Sentaurus.SProcess.User Guide
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 1226 [warning: Documents this large are best viewed by clicking the View PDF Link!]
- Return to Front Page
- Sentaurus™ Process User Guide
- Contents
- About This Guide
- Chapter 1 Getting Started
- Overview
- Setting Up the Environment
- Starting Sentaurus Process
- Using a Command File
- Example: 1D Simulation
- Defining Initial 1D Grid
- Defining Initial Simulation Domain
- Initializing the Simulation
- Choosing Process Models and Parameters
- Setting Up a Meshing Strategy
- Growing Screening Oxide
- Measuring Oxide Thickness
- Depositing Screening Oxide
- Tcl Control Statements
- Implantation
- Saving the As-Implanted Profile
- Thermal Annealing, Drive-in, Activation, and Screening Oxide Strip
- Example: 2D Simulation
- Defining Initial Structure and Mesh Refinement
- Implanting Boron
- Growing Gate Oxide
- Defining Polysilicon Gate
- Polysilicon Reoxidation
- Saving Snapshots
- Remeshing for LDD and Halo Implants
- Implanting LDD and Halo
- Forming Nitride Spacers
- Remeshing for Source/Drain Implants
- Implanting Source/Drain
- Transferring to Device Simulation
- Extracting 1D Profiles
- Adaptive Meshing: 2D npn Vertical BJT
- Full-Text Versions of Examples
- Chapter 2 The Simulator Sentaurus Process
- Overview
- Interactive Graphics
- Command-Line Options
- Environment Variables
- File Types Used in Sentaurus Process
- Syntax for Creating Input Command Files
- Default Simulator Settings: SPROCESS.models File
- Compatibility With Previous Releases
- Parameter Database
- Viewing the Defaults: Parameter Database Browser
- Creating and Loading Structures and Data
- References
- Chapter 3 Ion Implantation
- Overview
- Coordinate System
- Analytic Implantation
- Primary Distribution Functions
- Screening (Cap) Layer-dependent Moments
- Lateral Straggle
- Analytic Damage: Hobler Model
- Datasets
- Tables
- Multilayer Implantations
- Lateral Integration
- Point-Response Interface
- Analytic Damage and Point-Defect Calculation
- Backscattering Algorithm
- Multiple Implantation Steps
- Profile Reshaping
- Ge-dependent Analytic Implantation
- Analytic Molecular Implantation
- Performing 1D or 2D Analytic Implantation in 3D Mode
- Implantation on (110)/(111) Wafers Using (100) Implant Tables
- Monte Carlo Implantation
- Running Sentaurus MC or Crystal-TRIM
- Structure of Target Material
- Sentaurus MC Physical Models
- Crystal-TRIM Physical Models
- Molecular Implantations
- MC Implantation into Polysilicon
- MC Implantation into Compound Materials with Molar Fractions
- MC Implantation into Silicon Carbide
- Recoil Implantation
- Plasma Implantation
- MC Implantation Damage and Point-Defect Calculation
- Statistical Enhancement
- Datasets
- Boundary Conditions and Domain Extension
- Smoothing Implantation Profiles
- Automatic Extraction of Implant Moments
- Loading External Profiles
- Multithreaded Parallelization of 3D Analytic Implantation
- Multithreaded Parallelization of Sentaurus MC Implantation
- References
- Chapter 4 Diffusion
- Overview
- Basic Diffusion
- General Formulation
- Transport Models
- Solid Phase Epitaxial Regrowth Model
- Flash or Laser Anneal Model
- Diffusion in Polysilicon
- Dopant Diffusion in SiGe
- Diffusion in III–V Compounds
- Pressure-dependent Defect Diffusion
- Electron Concentration
- Epitaxy
- Other Effects on Dopant Diffusion
- Dopant Activation and Clustering
- Dopant Active Model: None
- Dopant Active Model: Solid
- Dopant Active Model: Precipitation
- Dopant Active Model: Transient
- Dopant Active Model: Cluster
- Dopant Active Model: NeutralCluster
- Dopant Active Model: FVCluster
- Dopant Active Model: Equilibrium
- Dopant Active Model: BIC
- Dopant Active Model: ChargedCluster
- Dopant Active Model: ComplexCluster
- Dopant and Dopant-Defect Cluster Initialization
- Dopant Trapping at EOR Defects
- Defect Clusters
- Ion Implantation to Diffusion
- Initializing Solution Variables
- Boundary Conditions
- Periodic Boundary Condition
- Boundary Conditions at Moving Interfaces
- Common Dopant and Defect Dataset Names
- References
- Chapter 5 Atomistic Kinetic Monte Carlo Diffusion
- Overview
- KMC Method
- Operating Modes
- Simulation Domain
- Internal Grid
- Randomization
- Boundary Conditions
- Parallelism
- Estimating CPU Time
- Atomistic Diffusion Simulation with Sentaurus Process KMC
- Particles
- Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
- Damage Accumulation Model: Amorphous Pockets
- Extended Defects
- Amorphization and Recrystallization
- Impurity Clusters
- Fermi-Level Effects: Charge Model
- Sentaurus Process KMC Approach
- Assumptions
- Formation Energies for Charged Species
- Binding Energies for Particles
- Binding Energies for Impurity Clusters
- Temperature Dependency
- Charge Attractions and Repulsions
- Fermi-Level Computation
- Updating Charged States
- Electric Drift
- Bandgap Narrowing
- Charge Model and Boron Diffusion Example
- Charge Model and Arsenic Diffusion Example
- Interfaces and Surfaces
- Oxidation
- Epitaxial Deposition
- Including New Impurities
- Reports
- Extracting KMC-related Information
- Transferring Fields from KMC to Continuum Information: deatomize
- Adding and Obtaining Defects in Simulations: add, defects.add, and defects.write
- Using the Sentaurus Process Interface
- Calling Directly the Sentaurus Process KMC Kernel
- Inquiring about KMC Profiles, Histograms, and Defects
- Common Dopant and Point-Defect Names
- Advanced Calibration for Sentaurus Process KMC
- References
- Chapter 6 Alagator Scripting Language
- Chapter 7 Advanced Calibration
- Chapter 8 Oxidation and Silicidation
- Chapter 9 Computing Mechanical Stress
- Overview
- Material Models
- Plane Stress Analysis
- Equations: Global Equilibrium Condition
- Boundary Conditions
- Time Step Control
- Stress-causing Mechanisms
- Saving Stress and Strain Components
- References
- Chapter 10 Mesh Generation
- Overview
- Mesh Refinement
- Mesh Settings
- Controlling Mesh during Oxidation
- UseLines: Keeping User-defined Mesh Lines
- Data Interpolation
- Troubleshooting
- Chapter 11 Structure Generation
- Overview
- Etching and Deposition Types and Options
- The mask and photo Commands
- Geometry Transformations
- MGOALS Interface
- Sentaurus Structure Editor Interface
- Sentaurus Topography Interface
- Examples
- Using Polygon and Rectangle Mask in 2D Simulation
- 3D Etching after 2D LOCOS Simulation (Sentaurus Structure Editor Interface)
- Using Layout File for 3D Etching (Sentaurus Structure Editor Interface)
- 3D Trench Etching, Sloped Sidewall with Predefined Angle (Sentaurus Structure Editor Interface)
- 3D Etching after 2D LOCOS Simulation using MGOALS
- Structure Assembly in MGOALS
- Polygon Creation and Insertion in MGOALS2D
- Polyhedron Creation and Insertion in MGOALS
- References
- Chapter 12 ICWBEV Plus Interface for Layout-driven Simulations
- Overview
- ICWBEV Plus Introduction for TCAD Users
- Files Relevant to ICWBEV Plus–TCAD Sentaurus
- ICWBEV Plus Batch Mode and Macros
- TCAD Layout Reader of Sentaurus Process
- Loading the TCAD Layout
- Finding Simulation Domains
- Finding Layer Names and Layer IDs
- Selecting the Simulation Domain
- Loading a GDSII Layout
- Finding Domain Dimensions
- Finding Bounding Box of Domain
- Interface with line Commands
- Creating Masks
- Layout-driven Meshing
- Layout-driven Contact Assignment
- Aligning Wafer and Simulation Domain
- Additional Query Functions
- Chapter 13 Extracting Results
- Overview
- Saving Data Fields
- Selecting Fields for Viewing or Analysis
- Obtaining 1D Data Cuts
- Examples
- Determining the Dose: Layers
- Extracting Values and Level Crossings: interpolate
- Extracting Values during diffuse Step: extract
- Fitting Routines: FitLine, FitArrhenius, FitPearson, and FitPearsonFloor
- Resistivity
- Sheet Resistance
- References
- Chapter 14 Numerics
- Appendix A Commands
- Syntax Conventions
- alias
- ambient
- ArrBreak
- Arrhenius
- beam
- bound
- Compatibility
- contact
- contour
- CutLine2D
- define
- defineproc
- DeleteRefinementboxes
- deposit
- diffuse
- doping
- element
- Enu2G
- Enu2K
- equation
- etch
- exit
- extract
- fbreak
- fcontinue
- fexec
- fproc
- fset
- gas_flow
- graphics
- grid
- help
- icwb
- icwb.contact.mask
- icwb.create.all.masks
- icwb.create.mask
- icwb.refine.mask
- implant
- init
- insert
- integrate
- interface
- interpolate
- KG2E
- KG2nu
- kmc
- KMC2PDE
- layers
- line
- line_edge_roughness
- load
- LogFile
- mask
- mater
- math
- mgoals
- optimize
- paste
- pdbDelayDouble
- pdbdiff
- pdbDopantLike
- pdbExprDouble
- pdbGet and Related Commands
- pdbIsAvailable
- pdbLike
- pdbSet and Related Commands
- pdbUnSet-related Commands
- PDE2KMC
- photo
- plot.1d
- plot.2d
- plot.tec
- plot.xy
- point
- point.xy
- polygon
- polyhedron
- PowerDeviceMode
- print.1d
- print.data
- profile
- RangeRefineboxes
- reaction
- refinebox
- region
- sde
- select
- SetAtomistic
- SetDFISEList
- SetDielectricOxidationMode
- SetFastMode
- setMobilityModel
- SetPlxList
- SetTDRList
- SetTemp
- SetTS4ImplantMode
- SetTS4MechanicsMode
- SetTS4OxidationMode
- SetTS4PolyMode
- SheetResistance
- simDelayDouble
- simGetBoolean
- simGetDouble
- simSetBoolean
- simSetDouble
- slice
- smooth
- solution
- sptopo
- stdiff
- strain_profile
- stressdata
- StressDependentSilicidation
- strip
- struct
- substrate_profile
- tclsel
- temp_ramp
- term
- topo
- transform
- transform.refinement
- translate
- UnsetAtomistic
- UnsetDielectricOxidationMode
- update_substrate
- WritePlx

Sentaurus™ Process User
Guide
Version I-2013.12, December 2013

ii Sentaurus™ Process User Guide
I-2013.12
Copyright and Proprietary Information Notice
Copyright © 2013 Synopsys, Inc. All rights reserved. This software and documentation contain confidential and proprietary
information that is the property of Synopsys, Inc. The software and documentation are furnished under a license agreement and
may be used or copied only in accordance with the terms of the license agreement. No part of the software and documentation may
be reproduced, transmitted, or translated, in any form or by any means, electronic, mechanical, manual, optical, or otherwise, without
prior written permission of Synopsys, Inc., or as expressly provided by the license agreement.
Destination Control Statement
All technical data contained in this publication is subject to the export control laws of the United States of America.
Disclosure to nationals of other countries contrary to United States law is prohibited. It is the reader’s responsibility to
determine the applicable regulations and to comply with them.
Disclaimer
SYNOPSYS, INC., AND ITS LICENSORS MAKE NO WARRANTY OF ANY KIND, EXPRESS OR IMPLIED, WITH
REGARD TO THIS MATERIAL, INCLUDING, BUT NOT LIMITED TO, THE IMPLIED WARRANTIES OF
MERCHANTABILITY AND FITNESS FOR A PARTICULAR PURPOSE.
Trademarks
Synopsys and certain Synopsys product names are trademarks of Synopsys, as set forth at
http://www.synopsys.com/Company/Pages/Trademarks.aspx.
All other product or company names may be trademarks of their respective owners.
Synopsys, Inc.
700 E. Middlefield Road
Mountain View, CA 94043
www.synopsys.com
Sentaurus™ Process User Guide iii
I-2013.12
Contents
About This Guide xxxi
Audience . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xxxii
Related Publications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xxxii
Typographic Conventions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xxxii
Customer Support . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xxxiii
Accessing SolvNet. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xxxiii
Contacting Synopsys Support . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xxxiii
Contacting Your Local TCAD Support Team Directly. . . . . . . . . . . . . . . . . . . . . xxxiv
Acknowledgments. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xxxiv
Chapter 1 Getting Started 1
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
Setting Up the Environment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
Starting Sentaurus Process . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
Starting Different Versions of Sentaurus Process . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
Using a Command File . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
Example: 1D Simulation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
Defining Initial 1D Grid . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 4
Defining Initial Simulation Domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
Initializing the Simulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
Choosing Process Models and Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
Setting Up a Meshing Strategy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
Growing Screening Oxide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 6
Measuring Oxide Thickness . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
Depositing Screening Oxide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 8
Tcl Control Statements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
Implantation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 9
Saving the As-Implanted Profile . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
Thermal Annealing, Drive-in, Activation, and Screening Oxide Strip . . . . . . . . . . . . 11
Example: 2D Simulation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
Defining Initial Structure and Mesh Refinement. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
Implanting Boron. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
Growing Gate Oxide . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
Defining Polysilicon Gate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
Working with Masks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
Polysilicon Reoxidation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 17
Saving Snapshots. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18

Contents
iv Sentaurus™ Process User Guide
I-2013.12
Remeshing for LDD and Halo Implants . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
Implanting LDD and Halo. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
Forming Nitride Spacers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
Remeshing for Source/Drain Implants . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 20
Implanting Source/Drain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
Transferring to Device Simulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
Remeshing for Device Simulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
Contacts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
Saving the Structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
Extracting 1D Profiles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
Adaptive Meshing: 2D npn Vertical BJT . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
Overview . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
Defining Initial Structure. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 26
Adaptive Meshing Settings . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 27
Buried Layer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 28
Epi Layer . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
Sinker Region . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
Base Region . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
Emitter Region. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
Backend . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
Full-Text Versions of Examples . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
1D NMOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
2D NMOS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
2D npn Vertical Bipolar. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 38
Chapter 2 The Simulator Sentaurus Process 43
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
Interactive Graphics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
Command-Line Options . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
Interactive Mode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 46
Fast Mode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
Terminating Execution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
Environment Variables . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
File Types Used in Sentaurus Process . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
Syntax for Creating Input Command Files . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
Tcl Input. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .50
Material Specification . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
Aliases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
Default Simulator Settings: SPROCESS.models File. . . . . . . . . . . . . . . . . . . . . . . . . . . . 53
Compatibility With Previous Releases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 54
Parameter Database. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 55

Contents
Sentaurus™ Process User Guide v
I-2013.12
Parameter Inheritance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
Materials in Parameter Database . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
Like Materials: Material Parameter Inheritance. . . . . . . . . . . . . . . . . . . . . . . . . . . 57
Interface Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
Regionwise Parameters and Region Name-handling. . . . . . . . . . . . . . . . . . . . . . . . . . 58
Viewing the Defaults: Parameter Database Browser . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
Starting the Parameter Database Browser . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
Browser PDB Functions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
PDB Preferences. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64
Viewing Parameters Stored in TDR Files . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65
Creating and Loading Structures and Data . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66
Understanding Coordinate Systems. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66
Wafer Coordinate System. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 66
Simulation Coordinate System (Unified Coordinate System) . . . . . . . . . . . . . . . . 67
Visualization Coordinate Systems . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68
Defining the Structure: The line and region Commands . . . . . . . . . . . . . . . . . . . . . . . 70
Creating the Structure and Initializing Data . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 71
Defining the Crystal Orientation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73
Automatic Dimension Control. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74
Saving and Visualizing Structures. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
Saving a Structure for Restarting the Simulation . . . . . . . . . . . . . . . . . . . . . . . . . . 76
Saving a Structure for Device Simulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
Saving Doping Information in SiC and GaN for Device Simulations . . . . . . . . . . 79
Saving 1D Profiles for Inspect . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
Saving 1D TDR Files from 2D and 3D Simulations . . . . . . . . . . . . . . . . . . . . . . . 79
The select Command (More 1D Saving Options) . . . . . . . . . . . . . . . . . . . . . . . . . 80
Loading 1D Profiles: The profile Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 80
Chapter 3 Ion Implantation 81
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 81
Selecting Models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 84
Dios or Default Tables . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
Taurus Tables . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
TSUPREM-4 Native Implant Tables . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
Multirotation Implantation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88
Energy Contamination Implantation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 88
Adaptive Meshing during Implantation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
Coordinate System . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
Coordinates for Implantation: Tilt and Rotation Angles . . . . . . . . . . . . . . . . . . . . . . . 89
2D Coordinate System. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 91
Contents
vi Sentaurus™ Process User Guide
I-2013.12
Analytic Implantation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 92
Primary Distribution Functions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
Gaussian Distribution: gaussian . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
Pearson Distribution: pearson. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
Pearson Distribution with Linear Exponential Tail: pearson.s. . . . . . . . . . . . . . . . 96
Dual Pearson Distribution: dualpearson. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
Point-Response Distribution: point.response . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
Screening (Cap) Layer-dependent Moments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98
Lateral Straggle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 99
Depth-dependent Lateral Straggle: Sentaurus Process Formulation . . . . . . . . . . 100
Depth-dependent Lateral Straggle: Dios Formulation . . . . . . . . . . . . . . . . . . . . . 100
Depth-dependent Lateral Straggle: Taurus Formulation . . . . . . . . . . . . . . . . . . . 101
Analytic Damage: Hobler Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 101
Datasets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
Tables. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
Implantation Table Library. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 104
File Formats . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 106
Multilayer Implantations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 111
Lateral Integration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113
Local Layer Structure in 2D. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113
Primary Direction and Scaling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 115
Point-Response Interface . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 116
Analytic Damage and Point-Defect Calculation . . . . . . . . . . . . . . . . . . . . . . . . . . . . 117
Implantation Damage . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 118
Point-Defect Calculation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 118
Backscattering Algorithm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
Multiple Implantation Steps. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
Preamorphization Implantation (PAI) Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . 121
CoImplant Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122
Profile Reshaping. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
Ge-dependent Analytic Implantation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 127
Analytic Molecular Implantation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 128
Molecular Implantation with Supplied Implant Tables . . . . . . . . . . . . . . . . . . . . 130
BF2 Implant . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 130
Damage Calculation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 131
Performing 1D or 2D Analytic Implantation in 3D Mode. . . . . . . . . . . . . . . . . . . . . 131
Implantation on (110)/(111) Wafers Using (100) Implant Tables. . . . . . . . . . . . . . . 132
Monte Carlo Implantation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 133
Running Sentaurus MC or Crystal-TRIM . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 133
Structure of Target Material . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 136
Composition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 136
Contents
Sentaurus™ Process User Guide vii
I-2013.12
Single-Crystalline Materials. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 137
Amorphous Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 139
Polycrystalline Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 139
Molar Fractions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 140
Sentaurus MC Physical Models. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 141
Binary Collision Theory. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 141
Electronic Stopping Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 148
Damage Accumulation and Dynamic Annealing . . . . . . . . . . . . . . . . . . . . . . . . . 149
Crystal-TRIM Physical Models. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155
Single-Crystalline Materials. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 155
Amorphous Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 156
Damage Buildup and Crystalline–Amorphous Transition . . . . . . . . . . . . . . . . . . 158
Internal Storage Grid for Implantation Damage. . . . . . . . . . . . . . . . . . . . . . . . . . 159
Molecular Implantations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 161
MC Implantation into Polysilicon . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 162
MC Implantation into Compound Materials with Molar Fractions. . . . . . . . . . . . . . 163
MC Implantation into Silicon Carbide. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 165
Recoil Implantation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 166
Plasma Implantation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 167
Simple Source. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168
Complex Source . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 168
Deposition of Material . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 169
Knock-on and Knock-off Effect. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 169
Conformal Doping . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 170
Other Plasma Implantation–related Parameters and Procedures . . . . . . . . . . . . . 170
MC Implantation Damage and Point-Defect Calculation . . . . . . . . . . . . . . . . . . . . . 172
Sentaurus MC Damage Calculation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 172
Crystal-TRIM: Damage Probability . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 173
Point Defects. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 174
Statistical Enhancement. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 176
Trajectory Splitting. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 176
Dose Split . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 177
Trajectory Replication . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 178
Datasets . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 179
Boundary Conditions and Domain Extension. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 180
Unified Implant Boundary Conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 181
Implant Boundary Conditions using PDB Commands . . . . . . . . . . . . . . . . . . . . . . . 181
Monte Carlo Implant . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 182
Analytic Implant. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 185
Smoothing Implantation Profiles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 186
Automatic Extraction of Implant Moments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 187

Contents
viii Sentaurus™ Process User Guide
I-2013.12
Required Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 188
Optional Parameters. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 188
Output Format . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 189
Utilities. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .189
Loading External Profiles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 190
Loading Files Using load.mc. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 190
Automated Monte Carlo Run. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 191
Multithreaded Parallelization of 3D Analytic Implantation . . . . . . . . . . . . . . . . . . . . . . 191
Multithreaded Parallelization of Sentaurus MC Implantation . . . . . . . . . . . . . . . . . . . . 192
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 193
Chapter 4 Diffusion 197
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 197
Basic Diffusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 198
Obtaining Active and Total Dopant Concentrations . . . . . . . . . . . . . . . . . . . . . . . . . 200
Transport Models. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 201
Recombination and Reaction Models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
Boundary Conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 203
Other Materials and Effects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 204
General Formulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 204
Transport Models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 205
ChargedReact Diffusion Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 206
React Diffusion Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 212
ChargedPair Diffusion Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 214
Pair Diffusion Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 216
ChargedFermi Diffusion Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 217
Fermi Diffusion Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 219
Constant Diffusion Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 220
NeutralReact Diffusion Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 220
Carbon Diffusion Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 221
Nitrogen Diffusion Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 222
Mobile Impurities and Ion-Pairing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 223
Solid Phase Epitaxial Regrowth Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 224
Level-Set Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 224
Phase Field Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 227
Flash or Laser Anneal Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 229
Dopant Diffusion in Melting Laser Anneal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 232
Guideline for Parameter Setting . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 233
Saving a Thermal Profile . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 234
Boundary Conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 235
Structure Extension . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 235
Contents
Sentaurus™ Process User Guide ix
I-2013.12
Intensity Models for Flash Anneal. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 236
Gaussian Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 236
Table Lookup Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 237
User-specified Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 238
Intensity Model for Scanning Laser. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 238
Control Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 240
Notes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 242
Diffusion in Polysilicon . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 242
Isotropic Diffusion Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 243
Grain Shape and the Grain Growth Equation. . . . . . . . . . . . . . . . . . . . . . . . . . . . 243
Diffusion Equations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 246
Anisotropic Diffusion Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 248
Diffusion in Grain Interiors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 248
Grain Boundary Structure. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 249
Diffusion along Grain Boundaries . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 249
Segregation Between Grain Interior and Boundaries. . . . . . . . . . . . . . . . . . . . . . 251
Grain Size Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 252
Surface Nucleation Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 253
Grain Growth . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 254
Interface Oxide Breakup and Epitaxial Regrowth . . . . . . . . . . . . . . . . . . . . . . . . 255
Dependence of Polysilicon Oxidation Rate on Grain Size. . . . . . . . . . . . . . . . . . 257
Boundary Conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 258
Boundary Conditions for Grain Growth Equation . . . . . . . . . . . . . . . . . . . . . . . . 258
Dopant Diffusion Boundary Conditions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 258
Dopant Diffusion in SiGe . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 260
Bandgap Effect . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 260
Potential Equation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 261
Effects on Point-Defect Equilibrium Concentrations . . . . . . . . . . . . . . . . . . . . . . . . 262
Effect of Ge on Point-Defect Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 263
Impact of Ge on Extended-Defect Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 263
Impact of Dopant Diffusivities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 263
SiGe Strain and Dopant Activation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 264
Germanium–Boron Pairing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 264
Initializing Germanium–Boron Clusters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 265
Diffusion in III–V Compounds. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 265
Material Conversion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 265
Physical Parameter Interpolation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 266
Dopant Diffusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 267
ChargedReact Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 267
Fermi Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 271
Constant Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 271
Contents
xSentaurus™ Process User Guide
I-2013.12
Activation Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 271
Point-Defect Diffusion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 272
Poisson Equation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 274
MoleFractionFields . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 274
Pressure-dependent Defect Diffusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 275
Electron Concentration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 276
Poisson Equation for Hetero-junctions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 278
Bandgap Narrowing. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 280
Epitaxy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 282
Using LKMC for Deposition Shape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 283
Epi Doping. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 284
Initialization of Dopant Clusters in Epi . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 284
Epi Auto-Doping . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 285
Epi Doping Using Resistivity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 286
Epi Growth Settings: Low-Temperature Epitaxy . . . . . . . . . . . . . . . . . . . . . . . . . . . 286
Simulating Facet Growth during Selective Epitaxy . . . . . . . . . . . . . . . . . . . . . . . . . 287
Controlling Where Facets Form . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 288
Time-stepping. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 288
Other Effects on Dopant Diffusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 289
Pressure-dependent Dopant Diffusion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 289
Diffusion Prefactors. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 290
High-Concentration Effects on Dopant Diffusion. . . . . . . . . . . . . . . . . . . . . . . . . . . 291
Hydrogen Effects on Dopant Diffusion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 291
Dopant Activation and Clustering. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 292
Dopant Active Model: None . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 292
Dopant Active Model: Solid . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 293
Dopant Active Model: Precipitation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 293
Initializing Precipitation Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 294
Dopant Active Model: Transient . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 296
Initializing Transient Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 298
Dopant Active Model: Cluster. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 299
Initializing Cluster Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 301
Dopant Active Model: NeutralCluster. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 301
Initializing NeutralCluster Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 303
Carbon Cluster . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 303
Nitrogen Cluster . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 304
Dopant Active Model: FVCluster . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 304
Initializing the FVCluster Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 306
Dopant Active Model: Equilibrium . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 306
Dopant Active Model: BIC . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 307
Initializing BIC Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 309
Contents
Sentaurus™ Process User Guide xi
I-2013.12
Dopant Active Model: ChargedCluster . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 309
Initializing ChargedCluster Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 312
Dopant Active Model: ComplexCluster . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 312
Initializing ComplexCluster Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 314
Dopant and Dopant-Defect Cluster Initialization . . . . . . . . . . . . . . . . . . . . . . . . . . . 315
Dopant Trapping at EOR Defects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 316
Initializing Dopant Trapping in EOR Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . 319
Defect Clusters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 319
Defect Cluster Model: None . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 320
Defect Cluster Model: Equilibrium . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 320
Defect Cluster Model: 311. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 320
Initializing 311 Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 327
Defect Cluster Model: Loop . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 328
Direct Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 328
Size-dependent Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 329
Initializing Loop Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 330
Defect Cluster Model: LoopEvolution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 331
Initializing LoopEvolution Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 332
Defect Cluster Model: FRENDTECH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 333
Initializing FRENDTECH Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 336
Defect Cluster Model: 1Moment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 337
Interstitial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 337
Vacancy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 339
Initializing 1Moment Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 341
Defect Cluster Model: 2Moment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 341
Interstitial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 341
Vacancy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 343
Initializing 2Moment Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 345
Defect Cluster Model: Full . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 346
Interstitial . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 346
Vacancy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 349
Initializing Full Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 352
Ion Implantation to Diffusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 353
Initializing Solution Variables . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 355
Boundary Conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 357
HomNeumann . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 357
Natural . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 358
Surface Recombination Model: PDependent . . . . . . . . . . . . . . . . . . . . . . . . . . . . 358
Surface Recombination Model: InitGrowth . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 360
Surface Recombination Model: Simple . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 360
Surface Recombination Model: Normalized . . . . . . . . . . . . . . . . . . . . . . . . . . . . 360

Contents
xii Sentaurus™ Process User Guide
I-2013.12
Modifying Point-Defect Equilibrium Values at Surface . . . . . . . . . . . . . . . . . . . 361
Segregation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 361
Surface Recombination Model: Default. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 362
Surface Recombination Model: PairSegregation . . . . . . . . . . . . . . . . . . . . . . . . . 362
Dirichlet . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 364
ThreePhaseSegregation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 365
Surface Recombination Model: Default. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 366
Surface Recombination Model: PairSegregation . . . . . . . . . . . . . . . . . . . . . . . . . 368
Trap . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .369
TrapGen . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 369
Continuous. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 369
Periodic Boundary Condition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 370
Boundary Conditions at Moving Interfaces . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 370
Enhanced and Retarded Diffusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 370
Conserving Dose . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 371
Common Dopant and Defect Dataset Names . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 371
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 377
Chapter 5 Atomistic Kinetic Monte Carlo Diffusion 381
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 381
KMC Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 382
Operating Modes. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 382
Atomistic Mode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 383
Restrictions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 383
Implant . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 384
Diffuse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 385
Nonatomistic Mode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 386
Atomistic/Nonatomistic Translation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 387
Sano Method. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 388
Simulation Domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 389
Recommended Domain Size . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 389
Internal Grid . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 390
Randomization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 392
Boundary Conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 392
Parallelism. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 393
How Parallelism Works. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 393
Estimating CPU Time . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 394
Atomistic Diffusion Simulation with Sentaurus Process KMC . . . . . . . . . . . . . . . . . . . 395
Units. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .396
Space Management . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 397
Materials and Space. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 398
Contents
Sentaurus™ Process User Guide xiii
I-2013.12
Supported Materials. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 399
Material Alloying. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 402
Point Defects. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 403
Ambiguous Alloying . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 403
Time Management . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 403
Simulation and CPU Times . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 404
Parallelism and CPU Time . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 406
Snapshots . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 407
Movie . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 407
Time Internal Representation and Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 408
Particles. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 408
Particle Types . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 408
Particles in Models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 410
Alias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 411
Colors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 411
Particles and Parameters. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 411
Undefining Particles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 414
Defect Types . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 414
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects . . . . . . . . . . . . 415
Interstitials and Vacancies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 415
Impurities. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 418
Migration (Diffusion) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 418
Breakup. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 419
Percolation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 421
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 421
Parameter Examples . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 422
Hopping Mode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 423
The short Mode. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 423
The long Mode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 423
The double Mode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 424
The longdouble Mode. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 424
Enabling and Disabling Interactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 424
Interaction Rules. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 425
Examples . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 426
Defining Nonstandard Interactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 427
Interaction Rules. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 427
Example . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 428
Stress Effects on Point Defects, Impurities, Dopants, and Impurity-Paired Point Defects
428
Migration Energy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 429
Binding Energy. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 429
Alloys. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 430
Contents
xiv Sentaurus™ Process User Guide
I-2013.12
Alloy Diffusion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 431
Alloy Effects. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 432
Introducing Alloys in the Simulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 432
Damage Accumulation Model: Amorphous Pockets . . . . . . . . . . . . . . . . . . . . . . . . . . . 432
Shape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 434
Growth . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 434
Recombination. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 434
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 435
Emission. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 436
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 437
Amorphous Pockets Life Cycle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 439
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 440
Interactions of Amorphous Pockets. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 440
Interaction with Point Defects: I and V . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 440
Interaction with Impurities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 441
Extended Defects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 442
{311} Defects (ThreeOneOne) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 442
Shape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 443
Capture . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 444
Emission . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 444
Recombination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 446
Interactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 446
Dislocation Loops . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 447
Shape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 447
Capture . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 448
Emission . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 448
Interactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 450
Voids . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 451
Shape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 452
Capture . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 453
Emission . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 453
Recombination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 454
Interactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 454
Amorphization and Recrystallization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 454
Amorphous Defects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 456
Material. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 456
Shape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 456
Growth . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 456
Recombination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 456
Diffusion in Amorphous Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 456
Direct diffusion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 457
Contents
Sentaurus™ Process User Guide xv
I-2013.12
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 457
Indirect Diffusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 457
Impurity Clusters in Amorphous Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 459
Recrystallization . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 460
KMC: Quasiatomistic Solid Phase Epitaxial Regrowth. . . . . . . . . . . . . . . . . . . . 460
LKMC: Fully Atomistic Modeling of Solid Phase Epitaxial Regrowth . . . . . . . 463
Defect Generation during SPER. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 467
Redistributing Damage. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 469
Impurity Sweep/Deposit. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 470
Impurity Clusters. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 472
Shape . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 473
Diffusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 474
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 474
Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 475
Growth . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 475
Initial Seeds . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 477
Percolation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 477
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 478
Emission. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 479
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 480
Recombination. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 481
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 481
Frank–Turnbull Mechanism . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 481
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 482
Complementary Recombination . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 483
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 484
Complementary Emission . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 484
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 485
Charge Dependency. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 485
Neutral Reactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 485
Nonneutral Reactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 485
Interactions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 487
Complex Impurity Clusters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 487
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 488
Setting Up Impurity Clusters in a Material . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 489
Fermi-Level Effects: Charge Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 490
Sentaurus Process KMC Approach . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 491
Assumptions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 491
Formation Energies for Charged Species . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 492
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 493
Binding Energies for Particles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 493
Contents
xvi Sentaurus™ Process User Guide
I-2013.12
Binding Energies for Impurity Clusters. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 493
Temperature Dependency . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 494
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 494
Charge Attractions and Repulsions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 495
Fermi-Level Computation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 495
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 496
Updating Charged States . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 497
Electronic Concentrations and Charge-State Ratios. . . . . . . . . . . . . . . . . . . . . . . 497
Mobile Particles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 498
Pairing and Breakup Reactions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 498
Electric Drift . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 499
Bandgap Narrowing. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 500
Narrowing due to Dopant Concentration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 500
Narrowing due to Strain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 501
Narrowing due to Presence of an Alloy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 504
Bandgap Narrowing Use . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 504
Charge Model and Boron Diffusion Example . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 505
Charge Model and Arsenic Diffusion Example. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 506
Interfaces and Surfaces . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 507
Different Interface Models. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 508
Interfaces for Self-Silicon Point Defects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 509
Capture . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 509
Emission . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 509
Stress. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 510
Alloys . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 511
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 511
Oxidation-enhanced Diffusion (OED) Model . . . . . . . . . . . . . . . . . . . . . . . . . . . 512
Interfaces for Impurities. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 514
Simple Material Side . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 514
Full Material Side . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 516
Oxidation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 518
Epitaxial Deposition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 519
Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 521
Including New Impurities . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 522
Impurities Diffusing without Pairing. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 525
Normal Diffusion . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 525
Diffusion without Pairing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 525
Reports . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 526
Models Used Report . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 526
Particle Distribution Report. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 527
Cluster Distribution Report . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 528
Contents
Sentaurus™ Process User Guide xvii
I-2013.12
Defect Activity Report. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 528
Interactions Report. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 530
PointDefect . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 530
AmorphousPocket . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 531
ThreeOneOne . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 531
Loop . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 531
ImpurityCluster. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 531
Interface . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 532
Event Report . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 532
PointDefect . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 532
AmorphousPocket . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 533
ThreeOneOne . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 534
Loop . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 534
ImpurityCluster. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 534
Amorphous Defects . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 535
Lattice Atoms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 535
Simple Materials. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 535
Extracting KMC-related Information . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 536
Transferring Fields from KMC to Continuum Information: deatomize . . . . . . . . . . 536
Smoothing Out Deatomized Concentrations . . . . . . . . . . . . . . . . . . . . . . . . . . . . 537
Adding and Obtaining Defects in Simulations: add, defects.add, and defects.write. 539
Using the Sentaurus Process Interface. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 541
The select, print, WritePlx, and plot Commands . . . . . . . . . . . . . . . . . . . . . . . . . 541
The init Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 542
The struct Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 542
The load Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 542
The deposit Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 542
The diffuse Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 543
Nonatomistic Mode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 543
Atomistic Mode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 543
Calling Directly the Sentaurus Process KMC Kernel . . . . . . . . . . . . . . . . . . . . . . . . 543
Writing and Displaying TDR Files with KMC Information . . . . . . . . . . . . . . . . 544
Inquiring about KMC Profiles, Histograms, and Defects . . . . . . . . . . . . . . . . . . . . . 547
The histogram Option. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 548
The profile Option . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 551
The supersaturation Option . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 554
The defects Option . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 555
The dose Option . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 557
The materials Option . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 559
The acinterface Option . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 560
Common Dopant and Point-Defect Names . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 560

Contents
xviii Sentaurus™ Process User Guide
I-2013.12
Advanced Calibration for Sentaurus Process KMC . . . . . . . . . . . . . . . . . . . . . . . . . . . . 565
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 566
Chapter 6 Alagator Scripting Language 571
Operators. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 571
Binary and Unary Operators . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 571
Simple Functions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 572
Differential Functions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 573
Special Functions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 573
The diag Operator. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 573
String Names . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 574
Solution Names and Subexpressions: Terms. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 574
Constants and Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 575
Alagator for Diffusion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 575
Basics. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 576
Setting Boundary Conditions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 578
Dirichlet Boundary Condition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 578
Segregation Boundary Condition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 578
Natural Boundary Condition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 579
Interface Traps . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 579
External Boundary Condition. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 580
Using Terms. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 580
Callback Procedures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 582
Callbacks during Execution of diffuse Command . . . . . . . . . . . . . . . . . . . . . . . . 583
Using Callback Procedures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 586
Setup Procedure: InitProc. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 587
Preprocessing and Postprocessing Data: diffPreProcess, UserDiffPreProcess,
diffPostProcess, UserDiffPostProcess . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 591
Complex Initialization Procedures: InitSolve and EquationInitProc . . . . . . . . . . 592
Diffusion Summary: pdb, TclLib, SPROCESS.models . . . . . . . . . . . . . . . . . . . . . .594
Alagator for Generic Growth . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 596
Basics. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 596
Epi Reactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 598
Callback Procedures . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 600
Setup Procedure: InitGrowth . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 602
Equation Procedure: EquationGrowthProc . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 603
Epitaxy Growth Rate: GrowthRateProc . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 605
Generic Growth Summary: pdb, TclLib, SPROCESS.models . . . . . . . . . . . . . . . . . 606
Modifying Diffusion Models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 608
UserAddEqnTerm and UserSubEqnTerm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 608
UserAddToTerm and UserSubFromTerm. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 609

Contents
Sentaurus™ Process User Guide xix
I-2013.12
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 610
Chapter 7 Advanced Calibration 611
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 611
Using Advanced Calibration. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 611
Additional Calibration by Users . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 612
Chapter 8 Oxidation and Silicidation 615
Oxidation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 615
Basic Oxidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 616
Temperature Cycles . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 616
Ambients and Gas Flows . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 617
Specifying Gas Flows. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 618
Computing Partial Pressures. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 619
In Situ Steam-generated Oxidation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 620
Oxidant Diffusion and Reaction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 620
Transition to Linear and Parabolic Rate Constants . . . . . . . . . . . . . . . . . . . . . . . 622
Massoud Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 623
Orientation-dependent Oxidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 624
Stress-dependent Oxidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 624
Trap-dependent Oxidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 626
Dopant-dependent Oxidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 627
Diffusion Prefactors . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 629
Oxidation with Dielectric on Top . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 630
N2O Oxidation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 630
SiC Oxidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 630
In Situ Steam-generated Oxidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 632
Silicide Models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 634
TiSi2 Growth Kinetics. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 634
TiSi2 Formation Reactions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 635
Tungsten-, Cobalt-, and Nickel-Silicide Models . . . . . . . . . . . . . . . . . . . . . . . . . . . . 637
Stress-dependent Silicidation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 637
Oxygen-retarded Silicidation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 638
Triple-Point Control. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 639
Dopants and Defects in Oxides and Silicides . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 640
Numerics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 640
Outer Time Loop . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 640
Inner Time Loop . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 641
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 642

