Differential Analyses With DSS Guide
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 22

Differential analyses with DSS
Hao Wu
[1em]Department of Biostatistics and Bioinformatics
Emory University
Atlanta, GA 303022
[1em] hao.wu@emory.edu
October 30, 2017
Contents
1 Introduction .............................. 2
2 Using DSS for differential expression analysis ........... 3
2.1 Input data preparation ...................... 3
2.2 Single factor experiment ..................... 4
2.3 Multifactor experiment ...................... 6
3 Using DSS for differential methylation analysis........... 7
3.1 Overview ............................. 7
3.2 Input data preparation ...................... 8
3.3 DML/DMR detection from two-group comparison ......... 9
3.4 DML/DMR detection from general experimental design ...... 14
3.4.1 Hypothesis testing in general experimental design ....... 14
3.4.2 Example analysis for data from general experimental design . . . 16
4 Session Info .............................. 21
Abstract
This vignette introduces the use of the Bioconductor package DSS (Dispersion Shrinkage
for Sequencing data), which is designed for differential analysis based on high-throughput
sequencing data. It performs differential expression analyses for RNA-seq, and differential

Differential analyses with DSS
methylation analyses for bisulfite sequencing (BS-seq) data. The core of DSS is a proce-
dure based on Bayesian hierarchical model to estimate and shrink gene- or CpG site-specific
dispersions, then conduct Wald tests for detecting differential expression/methylation.
1 Introduction
Recent advances in various high-throughput sequencing technologies have revolutionized ge-
nomics research. Among them, RNA-seq is designed to measure the the abundance of RNA
products, and Bisulfite sequencing (BS-seq) is for measuring DNA methylation. A fundamen-
tal question in functional genomics research is whether gene expression or DNA methylation
vary under different biological contexts. Thus, identifying differential expression genes (DEGs)
or differential methylation loci/regions (DML/DMRs) are key tasks in RNA-seq or BS-seq
data analyses.
The differential expression (DE) or differential methylation (DM) analyses are often based on
gene- or CpG-specific statistical test. A key limitation in RNA- or BS-seq experiments is that
the number of biological replicates is usually limited due to cost constraints. This can lead
to unstable estimation of within group variance, and subsequently undesirable results from
hypothesis testing. Variance shrinkage methods have been widely applied in DE analyses in
microarray data to improve the estimation of gene-specific within group variances. These
methods are typically based on a Bayesian hierarchical model, with a prior imposed on the
gene-specific variances to provide a basis for information sharing across all genes.
A distinct feature of RNA-seq or BS-seq data is that the measurements are in the form of
counts and have to be modeld by discrete distributions. Unlike continuous distributions (such
as Gaussian), the variances depend on means in these discrete distributions. This implies
that the sample variances do not account for biological variation, and shrinkage cannot be
applied on variances directly. In DSS, we assume that the count data are from the Gamma-
Poisson (for RNA-seq) or Beta-Binomial (for BS-seq) distribution. We then parameterize
the distributions by a mean and a dispersion parameters. The dispersion parameters, which
represent the biological variation for replicates within a treatment group, play a central role
in the differential analyses.
DSS implements a series of DE/DM detection algorithms based on the dispersion shrink-
age method followed by Wald statistical test to test each gene/CpG site for differential
expression/methylation. It provides functions for RNA-seq DE analysis for both two group
comparision and multi-factor design, BS-seq DM analysis for two group comparision, multi-
factor design, and data without biological replicate. Simulation and real data results show
that the methods provides excellent performance compared to existing methods, especially
when the overall dispersion level is high or the number of replicates is small.
2

Differential analyses with DSS
For more details of the data model, the shrinkage method, and test procedures, please read
[4] for differential expression from RNA-seq, [1] for differential methylation for two-group
comparison from BS-seq, [2] for differential methylation for data without biological replicate,
and [3] for differential methylation for general experimental design.
2 Using DSS for differential expression analysis
2.1 Input data preparation
DSS requires a count table (a matrix of integers) for gene expression values (rows are
for genes and columns are for samples). This is different from the isoform expression based
analysis such as in cufflink/cuffdiff, where the gene expressions are represented as non-integers
values. There are a number of ways to obtain the count table from raw sequencing data (fastq
file), here we provide some example codes using several Bioconductor packages (the codes
require installation of GenomicFeatures,Rsamtools, and GenomicRanges packages).
1. Sequence alignment. There are several RNA-seq aligner, for example, tophat or STAR.
Assume the alignment result is saved in a BAM file data.bam.
2. Choose a gene annotation. GenomicFeatures package provides a convenient way to
access different gene annotations. For example, if one wants to use RefSeq annotation
for human genome build hg19, one can use following codes:
> library(GenomicFeatures)
> txdb = makeTranscriptDbFromUCSC(genom="hg19",tablename="refGene")
> genes = genes(txdb)
3. Obtain count table based on the alignment results and gene annotation. This can be
done in several steps. First read in the BAM file using the Rsamtools package:
> bam=scanBam("data.bam")
Next, create GRanges object for the aligned sequence reads.
> IRange.reads=GRanges(seqnames=Rle(bam$rname), ranges=IRanges(bam$pos, width=bam$qwidth))
Finally, use the countOverlaps function in GenomicRanges function to obtain the read
counts overlap each gene.
> counts = countOverlaps(genes, IRange.reads)
3

