric configurations for each
23
10-6
10-5
10-4
10-3
10-2
10-1
0 50 100
|Vαβ|2 (eV2)
Time (fs)
|V32|2
FIG. 4. Cumulative average of squared electronic nonadiabatic coupling between 2nd and 3rd
excited state (V32) in porphyrin as a function of time.
excited state; and 3) the calculation of F(ωαβ ) using the Fourier transform as shown in
Equation 11. Each step is outlined in detail below, again using porphyrin as an example.
1. Phonons
First, phonon modes will be calculated within the harmonic approximation using Gaus-
sian09. This output, along with the excited state configurations, will be the input to calculate
Huang-Rhys factors for each mode between each excitation.
•Move to Porphyrin Example/FC DOS/Phonons. The porph phonons.com file acts as
input for a Gaussian calculation that will calculate all harmonic modes of the system.
Open the file to make sure all commands make sense - the addition of FREQ tells
Gaussian to calculate phonon modes of the system. Note that the atomic positions
have changed from the linear response calculations - this was just due to a symmetric
rotation done during their initial optimization. As long as NOSYMMETRY is included in
all calculations within this section, then this configuration is adequate.
•Submit the calculation using the job script, which should only take ten minutes or so
24

on two dozen or more processors. Once the job is finished, the porph phonons.out
file will include all frequencies of the system (search for the word Frequencies in the
file).
•If any of the frequencies are negative (beyond a few tens of cm−1, which could be
within the margin of error for the calculation), then this indicates that the geometric
configuration is not located at a stable minimum. Try nudging the atoms slightly
along the directions of the negative mode and rerun an optimization to try to push the
system to a stable ground state. Rerun the frequency calculation until all frequencies
are positive.
•This output file will serve as one of the inputs to calculate Huang-Rhys factors. The
second input will be the relaxed, excited state configurations.
2. Excited state geometry relaxation
In this step, six different calculations will be performed, a geometry relaxation for each
of the six LR-TDDFT excitations included in this example calculation.
•Move to Porphyrin Example/FC DOS/Excited States. In this folder, six folders la-
beled s1 to s6 exist. An optimization of the excited state configuration will be run
in each folder. At the end, output of each final configuration will be concatenated
together to serve as one input file for the Huang-Rhys factor calculations.
•Move into s1. Open the porph s1.com input file to understand its contents. All input
will be similar to the linear-response calculation except for two major differences:
–Root=1 has been included within the TD command - this tells Gaussian which
excited state to use for geometry optimization (since each excitation will have a
different charge density that will affect the nuclear configuration). This should
be 1 for the first excited state, 2 for the second excited state, and so on.
–OPT has been included in tandem with the TD command - this tells Gaussian to
perform a geometry optimization using the excited state charge density deter-
mined from the LR-TDDFT calculation.
25
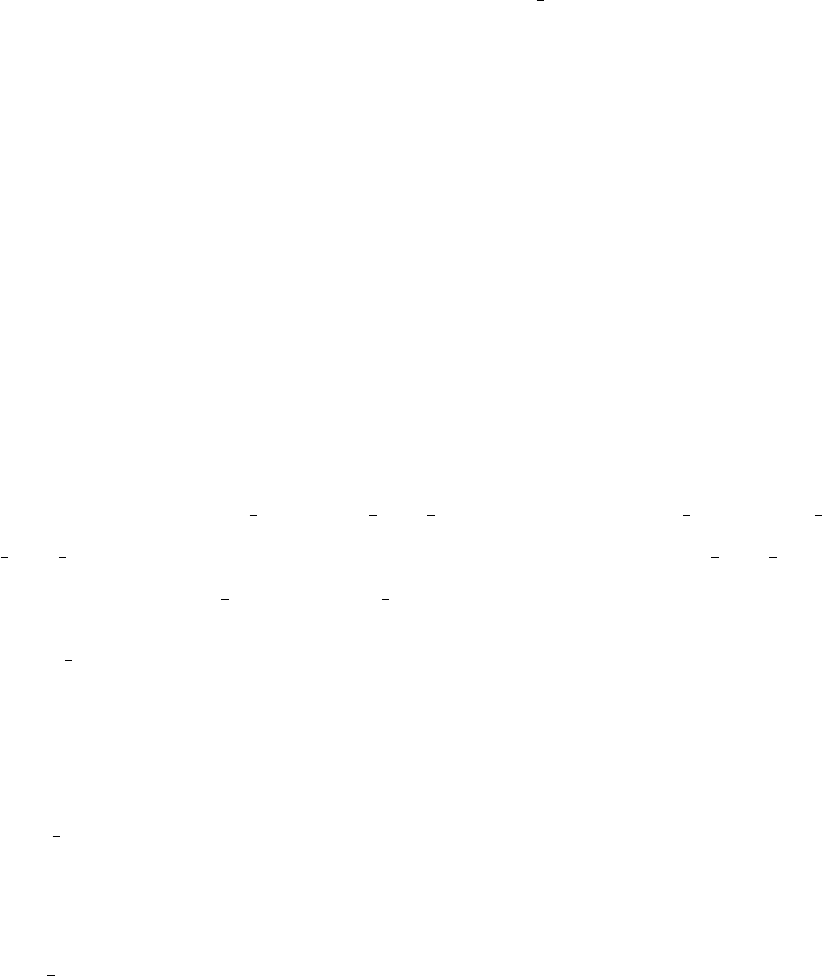
•Run this calculation to optimize the first excited state. These calculations can take
several hours to complete, so while waiting for it to finish, copy the input file and job
file into the other five folders, change the Root=X command to the relevant excited