Contents
xx Sentaurus™ Process User Guide
I-2013.12
Chapter 9 Computing Mechanical Stress 643
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 643
Material Models . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 644
Viscoelastic Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 645
Maxwell Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 645
Standard Linear Solid Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 646
Purely Viscous Materials. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 648
Shear Stress–dependent Viscosity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 648
Purely Elastic Materials. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 649
Anisotropic Elastic Materials. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 650
Cubic Crystal Anisotropy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 650
Orthotropic Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 651
Plastic Materials. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 653
Incremental Plasticity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 653
Deformation Plasticity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 655
Viscoplastic Materials . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 656
Anand Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 656
Power Law Creep . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 658
Swelling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 660
Mole Fraction–dependent Mechanical Properties . . . . . . . . . . . . . . . . . . . . . . . . . . . 661
Deprecated Syntax for Mole Fraction–dependent Mechanical Properties of Binary
Compounds. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 663
Temperature-dependent Mechanical Properties . . . . . . . . . . . . . . . . . . . . . . . . . . . . 664
Deprecated Syntax for Temperature-dependent Mechanical Properties . . . . . . . 665
Plane Stress Analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 665
Equations: Global Equilibrium Condition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 666
Boundary Conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 667
Example: Applying Boundary Conditions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 669
Pressure Boundary Condition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 670
Advanced Dirichlet Boundary Condition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 670
Periodic Boundary Condition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 670
Time Step Control. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 672
Stress-causing Mechanisms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 672
Stress Induced by Growth of Material. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 672
Densification-induced Stress . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 673
Selectively Switching Off Grid Movement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 673
Stress Caused by Thermal Mismatch. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 674
Lattice Mismatch . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 675
Using the Lattice Mismatch Model . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 677
Total Concentration Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 678
Reference Concentration Model. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 679

Contents
Sentaurus™ Process User Guide xxi
I-2013.12
Strained Deposition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 679
Edge Dislocation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 680
Intrinsic Stress . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 682
Stress Rebalancing after Etching and Deposition . . . . . . . . . . . . . . . . . . . . . . . . . . . 683
Automated Tracing of Stress History. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 683
Saving Stress and Strain Components . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 684
Description of Output Variables . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 684
Tracking Maximum Stresses . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 690
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 690
Chapter 10 Mesh Generation 693
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 693
Mesh Refinement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 694
Viewing Mesh Refinement. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 695
Static Refinement. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 695
Standard Refinement Boxes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 695
Interface Refinement Boxes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 696
Interface Offsetting Refinement Boxes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 696
Refinement Inside a Mask . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 697
Refinement Near Mask Edges . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 698
Adaptive Refinement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 699
Adaptive Refinement Criteria. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 700
Localizing Adaptive Meshing using refinebox Command. . . . . . . . . . . . . . . . . . 706
Examples . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 707
Adaptive Meshing during Diffusion. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 707
Adaptive Meshing during Implantation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 708
Tips for Adaptive Meshing. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 709
Default Refinement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 710
Refinement Box Manipulations: Using transform.refinement . . . . . . . . . . . . . . . . . 711
Mesh Settings . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 712
Controlling Mesh during Oxidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 714
TS4 Mesh Library . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 714
Control Parameters in TS4Mesh. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 715
Moving Mesh and Mechanics Displacements . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 717
Grid Spacing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 717
Grid Cleanup . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 717
Maximum-allowed Rate of Growth. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 718
Miscellaneous Tricks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 718
Meshing for 3D Oxidation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 719
MovingMesh . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 719
UseLines: Keeping User-defined Mesh Lines . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 722

Contents
xxii Sentaurus™ Process User Guide
I-2013.12
Using line Commands after init Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 723
Dimension within Current Spatial Dimension . . . . . . . . . . . . . . . . . . . . . . . . . . . 723
Dimension Greater Than Current Spatial Dimension. . . . . . . . . . . . . . . . . . . . . . 723
Creating More Than One Structure . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 724
The UseLines and transform Commands. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 725
The reflect Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 725
The stretch Command. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 725
The rotate Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 725
The translate Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 725
The cut Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 725
Examples . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 725
Testing line Commands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 725
Showing Clearing Lines for a New Structure. . . . . . . . . . . . . . . . . . . . . . . . . . . . 726
Data Interpolation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 727
Troubleshooting . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 727
Chapter 11 Structure Generation 731
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 731
Functionality . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 731
Etching . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 732
Deposition. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 732
Masks and Photoresist . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 732
Geometry Creation and Transformations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 732
Etching and Deposition Types and Options . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 733
Etching. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 733
Etching Tips . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 736
Etching Type: Isotropic . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 736
Etching Types: Anisotropic and Directional . . . . . . . . . . . . . . . . . . . . . . . . . . . . 737
Etching Types: Polygonal and CMP . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 740
Etching Type: Fourier. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 741
Etching Type: Crystallographic . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 744
Etching Type: Trapezoidal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 745
Etching Type: Piecewise Linear. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 749
Deposition . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 751
Mask Naming . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 752
Deposition Type: Isotropic. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 752
Deposition Types: Fill and Polygonal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 752
Deposition Type: Crystallographic. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 753
Deposition Type: Fourier . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 754
Deposition Type: Trapezoidal . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 755
Selective Deposition. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 756
Contents
Sentaurus™ Process User Guide xxiii
I-2013.12
Fields in Deposited Layers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 756
Stress Handling . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 757
Shape Library. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 757
PolyHedronSTI. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 758
PolyHedronSTIaccc . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 760
PolyHedronSTIaccv . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 761
PolyHedronCylinder. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 762
PolygonWaferMask . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 762
PolyHedronEpiDiamond . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 763
The mask and photo Commands. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 764
Photoresist Masks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 767
Boolean Masks. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 767
Line Edge Roughness Effect . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 769
Mirrored Boundary Conditions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 771
Geometry Transformations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 772
Refinement Handling during Transformation . . . . . . . . . . . . . . . . . . . . . . . . . . . 773
Contact Handling during Transformation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 773
The transform reflect Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 774
Refinement Handling during Reflection. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 774
The transform stretch Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 774
Refinement Handling during Stretch . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 775
The transform cut Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 775
Refinement Handling during Cut . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 776
The transform flip Command and Backside Processing . . . . . . . . . . . . . . . . . . . . . . 776
Refinement Handling during Flip. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 777
The transform rotate Command. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 777
Refinement Handling during Rotation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 778
The transform translate Command . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 778
MGOALS Interface. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 778
MGOALS Boundary-moving Algorithms. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 778
MGOALS Boundary-moving Parameters . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 780
MGOALS 3D Boundary-moving Algorithms . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 782
Summary of MGOALS Etching and Deposition Algorithms . . . . . . . . . . . . . . . . . . 783
MGOALS Backward Compatibility. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 784
Boundary Repair Algorithm . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 785
Inserting Segments in One Dimension . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 785
Inserting Polygons in Two Dimensions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 785
Inserting Polyhedra in Three Dimensions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 786
Reading Polyhedra from a TDR Boundary File . . . . . . . . . . . . . . . . . . . . . . . . . . 786
Creating a Rectangular Prism. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 787
Extruding a 2D Polygon . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 787

Contents
xxiv Sentaurus™ Process User Guide
I-2013.12
Creating a Polyhedron from Its Constituent Polygonal Faces . . . . . . . . . . . . . . . 788
Sentaurus Structure Editor Interface: External Mode. . . . . . . . . . . . . . . . . . . . . . 788
Inserting Polyhedra. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 789
Structure Assembly in MGOALS Mode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 790
Multithreading . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 790
Sentaurus Structure Editor Interface. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 791
Sentaurus Topography Interface . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 794
Sentaurus Topography . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 794
Sentaurus Topography 3D . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 796
Examples. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 797
Using Polygon and Rectangle Mask in 2D Simulation . . . . . . . . . . . . . . . . . . . . . . . 797
3D Etching after 2D LOCOS Simulation (Sentaurus Structure Editor Interface). . . 797
Using Layout File for 3D Etching (Sentaurus Structure Editor Interface) . . . . . . . . 799
3D Trench Etching, Sloped Sidewall with Predefined Angle (Sentaurus Structure Editor
Interface). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 803
3D Etching after 2D LOCOS Simulation using MGOALS. . . . . . . . . . . . . . . . . . . . 805
Structure Assembly in MGOALS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 807
Polygon Creation and Insertion in MGOALS2D . . . . . . . . . . . . . . . . . . . . . . . . . . . 809
Polyhedron Creation and Insertion in MGOALS . . . . . . . . . . . . . . . . . . . . . . . . . . . 812
Reading a TDR file. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 812
Extruding a 2D Polygon . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 813
Creating a Polyhedron using Polygons. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 814
Defining a Brick . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 815
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 816
Chapter 12 ICWBEV Plus Interface for Layout-driven Simulations 817
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 817
ICWBEV Plus Introduction for TCAD Users. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 818
Opening GDSII Layout Files. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 818
Graphical User Interface of ICWBEV Plus. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 819
Sentaurus Markups . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 820
Stretch Utility. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 822
Renaming Markups . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 824
Auxiliary Layers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 825
Editing Polygons . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 826
Resizing a Rectangle . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 826
Converting a Rectangle to a Polygon . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 827
Nonaxis-aligned Simulation Domains. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 827
Files Relevant to ICWBEV Plus–TCAD Sentaurus . . . . . . . . . . . . . . . . . . . . . . . . . . . . 828
Saving the Sentaurus Markup File. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 829
Contents of Sentaurus Markup File . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 830
Contents
Sentaurus™ Process User Guide xxv
I-2013.12
Reloading the Markup File . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 831
Saving the TCAD Layout File. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 832
Contents of TCAD Layout File . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 833
Reloading the TCAD Layout File . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 834
ICWBEV Plus Batch Mode and Macros. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 834
Starting ICWBEV Plus in Batch Mode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 834
ICWBEV Plus Macros. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 834
Tcl-based Macros for Layout Parameterization. . . . . . . . . . . . . . . . . . . . . . . . . . . . . 835
TCAD Layout Reader of Sentaurus Process. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 835
Loading the TCAD Layout . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 836
Finding Simulation Domains. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 836
Finding Layer Names and Layer IDs. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 836
Selecting the Simulation Domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 837
Loading a GDSII Layout . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 837
Finding Domain Dimensions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 838
Finding Bounding Box of Domain . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 838
Interface with line Commands. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 839
Creating Masks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 839
Layout-driven Meshing . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 841
Layout-driven Contact Assignment. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 842
Aligning Wafer and Simulation Domain. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 844
Additional Query Functions. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 846
Chapter 13 Extracting Results 849
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 849
Saving Data Fields . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 849
Selecting Fields for Viewing or Analysis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 850
Obtaining 1D Data Cuts . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 851
Examples. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 851
Determining the Dose: Layers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 853
Extracting Values and Level Crossings: interpolate. . . . . . . . . . . . . . . . . . . . . . . . . . . . 854
Extracting Values during diffuse Step: extract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 854
Optimizing Parameters Automatically. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 855
Fitting Routines: FitLine, FitArrhenius, FitPearson, and FitPearsonFloor. . . . . . . . . . . 856
Resistivity . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 857
Sheet Resistance . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 859
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 860
Chapter 14 Numerics 861
Overview. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 861

Contents
xxvi Sentaurus™ Process User Guide
I-2013.12
Setting Parameters of the Iterative Solver ILS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 862
Partitioning and Parallel Matrix Assembly. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 864
Matrix Size Manipulation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 867
Node and Equation Ordering . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 867
Time Integration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 868
Time-Step Control. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 869
Time-Step Control for PDEs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 869
Error Control for PDEs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 871
Time-Step Control for Mechanics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 871
Convergence Criteria . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 872
Time-Step Adjustment . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 873
Time-Step Cutback. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 874
References. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 875
Appendix A Commands 877
Syntax Conventions . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 877
Example of Command Syntax . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 878
Common Arguments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 879
alias . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 880
ambient . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 881
ArrBreak . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 883
Arrhenius. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 884
beam . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 885
bound. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 887
Compatibility . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 888
contact . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 889
contour . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 894
CutLine2D. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .896
define. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 897
defineproc . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 898
DeleteRefinementboxes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 900
deposit . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 901
diffuse . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 908
doping . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 917
element . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 919
Enu2G . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 920
Enu2K . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 921
equation. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 922
etch . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 923
exit. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 930
extract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 931
Contents
Sentaurus™ Process User Guide xxvii
I-2013.12
fbreak . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 933
fcontinue . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 933
fexec . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 934
fproc . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 934
fset. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 934
gas_flow . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 935
graphics . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 938
grid . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 940
help . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 950
icwb. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 951
icwb.contact.mask. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 954
icwb.create.all.masks . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 956
icwb.create.mask. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 957
icwb.refine.mask . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 959
implant . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 961
init . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 978
insert . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 982
integrate. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 985
interface. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 988
interpolate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 991
KG2E . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 993
KG2nu. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 994
kmc . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 995
KMC2PDE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1007
layers . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1008
line. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1010
line_edge_roughness. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1013
load . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1016
LogFile . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1019
mask . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1020
mater . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1025
math. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1027
mgoals . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1037
optimize. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1042
paste . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1046
pdbDelayDouble . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1048
pdbdiff. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1049
pdbDopantLike . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1050
pdbExprDouble . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1051
pdbGet and Related Commands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1052
pdbIsAvailable . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1054
Contents
xxviii Sentaurus™ Process User Guide
I-2013.12
pdbLike . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1055
pdbSet and Related Commands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1056
pdbUnSet-related Commands . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1059
PDE2KMC . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1060
photo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1061
plot.1d . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1063
plot.2d . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1066
plot.tec. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1070
plot.xy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1076
point . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1078
point.xy . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1080
polygon . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1082
polyhedron . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1086
PowerDeviceMode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1089
print.1d . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1090
print.data . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1092
profile . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1093
RangeRefineboxes . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1096
reaction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1099
refinebox . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1101
region . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1110
sde . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1114
select . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1117
SetAtomistic . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1121
SetDFISEList . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1122
SetDielectricOxidationMode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1124
SetFastMode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1126
setMobilityModel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1127
SetPlxList . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1128
SetTDRList . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1129
SetTemp . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1130
SetTS4ImplantMode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1131
SetTS4MechanicsMode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1132
SetTS4OxidationMode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1133
SetTS4PolyMode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1134
SheetResistance. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1135
simDelayDouble . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1136
simGetBoolean . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1137
simGetDouble . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1138
simSetBoolean . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1139
simSetDouble . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1140
Contents
Sentaurus™ Process User Guide xxix
I-2013.12
slice . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1141
smooth. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1144
solution . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1145
sptopo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1148
stdiff . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1149
strain_profile . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1150
stressdata . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1151
StressDependentSilicidation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1156
strip . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1157
struct . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1158
substrate_profile . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1163
tclsel . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1164
temp_ramp . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1166
term . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1173
topo . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1176
transform . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1177
transform.refinement. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1182
translate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1186
UnsetAtomistic . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1187
UnsetDielectricOxidationMode . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1189
update_substrate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1190
WritePlx . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .1191
Contents
xxx Sentaurus™ Process User Guide
I-2013.12

Sentaurus™ Process User Guide xxxi
I-2013.12
About This Guide
The Synopsys Sentaurus™ Process tool is an advanced 1D, 2D, and 3D process simulator
suitable for silicon and nonsilicon semiconductor devices. It features modern software
architecture and state-of-the-art models to address current and future process technologies.
Sentaurus Process simulates all standard process simulation steps, diffusion, implantation,
Monte Carlo (MC) implantation (Taurus MC or Crystal-TRIM), oxidation, etching, deposition,
and silicidation. Capabilities in 3D include meshing of 3D boundary files through the
MGOALS library, implantation through the Imp3D module from FhG Erlangen, mechanics
(stress and strain), diffusion, a limited capability for 3D oxidation, and an interface to
Sentaurus Structure Editor, which is the 3D geometry editing tool based on the ACIS solid
modeling library.
Sentaurus Process uses the Alagator scripting language that allows users to solve their own
diffusion equations. Alagator can be used to solve any diffusion equation including dopant,
defect, impurity, and oxidant diffusion equations. Simulation of 3D diffusion is handled exactly
as for 1D and 2D. Therefore, all the advanced models and user programmability available in
1D and 2D can be used in 3D. In addition, a set of built-in calibrated parameters is available
with Advanced Calibration.
The main chapters are:
■Chapter 1 describes how to run Sentaurus Process.
■Chapter 2 presents an overview of how Sentaurus Process operates.
■Chapter 3 presents the ion implantation technique used in Sentaurus Process.
■Chapter 4 provides information on the dopant and defect diffusion models and parameters.
■Chapter 5 describes atomistic kinetic Monte Carlo diffusion.
■Chapter 6 discusses the Alagator scripting language for solving diffusion equations.
■Chapter 7 provides details about using Advanced Calibration in Sentaurus Process.
■Chapter 8 describes the oxidation models.
■Chapter 9 describes the computation of mechanical stress.
■Chapter 10 describes the mesh algorithms and meshing parameters available in Sentaurus
Process.
■Chapter 11 discusses etching and deposition, and other geometry manipulations available
in Sentaurus Process.
■Chapter 12 presents strategies for using the ICWorkBench EVPlus–TCAD Sentaurus
interface.
■Chapter 13 presents strategies for analysing simulation results.

About This Guide
Audience
xxxii Sentaurus™ Process User Guide
I-2013.12
■Chapter 14 discusses numerics-related issues, time integration methods, and the linear
solvers used in Sentaurus Process.
■Appendix A lists the available commands, including descriptions, options, and examples.
Audience
This user guide is intended for users of the Sentaurus Process software package.
Related Publications
For additional information about Sentaurus Process, see:
■The TCAD Sentaurus release notes, available on SolvNet® (see Accessing SolvNet on
page xxxiii).
■Documentation available through SolvNet at https://solvnet.synopsys.com/DocsOnWeb.
Typographic Conventions
Convention Explanation
<> Angle brackets
{} Braces
[] Brackets
() Parentheses
Blue text Identifies a cross-reference (only on the screen).
Bold text Identifies a selectable icon, button, menu, or tab. It also indicates the name of a field or an
option.
Courier font
Identifies text that is displayed on the screen or that the user must type. It identifies the names
of files, directories, paths, parameters, keywords, and variables.
Italicized text Used for emphasis, the titles of books and journals, and non-English words. It also identifies
components of an equation or a formula, a placeholder, or an identifier.
Menu > Command Indicates a menu command, for example, File > New (from the File menu, select New).
NOTE: Identifies important information.

About This Guide
Customer Support
Sentaurus™ Process User Guide xxxiii
I-2013.12
Customer Support
Customer support is available through SolvNet online customer support and through
contacting the Synopsys support center.
Accessing SolvNet
SolvNet includes an electronic knowledge base of technical articles and answers to frequently
asked questions about Synopsys tools. SolvNet also gives you access to a wide range of
Synopsys online services, which include downloading software, viewing documentation, and
entering a call to the Synopsys support center.
To access SolvNet:
1. Go to the SolvNet Web page at https://solvnet.synopsys.com.
2. If prompted, enter your user name and password. (If you do not have a Synopsys user name
and password, follow the instructions to register with SolvNet.)
If you need help using SolvNet, click Help on the SolvNet menu bar.
Contacting Synopsys Support
If you have problems, questions, or suggestions, you can contact Synopsys support in the
following ways:
■Go to the Synopsys Global Support Centers site on www.synopsys.com. There you can find
e-mail addresses and telephone numbers for Synopsys support centers throughout the
world.
■Go to either the Synopsys SolvNet site or the Synopsys Global Support Centers site and
open a case online (Synopsys user name and password required).

About This Guide
Acknowledgments
xxxiv Sentaurus™ Process User Guide
I-2013.12
Contacting Your Local TCAD Support Team Directly
Send an e-mail message to:
■support-tcad-us@synopsys.com from within North America and South America.
■support-tcad-eu@synopsys.com from within Europe.
■support-tcad-ap@synopsys.com from within Asia Pacific (China, Taiwan, Singapore,
Malaysia, India, Australia).
■support-tcad-kr@synopsys.com from Korea.
■support-tcad-jp@synopsys.com from Japan.
Acknowledgments
Sentaurus Process is based on the 2000 and 2002 releases of FLOOPS written by
Professor Mark Law and coworkers at the University of Florida. Synopsys acknowledges the
contribution of Professor Law and his advice in the development of Sentaurus Process. For
more information about TCAD at the University of Florida, visit
http://www.swamp.tec.ufl.edu.
Sentaurus Process Kinetic Monte Carlo is based on DADOS written by Professor Martin Jaraiz
and coworkers at the University of Valladolid, Spain. Synopsys acknowledges Professor Jaraiz’
contribution and advice. For more information, visit http://www.ele.uva.es/~simulacion/
KMC.htm.
Sentaurus™ Process User Guide 1
H-2013.03
CHAPTER 1 Getting Started
This chapter describes how to run Sentaurus Process and guides you
through a series of examples.
This chapter is not a comprehensive reference but is intended to introduce some of the more
widely used features of Sentaurus Process in a realistic context. For new users, the sections
Interactive Mode on page 46, Syntax for Creating Input Command Files on page 50, and
Creating the Structure and Initializing Data on page 71 would be useful to refer to while
reading this chapter. For more advanced users who need to adjust model parameters, Like
Materials: Material Parameter Inheritance on page 57 would be useful. For the TCAD
Sentaurus Tutorial and examples, go to:
$STROOT/tcad/$STRELEASE/Sentaurus_Training/index.html
where STROOT is an environment variable that indicates where the Synopsys TCAD
distribution has been installed, and STRELEASE indicates the Synopsys TCAD release number.
Overview
Sentaurus Process is a complete and highly flexible, multidimensional, process modeling
environment. With its modern software architecture and extensive breadth of capabilities,
Sentaurus Process is a state-of-the-art process simulation tool. Calibrated to a wide range of
the latest experimental data using proven calibration methodology, Sentaurus Process offers
unique predictive capabilities for modern silicon and nonsilicon technologies.
Sentaurus Process accepts as input a sequence of commands that is either entered from
standard input (that is, at the command prompt) or composed in a command file. A process
flow is simulated by issuing a sequence of commands that corresponds to the individual
process steps. In addition, several commands allow you to select physical models and
parameters, grid strategies, and graphical output preferences, if required. You should place
parameter settings in a separate file, which is sourced at the beginning of input files using the
source command.
In addition, a special language (Alagator) allows you to describe and implement your own
models and diffusion equations.
1: Getting Started
Setting Up the Environment
2Sentaurus™ Process User Guide
H-2013.03
Setting Up the Environment
The STROOT environment variable is the TCAD Sentaurus root directory, and you must set this
variable to the installation directory of TCAD Sentaurus. The STRELEASE environment
variable can be used to specify the release of the software to run, for example, H-2013.03. If
STRELEASE is not set, the default version is used which is usually the last version installed.
To set the environment variables:
1. Set the TCAD Sentaurus root directory environment variable STROOT to the TCAD
Sentaurus installation directory, for example:
* Add to .cshrc
setenv STROOT <Sentaurus directory>
* Add to .profile, .kshrc, or .bashrc
STROOT=<Sentaurus directory>; export STROOT
2. Add the <Sentaurus directory>/bin directory to the user path.
For example:
* Add to .cshrc:
set path=(<Sentaurus directory>/bin $path)
* Add to .profile, .kshrc, or .bashrc:
PATH=<Sentaurus directory>/bin:$PATH
export PATH
Starting Sentaurus Process
You c an run S entauru s Pro ce ss in either the interactive mode or batch mode. In the interactive
mode, a whole process flow can be simulated by entering commands line-by-line as standard
input. To start Sentaurus Process in the interactive mode, enter the following on the command
line:
> sprocess
Sentaurus Process displays version and host information, followed by the Sentaurus Process
command prompt. You now can enter Sentaurus Process commands at the prompt:
sprocess>
1: Getting Started
Using a Command File
Sentaurus™ Process User Guide 3
H-2013.03
This is a flexible way of working with Sentaurus Process to test individual process steps or
short sequences, but it is inconvenient for long process flows. It is more useful to compile the
command sequence in a command file, which can be run in batch mode or inside Sentaurus
Workbench.
To run Sentaurus Process in batch mode, load a command file when starting Sentaurus Process,
for example:
> sprocess input.cmd
Starting Different Versions of Sentaurus Process
You can s elect a sp ecifi c re le ase and ver sion number of Sentaurus Process using the -rel and
-ver options:
> sprocess -rel <rel_number> -ver <version_number>
For example:
> sprocess -rel H-2013.03
The command:
> sprocess -rel H-2013.03 -ver 1.2 nmos_fps.cmd
starts the simulation of nmos_fps.cmd
using the 1.2 version of Release H-2013.03 as long as
this version is installed.
Using a Command File
As an alternative to entering Sentaurus Process commands line-by-line, the required sequence
of commands can be saved to a command file, which can be written entirely by users or
generated using Ligament. To save time and reduce syntax errors, you can copy and edit
examples of command files in this user guide or use Ligament to create a template.
If a command file has been prepared, run Sentaurus Process by typing the command:
sprocess <command_filename>
Alternatively, you can automatically start Sentaurus Process through the Scheduler in
Sentaurus Workbench. By convention, the command file name has the extension .cmd. (This is
the convention adopted in Sentaurus Workbench.)

1: Getting Started
Example: 1D Simulation
4Sentaurus™ Process User Guide
H-2013.03
The command file is checked for correct syntax and then the commands are executed in
sequence until the simulation is stopped by the command exit or the end of the file is reached.
Since Sentaurus Process is written as an extension of the tool command language (Tcl), all Tcl
commands and functionalities (such as loops, control structures, creating and evaluating
variables) are available in the command files. This results in some limitations in syntax control
if the command file contains complicated Tcl commands. Syntax-checking can be switched off
with the command-line option -n, for example:
sprocess -n inputfile
Sentaurus Process ignores character strings starting with # (although Sentaurus Workbench
interprets # as a special character for conditional statements). Therefore, this special character
can be used to insert comments in the simulation command file.
A file with the extension .log is created automatically whenever Sentaurus Process is run from
a command line, that is, outside the Sentaurus Workbench environment. This file contains the
run-time output, which is generated by Sentaurus Process and is sent to standard output. When
Sentaurus Process is run by using a command file <root_filename>_fps.cmd, the output
file is named <root_filename>_fps.log.
When Sentaurus Process is run in Sentaurus Workbench, no log file is created. Instead, the file
<root_filename>_fps.out is generated as a copy of the standard output. For a complete
list of all commands, see Appendix A on page 877.
Example: 1D Simulation
Many widely used process and control commands are introduced in the context of a nominal
n-channel MOSFET process flow. The MOSFET structure is simulated in 1D and
2D, and the processing of the isolation is excluded.
In this section, a simple 1D process simulation is performed.
Defining Initial 1D Grid
The initial 1D grid is defined with the line command:
line x location=0.0 spacing= 1<nm> tag=SiTop
line x location= 10<nm> spacing= 2<nm>
line x location= 50<nm> spacing= 10<nm>
line x location=300<nm> spacing= 20<nm>
line x location=0.5<um> spacing= 50<nm>
line x location=2.0<um> spacing=0.2<um> tag=SiBottom
0.18 µm

1: Getting Started
Example: 1D Simulation
Sentaurus™ Process User Guide 5
H-2013.03
The first argument of the line specifies the direction of the grid. For 1D, this is always x.
The grid spacing is defined by pairs of the location and spacing keywords. The keyword
spacing defines the spacing between two grid lines at the specified location. Sentaurus
Process expands or compresses the grid spacing linearly in between two locations defined in
the line command.
NOTE Units in Sentaurus Process can be specified explicitly by giving the units
in angle brackets. For most cases, the default unit of length is
micrometer. Therefore, the statements location=2.0<um> and
location=2.0 are equivalent. In this section, units are given
explicitly.
You c an label a line w ith th e tag keyword for later use in the region command.
Defining Initial Simulation Domain
The initial simulation domain is defined with the region command:
region Silicon xlo=SiTop xhi=SiBottom
The keyword Silicon specifies the material of the region. The keywords xlo and xhi take
tags as arguments, which are defined in the line command.
NOTE For 2D and 3D, the additional keywords ylo, yhi, zlo, and zhi are
used to define rectangular or cuboidal regions. In general, the initial
simulation domain can consist of several regions.
Initializing the Simulation
The simulation is initialized with the init command:
init concentration=1.0e15<cm-3> field=Boron
Here, the initial boron concentration in the silicon wafer (as defined in the previous region
command) is set to .
1015 cm 3–

1: Getting Started
Example: 1D Simulation
6Sentaurus™ Process User Guide
H-2013.03
Choosing Process Models and Parameters
The set of physical models and parameters to be used is declared with the
AdvancedCalibration command:
AdvancedCalibration I-2013.12
This command loads the Advanced Calibration set of models and parameters. This is
recommended for accurate process simulation of all silicon and germanium technologies. For
more information about the Advanced Calibration models and parameters, refer to the
Advanced Calibration for Process Simulation User Guide.
Setting Up a Meshing Strategy
The initial grid is valid until the first command that changes the geometry, such as oxidation,
deposition, and etching. For these steps, a remeshing strategy must be defined.
The Sentaurus Mesh meshing engine tries to preserve the initial mesh as much as possible and
only modifies the mesh in the new layers and in the vicinity of the new interfaces.
To define a remeshing strategy, use:
pdbSet Grid SnMesh min.normal.size 0.003
pdbSet Grid SnMesh normal.growth.ratio.2d 1.4 ;# this is for 1D and 2D
where:
■The command pdbSet is used to set the parameter value in parameter database (PDB).
■The parameter min.normal.size determines the grid spacing of the first layer starting
from the interface in micrometers.
■The parameter normal.growth.ratio.2d determines how fast the grid spacing can
increase from one layer to another. This parameter is unitless.
■The semicolon hash mark (; #) indicates the end of the command line and starts the inline
comments.
Growing Screening Oxide
The 1D process simulation is started by thermally growing a thin layer of sacrificial screening
oxide:
gas_flow name=O2_1_N2_1 pressure=1<atm> flowO2=1.2<l/min> flowN2=1.0<l/min>
diffuse temperature=900<C> time=40<min> gas_flow=O2_1_N2_1
1: Getting Started
Example: 1D Simulation
Sentaurus™ Process User Guide 7
H-2013.03
The gas_flow statement is used to specify the gas mixture. The name keyword defines a
gas_flow record for later use in a diffuse command. The pressure of the ambient gas is set
to 1 atm, and the flows of oxygen and nitrogen are set to 1.2 l/minute and 1.0 l/minute,
respectively.
NOTE Other gas flow parameters, such as ambient gases and partial pressures,
can be defined as well (see gas_flow on page 935 for details).
The thermal oxidation step is started with the diffuse command. Here, the wafer is exposed
to the oxidizing gases, defined in the gas_flow statement, for 20 minutes at an ambient
temperature of .
NOTE More options, such as temperature ramps and numeric parameters, are
available (see Oxidation on page 615 for details).
Sentaurus Process prints information about the progress of the oxidation step:
Anneal step: Time=40min, Ramp rate=0C/s, Temperature=900.0C
Temperature > minT. Diffusion: On Reaction: On Assembly: Serial
SProcess parallel assembly thread count = 1
Reaction : 0s to 0.0001s step : 0.0001s temp: 900.0C
SProcess Pardiso thread count = 1
Mechanics: 0s to 0.0001s step : 0.0001s temp: 900.0C
--- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --
Initializing:
--- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --
Initialization is done.
--- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --- --
Diffusion: 0s to 0.0001s step (d): 0.0001s temp: 900.0C
Reaction : 0.0001s to 0.0001712s step : 7.125e-05s temp: 900.0C
Mechanics: 0.0001s to 0.0001712s step : 7.125e-05s temp: 900.0C
Diffusion: 0.0001s to 0.0001712s step (d): 7.125e-05s temp: 900.0C
Reaction : 0.0001712s to 0.0002387s step : 6.741e-05s temp: 900.0C
Mechanics: 0.0001712s to 0.0002387s step : 6.741e-05s temp: 900.0C
Diffusion: 0.0001712s to 0.0002387s step (d): 6.741e-05s temp: 900.0C
...
Reaction : 37.29min to 40min step : 2.714min temp: 900.0C
Mechanics: 37.29min to 40min step : 2.714min temp: 900.0C
Diffusion: 37.29min to 40min step (d): 2.714min temp: 900.0C
Elapsed time for diffuse 41.34s
900°C

1: Getting Started
Example: 1D Simulation
8Sentaurus™ Process User Guide
H-2013.03
Measuring Oxide Thickness
To measure the thickness of the thermally grown oxide, use:
select z=1
layers
The select command chooses a quantity for postprocessing. Selecting 1 is a way to obtain
the material thicknesses.
The layers command prints a list of regions with their respective top and bottom coordinates.
This command also gives the integral over the selected quantity in each region. Having selected
1, the integral equals the thickness (in units of cm):
{ Top Bottom Integral Material }
{ -6.178796082035e-03 3.676329713272e-03 9.855125795306e-07 Oxide }
{ 3.676329713272e-03 2.000000000000e+00 1.996323670287e-04 Silicon }
Here, 3.67 nm of silicon was consumed in the thermal oxidation process, and the final oxide
thickness is 9.85 nm.
NOTE Internally, Sentaurus Process uses centimeters (cm) as the unit for
length.
Selecting boron, the output of layers command would look like:
{ Top Bottom Integral Material }
{ -6.178796082035e-03 3.676329713272e-03 3.012697967871e+09 Oxide }
{ 3.676329713272e-03 2.000000000000e+00 1.969873116640e+11 Silicon }
The integral boron concentration in the silicon layer is:
(1)
which is consistent with the specified wafer doping.
Depositing Screening Oxide
A faster alternative to the simulation of the oxide growth is to deposit an oxide layer and to
simulate afterwards a thermal cycle to account for the thermal budget during the oxidation.
This is an efficient way to emulate the creation of the screen oxide if oxidation-enhanced
diffusion (OED) and the silicon consumption during the oxidation are not important.
1.97 11
×10 cm 2–115
×10 cm 3–24–
×10 cm 3.67 7–
×10 cm–()=

1: Getting Started
Example: 1D Simulation
Sentaurus™ Process User Guide 9
H-2013.03
To deposit a 10 nm layer of screening oxide and perform a thermal cycle in an inert
environment, use:
deposit Oxide type=isotropic thickness=10.0<nm>
diffuse temperature=900<C> time=40<min>
The diffuse command assumes an inert environment if no gas flow is specified.
When you want to omit the oxide growth but OED is not negligible, specification of a reacting
ambient together with the following flag:
pdbSetBoolean Grid Reaction.Modify.Mesh 0
switches on OED without applying velocities to the mesh nodes. This is often used in three
dimensions.
Tcl Control Statements
Tcl constructs can be freely used in the command file of Sentaurus Process. (For an
introduction to Tcl, refer to the Tool Command Language module in the TCAD Sentaurus
Tutorial.)
The following code segment simulates oxidation or performs a deposition depending on the
value of the Tcl variable SCREEN:
set SCREEN Grow
if { $SCREEN == "Grow" } {
#--- Growing screening oxide -----------------------------------------
gas_flow name=O2_1_N2_1 pressure=1<atm> flowO2=1.2<l/min> flowN2=1.0<l/min>
diffuse temperature=900<C> time=40<min> gas_flow=O2_1_N2_1
} else {
#--- Depositing screening oxide --------------------------------------
deposit Oxide type=isotropic thickness=10.0<nm>
diffuse temperature=900<C> time=40<min>
}
Implantation
To implant arsenic with an energy of 50 keV, a dose of , an implant tilt of , and
a wafer rotation , use:
implant Arsenic energy=50<keV> dose=1e14<cm-2> tilt=7<degree> \
rotation=0<degree>
1014 cm 2–
7°
0°

1: Getting Started
Example: 1D Simulation
10 Sentaurus™ Process User Guide
H-2013.03
where “\” immediately followed by a new line (without any space in between) is used to
continue a command line. Sentaurus Process reports:
Species = Arsenic
Dataset = Arsenic
Energy = 30keV
Dose (WaferDose) = 1e+14/cm2
BeamDose = 1.0075e+14/cm2
Tilt = 7deg
Rotation = 0deg
Temperature = 300.00K
Total implant time: 0.61sec
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
Dose in: Silicon_1 Oxide_1 Total
Silicon Oxide
Boron 1.9699e+11 3.0127e+09 2.0000e+11
Arsenic 9.9703e+13 2.7722e+12 1.0247e+14
Int 9.4629e+07 7.8031e+02 1.1463e+08
Vac 8.9179e+09 1.3391e+06 8.9393e+09
ICluster 2.2353e+07 9.8551e+00 4.2353e+07
O2 1.9963e-04 2.6215e+10 3.6215e+10
B4 3.0629e-10 0.0000e+00 3.0629e-10
- - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - - -
The report shows that due to the nonzero tilt angle, Sentaurus Process adapted the beam dose
so that the total dose deposited on the wafer is as specified. The slice angle denotes the angle
between the simulation plane and the normal to the wafer flat. By default, the simulation
domain is parallel to the wafer flat.
The report shows the integrated doping concentrations for each species and region.
Saving the As-Implanted Profile
To save the as-implanted profile, use:
SetPlxList { BTotal Arsenic_Implant }
WritePlx 1DasImpl.plx
The SetPlxList command defines which solution variables are to be saved in the .plx file.
Here, only the total (chemical) boron and the as-implanted arsenic concentrations are saved. If
the SetPlxList command is omitted, all available solutions are saved in the .plx file by
default.

