SORIN CRM VR9250 Implantable cardioverter defibrillator User Manual
SORIN CRM Implantable cardioverter defibrillator Users Manual
Users Manual
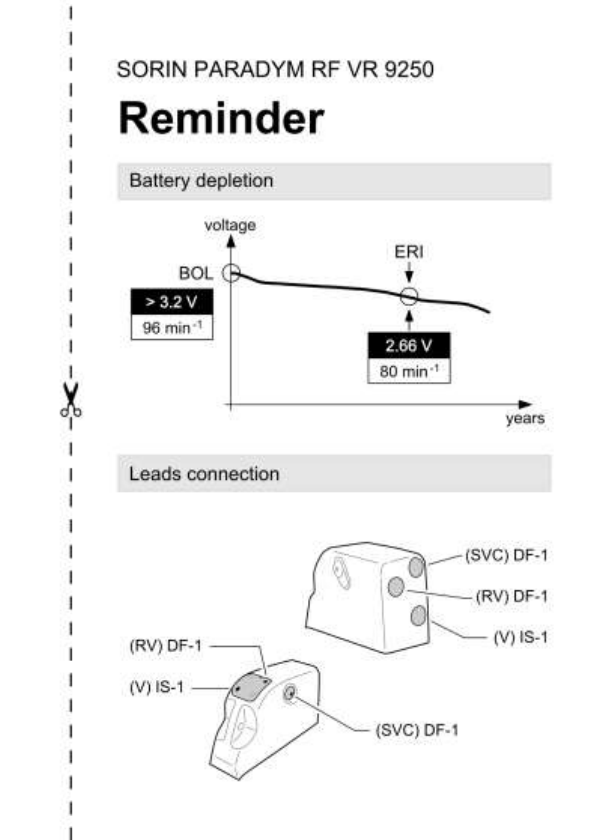
TABLE OF CONTENTS
1. General description .................................................................... 6
2. Indications ................................................................................... 6
3. Contraindications ....................................................................... 6
4. Warnings and precautions ........................................................ 7
4.1. Risks related to medical environment ........................... 8
4.2. Sterilization, storage and handling .............................. 10
4.3. Implantation and device programming ........................ 11
4.4. Lead evaluation and lead connection .......................... 12
4.5. Generator explant and disposal ................................... 13
5. Adverse events ......................................................................... 14
5.1. Defender study ............................................................... 14
5.2. Table 1: Summary of European Clinical
Complications ................................................................ 15
5.3. Table 2: Summary of European Clinical
Complications ................................................................ 16
6. Clinical studies ......................................................................... 18
6.1. Defender study ............................................................... 18
7. Patient selection and treatment .............................................. 23
7.1. Individualization of treatment ....................................... 23
7.2. Specific patient populations ......................................... 24
8. Patient counselling information .............................................. 25
9. Conformance to standards ...................................................... 25
10. Physician guidelines ................................................................ 29
10.1. Physician training .......................................................... 29
10.2. Directions for use........................................................... 29
10.3. Maintaining device quality ............................................ 29
11. Patient information ................................................................... 30

12. How supplied ............................................................................ 30
12.1. Sterility ............................................................................ 30
12.2. Warranty and replacement policy ................................ 30
13. Device description.................................................................... 31
14. Implant procedure .................................................................... 33
14.1. Necessary equipment .................................................... 33
14.2. Packaging ....................................................................... 34
14.3. Optional equipment ....................................................... 34
14.4. Before opening the package ......................................... 34
14.5. Prior to implantation ...................................................... 35
14.6. Device placement ........................................................... 35
14.7. Choosing the type of lead ............................................. 36
14.8. Measurement of thresholds at implant ........................ 37
14.9. Lead connection............................................................. 37
14.10. Device implantation ....................................................... 38
14.11. Tests and programming ................................................ 39
15. Special modes .......................................................................... 39
15.1. Safety mode (nominal values) ...................................... 39
15.2. Magnet mode .................................................................. 40
15.3. Response in the presence of interference .................. 40
15.4. Detection characteristics in the presence of
electromagnetic fields ................................................... 41
15.5. Protection against short-circuits .................................. 41
16. Main functions .......................................................................... 42
16.1. Automatic lead measurements ..................................... 42
16.2. Ventricular tachyarrhythmia management .................. 42
16.3. Pacing ............................................................................. 42
16.4. Sensing ........................................................................... 43
16.5. Follow-up functions ....................................................... 43
16.6. Remote Monitoring function ......................................... 44

17. Patient follow-up ....................................................................... 47
17.1. Follow-up recommendations ........................................ 47
17.2. Holter function ................................................................ 48
17.3. Elective Replacement Indicator (ERI) .......................... 49
17.4. Explantation .................................................................... 50
17.5. Defibrillator identification ............................................. 51
18. Physical characteristics .......................................................... 52
18.1. Materials used ................................................................ 52
19. Electrical characteristics ......................................................... 53
19.1. Table of delivered shock energy and voltage ............. 54
19.2. Battery ............................................................................. 55
19.3. Longevity ........................................................................ 55
20. Programmable parameters ...................................................... 57
20.1. Antibradycardia pacing ................................................. 57
20.2. Ventricular tachyarrhythmia detection ........................ 60
20.3. Ventricular tachyarrhythmia therapies ........................ 61
20.4. Remote alerts and warnings ......................................... 67
21. Non programmable parameters .............................................. 68
22. Limited warranty ....................................................................... 69
22.1. Article 1 : Terms of limited warranty ............................ 69
22.2. Article 2 : Terms of replacement .................................. 71
23. Patents ....................................................................................... 72
24. Explanation of symbols ........................................................... 73

6 – US-ENGLISH
1. GENERAL DESCRIPTION
PARADYM RF VR 9250 is an implantable single-chamber
cardioverter defibrillator. It is equipped with an accelerometer to allow
adaptation of pacing to suit the patient’s activity.
2. INDICATIONS
This device is indicated for use in patients who are at high risk of
sudden cardiac death due to ventricular tachyarrhythmias and who
have experienced one of the following situations:
― Survival of at least one episode of cardiac arrest (manifested by
the loss of consciousness) due to ventricular tachyarrhythmia,
― Recurrent, poorly tolerated sustained ventricular tachycardia (VT).
NOTE: The clinical outcome for hemodynamically stable VT patients is not
fully known. Safety and effectiveness studies have not been conducted.
3. CONTRAINDICATIONS
Implantation of PARADYM RF VR 9250 is contraindicated in
patients:
― whose ventricular tachyarrhythmias may have transient or
reversible causes such as: acute myocardial infarction, digitalis
intoxication, drowning, electrocution, electrolyte imbalance,
hypoxia, sepsis, or unstable ischemic episodes,
― who present incessant tachyarrhythmia,
― who have an internal pacemaker,
― whose primary disorder is bradyarrhythmias, or atrial
tachyarrhythmias.