state, and run the optimization calculations for these as well.
•Note: Make sure NOSYMMETRY is included in all calculations! If not, the molecule could
rotate during optimization, leading to unphysically large displacements in atoms and
nonsensical Huang-Rhys factors!
•Once all jobs have finished, check each output file (porph sX.out) to make sure the
geometry has relaxed to a minimum-energy configuration meeting the force criteria
set within Gaussian09. If any do not meet relaxation criteria, resubmit the job using
the same chk file and included GEOM=Check as an input command. This will instruct
Gaussian to read the last relaxed configuration from the chk file and continue opti-
mization.
3. Huang-Rhys factors
With phonon modes and eigenvectors as well as all ground and excited state configura-
tions, the next step will calculate the Huang-Rhys factors (Sk
αβ, Equation 12) between all
combinations of ground and excited states.
•Copy the contents of Codes/FC DOS/Huang Rhys Factors into Porphyrin Example/FC DOS/
Huang Rhys Factors. These files include the code that calculates Sk
αβ (Huang Rhys Factors.cpp),
and two input files (energy pop and input fcf):
–energy pop lists the minimum and maximum indices of all states included
(ground state starts as 1). The rest of the rows then include three columns:
1) state index; 2) state energy (in eV); and 3) occupancy (2 for occupied, 0 for
unoccupied). Fill in this file using results from the linear response calculation.
–input fcf provides input to calculate Sk
αβ, mainly atom number and mass to
calculate the reduced mass:
∗natoms indicates the total number of atoms.
∗n types indicates the total number of atom types.
26

∗The first two lines must contain natoms and n types (one on each line).
There should then be a subsequent input line for each atom type. For each
line, input the atomic symbol, atomic number, mass, and number of this
atom type, in this order (see example input file for porphyrin example).
•In addition, three more input files must be created from the Gaussian calculations in
the previous two steps:
–First, copy the output file from the phonon calculation from porph phonons.out
to this directory and rename it gauss out freq.
–Second, extract all phonon energies from the phonon output file and list them in
a separate file called modes cm in ascending order (the same order listed in the
Gaussian output so they coincide with the correct eigenvectors). Use grep or awk
to do this quickly.
–Third, concatenate the final atomic configurations from each output file of the
ground state calculation (taken from the phonon calculation) and each of the
excited state geometry relaxation calculations. Do this by deleting all lines from
each output file except for the final configuration, keeping the top four lines of
the header that include the dashed lines and thet text Center Number, Atomic
Number, Atomic Type, and Coordinates (Angstroms). See an example in
the sample output folder to better understand which part of the output to keep.
Once each of these configurations have been concatenated, in order of ascending
states, into one file, copy it to the Huang Rhys Factors folder and rename it
gauss out coord.
•Compile and run the code with the above input files in the same directory. This will
project atomic displacements between excited states onto mode eigenvectors and find
reduced masses in order to calculate Sk
αβ as in Equation 12. The following output files
will be created:
–output HRF matrix contains the Sk
αβ matrix that will be used as input in the
next section to calculate F(ωαβ).