1: Getting Started
Example: 1D Simulation
Sentaurus™ Process User Guide 11
H-2013.03
Besides the file name, here 1DasImpl.plx, the WritePlx command also accepts a material
specifier, which restricts the plot to the given material. For 2D and 3D structures, the x-, y-, or
z-coordinates of the 1D cutline must be given.
Figure 1 As-implanted arsenic profiles and background boron concentration
Figure 1 shows the as-implanted arsenic profiles and the background boron concentration. The
black vertical line marks the oxide–silicon interface. Note the boron depletion at the interface,
which is caused by boron segregation during the oxide growth.
Figure 1 is generated by loading the .plx file into Inspect with:
> inspect 1DasImpl.plx
Thermal Annealing, Drive-in, Activation, and Screening
Oxide Strip
To anneal the damage during implantation, or to drive the dopants deeper into the substrate, or
to activate the implanted dopants in an inert environment, use:
diffuse temperature=1000<C> time=30<min>
strip Oxide
SetPlxList { BTotal BActive AsTotal AsActive }
WritePlx 1Danneal.plx
Here, the structure is annealed at a constant temperature of for 30 minutes. The
annealing is performed in an inert gas because no particular environment is specified.
1000°C
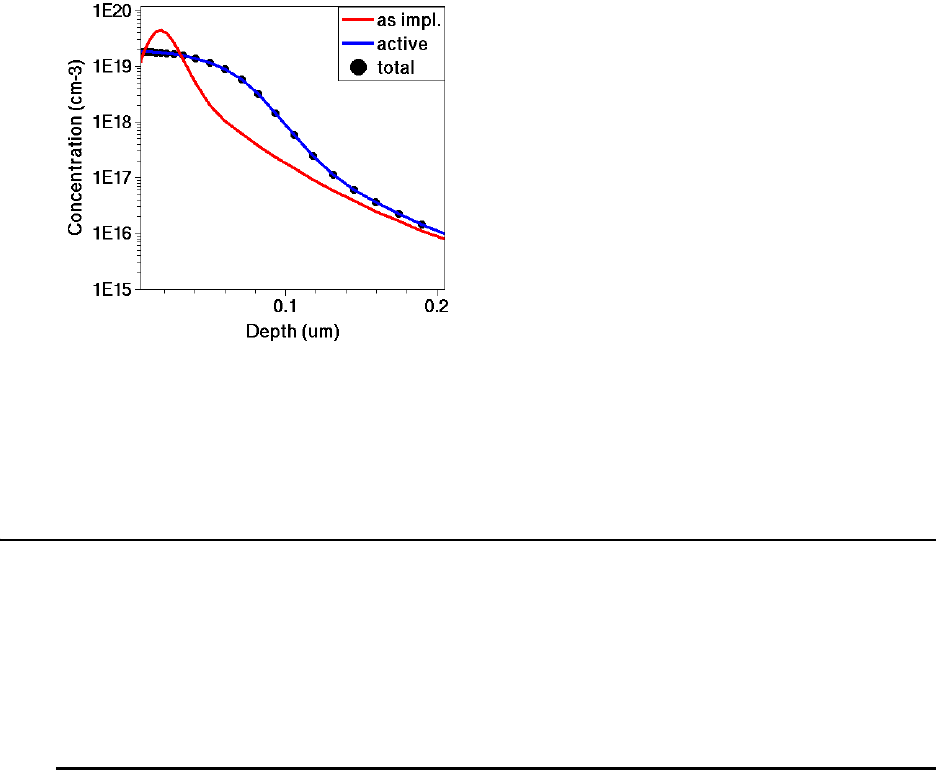
1: Getting Started
Example: 2D Simulation
12 Sentaurus™ Process User Guide
H-2013.03
The annealed profiles are written to the file 1Danneal.plx. The total (chemical)
concentration of boron and arsenic, as well as the respective electrically active (substitutional)
concentrations are saved.
Figure 2 Comparison of as-implanted and annealed arsenic profiles
Figure 2 compares the as-implanted and the annealed arsenic profiles. It is generated by
loading both .plx files into Inspect with:
> inspect 1DasImpl.plx 1Danneal.plx
Example: 2D Simulation
Many widely used process and control commands are introduced in the context of a nominal
n-channel MOSFET process flow. The MOSFET structure is simulated in 2D, and
the processing of the isolation is excluded. A simplified treatment is presented using only
default parameters and models.
Defining Initial Structure and Mesh Refinement
The command math coord.ucs is used to switch on the unified coordinate system (UCS).
Using the UCS is recommended because the default behavior is to rotate the structure when
saving and loading to the DF–ISE coordinate system. With the UCS, the structure is not
rotated. Therefore, the axes in Tecplot SV match the axes in the Sentaurus Process command
file. It is recommended to insert this as the first command in the command file.
The line command is used to:
■Define the initial size of the structure.
0.18 µm
1: Getting Started
Example: 2D Simulation
Sentaurus™ Process User Guide 13
H-2013.03
■Subdivide the structure.
Mesh refinement starts from the user-defined subdivisions; therefore, the specification of lines
helps to compartmentalize mesh refinement. In turn, compartmentalization of the mesh
prevents moving boundaries, and therefore, moving mesh refinement from affecting
geometrically static areas. Whenever mesh lines move, interpolation must be used to obtain
new field values, such as dopant concentrations, and this introduces errors in the simulation.
During the polysilicon reoxidation step, the oxide–silicon and oxide–polysilicon boundaries
move, and this interface movement may cause mesh lines to move. This could be prevented by
inserting lines as follows:
line x location= 0.0
line x location= 3.0<nm> ;# just deeper than reox in silicon
line x location= 10.0<um>
line y location= 0.0
line y location= 85.0<nm> ;# just deeper than reox in poly
line y location= 0.4<um>
To minimize this effect, the silicon and polysilicon regions are isolated from the moving
interfaces by introducing lines immediately inside the final oxide depth in both regions as
shown in Figure 3.
Figure 3 Final structure showing placement of user-defined lines: these lines are used to
isolate silicon and polysilicon regions from boundary movement at the oxide
interfaces
Sentaurus Process uses coordinate systems such that 1D, 2D, and 3D simulations are
consistent. Independent of the current simulation dimension, the positive x is into the wafer; y
is positive to the right, and z is positive out of the page.
NOTE By default, the simulation dimension is promoted only when necessary.
Therefore, until a mask is introduced, the simulation remains in 1D.
Similarly, when going from 2D to 3D, until a 3D mask is introduced
Y
X
0 0.1 0.2 0.3
-0.1
0
0.1
User-defined mesh lines
1: Getting Started
Example: 2D Simulation
14 Sentaurus™ Process User Guide
H-2013.03
(one that varies in the z-direction in the defined simulation domain), the
simulation remains in 2D.
The initial simulation domain is defined with the region command. Many, if not most,
simulations start with a block of silicon. The shorthand for this situation is to define a region
of silicon that spans all defined lines:
region Silicon
The region command also can be used to define a new region between specified lines. To limit
the size of the region to be less than all defined lines, the lines must be given a tag with the tag
parameter. These tags are used in the region command with the xlo, xhi, ylo, yhi, zlo,
and zhi parameters.
Finally, the initial mesh and background doping is specified using the init command as
follows:
init concentration=1.0e+15<cm-3> field=Phosphorus wafer.orient=100
Here, an n-doped substrate with a phosphorus concentration of is used. The wafer
orientation is set to 100, which is the default.
The Advanced Calibration set of physical models and parameters is loaded (this is the
recommended choice for accurate process simulation):
AdvancedCalibration I-2013.12
Usually, localized refinement is defined by introducing refinement boxes. This strategy
prevents excessive mesh that can result if mesh refinement is based solely on the line
command (with the spacing parameter). Lines specified with the line command run the
entire length (or breadth or depth) of the structure.
The refinement boxes can be inserted at any time during the simulation. The simplest form of
the refinement box, used in this example, consists of minimum and maximum coordinates
where the refinement box is valid and local maximum mesh spacing in the x-, y- and
z-directions. A refinement box specified for a 2D simulation will be applied to 1D if it is valid
for y = 0.0. Similarly a 3D refinement box will be applied if it covers z = 0.0.
The following refinement boxes specify refinement only in the x-direction for the 1D part of
the simulation:
#--- Refinement in vertical direction ---------------------------------
refinebox clear ;# remove all default refinement
refinebox min = 0 max = 50.0<nm> xrefine = {2.0<nm> 10.0<nm>}
refinebox min = 50.0<nm> max = 2.0<um> xrefine = {10.0<nm> 0.1<um> 0.2<um>}
refinebox min = 2.0<um> max = 10.0<um> xrefine = {0.2<um> 2.0<um>}
1015 cm 3–

1: Getting Started
Example: 2D Simulation
Sentaurus™ Process User Guide 15
H-2013.03
The other type of refinement box used in this example is the interface refinement type. Interface
refinement is a graded refinement that is refined near an interface in the perpendicular direction
and relaxed away from the interface. Using the refinebox command, you can specify
interface refinement using the interface.materials or interface.mat.pairs
parameter:
■Use interface.materials to indicate refinement will occur at all interfaces to the
specified materials.
■Use interface.mat.pairs to choose interface refinement only at specific material
interfaces.
#--- Interface refinement ---------------------------------------------
refinebox interface.materials = { PolySilicon Silicon }
For more details on mesh refinement, see Mesh Refinement on page 694.
Implanting Boron
First, three sets of boron implants are performed:
implant Boron dose=2.0e13<cm-2> energy=200<keV> tilt=0 rotation=0
implant Boron dose=1.0e13<cm-2> energy= 80<keV> tilt=0 rotation=0
implant Boron dose=2.0e12<cm-2> energy= 25<keV> tilt=0 rotation=0
The first high-energy implant creates the p-well, the second medium-energy implant defines a
retrograde boron profile to prevent punch-through, and the third low-energy implant is for a Vt
adjustment.
Growing Gate Oxide
The gate oxide is grown at a temperature of for 10 minutes in pure oxygen using:
diffuse temperature=850<C> time=10.0<min> O2
select z=Boron
layers
The layers command shows that the thickness of the grown oxide is 3.2 nm:
{ Top Bottom Integral Material }
{ -2.500551327519e-03 7.862861879285e-04 1.247399405710e+10 Oxide }
{ 7.862861879285e-04 1.000000000000e+01 3.197435354292e+13 Silicon }
For details, see Measuring Oxide Thickness on page 8.
850°C

1: Getting Started
Example: 2D Simulation
16 Sentaurus™ Process User Guide
H-2013.03
Defining Polysilicon Gate
The polysilicon gate is created using:
deposit PolySilicon type=isotropic thickness=0.18<um>
mask name=gate_mask left=-1 right=90<nm>
etch PolySilicon type=anisotropic thickness=0.2<um> mask=gate_mask
etch Oxide type=anisotropic thickness=0.1<um>
First, of polysilicon is deposited over the entire structure. The keyword
type=isotropic means that the layer is grown equally in all directions, but since the
simulation is in 1D, it would be the same as type=anisotropic.
A mask is defined to protect the gate area with the mask command. In this project, only half of
the transistor is simulated. Therefore, the left edge of the gate mask is unimportant. In general,
you should run the mask over the sides of the simulation to prevent round-off errors that could
prevent complete mask coverage. The name gate_mask is associated with this mask for later
reference.
The first etch command refers to the previously defined mask and, therefore, only the exposed
part of the polysilicon is etched. The requested etching depth is larger than the
deposited layer. This overetching ensures that no residual islands remain. The etching is
specified to be anisotropic, that is, the applied mask is transferred straight down, without any
undercut.
The second etch statement does not refer to any masks. However, the polysilicon naturally
acts as a mask for this selective etching process. Again, a considerable overetching is specified.
Working with Masks
Masks must be defined before they are used. For example, ex_mask blocks processing from
–1 to 2 and from 4 to 20 :
mask clear
mask name=ex_mask segments = { -1.0<um> 2.0<um> 4.0<um> 20.0<um> }
segments
specifies a list of coordinates of mask segments. Several mask segments can be
specified at the same time. The first coordinate defines the beginning of a segment; the second
defines the end of the segment; the third defines the beginning of the segment; and so forth. In
3D simulations, mask segments are extended across the entire structure in the z-direction.
0.18 µm
0.2 µm()
µm
µm
1: Getting Started
Example: 2D Simulation
Sentaurus™ Process User Guide 17
H-2013.03
Masks can be inverted using the negative option. For example, etch_mask prevents
processing from 2 to 4 :
mask clear
mask name=etch_mask segments = { -1.0<um> 2.0<um> 4.0<um> 20.0<um> } negative
Commands that use masking include etch, photo, and deposit.
Polysilicon Reoxidation
To release stresses, a thin oxide layer is grown on the polysilicon before the spacer formation:
diffuse temperature=900<C> time=10.0<min> O2
In all diffusion steps, Sentaurus Process automatically deposits a thin native oxide layer before
starting oxidation. This layer is always present on silicon exposed to air and quickly forms on
newly created interfaces.
Figure 4 Polysilicon reoxidation
During oxidation, mesh movement is controlled by the TSUPREM-4 mesh library in 2D. In 1D
and 3D, it is controlled by an internal moving-boundary mesh algorithm. Both of these moving-
boundary algorithms perform local atomic mesh operations (element removal, edge splitting,
edge flipping, and so on) which leave the rest of the mesh untouched. Mesh points are moved
with the material to maintain dopant dose conservation and the dopant segregation condition at
oxide–silicon and oxide–polysilicon interfaces. Figure 5 shows a close-up of the mesh after the
µm
0 0.05 0.1 0.15
-0.15
-0.1
-0.05
0
1: Getting Started
Example: 2D Simulation
18 Sentaurus™ Process User Guide
H-2013.03
polysilicon reoxidation step has been performed. Note that the mesh in the brown oxide layer
follows the growth contours.
Figure 5 Mesh in thin oxide layer and in adjacent polysilicon and silicon
Saving Snapshots
To save a snapshot of the current structure, the struct command is used. For example:
struct tdr= NMOS4
The keyword tdr specifies that the snapshot is saved in the TDR file format. The argument
specifies the stem used for the file name. Here, the file NMOS4_fps.tdr is created. The figures
in this section were generated from such snapshots.
For more information about the TDR format, refer to the Sentaurus™ Data Explorer User
Guide.
Remeshing for LDD and Halo Implants
Next, the LDD and halo implants are performed. Before that, however, the mesh must be
refined to properly capture the implant. The previously defined refinement boxes specified
vertical refinement with the xrefine parameter.
Now, lateral refinement is required to resolve the source and drain extensions (also known as
low-doped drain (LDD)) as well as the halo implants. This is accomplished by introducing a
new refinebox command that specifies:
■Lateral refinement using the yrefine parameter.
■Additional vertical refinement using the xrefine parameter.
0.080.085 0.09 0.095
-0.01
-0.005
0
0.005
1: Getting Started
Example: 2D Simulation
Sentaurus™ Process User Guide 19
H-2013.03
NOTE When specifying multiple overlapping refinement, the most refined
specification (smallest edge length) wins.
refinebox silicon min= {0.0 0.045<um>} max= {0.1<um> 0.125<um>} \
xrefine= 0.01<um> yrefine= 0.01<um>
grid remesh
The min and max keywords take x-, y-, and z-coordinates. Not all coordinates must be
specified. For example, if only one number is given for minimum, it means that refinement
applies to all y- and z-coordinates less than the max coordinate.
NOTE The refinebox command only specifies a refinement criterion, but the
mesh is not changed. The grid remesh command forces a remesh.
Figure 6 A combination of overlapping refinement boxes is used to define a finer mesh for
LDD and halo; if multiple criteria overlap, the finest mesh specification wins
Implanting LDD and Halo
The LDD and halo implants are performed using:
#--- LDD implantation -------------------------------------------------
implant Arsenic dose=4e14<cm-2> energy=10<keV> tilt=0 rotation=0
#--- Halo implantation: Quad HALO implants ----------------------------
implant Boron dose=1.0e13<cm-2> energy=20<keV> tilt=30<degree> \
rotation=0 mult.rot=4
diffuse temperature=1050<C> time=5.0<s>
0.05 0.1 0.15
-0.06
-0.04
-0.02
0
0.02
0.04
0.06
0.08

1: Getting Started
Example: 2D Simulation
20 Sentaurus™ Process User Guide
H-2013.03
The LDD implant uses a high dose of and a relatively low energy of 10 keV.
The halo is created by a quad implant using the mult.rot parameter, that is, the implant is
performed in four steps. Each step is separated in rotation by 360/4 = starting with the
specified rotation of 0. This is performed to ensure that the boron penetrates well into the
channel at the tips of the source–drain extensions. Again, a relatively high total dose of
is used.
The implants are activated with a short thermal cycle or rapid thermal anneal (RTA).
Forming Nitride Spacers
The nitride spacers are formed using:
#--- Nitride spacer ---------------------------------------------------
deposit Nitride type=isotropic thickness=60<nm>
etch Nitride type=anisotropic thickness=84<nm> isotropic.overetch=0.01
etch Oxide type=anisotropic thickness=10<nm>
First, a uniform, 60-nm thick layer of nitride is deposited over the entire structure. The keyword
type=isotropic ensures that the growth rate of the layer is the same in all directions. Then,
the nitride is etched again; however, now an anisotropic etching is used. This means that the
nitride deposited on the vertical sides of the gate is not fully removed and can serve as masks
for the source/drain implants. For this step, an isotropic overetch is specified. Specifying a
fraction of the etch thickness, 0.01 implies a 1% isotropic component. This is needed because
the oxide formed during poly oxidation has a nonvertical sidewall. Without the small
isotropic.overetch, a small nitride residual would remain. Finally, the thin oxide layer
grown during the poly reoxidation step is removed.
Remeshing for Source/Drain Implants
Next the source/drain implants are performed. However, before that, the mesh is refined again.
refinebox Silicon min= {0.04<um> 0.11<um>} max= {0.18<um> 0.4<um>} \
xrefine= 0.01<um> yrefine= {0.02<um> 0.05<um>}
grid remesh
This refinement box ensures that the grid is fine enough in the vertical direction to resolve the
junction depth.
410
14
× cm 2–
90°
110
14
× cm 2–

1: Getting Started
Example: 2D Simulation
Sentaurus™ Process User Guide 21
H-2013.03
Implanting Source/Drain
The source and drain regions are created using:
implant Arsenic dose=5e15<cm-2> energy=40<keV> tilt=7<degree> \
rotation=-90<degree>
diffuse temperature=1050<C> time=10.0<s>
To ensure a low resistivity of the source and drain regions, this implant step uses a very high
dose of . A tilt of is used to reduce channeling and a rotation of
ensures that the plane of incident is parallel to the gate stack, such that the tilt angle does
not lead to asymmetry between the source and drain.
Transferring to Device Simulation
To transfer from process simulation to device simulation, the normal steps are:
■The structure bottom is cropped.
■The full transistor is created by reflecting about the symmetry plane.
■A new mesh strategy is specified appropriate for device simulation.
■Contacts are specified.
■The struct command is called which remeshes and saves the structure.
Remeshing for Device Simulation
The following example shows the standard technique used to produce a structure and mesh
appropriate for device simulation. First, the structure bottom is truncated; then a new mesh
strategy is introduced:
#--Remove bottom of structure------------------------------------------
transform cut location= 1.00 down
#--Change refinement strategy and remesh-------------------------------
refinebox clear
line clear
pdbSet Grid Adaptive 1
pdbSet Grid AdaptiveField Refine.Abs.Error 1e37
pdbSet Grid AdaptiveField Refine.Rel.Error 1e10
pdbSet Grid AdaptiveField Refine.Target.Length 100.0
pdbSet Grid SnMesh DelaunayType boxmethod
refinebox name= Global refine.min.edge= {0.01 0.01} \
510
15
× cm 2–
7°
90–°
7°
1: Getting Started
Example: 2D Simulation
22 Sentaurus™ Process User Guide
H-2013.03
refine.max.edge= {0.1 0.1} refine.fields= { NetActive } \
def.max.asinhdiff= 0.5 adaptive
refinebox name= SiGOX min.normal.size= 0.2<nm> normal.growth.ratio= 1.4 \
max.lateral.size= 5.0<nm> min= {-0.01 -0.1} max= {0.01 0.1} \
interface.materials= {Silicon}
refinebox name= GDpn1 min= {0.0 0.04} max= {0.06 0.1} xrefine= 0.005 \
yrefine= 0.005 silicon
refinebox name= TopActive min= {0.0 0.0} max= {0.3 0.4} \
refine.min.edge= {0.02 0.02} refine.max.edge= {0.05 0.05} \
refine.fields= { NetActive } def.max.asinhdiff= 0.5 \
adaptive silicon
grid remesh
#--Reflect ---------------------------------------------------------
transform reflect left
The new mesh strategy uses a combination of interface refinement, fixed boxwise refinement,
and adaptive refinement on dopants.
Contacts
Next, contacts are added to the structure using the contact command. These contacts are
added to structure files upon writing. They are not present in the internal Sentaurus Process
structure, but are added only as required when writing the structure. There are two types of
contact specification:
■Box: For these contacts, you specify a box and a material, and all interfaces of that material
that are inside the box become the contact.
■Point: For this contact, you specify a point inside a chosen region. The chosen region is
removed, and all interfaces between the chosen region and bulk materials become part of
the contact.
In the following example, only box-type contacts are used:
#--- Contacts ---------------------------------------------------------
contact name= "substrate" bottom Silicon
contact name= "source" box Silicon adjacent.material= Gas \
xlo= 0.0 xhi= 0.005 ylo= -0.4 yhi= -0.2
contact name= "drain" box Silicon adjacent.material= Gas \
xlo= 0.0 xhi= 0.005 ylo= 0.2 yhi= 0.4
1: Getting Started
Example: 2D Simulation
Sentaurus™ Process User Guide 23
H-2013.03
contact name= "gate" box PolySilicon xlo= -0.181 xhi= -0.05 \
ylo= -0.088 yhi= 0.088
Saving the Structure
To save the structure, use:
struct tdr=NMOS !Gas
The file NMOS_fps.tdr is created with contacts and can be loaded into Sentaurus Device to
obtain device electrical characteristics.
Figure 7 Final structure showing contacts and refinement appropriate for device simulation
Extracting 1D Profiles
You can save 1D profiles at any point in the process flow using:
SetPlxList {BTotal NetActive}
WritePlx NMOS_channel.plx y=0.0 silicon
as well as:
struct tdr=NMOS_channel.tdr y=0.0
For details, see Saving the As-Implanted Profile on page 10.
-0.4 -0.3 -0.2 -0.1 0 0.1 0.2 0.3 0.4
-0.2
-0.1
0
0.1
0.2

1: Getting Started
Adaptive Meshing: 2D npn Vertical BJT
24 Sentaurus™ Process User Guide
H-2013.03
Adaptive Meshing: 2D npn Vertical BJT
A simple 2D npn vertical bipolar transistor example is introduced to show how the adaptive-
meshing capabilities in Sentaurus Process can be used to ease mesh setup and allow for mesh
evolution during dopant diffusion. For examples, see 2D npn Vertical Bipolar on page 38.
For all the applications involving long thermal diffusion steps or simulations of relatively large
structures (in which doping profiles may evolve greatly), using static mesh criteria is
impracticable because it requires using a fine mesh in many parts of the simulation domain.
Moreover, the placement of the refinement boxes is not straightforward because often the
location of gradients and junctions at the end of the thermal steps is not precisely known. For
such purposes, adaptive meshing could be used. Using this feature, you only have to define
some refinement criteria, more or less stringent depending on the level of accuracy required.
The meshing engine checks the mesh and decides automatically where, when, and if the mesh
needs to be refined.
Overview
Adaptive meshing can be switched on globally with:
pdbSet Grid Adaptive 1
which creates a default adaptive box covering the entire structure.
Adaptive refinement parameters can be set in the following ways:
■Fieldwise in the PDB with the pdbSet command
■Boxwise as parameters of the refinebox command
■Materialwise, specifying a material in a box definition
■Regionwise, specifying a region in a box definition
To prevent the number of mesh points from growing too large, switch off the keep.lines
option (which is switched on by default in silicon) when using adaptive meshing:
refinebox !keep.lines
Many different refinement criteria have been implemented in Sentaurus Process for flexibility
in handling different types of field and structure. For a complete list and detailed descriptions
of the refinement criteria, see Adaptive Refinement Criteria on page 700.
1: Getting Started
Adaptive Meshing: 2D npn Vertical BJT
Sentaurus™ Process User Guide 25
H-2013.03
The criteria in the following example are the most commonly used and are referred to as
relative difference and local dose error. Each computes the so-called desired edge length
(DEL), which is defined formally as:
DEL = min (l
12
* MaxError/Error)
where l12 is the length of the edge between two mesh points 1 and 2. Error (computed
internally) is the error between points 1 and 2, and MaxError (set by users) is the maximum
allowable error. The right-hand side of the expression is computed over all the fields that can
be refined (by default, all the solution variables): the minimum value is the DEL for the
corresponding criterion. The expression for Error and the name and the meaning of
MaxError vary from criterion to criterion. For the relative difference criterion, these quantities
have the form:
Error = 2*|C
1
- C
2
|/(C
1
+ C
2
+ alpha)
MaxError = R
f
where and are the concentration of the field in points 1 and 2, respectively, is the
relative error that sets the maximum-allowed change of the field across an edge, and alpha is
the absolute error, a type of cutoff threshold below which refinement is smoothed out. They can
be set in the PDB as follows:
pdbSet Grid Boron Refine.Abs.Error 1e15
pdbSet Grid Boron Refine.Rel.Error 0.5
or in the refinebox commands as:
refinebox name=Active refine.fields= {Boron Arsenic} \
rel.error= {Boron=0.5 Arsenic=0.5} abs.error= {Boron=2e15 Arsenic=1e16} \
Adaptive min= {-1.0 -0.1} max= {2.0 16.0}
For the definition of Error and MaxError for the local dose error criterion, see Local Dose
Error Criteria on page 703.
All the edges are compared to DEL to check the percentage of long edges by using the
following additional parameter:
pdbSetDoubleArray Grid Refine.Factor {X 2.0 Y 2.0}
These coefficients can be set directionwise and act in the following way: An edge is defined as
long when it is larger than Refine.Factor*DEL for at least one of the selected refinement
criteria. When the percentage of long edges is larger than certain values, adaptive refinement
is actually triggered. This value can be set as:
pdbSet Grid Refine.Percent 0.01
When adaptive meshing is switched on, it automatically affects refinement whenever a mesh is
generated (such as after geometry-changing operations). During the diffuse command, the
C1
C2
Rf

1: Getting Started
Adaptive Meshing: 2D npn Vertical BJT
26 Sentaurus™ Process User Guide
H-2013.03
mesh is checked after a certain number of steps that can be separately set depending on the
nature of the diffusion step:
pdbSet Diffuse Compute.Regrid.Steps 10 ;# during inert annealings
pdbSet Diffuse Growth.Regrid.Steps -1 ;# during oxidation and silicidation
pdbSet Diffuse Epi.Regrid.Steps -1 ;# during epitaxy
When the number of long edges is larger than Refine.Percent, remeshing is performed. The
mesh quality check can be omitted by setting:
pdbSet Grid Refinement.Check 0
which can save some CPU time when performing simulations on large meshes, where the mesh
checking is time consuming.
NOTE Formally, the adaptive-meshing feature consists of field-based and
implant-based adaptation. There is a small difference in the way
refinement criteria are applied. For details, see Adaptive Meshing
during Implantation on page 708 and Interval Refinement on page 704.
However, as the two modules use the same parameters, you do not need
to define them twice.
NOTE Adaptive-meshing syntax to set up parameters is the same in any
dimension.
The relative error criterion is effective in refining doping profiles in steep gradient regions. In
the vicinity of maxima and minima, the profiles are almost flat and some loss of accuracy may
occur there. Further reduction of Rel.Error would increase significantly the number of
points in the steep slope with negligible improvements at the peaks. In that case, the max dose
loss criterion can be used more effectively. This explains why the combination of these two
criteria provides an optimum adaptive-remeshing strategy.
Defining Initial Structure
The command math coord.ucs is used to switch on the unified coordinate system (UCS).
Using the UCS is recommended because the default behavior is to rotate the structure when
saving and loading to the DF–ISE coordinate system. With the UCS, the structure is not
rotated. Therefore, the axes in Tecplot SV match the axes in the Sentaurus Process command
file. It is recommended to insert this as the first command in the command file.
The line commands are used to compartmentalize the structure according to the meshing
strategy described in the previous example:
line x loc= 2.0<um>
line x loc= 4.0<um> tag=SubTop

1: Getting Started
Adaptive Meshing: 2D npn Vertical BJT
Sentaurus™ Process User Guide 27
H-2013.03
line x loc= 6.0<um>
line x loc= 10.0<um> tag=SubBottom
line y loc= 0.0<um> tag=SubLeft
line y loc=1.5<um>
line y loc=2.5<um>
line y loc=8<um>
line y loc=13<um>
line y loc=22<um>
line y loc=24<um>
line y loc=30.0<um> tag=SubRight
Along the x-axis, few lines are specified: the two tagged ones are needed to define the initial
silicon substrate. The other two lines are defined to have uniform spacing within the box
defined to refine the buried layer. Along the y-axis, more lines are defined because a coarse
initial mesh would degrade the quality of the mesh resulting from adaptation during
implantation. These lines are set corresponding to the mask edges: This information is usually
known to users, especially if the simulation starts from a layout, and the process flow is set up
in Ligament.
Adaptive Meshing Settings
As previously mentioned, adaptive parameters can be set in different ways, which lead to
different refinement strategies:
pdbSet Grid Adaptive 1
pdbSet Grid AdaptiveField Refine.Abs.Error 1e25
pdbSet Grid AdaptiveField Refine.Rel.Error 2.0
pdbSet Grid Damage Refine.Min.Value 1e25
pdbSet Grid Damage Refine.Max.Value 1e25
pdbSet Grid Damage Refine.Target.Length 1
Here the following strategy is used:
■The default relative difference–type refinement is switched off by setting high values for
absolute and relative errors and for the interval damage refinement.
■When parameters are set for AdaptiveField, they are applied to all the existing fields
that can be refined.
■Actual refinement will be then controlled in specific regions by using refineboxes.
Three refinement boxes are defined as the structure and the process flow clearly identifies three
main significant areas: buried layer, collector region, and base-emitter region:
refinebox name=BL refine.fields= {Antimony Phosphorus} \
rel.error= {Antimony=0.6 Phosphorus=0.6} \

1: Getting Started
Adaptive Meshing: 2D npn Vertical BJT
28 Sentaurus™ Process User Guide
H-2013.03
abs.error= {Antimony=1e16 Phosphorus=1e16} Adaptive min= {2.0 -0.1} \
max= {10.1 30.1} refine.min.edge= {0.2 0.4} max.dose.error= {Antimony=1e8} \
The min and max parameters set an xy pair of coordinates to define the extent of the box. The
keyword all means that refinement must be applied to all materials. When using a material
name, refinement is applied to the specified material only.
NOTE More than one adaptive type can be specified in the same box. In the BL
box, the relative difference and local dose loss criteria are selected by
specifying the parameters rel.error or abs.error and
max.dose.error, respectively.
refinebox name=Sinker refine.fields= {Phosphorus Arsenic} \
rel.error= {Phosphorus=0.5 Arsenic=0.5} \
abs.error= {Phosphorus=5e15 Arsenic=1e16} Adaptive min= {-1.0 16} \
max= {2.0 30.1} refine.min.edge= {0.1 0.2}
refinebox name=Active refine.fields= {Boron Arsenic} \
rel.error= {Boron=0.5 Arsenic=0.5} abs.error= {Boron=2e15 Arsenic=1e16} \
Adaptive min= {-1.0 -0.1} max= {2.0 16.0} refine.min.edge= {0.025 0.05} \
The BL box is defined to refine the buried layer: a high level of accuracy is not required here
and the values are more relaxed than in the other boxes. The refine.min.edge parameter
adds the additional directionwise constraint not to refine edges below the specified values
(units in micrometers).
The Sinker box is defined to refine the n-doped collector region, which contacts the buried
layer. More restrictive values are used in it.
The Active box is used to refine the base–emitter region. Higher accuracy is required here to
properly catch the base length, which all the main electrical parameters of the device are a
function of:
pdbSet Diffuse Compute.Regrid.Steps 10
pdbSet Grid Refine.Percent 0.01
According to these last two commands, the mesh is checked every 10 diffusion steps in inert
annealings, and remeshing is performed if there are more than 0.01% of long edges.
Buried Layer
The buried layer is obtained with high-energy and high-dose antimony implantation:
deposit material= {Oxide} type=isotropic time=1 rate= {0.025}
implant Antimony dose=1.5e15<cm-2> energy=100<keV>
etch material= {Oxide} type=anisotropic time=1 rate= {0.03}

1: Getting Started
Adaptive Meshing: 2D npn Vertical BJT
Sentaurus™ Process User Guide 29
H-2013.03
Before the implantation, 25 nm of a screening oxide is deposited. Here an alternate syntax is
used to specify the deposit material. The deposited oxide thickness is determined by the
product of rate and time. The implantation is performed with default angles (tilt of and
rotation of ). After the implantation, the oxide is etched to clean the surface and to prepare
it for the subsequent epi step.
Epi Layer
For speed and simplicity, an epitaxial regrowth step is not performed here. Instead, a simpler
deposition of a silicon layer with arsenic concentration is followed by a
diffusion step:
deposit material= {Silicon} type=isotropic time=1 rate= {4.0} Arsenic \
concentration=1e15<cm-3>
diffuse temp=1100<C> time=60<min> maxstep=4<min>
The maximum diffusion step is limited to 4 minutes to avoid having too much diffusion
between two subsequent adaptive remeshing steps. An alternative would be to reduce
Compute.Regrid.Steps, but this would lead to numerous remeshings at the beginning of
the annealing when the time step is small.
The following sections describe the process steps to create sinker, base, and emitter regions. At
the end of each group of steps, results are saved in TDR files.
Sinker Region
This is the beginning of the 2D simulation. A 50-nm screening oxide is deposited before the
phosphorus implantation to contact the buried layer. The Sinker mask protects the silicon area
where the base will be created. The Photo command is used to deposit the photoresist (mask
definition not shown here). The subsequent annealing is long (5 hours). For this reason, the
maximum time step is allowed to increase up to 8 minutes.
Figure 8 on page 30 shows the doping concentration distribution at this point of the simulation:
deposit material= {Oxide} type=isotropic time=1 rate= {0.05}
photo mask=Sinker thickness=1
implant Phosphorus dose=5e15<cm-2> energy=200<keV>
strip Resist
diffuse temp=1100<C> time=5<hr> maxstep=8<min>
struct tdr=vert_npn2
7°
90°
115
×10 cm 3–
1: Getting Started
Adaptive Meshing: 2D npn Vertical BJT
30 Sentaurus™ Process User Guide
H-2013.03
Figure 8 Doping concentration after phosphorus implantation and diffusion to contact
antimony buried layer
Base Region
The p-doped base region is created with a dose of implanted boron followed by
a 35-minute inert annealing:
photo mask=Base thickness=1
implant Boron dose=1e14<cm-2> energy=50<keV>
strip Resist
diffuse temp=1100<C> time=35<min> maxstep=4<min>
struct tdr=vert_npn3
Emitter Region
The highly n-doped emitter region is created with a dose of implanted arsenic
followed by a 25-minute inert annealing. Emitter mask is designed such that arsenic is
implanted also in the sinker region to increase the doping concentration at the collector contact.
In addition to a TDR file, 1D profiles are extracted. Figure 9 on page 31 shows the final doping
distribution:
photo mask=Emitter thickness=1
implant Arsenic dose=5e15<cm-2> energy=55<keV> tilt=7 rotation=0
strip Resist
diffuse temp=1100<C> time=25<min> maxstep=4<min>
struct tdr=vert_npn4
0 5 10 15 20 25 30
0
5
10
114
×10 cm 2–
515
×10 cm 2–
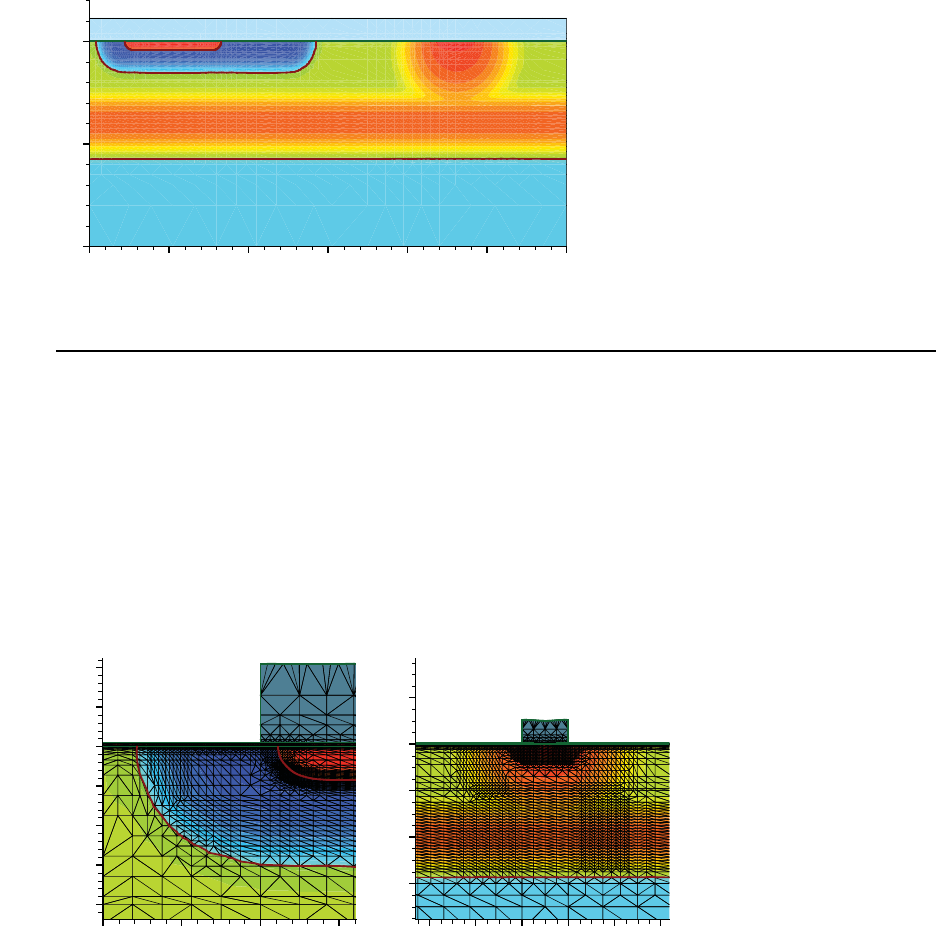
1: Getting Started
Adaptive Meshing: 2D npn Vertical BJT
Sentaurus™ Process User Guide 31
H-2013.03
SetPlxList {BTotal SbTotal AsTotal PTotal}
WritePlx Final.plx y=5.0
WritePlx Sinker.plx y=23.0
Figure 9 Final doping distribution
Backend
The real backend steps are not simulated here. A sequence of masked etching and deposition
steps are used to define emitter, base, and collector contacts:
etch material= {Oxide} type=anisotropic time=1 rate= {0.055} mask=Contact
deposit material= {Aluminum} type=isotropic time=1 rate= {1.0}
etch material= {Aluminum} type=anisotropic time=1 rate= {1.1} mask=Metal
struct tdr=vert_npn5
Figure 10 shows some details of the final mesh.
Figure 10 Details of final mesh: (left) the emitter–base region and (right) the buried layer
with collector contact
0 5 10 15 20 25 30
0
5
10
0 1 2 3
-1
-0.5
0
0.5
1
1.5
2
1820 22 24 26 28
-2
0
2
4
6

1: Getting Started
Full-Text Versions of Examples
32 Sentaurus™ Process User Guide
H-2013.03
The relative difference criterion refines the doping profiles, not the junctions. Obviously, if the
profiles are reproduced correctly, the junctions also will be in the right place. To obtain a
junction-like refinement with the relative difference criterion, set abs.error close to the
doping level of the less-doped side of the junction. A more effective way is to select
NetDoping as the field to be refined and apply to it the inverse hyperbolic sine (asinh)
difference criterion (for details, see Inverse Hyperbolic Sine (asinh) Difference Criteria on
page 702).
Full-Text Versions of Examples
The following full-text versions of the examples allow convenient electronic copying of text
into Sentaurus Process command files.
1D NMOS
# 1D Grid definition
#-------------------
line x location=0.0 spacing= 1<nm> tag=SiTop
line x location= 10<nm> spacing= 2<nm>
line x location= 50<nm> spacing= 10<nm>
line x location=300<nm> spacing= 20<nm>
line x location=0.5<um> spacing= 50<nm>
line x location=2.0<um> spacing=0.2<um> tag=SiBottom
# Initial simulation domain
#--------------------------
region Silicon xlo=SiTop xhi=SiBottom
# Initialize the simulation
#--------------------------
init concentration=1.0e15<cm-3> field=Boron
# Set of physical models and parameters
# ----------------------------------------------
AdvancedCalibration 2013.12
# Settings for automatic meshing in newly generated layers
#---------------------------------------------------------
pdbSet Grid SnMesh min.normal.size 0.003

