Manual
Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 20

GMXPBSA 2.1: a GROMACS tool to perform MM/PBSA and computational alanine
scanning
C. Paissonia, D. Spiliotopoulosa,b, G. Muscoa, A. Spitaleria,c,*
a Biomolecular NMR Unit, S. Raffaele Scientific Institute , via Olgettina 58, Milan 20132, Italy
b Present address: Computational Structural Biology Biochemisches Institut Universität Zürich, Winterthurerstrasse
190, CH- 8057 Zürich, Switzerland.
c Drug Discovery and Development, Istituto Italiano di Tecnologia, Via Morego 30, Genoa 16163, Italy.
* Corresponding author at: Drug Discovery and Development, Istituto Italiano di Tecnologia, Via Morego, 30, Genoa
16163, Italy. E-mail address: andrea.spitaleri@iit.it
1. Introduction
MM/PBSA is a versatile method to calculate the binding free energies of a protein–ligand complex
[1]. It incorporates the effects of thermal averaging with a force field/continuum solvent model to
post-process a series of representative snapshots from MD trajectories. MM/PBSA has been
successfully applied to compute the binding free energy of numerous protein–ligand interactions [2-
5]. The method expresses the free energy of binding as the difference between the free energy of the
complex and the free energy of the receptor plus the ligand (end-state method). This difference is
averaged over a number of trajectory snapshots [6]. Of note, the MM/PBSA approach allows for a
rapid estimation of the variation in the free energy of binding, with the caveat that generally it does
not reproduce the absolute binding free energy values. Nevertheless, it usually exhibits good
correlations with experiments, thus representing a fair compromise between efficiency and efficacy
for the calculation and comparison of binding free energy variations. The theory underlying
MM/PBSA approach has been described previously [6]. Briefly, the binding free energy of a protein
molecule to a ligand molecule in solution is defined as:
ΔGbinding = Gcomplex – (Gprotein + Gligand) (1)
A MD simulation is performed to generate a thermodynamically weighted ensemble of structures.
The free energy term is calculated as an average over the considered structures:
<G> = <EMM> + <Gsolv> – T<SMM> (2)
The energetic term EMM is defined as:
EMM = Eint + Ecoul + ELJ (3)
where Eint indicates bond, angle, and torsional angle energies, and Ecoul and ELJ denote the
intramolecular electrostatic and Lennard-Jones energies, respectively.
The solvation term Gsolv in Eq. 4 is split into polar Gpolar and nonpolar contributions, Gnonpolar:
Gsolv = Gpolar + Gnonpolar (4)

GMXPBSA 2.1 calculates Gpolar and Gnonpolar with Adaptive Poisson-Boltzmann Solver (APBS)
program [7].
The polar contribution Gpolar refers to the energy required to transfer the solute from a continuum
medium with a low dielectric constant (ε=1) to a continuum medium with the dielectric constant of
water (ε=80). Gpolar is calculated using the non linearized or linearized Poisson Boltzmann equation.
The nonpolar contribution Gnonpolar is considered proportional to the solvent accessible surface area
(SASA):
Gnonpolar = γ SASA + β (5)
where γ = 0.0227 kJ mol–1 Å–2 and β = 0 kJ mol–1 [8]. The dielectric boundary is defined using a
probe of radius 1.4 Å.
Herein, we present an updated and revised version of the tool, GMXPBSA 2.1 (Fig. 1). We have
introduced in GMXPBSA 2.1 the following improvements with respect to the previous version [11]:
control of the input and output options;
automatic setup and a posteriori CAS calculations;
CAS calculations on a single residues or on a set of residues simultaneously;
handling of multiple protein-ligands MD simulations to allow comparisons between
different ligands;
handling of multiple protein-ligands MD simulations to allow comparisons (e.g. between
wild-type complex and non-alanine mutants);
handling of APBS calculations on a multi core system (distributed calculations in cluster).
possibility to use custom van der Waals radii;
check and restart of the failed MM/PBSA calculations;
statistical analysis of the results.
2. Program usage
2.1 GMXPBSA 2.1 calculation workflow
GMXPBSA 2.1 is a user-friendly suite of Bash/Perl scripts that efficiently streamlines the set up
procedure and the calculation of binding free energies for an ensemble of complex structures
generated by GROMACS MD engine. The program workflow, (Figures 1 and 2) consists of three
different sequential steps comprising:
1. gmxpbsa0.sh:
In this step, the tool exploits the gmxpbsa0.sh script to setup the system and to perform preliminary
calculations including:

