Manual
User Manual: Pdf
Open the PDF directly: View PDF ![]() .
.
Page Count: 115 [warning: Documents this large are best viewed by clicking the View PDF Link!]
- Preface
- 1 Introduction
- 2 Input for DFTB+
- 3 Transport calculations
- 4 Output of DFTB+
- 5 modes
- 6 Waveplot
- A The HSD format
- B Unit modifiers
- C Description of the gen format
- D Atomic spin constants
- E Dispersion constants
- F Publications to cite
- Index
DFTB+XT
open software package for quantum nanoscale modeling*
USER MANUAL
Version 1.01, Release January 18, 2018
*http://quantranspro.org/dftb+xt/
2
Contents
Preface 7
1 Introduction 9
2 Input for DFTB+11
2.1 Main input ....................................... 12
2.2 Geometry ....................................... 12
2.2.1 Explicit geometry specification ........................ 12
2.2.2 GenFormat{} ................................. 13
2.2.3 NoGeometry{} ................................ 13
2.3 Driver ......................................... 14
2.3.1 SteepestDescent{} .............................. 14
2.3.2 ConjugateGradient{} ............................. 16
2.3.3 gDIIS{} .................................... 17
2.3.4 SecondDerivatives{} ............................. 17
2.3.5 VelocityVerlet{} ............................... 18
2.3.6 Socket{} ................................... 24
2.4 Hamiltonian ...................................... 25
2.4.1 Mixer ..................................... 30
2.4.2 SpinPolarisation ............................... 32
2.4.3 SpinOrbit ................................... 36
2.4.4 Eigensolver .................................. 36
2.4.5 Filling ..................................... 37
2.4.6 SlaterKosterFiles ............................... 38
2.4.7 KPointsAndWeights ............................. 39
2.4.8 OrbitalPotential ................................ 41
2.4.9 ElectricField ................................. 41
2.4.10 Dispersion .................................. 43
2.4.11 DFTB3 .................................... 47
2.4.12 Differentiation ................................ 48
2.4.13 ForceEvaluation ............................... 49
2.5 Options ........................................ 49
2.6 Analysis ........................................ 50
2.7 ExcitedState ...................................... 52
2.7.1 Casida ..................................... 53
2.8 ParserOptions ..................................... 54
3
4CONTENTS
3 Transport calculations 57
3.1 Definition of the geometry .............................. 57
3.2 Transport ....................................... 58
3.2.1 Device{} ................................... 59
3.2.2 Contact{} .................................. 59
3.2.3 Task = ContactHamiltonian{} ....................... 60
3.2.4 Task = UploadContacts{} .......................... 62
3.3 GreensFunction .................................... 62
3.4 Contour integration .................................. 64
3.5 Poisson solver ..................................... 65
3.5.1 Boundary Conditions ............................. 67
3.5.2 Electrostatic Gates .............................. 69
3.6 Model Hamiltonians ................................. 70
3.7 Elastic dephasing ................................... 70
3.7.1 Büttiker probes ................................ 71
3.7.2 Vibronic dephasing .............................. 71
3.8 Application to STM spectroscopy .......................... 72
3.9 Parallelizations .................................... 72
3.10 Analysis ........................................ 72
3.11 TunnelingAndDos ................................... 73
3.12 Troubleshooting .................................... 73
4 Output of DFTB+75
4.1 hamsqrN.dat, oversqr.dat ............................... 75
4.2 hamrealN.dat, overreal.dat .............................. 75
4.3 eigenvec.out, eigenvec.bin .............................. 76
4.4 charges.bin ...................................... 77
4.5 md.out ......................................... 77
4.6 Excited state results files ............................... 77
4.6.1 ARPACK.DAT ................................ 77
4.6.2 COEF.DAT .................................. 77
4.6.3 EXC.DAT ................................... 78
4.6.4 SPX.DAT ................................... 78
4.6.5 TDP.DAT ................................... 79
4.6.6 TRA.DAT ................................... 79
4.6.7 TEST_ARPACK.DAT ............................ 79
4.6.8 XCH.DAT .................................. 79
4.6.9 XplusY.DAT ................................. 79
4.6.10 XREST.DAT ................................. 80
5MODES 81
5.1 Input for MODES ................................... 81
5.1.1 Hessian{} ................................... 82
5.1.2 DisplayModes{} ............................... 82
6 WAVEPLOT 83
6.1 Input for WAVEPLOT ................................. 83
6.1.1 Options .................................... 84
6.1.2 Basis ..................................... 88
6.1.3 ParserOptions ................................. 90
CONTENTS 5
A The HSD format 91
A.1 Scalars and list of scalars ............................... 92
A.2 Methods and property lists .............................. 93
A.3 Modifiers ....................................... 94
A.4 File inclusion ..................................... 94
A.5 Processing ....................................... 95
A.6 Extended format .................................... 95
B Unit modifiers 99
C Description of the gen format 101
D Atomic spin constants 103
E Dispersion constants 105
F Publications to cite 107
Index 112
6CONTENTS
Preface
DFTB+XT is a general open source software package for
• fast atomistic electronic structure and molecular dynamics simulations
• model and atomistic quantum transport at nanoscale
• many-body nonequilibrium phenomena
• material and device modeling
DFTB+XT is an extended version of the DFTB+code.
DFTB+XT package [1] is based on the DFTB+[2,3] source code. Additionally, it suggests a
number of new features, which are in the testing phase and may be included into the DFTB+release
later. The extended functionality of DFTB+XTis mainly focused on many-body quantum transport
and applications in nanoscience, material and device modeling.
In this version we present the following new features.
• Model Hamiltonians for transport calculations
We introduced the possibility to read model Hamiltonians from external data files and use it
with or without a geometry structure. This is especially important for many-body quantum
transport problems. See Sec. 3.6.
• Elastic dephasing
Two models of elastic dephasing ("Büttiker probe" and "vibronic dephasing") can be used
now to include the dephasing and dissipation beyond the coherent Green function method.
Thus, we made a new step towards realistic material and device modeling. See Sec. 3.7.
• Application to STM spectroscopy
We added new options to simplify and make faster the calculation of currents for systems with
changeable geometry (like the STM setup). We also supply the python scripts for modeling
of the scanning process over tip position and voltage. See Sec. 3.8.
7
8CONTENTS
Chapter 1
Introduction
The code DFTB+is the Fortran 2003 successor of the old DFTB code, which implements the
density functional based tight binding approach [4]. The code has been completely rewritten from
scratch and extended with various features.
The main features of DFTB+are:
• Non-scc and scc calculations (with an expanded range of SCC accelerators)
–Cluster/molecular systems
–Periodic systems (arbitrary k-point sampling, band structure calculations, etc.)
• l-shell resolved calculations possible
• Spin polarised calculations (both collinear and non-collinear spin)
• Geometry and lattice optimisation
• Geometry optimisation with constraints (in xyz-coordinates)
• Molecular dynamics (NVE, NPH, NVT and NPT ensembles)
• Numerical vibrational mode calculations
• Finite temperature calculations
• Dispersion corrections (van der Waals interactions)
• Ability to treat f-electrons
• Linear response excited state calculations for cluster/molecular systems
• Geometry optimisation and molecular dynamics in singlet and triplet excited states of spin-
free molecules
• LDA+U/pSIC extensions
•L·Scoupling
• 3rd order on-site corrections (improved H-bonding)
• QM/MM coupling with external point charges (smoothing possible)
9
10 CHAPTER 1. INTRODUCTION
• OpenMP parallelisation
• Automatic code validation (autotest system)
• New user friendly, extensible input format (HSD or XML)
• Dynamic memory allocation
• Additional tool for generating cube files for charge distribution, molecular orbitals, etc.
(Waveplot)
Chapter 2
Input for DFTB+
DFTB+can read two formats, either XML or the Human-friendly Structured Data format (HSD).
If you are not familiar with the HSD format, a detailed description is given in appendix A. The
input file for DFTB+must be named dftb_in.hsd or dftb_in.xml. The input file must be present
in the working directory. To prevent ambiguity, the parser refuses to read any input if both files are
present. After processing the input, DFTB+creates a file of the parsed input, either dftb_pin.hsd
or dftb_pin.xml. This contains the user input as well as any default values for unspecified options.
The file also contains the version number of the current input parser. You should always keep
this file, since if you want to exactly repeat your calculation with a later version of DFTB+, it is
recommended to use this file instead of the original input. (You must of course rename dftb_pin.hsd
into dftb_in.hsd or dftb_pin.xml into dftb_in.xml.) This guarantees that you will obtain the same
results, even if the defaults for some non specified options have been changed. The code can also
produce dftb_pin.xml from dftb_in.hsd or vice versa if required (see section 2.8).
The following sections list properties and options that can be set in DFTB+input. The first column
of each of the tables of options specifies the name of a property. The second column indicates
the type of the expected value for that property. The letters “l”, “i”, “r”, “s”, “p”, “m” stand for
logical, integer, real, string, property list and method type, respectively. An optional prefixing
number specifies how often (if more than once) this type must occur. An appended “+” indicates
arbitrary occurrence greater than zero, while “*” allows also for zero occurrence. Alternative types
are separated by “|”. Parentheses serve only to delimit groups of settings.
Sometimes a property is only interpreted on meeting some condition(s). If this is the case, the the
third column gives details of the requirement(s). The fourth column contains the default value for
the property. If no default value is specified (“-”), the user is required to assign a value to that
property. The description of the properties immediately follows the table. If there is also a more
detailed description available for a given keyword somewhere else, the appropriate page number
appears in the last column.
Some properties are allowed to carry a modifier to alter the provided value (e.g. converting between
units). The possible modifiers are listed between brackets ([]) in the detailed description of the
property. If the modifier is a conversion factor for a physical unit, only the unit type is indicated
(length, energy, force, time, etc.). A list of the allowed physical units can be found in appendix B.
11

12 CHAPTER 2. INPUT FOR DFTB+
2.1 Main input
The input file for DFTB+(dftb_in.hsd/dftb_in.xml) must contain the following property defini-
tions:
Name Type Condition Default Page
Geometry p|m -12
Hamiltonian m-25
Additionally optional blocks of definitions may be present:
Name Type Condition Default Page
Driver m{} 14
Options p{} 49
Analysis p{} 50
ExcitedState p{} 52
ParserOptions p{} 54
Geometry Specifies the geometry for the system to be calculated. See p. 12.
Hamiltonian Configures the Hamiltonian and its options. See p. 25.
Driver Specifies a geometry driver for your system. See p. 14.
Options Various global options for the run. See p. 49.
Analysis Post-run analysis and properties options. See p. 50.
ExcitedState Calculations in excited state of the system. See p. 52.
ParserOptions Various options affecting the parser only. See p. 54.
2.2 Geometry
The geometry can be specified either directly by passing the appropriate list of properties or by
using the GenFormat{} method. It is also possible to make model calculations without geometry
with NoGeometry{} option.
2.2.1 Explicit geometry specification
If the geometry is being specified explicitly, the following properties can be set:
Periodic lNo
LatticeVectors 9r Periodic = Yes -
TypeNames s+ -
TypesAndCoordinates (1i3r)+ -
Periodic Specifies if the system is periodic in all 3 dimensions or is to be treated as a cluster. If set
to Yes, property LatticeVectors{} must be also specified.
LatticeVectors [length]The x,yand zcomponents of the three lattice vectors if the system is
periodic.
2.2. GEOMETRY 13
TypeNames List of strings with the names of the elements, which appear in your geometry.
TypesAndCoordinates [relative|length]For every atom the index of its type in the TypeNames
list and its coordinates. If for a periodic system (Periodic = Yes) the modifier relative is
specified, the coordinates are interpreted in the coordinate system of the lattice vectors.
Example: Geometry of GaAs:
Geometry = {
TypeNames = { "Ga" "As" }
TypesAndCoordinates [Angstrom] = {
1 0.000000 0.000000 0.000000
2 1.356773 1.356773 1.356773
}
Periodic = Yes
LatticeVectors [Angstrom] = {
2.713546 2.713546 0.
0. 2.713546 2.713546
2.713546 0. 2.713546
}
}
2.2.2 GenFormat{}
You can use the generic format to specify the geometry (see appendix C). The geometry specifica-
tion for GaAs would be the following:
Geometry = GenFormat {
2 S
Ga As
1 1 0.000000 0.000000 0.000000
2 2 1.356773 1.356773 1.356773
0.000000 0.000000 0.000000
2.713546 2.713546 0.
0. 2.713546 2.713546
2.713546 0. 2.713546
}
It is also possible to include the gen-formatted geometry from a file:
Geometry = GenFormat {
<<< "geometry.gen"
}
2.2.3 NoGeometry{}
The NoGeometry{} option is set by
14 CHAPTER 2. INPUT FOR DFTB+
Geometry = NoGeometry{}
it is used for model calculations without geometry, in this case the Hamiltonian is red from file. See
Sec. 3.6.
2.3 Driver
The driver is responsible for changing the geometry of the input structure during the calculation.
Currently the following methods are available:
{} Static calculation with the input geometry.
SteepestDescent{} Geometry optimisation by moving atoms along the acting forces. See p. 14.
ConjugateGradient{} Geometry optimisation using the conjugate gradient algorithm. See p. 16.
gDIIS{} Geometry optimisation using the modified gDIIS method. See p. 17.
SecondDerivatives{} Calculation of the second derivatives of the energy (the Hessian). See
p. 17.
VelocityVerlet{} Molecular dynamics with the velocity Verlet algorithm. See p. 18.
Socket{} Hands over control to an external program via a socket interface. See p. 24.
2.3.1 SteepestDescent{}
MovedAtoms (i|s)+ 1:-1
MaxForceComponent r1e-4
MaxSteps i200
StepSize r100.0
OutputPrefix s"geo_end"
AppendGeometries lNo
Constraints (1i3r)* LatticeOpt = No {}
LatticeOpt lPeriodic = Yes No
FixAngles lPeriodic = Yes, LatticeOpt = Yes No
FixLengths 3l FixAngles = Yes No No No
Isotropic lPeriodic = Yes, LatticeOpt = Yes No
Pressure rPeriodic = Yes, LatticeOpt = Yes 0.0
MaxAtomStep rMovedAtoms 6=None{} 0.2
MaxLatticeStep rPeriodic = Yes, LatticeOpt = Yes 0.2
ConvergentForcesOnly lSCC = Yes Yes
MovedAtoms Indices of the atoms which should be moved. The atoms can be specified as a
mixture of a list of atoms, ranges of atoms and/or the species of atoms. Index ranges are
specified as start:end (without white space as one word!), which inclusively selects all atoms
between start and end.
MovedAtoms = 1:6
# equivalent to MovedAtoms = { 1 2 3 4 5 6 }

2.3. DRIVER 15
Negative indices can be used to count backwards from the last atom (-1 = last atom, -2 =
penultimate atom, etc.):
MovedAtoms = 1:-1 # Move all atoms including the last
Species names can be used to select all atoms belonging to a given species:
MovedAtoms = Ga # select all Ga atoms
Various specifiers can be combined together:
# Move atoms 1, 2, 3, all Ga atoms, and the last two atoms.
MovedAtoms = 1:3 Ga -2:-1
MaxForceComponent [force]Optimisation is stopped, if the force component with the maximal
absolute value goes below this threshold.
MaxSteps Maximum number of steps after which the optimisation should stop (unless already
stopped by achieving convergence). Setting this value as -1 runs a huge() number of itera-
tions.
StepSize [time]Step size (δt) along the forces. The displacement δxialong the ith coordinate is
given for each atom as δxi=fi
2mδt2, where fiis the appropriate force component and mis the
mass of the atom.
OutputPrefix Prefix of the geometry files containing the final structure.
AppendGeometries If set to Yes, the geometry file in the XYZ-format will contain all the geome-
tries obtained during the optimisation (instead of containing only the last geometry).
Constraints Specifies geometry constraints. For every constraint the serial number of the atom is
expected followed by the x,y,zcomponents of a constraint vector. The specified atom is not
allowed to move along the constraint vector. If two constraints are defined for the same atom,
the atom will only by able to move normal to the the plane containing the two constraining
vectors.
Example:
Constraints = {
# Atom one can only move along the z-axis
1 1.0 0.0 0.0
1 0.0 1.0 0.0
}
LatticeOpt Allow the lattice vectors to change during optimisation. MovedAtoms can be option-
ally used with lattice optimisation if the atomic coordinates are to be co-optimised with the
lattice.1
FixAngles If optimising the lattice, allow only the lengths of lattice vectors to vary, not the angles
between them. For example if your lattice is orthorhombic, this option will maintain that
symmetry during optimisation.
1This is functional but not very efficient at the moment.
16 CHAPTER 2. INPUT FOR DFTB+
FixLengths If optimising the lattice with FixAngles =Yes, allow only the lengths of the specified
lattice vectors to vary.
Example:
Driver = ConjugateGradient {
LatticeOpt = Yes
FixAngles = Yes # Fix angles between lattice vectors
FixLengths = {Yes Yes No} # Allow only lat. vector 3 to change length
}
Isotropic If optimising the lattice, allow only uniform scaling of the unit cell. This option is
incompatible with FixAngles.
Pressure [pressure]If optimising the lattice, set the external pressure, leading to a Gibbs free
energy of the form G=E+PV −T S being printed as well (the included entropy term is only
the contribution from the electrons, therefore this is not the full free energy).
MaxAtomStep Sets the maximum possible line search step size for atomic relaxation.
MaxLatticeStep Sets the maximum possible line search step size for lattice optimisation. For
FixAngles or Isotropic calculations this is as a fraction of the lattice vectors or the volume
respectively.
ConvergentForcesOnly If using an SCC calculation, this option controls whether the geometry
optimisation will prematurely stop (= Yes) if the SCC cycle does not converge at any geomet-
ric step.
2.3.2 ConjugateGradient{}
MovedAtoms (i|s)+ 1:-1
MaxForceComponent r1e-4
MaxSteps i200
OutputPrefix s"geo_end"
AppendGeometries lNo
Constraints (1i3r)* {}
LatticeOpt lPeriodic = Yes No
FixAngles lPeriodic = Yes, LatticeOpt = Yes No
Isotropic lPeriodic = Yes, LatticeOpt = Yes No
Pressure rPeriodic = Yes 0.0
MaxAtomStep rMovedAtoms 6=None{} 0.2
MaxLatticeStep rPeriodic = Yes, LatticeOpt = Yes 0.2
ConvergentForcesOnly lSCC = Yes Yes
See previous subsection for the description of the properties.

2.3. DRIVER 17
2.3.3 gDIIS{}
Alpha r0.1
Generations i8
MovedAtoms (i|s)+ 1:-1
MaxForceComponent r1e-4
MaxSteps i200
OutputPrefix s"geo_end"
AppendGeometries lNo
Constraints (1i3r)* {}
LatticeOpt lPeriodic = Yes No
FixAngles lPeriodic = Yes, LatticeOpt = Yes No
Isotropic lPeriodic = Yes, LatticeOpt = Yes No
Pressure rPeriodic = Yes 0.0
MaxLatticeStep rPeriodic = Yes, LatticeOpt = Yes 0.2
ConvergentForcesOnly lSCC = Yes Yes
Specific properties for this method are:
Alpha Initial scaling parameter to prevent the iterative space becoming exhausted (this is dynami-
cally adjusted during the run).
Generations Number of generations to consider for the mixing.
See previous subsection for the description of the other properties.2
2.3.4 SecondDerivatives{}
Calculates the second derivatives of the energy (currently only using a numerical differentiation of
the forces). The derivatives matrix is written out for the i,jand kdirections of atoms 1...nas
∂2E
∂xi1∂xi1
∂2E
∂xj1∂xi1
∂2E
∂xk1∂xi1
∂2E
∂xi2∂xi1
∂2E
∂xj2∂xi1
∂2E
∂xk2∂xi1... ∂2E
∂xkn∂xkn
into hessian.out
Note: for supercell calculations, the derivatives are obtained at the q=0 point, irrespective of the
k-point sampling used.
Important: In order to get accurate results for the second derivatives (and the resulting frequen-
cies) you must set a smaller self-consistent tolerance than the default value in the Hamiltonian{}
section. We suggest SCCTolerance = 1e-7 or better. A less accurate tolerance can yield nonphysical
vibrational frequencies.
Atoms i+|m 1:-1
Delta r1e-4
Atoms Index of the atoms for which to calculate the second derivatives. The atoms can be specified
via indices, index ranges and species. (See MovedAtoms in section 2.3.1.)
2This approach is distinct from section 2.4.1, but uses a related algorithm based on Ref. [5] and comments from
P.R.Briddon.
18 CHAPTER 2. INPUT FOR DFTB+
Delta Step size for numerical differentiation of forces to get the second derivatives of the energy
with respect to atomic coordinates.
2.3.5 VelocityVerlet{}
The code propagates atomic motion using velocity Verlet dynamics with optional thermostats or
barostats to control the temperature and/or pressure. Information is printed out during the simulation
every MDRestartFrequency steps, and logged in the file md.out (see appendix 4.5).
MovedAtoms (i|s)+ 1:-1
Steps i-
TimeStep r-
KeepStationary lYes
Thermostat m-19
OutputPrefix s"geo_end"
MDRestartFrequency i1
Velocities (3r)* -
Barostat mPeriodic = Yes - 21
ConvergentForcesOnly lSCC = Yes Yes
Xlbomd pXlbomdFast not set 22
XlbomdFast pXlbomd not set 22
Masses p24
MovedAtoms List of atoms to move during the MD. (See more detailed description on page 14.)
Steps Number of MD steps to perform. In the case of a thermostat using a TemperatureProfile{}
the number of steps is calculated from the profile.
KeepStationary Remove translational motion from the system.
TimeStep [time]Time interval between two MD steps.
Thermostat Thermostating method for the MD simulation. See p. 19.
OutputPrefix Prefix of the geometry files containing the final structure.
MDRestartFrequency Interval that the current geometry and velocities are written to the XYZ
format geometry file and md.out (see section 4.5). In the case of SCC MD runs, the charge
restart information is also written at this interval overriding RestartFrequency (see section 2.5).
Velocities [velocity]Specified atomic velocities for all the atoms of the given structure (including
“velocities” for any stationary atoms, which are silently ignored). This option can be used
to restart an MD run, but make sure the geometry is consistent with the specified velocities.
The easiest way to do this is to copy both from the same iteration of the XYZ file produced
in the previous run (Note: the velocities printed in the XYZ files are specified in Å/ps, so
this should be set in the input). If restarting an SCC MD run, we strongly suggest you use
ReadInitialCharges, and in particular read charges for the geometry which you use to restart
(iterations at which charges are written to disc are marked in the XYZ file, with the most
recent being present in charges.bin).
Barostat Berendsen method barostat for the MD simulation. See p. 21.

2.3. DRIVER 19
ConvergentForcesOnly If using an SCC calculation, this option controls whether the molecular
dynamics will prematurely stop (= Yes) if the SCC cycle does not converge at any geometric
step. If the option is set to False, forces will be calculated using the non-converged charges
and the molecular dynamics continues. In this case you should consider using ForceEvalua-
tion = ’Dynamics’ (or ForceEvaluation = ’DynamicsT0’) in the Dftb block as it gives more
accurate forces for non-converged charges.
Xlbomd If present, extended Lagrangian type molecular dynamics is applied to speed up the sim-
ulation. For further options within the Xlbomd block see p. 22.
Masses If present, over-ride the atomic masses from the Slater-Koster files. See p. 24
Thermostat
None{} No thermostating during the run, only the initial velocities are set according to either a
given temperature or velocities, hence an NVE ensemble should be achieved for a reasonable time
step.
InitialTemperature r-
InitialTemperature [energy]Starting velocities for the MD will be created according the Max-
well-Boltzmann distribution at the specified temperature. This is redundant in the case of
specified initial velocities.
Andersen{} Andersen thermostat [6] sampling an NVT ensemble.
Note: Andersen thermostating has a reputation for suppressing diffusion and also prevents accu-
mulation of data for dynamical properties.
Temperature r|m -
ReselectProbability r-
ReselectIndividually l-
AdaptFillingTemp lNo
Temperature [energy]Target temperature of the thermostat. It can be either a real value, spec-
ifying a constant temperature through the entire run or the TemperatureProfile{} method
specifying a changing temperature. (See p. 21.)
ReselectProbability Probability for re-selecting velocities from the Maxwell-Boltzmann distri-
bution.
ReselectIndividually If Yes, each atomic velocity is re-selected individually with the specified
probability. Otherwise all velocities are re-selected simultaneously with the specified proba-
bility.
AdaptFillingTemp If Yes, the temperature of the electron filling is always set to the current tem-
perature of the thermostat. (The appropriate tag for the temperature of the electron filling is
ignored.)
Berendsen{} Berendsen thermostat [7] samples something like an NVT ensemble (but without
the correct canonical fluctuations, be aware of the “flying ice cube” problem before using this
thermostat [8]).