1: Getting Started
Full-Text Versions of Examples
Sentaurus™ Process User Guide 33
H-2013.03
pdbSet Grid SnMesh normal.growth.ratio.2d 1.4 ;# this is for 1D and 2D
set SCREEN Grow
if { $SCREEN == "Grow" } {
# Growing screening oxide
#------------------------
gas_flow name=O2_1_N2_1 pressure=1<atm> flowO2=1.2<l/min> flowN2=1.0<l/min>
diffuse temperature=900<C> time=40<min> gas_flow=O2_1_N2_1
# Measuring the oxide thickness
#------------------------------
select z=1
layers
} else {
# Depositing screening oxide
#---------------------------
deposit material= {Oxide} type=isotropic time=1.0 rate= {0.01}
diffuse temperature=900<C> time=40<min>
}
# Implanting Arsenic
#-------------------
implant Arsenic energy=30<keV> dose=1e14<cm-2> tilt=7<degree> \
rotation=0<degree>
# Plotting out the "as implanted" profile
#----------------------------------------
SetPlxList { BTotal Arsenic_Implant }
WritePlx 1DasImpl.plx
# Thermal annealing
#------------------
diffuse temperature=1000<C> time=30<min>
strip Oxide
SetPlxList { BTotal BActive AsTotal AsActive }
WritePlx 1Danneal.plx
2D NMOS
#----------------------------------------------------------------------
# 2D nMOSFET (0.18um technology)
#----------------------------------------------------------------------
math coord.ucs
1: Getting Started
Full-Text Versions of Examples
34 Sentaurus™ Process User Guide
H-2013.03
pdbSet Oxide Grid perp.add.dist 1e-7
#--- Specify lines for outer boundary and to separate moving boundaries
# from the rest of the structure------------------------------------
line x location= 0.0
line x location= 3.0<nm> ;# just deeper than reox in silicon
line x location= 10.0<um>
line y location= 0.0
line y location= 85.0<nm> ;# just deeper than reox in poly
line y location= 0.4<um>
#--- Silicon substrate definition -------------------------------------
region silicon
#--- Initialize the simulation ----------------------------------------
init concentration=1.0e+15<cm-3> field=Phosphorus
# Set of physical models and parameters
# ------------------------
AdvancedCalibration 2013.12
#--- Refinement in vertical direction ---------------------------------
refinebox clear
refinebox min = 0 max = 50.0<nm> xrefine = {2.0<nm> 10.0<nm>}
refinebox min = 50.0<nm> max = 2.0<um> xrefine = {10.0<nm> 0.1<um> 0.2<um>}
refinebox min = 2.0<um> max = 10.0<um> xrefine = {0.2<um> 2.0<um>}
#--- Interface refinement ---------------------------------------------
refinebox interface.materials = { PolySilicon Silicon }
#--- Sentaurus Mesh settings for automatic meshing in newly generated layers -
pdbSet Grid SnMesh min.normal.size 1.0e-3 ;# in micrometers
pdbSet Grid SnMesh normal.growth.ratio.2d 1.4 ;# used in 1D and 2D
#--- Create starting mesh from lines and refinement
grid remesh
#--- p-well, anti-punchthrough & Vt adjustment implants ---------------
implant Boron dose=2.0e13<cm-2> energy=200<keV> tilt=0 rotation=0
implant Boron dose=1.0e13<cm-2> energy= 80<keV> tilt=0 rotation=0
implant Boron dose=2.0e12<cm-2> energy= 25<keV> tilt=0 rotation=0
#--- p-well: RTA of channel implants ----------------------------------
diffuse temperature=1050<C> time=10.0<s>
1: Getting Started
Full-Text Versions of Examples
Sentaurus™ Process User Guide 35
H-2013.03
#--- Saving structure -------------------------------------------------
struct tdr=NMOS1 FullD; # p-Well
#--- Gate oxidation ---------------------------------------------------
diffuse temperature=850<C> time=10.0<min> O2
select z=Boron
layers
struct tdr=NMOS2 FullD; # GateOx
#--- Poly gate deposition ---------------------------------------------
deposit poly type=isotropic thickness=0.18<um>
#--- Poly gate pattern/etch -------------------------------------------
# MGoals settings for etch/depo
mgoals accuracy=2e-5
mask name=gate_mask segments = { -1 90<nm> }
etch poly type=anisotropic thickness=0.2<um> mask=gate_mask
etch oxide type=anisotropic thickness=0.1<um>
struct tdr=NMOS3 ; # PolyGate
#--- For graphics, first run "tecplot_sv -s:ipc" and uncomment
# the next line before running this file
# graphics on
#--- Poly reoxidation -------------------------------------------------
diffuse temperature=900<C> time=10.0<min> O2
struct tdr=NMOS4 ; # Poly Reox
#--- LDD implantation -------------------------------------------------
refinebox silicon min= {0.0 0.045<um>} max= {0.1<um> 0.125<um>} \
xrefine= 0.01<um> yrefine= 0.01<um>
grid remesh
implant Arsenic dose=4e14<cm-2> energy=10<keV> tilt=0 rotation=0
SetPlxList { BTotal Arsenic_Implant }
WritePlx 1DasImpl.plx y= 0.25<um>
diffuse temperature=1050<C> time=0.1<s> ; # Quick activation
struct tdr=NMOS5 ; # LDD Implant
#--- Halo implantation: Quad HALO implants ----------------------------
implant Boron dose=1.0e13<cm-2> energy=20<keV> \
tilt=30<degree> rotation=0 mult.rot=4
#--- RTA of LDD/HALO implants -----------------------------------------
diffuse temperature=1050<C> time=5.0<s>
1: Getting Started
Full-Text Versions of Examples
36 Sentaurus™ Process User Guide
H-2013.03
struct tdr=NMOS6 ; # Halo RTA
#--- Nitride spacer ---------------------------------------------------
deposit nitride type=isotropic thickness=60<nm>
etch nitride type=anisotropic thickness=84<nm> isotropic.overetch=0.01
etch oxide type=anisotropic thickness=10<nm>
struct tdr=NMOS7 ; # Spacer
#--- N+ implantation --------------------------------------------------
refinebox silicon min= {0.04<um> 0.11<um>} max= {0.18<um> 0.4<um>} \
xrefine= 0.01<um> yrefine= {0.02<um> 0.05<um>}
grid remesh
implant Arsenic dose=5e15<cm-2> energy=40<keV> \
tilt=7<degree> rotation=-90<degree>
SetPlxList { BTotal Arsenic_Implant }
WritePlx 1DasImpl2.plx y= 0.25<um>
#---- N+ implantation & final RTA -------------------------------------
diffuse temperature=1050<C> time=10.0<s>
struct tdr=NMOS8 ; # S/D implants
# - 1D cross sections
SetPlxList {BTotal NetActive}
WritePlx NMOS_channel.plx y=0.0 silicon
SetPlxList {AsTotal BTotal NetActive}
WritePlx NMOS_ldd.plx y=0.1 silicon
SetPlxList {AsTotal BTotal NetActive}
WritePlx NMOS_sd.plx y=0.35 silicon
#----------------------------------------------------------------------#
#Transfer to device simulation
#----------------------------------------------------------------------#
#--Remove bottom of structure------------------------------------------
transform cut location= 1.00 down
#--Change refinement strategy and remesh-------------------------------
refinebox clear
line clear
pdbSet Grid Adaptive 1
pdbSet Grid AdaptiveField Refine.Abs.Error 1e37
pdbSet Grid AdaptiveField Refine.Rel.Error 1e10
1: Getting Started
Full-Text Versions of Examples
Sentaurus™ Process User Guide 37
H-2013.03
pdbSet Grid AdaptiveField Refine.Target.Length 100.0
pdbSet Grid SnMesh DelaunayType boxmethod
refinebox name= Global \
refine.min.edge= {0.01 0.01} refine.max.edge= {0.1 0.1} \
refine.fields= { NetActive } def.max.asinhdiff= 0.5 adaptive
refinebox name= SiGOX \
min.normal.size= 0.2<nm> normal.growth.ratio= 1.4 \
max.lateral.size= 5.0<nm> min= {-0.01 -0.1} max= {0.01 0.1} \
interface.materials= {Silicon}
refinebox name= GDpn1 \
min= {0.0 0.04} max= {0.06 0.1} xrefine= 0.005 yrefine= 0.005 \
silicon
refinebox name= TopActive \
min= {0.0 0.0} max= {0.3 0.4} \
refine.min.edge= {0.02 0.02} refine.max.edge= {0.05 0.05} \
refine.fields= { NetActive } def.max.asinhdiff= 0.5 \
adaptive silicon
grid remesh
#--- Reflect ---------------------------------------------------------
transform reflect left
#--- Contacts ---------------------------------------------------------
contact name= "substrate" bottom Silicon
contact name= "source" box Silicon adjacent.material= Gas \
xlo= 0.0 xhi= 0.005 ylo= -0.4 yhi= -0.2
contact name= "drain" box Silicon adjacent.material= Gas \
xlo= 0.0 xhi= 0.005 ylo= 0.2 yhi= 0.4
contact name= "gate" box PolySilicon \
xlo= -0.181 xhi= -0.05 ylo= -0.088 yhi= 0.088
#--- Final ---------------------------------------------------------
struct tdr=NMOS !Gas

1: Getting Started
Full-Text Versions of Examples
38 Sentaurus™ Process User Guide
H-2013.03
2D npn Vertical Bipolar
# 2D NPN Vertical Bipolar Transistor
#-----------------------------------
math coord.ucs
line x loc= 2.0<um>
line x loc= 4.0<um> tag=SubTop
line x loc= 6.0<um>
line x loc= 10.0<um> tag=SubBottom
line y loc= 0.0<um> tag=SubLeft
line y loc=1.5<um>
line y loc=2.5<um>
line y loc=8<um>
line y loc=13<um>
line y loc=22<um>
line y loc=24<um>
line y loc=30.0<um> tag=SubRight
# Diffuse settings to speed up simulation
#----------------------------------------
pdbSet Diffuse IncreaseRatio 8.0
pdbSet Diffuse ReduceRatio 0.5
# Mesh settings
#--------------
mgoals normal.growth.ratio=2.0 accuracy=2e-5 min.normal.size=10<nm> \
max.lateral.size=30.0<um> minedge=1e-5
pdbSet Grid Adaptive 1
pdbSet Grid AdaptiveField Refine.Abs.Error 1e25
pdbSet Grid AdaptiveField Refine.Rel.Error 2.0
pdbSet Grid Damage Refine.Min.Value 1e25
pdbSet Grid Damage Refine.Max.Value 1e25
pdbSet Grid Damage Refine.Target.Length 1
pdbSet Diffuse Compute.Regrid.Steps 10
pdbSet Grid Refine.Percent 0.01
refinebox interface.mat.pairs= {Silicon Oxide}
refinebox name=BL refine.fields= {Antimony Phosphorus} \
rel.error={Antimony=0.6 Phosphorus=0.6} \
1: Getting Started
Full-Text Versions of Examples
Sentaurus™ Process User Guide 39
H-2013.03
abs.error= {Antimony=1e16 Phosphorus=1e16} \
Adaptive min= "2.0 -0.1" max= "10.1 30.1" \
refine.min.edge= {0.2 0.4} max.dose.error= {Antimony=1e8}
refinebox name=Sinker refine.fields= {Phosphorus Arsenic} \
rel.error= {Phosphorus=0.5 Arsenic=0.5} \
abs.error= {Phosphorus=5e15 Arsenic=1e16} \
Adaptive min= {-1.0 16} max= {2.0 30.1} refine.min.edge= {0.1 0.2}
refinebox name=Active refine.fields= {Boron Arsenic} \
rel.error= {Boron=0.5 Arsenic=0.5} \
abs.error= {Boron=2e15 Arsenic=1e16} \
Adaptive min= {-1.0 -0.1} max= {2.0 16.0} \
refine.min.edge= {0.025 0.05}
# Masks definition
#-----------------
mask name=Sinker segments= {-1 22 24 35} negative
mask name=Base segments= {-1 1.5 13 35} negative
mask name=Emitter segments= {-1 2.5 8 22 24 35} negative
mask name=Contact segments= {-1 3.5 7 10 12 22.5 23.5 35}
mask name=Metal segments= {-1 2 8 9 13 22 24 35} negative
# Creating initial structure
#---------------------------
region Silicon xlo=SubTop xhi=SubBottom ylo=SubLeft yhi=SubRight
init concentration=1e+15<cm-3> field=Boron
# Set of physical models and parameters
# ------------------------
AdvancedCalibration 2013.12
# Buried layer
#-------------
deposit material= {Oxide} type=isotropic time=1 rate= {0.025}
implant Antimony dose=1.5e15<cm-2> energy=100<keV>
etch material= {Oxide} type=anisotropic time=1 rate= {0.03}
# Epi layer
#----------
deposit material= {Silicon} type=isotropic time=1 rate= {4.0} \
Arsenic concentration=1e15<cm-3>
diffuse temp=1100<C> time=60<min> maxstep=4<min>
1: Getting Started
Full-Text Versions of Examples
40 Sentaurus™ Process User Guide
H-2013.03
struct tdr=vert_npn1
SetPlxList {BTotal SbTotal AsTotal PTotal}
WritePlx Buried.plx
# Sinker
#-------
deposit material= {Oxide} type=isotropic time=1 rate= {0.05}
photo mask=Sinker thickness=1
implant Phosphorus dose=5e15<cm-2> energy=200<keV>
strip Resist
diffuse temp=1100<C> time=5<hr> maxstep=8<min>
struct tdr=vert_npn2
# Base
#-----
photo mask=Base thickness=1
implant Boron dose=1e14<cm-2> energy=50<keV>
strip Resist
diffuse temp=1100<C> time=35<min> maxstep=4<min>
struct tdr=vert_npn3
# Emitter
#--------
photo mask=Emitter thickness=1
implant Arsenic dose=5e15<cm-2> energy=55<keV> tilt=7 rotation=0
strip Resist
diffuse temp=1100<C> time=25<min> maxstep=4<min>
struct tdr=vert_npn4
SetPlxList {BTotal SbTotal AsTotal PTotal}
WritePlx Final.plx y=5.0
WritePlx Sinker.plx y=23.0
# Back end
#---------
etch material= {Oxide} type=anisotropic time=1 rate= {0.055} mask=Contact
deposit material= {Aluminum} type=isotropic time=1 rate= {1.0}
etch material= {Aluminum} type=anisotropic time=1 rate= {1.1} mask=Metal
1: Getting Started
Full-Text Versions of Examples
Sentaurus™ Process User Guide 41
H-2013.03
struct tdr=vert_npn5
exit
1: Getting Started
Full-Text Versions of Examples
42 Sentaurus™ Process User Guide
H-2013.03
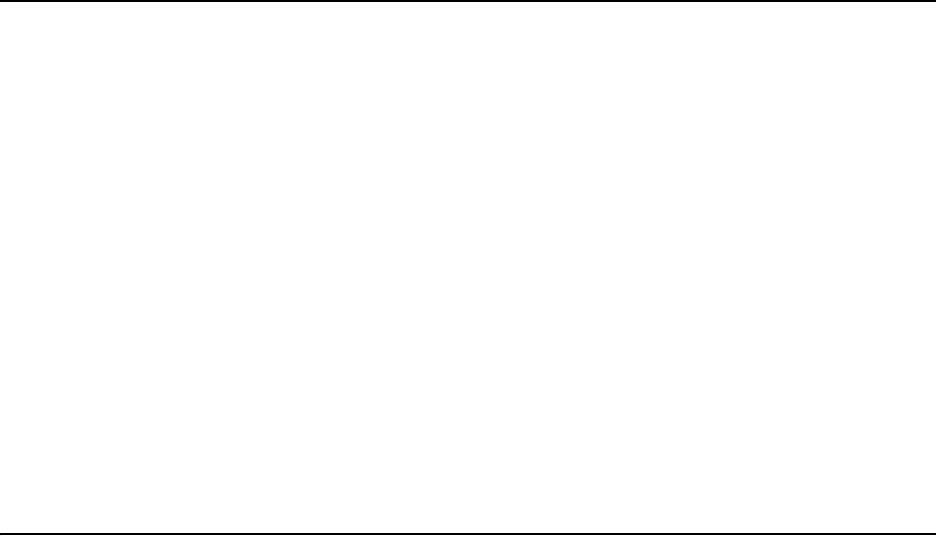
Sentaurus™ Process User Guide 43
H-2013.03
CHAPTER 2 The Simulator Sentaurus Process
This chapter provides an overview of how Sentaurus Process
operates.
The syntax and features of the command file are described, followed by an overview of the
Sentaurus Process parameter database, which contains all of the model parameters and
technical details regarding the running of the tool.
For new users, see Syntax for Creating Input Command Files on page 50, Creating and
Loading Structures and Data on page 66, and Interactive Mode on page 46. For advanced users
who need to adjust model parameters, see Parameter Database on page 55. For the TCAD
Sentaurus Tutorial and examples, go to:
$STROOT/tcad/$STRELEASE/Sentaurus_Training/index.html
where STROOT is an environment variable that indicates where the Synopsys TCAD
distribution has been installed, and STRELEASE indicates the Synopsys TCAD release number.
Overview
To familiarize users with the different formatting used in this documentation, input commands
from either a command file or the command line are presented this way:
sprocess -v
An example of output from Sentaurus Process is:
****************************************************************************
*** Sentaurus Process ***
*** Version H-2013.03 ***
*** (1.5, amd64, linux) ***
*** ***
*** Copyright (C) 1993-2002 ***
*** The board of regents of the University of Florida ***
*** Copyright (C) 1994-2013 ***
*** Synopsys, Inc. ***
*** ***
*** This software and the associated documentation are confidential ***
*** and proprietary to Synopsys, Inc. Your use or disclosure of this ***
*** software is subject to the terms and conditions of a written ***
*** license agreement between you, or your company, and Synopsys, Inc. ***

2: The Simulator Sentaurus Process
Interactive Graphics
44 Sentaurus™ Process User Guide
H-2013.03
****************************************************************************
Compiled Fri Jan 25 00:56:50 PDT 2013 on tcadamd12
Started at: Wed Jan 16 09:44:59 2013 (PDT)
User name: iavci
Host name: tcadintel1
PID: 12010
Architecture: x86_64
Operating system: Linux rel. 2.6.9-55.ELsmp ver. #1 SMP Fri Apr 20 16:36:54 EDT
2007
Interactive Graphics
There are two options for interactive graphics in Sentaurus Process:
■An X-Windows-based graphical display (which will be phased out in future releases)
■An interface to Tecplot SV (which will eventually replace the X-Windows display)
The interface of Tecplot SV is available on all platforms and can be used in 1D, 2D, and 3D.
The interface can be started with the simple command graphics on. The X-Windows-based
viewer is launched with either the plot.1d or plot.2d command (see plot.1d on page 1063
and plot.2d on page 1066). When the graphics command is used, graphical updating is
performed automatically.
The Sentaurus Process–Tecplot SV interface is designed to minimize the effects of the start-up
time of Tecplot SV. The usual mode of operation is to have one Tecplot SV window, which has
interprocess communication (IPC) enabled, and to start and stop Sentaurus Process many
times.
Because of the variability in user environments, automated start-up of Tecplot SV from inside
Sentaurus Process is not reliable. Therefore, to use the Sentaurus Process–Tecplot SV
interface, you must first start an IPC-enabled Tecplot SV from the UNIX command line before
starting the Sentaurus Process–Tecplot SV interface from within Sentaurus Process. To start an
IPC-enabled Tecplot SV, issue the following from the UNIX command line:
unix> tecplot_sv -s:ipc
Each time Sentaurus Process is started, it connects to the Tecplot SV window opened by the
above command and creates a new frame where the graphical output is sent. The name of the
frame contains the process ID, the user name, and the name of the computer where Sentaurus
Process is run.
NOTE It is not necessary that the computer where Tecplot SV is launched is the
same as the computer where Sentaurus Process is run, but the home
2: The Simulator Sentaurus Process
Command-Line Options
Sentaurus™ Process User Guide 45
H-2013.03
directory of the user should be the same on both computers (using NFS
or similar networking file-sharing).
In addition, it is possible to have multiple Sentaurus Process jobs sending graphics output to a
single Tecplot SV for comparing multiple simulations in real time. For more information, see
Tecplot SV User Guide, Launching or Connecting to Tecplot SV on page 13).
NOTE There is a convenient control mechanism built into Tecplot SV located
in a dialog box, which is displayed by selecting View > Sentaurus
Interface. In the dialog box, buttons allow you to pause and continue
Sentaurus Process so that the graphics can be more closely examined
when the structure or data in Sentaurus Process changes rapidly.
It is sometimes convenient to use the fbreak command when using interactive graphics. This
command pauses Sentaurus Process in the input command file where the fbreak command
occurs, allowing adjustments to be made to the display settings such as mesh on or off,
selection of field to view, and range of color scale. The fbreak command puts Sentaurus
Process into interactive mode and the command prompt ‘sprocess>’ appears in the terminal
window from which Sentaurus Process was run. After adjustments to the graphics have been
made, the command fcontinue can be entered, which will resume Sentaurus Process
execution.
In Sentaurus Workbench or batch mode (that is, sprocess -u or sprocess -b), the
commands fbreak and fcontinue have no effect. Therefore, these commands can be placed
in a Sentaurus Workbench project.
Command-Line Options
Table 1 lists the command-line options that are available in Sentaurus Process.
Table 1 Command-line options
Option Short name Function
--batchMode -b
Switch off graphics.
--diff NA
Diff mode. To see differences in data and Sentaurus
Process parameter settings between two TDR files.
Interpolation is used to compare results from different
meshes. Usage:
sprocess --diff <file1> <file2>
where <file1> and <file2> are TDR files.
--FastMode -f
Generate structure, no diffusion, no Monte Carlo
implantation, no partial differential equation (PDE) solve,
and so on.

2: The Simulator Sentaurus Process
Command-Line Options
46 Sentaurus™ Process User Guide
H-2013.03
Interactive Mode
Sentaurus Process runs in interactive mode if no command file is given. In this mode,
commands can be entered (at the command prompt) line-by-line and are executed immediately.
It is useful to run Sentaurus Process in the interactive mode for the following reasons:
■When debugging Tcl code, the program does not quit if a Tcl error is found. The error is
displayed and you are prompted again for input. You can source a command file repeatedly
if required.
■To easily obtain pdb parameter names and defaults with the pdbGet command.
■To print the list of built-in functions with the help command, and to print the list of Tcl
procedures with the info procs command.
■To obtain command parameter names and defaults for any built-in command by using the
params flag available in all built-in functions.
Another use of the interactive mode is to pause the simulation using the fbreak command.
When the simulation is paused in interactive mode, the state of the simulator can be queried
using a number of commands including grid, mater, select, and so on. Pausing the
simulation can also be useful when using interactive graphics as described in Interactive
Graphics on page 44.
--GENESISeMode -u
Switch off log file creation.
--home <directory> -o <directory>
Set SPHOME to <directory>.
--noSyntaxCheck -n
Switch off syntax check.
--pdb -p
Run Parameter Database Browser showing parameters as
they are set during run-time. Include default parameters
and parameters from the input command file if specified.
--ponly
Same as --pdb, but only shows parameters set in input
command file; does not show default parameters.
--quickSyntaxCheck -q
Only check syntax of branches that are true.
--syntaxCheckOnly -s
Only check syntax, no execution.
NA
-v
Print header with version number.
NA
-h
Print use and command-line options.
NA
-x
Test floating-point exception handling.
NA
-X
Switch off floating-point exception catching.
Table 1 Command-line options
Option Short name Function

2: The Simulator Sentaurus Process
Environment Variables
Sentaurus™ Process User Guide 47
H-2013.03
Fast Mode
When working on a new process flow, it is particularly useful to run Sentaurus Process a few
times using the fast mode (-f command-line option). Developing a new process flow can be
complex, involving many etch, deposit, and photo steps, some with masks; sometimes
adjustments are required. In the fast mode, all diffusion, Monte Carlo implantation, and 3D
remeshing commands are ignored. Only process commands for structure generation and
analysis are performed. In this mode, when in three dimensions, all struct commands will
only write a boundary into the TDR file, since the simulation mesh is not synchronized with
the modified structure.
Terminating Execution
You can t erm in ate a ru nnin g Se nta ur us Proce ss job in several ways. In some cases, the
termination will take time or will fail for other reasons. The most fail-safe method is to use the
UNIX command:
kill -9 <process_id>
where <process_id> is the process ID number of the running Sentaurus Process job which
can be obtained with the UNIX ps command. This sends a signal SIGKILL to the
corresponding Sentaurus Process job, which will cause the job to terminate immediately.
If Sentaurus Process is run directly from a UNIX shell, usually you can terminate the run by
using shortcut keys. The key sequence is interpreted by the shell command, which sends a
signal to the job in the foreground. Usually, Ctrl+C sends a SIGINT signal and Ctrl+\
(backslash) sends a SIGQUIT signal. The running Sentaurus Process job catches all SIGINT
signals and waits for three signals to be caught (in case it was typed accidentally) before
terminating itself. However, Sentaurus Process does not catch the SIGQUIT signal, so this
signal will typically cause Sentaurus Process to terminate immediately.
Because the exact behavior may depend on your UNIX shell, the operating system, and the
local configuration, refer to the manual for the UNIX shell you are running or contact your
local systems administrator for more information.
Environment Variables
The Sentaurus Process binary relies on a number of supporting files found using the
environment variables SPHOME and SCHOME. To change default models and parameters without
modifying the installed Sentaurus Process files, copy the default SPHOME and SCHOME

2: The Simulator Sentaurus Process
File Types Used in Sentaurus Process
48 Sentaurus™ Process User Guide
H-2013.03
directories and set the environment variables (SPHOME and SCHOME) to the location of the
modified directories.
By default, SPHOME and SCHOME are set based on the Synopsys standard environment variables
STROOT and STRELEASE, and by the version number of Sentaurus Process using:
SPHOME
= $STROOT/tcad/$STRELEASE/lib/sprocess-<version number>
SCHOME = $STROOT/tcad/$STRELEASE/lib/score-<version number>
The SPHOME directory has two major subdirectories, TclLib and ImpLib, where:
■The directory $SPHOME/TclLib contains all the default model selections in a file
SPROCESS.models.
■The Tcl files are located in directory $SPHOME/TclLib and $SCHOME/TclLib.
■The subdirectory $SCHOME/Params contains the Sentaurus Process parameter database
(see Parameter Database on page 55).
■The subdirectory $SPHOME/ImpLib contains all the implant tables.
File Types Used in Sentaurus Process
The main file types used in Sentaurus Process are:
■Sentaurus Process command file (*.cmd)
This file, which is the main input file type for Sentaurus Process, contains all the process
steps and can be edited. It is referred to as the command file or input file.
■Log file (*.log)
This file is generated by Sentaurus Processs during a run. It contains information about
each processing step, and the models and values of physical parameters used in it. The
amount of information written to the log file can be controlled by the info parameter,
which is available in nearly every command and the global default info level, 0, can be
changed with pdbSet InfoDefault <level>. The higher the info level, the more
information is logged, but it is not recommended to use <level> > 2 for normal use
because many normally unnecessary operations are performed for higher info levels which
can slow execution.
■TDR boundary file (*_bnd.tdr)
This format stores the boundaries of the structure without the bulk mesh or fields. This file
can be used as the structure file for the meshing engine Sentaurus Mesh and can be loaded
into Tecplot SV for viewing. The name of a TDR boundary file can be specified in the tdr
parameter of the init command of Sentaurus Process, and then the loaded boundary will
be meshed using the MGOALS meshing library.
2: The Simulator Sentaurus Process
File Types Used in Sentaurus Process
Sentaurus™ Process User Guide 49
H-2013.03
■TDR grid and doping file (*_fps.tdr)
TDR files can be used to split and restart a simulation. Such restart files are saved in the
struct tdr=filename command because restarting requires interface data, parameter
and command settings, mesh ordering information as well as bulk grid and data. If
either !pdb or !interfaces is specified in the struct command, the TDR file will not
be suitable for restarting. The TDR file can be loaded into Sentaurus Process in the init
command, but the results of the subsequent simulation steps might differ in the simulation
with the split and restart compared to a simulation of the entire flow in one attempt. TDR
files store the following types of information:
• Geometry of the device and the grid.
•Distribution of doping and other datasets in the device.
•The internal structure of the mesh in Sentaurus Process required to restore the
simulation mesh to the same state in memory that is present at the time of saving the
file. Restart files store coordinates and field values without scaling them to DF–ISE
units; files that cannot be restarted store coordinates and field values scaled to DF–ISE
units.
• Finally, by default, Sentaurus Process stores all changes to the parameter database
made after initial loading the database and all commands that create objects later
referenced, such as refinement boxes and masks in the TDR file. A TDR file can be
either reloaded into Sentaurus Process to continue the simulation or be loaded into
Tecplot SV for visualization.
The parameter settings stored in a TDR file can be viewed using pdbBrowser
-nopdb -tdr <tdrfile> (see Viewing Parameters Stored in TDR Files on page 65
for details).
For more information about the TDR file format, refer to the Sentaurus™ Data Explorer
User Guide.
■DF–ISE doping and refinement file (*_msh.cmd)
This file stores doping and mesh refinement commands and, along with the boundary file,
it is used as input for the Synopsys meshing engines. This file is usually saved by the user
at the end of a simulation.
■DF–ISE file (*.plx)
This DF–ISE file format is used for saving 1D distributions of the doping concentration or
other fields in a specified 1D cross section. This file can be viewed by loading it into
Inspect.

2: The Simulator Sentaurus Process
Syntax for Creating Input Command Files
50 Sentaurus™ Process User Guide
H-2013.03
Syntax for Creating Input Command Files
This section is intended for users who want to create input command files manually, that is,
outside the Ligament environment. It is important to remember that Sentaurus Process is
written as an extension of the tool command language (Tcl). This means that the full capability
and features of Tcl are available in the input command files as well as the interactive mode of
Sentaurus Process.
Standard Tcl syntax must be followed; for example, a hash symbol (#) at the beginning of a
line denotes a comment and the dollar sign ($) is used to obtain the value of a variable. Major
features of Tcl include for loops, while loops, and if then else structures, switch
statements, file input and output, sourcing external files, and defining procedures (functions).
Var ia bles ca n b e n um ber s, s tr ing s, l ists , or arrays. Refer to the literature for more
information [1].
Before execution of the command file takes place, the syntax of the file is checked. This is
accomplished by first modifying the command file so that all branches of control structures
such as if, then else, and switch commands are executed. In addition, a special flag is set
so that no structure operations or operations that depend on the structure are performed. This
allows the syntax check to run quickly, but thoroughly. Sometimes, the modifications made to
the command file during syntax checking interfere with the definition or redefinition of Tcl
variables, generating a false syntax error. In these cases, switch off syntax checking for part of
a command file using the special CHECKOFF and CHECKON commands:
# Skip syntax check for part of command file
# The CHECKOFF/CHECKON commands must start at the beginning of the line
# and be the only command on the line
CHECKOFF
if { $mode } {
array set arr $list1
} else {
set arr $list2 ;# error only if both branches are executed
}
CHECKON
# further commands are syntax checked
Tcl Input
Sentaurus Process has been designed to optimize the use of the Tcl. Some examples of this
interaction include:
■Command parameter values are evaluated with Tcl. For example, expr can appear in the
value of an expression, that is, parameter=[expr $pp/10.0] is valid Sentaurus Process
2: The Simulator Sentaurus Process
Syntax for Creating Input Command Files
Sentaurus™ Process User Guide 51
H-2013.03
syntax. This particular expression sets the parameter parameter to the value of pp/10 if
the Tcl variable pp was previously defined with the Tcl set command.
■Tcl expressions may appear in model parameter values in the parameter database. In some
cases, Sentaurus Process parameters are set with Tcl commands to be a function of other
parameters.
■Sentaurus Process contains many callback procedures, which can be redefined by users to
provide flexibility. For example, a callback procedure is used to initialize defects after
implantation.
■Many modular built-in functions are available for postprocessing, which can be combined
into a Tcl script to create powerful analytic tools.
■There are special Sentaurus Process versions of set (fset) and proc (fproc), which are
stored in TDR files. When simulations are restarted using a TDR file, the settings given by
fset and fproc from the previous simulation will be available.
Other syntax rules to consider when writing input command files are:
■One command is entered on one line only. There are two exceptions to this rule:
•A backslash (\) is used to extend a command on to multiple lines if it appears as the last
character on the line.
• If there is an opening brace, Tcl will assume the command has not finished until the
line containing the matching closing brace.
■Command parameters have the following form:
• Boolean parameters are true if the name appears on the line. They are false if they are
preceded by an exclamation mark (!).
•Parameters that are of type integer or floating point must appear as
parameter=value pairs.
• String parameters are enclosed, in general, in double quotation marks (""), for
example, parameter="string value".
• Lists can appear enclosed in double quotation marks or braces, for example,
parameter= { item1 item2 ... } or parameter= " item1 item2 ...".
It is necessary to have a space between the equal sign and the opening brace.
NOTE It is important to separate the equal sign from the parameter value by a
space because Tcl delimiters such as ‘"’ and ‘{’ are ignored if they
appear in the middle of a string. Sentaurus Process can handle no space
between an equal sign and a double quotation mark, but it cannot correct
the case where there is no space between an equal sign and an opening
brace.

2: The Simulator Sentaurus Process
Syntax for Creating Input Command Files
52 Sentaurus™ Process User Guide
H-2013.03
Material Specification
Materials are specified the same way for all commands that require a material parameter. For
a bulk material, specify only one material. For an interface material, specify two materials: one
with a slash (/) and one without a slash.
Some examples are:
oxide ;# This command applies to oxide.
silicon /oxide ;# This command applies to the Si-SiO2 interface
The complete list of materials available can be found in the file:
$STROOT/tcad/$STRELEASE/lib/score-<version number>/TclLib/tcl/Mater.tcl
In that file, the lines that contain mater add create a material. For more information about
creating new materials, see mater on page 1025.
NOTE Materials present in the Mater.tcl file do not necessarily have
parameters in the parameter database. Attention must be paid to
initializing parameters for a new material.
Aliases
Sentaurus Process allows more control over the names of command parameters and
abbreviations of parameter names. These aliases only apply to parameters of built-in Sentaurus
Process commands, and the pdbSet and pdbGet family of commands.
This permits clarity and uniformity to commonly used names. Another benefit is that it is easier
to maintain backward compatibility for parameter names while not restricting future parameter
names that could conflict with common abbreviations (that is, V could refer to either vacancy
or void).
An explicit list of allowed aliases is maintained in the $SCORE/TclLib directory (see
Environment Variables on page 47 for information about how the location of the TclLib
directory is determined). The alias command is used to view and extend the list of allowed
aliases.
To print the list of aliases:
sprocess> alias -list

2: The Simulator Sentaurus Process
Default Simulator Settings: SPROCESS.models File
Sentaurus™ Process User Guide 53
H-2013.03
To view the alias of a parameter name, for example, Vac:
sprocess> alias Vac
Vacancy
If an alias does not exist, the same parameter name is returned:
sprocess> alias NotAParam
NotAParam
To create a new alias for a parameter name, for example, the alias Vaca for the parameter
Vacancy:
sprocess> alias Vaca
Vaca
sprocess> alias Vaca Vacancy
sprocess> alias Vaca
Vacancy
Default Simulator Settings: SPROCESS.models File
Sentaurus Process starts a simulation by reading the SPROCESS.models file in the $SPHOME/
TclLib directory. This file defines various default parameters and directories used during the
simulation such as setting:
■The path for Tcl library files
■The path for Advanced Calibration Tcl library files
■The path for implant tables
■Default material names
■The math parameters for 1D, 2D, and 3D oxidation and diffusion simulations
■Default solution names
■Default diffusion callback procedures
■Default oxidation or silicidation reactions
■Default oxidation or silicidation solution callback procedures
■Default epitaxial growth callback procedures
The SPROCESS.models file is read once at the beginning of the simulation. You can override
any of the default parameters after the file is read.

2: The Simulator Sentaurus Process
Compatibility With Previous Releases
54 Sentaurus™ Process User Guide
H-2013.03
Compatibility With Previous Releases
Occasionally, the default parameter and model settings change in Sentaurus Process to ensure
that the default behavior gives robust, accurate, and computationally efficient results on current
production technologies. Usually, when new models and algorithms are developed, they are
optional. After some experience is gained, the default can be changed to take advantage of the
new model or algorithm.
The old model and algorithm settings are collected into a file for each release and are available
so that you can recover results from previous releases. Each file contains only those parameter
changes that occurred for that particular release, so that if the release specified in the
Compatibility command is older than the most recent release, the most recent release
parameters are set first, followed by older releases in reverse chronological order.
For example, the command Compatibility F-2011.09 issued for Version H-2013.03 will
first apply parameters consistent with H-2013.03, then parameters consistent with G-2012.06,
and finally parameters consistent with F-2011.09. Aliases are available for the release name so
you do not need to know the release foundation letter. For example, 2012.06 can be used
instead of G-2012.06.
The files with the compatibility parameter settings are stored in $STROOT/tcad/
$STRELEASE/lib/sprocess/TclLib/Compatibility. These files are a useful list of all
default parameter changes for each release.
NOTE As a result of the repair of code flaws and because of numeric accuracy
limitations, exact reproduction of results from previous releases is not
always possible.
NOTE If the Compatibility command is used, it must be the first command
in an input file so that all subsequent commands that depend on the
defaults take into account the compatibility setting.
For example:
# Apply defaults of the 2012.06 release (first line of input file)
Compatibility 2012.06
NOTE Default parameter and algorithm settings of the tools Sentaurus Mesh,
Sentaurus Structure Editor, and the MGOALS library are not changed
by the Compatibility command. For MGOALS library backwards
compatibility, see Summary of MGOALS Etching and Deposition
Algorithms on page 783. To obtain backwards compatible default
parameters and settings for Sentaurus Mesh and Sentaurus Structure

2: The Simulator Sentaurus Process
Parameter Database
Sentaurus™ Process User Guide 55
H-2013.03
Editor, see the backwards compatibility mechanisms described for those
tools in the corresponding manual sections.
Parameter Database
The Sentaurus Process parameter database stores all Sentaurus Process material and model
parameters as well as global information needed for save and reload capabilities. There is a
hierarchical directory tree inside the Params directory, which stores the default values. (To
locate the Params directory, see Environment Variables on page 47.)
Data is retrieved by using the pdbGet command and is set by using the pdbSet command. The
pdbGet and pdbSet commands are checked for correctness of syntax and they print the
allowed parameter names if a mistake is made. These commands are used to obtain and set all
types of data stored in the database: Boolean, string, double, double array, and switch.
The higher level pdbSet and pdbGet commands call lower-level type-specific commands
(pdbGetSwitchString, pdbGetDoubleArray, pdbGetString, pdbGetDouble,
pdbGetSwitch, pdbGetBoolean, pdbSetDoubleArray, pdbSetString,
pdbSetBoolean, pdbSetDouble, and pdbSetSwitch) that are not checked for errors and,
therefore, are not recommended for typical use. These commands have a slight performance
advantage and are used internally.
You c an se t s om e p ar ameter s in a reg io n-sp ecific manner. Regions can be named with the
region and deposit commands and, if region-specific parameters exist, they will override
the material-specific parameters if any. However, there are many circumstances where this will
not give the desired behavior. In that case, you must create a new material that inherits its
parameters from an existing material. Then, you must change the material properties of the new
material as needed. For more information, see Like Materials: Material Parameter Inheritance
on page 57.
Inside the Params directory are subdirectories that define the highest level nodes in the
database. Inside each subdirectory is a file Info, which contains parameters of that level. In
addition, directories in the database have named files that contain parameters, which are under
the node defined by the file name. For example, in the Params database, there is a directory
called Silicon, which contains a file Info. The parameters inside Info are located under the
Silicon node. As another example, inside the Silicon directory is another file
Interstitial that contains parameters under the Interstitial node, which is under the
Silicon node.
Inside the files of the parameter database are commands that set database parameters. The
commands have the form:
array set $Base { <NAME> { <TYPE> <VALUE> } }
2: The Simulator Sentaurus Process
Parameter Database
56 Sentaurus™ Process User Guide
H-2013.03
where:
■<NAME> is the parameter name.
■<TYPE> is one of Boolean, String, Double, DoubleArray, or Switch.
■<VALUE> is a Tcl expression that sets the default value.
It is often necessary to enclose the <VALUE> expression in braces. Some Tcl procedures have
been created to increase the usefulness of <VALUE> expressions. For example, in many places
in the database, the built-in function Arrhenius is used to set the value of a parameter.
Parameters that contain a Tcl function are evaluated at each diffusion time step so that
temperature-dependent parameters will update correctly during a temperature ramp. It is
important to remember that the Arrhenius function uses the global Tcl variable for
temperature, which defaults to room temperature.
If you start Sentaurus Process and call the pdbGet command of a parameter that contains an
Arrhenius function, it will return the value of that parameter at room temperature. The
temperature can be changed with the SetTemp function. Subsequent calls to the Arrhenius
command through pdbGet return values based on the given temperature. In addition, the
diffusion command changes the global temperature for each time step, and the temperature
after diffusion will be same as the temperature in the last diffusion time step.
Other functions that appear in the pdb parameters are DiffLimit, which calculates a
diffusion-limited reaction rate given the diffusivity of the two reacting species, and pdbGet*
functions, which allow parameters to be set as a function of other parameters.
For the DoubleArray type, a Tcl list is set that is ordered pairwise:
{key1 value1 key2 value2 ...} where the parameter setting for key1 is value1.
Material parameters can be stored under the known region name. To set and obtain the
parameter value, use the region name instead of the material name. If the parameter is not found
under the region name, it is taken from the material of that region.
Sentaurus Process writes directly to the parameter database in a number of ways. Mostly this
is performed to save information for save and reload capabilities using the TDR format. Data
written by the program into the parameter database is not available within the default Params
directory or the Parameter Database Browser (PDB), but can be read using the pdbGet
command.
For information about the TDR format, refer to the Sentaurus™ Data Explorer User Guide.

