1 LAMMPS Short Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 66
- LAMMPS SHORT MANUAL
- Slide Number 2
- Slide Number 3
- Slide Number 4
- Slide Number 5
- Slide Number 6
- Slide Number 7
- Slide Number 8
- Slide Number 9
- Slide Number 10
- Slide Number 11
- Slide Number 12
- Slide Number 13
- Slide Number 14
- Slide Number 15
- Slide Number 16
- Slide Number 17
- Slide Number 18
- Slide Number 19
- Slide Number 20
- Slide Number 21
- Slide Number 22
- Slide Number 23
- Slide Number 24
- Slide Number 25
- Slide Number 26
- Slide Number 27
- Slide Number 28
- Slide Number 29
- Slide Number 30
- Slide Number 31
- Slide Number 32
- Slide Number 33
- MSLAB for LAMMPS on MIGALE
- Custom LAMMPS installation
- CUSTOM LAMMPS JOB
- Slide Number 37
- Slide Number 38
- MSLAB custom installation
- custom PIZZA.py on MIGALE
- PIZZA.py custom installation
- CUSTOM PIZZA TEST
- Slide Number 43
- PIZZA with GUI
- LAMMPS EXAMPLES
- BEAD-SPRING POLYMER MELT FENE: Finite Extendible Nonlinear Elastic Model (here: 2880 beads, 2715 bonds)
- BEAD-SPRING POLYMER MELT FENE: Finite Extendible Nonlinear Elastic Model (here: 2880 beads, 2715 bonds)
- BEAD-SPRING POLYMER MELT
- Slide Number 49
- ALL ATOM SIMULATION EXAMPLE 1: CVFF (no warnings), shrink boundary conditions
- ALL ATOM SIMULATION EXAMPLE 2: CVFF (warnings), shrink boundary conditions
- ALL ATOM SIMULATION EXAMPLE 3: CFF (warnings), periodic boundary conditions, NPT
- MISCELLANEOUS
- LEARNING PYTHON
- LEARNING PYTHON
- LEARNING PYTHON
- LEARNING MATLAB
- Slide Number 58
- Slide Number 59
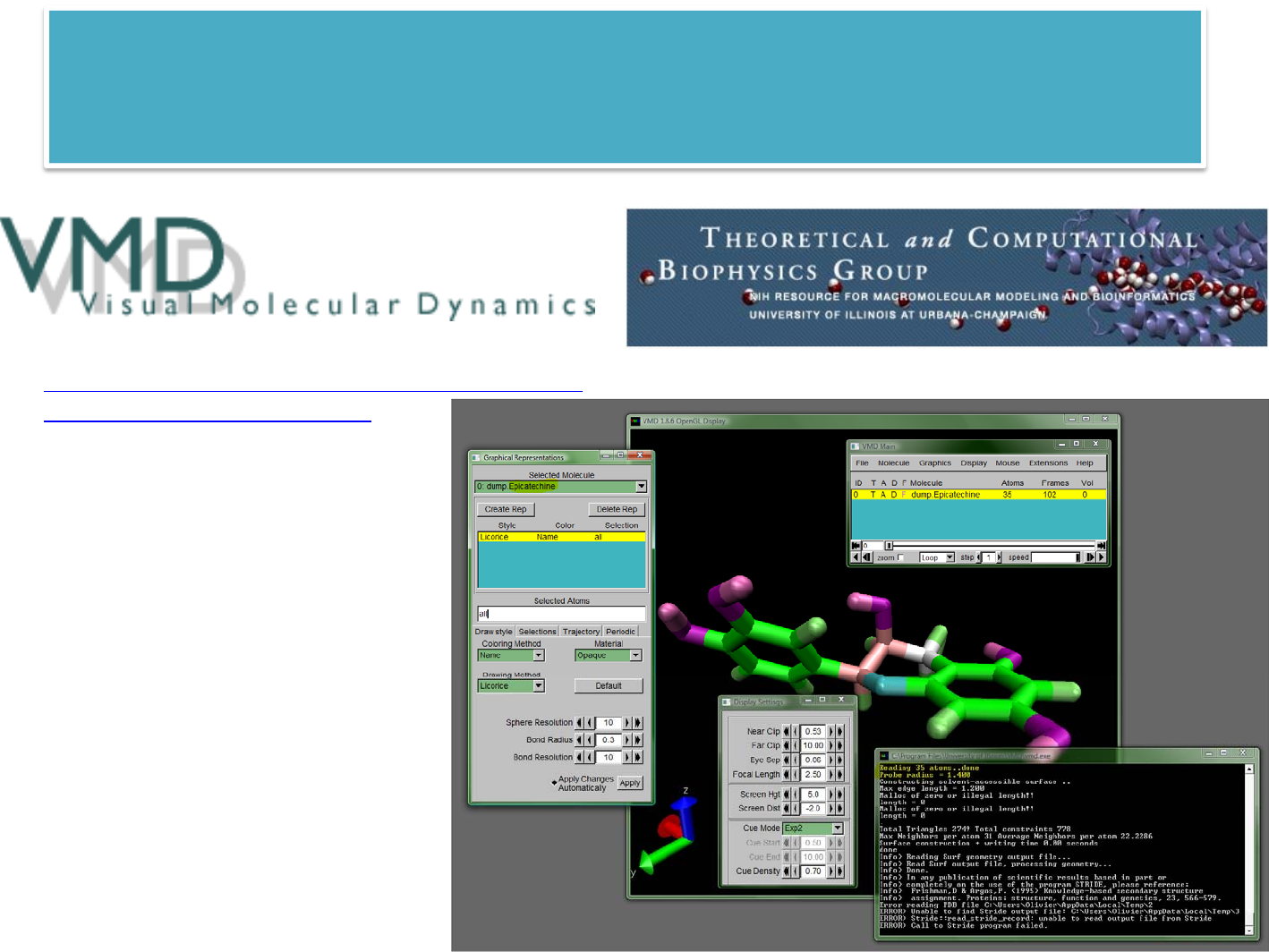
- VISUALIZATION OF DUMP FILES
- WIN2LINUX Solutions
- WIN2LINUX Solutions
- WIN2LINUX Solutions
- MAKING LAMMPS
- $PROJECT\make\lammps-31Jan08\src\MAKE\Makefile.g++_lam_all_100208
- Slide Number 66


LAMMPS
LARGE
SCALE
ATOMIC
MOLECULAR
MASSIVELY
PARALLEL
SIMULATOR
INIT
ATOMDEFINITION
FORCEFIELDS
SETTINGS
FIX
COMPUTE
ACTIONS
OUTPUTS
LAMMPS is a molecular dynamics program from Sandia National
Laboratories. LAMMPS makes use of MPI for parallel communication and is a
free open‐source code, distributed under the terms of the GNU General
Public License.
LAMMPS was originally developed under a Cooperative Research and Development Agreement
(CRADA) between two laboratories from United States Department of Energy and three other
laboratories from private sector firms. It is currently maintained and distributed by researchers at the
Sandia National Laboratories.
For computational efficiency LAMMPS uses neighbor lists to keep track of nearby particles. The lists
are optimized for systems with particles that are repulsive at short distances, so that the local
density of particles never becomes too large.
On parallel computers, LAMMPS uses spatial‐decomposition techniques to partition the simulation
domain into small 3d sub‐domains, one of which is assigned to each processor. Processors
communicate and store "ghost" atom information for atoms that border their sub‐domain.
LAMMPS is most efficient (in a parallel computing sense) for systems whose particles fill a 3D
rectangular box with approximately uniform density.
Features
http://lammps.sandia.gov/
http://lammps.sandia.gov/doc/Manual.html 2
PRINCIPLES
1)Initialization
2)Atom definition
3)Settings
4)Run
units,dimension,boundary,atom_style,atom_modify.
read_data,read_restart,lattice,region,create_box,create_atoms
pair_coeff,bond_coeff,angle_coeff,dihedral_coeff,improper_coeff,
kspace_style,dielectric,special_bonds
neighbor,neigh_modify,group,timestep,reset_timestep,run_style,
min_style,min_modify.
compute,compute_modify,variable
run,minimize
3

SCRIPT
1)Initialization
2)Atom definition
3)Settings
4)Run
#3dLennard‐Jonesmelt
units lj
atom_style atomic
lattice fcc 0.8442
region boxblock020020020
create_box 1box
create_atoms 1
mass 11.0
velocity allcreate3.087287
pair_style lj/cut 2.5
pair_coeff 111.01.02.5
neighbor 0.3bin
neigh_modify every20delay0checkno
fix 1allnve
dump idallatom 10dump.melt
thermo 50
run 250 4
INIT
atom_modify
ATOM_STYLE
boundary
dimension
newton
processors
units
atom_style styleargs
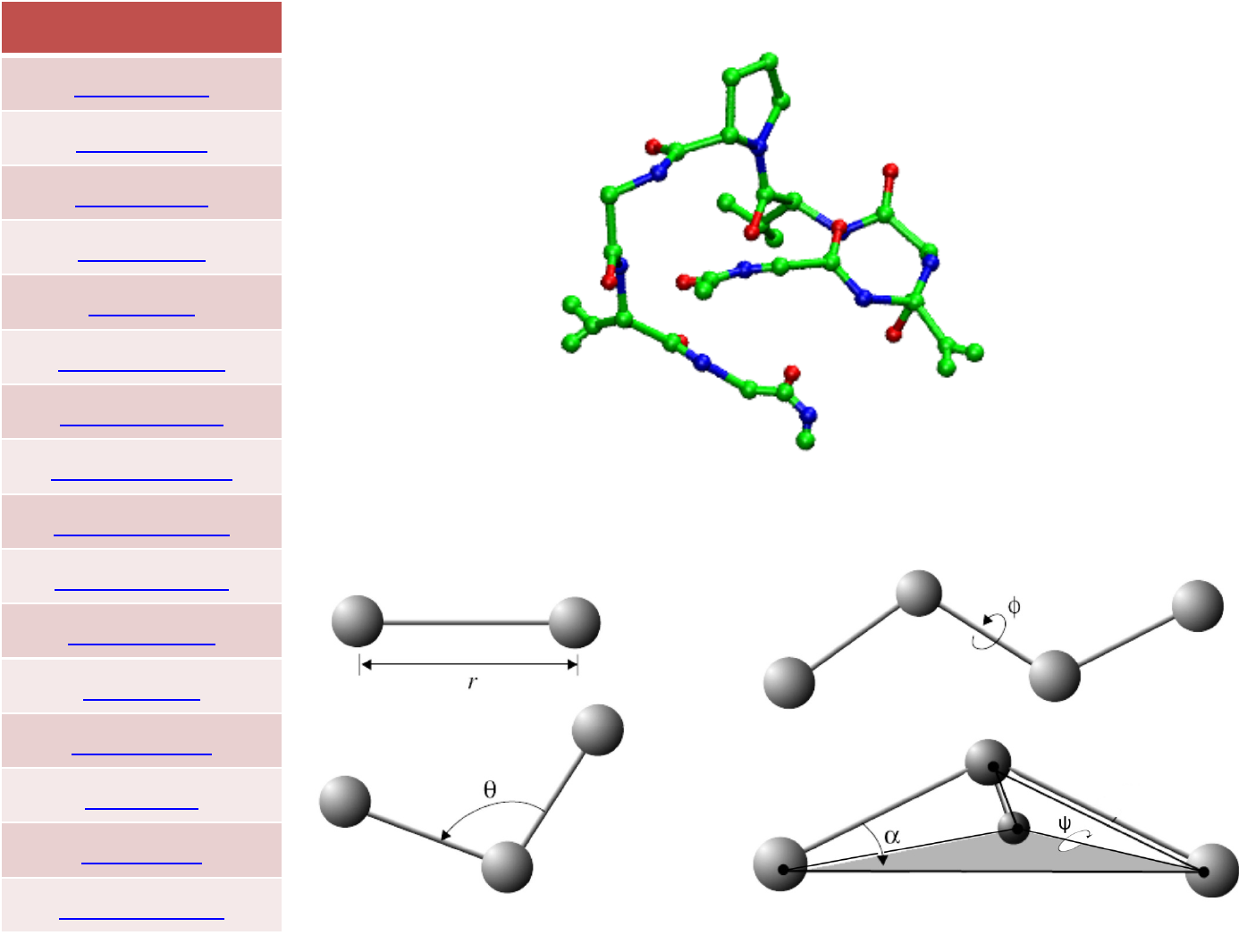
angle =bondsandangles‐ e.g.bead‐spring polymers with stiffness
atomic =only thedefaultvalues
bond =bonds‐ e.g.bead‐spring polymers
charge =charge
dipole =chargeanddipole moment
dpd =defaultvalues,also communicates velocities
ellipsoid =quaternionforparticle orientation,angular velocity/momentum
full =molecular +charge‐ e.g.biomolecules,charged polymers
granular =granular atoms with rotational properties
molecular =bonds,angles,dihedrals,impropers ‐e.g.all‐atom polymers
5

INIT
atom_modify
atom_style
boundary
dimension
newton
processors
UNITS
units lj stylereal
distance=sigma
time=tau
mass=one
energy =epsilon
velocity =sigma/tau
force=epsilon/sigma
temperature =reduced LJ
temperature
pressure=reduced LJpressure
charge=reduced LJcharge
dipole =reduced LJdipole
moment
electric field =force/charge
distance=Angstroms
time=femtoseconds
mass=grams/mole
energy =Kcal/mole
velocity =Angstroms/femtosecond
force=Kcal/mole‐Angstrom
temperature =degrees K
pressure=atmospheres
charge=multipleofelectron
charge(+1.0is aproton)
dipole =charge*Angstroms
electric field =volts/Angstrom
7

ATOMDEFINITION
create_atoms
create_box
lattice
READ_DATA
read_restart
region
replicate
read_data file
atoms =#ofatoms insystem
bonds =#ofbondsinsystem
angles =#ofanglesinsystem
dihedrals =#ofdihedrals insystem
impropers =#ofimpropers insystem
atom types =#ofatom typesinsystem
bondtypes =#ofbondtypesinsystem
angletypes =#ofangletypesinsystem
dihedral types =#ofdihedral typesinsystem
improper types =#ofimproper typesinsystem
xlo xhi =simulationboxboundaries inxdimension
ylo yhi =simulationboxboundaries inydimension
zlo zhi =simulationboxboundaries inzdimension
xy xz yz =simulationboxtiltfactors fortriclinic domain 8

ATOMDEFINITION
create_atoms
create_box
lattice
READ_DATA
read_restart
region
replicate
LAMMPS Description (1st line of file)
100 atoms (this must be the 3rd line, 1st 2 lines are ignored)
95 bonds (# of bonds to be simulated)
50 angles (include these lines even if number = 0)
30 dihedrals
20 impropers
5 atom types (# of nonbond atom types)
10 bond types (# of bond types = sets of bond coefficients)
18 angle types
20 dihedral types (do not include a bond, angle,dihedral,improper type
2 improper types line if number of bonds,angles,etc is 0)
-0.5 0.5 xlo xhi (for periodic systems this is box size,
-0.5 0.5 ylo yhi for non-periodic it is min/max extent of atoms)
-0.5 0.5 zlo zhi (do not include this line for 2-d simulations)
Masses
1 mass
...
N mass (N = # of atom types)
Pair Coeffs Nonbond Coeffs (in old versions)
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of atom types)
Bond Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of bond types)
Angle Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of angle types) 9

ATOMDEFINITION
create_atoms
create_box
lattice
READ_DATA
read_restart
region
replicate
Dihedral Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of dihedral types)
Improper Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of improper types)
BondBond Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of angle types)
BondAngle Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of angle types)
MiddleBondTorsion Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of dihedral types)
EndBondTorsion Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of dihedral types)
AngleTorsion Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of dihedral types) 10

ATOMDEFINITION
create_atoms
create_box
lattice
READ_DATA
read_restart
region
replicate
AngleAngleTorsion Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of dihedral types)
BondBond13 Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of dihedral types)
AngleAngle Coeffs
1 coeff1 coeff2 ...
...
N coeff1 coeff2 ... (N = # of improper types)
Atoms
1 molecule-tag atom-type q x y z nx ny nz (nx,ny,nz are optional -
... see "true flag" input command)
...
N molecule-tag atom-type q x y z nx ny nz (N = # of atoms)
Velocities
1 vx vy vz
...
N vx vy vz (N = # of atoms)
Bonds
1 bond-type atom-1 atom-2
...
N bond-type atom-1 atom-2 (N = # of bonds)
Angles
1 angle-type atom-1 atom-2 atom-3 (atom-2 is the center atom in angle)
...
N angle-type atom-1 atom-2 atom-3 (N = # of angles) 11