Differential analyses with DSS
There are other ways to obtain the counts, for example, using QuasR or easyRNASeq Biocon-
ductor package. Please refer to the package vignettes for more details.
2.2 Single factor experiment
In single factor RNA-seq experiment, DSS also requires a vector representing experimental
designs. The length of the design vector must match the number of columns of the count
table. Optionally, normalization factors or additional annotation for genes can be supplied.
The basic data container in the package is SeqCountSet class, which is directly inherited
from ExpressionSet class defined in Biobase. An object of the class contains all necessary
information for a DE analysis: gene expression values, experimental designs, and additional
annotations.
A typical DE analysis contains the following simple steps.
1. Create a SeqCountSet object using newSeqCountSet.
2. Estimate normalization factor using estNormFactors.
3. Estimate and shrink gene-wise dispersion using estDispersion
4. Two-group comparison using waldTest.
The usage of DSS is demonstrated in the simple simulation below.
1. First load in the library, and make a SeqCountSet object from some counts for 2000
genes and 6 samples.
> library(DSS)
> counts1=matrix(rnbinom(300, mu=10, size=10), ncol=3)
> counts2=matrix(rnbinom(300, mu=50, size=10), ncol=3)
> X1=cbind(counts1, counts2) ## these are 100 DE genes
> X2=matrix(rnbinom(11400, mu=10, size=10), ncol=6)
> X=rbind(X1,X2)
> designs=c(0,0,0,1,1,1)
> seqData=newSeqCountSet(X, designs)
> seqData
SeqCountSet (storageMode: lockedEnvironment)
assayData: 2000 features, 6 samples
element names: exprs
protocolData: none
phenoData
sampleNames: 1 2 ... 6 (6 total)
4
Differential analyses with DSS
varLabels: designs
varMetadata: labelDescription
featureData: none
experimentData: use 'experimentData(object)'
Annotation:
2. Estimate normalization factor.
> seqData=estNormFactors(seqData)
3. Estimate and shrink gene-wise dispersions
> seqData=estDispersion(seqData)
4. With the normalization factors and dispersions ready, the two-group comparison can
be conducted via a Wald test:
> result=waldTest(seqData, 0, 1)
> head(result,5)
geneIndex muA muB lfc difExpr stats pval
35 35 6.000000 72.79487 -2.422688 -66.79487 -6.106274 1.019840e-09
60 60 4.333333 59.94872 -2.526259 -55.61538 -6.063770 1.329677e-09
12 12 7.666667 74.92308 -2.223052 -67.25641 -5.965320 2.441553e-09
99 99 7.666667 70.97436 -2.169278 -63.30769 -5.885008 3.980348e-09
52 52 7.333333 62.79487 -2.089416 -55.46154 -5.749004 8.977052e-09
local.fdr fdr
35 1.469796e-05 1.469796e-05
60 1.474650e-05 1.469796e-05
12 2.016697e-05 1.704275e-05
99 2.580658e-05 1.994590e-05
52 3.904835e-05 2.802468e-05
A higher level wrapper function DSS.DE is provided for simple RNA-seq DE analysis in a
two-group comparison. User only needs to provide a count matrix and a vector of 0’s and
1’s representing the design, and get DE test results in one line. A simple example is listed
below:
> counts = matrix(rpois(600, 10), ncol=6)
> designs = c(0,0,0,1,1,1)
> result = DSS.DE(counts, designs)
> head(result)
5
Differential analyses with DSS
geneIndex muA muB lfc difExpr stats pval
68 68 6.255892 11.963636 -0.6124003 -5.707744 -2.044332 0.04092073
19 19 5.973064 10.403030 -0.5213912 -4.429966 -1.703204 0.08852996
33 33 8.956229 5.072727 0.5287891 3.883502 1.651242 0.09868912
22 22 7.164983 11.860606 -0.4778522 -4.695623 -1.644890 0.09999251
55 55 7.481481 12.218182 -0.4659086 -4.736700 -1.622868 0.10461760
7 7 7.474747 11.796970 -0.4330729 -4.322222 -1.502718 0.13291190
local.fdr fdr
68 0.3178875 0.3178875
19 0.5192850 0.4198830
33 0.7003377 0.4727321
22 0.5619836 0.4443812
55 0.5788054 0.4572273
7 0.6804533 0.5543755
2.3 Multifactor experiment
DSS provides functionalities for dispersion shrinkage for multifactor experimental designs.
Downstream model fitting (through genearlized linear model) and hypothesis testing can be
performed using other packages such as edgeR, with the dispersions estimated from DSS.
Below is an example, based a simple simulation, to illustrate the DE analysis of a crossed
design.
1. First simulate data for a 2x2 crossed experiments. Note the counts are randomly
generated.
> library(DSS)
> library(edgeR)
> counts=matrix(rpois(800, 10), ncol=8)
> design=data.frame(gender=c(rep("M",4), rep("F",4)), strain=rep(c("WT", "Mutant"),4))
> X=model.matrix(~gender+strain, data=design)
2. make SeqCountSet, then estimate size factors and dispersion
> seqData=newSeqCountSet(counts, as.data.frame(X))
> seqData=estNormFactors(seqData)
> seqData=estDispersion(seqData)
3. Using edgeR’s function to do glm model fitting, but plugging in the estimated size
factors and dispersion from DSS.
6