2: The Simulator Sentaurus Process
Parameter Database
Sentaurus™ Process User Guide 57
H-2013.03
Parameter Inheritance
The parameter database has a parameter inheritance feature where parameters at a certain level
or node can inherit the parameters from another node at the same level. The inherited
parameters can be overwritten with new values. Inheritance is indicated by the presence of a
special parameter named Like. In one of the parameter database files, the Like parameter is
specified as follows:
array set $Base {Like <Node>}
which means that parameters at the level of the file inherit parameters from <Node>, which
should be another node at the same level. For example, the file:
Params/Silicon/Arsenic/Info
contains the line array set array set $Base {Like Dopant}, which indicates that
Arsenic in Silicon should inherit the common parameters of all Dopant species in
Silicon. Other parameters specified in that file indicate parameter settings specific to
Arsenic in Silicon.
It is also possible to use inheritance to create new parameters from the command line using the
pdbLike command (see pdbLike on page 1055 ). For example, assuming MyBoron is defined
as solution:
pdbLike Silicon MyBoron Boron
inherits Boron parameters including user-defined and callback parameters for MyBoron in
silicon. The new parameters are used to set up diffusion equations for MyBoron.
Materials in Parameter Database
Like Materials: Material Parameter Inheritance
The parameters of a material can be inherited from the parameters of another material using
the special Like parameter in the PDB. When this is the case, the two materials are referred to
as like materials. This can be used to specify different settings in different regions. First, a new
material is created and made to be like an existing material using:
mater add name = <NewMat> new.like = <ExistingMat>
where:
■<NewMat> is the name of the material being created.
■<ExistingMat> is the name of the material whose parameters will be inherited.

2: The Simulator Sentaurus Process
Parameter Database
58 Sentaurus™ Process User Guide
H-2013.03
NOTE It is important to use the mater command instead of directly creating
the Like parameter because the mater command will make all
interfaces to <NewMat> Like the appropriate interface to
<ExistingMat>.
NOTE Reaction specifications, such as oxidation, silicidation, and epitaxy, are
not stored in the PDB. Therefore, for a new material to react, a new
reaction command must be issued (see reaction on page 1099).
Interface Parameters
When using the PDB commands and the Alagator language, interfaces are specified as a pair
of materials separated by an underscore (_), for example, Gas_Oxide and Oxide_Silicon.
The official name follows alphabetic order, and the first letter is capitalized. However, aliases
are provided that allow their order to be reversed; some shorter names are allowed; and all
lowercase is generally available.
As an example of setting an interface parameter, the following command sets the numeric
tolerance Abs.Error at the gas–silicon interface to 1e3:
pdbSet Gas_Silicon Vac Abs.Error 1e3
Regionwise Parameters and Region Name-handling
Many parameters in the parameter database can be specified regionwise including parameters
related to meshing, parameters for both analytic implantation and MC implantation, and
mechanics parameters. Those parameters used by Alagator as part of equations and terms,
however, cannot be specified regionwise: this includes all dopant diffusion parameters and all
oxidation and silicidation parameters. For the rest of the parameters, internally, the program
checks if there is a regionwise specification of the parameter; if not, the materialwise
specification is used.
The name of regions can be specified with the region command and deposit command;
however, the name should not contain an underscore (_) or a period (.) because these characters
have special meaning. During the course of the simulation, geometric operations such as etch
and reflect can split regions in two. If this happens, the history of the region is maintained
through its name. For example, if a region is originally named layer1 and it is etched into two
pieces, they will be named layer1.1 and layer1.2 according to rules given below.
These two regions will inherit the parameters of layer1. Furthermore, parameters for
layer1.1 and layer1.2 also can be specified separately. If a subsequent step such as a
deposit reunites layer1.1 and layer1.2, the region will be given the name layer1.
Conversely, if layer1.1 is split into two regions, the regions will be named layer1.1.1 and
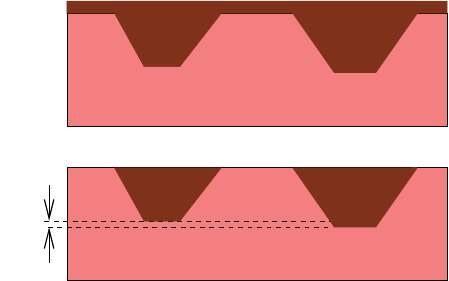
2: The Simulator Sentaurus Process
Parameter Database
Sentaurus™ Process User Guide 59
H-2013.03
layer1.1.2, and so on. In this way, regionwise parameter specification is preserved for the
life of the region or its parts.
The numbering of split regions is performed according to the spatial location of the pieces. The
lowest point of each piece to be renamed is found (in the coordinate system of Sentaurus
Process, this would be the largest x-coordinate). To avoid numeric noise, the coordinates are
compared with a specified epsilon given by pdbGet Grid RenameDelta (hereafter, referred
to as RN). If the x-coordinates of the pieces to be renamed are not within RN of each other, the
regions are ordered from lowest to highest, that is, from the highest x-coordinate to the lowest.
If any piece has its lowest coordinate within RN, its y-coordinate is compared, that is, from the
lowest coordinate to the highest.
For example, in Figure 11, layer1 is split into two regions and the quantity deltax is less
than RN, so the region on the left is given the name layer1.1 and the region on the right is
given the name layer1.2. If deltax had been greater than RN, the region on the right would
have been given the name layer1.1 because it would have been considered lower than the
region on the left. Similarly, in three dimensions, first x and y are compared, and if they are
both within RN, z is used for ordering, that is, from the lowest coordinate to the highest.
Figure 11 Illustration of region-naming rules
You c an ap pl y t he ab ove operat io n to the whole structure with grid rename. In this case, all
the regions are renamed similarly to the above rules but, instead of the root being chosen by the
user, all regions of the same material have the root given by the names of the materials and the
extension is _<n> where <n> is the region number, for example Silicon_1, Silicon_2, and
so on. This should only be used as a postprocessing step because all region-specific parameters
no longer apply when the name of a region has changed.
For example, if two oxide layers are grown, one with steam (if it is the first oxide region, its
name would be Oxide_1) and one from pure O2 (which would be Oxide_2 if it were the
second oxide region), they can have different densities. This can be considered in an MC
implantation using:
pdbSetDouble Oxide_1 MassDensity <wet oxide density>
layer1
deltax layer1.1 layer1.2

2: The Simulator Sentaurus Process
Viewing the Defaults: Parameter Database Browser
60 Sentaurus™ Process User Guide
H-2013.03
pdbSetDouble Oxide_2 MassDensity <dry oxide density>
where <wet oxide density> and <dry oxide density> would be replaced with values
given in g/cm3.
Viewing the Defaults: Parameter Database Browser
The Parameter Database Browser (PDB) is a graphical representation of the Sentaurus Process
parameter database that allows you to view and edit parameters. The PDB has three distinct
areas (see Figure 12 on page 61):
■Parameter hierarchy overview in a tree structure representation.
■Parameter information in a spreadsheet representation. The columns are:
•Parameter
•Type
•Value
•Unit
•Evaluate
• Comment
•Tool
•Info Level (hidden by default)
■Graphic window to plot parameter dependence on the temperature.
The status bar has three indicators that show:
■The temperature used in temperature-dependent functions such as Arrhenius.
■The temperature point set for the x-axis.
■The x-coordinate and y-coordinate of the pointer in the graphic window.
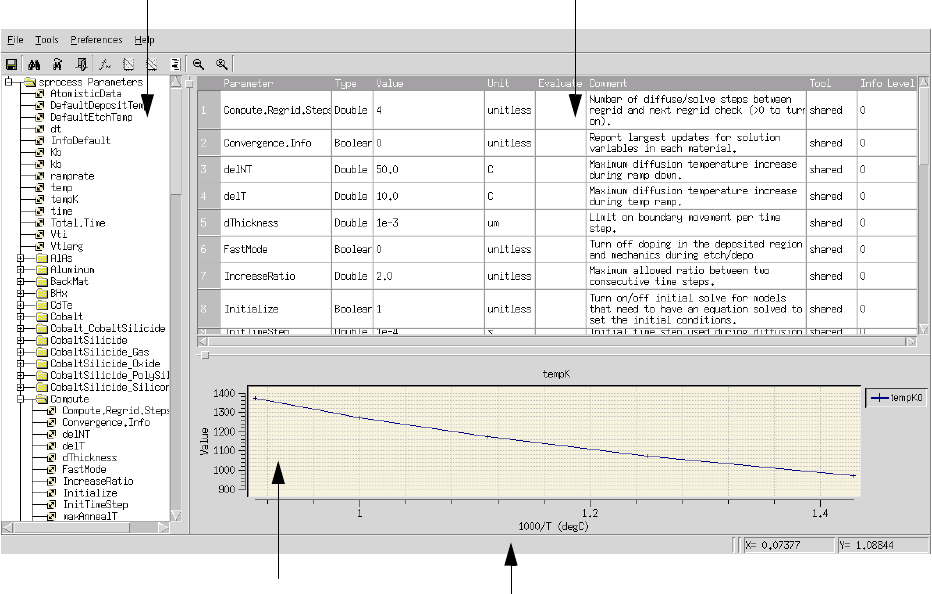
2: The Simulator Sentaurus Process
Viewing the Defaults: Parameter Database Browser
Sentaurus™ Process User Guide 61
H-2013.03
Figure 12 Parameter Database Browser
Spreadsheet
Representation
Tree Structure
Representation
Graphic Window Status Bar

2: The Simulator Sentaurus Process
Viewing the Defaults: Parameter Database Browser
62 Sentaurus™ Process User Guide
H-2013.03
Starting the Parameter Database Browser
To start the PDB from the command line, enter:
pdbBrowser
This searches for the database in the same location as Sentaurus Process. You can set the
environment variables SPHOME and SCHOME to change the location of the parameter database
for the PDB and Sentaurus Process (see Environment Variables on page 47 for details). To view
parameters in an input file merged with defaults, use:
sprocess --pdb <input command file>
or to view only the parameters specified as input in a command file, use:
sprocess --ponly <input command file>
Browser PDB Functions
The following functions are available:
Figure 13 Find dialog box
Export Tree Saves the whole parameter database into a specified file in the tab-
delimited format. The fields of the file are Parameter Name, Type,
Value Evalua tion , Original Value, and Comments.
Find and Find Next Matches the pattern entered against parameter names according to the
selected options. Patterns can include regular Tcl expressions. The
match is highlighted when found (see Figure 13).
Goto Line Highlights a table row or tree node that corresponds to the number
entered.
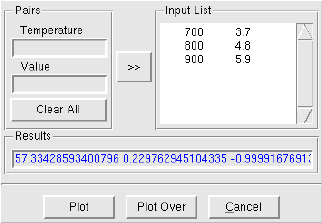
2: The Simulator Sentaurus Process
Viewing the Defaults: Parameter Database Browser
Sentaurus™ Process User Guide 63
H-2013.03
NOTE Yo u c an z oo m b y d ra gging t he mou se . To z oom o ut , u se th e m idd le
mouse button, or click the Zoom Out and Zoom Off buttons.
NOTE To display a shortcut menu, right-click a parameter for plotting and
evaluation in the tree and table areas.
Figure 14 Arrhenius Fit dialog box
Plot (Applies only to parameters of type double and double array.) Plots
the dependency of the selected parameter on the temperature in
logarithmic coordinates versus 1/T. The default set of temperature
values is {700.0 800.0 900.0 1000.0 1100.0}. The resulting
graphs are displayed in the graphic window; otherwise, an error
message is displayed.
Plot Over The same as Plot but it does not clear the graphic window of previous
graphs.
Evaluate Evaluates the value of the selected parameter and displays the result
in the Evaluate column of the table. Values can contain Tcl
expressions.
Edit Opens the appropriate database file with an editor regardless of the
user write-permissions, but the standard installation will switch off
write permissions for the database. The default editor, SEdit, can be
changed. The PDB Browser is updated upon file saving.
Parameter
Information
Double-clicking a nonempty table row allows you to view the
corresponding parameter information in a separate window. To close
the window, click the Close button.
Arrhenius Fit Finds the best prefactor and energy for an Arrhenius fit of a given
profile, taken from the list of temperature–value pairs. The results can
be plotted in the graphic window.
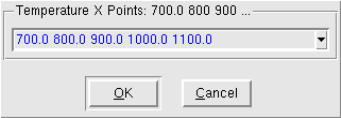
2: The Simulator Sentaurus Process
Viewing the Defaults: Parameter Database Browser
64 Sentaurus™ Process User Guide
H-2013.03
PDB Preferences
The PDB allows you to reset the default settings for the following values by using the
Preferences menu, shortcut keys, or shortcut menu of the graphic window:
Figure 15 Reset Temperature Points dialog box
Preferences > Font > Family
Changes the font family.
Preferences > Font > Size
Changes the font size.
Preferences > Cursor
Changes the style of the pointer.
Preferences > Graph > Set Temperature
The global temperature used in the temperature-dependent functions;
the default is 1000.0.
Preferences > Graph > Reset X Points
The x-axis temperature point set; the default set is
{700.0 800.0 900.0 1000.0 1100.0}.
Preferences > Graph > Data Point Symbol
Node Tip: hide / show.
Preferences > Info Level
Shows or hides the Info Level column of the table.
Preferences > Editor > Change Editor
Resets the default editor.

2: The Simulator Sentaurus Process
Viewing the Defaults: Parameter Database Browser
Sentaurus™ Process User Guide 65
H-2013.03
Viewing Parameters Stored in TDR Files
Parameters stored in TDR files can be viewed using the pdbBrowser command run from the
UNIX command line instead of through Sentaurus Process. By default, the PDB reads
parameters from the Sentaurus Process database directory (which can be changed with the
SPHOME and SCHOME environment variables). In addition, parameters stored in a TDR file can
be read in using the -tdr <filename> option of the PDB. Parameters that appear in the
database are overwritten by those contained in the TDR file, so the resultant parameter set will
be the same as if Sentaurus Process had read in the file. On the other hand, it is also useful to
know which parameters are only in the TDR file. To read only those parameters, the database
reading can be switched off using the -nopdb command-line option.
For example:
> pdbBrowser -tdr n10_fps.tdr
reads the Sentaurus Process PDB and then reads parameters from n10_fps.tdr file
overwriting values contained in the database.
For example:
> pdbBrowser -nopdb -tdr n10_fps.tdr
reads only the parameters in n10_fps.tdr file.
Preferences > Editor > Reset Update Time
Resets the update interval.
Preferences > Graph > X Scale
Resets the scale to logarithmic or linear.
Preferences > Graph > Y Scale
Resets the scale to logarithmic or linear.
Tools > Info Level Chooses which parameters to display ranging from basic parameters
to all parameters.
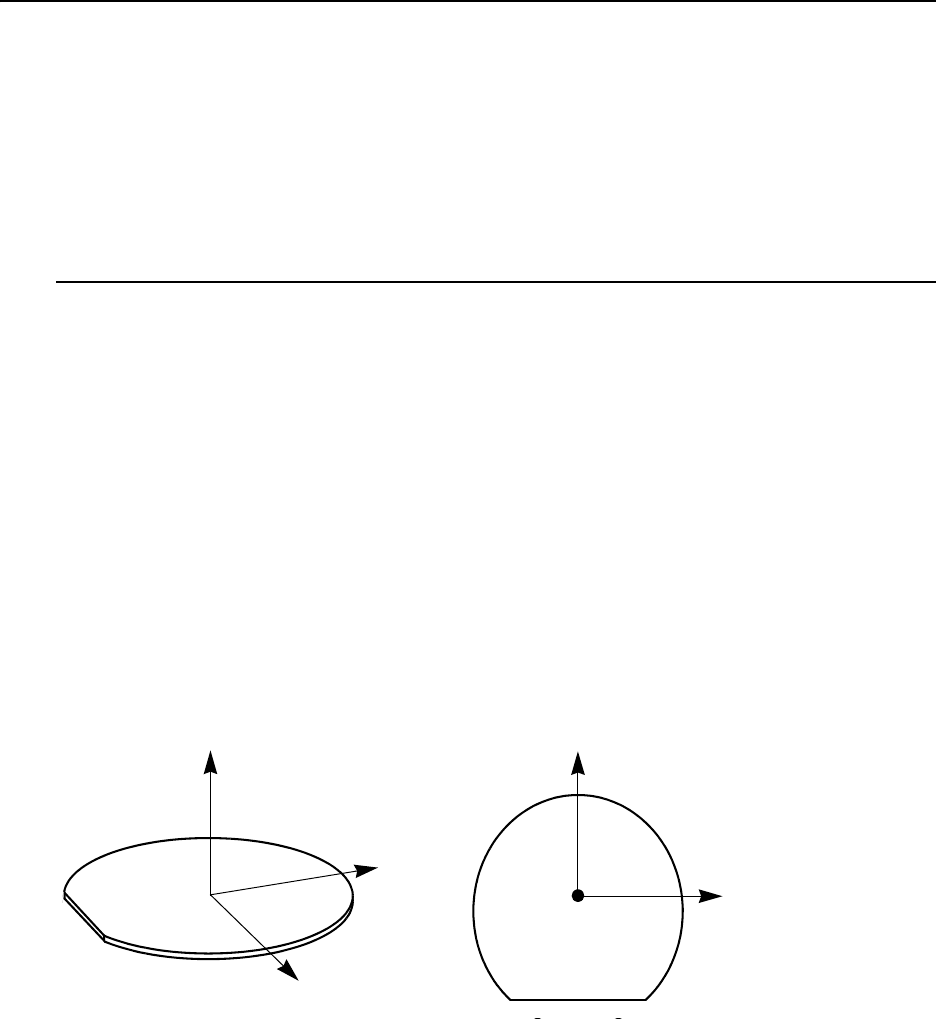
2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
66 Sentaurus™ Process User Guide
H-2013.03
Creating and Loading Structures and Data
The first step in most simulations is either to load an existing structure or to create a new one.
New structures are created through a combination of the line, region, and init commands.
The initial mesh is a tensor-product mesh where the density of lines is specified in the line
command, and the regions are defined by specifying tags in the line commands and defined
in the region command. The initial regions are always defined as axis-aligned rectangles in
2D and axis-aligned bricks in 3D.
Understanding Coordinate Systems
Sentaurus Process and related tools use different coordinate systems. The most commonly
encountered coordinate systems include wafer coordinates, simulation coordinates, and
visualization coordinates.
Wafer Coordinate System
The wafer coordinate system is fixed with respect to the wafer flat or notch, and is used to
define the relationship of all other coordinate systems to the physical wafer. The wafer
coordinate system is shown in Figure 16.
The wafer x- and y-axes form a naturally oriented coordinate system when the wafer is drawn
with the flat pointing down as shown in Figure 16. This coordinate system is used for layout
information, such as mask locations, and for setting a cutline using the CutLine2D command.
Figure 16 Wafer coordinate system
ZW
XW
YW
YW
ZW
XW
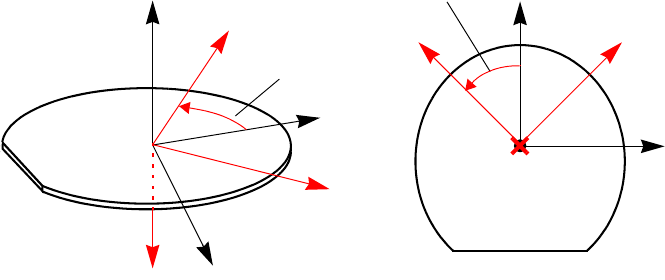
2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
Sentaurus™ Process User Guide 67
H-2013.03
Simulation Coordinate System (Unified Coordinate System)
The simulation coordinate system is used to define the mesh for the simulation. All coordinates
that are specified with respect to the mesh are given in simulation coordinates. This includes
all coordinates that are given in the Sentaurus Process command file.
The simulation coordinate system has the x-axis pointing into the wafer and the y-axis rotated
with respect to the wafer y-axis. The simulation coordinate system is shown in Figure 17.
Simulations in 1D use only the x-axis. Simulations in 2D use only the x- and y-axes.
Figure 17 Simulation coordinate system (slice.angle = 45)
The rotation of the simulation axes with respect to the wafer axes is given by the slice.angle
parameter of the init command. The slice angle is measured from the wafer y-axis to the
simulation y-axis with positive angles counterclockwise about the wafer z-axis.
ZW
XW
YW
YW
XS
YSZS
XW
slice.angle
YS
ZS
XS
slice.angle
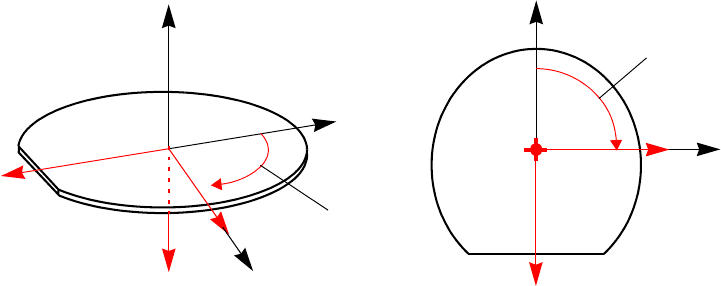
2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
68 Sentaurus™ Process User Guide
H-2013.03
The default value of slice.angle is set to . This causes the simulation y-axis to match
the wafer x-axis, which is the usual cut direction through the layout for 2D simulations. The
default simulation coordinate system is shown in Figure 18.
Figure 18 Default simulation coordinate system (slice.angle = –90)
Visualization Coordinate Systems
Two systems can be used for visualization:
■The unified coordinate system (UCS)
■The DF–ISE coordinate system
Unified Coordinate System
To use the UCS, specify:
math coord.ucs
This system of coordinates is explained in Simulation Coordinate System (Unified Coordinate
System) on page 67.
NOTE The UCS is the recommended way of visualization and may become the
default in the future.
DF–ISE Coordinate System
The DF–ISE coordinate system is the default for visualizing TDR files. It can be set explicitly
by:
math coord.dfise
90°–
ZW
YS XW
YW
XS
ZS
slice.angle
YW
XW
slice.angle
ZS
XSYS
2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
Sentaurus™ Process User Guide 69
H-2013.03
The DF–ISE coordinate system is used by the DF–ISE and some TDR file formats as well as
Tecplot SV. Unlike the simulation coordinate system in which the x-axis points into the wafer
for 1D, 2D, and 3D, the visualization coordinate system has different axis conventions for 1D,
2D, and 3D. Figure 19 shows the relationship between simulation coordinates and DF–ISE
coordinates.
Figure 19 Simulation and DF–ISE coordinate systems
The only difference between UCS coordinates and DF–ISE coordinates is that different
conventions are used to label the axes. Sentaurus Process automatically converts the axis labels
when reading and writing DF–ISE or TDR files.
The relationship between DF–ISE coordinates and UCS coordinates shown in Figure 19
applies to all values of slice.angle. In other words, the DF–ISE system is not fixed with
respect to the wafer system. It always has the same rotation with respect to the wafer coordinate
system as the simulation coordinate system.
Figure 20 on page 70 shows the relationship between simulation coordinates and visualization
coordinates.
Simulation Coordinates
x
x
x
y
yz
DF-ISE Coordinates
x
y
x
x
y
z
1D
2D
3D
2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
70 Sentaurus™ Process User Guide
H-2013.03
Figure 20 Simulation and visualization coordinate systems
The tensor components, for example, mechanical stresses, in 2D and 3D are the same in UCS
coordinates. For a conversion table for the stress components in 2D and 3D DF–ISE coordinate
systems, see Chapter 9 on page 643. This also applies to other second-order symmetric tensors.
Defining the Structure: The line and region Commands
The line and region commands are used together to define the structure. In the init
command, the structure is actually formed. Care must be taken when creating a structure
because there are few checks for errors.
These rules must be followed to obtain a valid structure:
■If this is not the first structure being created in a command file, the command line clear
must be issued to remove line commands and stored mesh ticks.
■Line locations must be given in increasing order.
■The region boundaries are defined by tagged lines. Tagged lines are created with the line
command where the parameter tag has been set (as well as the location parameter).
■At least one region command must be given to define the substrate.
■Regions must have a material specification, except for the substrate case described
below.
■Regions must have the same dimensionality as the line commands used (that is, if line y
is given, a 2D region is expected with ylo and yhi set).
Simulation Coordinates
x
x
x
y
yz
Visualization Coordinates
x
y
x
x
y
z
1D
2D
3D

2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
Sentaurus™ Process User Guide 71
H-2013.03
■The spacing parameter is used to create lines between user-defined lines, so that not every
line must be specified in the command file. Sentaurus Process smoothly grades the line
density between user-defined lines to match as closely as possible the spacing at each user-
defined line. In addition, there will be lines at locations given by the location parameter
of the line command. By default, the spacing parameter is extremely large, so that if it
is not set, only lines given by the location parameter will be in the mesh.
■The *lo parameter refers to the lowest coordinate value, that is, the location of the line
corresponding to the xlo tag must be less than the coordinate corresponding to the xhi tag.
■The region command can be used to tag a region as a substrate in two ways:
•If the region is being defined with the material name and the parameters *hi and *lo,
the Boolean keyword substrate will tag this region as the substrate.
•If the structure is being loaded from a previously saved file, the command:
region name=<region_name> substrate
will tag the region with region_name as the name of the substrate. This is the only
occasion when the region command will be called after the init command.
Considerations when creating structures are:
■For 2D and 3D simulations, it is advantageous to create a coarse mesh in the lateral (that
is, y- or z-directions) because lines created with the line command run all the way through
the structure. Often, finer spacing in the y- or z-direction is needed near the surface;
whereas, further in the bulk, a coarser spacing is required (to minimize the size of the
problem).
■When MGOALS is used for etching and deposition, or meshing, it automatically creates a
local refinement near interfaces that does not run the length of the structure.
■To specify refinement boxes, use the refinebox command. Control over the meshing
parameters for MGOALS is explained in Chapter 11 on page 731.
Creating the Structure and Initializing Data
The init command is used to create the structure. If the line and region commands have
been given to create a structure from the beginning, the init command does not require any
options. It will take the structure definition and create a new structure.
Many process steps such as etching, deposition, diffusion, and implantation require a gas mesh.
By default, Sentaurus Process does not add a gas mesh during the init command, but delays
creating the gas mesh until it is needed. To add the gas mesh immediately, use the command:
pdbSet Grid AddGasMesh 1
2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
72 Sentaurus™ Process User Guide
H-2013.03
NOTE The parameter must be set before the init command to generate the
gas mesh during the init command.
There are several ways to initialize fields at the time the initial structure is created from line
and region commands:
■To initialize data everywhere in the structure, a field specification can be given in the init
command.
■To initialize data in one particular region only, a field specification is given in the region
command.
In both the init and region commands, the field parameter specifies the name of the data
field that will be created and either the concentration parameter or the resistivity
parameter is used to specify the value created. Although initialization was intended for
dopants, a field with any name can be initialized with the concentration parameter.
However, it will create a field with nodal values and, because stresses are computed on
elements, it should not be used for initializing stress values (use the stressdata command or
the select command for this). The resistivity parameter only works for fields that have
the resistivity parameters set (which by default are only As, B, P, Sb, and In in silicon).The
init command also is used to read a structure from a file. In this case, the parameter name
serves as the type specification, and the value of the parameter is the file name or root name (in
the case of DF–ISE), for example:
init dfise=file ;# Read 'file.grd' and 'file.dat'.
# If not available try 'file.grd.gz' and 'file.dat.gz'
init dfise=file.grd ;# Only read a structure (no data) from 'file.grd'
# or 'file.grd.gz'
init tdr=file ;# Read a geometry, data, and pdb parameters from
# 'file.tdr'.
init bnd=file ;# Read a bnd file and mesh it.
The TDR format is used to restart a simulation by default when creating ‘splits’ in Sentaurus
Workbench. This format stores all pdb parameter settings as well as numerous other settings
coming from commands (see Saving a Structure for Restarting the Simulation on page 76).
The bnd parameter is used to load boundaries that are then meshed by MGOALS. In this case,
the structure is meshed with the constrained Delaunay mesher, which makes use of lines
coming from the line command.
The init command is used to specify the principal wafer orientation (wafer.orient), the
lateral crystal orientation of the wafer flat or notch (flat.orient), and the slice.angle
for the implant command, that is angle:
init wafer.orient= {<i> <j> <k>} flat.orient= {<i> <j> <k>} slice.angle=<n>
where <i>, <j>, and <k> are the crystallographic (Miller) indices. For more information about
the wafer orientation and the slice angle, see 2D Coordinate System on page 91.
2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
Sentaurus™ Process User Guide 73
H-2013.03
You al so can se t the slice.angle by using a 2D cutline, for example:
init slice.angle= [CutLine2D <x1> <y1> <x2> <y2>]
The first two values define the start point, and the third and fourth values define the endpoint
in the wafer plane. The two points are defined in the wafer coordinate system (see
Understanding Coordinate Systems on page 66).
Defining the Crystal Orientation
The crystal orientation of the wafer is established by specifying the Miller indices of the wafer
surface and the wafer flat. The wafer.orient and flat.orient parameters of the init
command specify the Miller indices of the wafer z- and y-axes, respectively. The wafer surface
orientation (z-axis) is set using wafer.orient= {<i> <j> <k>} where <i>, <j>, and <k>
are the crystallographic (Miller) indices. The flat orientation (y-axis) can be set arbitrarily, but
it must be orthogonal to the wafer surface orientation. The default surface orientation is 100
and the default flat orientation is a 110 direction for all values of wafer.orient.
NOTE The wafer.orient and flat.orient parameters of the init
command apply to orthorhombic crystal systems only (such as silicon).
For information on how to change the crystal orientation in hexagonal
systems, see MC Implantation into Silicon Carbide on page 165. Setting
info=2 in the implant command confirms user-defined settings for
each region in the log file only.
Table 2 lists the crystallographic directions of the wafer axes for the most common
crystallographic orientations of the wafer as shown in Figure 16 on page 66.
Sentaurus Process also allows you to define different crystal orientations for different regions
by using the commands:
pdbSetDoubleArray <region_name> crystal.orient <double array>
pdbSetDoubleArray <region_name> flat.orient <double array>
Ta bl e 2 M i l le r i n d i ce s o f wa fer a xe s fo r e a c h va lu e o f wa fer.o r i en t
(wafer axes are defined in Figure 16)
Wafer orientation XW Y
W Z
W
100
110[]
110[]
001[]
110
001[]
110[]
110[]
111
112[]
110[]
111[]

2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
74 Sentaurus™ Process User Guide
H-2013.03
To facilitate simulations of hybrid orientation technology (HOT), Sentaurus Process predefines
three materials (Silicon, Silicon110, and Silicon111) for crystalline silicon. These
materials have exactly the same properties, except for the default crystal orientations that are
<100>, <110>, and <111> for Silicon, Silicon110, and Silicon111, respectively.
Automatic Dimension Control
The maximum dimension of a simulation is determined by the specified line commands;
line x commands define the extensions in the vertical direction and are required for 1D, 2D,
and 3D simulations. If, in addition, line y commands are specified, the maximum dimension
of the simulation will be at least 2D and, if also line z commands are specified, the maximum
dimension of the simulation will be three dimensions. By default, Sentaurus Process delays the
creation of a full-dimensional structure until it becomes necessary. This means that if you
specify a 2D structure where all regions span the entire simulation domain in the y-direction,
Sentaurus Process will create a 1D structure.
When a 2D or 3D mask is used in an etch, a deposit, or a photo command, Sentaurus
Process automatically extrudes the structure and the mesh into the appropriate dimension and
copies the data. This delay of creating a full-dimensional structure can be switched off in the
init command by using the option !DelayFullD. To increase the dimension manually, use
the grid command. If a 2D structure is required, that is, both the line x and line y
commands but no line z commands have been specified, grid 2D or grid FullD will
cause a 2D structure to be created.
Similarly, if line x, line y, and line z commands have been specified, grid 2D can be
used to extrude a 1D structure to two dimensions, and a 1D or 2D structure is extruded to three
dimensions using grid 3D or grid FullD. This functionality also can be used to increase
the dimension of structures loaded from files. After the structure has been loaded, line
commands can be issued and the dimension of the structure will increase automatically when
necessary or manually using the grid command.
Sentaurus Process does not provide a facility to reduce the dimension of a simulation.
When structures are saved to DF–ISE files or TDR files (other than TDR restart files), the
current maximum dimension as specified with line commands is used by default in the file.
The dimension of the simulation itself is not affected. To save files in the current dimension,
the !FullD parameter of the struct command can be used (see Saving and Visualizing
Structures on page 75). TDR restart files are always saved in the dimension currently used in
the simulation.

2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
Sentaurus™ Process User Guide 75
H-2013.03
Saving and Visualizing Structures
Sentaurus Process supports two file formats for reading and writing structures: TDR and the
older DF–ISE. Both the TDR and DF–ISE formats allow saving the structure geometry with
and without the bulk mesh and data, and with contacts. These files contain simply connected
regions to operate smoothly with other Synopsys TCAD tools. One important option available
for saving TDR and DF–ISE files is to omit saving gas regions because this may cause
problems for other tools.
The TDR format allows for the saving and loading of geometry and data information along
with pdb parameters. For more information about file types and standard file extensions, see
File Types Used in Sentaurus Process on page 48.
The TDR format is the preferred file format over the DF–ISE format. TDR files can be used to
split a simulation, and restart and continue the simulation as if no file save or file load was
performed. Besides the simulation grid and data, additional information is stored to facilitate
such a restart. Only TDR files provide such restart capability; simulation results will differ if a
simulation is performed in one contiguous run compared to saving and loading the intermediate
state into _bnd.tdr or DF–ISE files.
Setting the parameter math coord.<coord name> configures whether the visualization
coordinates will be identical to the simulation ones (when using coord.ucs) or will follow
the DF–ISE criteria (when using coord.dfise).
When using DF–ISE, it is important to understand the difference between the simulation
coordinates used by Sentaurus Process and the coordinates seen in Tecplot SV. For Sentaurus
Process, the positive x-direction always points into the substrate in 1D, 2D, and 3D. TDR and
Tecplot SV have different axis directions in 1D, 2D, and 3D. With coord.dfise, Sentaurus
Process rotates the structure into the DF–ISE coordinate system when saving the structure and
rotates the structure back when reading it.
Figure 19 on page 69 shows the relation between the UCS and DF–ISE coordinates. The
exposed surface of the substrate is oriented upwards; that is, the ‘up’ direction is always in the
negative x-direction in the UCS.
To select the fields stored in TDR files, use the SetTDRList command. Each field name in the
SetTDRList command is added to the list of fields, which are usually saved (if the field is
present in the structure). This command also takes as arguments the macro parameters
Dopants and Solutions, and their negative counterparts !Solutions and!Dopants.
Solutions refers to variables of partial differential equations (PDEs). The solution variables
must be stored in a TDR file if that file is to be used to continue a simulation. The parameter
Dopants refers to the total and active dopant concentration fields. By default, TDR files are
saved with both Solutions and Dopants names in SetTDRList. However, this requires
2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
76 Sentaurus™ Process User Guide
H-2013.03
many fields to be stored in the TDR files and, sometimes, it is more convenient to have fewer
fields.
To do this, set !Solutions in SetTDRList, which unselects all fields. Then, specify the field
names to be stored in the TDR file (see SetAtomistic on page 1121 for saving KMC fields).
Saving a Structure for Restarting the Simulation
When saving files using the TDR format, the current state of the parameter database is, by
default, saved in the file. The parameter database contains all of the information necessary to
restart a simulation including:
■Model settings
■Parameter settings
■Mesh settings from the mgoals command
■Refinement boxes from the refinebox command
■Temperature ramps from the temp_ramp command
■Gas flow specifications from the gas_flow command
■Line specifications from the line command
■Region specifications from the region command
■Reaction specifications from the reaction command
■Specifications for point, polygon, polyhedron
■Doping specifications with the doping command
■User materials created with the mater command
■Contact definitions created with the contact command
■Mask definitions created with the mask command
■Solution commands can be optionally stored using the store parameter of the solution
command
■Term commands can be optionally stored using the store parameter of the term
command
■Global Tcl variables can be stored with fset
■Tcl procedures can be stored using fproc
By default, when loading a TDR file, the changes in the parameter database are read in from
the TDR file and are applied. For information about the TDR format, refer to the Sentaurus™
Data Explorer User Guide.
2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
Sentaurus™ Process User Guide 77
H-2013.03
When saving a TDR file, the coordinate system used for visualization is also included in the
file and is used by Tecplot SV when opening it. The visualization coordinate system can be
changed using the math pdb command.
For the UCS, use:
math pdb coord.ucs
For the DF–ISE coordinate system, use:
math pdb coord.dfise
Saving a Structure for Device Simulation
In general, there are three main steps to saving a structure appropriate for device simulation:
1. Define contacts.
2. Remesh the structure with appropriate refinement for device simulation.
3. Save the structure with contacts and with Delaunay weights.
Contacts are defined using the contact command. There are two main ways to define
contacts, either:
■Using a box where the contact is created at the intersection of a material interface and a box.
■Using a point contact in which a region is specified by giving a point inside the region; then
all boundaries of this region become a contact.
The contact is given a name and, if the command is executed multiple times with the same
contact and the add parameter, the contact will include all parts specified. There are also
options for creating a contact on the outer boundaries and so on. For more information, see
contact on page 889.
Remeshing the structure is needed to create a mesh that is better suited to device simulation.
Typically, this means discarding process-based refinements, creating a very fine mesh under
the channel, and refining on the p-n junction. A typical sequence of steps is:
■Clear the process mesh:
refinebox clear
line clear
■Reset default settings for adaptive meshing:
pdbSet Grid AdaptiveField Refine.Abs.Error 1.e37
pdbSet Grid AdaptiveField Refine.Rel.Error 1e10
pdbSet Grid AdaptiveField Refine.Target 100.0
2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
78 Sentaurus™ Process User Guide
H-2013.03
■Set high-quality Delaunay meshes:
pdbSet Grid SnMesh DelaunayType boxmethod
■Set mesh spacing near interfaces:
mgoals min.normal.size=<n> normal.growth.ratio=<n>
■Set which interfaces will have interface refinement:
refinebox interface.materials= {Silicon}
■Specify adaptive refinement:
pdbSet Grid Adaptive 1
■Specify lines if necessary:
line y loc= $Ymin+0.001
line z loc= $Zmin+0.001
■Specify refinement boxes, for example:
refinebox min= {<n> <n> <n>} max= {<n> <n> <n>} \
xrefine= <n> yrefine=<n> zrefine=<n> ;# gate refinement
refinebox refine.fields=NetActive max.asinhdiff= {NetActive= 1.0} \
refine.min.edge=<n> Silicon ;# adaptive refinement on NetActive
■If using the IC WorkBench EV Plus interface, it may be useful to consider using the
icwb.refine.mask command (see Chapter 12 on page 817 and icwb.refine.mask on
page 959 for more information).
In these steps, <n> are coordinates or edge lengths in micrometers, and
normal.growth.ratio is a unitless ratio.
To save the structure, use the command struct tdr=<filename> !Gas. This command
causes a remesh if necessary, stores any contacts that have been defined previously, and
includes fields required for device simulation.
Delaunay weights can be saved in the structure intended for device simulation by setting these
parameters before generating the mesh:
pdbSet Grid SnMesh StoreDelaunayWeight 1
pdbSet Grid Contact.In.Brep 1
The first parameter StoreDelaunayWeight creates the field variable Delaunay–Voronoï
weight (DelVorWeight) that is used in the weighted box method in Sentaurus Device. The
second parameter Contact.In.Brep switches on an experimental feature that creates
contacts in the boundary representation (brep) and prevents changes to the mesh that can
locally invalidate the Delaunay weight.
2: The Simulator Sentaurus Process
Creating and Loading Structures and Data
Sentaurus™ Process User Guide 79
H-2013.03
Saving Doping Information in SiC and GaN for Device Simulations
Basic process simulation capabilities such as etching, deposition, and implantation with Monte
Carlo are available for multicomponent materials, for example, silicon carbide (SiC) and
gallium nitride (GaN). However, there are no activation models for dopants in these materials.
To create active doping concentration fields that are equal to their associated total fields, when
saving a file for transfer to device simulation, use the diffuse command with zero time, for
example:
diffuse time=0 temperature=900
struct tdr= <file name>
Saving 1D Profiles for Inspect
To store .plx files, use the WritePlx command. The command SetPlxList selects the
fields to be stored in the .plx file. The command SetPlxList is similar to the
SetDFISEList command, except that no fields are selected by default. Only the field names
specified in SetPlxList are stored in the .plx file (see SetPlxList on page 1128 and WritePlx
on page 1191).
Saving 1D TDR Files from 2D and 3D Simulations
The command struct also saves a 1D TDR file if the proper cutting coordinates are specified.
In 2D, only one cutting coordinate is needed (either x or y; coordinate z makes no sense here).
In 3D, the command saves the intersection of the planes specified by two cutting coordinates
(for example, specifying x and z will save the y line containing those x- and z-coordinates). In
addition to storing the mesh and data, these files save any contacts that apply at the cut point,
so that the file can be loaded into Sentaurus Device for electrical analysis. This file can be
visualized with Tecplot SV.
For example, in a 2D simulation, the following command:
struct tdr=filename y=0.5
picks up all the x-coordinates with y=0.5 and saves them in a 1D TDR file.
In addition, in a 3D simulation, the following command:
struct tdr=filename x=0.2 z=0.1
saves the y-coordinates with x=0.2 and z=0.1 as a 1D TDR file.
For more information, see struct on page 1158.