20 CHAPTER 2. INPUT FOR DFTB+
Temperature r|m -
CouplingStrength rTimescale not set -
Timescale rCouplingStrength not set -
AdaptFillingTemp lNo
Temperature [energy]Target temperature of the thermostat. It can be either a real value specifying
a constant temperature through the entire run or the TemperatureProfile{} method specifying
a changing temperature. (See p. 21.)
CouplingStrength Dimensionless coupling strength for the thermostat (given as ∆t/τtin the orig-
inal paper where ∆tis the MD time step). Alternatively Timescale[time]can be set directly
as the characteristic length of time to damp the temperature towards the target temperature.
The CouplingStrength and Timescale options are mutually exclusive.
AdaptFillingTemp If Yes, the temperature of the electron filling is always set to the current tem-
perature of the thermostat. (The appropriate tag for the temperature of the electron filling is
ignored.)
NoseHoover{} Nosé-Hoover chain thermostat [9] sampling an NVT ensemble, currently with the
chain coupled to all of the atoms in the system.
Temperature r|m -
CouplingStrength r-
ChainLength i3
Order i3
IntegratorSteps i1
Restart m
AdaptFillingTemp lNo
Temperature [energy]Target temperature of the thermostat. It can be either a real value, spec-
ifying a constant temperature through the entire run or the TemperatureProfile{} method
specifying a changing temperature. (See p. 21, but note that profiles are not well tested with
this thermostat yet.)
CouplingStrength [Frequency]Frequency of oscillation of the thermostating particles (see sec-
tion 2.5 of Ref. [9]). This is typically related to the highest vibrational mode frequency of the
system.
ChainLength Number of particles in the thermostat chain.
Order and IntegratorSteps See section 4.3 of Ref. [9]).
Restart Specifies the internal state of the thermostat chain, using three keywords (all three must
be present): x{},v{} and g{} containing the internal chain positions, velocities and forces
respectively (these are provided in md.out). See also section 2.3.5.
AdaptFillingTemp If Yes, the temperature of the electron filling is always set to the current tem-
perature of the thermostat. (The appropriate tag for the temperature of the electron filling is
ignored.)

2.3. DRIVER 21
TemperatureProfile{} Specifies a temperature profile during molecular dynamics. It takes as
argument one or more lines containing the type of the annealing (string), its duration (integer), and
the target temperature (real), which should be achieved at the end of the given period. For example:
Temperature [Kelvin] = TemperatureProfile { # Temperatures in K
constant 1 10.0 # Setting T=10 K for the 0th MD-step
linear 500 600.0 # Linearly rising T in 500 steps up to T=600 K
constant 2000 600.0 # Constant T through 2000 steps
exponential 500 10.0 # Exponential decreasing in 500 steps to T=10 K
}
The annealing method can be constant,linear or exponential, with the duration of each stage of the
anneal specified in steps of the driver containing the thermostat. The starting temperature for each
annealing period is the final target temperature of the previous period, with the last step of each stage
being at the target temperature. Since the initial stage in the temperature profile has no previous step,
the default starting temperature is set to 0, but no actual calculation for that temperature is made.
In order to start the calculation from a finite temperature for the first annealing period, a constant
profile temperature stage with the duration of only one step should be specified as the first step (see
the example). The temperatures of the stages are specified in atomic units, unless the Temperature
keyword carries a modifier, as in the example above.
Barostat
Berendsen barostat [7] samples something like an NPH ensemble for MD (but without the correct
fluctuations). Options are provided for either isotropic or cell shape changing pressure control. This
can also be used in tandem with a thermostat (p. 19) for the NPT ensemble. If the barostat is used,
a partial Gibbs free energy is reported in code output, of the form
G=E+PV −T Selectronic.
This does not include structural entropy, only any electronic entropy. For barostated constant energy
simulations (no thermostat in use), the conserved quantity is the sum of the kinetic and Gibbs partial
energies.
Pressure r-
Coupling rTimescale not set -
Timescale rCoupling not set -
Isotropic lYes
Pressure [pressure]Sets the external target pressure.
Coupling Coupling strength for the barostat (given as β∆t/τpin the original paper where ∆tis
the MD time step and βthe compressibility, so the coupling is technically dimensioned as
reciprocal pressure, but this is usually ignored). Alternatively Timescale[time]can be set
directly (β/τp) as the characteristic length of time to damp the pressure. Typically, βis
assumed to be either the experimental value or ∼1, and Ref. [7] chooses the time scale to be
around 10–100 fs. The Coupling and Timescale options are mutually exclusive.
Isotropic Should isotropic scaling of the unit cell be used, or can the cell shape vary? There is a
slight inconsistency between the standard forms of these scalings in the literature, which is
reproduced here, in brief the isotropic case scales the cell volume by a factor proportional to
the differences in the instantaneous and expected pressures (i.e., the cube of the cell vectors),
while the anisotropic case changes the cell vectors proportional to the difference instead.

22 CHAPTER 2. INPUT FOR DFTB+
Extended Lagrangian Born-Oppenheimer dynamics
For several systems Born-Oppenheimer molecular dynamics simulations can be significantly sped
up by using the extended Lagrangian formalism described in Ref. [10]. The XLBOMD integrator
can be used in two different modes:
• Conventional XLBOMD scheme (Xlbomd): The extended Lagrangian is used to predict the
input charge distribution for the next time step, instead of taking charges that were converged
for the geometry in the previous time step. The predicted starting charges should then require
fewer SCC iterations to converge.
• Fast XLBOMD scheme, XlbomdFast (one diagonalisation per time step): The extended La-
grangian is used to predict the population for each time step. This predicted population is
then used to build the Hamiltonian, but in contrast to the conventional XLBOMD scheme,
there is no self consistent cycle – the forces are calculated immediately after the diagonali-
sation of the first Hamiltonian. The fast XLBOMD method usually only works for systems
without SCC instabilities (e.g. wider gap insulators or molecules without degenerate states).
See Ref. [10] for details.
The extended Lagrangian dynamics can be activated by specifying either the Xlbomd or the
XlbomdFast option block. Both methods offer the following options:
IntegrationSteps i5
PreSteps i0
IntegrationSteps Number of time steps used for determining the population for the next time
step. Currently, only integration schemes for 5, 6 or 7 steps are implemented.
PreSteps Number of molecular dynamics time steps before the XLBOMD integration becomes
activated.
Note: At the step where the XLBOMD integrator becomes active, it is initialised with results
of several subsequent converged MD steps, so a further IntegrationSteps + 1 steps will be car-
ried out before the extended Lagrangian dynamics starts to predict the charges and accelerate
the calculation.
The conventional Xlbomd block has the following specific options in addition to the common XL-
BOMD settings above:
MinSccIterations i1
MaxSccIterations i200
SccTolerance r1e-5
MinSccIterations Minimum number of SCC iterations to perform at each time step during the
extended Lagrangian dynamics.
MaxSccIterations Maximum number of SCC iterations to perform at each step in the extended
Lagrangian dynamics. If this number of SCC iterations have been reached the forces will be
calculated and the MD advances to the next time step. See the note in section 2.4.7 regarding
non-convergent k-point sampling.

2.3. DRIVER 23
SccTolerance SCC convergence tolerance during the extended Lagrangian dynamics. Once this
tolerance has been achieved the SCC cycle will stop and the forces will be calculated. You can
use this parameter to override the SccTolerance parameter in the DFTB block for time steps
where the extended Lagrangian integrator is active (This way, you can calculate populations
with tight SCC tolerance when initialising the XLBOMD integrator, then use a less strict SCC
tolerance once the integrator is active).
The XlbomdFast block allows has the following specific options in addition to the common XL-
BOMD settings above:
TransientSteps i10
Scale r1.0
TransientSteps Enables a smoother transition between Born-Oppenheimer and extended Lagrangian
dynamics by carrying out intermediate additional steps with full SCC convergence, during
which the converged population and the one predicted by the extended Lagrangian integrator
are averaged.
Scale Scaling factor for the predicted charge densities ∈(0,1]. The optimal value is system de-
pendent. One should take the highest possible value that still produces stable dynamics (good
conservation of energy).
Example for conventional XLBOMD:
Xlbomd {
IntegrationSteps = 6
MinSccIterations = 2
MaxSccIterations = 200
SccTolerance = 1e-6
}
Fast (SCC-free) XLBOMD with one diagonalisation per time step:
XlbomdFast {
PreSteps = 5
TransientSteps = 10
IntegrationSteps = 5
Scale = 0.5
}
Points to be aware of:
• The extended Lagrangian (especially in the fast mode) needs special force evaluation giving
more accurate forces for non-convergent charges. Therefore you must set the ForceEvaluation
option to ’Dynamics’ (for simulations with finite electronic temperature) or to ’DynamicsT0’
(for simulations at 0 K electronic temperature) in the DFTB block (see p. 49).
• The extended Lagrangian implementation only works for the (N,V,E)ensemble so far, so
neither thermostats nor barostats are allowed.
• The extended Lagrangian implementation currently cannot be used for spin-polarised or spin-
orbit systems, or when electron filling methods other than Fermi{} filling (see p. 37) are used.

24 CHAPTER 2. INPUT FOR DFTB+
Masses
Provides values of atomic masses for specified atoms, ranges of atoms or chemical species. This is
useful for example to set isotopes for specific atoms in the system.
Mass p
Any atoms not given specified masses will use the default values from the appropriate homonuclear
Slater-Koster file. An example is given below
Masses {
Mass {
Atoms = 1:2
MassPerAtom [amu] = 2.0
}
}
where Atoms specifies the atom or atoms which each have a mass of MassPerAtom assigned.
2.3.6 Socket{}
The code tries to connect to a socket interface to receive control instructions from an external driver
code.
File sHost not set -
Prefix sHost not set “/tmp/ipi_” for Protocol = i-PI{}
Host sFile not set -
Port iFile is set -
Verbosity i0
Protocol mi-PI{}
MaxSteps i200
File Name of UNIX style file socket to connect to.
Prefix Prefix to the file name, in the case of i-PI dialogue, the defaults to the path and file start that
i_PI expects.
Host Name or ip address of internet host to connect to (“localhost” also possible).
Port Port of host to connect to.
Verbosity Level of port traffic to document.
Protocol Choice of message protocol over the socket connection (only communication with i-PI [11]
is currently supported).
MaxSteps Number of geometry steps before termination of the DFTB+instance. Setting this
value as -1 runs a huge() number of iterations.
Examples
First an ip address connection:
2.4. HAMILTONIAN 25
Driver = Socket {
Host = localhost
Port = 21012 # port number
Verbosity = 0 # minimal verbosity
Protocol = i-PI {} # i-PI interface
MaxSteps = -1 # Run indefinitely
}
Then a UNIX socket via the /tmp file system
Driver = Socket {
File = "dftb" # The protocol defines a default path in this case
Protocol = i-PI {} # i-PI interface
MaxSteps = 1000 # Terminate this instance after 1000 steps
}
2.4 Hamiltonian
For calculations without geometry if Geometry = NoGeometry{}, the type of the Hamiltonian
must be set to Model{}:
Hamiltonian = Model{}
The properties of the Model{} method are discussed in Sec. 3.6.
Currently only a DFTB Hamiltonian is implemented for ab initio atomistic calculations, so you
must set Hamiltonian = DFTB{} or Hamiltonian = Model{}.

26 CHAPTER 2. INPUT FOR DFTB+
The DFTB{} method may contain the following properties:
SCC lNo
SCCTolerance rSCC = Yes 1e-5
MaxSCCIterations iSCC = Yes 100
EwaldParameter rPeriodic = Yes SCC = Yes 0.0
OrbitalResolvedSCC lSCC = Yes No
Mixer mSCC = Yes Broyden{} 30
MaxAngularMomentum p-
Charge r0.0
SpinPolarisation mSCC = Yes {} 32
SpinConstants pSpinPolarisation 6={} - 34
ShellResolvedSpin lOrbitalResolvedSCC = No No
SpinOrbit mSpinPolarisation 6=Colinear{} {} 36
Eigensolver mRelativelyRobust{} 36
Filling mFermi{} 37
SlaterKosterFiles p|m -38
OldSKInterpolation lNo
PolynomialRepulsive p|m {}
KPointsAndWeights (4r)+|m Periodic = Yes - 39
OrbitalPotential mSpinPolarisation 6={} {} 41
ReadInitialCharges lSCC = Yes No
InitialCharges pSCC = Yes {}
ElectricField pSCC = Yes {} 41
Dispersion m{} 43
DampXH lSCC = Yes No 47
DampXHExponent rDampXH = Yes -
ThirdOrder lSCC = Yes No
ThirdOrderFull lSCC = Yes No 47
HubbardDerivs pThirdOrder(Full) = Yes -
Differentiation mFiniteDiff 48
ForceEvaluation s"Legacy"
CustomisedHubbards p
Dephasing p70
SCC If set to Yes, a self consistent charge (SCC) calculation is made.
SCCTolerance Stopping criteria for the SCC. Specifies the tolerance for the maximum difference
in any charge between two SCC cycles.
MaxSCCIterations Maximal number of SCC cycles to reach convergence. If convergence is not
reached after the specified number of steps, the program stops. However in cases where the
calculation is not for a static structure (so Driver 6={}), this behaviour can be overridden
using ConvergentForcesOnly.
EwaldParameter Sets the dimensionless parameter αin the Ewald electrostatic summation for
periodic calculations. This controls the fraction of the Ewald summation occurring in real
and reciprocal space. Setting it to zero or negative activates an automatic determination of
this parameter (default and recommended behaviour). Setting it positive forces the code to
use the supplied value. This is useful for very asymmetrical unit cells (typically a slab or
nanowire with a huge surrounding vacuum region) since the memory demand of DFTB+can
2.4. HAMILTONIAN 27
increase dramatically in these cases (due to storage of a long range real space neighbour list).
To determine a suitable value of αfor such a cell, you should initially reduce the vacuum
region and run a test calculation, looking for the value of the automatically chosen Ewald
parameter in the standard output. This is then a suitable choice for the original cell with the
large vacuum region.
OrbitalResolvedSCC If set to Yes, all distinct Hubbard Uvalues for the different atomic angular
momenta shells are used, when calculating the SCC contributions. Otherwise, the value
supplied for the s-shell is used for all angular momenta. Please note, that the old standard
DFTB code was not orbitally resolved, so that only the Hubbard Ufor the s-shell was used.
Please check the documentation of the SK-files you intend to use as to whether they are
compatible with an orbitally resolved SCC calculation (many of the biological files do not
use orbitally resolved charges), before you switch this option to Yes. Even if the Hubbard U
values for different shells are the same in the SK-files, this flag would still effect your results,
since when it is set to Yes, any charge transfer between atomic shells will change the energy
of the system compared to when it is set to set to No.
Mixer Mixer type for mixing the charges in an SCC calculation. See p. 30.
MaxAngularMomentum Specifies the highest angular momentum for each atom type. All or-
bitals up to that angular momentum will be included in the calculation. Several main-block
elements require d-orbitals, check the documentation of the SK-files you are using to deter-
mine if this is necessary. Possible values for the angular momenta are s,p,d,f.
Example:
MaxAngularMomentum = {
Ga = "p" # You can omit the quotes around the
As = "p" # orbital name, if you want.
}
By using the SelectedShells method it is also possible to combine shells from different Slater-
Koster files together to treat atoms containing multiple shells with the same angular momen-
tum. The shells to be picked from a particular atom type should be listed without any sep-
arating characters. The list of shells of different atom types should be separated by white
spaces.
Example:
# Defining sps* basis for Si and C by combining sp and s shells from
# Si and Si2, and C and C2, resp.
MaxAngularMomentum = {
Si = SelectedShells { "sp" "s" } # Si atom with sps* basis
C = SelectedShells { "sp" "s" } # C atom with sps* basis
}
# Note, that you have to modify the Slater-Koster file definition accordingly
SlaterKosterFiles = {
Si-Si = "Si-Si.skf" "Si-Si2.skf" "Si2-Si.skf" "Si2-Si2.skf"
Si-C = "Si-C.skf" "Si-C2.skf" "Si2-C.skf" "Si2-C2.skf"
C-Si = "C-Si.skf" "C-Si2.skf" "C2-Si.skf" "C2-Si2.skf"
C-C = "C-C.skf" "C-C2.skf" "C2-C.skf" "C2-C2.skf"
}
28 CHAPTER 2. INPUT FOR DFTB+
If for a given atomic type you pick orbitals from more than one species, you must specify
an appropriate combinations of file names for the Slater-Koster tables in SlaterKosterFiles{}.
For every atom type combination nSK1 ×nSk2 Slater-Koster files must be defined, where nSK1
and nSK2 are the number species combined to build up the shells of the two interacting atoms.
The file names must be ordered with respect to the interacting species, so that the name
for the second interacting species is changed first. Above you see an example, where an
extended basis with an s∗-orbital was generated by introducing the new species "Si2" and
"C2", containing the appropriate s∗-orbital for Si and C, resp., as only orbitals.
In the case of multiple Slater-Koster files for a certain interaction, the repulsive data is read
from the first specified file (e.g. Si-Si.skf,Si-C.skf,C-Si.skf and C-C.skf in the example
above). The repulsive interactions in the other files are ignored. The mass for a certain
species is read from the first SK-file for its homo-nuclear interaction.
Non-minimal basis Slater-Koster data may be directly defined in the SK-files in future.
Charge Total charge of the system in units of the electron charge. Negative values mean an ex-
cess of electrons. If the keyword FixedFermiLevel is present (see section 2.4.5), then value
specified here will be ignored.
SpinPolarisation Specifies if and how the system is spin polarised. See p. 32.
SpinConstants Specifies the atom type specific constants needed for the spin polarised calcula-
tions, in units of Hartree. See p. 34.
SpinOrbit Specifies if the system includes Russel-Saunders coupling. See p. 36
Eigensolver Specifies which eigensolver to use for diagonalising the Hamiltonian. See p. 36.
Filling Method for occupying the one electron levels with electrons. See p. 37.
SlaterKosterFiles Name of the Slater-Koster files for every atom type pair combination. See 38.
OldSKInterpolation If set to Yes (strongly discouraged), the look-up tables for the overlap and
non-scc Hamiltonian contribution are interpolated with the same algorithm as in the old
DFTB code. Please note, that the new method uses a smoother function, is more systematic,
and yields better derivatives than the old one. This option is present only for compatibility
purposes, and may be removed in the future.
PolynomialRepulsive Specifies for each interaction, if the polynomial repulsive function should
be used. for every pairwise combination of atoms it should contain a logical value, where Yes
stands for the use of a polynomial repulsive function and No for a spline. If a specific pair of
species is not specified, the default value No is used.
Example:
# Use the polynomial repulsive function for Ga-Ga and As-As interactions
# in GaAs
PolynomialRepulsive = {
Ga-Ga = Yes
Ga-As = No
# As-Ga unspecifed, therefore per default set to No
As-As = Yes
}
2.4. HAMILTONIAN 29
If you want to apply the same setting for all species pairs, you can specify the appropriate
logical value as argument of the SetForAll keyword:
# Using polynomial repulsive functions for all interactions in GaAs
PolynomialRepulsive = SetForAll { Yes }
KPointsAndWeights [relative|absolute] Contains the special k-points to be used for the Brillouin-
zone integration. See p. 39. For automatically generated k-point grids the modifier should
not be set.
OrbitalPotential Specifies which (if any) orbitally dependant contributions should be added to
the DFTB energy and band structure. See p. 41.
ReadInitialCharges If set to Yes the first Hamiltonian is constructed by using the charge informa-
tion read from the file charges.bin.
InitialCharges Specifies initial net charges, either for all atoms or for only selected ones. In order
to specify it for all atoms, use the keyword AllAtomCharges and supply the net charge for
every atom as a list of real values:
InitialCharges = {
AllAtomCharges = { # Specifies net charge for each atom in an H2O molecule
-0.88081627 # charge for atom 1 (O)
0.44040813 # charge for atom 2 (H1)
0.44040813 # charge for atom 3 (H2)
}
}
Alternatively you can specify charges individually on atoms or species using the AtomCharge
keyword. For every AtomCharge declaration you must provide a net charge and the list of
atoms, which should be initialised to that net charge. (You can use the same format for the
list of atoms, as described at the MovedAtoms keyword in the section for SteepestDescent):
InitialCharges = { # Specifying charge for various species
AtomCharge = {
Atoms = H
ChargePerAtom = 0.44040813
}
AtomCharge {
Atoms = O
ChargePerAtom = -0.88081627
}
}
Net charge on atoms not appearing in any AtomCharge specification is set to be zero.
ElectricField Specifies an external electric field, arising currently from either an applied field or a
distribution of electrostatic charges. See p. 41.
Dispersion Specifies which kind of dispersion correction to apply. See p. 43.
Differentiation Specifies how to calculate finite difference derivatives in the force routines. See
p. 48.

30 CHAPTER 2. INPUT FOR DFTB+
ForceEvaluation Decides which expressions are used to calculate the ground state electronic
forces. See p. 49.Note: all methods give the same final forces when the charges are well
converged. For non-converged charges however the resulting forces can differ considerably
between methods.
CustomisedHubbards Enables overriding of the Hubbard U values for given species. If the option
OrbitalResolvedScc has been set to Yes, you need to specify one Hubbard U value for each
atomic shell in the basis of the given atom type, otherwise only one atomic value is required.
For all species not specified in this block, the value(s) found in their respective Slater-Koster
files will be used.
Warning: This option is for experts only! Overriding values stored in the SK-files may result
in inconsistent results. Make sure you understand the consequences when using this option.
Example:
CustomisedHubbards {
Si = 0.32 0.24
}
Dephasing Two models of elastic dephasing ("Büttiker probe" and "vibronic dephasing") can
be used now to include the dephasing and dissipation beyond the coherent Green function
method. Thus, we made a new step towards realistic material and device modeling. See
Sec. 3.7.
2.4.1 Mixer
DFTB+currently offers the charge mixing methods Broyden{} (Broyden-mixer), Anderson{} (Anderson-
mixer), DIIS{} (DIIS-accelerated simple mixer) and Simple{} (simple mixer).
Broyden{}
Minimises the error function
E=ω2
0
G(m+1)−G(m)
+
m
∑
n=1
ω2
n
n(n+1)−n(n)
|F(n+1)−F(n)|+G(m+1)F(n+1)−F(n)
|F(n+1)−F(n)|
2
,
where G(m)is the inverse Jacobian, n(m)and F(m)are the charge and charge difference vector in
iteration m. The weights are given by ω0and ωm, respectively. The latter is calculated as
ωm=c
√F(m)·F(m), (2.1)
cbeing a constant coefficient [12].
The Broyden{} method can be configured using following properties:
MixingParameter r0.2
InverseJacobiWeight r0.01
MinimalWeight r1.0
MaximalWeight r1e5
WeightFactor r1e-2

2.4. HAMILTONIAN 31
MixingParameter Mixing parameter.
InverseJacobiWeight Weight for the difference of the inverse Jacobians (ω0).
MinimalWeight Minimal allowed value for the weighting factors ωm.
MaximalWeight Maximal allowed value for ωm.
WeightFactor Weighting factor cfor the calculation of the weighting factors ωmin (2.1).
Note: As the Broyden-mixer stores a copy of the mixed quantity for each SCC iteration at a given
geometry, you may consider to choose a different mixer with lower memory requirements, if your
system needs density matrix mixing (e.g. DFTB+U), is large and needs a high number of SCC-
iterations (MaxSCCIteration).
Anderson{}
Modified Anderson mixer [13].
MixingParameter r0.05
Generations i4
InitMixingParameter r0.01
DynMixingParameters (2r)* {}
DiagonalRescaling r0.01
MixingParameter Mixing parameter.
Generations Number of generations to consider for the mixing. Setting it too high can lead to
linearly dependent sets of equation.
InitMixingParameter Simple mixing parameter used until the number of iterations is greater or
equal to the number of generations.
DynMixingParameters Allows specification of different mixing parameters for different levels
of convergence during the calculation. These are given as a list of tolerances and the mixing
factor to be used below this level of convergence. If the loosest specified tolerance is reached,
the appropriate mixing parameter supersedes that specified in MixingParameter.
DiagonalRescaling Used to increase the diagonal elements in the system of equations solved by
the mixer. This can help to prevent linear dependencies occurring during the mixing process.
Setting it to too large a value can prevent convergence. (This factor is defined in a slightly
different way from Ref. [13]. See the source code for more details.)
Example:
Mixer = Anderson {
MixingParameter = 0.05
Generations = 4
# Now the over-ride the (previously hidden) default old settings
InitMixingParameter = 0.01
DynMixingParameters = {
1.0e-2 0.1 # use 0.1 as mixing if more converged that 1.0e-2
1.0e-3 0.3 # again, but 1.0e-3

32 CHAPTER 2. INPUT FOR DFTB+
1.0e-4 0.5 # and the same
}
DiagonalRescaling = 0.01
}
DIIS{}
Direct inversion of the iterative space is a general method to acceleration iterative sequences. The
current implementation accelerates the simple mix process.
InitMixingParameter r0.2
Generations i6
UseFromStart lYes
MixingParameter Mixing parameter.
Generations Number of generations to consider for the mixing.
UseFromStart Specifies if DIIS mixing should be done right from the start, or only after the
number of SCC-cycles is greater or equal to the number of generations.
Simple{}
Constructs a linear combination of the current input and output charges as (1−x)qin +xqout.
MixingParameter r0.05
MixingParameter Coefficient used in the linear combination.
2.4.2 SpinPolarisation
In an SCC calculation, the code currently supports three different choices for spin polarisation;
non-SCC calculations are not spin polarised.
No spin polarisation ({})
No spin polarisation contributions to the energy or band-structure.
Colinear{}
Colinear spin polarisation in the zdirection. The following options can be specified:
UnpairedElectrons r0
RelaxTotalSpin lNo
InitialSpins p{}
UnpairedElectrons Number of unpaired electrons. This is kept constant during the run, unless
the RelaxTotalSpin keywords says otherwise.