US-ENGLISH – 7
4. WARNINGS AND PRECAUTIONS
The patient should be warned of the potential risks of defibrillator
malfunction if he is exposed to external magnetic, electrical, or
electromagnetic signals.
These potential interference sources may cause conversion to
inhibited mode (because of noise detection), erratic delivery of VT or
VF therapies, nominal programming, or much more rarely,
irreversible damage to the device’s circuits.
The main sources of high magnitude electromagnetic interference
are: powerful radiofrequency equipment (radar), industrial motors
and transformers, arc-welding equipment, high power loudspeakers.
Resuscitation Availability: Do not perform device testing unless an
external defibrillator and medical personnel skilled in
cardiopulmonary resuscitation (CPR) are readily available.
Electrical Isolation: Do not permit the patient to contact grounded
equipment that could produce hazardous leakage current. Ensuing
arrhythmia induction could result in the patient’s death.
Disable the ICD During Handling: Program Shock Therapy to OFF
during surgical implant and explant or post mortem procedures. The
device can deliver a serious high energy shock should accidental
contact be made with the defibrillation electrodes.
Antitheft gates: Since antitheft devices at the entrance to stores are
not subject to any safety standards, it is advisable to spend as little
time as possible in their vicinity.
Airport detection systems: Since airport detection systems are not
subject to any safety standards, it is advisable to spend as little time
as possible in their vicinity.
High voltage power transmission lines: High voltage power
transmission lines may generate enough EMI to interfere with
defibrillator operation if approached too closely.
8 – US-ENGLISH
Communication equipment: Communication equipment such as
microwave transmitters, linear power amplifiers, or high-power
amateur transmitters may generate enough EMI to interfere with
defibrillator operation if approached too closely.
Home appliances: Home appliances that are in good working order
and properly grounded do not usually produce enough EMI to
interfere with defibrillator operation. There are reports of device
disturbances caused by electric hand tools or electric razors used
directly over the device implant site.
CAUTION: Do not tap sharply on the ICD can after implant, because
the ICD's sensing circuits can detect this as R-waves, and such
oversensing could result in inappropriate pacing, inhibition, or
therapy. Normal activities after implant do not result in such
oversensing.
4.1. RISKS RELATED TO MEDICAL ENVIRONMENT
It is advisable to carefully monitor defibrillator operation prior to and
after any medical treatment during which an electrical current from an
external source passes through the patient's body.
Magnetic Resonance Imaging: MRI is strictly contraindicated in
cardiac defibrillator patients.
A radio frequency ablation: A radio frequency ablation procedure in
a patient with a generator may cause device malfunction or damage.
RF ablation risks may be minimized by: 1. Programming Shock
Therapy and ATP to OFF. 2. Avoiding direct contact between the
ablation catheter and the implanted lead or generator. 3. Positioning
the ground, placing it so that the current pathway does not pass
through or near the device, i.e. place the ground plate under the
patient’s buttocks or legs. 4. Having external defibrillation equipment
available.
US-ENGLISH – 9
Electrocautery or diathermy device: Diathermy and electrocautery
equipment should not be used. If such devices must be used:
1. Keep the current path and ground plate as far away from the
device and the lead as possible (a minimum of 15 cm [six inches]).
2. Before procedure, deactivate ATP and shock therapies. 3. During
the procedure, keep the electrocautery device as far as possible from
the cardiac defibrillator. Set it at minimum intensity. Use it briefly.
4. After the procedure, check for proper implant function. The device
should never be exposed directly to the diathermy source.
External defibrillation: PARADYM RF VR 9250 is protected from
external defibrillation shocks. Before external defibrillation, deactivate
ATP and shock therapies. During external defibrillation, it is advisable
to avoid placing the defibrillating paddles directly over the casing or
over the lead. The defibrillating paddles should preferably be placed
in an anteroposterior position. Avoid any direct contact between the
defibrillation paddles and the conductive parts of the implanted leads
or casing of the implanted device. After external defibrillation, check
for proper device function.
Radiation therapy: Avoid exposure to ionizing radiation. Betatrons
are contraindicated. If high doses of radiation therapy cannot be
avoided, the defibrillator should be protected from direct exposure
with a screen. ATP and shock therapies should be disabled during
exposure and proper device function should be checked regularly
afterwards. Resulting damage may not be immediately detectable. If
irradiation of tissues close to the implantation site is necessary, it is
recommended that the cardiac defibrillator be moved. As a safety
measure, an external defibrillator should be immediately available.
Lithotripsy: Lithotripsy may permanently damage the device if this
one is at the focal point of the lithotripsy beam. If lithotripsy must be
used, keep the defibrillator at least 2.5 to 5 cm (1-2 inches) away
from the focal point of the lithotripsy beam.
Diagnostic ultrasound (echography): The defibrillator is not
affected by ultrasound imaging devices.
10 – US-ENGLISH
Scales with body fat monitors and electronic muscle stimulators:
A patient with an implanted PARADYM RF VR 9250 should not use
these devices.
4.2. STERILIZATION, STORAGE AND HANDLING
Resterilization: Do not resterilize and re-implant explanted ICDs.
"Use Before" Date: A "Use Before" date is printed on the outer storage
package and on the sterile package. Do not implant the device after this
date because the battery may have reduced longevity and sterility may be
affected. It should be returned to Sorin CRM.
If Package Is Damaged: Do not use the device or accessories if the
packaging is wet, punctured, opened or damaged because the
integrity of the sterile packaging may be compromised. Return the
device to the manufacturer.
Device Storage: Store the device in a clean area, away from
magnets, kits containing magnets, and sources of electromagnetic
interference to avoid device damage. Store the device between 0 -
50 °C (32 - 122 °F). Temperatures outside the specified range may
damage the device.
Equilibration: Allow the device to reach room temperature before
programming or implanting the device because rapid temperature
changes may affect initial device function.
US-ENGLISH – 11
4.3. IMPLANTATION AND DEVICE PROGRAMMING
Use only a Sorin CRM programmer to communicate with the device.
Do not position any magnet over the ICD; this suspends
tachyarrhythmia detection and treatment.
Replace the device when the programmer displays an ERI* (defined
by a battery voltage of 2.66 ± 0.01 V or a magnet rate lower than or
equal to 80 bpm).
Program device parameters such as sensitivity threshold and VT and
VF detection intervals as specified in the device manuals.
Lead System: Do not use a lead system other than those with
demonstrated compatibility because undersensing cardiac activity
and failure to deliver necessary therapy may result.
In situations where an ICD and a pacemaker are implanted in the
same patient, interaction testing should be completed. If the
interaction between the ICD and the pacemaker cannot be resolved
through repositioning of the leads or reprogramming of either the
pacemaker or the ICD, the pacemaker should not be implanted (or
should be explanted if previously implanted).
Failure to properly insert the torque wrench into the perforation at an
angle perpendicular to the connector receptacle may result in
damage to the sealing system and its self-sealing properties.
A safety margin of at least 10 J in the defibrillation threshold (DFT) is
recommended. Carefully confirm that true ventricular fibrillation has
been induced because the DFT for ventricular tachycardia or flutter
may be lower.
The defibrillator should be implanted with the engraved side facing
outwards in order to facilitate telemetric communication with the
programming head and to display the radiographic identification correctly.
*: corresponds to Recommended Replacement Time (RRT) / End of
Service (EOS) as referred in the EN45502-2-2 standard.
12 – US-ENGLISH
4.4. LEAD EVALUATION AND LEAD CONNECTION
PARADYM RF VR 9250 has two DF-1 and one IS-1 connector ports.
IS-1 refers to the international standard whereby leads and
generators from different manufacturers are assured a basic fit (ISO
5841-1:2000). DF-1 refers to the international standard for
defibrillation lead connectors (ISO 11318:2002).
Do not tie a ligature directly to the lead body, tie it too tightly, or
otherwise create excessive strain at the insertion site as this may
damage the lead. Use the lead stabilizer to secure the lead lateral to
the venous entry site.
Do not immerse the lead in mineral oil, silicone oil, or any other
liquid.
Do not grip the lead with surgical instruments.
Do not use excessive force or surgical instruments to insert a stylet
into a lead.
Use ventricular transvenous leads with caution in patients with either
a mechanical or bioprosthetic tricuspid valvular prosthesis.
Use the correct suture sleeve (when needed) , to immobilize the lead
and protect it against damage from ligatures.
Never implant the system with a lead system that has a measured
shock impedance of less than 30 ohms. A protection circuit in the
defibrillator prevents shock delivery when impedance is too low. If the
shock impedance is less than 30 ohms, reposition the lead system to
allow a greater distance between the electrodes.
Do not kink leads. Kinking leads may cause additional stress on the
leads, possibly resulting in lead fracture.
Do not insert a lead connector pin into the connector block without
first visually verifying that the setscrews are sufficiently retracted. Do
not tighten the setscrews unless a lead connector pin is inserted
because it could damage the connector block.
US-ENGLISH – 13
Lead electrodes in contact during a cardioversion or defibrillation
therapy will cause current to bypass the heart, possibly damaging the
ICD and the lead. While the ICD is connected to the lead, make sure
that the metal portions of any electrodes do not touch each other.
If a pacing lead is abandoned rather than removed, it must be capped to
ensure that it is not a pathway for currents to or from the heart.
If a thoracotomy is required to place epicardial patches, it should be
done during a separate procedure to reduce the risk of morbidity and
mortality.
Do not place the patch lead over nerve tissue as this may cause
nerve damage.
Place the patch lead with the conducting coil side facing the heart to
ensure delivery of energy to the heart.
Place the sutures well outside the coil of the patch lead or in the area
between the coils to avoid possible coil fracture.
If countershock is unsuccessful using external paddles, adjust the
external paddle position (e.g., anterior-lateral to anterior-posterior)
and be sure that the external paddle is not positioned over the patch.
Do not fold, alter, or remove any portion of the patch as it may
compromise electrode function or longevity.
If a header port is unused on the generator, the port must be plugged
to protect the generator.
4.5. GENERATOR EXPLANT AND DISPOSAL
Interrogate the device, and program shock therapy off prior to
explanting, cleaning or shipping the device to prevent unwanted
shocks.
Return all explanted generators and leads to the manufacturer.
Never incinerate the device due to the potential for explosion.
The device must be explanted before cremation.

14 – US-ENGLISH
5. ADVERSE EVENTS
Clinical data presented in this section are from the Defender study.
PARADYM RF VR 9250 is similar in design and function to the
Defender devices. The data provided are applicable to
PARADYM RF VR 9250.
5.1. DEFENDER STUDY
Clinical study of Defender IV DR 612 included 60 devices implanted
in 60 patients, 38 in Europe (37 patients followed for a minimum of 3
months), and 22 in the U.S. (IDE G970282/S15) with a total device
exposure of 228.7 and 30.3 device months, respectively. No deaths,
serious adverse experiences or complications were judged to be
device-related, as determined by the investigator. The following
tables summarize the safety data for this study.
There was 1 death in the study that was classified as arrhythmic. The
cause of death was recurrent VT/VF which occurred 19 days post
implant.
In the following tables, complications are defined as adverse device
effect, which cannot be treated or resolved by simple adjustments
(e.g. reprogramming) and requires intervention.
NOTE: The company classified as complications those adverse
device effects that were treated with surgery or with external
defibrillation of a ventricular cardiac event.
Observations are defined as symptomatic or asymptomatic clinical
events with potential adverse device effects that do not require
intervention or can be corrected by simple adjustments.
NOTE: The company classified as observations those adverse
device effects that were treated with programming changes,
medication, or other method that was not classified as a
complication.

US-ENGLISH – 15
Two of the 38 Defender IV DR 612 patients in Europe (37 patients
followed for a minimum of 3 months) experienced a total of three
complications, including device failures and replacements. Fourteen
of the 38 Defender IV DR 612 patients experienced a total of 18
observations. Complications and observations are reported in Tables
1 and 2. It should be noted that a patient can have more than one
observation or complication. There were no observations or
complications in the U.S.
5.2. TABLE 1: SUMMARY OF EUROPEAN
CLINICAL COMPLICATIONS
(Including Device Failures and Replacements)
All complications, 2 of 38 Defender IV DR 612 patients in Europe
Event
# of
Patients
% of
Patients
# of
Events
Events/100
Device-
Years*
Hematoma
1
2.6
1
5.2
Ventricular lead
migration/dislodgment
2
5.3
2
10.5
* There were 228.7 device months in this study.

16 – US-ENGLISH
5.3. TABLE 2: SUMMARY OF EUROPEAN
CLINICAL COMPLICATIONS
(Including Patient Complaints)
All complications, 14 of 38 Defender IV DR 612 patients in Europe
Event
# of
Patients*
% of
Patients
# of
Events
Events/100
Device-
Years**
Change in
ventricular sensing
threshold
1
2.6
1°
5.2
Device reset***
1
2.6
1°
5.2
Inappropriate
therapy for EMI
1
2.6
1°
5.2
Pneumothorax
1
2.6
1°
5.2
Pocket hematoma
2
5.3
2°
10.5
Pocket
infection/hematoma
1
2.6
1°
5.2
Pocket infection
from previous
pacemaker
1
2.6
1°
5.2
Prolonged implant
procedure
1
2.6
1
5.2
Sensor acceleration
during telemetry***
1
2.6
1
5.2
Shock for VT in VF
Zone
1
2.6
1°
5.2
US-ENGLISH – 17
Event
# of
Patients*
% of
Patients
# of
Events
Events/100
Device-
Years**
Slow VT not
converted by ATP
therapy
1
2.6
2°
10.5
Unsatisfactory
sensing threshold
test***
2
5.3
2
10.5
Ventricular
oversensing
3
7.9
3
15.7
* A patient can have more than one observation.
** There were 228.7 device months in this study.
***These observations would not have happened with the currently
marketed device and programmer.
°Investigator indicated that Defender IV DR did not cause or
contribute to the event.