–output deltaR provides the configurational difference between each ground and
excited state configuration, both in sum and for each atom.
27
10-2
10-1
100
0 1000 2000 3000
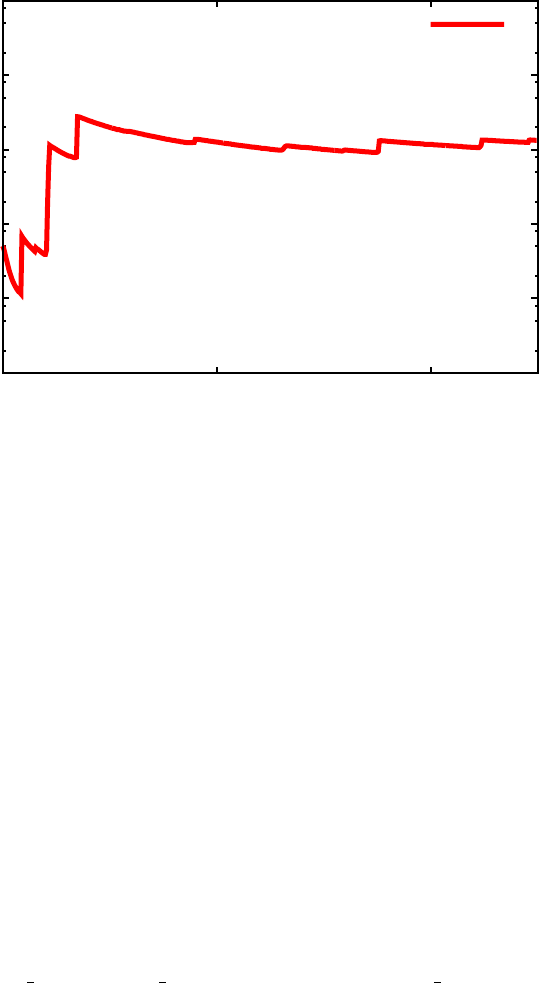
Sαβ (unitless)
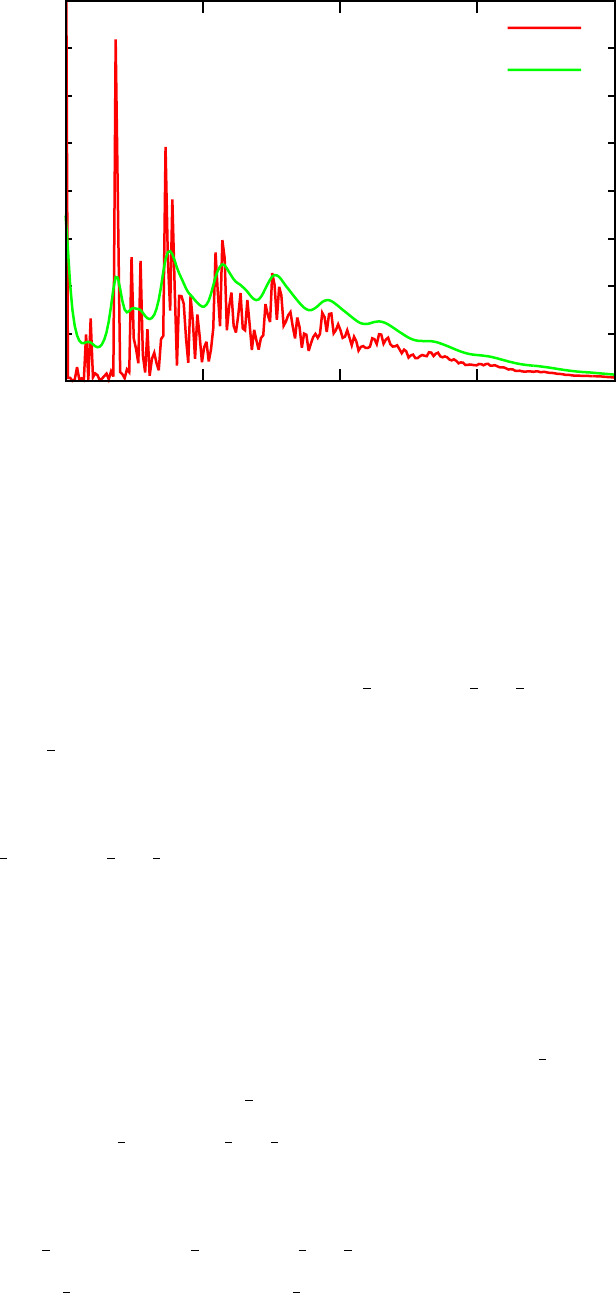
Frequency (cm-1)
Sk
32
FIG. 5. Huang-Rhys factors (Sk
αβ) as a function of phonon mode energy coupling the 2nd and 3rd
excited staters in porphyrin.