ATOMDEFINITION
create_atoms
create_box
lattice
READ_DATA
read_restart
region
replicate
Dihedrals
1 dihedral-type atom-1 atom-2 atom-3 atom-4 (atoms 2-3 form central bond)
...
N dihedral-type atom-1 atom-2 atom-3 atom-4 (N = # of dihedrals)
Impropers
1 improper-type atom-1 atom-2 atom-3 atom-4 (atom-2 is central atom)
...
N improper-type atom-1 atom-2 atom-3 atom-4 (N = # of impropers)
comments
blank lines are ignored
lines starting with a # are echoed into the log file
for commands, everything on a line after the last
parameter is ignored
12
FORCEFIELD
angle_coeff
angle_style
bond_coeff
bond_style
dielectric
dihedral_coeff
dihedral_style
improper_coeff
improper_style
kspace_modify
kspace_style
pair_coeff
pair_modify
pair_style
pair_write
special_bonds
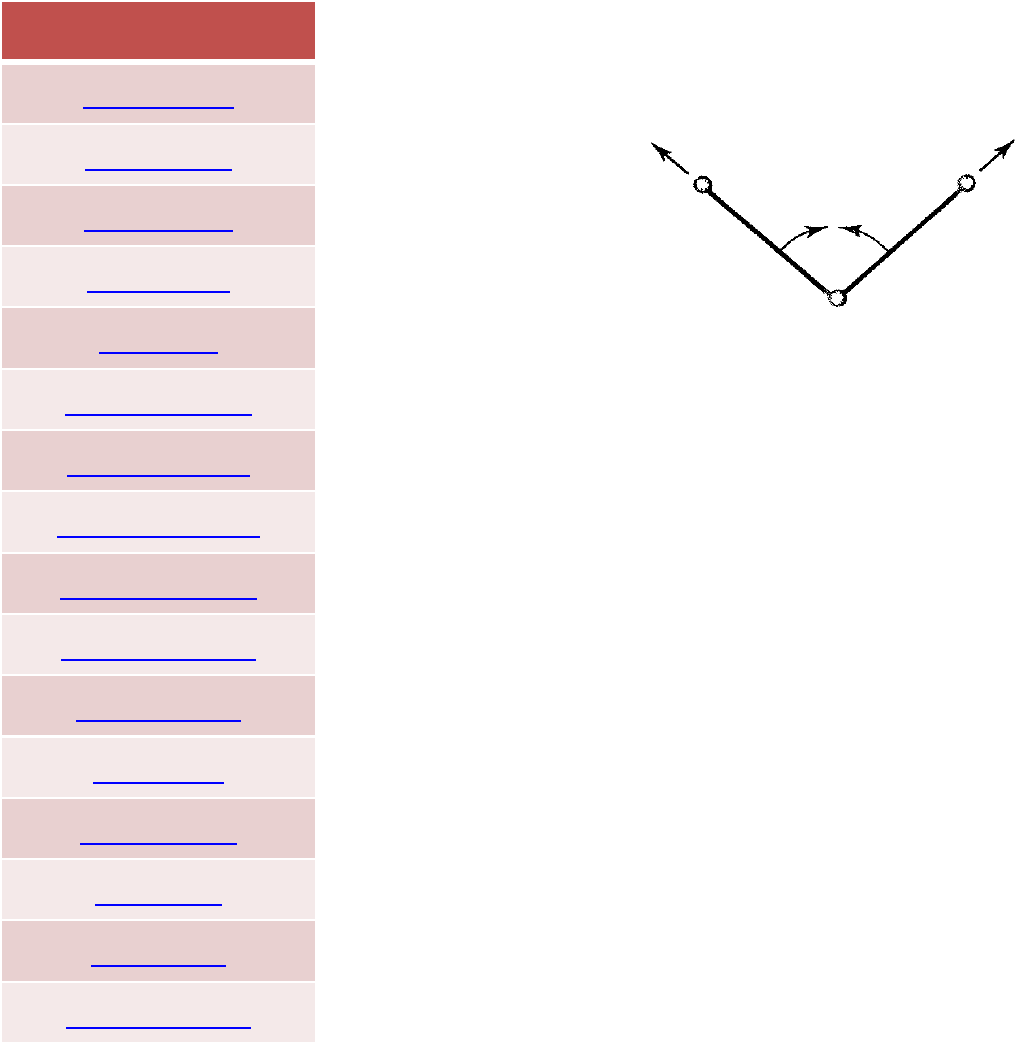
The presence of cross terms in a force field reflects coupling between
the internal coordinates. For example, as a bond angle is decreased it
is found that the adjacent bonds stretch to reduce the interaction
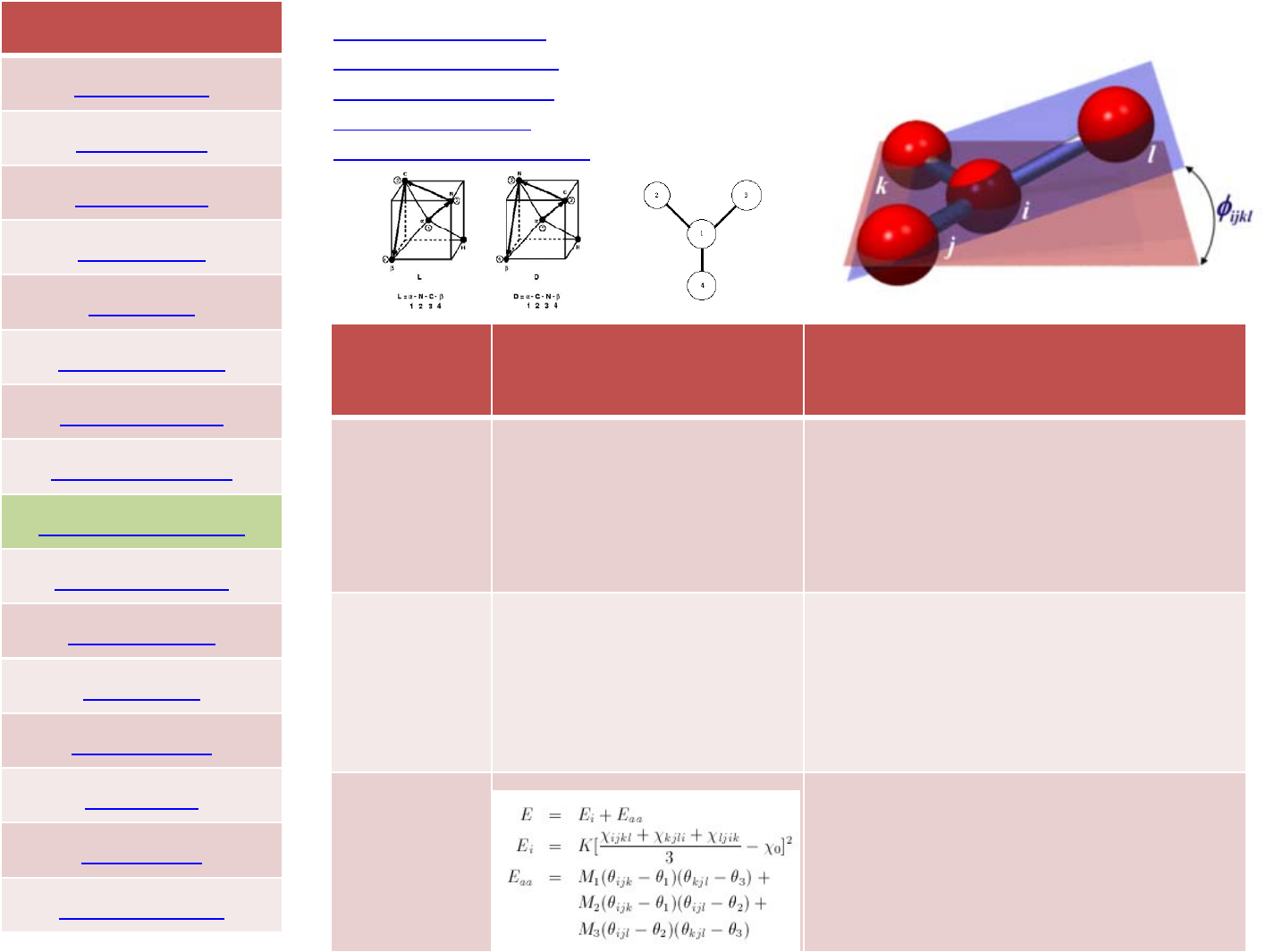
between the 1,3 atoms, as illustrated in Figure. Cross terms were
found to be important in force fields designed to predict vibrational
spectra that were the forerunners of molecular mechanics force
fields, and so it is not surprising that cross terms must often be
included in a molecular mechanics force field to achieve optimal
performance.
One should in principle include cross terms between all contributions
to a force field. However, only a few cross terms are generally found
to be necessary in order to reproduce structural properties accurately;
more may be needed to reproduce other properties such as
vibrational frequencies, which are more sensitive to the presence of
such terms,.
Crossterms:class1,2and3forcefields
14
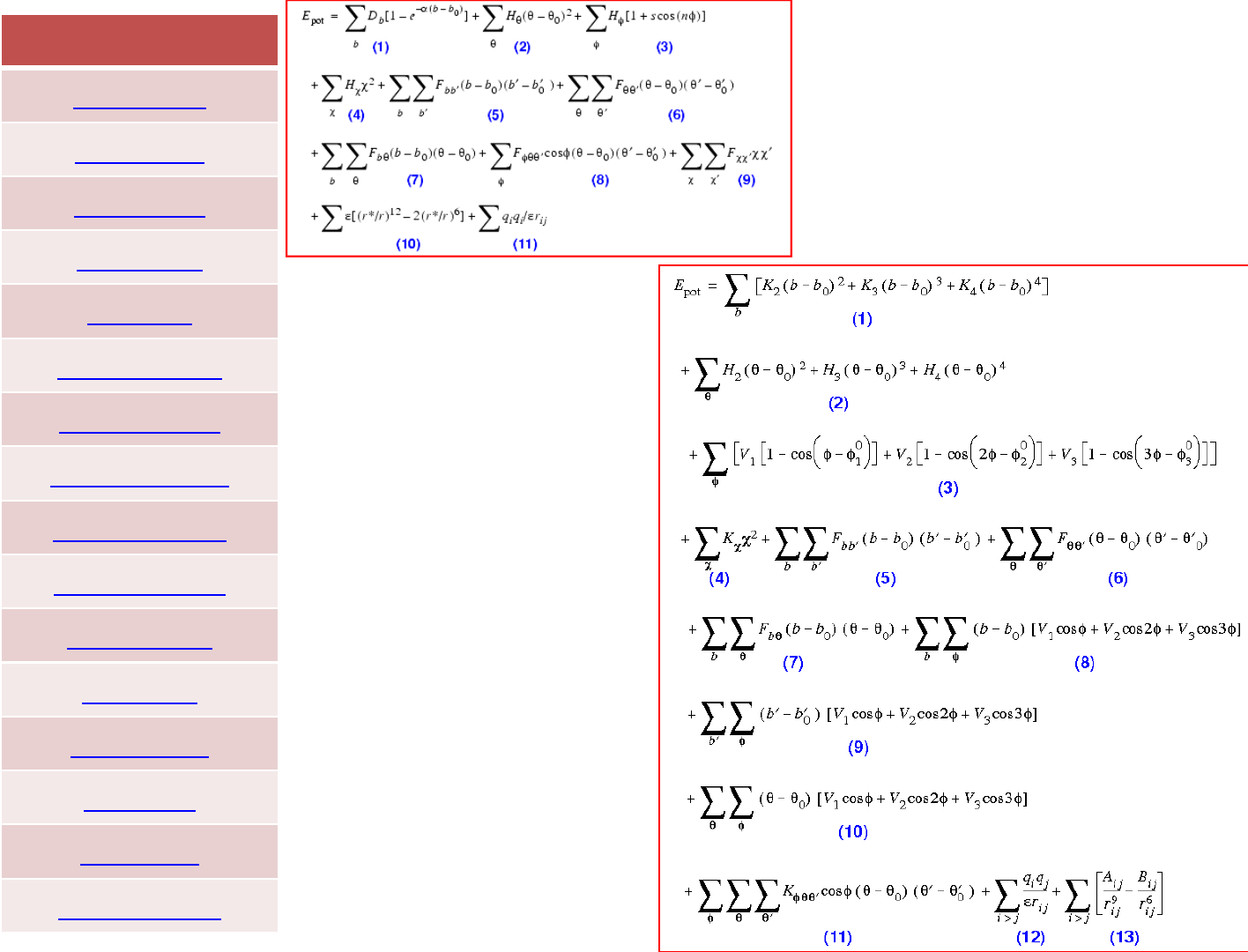
FORCEFIELD
angle_coeff
ANGLE_STYLE
bond_coeff
bond_style
dielectric
dihedral_coeff
dihedral_style
improper_coeff
improper_style
kspace_modify
kspace_style
pair_coeff
pair_modify
pair_style
pair_write
special_bonds
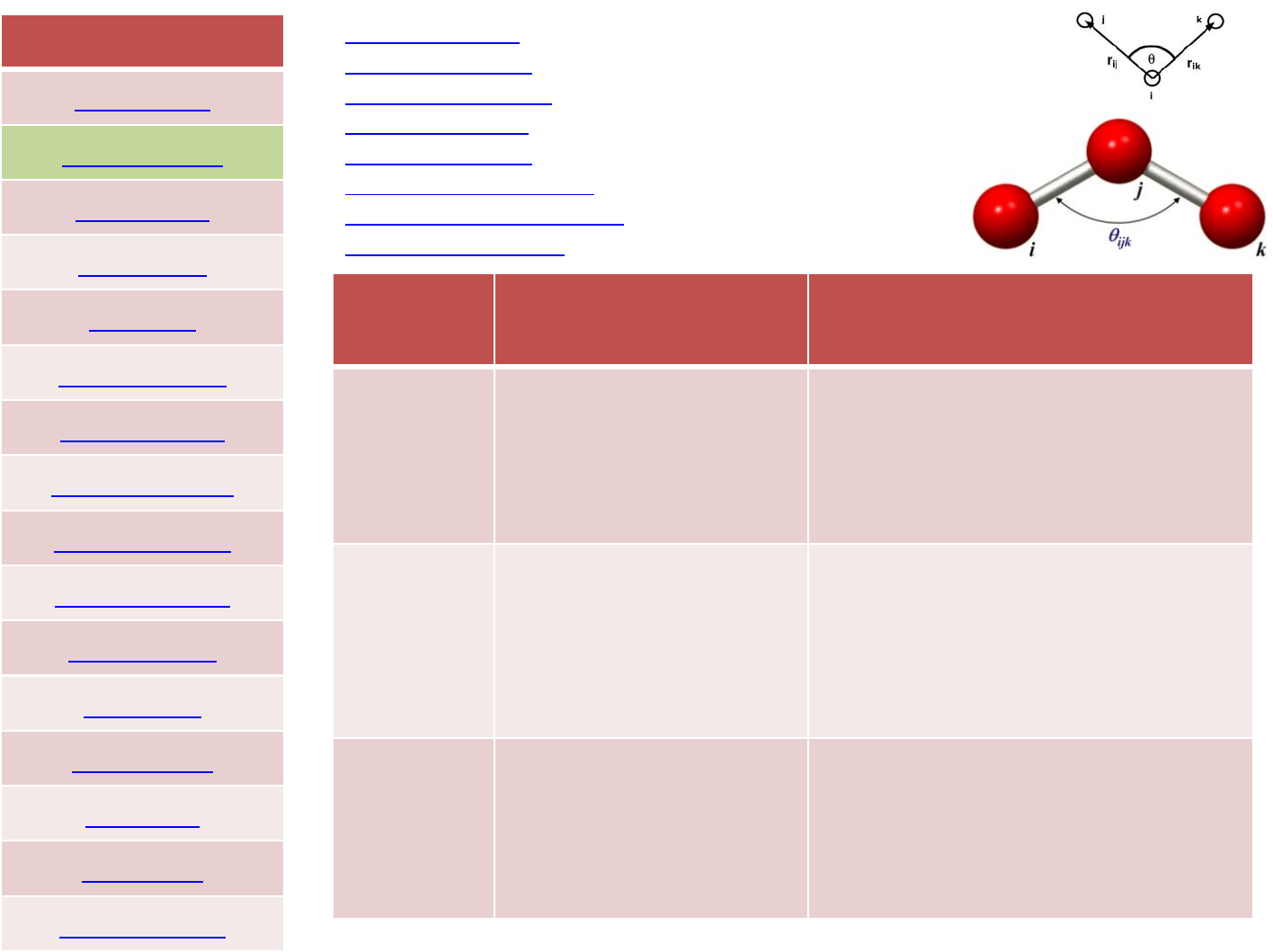
angle_style none ‐turn offangleinteractions
angle_style hybrid ‐define multiplestylesofangleinteractions
angle_style charmm ‐CHARMMangle
angle_style class2 ‐COMPASS(class2)angle
angle_style cosine ‐cosine anglepotential
angle_style cosine/delta ‐difference ofcosines anglepotential
angle_style cosine/squared ‐cosine squared anglepotential
angle_style harmonic ‐harmonic angle
style Expression Examples
harmonic K(energy/radian^2)
theta0(degrees)
angle_style harmonic
angle_coeff 1300.0107.0
Charmm K(energy/radian^2)
theta0(degrees)
K_ub (energy/distance^2)
r_ub (distance)
angle_style charmm
angle_coeff 1300.0107.050.03.0
class2 angle_style class2
angle_coeff *75.0
()
2
0
EK
θ
θ
=−
()()()
()( )
()
()
()
()
234
203040
12
11 02 2 0
abbba
a
bb ij jk
ba ij jk
EE E E
EK K K
EMr rr r
ENrr Nrr
θθ θθ θθ
θ
θθθ
=
=
++
=−+−+−
−−
=−−+ −−
() ( )
22
0UB UB
EK K rr
θθ
=−+ −
16
FORCEFIELD
angle_coeff
angle_style
bond_coeff
BOND_STYLE
dielectric
dihedral_coeff
dihedral_style
improper_coeff
improper_style
kspace_modify
kspace_style
pair_coeff
pair_modify
pair_style
pair_write
special_bonds
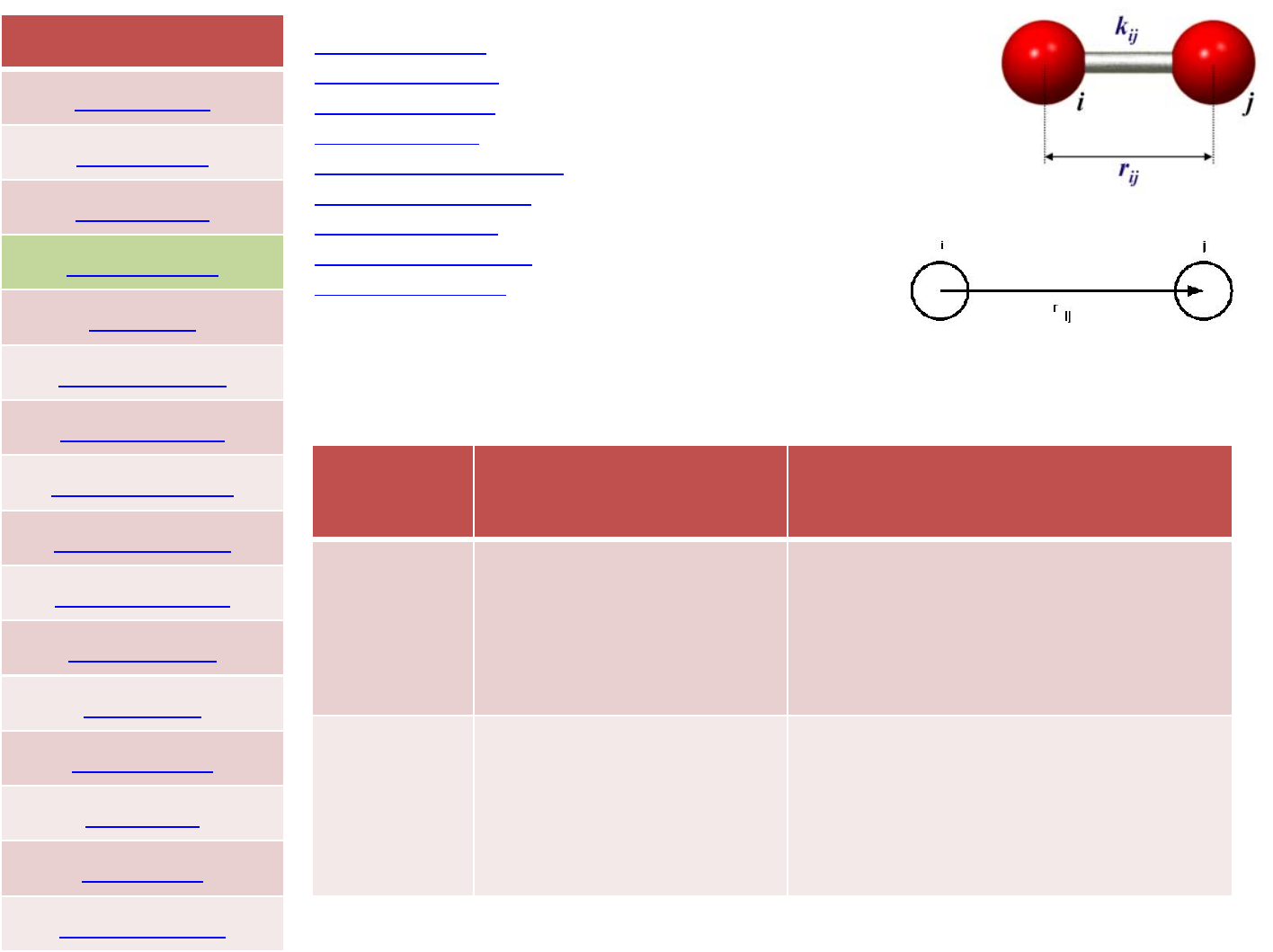
style Expression Examples
harmonic K(energy/distance^2)
r0(distance)
bond_style harmonic
bond_coeff 580.01.2
class2 r0(distance)
K2(energy/distance^2)
K3(energy/distance^2)
K4(energy/distance^2)
bond_style class2
bond_coeff 11.0100.080.080.0
()
2
0
EKrr=−
bond_style none ‐turn offbonded interactions
bond_style hybrid ‐define multiplestylesofbondinteractions
bond_style class2 ‐COMPASS(class2)bond
bond_style fene ‐FENE(finite‐extensiblenon‐linear elastic)bond
bond_style fene/expand ‐FENEbondswith variablesizeparticles
bond_style harmonic ‐harmonic bond
bond_style morse ‐Morsebond
bond_style nonlinear ‐nonlinear bond
bond_style quartic ‐breakable quartic bond
() () ()
234
20 30 40
K
Krr Krr Krr=−+−+−
17