18 – US-ENGLISH
6. CLINICAL STUDIES
Clinical data presented in this section are from the Defender study.
PARADYM RF VR 9250 is similar in design and function to the
Defender devices. The data provided are applicable to
PARADYM RF VR 9250.
6.1. DEFENDER STUDY
Objectives: The primary objectives of this study were to
demonstrate a complication free rate (CFR) comparable to that of
historical controls, to demonstrate, using a chronotropic assessment
exercise protocol (CAEP), a rate response proportional to and
appropriate for the level of exercise, and to evaluate and report the
incidence of adverse events.
Materials: Each patient received one Defender IV DR 612
defibrillator, an atrial pacing and sensing lead, and a Medtronic,
Angeion, or Biotronik defibrillation lead in the U.S. or any
commercially available defibrillator lead outside the U.S.
Methods: Investigators selected patients who survived at least one
episode of cardiac arrest (manifested by loss of consciousness)
presumably due to a ventricular tachyarrhythmia or exhibited
recurrent, poorly tolerated, sustained ventricular tachycardia (VT).
The protocol required evaluation of performance and adverse events
at pre-discharge, one month, three months, six months, and (in the
U.S.) every three months thereafter. At the one-month visit, eligible
patients performed a chronotropic assessment exercise protocol
(CAEP) maximal exercise test.

US-ENGLISH – 19
Study Population. The table below summarizes inclusions.
Region
Date of
first
implant
Date of
last
implant
Data cut-
off date
Number
of
centers
Number
of
patients
US
14-Dec-
99
08-Mar-
00
14-Mar-
00
6
22
Europe
04-May-
99
26-Jul-99
14-Apr-
00
11
38
All
04-May-
99
08-Mar-
00
14-Apr-
00 (Eur),
14-Mar-
00 (US)
17
60
Complication-free rate
Only European patients followed for at least 3 months:
Symbol
Parameter
Defender IV DR 612
N
Overall number of
patients
37
Pe*N
Number of successes
35
Pe
Observed
experimental
proportion
0.95
Ps
Null hypothesis
success rate
0.76
ES
Estimated standard
error of Pe
0.04
z´
Test statistic (1)
4.75
p
Associated p-value
< 0,0001
(1) Statistical test: z´ = (Pe-Ps)/SE where SE = sqrt(Pe(1-Pe)/N)
20 – US-ENGLISH
Rate response
European patients only:
GROUP
Number
of
patients
included
Mean
slope
%SRR
on
%MR
STD of
slopes
%SRR
on
%MR
SE of
mean
slope
%SRR
on
%MR
Lower
95% CI
Upper
95% CI
Europe
20
0.77
0.17
0.04
0.69
0.84
Small
Centers
9
0.79
0.18
0.06
0.67
0.91
Large
Centers
11
0.75
0.15
0.05
0.66
0.84
Males
17
0.77
0.16
0.04
0.70
0.85
Females
3
0.73
0.22
0.13
0.47
0.98
SRR: Sensor Rate Reserve
MR: Metabolic Reserve
STD: Standard Deviation
SE: Standard Error
CI: Confidence Interval

US-ENGLISH – 21
Adverse events
Event US
(N=22)
Number of
events*
Number of
patients
Percent of
patients
Intent to treat
but did not
0
0
0.0
Non-device
related death
0
0
0.0
Explant
0
0
0.0
Complication
0
0
0.0
Observation
0
0
0.0
Serious non-
related other
than death
1
1
4.5
Event Europe
(N=38)
Number of
events*
Number of
patients
Percent of
patients
Intent to treat
but did not
0
0
0.0
Non-device
related death
1
1
2.6
Explant
1
1
2.6
Complication
3
2
5.3
Observation
18
14
36.8
Serious non-
related other
than death
12
7
18.4

22 – US-ENGLISH
Event All
(N=60)
Number of
events*
Number of
patients
Percent of
patients
Intent to treat
but did not
0
0
0.0
Non-device
related death
1
1
1.7
Explant
1
1
1.7
Complication
3
2
3.3
Observation
18
14
23.3
Serious non-
related other
than death
13
8
13.3
* A patient can have more than one complication, observation, or
serious adverse event, not device-related.
Device Failures and Replacements: No device failures or
replacements occurred with Defender IV DR 612 during the study.

US-ENGLISH – 23
7. PATIENT SELECTION AND TREATMENT
7.1. INDIVIDUALIZATION OF TREATMENT
Exercise stress testing: If the patient’s condition permits, use
exercise stress testing to:
Determine the maximum rate of the patient’s normal rhythm,
Identify any supraventricular tachyarrhythmias,
Identify exercise-induced tachyarrhythmias.
The maximum exercise rate or the presence of supraventricular
tachyarrhythmias may influence selection of programmable
parameters. Holter monitoring or other extended ECG monitoring
also may be helpful.
CAUTION: To avoid inappropriate therapy during an exercise stress
test, do not reprogram any parameter during the test. When a
parameter is reprogrammed, "Discrimination" algorithm forces
acceleration to "ventricular". During conducted sinus tachycardia
within the programmed Tachy zone, the device detects a 1:1 fast
rhythm. Assuming that acceleration was set to ventricular by
reprogramming, the device may identify this as a VT, and may
immediately apply the corresponding therapy.
Electrophysiologic (EP) testing: EP testing may be useful for ICD
candidates. EP testing may identify the classifications and rates of all the
ventricular and atrial arrhythmias, whether spontaneous or during EP
testing.
Drug resistant supraventricular tachyarrhythmias (SVTs): Drug
resistant supraventricular tachyarrhythmias (SVTs) may initiate
frequent unwanted device therapy. A careful choice of programming
options is necessary for such patients.
24 – US-ENGLISH
Antiarrhythmic drug therapy: If the patient is being treated with
antiarrhythmic or cardiac drugs, the patient should be on a
maintenance drug dose rather than a loading dose at the time of ICD
implantation. If changes to drug therapy are made, repeated
arrhythmia inductions are recommended to verify ICD detection and
conversion. The ICD also may need to be reprogrammed.
Changes in a patient’s antiarrhythmic drug or any other medication
that affects the patient’s normal cardiac rate or conduction can affect
the rate of tachyarrhythmias and/or efficacy of therapy.
Direct any questions regarding the individualization of patient therapy
to Sorin CRM’s representative.
7.2. SPECIFIC PATIENT POPULATIONS
Pregnancy: If there is a need to image the device, care should be
taken to minimize radiation exposure to the fœtus and the mother.
Nursing Mothers: Although appropriate biocompatibility testing has
been conducted for this implant device, there has been no
quantitative assessment of the presence of leachables in breast milk.
Pediatric Patients: This device has not been studied in patients
younger than 18 years of age.
Geriatric Patients: Most of the patients receiving this device in
clinical studies were over the age of 60 years.
Handicapped and Disabled Patients: Special care is needed in
using this device for patients using an electrical wheel chair or other
electrical (external or implanted) devices.

US-ENGLISH – 25
8. PATIENT COUNSELLING INFORMATION
The physician should consider the following points in counselling the
patient about this device:
Persons administering CPR may experience tingling on the
patient’s body surface when the patient’s ICD system delivers a
shock.
Advise patients to carry Sorin CRM ID cards and/or ID bracelets
documenting their ICD system.
9. CONFORMANCE TO STANDARDS
This device was developed in conformance with all or parts of the
following standards:
EN 45502-1: 1998 – Active implantable medical devices.
General requirements for safety, marking and information to be
provided by the manufacturer.
EN 45502-2-1: 2003 - Active implantable medical devices.
Part 2-1: Particular requirements for active implantable medical
devices intended to treat bradyarrhythmia (cardiac pacemakers).
EN 45502-2-2: 2008 – Active implantable medical devices.
Part 2-2: Particular requirements for active implantable medical
devices intended to treat tachyarrhythmia (includes implantable
defibrillators).
ISO 5841-3: 2000 Low profile connectors (IS1) for implantable
pacemakers.
ISO 11318 (DF-1): Cardiac defibrillator: connector assembly for
implantable defibrillators - Dimensional and test requirements,
August 2002.
ANSI/AAMI PC69:2007 Active implantable Medical Devices -
Electromagnetic compatibility - EMC test protocols for implantable
cardiac pacemakers and implantable Cardioverter Defibrillators.
26 – US-ENGLISH
IEC 60601-1-2 (2007): Electromagnetic compatibility - Medical
electrical equipment. General requirements for basic safety and
essential performance - Collateral standard
EN 50371 (2002) : Generic standard to demonstrate the
compliance of low power electronic and electrical apparatus with
the basic restrictions related to human exposure to
electromagnetic fields (10 MHz - 300 GHz)
EN 301 489-1 (v1.8.1) & EN 301 489-27 (v1.1.1): Electromagnetic
compatibility and Radio spectrum Matters (ERM);
Electromagnetic Compatibility (EMC) standard for radio
equipment and services - Part 1 : Technical Requirements and
Part 27: Specific conditions for Ultra Low Power Active Medical
Implants (ULP-AMI) and related peripheral devices (ULP-AMI-P)
EN 301839-1 (v1.3.1) & EN 301839-2 (v1.2.1): Electromagnetic
compatibility and Radio spectrum Matters (ERM); Short Range
Devices (SRD); Ultra Low Power Active Medical Implants (ULP-
AMI) and Peripherals (ULP-AMI-P) operating in the frequency
range 402 MHz to 405 MHz; Part 1: Technical characteristics and
test methods and Part 2: Harmonized EN covering essential
requirements of Article 3.2 of the R&TTE Directive
EN 62311 (2008) : Assessment of electronic and electrical
equipment related to human exposure restrictions for
electromagnetic fields (0Hz to 300 GHz)
EN 62209-2 (2010) : Human exposure to radio frequency fields
from hand-held and body-mounted wireless communication
devices – Human models, instrumentation and procedures – Part
2: Procedure to determine the specific absorption rate (SAR) for
wireless communication devices used in close proximity to the
human body (frequency range of 30MHz to 6 GHz)
This information should not be used as a basis of comparisons
among devices since different parts of the standards mentioned may
have been used.