–output HRF info contains a list of phonon mode index, energy, absolute displace-
ment along this mode, Sk
αβ, and reorganization energy for each state combination.
This can be helpful for plotting Huang-Rhys factors and finding dominant modes
coupled to a given transition. For example, Figure 5 plots the Huang-Rhys fac-
tors Sk
23 (corresponding to Bands 3 and 4 in the output, since the ground state
is indexed as 1).
–output dos unweighted provides a phonon density of states with 25 meV smear-
ing.
–output phonon lists phonon mode energies and reduced masses.
The key output file here is output HR matlab, as it contains all Sk
αβ elements necessary
to construct G(t) in Equation 11 and calculate F(ωαβ) for each possible transition. This
will be done in the next section, after which the phonon-induced relaxation rates can be
calculated.
28

4. Franck-Condon-weighted density of states
The output HR matlab,modes cm, and energy pop files will be used as input to calculate
the matrix elements of F(ωαβ ) elements for each transition using a Matlab code (see Section
II D).
•Copy output HR matlab,modes cm, and energy pop from Porphyrin Example/FC DOS/
Huang Rhys Factors to Porphyrin Example/FC DOS/FC DOS Calc.
•Copy the Matlab code in Codes/FC DOS/FC DOS Calc to the same folder. The code,
FC DOS Calculation.m, will perform three major functions: 1) construct G(t) in Equa-
tion 11 using Sk
αβ and phonon modes ωkprovided as input from output HR matlab
and modes cm; 2) perform the inverse Fourier transform to construct the F(ω) function
for each transition; and 3) fit a cubic spline to each F(ω) and pick out the value of the
function at the corresponding transition energy ωαβ,F(ωαβ). A matrix of the same
size as Vαβ will be created with the values of F(ωαβ ) to be used as input in the final
section to calculate phonon-induced relaxation dynamics.
•Open the file to understand the input section (and rest of code if desired):
–e min and e max indicate the first and last excited state indices. Remove the row
containing these values in energy pop.
–energy pop loads in the system’s energy levels from file.
–HR factors loads in the system’s Huang-Rhys factors from the file created in the
previous section.
–modes cm loads in the system’s phonon modes from the file created for the previ-
ous section. These modes are then converted to units of eV and angular frequency.
–IMPORTANT: The Fourier transform in this code should result in a lineshape whose
real part is always positive. In Matlab, this only occurs if enough timesteps and
a short enough unit time step is used to exactly capture the period of all normal
mode frequencies. Unfortunately, this can be prohibitively costly if using the
modes with their default floating point representation. Thus, the code converts
the modes to units of inverse femtosecond, and then rounds to three significant
29
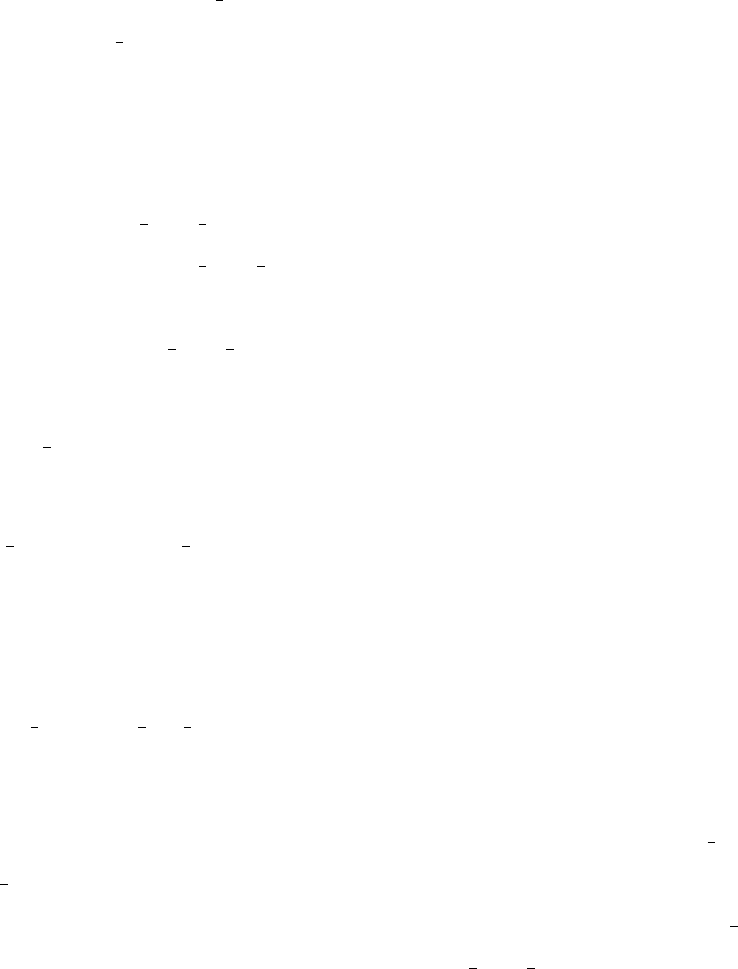
digits by default. This usually works well for the default 10,000 timesteps in the
code, however a user can test and change these parameters. Just make sure to