FORCEFIELD
angle_coeff
angle_style
bond_coeff
bond_style
dielectric
dihedral_coeff
DIHEDRAL_STYLE
improper_coeff
improper_style
kspace_modify
kspace_style
pair_coeff
pair_modify
pair_style
pair_write
special_bonds
dihedral_style none ‐turn offdihedral interactions
dihedral_style hybrid ‐define multiplestylesofdihedral interactions
dihedral_style charmm ‐CHARMMdihedral
dihedral_style class2 ‐COMPASS(class2)dihedral
dihedral_style harmonic ‐harmonic dihedral
dihedral_style helix ‐helix dihedral
dihedral_style multi/harmonic ‐multi‐harmonic dihedral
dihedral_style opls ‐OPLSdihedral
style Expression Examples
harmonic K(energy)
d(+1or‐1)
n(integer >=0)
dihedral_style harmonic
dihedral_coeff 180.012
Charmm K(energy)
n(integer>=0)
d(integervalueofdegrees)
weightingfactor(0.0to1.0)
dihedral_style charmm
dihedral_coeff 1120.01600.5
class2 dihedral_style class2
dihedral_coeff 110075100708060
()
1cosEK d n
φ
=
⋅+⋅ ⋅
⎡
⎤
⎣
⎦
(
)
1cosEK n d
φ
=⋅+ ⋅−
⎡
⎤
⎣
⎦
18
FORCEFIELD
angle_coeff
angle_style
bond_coeff
bond_style
dielectric
dihedral_coeff
dihedral_style
improper_coeff
IMPROPER_STYLE
kspace_modify
kspace_style
pair_coeff
pair_modify
pair_style
pair_write
special_bonds
improper_style none ‐turn offimproper interactions
improper_style hybrid ‐define multiplestylesofimproper interactions
improper_style class2 ‐COMPASS(class2)improper
improper_style cvff ‐CVFFimproper
improper_style harmonic ‐harmonic improper
style Expression Examples
harmonic K(energy/radian^2)
X0(degrees)
improper_style harmonic
improper_coeff 1100.00
CVFF K(energy)
d(+1or‐1)
n(0,1,2,3,4,6)
improper_stylecvff
improper_coeff180.0‐14
class2 improper_style class2
improper_coeff 1100.00
()
2
0
EK
χ
χ
=−
(
)
1cosEK d n
φ
=
⋅+⋅ ⋅
⎡
⎤
⎣
⎦
19
FORCEFIELD
angle_coeff
angle_style
bond_coeff
bond_style
dielectric
dihedral_coeff
dihedral_style
improper_coeff
improper_style
kspace_modify
kspace_style
pair_coeff
pair_modify
PAIR_STYLE
pair_write
special_bonds
pair_style lj/charmm/coul/charmm ‐CHARMMpotential with cutoff Coulomb
pair_style lj/charmm/coul/charmm/implicit ‐CHARMMforimplicit solvent
pair_style lj/charmm/coul/long ‐CHARMMwith long‐rangeCoulomb
pair_style lj/charmm/coul/long/opt ‐optimized versionofCHARMMwith long‐rangeCoulomb
pair_style lj/class2 ‐COMPASS(class2)forcefield with noCoulomb
pair_style lj/class2/coul/cut ‐COMPASSwith cutoff Coulomb
pair_style lj/class2/coul/long ‐COMPASSwith long‐rangeCoulomb
pair_style lj/cut ‐cutoff Lennard‐Jonespotential with noCoulomb
pair_style lj/cut/opt ‐optimized versionofcutoff LJ
pair_style lj/cut/coul/cut ‐LJwith cutoff Coulomb
pair_style lj/cut/coul/debye ‐LJwith Debyescreeningadded toCoulomb
pair_style lj/cut/coul/long ‐LJwith long‐rangeCoulomb
pair_style lj/cut/coul/long/tip4p ‐LJwith long‐rangeCoulombforTIP4Pwater
pair_style lj/expand ‐Lennard‐Jonesforvariablesizeparticles
pair_style lj/smooth ‐smoothed Lennard‐Jonespotential
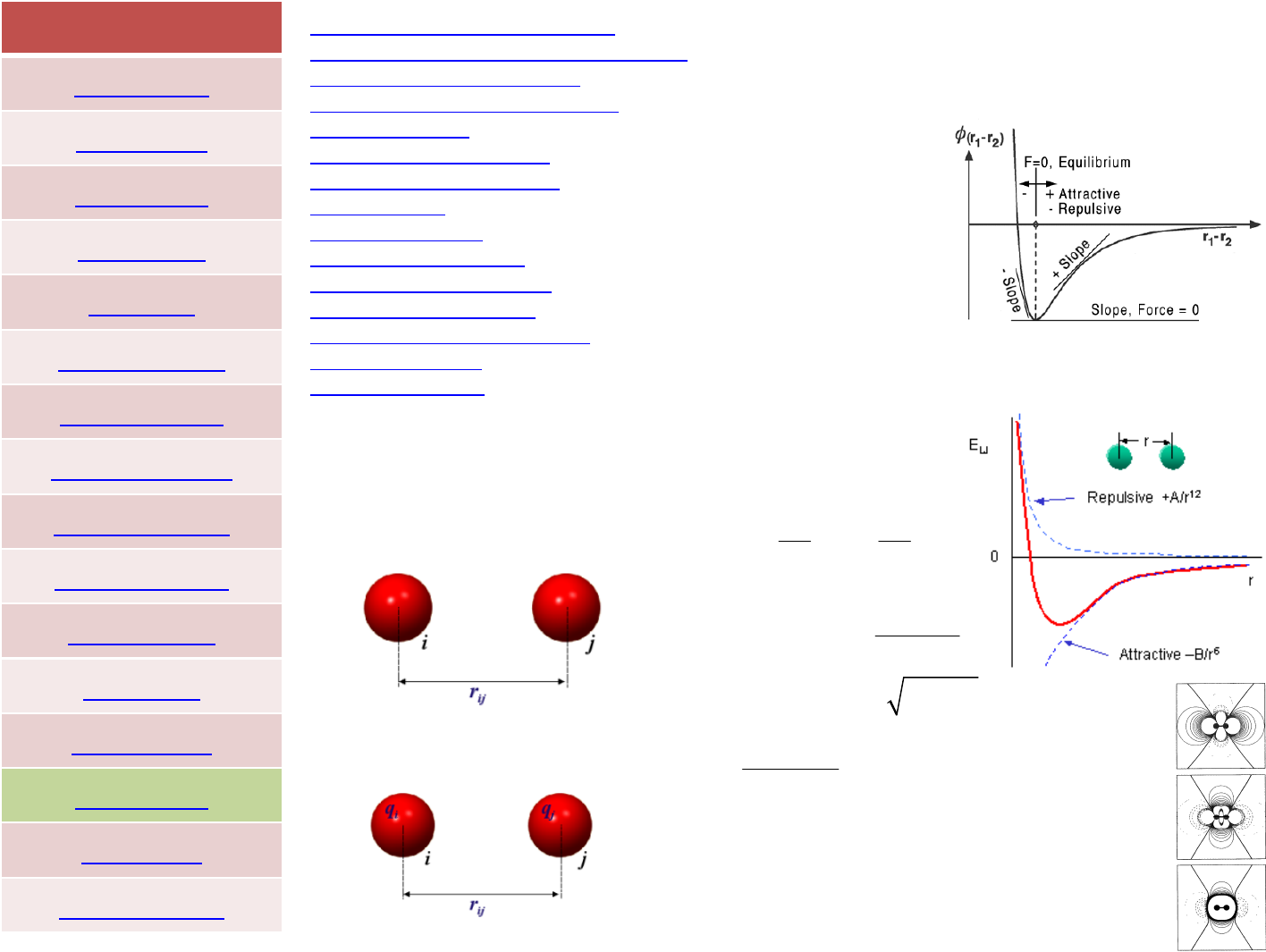
Van‐der‐Waals with
Lorentz—Berthelot mixing rule
12 6
4
av.diameter 2
av.welldepth
ij ij
ij
ij ij
ii ij
ij
ij ii ij
Err
σσ
ε
σσ
σ
ε
εε
⎡
⎤
⎛⎞⎛⎞
⎢
⎥
=−
⎜⎟⎜⎟
⎜⎟⎜⎟
⎢
⎥
⎝⎠⎝⎠
⎣
⎦
+
==
=
=+
Coulomb
0
12 2 1 2
0
R
4
8.8542·10 C N m
1 for a vacuum by definition
ij
Rij
qq
Er
πε ε
ε
ε
−−−
=
=
=
20

FORCEFIELD
angle_coeff
angle_style
bond_coeff
bond_style
dielectric
dihedral_coeff
dihedral_style
improper_coeff
improper_style
kspace_modify
kspace_style
pair_coeff
pair_modify
PAIR_STYLE
pair_write
special_bonds
pair_style hybrid ‐multiplestylesofpairwise interactions
pair_style hybrid/overlay ‐multiplestylesofsuperposed pairwise interactions
pair_style airebo ‐AI‐REBOpotential
pair_style buck ‐Buckinghampotential
pair_style buck/coul/cut ‐Buckinghamwith cutoff Coulomb
pair_style buck/coul/long ‐Buckinghamwith long‐rangeCoulomb
pair_style colloid ‐integrated colloidal potential
pair_style coul/cut ‐cutoff Coulombic potential
pair_style coul/debye ‐cutoff Coulombic potential with Debyescreening
pair_style coul/long ‐long‐rangeCoulombic potential
pair_style dipole/cut ‐pointdipoles with cutoff
pair_style dpd ‐dissipativeparticle dynamics (DPD)
pair_style eam ‐embedded atom method (EAM)
pair_style eam/opt ‐optimized versionofEAM
pair_style eam/alloy ‐alloy EAM
pair_style eam/alloy/opt ‐optimized versionofalloy EAM
pair_style eam/fs ‐Finnis‐SinclairEAM
pair_style eam/fs/opt ‐optimized versionofFinnis‐SinclairEAM
pair_style gayberne ‐Gay‐Berneellipsoidal potential
pair_style gran/hertzian ‐granular potential with Hertizain interactions
pair_style gran/history ‐granular potential with history effects
pair_style gran/no_history ‐granular potential without history effects
pair_style lubricate ‐hydrodynamic lubrication forces
pair_style meam ‐modified embedded atom method (MEAM)
pair_style morse ‐Morsepotential
pair_style morse/opt ‐optimized versionofMorsepotential
pair_style resquared ‐Everaers RE‐Squared ellipsoidal potential
pair_style soft ‐Soft(cosine)potential
pair_style sw ‐Stillinger‐Weber3‐bodypotential
pair_style table ‐tabulated pairpotential
pair_style tersoff ‐Tersoff 3‐bodypotential
pair_style yukawa ‐Yukawa potential
21