US-ENGLISH – 27
Sorin CRM declares that this device is in conformity with the
essential requirements of Directive 1999/5/EC on Radio and
Telecommunications Terminal Equipment, with the mutual
recognition of their conformity (R&TTE).
Federal Communication Commission Interference Statement 47
CFR Section 15.19 and 15.105(b)
- The FCC product ID is YSGVR9250.
This equipment has been tested and found to comply with the limits for a
Class B digital device, pursuant to Part 15 of the FCC Rules. These limits
are designed to provide reasonable protection against harmful
interference in a residential installation. This equipment generates uses
and can radiate radio frequency energy and, if not installed and used in
accordance with the instructions, may cause harmful interference to radio
communications. However, there is no guarantee that interference will not
occur in a particular installation.
This device complies with Part 15 of the FCC Rules. Operation is subject
to the following two conditions: (1) This device may not cause harmful
interference, and (2) this device must accept any interference received,
including interference that may cause undesired operation.
FCC Interference Statement 47 CFR Section 15.21 - No Unauthorized
Modifications
CAUTION: This equipment may not be modified, altered, or changed in
any way without signed written permission from SORIN. Unauthorized
modification may void the equipment authorization from the FCC and will
void the SORIN warranty.
Identification of the equipment according Section 95.1217(a)
This transmitter is authorized by rule under the Medical Device
Radiocommunication Service (in part 95 of the FCC Rules) and must not
cause harmful interference to stations operating in the 400.150-406.000
MHz band in the Meteorological Aids (i.e., transmitters and receivers
28 – US-ENGLISH
used to communicate weather data), the Meteorological Satellite, or the
Earth Exploration Satellite Services and must accept interference that
may be caused by such stations, including interference that may cause
undesired operation. This transmitter shall be used only in accordance
with the FCC Rules governing the Medical Device Radiocommunication
Service. Analog and digital voice communications are prohibited.
Although this transmitter has been approved by the Federal
Communications Commission, there is no guarantee that it will not
receive interference or that any particular transmission from this
transmitter will be free from interference.
IC Requirements for canada
- The IC product ID is 10270A-VR9250
This class B digital apparatus meets all requirements of the Canadian
Interference- causing equipment regulations.
This device complies with Industry Canada licence-exempt RSS
standard(s). Operation is subject to the following two conditions: (1) this
device may not cause interference, and (2) this device must accept any
interference, including interference that may cause undesired operation of
the device.
Under Industry Canada regulations, this radio transmitter may only
operate using an antenna of a type and maximum (or lesser) gain
approved for the transmitter by Industry Canada. To reduce
potential radio interference to other users, the antenna type and its gain
should be so chosen that the equivalent isotropically radiated power
(e.i.r.p.) is not more than that necessary for successful communication.
This device may not interfere with stations operating in the 400.150–
406.000 MHz band in the Meteorological Aids, Meteorological Satellite,
and Earth Exploration Satellite Services and must accept any interference
received, including interference that may cause undesired operation.

US-ENGLISH – 29
10. PHYSICIAN GUIDELINES
10.1. PHYSICIAN TRAINING
Physicians should be familiar with sterile pulse generator implant
procedure and familiar with follow-up evaluation and management of
patients with an implantable defibrillator (or referral to such a physician).
10.2. DIRECTIONS FOR USE
ICD operating characteristics should be verified at the time of
implantation and recorded in the patient file. Complete the Patient
Registration Form and return it to Sorin CRM, as it provides
necessary information for warranty purposes and patient tracking.
Additional programming instructions can be found by accessing
Online Help (click the “?” on the screen) on the Sorin CRM dedicated
programmer. Paper copies of Online Help can be obtained by
contacting your Sorin CRM representative.
10.3. MAINTAINING DEVICE QUALITY
This device is FOR SINGLE USE ONLY. Do not resterilize and
reimplant explanted ICDs.
Do not implant the device when:
It has been dropped on a hard surface because this could have
damaged pulse generator components.
Its sterility indicator within the inner package is not green,
because it might not have been sterilized.
Its storage package has been pierced or altered, because this
could have rendered it non-sterile.

30 – US-ENGLISH
It has been stored or transported outside the environmental
temperature limits: 32 °F (0 °C) to 122 °F (50 °C) as an electrical
reset condition may occur.
"Use Before" date has expired, because this can adversely affect
pulse generator longevity or sterility.
11. PATIENT INFORMATION
Information for the patient is available in the patient booklet,
contained in the outer storage package. Additional copies can be
obtained by contacting your Sorin CRM representative or on the
Sorin CRM's web site: http://www.sorin.com.
This information should be given to each patient with their first ICD
and offered to the patient on each return visit or as deemed
appropriate.
12. HOW SUPPLIED
12.1. STERILITY
The PARADYM RF defibrillators are supplied one per package in a
sterile package.
12.2. WARRANTY AND REPLACEMENT POLICY
Sorin CRM warrants its defibrillators. Refer to the section "Warranty"
for additional information. Please see the following labelling sections
for information concerning the performance of this device:
Indications, Contraindications, Warnings and Precautions, and
Adverse Events.

US-ENGLISH – 31
13. DEVICE DESCRIPTION
The PARADYM RF VR system includes the model 9250 ICD device
and programming system. The programming system includes the
Sorin CRM Orchestra Plus programmer with the SMARTVIEW
programming software connected to a CPR3 programming head. The
programming system is configured and furnished by Sorin CRM.
The PARADYM RF VR 9250 can serve as a defibrillation electrode
(active housing) with a total surface area of 76 cm².
The PARADYM RF VR 9250 is designed to recognize and treat slow
or fast VT and VF by continuously monitoring ventricular activity to
identify persistent ventricular arrhythmias and to deliver appropriate
therapies. PARADYM RF VR 9250 features DISCRIMINATION
algorithm, which is specifically designed to differentiate ventricular
tachycardias from fast rhythms of supraventricular origin.
DISCRIMINATION continuously monitors R-R interval stability,
searches for long cycles and evaluates sudden onset.
In addition to the advanced detection scheme, PARADYM RF VR 9250
offers programmable single-chamber pacing therapy with or without
rate-responsive capabilities using an acceleration sensor.
PARADYM RF VR 9250 offers tiered therapy. Therapies can be
programmed independently in each zone:
in the Slow VT and VT zones: two ATP programs, up to
two shocks with programmable energy and up to four
shocks with maximum energy can be programmed;
in the VF zone: one ATP program, up to two shocks with
programmable energy and up to four shocks with
maximum energy can be programmed.
When the rhythm changes from one zone to another, the device
delivers the therapy programmed in this zone, starting with the same
or more aggressive program for the area. The ATP program in the
VF zone will only be applied if the VT coupling interval is longer than
the programmed fast VT cycle length.
32 – US-ENGLISH
The PARADYM RF VR 9250 offers biphasic shocks with a maximum
stored energy of 42 J. The shock configuration (electrodes used to
apply the shock) can be chosen by programming one of the following
combinations: can and one coil, can and 2 coils, 2 coils only.
Other features are as follows:
Automatic ventricular sensitivity control
Non-committed shocks
Electrophysiological studies (EPS) with real-time markers or
electrograms:
Programmer-controlled VT induction sequences,
Programmer-controlled VF inductions (30 Hz rapid pacing
or shock on T),
Programmable electrogram vectors ( EGM V, RVcoil-
CAN, SVC-CAN, RVcoil-SVC)
Real-time annotations displayed with the markers and
indicating the majority rhythm,
Manual ATP sequences,
Manual shocks.
Rescue shock
Follow-up tests:
Pacing lead impedance,
Coil impedance,
Capacitor charge time,
Pacing threshold tests.
Data storage:
Therapy History Report,
Statistics (pace/sense, therapy, shocks, and battery
voltage),
Up to 14 complete Holter records with event logs, marker
channel notation, and electrogram records.