always check that the real part of the Fourier transform is positive!
– IMPORTANT:num ts indicates the number of time steps used to construct
G(t), and f s indicates the sampling frequency used for the Fourier transform
to calculate F(ωαβ). Again, make sure these choice of values gives only positive
values for the real part of the Fourier transform performed later in the code.
Values of 10,000 and 10 Hz (default) should suffice for most cases.
–Outputs: 1) x axis ev provides the x-axis in units of eV for plotting F(ω) for
each transition; 2) x axis cm provides the same in units of cm−1; 3) lineshapes
plots F(ω) in columns corresponding to each transition in the same order they
appear in output disp norm from the Huang-Rhys factor calculations. Use either
of the previous axis files to plot against; and 4)
–temp smearing provides the temperature used in calculating the Bose-Einstein
distribution.
–l smearing and g smearing provide broadening parameters in eV for optional
Lorentzian and Gaussian smearing, respectively. Traditionally, the spectrum
should be broadened at least using Lorentzian lineshapes to account for dephasing
times. Gaussian smearing can also be used to account for thermal effects.
–fcf weighted dos values.txt contains the F(ωαβ ) matrix used as input for the
next section.
•After understanding the input and code, run the Matlab code using num ts=10000
and f s=10. The 12th column of the lineshapes output contains F(ω) for the S3-S2
transition. Rerun the code after turning on Lorentzian smearing (smearing l = 1).
Plot F(ω) with and without smearing using the x axis ev file as the x-axis input.
Compare results to those shown in Figure 6. Applying smearing makes the end result
more realistic when considering homogeneous broadening from electronic dephasing.
•Note that the transition energy for the S3-S2transition is about 0.78 eV - the code
then uses the cubic spline fitting to evaluate F(ω) at this energy value, which ends up
30
0
5
10
15
20
25
30
35
40
0 0.25 0.5 0.75 1
F(ω)
Energy (eV)
No smearing
25 meV Lorentzian
FIG. 6. The F(ω) function for the S3-S2transition with and without 25 meV Lorentzian smearing.
F(ωαβ) is evaluated at the transition energy, which is around 0.78 eV.
being F(ω32)≈2.5 (remember, S0is the ground state, so F(ω32) corresponds to the
4th row and 3rd column of the matrix in fcf weighted dos values).
•Try testing ts int in the same way - increasing this variable will decrease the maxi-
mum energy for which F(ω) is calculated.
Save the fcf weighted dos values.txt output file from the Lorentzian smearing calcu-
lation. This will be used as input in the final section to calculate relaxation rates.
D. Phonon-induced relaxation dynamics
The final step will gather excitation energy information (energy pop, the nonadiabatic
electronic coupling matrix (Vαβ,final mat), and the Franck-Condon-weighted density of
states matrix (F(ωαβ,fcf weighted dos values) in the same directory and run a final
Matlab code to calculate phonon-induced relaxation rates over time.