Differential analyses with DSS
> fit.edgeR <- glmFit(counts, X, lib.size=normalizationFactor(seqData),
+ dispersion=dispersion(seqData))
4. Using edgeR’s function to do hypothesis testing on the second parameter of the model
(gender).
> lrt.edgeR <- glmLRT(glmfit=fit.edgeR, coef=2)
> head(lrt.edgeR$table)
logFC logCPM LR PValue
1 -0.2062103 21.32223 0.23420850 0.6284207
2 -0.4818837 21.22234 1.32917575 0.2489519
3 -0.2443803 21.17545 0.32698607 0.5674392
4 0.1101438 21.23479 0.06787492 0.7944564
5 -0.0988723 21.28219 0.04780051 0.8269357
6 -0.2587670 20.97309 0.31941263 0.5719608
3 Using DSS for differential methylation analysis
3.1 Overview
To detect differential methylation, statistical tests are conducted at each CpG site, and
then the differential methylation loci (DML) or differential methylation regions (DMR) are
called based on user specified threshold. A rigorous statistical tests should account for
biological variations among replicates and the sequencing depth. Most existing methods
for DM analysis are based on ad hoc methods. For example, using Fisher’s exact ignores
the biological variations, using t-test on estimated methylation levels ignores the sequencing
depth. Sometimes arbitrary filtering are implemented: loci with depth lower than an arbitrary
threshold are filtered out, which results in information loss
The DM detection procedure implemented in DSS is based on a rigorous Wald test for beta-
binomial distributions. The test statistics depend on the biological variations (characterized
by dispersion parameter) as well as the sequencing depth. An important part of the algorithm
is the estimation of dispersion parameter, which is achieved through a shrinkage estimator
based on a Bayesian hierarchical model [1]. An advantage of DSS is that the test can be
performed even when there is no biological replicates. That’s because by smoothing, the
neighboring CpG sites can be viewed as “pseudo-replicates", and the dispersion can still be
estimated with reasonable precision.
7
Differential analyses with DSS
DSS also works for general experimental design, based on a beta-binomial regression model
with “arcsine” link function. Model fitting is performed on transformed data with generalized
least square method, which achieves much improved computational performance compared
with methods based on generalized linear model.
DSS depends on bsseq Bioconductor package, which has neat definition of data structures
and many useful utility functions. In order to use the DM detection functionalities, bsseq
needs to be pre-installed.
3.2 Input data preparation
DSS requires data from each BS-seq experiment to be summarized into following information
for each CG position: chromosome number, genomic coordinate, total number of reads, and
number of reads showing methylation. For a sample, this information are saved in a simple
text file, with each row representing a CpG site. Below shows an example of a small part of
such a file:
chr pos N X
chr18 3014904 26 2
chr18 3031032 33 12
chr18 3031044 33 13
chr18 3031065 48 24
One can follow below steps to obtain such data from raw sequence file (fastq file), using
bismark (version 0.10.0, commands for newer versions could be different) for BS-seq align-
ment and count extraction. These steps require installation of bowtie or bowtie2,bismark,
and the fasta file for reference genome.
1. Prepare Bisulfite reference genome. This can be done using the bismark_genome_preparation
function (details in bismark manual). Example command is:
bismark_genome_preparation -path_to_bowtie /usr/local/bowtie/ -verbose /path/to/refgenomes/
2. BS-seq alignment. Example command is:
bismark -q -n 1 -l 50 -path_to_bowtie /path/bowtie/ BS-refGenome reads.fastq
This step will produce two text files reads.fastq_bismark.sam and reads.fastq_bismark_SE_report.txt.
3. Extract methylation counts using bismark_methylation_extractor function:
bismark_methylation_extractor -s -bedGraph reads.fastq_bismark.sam. This will
create multiple txt files to summarize methylation call and cytosine context, a bedGraph
file to display methylation percentage, and a coverage file containing counts infor-
mation. The count file contain following columns:chr, start, end, methylation%,
count methylated, count unmethylated. This file can be modified to make the input
file for DSS.
8