FORCEFIELD
angle_coeff
angle_style
bond_coeff
bond_style
dielectric
dihedral_coeff
dihedral_style
improper_coeff
improper_style
kspace_modify
kspace_style
pair_coeff
pair_modify
PAIR_STYLE
pair_write
special_bonds
style Expression Examples
lj/cut pair_style lj/cut2.5
pair_coeff 1111.12.8
lj/cut/coul
/cut
pair_style lj/cut/coul/cut10.08.0
pair_coeff 11100.03.59.0
lj/class2 pair_style lj/class210.0
pair_coeff 11100.03.59.0
lj/class2/c
oul/cut
pair_style lj/class2/coul/cut
10.08.0
pair_coeff 11100.03.59.0
12 6
4
vdw C
EE rr
rr
σσ
ε
⎡⎤
⎛⎞ ⎛⎞
=
=−<
⎢⎥
⎜⎟ ⎜⎟
⎝⎠ ⎝⎠
⎢⎥
⎣⎦
ij
vdw C
qq
E
EC rr
r
ε
=
+<
96
23
for
vdw
C
EE rr
rr
σσ
ε
⎡
⎤
⎛⎞ ⎛⎞
== −
⎢
⎥
⎜⎟ ⎜⎟
⎝⎠ ⎝⎠
⎢
⎥
⎣
⎦
<
for
ij
vdw
C
qq
EE Cr
rr
ε
=+
<
epsilon(energy units)
sigma(distanceunits)
cutoff (distanceunits)
cutoff2(distanceunits)
22

FORCEFIELD
angle_coeff
angle_style
bond_coeff
bond_style
dielectric
dihedral_coeff
dihedral_style
improper_coeff
improper_style
kspace_modify
kspace_style
pair_coeff
pair_modify
pair_style
pair_write
special_bonds
pair coeff 1 2 1.0 3.45 10.0 (pair style lj/cutoff)
pair coeff 1 2 1.0 3.45 8.0 10.0 (pair style lj/smooth)
pair coeff 1 2 1.0 3.45 2.0 10.0 (pair style lj/shift)
pair coeff 1 2 1.0 30.0 2.5 (pair style soft)
pair coeff 1 2 1.0 3.45 10.0 (pair style class2/cutoff)
pair coeff 1 2 1.0 3.45 1.0 3.45 (pair style lj/charmm)
pair coeff 1 2 1.0 3.45 12.0 10.0 (pair style expo_6/cutoff)
pair coeff 1 2 1.0 3.45 12.0 10.0 12. 0 (pair style expo_6/spline)
pair coeff 1 2 1.0 3.45 12.0 10.0 (pair style expo_6/smooth)
special bonds amber
special bonds 0.0 0.0 0.5
pppm mesh 32 32 64
pppm order 5
dielectric 1
angle coeff 1 30.0 108.0 (angle style harmonic)
angle coeff 1 30.0 108.0 30.0 2.5 (angle style charmm)
angle coeff 1 30.0 108.0 (angle style cosharmonic)
bond coeff 1 100.0 3.45 (bond style harmonic)
bond coeff 1 30.0 1.5 1.0 1.0 (bond style fene/standard )
bond coeff 1 30.0 1.5 1.0 1.0 0.2 (bond style fene/shift)
bond coeff 1 28.0 0.748308 0.166667 (bond style nonlinear)
dihedral coeff 1 10.0 1 3 (dihedral style harmonic)
dihedral coeff 1 2.0 2.0 2.0 2.0 2.0 (dihedral style multiharmonic)
dihedral coeff 1 2.0 5 180.0 0.5 (dihedral style charmm)
dihedral coeff 1 2.0 1 3.0 (dihedral style dreiding)
improper coeff 1 20.0 0.0 (improper style harmonic)
improper coeff 1 20.0 10.0 (improper style cvff)
EXAMPLES
(whenused,mustappearafter"readdata"or"readrestart"command)
23
SETTINGS
communicate
dipole
group
mass
min_modify
min_style
neigh_modify
NEIGHBOR
reset_timestep
run_style
set
shape
timestep
velocity
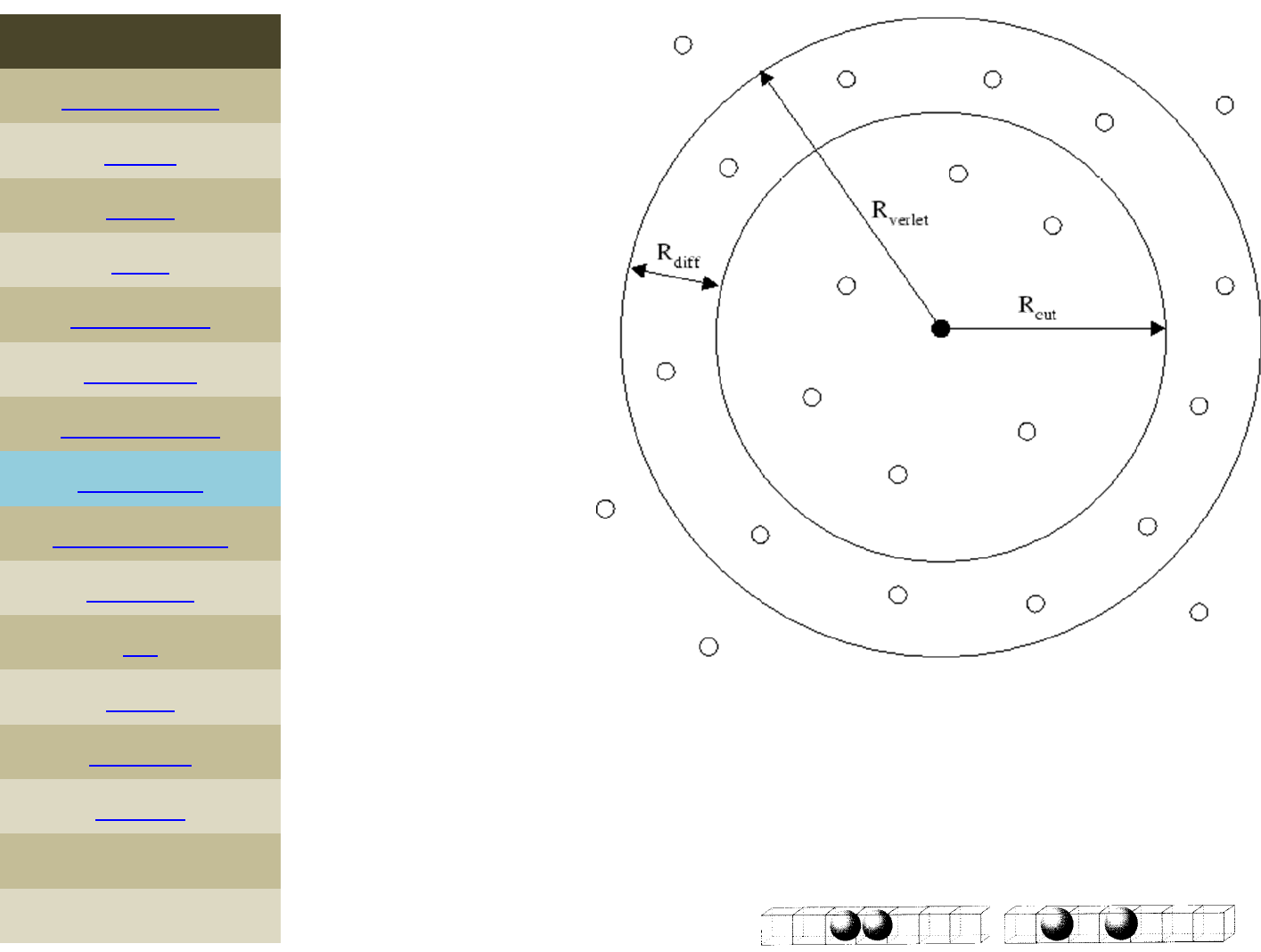
neighbor skinstyle
neighbor0.3bin
neighbor2.0nsq
This command sets parameters that affect the building of the
pairwise neighbor list. All atom pairs within a cutoff distance equal
tothetheirforcecutoffplustheskin distance are stored in the list.
Typically, the larger the skin distance, the less often neighbor lists
need to be built, but more pairs must be checked for possible force
interactions every timestep. 24
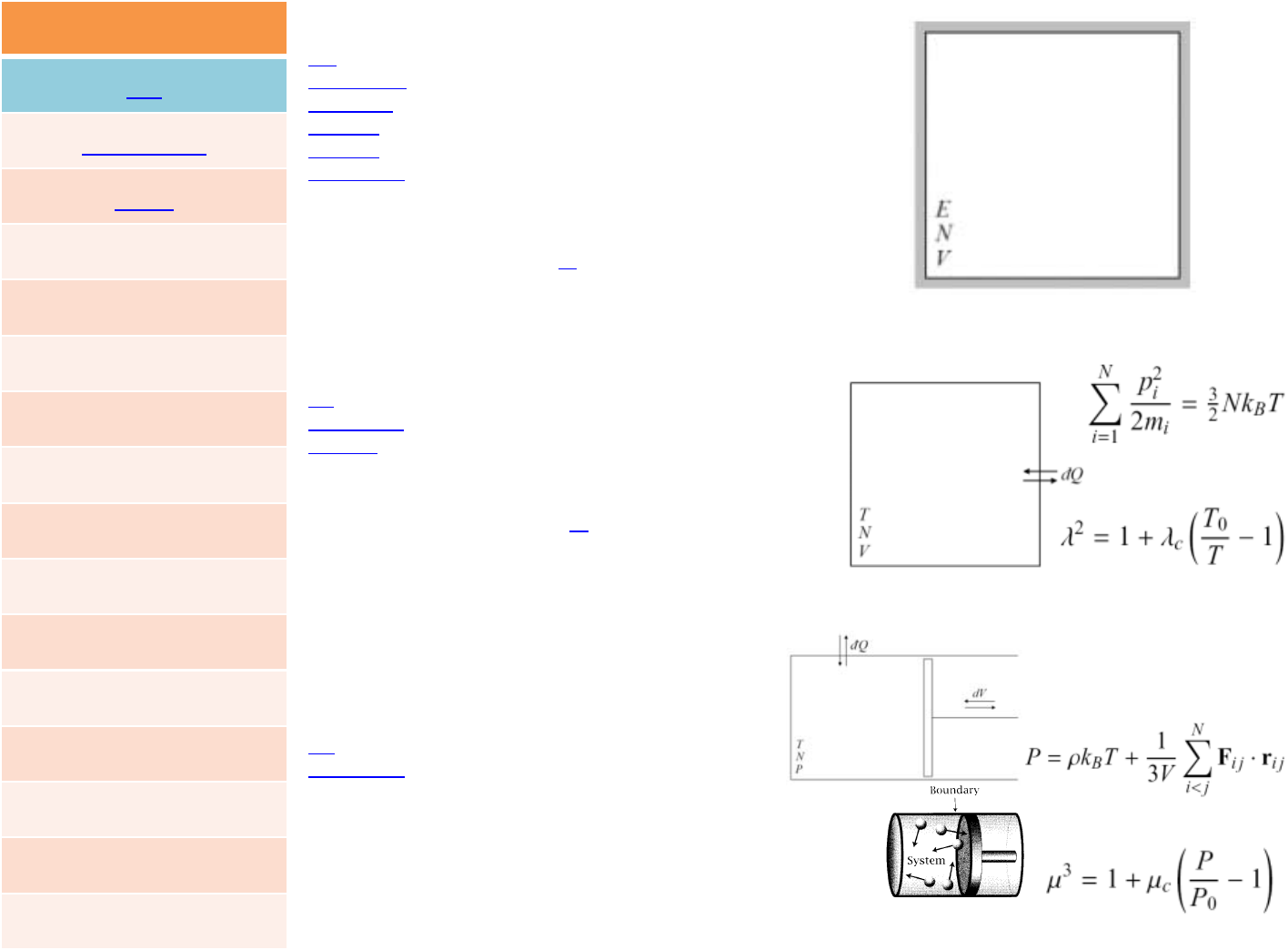
FIX
FIX
fix_modify
unfix
fix idallnve
nve ‐constantNVEtimeintegration
nve/asphere ‐NVTforaspherical particles
nve/dipole ‐NVEforpointdipolarparticles
nve/gran ‐NVEforgranularparticles
nve/limit ‐NVEwithlimitedsteplength
nve/noforce ‐NVEwithoutforces(vonly)
nvt ‐constantNVTtimeintegrationviaNose/Hoover
nvt/asphere ‐NVTforaspherical particles
nvt/sllod ‐NVTforNEMDwithSLLODequations
npt ‐constantNPTtimeintegrationviaNose/Hoover
npt/asphere ‐NPTforaspherical particles
fix idallnvt
fix idallnpt
fix IDgroup‐IDnpt Tstart Tstop Tdamp p‐styleargs keywordvalue...
xyz args =Pstart Pstop Pdamp
Pstart,Pstop =desiredpressureatstart/endofrun(pressureunits)
Pdamp =pressuredampingparameter(timeunits)
xy oryz orxz oraniso args =Px_start Px_stop Py_start Py_stop Pz_start Pz_stop Pdamp
Px_start,Px_stop,...=desiredpressureinx,y,z atstart/endofrun(pressureunits)
Pdamp =pressuredampingparameter(timeunits)
fix IDgroup‐IDnvt Tstart Tstop Tdamp keywordvalue...
ID,group‐IDaredocumentedinfix command
nvt =stylenameofthisfixcommand
Tstart,Tstop =desiredtemperatureatstart/endofrun
Tdamp =temperaturedampingparameter(timeunits)
zeroormorekeyword/valuepairsmaybeappended
keyword=drag
fix IDgroup‐IDnve
ID,group‐IDaredocumentedinfix command
nve =stylenameofthisfixcommand
26
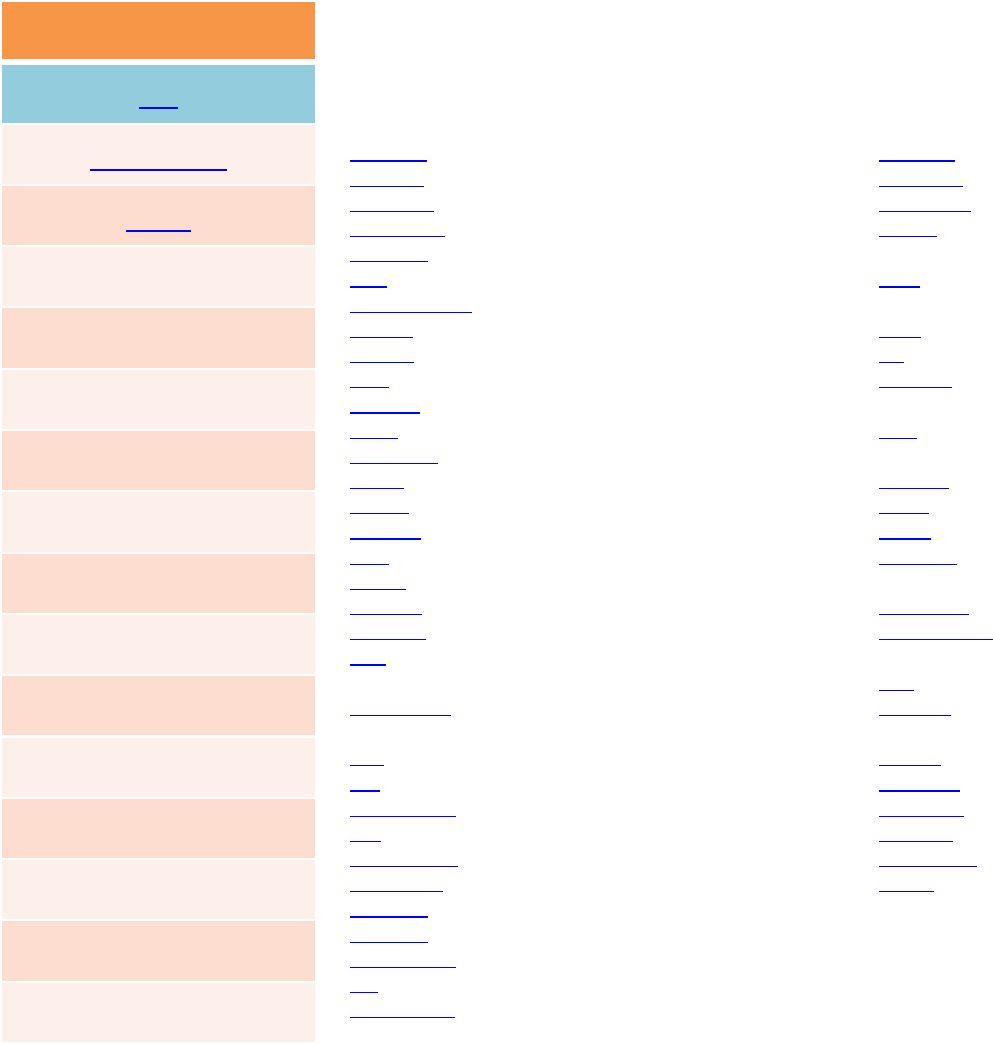
FIX
FIX
fix_modify
unfix
fix IDgroup‐IDstyleargs
addforce ‐addaforcetoeachatom
aveforce ‐addanaveragedforcetoeachatom
ave/atom ‐computeper‐atomtime‐averagedquantities
ave/spatial ‐outputper‐atomquantitiesbylayer
ave/time ‐outputtime‐averagedcomputequantities
com ‐computeacenter‐of‐mass
coord/original ‐storeoriginalcoords ofeachatom
deform ‐changethesimulationboxsize/shape
deposit ‐addnewatomsaboveasurface
drag ‐dragatomstowardsadefinedcoordinate
dt/reset ‐resetthetimestep basedonvelocity,forces
efield ‐imposeelectricfieldonsystem
enforce2d ‐zerooutz‐dimensionvelocityandforce
freeze ‐freezeatomsinagranularsimulation
gravity ‐addgravitytoatomsinagranularsimulation
gyration ‐computeradiusofgyration
heat ‐add/subtractmomentum‐conservingheat
indent ‐imposeforceduetoanindenter
langevin ‐Langevin temperaturecontrol
lineforce ‐constrainatomstomoveinaline
msd ‐computemean‐squareddisplacement(i.e.diffusion
coefficient)
momentum ‐zerothelinearand/orangularmomentumof
agroupofatoms
nph ‐constantNPHtimeintegrationviaNose/Hoover
npt ‐constantNPTtimeintegrationviaNose/Hoover
npt/asphere ‐NPTforaspherical particles
nve ‐constantNVEtimeintegration
nve/asphere ‐NVTforaspherical particles
nve/dipole ‐NVEforpointdipolarparticles
nve/gran ‐NVEforgranularparticles
nve/limit ‐NVEwithlimitedsteplength
nve/noforce ‐NVEwithoutforces(vonly)
nvt ‐constantNVTtimeintegrationviaNose/Hoover
nvt/asphere ‐NVTforaspherical particles
nvt/sllod ‐NVTforNEMDwithSLLODequations
orient/fcc ‐addgrainboundarymigrationforce
planeforce ‐constrainatomstomoveinaplane
poems ‐constrainclustersofatomstomoveas
coupledrigidbodies
pour ‐pournewatomsintoagranularsimulation
domain
print ‐printtextandvariablesduringasimulation
rdf ‐computeradialdistributionfunctions
recenter ‐constrainthecenter‐of‐masspositionofa
groupofatoms
rigid ‐constrainoneormoreclustersofatomsto
moveasarigidbody
setforce ‐settheforceoneachatom
shake ‐SHAKEconstraintsonbondsand/orangles
spring ‐applyharmonicspringforcetogroupofatoms
spring/rg ‐springonradiusofgyrationofgroupof
atoms
spring/self ‐springfromeachatomtoitsorigin
temp/rescale ‐temperaturecontrolbyvelocity
rescaling
tmd ‐guideagroupofatomstoanewconfiguration
viscosity ‐Muller‐Plathe momentumexchangefor
viscositycalculation
viscous ‐viscousdampingforgranularsimulations
wall/gran ‐frictionalwall(s)forgranularsimulations
wall/lj126 ‐Lennard‐Jones12‐6wall
wall/lj93 ‐Lennard‐Jones9‐3wall
wall/reflect ‐reflectingwall(s)
wiggle ‐oscillatewallsandfrozenatoms
27