2.4. HAMILTONIAN 33
RelaxTotalSpin If set to Yes, a common Fermi-level is used for both spin channels, so that the
total spin polarisation can change during run. In this case, the spin polarisation specified
using the UnpairedElectrons keyword is only applied at initialisation. If set to No (default),
the initial spin polarisation is kept constant during the entire run.
InitialSpins Optional initialisation for spin patterns. If this keyword is present, the default code
behaviour is that the initial input charge distribution has a magnetisation of 0. Otherwise if it
is not present, the initial input charge distribution has a magnetisation matching the number
of UnpairedElectrons keyword.
The initial spin distribution for the input charges can be set by specifying the spin polarisation
of atoms. For atoms without an explicit specification, a spin polarisation of zero is assumed.
The InitialSpins property block must contain either the AllAtomSpins keyword with a list of
spin polarisation values for every atom, or one or more AtomSpin blocks giving the spin for
a specific group of atoms using the following keywords:
Atoms (i|s)+ -
SpinPerAtom r-
Atoms Atoms to specify an initial spin value. The atoms can be specified via indices, index
ranges and species. (See MovedAtoms in section 2.3.1.)
SpinPerAtom Initial spin polarisation for each atom in this InitialSpins block.
For atoms not appearing in any of the SpinPerAtom block, an initial spin polarisation of 0 is
set.
Example (individual spin setting):
SpinPolarisation = Colinear {
UnpairedElectrons = 0.0
InitialSpins = {
AtomSpin = {
Atoms = 1:2
SpinPerAtom = -1.0
}
AtomSpin = {
Atoms = 3:4
SpinPerAtom = +1.0
}
}
}
Example (setting all spins together):
SpinPolarisation = Colinear {
UnpairedElectrons = 0.0
InitialSpins = {
AllAtomSpins = { -1.0 -1.0 1.0 1.0 } # Atoms 1,2: -1.0, atoms 3,4: 1.0
}
}
NonColinear{}
Non-collinear spin polarisation with arbitrary spin polarisation vector on every atom. The only
option allowed is to set the initial spin distribution:

34 CHAPTER 2. INPUT FOR DFTB+
InitialSpins p{}
InitialSpins Initialisation of the x,yand zcomponents of the initial spins for atoms. The default
code behaviour is an initial spin polarisation of (0 0 0) for every atom.
The initial spin distribution can be set by specifying the spin polarisation vector on all atoms
using the AllAtomSpins keyword or by using one or more AtomSpin blocks with the following
options:
Atoms (i|s)+ -
SpinPerAtom (3r)+ -
Atoms Atoms to specify an initial spin vector. The atoms can be specified via indices, index
ranges and species. (See MovedAtoms in section 2.3.1.)
SpinPerAtom Initial spin polarisation for each atom in this InitialSpins block.
For atoms not appearing in any of the SpinPerAtom block, the vector (0 0 0) is set.
Please note, that in contrast to the collinear case, in the non-collinear case you must specify
the spin vector (3 components!) for the atoms.
Example:
SpinPolarisation = NonColinear {
InitialSpins = {
# Setting the spin for all atoms in the system
AllAtomSpins = {
0.408 -0.408 0.816
0.408 -0.408 0.816
-0.408 0.408 -0.816
-0.408 0.408 -0.816
}
}
}
Example:
SpinPolarisation = NonColinear {
InitialSpins = {
AtomSpin = {
Atoms = 1:2
SpinPerAtom = 0.408 -0.408 0.816
}
AtomSpin = {
Atoms = 3:4
SpinPerAtom = -0.408 0.408 -0.816
}
}
}
SpinConstants
For each atomic species in the calculation the spin coupling constants for that atom must be speci-
fied.
2.4. HAMILTONIAN 35
When OrbitalResolvedSCC =No, an extra keyword in this block controls whether the spin constants
are resolved by shell or are identical for all shells: ShellResolvedSpin, defaulting to the same value
as OrbitalResolvedSCC.
When shell resolved spin constants are specified, they must be ordered with respect to the pairs of
shells they couple, such that the index for the second shell increases faster. For an spd-basis, that
gives the following ordering:
wss,wsp,wsd ,...,wps,wpp,wpd ,...,wds,wd p,wdd ,...
Example (GGA parameters for H2O):
SpinConstants = {
O={
# Wss Wsp Wps Wpp
-0.035 -0.030 -0.030 -0.028
}
H={
# Wss
-0.072
}
}
Several standard values of atomic spin constants are given in appendix D. Constants calculated with
the same density functional as the SK-files should be used. This input block may be moved to the
SK-data definition files in the future.
When using the SelectedShells method for the keyword MaxAngularMomentum, the spin constants
are listed as an array of values running over SK1SK2... in the same order as listed for SlaterKoster-
Files.
SpinConstants = { # not real values, only an example
Si = {
# Wss Wsp Wss*
-0.035 -0.030 -0.01
# Wps Wpp Wps*
-0.030 -0.037 -0.02
# Ws*s Ws*p Ws*s*
-0.01 -0.02 -0.01
}
For cases where ShellResolvedSpin =No, the spin constant for the the highest occupied orbital of
each atom should be supplied: Example (GGA parameters for H2O):
SpinConstants = {
O={
#Wpp
-0.028
}
H={
36 CHAPTER 2. INPUT FOR DFTB+
# Wss
-0.072
}
}
2.4.3 SpinOrbit
If present, specifies that L·Scoupling should be included for a calculation. Currently spin unpo-
larised and non-collinear spin polarisation are supported, but not collinear spin polarisation. For
every atomic species present in the calculation the spin-orbit coupling constants, ξ, for that atom
must be specified for all shells present. The constants must be ordered with respect to the list of
shells for the given atom.
In the case that the spin-orbit constant for an sorbital has been set to be a non-zero value the code
prints a warning. For periodic systems, use of this keyword automatically prevents the folding by
inversion normally used in SupercellFolding{}, but manually specified KPointsAndWeights should
not be reduced by inversion.
Example (GaAs):
SpinOrbit = {
Ga [eV] = {0.0 0.12 0.0} # s p d shells
As [eV] = {0.0 0.32703} # s p shells
}
The additional option in this block, Dual, sets whether to use a block population for the local spin
matrices consistent with the dual populations of Han et al. [14] or the conventional on-site part of the
single particle density matrix. The default value of this option is Yes, also giving extra information
regarding atomic orbital moments in the detailed output.
2.4.4 Eigensolver
Currently the following LAPACK 3.0 [15] eigensolver methods are available:
•QR{}
(QR decomposition based solver)
•DivideAndConquer{}
(this requires about twice the memory of the other solvers)
•RelativelyRobust{}
(using the subspace form but calculating all states)
None of them needs any parameters or properties specified.
Example:
Eigensolver = DivideAndConquer {}

2.4. HAMILTONIAN 37
2.4.5 Filling
There are currently two types of filling supported (see below). Both have common options:
Temperature rAdaptFillingTemp = No 0.0
IndependentKFilling lPeriodic = Yes No
FixedFermiLevel (1|2)r -
Temperature [energy]Electron temperature in energy units. This property is ignored for ther-
mostated MD runs, if the AdaptFillingTemp property of the thermostat has been set to Yes
(See p. 19).
IndependentKFilling Causes the occupation of the eigenstates to be independently determined
for each k-point, thus preventing electron transfer between the k-points. Please note that the
value for the Fermi level printed out by the code is meaningless in that case, since there is no
common Fermi level for all k-points. This option is incompatible with use of the FixedFer-
miLevel keyword.
FixedFermiLevel [energy]Can be used to fix the Fermi-level (total chemical potential, µ) of the
electrons in the system. For collinear spin polarisation, values for up and down spin channels
are required. Otherwise only a single global chemical potential is required. If this option is
present, the total charge and the total spin of the system are not conserved. (Settings in the
options Charge and UnpairedElectrons will be ignored.)
Fermi{}
Fills the single particle levels according to a Fermi distribution. When using a finite temperature,
the Mermin free energy (which the code prints) should be used instead of the total energy. This is
given by E−T S, where the electron entropy Sis used.
Example:
Filling = Fermi {
Temperature [K] = 300
}
MethfesselPaxton{}
Produces a Fermi-like distribution but with much lower electron entropy [16]. This is useful for
systems that require high electron temperatures (for example when calculating metallic systems).
There is an additional option for this type of filling:
Order i2
Order Order of the Methessel-Paxton scheme, the order must be greater than zero, and the 1st
order scheme is equivalent to Gaussian filling.
Note: Due to the non-monotonic behaviour of the Methfessel-Paxton filling function, the position of
the Fermi-level is not necessary unique for a given number of electrons. Therefore, different fillings,
band entropies, and Mermin free energies may result, depending which one has been found by the
Fermi-level search algorithm. The differences, however, are usually not physically significant.

38 CHAPTER 2. INPUT FOR DFTB+
2.4.6 SlaterKosterFiles
There are two different ways to specify the Slater-Koster files for the atom type pairs, explicit
specification and using the Type2FileNames{} method.
Explicit specification
Every pairwise permutation atomic types, connected by a dash, must occur as a property with the
name of the corresponding file as an assigned value.
Example (GaAs):
SlaterKosterFiles = {
Ga-Ga = "./Ga-Ga.skf"
Ga-As = "./Ga-As.skf"
As-Ga = "./As-Ga.skf"
As-As = "./As-As.skf"
}
If you treat shells from different species as shells of one atom by using the SelectedShells{} key-
word in the MaxAngularMomentum{} block, you have to specify more than one file name for cer-
tain species pairs. (For details see the description about the MaxAngularMomentum{} keyword.)
Type2FileNames{}
You can use this method to generate the name of the Slater-Koster files automatically using the
element names from the input geometry. You have to specify the following properties:
Prefix s""
Separator s""
Suffix s""
LowerCaseTypeName lNo
Prefix Prefix before the first type name, usually the path.
Separator Separator between the type names.
Suffix Suffix after the name of the second type, usually extension.
LowerCaseTypeName If the name of the types should be converted to lower case. Otherwise
they are used in the same way, as they were specified in the geometry input.
Example (for producing the same file names as in the previous section):
SlaterKosterFiles = Type2FileNames {
Prefix = "./"
Separator = "-"
Suffix = ".skf"
LowerCaseTypeName = No
}
The Type2FileNames method can not be used if an extended basis was defined with the Selected-
Shells method.
2.4. HAMILTONIAN 39
2.4.7 KPointsAndWeights
The k-points for the Brillouin-zone integration can either be specified explicitly or using the KLines{}
or the SupercellFolding{} methods. If the latter is used the KPointsAndWeights keyword is not al-
lowed to have a modifier.
Explicit specification
For every k-point four real numbers must be specified: The coordinates of the given k-point followed
by its weight. By default, the coordinates are specified in fractions of the reciprocal lattice vectors.
If the modifier absolute is set for the KPointsAndWeights keyword, absolute k-point coordinates in
atomic units are instead expected. The sum of the k-point weights is automatically normalised by
the program.
KPointsAndWeights = { # 2x2x2 MP-scheme
0.25 0.25 0.25 1.0
0.25 0.25 -0.25 1.0
0.25 -0.25 0.25 1.0
0.25 -0.25 -0.25 1.0
}
SupercellFolding{}
This method generates a sampling set containing all the special k-points in the Brillouin zone related
to points that would occur in an enlarged supercell repeating of the current unit cell. If two k-points
in the BZ are related by inversion, only one (with double weight) is used (in the absence of spin-orbit
coupling this is permitted by time reversal symmetry). The SupercellFolding{} method expects 9
integers and 3 real values as parameters:
n11 n12 n13
n21 n22 n23
n31 n32 n33
s1s2s3
The integers ni j specify the coefficients used to build the supercell vectors Aifrom the original
lattice vectors aj:
Ai=
3
∑
j=1
ni j aj.
The real values, si, specify the point in the Brillouin-zone of the super lattice, in which the folding
should occur. The coordinates must be given in relative coordinates, in the units of the reciprocal
lattice vectors of the super lattice.
The original l1×l2×l3Monkhorst-Pack sampling [17] for cubic lattices corresponds to a uniform
extension of the lattice:
l10 0
0l20
0 0 l3
s1s2s3
where siis 0.0, if liis odd, and siis 0.5 if liis even. For the 2×2×3 scheme, you would write for
example
40 CHAPTER 2. INPUT FOR DFTB+
# 2x2x3 MP-scheme according original paper
KPointsAndWeights = SupercellFolding {
200
020
003
0.5 0.5 0.0
}
To use k-points for hexagonal lattices which are consistent with the erratum to the original paper
[18], you should set the shift for the unique “c” direction, s3, in the same way as in the original
scheme. The s1and s2shifts should be set to be 0.0 independent of whether l1and l2are even or
odd. So, for a 2 ×3×4 sampling you would have to set
# 2x3x4 MP-scheme according modified MP scheme
KPointsAndWeights = SupercellFolding {
200
030
004
0.0 0.0 0.5
}
It is important to note that DFTB+does not take the symmetry of your system explicitly into
account. For small high symmetric systems with a low number of k-points in the sampling this
could eventually lead to unphysical results. (Components of tensor properties–e.g. forces–could be
finite, even if they must vanish due to symmetry reasons.) For those cases, you should explicitly
specify k-points with the correct symmetry.
KLines{}
This method specifies k-points lying along arbitrary lines in the Brillouin zone. This is useful when
calculating the band structure for a periodic system. (In that case, the charges should be initialised
from the saved charges of a previous calculation with a proper k-sampling. Additionally for SCC
calculations the number of SCC cycles should be set to 1, so that only one diagonalisation is done
using the initial charges.)
The KLines{} method accepts for each line an integer specifying the number of points along the
line segment, and 3 real values specifying the end point of the line segment. The line segments do
not include their starting points but their end points. The starting point for the first line segment
can be set by specifying a (zeroth) segment with only one point and with the desired starting point
as end point. The unit of the k-points is determined by any modifier of the KPointsAndWeights
property. (Default is relative coordinates.)
Example:
KPointsAndWeights [relative] = KLines {
1 0.5 0.0 0.0 # Setting (and calculating) starting point 0.5 0.0 0.0
10 0.0 0.0 0.0 # 10 points from 0.5 0.0 0.0 to 0.0 0.0 0.0
10 0.5 0.5 0.5 # 10 points from 0.0 0.0 0.0 to 0.5 0.5 0.5
1 0.0 0.0 0.0 # Setting (and calculating) a new starting point
10 0.5 0.5 0.0 # 10 points from 0.0 0.0 0.0 to 0.5 0.5 0.0
}

2.4. HAMILTONIAN 41
Note: Since this set of k-points probably does not correctly integrate the Brillouin zone, the default
value of MaxSccIterations is set to be 1 in this case.
2.4.8 OrbitalPotential
Currently the FLL (fully localised limit) and pSIC [19] (pseudo self interaction correction ) forms
of the LDA+U corrections [20] are implemented. These potentials effect the energy of states on
designated shells of particular atoms, usually increasing the localisation of states at these sites. The
FLL potential lowers the energy of occupied states localised on the specified atomic shells while
raising the energy of unoccupied states. The the pSIC potential corrects the local part of the self-
interaction error and so lowers the energy of occupied states (see Ref. [19] for a discussion of the
relation between these two potentials, and possible choices for the UJ constant). These particular
corrections are most useful for lanthanide/actinide fstates and some localised dstates of transition
metals (Ni3dfor example).
The Functional option chooses which correction to apply, followed by a list of the specific correc-
tions, listed as an atomic species and the shells on that atom followed by the U−Jconstant for that
block of shells.
OrbitalPotential = {
Functional = {FLL}
Si = {
Shells = {1 2} # sp block on the atom
UJ = 0.124
}
}
2.4.9 ElectricField
This tag contains the specification for an external electric field. Electric fields can only be specified
for SCC calculations. You can apply the electric field of point charges3and/or a homogeneous
external field (which may change harmonically in time). The ElectricField block can currently
contain either one or more PointCharges blocks and potentially an External block.
PointCharges
The specification for PointCharges has the following properties:
CoordsAndCharges (4r)+ -
GaussianBlurWidth rPeriodic = No 0.0
CoordsAndCharges [length]Contains the coordinates and the charge for each point charge (four
real values per point charge). A length modifier can be used to alter the units of the coordi-
nates. The charge must be specified in proton charges. (The charge of an electron is -1.)
If you read in a huge number of external charges the parsing time to process this data could be
unreasonably long. You can avoid this by including the coordinates and the charges directly
from an external file via the DirectRead{} method (see the example in the next paragraph).
3Only in calculations with fixed lattice constants.

42 CHAPTER 2. INPUT FOR DFTB+
Please note that when using this method the program will only read the specified number of
records from the external file, and ignores any additional data (so do not leave comments in
the external file for example). The external file should contain only one record (3 coordinates
and 1 charge) per line.
GaussianBlurWidth [length]Specifies the half width σof the Gaussian charge distribution, which
is used to delocalise the point charges. The energy of the coulombic interaction ECbetween
the delocalised point charge Mwith charge QMand the atom Awith charge qAis weighted by
the error function as
EC(A,M) = qAQM
rAM
erfhrAM
σi,
where rAM is the distance between the point charge and the atom.
This delocalisation can only be used for non-periodic systems. A length modifier can be used
to specify the unit for σ.
Example:
ElectricField = {
# 1st group of charges, with Gaussian delocalisation
# We have 100000 charges, therefore we choose the fast reading method.
PointCharges = {
GaussianBlurWidth [Angstrom] = 3.0
CoordsAndCharges [Angstrom] = DirectRead {
Records = 100000
File = "charges.dat"
}
}
# 2nd group of charges, no delocalisation (sigma = 0.0)
PointCharges = {
CoordsAndCharges [Angstrom] = {
3.3 -1.2 0.9 9.2
1.2 -3.4 5.6 -3.3
}
}
}
External
Specifies a homogeneous external electric field. In the case of periodic calculations, a saw-tooth
potential is currently used, hence it is up to the user to guarantee that there is a vacuum region
isolating periodic copies of the system along the applied field direction. We suggest that you place
the structure in the ‘middle’ of the unit cell if possible, to reduce the chances of atoms approaching
cell boundaries along the direction of the applied electric field. The code will halt if atoms interact
with periodic images of the unit cell along the direction of the electric field.
The External field keyword has the following options
Strength r-
Direction 3r
Frequency r molecular dynamics used 0.0
Phase iGeometry step offset 0

2.4. HAMILTONIAN 43
Strength [Electric field strength]Specified strength of the applied field.
Direction Vector direction of the applied field (the code normalises this vector). In the case of pe-
riodic calculations, currently the system must not be continuous in this direction (see above).
Frequency [Frequency]If using molecular dynamics, the field can be time varying with this fre-
quency.
Phase Initial field phase in units of geometry steps, this is needed if restarting an MD run in an
external field to give the offset in phase of the field after the specified number of steps from
the old calculation. The applied field is of the form
E0sin(ω∆t(step +phase))
where E0is the field vector specified by Strength and Direction,ωis the angular Frequency
and step is the current MD-step in the simulation, using the MD TimeStep of ∆t(see section
2.3.5).
2.4.10 Dispersion
The Dispersion block controls whether DFTB interactions should be empirically corrected for van
der Waals interactions, since DFTB (and SCC-DFTB) does not include these effects. Currently,
three different dispersion correction schemes are implemented (for the detailed description of the
methods see the following subsections):
•LennardJones: Dispersion is included via a Lennard-Jones potential between each pair of
atoms. The parameters for the potential can either be entered by the user or the program can
automatically take the parameters from the Universal Force Field (UFF) [21].
•SlaterKirkwood: The dispersion interaction between atoms is taken from a Slater-Kirkwood
polarisable atomic model [22].
•DftD3: Dispersion is calculated as in the dftd3 code [23,24].
LennardJones
The Lennard-Jones dispersion model in DFTB+follows the method of Ref. [25], using the follow-
ing potential:
Ui j(r) = di j −2ri j
r6+ri j
r12r>=r0
Ui j(r) = U0+U1r5+U2r10 r<r0
where r0is the distance at which the potential turns from repulsive to attractive. The parameters di j
and ri j are built from atomic parameters di,djand ri,rjvia the geometrical mean (di j =pdidj,
ri j =√rirj). The parameters U0,U1,U2ensure a smooth functional form at r0.
The parameters riand dican either be taken from the parameters of the UFF [21] (as in Ref. [25])
or can be specified manually for each species.
Example using UFF parameters:
44 CHAPTER 2. INPUT FOR DFTB+
Dispersion = LennardJones {
Parameters = UFFParameters {}
}
Example using manually specified parameters:
Dispersion = LennardJones {
Parameters {
H {
Distance [AA] = 2.886
Energy [kcal/mol] = 0.044
}
O {
Distance [AA] = 3.500
Energy [kcal/mol] = 0.060
}
}
}
The UFF provides dispersion parameters for nearly every element of the periodic table, therefore
it can be used for almost all systems “out of the box”. The parameters are also independent of the
atomic coordination number, allowing straight forward geometry relaxation or molecular dynamics
(unlike the current implementation of Slater-Kirkwood type dispersion).
SlaterKirkwood
A Slater-Kirkwood type dispersion model is also implemented in DFTB+as described in Ref. [22].4
This model requires atomic polarisation values, van der Waals radii and effective charges for every
atom in your system. These parameters are dependent on the coordination of each atom, hence
values for different atoms of the same species may vary depending on local environment. You can
supply these parameters for the atoms in either of two ways, both using the PolarRadiusCharge tag.
The first option is to specify the values within the PolarRadiusCharge environment by providing
three real values (polarisability, van der Waals radius, effective charge) for each atom separately.
Example:
Dispersion = SlaterKirkwood {
# Using Angstrom^3 for volume, Angstrom for length and default
# unit for charge (note the two separating commas between the units)
PolarRadiusCharge [Angstrom^3,Angstrom,] = {
# Polar Radius Chrg
0.560000 3.800000 3.150000 # Atom 1: O
0.386000 3.500000 0.800000 # Atom 2: H
0.386000 3.500000 0.800000 # Atom 3: H
}
}
4Please note, that Ref. [22] contains two typos: equation (7) should be read Cαβ
6=2Cα
6Cβ
6pαpβ
p2
αCβ
6+p2
βCα
6
, in equation (9) the
contribution from the dispersion should be Edis =−1
2∑αβ f(Rαβ )Cαβ
6(Rαβ )−6. This option is also currently incompat-
ible with lattice optimisation and the use of barostats.
2.4. HAMILTONIAN 45
Alternatively you can provide values for each atomic species in your system, but must supply dif-
ferent values for different coordination numbers using the HybridDependentPol{} keyword. The
code needs specific parameters for each type of atom in environments with 0, 1, 2, 3, 4 or >5 neigh-
bours. DFTB+then picks the appropriate values for each atom based on their coordination in the
starting geometry. Two atoms are considered to be neighbours if their distance is less than the sum
of their covalent radii, hence you need to supply the covalent radii for each atomic species using the
CovalentRadius keyword. This is then followed by a HybridPolarisations block containing a list of
six values for atomic polarisabilities then six van der Waals radii and finally a single hybridisation
independent effective charge for that atomic species.
Example:
Dispersion = SlaterKirkwood {
PolarRadiusCharge = HybridDependentPol {
O={
CovalentRadius [Angstrom] = 0.8
HybridPolarisations [Angstrom^3,Angstrom,] = {
# Atomic polarisabilities 0-5 van der Waals radii 0-5 chrg
0.560 0.560 0.560 0.560 0.560 0.560 3.8 3.8 3.8 3.8 3.8 3.8 3.15
}
}
H={
CovalentRadius [Angstrom] = 0.4
HybridPolarisations [Angstrom^3,Angstrom,] = {
# Atomic polarisabilities 0-5 van der Waals radii 0-5 chrg
0.386 0.396 0.400 0.410 0.410 0.410 3.5 3.5 3.5 3.5 3.5 3.5 0.8
}
}
}
}
Warning: For both methods of specifying the Slater-Kirkwood dispersion model the code keeps the
dispersion parameters fixed for each atom during the entire calculation. Even if the geometry (and
therefore the hybridisation) of atoms changes significantly during the calculation, the parameters are
unchanged. Therefore if atoms are able to move during your calculation (geometry relaxation or
molecular dynamics) you should always check whether the coordination of your atoms has changed
during the run.
Furthermore, when using the HybridDependentPol{} method we suggest that you first set the
StopAfterParsing keyword in the ParserOptions block to Yes (see p. 54) and inspect the gener-
ated polarisabilities, radii and charges for every atom in the dftb_pin.hsd file. If fine tuning of the
generated values turns out to be necessary, you should replace the hybrid dependent specification in
the input file with corrected atom specific values based on dftb_pin.hsd.
In order to find suitable parameters for the Slater-Kirkwood model, you should consult Ref. [22]
and further references therein. Appendix Econtains values which have already been used by some
DFTB-users for a few elements.
DftD3
The DFT-D3 dispersion correction in DFTB+is an implementation of the method used in the code
’dftd3’ by Stefan Grimme and coworkers. It is based on the ansatz described in Refs. [23] and [24].