Differential analyses with DSS
A typical DML detection contains two simple steps. First one conduct DM test at each CpG
site, then DML/DMR are called based on the test result and user specified threshold.
3.3 DML/DMR detection from two-group comparison
Below are the steps to call DML or DMR for BS-seq data in two-group comparison setting.
1. Load in library. Read in text files and create an object of BSseq class, which is defined in
bsseq Bioconductor package. This step requires bsseq Bioconductor package. BSseq
class is defined in that package.
> library(DSS)
> require(bsseq)
> path <- file.path(system.file(package="DSS"), "extdata")
> dat1.1 <- read.table(file.path(path, "cond1_1.txt"), header=TRUE)
> dat1.2 <- read.table(file.path(path, "cond1_2.txt"), header=TRUE)
> dat2.1 <- read.table(file.path(path, "cond2_1.txt"), header=TRUE)
> dat2.2 <- read.table(file.path(path, "cond2_2.txt"), header=TRUE)
> BSobj <- makeBSseqData( list(dat1.1, dat1.2, dat2.1, dat2.2),
+ c("C1","C2", "N1", "N2") )[1:1000,]
> BSobj
An object of type 'BSseq' with
1000 methylation loci
4 samples
has not been smoothed
All assays are in-memory
2. Perform statistical test for DML by calling DMLtest function. This function basically
performs following steps: (1) estimate mean methylation levels for all CpG site; (2)
estimate dispersions at each CpG sites; (3) conduct Wald test. For the first step, there’s
an option for smoothing or not. Because the methylation levels show strong spatial
correlations, smoothing can help obtain better estimates of mean methylation when
the CpG sites are dense in the data (such as from the whole-genome BS-seq). However
for data with sparse CpG, such as from RRBS or hydroxyl-methylation, smoothing is
not recommended.
To perform DML test without smoothing, do:
> dmlTest <- DMLtest(BSobj, group1=c("C1", "C2"), group2=c("N1", "N2"))
Estimating dispersion for each CpG site, this will take a while ...
9

Differential analyses with DSS
> head(dmlTest)
chr pos mu1 mu2 diff diff.se stat
1 chr18 3014904 0.3817233 0.4624549 -0.08073162 0.24997034 -0.3229648
2 chr18 3031032 0.3380579 0.1417008 0.19635711 0.11086362 1.7711592
3 chr18 3031044 0.3432172 0.3298853 0.01333190 0.12203116 0.1092500
4 chr18 3031065 0.4369377 0.3649218 0.07201587 0.10099395 0.7130711
5 chr18 3031069 0.2933572 0.5387464 -0.24538920 0.13178800 -1.8619996
6 chr18 3031082 0.3526311 0.3905718 -0.03794068 0.07847999 -0.4834440
phi1 phi2 pval fdr
1 0.300542998 0.01706260 0.74672190 0.9985094
2 0.008911745 0.04783892 0.07653423 0.6792127
3 0.010409029 0.01994821 0.91300423 0.9985094
4 0.010320888 0.01603200 0.47580174 0.9985094
5 0.012537553 0.02320887 0.06260315 0.6158797
6 0.007665696 0.01145531 0.62878051 0.9985094
To perform statistical test for DML with smoothing, do:
> dmlTest.sm <- DMLtest(BSobj, group1=c("C1", "C2"), group2=c("N1", "N2"), smoothing=TRUE)
Smoothing ...
Estimating dispersion for each CpG site, this will take a while ...
User has the option to smooth the methylation levels or not. For WGBS data, smooth-
ing is recommended so that information from nearby CpG sites can be combined to
improve the estimation of methylation levels. A simple moving average algorithm is
implemented for smoothing. In RRBS since the CpG coverage is sparse, smoothing
might not alter the results much. If smoothing is requested, smoothing span is an
important parameter which has non-trivial impact on DMR calling. We use 500 bp as
default, and think that it performs well in real data tests.
3. With the test results, one can call DML by using callDML function. The results DMLs
are sorted by the significance.
> dmls <- callDML(dmlTest, p.threshold=0.001)
> head(dmls)
chr pos mu1 mu2 diff diff.se stat
450 chr18 3976129 0.01027497 0.9390339 -0.9287590 0.06544340 -14.19179
451 chr18 3976138 0.01027497 0.9390339 -0.9287590 0.06544340 -14.19179
638 chr18 4431501 0.01331553 0.9430566 -0.9297411 0.09273779 -10.02548
10