check of the required input files and directories;
extraction of the frames of the complex from the MD simulations, subsequently split in the
protein and the ligand components by the GROMACS tools;
calculation of the Coulomb energy contributions using either GROMACS tools or the
“coulomb” program available in the APBS suite, and Lennard-Jones term using
GROMACS.
If the computational alanine scanning (CAS) calculation is required, the script performs alanine
mutations on the defined residues on every single extracted frames removing the side chains atoms
of the target residues up to the beta C atom (CB atom) and then recalculating the Coulomb and the
Lennard-Jones energy contributions of the structure containing the alanine mutant. It also generates
the grid and the input to perform the APBS calculations for each frame of the simulation. The latter
task is critical, since deletion of artefacts in the MM/PBSA calculation requires an exact matching
of the grid setup between all the system components (complex, protein and ligand).
2. gmxpbsa1.sh:
In this step, the gmxpbsa1.sh script computes the solvation polar and nonpolar energy contributions
using APBS program. These calculations can be distributed on a cluster or on a multi core
workstation.
3. gmxpbsa2.sh:
In this last step, the gmxpbsa2.sh script combines for all the frames the single terms, <EMM> and
<Gsolv> respectively, in order to calculate the final binding free energy value. It also checks and tries
to fix errors and/or failures occurring in the preceding step 2 (APBS calculations). Statistical
analysis is also performed computing average and standard error (SE). The SE is calculated as
follows: SE = σ/√N, where σ is the standard deviation and N is the number of structures (MD
frames) used in the calculation. The average Coulomb and Lennard-Jones values, the polar and
nonpolar solvation terms are calculated along each trajectory. If a value differs from the average
more than two standard deviations it is considered as an outlier and the corresponding frame is
excluded from the final calculation. However, it is always possible to check for outlier frames, since
their reference-numbers are stored in the WARNING.dat file.
2.2 Installation and execution of the program
Once the source code of the program GMXPBSAtool.tar.gz has been downloaded the user should
perform the following steps:
1. extract the source code in a user defined location, e.g.. /home/myprogram/, by typing tar

zxvf GMXPBSAtool.tar.gz; set the GMXPBSAHOME environment variable in bash: export
GMXPBSAHOME=/home/myprogram/GMXPBSAtool; change the /home/myprogram to
whatever directory is appropriate for your machine; verify write permissions in the directory
tree, and execute permissions for the gmxpbsa0.sh, gmxpbsa1.sh and gmxpbsa2.sh scripts.
$GMXPBSAHOME should be also added to the PATH.
2. In order to perform MM/PBSA calculations, the user has to run the tool by typing
$GMXPBSAHOME/<script>, where <script> can be either gmxpbsa0.sh, or gmxpbsa1.sh
or gmxpbsa2.sh , according to the calculation step (see section 2.4). Each script will read the
INPUT.dat file to perform the MM/PBSA calculation. For instance, if the INPUT.dat file and
the directory containing the simulations are located in /home/mysimulations, the user will
type $GMXPBSAHOME/<script> in the aforementioned directory. See section 3 for further
details.
In order to test the correctness of GMXPBSA 2.1 installation, the tool is distributed with the
examples (with shortened trajectories) presented in Section 4 to test the correctness of the
installation.
2.3 Input files preparation
In order to perform binding free energy calculations on a ligand-receptor system (where the ligand
can be either a protein, a peptide or a small molecule), the user needs first to perform a MD
simulation using GROMACS engine 4.5 or later versions. Before starting any GMXPBSA 2.1
calculations, the user should verify the convergence of MD simulations, as lack of convergence
might strongly compromise the reliability of the MM/PBSA results, as pointed out in [11]. Along
with simulations data, the user should edit the INPUT.dat file, defining all the options on the
binding free energy calculations (see section 2.4).
For each system under investigation MM/PBSA calculations require the following input files:
1. the trajectory file describing the dynamic of the complex (name_xtc in the INPUT.dat). We
encourage the user to strip off the water from the trajectory to speed up calculations. The
possible artefacts deriving from periodic boundary condition (pbc) should be removed from
the trajectory, using the trjconv GROMACS tool (-pbc whole or -pbc nojump or -pbc res is
usually sufficient). The latter step is fundamental before carrying out the MM/PBSA
calculations in order to remove the presence of possible broken molecules. The processed
trajectory can be checked using a molecular visualizer before performing GMXPBSA
calculations.
2. the portable binary run input file (name_tpr in the INPUT.dat). This file contains the
information on mass, charges and force field parameters used in the MD simulations.

3. the index file, with mandatory name index.ndx. This file contains the groups used in the
simulations. Three groups are compulsory in order to run GMXPBSA 2.1: the complex,
containing the atoms index of the complex (union of the receptor and ligand atoms), the
receptor, containing the atoms index of the receptor, and the ligand, containing the atoms
index of the ligand. The three group index names can be chosen by the user.
The three files are placed in a directory, whose name will be referred to as root in the INPUT.dat
file. Additional files should be present in the root directory in case the MD simulation has been
carried out using either a custom GROMACS force field (e.g. including modified amino acid) or
custom topologies (i.e. ligand). See section 2.4.2 for further details (keyword use_nonstd_ff and
use_topology, respectively). When handling different trajectories, the user should create different
root-directories, one for each simulation, and define the name of these directories in the
root_multitrj variable contained in the INPUT.dat file. The tool will then automatically perform the
MM/PBSA calculations on all the systems defined in the root_multitrj directories. In order to cancel
out artefacts for each system (i.e. for each directory) an identical grid setup in the PBSA calculation
will be defined.
2.4 INPUT.dat file
The INPUT.dat is the macro file in ASCII format, through which the user can define several options
to perform binding free energy calculations. It contains 7 different sections, in which the user has to
define the mandatory keywords with the options described in the following chapters and
summarized in Table 1.
2.4.1. GENERAL
In this section the user defines the molecular system and the environment path as follows.
root: the name of the directory that contains the input files necessary to calculate the binding
free energy (trajectory, index, tpr and custom force field or topology files).
multitrj: if set to y more than one system will be analysed. If set to n only the directory
named root will be considered.
root_multitrj: this variable is considered only if multitrj is set to y. It is the list containing the
names of the directories that will be analysed (e.g if the user aims to analyse the directories
Name1, Name2 and Name3, the command should be set to: root_multitrj Name1 Name2
Name3).
run: can be either a string or an integer. For example, if the chosen option is “1” the program
will create the RUN1_root directory. In this directory GMXPBSA 2.1 will carry out all the
calculations and store the corresponding output and an input-reminders. This might be