46 CHAPTER 2. INPUT FOR DFTB+
Note: the DFTB+binary must be compiled with the DFT-D3 library enabled to use this feature.
This dispersion correction for DFTB adds a contribution to the general Kohn-Sham-like energy
EDFTB-D3 =EDFTB +Edisp
with EDFTB being the DFTB total energy and Edisp the dispersion energy. The latter contains two-
body and optional three-body contributions:
Edisp =E(2)
disp +E(3)
disp
The form of the two-body contribution can change depending on the chosen damping factor:
• Becke-Johnson damping function:
E(2)
disp =−1
2∑
A6=B
∑
n=6,8
sn
CAB
n
rn
AB +f(RAB
0)
with
f(RAB
0) = a1RAB
0+a2.
• Zero-damping (dispersion at short distances is damped to zero):
E(2)
disp =−1
2∑
A6=B
sn
CAB
n
rn
AB
fd,n(rAB)
with
fd,n=1
1+6(rAB/(sr,nRAB
0))−αn
In order to adjust the dispersion for various energy functionals, the choice of s6,s8and the damping
parameters a1and a2(for Becke-Johnson-damping) or sr,6and α6(for zero damping) are treated as
functional-dependent values. All other parameters are fixed based on these parameters.
As the DFTB energy functional is largely determined by the underlying parameterisation (the Slater-
Koster-files) and the chosen DFTB model (e.g. non-scc, scc, 3rd order, etc.), there are no universal
parameter choices which can be used with all settings. The dftd3-program contains optimised
parameters for the DFTB3 functional, described in section 2.4.11. The DFTB3 appropriate values
are given for the Becke-Johnson-damping function, and are used as default values in the DFTB+
parser.
Warning: Please note, that these default values have only been tested for the DFTB3 model. If you
use any other DFTB-model, make sure that the parameters still give reasonable results, and adjust
them if needed.
Example using default parameters:
ThirdOrderFull = Yes
DampXH = Yes
Dispersion = DftD3 {}
Example using adjusted parameters with Becke-Johnson damping:

2.4. HAMILTONIAN 47
Dispersion = DftD3 {
Damping = BeckeJohnson {
a1 = 0.5719
a2 = 3.6017
}
s6 = 1.0
s8 = 0.5883
}
Example using zero-damping:
Dispersion = DftD3 {
Damping = ZeroDamping {
sr6 = 0.7461
alpha6 = 14.0
}
s6 = 1.0
s8 = 3.209
}
Apart from the functional dependent dispersion parameters, you can also adjust the additional pa-
rameters as shown below. The default values for these parameters are taken to be the same as in the
dftd3 code.
Cutoff r√9000
CutoffCN r 40
Threebody lNo
Cutoff [length]Cutoff distance when calculating two-body interactions.
CutoffCN [length]Cutoff distance when calculating three-body interactions.
Threebody Whether three-body contributions should be included in the dispersion interactions.
2.4.11 DFTB3
If you would like to use what is called “DFTB3” in some publication(s) [26], this group of options
include the relevant modifications to the SCC Hamiltonian and energy. To enable the DFTB3 model
you will need to set ThirdOrderFull = Yes,DampXH = Yes and set the exponents in DampXHEx-
ponent .
DampXH If set to Yes the short range contribution to the SCC interaction between atoms Aand B
is damped by the factor
e−UAl +UBl
2ζ
r2
AB
provided that at least one of the two atoms is hydrogen [26,27]. (UAl and UBl are the Hubbard
Us of the two atoms for the l-shell, rAB is the distance between the atoms.) An atom is
considered to be a hydrogen-like atom, if its mass (stored in the appropriate homonuclear SK-
file) is less than 3.5 amu. The parameter ζcan be set with the keyword DampXHExponent.

48 CHAPTER 2. INPUT FOR DFTB+
DampXHExponent Sets the parameter ζfor the short range damping. (See keyword DampXH
above.)
ThirdOrder If set to Yes the on-site 3rd order correction [27] is switched on. This corrects the
SCC-Hamiltonian with the derivatives of the Hubbard U parameters, which you have to spec-
ify for every element in HubbardDerivs. This correction only alters the on-site elements and is
only maintained for backward compatibility. You should use the full version ThirdOrderFull
instead.
ThirdOrderFull If set to Yes the full 3rd order correction [26] is switched on. This corrects the
SCC-Hamiltonian with the derivatives of the Hubbard U parameters, which you have to spec-
ify for every element in HubbardDerivs.
HubbardDerivs Derivatives of the Hubbard U for the 3rd order correction (on-site or full). For
every element the appropriate parameter (in atomic units) must be specified. If you use shell
resolved SCC (with full 3rd order), you must specify a list of derivatives for every element,
with one Hubbard U derivative for each shell of the given element.
Hamiltonian = DFTB {
:
ThirdOrder = Yes
HubbardDerivs {
O = -0.14
H = -0.07
}
:
}
2.4.12 Differentiation
Calculations of forces currently require the numerical derivatives of the overlap and non-self-
consistent Hamiltonian. This environment controls how these derivatives are evaluated.
Note: In earlier DFTB+ versions (up to version 1.2), differentiation was done using finite
difference derivatives with a step size of 0.01 atomic units. If you want to reproduce old
results, choose the FiniteDiff method and set the step size explicitly to this value.
FiniteDiff{}
Finite difference derivatives with a specified step size
Delta repsilon1/4
Delta [length]Step size
Richardson{}
Extrapolation of finite difference via Richardson’s deferred approach to the limit (in principle
the most accurate of the currently available choices).
2.5. OPTIONS 49
2.4.13 ForceEvaluation
Chooses the method for evaluating the electronic contribution to the forces.
’traditional’ Uses the “traditional” DFTB-force expression, given for example, in Ref. [28].
’dynamics’ Force expression from Ref. [10]. This choice should be used if forces are being
calculated with non-converged charges (e.g. when doing XLBOMD dynamics). Note:
this force expression is only compatible with the Fermi filling (see keyword Filling,
p. 37.)
’dynamicsT0’ Simplified dynamic force expression valid for electronic temperature T=0 K
[10]. This choice should be used if forces are calculated with non-converged charges
and the electronic temperature is zero (e.g. when doing XLBOMD dynamics at T=0 K).
Note: that XLBOMD calculations (Section 2.3.5) are not able to use the ’traditional’ forces.
Example:
ForceEvaluation = ’dynamics’
2.5 Options
This block collects some global options for the run.
WriteAutotestTag lNo
WriteDetailedXML lNo
WriteResultsTag lNo
WriteDetailedOut lYes
RestartFrequency iDriver = {}, SCC = Yes 20
RandomSeed i0
MinimiseMemoryUsage lNo
ShowFoldedCoords lPeriodic = Yes No
WriteHS lNo
WriteRealHS lNo
Verbosity l51
WriteAutotestTag Turns the creation of the autotest.tag file on and off. (This file can get quite
big and is only needed for the autotesting framework.)
WriteDetailedXML Turns the creation of the detailed.xml file on and off. (The detailed.xml file
is needed among others by the waveplot utility for visualising molecular orbitals.)
WriteResultsTag Turns the creation of the results.tag file on and off. (That file is used by several
utilities processing the results of DFTB+.)
WriteDetailedOut Controls the creation of the file detailed.out. Since this contains the detailed
information about the last step of your run, you shouldn’t turn it off without good reasons.
RestartFrequency Specifies the interval at which charge restart information should be written to
disc for static SCC calculations. Setting it to 0prevents the storage of restart information. If
running an MD calculation, see also section 2.3.5 regarding MDRestartFrequency.
50 CHAPTER 2. INPUT FOR DFTB+
RandomSeed Sets the seed for the random number generator. The value 0causes random initial-
isation. (This value can be used to reproduce earlier MD calculations by setting the initial
seed to the same value.)
MinimiseMemoryUsage Tries to minimise memory usage by storing various matrices on disc
instead of keeping them in memory. Set it to Yes to reduce the memory requirement for
calculations with many k-points or spin polarisation.
ShowFoldedCoords Print coordinates folded back into the central cell, so if an atom moves out-
side the central cell it will reappear on the opposite side. The default behaviour is to use
unfolded coordinates in the output. (Please note, that this option only influences how the
coordinates are printed and written, it does not change the way, periodic systems are treated
internally.)
WriteHS Instructs the program to build the square Hamiltonian and overlap matrices and write
them to files. The output files are hamsqrN.dat and oversqr.dat, where Nenumerates the
spin channels. For a detailed description of the file format see p. 75.
Note: If either of the options WriteHS or WriteRealHS are set to Yes, the program only builds
the matrices, writes them to disc and then stops immediately. No diagonalisation, no SCC-
cycles or geometry optimisation steps are carried out. You can use the ReadInitialCharges
option to build the Hamiltonian with a previously converged charge distribution.
WriteRealHS Instructs the program to build the real space (sparse) Hamiltonian and overlap ma-
trices and write them to files. The output files are hamreal.dat and overreal.dat. For a detailed
description of the file format see p. 75.
Note: If either of the options WriteHS or WriteRealHS are set to Yes, the program only builds
the matrices, writes them to disc and then stops immediately. No diagonalisation, no SCC-
cycles or geometry optimisation steps are carried out. You can use the ReadInitialCharges
option to build the Hamiltonian with a previously converged charge distribution.
Verbosity This parameter controls the amount of output messages globally and takes values rang-
ing from 1 to 100.
2.6 Analysis
This block collects some options to analyse the results of the calculation and/or calculate properties.
AtomResolvedEnergies lNo
MullikenAnalysis lYes
ProjectStates m{}
Localise m{}
WriteEigenvectors lNo
EigenvectorsAsTxt lNo WriteEigenvectors = Yes
WriteBandOut lYes
CalculateForces lNo
AtomResolvedEnergies Specifies whether the contribution of the individual atoms to the total
energies should be displayed or not.
MullikenAnalysis If Yes, the results of a Mulliken analysis of the system is given.

2.6. ANALYSIS 51
ProjectStates
ProjectStates evaluates the Mulliken projection of electronic states onto specific regions of
the system being modelled (partial density of states – PDOS). The format of the projected
data files is similar to band.out, but the second column is the fraction of the state within that
region, instead of its occupation number (for non-collinear and spin-orbit calculations, three
additional columns for the magnetisation of the state are also given).
Each region for projection is specified within a Region{} block, with the following options
Atoms (i|s)+ -
ShellResolved lNo
OrbitalResolved lNo
Label s"regioni"
ShellResolved Project onto separate atomic shells of the region. These are taken in order
of increasing shell number of the atoms. ShellResolved = Yes is only allowed, if all the
selected atoms are of the same type.
OrbitalResolved Project onto separate atomic orbitals of the region. These are taken in
order of increasing shell number of the atoms. As with ShellResolved, this only allowed,
if all the selected atoms are of the same type.
Atoms Specification of the atoms over which to make the projection.. Atoms are specified
in the same way as MovedAtoms in section 2.3.1.)
Label Prefix of the label for the resulting file of data for this region. The default is “re-
gioni.out” where iis the number of the region in the input. In the case that ShellResolved
=Yes, the shell index is appended, so that files with names “Label.j.out” are written. For
OrbitalResolved = Yes, the shell and then m-value is appended, so that files with names
“Label.j.m.out” are written.
Examples:
ProjectStates = {
Region = { # first region
Atoms = 23:25 27 # atoms 23, 24, 25 and 27
}
Region = {
Atoms = N # All nitrogen atoms
ShellResolved = Yes # s and p shells separated instead of atomic PDOS
Label = "N" # files N.1.out and N.2.out for s and p states
}
}
Localise
Convert the single particle states of the calculation to localised orbitals via a unitary transfor-
mation. Localised orbitals span the same states as the occupied orbitals, so are equivalent to
the usual valence band states, but are more localised in space. Currently only PipekMezey
localisation is supported (but not for non-collinear or spin-orbit calculations).
Pipek-Mezey [29] localisation transforms the occupied orbitals such that the square of the
Mulliken charges for each orbital is maximised. The resulting localised states are output as

52 CHAPTER 2. INPUT FOR DFTB+
localOrbs.out and localOrbs.bin following the format given in appendix 4.3 for eigenvec.out
and eigenvec.bin.
Tollerance r1E-4
MaxIterations i100
Tollerance Cut off for rotations in the localisation process.
MaxIterations Maximum number of total sweeps to perform.
For systems with non-gamma-point k-points, no further options are available.
Analysis = {
Localise = {
PipekMezey = Yes # Default options otherwise
}
}
For molecular and gamma point periodic calculations there are two implementations avail-
able, Dense =Yes will use the O(n4)scaling conventional algorithm, while Dense =No, uses
the default sparse method which may have better scaling properties.
Dense lNo
SparseTollerances r+ Dense = No 1E-1 1E-2 1E-6 1E-12
Dense Selects the conventional method (Yes) using Jacobi sweeps over all orbital pairs or
(No) uses the default sparse method.
SparseTollerances The sparse method introduces support regions during evaluation to in-
crease performance, and these requires a set of tolerances to determine the regions to be
used (these are listed in decreasing order, i.e., with tighter tolerances as the localisation
proceeds).
WriteEigenvectors Specifies, if eigenvectors should be printed in eigenvec.bin. For a description
of the file format see p. 76.
EigenvectorsAsTxt If eigenvectors are being written, specifies if a text version of the data should
be printed in eigenvec.out. For a description of the file format see p. 76.
WriteBandOut Controls the creation of the file band.out which contains the band structure in a
more or less human friendly format.
CalculateForces If Yes, forces are reported, even if not needed for the actual calculation (e.g.
static geometry calculation).
2.7 ExcitedState
This block collects some options to calculate in the excited state.
Casida pSCC = Yes {}

2.7. EXCITEDSTATE 53
2.7.1 Casida
This tag contains the specifications for a time-dependent DFTB calculation, based on linear re-
sponse theory [30].
Note: the DFTB+binary must be compiled with linear response calculations enabled to make use
of these features (the ARPACK [31] library or ARPACK-ng [32] is required).
The calculation of vertical excitation energies and the corresponding oscillator strengths as well as
excited state geometry optimisation can be performed with these options, details of the resulting
output files are given in appendix 4.6. Linear response theory is currently implemented only for
the SCC-DFTB level of theory and molecular systems.5Excitations can be calculated for fractional
occupations and collinear spin-polarisation, but forces (and hence geometry optimisation or MD)
are only available for spin-unpolarised systems with no fractional occupations. The specifications
for this block have the following properties:
NrOfExcitations i-
StateOfInterest i0
Symmetry sSpinPolarisation ={} -
EnergyWindow rFORTRAN HUGE()
OscillatorWindow r-1
WriteTransitions lNo
WriteSPTransitions lNo
WriteMulliken lStateOfInterest 6=0 No
WriteCoefficients lStateOfInterest 6=0 No
WriteEigenvectors lStateOfInterest 6=0 No
TotalStateCoeffs lWriteEigenvectors | WriteCoefficients = YesNo
WriteXplusY lNo
WriteTransitionDipole lNo
WriteStatusArnoldi lNo
TestArnoldi lNo
NrOfExcitations Specifies the number of vertical excitation energies to be computed for every
symmetry (singlet or triplet). It is recommended that a value slightly greater than the actual
number of the states of interest is specified (the eigenvalue solver may not converge to the
right roots otherwise).
StateOfInterest Specifies the target excited state or states that should be calculated. These are
numbered from the first (lowest) excited state as 1, and so on. If the absorption spectrum at
a given geometry is required (i.e., a single-point calculation), this parameter should be set to
zero (default) and the Driver section (2.3) should be left empty (forces will not be available).
A value less than 0 requests that the state with the largest dipole transition moment be found
(again a single-point calculation).
Symmetry Specifies the spin symmetry of the excited states being computed: “singlet”, “triplet”
or “both”. This tag is only applicable for spin restricted calculation. For calculations in the
“triplet” or “both” cases, SpinConstants must be supplied (see p. 34).
EnergyWindow [energy]Energy range above the last transition at NrOfExcitations to be included
in excited state spectrum calculation.
5Excitation energies can also be calculated for gamma point periodic systems, but will be incorrect for delocalised
excitations or for charge transfer-type excited states.

54 CHAPTER 2. INPUT FOR DFTB+
OscillatorWindow [Dipole moment]Screening cut-off below which single particle transitions are
neglected in excitation spectra calculations. This selects from states above the top of the
EnergyWindow (if present). This keyword should not be used if calculating forces or other
excited state properties.
WriteTransitions If set to Yes, the file TRA.DAT is created. This file contains a description of
each requested excited state in terms of its single-particle transitions.
WriteSPTransitions If set to Yes, the file SPX.DAT is created, which contains the spectrum at
the uncoupled DFTB level (i.e. the single-particle excitations).
WriteMulliken If set to Yes, the files XCH.DAT and XREST.DAT are created. The former contains
atom-resolved Mulliken (net) charges for the excited state of interest, the latter the excited-
state dipole moment of the state.
WriteCoefficients If set to Yes, the file COEF.DAT is created. This file contains the complex
eigenvectors (molecular orbital coefficient) for the excited state of interest. They are derived
from the relaxed excited state density matrix.
WriteEigenvectors If set to Yes, the file excitedOrbs.bin is created. This file contains the natural
orbitals for the specified excited state.
TotalStateCoeffs Option to control the data written for either WriteCoefficients or WriteEigen-
vectors. If set to No the total charge density of the output orbitals corresponds to the change
in charge from the ground to excited state. If set to Yes instead it corresponds to the total
charge density in the excited state.
WriteXplusY If set to Yes, the file XplusY.DAT is created. This file contains the RPA vector
(X+Y)IΣ
ia for all excited states (c.f., Eqn. (18) in Ref. [33]).
WriteTransitionDipole If set to Yes, the file TDP.DAT is created. This file contains the Mulliken
transition dipole for each excited state.
WriteStatusArnoldi If set to Yes, the file ARPACK.DAT is created, which allows the user to
follow the progress of the Arnoldi diagonalisation.
TestArnoldi If set to Yes, the file TEST_ARPACK.DAT is created, which gives data on the quality
of the resulting eigenstates.
2.8 ParserOptions
This block contains the options, which are effecting only the behaviour of the HSD/XML parser
and are not passed to the main program.
ParserVersion i current input version
WriteHSDInput lYes
WriteXMLInput lNo
IgnoreUnprocessedNodes lNo
StopAfterParsing lNo
ParserVersion Version number of the input parser, which the input file was written for. If you are
using an input file, which was created for an older version of DFTB+, you should set it to
the parser version number of that code version. (The parser version number is printed at the
2.8. PARSEROPTIONS 55
beginning of the program run to the standard output.) DFTB+internally converts the input
to its current format. The processed input (written to dftb_pin.hsd) is always in the current
format, and the ParserVersion property in it is always set to be the current parser version.
WriteHSDInput Specifies, if the processed input should be written out in HSD format. (You
shouldn’t turn it off without really good reasons.)
WriteXMLInput Specifies, if the processed input should be written out in XML format.
IgnoreUnprocessedNodes By default the code stops if it detects unused or erroneous keywords
in the input, which probably indicates error(s) in the input. This dangerous flag suspends
these checks. Use only for debugging purposes.
StopAfterParsing If set to Yes, the parser stops after processing the input and written out the
processed input to the disc. It can be used to make sanity checks on the input without starting
an actual calculation.
56 CHAPTER 2. INPUT FOR DFTB+
Chapter 3
Transport calculations
Non-equilibrium Green’s function calculations are now possible with DFTB+. Within this formal-
ism it is possible to treat quantum mechanical systems with open boundary conditions and therefore
quantum transport. A specific new Transport{} block has been added to control the transport prob-
lem. The NEGF solver parameters can be controlled within the Eigensolver section, using the
keyword GreensFunction{}. Finally the real-space Poisson solver parameters have been organized
within a new section, Electrostatics, using the keyword Poisson{}. The default value for the elec-
trostatic calculations is the usual γ-functional which contains the Hartree and local XC potential.
3.1 Definition of the geometry
The input geometry for transport calculations is a little tricky. In comparison to cluster or supercell
calculations the geometry for transport calculation must also contain information about the contacts.
The contacting leads are actually semi-infinite structures, supporting travelling waves. No stationary
current is possible in a finite structure, as travelling waves can only exist in open systems. The
structure is partitioned into a device region and two or more contact regions.
Rules to build a valid input structure:
1. All device atoms must come first.
2. Each contact must comprise two subsequent unit cells called principal layers (PLs). The two
PLs together give all information about the contact structure and in the following are referred
generally as ’contact’.
3. A PL is a unit cell of the contacting lead that has interactions only with nearest neighbour
PLs in tight-binding terms.
4. The ordering of the atoms within the two PLs must be consitent in the sense that the two PLs
must be exact periodic replica of each other: If each PL comprise N atoms, atom M in the
first PL must have a corresponding identical atom in the second PL at position M+N.
5. The first PL should be always the one closer to the device region.
6. All blocks should be contiguous in the structure and each atom must belong to one and only
one region.
57

58 CHAPTER 3. TRANSPORT CALCULATIONS
7. The geometry can be defined as a cluster or a supercell. In the first case is it understood that
the contacts are just one-dimensional wire leads.
8. If a structure is defined as supercell, only the lattice vectors transverse to the transport direc-
tion are meaningful. The periodicity specified along the transport direction is dummy.
9. For each contact the periodicity along the transport direction is deduced from the separation
between the two PLs (as the coordinate difference r(M+N)−r(M)). We refer to this as
contact direction.
10. All lattice vectors (including the periodicity vector of the contacts) must be aligned to one
of the cartesian axes x, y or z. In practice only rectangular cells are currently allowed in
transport calculations.
Note: Currently the code makes only a consistency check on the definition of the two PLs (rule 4),
namely it checks whether the two contact PLs are really shifted copies of each other. The code does
not check if the device regions are consistently defined (rules 1and 6), if the PL defined are really
PLs (rule 3) and does not check if the first PL defined is really the one closest to the device (rule 5).
The code checks rules 8,9and 10. The check is performed on the atomic coordinates, such that
R2
i+N=R1
i+v∀i∈PL (3.1)
where R2
iare atomic coordinates of atoms in the second PL, R1
iare atomic coordinates of atoms in
the first PL and vis the contact lattice vector. The equality is verified within an accuracy that can
be set by the user (see below).
Please take your time to build up structures and cross-check them. Also consider to look at the
examples distributed with the code. The input structure is often the first suspect when there’s some
problem in the calculation.
3.2 Transport
DFTB+allows for NEGF calculations on different levels. It is possible to calculate linear response
transmission within Landauer formalism, charge density out of equilibrium with self-consistent
schemes, local currents etc. The Transport section groups the information needed whenever open
boundary conditions are used. It contains the description of the partitioning of the system into
device and contact regions and additional contact information needed to calculate the associated
Self Energies. The transport block contains the following properties:
Device p-59
Contact p-59
Task mUploadContacts 60
Example:
Transport {
Device {
AtomRange = 1 8
}
Contact {
Id = "source"

3.2. TRANSPORT 59
AtomRange = 9 24
}
Contact {
Id = "drain"
AtomRange = 25 40
}
Task = ContactHamiltonian
}
3.2.1 Device{}
The Device blocks contains the following properties:
Name Type Condition Default Page
AtomRange 2i -60
FirstLayerAtoms i+ 1
ContactPLs i+ Geometry = NoGeometry{} 1 1
AtomRange defines the first and last atom of the device region.
FirstLayerAtoms defines the first atom of PLs in the device region. By default there is only one
layer (the entire device region). Alternatively the user can manually reorder and partition the
structure into layers for efficient GF calculations.
Differently from the contact PLs, the device layers do not need to represent unit cell repeti-
tions. The device geometry must be manually ordered in such a way that all the atoms within
each layer are contiguous and adjacent layers are placed next to each other. This ensures that
the constructed Hamiltonian and Overlap are block tri-diagonal. Refer to [3] for a description
of the iterative algorithm.
ContactPLs are the indices of PLs coupled to every electrode (e.g. 1 7 means the first contact is
coupled to the PL number 1, the second contact – to the PL number 7). Every contact can be
coupled to only one of PLs. This property is required for model calculations.
3.2.2 Contact{}
The contact block contains the following properties:
Id s
AtomRange 2i
ShiftAccuracy r1e-8
FermiLevel r
Potential r0.0
WideBand lNo
LevelSpacing rWideBand = Yes 20.0
The sections Device and Contact are used to define the atomic range of each region. The user can
also assign a label (Id) to each contact that can be used later for cross referencing. In the section
Contact the user can add a keyword that specifies the accuracy for the internal check of the PLs.
Id Assign a label to each contact.