FIX
fix
fix_modify
unfix assign fix 1atom 200
assign fix 1molecule 50
assignfix 1type2
assignfix 1region0.01.0INFINF0.01.0
assignfix 1bondtype 4
assignfix 1remainder
fix style none
fix style 1setforce 0.0NULL0.0
fix style 1addforce 1.00.00.0
fix style 1aveforce 1.00.00.0
fix style 1rescale300.0300.010020.00.5
fix style 1hoover/drag50.050.00.001
fix style 1langevin50.050.00.0112345111
fix style 1springforce 10.0NULLNULL 1.0
fix style 1dragforce 10.0‐5.0NULL2.01.0
fix style 1shake30.001100
EXAMPLESOFCONSTRAINTS
(whenused,mustappearafter"readdata"or"readrestart"command)
28

COMPUTES
COMPUTE
compute_modify
uncompute
centro/atom ‐centro‐symmetry parameter foreach atom
coord/atom ‐coordinationnumber foreach atom
displace/atom ‐displacement ofeach atom
group/group ‐energy/forcebetween two groupsofatoms
ke/atom ‐kinetic energy foreach atom
pe ‐potential energy
pe/atom ‐potential energy foreach atom
pressure ‐totalpressureandpressuretensor
reduce ‐combineper‐atom quantities into asingleglobalvalue
rotate/dipole ‐rotational energy ofdipolar atoms
rotate/gran ‐rotational energy ofgranular atoms
stress/atom ‐stresstensor foreach atom
temp ‐temperature ofgroupofatoms
temp/asphere ‐temperature ofaspherical particles
temp/deform ‐temperature excluding boxdeformation velocity
temp/dipole ‐temperature ofpointdipolar particles
temp/partial ‐temperature excluding oneormoredimensionsofvelocity
temp/ramp ‐temperature excluding ramped velocity component
temp/region ‐temperature ofaregion ofatoms
compute IDgroup‐IDstyleargs
compute1alltemp
computenewtemp flowtemp/partial110
compute3allke/atom
29
ACTIONS
delete_atoms
delete_bonds
displace_atoms
displace_box
minimize
RUN
temper
run Nkeywordvalues
N=#oftimesteps
zeroormorekeyword/valuepairsmaybeappended
keyword=upto orstart orstop orpre orpost orevery
run10000run1000000upto
run100start0stop1000
run1000prenopostyes
run100000start0stop1000000every1000print"ProteinRg =$r“
run100000every1000NULL
minimize tolerance maxiter maxeval
30
OUTPUT
DUMP
dump_modify
restart
thermo
thermo_modify
thermo_style
undump
write_restart
dump IDgroup‐IDstyleNfileargs
ID=user‐assignednameforthedump
group‐ID=IDofthegroupofatomstobedumped
style=atom orbond ordcd orxtc orxyz orcustom
N=dumpeverythismanytimesteps
file=nameoffiletowritedumpinfoto
args =listofargumentsforaparticularstyle
dumpmyDump allatom 100
dump.atom dump2subgroup atom 50
dump.run.bindump4aallcustom100
dump1allxtc 1000file.xtc
31

OUTPUT
dump
dump_modify
restart
THERMO
thermo_modify
thermo_style
undump
write_restart
thermo_style styleargs
style=one ormulti orgranular orcustom
args =listofargumentsforaparticularstyle
one args = none
multi args = none
granular args = none
custom args = list of attributes
possible attributes = step, atoms, cpu, temp, press,
pe, ke, etotal, enthalpy,
evdwl, ecoul, epair, ebond, eangle, edihed, eimp,
emol, elong, etail,
vol, lx, ly, lz, xlo, xhi, ylo, yhi, zlo, zhi,
pxx, pyy, pzz, pxy, pxz, pyz
drot, grot,
c_ID, c_ID[n], f_ID, f_ID[n], v_name
step = timestep
atoms = # of atoms
cpu = elapsed CPU time
temp = temperature
press = pressure
pe = total potential energy
ke = kinetic energy
etotal = total energy (pe + ke)
enthalpy = enthalpy (pe + press*vol)
evdwl = VanderWaal pairwise energy
ecoul = Coulombic pairwise energy
epair = pairwise energy (evdwl + ecoul + elong + etail)
ebond = bond energy
eangle = angle energy
edihed = dihedral energy
eimp = improper energy
emol = molecular energy (ebond + eangle + edihed + eimp)
elong = long-range kspace energy
etail = VanderWaal energy long-range tail correction
vol = volume
lx,ly,lz = box lengths in x,y,z
xlo,xhi,ylo,yhi,zlo,zhi = box boundaries
pxx,pyy,pzz,pxy,pxz,pyz = 6 components of pressure tensor
drot = rotational energy of dipolar atoms
grot = rotational energy of granular atoms
c_ID = global scalar value calculated by a compute with ID
c_ID[N] = Nth component of global vector calculated by a compute with ID
f_ID = global scalar value calculated by a fix with ID
f_ID[N] = Nth component of global vector calculated by a fix with ID
v_name = global valuecalculatedbyanequal‐stylevariablewithname32

TOOLS DESCRIPTION
amber2lammps Pythonscriptsforconvertingfilesback‐and‐forthbetweentheAMBERMDcodeandLAMMPS
binary2txt convertsoneormorebinaryLAMMPSdumpfileintoASCIItextfiles
ch2lmp containstoolsforconvertingfilesback‐and‐forthbetweentheCHARMMMDcodeandLAMMPS
chain LAMMPSdatafilecontainingbead‐springpolymerchainsand/ormonomersolventatoms
data2xmovie convertsaLAMMPSdatafileintoasnapshotsuitableforvisualizingwiththexmovie tool
eam generate convertsananalyticformulaintoatabulatedembeddedatommethod(EAM) setfl potentialfile
lmp2arc convertingLAMMPSoutputfilestotheformatforAccelrys's InsightMDcode
lmp2cfg toolforconvertingLAMMPSoutputfilesintoaseriesof*.cfgfileswhichcanbereadintotheAtomEye
visualizer
lmp2traj toolforconvertingLAMMPSoutputfilesinto3analysisfiles
matlab /MSLAB severalMATLAB scriptsforpost‐processingLAMMPSoutput
micelle2d createsaLAMMPSdatafilecontainingshortlipidchainsinamonomersolution
msi2lmp toolforcreatingLAMMPSinputdatafilesfromAccelrys's InsightMDcode(formerlyMSI/Biosysm andits
DiscoverMDcode)
pymol_asphere toolforconvertingaLAMMPSdumpfilethatcontainsorientationinfoforellipsoidalparticlesintoaninput
fileforthePyMol visualizationpackage
restart2data convertsabinaryLAMMPSrestartfileintoanASCIIdatafile
thermo_extract readsoneofmoreLAMMPSlogfilesandextractsathermodynamicvalue(e.g.Temp,Press)
vim ,xmovie X‐basedvisualizationpackagethatcanreadLAMMPSdumpfilesandanimatethem 33

MODMOL25‐27Feb 2008,Jouy‐en‐Josas
olivier.vitrac@agroparistech.fr
Thisdocumentisbasedonourowncontribution.
Theresultisfreelydistributablewithoutguaranteeorwarranteeofanykind.
# MSLAB starts within $PROJECT
# example of line to be added/updated in ~/.bashrc
export PROJECT="~/project"

CustomLAMMPSinstallation
Important definitions for LAMMPS in this ./.bashrc (use LAMRC to see the related HELP)
Installation parameters
=======================
topaze: open a ssh connection on topaze
see also: GENKEY, EXPORTKEY (to make it possible a SSH connection without password on cluster
nodes)
$PROJECT: main directory of LAMMPS projects (TO BE CHECKED with "echo $PROJECT")
LAMMPS scripts must be located in $PROJECT (default) or in its subdirectories (e.g. examples)
LAMMPS jobs are automatically stored in $PROJECT/XXXXX (where X=[A-Z0-9])
see also: LSLAM, CDLAM, TREELAM
Included directories
bin/ LAMMPS executables (several versions are available)
executions with MPICH should be avoided, LAM/MPI must be preferred as it is
integrated with SUN GRID ENGINE
bin/tools/ other executables
doc/ documentation
make/ make directory (for advanced users)
examples/ examples
pizza/ PYTHON interface to LAMMPS (see PIZZA)
installation file has been modified to match $PROJECT
NB: PIZZA works on LINUX and WINDOWS XP/Vista
MSLAB (Matlab with the toolbox MS) requires:
> a directory $PROJECT/../codes/MS where all MS functions are located
> a script $PROJECT/startms.m where the GLOBAL variables LAMMPS and PATHPROJECT are defined
> The variable LAMMPS must be updated to your needs (e.g. define your e-mail).
NB: MSLAB works on LINUX and WINDOWS XP/Vista
FILE:~/.bashrc
35

CUSTOMLAMMPSJOB
Typical job $PROJECT/XXXXX
===========================
Each job directory contains initially:
a LAMMPS executable: e.g. lmp_g++_lam_all_100208 (last version 10/02/08 for LAM/MPI, including all
modules)
a LAMMPS input file: e.g. long.in.lj
additional data files: e.g. data.micelle (see LAMJOB to add user files)
3 shell scripts:
run.sh: submit the job on the cluster via SUN GRID ENGINE
manual queueue and execution on the cluster via: $PROJECT/XXXXX/run.sh (or ./run.sh)
lammpsscript.sh: (e.g. long.in.sh) main launcher via MPIRUN (can be used direclty on TOPAZE)
manual execution on TOPAZE via: $PROJECT/XXXXX/lammpsscript.sh (or ./lammpsscript.sh)
prior a manual execution on TOPAZE, an active lamboot is required and subsequently a
lamhalt (lamboot and lamhalt lines in the scrip are inactivated by default in the script
since they are managed automatically by the SUN GRID ENGINE)
mpi.sh: MPI argument (to used only by lammpsscript.sh)
After/during execution, several files are created:
jobid: jod id to be used with qstat -j jobid
project.log: log file
project.out: standard output (STDOUT)
project.err: standard error (STDERR)
lammps.log: additional log (default)
see also: MSLAB, PIZZA, RMLAMJOB, CLLAMJOB, LAMAN, TREELAM, PSSEARCH
FILE:~/.bashrc
36
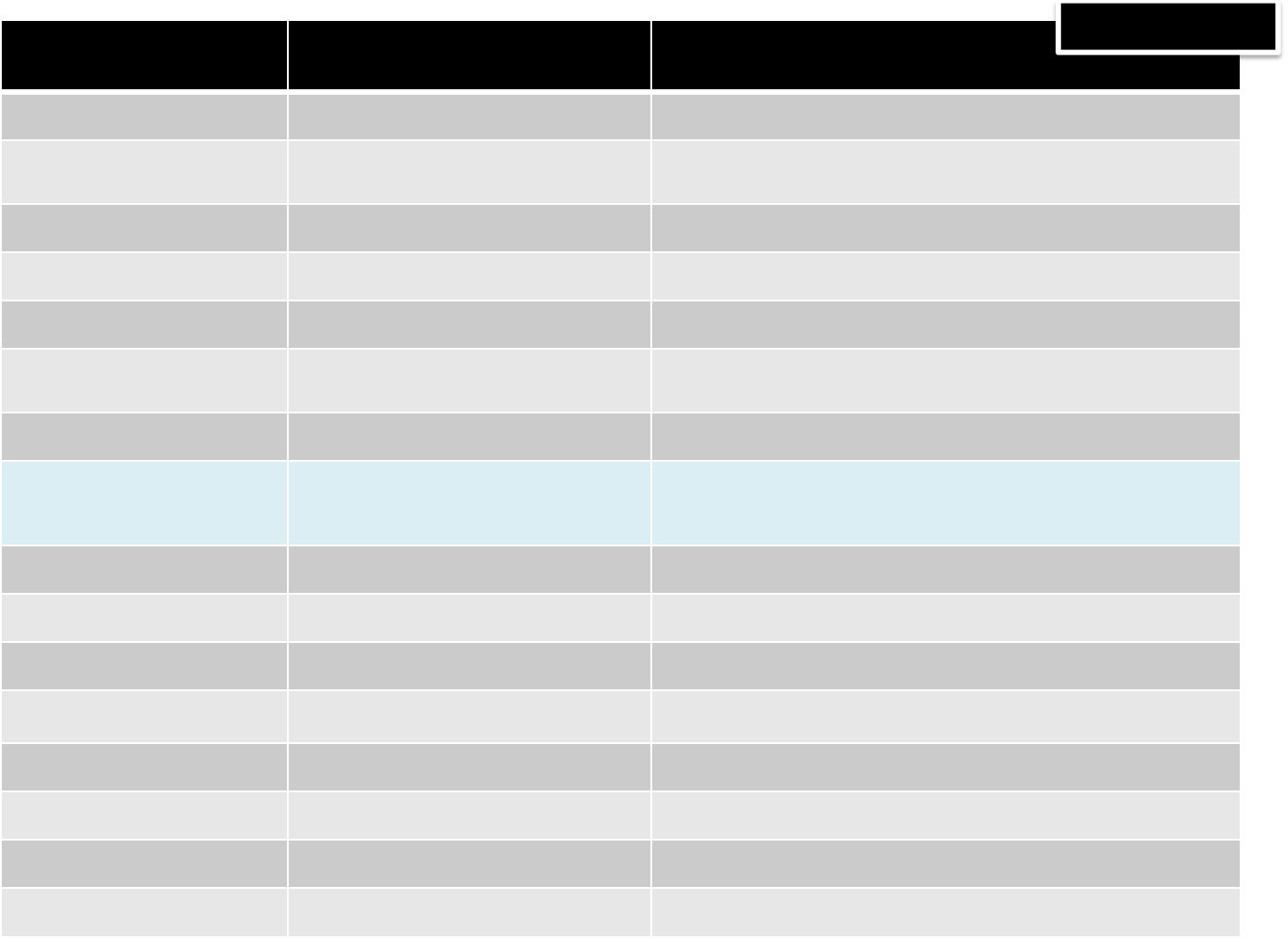
BASHWRAPPERS DESCRIPTION USAGE
cdlam cd intoaLAMMPSdir/path cdlam localpath
cllamjob orcllammpsjob cleanLAMMPSjobsviaMSLAB ccllamjob jobname1[jobname2][jobname3][jobname4]...
exportkey exportRSAkeyonthecluster exportkey firstnode [lastnode]
genkey RSAgenkey (requireforSSH) genkey
killsearch killaprocessonallnodes killsearch processname [username][firs
msi2lmp convertaMSIprojectintoaLammps
data file msi2lmpmsiproject [class][ff][print][ffpath]
laman helponLAMMPSscriptcommand laman [lammps_command]
lamjob,lammpsjob Prepare/queue/runaLAMMPSjobvia
MSLABon thecluster
lamjob mylammpsscript cmd[numproc][jobname][jobpath]
[jobpath][datafile1][datafile2]…
cmd =run,scriptorrunone
lamrc orlammpsrc displayageneral helpvia.bashrc lamrc
lslam ls intoaLAMMPSdir/path lslam localpath
mslab matlab withMS(textmode) mslab,see"helpMS"fordetailedfunct
pizza PYTHONextensionforLAMMPS
(custominstallation) pizza
pssearch searchaprocessonallnodes pssearch processname [username][firstn
rmlamjob orrmlammpsjob remove LAMMPSjobsviaMSLAB rmlamjob jobname1[jobname2][jobname3][jobname4]...
treelam tree allLAMMPSprojects treelam,treelam long,treelam short
ulist alljobsforthecurrent user u
FILE:~/.bashrc
37