Differential analyses with DSS
639 chr18 4431511 0.01327049 0.9430566 -0.9297862 0.09270080 -10.02997
710 chr18 4564237 0.91454619 0.0119300 0.9026162 0.05260037 17.15988
782 chr18 4657576 0.98257334 0.0678355 0.9147378 0.06815000 13.42242
phi1 phi2 pval fdr postprob.overThreshold
450 0.052591567 0.02428826 0 0 1
451 0.052591567 0.02428826 0 0 1
638 0.053172411 0.07746835 0 0 1
639 0.053121697 0.07746835 0 0 1
710 0.009528898 0.04942849 0 0 1
782 0.010424723 0.06755651 0 0 1
By default, the test is based on the null hypothesis that the difference in methylation
levels is 0. Alternatively, users can specify a threshold for difference. For example, to
detect loci with difference greater than 0.1, do:
> dmls2 <- callDML(dmlTest, delta=0.1, p.threshold=0.001)
> head(dmls2)
chr pos mu1 mu2 diff diff.se stat
450 chr18 3976129 0.01027497 0.9390339 -0.9287590 0.06544340 -14.19179
451 chr18 3976138 0.01027497 0.9390339 -0.9287590 0.06544340 -14.19179
638 chr18 4431501 0.01331553 0.9430566 -0.9297411 0.09273779 -10.02548
639 chr18 4431511 0.01327049 0.9430566 -0.9297862 0.09270080 -10.02997
710 chr18 4564237 0.91454619 0.0119300 0.9026162 0.05260037 17.15988
782 chr18 4657576 0.98257334 0.0678355 0.9147378 0.06815000 13.42242
phi1 phi2 pval fdr postprob.overThreshold
450 0.052591567 0.02428826 0 0 1
451 0.052591567 0.02428826 0 0 1
638 0.053172411 0.07746835 0 0 1
639 0.053121697 0.07746835 0 0 1
710 0.009528898 0.04942849 0 0 1
782 0.010424723 0.06755651 0 0 1
When delta is specified, the function will compute the posterior probability that the
difference of the means is greater than delta. So technically speaking, the threshold
for p-value here actually refers to the threshold for 1-posterior probability, or the local
FDR. Here we use the same parameter name for the sake of the consistence of function
syntax.
11

Differential analyses with DSS
4. DMR detection is also Based on the DML test results, by calling callDMR function.
Regions with many statistically significant CpG sites are identified as DMRs. Some
restrictions are provided by users, including the minimum length, minimum number of
CpG sites, percentage of CpG site being significant in the region, etc. There are some
post hoc procedures to merge nearby DMRs into longer ones.
> dmrs <- callDMR(dmlTest, p.threshold=0.01)
> head(dmrs)
chr start end length nCG meanMethy1 meanMethy2 diff.Methy areaStat
27 chr18 4657576 4657639 64 4 0.506453 0.318348 0.188105 14.34236
Here the DMRs are sorted by “areaStat", which is defined in bsseq as the sum of the
test statistics of all CpG sites within the DMR.
Similarly, users can specify a threshold for difference. For example, to detect regions
with difference greater than 0.1, do:
> dmrs2 <- callDMR(dmlTest, delta=0.1, p.threshold=0.05)
> head(dmrs2)
chr start end length nCG meanMethy1 meanMethy2 diff.Methy areaStat
31 chr18 4657576 4657639 64 4 0.5064530 0.3183480 0.188105 14.34236
19 chr18 4222533 4222608 76 4 0.7880276 0.3614195 0.426608 12.91667
Note that the distribution of test statistics (and p-values) depends on the differences in
methylation levels and biological variations, as well as technical factors such as coverage
depth. It is very difficulty to select a natural and rigorous threshold for defining DMRs.
We recommend users try different thresholds in order to obtain satisfactory results.
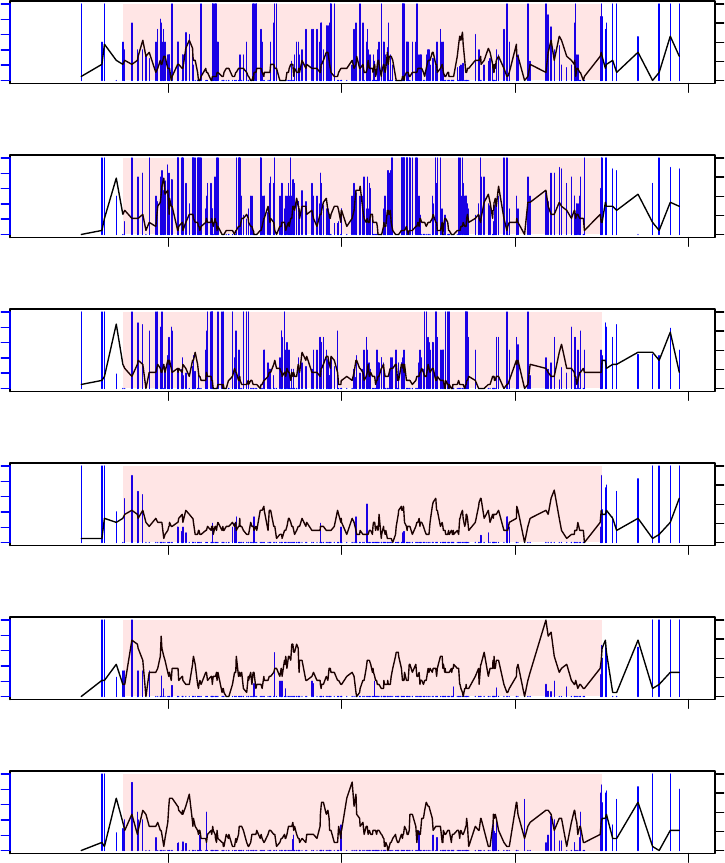
5. The DMRs can be visualized using showOneDMR function, This function provides more
information than the plotRegion function in bsseq. It plots the methylation percent-
ages as well as the coverage depths at each CpG sites, instead of just the smoothed
curve. So the coverage depth information will be available in the figure.
To use the function, do
> showOneDMR(dmrs[1,], BSobj)
The result figure looks like the following. Note that the figure below is not gener-
ated from the above example. The example data are from RRBS experiment
so the DMRs are much shorter.
12
Differential analyses with DSS
C1
32930000 32931000 32932000 32933000
0.0 0.6
chr21
methyl%
0 10 19
read depth
C2
32930000 32931000 32932000 32933000
0.0 0.6
chr21
methyl%
0 10 19
read depth
C3
32930000 32931000 32932000 32933000
0.0 0.6
chr21
methyl%
0 10 19
read depth
N1
32930000 32931000 32932000 32933000
0.0 0.6
chr21
methyl%
0 10 19
read depth
N2
32930000 32931000 32932000 32933000
0.0 0.6
chr21
methyl%
0 10 19
read depth
N3
32930000 32931000 32932000 32933000
0.0 0.6
chr21
methyl%
0 10 19
read depth
13