60 CHAPTER 3. TRANSPORT CALCULATIONS
AtomRange Defines the first and last atom of the device region. Note that the contacts should be
defined in order of increasing atomic range.
ShiftAccuracy [length]It can be used to set the absolute accuracy used to check the PL consistency
(see above). The default is 10−5atomic units. Please be aware that using a large values
may hide errors due to a non consistent definition of the contacts, therefore it should not be
modified.
FermiLevel [energy]Specifies the contact Fermi levels.
Potential [energy]Specifies the electrostatic potential applied to each contact. The natural units
of this quantity are potential energy (e.g., V). They can be loosely identified with eV or a.u.
since in both these units the electronic charge is practically defined as e=1.
WideBand Use the wide band approximation for the contact. If set to Yes, the surface green’s
function of the contact is not explicitly calculated but rather assumed to be local and constant
according to a specified density of states.
LevelSpacing [energy]Specify the inverse of the density of states per atom to be used in the Wide
Band approximation. As an example, the DOS of gold at the Fermi level is 0.05ev−1atom−1,
which corresponds to an energy spacing of 20 Ev '0.735 Hartree (the default value).
3.2.3 Task = ContactHamiltonian{}
The Task option is used to define which type of calculation should be performed. Before an SCC
transport calculation it is necessary to compute some equilibrium properties of the contacts, by
running a periodic boundary conditions DFTB calculation.This necessary step must be carried sep-
aratley for each contact and can be done by specifying setting Task=ContactHamiltonian as in the
following example
Task = ContactHamiltonian {
ContactId = source
ContactSeparation [Angstrom] = 50.0
}
When Task=ContactHamiltonian the following options can be defined
ContactId s
ContactSeparation r1e3
ContactId Id of the contact to be calculated.
ContactSeparation [length]Dummy separation in transverse direction (see following explana-
tion).
The contact calculation computes the bulk Hamiltonian and self-consistent charges for each contact.
This is a usual DFTB+calculation for which appropriate parameters must be included in the input
file. For supercell structures the calculation of the contact is performed using corresponding super-
cells in which the transverse lattice vectors are those specified in the Geometry tag and the lattice
vector along the contact direction is deduced from the PL separation (rule 9). If the structure is de-
fined as a cluster, the contact calculation is performed for a supercell in which the contact is treated
3.2. TRANSPORT 61
as one-dimensional wire. However, since DFTB+does not support pure one- and two-dimensional
calculations, dummy lattice vectors are defined for the two remaining directions. The default value
for these lattice vectors is 1000 a.u. (527 Å), which should guarantee sufficient wire-wire distance
to avoid Coulomb interactions. The user can specify and alternative contact separation using the
keyword ContactSeparation placed in the ContactHamiltonian block. Each contact computation
produces one output file called shiftcont_ContactId.dat storing energy shifts and Mulliken charges
that must be present in the working folder in all subsequent transport calculations.
Note that during the contact calculation you will need to perform a k-point integration. Whenever
the system is defined as a cluster, DFTB+will automatically extract the periodicity vectors from
the geometry such that the first reciprocal vector will correspond to the transport direction. There-
fore you must specify the k-point sampling for the periodic calculation by sampling along the first
reciprocal lattice vector. As an example, if the structure is defined as a cluster (i.e., 1-dimensional
wire leads), the source contact calculation will have an input file similar to:
...
Task = ContactHamiltonian {
ContactId = source
}
...
Hamiltonian = DFTB {
...
KpointsAndWeights = SupercellFolding {
8 0 0 # Regardless of transport direction
0 1 0
0 0 1
0.5 0.0 0.0
}
}
On the other hand, if your structure is defined as a supercell (as an example, a molecule with bulk
contacts) and the transport direction is along y, your the source contact calculation will have an
input file similar to:
...
Task = ContactHamiltonian {
ContactId = source
}
...
Hamiltonian = DFTB {
...
KpointsAndWeights = SupercellFolding {
4 0 0 # Folding in parallel direction
0 8 0 # Folding in transport direction
0 0 4 # Folding in parallel direction
0.5 0.5 0.5
}
}
62 CHAPTER 3. TRANSPORT CALCULATIONS
This could seem confusing, but the underlining reasons is that in the cluster calculation the recipro-
cal lattice is set up by the code itself, while in the periodic calculation is set up by the user who can
chose any arbitrary direction. Refer to the transport cookbook and to the distributed examples for
further clarification.
3.2.4 Task = UploadContacts{}
After the contact calculations, it is possible to perform actual transport calculations. This is activated
simply specifying Task = UploadContacts, without additional options. In order to set a proper
transport calculation the user should also define the FermiLevel and Potential to each contact.
Transport {
Device {
AtomRange = 1 8
}
Contact {
Id = "source"
AtomRange = 9 24
FermiLevel [eV] = -8.4123
Potential = 0.0
}
Contact {
Id = "drain"
AtomRange = 25 40
FermiLevel [eV] = -8.4123
Potential = 1.0
}
Task = UploadContacts
}
Note: During the transport calculation you will not need to set up the k-point integration when the
structure is defined as a cluster, just as in a regular DFTB+calculation.
3.3 GreensFunction
In order to activate Green’s functions calculations the user must define the keyword EigenSolver =
GreensFunction in the Hamiltonian section. The Green’s function section describes all the parame-
ters needed by the Green’s function (GF) solver. The GF solver, either under equilibrium (no bias
applied) or under non-equilibrium conditions, builds up the density-matrix of the device region.
Strictly speaking the GF does not solve for the eigenstates of the open system, however it logically
substitutes the traditional construction of the density matrix from the eigenstates of the system,
obtained after the diagonalization step. The density matrix can be used to compute any physical
observable by ’tracing’ with the appropriate operator. In particular it is possible to calculate the
Mulliken charges necessary for the DFTB self-consistent loop. Therefore the usual DFTB+self-
consistent calculations can be driven using the GF solver. The Green’s function section contains
important parameters used by the solver. The following table describes these parameters.

3.3. GREENSFUNCTION 63
Name Type Condition Default Page
Delta r1e-5
ContourPoints 2i 20 20
LowestEnergy r-2.0
FermiCutoff i10
EnclosedPoles i3
RealAxisStep rRealAxisPoints=undefined 6.65e-4
RealAxisPoints rRealAxisStep=undefined
SaveSurfaceGFs lYes
ReadSurfaceGFs lNo
FirstLayerAtoms i+ Transport = undefined 1
FermiLevel rTransport = undefined
LocalCurrents lNo
Note: For efficient GF calculation the device region must be partitioned into layers whose funda-
mental property is to interact with nearest-neighbour layers only.
Delta [energy][energy]A small positive imaginary delta used in the GF definition.
ContourPoints The number of points along the complex contour integration of the GF along the
contour segments Cand L(see contour integration).
LowestEnergy [energy][energy]The initial energy from which the integration starts. It should be
low enough to ensure that all the electronic states are correctly included in the integration.
The default is -2.0 Hartree (see contour integration).
FermiCutoff Integer number setting the Fermi distribution cutoff in units of kT . It is read only if
the Fermi distribution temperature is greater than 0 (see contour integration).
EnclosedPoles The number of Poles enclosed in the contour. It is meaningful only in finite tem-
perature calculations (see contour integration).
RealAxisStep [energy]The energy step along the real axis integration for non-equilibrium calcu-
lations. Note: RealAxisStep and RealAxisPoints can not be both defined at the same time.
RealAxisPoints The number of points along the real axis integration needed in non-equilibrium
calculations. The default depends on the electronic temperature and bias. Note: RealAxisStep
and RealAxisPoints can not be both defined at the same time.
SaveSurfaceGFs As the SCC cycle usually needs to repeat the calculation of the Green’s function
at given energy points and as the surface Green functions do not change during the SCC cycle,
this flag allows for saving the surface Green functions to disk and save computational time
on every SCC cycle beyond the first.
ReadSurfaceGFs Allow to load surface Green’s function from file also at the first SCC cycle.
Note that this operation only makes sense if the energy integration points are identical to the
calculation used to generate the surface Green’s function files. The code do not verify whether
this condition s fulfilled. In general there is no need to modify this default for ReadSurfaceGS
and SaveSurfaceGS.
Note: the Green solver can be used also to calculate the density matrix when there is no
open buondary conditions, for example to take advantage of the iterative scheme in quasi-
1d systems. In this case, a Transport block is not defined and therefore FirstlayerAtoms{}

64 CHAPTER 3. TRANSPORT CALCULATIONS
should be defined here. Also, a Fermi level of the system must be known and provided to fill
up the electronic states.
FirstLayerAtoms As described in Device block. Can be specified only if no Transport block
exists.
FermiLevel [energy]The Fermi level used by the Green solver to fill up the electronic states.
LocalCurrents if set to Yes, local bond-currents are computed using the non-equilibrium density
matrix. This task is currently limited to non-periodic systems. The output is placed in a file
lcurrent_u.dat (or lcurrent_d.dat dending on spin). The files are arranged in a table in order
of increasing neighbour distance,
Atom(i) x y z nNeighbors j1 Ii,j1j2 Ii,j2j3 Ii,j3...
This file can be processed using the small code flux provided in tools/transport that helps in
building plots for jmol.
GreensFunction section example:
Eigensolver = GreensFunction {
FirstLayerAtoms = 1 61 92 145
Delta [eV] = 1e-4
ContourPoints = 20 20
RealPoints = 55
LowestEnergy [eV] = -60.0
FermiCutoff = 10
EnclosedPoles = 3
}
Note: in order to solve the self-consistent NEGF transport problem, the GreensFunction Eigen-
solver must be used. However, the TunelingAndDos{} section can be used to calculate the trans-
mission coefficients according to Landauer formula in a non self-consistent manner. In this case
an Eigensolver is not needed as the charge density doesn’t need to be calculated. To run such a
calculation, Eigensolver = TransportOnly must be set and no electrostatics must be calculated (i.e.
the Electrostatics = Poisson should not be declared).
3.4 Contour integration
Much of the computational work is in the integration of the energy resolved density matrix, repre-
sented via the NEGF matrix. The integration is efficiently performed with a complex contour inte-
gration and a real axis integration, as shown in Figure 3.1 and discussed in references [34,35,3].
All integrations are performed with Gaussian quadratures and the number of points must be spec-
ified manually. The complex contour integration is subdivided into two sections: the first section
is an arc of circle, C, that can be computed with few integration points (default 20); the second
section is a line that intersects the contour and runs parallel to the real axis at a distance that de-
pends on the number of poles of the Fermi function enclosed within the contour. Usually a good
choice for the number of poles is between 3 and 5 (the default is 3). The poles are placed at the
complex points zm=EF+i(2m+1)πkT and, therefore, are separated from each other by 2πkT .
At T=300 K this corresponds to a separation of 156 meV. It should be noted that, as the temper-
ature decreases, the separation between poles reduces. This makes the contour integration harder
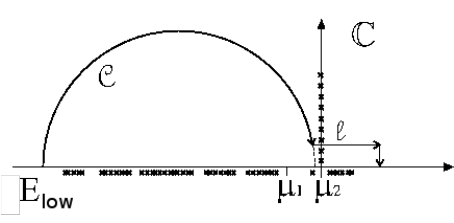
3.5. POISSON SOLVER 65
Figure 3.1: Contour integration in the complex plane for the Green’s fuctions. The crosses repre-
sent poles of either Gror the Fermi function.
as it needs to walk across two singularities. At very low temperatures, T=10 K, the separation
is 5.2 meV. Below this temperature the contour integration is treated as T=0 in order to avoid
numerical inaccuracies. The integration along the segment Lextends up to Re[z] = EF+nkT ,
where nis an integer number specified by the keyword FermiCutoff and has a default value of
10. In the limit T=0 K the poles collapse into a non-analytic cut and the contour needs to be
changed such that the second section of the complex contour becomes the arc of circle closing on
the real axis. Finally, the real axis integration extends between the lowest and highest chemical
potentials. The number of quadrature points should depend on the bias itself and can be set using
RealAxisPoints or RealAxisStep. The default value is 1 pt/0.018 eV (actually 1500 pt/1 H). In
finite temperature calculations the segment is extended to include the Fermi cutoff by nkT on both
sides (µ1−nkT,µ2+nkT ). In this case the number of quadrature points are increased by assuming
the same point density defined in the range (µ1,µ2). Example: for a bias of 0.2 V, the default num-
ber of points is 0.2·1500/27.21139 =11. At T=300 K the interval is increased by 0.26 eV on
both sides, therefore 0.26·1500/27.21139 =14.33 which is truncated to 14 points, leading to a total
of 38 points along the real axis. The use of the keyword RealAxisStep is usually more convenient
because it ensures a consistent real axis integrations during, for example, a bias sweep.
Note: The GF solver can be used also for calculations other than the transport context. In case
the position of the Fermi Energy is known with good accuracy the density matrix solver based on
the GF can be used to compute the electronic properties of clusters and supercells. The recursive
algorithm may be an efficient solution to large problem and could be efficient for systems having
an elongated 1D shape.
3.5 Poisson solver
The Poisson solver can not be used at the moment together with Model NoGeometry calculations
and is not used for Non-SCC calculations, but it is a fundamental part of the non-equilibrium SCC
transport calculations and must be declared whenever a NEGF calculation is performed using Elec-
trostatics = Poisson. Under non-equilibrium conditions the self-consitent potential of the KS equa-
tions cannot be solved using the efficient γ-functional, but requires the definition of appropiate
boundary conditions for the potentials imposed on the contacts. However, since the γ-functional is
formally identical to a pure Hartree potential, it can be obtained in real space by solving a Poisson
solver. The Poisson equation is solved in a box with hexahedral prism shape. This restriction is
imposed by the solver employed. This restricts calculations of supercell structures to orthorhombic
super-lattices. An additional restriction is that the box sides must be aligned with the Cartesian
axes, x, y, z.

66 CHAPTER 3. TRANSPORT CALCULATIONS
Name Type Condition Default Page
PoissonBox 3r
MinimalGrid 3r 0.5 0.5 0.5
PoissonAccuracy r1e-7
AtomDensityTolerance r1e-6
CutoffCheck lYes
Verbosity i51
SavePotential lNo
PoissonAccuracy r1e-6
MaxPoissonIterations i60
BuildBulkPotential lYes 67
ReadOldBulkPotential lYes 67
OverrideDefaultBC mnone{} 67
OverrideBulkBC mnone{} 67
BoundaryRegion mglobal{} 67
Gate mnone{} 69
MaxParallelNodes mnone{} 72
PoissonBox [length]Dimension of the Poisson box along directions x, y and z.
MinimalGrid [length]The minimal requested grid spacing along x, y and z. The actual grid spac-
ing chosen by the multigrid will be lower than this. charge densities.
AtomDensityTolerance In order to calculate the potential, the Mulliken charges are projected on
the real space grid. This parameter defines the cutoff after which the charge is considered
vanish (i.e., the space extension of the projected charge). The default is 1e-6. Note that the
contacts must be at least twice the length of the extent of a projected Mulliken charge. If this
conditions is not fullfilled and is set to Yes, the code will exit with an error message. Setting
this parameter to a lower value could allow to define shorter contacts in some cases. However
this could lead to relevant error in the potential hence to spurious reflections, therefore it
should be left to default value or changed very carefully.
CutoffCheck If set to No, consistency between contact length and charge extension is not veri-
fied (see above section). The default is No. As for AtomDensityTolerance, this parameter
shouldn’t be touched unless you know exactly what you’re doing.
Verbosity This parameter controls the amount of output messages and takes values ranging from
1 to 100.
SavePotential Save the potential to file.
PoissonAccuracy Defines the accuracy for the approximate solution of the Poisson equation (de-
fault value 10−6).
MaxPoissonIterations Defines the maximum amounts of iterations for the solver.
Note: The Poisson Box can be specified using the keyword PoissonBox. In calculations in which
the two contacts face each other along the same axis, setting the box-size along this axis has no
effect because the code needs to adjust the correct size internally. This keyword is redundant (and
should not be specified) when the system is periodic, since in this case the Poisson box is taken
from the supercell lattice vectors.
3.5. POISSON SOLVER 67
Numerical error in the potential will results in spurious discontinuities at the contact-device inter-
faces. The default tolerances should do the job in most cases.
This is a tipical example of the whole Poisson block specification. Some of the keywords are
described in the next subsections.
Example:
Electrostatics = Poisson {
PoissonBox [Angstrom] = 20.0 20.0 20.0
MinimalGrid [Angstrom] = 0.3 0.3 0.3
SavePotential = No
BuildBulkPotential = Yes
ReadOldBulkPotential = No
BoundaryRegion = Global {}
PoissonAccuracy = 1e-7
Gate = Planar{
GateLength_l [Angstrom] = 10.0
GateLength_t [Angstrom] = 20.0
GateDistance [Angstrom] = 7.0
GatePotential [eV] = 1.0
}
}
3.5.1 Boundary Conditions
The Poisson equation is solved imposing special boundary conditions (BC) on the six faces of the
Poisson Box. In basic transport calculations, comprising two contacts placed along the same axis,
the BCs are choosen as follows:
Dirichlet Fixed potentials on the two contact faces with values defined by the applied potentials
(see UploadContacts,ContactPotentials).
Neumann Zero normal field on the remaining 4 lateral box faces.
In periodic supercells the BCs are: Dirichlet (fixed potentials) on the two contact faces with values
defined by the applied potentials (see UploadContacts,ContactPotentials) and Periodic on the
remaining 4 lateral box faces.
In some specific cases Neumann BCs can be set on one contact. In order to do so it is necessary to
use OverrideDefaultBC (see below).
Device and contact potentials should smoothy join at the interface. In order to reach this goal
the code computes the Bulk potential of each contact and uses the result as a BC on the contact
face of the Poisson box. This is useful when the contact potential is not uniform due to charge
rearrangments. The external applied contact potential is added to the bulk potential. The user can
deactivate this calculation with the keyword BuildBulkPotential.
Note: The bulk potential is computed on a special box that has ’lateral’ sizes copied from the device
box, and has the size of one PL along the contact direction. The BCs are –so to speak– inherited
from the device region. In particular:
68 CHAPTER 3. TRANSPORT CALCULATIONS
1. Along the contact direction periodic BCs are imposed.
2. On the other four faces the BCs are copied from the device region.
3. The user can override this setting using OverrideBulkBC (see below).
4. When all four faces inherit Neumann BC (default for the device region), these are ALL inter-
nally changed to Dirichlet, because the solver cannot handle this situation that gives rise to a
singular matrix.
BuildBulkPotential (default: Yes) is used to calculate the electrostatic potential of the contacts
and the result is used as a Dirichlet boundary condition on the contact face (superimposed to
the contact potential).
ReadOldBulkPotential Read a previously computed bulk potential from hard-disk.
BoundaryRegion Specifies how the Dirichlet boundary conditions are treated on each contact face
of the Poisson box. It can be Global,Square or Circle.Global means that the BC is applied to
the entire face of the box, whereas the other keywords imply that the Dirichlet BC are applied
on a cross-section projected on the contact face. This is useful for instance when handling
nanowire contacts, for which it is not really consistent to impose a constant potential on the
whole face of the Poisson box.
BufferLength [length]can be used to set the size of the boundary region beyond the atomistic
size which is determined as the minimal circle or qquare containg all atoms of the contact
cross-section.
Example:
BoundaryRegion = Circle {
BufferLength [Angstrom] = 3.0
}
In some special case it might be necessary to override the default BCs applied by the code on
the Poisson equation. Currently this can be done using the keywords: OverrideDefaultBC and
OverrideBulkBC.
OverrideDefaultBC block is used to override the BCs described above. It can be used to force
Dirichlet or Neumann BCs along some specified directions or on one of the four lateral faces
of the Poisson box.
Boundaries is used to specify on which face different BCs must be imposed. Assuming contacts
along z, the keyword can be set any of xmin, xmax, x, ymin, ymax, y.
OverrideDefaultBC = Dirichlet {
Boundaries = xmin
}
For instance setting Dirichlet BC on Boundaries = xmin imposes φ(x,y,z) = 0 on the face placed
at x=xmin,boundaries = xmax imposes φ(x,y,z) = 0 on the face placed at x=xmax. When
Dirichlet needs to be forced on both faces, it is possible to use either boundaries = xmin,xmax or
3.5. POISSON SOLVER 69
simply boundaries = x. The same syntax can be used to impose conditions on more faces, using
boundaries = x,y or boundaries = x,ymin.
A similar strategy can be used to impose different boundary conditions on the contacts. For instance,
a Neumann BC can be set on one contact face using
OverrideDefaultBC = Neumann {
Boundaries = zmin
}
Note that the user should know which face of the Poisson Box corresponds to the desired contact.
Furthermore, if the user sets Neumann at all contacts the Poisson solver will not converge (singular
matrix) unless Dirichlet is imposed somewhere else (e.g., a gate potential is present).
It is also possible to override default BCs when computing the bulk potential.
OverrideBulkBC block is used to override bulk BC usually copied from the device region.
Boundaries has the same meaning and syntax as in OverrideDefaultBC.
OverrideBulkBC = Neumann {
Boundaries = x, y
}
3.5.2 Electrostatic Gates
The option Gate can be used to specify an electrostatic gate. Currently the gate type Planar and
Cylindrical are allowed. Restrictions. The planar gate must be placed with its face parallel to the
x-z plane, i.e., the gate direction must be along y. At the same time the transport direction should
be placed along the z-axis. The latter is not really a restriction but it gives meaning to ’longitudinal’
and ’transverse’ in the geometrical definitions of the gate lengths. Example:
Gate = Planar {
GateLength_l [Angstrom] = 20.0
GateLength_t [Angstrom] = 20.0
GateDistance [Angstrom] = 7.0
GatePotential [eV] = 1.0
}
Gate = Cylindrical {
GateLength [Angstrom] = 10.0
GateRadius [Angstrom] = 7.0
GatePotential [eV] = 1.0
}
The various options for the gates have the following meanings:
GateLength_l [length]Sets the gate length along the transport direction (assumed to be z). The
gate is centered in the device region.