•Copy energy pop and fcf weighted dos values from the FC-DOS calculations
into Porphyrin Example/Relaxation Dynamics. Also, copy the final output ma-
31

trix contaning Vαβ from Section ?? into the same directory (here it is referred
to as final mat). Finally, copy Phonon Induced Dynamics LRTDDFT RDM.m from
Codes/Relaxation Dynamics into this directory as well.
•This final code solves the Redfield equations given in Equation 1 using relaxation rates
defined in 7. For each transition, kαβ is calculated using the Vαβ and F(ωαβ) matrix
elements, and the resulting phonon-induced relaxation rates are determined. If this
Matlab code is run with visualization enabled, then the output will be a graph of
the an excited population’s energy as a function of time as described in Section II E.
The population of a given state can then be fit to an exponential decay to determine
excited state lifetimes.
•Open the code to understand the input:
–energy pop loads in the ground and excited state energies.
–Vab2 loads in the thermally averaged, squared nonadiabatic electronic coupling
matrix (Vαβ).
–field loads in the Franck-Condon-weighted density of states matrix (F(ωαβ)).
–forME calculates the relaxation rates (kαβ) and converts them to units of ps−1.
–Omin and Omax are the minimum and maximum state index (ground state is 1).
–inib indicates the initial excitation - phonon-induced relaxation dynamics will
inititiate witht the entire excitation in this state.
–time indicates the time (x-axis) grid along which to calculate energies of the
excited state as a function of time. Over time, electron-phonon interactions will
reduce the energy of the initially excited state until it reaches the ground state.
Each of the possible choices for time provides a different range, from 200 ps up to
100 ns. The choice of range depends on the system being studied. Here, choose
the first range that goes up to 200 ps.
–Egrid provides the energy range to visualize state dynamics - make sure this
range encompasses all excited state energies included in energy pop.
•Since the Soret band corresponds to excited states 3 and 4, this correponds to states
4 and 5 in energy pop. We will begin with a photoexcited electron in the higher of
32
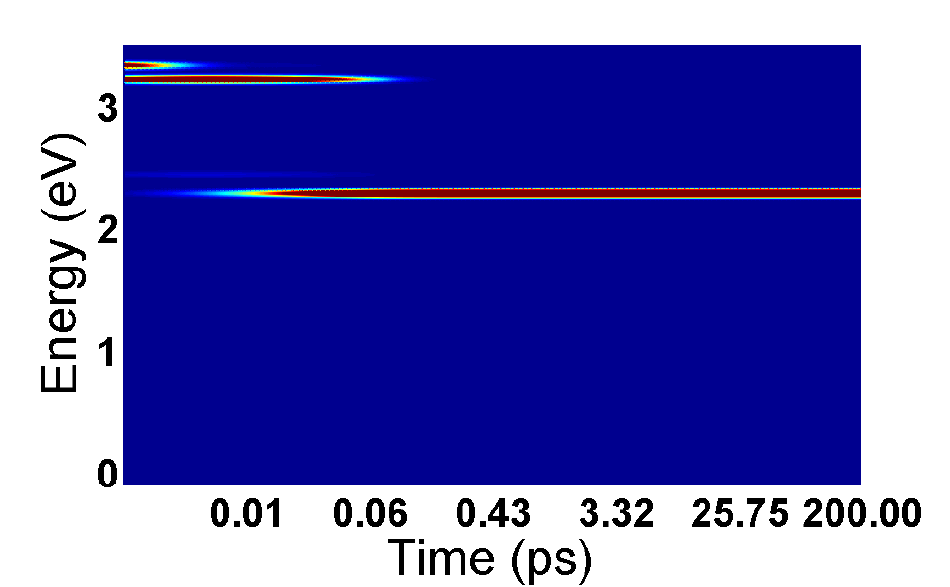
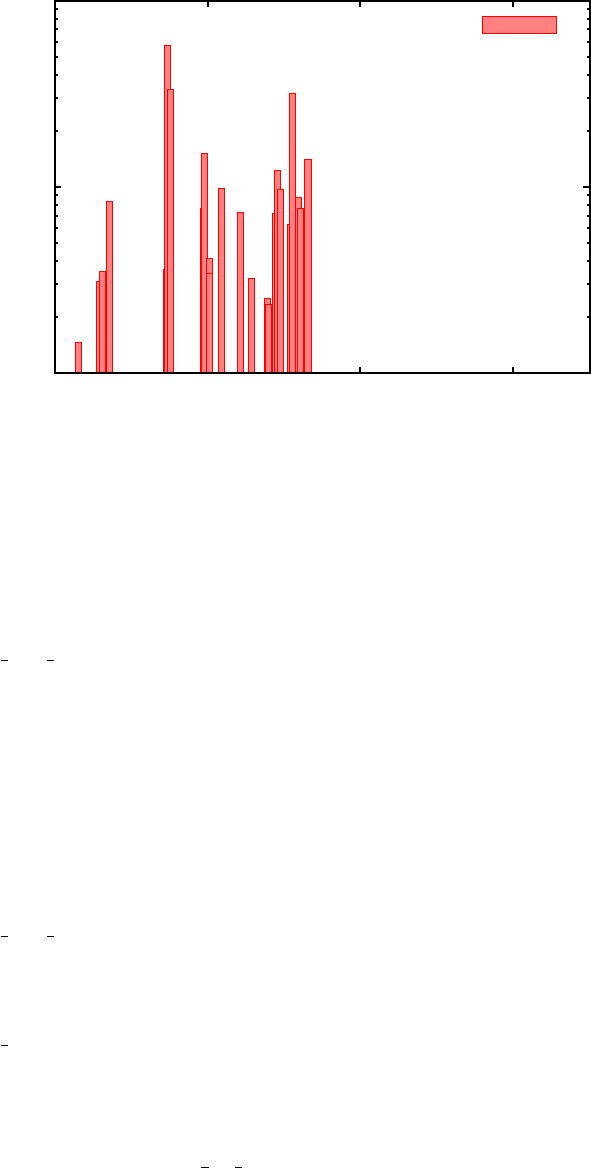