MSLAB FUNCTIONS DESCRIPTION
cllammpsjob cleanaorseveralLAMMPSjobs(removetheLAMMPSexecutable)
lamdumpread readLAMMPSdumpfile(allfieldsareidentifiedafterthekeyword'ITEM:'andthefollowing
valuesareassumedtobenumerical)
readlog LAMMPSlogfiles
lammpsjob createandlaunchaLAMMPSjobonthecluster(asdefinedinglobalvariables:PROJECTPATHand
LAMMPS)
lmp2cfg LAMMPSdumpfiletoExtendedCFGFormat(Novelocity)tobeused
msi2lmp convertsaMSIproject (MSIproject.car,MSIproject.mdf)intoaLAMMPSdatafile
readdump_all alltimesteps fromaLAMMPSdumpfile
readdump_one LAMMPSdumpfileonetimestep atatime
readlog LAMMPSlogfiles
readrdf toreadRadialDistributionFuntion outputfromLAMMPS
rmlammpsjob removeaorseveralLAMMPSjob(s)(deletetheentiredirectoriescreatedbyLAMMPSJOB)
scandump toscanLAMMPSdumpfile
MS(Molecular Studio)Toolbox forMatlab include 254functions
Maincontributor:O.Vitrac
LINUXCAPAPLE,WIN32/64COMPATIBLE
PATH:$PROJECT/../codes/MS
38

MSLABcustominstallation
% STARTMS (PATH: $PROJECT/startms.m) – MATLAB SCRIPT
% MINIMUM setup configuration for MS on UNIX machines
% It works also on WINBOXES (Note: MS was designed on WIN32 machines)
%
% This script defines two global variables PROJECTPATH (string) and LAMMPS (structure)
% A new user must update PROJECTPATH and possibly some fields in LAMMPS
%
% PROJECTPATH: defines the main directory for all LAMMPS projects
% When the environment variable $PROJECT exist, its value is assigned to PROJECTPATH (default behavior)
% If not the script definition is used.
%
% LAMMPS: object used to generate all required BASH scripts to submit a job instance
% (with MPIRUN or not, with SUN GRID ENGINE or not)
% In future versions, this variable will be replaced by a single XML file.
% Customizable fields in LAMMPS include:
% LAMMPS.bin: bin/ directory in PROJECTPATH (all required binaries are assumed to be located there)
% NB: Tools are expected to be located in bin/tools (see MSI2LMP)
% Makefiles are located in PROJECTPATH/make
% LAMMPS.lammps: filename of LAMMPS executable (several versions of LAMMPS can be used according to
MPI, compilation options...)
% LAMMPS.maxnumproc: max number of processors, which can be invoked (theoretically 1000, 16 could be an
acceptable value on MIGALE)
% LAMMPS.email: contact e-mail (use to send e-mails during main job events)
%
% The local variable localMS define the relative path from the location of startms, where MS is installed.
% By default, MS is outside PROJECT (safe behavior). Update its content to your need.
FILE:$PROJECT/startms.m
39

MODMOL25‐27Feb 2008,Jouy‐en‐Josas
olivier.vitrac@agroparistech.fr
Thisdocumentisbasedonourowncontribution.
Theresultisfreelydistributablewithoutguaranteeorwarranteeofanykind.
# PIZZA and MSLAB starts within $PROJECT
# example of line to be added/updated in ~/.bashrc
export PROJECT="~/project"

PIZZA.pycustominstallation
#!/usr/local/bin/python –I
# Pizza.py toolkit, www.cs.sandia.gov/~sjplimp/pizza.html
...
...
# modules needed by pizza.py
import sys, commands, os, string, exceptions, glob, refrom time import clock
# Customization by O. Vitrac
boxname = os.name
if boxname.find("nt") >=0:
PIZZAROOT = os.path.normpath("C:\Data\Olivier\INRA\Codes\mslab\pizza")
print "NT system"
else:
PIZZAROOT = os.path.normpath(os.path.join(os.environ.get('PROJECT'),'pizza'))
print "assume a LINUX machine"
os.chdir(PIZZAROOT)
print "current path: %s (default directory for data)" % os.getcwd()
print "available functions in src:\n\n %s\n" % os.listdir(os.path.normpath(os.path.join(PIZZAROOT,'src')))
...
...
# ALL SCRIPTS ARE NOW LAUNCHED FROM PIZZAROOT: $PROJECT/pizza
# Pizza.py toolkit, www.cs.sandia.gov/~sjplimp/pizza.html
# Steve Plimpton, sjplimp@sandia.gov, Sandia National Laboratories...
...
import osPIZZA_TOOLS = [os.path.normpath(os.path.join(os.getcwd(),'src'))]
PIZZA_SCRIPTS = [os.path.join(os.getcwd(),'scripts'),os.path.join(os.getcwd(),'examples')]
PIZZA_EXCLUDE = ["pizza", "DEFAULTS", "vizinfo"]
FILE:$PROJECT/pizza/src/pizza.py
FILE:$PROJECT/pizza/src/DEFAULTS.py
41

CUSTOMPIZZATEST
# simple test of chain tool
# creates tmp.data.chain file (see test_chain.py)
c = chain(500,0.7,1,1,2)
c.seed = 54321
c.build(25,10)
c.mtype = 2
c.btype = 2
c.blen = 1.5
c.dmin = 1.2
c.id = "end1"
c.build(10,25)
c.write("tmp.data.chain")
print "all done ... type CTRL-D to exit Pizza.py"
42
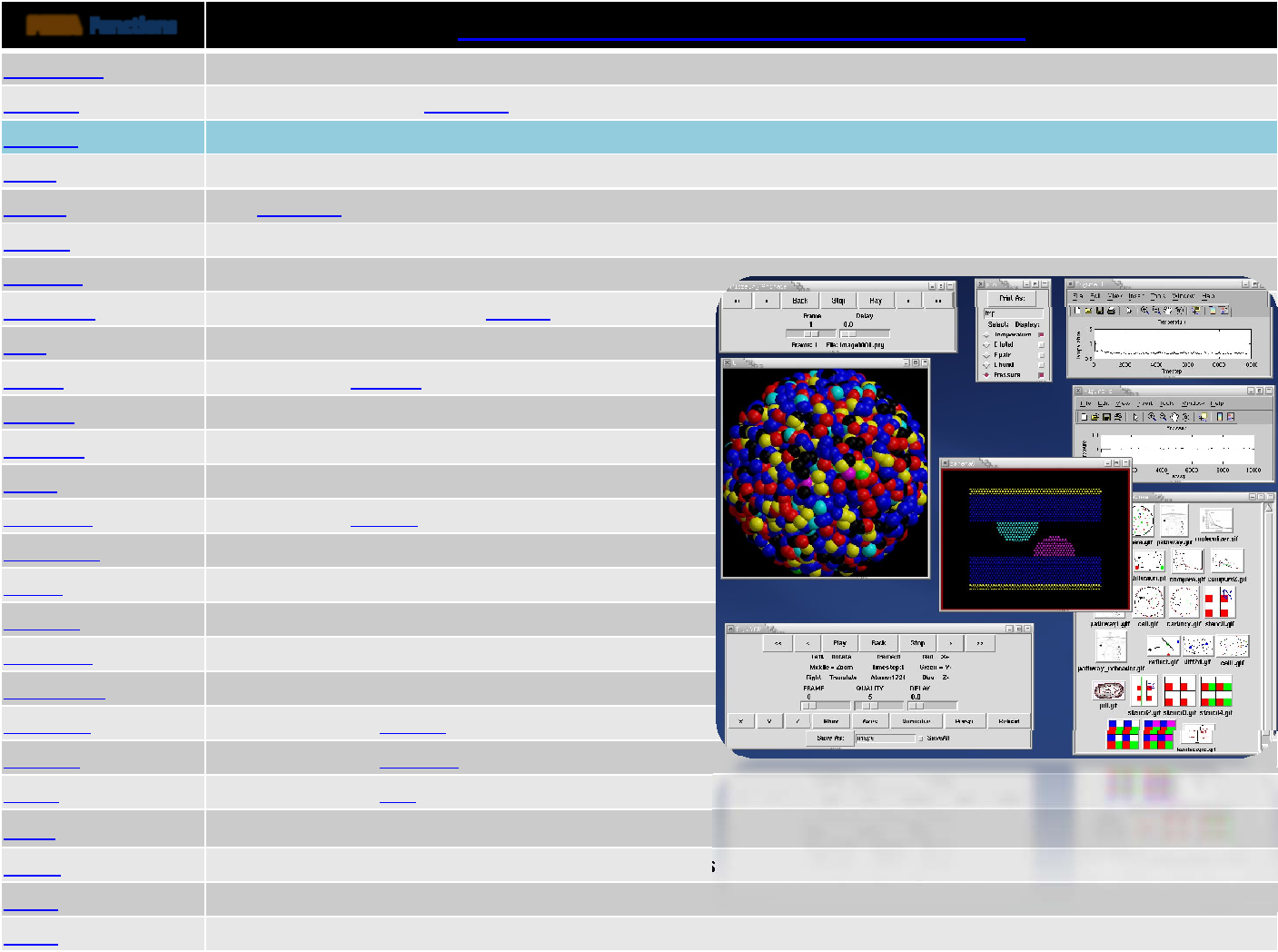
PIZZA Functions http://www.cs.sandia.gov/~sjplimp/pizza.html
animate.py Animateaseriesofimagefiles
cdata.py Read,write,manipulateChemCell datafiles
chain.py Createbead‐springchainsforLAMMPSinput
cfg.py ConvertLAMMPSsnapshotstoCFGformat
clog.py ReadChemCell logfilesandextractspeciesdata
data.py Read,write,manipulateLAMMPSdatafiles
dump.py Read,write,manipulate dumpfilesandparticle attributes
ensight.py ConvertLAMMPSsnapshotstoEnsight format
gl.py 3dinteractivevisualizationviaOpenGL
gnu.py Create plotsviaGnuPlot plotting program
histo.py Particledensityhistogramfromadump
image.py Viewandmanipulateimages
log.py ReadLAMMPSlogfilesandextractthermodynamicdata
matlab.py CreateplotsviaMatLab numericalanalysisprogram
mdump.py Read,write,manipulatemeshdumpfiles
pair.py ComputeLAMMPSpairwiseenergies
patch.py CreatepatchyLennard‐JonesparticlesforLAMMPSinput
pdbfile.py Read,writePDBfilesincombowithLAMMPSsnapshots
plotview.py Plotmultiplevectorsfromadataset
rasmol.py 3dvisualizationviaRasMol program
raster.py 3dvisualization viaRaster3d program
svg.py 3dvisualizationviaSVG files
vcr.py VCR‐styleGUIfor3dinteractiveOpenGLvisualization
vec.py Createnumericvectorsfromcolumnsinfileorlistofvecs
vtk.py ConvertLAMMPSsnapshotstoVTKformat
xyz.py Convert LAMMPSsnapshots toXYZformat43

PIZZAwith GUI
#ADDtheexportlinetoyour bash ortypeit:
$exportPYTHONPATH="/usr/local/src/public/PyOpenGL‐
3.0.0a6/:/usr/local/src/public/modules‐python/Imaging‐
1.1.6/PIL/:/usr/local/public/python‐2.4.3/lib/python2.4/site‐
packages/Numeric/:/usr/local/genome/mgcat1.24‐linux32‐
py23/:/usr/local/src/public/ctypes‐1.0.2/build/lib.linux‐x86_64‐
2.3/:/usr/local/public/python‐2.4.3/lib/python2.4/site‐packages/setuptools‐0.6c7‐
py2.4.egg/:/usr/local/src/public/Togl‐1.7/"
$\pizza#\isusedtooverride thecurrent alias
ssh –Yusername@topaze.jouy.inra.fr
Check:echo $DISPLAY 44

MODMOL25‐27Feb 2008,Jouy‐en‐Josas
olivier.vitrac@agroparistech.fr
Thisdocumentgathersseveralfreelyavailablesources.
Theresultisfreelydistributablewithoutguaranteeorwarranteeofanykind.

BEAD‐SPRINGPOLYMERMELT
FENE:FiniteExtendibleNonlinearElasticModel(here:2880beads,2715bonds)
$PROJECT/bin/tools/chain <$PROJECT/examples/example.def.chain
>$PROJECT/chain/data.chain
Polymer chain definition
0.8442 rhostar
592984 random # seed (8 digits or less)
2 # of sets of chains (blank line + 6 values for each set)
0 molecule tag rule: 0 = by mol, 1 = from 1 end, 2 = from 2
ends
160 number of chains
16 monomers/chain
1 type of monomers (for output into LAMMPS file)
1 type of bonds (for output into LAMMPS file)
0.97 distance between monomers (in reduced units)
1.02 no distance less than this from site i-1 to i+1 (reduced
unit)
5 number of chains
64 monomers/chain
2 type of monomers (for output into LAMMPS file)
2 type of bonds (for output into LAMMPS file)
1.05 distance between monomers (in reduced units)
1.12 no distance less than this from site i-1 to i+1 (reduced
unit) FILE:$PROJECT/examples/example.def.chain
46