US-ENGLISH – 33
The connector head has three ports: ventricular bipolar pace/sense
and two ports for RV and SVC defibrillation coils. The pace/sense
port is compatible with the IS-1 standard and both defibrillation ports
are compatible with the DF-1 standard. Distal lead terminal
connections are secured with set-screws accessed via self-sealing
silicone plugs. All lead connections pass through the header into the
device via feedthroughs.
Programming System: The Sorin CRM programmer is used in
conjunction with specific programmer software to interrogate and
program the implanted device at implant and during patient follow-up
procedures.
Remote Monitoring: The PARADYM RF VR 9250 is also equipped
with the RF wireless technology which enables to remotely monitor
the patients who have the Sorin CRM SMARTVIEW Monitor installed
at home.
14. IMPLANT PROCEDURE
14.1. NECESSARY EQUIPMENT
Implantation of PARADYM RF VR 9250 requires the following
equipment:
Sorin CRM dedicated programmer, equipped with the
SMARTVIEW software interface and with the programming head,
pacing system analyser, as well as its sterile connecting cables,
to evaluate the pacing and sensing thresholds,
a ventricular pacing and defibrillation lead,
physiological signal monitor capable of displaying simultaneously
the surface ECG and arterial pressure,
an external defibrillator with sterile external paddles,
sterile cover for the telemetry head.
34 – US-ENGLISH
14.2. PACKAGING
Contents
The PARADYM RF VR 9250 and its accessories are ethylene oxide
sterilized and hermetically sealed in two-ply clear packaging meeting
international requirements.
The sterile packaging contains a defibrillator, one screwdriver, and
an insulating plug for the DF-1 defibrillation connector.
The non-sterile items contained in the outer storage package are the
implant manual, the ICD Registration Form and its envelope, the
patient booklet, the ICD ID card and 12 identification labels.
Once delivered, PARADYM RF VR 9250 is programmed to as-
shipped values that are different from nominal values (see Chapter
“Programmable Parameters” for details).
14.3. OPTIONAL EQUIPMENT
The following equipment may be required during implantation
of PARADYM RF VR 9250:
sterile water to clean traces of blood. Any parts cleaned with
sterile water must be thoroughly dried.
mineral oil to lubricate if necessary
a lead cap to isolate a lead which is not used
14.4. BEFORE OPENING THE PACKAGE
Before opening the package, check the "Use Before" date printed on
the labels on the box and on the sterile package. Defibrillators that
have not been implanted before that date should be returned to
Sorin CRM.
Devices MUST NOT be interrogated and programmed within the
vicinity of other devices.
US-ENGLISH – 35
Also check the integrity of the sterile package. The sterility of the
contents is no longer guaranteed if the package has been pierced or
altered. If the defibrillator is no longer sterile, it should be returned in
its packaging to Sorin CRM. Any re-sterilization of the unit is at the
discretion of Sorin CRM.
14.5. PRIOR TO IMPLANTATION
Use the programmer to verify the defibrillator can be interrogated
before implantation.
Verify all shock therapies are disabled in order to avoid accidental
discharge during implantation.
It is not advisable to program the Smoothing function before
implantation, since the defibrillator may detect noise and pace at a
rate higher than the programmed basic rate.
CAUTION: Do not shake or tap sharply on the ICD package with the
ICD inside, because the ICD's sensing circuits can interpret this as
R-waves and record these as an arrhythmia episode. If unusual
shaking or tapping of the package results in a stored arrhythmia
episode, erase the recording before using the ICD.
14.6. DEVICE PLACEMENT
The pocket should be prepared in the left pectoral position, either
subcutaneously or submuscularly. Subcutaneous device implantation
is recommended for optimal RF communication efficacy.
Implantation in an abdominal position is not advisable.
In its final position, the defibrillator should be no more than 4 cm
below the skin surface.
36 – US-ENGLISH
14.7. CHOOSING THE TYPE OF LEAD
The defibrillator should be connected to:
one ventricular defibrillation lead with sensing/pacing bipolar
electrodes, and one or two defibrillation electrodes.
The choice of leads and their configuration is left to the implanting
physician’s judgment.
Connectors: The bipolar pacing/sensing connector is compliant with
the IS-1 standard and the defibrillation connectors are compliant with
the DF-1 standard.
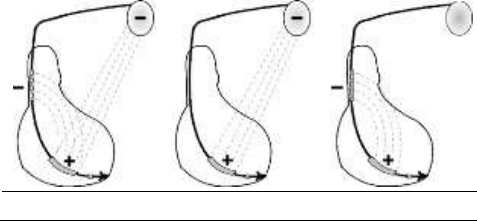
Shock configuration (+ -> -)
The shock configuration is the energy pathway between the
defibrillation electrodes. If an atrial coil is present, the shock
configuration can be programmed for bi-directional shocks.
Programming: When active case and SVC are both programmed to
Yes, the shock configuration can be programmed to: 1. RV to Case
(or Case to RV), 2. RV to SVC (or SVC to RV), 3. RV to Case+SVC
(or Case+SVC to RV).
The polarity of shock is determined by the parameter itself.
RV to Case+SVC
RV to Case
RV to SVC
US-ENGLISH – 37
14.8. MEASUREMENT OF THRESHOLDS AT
IMPLANT
Pacing and sensing thresholds should be measured at implant.
Pacing threshold: Acute thresholds should be lower than 1 V (or
2 mA) for a 0.35 ms pulse width.
Sensing threshold: For proper ventricular sensing, the amplitude of
the R-wave should be greater than 5 mV.
Pacing impedance measurement: Ventricular pacing impedance
should range from 200 to 3000 ohms (refer to the lead
characteristics, especially if high impedance lead is used).
14.9. LEAD CONNECTION
The lead must be connected to the corresponding connector port.
The position of each connector is indicated on the casing.
CAUTION: Tighten only the distal insert.
To connect each lead, proceed as follows:
1. Clean the lead terminal pins thoroughly, if necessary (device
replacement).
2. Lubricate the lead terminal pins with sterile water, if necessary.
3. Do not insert a lead connector pin into the connector block
without first visually verifying that the lead port is not filled with
any obstacle.
4. Insert the screwdriver into the pre-inserted screw socket of the
appropriate port (in order to allow excess air to bleed out and to
make the insertion of the lead pin easier).
5. Insert the lead pin all the way into the port (check that the pin
protrudes beyond the distal insert).
6. Tighten, check the tightness and ensure the lead pin still
protrudes beyond the distal insert, and did not move.
38 – US-ENGLISH
CAUTION: 1. One single set screw is located on the side of the
connection header. 2. Do not tighten the pre-inserted screws when
there is no lead (this could damage the connector). 3. Do not loosen
the screws before inserting the connector (subsequent risk of being
unable to reinsert the screw). 4. Removing the screwdriver: to avoid
all risk of loosening screws during removal, hold the screwdriver by
its metal part and not by the handle. 5. When mineral oil or sterile
water is used to make lead insertion easier, the screwdriver should
remain inserted into the pre-inserted screw socket when checking the
tightness. As a matter of fact, when the lead port is filled with a liquid,
the physics piston effect can give the feeling the lead is properly
tightened.
NOTE: To optimise cardioversion/defibrillation shocks, electrodes
must be positioned so that the electric field between anode (s) and
cathode covers the largest myocardial mass. In normal conditions,
the anode and cathode are adequately separated. In case of a short-
circuit, the shock may be aborted to prevent damaging the
defibrillator.
In the case of an external defibrillation shock delivered to the patient,
always check the programming and functioning of the device, in
particular its capacity to deliver shocks.
14.10. DEVICE IMPLANTATION
PARADYM RF VR 9250 should be implanted with the engraved side
facing outwards for optimal communication with the programming
head and radiographic identification.
Place the device in the pocket. Once in place, the defibrillator should
be no more than 4 cm below the skin surface.
Carefully wind excess lead and place in a separate pocket to the side
of the defibrillator.
It is recommended to not place any excess wire between the can and
the heart.

US-ENGLISH – 39
Suture the casing connector to the muscle using the hole provided
for this purpose, in order to avoid potential migration of the device
into the pectoral muscle.
14.11. TESTS AND PROGRAMMING
During the implant testing procedure, it is recommended that a
security margin of at least 10 J be demonstrated between the
effective shock energy and maximum programmable energy.
Enable shock therapies, then program the defibrillator.
Verify that the defibrillation lead impedance for each shock delivered
ranges from 30 to 150 ohms. Check the lead connection if the values
are outside these boundaries.
Save the programming data on the programmer’s hard disk and on
an external storage device (if desired).
15. SPECIAL MODES
15.1. SAFETY MODE (NOMINAL VALUES)
Nominal values may be rapidly restored by pressing the following
button on the programming head or programmer keyboard:
or via the "Emergency" button on the SMARTVIEW screen.
In safety mode, the defibrillator operates with the parameters
underlined in the table of programmable parameters.

40 – US-ENGLISH
15.2. MAGNET MODE
When the magnet is applied:
antiarrhythmia functions are inhibited (detection of rhythm
disturbances, charging, and therapy),
pacing amplitude is set to 6 V,
pulse width is set to maximum,
pacing rate is set to the magnet rate,
the following functions are disabled: Smoothing, Rate Response.
When the magnet is removed:
the sensor rate is forced to the basic rate,
arrhythmia detection algorithms and sequential therapies are
reinitialized,
therapies start with the least aggressive program for each area.
The other parameters remain at their programmed value.
The magnet rate values are as follow:
Magnet rate (bpm)
96
94
91
89
87
85
Magnet period (ms)
625
641
656
672
688
703
Magnet rate (bpm)
83
82
80
78
77
Magnet period (ms)
719
734
750
766
781
15.3. RESPONSE IN THE PRESENCE OF
INTERFERENCE
If the defibrillator senses electrical noise at a frequency above 16 Hz, it
switches to an asynchronous mode at the basic rate. The programmed
mode is restored as soon as the noise is no longer detected.
Ventricular pacing is also inhibited by ventricular noise. It can be
restored by setting the parameter V pacing on noise to Yes.
US-ENGLISH – 41
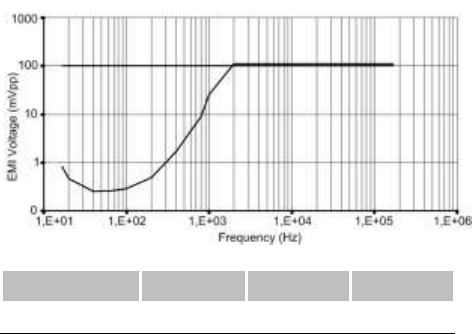
15.4. DETECTION CHARACTERISTICS IN THE
PRESENCE OF ELECTROMAGNETIC FIELDS
Per Clause 27.4 of Standard EN 45502-2-2, detection in the
presence of electromagnetic fields is characterized as follows:
Differential mode:
Common mode rejection ratio:
16.6 Hz
50 Hz
60 Hz
Ventricular channel
≥ 69 dB
≥ 69 dB
≥ 69 dB
15.5. PROTECTION AGAINST SHORT-CIRCUITS
The defibrillator can undergo a short-circuit if the anode and cathode
are not adequately separated.
In this case, the shock is aborted to prevent damaging the
defibrillator and a warning will indicate that a short circuit (shock
impedance < 20 ohms) was detected during the last shock.