FIG. 7. Excited state energy as a function of time for porphyrin molecule with initial excitation
to Soret band (3.33 eV).
the two states, so set inib=5.
•Make sure the names of all input files match what the Matlab code will look for,
and then run the code. If visualization is enabled, the output should look like Figure
7, where darker red colors indicate more population at that energy at a given time.
This graph just visualizes the PPe output file, which can be visualized using any other
software as well.
From these results, one can see that an initial excitation to the top Soret band (3.33 eV),
leads to fast relaxation to the lower Soret band (3.22 eV) in under 10 fs. From there, it
takes about 30 fs to relax to the lowest excited state at 2.28 eV. This 30 fs Soret lifetime
agrees extremely well with experimental results that predict the relaxation rate to be under
50 fs8, supporting the accuracy of the present method. The excitation stays in this state for
over 200 ps - if the time range were extended, nonradiative relaxation to the ground state
would be seen in around 10 ns.
33

Congratulations! This example illustrates all the major tools included in this method
- although porphyrin only contains 38 atoms, this method can be applied to systems with
hundreds of atoms and is still computationally feasible.
Good luck and please provide feedback about the method and manual at jptrinas-
tic@gmail.com.
1J. Trinastic, I.-H. Chu, and H.-P. Cheng, J Phys Chem C , under review (2015).
2V. May and O. K¨uhn, Charge and energy transfer dynamics in molecular systems (John Wiley
& Sons, 2008).
3M. Khalil, N. Demirdven, and A. Tokmakoff, J. Chem. Phys. 121, 362 (2004).
4O. V. Prezhdo and P. J. Rossky, J. Chem. Phys. 107, 5863 (1997).
5I. Tavernelli, B. F. E. Curchod, A. Laktionov, and U. Rothlisberger, J. Chem. Phys. 133,
194104 (2010).
6I. Tavernelli, E. Tapavicza, and U. Rothlisberger, J. Chem. Phys. 130, 124107 (2009).
7M. E. Casida, Journal of Molecular Structure: THEOCHEM 914, 3 (2009).
8A. Marcelli, P. Foggi, L. Moroni, C. Gellini, and P. R. Salvi, The Journal of Physical Chemistry
A112, 1864 (2008).
9K. Y. Yeon, D. Jeong, and S. K. Kim, Chemical Communications 46, 5572 (2010).
10 Y. He, Y. Xiong, Q. Zhu, and F. Kong, Acta Physico-Chimica Sinica 15, 636 (1999).
34