useful when different runs with different parameters for either Molecular Mechanics (MM)
or solvation terms (PBSA) analysis are performed.
RecoverJobs: can be set to n or y. If it is set to y, during the third calculation step
GMXPBSA 2.1 will try to recover APBS failed jobs and will try to re-run them; if it is set to
n failed jobs are neglected in the final statistical analysis.
backup: can be set to n or y. If it is set to y (default), GMXPBSA 2.1 will copy the
RUN1_root in backup_RUN1_root before analysis and merging of l the energetic terms
(MM and PBSA) during the gmxpbsa2.sh step. This is useful when: i) problems arise in the
final analysis, ii) the user wants to repeat the analysis.
Cpath: the full path of the APBS “coulomb” tool. If the variable coulomb is set to coul and
no path is defined, GMXPBSA 2.1 will try to locate the binary program only if it is present
in the user's path environment.
Apath: the full path of the apbs program from the APBS suite. The user can skip this option
in case the apbs program, which is the executive PBSA solver of APBS suite, is present in
the path environment.
Gpath: the full path of the GROMACS binary tools. The user can skip this option if the
GROMACS binary tools path is present in the path environment.
2.4.2 FORCE FIELD
To calculate the Coulomb and Lennard-Jones energy contributions GMXPBSA 2.1 performs a short
energy minimization on each frame extracted from the MD simulation, that requires a GROMACS
force field. GMXPBSA 2.1 provides three options, ffield, use_nonstd_ff and use_topology.
ffield: it can be an integer representing the force field used in MD simulations (the list of
the force field can be visualized typing in the bash shell the GROMACS tool “pdb2gmx”).
The user should set the identical force field used in the MD simulations (Figure 3A). It is
possible to perform the CAS calculations. See example 1, hMdmd2-p53 complex (section
4.1).
use_nonstd_ff: it can be either n or y . If n, GMXPBSA 2.1 will use standard force field
parameters as reported in GROMACS, defined in the previous ffield keyword. If y, the
complex can be described by a modified force field in GROMACS, the user should add in
the root directory the force field files along with the trajectory, index and tpr file. The
custom force field should have the GROMACS 4.5 or later version format, with all the
parameters files placed in a directory, i.e. amber99sb.ff or oplsaa.ff. Moreover, the custom
residuetypes.dat file, which includes all the modified amino acids, should be present in the
root directory (Figure 3B). For details, please refer to the GROMACS manual. It is

possible to perform CAS calculations. See example 2, PHD-H3 complex (section 4.2).
use_topology: it can be either n or y. If n, GMXPBSA 2.1 will use either the standard force
field from the GROMACS package, defined in the previous ffield keyword, or the custom
force field placed in the root directory. If y, GMXPBSA 2.1 will perform the calculations
using the user defined custom topologies, namely receptor.itp and ligand.itp files. In this
case, the root directory should contain the topology file named topol.top (Figure 3C). This
file retrieves the receptor and ligand itp files. If use_topology is set to y, it is not possible to
perform CAS, since the modification of the topology files could lead to errors in the
modified topology. When the MM/PBSA calculations are performed on different
trajectories, each root directory should contain the proper topologies. For details see
example 3, Trypsin-Benzamidine complex, described in section 4.3.
itp_receptor: receptor topology file name. Receptors with more than one chain, require
repetition of the keyword for all the chains.
itp_ligand: ligand topology file name.
2.4.3 GROMACS
The users can define the name of the complex, receptor and ligand as defined in the index.ndx file.
They can also set different options for the Molecular Mechanics (MM) analysis in the MM/PBSA
calculation, as explained subsequently.
name_xtc: name of the trajectory file in GROMACS format.
name_tpr: name of the binary tpr file in GROMACS format.
complex: name of the complex in the index.ndx file.
receptor: name of the receptor in the index.ndx file.
ligand: name of the ligand in the index.ndx file.
multichain: Useful if in the pdbs extracted from the trajectory there are more than one
chains.The option "multichain" must be used ONLY if the string TER is not present at the
end of each chain in the comp/receptor pdb files.
protein_alone: it can be either y or n, depending on whether the user wants perform an
energetic estimation of a free protein and to study the CAS mutations. Default is n.
itp_protein: the name of the itp file of the protein in case “use_topology=y”.
skip: any integer no lower than 1. If skip is set to the integer N, a structure will be extracted
from the trajectory every N frames to be used for the subsequent analysis. In order to have
statistical reliability of the calculations, we suggest to use at least 100 frames. For instance,
in a trajectory of 1000 frames, if skip = 10, GMXPBSA 2.1 will extract 100 frames that are