70 CHAPTER 3. TRANSPORT CALCULATIONS
GateLength_t [length]Sets the gate transverse to the transport direction (assumed to be x). The
gate is centered in the device region.
GateDistance [length]Sets the distance of the gate from the center axis of the device region.
GatePotential [energy]Sets the potential applied to the gate.
GateRadius [length]For cylindrical gate, sets the distance of the gate from the center axis or gate
radius.
Note that the gate option has not be tested tharoughly and may still contain bugs. Please report to
the developers any problem encountered.
Developments. In forthcoming releases also double gates will be possible. Similarly to a usual
DFTB+calculation, the output from a Transport calculation will be generated in the detailed.out
and detailed.xml. These files are self-documenting, i.e. you will find a human-readable description
of the output data in the files themselves. After a transport calculation, the files will contain the
transmission coefficient for every energy point and for every k point and the local density of states
for every energy point and projection ranges specified in input. They will also contain the total
current and the partial current for every k point. In multiterminal calculation, this data will be
written for every terminal couple.
3.6 Model Hamiltonians
To use the external Hamiltonian without geometry if Geometry = NoGeometry{}, the type of
the Hamiltonian must be set to Model{}:
Hamiltonian = Model{}
The Model{} method may contain the following properties:
Name Type Condition Default Page
NumStates iNo default!
HamiltonianFile sH.mtr
NumStates Is a full number of states in the Hamiltonian. Required for model calculations without
geometry.
HamiltonianFile [energy]The name of the data file with the Hamiltonian, saved as an array of
real numbers.
Model Hamiltonians are used at the moment only for transport calculations. It is important to add
the property ContactPLs{} to the Device{} section (see Sec. 3.2.1).
3.7 Elastic dephasing
To switch on the dephasing effects, the group Dephasing{} must be included into the Hamiltonian
group. The Dephasing{} group may contain the following methods/properties:

3.7. ELASTIC DEPHASING 71
Name Type Condition Default Page
BuettikerProbes mnone{}
VibronicElastic mnone{}
3.7.1 Büttiker probes
BuettikerProbes switches on the Büttiker probe dephasing. There are two methods: ZeroPo-
tential{} and ZeroCurrent{}. The first one describes physical conducting environment with
fixed (at the moment zero) electrical potential. The second is for the Büttiker Probe Model
of dephasing, when the zero-current condition is fulfilled by artificial adjustment of local
electrical potentials.
BuettikerProbes = ZeroPotential {
Coupling [eV] = constant { 0.01 }
}
BuettikerProbes = ZeroCurrent {
Coupling [eV] = constant { 0.01 }
}
Coupling [energy]describes coupling to the environment. The method constant{} couples all
states equivalently. The other possible methods are AtomCoupling{} – with different cou-
plings for different atoms, and AllOrbitals{} – with different couplings for all atomic orbitals.
For the model calculations these two options are equivalent.
Coupling [eV] = AtomCoupling {
AtomList { Atoms = 1
Value = 0.1
}
AtomList { Atoms = 2 3
Value = 0.01
}
AtomList { Atoms = 4 5
Value = 0.03
}
}
Coupling [eV] = AllOrbitals { 0.1 0.1 0.1 ... }
3.7.2 Vibronic dephasing
VibronicElastic switches on the elastic vibronic dephasing method. There is only one method
local{} available at the moment. The Coupling{} method can get the same values as for
Büttiker probe dephasing. There are several additional options: AtomBlock (default=.false.),
which changes the way of coupling between electrons and phonons; MaxNumIter – the max-
imum number of iterations (default=100); Mixing – control of mixing in the self-consistent
cycle (default = 0.05); Tolerance – control of the convergency tolerance (default = 0.001).

72 CHAPTER 3. TRANSPORT CALCULATIONS
VibronicElastic = local {
AtomBlock = Yes
Coupling [eV] = Constant {0.01}
MaxNumIter = 1000
Mixing = 0.5
Tolerance = 0.001
}
3.8 Application to STM spectroscopy
3.9 Parallelizations
The code has been parallelised in two main parts. The Non-equilibrium Green’s functions are
computed by distributing the energy points along the countour and real axis calculations. Contour
and real axis integrations are independent and separately distributed. Load balancing has to be taken
care by the user. For instance if ContourPoints = {20 20} and RealAxisPoints = 60, by setting 10
MPI nodes, each node will handle 4 points along the contour and 6 points along the real axis.
Mixed OpenMP/MPI calculations are possible. When compiling dftb+ the user should link against
threaded mkl, rather than sequential. Numerical experiments show that best performance on mul-
ticore CPUs is generally obtained by running independent MPI processes on physical sockets, ex-
ploiting OpenMP multithreading on each socket. For instance NEGF can exploit threaded matrix-
matrix products. The user can experiment by setting the environment variable OMP_NUM_THREADS.
The Poisson solver has not been parallelized yet. However the efficient multigrid solver does not
typically represent a bottleneck. Currently the assembly of the charge density on the real-space grid
and the projection of the potential on the atoms have been parallelized. Since gather of the charge
density on each node can easily hit communication bottlenecks the user can use the parameter Max-
ParallelNodes to control distributions of these calculations. The default is MaxParallelNodes=1,
that can be increased until speedups are observed.
MaxParallelNodes i1
3.10 Analysis
The Analyis block is used to specify post-scf calculations such as tunneling or projected DOS.
Analysis{
TunnelingAndDOS{
EnergyRange [eV] = {-5.0 -3.0}
EnergyStep [eV] = 0.02
}
}

3.11. TUNNELINGANDDOS 73
3.11 TunnelingAndDos
This method block can be specified in Analysis{} and it is used to calculate the transmission by
means of Caroli formula, the current by means of Landauer formula and the Density of States from
the spectral function. This block can only be specified if an open boundary system has been defined
in Transport{}.
EnergyRange 2r
EnergyStep r
TerminalCurrents p
Region p73
WriteTunn lYes
WriteLDOS lYes
EnergyRange [energy]Contains the energy range over which the transmission function and local
density of states are computed.
EnergyStep [energy]Is the energy sampling step.
TerminalCurrents{} in multiterminal configurations is used to define the terminal across which
current must be computed. The terminal pairs are defined by using the keyword EmitterCol-
lector. example:
TerminalCurrents{
EmitterCollector = {"source" "drain"}
EmitterCollector = {"source" "gate"}
}
The block TerminalCurrents may be omitted since the code automatically sets all possible
independent combinations for the terminal currents. For example in a 4-contact calculations
the currents are 1-2, 1-3, 1-4, 2-3, 2-4, 3-4.
Region{} This block defines atomic ranges or orbitals where the local density of states is calcu-
lated projected. The definition in the block follow the same syntax as a DFTB+calculation
without transport.
WriteTunn The transmission coefficients are written also to a separate file for quick reference. If
set to No, the transmission coefficient are only written to DFTB+output files (detailed.out
and detailed.xml, autotest.tag).
WriteLDOS same as above, for the density of states.
3.12 Troubleshooting
DFTB+transport machinery is designed to calculate transport in structures with a large number
of atoms. To take full advantage of the iterative algorithm, be sure that the system is correclty
partitioned in Principal Layers, as described in Transport and Green’s solver sections. Be aware
that a wrong partitioning will lead to wrong results. If you’re not completely confident, you can run
a calculation on a test system with and without partitioning. The results should be the same.
74 CHAPTER 3. TRANSPORT CALCULATIONS
On some systems, a Segmentation Fault error could occur while running relatively large structures.
This could happen because the stack memory limit has been exceeded. You can troubleshoot this
setting a higher limit for the stack memory. In bash you can remove stack memory limitation with
the command line ulimit -s unlimited.
Chapter 4
Output of DFTB+
This chapter contains the description of some of the output files of DFTB+where the output format
is not self documenting. Unless indicated otherwise, numbers in the output files are given in atomic
units (with Hartree as the energy unit).
4.1 hamsqrN.dat, oversqr.dat
The files hamsqrN.dat and oversqr.dat contain the square (folded) Hamiltonian and overlap matri-
ces. The number Nin the filename hamrealN.dat indicates the spin channel. For spin unpolarised
calculation it is 1, for spin polarised calculation it is 1 and 2 for spin-up and spin-down, respectively
while for non-collinear spin it is charge, x,yand zfor 1, 2, 3 and 4. Spin orbit is not currently
supported for this option.
Only non-comment lines (lines not starting with "#") are documented:
• Flag for signalling if matrix is real (REAL), number of orbitals in the system (NALLORB),
number of kpoints (NKPOINT). For non-periodic (cluster) calculations, the number of kpoints
is set to 1.
• For every k-point:
–Number of the k-point. For molecular (non-periodic) calculations only 1 k-point is
printed.
–The folded matrix for the given k-point. It consists of NALLORB lines ×NALLORB
columns. If the matrix is not complex (REAL is F), every column contains two numbers
(real and imaginary part).
The files are produced if requested by WriteHS =Yes (see section 2.5).
4.2 hamrealN.dat, overreal.dat
The files hamrealN.dat and overreal.dat contain the real space Hamiltonian and overlap matrices.
The number Nin the filename hamrealN.dat indicates the spin channel. For spin unpolarised cal-
culation it is 1, for spin polarised calculation it is 1 and 2 for spin-up and spin-down, respectively,
75
76 CHAPTER 4. OUTPUT OF DFTB+
while for non-collinear spin it is charge, x,yand zfor 1, 2, 3 and 4. Spin orbit is not currently
supported for this option.
Note: The sparse format contains only the "lower triangle" of the real space matrix. For more details
about the format and how to obtain the upper triangle elements, see reference [2]. Also note, that
for periodic systems the sparse format is based on the folded coordinates of the atoms, resulting in
translation vectors (ICELL) which look surprising at first glance.
Only non-comment lines (lines not starting with "#") are documented:
• Number of atoms in the system (NATOM)
• For every atom:
–Atom number (IATOM), number of neighbours including the atom itself (NNEIGH),
number of orbitals on the atom (NORB)
• For every neighbour of every atom:
–Atom number (IATOM1), neighbour number (INEIGH), corresponding image atom to
the neighbour in the central cell (IATOM2F), coefficients of the translation vector be-
tween the neighbour and its corresponding image (ICELL(1),ICELL(2),ICELL(3)).
Between the coordinates of the neighbour rINEIGH and the image atom rIATOM2F the
relation
rINEIGH =rIATOM2F +
3
∑
i=1
ICELL(i)ai
holds, where aiare the lattice vectors of the supercell.
–The corresponding part of the sparse matrix. The data block consists of NORB(IAT1)
lines and NORB(IAT2F) columns.
The files are produced if requested by WriteRealHS =Yes (see section 2.5).
4.3 eigenvec.out, eigenvec.bin
These files contain the eigenvectors from the Hamiltonian, stored either as plain text (eigenvec.out)
or in the native binary format of your system (eigenvec.bin).
The plain text format file eigenvec.out contains a list of the values of the components of each
eigenvector for the basis functions of each atom. The atom number in the geometry, its chemical
type and the particular basis function are listed, followed by the relevant value from the current
eigenvector and then the Mulliken population for that basis function for that level. The particular
eigenvector, k-point and spin channel are listed at the start of each set of eigenvector data. In the
case of non-collinear spin, the format is generalised for spinor wavefunctions. Complex coefficients
for both the up and down parts of the spinors are given (instead of single eigenvector coefficient)
followed by four values – total charge, then (x,y,z)magnetisation.
The binary format file eigenvec.bin contains the (unique) runId of the DFTB+ simulation which
produced the output followed by the values of the eigenvectors. The eigenvector data is ordered so
that the individual components of the current eigenvector are stored, with subsequent eigenvectors
for that k-point following sequentially. All k-points for the current spin channel are printed in this
order, followed by the data for a second channel if spin polarised.
4.4. CHARGES.BIN 77
The files are produced if requested by setting WriteEigenvectors =Yes, with EigenvectorsAsTxt
being also required to produce the plain text file (see section 2.6 for details).
4.4 charges.bin
The file charges.bin contains the orbitally-resolved charges for each atom, ordered as the charges on
each orbital of an atom for a given spin channel, then each spin channel and finally over each atom.
In later versions of DFTB+this format includes a check sum for the total charge and magnetisation.
In the case of orbital potentials (p. 41) the file also contains extra population information for the
occupation matrices.
This file is produced as part of the mechanism to restart SCC calculations, see sections 2.5 and 2.3.5.
4.5 md.out
This file is only produced for VelocityVerlet{} calculations (See p. 18). It contains a log of infor-
mation generated during MD calculations, and appended every MDRestartFrequency steps. In the
case of small numbers of atoms and long MD simulations it may be useful to set WriteDetailedOut
to No and examine the information stored in this file instead.
4.6 Excited state results files
Several files are produced during excited state calculations depending on the particular settings from
section 2.7.
Note: in the case of degeneracies, the oscillator strengths depend on arbitrary phase choices made
by the ground state eigensolver. Only the sum over the degenerate contributions is well defined
for most single particle transition properties, and label ordering of states may change if changing
eigensolver or platform. For the excited state, properties like the intensities for individual excita-
tions in degenerate manifolds again depend on phase choices made by both the ground and excited
eigensolvers.
4.6.1 ARPACK.DAT
Internal details of the ARPACK solution vectors, see the ARPACK documentation [31] for details.
4.6.2 COEF.DAT
Data on the projection of this specific excited state onto the ground state orbitals. For the specific
exited state, the (complex) decomposition of its single particle states onto the ground state single
particle levels, together with its fractional contribution to the full excited state are given.
General format:
78 CHAPTER 4. OUTPUT OF DFTB+
T F Legacy flags
1 1.9999926523 2.0000000000 level 1, fraction of total WF, 2.0
-0.1944475716 0.0000000000 -0.1196876988 0.0000000000 .... real then imaginary projection of level 1
onto ground state 1, then ground state 2, etc.
-0.1196876988 0.0000000000 -0.1944475703 0.0000000000 ....
.
.
.
2 1.9999866161 2.0000000000 level 2
-0.2400145188 0.0000000000 -0.1767827333 0.0000000000 .... real then imaginary projection of state 2
.
.
.
4.6.3 EXC.DAT
Excitations data including the energies, oscillator strength, dominant Kohn-Sham transitions and
the symmetry.
Example first few transitions for C4H4:
w [eV] Osc.Str. Transition Weight KS [eV] Sym.
=========================================
5.551 0.5143882 11 -> 12 1.000 4.207 S
5.592 0.0000000 10 -> 12 1.000 5.592 S
Two examples of singlet transitions with energies of 5.551 and 5.592 eV. The first is dipole allowed,
the second not. In both cases they are transitions primarily (weight of 1.000) to single particle state
12, and are of singlet character (“S”).
In the case of spin-polarised calculations, an additional column of values are given instead of the
symmetry, showing the level of spin contamination in the state (labelled as D<S*S>), with typically
states where a magnitude of less than 0.5 is usually considered reliable [36].
4.6.4 SPX.DAT
Single particle excitations (SPX) for transitions between filled and empty single particle states of the
ground state. These are given in increasing single particle energy and show the oscillator strength
and index of the Kohn-Sham-like states that are involved.
# w [eV] Osc.Str. Transition
============================
1 5.403 0.2337689 15 -> 16
2 5.403 0.2337689 14 -> 16
3 5.403 0.2337689 15 -> 17
4.6. EXCITED STATE RESULTS FILES 79
4 5.403 0.2337689 14 -> 17
5 6.531 0.0000000 13 -> 16
6 6.531 0.0000000 12 -> 16
4.6.5 TDP.DAT
Detail of the magnitude and direction of the transition dipole from the ground to excited states.
4.6.6 TRA.DAT
Decomposition of the transition from the ground state to the excited states. The energy and spin
symmetry are given together with the contributions from each of the single particle transitions.
4.6.7 TEST_ARPACK.DAT
Tests on the quality of the eigenvalues and vectors returned by ARPACK. For the ith eigen-pair, the
eigenvalue deviation corresponds to the deviation from (hxi|H|xii−εi), The eigen-vector deviation
is a measure of rotation of the vector under the action of the matrix: |(H|xii−εi|xii)|2, the nor-
malisation deviation is hxi|xii−1 and finally largest failure in orthogonality to other eigenvectors is
given.
Example:
State Ei deviation Evec deviation Norm deviation Max non-orthog
1 -0.19428903E-15 0.80601119E-15 0.19984014E-14 0.95562226E-15
2 0.27755576E-16 0.85748374E-15 0.48849813E-14 0.36924443E-15
3 -0.12490009E-15 0.88607302E-15 0.88817842E-15 0.60384195E-15
4.6.8 XCH.DAT
Net charges on atoms in the specified excited state. The top line contains the symmetry (Singlet or
Triplet) and the number of the excited state. The next line is the number of atoms in the structure
followed by some header text. Then on subsequent lines the number of each atom in the structure
and its net charge are printed.
4.6.9 XplusY.DAT
Expert file with the RPA (X+Y)IΣ
ia data for all the calculated excited states.
Line 1: number of single particle excitations and the number of calculated excited states
Line 2: Level number 1, nature of the state (S, T, U or D) then excitation energy (in Hartree)
Line 3: expansion in the KS single particle transitions
.
.
.
Line 2: Level number 2, nature of the state (S, T, U or D) then excitation energy (in Hartree)
80 CHAPTER 4. OUTPUT OF DFTB+
4.6.10 XREST.DAT
Net dipole moment of the specified excited state in units of Debye.

Chapter 5
MODES
The MODES program calculates vibrational modes using data created by DFTB+.
5.1 Input for MODES
The input file for MODES must be named modes_in.hsd and should be a Human-friendly Structured
Data (HSD) formatted file (see Appendix A). The program can read the input in XML instead
of HSD format if the input file is modes_in.xml. The input file must be present in the working
directory. As with DFTB+to prevent ambiguity, the parser refuses to read in any input if two types
of input file are present.
The table below contains the list of the properties, which must occur in the input file modes_in.hsd:
Name Type Condition Default Page
Geometry p|m -12
Hessian p{} 82
SlaterKosterFiles p|m -
Additionally optional definitions may be present:
Name Type Condition Default Page
DisplayModes p-82
Atoms i+|m 1:-1
WriteHSDInput lYes
WriteXMLInput lNo
Geometry Specifies the geometry for the system to be calculated. See p. 12.
Hessian Contains the second derivatives matrix of the system energy with respect to atomic posi-
tions. See p. 82.
SlaterKosterFiles Name of the Slater-Koster files for every atom type pair combination. See
p. 38.
DisplayModes Optional settings to plot the eigenmodes of the vibrations. See p. 82.
Atoms Optional list of atoms, ranges of atoms and/or the species of atoms for which the Hessian
has been supplied. This must be equivalent to the setting you used for MovedAtoms in your
81

82 CHAPTER 5. MODES
DFTB+input when generating the Hessian.
WriteHSDInput Specifies, if the processed input should be written out in HSD format. (You
shouldn’t turn it off without good reason.)
WriteXMLInput Specifies, if the processed input should be written out in XML format.
5.1.1 Hessian{}
Contains the second derivatives of the energy supplied by DFTB+, see p. 17 for details of the
options to generate this data. The derivatives matrix must be stored as the following order: For the
i,jand kdirections of atoms 1...nas
∂2E
∂xi1∂xi1
∂2E
∂xj1∂xi1
∂2E
∂xk1∂xi1
∂2E
∂xi2∂xi1
∂2E
∂xj2∂xi1
∂2E
∂xk2∂xi1... ∂2E
∂xkn∂xkn
Note: for supercell calculations, the modes are currently obtained at the q=0 point, irrespective of
the k-point sampling used.
5.1.2 DisplayModes{}
Allows the eigenvectors of the system to be plotted out if present
PlotModes i+|m 1:-1
Animate lYes
XMakeMol lYes
PlotModes Specifies list of which eigenmodes should be plotted as xyz files. Remember that
there are 3Nmodes for the system (including translation and rotation).
Animate Produce separate animation files for each mode or a single file multiple modes where the
mode vectors are marked for each atom.
XMakeMol Adapt xyz format output for XMakeMol dialect xyz files.
Chapter 6
WAVEPLOT
The WAVEPLOT program is a tool for the visualisation of molecular orbitals. Based on the files cre-
ated by a calculation performed by DFTB+it is capable of producing three dimensional information
about the charge distribution. The information is stored as cube files, which can be visualised with
many common graphical tools (e.g. VMD or JMOL).
The user controls WAVEPLOT through an input file, choosing which orbitals and charge distributions
should be plotted for which spatial region. Since the information about the shape of the basis
functions is usually not contained in the Slater-Koster files, the coefficients and exponents for the
Slater type orbitals must be entered by the user as part of the input file.
The WAVEPLOT tool offers the following plotting capabilities:
• Total charge density.
• Total spin polarisation.
• Difference between the total charge density and the density obtained by the superposition of
the neutral atomic densities (visualisation of the charge shift).
• Electron density for individual levels.
• Real and imaginary part of the wavefunctions for individual levels.
6.1 Input for WAVEPLOT
The input file for WAVEPLOT must be named waveplot_in.hsd and should be a Human-friendly
Structured Data (HSD) formatted file (see Appendix A) The program can read the input in XML
instead of HSD format if the input file is waveplot_in.xml. The input file must be present in the
working directory. To prevent ambiguity, the parser refuses to read in any input if both files are
present.
The table below contains the list of the properties, which must occur in the input file waveplot_in.hsd:
83
84 CHAPTER 6. WAVEPLOT
Name Type Condition Default Page
Options p-84
DetailedXML s-
EigenvecBin s-
GroundState sYes
Basis p-88
Options Contains the options for WAVEPLOT. See p. 84.
DetailedXML Specifies the name of the file, which contains the detailed XML output of the
DFTB+calculation (presumably detailed.xml).
EigenvecBin Specifies the name of the file, which contains the eigenvectors in binary format (pre-
sumably eigenvec.bin).
GroundState Read ground or excited state occupation data from the detailed XML output.
Basis Contains the definition of the Slater-type orbitals which were used as basis in the DFTB+
calculation. At the moment, due to technical reasons this information has to be entered by
the user per hand. In a later stage, it will be presumably read in by WAVEPLOT automatically.
See p. 88.
Additionally optional definitions may also be present:
Name Type Condition Default Page
ParserOptions p{} 90
6.1.1 Options
This property contains the options (as a list of properties), which the user can set, in order to
influence the behaviour of WAVEPLOT. The following properties can be specified:
PlottedRegion p|m -86
NrOfPoints 3i -
PlottedKPoints i+|m periodic system -
PlottedLevels i+|m -
PlottedSpins i+|m -
TotalChargeDensity lNo
TotalSpinPolarisation lNo
TotalChargeDifference lNo
TotalAtomicDensity lNo
ChargeDensity lNo
RealComponent lNo
ImagComponent l complex wavefunction No
FoldAtomsToUnitCell l periodic system No
FillBoxWithAtoms lNo
NrOfCachedGrids i-1
Verbose lNo
RepeatBox 3i {1 1 1}
ShiftGrid lYes
6.1. INPUT FOR WAVEPLOT 85
PlottedRegion Regulates the region which should be plotted. See p. 86.
NrOfPoints Specifies the resolution of the equidistant grid on which the various quantities should
be calculated. The three integers represent the number of points along the three vectors of the
parallelepiped specifying the plotted region. The number of all calculated grid points is the
product of the three integers.
Example:
NrOfPoints = { 50 50 50 } # 125 000 grid points
PlottedKPoints The list of integers specified here represent the k-points, in which the molecular
orbitals should be plotted. The first k-point in the original DFTB+calculation is represented
by "1". The order of the specified k-points does not matter. You can also use the specifica-
tion of the form from:to to specify ranges. (For more details on range specification, see the
MovedAtoms keyword in the DFTB+manual.) The actual list of molecular orbitals to plot
is obtained by intersecting the specifications for PlottedKPoints,PlottedLevels and Plotted-
Spins. The option is ignored if the original calculation was not periodic.
Example:
PlottedKPoints = 1 3 5 # The 1st, 3rd and 5th k-point is plotted
PlottedLevels The list of integers specified here represent the states, which should be plotted. The
first (lowest lying) state in the original DFTB+calculation is represented by "1". The order
of the specified states does not matter. You can also use the specification of the form from:to
to specify ranges. (For more details on range specification, see the MovedAtoms keyword in
the DFTB+manual.) The actual list of molecular orbitals to plot is obtained by intersecting
the specifications for PlottedKPoints,PlottedLevels and PlottedSpins.
Example:
PlottedLevels = 1:-1 # All levels plotted
PlottedSpins The list of integers specified here represent the spins, for which the molecular or-
bitals should be plotted. The first spin in the original DFTB+calculation is represented by
"1". The order of the specified spins does not matter. You can also use the specification of the
form from:to to specify ranges. (For more details on range specification, see the MovedAtoms
keyword in the DFTB+manual.) The actual list of molecular orbitals to plot is obtained by
intersecting the specifications for PlottedKPoints,PlottedLevels and PlottedSpins.
Example:
PlottedSpins = 1 2 # Both spin-up and spin-down plotted
ChargeDensity If true, the absolute square of the wavefunction is plotted for the selected molec-
ular orbitals.
RealComponent If true, the real component of the wavefunction is plotted for the selected molec-
ular orbitals.
ImagComponent If true, the imaginary component of the wavefunction is plotted for the selected
molecular orbitals. This option is only parsed, if the wavefunctions in the DFTB+calculation
were complex.
86 CHAPTER 6. WAVEPLOT
TotalChargeDensity If true, the total charge density of the system is plotted.
TotalSpinPolarisation If true, the total spin polarisation of the system (difference of the spin up
and spin down densities) is plotted. This option is only interpreted if the processed DFTB+
calculation was spin polarised.
TotalChargeDifference If true, the difference between the total charge density and the charge
density obtained by superposing the neutral atomic densities is plotted.
TotalAtomicDensity If true, the superposed neutral atomic densities are plotted.
FoldAtomsToUnitCell If true, the atoms are folded into the parallelepiped unit cell of the crystal.
FillBoxWithAtoms If true, the geometry is extended by those periodic images of the original
atoms, which falls in the plotted region or on its borders. It sets FoldAtomsToUnitCell to Yes.
NrOfCachedGrids Specifies how many grids should be cached at the same time. The value -1
stands for as many as necessary to be as fast as possible. Since the plotted grids could even-
tually become quite big, you should set it to some positive non-zero value if you experience
memory problems.
Example:
NrOfCachedGrids = 5 # Maximal 5 cached grids
RepeatBox The three integers specify how often the plotted region should be repeated in the gen-
erated cube files. Since repeating the grid is not connected with any extra calculations, this
is a cheap way to visualise a big portion of a solid. You want probably set the FillBoxWith-
Atoms option to Yes to have the atoms also repeated (otherwise only the plotted function is
repeated). In order to obtain the correct picture, you should set the plotted region to be an
integer multiple of the unit cell of the crystal. Please note, that the phase of the wavefunctions
in the repeated cells will be incorrect, except in the Γ-point.
Example:
RepeatBox = { 2 2 2 } # Visualising a 2x2x2 supercell
ShiftGrid Whether the grid should be shifted, so that the specified origin lies in the middle of a
cell and the grid fills out the specified plotted region symmetrically. The default is Yes. If set
to No, the specified grid origin will be at the edge of a cell.
Verbose If true, some extra messages are printed out during the calculation.
PlottedRegion
Specifies the region, which should be included in the plot. You can specify it explicitly (as property
list), or let WAVEPLOT specify it automatically using either the UnitCell{} or the OptimalCuboid{}
methods.