Differential analyses with DSS
3.4 DML/DMR detection from general experimental design
In DSS, BS-seq data from a general experimental design (such as crossed experiment, or
experiment with covariates) is modeled through a generalized linear model framework. We
use “arcsine” link function instead of the typical logit link for it better deals with data
at boundaries (methylation levels close to 0 or 1). Linear model fitting is done through
ordinary least square on transformed methylation levels. Variance/covariance matrices for
the estimates are derived with consideration of count data distribution and transformation.
3.4.1 Hypothesis testing in general experimental design
In a general design, the data are modeled through a multiple regression thus there are multiple
regression coefficients. In contrast, there is only one parameter in two-group comparison
which is the difference between two groups. Under this type of design, hypothesis testing
can be performed for one, multiple, or any linear combination of the parameters.
DSS provides flexible functionalities for hypothesis testing. User can test one parameter in
the model through a Wald test, or any linear combination of the parameters through an
F-test.
The DMLtest.multiFactor function provide interfaces for testing one parameter (through
coef parameter), one term in the model (through term parameter), or linear combinations of
the parameters (through Contrast parameter). We illustrate the usage of these parameters
through a simple example below. Assume we have an experiment from three strains (A, B,
C) and two sexes (M and F), each has 2 biological replicates (so there are 12 datasets in
total).
> Strain = rep(c("A", "B", "C"), 4)
> Sex = rep(c("M", "F"), each=6)
> design = data.frame(Strain,Sex)
> design
Strain Sex
1 A M
2 B M
3 C M
4 A M
5 B M
6 C M
7 A F
8 B F
14
Differential analyses with DSS
9 C F
10 A F
11 B F
12 C F
To test the additive effect of Strain and Sex, a design formula is Strain+Sex, and the
corresponding design matrix for the linear model is:
> X = model.matrix(~Strain+ Sex, design)
> X
(Intercept) StrainB StrainC SexM
1 1 0 0 1
2 1 1 0 1
3 1 0 1 1
4 1 0 0 1
5 1 1 0 1
6 1 0 1 1
7 1 0 0 0
8 1 1 0 0
9 1 0 1 0
10 1 0 0 0
11 1 1 0 0
12 1 0 1 0
attr(,"assign")
[1]0112
attr(,"contrasts")
attr(,"contrasts")$Strain
[1] "contr.treatment"
attr(,"contrasts")$Sex
[1] "contr.treatment"
Under this design, we can do different tests using the DMLtest.multiFactor function:
•If we want to test the sex effect, we can either specify coef=4,coef="SexM", or
term="Sex". Notice that when using character for coef, the character must match
the column name of the design matrix, cannot do coef="Sex". It is also important to
note that using term="Sex" only tests a single paramter in the model because sex only
has two levels.
15