equally distributed along the simulation.
min: it can be either y or n, depending on whether the user does or does not perform the
energy minimization, respectively. Energy minimization will be performed on each frame
before calculating the Coulomb/ Lennard-Jones contributions.
double_p: it can be either y or n, depending on whether double precision in energy
minimization is required, respectively.
read_vdw_radii: it can be either y or n. In case of y GMXPBSA 2.1 requires the presence of
the vdwradii.dat file (containing the Van der Waals radii) in the root directory. In this case
the “editconf” gromacs tool will use this file to generate the pqr files and will not compute
the radii based on the force field. The default option is n, however, care should be taken
when using this option, as the definition of the Van der Waals radii might influence the
MM/PBSA results.
coulomb: GMXPBSA 2.1 can calculate the coulomb energy term either using the APBS
“coulomb” tool (option coul) or using GROMACS (option gmx). By default it is set to coul.
2.4.4 APBS
The following section allows the user to define the options for the polar (PB) and nonpolar (SA)
solvation terms in MM/PBSA calculations. For details, please refer to the APBS manual
(http://www.poissonboltzmann.org/apbs/).
linearized: APBS can calculate the solvation energy by solving the linearized (option y) or
nonlinear (option n) Poisson-Boltzmann equation. By default it is set to n.
precF: can be a digit (either 0, 1, 2 or 3), which controls the size of the grid generated during
the APBS calculations (from 0 to 3 the grid spacing is decreased, resulting in more
expensive calculations ). By default it is set to 1.
temp: indicates the temperature at which the APBS calculations are performed. By default it
is set to 293 K.
bcfl: specifies the type of boundary conditions used to solve the Poisson-Boltzmann
equation. It can be either sdh, mdh or focu. By default it is set to mdh.
pdie: defines the dielectric constant of the biomolecule. This is usually a value between 2 to
20, lower values consider only electronic polarization and higher values consider additional
polarization due to intramolecular motion. By default it is set to 2.
extraspace: 5, quantity to add (A°) for each side to get fine-grid dimensions.
coarsefactor: 1.7, factor to get coarse-grid dimensions.
grid_spacing: 0.5 fine mesh spacing

sdie: 80
chgm: spl2
srfm: smol
srad: 1.4
swin: 0.3
sdens: 10.0
calcforce: no
ion_ch_pos: 1 positive ion charge in electron units
ion_rad_pos: 2.0 positive ion radius
ion_conc_pos: 0.15 positive ion concentration
ion_ch_neg: -1 negative ion charge in electron units
ion_rad_neg: 2.0 negative ion radius
ion_conc_neg: 0.15 negative ion concentration
Hsrfm: sacc srfm for non-polar calculations
Hpress : 0.00 press for non-polar calculations
Hgamma: 0.0227 gamma for non-polar calculations
Hdpos: 0.20 dpos for non-polar calculations
Hcalcforce: total calcforce for non-polar calculations
Hxgrid: 0.1xgrid for non-polar calculations
Hygrid: 0.1ygrid for non-polar calculations
Hzgrid : 0.1zgrid for non-polar calculations
2.4.5 DISTRIBUTED CALCULATIONS
The user can define the different options for calculations submission to a cluster facility. Since
energy calculations for each frame are independent, they can be easily parallelized in a distributed
fashion assigning single frames to the available processors. MM/PBSA calculations can be
performed either in a workstation exploiting one single core or multi core, or in a cluster exploiting
the PBS/TORQUE queue system. The latter option is useful for the analysis of a large amount of
frames, whereby GMXPBSA 2.1 submits to a batch queue a series of jobs carrying out MM/PBSA
calculations. The number of submitted jobs depends on the mnp keyword and on the total number of
frames (total_frames) that are analysed. GMXPBSA 2.1 calculates the number of total jobs to be
submitted according to the following rule: total_number_of_jobs = total_frames/mnp. For instance,

if the user sets mnp to20 and the trajectory contains 1000 frames, the total_number_of_jobs will be
50, whereby each job will contain 20 PBSA calculations. GMXPBSA 2.1 will then submit to the
queue these 50 jobs, monitoring the status (Running, Queue, Hold), checking the completion of
each job, and verifying the end of the calculations (jobs failed and/or successful completed). In case
of failed jobs, the program will recover them (if RecoverJobs is set to y) and resubmit to the queue
as previously described. In case of further failures, GMXPBSA 2.1 will print a warning in a log file,
and the failed frames will be excluded from the final MM/PBSA calculations. Finally, if cluster is
set to n and mnp is bigger than 1, GMXPBSA 2.1 will use the requested processors without using
the PBS system.
cluster: when the option is set to y GMXPBSA 2.1 performs calculations on a cluster in a
distributed fashion taking advantage of the PBS queue manager and divides frames across
all processors thus speeding up calculations.
Q: defines the name of the queue that is used for solvation energy calculations (this is
necessary only if cluster is set to y).
budget_name: the name of the user account in the computing facilities
walltime: the total maximum wall-clock time during which this job can run (note than 800
means seconds, 80:00 means minutes and seconds and 1:00:00 means hours, minutes and
seconds).
mnp: defines the maximum number of processors used during GMXPBSA 2.1 calculations.
We highly recommend to use numbers > 1 in workstations bearing multi core only when a
large amount of physical memory is available (at least 2Gb).
nodes: 1
mem: 5GB
When performing the calculations in a cluster with the PBS queue manager, the job are submitted
as: PBS -l select:$mnp:ncpus:$nodes:mem=$mem.
2.4.6 OUTPUT
The scripts always generate an output file in ASCII format, the user can chose an option that
generates the output file in PDF format.
pdf: generates a PDF file report as output (y or n). To set pdf to y it is necessary to have
installed LaTeX.
2.4.7 COMPUTATIONAL ALANINE SCANNING
In this section, the user can perform CAS calculations a posteriori on the trajectory. In this case, the
user should specify which residues (ligand and/or receptor) should be modified in alanine.