6.1. INPUT FOR WAVEPLOT 87
Explicit specification Specifies origin and box size explicitly.
Origin 3r -
Box 9r -
Origin [length]Specifies the xyz coordinates of the origin as three real values.
Box [length]Specifies the three vectors which span the parallelepiped of the plotted region. The
vectors are specified sequentially (a1xa1ya1za2xa2ya2za3xa3ya3z). You are allowed to
specify an arbitrary parallelepiped with nonzero volume here. Please note, however, that
some visualisation tools only handles cube files with cuboid boxes correctly.
Example:
PlottedRegion = {
Origin = { 0.0 0.0 0.0 }
Box [Angstrom] = {
12.5 12.5 -12.5
12.5 -12.5 12.5
-12.5 12.5 12.5
}
}
UnitCell{} For the periodic geometries, this method specifies the plotted region to be spanned
by the three lattice vectors of the crystal. The origin is set to (0 0 0). For cluster geometries, the
smallest cuboid containing all atoms is constructed. For a cluster geometry the UnitCell{} object
may have the following property:
MinEdgeLength r1.0
MinEdgeLength [length]Minimal side length of the cuboid, representing the plotted region. This
helps to avoid cuboids with vanishing edge lengths (as it would be the case for a linear
molecule).
Example:
PlottedRegion = UnitCell {
MinEdgeLength [Bohr] = 2.0
}
OptimalCuboid{} Specifies the plotted region as a cuboid, which contains all the atoms and
enough space around them, that no wavefunctions are leaking out of the cuboid. This object does
not have any parameters.
Example:
PlottedRegion = OptimalCuboid {}

88 CHAPTER 6. WAVEPLOT
6.1.2 Basis
The basis definition is done by specifying the following properties:
Resolution r-
ElementName1 p-89
ElementName2 p-89
.
.
.
Resolution Specifies the grid distance used for discretising the radial wavefunctions. Setting it
too small, causes a long initialisation time for WAVEPLOT. Setting it too high causes a very
coarse grid with bad mapping and inaccurate charges. Values around 0.01 seem to work fine.
(Units must be in Bohr.)
ElementName1 Basis for the first atom type. The name of this property is the name of that atom
type.
ElementName2 Basis for the second atom type. The name of this property is the name of that atom
type.
Before describing the properties (and their sub-properties) in detail, the full basis definition for
carbon (sp) and hydrogen (s) should be presented as example:
Basis = {
Resolution = 0.01
C = { # Basis of the C atom
AtomicNumber = 6
Orbital = { # 2s orbital
AngularMomentum = 0
Occupation = 2
Cutoff = 4.9
Exponents = { 6.00000 3.00000 1.50000 }
Coefficients = {
1.050334389886e+01 2.215522018905e+01 9.629635264776e+00
-4.827678012828e+01 -5.196013014531e+00 -2.748085126682e+01
3.072783267234e+01 -1.007000163584e+01 8.295975876552e-01
}
}
Orbital = { # 2p orbital
AngularMomentum = 1
Occupation = 2
Cutoff = 5.0
Exponents = { 6.00000 3.00000 1.50000 }
Coefficients = {
-2.659093036065e+00 -6.650979229497e+00 -1.092292307510e+01
2.190230021657e+00 -9.376762008640e+00 -5.865448605778e-01
8.208019468802e+00 -2.735743196612e+00 2.279582669709e-01
}
}
}
H = { # Basis for the H atom

6.1. INPUT FOR WAVEPLOT 89
AtomicNumber = 1
Orbital = { # 1s orbital
AngularMomentum = 0
Occupation = 1
Cutoff = 4.2
Exponents = { 2.00000 1.00000 }
Coefficients = {
1.374518455977e+01 1.151435212242e+01 2.072671588012e+00
-1.059020844305e+01 3.160957468828e+00 -2.382382105798e-01
}
}
}
}
Basis for an atom type
The actual basis for every atom type is specified as a property with the name of that type:
AtomicNumber i-
Orbital p-89
.
.
.
AtomicNumber The atomic number of the species. This is not needed in the actual calculations,
but for creating proper cube-files.
Orbital Contains the parameters of the orbitals. For every orbital a separate Orbital block must be
created. See below.
Orbital For every orbital there is an orbital block which specifies the radial wavefunction. Thereby
the following properties must be used:
AngularMomentum i-
Occupation r-
Cutoff r-
Exponents r+ -
Coefficients r+ -
AngularMomentum Angular momentum of the current orbital. (s– 0, p– 1, d– 2, f– 3)
Occupation Occupation of the orbital in the neutral ground state. (Needed to obtain the right
superposed atomic densities.)
Cutoff Cutoff for the wave function. You should choose a value, where the value of 4πr2|R(r)|2
drops below 10−4or 10−5.R(r)is the radial part of the wave function. If you do not have
the possibility to visualise the radial wave function, take the half of the longest distance, for
which an overlap interaction exists in the appropriate homonuclear Slater-Koster file. (Value
must be entered in Bohr.)
Exponents The radial wave function with angular momentum lhas the form:
Rl(r) =
nexp
∑
i=1
npow
∑
j=1
ci j rl+j−1e−αir(6.1)

90 CHAPTER 6. WAVEPLOT
This property defines the multiplication factors in the exponent (αi).
Coefficients This property contains the coefficients ci j as defined in equation (6.1). The sequence
of the coefficients must be as follows:
c11 c12 ... c1npow c21 c22 ... c2npow ...
6.1.3 ParserOptions
This block contains the options, which are effecting only the behaviour of the HSD/XML parser
and are not passed to the main program.
IgnoreUnprocessedNodes lNo
StopAfterParsing lNo
IgnoreUnprocessedNodes By default the code stops if it detects unused or erroneous keywords
in the input. This dangerous flag suspends these checks. Use only for debugging purposes.
StopAfterParsing If set to Yes, the parser stops after processing the input and written out the
processed input to the disc. It can be used to make sanity checks on the input without starting
an actual calculation.
Appendix A
The HSD format
The Human-friendly Structured Data (HSD) format is a structured input format, which can be bi-
jectively mapped onto a subset of the XML-language. Its simplified structure and notation should
make it a more convenient user interface than reading and writing XML tags. This section contains
a brief overview of the most important aspects of this format.
An input file in the HSD format consists basically of property assignments of the form
Property = value
where the value value was assigned to the property Property. The value must be one of the following
types (detailed description of each follows later on):
• Scalar, such as
–integer
–real
–logical
–string
• list of scalars
• method
• list of further property assignments
An unquoted hash mark (#) is interpreted as the start of a comment, everything after it, up to the
end of the current line, is ignored by the parser (hash marks inside of quotes are taken as literals not
comments):
# Entire line with comment
Prop1 = "hell#oo" # Note, that the first hashmark is quoted!
The name of the properties, the methods and the logical values are case insensitive, so the assign-
ments
91
92 APPENDIX A. THE HSD FORMAT
Prop1 = 12
prOP1 = 12
Prop2 = Yes
Prop2 = YES
are pairwise identical. Quoted strings (specified either as a value for a property or as a file name),
however, are case sensitive.
Due to technical issues, the maximal line length is currently limited to 1024 characters. Lines longer
than this are chopped without warning.
If a property, which should only appear once, is defined more than once, the parser uses the first def-
inition and ignores all the other occurrences. Thus specifying a property in the input a second time,
does not override the first definition. (For advanced use the HSD syntax also offers the possibility
of conditional overriding/extending of previous definitions. For more details see A.6.)
A.1 Scalars and list of scalars
The following examples demonstrate the assignments with scalar types:
SomeInt = 1
SomeInt2 = -3
SomeRealFixedForm = 3.453
SomeRealExpForm = 2.12e-45
Logical1 = Yes
Logical2 = no
SomeString = "this is a string value"
As showed above, real numbers can be entered in either fixed or exponential form. The value for
logical properties can be either Yes or No (case insensitive). Strings should always be enclosed in
quotation marks, to make sure that they are treated as one string and that they are not interpreted by
the parser:
String1 = "quoted string"
String2 = this value is actually a list of 9 strings # list of strings!
String3 = "Method { ;" # This is a string assignment
String4 = Method { # This is syntactically incorrect, since
# it tries to assign a method to String4
A list of scalars is created by sequentially writing the scalars separated by one or more spaces:
PlottedLevels = 1 2 3
Origin = 0.0 0.0 0.0
ConfirmItTwice = Yes Yes
SpeciesNames = "Ga" "As"
The assignments statements are usually terminated by the end of the line. If the list of the assigned
values goes over several lines, it must be entered between curly (brace) brackets. In that case,
instead of the line end, the closing bracket will signal the end of the assignment. It is allowed to put
a list of scalars in curly brackets, even if it is only one line long.
A.2. METHODS AND PROPERTY LISTS 93
PlottedLevels = {
123
}
Origin = { 0.0
0.0 0.0 }
Short = { 1 2 3 }
If you want to put more than one assignment in a line, you have to separate them with a semi-colon:
Variable = 12; Variable2 = 3.0
If a property should be defined as empty, either the empty list must be assigned to it or it must be
defined as an empty assignment terminated by a semi-colon:
EmptyProperty = {}
EmptyProperty2 = ;
Please note, that explicitly specifying a property to be empty is not the same as not having specified
it at all. In the latter case, the parser substitutes the default value for that property (if there is a
default for it), while in the first case it interprets the property to be empty. If a property without
default value is not specified, the parser stops with an appropriate error message.
A.2 Methods and property lists
Besides the scalar values and the list of scalars, the right hand side of an assignment may also con-
tain a method, which itself may contain one or more scalar values or further property assignments
as parameters:
Diagonaliser = LapackDAC {} # Method without further params
PlottedLevels = Range { 1 3 } # Range needs two scalar params
PlottedRegion = UnitCell { # UnitCell needs a property list
MinEdgeLength = 1.0 # as parameter
SomeOtherProperty = Yes
}
The first assignment above is an example, where the method on the right hand side does not need
any parameters specified. Please note, that even if no parameters are required, the opening and
closing brackets after the method are mandatory. If the brackets are missing, the parser interprets
the value as a string.
In the second assignment, the method Range needs only two integers as parameters, while for
the method UnitCell several properties must be specified. A method may contain either nothing
or scalars or property assignments, but never scalars and property assignments together. So the
following assignment would be invalid:
InvalidSpecif = SomeMethod {
123
Property1 = 12
"Some strings here"
}
94 APPENDIX A. THE HSD FORMAT
Very often a value for the property is represented by a list of further property assignments (as above,
but without naming an explicit method beforehand). In that case, the property assignments must be
put between curly brackets (property list):
Options = {
SubOption1 = 12
Suboption2 = "string"
}
A.3 Modifiers
Each property may carry a modifier, which changes the interpretation of the assigned value:
LatticeConstant [Angstrom] = 12.23
Here, the property LatticeConstant possesses the Angstrom modifier, so the specified value will be
interpreted to be in Ångström instead of the default length unit. Specifying a modifier for a property
which is not allowed to carry one leads to parsing error.
The syntax of the HSD format also allows methods (used as values on the right hand side of an
assignment) to carry modifiers, but this is usually not used in the current input structures.
Sometimes, the assigned value to a property contains several values with different units, so that more
than one modifiers can be specified. In that case, the modifiers must be separated by a comma.
VolumeAndChargePerElement [Angstrom^3,au] = {
1.2 0.3 # first element
4.2 0.1 # second element
}
You have to specify either no modifier or all modifiers. If you want specify the default units for
some of the quantities, you can omit the name of the appropriate modifier, but you must include the
separating comma:
# Specifying the default unit for the charge. Note the separating comma!
VolumeAndChargePerElement [Angstrom^3,] = {
1.2 0.3 # first element
4.2 0.1 # second element
}
Specifying not enough or too many modifiers leads to parser error.
A.4 File inclusion
It is possible to include files in an HSD-formatted input by using the <<< and <<+ operators.
The former includes the specified file as raw text without parsing it, while latter parses the included
text:
A.5. PROCESSING 95
Geometry = GenFormat {
<<< "geo_start.gen"
}
Basis = {
<<+ "File_containing_the_property_definitons_for_the_basis"
}
The file included with the <<+ operator must be a valid HSD document in itself.
A.5 Processing
After having parsed and processed the input file, the parser writes out the processed input to a
separate file in HSD format. This file contains the internal representation for all properties, which
can be specified by the user. In particular, all default values are explicitly set and all automatic
definitions (e.g. ranges) are converted to their internal representations.
Assuming the following example as input
# Lattice contant specified in Angstrom.
# Internal representation uses Bohr, so it will be converted.
LatticeConstant [Angstrom] = 12.0
# This property is not set, as its commented out, so the
# default value will be set for this (let’s assume, it’s Yes)
#DoAProperJob = No
# Plotted levels specified as a range with parameter 1:3.
# This will be replaced by an explicit listing of the levels
PlottedLevels = { 1:3 }
the parsed and processed input (written to a special file) should look something like
LatticeConstant = 22.676713499923075
DoAProperJob = Yes
PlottedLevels = {
123
}
If you want to reproduce your calculation later, you should use this processed input. It should give
you identical results, even if the default setting for some properties had been changed in the code.
Since the HSD format is mapped by the parser internally to an XML tree, most codes using this
format allow (or hopefully will allow) to dumping out of the processed input in the XML format
as well, and to use that later as an input, instead of the HSD formatted input. This is practical for
people preferring to work with XML or if the input should be automatically generated by a script.
A.6 Extended format
As stated earlier, if a property, which should be defined only once, occurs more than once in the
input, the parser uses per default the first definition and ignores all the others. Sometimes this is not

96 APPENDIX A. THE HSD FORMAT
the desired behaviour, therefore, the HSD format also offers the possibility to override properties
that were set earlier. This feature can be very useful for scripts which are generate HSD input based
on some user provided template. By just appending a few lines to the end of the user provided input
the scripts can make sure that certain properties are set correctly. Thus, the script can modify the
user input, without having to parse it at all.
The parser builds internally an XML DOM-tree from the HSD input. For every property or method
name an XML tag with the same name (but lowercased) is created, which will contain the value of
the property or the method. If the value contains further properties or methods, new XML tags are
created inside the original one. Shortly, the HSD input is mapped on a tree, whereas the assignment
and the containment (equal sign and curly brace) are turned into a parent-child relationships.1As
an example an HSD input and the corresponding XML-representation is given below:
Level0Elem1 = 1
Level0Elem2 = { 1 2 3 }
Level0Elem3 = {
Level1Elem1 = 12
Level1Elem2 = Level2Elem1 {
Level3Elem1 = "abcd"
Level3Elem2 = {
Level4Elem1 = 12
}
}
}
<level0elem1>1</level0elem1>
<level0elem2>1 2 3</level0elem2>
<level0elem3>
<level1elem1>12</level1elem1>
<level1elem2>
<level2elem1>
<level3elem1>"abcd"</level3elem1>
<level3elem2>
<level4elem1>12</level4elem1>
</level3elem2>
</level2elem1>
</level1elem2>
</level0elem3>
By prefixing property and method names, the default behaviour of the parser can be overridden.
Instead of creating a new tag (on the current encapsulation level) with the appropriate name, it will
look for the first occurrence of the given tag and will process that one. Depending of the prefix
character, the tag is processed in the following ways:
+: If the tag exists already, it’s value is modified, otherwise the parser stops.
?: If the tag exists already, it’s value is modified, otherwise the parser ignores the prefixed HSD
construct.
*: If the tag exists already, it’s value is modified, otherwise it is created (and then it’s value is
modified).
/: If the tag does not exist yet, it is created and modified, otherwise the prefixed HSD construct is
ignored.
!: The tag is newly created and modified. If it exists already, the old occurrence is deleted first.
The way the value of the tag is going to be modified, is ruled by the constructs inside the prefixed
property or method name. If the parser finds non prefixed constructs here, the appropriate tags are
just added, otherwise the behaviour is determined by the rules above, just acting one level deeper in
the tree. The following examples should make this a little bit more clear.
1In the internal tree representation of the HSD input there is no difference between properties and methods, both are
just elements capable to contain some value or further elements. The differentiation in the HSD input is artificial and is
only for human readability (equal sign after property names, curly brace after method names),
A.6. EXTENDED FORMAT 97
• Changing the value of Level0Elem1 to 3. If the element does not exist, it should be created
with the value 3.
!Level0Elem1 = 3
• Changing the value of Level0Elem3/Level1Elem1 to 21 (the slash indicates the parent-child
relationship). If the element does not exist, stop with an error message:
# Make sure the containing element exists. If yes, go inside, otherwise die.
+Level0Elem3 = {
# Set the value of Level1Elem1 or die, if it does not exist.
+Level1Elem1 = 21
}
Please note, that each tag in the path must be prefixed. Using the following construct instead
of the original one
# Not prefixed, so it creates a new tag with empty value
Level0Elem3 = {
# The new tag doesn’t contain anything, so the parser stops here
+Level1Elem1 = 21
}
would end with an error message. Since Level0Elem1 is not prefixed here, a tag is created for
it with an empty value (no children). It does not matter, whether the tag already existed before
or not, a new tag is created and appended as the last element (last child) to the current block.
Then the parser is processing its value. Due to the +Level1Elem1 directive it is looking for a
child tag <level1elem1>. Since the tag was newly created, it does not contain any children,
so the parser stops with an error message.
• Create a new tag Level1Elem3 inside Level0Elem3 with some special value. If the tag already
exists, replace it.
# Modifing the children of Level0Elem3 or dying if not present
+Level0Elem3 = {
# Replacing or if not existent creating Level1Elem3
!Level1Elem3 = NewBlock {
NewValue1 = 12
}
This example also shows, that the value for the new property can be any arbitrary complex
HSD construct.
• Provide a default value "string" for Level0Elem3/Level1Elem2/Level2Elem1/Level3Elem1.
If the tag is already present do not change its value.
# Modify Level0Elem3 or create it if non-existent
*Level0Elem3 = {
# Modify Level1Elem2 and Level2Elem1 or create them if non-existent
*Level1Elem2 = *Level2Elem1 {
# Create Level3Elem1 if non-existent with special value.
/Level3Elem1 = "string"
}
}
98 APPENDIX A. THE HSD FORMAT
• If Level0Elem3/Level1Elem2 has the value Level2Elem1, make sure that Level3Elem1 in it
exists, and has "" as value. If Level1Elem2 has a different value, do not change anything.
# If Level0Elem3 is present, process it, otherwise skip this block
?Level0Elem3 = {
# The same for the next two containers
?Level1Elem2 = ?Level2Elem1 {
# Create or replace Level3Elem1
!Level3Elem1 = ""
}
}