42 – US-ENGLISH
16. MAIN FUNCTIONS
16.1. AUTOMATIC LEAD MEASUREMENTS
Automatic pacing lead impedance measurement: A lead
impedance measurement is automatically performed on the lead
every 6 hours. The daily mean impedance is stored.
Automatic coil impedance measurement: A coil impedance
measurement is automatically performed on RV and SVC coils once
a week. The result is stored in the device memory.
16.2. VENTRICULAR TACHYARRHYTHMIA
MANAGEMENT
Searching for a long cycle (Stability+): Additional arrhythmia
classification criterion to improve identification of atrial fibrillation and
avoid inappropriate shocks.
Fast VT treatment: Applies detection criteria on fast ventricular
tachycardiathat are different from those of the VT zone, as well as
different therapies. The fast VT zone is included in the VF zone: its
lower limit is determined by the programmed value for the VF zone
and its upper limit by the programmed value for the fast VT zone.
Polarity alternation on Max:shock Reverses the programmed polarity
of every second shock set at maximum energy. The number, type, and
energy of shocks is independently programmable by detection zone.
16.3. PACING
BTO (Brady Tachy Overlap): Allows pacing in the slow VT zone,
without affecting detection specificity.
Post-shock mode: After any automatic shock therapy, the post-
shock mode makes it possible to apply different pacing parameters.
US-ENGLISH – 43
16.4. SENSING
Automatic Refractory Periods: Optimize sensing and make the
implant progamming easier. These periods are composed of a
minimal Refractory Period and a triggerable Refractory Period. The
duration of the refractory periods lengthens automatically as needed.
Protection against noise: Allows the distinction between ventricular
noise and ventricular fibrillation. If the device senses ventricular noise, the
ventricular sensitivity is decreased until noise is no longer detected.
Ventricular pacing can be inhibited to avoid a potential paced T-wave.
Automatic sensitivity control: Optimizes arrhythmia detection and
avoids late detection of T-waves and over-detection of wide QRS
waves. The device automatically adjusts the sensitivities based on
the ventricular sensing amplitude. In case of arrhythmia suspicion or
after a paced event, the programmed ventricular sensitivity will be
applied. The minimum ventricular sensitivity threshold is 0.4 mV
(minimum programmable value).
16.5. FOLLOW-UP FUNCTIONS
Storage of memory data: AIDA+ (Automatic Interpretation for
Diagnosis Assistance) software provides access up to 6 months of
patient follow-up with day by day data collection, or up to 24 hours
with hourly data collection. Episodes of ventricular tachyarrhythmia
are recorded with the programmable EGM channels: either by
selecting up to two traces, or by selecting "V-Double" which enables
a one-channel recording that is twice as long.
Alerts / Warnings: The device routinely performs security self-
checks and technical measurements to ensure system integrity.
When system integrity is found to be at risk outside a follow-up, alerts
are stored in the device memory. When system integrity is found to
be at risk during a follow-up, the information is managed as a
warning (pop-up message) to notify immediately the user.
44 – US-ENGLISH
For example, the following types of event can trigger a warning or an
alert: technical problem during a shock, pacing lead impedance or
coil impedance measurements out-of-range, battery depletion,…
16.6. REMOTE MONITORING FUNCTION
Remote monitoring enables the automatic remote transmission of
implant data to the physician thanks to the wireless Radio Frequency
(RF) communication ability of the implant in order to provide a
comprehensive report to the physician about device functioning and
patient cardiac status without having the patient physically in the
clinic.
The data is transmitted from the implant and the SMARTVIEW
monitor, a small transmitter placed in the patient home.
Implant data are first transmitted to the SMARTVIEW monitor via RF.
Data are then rooted through the phone network to an internet
website. This website is responsible for transforming the implant data
into a comprehensive report that can be consulted by the physician.
SMARTVIEW Monitor
The SMARTVIEW monitor is a small device equipped with an RF
transmission module to communicate with the implant and a modem
to export data through the internet.
The SMARTVIEW monitor is delivered to the patient who has to
install it at home. Preferably the SMARTVIEW monitor will be placed
on the nightstand of the patient, as close as possible to the side of
the bed the patient usually sleeps. The SMARTVIEW monitor shall
be connected to the phone network and the power plug. Regular
transmissions are done during the night when the patient is asleep
next to the SMARTVIEW monitor without any intervention from the
patient.
US-ENGLISH – 45
Transmission trigger
There are 3 different triggers for a remote transmission:
the remote follow-up transmission is scheduled by the physician
to occur regularly (according to the programming).
the alert transmission will take place when the implant has
recorded an abnormal event. The list of abnormal event is
available in a following paragraph. Alert conditions are checked
daily.
the on-demand follow-up transmission is triggered by the patient
himself through the use of a specific button on the
remote-monitor.
Data transmitted
The data transmitted are identical to the data available during a
standard interrogation with the Orchestra Plus programmer.
All counters, histograms, IEGMs and diagnosis available in the
device are transmitted containing (not exhaustive list):
programmed parameters
Information on patient and system implanted
battery status
lead status (brady leads and defibrillation coils)
pacing counters and mean heart rate (brady)
atrial and ventricular arrhythmia counters and episodes
ventricular therapy counters
heart failure monitoring
Data are presented in the form of 2 reports to the physician: the first
one contains a summary of major counters, histograms, warnings
and diagnosis. The second one presents the 3 most important IEGM
episodes automatically selected based on the degree of severity for
the patient.
46 – US-ENGLISH
User website
On the website, the physician is able to:
consult and schedule the remote follow-ups of their patient
configure additional ways of being notified of alerts (for instance
by SMS, fax or e-mail)
consult, print and export patient reports
Alert system
The following set of alert trigger can be independently programmed
ON/OFF by the physician using the Orchestra Plus programmer and
can trigger an alert transmission:
Reset of the device
ERI reached
Low or high impedance (A, RV, LV)
Abnormal coil impedance (shock lead)
Low or High shock impedance
Long charge time
Inefficient high energy shock
All shocks programmed OFF
Shock treated VT/VF
Lack of V pacing in CRT device
Suspicion of noise on the V lead
Fast V rate during AF
WARNINGS
The use of remote monitoring does not replace regular follow-up.
Therefore, when using remote monitoring, the time period between
follow-ups visits may not be extended.
When ERI mode is reached, this information is transmitted via the
remote monitoring facility and then the remote-monitoring is switched
off to preserve battery life.

US-ENGLISH – 47
17. PATIENT FOLLOW-UP
17.1. FOLLOW-UP RECOMMENDATIONS
Before the patient is discharged and at each subsequent follow-up
visit, it is advisable to:
check the occurrence of system warnings
check the battery status,
check the integrity of the pacing and defibrillation leads,
check for proper sensing (sensitivity) and pacing ; set the pacing
amplitude to twice the pacing threshold,
interrogate the implant memories (AIDA+),
check the efficacy of the therapies delivered,
keep a printout of programmed parameters, test results, and
memory data,
reset the memory data and statistics.
These operations should be performed by medical personnel in an
appropriate care unit, with resuscitation equipment present.
It is recommended that a routine follow-up examination be done one
month after discharge, and then every three months until the device
nears the replacement date.
After a device reset, the magnet rate is equal to 87 ppm; it will be
updated within the next 24 hours.
Refer to the online help for a description of displayed warning, and the
necessity to contact Sorin CRM for an evaluation.
Implant software upgrade: In case a new implant software is
downloaded in the device memory through the programmer, a
warning message could be displayed by the programmer to inform
the user and give the proper instructions to follow.
48 – US-ENGLISH
17.2. HOLTER FUNCTION
The Holter records 14 tachyarrhythmia episodes as well as the
therapy history.
STORED EPISODES
PARADYM RF VR 9250 stores 14 episodes (VF, VT, Slow VT,
SVT/ST, nonsustained).
For each episode four levels of details are presented:
Tachogram
Event log for the entire episode:
"Discrimination" analysis for each majority,
Delivered therapies,
Markers: Ventricular markers, sensed, paced and in relative
refractory periods,
EGM: onset and detection of the arrhythmia, on two therapies,
and the return to slow rhythm by recording electrogram.
Therapy history: For each arrhythmia detection, each therapy
delivered (either automatically or during an electrophysiological
study) and at the end of each arrhythmia, PARADYM RF VR 9250
records the type of majority rhythm, the number of ATP sequences
delivered, the energy and the number of shocks delivered..

US-ENGLISH – 49
17.3. ELECTIVE REPLACEMENT INDICATOR (ERI)
Elective Replacement Indicators (ERI)(1) are:
magnet rate equal to 80 ± 1 min-1 or
battery voltage equal to 2.66 V ± 0.01 V
CAUTION: The defibrillator should be replaced as soon as the
Elective Replacement Indicator (ERI) point is reached.
Between the ERI and the EOL (End of Life)(2), PARADYM RF VR 9250
can still function for:
9.7 months (100% pacing in VVI mode, 500 ohms, with as-
shipped settings), and deliver 7 shocks at 34 J or
6.4 months (0% pacing, sensor OFF, one 42 J shock every
2 weeks).
Once the Elective Replacement Indicator (ERI) point has been
reached, the device operates normally, except that the charge time
increases. Under normal conditions (and without programmer use)
the charge times are as follows:
Shock energy
Charge time (sec)
BOL
42 J
10 (± 2)
ERI
42 J
13 (± 3)
(1) Elective Replacement Indicators (ERI) corresponds to
Recommended Replacement Time (RRT) as referred in the EN45502-
2-2 standard.
(2) End of Life (EOL) corresponds to End of Service (EOS) as
referred in the EN45502-2-2 standard.
50 – US-ENGLISH
17.4. EXPLANTATION
The defibrillator should be explanted in the following cases:
The Elective Replacement Indicator (ERI) point is reached
Confirmed malfunction
Burial of the patient (for environmental reasons, the local regulation
may require the explantation of the devices containing a battery
supply)
Cremation of the patient (the defibrillator may explode if placed in
an incinerator)
The explanted defibrillator should not be reused in another patient.
All explanted defibrillators should be returned to Sorin CRM, carefully
cleaned of all traces of contamination. This may be done by
immersing them in an aqueous sodium hypochlorite containing at
least 1% chlorine, followed by rinsing copiously with water.
The defibrillator should be protected against mechanical impact and
the temperature variations that may occur during shipping.
Before explantation, it is advisable to:
print out all programmed parameters, statistics and Holter function
report,
save Patient data on floppy disk or hard disk,
disable shock therapies (VT and VF) to avoid any risk of untimely
shock.
US-ENGLISH – 51
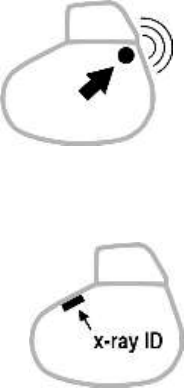
17.5. DEFIBRILLATOR IDENTIFICATION
The defibrillator can be interrogated and programmed via telemetry,
using the programming head interfaced with the Sorin CRM
dedicated programmer.
Position the programming head over the telemetry antenna located in
the upper part of the device, in order to communicate effectively via
telemetry (see diagram below).
The device can be non-invasively identified as follows:
1. Take an X-ray to identify the name of the manufacturer and
model, printed on the device (x-ray ID is SDB : S = SORIN; D =
Defibrillator; B = PARADYM RF VR 9250).
2. Interrogate the device using the Sorin CRM dedicated
programmer. The model and serial number of the device are
automatically displayed. The first figure in the serial number
corresponds to the last figure in the year of manufacture.

52 – US-ENGLISH
18. PHYSICAL CHARACTERISTICS
Dimensions
69.5 x 73.4 x 11 mm
Weight
95 g
Volume
38.6 cm3
Active surface area of casing
76 cm2
Connector
IS-1, DF-1
18.1. MATERIALS USED
Active surface area of casing
99% pure titanium
Connectors
Polyurethane* and silicone elastomer*
DF-1 insulating plug
silicone elastomer*
*Medical-grade materials that have undergone "in vitro" and "in vivo"
qualifications.
US-ENGLISH – 53
19. ELECTRICAL CHARACTERISTICS
Ventricular input impedance
80 kilohms ± 30 %
D.C. capacitance
148 µF ± 8 %
Capacitor formation
No formation required
Rate limit
192 min-1 ± 10 min-1
Pacing waveform
Defibrillation waveform
54 – US-ENGLISH
19.1. TABLE OF DELIVERED SHOCK ENERGY
AND VOLTAGE
The relationship between stored energies, maximum voltages and
delivered energies (at 37 °C, 50 ohm load) for the minimum, low,
mean and maximum programmed energy values is as follows:
Stored energy (J)
0.5
10
20
34
42
V1 (Volt)
75
341
483
631
702
V2 (Volt)
37
173
245
318
353
Delivered E: Phase 1 (J)
0.31
7.0
14.0
23.9
29.6
Delivered E: Phase 2 (J)
0.08
1.8
3.6
6.1
7.5
Delivered E: Total (J)
0.4
8.8
17.6
30.0
37.1
Tolerances are 12% for voltage (25% at 0.5 J) and 30% for energy.