BEAD‐SPRINGPOLYMERMELT
FENE:FiniteExtendibleNonlinearElasticModel(here:2880beads,2715bonds)
# PIZZA-PYTHON SCRIPT using the method CHAIN
# Such script is equivalent to
# $PROJECT/bin/tools/chain <$PROJECT/examples/example.def.chain
# >$PROJECT/chain/data.chain
N = 2880 # total number of monomers
rhostar = 0.8442 # density
#c = chain(N,rhostar) #setup box with N monomers at reduced density rho
c = chain(N,rhostar,1,1,1) #x,y,z = aspect ratio of box (def = 1,1,1)
c.seed = 592984 #set random # seed (def = 12345)
c.mtype = 1 #set type of monomers (def = 1)
c.btype = 1 #set type of bonds (def = 1)
c.blen = 0.97 #set length of bonds (def = 0.97)
c.dmin = 1.02 #set min dist from i-1 to i+1 site (def = 1.02)
c.build(160,16) #create 160 chains, each of length 16
c.mtype = 2 #set type of monomers (def = 1)
c.btype = 2 #set type of bonds (def = 1)
c.blen = 1.05 #set length of bonds (def = 0.97)
c.dmin = 1.12 #set min dist from i-1 to i+1 site (def = 1.02)
c.build(5,64) #create 5 chains, each of length 64
c.write("data.chain") #write out all built chains to LAMMPS data file
FILE:$PROJECT/pizza/examples/example_chain_data.py
pizza
47
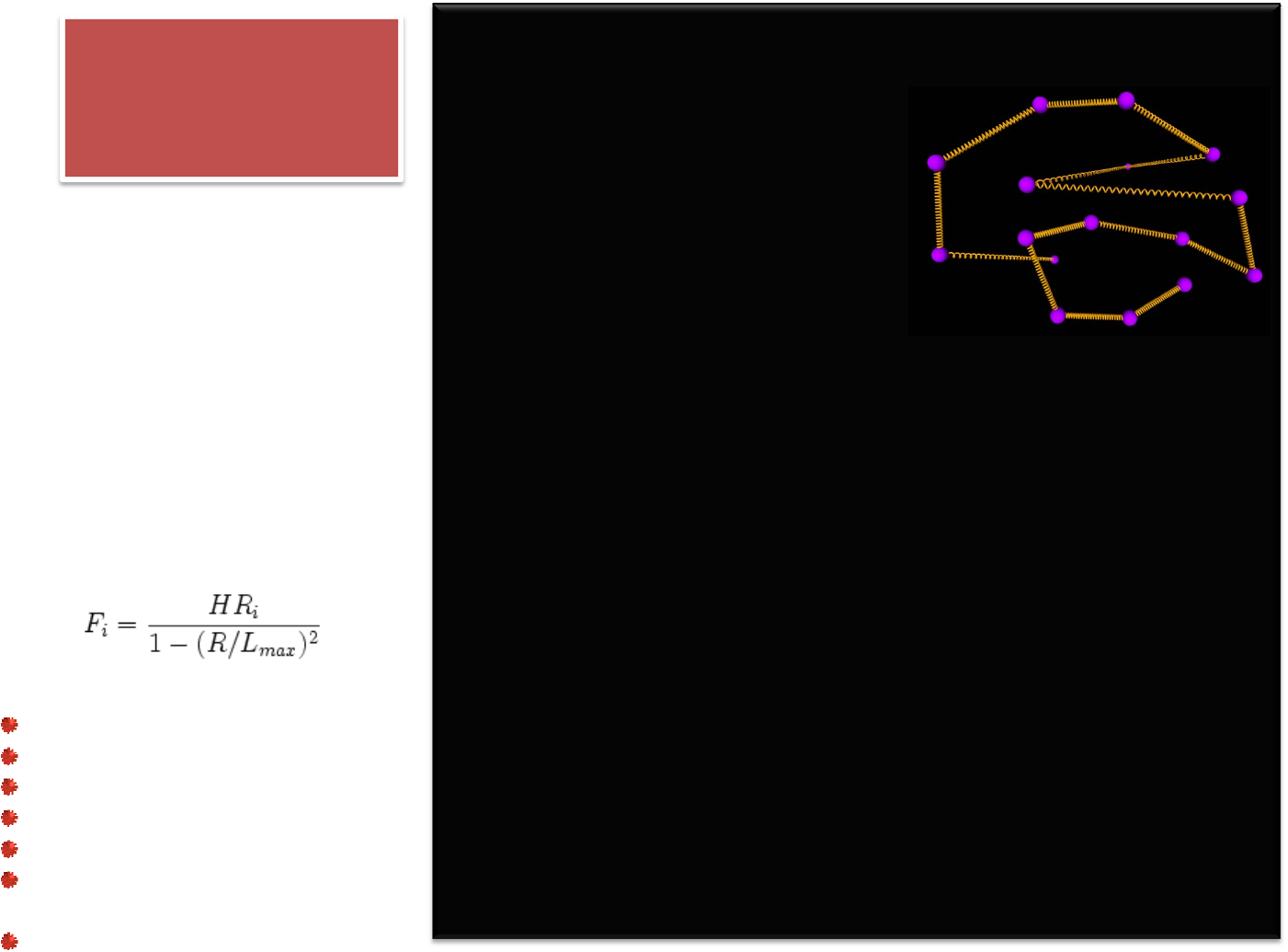
BEAD‐SPRING
POLYMERMELT
# SCRIPT derived from FENE beadspring benchmark
units lj
atom_style bond
special_bonds 0.0 1.0 1.0
read_data data.chain
neighbor 0.4 bin
neigh_modify every 1 delay 1
bond_style fene
bond_coeff 1 30.0 1.5 1.0 1.0
bond_coeff 2 30.0 1.5 1.0 1.0
pair_style lj/cut 1.20
pair_modify shift yes
pair_coeff 1 1 1.0 1.0 1.12
pair_coeff 2 2 1.5 1.1 1.20
fix 1 all nve
fix 2 all langevin 1.0 1.0 10.0 904297
thermo 100
timestep 0.012
run 10000
160+5‐merchainsandFENEbonds:
2,880monomersfor104timesteps
reduceddensity0.8442(liquid)
forcecutoffof2^(1/6)sigma
neighborskin=0.4sigma
neighbors/atom=5(withinforce
cutoff)
NVEtimeintegration
FENE standsforthefinitely
extensiblenonlinearelastic
modelofalong‐chained
polymer.Itsimplifiesthechain
ofmonomersbyconnectinga
sequenceofbeadswith
nonlinearsprings.Thespring
forcelawisgovernedby
inverseLangevin functionor
approximatedbytheWarner's
relationship:
48

# DEMO: bidisperse (advanced ex.)
# 26/02/08
echo $PROJECT # current queue
lslam examples # ls available templates
# PROJECT 1) BOX CREATION AND MINIMIZATION
#==========================================
lamjob examples/in.bidisperse.relax script 1test.birelax []
def.chain.bidisperse in.bidisperse.nvt # copy templates, generate scripts
cdlam test.birelax # cd into the new project
$PROJECT/bin/tools/chain <def.chain.bidisperse >data.chain.bidisperse
# generate chains on a lattice
nano in.bidisperse.relax # edit the relaxation script (no change required)
./run.sh # launch/submit the job on a single proc
u# check your job
# STEP 2) NVT DYNAMICS
#=====================
echo $QUEUE # current queue
export QUEUE="stage.q“ # change queue (empty assign the default “long.q”)
lamjob test.birelax/in.bidisperse.nvt run 4test.binvt []
bidisperse.relax.restart.10000 # generate/submit the dynamics on 4 procs
u# check your job 49
FILE:$PROJECT/examples/bidisperse.demo

ALLATOMSIMULATION
EXAMPLE1:CVFF(nowarnings),shrink boundary conditions
units real
atom_style full
boundary s s s
pair_style lj/cut 10.0
bond_style harmonic
angle_style harmonic
dihedral_style harmonic
improper_style none
read_data decane.lammps05
neighbor 2.0 bin
neigh_modify delay 5
timestep 2.0
thermo_style multi
thermo 50
msi2lmp $PROJECT/examples/decane I CVFF
lamjob examples/in.decane script 1 decane [] decane.lammps05
decane
fix 1 all nvt
298.0 298.0 100.0
dump 1 all atom
100 dump.decane
minimize 1.0e-4 100
1000
run 10000
FILE:$PROJECT/examples/in.decane 50
ALLATOMSIMULATION
EXAMPLE2:CVFF(warnings),shrink boundary conditions
msi2lmp $PROJECT/examples/Epicatechin I cvff
lamjob examples/in.Epicatechin script 1 Epicatechin []
Epicatechin.lammps05
Epicatechin
units real
atom_style full
boundary s s s
pair_style lj/cut 10.0
bond_style harmonic
angle_style harmonic
dihedral_style harmonic
improper_style cvff
read_data
Epicatechin.lammps05
neighbor 2.0 bin
neigh_modify delay 5
timestep 2.0
thermo_style multi
thermo 50
fix 1 all nvt
298.0 298.0 100.0
dump 1 all atom
100 dump.decane
minimize 1.0e-4 100
1000
run 10000
FILE:$PROJECT/examples/in.Epicatechin
51

ALLATOMSIMULATION
EXAMPLE3:CFF(warnings),periodic boundary conditions,NPT
msi2lmp $PROJECT/examples/BHT/ethanol_BHTx1 II cff91
lamjob examples/BHT/in.ethanol_BHTx1 script 1 BHT []
ethanol_BHTx1.lammps05
BHTin200
molecules
ofethanol
units real
atom_style full
boundary p p p
pair_style lj/cut/coul/cut 10.0
8.0
bond_style class2
angle_style class2
dihedral_style class2
improper_style class2
read_data
ethanol_BHTx1.lammps05
neighbor 2.0 bin
neigh_modify delay 5
timestep 1.0
thermo_style multi
thermo 50
fix 1 all npt
298.0 298.0 100.0 xyz 1.0
1.0 1.0
dump 1 all atom
1000 dump.ethanol_BHTx1
run 10000
FILE:$PROJECT/examples/BHT/in.ethanol_BHTx1
52

MODMOL25‐27Feb 2008,Jouy‐en‐Josas
olivier.vitrac@agroparistech.fr
Thisdocumentgathersseveralfreelyavailablesources.
Theresultisfreelydistributablewithoutguaranteeorwarranteeofanykind.
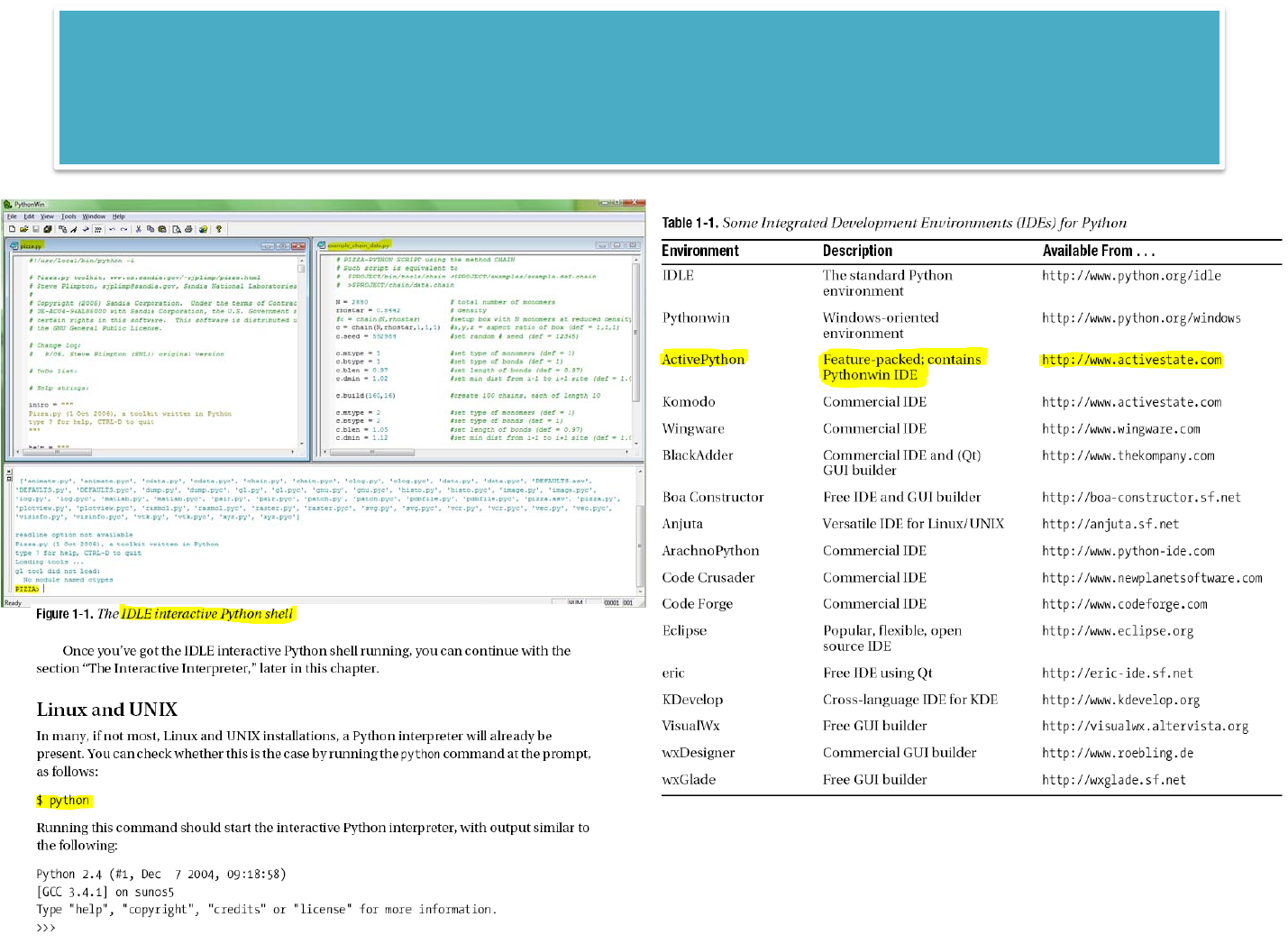
LEARNINGPYTHON
54

LEARNINGPYTHON
55
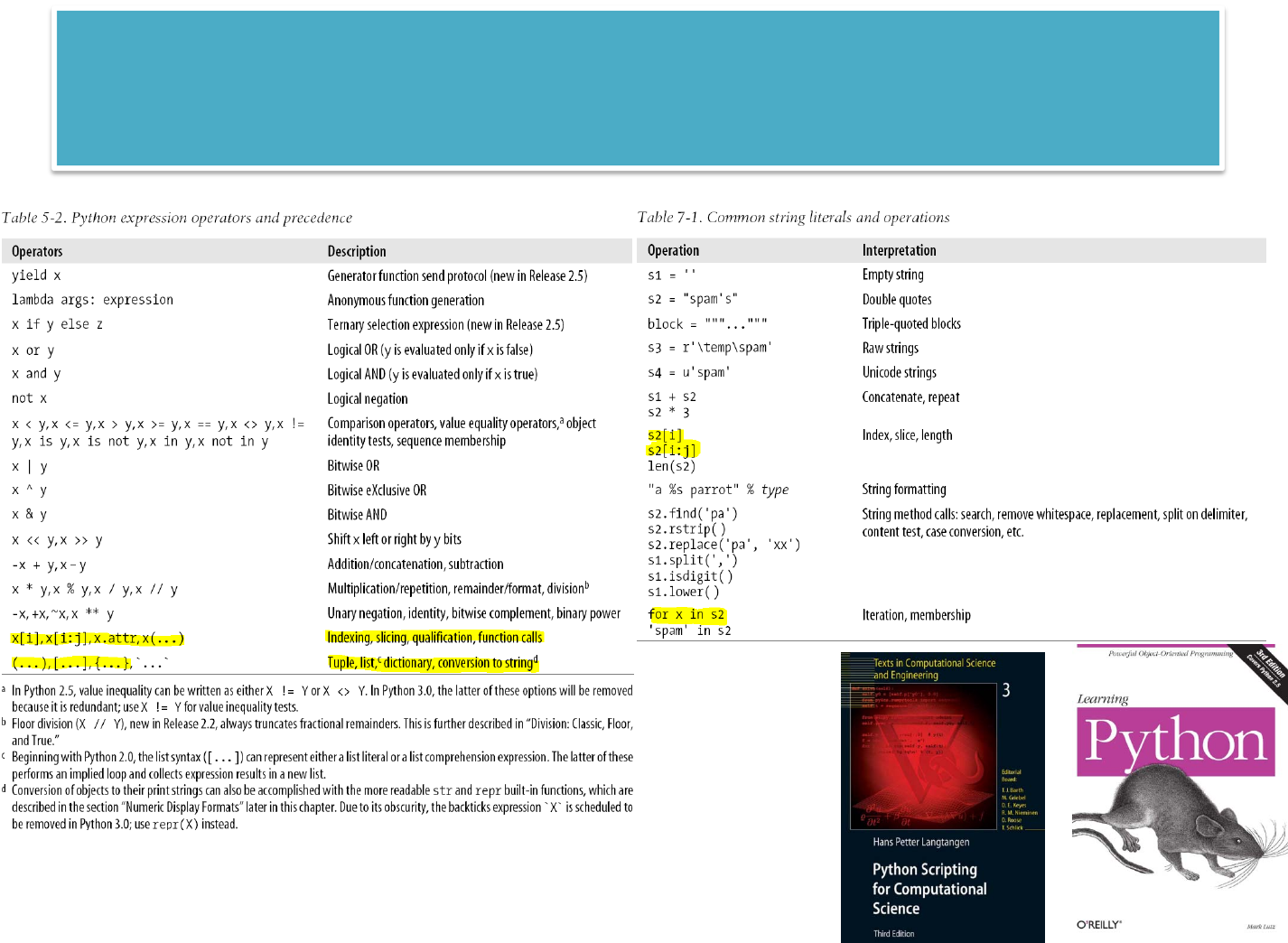
LEARNINGPYTHON
56
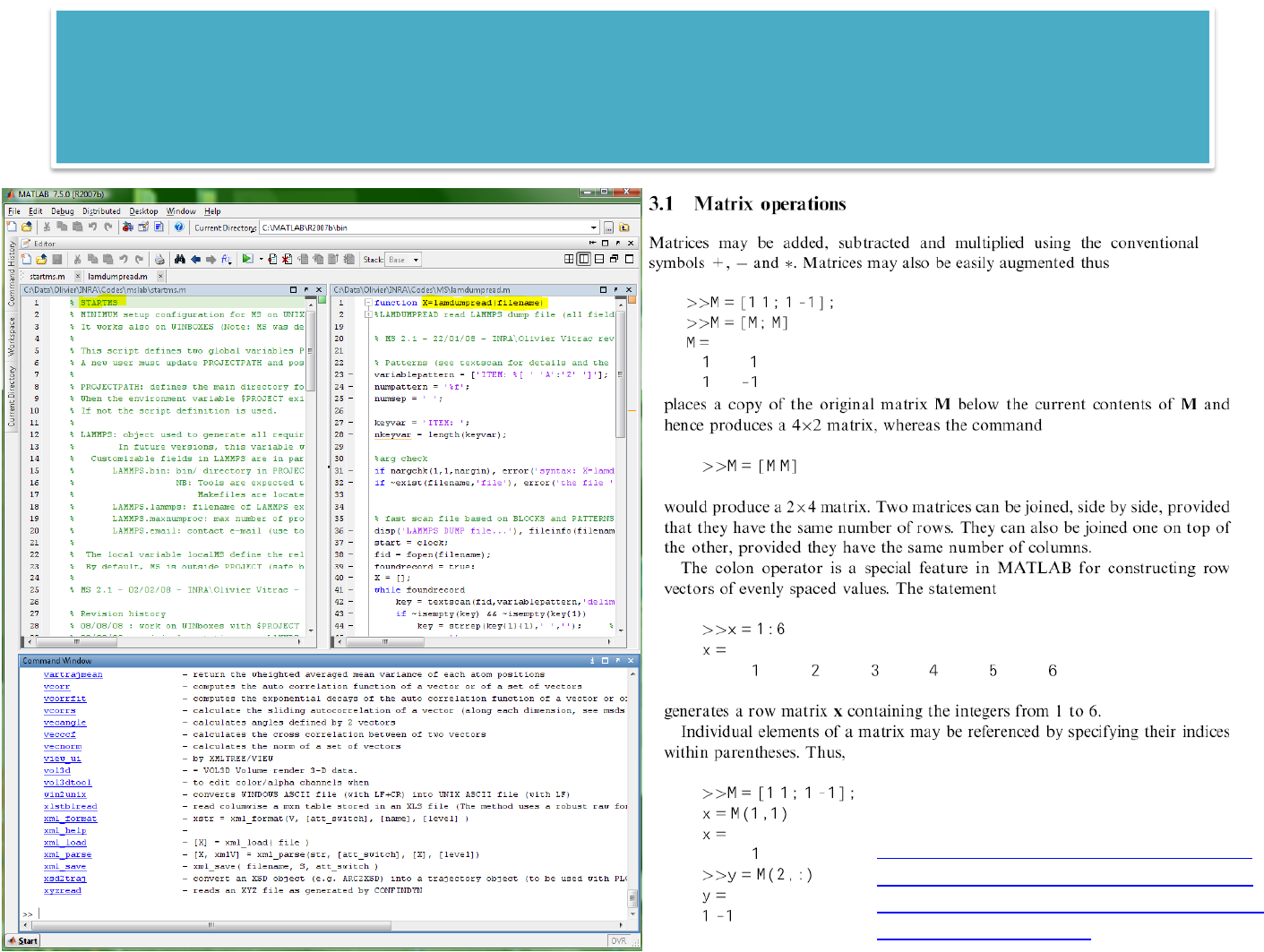