cas: if the y option is selected, GMXPBSA 2.1 will perform CAS
The setting for the mutation should contain the following string:
MUTATION root directory residue_number residue_name [receptor or lig] mutation_name.
The keywords receptor or lig are mandatory. GMXPBSA 2.1 automatically creates in the
RUN1_root directory a new directory for each mutation
( (root_MUTATION_mutation_name).
In the following, we present some syntax examples of the INPUT.dat file (Table 2):
a. “MUTATION PHD 9 ASP receptor ASP9ALA”, indicates that GMXPBSA 2.1 mutates the
residue ASP9 of the receptor in ALA.
b. “MUTATION PHD 8 ARG lig ARG8ALA”, indicates that GMXPBSA 2.1 mutates the
residue ARG8 of the peptide (lig) in ALA.
c. “MUTATION PHD 9 ASP receptor RES_9-11”, “MUTATION PHD 10 GLU receptor
RES_9-11”, “MUTATION PHD 11 CYZ receptor RES_9-11”, indicates that GMXPBSA
2.1 mutates the residues in the range 9-11 to ALA of the receptor.
It is possible to simultaneously apply all the above combinations in the CAS study, in this case
GMXPBSA 2.1 will create for each mutation name a directory.
3. Calculations steps of GMXPBSA 2.1
In the subsequen section we present the three calculation steps performed by GMXPBSA 2.1, a
summary of the main calculations and of the main output files generated by the scripts. Further
details are reported in the Supporting Material.
3.1 Calculations of Molecular Mechanics terms: gmxpbsa0.sh
In this first step GMXPBSA 2.1 calculates the Molecular Mechanics term (MM) of the MM/PBSA
approach, including Coulomb and Lennard-Jones terms. In order to perform the MM/PBSA
calculations, each GMXPBSA 2.1 script should read the INPUT.dat file. For instance, if the input
files (npt.xtc, npt.tpr and index.ndx and possible additional files) are placed in
/home/mysimulations/MD, the corresponding INPUT.dat file should be placed in
/home/mysimulations (working directory). The root keyword in INPUT.dat will be MD. In the
working directory (/home/mysimulations) the user will run the script:
$GMXPBSAHOME/gmxpbsa0.sh
to calculate the Coulomb and Lennard-Jones energies, the so called EMM terms. In this step
GMXPBSA 2.1 will also generate the grid needed to perform the APBS calculations in the second
workflow step. By default the grid is generated as follows (Figure 4):
fine grid: 10 Å added in each direction from the extreme coordinates of the complex;

coarse grid: 1.7 time larger than the fine grid.
The grid spacing is automatically set to an upper limit of 0.5 Å. Setting run keyword in INPUT.dat
to 1, GMXPBSA 2.1 will create RUN1_MD (RUNrun_root). At the end of the calculations, the
RUN1_MD directory will contain several output files (described in detail in the Supporting
Material). In particular, the RUNrun_root will be organized in three sub-folders:
1. STORED_FILES, containing all the files used during the Coulomb and Lennard-Jones
energy calculations;
2. APBS_CALCULATIONS, containing all the input files necessary for APBS. In this
directory, during the second step of the calculation APBS will generate also the
corresponding output;
3. SUMMARY_FILES for each analysed frame, a strun.rep file is generated, where n is the
number of generated frames (n = [Numer of total frames]/skip). These files contain all the
energy contributions (Coulomb and Lennard-Jones) of each frame.
In case of error or failure occurring in this step, GMXPBSA 2.1 will stop and will report the
possible failure causes in the STD_ERR0 file, that is present in each RUNrun_root.
3.2 Calculations of Solvation energy terms: gmxpbsa1.sh
In this step the PB and SA solvation energy terms of MM/PBSA are calculated. These contributions
are calculated typing the following command in the working directory (i.e. /home/mysimulations):
$GMXPBSAHOME/gmxpbsa1.sh
These calculations are often computationally expensive. Depending on the system size and frame
numbers they might require hours to finish; e.g. a system composed by 70 amino acids requires 5
min/frame calculation time on a single core.
The APBS calculations are performed by default at a NaCl concentration of 0.15 M and at a
temperature of 293 K, however these parameters can be easily changed by the user in the
INPUT.dat file. At the end of the calculations, the RUNrun_root directory (i.e. RUN1_MD) will
contain several output files (described in detail in the Supporting Material). The output file of each
APBS calculation will be stored in the SUMMARY_FILES folder.
3.3 Calculations of MM/PBSA binding free energy: gmxpbsa2.sh
This is the last step of the tool that combines all the energetic terms to compute the MM/PBSA
binding free energy value. To this aim it is sufficient to type in the working directory:
$GMXPBSAHOME/gmxpbsa2.sh
This script calculates the average and standard error of the MM/PBSA binding free energy values
deriving from the extracted frames. Before calculating the MM/PBSA value, the script will try to