Differential analyses with DSS
•If we want to test the effect of Strain B versus Strain A (this is also testing a single
parameter), we do coef=2 or coef="StrainB".
•If we want to test the whole Strain effect, it becomes a compound test because Strain
has three levels. We do term="Strain", which tests StrainB and StrainC simulta-
neously. We can also make a Contrast matrix L as following. It’s clear that testing
LTβ= 0 is equivalent to testing StrainB=0 and StrainC=0.
> L = cbind(c(0,1,0,0),c(0,0,1,0))
> L
[,1] [,2]
[1,] 0 0
[2,] 1 0
[3,] 0 1
[4,] 0 0
•One can perform more general test, for example, to test StrainB=StrainC, or that
strains B and C has no difference (but they could be different from Strain A). In this
case, we need to make following contrast matrix:
> matrix(c(0,1,-1,0), ncol=1)
[,1]
[1,] 0
[2,] 1
[3,] -1
[4,] 0
3.4.2 Example analysis for data from general experimental design
1. Load in data distributed with DSS. This is a small portion of a set of RRBS experiments.
There are 5000 CpG sites and 16 samples. The experiment is a 2design (2 cases and
2 cell types). There are 4 replicates in each case:cell combination.
> data(RRBS)
> RRBS
An object of type 'BSseq' with
5000 methylation loci
16 samples
has not been smoothed
All assays are in-memory
16
Differential analyses with DSS
> design
case cell
1 HC rN
2 HC rN
3 HC rN
4 SLE aN
5 SLE rN
6 SLE aN
7 SLE rN
8 SLE aN
9 SLE rN
10 SLE aN
11 SLE rN
12 HC aN
13 HC aN
14 HC aN
15 HC aN
16 HC rN
2. Fit a linear model using DMLfit.multiFactor function, include case, cell, and case:cell
interaction.
> DMLfit = DMLfit.multiFactor(RRBS, design=design, formula=~case+cell+case:cell)
Fitting DML model for CpG site:
3. Use DMLtest.multiFactor function to test the cell effect. It is important to note that
the coef parameter is the index of the coefficient to be tested for being 0. Because
the model (as specified by formula in DMLfit.multiFactor) include intercept, the cell
effect is the 3rd column in the design matrix, so we use coef=3 here.
> DMLtest.cell = DMLtest.multiFactor(DMLfit, coef=3)
Alternatively, one can specify the name of the parameter to be tested. In this case, the
input coef is a character, and it must match one of the column names in the design
matrix. The column names of the design matrix can be viewed by
> colnames(DMLfit$X)
[1] "(Intercept)" "caseSLE" "cellrN" "caseSLE:cellrN"
17

Differential analyses with DSS
The following line also works. Specifying coef="cellrN" is the same as specifying
coef=3.
> DMLtest.cell = DMLtest.multiFactor(DMLfit, coef="cellrN")
Result from this step is a data frame with chromosome number, CpG site position, test
statistics, p-values (from normal distribution), and FDR. Rows are sorted by chromo-
some/position of the CpG sites. To obtain top ranked CpG sites, one can sort the data
frame using following codes:
> ix=sort(DMLtest.cell[,"pvals"], index.return=TRUE)$ix
> head(DMLtest.cell[ix,])
chr pos stat pvals fdrs
1273 chr1 2930315 5.280301 1.289720e-07 0.0006448599
4706 chr1 3321251 5.037839 4.708164e-07 0.0011770409
3276 chr1 3143987 4.910412 9.088510e-07 0.0015147517
2547 chr1 3069876 4.754812 1.986316e-06 0.0024828953
3061 chr1 3121473 4.675736 2.929010e-06 0.0029290097
527 chr1 2817715 4.441198 8.945925e-06 0.0065858325
Below is a figure showing the distributions of test statistics and p-values from this
example dataset
> par(mfrow=c(1,2))
> hist(DMLtest.cell$stat, 50, main="test statistics", xlab="")
> hist(DMLtest.cell$pvals, 50, main="P values", xlab="")
test statistics
Frequency
−4 −2 0 2 4
0 100 200 300
P values
Frequency
0.0 0.2 0.4 0.6 0.8 1.0
0 50 150 250 350
4. DMRs for multifactor design can be called using callDMR function:
18