58
59
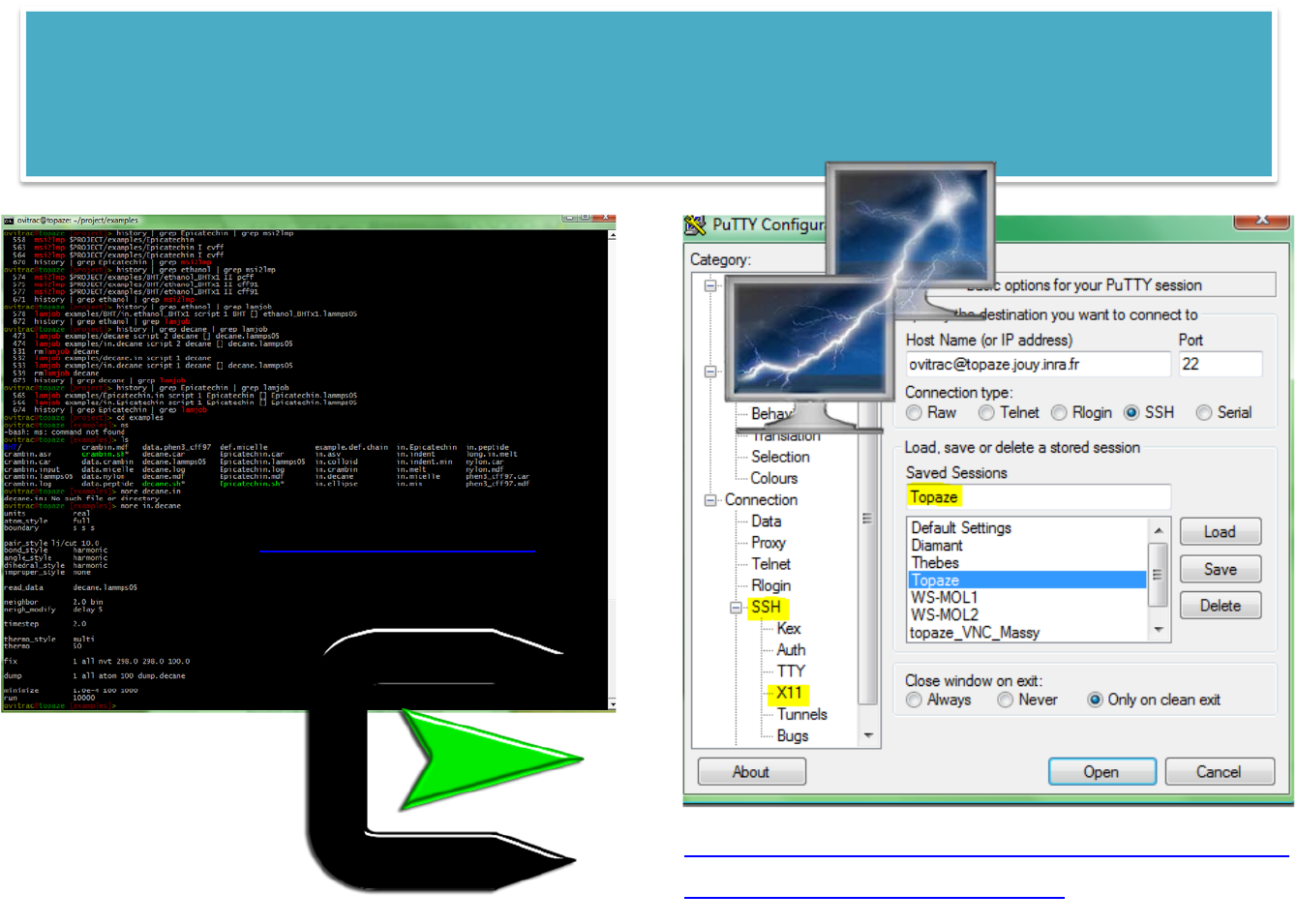
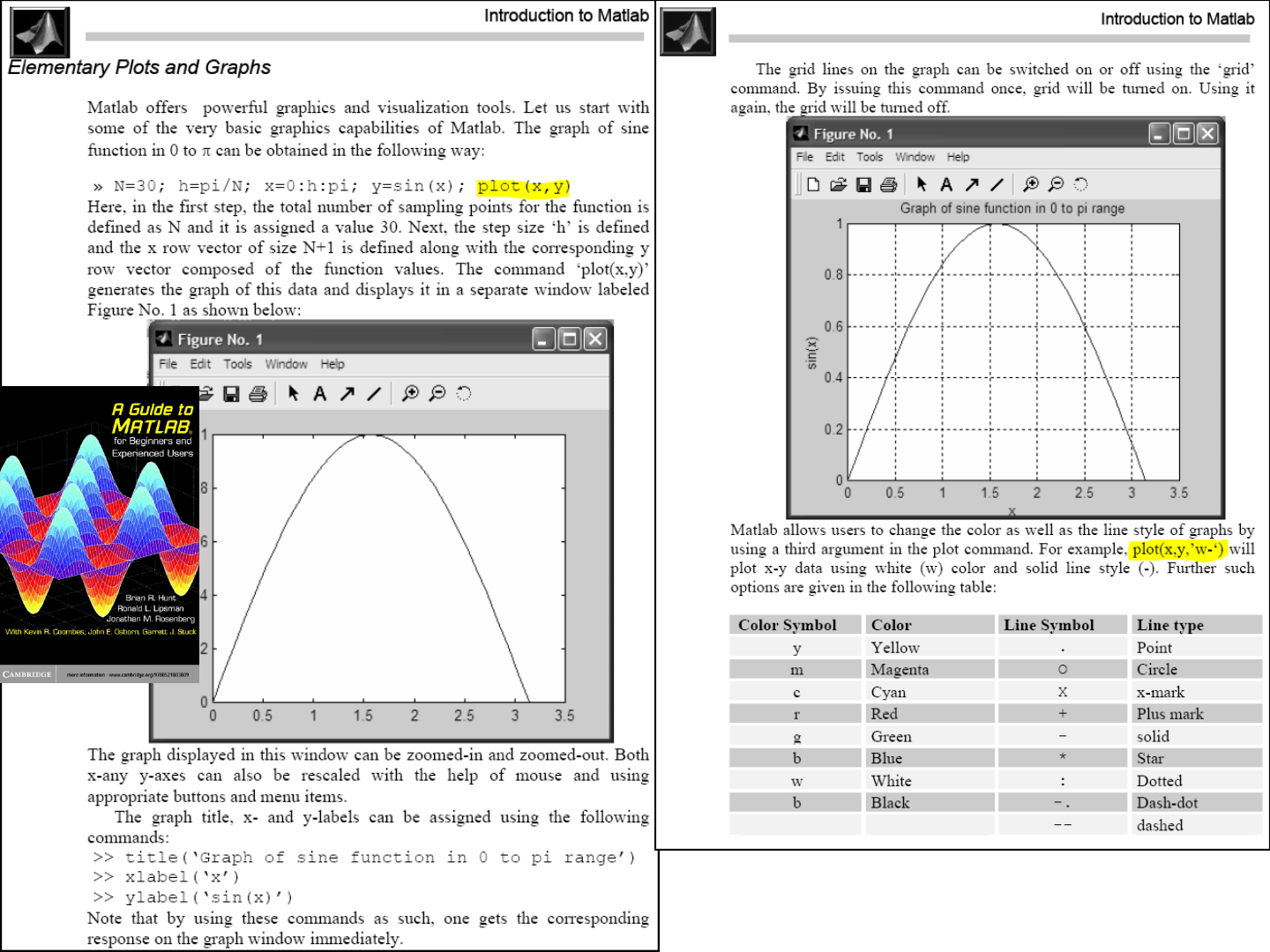
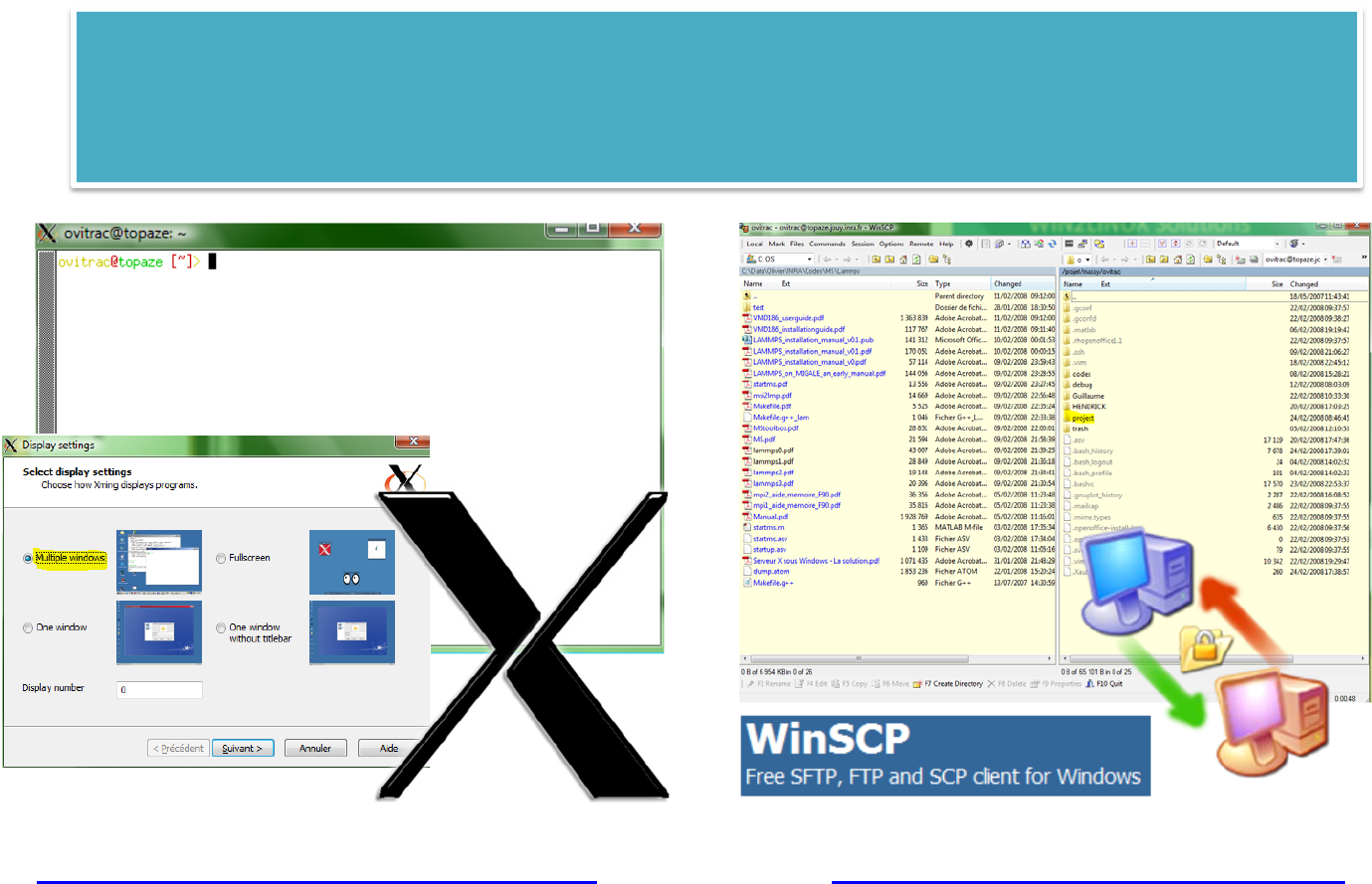
WIN2LINUXSolutions
#Cygwin withSSHD:requireszlib,tcpwrappers,openssh
#Install
mkpasswd ‐l>/etc/passwd
mkgroup ‐l>/etc/group
exit
#Relogin
which sshd
ssh‐host‐config‐y
cygrunsrv ‐Ssshd
sc descriptionsshd
#create publickey
ssh‐keygen ‐tdsa
#send key
cat.ssh/*.pub|ssh ovitrac@topaze.jouy.inra.frtee‐a.ssh/authorized_keys
#autologin
ssh ovitrac@topaze.jouy.inra.fr
http://www.chiark.greenend.org.uk/~sgtat
ham/putty/download.html
(free)
http://cygwin.com/
(free)
61

MODMOL25‐27Feb 2008,Jouy‐en‐Josas
olivier.vitrac@agroparistech.fr
Thisdocumentgathersseveralfreelyavailablesources.
Theresultisfreelydistributablewithoutguaranteeorwarranteeofanykind.

$PROJECT\make\lammps‐
31Jan08\src\MAKE\Makefile.g++_lam_all_100208
•#g++=RedHat Linuxbox,g++,LAM,FFTW
•#INRA\OlivierVitrac13/02/08(forLAMMPS10/02/08)
•#
•#Compilationis ok
•#>>TESTEXAMPLE:
•#>>cd$PROJECT/../testlam;./lmp_g++_lam<in.lj
•#
•#>>BUGREPORT
•#TheMPIRUNhangs.
•#cd$PROJECT/../testlam;lamboot;mpirun ‐np 1
lmp_g++_lam <in.lj
•
•
•SHELL=/bin/sh
•#.IGNORE:
•
•#System‐specific settings
•CC=g++
•CCFLAGS=‐g‐O‐I/usr/local/public/lam/include \
•‐I/usr/include ‐DFFT_FFTW‐DLAMMPS_GZIP‐
DMPICH_IGNORE_CXX_SEEK
•DEPFLAGS=‐M
•LINK=g++
•LINKFLAGS=‐g‐O‐L/usr/local/public/lam/lib\
•‐L/usr/lib64
•USRLIB=‐lfftw ‐llammpio ‐llammpi++‐llamf77mpi‐lmpi ‐
llam
•SYSLIB=‐lpthread ‐ldl
•ARCHIVE=ar
•ARFLAGS=‐rc
•SIZE=size
•
•#Linktarget
•$(EXE):$(OBJ)
•$(LINK)$(LINKFLAGS)$(OBJ)$(USRLIB)$(SYSLIB)‐o$(EXE)
•$(SIZE)$(EXE)
•
•#Librarytarget
•lib:$(OBJ)
•$(ARCHIVE)$(ARFLAGS)$(EXE)$(OBJ)
•
•#Compilationrules
•%.o:%.cpp
•$(CC)$(CCFLAGS)‐c$<
•%.d:%.cpp
•$(CC)$(CCFLAGS)$(DEPFLAGS)$<>$@
•
•#Individual dependencies
•DEPENDS=$(OBJ:.o=.d)
•include $(DEPENDS)
65

66