2: The Simulator Sentaurus Process
References
80 Sentaurus™ Process User Guide
H-2013.03
The select Command (More 1D Saving Options)
The select command is a versatile command for many operations such as viewing results,
postprocessing, and initializing or changing datasets. The basic command is:
select z=<expression>
where <expression> is an Alagator expression (see Chapter 6 on page 571). A simple
example of an <expression> is the name of a data field such as Potential and VTotal.
The value of the expression is stored in the selected field.
This selected field can be viewed with print.data or print.1d, for example, or the
integrated values can be obtained using the layers command. The select command can also
be used to set an existing data field or create a new data field, for example:
select z=1.0 name=MyDataField ;# create a new datafield named MyDataField
# and set it to 1.0 (everywhere)
select z= 0.1*Vacancy name=Void store ;# Set Void equal to 0.1*Vacancy
Loading 1D Profiles: The profile Command
The profile command is used to load a 1D profile into 1D, 2D, or 3D structures. The file to
be read should contain one x-coordinate data pair per line. Both linear (using the linear
parameter) and logarithmic interpolation (default) are available. Profiles are loaded by using:
profile infile = file.dat name = Boron
Sentaurus Process reads the file file.dat and sets the field Boron accordingly.
References
[1] B. B. Welch, Practical Programming in Tcl & Tk, Upper Saddle River, New Jersey:
Prentice Hall PTR, 3rd ed., 2000.

Sentaurus™ Process User Guide 81
H-2013.03
CHAPTER 3 Ion Implantation
The chapter presents the ion implantation technique used in
Sentaurus Process.
Overview
Ion implantation is one of the most widely used processing techniques to introduce impurity
atoms into semiconductor materials. In Sentaurus Process, either analytic functions or the
Monte Carlo (MC) method is used to compute the distribution of implanted ions and the
implantation damage. Analytic implantation models use the simple Gaussian and Pearson as
well as the advanced dual Pearson functions. The implantation damage with analytic models is
calculated according to the Hobler model [1]. The MC method uses a statistical approach to the
calculation of the penetration of implanted ions into the target and accumulation of crystal
damage based on the binary collision approximation [2].
Analytic implantation simulates the spatial distribution of the implanted ions based on the
selected distribution function, which is described by moments. The distribution moments
depend on the ionic species, implantation energy, dose, and tilt and rotation angles. Sets of
moments for a given range of implantation parameters are provided in the form of lookup
tables. Sentaurus Process can use implantation tables in the Dios format, TSUPREM-4
formats, and the Taurus Process table format. The implantation data available includes the
default tables [3], the Advanced Calibration tables [4], the Taurus table set [5], and the original
Tasch tables [6].
Sentaurus Process handles 1D, 2D, and 3D geometries for both analytic implantation
simulations and MC simulations. The algorithms for analytic implantation are an integral part
of Sentaurus Process; whereas, MC simulations are performed with the binary collision code
Sentaurus MC [7] or Crystal-TRIM [8].
Analytic ion implantation is performed using the implant command:
implant <dopant> [energy=<n>] [dose=<n>] [tilt=<n>] [rotation=<n>]
Sentaurus Process simulates an analytic implantation step producing output such as:
---------------------------------------------------------------- implant -----
implant energy=35.00<keV> dose=1.00e.+14<cm-2> tilt=7.00<degree>
rotation=-90.00<degree> Boron
------------------------------------------------------------------------------
Species = Boron
3: Ion Implantation
Overview
82 Sentaurus™ Process User Guide
H-2013.03
Dataset = Boron
Energy = 35keV
Dose (WaferDose) = 1e+14/cm2
BeamDose = 1.0075e+14/cm2
Tilt = 7deg
Tilt2D = 7deg
Rotation = -90deg
Slice angle = -90deg
Temperature = 300.00K
For a description of the analytic implantation mode, see Analytic Implantation on page 92.
To switch from analytic implantation to MC implantation with Sentaurus MC, use the logical
switch sentaurus.mc (or its alias tmc):
implant <dopant> [energy=<n>] [dose=<n>] [tilt=<n>] [rotation=<n>]
[sentaurus.mc]
To switch from analytic implantation to MC implantation with Crystal-TRIM, use the logical
switch crystaltrim (or its alias ctrim):
implant <dopant> [energy=<n>] [dose=<n>] [tilt=<n>] [rotation=<n>]
[crystaltrim]
If the cascades switch is used in addition to sentaurus.mc or crystaltrim, the MC
implantation is run in the full-cascade mode. For a description of Sentaurus MC and the
Crystal-TRIM mode, see Monte Carlo Implantation on page 133.
An external profile can be loaded using the load.mc switch:
implant <dopant> [energy=<n>] [dose=<n>] [tilt=<n>] [rotation=<n>]
[load.mc] [file=<c>]
A TDR file must be specified with the file selector. load.mc works with files created by
either Sentaurus MC or Crystal-TRIM. For a full description of the file-loading mode, see
Loading External Profiles on page 190.
The implantation energy in the implant <dopant> facility is given in keV by default. The
implantation dose has two modes:
■The wafer dose (WaferDose), which refers to the expected dose in the structure after the
implantation is finished. This dose is measured in ions per .
■Alternatively, the implantation dose can mean the beam dose (BeamDose).
In the wafer dose mode, the final implanted dose does not depend on the wafer orientation with
respect to the ion beam. In the beam dose mode, the final implanted dose may change as tilt
and rotation angles change. For a discussion of the meaning and implications of the tilt and
rotation angles, see Coordinate System on page 89. All angles are measured in degrees.
cm2
3: Ion Implantation
Overview
Sentaurus™ Process User Guide 83
H-2013.03
The mode of the implant dose can be specified with the following pdb switches:
pdbSet ImplantData DoseControl {Default WaferDose BeamDose}
The default value of DoseControl switch is Default, in which case, the mode of implant
dose is chosen automatically based on the implant table format. If the currently selected
implant tables are in Taurus/TSUPREM-4 format, the beam dose mode is used automatically.
Otherwise, the wafer dose mode is applied. If the DoseControl switch is set to WaferDose,
the wafer dose mode is used for all implantations regardless of table formats, likewise for
BeamDose.
NOTE To obtain consistent results and prevent unexpected dose mode, it is
strongly recommended to always set the DoseControl parameter at
the start of command files to either WaferDose or BeamDose.
To override these global settings, use the logical switch beam.dose in the implant
command:
implant <dopant> [dose=<n>] [beam.dose !beam.dose]
NOTE The main parameters for the implant statements energy, dose, tilt,
and rotation must always be specified. Otherwise, default values are
chosen that may not reflect the assumed process conditions.
In addition to energy, dose, tilt, and rotation, you can specify the implant temperature
and the dose rate. Temperature and current are recognized as parameters by the format moment
tables of Taurus Process.
If the structure is completely covered by photoresist, you can omit an implantation step by
using the following pdb command:
pdbSet ImplantData ResistSkip 1
By default, it is not omitted.
The amount of information printed to the log file and displayed is controlled by the parameter
info in the implant command. The value of info must be set to an integer value between 0
and 2. The higher the value, the more detailed information is printed to the log file and
displayed. Output messages with an information level less than 3 can be easily understood by
typical users.
NOTE Messages with info=3 or more are better understood by users with
greater knowledge of the Sentaurus Process implantation code and is
reserved for debugging.

3: Ion Implantation
Overview
84 Sentaurus™ Process User Guide
H-2013.03
Selecting Models
The implanted species must be a previously initialized species. To initialize an implantation
species, use the implant species=<dopant> facility, that is:
implant species=<dopant> <material> [imp.table=<file>] [model] [damage]
NOTE This command does not perform an implantation step. It is distinguished
from the standard use of the implant command by the keyword
species (or tables).
Here, dopant can be any name, while material should be an initialized material (see
Material Specification on page 52). To select the implantation table file, containing moments
for the primary and lateral distributions, use the keyword imp.table. The <model> switch
selects the implant model. The available choices are discussed in Primary Distribution
Functions on page 94. The following models are available:
■Gaussian distribution: gaussian
■Single Pearson distribution: pearson
■Single Pearson distribution with linear exponential tail: pearson.s
■Dual Pearson distribution: dualpearson
■External distribution: point.response
To switch on damage calculation, use the damage flag.
The following command, for example, changes the default implantation table for boron in
silicon to my_table.tab and the implant model to pearson. It also switches off the damage
calculation for boron in silicon:
implant species=Boron Silicon imp.table=my_table.tab pearson !damage
At the beginning of a Sentaurus Process run, all species are initialized automatically using the
implant species=<dopant> facility.
Table 3 lists the species that are supported and recognized in a Sentaurus Process run.
Table 3 Overview of default species initialized by Sentaurus Process
Atomic species Molecular Description
Aluminum, Antimony, Arsenic,
Boron, Carbon, Fluorine,
Gallium, Germanium, Indium,
Nitrogen, Phosphorus,
Silicon
AsH2,BF2,
B10H14,
B18H22, BCl2,
C2B10H12,
C2B10H14,PH2
Used in analytic and MC implantation. Implant tables
are available for atomic species and molecular BF2.
For other molecular species, implantation is performed
based on the tables for primary dopant species (As, B,
or P).

3: Ion Implantation
Overview
Sentaurus™ Process User Guide 85
H-2013.03
You ca n ove rw rite o r ex te nd th es e set ti ngs at any time during a Sentaurus Process run. There
are three principal ways to change the initial settings. With the previously described command,
you can change the settings for one pair of dopant species and material. To overwrite the
settings for one particular dopant species in all materials, use:
implant [species=<dopant>] tables=<name>
The <name> string selects a set of tables and model switches. Internally, Sentaurus Process
executes a set of implant species=<dopant> <material> commands, which set the
implant parameters for one pair of dopant species and material, respectively.
NOTE The keyword tables=<name> does not refer to a particular table or
table name. It sets all tables and model switches for the species in all
materials using a Tcl procedure.
The possible choices for <name> are discussed in Tables on page 104. The following settings
are available:
■Mixed dual Pearson and single Pearson tables: Default
■Taurus Process table set: Taurus
■University of Texas tables: Tasch
■Single Pearson tables used in Dios: Dios
■TSUPREM-4 native implant tables: TSuprem4
Dios or Default Tables
For example, the following command changes all implant specifications for the species boron
from the default to the Dios implantation tables and models:
implant species=Boron tables=Dios
If the above command is given without the keyword species, that is:
implant tables=<name>
Argon, Beryllium, Bromine,
Cadmium, Chlorine, Helium,
Hydrogen, Iodine, Iron,
Krypton, Lead, Magnesium,
Neon, Oxygen, Selenium,
Sulfur, Tellurium, Tin,
Titanium, Xenon, Zinc
No implantation tables are available. Analytic
implantation will abort. Recommended for use in MC
only.
Table 3 Overview of default species initialized by Sentaurus Process
Atomic species Molecular Description
3: Ion Implantation
Overview
86 Sentaurus™ Process User Guide
H-2013.03
the implant tables and model switches are overwritten for all species in all materials. The
default setting for <name> is Default and the command:
implant tables=Default
is equivalent to the (default) initialization of all species and models at the beginning of each
Sentaurus Process run.
Taur us Tabl e s
The command:
implant tables=Taurus [data.suf=<suffix>] [dam.suf=<suffix>]
switches to the Taurus mode. This means that Sentaurus Process uses the same moment tables
as the Taurus Process implant library in TSUPREM-4. The file names for Taurus tables are
conventionally named as <ion>_in_<material>_<suffix> and
<ion>_damage_in_<material>_<suffix> for implant data and damage data,
respectively. The default suffix is standard for both implant data and damage data. The
optional parameters data.suf and dam.suf can be used to change the default suffix for
implant data and damage data, respectively. By using different suffices, different tables for the
same species/material combination can coexist in the same directory.
In addition, if tables=Taurus was specified, several models are switched on that are not used
by default. These models are:
■Beam dose control: beam.dose (see Overview on page 81)
■Proportional range scaling: range.sh (see Multilayer Implantations on page 111)
■Effective channelling suppression: eff.channeling.suppress (see Screening (Cap)
Layer-dependent Moments on page 98)
■Profile reshaping: profile.reshaping (see Profile Reshaping on page 125)
■Preamorphization implants (PAI): pai (see Preamorphization Implantation (PAI) Model
on page 121)
NOTE This does not give the same results as TSUPREM-4; however, the
results are similar.
TSUPREM-4 Native Implant Tables
Sentaurus Process also can read implant tables in TSUPREM-4 native format. To select native
TSUPREM-4 implant tables, use the command:
implant [species=<c>] tables=TSuprem4 [ts4.prefix=<c>]
3: Ion Implantation
Overview
Sentaurus™ Process User Guide 87
H-2013.03
If species is specified, TSUPREM-4 implant tables are applied to this particular species only.
If species is not specified, implant tables are applied to all TSUPREM-4 supported species,
which include antimony, arsenic, BF2, boron, fluorine, indium, fluorine, and phosphorus.
The name of the TSUPREM-4 native implant table conventionally uses the species name or the
species name with a prefix. Eight different implant tables in silicon are distinguished by a
prefix. For example, chboron (which means channeling boron) is one of the boron implant
tables in silicon.
The parameter ts4.prefix takes one of the followings values: default, none, le, ch,
dual, ut, tr, or scr:
■The default value is ts4.prefix=default, which selects TSUPREM-4 default implant
tables, that is, antimony, fluorine, chboron, dual.ars, dual.pho, dual.bf2, and
tr.indium for antimony, fluorine, boron, arsenic, phosphorus, BF
2, and indium,
respectively.
■If ts4.prefix=none, no prefix is added, so the TSUPREM-4 implant tables (antimony,
boron, and so on) are used for antimony, boron, and so on, respectively. If the
corresponding table for a species in a material is not available, the default table is used.
■If ts4.prefix=le or ch, then le<species> or ch<species> tables are selected, for
example, leboron or chboron for boron implantation. If the corresponding table for a
species in a material is not available, the default table is used.
■If ts4.prefix=dual, ut, tr, or scr, then <prefix>.<species> tables are selected,
for example, dual.boron, ut.boron, tr.boron, and scr.boron for boron
implantation. If the corresponding table for a species in a material is not available, the
default table is used.
You also can use your own TSUPREM-4 native-formatted implant tables by using the
following command:
implant species=<dopant> <material> imp.table=<file> ts4.species=<name>
ts4.material=<name>
imp.table specifies the file name (which should have the file-name extension .ts4) that
contains implant moment tables in TSUPREM-4 format, such as mys4imp0.ts4. If the file is
in the same directory where Sentaurus Process is being run, then only the name of the file is
needed for imp.table; otherwise, the full path is required. ts4.species specifies the
TSUPREM-4 table name for the dopant, which is one of the predefined impurity names in the
implant data file. For example, in the standard s4imp0, the valid names for boron implant are
boron, leboron, chboron, ut.boron, tr.boron, and scr.boron. ts4.material
specifies the material name used in TSUPREM-4, which is one of the predefined material
names in the implant data file. For example, in the standard s4imp0, the material names
include silicon, polysilicon, oxide, nitride, and so on.

3: Ion Implantation
Overview
88 Sentaurus™ Process User Guide
H-2013.03
If not specified, ts4.species and ts4.material default to the species name (for example,
Boron) and the material name (for example, Silicon) used in Sentaurus Process,
respectively.
NOTE Ensure that these names match exactly the names in the TSUPREM-4
implant data file. While these names are not case sensitive, they cannot
be abbreviated. For example, while ts4.material=Polysilicon is
acceptable; ts4.material=poly will result in an error.
Multirotation Implantation
The simulation of multirotation implantations for both the MC and analytic methods is
controlled by the integer parameter mult.rot=<n>. If mult.rot is set to a number higher
than 1, an implantation with a revolving ion beam is simulated. Starting with the user-defined
rotation angle, Sentaurus Process performs mult.rot implantations with the same energy
and tilt in one implant command.
The rotation angle is incremented by ( )/mult.rot and, for each implantation step, the
dose is the 1/mult.rot-th part of the user-specified dose.
Energy Contamination Implantation
Sentaurus Process has a built-in feature for implantation with energy contamination, in which
a fraction of the nominal dose has a different energy than the specified energy. To perform an
energy contamination implantation, you must specify the parameter contamination in the
implant command. The syntax is:
implant dose=<n1> energy=<n2>
contamination= {energy=<n3> dose.fraction=<fraction>} ...
Then, Sentaurus Process treats the implantation as two separate implantations in the following
order:
implant dose=<n1*fraction> energy=<n3> ...
implant dose=<n1*(1-fraction)> energy=<n2> ...
360°
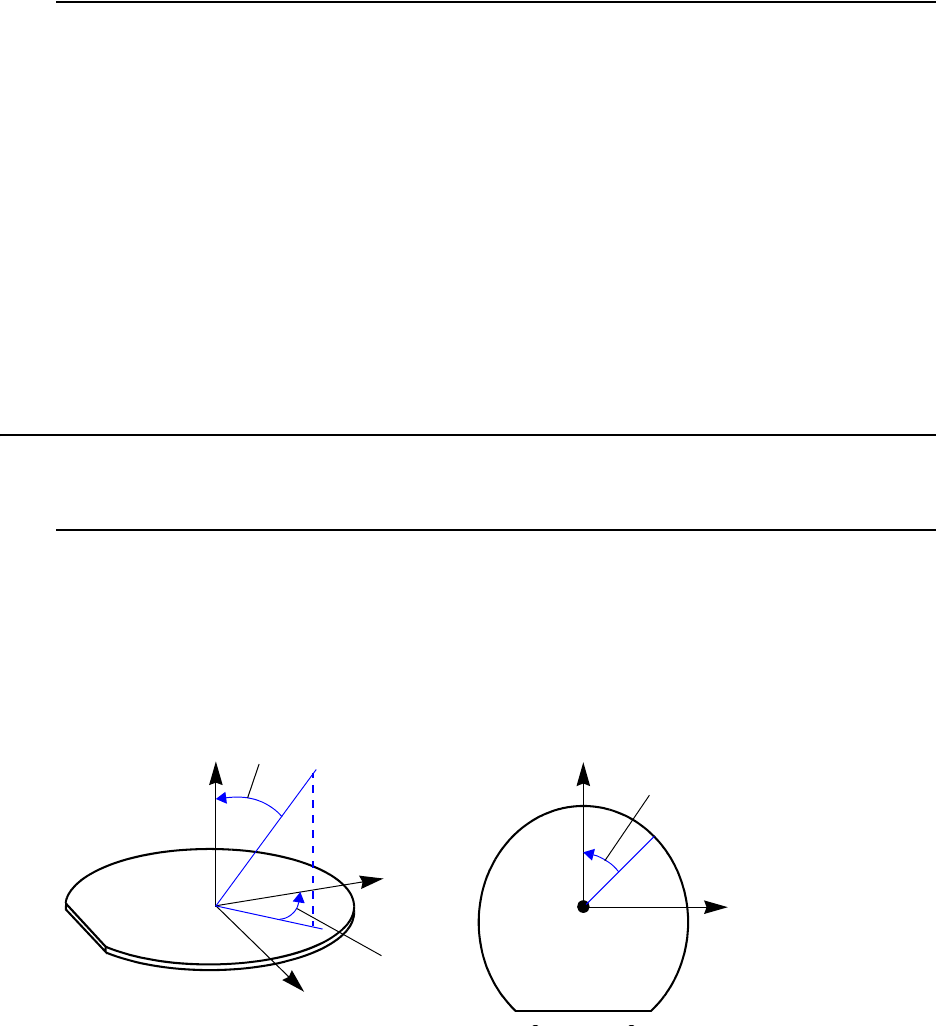
3: Ion Implantation
Coordinate System
Sentaurus™ Process User Guide 89
H-2013.03
Adaptive Meshing during Implantation
Adaptive meshing during implantation is active whenever adaptive meshing is switched on,
that is, pdbGet Grid Adaptive returns a 1. It also can be switched on by specifying the
parameter Adaptive in the implant command. For details, see Adaptive Meshing during
Implantation on page 708.
Since generally analytic implantation and MC implantation produce similar results, MC
implantation can take advantage of this similarity for adaptive meshing. When adaptive
meshing is active, MC implantation will first call analytic implantation for mesh refinement.
At the end of analytic implantation, the concentration generated by the analytic implantation is
discarded, while the new mesh is used for MC implantation. If the analytic implantation fails
for whatever reasons (such as no implant tables for certain materials), Sentaurus Process issues
a warning, and the MC implantation proceeds with the original mesh.
Coordinate System
Coordinates for Implantation: Tilt and Rotation Angles
Regardless of whether a simulation is 1D, 2D, or 3D, the direction of the ion beam is defined
relative to the wafer coordinate system (see Figure 16 on page 66) by the values of the tilt and
rotation parameters of the implant command. Figure 21 shows the tilt and rotation angles in
the wafer coordinate system.
Figure 21 Tilt and rotation angles for implantation; beam angle shown corresponds
to tilt = 20 and rotation = 45
The tilt and rotation angles are measured from the ion beam to the wafer z-axis and wafer y-
axis, respectively. In this definition, the tilt angle is always positive, and between (inclusive)
ZW
XW
YW
YW
XW
rotation
rotation
tilt ion beam
0°
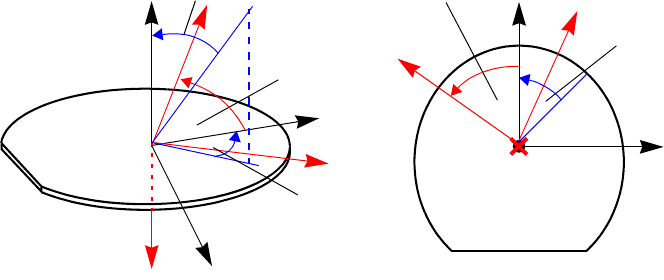
3: Ion Implantation
Coordinate System
90 Sentaurus™ Process User Guide
H-2013.03
and . However, for convenience, a negative tilt angle is allowed, and it is converted
automatically to a positive tilt by adding to the specified rotation angle. The rotation
angle is positive when the beam is rotated in the clockwise direction about the wafer z-axis,
and it is negative when it is counterclockwise.
Since the tilt and rotation angles are measured with respect to the wafer axes, the direction of
the beam in the simulation coordinate system depends on the slice angle.
Figure 22 shows the relationship between wafer coordinates, simulation coordinates, and the
beam direction.
Figure 22 Tilt and rotation angles for implantation; angles shown correspond to tilt = 20,
rotation = 45, and slice.angle = 60
The default values of tilt and rotation are and , respectively; in other words, by
default the incident ion beam is directed parallel to the wafer flat tilted away from the wafer x-
axis. For the default slice angle of , this corresponds to an ion beam in the simulator xy
plane, tilted away from the simulator y-axis. In a 2D simulation, the default ion beam comes
from the left side.
Figure 23 on page 91 shows the projection into the wafer plane of the direction from which the
beam strikes the wafer for tilt > 0 and various rotation angles.
The default simulation coordinate system (slice.angle = -90) is also shown.
90°
180°
ZW
XW
YW
YW
XS
YS
ZS
XW
slice.angle
YS
ZS
XS
slice.angle
rotation
tilt
rotation
ion beam
7°
90°–
90°–
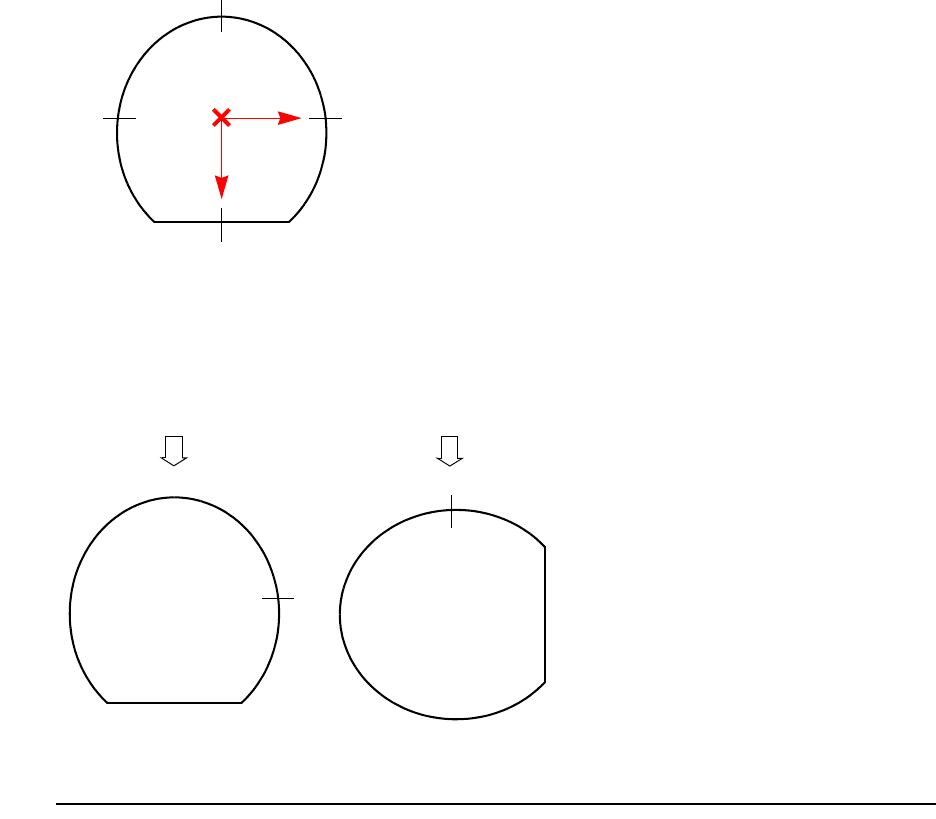
3: Ion Implantation
Coordinate System
Sentaurus™ Process User Guide 91
H-2013.03
Figure 23 Implant rotation directions for positive tilt
Figure 24 shows clearly that the orientations shown in Figure 23 are consistent with the
conventions defined in Figure 21 on page 89. A rotation of corresponds to rotating the
wafer a quarter turn counterclockwise.
Figure 24 Rotating wafer and fixed beam direction
2D Coordinate System
In a 2D simulation, the orientation of the 2D simulation plane with respect to the wafer
coordinate system must be defined. The angle between the 2D simulation plane and the y-axis
is set by the slice.angle. The default value is , which orients the 2D simulation plane
parallel to the wafer flat. The transformed y-axis (ys) is the y-axis in the 2D simulation plane.
0o
–90o90o
180o
ZS
XS
YS
90°
90o
rotate wafer +90o
90o
ion beam ion beam
90°–

3: Ion Implantation
Analytic Implantation
92 Sentaurus™ Process User Guide
H-2013.03
There are two ways to set slice.angle in the init command:
■To set directly, use:
init slice.angle=<n>
■To set by specifying a 2D cutline, use:
init slice.angle=[CutLine2D <x1> <y1> <x2> <y2>]
The <x1> and <y1> define the start point, and <x2> and <y2> define the end point in the wafer
plane. The two points are in the wafer coordinate system (for more information on coordinate
systems, see Understanding Coordinate Systems on page 66).
In general, the tilt projected to the 2D simulation plane is different from the tilt value. It is
given by the geometric relation:
(2)
The angle tilt2D can be found in the output of Sentaurus Process and can be negative
depending on the rotation angle and slice angle.
The tilt value defines the relation between the wafer dose (dose), which is given at the
command line by default and the dose, which would have to be specified in the beam-dose
mode to obtain the same final implanted dose, that is:
(3)
BeamDose2D as it appears in the Sentaurus Process output is defined using tilt2D, that is:
(4)
Analytic Implantation
Analytic implantation is performed using empirical point-response distributions. Point-
response distributions are generated using the method of moments. The moments representing
the primary and lateral point-response functions are taken from implantation tables.
For the purposes of 2D simulations based on analytic functions, an ion beam incident at the
point is assumed to generate a distribution function .
tilt2D()cos tilt()cos
2tilt
()cos 2tilt
()sin 2rotation+slice.angle
()cos⋅+
-----------------------------------------------------------------------------------------------------------------------------------------------------------=
BeamDose dose
tilt
()cos
--------------------------=
BeamDose2D dose
tilt2D()cos
---------------------------=
ξη,()
Fx y ξη,,,()
3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 93
H-2013.03
To calculate the concentration of the implanted species at a point of the simulation
domain, the superposition of all distribution functions of all possible points of incidence
must be computed:
(5)
where is the total dose per exposed area and is the doping profile.
Figure 25 Point-response distribution for a particle incident at the point ( ) at the
surface; intervals are used for lateral integration at the point (x, y); shaded
regions to left and right mark the lateral extension elements
The 2D distribution functions are always assumed to be given as a product of two 1D
distribution functions orthogonal to each other: a primary distribution function and a
lateral distribution function :
(6)
To perform the computation of the convolution integral in 2D, Sentaurus Process uses a set of
lateral intervals perpendicular to the projected ion beam. A local 1D layer structure is
computed in each interval. The spacing and width of these intervals depend on the complexity
of the exposed gas surface.
In 3D, Sentaurus Process uses a slightly different algorithm. The point-response function is a
3D function. The lateral function is also used in the third direction:
(7)
assuming an axially symmetric point-response function. The lateral integration is performed in
the plane perpendicular to the ion beam. For each point in the lateral integration plane, again,
a local 1D layer structure is computed.
xy,()
ξs()ηs()(,)Γgas
∈
Cx,y() NdFxyξs()ηs(),, ,()sd
Γgas
=
Nd
Cxy,()
(ξ,η)
x
y
(x,y)
ξη,
fpx()
flx()
Fxyξη,,,()fpxξs()–()flyηs()–()⋅=
flx()
Fxyzξη Θ,,,, ,()fpxξs()–()flyηs()–()flzΘs()–()⋅⋅=

3: Ion Implantation
Analytic Implantation
94 Sentaurus™ Process User Guide
H-2013.03
Primary Distribution Functions
Primary distribution functions can be set for a dopant/material combination using:
implant species=<dopant> <material>
[{gaussian pearson pearson.s dualpearson point.response}]
The previous model selection is used if no selection for <model> is made. The primary
distribution is used to represent the point-response function in 1D or the vertical point-response
in 2D and 3D. Point-response functions are characterized by moments.
The first moment, the projected range , is defined as:
(8)
while the higher moments are defined as:
(9)
The standard deviation , the skewness , and the kurtosis are defined as:
(10)
(11)
(12)
Gaussian Distribution: gaussian
(13)
(14)
(15)
Rp
Rpxfx() xd⋅⋅
∞–
∞
=
mi
mixR
p
–()
ifx() xd⋅⋅
∞–
∞
=
σ
γ
β
σm2
=
γm3
σ3
------=
βm4
σ4
------=
fpx() 1
2πσ
-------------- xR
p
–()
2
2σ2
----------------------–
exp=
γ0=
β3=

3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 95
H-2013.03
Pearson Distribution: pearson
The Pearson distributions are the solution to the following differential equations:
(16)
(17)
Different types of Pearson distribution are distinguished by different values of and :
(18)
(19)
(20)
The Pearson–IV distribution is given by:
(21)
Sentaurus Process automatically switches between the Pearson–IV, Pearson–V, and Pearson–
VI distribution functions depending on the conditions for and given in Eq. 18 to Eq. 20.
The factor is chosen to fulfill the normalization condition:
(22)
yd
dfy() ya–
b0b1y⋅b2y2
⋅++
----------------------------------------------fy()⋅, yxR
p
–==
ab
1γσ β 3+()⋅⋅
A
--------------------------------–==
b0σ24β3γ2
–()⋅
A
------------------------------------–=
b22β3γ2
–6–
A
------------------------------–=
A10β12γ2
–18–=
γ
β
0γ32<<
β48 39γ26γ24+()
32⁄
++
32 γ2
–
-------------------------------------------------------------> Type IV
0γ32<<
β48 39γ26γ24+()
32⁄
++
32 γ2
–
-------------------------------------------------------------= Type V
0γ32<<
3 1.5 γ⋅+β48 39γ26γ24+()
32⁄
++
32 γ2
–
-------------------------------------------------------------
<< Type VI
fpy() Kb
2y2b1yb
0
++
1
2b2
--------
b1
b2
----- 2a+
4b2b0b1
2
–
------------------------------–2b2yb
1
+
4b2b0b1
2
–
------------------------------
atan
exp⋅=
γ
β
K
fpx()xd
∞–
∞
1=

3: Ion Implantation
Analytic Implantation
96 Sentaurus™ Process User Guide
H-2013.03
Pearson Distribution with Linear Exponential Tail: pearson.s
A linear exponential tail is added to the Pearson distribution. This is performed in an attempt
to describe more accurately the profile tails for some implantations, that is:
(23)
where is the Pearson distribution, is a transition function, and is the exponential tail.
The decay length of the exponential tail is give by the parameter :
(24)
The constants and are computed from the continuity conditions:
(25)
and:
(26)
NOTE Exponential tail distributions are available with the Dios tables.
However, care is required when using the exponential tail for
implantation with large tilt angles. The -fit in these tables was
performed for a standard tilt in amorphous materials and does not
apply to large tilt angles or strong channeling conditions.
fpx()
Ppx()0xx
max
<≤,
Pvx()xmax x<xa
≤,
Plx()xax∞<≤,
=
Pp
Pv
Pi
lexp
xmax :Pmax Ppxmax
():= maxPpx()=
xaxmax
≥:PaPpxa
():= 1
2
---Ppxmax
()=
Pvx() Ppxmax
() A1xx
max
–()
2Bx x
max
–()
3
+()exp⋅=
Plx() Ppx() xx
max
–
lexp
--------------------Ppxmax
() xx
max
–
lexp
--------------------–
exp⋅⋅+=
A1
B
Plxa
() Pvxa
()=
xd
dPlx()
xaxd
dPvx()
xa
P′a
=:=
A1
3Pa
Pmax
------------
ln
xaxmax
–()
2
------------------------------P′a
Paxaxmax
–()
----------------------------------–=
B
2Pa
Pmax
------------
ln–
xaxmax
–()
3
------------------------------P′a
Paxaxmax
–()
2
------------------------------------+=
lexp
7°
3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 97
H-2013.03
Dual Pearson Distribution: dualpearson
The most advanced primary distributions are available with the dual Pearson function [9],
which can be chosen with the switch dualpearson. The dual Pearson model includes a
superposition of two Pearson functions:
(27)
The head and tail functions are two independent Pearson functions. The head function accounts
for the profile of ions that do not channel (nonchanneling or amorphous part). The tail function
accounts for the channeled ions that form the characteristic tail in the implantation profile.
A dualpearson function is characterized by nine parameters: the two sets of four Pearson
parameters and the ratio between the amorphous and channeling doses. These parameters are
usually taken from moment table files.
You can s et t he i ndivi du al m ome nts di re ctly in the Sentaurus Process command line, for
example:
implant species=<dopant> <material> [rp=<n>] [stdev=<n>] [gamma=<n>]
[beta=<n>] [rp2=<n>] [stdev2=<n>] [gamma2=<n>] [beta2=<n>] [ratio=<n>]
[lat.stdev=<n>] [lat.stdev2=<n>]
This overwrites the parameters found in the specified implant table. Using this facility, it is also
possible to force the Pearson distributions in the dualpearson and pearson models to
behave like a Gaussian distribution, for example:
implant species=Boron Silicon pearson gamma=0 beta=3
The first statement sets the implantation model to a Pearson distribution. The parameters are
read from the default table. The skewness and kurtosis are set according to Eq. 14 and Eq. 15,
overwriting the values found in the table. This results in a Gaussian distribution for the function
characterizing the amorphous part of the profile.
You can en able or di sa bl e indivi du al m ome nts us in g <moment>.isset pdb switches, where
<moment> is the name of the moments such as rp, stdev, rp2, stdev2, and so on. For
example:
pdbSetBoolean Silicon Boron rp.isset 0
This command would disable a user set value for rp.
fpx() ratio fhead x() 1ratio–()ftail x()⋅+⋅=

3: Ion Implantation
Analytic Implantation
98 Sentaurus™ Process User Guide
H-2013.03
NOTE All moments set at the command line are ignored after a new implant
table is selected, or an implant table has been specified again using the
implant species=<species> <material> imp.file=<name>
command. In this case, the moments from this new implant table will
then be used, regardless of which moments have been set previously at
the command line.
Point-Response Distribution: point.response
See Point-Response Interface on page 116.
Screening (Cap) Layer-dependent Moments
Cap layer–dependent implant tables are used to describe correctly the screening of the ion
beam in the structure. The implant moments in a particular region generally depend on the
combined thickness of all layers above this region. The moments are parameterized with
respect to the effective thickness , which is defined as:
(28)
that is, as the sum over the thicknesses of all layers above the current layer multiplied by
corresponding efficiency factors . These factors can be set for a particular material and a
species using:
implant species=Boron Oxide eff.caplayer.thick 1.0
The default value is 1 for all materials other than silicon, where it is set to zero. Therefore,
silicon layers are effectively not included in the total effective cap layer thickness.
If the implant table for a specific <material>/<species> combination does not contain an
explicit cap layer dependence, the effective channeling suppression model is used. This model
suppresses the channeling tail by multiplying the channeling part in Eq. 27 by a factor
calculated according to:
(29)
ti
cap
ti
cap tj fj
eff
j
ji<
=
fj
eff
rsuppress
rsuppress 1
1
2
---CR
phead,
()
CR
phead,
()CR
ptail,
()+
--------------------------------------------------------
⋅MinRatio
σ
--------------------------------
Exponent–
+
-----------------------------------------------------------------------------------------------------------------------------------------------=
3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 99
H-2013.03
where and are the peak concentrations of the unscaled profile, and
is defined as:
(30)
using the values of for the amorphous (head) part of the profile for all layers above the
present layer. The amorphous part of the profile is multiplied by to conserve the
total dose.
MinRatio is the minimum value of the ratio . The parameters MinRatio and Exponent
can be set in the parameter database, that is:
pdbSet <material> <species> MinRatio
pdbSet <material> <species> Exponent
The model is applied only for values of greater than MinRatio and effective cap layer
thickness greater than 2.1 nm. The effective channeling suppression model can be switched on
using:
implant species=<species> <material> [eff.channeling.suppress]
The model remains inactive for explicitly cap layer–dependent implant tables.
NOTE This model is switched off by default and is switched on in the Taurus/
TSUPREM-4 mode.
Lateral Straggle
The lateral straggling of the distribution of implanted ions is specified by defining a lateral
distribution function, which is a Gaussian distribution with a lateral standard deviation :
(31)
In general, the lateral standard deviation depends on the vertical depth of the profile. The depth
dependence can be switched on or off for a particular combination of dopant species and
material using the flag depth.dependent:
implant species=<dopant> <material> [depth.dependent]
CR
phead,
()
CR
ptail,
()
σ
σti
Rpi
-------
i
=
Rp
i
1rsuppress
–
σ
σ
σl
flxy,() 1
2πσ
lx()
------------------------y2
2σl
2x()
-----------------–
exp=
3: Ion Implantation
Analytic Implantation
100 Sentaurus™ Process User Guide
H-2013.03
The lateral standard deviation can also be set in the command line using the keyword
lat.stdev:
implant species=<dopant> <material> [lat.stdev=<n>] [lat.stdev2=<n>]
where lat.stdev2 sets the lateral standard deviation for the tail function. If either
lat.stdev or lat.stdev2 is set, Sentaurus Process switches to the depth-independent
lateral straggling. All depth.dependent switches are ignored in this case.
An additional scaling factor for both the depth-dependent and depth-independent lateral
standard deviation can be used to vary the lateral straggling:
implant species=<dopant> <material> [lat.scale=<n>] [lat.scale2=<n>]
Depth-dependent Lateral Straggle: Sentaurus Process Formulation
If a TSUPREM-3-compatible implantation table is used (.s3), the depth-dependent lateral
standard deviation is calculated according to:
(32)
where lstdev and lv are parameters taken from the implantation table. There are two
independent sets of parameters for the two Pearson functions in the dualpearson model. This
formulation also is used with the Tasch implantation tables.
Depth-dependent Lateral Straggle: Dios Formulation
If a Dios-compatible implantation table is used, the depth-dependent lateral standard deviation
is calculated using a vector of five parameters p1, p2, ..., p5. The following formula is
applied [10]:
(33)
There is only one set of these parameters in each table entry. In the case of the dualpearson
implant model (see Eq. 33), the same set of parameters p1, p2, ... p5 together with the
standard deviation of the first Pearson function described by stdev is applied to both the
amorphous and the channeling part of the distribution.
σlx() lstdev x
lstdev lv⋅
------------------------------–
exp⋅=
σlx() stdev max 0.01
p1 p2 x⋅
Rp
------------- p3+
⋅
exp p1 p4 x⋅
Rp
------------- p5+
⋅
exp+
log
p1
-----------------------------------------------------------------------------------------------------------------------------------------
,
⋅=