Appendix B
Unit modifiers
The DFTB+code uses internally atomic units (with Hartree as the energy unit). The value of every
numerical property in the input is interpreted to be in atomic units (au), unless the property carries
a modifier.
The allowed modifiers and the corresponding conversion factors are given below.1(The modifiers
are case insensitive).
Length:
Angstrom,AA (for Ångström) 0.188972598857892E+01
Meter,m0.188972598857892E+11
pm 0.188972598857892E-01
Bohr,au 1.000000000000000E+00
Mass:
amu 0.182288848492937E+04
au 1.000000000000000E+00
da 0.182288848492937E+04
dalton 0.182288848492937E+04
Volume:
Angstrom∧3,AA∧30.674833303710415E+01
meter∧3,m∧30.674833303710415E+31
pm∧30.674833303710415E-05
bohr∧3,au 1.000000000000000E+00
Energy:
Rydberg,Ry 0.500000000000000E+00
Electronvolt,eV 0.367493245336341E-01
kcal/mol 0.159466838598749E-02
Kelvin,K0.316681534524639E-05
cmˆ-1 0.455633507361033E-05
Joule,J0.229371256497309E+18
Hartree,Ha,au 1.000000000000000E+00
1The conversion factors listed here were calculated with double precision on i686-linux architecture. Depending on
your architecture, the values used there may deviate slightly.
99
100 APPENDIX B. UNIT MODIFIERS
Force:
eV/Angstrom,eV/AA 0.194469064593167E-01
Joule/meter,J/m 0.121378050512919E+08
Hartree/Bohr,Ha/Bohr,au 1.000000000000000E+00
Time:
femtosecond,fs 0.413413733365614E+02
picosecond,ps 0.413413733365614E+05
second,s0.413413733365614E+17
au 1.000000000000000E+00
Charge:
Coulomb,C0.624150947960772E+19
au,e1.000000000000000E+00
Velocity:
au 1.000000000000000E+00
m/s 0.457102857516272E-06
pm/fs 0.457102857516272E-03
AA/ps 0.457102857516272E-04
Pressure:
Pa 0.339893208050290E-13
au 1.000000000000000E+00
Frequency:
Hz 0.241888432650500E-16
THz 0.241888432650500E-04
cmˆ-1 0.725163330219952E-06
au 1.000000000000000E+00
Electric field strength:
v/m 0.194469063788953E-11
au 1.000000000000000E+00
Dipole moment:
CoulombMeter,Cm 0.117947426715764E+30
Debye 0.393430238326893E+00
au 1.000000000000000E+00
Appendix C
Description of the gen format
The general (gen) format can be used to describe clusters and supercells. It is based on the xyz
format introduced with xmol, and extended to periodic structures. Unlike some earlier implemen-
tations of gen, the format should not include any neighbour mapping information.
The first line of the file contains the number of atoms, n, followed by the type of geometry. Cfor
cluster (non-periodic), Sfor supercell in Cartesian coordinates or Ffor supercell in fractions of the
lattice vectors. The supercells are periodic in 3 dimensions.
The second line contains the chemical symbols of the elements present separated by one or more
spaces. The following nlines contain a list of the atoms. The first number is the atom number in the
structure (this is currently ignored by the program). The second number is the chemical type from
the list of symbols on line 2. Then follow the coordinates. For Sand Cformat, these are x,y,zin
Å, but for Fthey are fractions of the three lattice vectors.
If the structure is a supercell, the next line after the atomic coordinates contains the coordinate
origin in Å (this is ignored by the parser). The last three lines are the supercell vectors in Å. These
four lines are not present for clusters.
Example: Geometry of GaAs with 2 atoms in the fractional supercell format
2 F
Ga As
1 1 0.0 0.0 0.0
2 2 0.25 0.25 0.25
0.000000 0.000000 0.000000
2.713546 2.713546 0.
0. 2.713546 2.713546
2.713546 0. 2.713546
101
102 APPENDIX C. DESCRIPTION OF THE GEN FORMAT

Appendix D
Atomic spin constants
These are suggested values for some atomic spin constants (Wvalues) as given in reference [37],
only the first two decimal places of the finite spin constants are numerically significant. These
constants may eventually be included in the Slater-Koster files directly. Check the documentation
of the Slater-Koster files required for a calculation to decide whether to use the LDA or PBE-GGA
spin constants.
W LDA PBE
s p d s p d
Hs-0.064 -0.072
Cs-0.028 -0.024 -0.031 -0.025
p-0.024 -0.022 -0.025 -0.023
Ns-0.030 -0.026 -0.033 -0.027
p-0.026 -0.025 -0.027 -0.026
Os-0.032 -0.028 -0.035 -0.030
p-0.028 -0.027 -0.030 -0.028
Si s-0.018 -0.013 0.000 -0.020 -0.015 0.000
p-0.013 -0.012 0.000 -0.015 -0.014 0.000
d0.000 0.000 -0.019 0.002 0.002 -0.032
Ss-0.019 -0.016 0.000 -0.021 -0.017 0.000
p-0.016 -0.014 0.000 -0.017 -0.016 0.000
d0.000 0.000 -0.010 0.000 0.000 -0.080
Fe s-0.013 -0.009 -0.003 -0.016 -0.012 -0.003
(3d74s1)p-0.009 -0.011 -0.001 -0.012 -0.029 -0.001
d-0.003 -0.001 -0.015 -0.003 -0.001 -0.015
Ni s-0.009 -0.009 -0.003 -0.016 -0.012 -0.003
p-0.009 -0.010 -0.001 -0.012 -0.022 -0.001
d-0.003 -0.001 -0.017 -0.003 -0.001 -0.018
103
104 APPENDIX D. ATOMIC SPIN CONSTANTS

Appendix E
Dispersion constants
The following table contains recommended dispersion constants for some elements with the Slater-
Kirkwood dispersion model (see p. 44). The values have been tested for biological systems, C, N,
O and H predominantly for DNA [22]. If you would like to calculate different systems or you’re
looking for other elements, check references [38] and [39]. The values of the atomic polarisabilities
and cutoffs are given for zero, one, two, three, four and more than four neighbors.
Element Polarisability [Å3] Cutoff [Å] Chrg Note
O 0.560 0.560 0.000 0.000 0.000 0.000 3.8 3.8 3.8 3.8 3.8 3.8 3.15
N 1.030 1.030 1.090 1.090 1.090 1.090 3.8 3.8 3.8 3.8 3.8 3.8 2.82
C 1.382 1.382 1.382 1.064 1.064 1.064 3.8 3.8 3.8 3.8 3.8 3.8 2.50
H 0.386 0.386 0.000 0.000 0.000 0.000 3.5 3.5 3.5 3.5 3.5 3.5 0.80
P 1.600 1.600 1.600 1.600 1.600 1.600 4.7 4.7 4.7 4.7 4.7 4.7 4.50 PO4only
S 3.000 3.000 3.000 3.000 3.000 3.000 4.7 4.7 4.7 4.7 4.7 4.7 4.80 S, not SO2
105
106 APPENDIX E. DISPERSION CONSTANTS
Appendix F
Publications to cite
The following publications should be considered for citation, if you are publishing any results cal-
culated by using DFTB+.
DFTB+code [2]
non-SCC DFTB [40], [41]
SCC DFTB [28]
Collinear spin polarisation [42]
Non-collinear spin polarisation [43]
Spin orbit coupling [43]
QM/MM coupling (external charges) [44], [45]
Van der Waals interaction (dispersion) [22]
DFTB+U [19]
3rd order corrections [27]
linear-response TD-DFTB [30]
107
108 APPENDIX F. PUBLICATIONS TO CITE
Bibliography
[1] D. A. Ryndyk, “DFTB+XT open software package for quantum nanoscale modeling,”,
http://quantranspro.org/dftb+xt/. 7
[2] B. Aradi, B. Hourahine, and T. Frauenheim, “DFTB+, a Sparse Matrix-Based Implementation
of the DFTB Method,” J. Phys. Chem. A 111, 5678 (2007), http://dftbplus.org/. 7,76,107
[3] A. Pecchia, G. Penazzi, L. Salvucci, and A. D. Carlo, “Non-equilibrium Green’s functions
in density functional tight binding: method and applications,” New Journal of Physics 10,
065022 (2008). 7,59,64
[4] T. Frauenheim, G. Seifert, M. Elstner, T. Niehaus, C. Kohler, M. Amkreutz, M. Sternberg, Z.
Hajnal, A. Di Carlo, and S. Suhai, “Atomistic simulations of complex materials: ground-state
and excited-state properties,” J. Phys. Cond. Matter 14, 3015–3047 (2002). 9
[5] A. Kovalenko, S. Ten-no, and F. Hirata, “Solution of three-dimensional reference interaction
site model and hypernetted chain equations for simple point charge water by modified method
of direct inversion in iterative subspace,” J. Comp. Chem. 20, 928–936 (1999). 17
[6] H. C. Andersen, “Molecular dynamics at constant pressure and/or temperature,” J. Chem.
Phys. 72, 2384 (1980). 19
[7] H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. Dinola, and J. R. Haak,
“Molecular-Dynamics with Coupling to an External Bath,” J. Chem. Phys. 81, 3684–3690
(1984). 19,21
[8] S. C. Harvey, R. K. Z. Tan, and T. E. Cheatham, “The flying ice cube: Velocity rescaling in
molecular dynamics leads to violation of energy equipartition,” J. Comp. Chem. 19, 726–740
(1998). 19
[9] G. J. Martyna, M. E. Tuckerman, D. J. Tobias, and M. L. Klein, “Explicit reversible integrators
for extended systems dynamics,” Molecular Phys. 87, 1117–1157 (1996). 20
[10] B. Aradi, A. M. N. Niklasson, and T. Frauenheim, “Extended Lagrangian Density Functional
Tight-Binding Molecular Dynamics for Molecules and Solids,” J. Chem. Theory Comput. 11,
3357–3363 (2015). 22,49
[11] M. Ceriotti, J. More, and D. E. Manolopoulos, “i-PI: A Python interface for ab initio path
integral molecular dynamics simulations,” Computer Phys. Comm. 185, 1019–1026 (2014).
24
[12] D. D. Johnson, “Modified Broyden’s method for accelerating convergence in self consistent
calculations,” Phys. Rev. B 38, 12807 (2003). 30
109
110 BIBLIOGRAPHY
[13] V. Eyert, “A Comparative Study on Methods for Convergence Acceleration of Iterative Vector
Sequences,” J. Comp. Phys. 124, 271 (1996). 31
[14] M. J. Han, T. Ozaki, and J. Yu, “O(N) LDA+Uelectronic structure calculation method based
on the nonorthogonal pseudoatomic orbital basis,” Phys. Rev. B 73, 045110 (2006). 36
[15] E. Anderson et al.,LAPACK Users’ Guide, 3rd ed. (Society for Industrial and Applied Math-
ematics, Philadelphia, PA, 1999). 36
[16] M. Methfessel and A. T. Paxton, “High-precision sampling for Brillouin-zone integration in
metals,” Phys. Rev. B 40, 3616 (1989). 37
[17] H. J. Monkhorst and J. D. Pack, “Special points for Brillouin-zone integrations,” Phys. Rev. B
13, 5188 (1976). 39
[18] H. J. Monkhorst and J. D. Pack, “"Special points for Brillouin-zone integrations"–a reply,”
Phys. Rev. B 16, 1748 (1977). 40
[19] B. Hourahine, S. Sanna, B. Aradi, C. Köhler, T. Niehaus, and T. Frauenheim, “Self-Interaction
and Strong Correlation in DFTB,” J. Phys. Chem. A 111, 5671 (2007). 41,107
[20] A. G. Petukhov, I. I. Mazin, L. Chioncel, and A. I. Lichtenstein, “Correlated metals and the
LDA+U method,” Phys. Rev. B 67, 153106–4 (2003). 41
[21] A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III, and W. M. Skiff, “UFF, a full
periodic table force field for molecular mechanics and molecular dynamics simulations,” J.
Am. Chem. Soc. 114, 10024–10035 (1992). 43
[22] M. Elstner, P. Hobza, T. Frauenheim, S. Suhai, and E. Kaxiras, “Hydrogen bonding and stack-
ing interactions of nucleic acid base pairs: a density-functional-theory based treatment,” J.
Chem. Phys. 114, 5149 (2001). 43,44,45,105,107
[23] S. Grimme, J. Antony, S. Ehrlich, and H. Krieg, “A consistent and accurate ab initio
parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-
Pu,” J. Chem. Phys. 132, 154104 (2010). 43,45
[24] S. Grimme, S. Ehrlich, and L. Goerigk, “Effect of the Damping Function in Dispersion Cor-
rected Density Functional Theory,” J. Chem. Phys. 32, 1456–1465 (2011). 43,45
[25] L. Zhechkov, T. Heine, S. Patchkovskii, G. Seifert, and H. A. Duarte, “An Efficient a Posteriori
Treatment for Dispersion Interaction in Density-Functional-Based Tight Binding,” J. of Chem.
Theory Comput. 1, 841–847 (2005). 43
[26] M. Gaus, Q. Cui, and M. Elstner, “DFTB3: Extension of the Self-Consistent-Charge Density-
Functional Tight-Binding Method (SCC-DFTB),” J. of Chem. Theory Comput. 7, 931–948
(2011). 47,48
[27] Y. Yang, H. Yu, D. York, Q. Cui, and M. Elstner, “Extension of the Self-Consistent-Charge
Density-Functional Tight-Binding Method: Third-Order Expansion of the Density Functional
Theory Total Energy and Introduction of a Modified Effective Coulomb Interaction,” J. Phys.
Chem. A 111, 10861 (2007). 47,48,107
[28] M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai, and
G. Seifert, “Self-consistent-charge density-functional tight-binding method for simulations of
complex materials properties,” Phys. Rev. B 58, 7260 (1998). 49,107
BIBLIOGRAPHY 111
[29] J. Pipek and P. G. Mezey, “A fast intrinsic localization procedure applicable for ab initio and
semiempirical linear combination of atomic orbital wave functions,” J. Chem. Phys. 90, 4916
(1989). 51
[30] T. A. Niehaus, S. Suhai, F. Della Sala, P. Lugli, M. Elstner, G. Seifert, and T. Frauenheim,
“Tight-binding approach to time-dependent density-functional response theory,” Phys. Rev. B
63, 085108 (2001). 53,107
[31] R. B. Lehoucq, D. C. Sorensen, and C. Yang, “ARPACK Users Guide: Solution of Large Scale
Eigenvalue Problems by Implicitly Restarted Arnoldi Methods.,”, 1997. 53,77
[32] https://github.com/opencollab/arpack-ng. 53
[33] D. Heringer, T. A. Niehaus, M. Wanko, and T. Frauenheim, “Analytical excited state forces
for the time-dependent density-functional tight-binding method.,” J. Comp. Chem. 28, 2589
(2007). 54
[34] A. Pecchia and A. D. Carlo, “Atomistic theory of transport in organic and inorganic nanos-
tructures,” Reports on Progress in Physics 67, 1497 (2004). 64
[35] A. Di Carlo, A. Pecchia, L. Latessa, T. Frauenheim, and G. Seifert, in Introducing Molecular
Electronics, G. Cuniberti, K. Richter, and G. Fagas, eds., (Springer Berlin Heidelberg, Berlin,
Heidelberg, 2005), pp. 153–184. 64
[36] A. D. Garcia, Ph.D. thesis, Universität Bremen, 2014, http://elib.suub.uni-
bremen.de/edocs/00103868-1.pdf. 78
[37] C. Köhler, Ph.D. thesis, Department Physik der Fakultät fur Naturwissenschaften an der Uni-
versität Paderborn, 2004, http://ubdata.uni-paderborn.de/ediss/06/2004/koehler/. 103
[38] K. J. Miller, “Additivity methods in molecular polarizability,” J. Am. Chem. Soc. 112, 8533
(1990). 105
[39] Y. K. Kang and M. S. Jhon, “Additivity of atomic static polarizabilities and dispersion coeffi-
cients,” Theoretica Chimica Acta 61, 41 (1982). 105
[40] D. Porezag, T. Frauenheim, T. Köhler, G. Seifert, and R. Kaschner, “Construction of tight-
binding-like potentials on the basis of density-functional theory: Application to carbon,” Phys.
Rev. B 51, 12947 (1995). 107
[41] G. Seifert, D. Porezag, and T. Frauenheim, “Calculations of molecules, clusters, and solids
with a simplified LCAO-DFT-LDA scheme,” Int. J. Quant. Chem. 58, 185 (1996). 107
[42] C. Köhler, G. Seifert, and T. Frauenheim, “Density-Functional based calculations for Fe(n),
(n≤32),” Chem. Phys. 309, 23 (2005). 107
[43] C. Köhler, T. Frauenheim, B. Hourahine, G. Seifert, and M. Sternberg, “Treatment of Collinear
and Noncollinear Electron Spin within an Approximate Density Functional Based Method,” J.
Phys. Chem. A 111, 5622 (2007). 107
[44] Q. Cui, M. Elstner, T. Frauenheim, E. Kaxiras, and M. Karplus, “Combined self-consistent
charge density functional tight-binding (SCC-DFTB) and CHARMM,” J. Phys. Chem. B 105,
569 (2001). 107
[45] W. Han, M. Elstner, K. J. Jalkanen, T. Frauenheim, and S. Suhai, “Hybrid
SCC-DFTB/Molecular Mechanical Studies of H-Bonded Systems and of N-acetyl-(L-Ala)n-
N’-Methylamide Helices in Water Solution,” Int. J. Quant. Chem. 78, 459 (2000). 107
Index
ContactPLs,59
Dephasing,26
Verbosity,49
AdaptFillingTemp,19,20
AllAtomCharges,29
AllAtomSpins,33,34
Alpha,17
Analyis,72
Analysis,12
AngularMomentum,89
Animate,82
AppendGeometries,14,16,17
ARPACK.DAT, 77
Atom list, 14
AtomCharge,29
AtomDensityTolerance,66
AtomicNumber,89
AtomRange,59
AtomResolvedEnergies,50
Atoms,17,24,33,34,51,81
AtomSpin,33,34
band structure calculation, 40
Barostat,18
Basis,84
BoundaryRegion,66
Box,87
Brillouin-zone sampling, 39
BuettikerProbes,71
BuildBulkPotential,66,67
CalculateForces,50
Casida,52
ChainLength,20
Charge,26
ChargeDensity,84
charges.bin, 77
Circle,68
COEF.DAT, 77
Coefficients,89
Constraints,14,16,17
Contact,58
Contact{},59
ContactHamiltonian,61
ContactId,60
ContactSeparation,60
ContourPoints,63
ConvergentForcesOnly,14,16–18,26
CoordsAndCharges,41
Coupling,21
CouplingStrength,20
CovalentRadius,45
CustomisedHubbards,26
Cutoff,47,89
CutoffCheck,66
CutoffCN,47
Cylindrical,69
DampXH,26
DampXHExponent,26
Delta,17,48,63
Dense,52
DetailedXML,84
Device,58
Device{},59
DFTB+U, 41
DiagonalRescaling,31
Differentiation,26
Direction,42,43
DirectRead{},41
Dispersion,26
DisplayModes,81
Driver,12
DynMixingParameters,31
Eigensolver,26,57
eigenvec.bin, 76
eigenvec.out, 76
EigenvecBin,84
EigenvectorsAsTxt,50
ElectricField,26
Electrostatics,57
EnclosedPoles,63
EnergyRange,73
112
INDEX 113
EnergyStep,73
EnergyWindow,53
EwaldParameter,26
EXC.DAT, 78
ExcitedState,12
Exponents,89
External,41,42
FermiCutoff,63,65
FermiLevel,59,63
File,24
FillBoxWithAtoms,84
Filling,26
FirstLayerAtoms,59,63
FixAngles,14,16,17
FixedFermiLevel,37
FixLengths,14
FoldAtomsToUnitCell,84
ForceEvaluation,26
Frequency,42,43
g{},20
Gate,66,69
GaussianBlurWidth,41
Generations,17,31,32
Geometry,12,81
Global,68
GreensFunction,62
GreensFunction{},57
GroundState,84
Hamiltonian,12
HamiltonianFile,70
hamreal.dat, 75
hamsqr.dat, 75
Hessian, 14,17,82
Hessian,81
Host,24
HubbardDerivs,26
HybridDependentPol{},45
HybridPolarisations,45
i-PI{},24
Id,59
IgnoreUnprocessedNodes,54,90
ImagComponent,84
IndependentKFilling,37
InitialCharges,26
InitialSpins,32,34
InitialTemperature,19
InitMixingParameter,31,32
IntegrationSteps,22
IntegratorSteps,20
InverseJacobiWeight,30
Isotropic,14,16,17,21
KeepStationary,18
KPointsAndWeights,26
Label,51
LatticeOpt,14,16,17
LatticeVectors,12
LevelSpacing,59
List of atoms, 14
LocalCurrents,63
Localise,50
localOrbs.bin, 52
localOrbs.out, 52
LowerCaseTypeName,38
LowestEnergy,63
Mass,24
Masses,18
MassPerAtom,24
MaxAngularMomentum,26
MaxAtomStep,14,16
MaxForceComponent,14,16,17
MaximalWeight,30
MaxIterations,52
MaxLatticeStep,14,16,17
MaxParallelNodes,66,72
MaxPoissonIterations,66
MaxSCCIterations,26
MaxSccIterations,22,41
MaxSteps,14,16,17,24
md.out, 77
MDRestartFrequency,18,77
MinEdgeLength,87
MinimalGrid,66
MinimalWeight,30
MinimiseMemoryUsage,49
MinSccIterations,22
Mixer,26
MixingParameter,30,31
Monkhorst-Pack scheme, 39
MovedAtoms,14,16–18
MullikenAnalysis,50
NPH ensemble, 21
NPT ensemble, 21
NrOfCachedGrids,84
NrOfExcitations,53
114 INDEX
NrOfPoints,84
NumStates,70
NVE ensemble, 19
NVT ensemble, 19,20
Occupation,89
OldSKInterpolation,26
Options,12,84
Orbital,89
OrbitalPotential,26
OrbitalResolved,51
OrbitalResolvedSCC,26
Order,20,37
Origin,87
OscillatorWindow,53
OutputPrefix,14,16–18
overreal.dat, 75
OverrideBulkBC,66
OverrideDefaultBC,66
oversqr.dat, 75
ParserOptions,12,84
ParserVersion,54
Periodic,12
Phase,42
PipekMezey,51
Planar,69
PlotModes,82
PlottedKPoints,84
PlottedLevels,84
PlottedRegion,84
PlottedSpins,84
PointCharges,41
Poisson{},57
PoissonAccuracy,66
PoissonBox,66
PolynomialRepulsive,26
Port,24
Potential,59
Prefix,24,38
Pressure,14,16,17,21
PreSteps,22
ProjectStates,50,51
Protocol,24
RandomSeed,49
ReadInitialCharges,26
ReadOldBulkPotential,66
ReadSurfaceGFs,63
ReadSurfaceGS,63
RealAxisPoints,63,65
RealAxisStep,63,65
RealComponent,84
Region,73
RelaxTotalSpin,32
RepeatBox,84
ReselectIndividually,19
ReselectProbability,19
Resolution,88
Restart,20
RestartFrequency,49
SavePotential,66
SaveSurfaceGFs,63
SaveSurfaceGS,63
Scale,23
SCC,18,26
SCCTolerance,26
SccTolerance,22
SelectedShells,27
Separator,38
ShellResolved,51
ShellResolvedSpin,26,35
ShiftAccuracy,59
ShiftGrid,84
ShowFoldedCoords,49
SlaterKosterFiles,26,81
SparseTollerances,52
SpinConstants,26,53
SpinOrbit,26
SpinPerAtom,33,34
SpinPolarisation,26
SPX.DAT, 78
Square,68
state resolved Mulliken population, 76
StateOfInterest,53
Steps,18
StepSize,14
StopAfterParsing,54,90
Strength,42,43
Suffix,38
Symmetry,53
Task,58,60
Task = ContactHamiltonian{},60
Task = UploadContacts,62
Task = UploadContacts{},62
TDP.DAT, 79
Temperature,19,20,37
TemperatureProfile{},18–20
TerminalCurrents,73
INDEX 115
TEST_ARPACK.DAT, 79
TestArnoldi,53
Thermostat,18
ThirdOrder,26
ThirdOrderFull,26
Threebody,47
Timescale,20,21
TimeStep,18,43
Tollerance,52
TotalAtomicDensity,84
TotalChargeDensity,84
TotalChargeDifference,84
TotalSpinPolarisation,84
TotalStateCoeffs,53
TRA.DAT, 79
TransientSteps,23
Transport,58,63,64
Transport{},57
TunnelingAndDos,73
TypeNames,12
TypesAndCoordinates,12
UnpairedElectrons,32
UseFromStart,32
v{},20
Velocities,18
Verbose,84
Verbosity,24,66
VibronicElastic,71
WeightFactor,30
WideBand,59
WriteAutotestTag,49
WriteBandOut,50
WriteCoefficients,53
WriteDetailedOut,49
WriteDetailedXML,49
WriteEigenvectors,50,53
WriteHS,49
WriteHSDInput,54,81
WriteLDOS,73
WriteMulliken,53
WriteRealHS,49
WriteResultsTag,49
WriteSPTransitions,53
WriteStatusArnoldi,53
WriteTransitionDipole,53
WriteTransitions,53
WriteTunn,73
WriteXMLInput,54,81
WriteXplusY,53
x{},20
XCH.DAT, 79
Xlbomd,18
XlbomdFast,18
XMakeMol,82
XplusY.DAT, 79
XREST.DAT, 80