recover failed jobs generated in the preceding step (if the variable RecoverJob is set to y). The
RUNrun_root directory will contain a series of files that are described in detail in Supporting
Material. To facilitate comparisons the file Compare_MMPBSA.dat generated in this step, contains
the average energy values plus the standard error SE (both the total energy and the single
contributions) of each system.
4 Examples
We run GMXPBSA 2.1 on three different systems in order to test its performance using different
INPUT.dat parameters. The first test was performed on cellular regulatory phosphoprotein p53 in
complex with oncoprotein Mdm2. This complex is considered as a reference system, as it has been
the first to be studied by the MM/PBSA approach [6]. This example requires to use the ffield
keyword in INPUT.dat. The second test was performed on the first PHD finger domain of
autoimmune regulator protein (AIRE) in complex with a 10 residue peptide corresponding to the N-
terminal tail of histone H3 [11]. In this example we perform MM/PBSA calculations on a system
bearing modified amino acids (use_nonstd_ff keyword in INPUT.dat). Finally, in the third example
we studied trypsin in complex with drug-like molecules (reversible competitive inhibitors
benzamidine and 1,3benzamidine [12]). In this example we used the use_topology keyword in
INPUT.dat. All the calculations have been carried out on a workstation Intel 3.30 GHz bearing 4 Gb
of RAM.
4.1. Example 1: p53 in complex with hMdm2 and CAS
We used GMXPBSA 2.1 to: i) calculate the binding free energy generated by the interaction
between p53 and hMdm2; ii) to perform CAS calculations. In this example we apply the standard
procedure, where the user gives as input files only the trajectory (xtc), the portable input binary (tpr)
and the index (ndx) files (Figure 3a). Before running GMXPBSA we performed 10ns of MD
simulation on the wild-type complex using amber99sb-ildn force field. In order to speed up the
calculations we removed the water molecules and the analysis was therefore performed on 1678
atoms. The final trajectory contained 1000 frames; in the INPUT.dat file we defined the following
parameters: ffield to 6 (amber99sb-ildn force field), skip 50 (20 frames to be considered),
linearized n, coulomb coul, mnp 1 and four alanine mutants on the 12-residue peptide of p53
defined in Table 3 (CAS calculations). The first step, gmxpbsa0.sh, required 3 minutes to extract all
the MM terms and to setup the grid for the PBSA calculation from 100 frames (20 frames for each
system, i.e. the wild type and the four alanine mutants). The second and third steps required 450
minutes and 5 seconds, respectively.

4.2 Example 2: AIRE-PHD1 in complex with histone H3 peptide and CAS
PHD fingers are Zn2+ binding domains consisting of 50–80 amino acids that form a two-stranded
antiparallel β-sheet followed by an α helix. The first PHD finger domain of AIRE recognizes the
unmodified tail of histone H3 to promote the expression of AIRE target genes [13]. We generated
10ns of trajectory of the PHD in complex with a histone peptide corresponding to the first 10
residues of histone H3 N-terminal tail (H3K4me0), using a custom oplsa force field, that was used
to define two Zn2+ coordinating residues, namely: CYM and HIZ, corresponding to an unprotonated
cysteine and a single protonated histidine, respectively. We also performed CAS calculations. We
stripped out the water molecules from the trajectory (final number of atoms: 1136). In the
INPUT.dat we defined the following parameters: use_nonstd_ff y, skip 1, linearized n, coulomb
coul, mnp 1 and two alanine mutants defined in Table 4 (CAS calculations). The PHD directory
contains the trajectory (xtc), the portable binary (tpr) and the index (ndx) files, the oplsaa.ff
directory and the residuetypes.dat file, which were used to carry out MD simulations (Figure 3b). In
this case we performed calculations on a total of 60 frames (20 frames for the wild type and 20 for
each of the mutants). The three steps required 2 minutes, 225 minutes and 2 seconds, respectively.
4.3 Example 3: Trypsin in complex with benzamidine ligands
In this example we used GMXPBSA 2.1 to compare the affinity of two ligands towards the same
receptor, to this aim we performed MM/PBSA calculations on trypsin in complex with benzamidine
and 1,3benzamidine ligands [12]. In this case, in order to carry out MD simulations followed by
MM/PBSA calculations, it was necessary to generate the topology file of the benzamidine ligands.
We exploited the Amber Antechamber program to calculate the ligand charges and the topology
parameter. Once we created the topology files for each ligand, ben.itp and ben2.itp, respectively
(see details in Supporting Material) we performed MD calculations (10ns) for each complex system.
Thereafter, we used GMXPBSA 2.1 to calculate the binding free energy of the benzamidine-trypsin
and 1,3benzamidine-trypsin complexes on the GROMACS trajectories. We stripped out of the water
molecules (final number of atoms: 3237 and 3239, respectively). We created two directories called
TRY and TRY2. In each directory we put the corresponding trajectory file (npt.xtc), portable input
binary file (npt.tpr), and index file with the complex, receptor and ligand group names (index.ndx),
and the complex, protein and ligand topologies, topol.top, trp.itp and ben.itp, in TRY1 and in TRY2
directories, respectively (Figure 3c). The main parameters in the INPUT.dat file were the following:
use_topology y, skip 100, linearized n, coulomb coul and mnp 1. Calculations were performed on a
total of 20 frames (10 frames for TRY and 10 for TRY2). The three steps required 1 minute, 80
minutes and 2 seconds, respectively. Computational alanine scanning cannot be performed in this
example since we are using the use_topology keyword.

Table 1. INPUT.dat file to run GMXPBSA 2.1
Keywords Value Note Default
root Name_of_Directory String -
run Any_Number Integer 1
multitrj “y” “n” Boolean n
root_multitrj* List of directory name String -
RecoverJobs “y” “n” Boolean y
backup “y” “n” Boolean y
Cpath Full_path String -
Apath Full_path String -
Gpath Full_path String -
use_topology “y” “n” Boolean -
itp_receptor* Name_of_topology String -
itp_ligand* Name_of_topology String -
use_nonstd_ff “y” “n” Boolean n
ffield* Number_of_ff_used_in_MD Integer -
name_xtc Name of GROMACS xtc String -
name_tpr Name of GROMACS tpr String -
complex Name_in_index_file String -
receptor Name_in_index_file String -
multichain “y” “n” Boolean n
protein_alone “y” “n” Boolean n
itp_protein Name_of_topology String -
ligand Name_in_index_file String -
skip Any_Digit Integer 1
min “y” “n” Boolean n
double_p “y” “n” Boolean n
read_vdw_radii “y” “n” Boolean n
coulomb “coul” “gmx” String gmx
linearized “y” “n” Boolean y
precF “0” “1” “2” “3” Integer 0
temp Temperature Integer 293
bcfl “sdh” “mdh” “focus” String mdh
pdie Any_Digit in [0:20] range Integer 2