3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 101
H-2013.03
Depth-dependent Lateral Straggle: Taurus Formulation
If a TSUPREM-4/Taurus-compatible implantation table is used, the depth-dependent lateral
standard deviation is calculated using two parameters:
(34)
The depth-independent standard deviation and the depth-dependent slope are read
from the moment table.
This formulation is compatible with the Dios formulation (see Depth-dependent Lateral
Straggle: Dios Formulation) for the following conditions: , . The
remaining parameters can be translated as follows:
(35)
Analytic Damage: Hobler Model
The damage distribution is calculated using Eq. 33 and Eq. 6, p. 93. The primary and lateral
distribution functions are taken from the literature [10]. The primary function consists of a
Gaussian function and an exponential tail, joined continuously with continuous first
derivatives. The distribution is normalized. The normalization factors are and , and
is the number of Frenkel defects per ion.
Three types of primary function are distinguished:
■Type 0
A simple Gaussian distribution with the primary range and the standard deviation :
(36)
■Type 1
For light ion species:
(37)
σlx() σ
01∆σ x
Rp
------1–
+
⋅=
σ0
∆σ
p4 0=
p1 p5⋅∞–=
σ0stdev p2 p3+()⋅=
∆σ p2 p2 p3+()⁄=
c1
c2
Nvac
Rp
σ
fpx() Nvacc1
xR
p
–()
2
2σ2
----------------------–
exp=
fpx()
Nvacc1x
l
--
, x x0
≤exp
Nvacc2
xR
p
–()
2
2σ2
----------------------–
, x x0
>exp
=
3: Ion Implantation
Analytic Implantation
102 Sentaurus™ Process User Guide
H-2013.03
where is the decay length of the exponential function. The joining point is calculated
by:
(38)
■Type 2
For heavier ions, the exponential tail is directed towards the bulk:
(39)
In this model, four parameters , , , and are required. These parameters were
obtained by MC simulations between 1 keV and 300 keV. If damage calculation is switched
on, that is, if:
implant species=<dopant> damage
has been set, Sentaurus Process generates these parameters using an internal lookup table,
which contains the original data available for boron, BF2, phosphorus, arsenic, and antimony
in silicon.
NOTE For some other species, the parameters of these original species are used
that are closest with respect to the atomic number in the periodic table
of elements. Nitrogen uses the boron parameters. Silicon and aluminum
use the phosphorus parameters. Germanium and gallium use the arsenic
parameters, and indium uses the antimony parameters. Damage
calculation is automatically switched off for any other species.
Type 0 is used for boron at energies E < 20 keV, phosphorus at E < 55 keV, and arsenic at
E>170keV. Type1 is applied to boron and phosphorus elsewhere, and Type 2 is applied to
arsenic at energies below 170 keV and antimony at all energies.
The lateral distribution is modeled using Eq. 33. The five lateral parameters p1, p2, ...,
p5 are provided in the internal lookup table.
An alternative to the internal lookup table is to load a table file similar to the implant tables.
The keyword for this is dam.table:
implant species=<dopant> <material> [dam.table=<name>]
l
x0
x0Rp
σ2
l
------–=
fpx()
Nvacc2
xR
p
–()
2
2σ2
----------------------–
, x x0
≤exp
Nvacc1
x
l
--
, x x0
>exp
=
Rp
σ
l
Nvac

3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 103
H-2013.03
This overwrites the internal lookup table for the above mentioned species using the parameters
from the table instead. In addition, it enables damage calculation for species other than the
original ones.
Datasets
Several datasets are used to store the as-implanted profile and the implantation damage. Point-
defect profiles are created at the end of the implantation step. Datasets with the ending
_Implant contain profiles generated during subsequent implant steps. These datasets are
deleted at the beginning of the next diffuse step.
Table 4 Datasets used in analytic implantation step
Dataset Description
Damage
Accumulative damage (damage history). At the end of an implant step, the
Damage_LastImp concentration is added using DFactor. This dataset is
deleted by the diffuse command.
Damage_LastImp
Damage created during the last implant step. This dataset is deleted at the end
of the implant step.
<dopant>
Accumulative density of the dopant concentration. At the end of an implant
step, the <dopant>_LastImp concentration is added to <dopant>.
<dopant>_Implant
Accumulative density of the dopant concentration. At the end of an implant
step, the <dopant>_LastImp concentration is added to
<dopant>_Implant. This dataset is deleted by the diffuse command.
<dopant>_LastImp
As-implanted dopant concentration that contains the profile generated during the
last implant step. This dataset is deleted at the end of the implant step.
Int_Implant
Accumulative interstitial profile updated at the end of an implant step.
Vac_Implant
Accumulative vacancy profile updated at the end of an implant step.
Int<component>_Implant
Accumulative interstitial profiles in multicomponent material with
DistinctDefects set to true, where <component> is the component
of the composition of the material. For example, in SiC, interstitial profiles
include IntSilicon_Implant and IntCarbon_Implant.
Vac<component>_Implant
Accumulative vacancy profiles in multicomponent material with
DistinctDefects set to true, where <component> is the component
of the composition of the material. For example, in SiC, interstitial profiles
include VacSilicon_Implant and VacCarbon_Implant.

3: Ion Implantation
Analytic Implantation
104 Sentaurus™ Process User Guide
H-2013.03
Tables
Implantation Table Library
The implantation table library is located at $SPHOME/ImpLib/.
Dios Tables
The subdirectory Dios/ contains the tables used by default in Dios. This tables can be made
the default tables for all species in Sentaurus Process by using:
implant tables=Dios
For arsenic, antimony, phosphorus, indium, germanium, gallium, nitrogen, and aluminum, the
data in these tables are taken from the literature [3]. The values for boron are obtained from
simulations with the 1D process simulator TESIM [11]. The values for energies MeV are
taken from the literature [2]. These tables provide moments that can be used with the Gaussian
and Pearson implantation models.
Taurus Tables
The directory Taurus/ contains the Taurus Process implant tables for boron, BF2, phosphorus,
germanium, indium, antimony, and arsenic. To select these tables as the default, use the
keyword Taurus:
implant tables=Taurus
The tables contain calibrated data from sub-keV to above 10 MeV. The calibration was
performed using both SIMS data and Taurus MC calculations [5].
Default Tables
The directory Default/ contains tables extracted from MC simulations with Crystal-
TRIM [4], which are tabulated in DIOS format. The data are available for arsenic, antimony,
BF2, boron, phosphorus, indium, and germanium in silicon, polysilicon, oxide, and nitride.
These tables provide moments that can be used with all implantation models including the
dualpearson model. For silicon, dual Pearson moments are available that depend on energy,
tilt, dose, and cap-layer thickness. For polysilicon, oxide, and nitride, single Pearson moments
are available that depend on energy and tilt only.
The tables cover different energy ranges. The tilt angles range from to , and the oxide
thickness ranges from 0 nm to 100 nm. There are tables for low, medium, and high doses for
all species except germanium where only one table for a medium to high dose is available.
1≥
0°
60°

3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 105
H-2013.03
Sentaurus Process selects the correct table depending on the implant dose. These tables
constitute most of the default tables used in Sentaurus Process.
The default tables used in Sentaurus Process are selected by using the command:
implant tables=Default
This implant command not only selects the tables from the Default/ directory for arsenic,
antimony, BF2, boron, phosphorus, indium, and germanium in silicon, polysilicon, oxide, and
nitride, but also selects the tables from the Taurus/ directory for carbon, fluorine, and
germanium in silicon, polysilicon, oxide, and nitride (see Table 5). For all other species and
materials, the respective Dios tables are used.
NOTE Outside the specified range, the Default implant tables may fall back
to the Dios tables. Therefore, near the boundaries of the Default
tables, inconsistent results may occur.
Ta bl e 5 D e fa u l t t a bl es
Species Table file Energy range [keV]
Arsenic
<material>As_1e12-5e13.tab
<material>As_1e13-8e14.tab
<material>As_2e14-6e15.tab
0.5–400
Antimony
<material>Sb_1e12-5e13.tab
<material>Sb_1e13-5e14.tab
<material>Sb_2e14-1e16.tab
1.5–600
BF2
<material>BF2_1e12-5e13.tab
<material>BF2_1e13-8e14.tab
<material>BF2_2e14-6e15.tab
0.5–400
Boron
<material>B_1e12-4e13.tab
<material>B_1e13-6e14.tab
<material>B_16e13-8e15.tab
0.2–517 (silicon)
0.2–480 (other materials)
Carbon
carbon_in_<material>_standard
0.2–400
Fluorine
fluorine_in_<material>_standard
0.2–400
Germanium
germanium_in_<material>_2007
0.6–800
Indium
<material>In_1e12-4e13.tab
<material>In_1e13-6e14.tab
<material>In_16e13-8e15.tab
1–400
Phosphorus
<material>P_1e12-4e13.tab
<material>P_1e13-6e14.tab
<material>P_16e13-8e15.tab
0.3–400
0.12–3000 (10.0 upgrade)
3: Ion Implantation
Analytic Implantation
106 Sentaurus™ Process User Guide
H-2013.03
Tasch Tables
The directory Tasch/ contains the University of Texas (UT) implant tables for boron, BF2,
phosphorus, and arsenic in silicon [12]. For all other materials and species, single Pearson
tables are available. The tables can be selected to be the default by using the keyword Tasch:
implant tables=Tasch
The tables cover different energy ranges. The boron table ibout1.s3 contains cap layer–
dependent implantation moments valid for thicknesses between 1.5–40 nm. The moments in all
other tables are cap-layer independent.
The valid range for the tilt is to and, for the rotation, the range is to . These
tables provide data to be used with all implant models.
The single Pearson tables provide only energy-dependent data covering the range between
10 keV and 1000 keV.
TSuprem4 Tables
The directory TSuprem4/ contains the TSUPREM-4 native implant tables, s4imp0.ts4, for
boron, BF2, phosphorus, indium, antimony, and arsenic. To select these tables as the default,
use the keyword TSuprem4 with an optional prefix:
implant tables=TSuprem4 [ts4.prefix=<c>]
These tables contain the original implant moments of TSUPREM-4.
File Formats
Sentaurus Process handles a variety of table formats. The table format of the implantation table
is automatically recognized by Sentaurus Process from the file extension.
Table 6 Tasch tables
Species Table file Energy range [keV]
Arsenic
iasut0.s3
7–180
BF2
ibfut0.s3
0.5–65
Boron
ibout0.s3
ibout1.s3
0.5–80
15–80
Phosphorus
iphut0.s3
15–180
0°
10°
0°
45°
3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 107
H-2013.03
Single-Pearson Table File Format: <file>.sp
This format provides the simplest table format that can be used with Sentaurus Process. It
contains energy-dependent entries for the moments to be used with the (single) pearson or
gaussian model.
NOTE These tables cannot be used with the dualpearson model.
The format of the table entries is:
*
energy rp stdev gamma beta lat.stdev
There is no dependence of the moments on dose, tilt, rotation, or cap layer thickness. Lines
with an asterisk in the first column are treated as comment lines. Missing or incomplete blocks
are not properly read when the file is parsed.
SUPREM-III Table File Format: <file>.s3
This format allows the handling of energy, dose, tilt, rotation, and cap-layer thickness–
dependent dual Pearson moments. A SUPREM-III implant table file consists of two sections:
one for the primary moments and one for lateral moments. Both sections start with a header,
which contains the parameter range covered by the table.
The header is organized as follows:
*Energies:
NumberOfEnergies energy1 energy2 ...
*Tilts:
NumberOfTilts tilt1 tilt2 ...
*Rotations:
NumberOfRotations rotation1 rotation2 ...
*Doses:
NumberOfDoses dose1 dose2 ...
*Thickness:
NumberOfThickness thickness1 thickness2 ...
NOTE The order of these entries must not be changed.
Lines with an asterisk in the first column are treated as comment lines. A table entry for a
particular combination of lookup parameters has the format:
*
rp stdev gamma beta rp2 stdev2 gamma2 beta2 ratio1
rp stdev gamma beta rp2 stdev2 gamma2 beta2 ratio2
...
rp stdev gamma beta rp2 stdev2 gamma2 beta2 ratio<NumberOfDoses>
3: Ion Implantation
Analytic Implantation
108 Sentaurus™ Process User Guide
H-2013.03
Each line contains eight dualpearson moments and the ratio as defined in Eq. 27, p. 97. The
entries are ordered increasingly with respect to cap layer thickness, energy, tilt, and rotation.
NOTE There is no automatic check of the ordering of the table entries.
The tables are for one species/material combination only. The cap-layer thickness, rp, rp2,
stdev, and stdev2 should be given in micrometers and the angles, in degrees. The energy
values must be specified in keV. No units must be specified in the tables.
The lateral part is organized in the same manner. Corresponding to the header information, the
entries are ordered in the same manner as in the primary part. Each entry has the format:
*
<void> lstdev lv lstdev2 lv2
The first item is void and can be used for information purposes. The parameters are used in
Eq. 32, p. 100 to calculate the depth-dependent lateral standard deviation. The units for
lstdev and lstdev2 are micrometers, whereas lv and lv2 are unitless.
Dios Table File Format: <file>.tab
The Dios table file format for implantation data files allows for dependencies on energy, dose,
tilt, rotation, and the cap-layer thickness. It provides the primary moments for all implantation
models including the dualpearson model. Parameters for depth-dependent lateral straggling
are available as well. The format of the table entries is:
# Look up parameters
material species thickness rotation tilt energy NumberOfFunctions
NumberOfDoses
# Primary moments
rp stdev <void> gamma beta lexp <void>
rp2 stdev2 <void> gamma2 beta2 lexp2 <void>
# Channeling table
dose ChannelingDose
dose ChannelingDose
...
# Lateral straggling
p1 p2 p3 p4 p5
Lines with a # character in the first column are treated as comment lines. Missing or incomplete
blocks are not read properly when the file is parsed.
The first block contains entries for the material and species names, cap-layer thickness, rotation
angle, tilt angle, and energy. The NumberOfFunctions defines the number of components of
the primary distribution function. A maximum of two functions are allowed. NumberOfDoses
defines the number of entries in the channeling table. Each entry consists of a dose and the
corresponding channeling dose. All doses are expected to be positive.
3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 109
H-2013.03
Figure 26 Piecewise linear nonchanneling and channeling dose for a dual Pearson profile
as represented in default channeling table
The ratio between the amorphous part and channeling part in Eq. 27, p. 97 is calculated from
the channeling table using:
(40)
where the value for ChannelingDose is interpolated linearly using the value of the implant
Dose.
The second block contains the moments for all the components of the primary distribution
function. Parameters, which by definition do not exist for the function the set describes, are
ignored.
NOTE Some entries are always ignored since they are not used in the implant
models of Sentaurus Process. For example, the last moment entry
(<void>) is always disregarded.
The third block contains the channeling table ordered with increasing dose, and the fourth
block contains parameters for the depth-dependent lateral straggling.
NOTE The entries must be increasingly ordered with respect to the cap-layer
thickness, rotation, tilt, and energy, so that the values for various
energies (but the same other three parameters) follow each other. All
data entries for the same material–dopant combination should follow
each other with no interruption by entries for another material–dopant
combination. The cap-layer thickness, rp, rp2, stdev, and stdev2
should be given in micrometers and the angles, in degrees. The energy
values must be specified in keV. No units must be specified in the tables.
1013 1014 10
15
To tal D os e
Channeling Dose
Nonchanneling Dose
Dose
1013
1014
ratio 1ChannelingDose
dose
--------------------------------------------–=
3: Ion Implantation
Analytic Implantation
110 Sentaurus™ Process User Guide
H-2013.03
Damage Table File Format: <file>.dam
The damage tables for the Hobler damage model are similar to the Dios table file format, which
allows for dependencies on energy, tilt, and rotation. The Hobler damage model table provides
the primary moments for the damage model. Moments for depth-dependent lateral straggling
are available as well. The format of the table entries is:
# Look up parameters
material species rotation tilt energy
# Primary moments
rp stdev decay nvac type
# Lateral straggling
p1 p2 p3 p4 p5
The syntax is the same as for the Dios table format. The item decay refers to the parameter ,
and the item nvac refers to the parameter in Analytic Damage: Hobler Model on
page 101. The item type refers to the type of Hobler model.
Taurus Table Format: <file>
The Taurus table format, which is the most general table format used in Sentaurus Process,
handles data for all implant and damage models. Implant table files in the Taurus format have
no file extension; that is, an implant table file without a file extension is considered to be in the
Taurus format. It contains a file header and a block of numeric data. The file header consists of
a list of names of the implant conditions. The names should be lowercase only. The following
names are recognized:
energy tilt rotation dose screen temperature current
The sequence of these names can be arbitrary. Some names from this list can be omitted. The
following units should be used for the implant conditions:
energy,[keV]
tilt,[degrees]
rotation,[degrees]
dose,[cm-2]
screen,[um]
temperature,[K]
current,[mA/cm2]
The numeric data consists of an arbitrary number of lines that form the lookup tables for
implant conditions and implant moments. Each line should contain a list of numeric values for
the implant conditions followed by the implant moments. The numeric values should be
separated by space.
l
Nvac

3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 111
H-2013.03
The number of the numeric values should be the same on each line. There should be at least
n+4 values per line for a gaussian profile, n+6 values per line for a pearson profile, and
n+13 values per line for a dualpearson profile, where n is the number of the implant
conditions specified in the file header.
The sequence of implant conditions should correspond exactly to the sequence of implant
condition names in the file header. The sequence of the implant moments in one line is fixed
as follows:
Any line that starts with a double slash // is considered a comment and is omitted. Always put
the double slash at the first position in the line.
If the requested set of implant conditions does not have an exact match in the lookup table, a
multidimensional linear interpolation is used. If a requested implant condition extends beyond
the range of the lookup table, the closest value from the lookup table is used.
If the lookup table contains several lines with identical sets of the implant conditions, only the
last set is used, and all the previous lines are discarded.
If a table contains data for the Hobler damage model, the following sequence of moments is
used:
rp stdev lat.stdev lat.slope gamma beta decay nvac
Multilayer Implantations
Point-response functions are valid only for a single material layer. For multiple layers of
different materials, the point-response functions must be combined in a way that corrects the
effect of the different stopping power in the covering layers. This must be performed for each
lateral interval taking into account the local layer sequence parallel to the ion beam. Two
algorithms are available in Sentaurus Process: numerical range scaling (NRS) [13] and dose-
matching [12]. Both algorithms calculate a shift applied to the primary point-response
function. Sentaurus Process also provides an option no, which switches off the matching. In
this case, is set to zero in all layers.
The matching algorithm can be selected globally by using the command:
pdbSet ImplantData MatchControl { no | range | dose }
Gaussian:
rp stdev lat.stdev lat.slope
Pearson:
gamma beta
Dual Pearson:
rp2 stdev2 lat.stdev2 lat.slope2 gamma2 beta2 ratio
δi
δi
3: Ion Implantation
Analytic Implantation
112 Sentaurus™ Process User Guide
H-2013.03
The default value of MatchControl is range. In addition, you can select locally the matching
algorithm with the keyword match in each implant command:
implant <dopant> [match={no range dose}]
The locally selected algorithm overwrites the one globally set in the PDB.
The NRS algorithm accounts for the different stopping power in different materials using the
ratio of the projected ranges of the materials.
Figure 27 NRS algorithm: the point-response function in the second material is shifted and
rescaled (left) due to existence of a layer with different stopping power (shaded
region); the new profile is combined from the point response in the first layer and
shifted point response in the second layer (right).
The shift in the -th layer is calculated according to:
(41)
where represents the thickness of the -th layer. The profiles are matched
according to:
(42)
where is a rescaling factor that satisfies the normalization condition. The point-response
function in the first layer is always used without a shift.
The proportional range shift model is used to shift the channeling portion of the implant profile
independent of the amorphous part. To calculate the shift of the channeling part, the shift of the
amorphous part is scaled by the ratio of the channeling range and the amorphous range:
(43)
δ2
Rp2+δ2Rp2
x
C(x)
x
C(x)
i
δitj1Rpi
Rpj
-------–
j1=
ji<
=
tjdjdj1–
–=
j
Cx() fp1x() for 0 xd
1
<<
αifpi xδi
–()⋅ for di1–xd
i
<<
=
αi
δitail,δihead,
Rpi tail,
Rpi head,
-----------------------
=

3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 113
H-2013.03
The proportional range scaling can be switched on with:
implant <species> [range.sh]
The default setting for this model is off. In the Taurus/TSUPREM-4 mode, the switch is set to
on.
The dose-matching algorithm can be selected with the option dose. The shift is calculated
according to the dose accumulated in the above layers:
(44)
where is the position of the top of the -th layer. The dose is the integral over the
primary point-response function.
Lateral Integration
Local Layer Structure in 2D
Local 1D layer structures are defined for a set of lateral intervals. These lateral intervals are
chosen perpendicular to the projection of the ion beam into the simulation plane as shown in
Figure 28 on page 115.
The width of the lateral intervals is controlled by several parameters set in the parameter
database. The default values can be changed by using:
pdbSet ImplantData LateralGridSpacing <n>
pdbSet ImplantData VerticalGridSpacing <n>
Starting from an initial grid, the intervals are bisected until a certain limit is reached. This limit
is set by LateralGridSpacing, which has the default value of . Then, the intervals
are bisected again until a certain vertical limit is reached. This limit is set by
VerticalGridSpacing with the default value of .
The lateral integration is limited to a certain range of intervals to the left and right of a mesh
node. This integration range depends on the maximum lateral standard deviation applied to the
structure.
δi
δidideff
–=
Dsofar fpx()xd
0
deff
=
di
i
Dsofar
0.01 µm
0.01 µm
3: Ion Implantation
Analytic Implantation
114 Sentaurus™ Process User Guide
H-2013.03
Control over the lateral integration is possible by setting the number of lateral standard
deviations used to set the integration range:
(45)
The value of can be set by using:
pdbSet ImplantData NumLateralStdev <n>
The default value is 5, which means that the total lateral integration width is 10 .
In 3D, the integration is performed over a square grid in the plane perpendicular to the ion
beam. The grid is centered about a mesh node. It has a fixed size and resolution. The size is
controlled by the parameter:
pdbSet ImplantData NumLateralStdev3D <n>
having the same meaning as the corresponding 2D parameter. The default value is 3.5. Each
interval is subdivided by a certain number of grid points. The subdivision can be set by using:
pdbSet ImplantData NumGridPoints3D <n>
so that the total number of grid points is:
(46)
The default value for NumGridPoints3D
is 4. Therefore, the total number of grid points is
784. The size of the integration grid is the parameter that limits the time performance of
analytic implantation in 3D.
The lateral intervals are expanded by a certain amount over the left and right boundaries of the
2D device to ensure flat profiles on the left and right sides. This extension depends on the
implantation tilt and the maximum lateral standard deviation. The maximum extension can
be controlled from the parameter database. The value can be changed by using:
pdbSet ImplantData MaxLateralExtension <n>
The default is set to . A similar extension is applied in three dimensions.
Cxy,() fpx()flxy',()y'd
yNσlmax,
–
yNσlmax,
+
=
N
σlmax,
2NumLateralStdev3D NumGridPoints3D⋅⋅()
2
1.5 µm
3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 115
H-2013.03
Primary Direction and Scaling
The interpretation of the range and lateral range parameters depends on the value of the
implantation parameter primary. This can be set by using:
implant <dopant> [primary={beam wafer}]
The option beam switches to the beam projection mode. In this case, the primary moments are
applied along the projection of the ion beam onto the simulation plane, and the lateral
integration is performed perpendicular to the projection of the ion beam. This is the default
mode in Sentaurus Process. The option wafer switches to the wafer normal mode. Here, the
primary distribution function and the moments are interpreted orthogonally to the wafer
surface.
Figure 28 Beam projection mode (left) and wafer normal mode (right) for analytic
implantation
An implant table can be declared angle-dependent or angle-independent by using:
implant species=<dopant> [angle.dependent]
Tilt-dependent and rotation-dependent data values extracted from SIMS measurements, or
user-specified range parameters are assumed to be angle dependent. On the other hand,
theoretical range parameters, such as those calculated by LSS theory, are assumed to be angle
independent.
For the same pair of tilt and rotation parameters, different projected tilt angles can be
observed in the 2D simulation plane. This angle is called tilt2D and depends on both
rotation and slice.angle. Profiles in quasi-1D parts of the structure away from mask
edges depend on the choice of slice.angle. Exactly the same 1D profiles can be observed
only for symmetric primary distribution functions like the ones used in the Gaussian model and
only if the primary and lateral standard deviations have the same value.
To ensure, at least approximately, that the same depth profiles are obtained for different rotation
angles, and for different dimensions, the range parameters for the primary distribution function
fl(y)
fl(y)fp(x)
fp(x)
3: Ion Implantation
Analytic Implantation
116 Sentaurus™ Process User Guide
H-2013.03
are scaled depending on tilt (for 1D and 3D), or tilt2D (for 2D). For example, the projected
range is scaled as follows:
(47)
For tilt implants, the integrated dopant profile depends on not only the primary range
parameters ( , , and so on), but also the lateral straggling ( ). To ensure that the same
depth profiles are obtained approximately for different rotation angles, and for different
dimensions, the primary standard deviation is scaled as follows:
(48)
Note that if the ratio is too large, the scaling of the primary standard deviation may not
be possible. In such a case, Sentaurus Process issues a warning message and continues by
assuming .
The scaling factor , which is used to scale and , is selected with respect to the values
of primary and angle.dependent, as shown in Table 7.
Point-Response Interface
This feature allows the use of externally generated point responses in analytic implantation. As
an alternative to using implant tables, it replaces the moment-based point-response
distributions. Only 1D primary distributions can be loaded with Sentaurus Process.
To use the point-response interface, the implant model must be changed to point.response,
that is:
implant spec=<dopant> <material> point.response file=<name> y.position=<n>
Ta bl e 7 S c a li ng fa c to r for t h e p r im ar y r a ng e
Dimension
primary !angle.dependent angle.dependent
1D
wafer
beam
2D
wafer
beam
3D
wafer
beam
Rp
Rp
′Rpsr
⋅=
Rp
σp
σl
σp′sr
2σp
2
⋅1sr
2
–()σ
l
2
⋅+=
σlσp
⁄
σ'pσp
=
sr
Rp
σp
srtilt()cos=
sr1=
srtilt()cos=
sr1=
srtilt()cos=
sr1=
sr
tilt()cos
tilt2D()cos
---------------------------=
sr
1
tilt2D()cos
---------------------------=
srtilt()cos=
sr1=
sr1=
sr
1
tilt()cos
---------------------------=

3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 117
H-2013.03
To revert to the moment-based point-response distribution, switch to one of the analytic
implantation functions, that is, to switch to the dual Pearson model for boron in silicon:
implant spec=Boron Silicon dualpearson
The default table setting can be used again since it has not been overwritten by the
point.response flag.
The external primary distribution function and the damage are read from a plx file. A separate
file can be selected for each dopant–material combination with the file selector and the above
command.
The 1D MC implantation run is started to generate the data if the file is not found. This run is
fully automated. The MC implantation model can be chosen by using the following command:
pdbSet MCImplant model {sentaurus.mc | crystaltrim}
The default MC model is sentaurus.mc. The 1D layer structure for this run consists of an
oxide layer on top and a layer of the specified material. The thickness of the oxide layer is
chosen as the total effective overlayer thickness at some point of reference at the gas surface of
the structure. The position of this point at the y-axis is specified with the y.position
parameter.
The integration routine treats the data as a continuous set to be used in the material as specified.
In the convolution integral computation, the zero of the x-axis is locally matched to the surface
of the structure.
The initial damage for the MC implantation simulation is taken from the damage already
present in the device along a line starting from the surface at the y.position normal to the
wafer surface.
The external profiles are interpreted as taken normal to the wafer surface. Therefore, the
direction of the primary distribution should be switched from beam to wafer (see Primary
Direction and Scaling on page 115). In addition, the multilayer matching method should be set
to dose-matching (see Multilayer Implantations on page 111), that is:
implant <dopant> primary=wafer match=dose
The value for the primary range is taken from the implant table if match=range is set.
Analytic Damage and Point-Defect Calculation
The analytic implantation facility can generate damage profiles that are stored in the dataset
Damage and interstitial and vacancy profiles that are stored in the datasets Int_Implant and
Vac_Implant, respectively.
Rp
3: Ion Implantation
Analytic Implantation
118 Sentaurus™ Process User Guide
H-2013.03
Implantation Damage
The damage to the crystal is calculated on the basis of analytic damage models. Sentaurus
Process calculates the damage using the model by Hobler and Selberherr [1]. Damage
calculation for a species in a material can be switched on using the logical switch damage:
implant species=<dopant> <material> [damage]
A damage profile is calculated if the damage switch is set and the moments are found in the
internal lookup table. Sentaurus Process can use the moments provided by Hobler [1] as
described in Analytic Damage: Hobler Model on page 101. At the end of an implantation step,
the damage for this step (damage_LastImp) is added to the Damage profile (damage history)
using:
(49)
where + indicates the total damage as the sum of new damage and existing damage.
The default value for the DFactor is 1, which can be changed in at the implant command
line or in the parameter database:
implant <dopant> [dfactor=<n>]
pdbSetDouble <material> <dopant> DFactor <n>
Point-Defect Calculation
Elemental Materials
The interstitial and vacancy profiles are calculated in a postprocessing step at the end of the
implant command. The model used to calculate point defects is selected with the
defect.model selector:
implant <dopant> [defect.model= {plus.one | effective.plus.n |
frenkel.pair | user.defined}]
The plus.one switch selects the ‘+1’ model to calculate the interstitial and vacancy profiles
from the as-implanted profile at a particular implantation step <dopant>_LastImp:
(50)
where IFactor and VFactor are material-dependent factors that can be set in the parameter
database. For example, for boron in silicon, this is performed by using:
pdbSet Silicon Boron IFactor <n>
Damage += DFactor Damage_LastImp⋅
Int_Implant += IFactor <dopant>_LastImp⋅
Vac_Implant += VFactor <dopant>_LastImp⋅
3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 119
H-2013.03
The internal default values are 1 for IFactor and zero for VFactor. This is motivated by a
simple lattice site balance argument: for each dopant atom that is assumed after implantation
on a lattice site, one free interstitial is produced. The global values for IFactor and VFactor
can be overwritten at the implant command line:
implant <dopant> <material> [ifactor=<n>] [vfactor=<n>]
The effective.plus.n model dynamically calculates an NFactor using an energy-
dependent and a dose-dependent fitting formula after Hobler [14]. The NFactor replaces the
IFactor in Eq. 50. This ‘+n’ model provides an improved way to calculate the interstitial
profile for heavy ions and low implant doses. Under these implant conditions, the NFactor can
significantly deviate from one [14].
A third model can be chosen with the selector frenkel.pair. Here, the interstitial and
vacancy profiles are calculated from the damage and dopant profiles resulting from the last
implantation step:
(51)
where <dopant>_LastImp term accounts for the extra interstitials coming from substituted
dopants. FPIFactor and FPVFactor can be set in the parameter database, and can be
overwritten by parameters fp.ifactor and fp.vfactor at the implant command line.
If crit.dose is defined, the given value of IFactor in the plus.one and damage models
for point defects is taken from:
(52)
The user.defined switch allows you to define your own algorithms to calculate interstitial
and vacancy profiles. It is expected that users will define the algorithm in the
UserPointDefectModel procedure. For example:
proc UserPointDefectModel { Species Name Energy Dose Model IFactor \
VFactor CDose } {
...
}
where Species is the name of the implanted species; Name is the name of the dopant; Energy
is the implant energy; Dose is the implant dose; Model is the implant model (for example,
tables or sentaurus.mc or crystaltrim); IFactor and VFactor are the interstitial and
vacancy factors; and CDose is the critical dose.
Int_Implant += FPIFactor Damage_LastImp IFactor <dopant>_LastImp⋅+⋅
Vac_Implant += FPVFactor Damage_LastImp⋅
IFactor IFactor min 1crit.dose
dose
----------------------------,
⋅=

3: Ion Implantation
Analytic Implantation
120 Sentaurus™ Process User Guide
H-2013.03
Multicomponent Materials
In multicomponent materials, such as silicon carbide (SiC), the material is composed of
different types of atom. When an impurity is implanted into SiC, both silicon and carbon lattice
atoms can be displaced, thereby forming silicon interstitials or carbon interstitials, and leaving
behind silicon-site or carbon-site vacancies. Instead of classifying them together as interstitials
or vacancies, as in silicon, Sentaurus Process provides a mechanism to distinguish different
types of interstitial or vacancy.
To generate distinct types of point defect in multiple-component materials, you must switch on
the DistinctDefects flag, for example:
pdbSetBoolean SiliconCarbide DistinctDefects 1
By default, this flag is true for SiC but false for other materials. As a result, instead of
Int_Implant and Vac_Implant, the generated point-defect datasets in SiC are
IntSilicon_Implant, IntCarbon_Implant, VacSilicon_Implant, and
VacCarbon_Implant.
In this model, the total point-defect concentration is computed in the same way as the elemental
material. The implantation parameters defect.model, ifactor, vfactor, fp.ifactor,
and fp.vfactor in the implant command still work. ifactor and vfactor are scaling
factors for interstitial profiles and vacancy profiles, respectively, in the plus.one defect
model; while fp.ifactor and fp.vfactor are scaling factors for interstitial profiles and
vacancy profiles, respectively, in the frenkel.pair defect model. The same Tcl procedure
CalcPlusNFactor calculates automatically the plus factors for the effective.plus.n
defect model.
Then, the individual point-defect concentration is computed by multiplying the total point-
defect concentration by the fraction of each component. The fraction of each component is, by
default, their stoichiometric weight, but it can be changed in the parameter database with the
parameters IFactor.Fraction and VFactor.Fraction. For example, in SiC:
pdbSet SiC Composition Component0 IFactor.Fraction <n>
pdbSet SiC Composition Component1 IFactor.Fraction <n>
pdbSet SiC Composition Component0 VFactor.Fraction <n>
pdbSet SiC Composition Component1 VFactor.Fraction <n>
Backscattering Algorithm
During the implantation, some particles may be backscattered and lost to the ambient. Analytic
implantation accounts for this effect by assuming that the portion of the distribution which
sticks out of the structure is backscattered from the surface, resulting in less dose implanted in
the structure. This backscattering model – the TS4 backscattering model – is switched off by
3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 121
H-2013.03
default. To switch on the model, either specify the logical switch ts4.backscattering in
the implant command or use the global switch:
pdbSet ImplantData TS4Backscattering 1
In addition to the TS4 backscattering model, Sentaurus Process uses an advanced integration
algorithm that accounts for particles backscattered from the surface. The lateral integration for
a mesh node also is performed over 1D intervals above the surface. The point response is taken
from the surface layer. The contributions from backscattered ions make a difference in the
profile of vertical mask edges. The mask example in Figure 29 illustrates the difference.
The backscattering algorithm is switched on by default. To switched off the algorithm, use
either the logical switch !backscattering in the implant command, or use the global
switch:
pdbSet ImplantData Backscattering 0
Figure 29 Boron implantation at a vertical mask edge (left) with backscattering
and (right) without backscattering; tilt angle is 0o and energy is 35 keV
Multiple Implantation Steps
Preamorphization Implantation (PAI) Model
A structure already has implantation-related damage by the time an implantation is performed.
This damage contributes to the suppression of the channeling tail. This applies to a series of
Y
-0.5
0
X
Y
-0.2 0 0.2 0.4
-0.5
0
2.8e+18
1.7e+15
1.0e+12
X
-0.2 0 0.2 0.4
2.8e+18
1.7e+15
1.0e+12
3: Ion Implantation
Analytic Implantation
122 Sentaurus™ Process User Guide
H-2013.03
implantations performed without intermediate anneals. In this case, an equivalent amorphous
thickness is extracted as:
(53)
where Damage denotes the preexisting implant damage in terms of Frenkel pairs and
PAIThreshold is a normalization parameter that can be specified in the parameter database:
pdbSet <material> <species> PAIThreshold <n>
The extracted equivalent amorphous thickness is added to the total amorphous layer thickness.
If the implant table contains screen (cap) layer-dependent data, the total amorphous thickness
is used as a parameter to select the implant moments as described in Screening (Cap) Layer-
dependent Moments on page 98. Otherwise, the profile reshaping model and the effective
channeling suppression model are used.
The integral over the preamorphizing damage assumes periodic boundary conditions for the
structure in 2D.
The PAI model can be switched on using:
implant <species> energy=<n> dose=<n> pai
NOTE The switch is off by default. The model is switched on for the Taurus
implant tables.
CoImplant Model
The fraction of the ions described by the second Pearson function is taken from implantation
tables, which have been created for single ion implantation steps. This treatment is acceptable
only for low-dose implantations, which create little crystal damage, but leads to a severe
overestimation of the ion channeling in successive implantations with medium and high doses.
Without a thermal annealing step in between several ion implantations, the crystal damage of
the first implantations remains present and reduces the ion channeling of the subsequent
implantations. The channeling tail is lowered. Besides the PAI model as previously mentioned,
analytic implantation provides the CoImplant (CI) model, which also takes this effect into
account. In contrast to the PAI model in which the implant moments are modified locally for
each cutline during the integration, the CI model modifies the channeling ratio globally for
each implant. The CI model is switched on using the command:
pdbSetBoolean ImplantData UseCoImplant 1
tieqv,
1
PAIThreshold
-------------------------------------------Damage xd
xi
j
ji≤
=
3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 123
H-2013.03
The CI model is switched on by default.
NOTE The CI model is active only for Default implant tables, and does not
affect any other implant tables.
The CI model considers damage produced by analytic or MC implantation steps. The damage
information is used in subsequent analytic implantation steps to estimate the channeling ratio.
Using a least-square fit, an equivalent dose is calculated. This dose is chosen as the dose
that would give the same amount of damage in one implant step (using the present species and
implant conditions) as the preexisting damage, that is:
(54)
This is used to calculate the channeling dose from the total channeling dose and the
equivalent channeling dose, that is:
(55)
The channeling dose is calculated from the total dose using the differential channeling dose
technique (see Figure 30). is the channeling dose corresponding to an implantation of
into undamaged silicon.
Figure 30 Channeling dose as a function of dose
In Figure 30, due to the creation of damage during implantation, the number of channeling ions
increases sublinearly with the total ion dose, and eventually saturates at very high total doses.
The damage from previous implantations is set equivalent to a dose . The dose of the
additional implantation is shifted and, consequently, the gradient / and, therefore,
the dual Pearson ratio are reduced.
Deq
damage Deq x()][damagepreexist
–()
2xd
min=
Dc
new
Dc
new Dc
total Dc
eq
–=
Dc
total
Dc
eq Dc
new
+
Dcnew
Dnew
Dceq
Deq Dose
Channeling Dose
Deq
Dc
new
Dnew
3: Ion Implantation
Analytic Implantation
124 Sentaurus™ Process User Guide
H-2013.03
Both and are stored in lookup tables. The channeling dose is used to
calculate the new ratio for the dualpearson model (see Primary Distribution Functions on
page 94).
This simple model is very accurate for mixed species implantations and works best in cases of
subsequent implantations with similar energies. The model is only available for the Default
implant mode.
The simplest way to calibrate the strength of de-channeling is given by using the effective
damage factor that scales the calculated equivalent dose:
pdbSetDouble ImplantData <species> EffDamFac <factor>
The default value is 1. Increased values lead to less channeling; lower values increase the
channeling of the species specified.
To calibrate the effective damage factor depending on parameters of the implantation, the
following procedure must be overwritten:
proc CI::coimp_dosesofar_calib { species energy dose tilt rotation } {
switch $species {
Boron { set cuc <expression_for_Boron> }
default { set cuc 1.0 }
}; # end of switch
return $cuc
}
Sometimes, it will be necessary to reset the accumulated damage field internally used by the
CI model. This can be achieved by using:
CI::Reset
To save and load the accumulated damage field, use:
CI::Save <filename>
CI::Load <filename>
The loaded damage is added to existing accumulated damage. The accumulated damage
produced by any implantation history can be checked with:
CI::Get_Damage_pdb Accumulated_Damage
This returns a list containing the vector describing the damage in the wafer on a logarithmic
depth scale. A zero vector means no damage history is seen by any follow-up table
implantation.
For more details about the CI model, see [15].
Dc
eq
Dc
total
Dc
new