US-ENGLISH – 55
19.2. BATTERY
Manufacturer
Greatbatch
Type
Quasar High Rate (QHR)
Model
GB 2593
Number of batteries
1
Total capacity
1964 mAh
Usable capacity
Between BOL and ERI: 1278 mAh.
Between BOL and EOL: 1675 mAh.
Voltage
BOL: 3.25 V. ERI: 2.66 V. EOL: 2.5 V.
19.3. LONGEVITY
The longevities mentioned below are calculated by taking into
account 6 months storage.
7.3 years
Pacing in VVI mode, 100%, 500 ohm, 3.5 V, 0.35 ms, 60 min-1,
one 42 J shock per quarter, sensor OFF
7.1 years
Pacing in VVI mode, 100%, 500 ohm, 3.5 V, 0.35 ms, 60 min-1,
one 42 J shock per quarter, sensor ON
9.1 years
Pacing in VVI mode, 1%, 500 ohm, 3.5 V, 0.35 ms, 60 min-1,
one 42 J shock per quarter, sensor OFF
8.5 years
Pacing in VVI mode, 15%, 500 ohm, 4.5 V, 0.50 ms, 60 min-1,
one 42 J shock per quarter, sensor OFF
9.3 years
0% pacing, one 42 J shock per quarter, sensor OFF
56 – US-ENGLISH
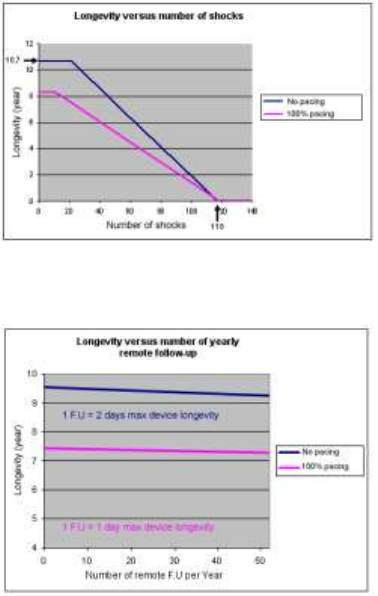
The mean longevity as a function of shocks delivered at maximum
energy, with and without pacing, is as follows:
The mean longevity as a function of yearly remote follow-ups(1), with
and without pacing, is as follows:
(1) An excessive number of remote follow-up can have a non-
negligible impact on device longevity.

US-ENGLISH – 57
20. PROGRAMMABLE PARAMETERS
measured at 37 °C under a 500 ohm load
Legend:
Value in bold: “as shipped” value
Underlined value: nominal value
20.1. ANTIBRADYCARDIA PACING
Basic parameters
Values
Mode
VVI-VVIR
Basic rate (min-1) (1)
From 30 to 90 by steps of 5;
60 (± 4 %)
Maximum rate (min-1)
From 100 to 145 by steps of 5;
120 (± 6 %)
Rate hysteresis (%)
0-5-10-20-35 (± 18 ms)
(1) The corresponding periods are (in ms): 2000-1714-1500-1333-1200-1091-
1000-923-857-800-750-706-667 ms.
58 – US-ENGLISH
Special features
Values
Smoothing
OFF-Very slow-Slow-Medium-Fast
Physical activity
Very low-Low-Medium-High-Very high
Pacing/Sensing
Values
Ventricular sensitivity (mV) (1)
From 0.4 to 4 by steps of 0.2;
0.4 (± 50 %)
Ventricular amplitude (V) (2)
1-1.5-2-2.5-3-3.5-4-4.5-5-6 (± 20 %)
Ventricular pulse width (ms)
0.12-0.25-0.35-0.5-0.6-0.75-0.85-1
(± 10 %)
(1) Values are measured using a positive and negative triangular signal of
2/13 ms.
(2) The correlation between the programmed amplitudes, the stored
amplitudes and the mid-pulse delivered amplitudes under a 500 ohm
load are given in the following table:
Programmed ampl. (V)
1
1.5
2
2.5
3
3.5
Mid-pulse delivered ampl. (V)
0.97
1.39
1.79
2.35
2.84
3.25
Stored amplitude (V)
1.14
1.63
2.1
2.76
3.33
3.82
Programmed ampl. (V)
4
4.5
5
6
Mid-pulse delivered ampl. (V)
3.58
4.23
4.47
5.37
Stored amplitude (V)
4.2
4.96
5.25
6.3
US-ENGLISH – 59
Post-shock mode
Values
Mode
OFF-VVI
Duration
10s-20s-30s-1min-2min-3min-4min-
5min
Basic rate (min-1)
From 50 to 90 by steps of 5;
60 (± 4 %)
V amplitude (V)
1-1.5-2-2.5-3-3.5-4-4.5-5-6 (± 20 %)
V pulse width (ms)
0.12-0.25-0.35-0.5-0.6-0.75-0.85-1
(± 10 %)
Sensitivity margins
Values
Ventricular post pacing margin (mV)
From 0 to 2 by steps of 0.2; 0.8
Response to noise
Values
Automatic sensitivity on noise
ON-OFF
V pacing on noise
ON-OFF

60 – US-ENGLISH
20.2. VENTRICULAR TACHYARRHYTHMIA
DETECTION
Therapy zones
Values
Slow VT detection zone (1)
Slow VT ON-Slow VT OFF
VT detection zone
VT ON-VT OFF
Fast VT / VF detection zone
Fast VT+VF ON-VF ON
Slow VT rate (lower limit) (min-1)
From 100 to 200 by steps of 5 ; 190
VT rate (lower limit) (min-1)
130-135-140-145-150-155-160-165-170-
175-180-185-190-195-200-210-220-230
VF rate (lower limit) (min-1)
150-155-160-165-170-175-180-185-190-
195-200-210-220-230-240
Fast VT rate (upper limit) (min-1)
155-160-165-170-175-180-185-190-195-
200-210-220-230-240-255
Slow VT persistence (cycles)
4-6-8-12-16-20-30-50-100-200
VT persistence (cycles)
4-6-8-12-16-20-30-50-100-200
VF persistence (cycles)
From 4 to 20 by steps of 1 ; 6
(1) The Slow VT zone should be programmed ON only if the VT zone is
programmed ON.

US-ENGLISH – 61
Detection criteria
Values
Slow VT and VT detection criteria
Rate Only-Stability-Stability+-
Stability/Acc-Stability+/Acc
Fast VT detection criteria
Rate+Stability-Rate Only
Majority: (X/Y), Y (cycles)
8-12-16
Majority: (X/Y), X (%)
65-70-75-80-90-95-100
Window of RR stability for Slow VT
and VT (ms)
30-45-65-80-95-110-125-125
Window of RR stability for fast VT (ms)
30-45-65
Prematurity acceleration (%)
6-13-19-25-31-38-44-50
Long cycle persistence extension
(cycles)
From 0 to 16 by steps of 1; 10
Long cycle gap (ms)
15-30-45-65-80-95-110-125-140-155-
170-190-205
20.3. VENTRICULAR TACHYARRHYTHMIA
THERAPIES
Common parameters
Values
Enable ATP therapy
Yes-No
Enable shock therapy
Yes-No
Polarity alternation (42J)
Yes-No
Atrial coil (SVC) present
Yes-No
Active case
Yes-No
Shock configuration (+ --> -)
Case to RV-SVC to RV-Case + SVC to RV-
RV to Case-RV to SVC-RV to Case + SVC
SVC exclusion (shock < 15J)
Yes-No

62 – US-ENGLISH
Therapy parameters in slow VT zone
ATP 1 program
Values
ATP program
OFF-Burst-Burst+Scan-Ramp-
Ramp+Scan
Number of sequences
1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Cycles in first sequence
1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Cycles added per sequence
0-1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Coupling interval (%)
50-55-60-65-70-75-80-85-90-95
Ramp decrement (per cycle) (ms)
0-4-8-12-16-20-30-40-50-60
Scan decrement (per sequence) (ms)
0-4-8-12-16-20-30-40-50-60
Time limit (min)
0.5-1-1.5-2-2.5-3-3.5-4
Minimum cycle length (ms)
95-110-125-140-155-170-190-205-220-
235-250-265-280-295-310

US-ENGLISH – 63
ATP 2 program
Values
ATP program
OFF-Burst-Burst+Scan-Ramp-
Ramp+Scan
Number of sequences
1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Cycles in first sequence
1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Cycles added per sequence
0-1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Coupling interval (%)
50-55-60-65-70-75-80-85-90-95
Ramp decrement (per cycle) (ms)
0-4-8-12-16-20-30-40-50-60
Scan decrement (per sequence) (ms)
0-4-8-12-16-20-30-40-50-60
Time limit (min)
0.5-1-1.5-2-2.5-3-3.5-4
Minimum cycle length (ms)
95-110-125-140-155-170-190-205-220-
235-250-265-280-295-310
Shock program
Values
Shock 1 (J)
OFF-0.5-0.8-1-1.3-1.5-2-2.5-3-3.5-4-5-
6-7-8-9- (± 30 %)
10-12-14-16-18-20-22-24-26-28-30-32-
34-42 (± 15 %)
Shock 2 (J)
OFF-0.5-0.8-1-1.3-1.5-2-2.5-3-3.5-4-5-
6-7-8-9- (± 30 %)
10-12-14-16-18-20-22-24-26-28-30-32-
34-42 (± 15 %)
Number of Max. Shock (42 J)
OFF-1-2-3-4

64 – US-ENGLISH
Therapy parameters in VT zone
ATP 1 program
Values
ATP program
OFF-Burst-Burst+Scan-Ramp-
Ramp+Scan
Number of sequences
1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Cycles in first sequence
1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Cycles added per sequence
0-1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Coupling interval (%)
50-55-60-65-70-75-80-85-90-95
Ramp decrement (per cycle) (ms)
0-4-8-12-16-20-30-40-50-60
Scan decrement (per sequence) (ms)
0-4-8-12-16-20-30-40-50-60
Time limit (min)
0.5-1-1.5-2-2.5-3-3.5-4
Minimum cycle length (ms)
95-110-125-140-155-170-190-205-220-
235-250-265-280-295-310