coarsefactor Float 1.7
grid_spacing Float 0.5
sdie Integer 80
chgm String spl2
srfm String smol
srad Float 1.4
swin Float 0.3
sdens Float 10.0
calcforce Boolean no
ion_ch_pos Integer 1
ion_rad_pos Float 2.0
ion_conc_pos Float 0.15
ion_ch_neg Float -1
ion_rad_neg Float 2.0
ion_conc_neg Float 0.15
Hsrfm String sacc
Hpress Float 0.0
Hgamma Float 0.0227
Hdpos Float 0.2
Hcalcforce String total
Hxgrid Float 0.1
Hygrid Float 0.1
Hzgrid Float 0.1
cluster “y” “n” Boolean y
QName_of_queue String -
budget_name Name_of_budget String -
walltime Any Digit Integer -
mnp Any_Digit Integer 1
pdf “y” “n” Boolean n
cas “y” “n” Boolean n
Table 2. Example of CAS parameters as defined in INPUT.dat file
Requested mutation Name of working directory
MUTATION PHD 9 ASP receptor ASP9ALA RUN1_PHD_ASP9ALA
MUTATION PHD 8 ARG lig ARG8ALA RUN1_PHD_ARG8ALA
MUTATION PHD 9 ASP receptor RES_9-11
MUTATION PHD 10 GLU receptor RES_9-11
RUN1_PHD_RES_9-11

MUTATION PHD 11 CYZ receptor RES_9-11
Table 3. CAS parameters as defined in INPUT.dat file of Example 1
Requested mutation Name of working directory
MUTATION P53 112 PHE lig PHE19ALA RUN1_P53_PHE112ALA
MUTATION P53 115 LEU lig LEU22ALA RUN1_P53_LEU115ALA
MUTATION P53 116 TRP lig TRP23ALA RUN1_P53_TRP116ALA
MUTATION P53 119 LEU lig LEU26ALA RUN1_P53_LEU119ALA
Table 4. CAS parameters as defined in INPUT.dat file of Example 2
Requested mutation Name of working directory
MUTATION PHD 9 ASP receptor ASP9ALA RUN1_PHD_ASP9ALA
MUTATION PHD 8 ARG lig ARG8ALA RUN1_PHD_ARG8ALA
Figure 1 Workflow diagram for GMXPBSA 2.1. Diagram describing the general GMXPBSA 2.1
workflow scheme. GMXPBSA 2.1 combines the GROMACS and APBS programs in order to use
the frames extracted from the molecular dynamics simulations and to calculate the binding free
energy.
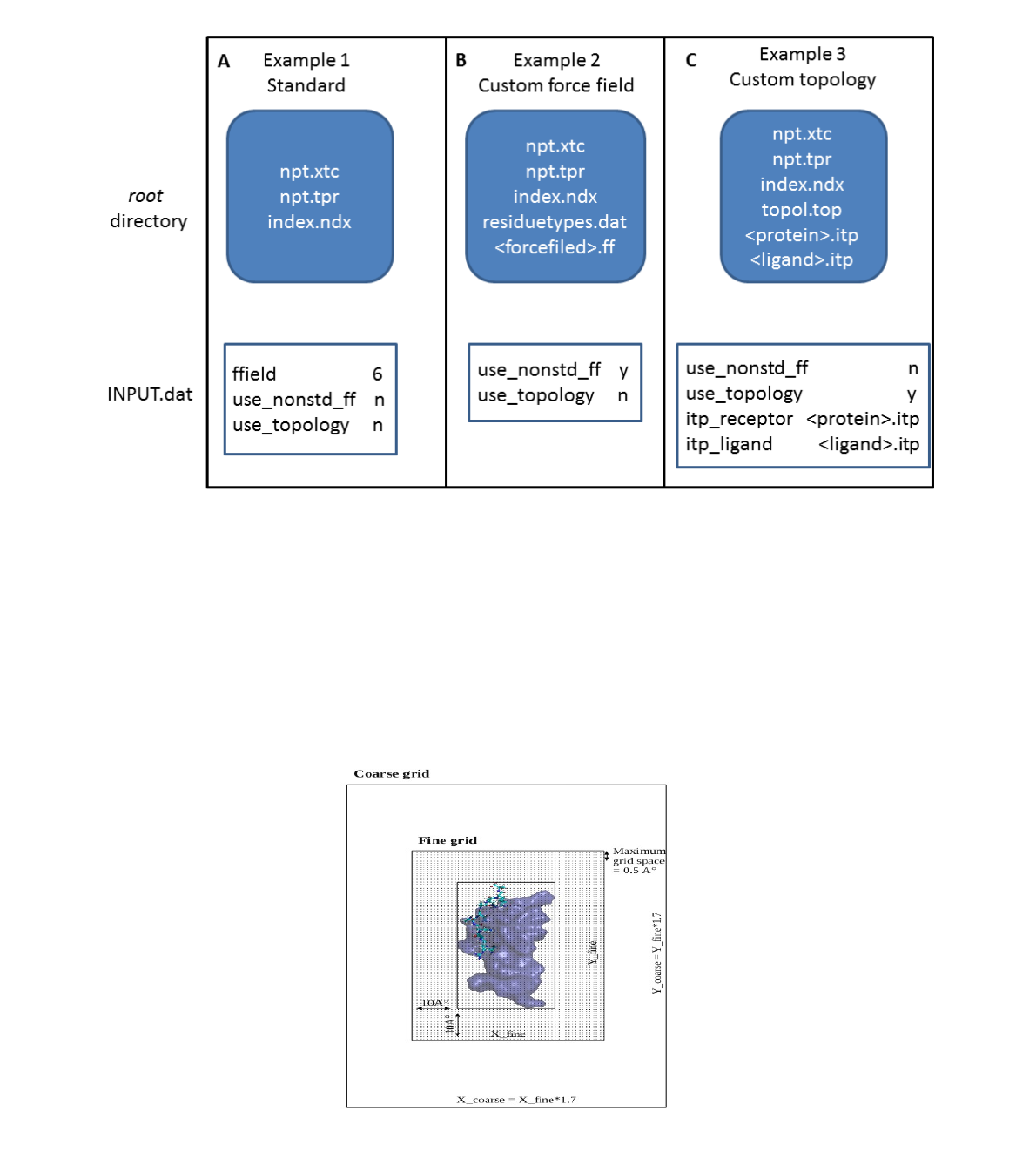
Figure 2 Schematic diagram of the three GMXPBSA 2.1 calculation steps Diagram showing the
input files used by GMXPBSA 2.1 and the output files generated during each MM/PBSA step.
Figure 3 Schematic diagram of the three GMXPBSA 2.1 examples. Diagram showing the input
files and the INPUT.dat parameters used by GMXPBSA 2.1 in each example.