Differential analyses with DSS
> callDMR(DMLtest.cell, p.threshold=0.05)
chr start end length nCG areaStat
33 chr1 2793724 2793907 184 5 12.619968
413 chr1 3309867 3310133 267 7 -12.093850
250 chr1 3094266 3094486 221 4 11.691413
262 chr1 3110129 3110394 266 5 11.682579
180 chr1 2999977 3000206 230 4 11.508302
121 chr1 2919111 2919273 163 4 9.421873
298 chr1 3146978 3147236 259 5 8.003469
248 chr1 3090627 3091585 959 5 -7.963547
346 chr1 3200758 3201006 249 4 -4.451691
213 chr1 3042371 3042459 89 5 4.115296
169 chr1 2995532 2996558 1027 4 -2.988665
Note that for results from for multifactor design, delta is NOT supported. This is
because in multifactor design, the estimated coefficients in the regression are based on
a GLM framework (loosely speaking), thus they don’t have clear meaning of methylation
level differences. So when the input DMLresult is from DMLtest.multiFactor,delta
cannot be specified.
5. More flexible way to specify a hypothesis test. Following 4 tests should produce the
same results, since ’case’ only has two levels. However the p-values from F-tests (using
term or Contrast) are slightly different, due to normal approximation in Wald test.
> ## fit a model with additive effect only
> DMLfit = DMLfit.multiFactor(RRBS, design, ~case+cell)
Fitting DML model for CpG site:
> ## test case effect
> test1 = DMLtest.multiFactor(DMLfit, coef=2)
> test2 = DMLtest.multiFactor(DMLfit, coef="caseSLE")
> test3 = DMLtest.multiFactor(DMLfit, term="case")
> Contrast = matrix(c(0,1,0), ncol=1)
> test4 = DMLtest.multiFactor(DMLfit, Contrast=Contrast)
> cor(cbind(test1$pval, test2$pval, test3$pval, test4$pval))
[,1] [,2] [,3] [,4]
[1,] 1 1 1 1
[2,] 1 1 1 1
[3,] 1 1 1 1
[4,] 1 1 1 1
19

Differential analyses with DSS
The model fitting and hypothesis test procedures are computationally very efficient. For
a typical RRBS dataset with 4 million CpG sites, it usually takes less than half hour. In
comparison, other similar software such as RADMeth or BiSeq takes at least 10 times longer.
20

Differential analyses with DSS
4 Session Info
> sessionInfo()
R version 3.4.2 (2017-09-28)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 16.04.3 LTS
Matrix products: default
BLAS: /home/biocbuild/bbs-3.6-bioc/R/lib/libRblas.so
LAPACK: /home/biocbuild/bbs-3.6-bioc/R/lib/libRlapack.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=C
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=en_US.UTF-8
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] splines stats4 parallel stats graphics grDevices utils
[8] datasets methods base
other attached packages:
[1] edgeR_3.20.0 limma_3.34.0
[3] DSS_2.26.0 bsseq_1.14.0
[5] SummarizedExperiment_1.8.0 DelayedArray_0.4.0
[7] matrixStats_0.52.2 GenomicRanges_1.30.0
[9] GenomeInfoDb_1.14.0 IRanges_2.12.0
[11] S4Vectors_0.16.0 Biobase_2.38.0
[13] BiocGenerics_0.24.0
loaded via a namespace (and not attached):
[1] Rcpp_0.12.13 compiler_3.4.2 plyr_1.8.4
[4] XVector_0.18.0 R.methodsS3_1.7.1 bitops_1.0-6
[7] R.utils_2.5.0 tools_3.4.2 zlibbioc_1.24.0
[10] digest_0.6.12 evaluate_0.10.1 lattice_0.20-35
[13] Matrix_1.2-11 yaml_2.1.14 GenomeInfoDbData_0.99.1
21

Differential analyses with DSS
[16] stringr_1.2.0 knitr_1.17 gtools_3.5.0
[19] locfit_1.5-9.1 rprojroot_1.2 grid_3.4.2
[22] data.table_1.10.4-3 rmarkdown_1.6 magrittr_1.5
[25] backports_1.1.1 scales_0.5.0 htmltools_0.3.6
[28] permute_0.9-4 BiocStyle_2.6.0 colorspace_1.3-2
[31] stringi_1.1.5 RCurl_1.95-4.8 munsell_0.4.3
[34] R.oo_1.21.0
References
[1] Hao Feng, Karen Conneely and Hao Wu. (2014). A bayesian hierarchical
model to detect differentially methylated loci from single nucleotide resolution
sequencing data. Nucleic Acids Research. 42(8), e69–e69.
[2] Hao Wu, Tianlei Xu, Hao Feng, Li Chen, Ben Li, Bing Yao, Zhaohui
Qin, Peng Jin and Karen N. Conneely. (2015). Detection of differentially
methylated regions from whole-genome bisulfite sequencing data without replicates.
Nucleic Acids Research. doi: 10.1093/nar/gkv715.
[3] Yongseok Park, Hao Wu. (2016). Differential methylation analysis for BS-seq
data under general experimental design.
Bioinformatics. doi:10.1093/bioinformatics/btw026.
[4] Hao Wu, Chi Wang and Zhijing Wu. (2013). A new shrinkage estimator for
dispersion improves differential expression detection in RNA-seq data.
Biostatistics. 14(2), 232–243.
22