US-ENGLISH – 65
ATP 2 program
Values
ATP program
OFF-Burst-Burst+Scan-Ramp-
Ramp+Scan
Number of sequences
1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Cycles in first sequence
1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Cycles added per sequence
0-1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Coupling interval (%)
50-55-60-65-70-75-80-85-90-95
Ramp decrement (per cycle) (ms)
0-4-8-12-16-20-30-40-50-60
Scan decrement (per sequence) (ms)
0-4-8-12-16-20-30-40-50-60
Time limit (min)
0.5-1-1.5-2-2.5-3-3.5-4
Minimum cycle length (ms)
95-110-125-140-155-170-190-205-220-
235-250-265-280-295-310
Shock program
Values
Shock 1 (J)
OFF-0.5-0.8-1-1.3-1.5-2-2.5-3-3.5-4-5-
6-7-8-9- (± 30 %)
10-12-14-16-18-20-22-24-26-28-30-32-
34-42 (± 15 %)
Shock 2 (J)
OFF-0.5-0.8-1-1.3-1.5-2-2.5-3-3.5-4-5-
6-7-8-9- (± 30 %)
10-12-14-16-18-20-22-24-26-28-30-32-
34-42 (± 15 %)
Number of Max. Shock (42 J)
OFF-1-2-3-4

66 – US-ENGLISH
Therapy parameters in fast VT / VF zone
ATP 1 program
Values
ATP program
OFF-Burst-Burst+Scan-Ramp-
Ramp+Scan
Number of sequences
1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Cycles in first sequence
1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Cycles added per sequence
0-1-2-3-4-5-6-7-8-9-10-11-12-13-14-15
Coupling interval (%)
50-55-60-65-70-75-80-85-90-95
Ramp decrement (per cycle) (ms)
0-4-8-12-16-20-30-40-50-60
Scan decrement (per sequence) (ms)
0-4-8-12-16-20-30-40-50-60
Time limit
10s-20s-30s-1min-1.5min-2min
Minimum cycle length (ms)
95-110-125-140-155-170-190-205-220-
235-250-265-280-295-310
Shock program
Values
Shock 1 (J)
OFF-0.5-0.8-1-1.3-1.5-2-2.5-3-3.5-4-5-
6-7-8-9- (± 30 %)
10-12-14-16-18-20-22-24-26-28-30-32-
34-42 (± 15 %)
Shock 2 (J)
OFF-0.5-0.8-1-1.3-1.5-2-2.5-3-3.5-4-5-
6-7-8-9- (± 30 %)
10-12-14-16-18-20-22-24-26-28-30-32-
34-42 (± 15 %)
Number of Max. Shock (42 J)
1-2-3-4
US-ENGLISH – 67
20.4. REMOTE ALERTS AND WARNINGS
General parameters
Values
RF communication (1)
ON-OFF
Remote alerts (1)
ON-OFF
(1) RF and Remote alerts are turned on automatically if Shocks are
programmed ON.
System Alerts
Values
Battery depletion – ERI
ON-OFF
Device reset
ON-OFF
Excessive charge time (>25s)
ON-OFF
System integrity
ON-OFF
Lead Alerts
Values
Abnormal lead impedance
ON-OFF
Abnormal lead low limit (Ohm)
200-250-300-350-400-450-500
Abnormal lead high limit (Ohm)
1500-1750-2000-2500-3000
Abnormal RV coil impedance
ON-OFF
Abnormal SVC coil impedance
ON-OFF
Abnormal Shock impedance (1)
ON-OFF
(1) Normal impedance range [20 Ohm-200 Ohm]

68 – US-ENGLISH
Clinical status
Values
V oversensing
ON-OFF
Therapy information
Values
Shock disabled
ON-OFF
Shocks delivered
OFF-All shocks-Inefficient shock-
Inefficient max shock
21. NON PROGRAMMABLE PARAMETERS
Ventricular refractory periods
Values
Post ventricular sensing
95 ms (± 16 ms)
Post ventricular pacing
220 ms (± 4 ms)
Therapies
Values
Waveform
Constant tilt (50% - 50%)
Stored energy for the Max. shock
42 J (± 15 %)
Pacing amplitude during ATP therapies
7 V (Actual value at 300 ms: 5.3 V)

US-ENGLISH – 69
22. LIMITED WARRANTY
The PARADYM RF implantable cardioverter defibrillator is the result
of highly advanced research and all components have been selected
after exhaustive testing.
Sorin CRM S.r.l. (identified as “Sorin CRM” hereafter) guarantees the
product PARADYM RF against any damage caused by component
failure or production defects during a period of four years after the
implantation date, and Sorin CRM commits itself to replace all
PARADYM RF devices according to the terms described in article 1
and described in article 2 of this section.
Sorin CRM makes no claim that the human body will not react
unsuitably to the implantation of the PARADYM RF device, or that
failure will never occur.
Sorin CRM does not guarantee the suitability of PARADYM RF in
defined types of patients; selection of the device is a medical
decision.
Sorin CRM shall not be held liable for any damage indirectly
associated with the PARADYM RF, whether as part of normal or
abnormal operation, nor damage from its explantation or
replacement.
Sorin CRM does not authorise anyone to modify these limited
warranty conditions.
22.1. ARTICLE 1 : TERMS OF LIMITED WARRANTY
1. The PARADYM RF implantable cardioverter defibrillator is only
guaranteed for one implantation.
2. The EURID/IAPM implant form must be sent to Sorin CRM within
30 days after implantation.
3. The PARADYM RF cardioverter defibrillator must be implanted
prior to the use-before date indicated on the packaging.
70 – US-ENGLISH
4. The limited guarantee only applies to suspect devices returned
to the manufacturer, carefully packed and accompanied by an
explantation report duly completed by the hospital or the doctor
and considered defective after analysis by Sorin CRM.
The device must be returned within the 30 days following
explantation to Sorin CRM.
Any device returned and replaced under the terms of this limited
warranty will become the exclusive property of Sorin CRM.
Any rights under the terms of this limited warranty will be
forfeited if the PARADYM RF device has been opened by
anyone other than Sorin CRM.
These rights will also be forfeited if the device has been
damaged by carelessness or accident.
This is the case especially if the device has been exposed to
temperatures above 50°C, to electrical abuse or to mechanical
shock, particularly as a result of being dropped. Consequently,
any expert opinion offered by a third party after the device has
been removed also nullifies the guarantee.
5. The limited warranty will be forfeited if it is proven that the device
has been misused or inadequately implanted, against the
physicians’manual recommendations of PARADYM RF.
6. The limited warranty does not include leads and other
accessories used for the implantation.
7. The replacement terms or conditions described in article 2
include all devices that shall be replaced within the limited
warranty period because of battery depletion, without any link to
a component failure or a production hazard. The device battery
longevity varies with the type and number of delivered therapies.
8. Legal requirements of jurisdictions where the PARADYM RF
device is distributed will supersede any warranty conditions
indicated in this manual that conflict with such laws.
US-ENGLISH – 71
22.2. ARTICLE 2 : TERMS OF REPLACEMENT
1. In case of PARADYM RF failure because of a component failure,
a production defect, or a conception error, occurring within two-
year period starting from the implantation date, Sorin CRM is
committed to:
replacing free of charge the explanted device by a Sorin CRM
device with equivalent features,
or issuing a replacement credit equal to the purchase price for
the purchase of any other Sorin CRM replacement device.
2. After a two-year period and up to 4 years after the implantation,
Sorin CRM, because of limited warranty terms, will issue a
replacement credit to the buyer of an amount equivalent to half
of the initial purchase price minus prorata temporis during this
two-years period.
3. In any case the credit issued by the limited warranty terms
cannot exceed the purchase price of a Sorin CRM replacement
device.

72 – US-ENGLISH
23. PATENTS
The PARADYM RF model described in this manual is covered by the
following US patents:
5 167 224, 5 226 415, 5 271 394, 5 312 451, 5 325 856, 5 339 820,
5 350 406, 5 411 533, 5 462 060, 5 513 645, 5 545 181, 5 558 097,
5 564 430, 5 591 218, 5 626 619, 5 645 574, 5 674 265, 5 697 960,
5 702 424, 5 702 426, 5 713 928, 5 741 315, 5 776 164, 5 776 165,
5 818 703, 5 836 980, 5 868 793, 5 891 170, 5 891 184, 5 899 931,
5 931 856, 5 935 153, 5 954 660, 5 978 708, 6 181 968, 6 230 058,
6 236 111, 6 251 703, 6 256 206, 6 307 261, 6 337 996, 6 397 105,
6 408 209, 6 487 451, 6 487 452, 6 505 068, 6 532 238, 6 556 866,
6 604 002, 6 622 039, 6 625 491, 6 711 441, 6 738 665, 6 830 548,
6 889 080, 6 898 845, 6 912 421, 6 937 898, 6 975 905, 7 065 402,
7 072 716, 7 076 297, 7 113 826, 7 142 924, 7 164 946, 7 251 526,
7 366 566, 7 400 921, 7 400 922, 7 953 483.
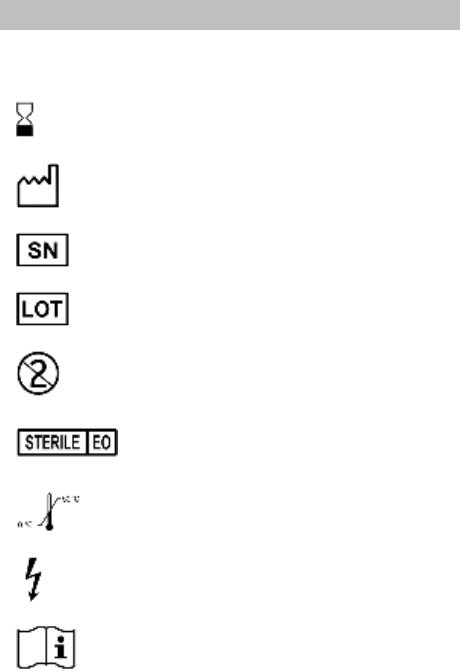
US-ENGLISH – 73
24. EXPLANATION OF SYMBOLS
The symbols on product labelling have the following meaning:
Use by
Date of manufacture
Serial number
Batch number
For single use only
Sterilised using ethylene oxide
Temperature limitation
High voltage
Consult instruction for use.
74 – US-ENGLISH
FCC ID YSGVR9250
IC : 10270A-VR9250
Last revision date of this implant manual: 2012-03