Figure 4 2D-schematic representation of the grid preparation in the apbs calculation. The
protein and the ligand are shown in blue surface and licorice, respectively. The fine grid is
generated adding 10 Å in each direction from the extreme coordinates of the complex. The coarse
grid is 1.7 times larger than the fine grid. In this scheme the z-axis is omitted for clarity.
REFERENCES
[1] MR Reddy, CR Reddy, RS Rathore, MD Erion, P Aparoy, RN Reddy, et al. Free Energy
Calculations to Estimate Ligand-Binding Affinities in Structure-Based Drug Design,
Curr.Pharm.Des. (2013).
[2] MK Gilson, HX Zhou. Calculation of protein-ligand binding affinities,
Annu.Rev.Biophys.Biomol.Struct. 36 (2007) 21-42.
[3] S Huo, I Massova, PA Kollman. Computational alanine scanning of the 1:1 human growth
hormone-receptor complex, J.Comput.Chem. 23 (2002) 15-27.
[4] IS Moreira, PA Fernandes, MJ Ramos. Protein-protein docking dealing with the unknown,
J.Comput.Chem. 31 (2010) 317-342.
[5] RT Bradshaw, BH Patel, EW Tate, RJ Leatherbarrow, IR Gould. Comparing experimental
and computational alanine scanning techniques for probing a prototypical protein-protein
interaction, Protein Eng.Des.Sel. 24 (2011) 197-207.
[6] KP Massova I. Computational alanine scanning to probe protein-protein interactions: A
novel approach to evaluate binding free energies, J. Am. Chem. Soc. 121 (1999) 8133-8143.
[7] NA Baker, D Sept, S Joseph, MJ Holst, JA McCammon. Electrostatics of nanosystems:
application to microtubules and the ribosome, Proc.Natl.Acad.Sci.U.S.A. 98 (2001) 10037-
10041.
[8] SP Brown, SW Muchmore. Large-scale application of high-throughput molecular
mechanics with Poisson-Boltzmann surface area for routine physics-based scoring of protein-
ligand complexes, J.Med.Chem. 52 (2009) 3159-3165.
[9] Bill R. Miller, III, T. Dwight McGee, Jr., Jason M. Swails, Nadine Homeyer, Holger
Gohlke, and Adrian E. Roitberg. MMPBSA.py: An Efficient Program for End-State Free
Energy
Calculations, J. Chem. Theory Comput. 8 (2012) 3314-3321.
[10] S Pronk, S Pall, R Schulz, P Larsson, P Bjelkmar, R Apostolov, et al. GROMACS 4.5: a
high-throughput and highly parallel open source molecular simulation toolkit, Bioinformatics.
29 (2013) 845-854.
[11] D Spiliotopoulos, A Spitaleri, G Musco. Exploring PHD fingers and H3K4me0
interactions with molecular dynamics simulations and binding free energy calculations:
AIRE-PHD1, a comparative study, PLoS One. 7 (2012) e46902.
[12] D Jiao, J Zhang, RE Duke, G Li, MJ Schnieders, P Ren. Trypsin-ligand binding free
energies from explicit and implicit solvent simulations with polarizable potential,
J.Comput.Chem. 30 (2009) 1701-1711.
[13] J Derbinski, J Gabler, B Brors, S Tierling, S Jonnakuty, M Hergenhahn, et al.
Promiscuous gene expression in thymic epithelial cells is regulated at multiple levels,
J.Exp.Med. 202 (2005) 33-45.

[14] T Hou, J Wang, Y Li, W Wang. Assessing the performance of the MM/PBSA and
MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular
dynamics simulations, J.Chem.Inf.Model. 51 (2011) 69-82.
[15] L Li, C Li, S Sarkar, J Zhang, S Witham, Z Zhang, et al. DelPhi: a comprehensive suite
for DelPhi software and associated resources, BMC Biophys. 5 (2012) 9-1682-5-9.