3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 125
H-2013.03
To choose the PAI or CI model, use the global switch DamageControl:
pdbSet ImplantData DamageControl {Default PAI CoImplant}
The default value of DamageControl is Default in which the PAI model is automatically
switched on when Taurus tables are used; whereas, the CI model is chosen when default tables
are used. If DamageControl is set to PAI, the PAI model is active for all subsequent
implantations regardless of which tables are used. If this switch is set to CoImplant, the CI
model is used for all subsequent implantations (note that the CI model only supports default
tables), and PAI is disabled even for Taurus tables.
NOTE Both the PAI and CI models are designed to take into account the pre-
existing damage. However, the PAI model modifies the implant
moments locally, while the CI model modifies the channeling ratio
globally. Generally, for a complex structure, the PAI model is more
accurate at the expense of longer computation time.
NOTE The CI model was designed to work on the same structure among a
sequence of implant steps. If the structure changes between implants,
erroneous results could occur.
NOTE To avoid double-counting the damage effect, when the PAI model is
active, the CI model is disabled automatically.
Profile Reshaping
Traditionally, it is believed that the first peak of the implanted profile in monocrystalline silicon
is due to random scattering and is described by the first Pearson distribution in the dual-Pearson
analytic model. The second peak (or hump) of the implanted distribution is attributed to ion
channeling and is described by the second Pearson distribution in the dual Pearson model.
This approach works well for implantations with tilt angles above approximately , where
the position and width of the first Pearson distribution do not change as a function of the screen
oxide thickness. However, for low tilt implantations (below ), the position and width of the
first Pearson distribution changes considerably (up to 50%) with the thickness of the screen
oxide.
Typically, for a low tilt implantation performed into bare silicon, the first Pearson distribution
shifts deeper into the substrate and is much wider than for a similar high tilt implantation. As
the screen oxide thickness increases, the projected range and the standard deviation of the first
Pearson distribution relax to their respective values at high tilt angles due to reduced
channeling. Physically, this means that, for a low tilt implantation, even the first peak contains
a considerable number of channeled ions. To model this effect, it is necessary to reshape both
4°
4°
3: Ion Implantation
Analytic Implantation
126 Sentaurus™ Process User Guide
H-2013.03
Pearsons in the dual Pearson model. This profile reshaping complements the reduction in
channeling fraction provided by the effective channeling suppression model.
For ions with explicit dependency on the screen oxide thickness in the implant tables, this
change in shape is addressed automatically. Otherwise, a shift is added to the projected range,
the standard deviation, and the lateral standard deviation of both Pearson distributions. The
shift is given by:
(56)
where:
■ is the shift for moment .
■ is the shift factor for moment .
■ is the cap layer thickness.
■ is the value of moment at high tilt value ( ).
■ is the value of moment at a given low tilt angle.
■For historical reasons, is the standard deviation of the first Pearson distribution.
However, this normalization quantity can be switched to the projected range by using the
command:
pdbSet ImplantData ProfileReshaping.Rp 1
The shift factor parameters of the profile reshaping model can be set in the parameter database,
that is:
pdbSet <material> <species> RangeFactor
pdbSet <material> <species> SigmaFactor
pdbSet <material> <species> ChannelingRangeFactor
pdbSet <material> <species> ChannelingSigmaFactor
The value of RangeFactor is used when calculating the shift of the projected range; the value
of SigmaFactor is used for both the standard deviation and the lateral standard deviation.
Setting a shift factor to zero effectively switches off this model for the respective moments.
Higher values of the shift factor lead to a faster transition from a low tilt profile to a high tilt
profile, with increasing amorphous layer thickness. By default, the shift factors are zero in all
materials except silicon.
The profile reshaping model can be switched on using:
implant <species> [profile.reshaping]
The model remains inactive for explicitly cap layer–dependent implant tables.
∆i1e
αit–
σ
----------
–
MHiMLi
–()=
∆i
i
αi
i
t
MHi
i
7°
MLi
i
σ

3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 127
H-2013.03
NOTE This model is switched off by default and is switched on in the Taurus/
TSUPREM-4 mode.
Ge-dependent Analytic Implantation
SiGe material technology is used widely in stress engineering to improve device performance
(such as mobility). In addition, the depth of the source/drain junctions in Si(1–x)Ge(x) can be
remarkably reduced with an increase of the Ge content, which results from, not only the
reduced boron diffusion for PMOS source/drain, but also the reduced projected range and
channeling in as-implant itself.
Since the average mass of the atomic nucleus of the target is heavier in SiGe than in pure
silicon, a scattering angle from a nuclear collision is larger. In addition, SiGe has a larger
electronic stopping power than silicon due to the higher electron density. Therefore, similar to
the PAI model, the Ge effect on implantation can be modeled by using similar models for
profile reshaping and effective channeling suppression.
In this model, the equivalent germanium thickness is first extracted:
(57)
where denotes the germanium concentration and GeThreshold is a normalization
parameter that can be specified in the parameter database:
pdbSet <material> <species> GeThreshold <n>
The following formulas are then used for the projected range reduction and the standard
deviation shift:
(58)
(59)
where:
Ge.RangePreFactor
Ge.RangeFactor
Ge.SigmaPreFactor
Ge.SigmaFactor
tGe, eqv 1
GeThreshold
--------------------------------------------CGe xd
=
CGe
∆RPGe,Ge.RangePreFactor–RP0,1Ge.RangeFactor–tGe, eqv
RP0,
----------------⋅
exp–
⋅⋅=
∆σGe Ge.SigmaPreFactor–σGe 0,1Ge.SigmaFactor–tGe, eqv
RP0,
----------------⋅
exp–
⋅⋅=

3: Ion Implantation
Analytic Implantation
128 Sentaurus™ Process User Guide
H-2013.03
can be specified respectively in the parameter database as:
pdbSet <material> <species> Ge.RangePreFactor <n>
pdbSet <material> <species> Ge.RangeFactor <n>
pdbSet <material> <species> Ge.SigmaPreFactor <n>
pdbSet <material> <species> Ge.SigmaFactor <n>
Similar formulas also exist for the channeling projected range and channeling standard
deviation shifts with the parameter names:
Ge.ChannelingRangePreFactor
Ge.ChannelingRangeFactor
Ge.ChannelingSigmaPreFactor
Ge.ChannelingSigmaFactor
Finally, the following formula is used for effective channeling suppression:
(60)
where Ge.Sup.Ratio and Ge.Sup.Exponent can be specified in the parameter database:
pdbSet <material> <species> Ge.Sup.Ratio <n>
pdbSet <material> <species> Ge.Sup.Exponent <n>
Analytic Molecular Implantation
Sentaurus Process allows implanting arbitrary molecular species (such as BF2 and B10H14).
The implantation can proceed with or without the implant tables for the molecular species. If
implant tables are not available for the molecular species, an approximate calculation of the
dopant distribution is performed based on the tables for primary dopant species. Therefore, the
only requirement for molecular implantation is that the implant data tables are available for the
primary dopant species (such as B, As, or P). The primary dopant species, for which the profile
is calculated, is specified with the dataset parameter in the implant command:
implant species=<molecule> dataset=<dopant>
To switch on the damage calculation in silicon for the molecular implant, use:
implant species=<molecule> Silicon damage
rGe rGe 0,
Ge.Sup.Ratio
Ge.Sup.Ratio tGe, eqv
RP0,
----------------+
-----------------------------------------------------------------------
Ge.Sup.Exponent
⋅=
3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 129
H-2013.03
In a molecule, the implant energy is shared by several atoms according to:
(61)
where is the energy of the -th species, is the atomic mass, and is the statistical
weight according to the stoichiometry of the molecule. The constituent and stoichiometry of
the molecule are defined in the PDB.
You c an de fin e n ew mo le cul ar s pec ies wi th pdb commands. For example, you can define
carborane as follows:
pdbSetString ImplantData Carborane Atom0 Name Boron
pdbSetDouble ImplantData Carborane Atom0 StWeight 10
pdbSetString ImplantData Carborane Atom1 Name Hydrogen
pdbSetDouble ImplantData Carborane Atom1 StWeight 14
Then to initialize the species, use the command:
implant species=Carborane dataset=Boron
After the above two steps are performed, you can use carborane like any other predefined
implant species. For example, use the following command to perform analytic implantation for
carborane:
implant
Carborane
energy=10 dose=1e14
NOTE The dose specified for molecular implantation is the dose for the
molecular species. In the above example, the implanted dose for
carborane is . Therefore, the boron dose is ,
and the hydrogen dose is .
For convenience, Sentaurus Process predefines the following molecular species: BF2 (BF2),
BCl2 (BCl2), B10H14 (B10H14), B18H22 (B18H22), C2B10H14 (C2B10H14), AsH2 (AsH2),
and PH2 (PH2).
Depending on whether the implant tables are supplied for the molecular species, analytic
molecular implantation will proceed in two different ways:
■With supplied implant tables
■Without supplied implant tables
Eienergy Mi
wjMj
j
-------------------⋅=
Ei
i
Mi
wj
114
×10 cm 2–
115
×10 cm 2–
1.4 15
×10 cm 2–
3: Ion Implantation
Analytic Implantation
130 Sentaurus™ Process User Guide
H-2013.03
Molecular Implantation with Supplied Implant Tables
If the implant tables are available for the molecular species (for example, BF2), the
implantation proceeds in the same way as the atomic species; in other words, the specified
energy and dose are used to look up the moments in the implant tables. No scaling is applied
to energy, dose, or the resulting profiles.
The implant tables can be specified for a molecular species with the command:
implant species=<molecule> <material> imp.table=<file> dam.table=<file>
The implant data files should be placed in the current working directory or the full path to the
file should be specified in imp.table.
Molecular Implantation without Supplied Implant Tables
If the implant tables are not available for the molecular species, Sentaurus Process performs an
approximate calculation of the dopant distribution using the implant tables for the primary
dopant species. The energy for the -th species (which is assumed to be the primary dopant
species) is calculated using Eq. 61.
Assuming that there are dopant atoms in a molecule, the molecular implantation is
equivalent to a single atomic species implantation with the energy and dose equal to and
(where dose is the molecular dose), respectively. and are used for
implant moments lookup. Then, the dopant distribution is calculated in the same way as atomic
implant.
BF2 Implant
BF2 is a special molecular species in analytic implant because both boron and fluorine
distributions are calculated in Sentaurus Process. By default, the fluorine profile is simply a
boron profile multiplied by two. However, you can turn on the following switch to enable the
separate calculation of fluorine distribution by using the fluorine implant tables:
pdbSet ImplantData TS4FluorineMode 1
When TS4FluorineMode is true, the fluorine profile will be computed using the fluorine
tables in the same way as in TSUPREM-4.
Ei
i
Ni
Ei
Nidose×
Ei
Nidose×

3: Ion Implantation
Analytic Implantation
Sentaurus™ Process User Guide 131
H-2013.03
Damage Calculation
If damage tables are not supplied for the molecular species, the damage also can be calculated
using the internal damage tables for the primary dopant species. The Boolean parameter
FullDamage can be used to control the amount of damage for the molecular species:
pdbSetBoolean ImplantData <molecule> FullDamage <bool>
If FullDamage is true, the calculated damage is multiplied by a scaling factor:
(62)
This damage scaling factor roughly takes into account the damage produced by all atomic
species (including the primary dopant species) and is consistent with the damage calculation
used in TSUPREM-4 for BF2 implantation.
Performing 1D or 2D Analytic Implantation in 3D Mode
Because analytic implantation performs lateral integration differently for one, two, and three
dimensions, it may result in slightly different profiles from vertical 1D cuts, even though the
same implant moments are used. In addition, for a 2D structure, the vertical 1D profiles also
may be different depending on the beam direction on the simulation plane or not, in other
words, depending on the rotation angles.
To obtain the same results in one, two, or three dimensions, or with different rotation angles,
Sentaurus Process provides an option to perform 1D or 2D implantation in 3D mode, in which
case, a 1D or 2D structure will first be extruded into a pseudo-3D structure. In other words,
only the surfaces and interfaces (not the bulk) will be extended in the y- or z-direction or both
directions, with the boundary conditions being taken into account. In the case of PAI, damage
integration is performed in a real 1D or 2D structure. Then, the lateral integration proceeds in
exactly the same way as in a 3D analytic implantation. This ensures consistent results for 1D,
2D, and 3D implantation.
To switch on this option, use either the Boolean parameter extrude in the implant command
or the global pdb switch:
pdbSet ImplantData Extrude 1
Si
wjMj
j
Mi
-------------------=

3: Ion Implantation
Analytic Implantation
132 Sentaurus™ Process User Guide
H-2013.03
Implantation on (110)/(111) Wafers Using (100) Implant
Tables
The Sentaurus Process software distribution typically includes a large set of implant tables for
Si(100) wafers, but it does not include any implant tables for Si(110) or Si(111) wafers.
However, you sometimes need to perform process simulations on (110) or (111) wafers. Since
the Si(100) implant moments cannot be used directly for Si(110) or Si(111) wafers, certain
transformations of implant moments are required to use these tables for Si(110) or Si(111)
wafers.
Since SIMS depth profiles are measured along the wafer normal direction, the extracted
implant moments also are obtained with respect to the wafer normal direction. On the other
hand, analytic implantation is usually calculated by using the beam direction as its primary
direction. If the beam direction is coincidental with the normal direction, the implant moments
can be used directly without modification. However, for tilt implantations, the beam direction
does not coincide with the wafer normal direction. In this case, implant moments to be applied
to the primary beam direction must be scaled, or transformed, to reproduce the 1D profiles in
the wafer normal direction. See Primary Direction and Scaling on page 115 for more details.
Essentially, the projected range is scaled as follows:
(63)
where is the scaling factor, and the primary standard deviation is scaled as follows:
(64)
To use Si(110) implant tables for Si(110) or Si(111) implantations, you must calculate the
corresponding angles on the (100) wafers from the specified implantation angles on the (110)
or (111) wafers. For typical implantations (for example, tilt=), these angles are very large.
When you know the corresponding angle on the (100) wafer, you can use Eq. 63 and Eq. 64 to
transform and . Eq. 63 works by simple geometry consideration. Eq. 64 works
reasonably well for small tilt implantations and, in theory, is accurate for isotropic amorphous
material. However, due to ion channeling, Eq. 64 may not be good under all situations.
Rp
Rp
′Rpsr
⋅=
sr
σp
σp′sr
2σp
2
⋅1sr
2
–()σ
l
2
⋅+=
7°
Rp
σp
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 133
H-2013.03
Therefore, the following options for scaling are provided:
■Case 0 (constant):
■Case 1 (linear):
■Case 2 (standard):
The default scaling is the same as the scaling (linear scaling). These cases can be
selected by using the command:
pdbSet ImplantData StdevScalingMode {0 | 1 | 2}
Monte Carlo Implantation
Running Sentaurus MC or Crystal-TRIM
Sentaurus Process is capable of the atomistic simulation of ion implantation using either the
Monte Carlo (MC) simulator Sentaurus MC, which is an improved multithreaded version of
Taurus MC [7], or Crystal-TRIM [8], which originated from the Transport of Ions in Matter
(TRIM) code [2]. MC implantation simulates ion implantation into single-crystalline materials
or into amorphous materials of arbitrary composition. In Sentaurus Process, to select MC
implantation at the command line, use:
implant <dopant> [crystaltrim | sentaurus.mc]
Alternatively, to select MC implantation as the default implantation model, use a global switch:
pdbSet ImplantData MonteCarlo 1
pdbSet MCImplant model [crystaltrim | sentaurus.mc]
When MonteCarlo is set to 1, Sentaurus Process performs all the implantations using one of
the selected MC models (crystaltrim
or
sentaurus.mc).
NOTE For simplicity, you could use the alias tmc instead of sentaurus.mc
or ctrim instead of crystaltrim. For example, you may initiate
Sentaurus MC implant with the following command:
implant <dopant> energy=<n> tmc
Fundamental implantation parameters, such as the implantation energy and dose, and the
orientation of the ion beam with respect to the substrate must be specified using energy, dose,
tilt, and rotation in the same way as for analytic implantation.
σp
σp′σ
p
=
σp
′σpsr
⋅=
σp′sr
2σp
2
⋅1sr
2
–()σ
l
2
⋅+=
σp
Rp
3: Ion Implantation
Monte Carlo Implantation
134 Sentaurus™ Process User Guide
H-2013.03
To run MC implantation in a full-cascade mode or improved BCA (iBCA) damage model, use
the cascades or iBCA switch:
implant <dopant> [ctrim | tmc] [cascades | iBCA]
or using a global switch:
pdbSet MCImplant cascades 1
pdbSet MCImplant iBCA 1
In the KMC mode, to specify the dose rate of the implantation, use the dose.rate parameter:
implant <dopant> [ctrim | tmc] [cascades | iBCA] [dose.rate=<n>]
If dose.rate is specified, it is assumed to be a uniform dose rate in units of cm–2/s. If it is not
specified, a Tcl procedure will be called:
proc DoseRate {dose} { ... }
which returns an implantation time as a function of implantation dose. By default, it is a
uniform dose rate; that is, DoseRate is a linear function of dose. However, you can specify
any monotonic function to take into account the particular implantation equipment setup or
scanning patterns.
During the implantation, pseudoparticles representing a part of the whole dose are started from
the start surface, which is constructed above the target, parallel to the wafer surface. For 2D
and 3D target geometries, the start surface is subdivided into segments of equal size for which
the required implantation dose is accumulated. The size of these segments can be controlled by
setting:
pdbSet MCImplant Intervals dy <n>
pdbSet MCImplant Intervals dz <n>
For 1D structures, no subdivision is performed. The number of pseudoparticles that will be
started per segment can be set in the parameter database or at the command line:
pdbSet MCImplant Particles <n>
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 135
H-2013.03
or:
implant <dopant> [crystaltrim | sentaurus.mc] [particles=<n>]
The default value is 1000. Increasing this number leads to better accuracy and an increase in
simulation time. Together with the sizes of the segments, this parameter determines the
statistical weight of each pseudoparticle.
Launching particles are assumed to be traveling along the direction as specified by the tilt and
rotation angles. However, there is usually a small angular divergence of the ion beam so that
the particles form a right circular cone in which particles are assumed to be uniformly
distributed. To specify BeamDivergence angle (the angle between the cone axis and the cone
surface), use:
pdbSet MCImplant BeamDivergence <n>
Parameters controlling the electronic and nuclear stopping as well as the damage accumulation
are available in the parameter database (see Parameter Database on page 55). You can set these
parameters in there.
If the information level is set to 1 or above, a progress report similar to the following will be
shown during the progress of implant:
implanted orig equiv active repl traject CPU time
particles traject classes segm OK fail step total
.............................................................................
1300( 5%) 84 4 26 1216 6 0.22 0.22
2600( 10%) 170 4 26 2430 9 0.21 0.43
3900( 15%) 262 4 26 3638 16 0.21 0.64
5200( 20%) 349 4 26 4851 24 0.30 0.94
6500( 25%) 437 4 26 6063 29 0.47 1.41
7800( 30%) 523 4 26 7277 33 0.28 1.69
...... ...... ......
22100( 85%) 1894 4 5 20206 131 3.15 7.27
23400( 90%) 2875 4 5 20525 272 5.20 12.47
24700( 95%) 3837 4 5 20863 394 4.75 17.22
24960( 96%) 4029 4 5 20931 419 0.99 18.21
25220( 97%) 4231 4 5 20989 453 1.13 19.34
25480( 98%) 4422 4 5 21058 472 0.90 20.24
25740( 99%) 4628 4 1 21112 483 0.93 21.17
26000(100%) 4888 4 0 21112 483 0.45 21.62
.............................................................................
Pseudo particles:
implanted : 26000
lost : 0 (0%)
Trajectories : 4888
Equivalence classes: 4

3: Ion Implantation
Monte Carlo Implantation
136 Sentaurus™ Process User Guide
H-2013.03
where:
■implanted particles: The total number of pseudoparticles implanted, which is, at the
end of the simulation, equal to the product of the specified number of particles and the
total number of segments of the implant surface. This number includes both the calculated
number of particles and the successfully replicated number of particles. The percentage of
already finished particles is also indicated in parentheses.
NOTE In a multithreaded implant, the thread ID is also shown before the
percentage. For example, 6500 (2: 25%) means that thread #2 has
implanted 6500 particles and finished 25%.
■orig traject: The original number of trajectories that are based on the physical
calculations.
■equiv classes: The number of equivalent classes in the current structure as detected by
probing ions. For more details on equivalent classes and probing ions, see Trajectory
Replication on page 178.
■active segm: The number of currently active segments of the implant surface. At the
beginning of the implant, this number is equal to the total number of start segments. This
number should decrease as the implant progresses. At the end of the implantation, the
number becomes zero as all segments have the required implant dose and become
deactivated.
■repl traject (OK and fail): The number of replicated trajectories. The number of
successfully replicated trajectories is shown in the OK column; whereas, the number of
unsuccessful trajectories is shown in the fail column.
■CPU time (step and total): This CPU time includes the time spent for the current step
and the total CPU time for the current implant.
For 2D structures, the progress of an ion implantation step can be graphically viewed using the
switch ion.movie, for example:
implant <dopant> [ctrim | tmc] [ion.movie]
Structure of Target Material
MC implantation simulates the motion of energetic particles in amorphous materials and
single-crystalline materials.
Composition
For each material, the composition is set in the parameter database. The composition can be
found in the <material> -> Composition entry. For each <n>-component of the material,
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 137
H-2013.03
the entry Component<n> gives the name and the stoichiometric weight, for example, for
GaAs:
GaAs -> Composition -> Component0 -> Name = Gallium
GaAs -> Composition -> Component0 -> StWeight = 1
GaAs -> Composition -> Component1 -> Name = Arsenic
GaAs -> Composition -> Component1 -> StWeight = 1
or for silicon nitride:
Nitride -> Composition -> Component0 -> Name = Silicon
Nitride -> Composition -> Component0 -> StWeight = 3
Nitride -> Composition -> Component1 -> Name = Nitrogen
Nitride -> Composition -> Component1 -> StWeight = 4
The composition for both single-crystalline and amorphous materials is set this way.
Single-Crystalline Materials
Lattice Structure
In the case of a single-crystalline material, the positions of target atoms are calculated based
on the lattice type. Crystal-TRIM supports zinc-blende (Zincblende) lattice only, while
Sentaurus MC supports several lattice types that include simple cubic (Sc), body-center cubic
(Bcc), face-center cubic (Fcc), zinc-blende (Zincblende), and hexagonal (Hexagonal)
lattices. To set the lattice type, use:
pdbSet <material> LatticeType [Sc | Bcc | Fcc | Zincblende | Hexagonal]
Lattice Constants
To change the lattice constant defined in the parameter database, use:
pdbSet <material> LatticeConstant <n>
For all lattice types, Sentaurus MC defines different lattice constants for three different axes.
To define the other two lattice constants, use:
pdbSet <material> LatticeConstant_b <n>
pdbSet <material> LatticeConstant_c <n>
If LatticeConstant_b and LatticeConstant_c are not defined, LatticeConstant is
used for all three axes. For a hexagonal lattice, LatticeConstant_b should be equal to
LatticeConstant.
3: Ion Implantation
Monte Carlo Implantation
138 Sentaurus™ Process User Guide
H-2013.03
Polytypes
For hexagonal systems, there may exist many different crystal structures due to the different
stacking sequence along the -axis, which is perpendicular to the plane formed by three -
axes ( , , and ). This is called polytypism. Four different polytypes are supported in
Sentaurus MC, and you can select them using the following command:
pdbSet <material> Polytype {2H 3C 4H 6H}
The default polytype for silicon carbide (SiC) is 4H. For more details, see MC Implantation
into Silicon Carbide on page 165.
Atomic Basis
The crystal structure consists of an atomic basis attached to the lattice points. A basis can be a
single atom or a group of atoms attached to each lattice point. In Sentaurus MC, for simple
crystals (such as a single-atom basis with simple cubic, face-centered cubic, or body-centered
cubic lattice and binary compounds with zinc-blende and hexagonal lattice), the undisturbed
positions of the lattice sites are constructed automatically using the information of the lattice
type, the polytype (if hexagonal lattice), and the lattice constants. For more complex crystal
structures, the positions of basis atoms should be specified with the pdb parameter
BasisVector. The units of basis vectors are lattice constants in three crystallographic axes.
For example, for zinc-blende silicon, the positions of two basis silicon atoms can be specified
as follows:
Silicon -> Composition -> Component0 -> Name = Silicon
Silicon -> Composition -> Component0 -> StWeight = 1
Silicon -> Composition -> Component0 -> BasisVector = {0 0 0 0.25 0.25 0.25}
For another example, NaCl has a face-centered cubic (Fcc) lattice with an atomic basis of two
atoms. The positions of Na and Cl can be specified as follows:
NaCl -> Composition -> Component0 -> Name = Sodium
NaCl -> Composition -> Component0 -> StWeight = 1
NaCl -> Composition -> Component0 -> BasisVector = {0 0 0}
NaCl -> Composition -> Component1 -> Name = Chloride
NaCl -> Composition -> Component1 -> StWeight = 1
NaCl -> Composition -> Component1 -> BasisVector = {0.5 0.5 0.5}
In Crystal-TRIM, the positions of lattice sites of the basic cell are set in the parameter database
in MCImplant -> Lattice -> Zincblende -> Cell0 in the natural coordinate system
of crystal. The unit is one-half of the lattice constant. The undisturbed positions of all lattice
sites of an ideal zinc blende–type crystal can be obtained from the basic cell by shifting the
atomic positions in the directions of the crystallographic axes. Therefore, for any given position
of the projectile, only the immediate crystalline environment is generated and rebuilt every
time the projectile moves out of the current crystalline cell.
c
a
a1
a2
a3
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 139
H-2013.03
The entry MCImplant -> Lattice -> Zincblende -> Cell1 is a complementary basic
cell and gives the configuration that is obtained by shifting Cell0 by one-half of the lattice
constant.
Thermal Vibrations
The thermal vibrations of the target atoms are important for the treatment of the motion of a
projectile in single-crystalline material. In MC implantation, only instantaneous thermal
displacements of target atoms from their ideal lattice sites are considered.
The displacements are assumed to obey a 3D Gaussian distribution with a root-mean-square
obtained by the Debye model. The Debye temperature is set in the parameter database and can
be changed with:
pdbSet <material> DebyeTemperature <n>
The default Debye temperature is 519 K for silicon.
The substrate temperature for the Debye model can be set by:
pdbSetDouble MCImplant Temperature <n>
The default substrate temperature is 300 K.
Amorphous Materials
The structure of an amorphous material is described in a simplified manner by assuming an
average interatomic distance in the target material.
Variable Mass Density
It is possible to use a dataset MassDensity as the mass density of an amorphous material:
pdbSetBoolean <material> VariableMassDensity 1
Using molar fractions (see Molar Fractions on page 140) is disabled in these materials.
Polycrystalline Materials
A polycrystalline material is characterized by its crystal orientation and grain size. Crystal
orientation (one of 100, 110, and 111) can be specified by using a material-specific command:
pdbSet <material> CrystalOrient <n>
3: Ion Implantation
Monte Carlo Implantation
140 Sentaurus™ Process User Guide
H-2013.03
There are two different ways to change the crystallinity (Amorphous, Crystalline, and
Polycrystalline) of a material. If parameter Crystallinity is available (which is true
for polysilicon) in the PDB, use this switch to set the crystallinity, for example:
pdbSet PolySilicon Crystallinity Polycrystalline
This command makes MC implantation models consider both crystal orientation and grain size
for polysilicon.
If Crystallinity does not exist for a material, use parameters Amorphous and Granular:
pdbSet <material> Amorphous 0
pdbSet PolySilicon Granular 1
The first command switches off the amorphous treatment, and the second command makes MC
implantation models consider the grain size.
For more details on ion implantation into polysilicon, see MC Implantation into Polysilicon on
page 162.
Molar Fractions
It is possible to define a compound material with a spatially-dependent molar fraction. For
example, for single-crystalline silicon, the following PDB entry:
array set $Base {BinaryCompounds {String {
{ SiliconGermanium GeTotal "GeTotal/[pdbGetDouble Si LatticeDensity]" }
}}}
specifies a binary compound Si1–xGex with the mole fraction of Ge calculated from the
germanium concentration (GeTotal) divided by the silicon lattice density. Due to more
computational demands, a minimum Ge concentration is required to trigger MC implantation
models to treat this material in a more sophisticated way. To specify this minimum
concentration, use the command:
pdbSet Silicon SiliconGermanium.MCmin 1e20
If the concentration of Ge in any of the mesh elements of silicon regions exceeds
, MC models treat silicon as a compound material. In this case, the average
charge and mass of the material is calculated individually for each mesh element. The lattice
constant, the nonlocal electron stopping power, and the Debye temperature are linearly
interpolated based on the mole fractions. For more details, see MC Implantation into
Compound Materials with Molar Fractions on page 163.
110
20
× cm 3–

3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 141
H-2013.03
Sentaurus MC Physical Models
The Sentaurus MC implantation model, which is an improved, multithreaded version of Taurus
MC, was designed to be generally accurate and predictive with minimum user calibrations for
most implant conditions. It has been calibrated from sub-keV to above 10 MeV, and for
different implant conditions including random implant direction, <100>, <111>, and <110>
channeling directions, with the same set of parameters for boron, phosphorus, and arsenic
implants [7]. It also is accurate for other implant species such as BF2, F, Al, Ge, In, and Sb
[7][16]. For a detailed discussion of the physical models in Sentaurus MC and an extensive
comparison with experimental SIMS profiles from sub-keV to above 10 MeV and with other
MC simulators, refer to the literature [7][16]. This section briefly outlines the pertinent theory
and models.
The calculation used in the Sentaurus MC model assumes that ions lose energy through two
processes:
■Nuclear scattering, where the nucleus of the ion elastically scatters off the nucleus of an
atom in the target. This interaction is based on the binary collision theory and is described
in the following section.
■Interaction of the ion with the electrons of the target atoms. This mechanism is inelastic
and does not alter the direction of the motion of the ion.
Therefore, the total change in energy of the ion after the -th collision is the sum of the nuclear
energy loss and the electronic energy loss :
(65)
Binary Collision Theory
Sentaurus MC implantation models the energy loss of nuclear collision according to the
classical binary scattering theory. The basic assumption of the mechanism for the energy loss
of nuclear collision is that the ion interacts with only one target atom at a time. This assumption
enables the use of the binary scattering theory from classical mechanics [17].
Consider a particle of mass and kinetic energy approaching a stationary particle with
mass . The impact parameter, , is the distance of closest approach if the particle is not
deflected and gives a convenient measure of how close the collision is. After collision, the first
particle deviates from its original course by an angle .
i
∆En
∆Ee
EiEi1–∆En
–∆Ee
–=
M1
E0
M2
b
θ

3: Ion Implantation
Monte Carlo Implantation
142 Sentaurus™ Process User Guide
H-2013.03
Energy Loss
It can be shown that the first particle loses kinetic energy:
(66)
where:
■ is the energy lost by particle 1.
■ is its energy before collision.
■ is the integral.
(67)
where is the inverse separation between the two particles. is the potential
between the two particles (assumed to be repulsive), and:
(68)
is the reduced energy in the center of mass coordinates.
The upper limit of the integral, , is the inverse distance of closest approach of the two
particles and is given by the solution to the equation:
(69)
Scattering Angle
The angle by which particle 1 is deflected is given by:
(70)
NOTE For , approaches zero.
∆En
E0
----------4M1M2
M1M2
+()
2
---------------------------- cos2bI()=
∆En
E0
I
Isd
1Vs()
Er
-----------– b2s2
–
-----------------------------------------
0
smax
=
s1r⁄=
Vs()
Er
E0
1M1M2
⁄+
----------------------------=
smax
1Vs
max
()
Er
-------------------– b2smax
2
–0=
θ
cosθ
1 0.5 1 M2
M1
-------+∆EnE0
⁄–
1∆EnE0
⁄–
------------------------------------------------------------=
∆EnE0
⁄1«
θ
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 143
H-2013.03
Dimensionless Form
Eq. 66 to Eq. 70 are the basic equations for classical two-body scattering. The scattering
integral, Eq. 67, can be cast into a dimensionless form by assuming the potential has the form:
(71)
where:
■ is the charge on particle 1.
■ is the charge on particle 2.
■ is the constant.
(72)
is an arbitrary function of , to be defined later, and is a unit of length. Taurus
MC uses the so-called universal screening length [18]:
(73)
and a dimensionless impact parameter:
(74)
and a dimensionless energy:
(75)
Using Eq. 71, Eq. 74, and Eq. 75 in the scattering integral Eq. 67 and making the substitution
gives:
(76)
From Eq. 66, the quantity of interest is , which becomes:
(77)
Therefore, using Eq. 77, can be evaluated in terms of the dimensionless variables
and , without reference to the charge or mass of a particular particle.
Vs() Z1Z2k1sg aus()=
Z1
Z2
k1
k1q2
4πε0
------------14.39495 10 7–
×keVµm==
ga
us()
aus
au
au0.8854 10 4–
×0.529
Z1
0.23 Z2
0.23
+()
---------------------------------- µm=
bnba
u
⁄=
εauEr
Z1Z2k1
-----------------=
s′aus=
I1
au
-----= s′d
1s′gs′()εbn
2s′2
–⁄–
-------------------------------------------------------
0
s′max
cos2bI()
cos2bI() cos2bn
s′d
1s′gs′()εbn
2s′2
–⁄–
-------------------------------------------------------
0
s′max
=
cos2bl()
bn
ε
3: Ion Implantation
Monte Carlo Implantation
144 Sentaurus™ Process User Guide
H-2013.03
Coulomb Potential
As an example of the above procedure, consider the Coulomb potential between two particles:
(78)
or:
(79)
In this case, . Then, from Eq. 77:
(80)
with:
(81)
from a solution of Eq. 69.
Then, the integral can be evaluated exactly, giving:
(82)
For a given impact parameter and incident energy , the dimensionless and can be
obtained from Eq. 74 and Eq. 75, giving from Eq. 82. Then, the energy loss due to
the collision is given by Eq. 66, and the angle at which particle 1 leaves the collision is given
by Eq. 70.
Universal Potential
For the simple form of the Coulomb potential used in the previous example, the scattering
integral can be solved analytically. For more realistic interatomic potentials, however, the
scattering integral cannot be evaluated analytically.
Vr() Z1Z2k1
r
-----------------=
Vs() Z1Z2k1s=
ga
us()1=
cos2bI() cos2bn
s′d
1s′ε bn
2s′2
–⁄–
------------------------------------------
0
s′max
=
s′max
14bn
2ε2
+1–
2εbn
2
------------------------------------------=
cos2bI() 1
14bn
2ε2
+
-----------------------=
b
E0
bn
ε
cos2bI()

3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 145
H-2013.03
For example, the universal potential [18] that is used in Sentaurus MC is:
(83)
An analytic solution does not exist since the upper limit of the integral in Eq. 67 is given by
Eq. 69, which becomes a transcendental equation with this potential. In Taurus MC, the
quantity , in its dimensionless form (Eq. 77), is numerically integrated for a wide
range of its parameters and . These results are stored in tables. Then, at each collision,
is obtained from these tables. This scheme eliminates the need to find for each
collision, minimizing the amount of arithmetic operations performed during the calculation of
the trajectory of an ion, while retaining accuracy.
Tables for the universal potential over a wide range of energies and impact parameters are
provided for immediate use in Taurus MC. These tables span the normalized energy range of
and the normalized impact parameter range . For , the
Coulombic form (Eq. 83) is used. Values of are not encountered for ion–atom
combinations of interest at energies above the energy at which the ion is assumed to have
stopped (5 eV). For values of , the ion is assumed to be undeflected.
Implantation into Amorphous Materials
This section describes how the binary scattering theory of the previous section is used to
calculate ion trajectories in an amorphous solid. Assume an ion with kinetic energy hits a
target with an angle with respect to the target normal. The surface of the target is assumed
to be at , with increasing vertically into the target. To set the incident energy in
the implant command, use the Energy parameter. To specify the incident angle in the
implant command, use the tilt parameter.
Given the atomic density for the target material, the mean atomic separation between
atoms in the target is . Between scattering events, the ion is assumed to travel a
distance:
(84)
As the ion enters the target material, it approaches the first target atom with impact parameter
, defined in the previous section. The probability of finding a target atom between and
is given by:
(85)
for .
Vr() Z1Z2k
r
---------------0.18175e3.1998ra
u
⁄–0.50986e0.94229r–au
⁄
0.28022e0.4029ra
u
⁄–0.028171e0.20162ra
u
⁄–
+
++
=
cos2bI()
bn
ε
cos2bI()
smax
10 5–ε100≤≤
0bn30≤≤
ε100>
ε10 5–
<
bn30>
E0
θ0
y0=
y
E0
θ0
Ndens
1Ndens
()⁄13⁄
L1Ndens
()⁄13⁄
=
b
b
bδb+
wb()δb2πNdens
23⁄b δb=
b1πNdens
23⁄
⁄<

3: Ion Implantation
Monte Carlo Implantation
146 Sentaurus™ Process User Guide
H-2013.03
If is a uniformly distributed random number between 0 and 1, the probability distribution
gives:
(86)
Given the above definitions, the algorithm for calculating the energy loss through nuclear
collisions experienced by the ion proceeds as follows:
■A random number between 0 and 1 is chosen.
■The normalized impact parameter for this collision is calculated from Eq. 74 and Eq. 86:
(87)
■The ion energy, , is normalized to:
(88)
from Eq. 68, p. 142 and Eq. 75, p. 143.
■Now, the value of can be obtained from the tables, and Eq. 66, p. 142 gives the
energy loss due to nuclear scattering:
(89)
where nucl.cor is an empirical nuclear-scattering correction factor with a default value
of 1.0, which can be changed in the parameter database by using:
pdbSetDouble <material> <dopant> nucl.cor <n>
This procedure is repeated for each collision event.
Implantation into Crystalline Materials
The binary collision calculation for crystalline materials proceeds in the same way as in the
amorphous case, except that the selection of the collision partners of the projectile with target
atoms is conducted in a more sophisticated manner.
Instead of using the density of the target material and a random number, Sentaurus MC
determines the collision partners based on the position of the projectile relative to the sites on
an idealized lattice. The algorithm for selecting the collision partners is based on
Rrand
bRrand
πNdens
23⁄
----------------=
b1
au
----- Rrand
πNdens
23⁄
----------------=
E0
εauE0
1M1M2
⁄+()Z1Z2k1
---------------------------------------------------=
cos2bI()
∆E0nucl.cor E⋅0 4M1M2
M1M2
+()
2
---------------------------- cos2bI()=
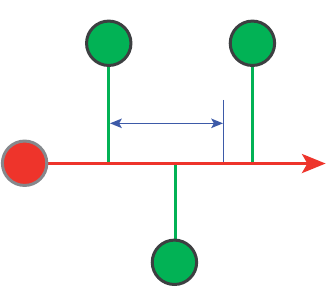
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 147
H-2013.03
MARLOWE [19]. Sentaurus MC implantation uses a sophisticated multibody collision
algorithm to simulate the collisions of well-channeled particles, as shown in Figure 31.
Figure 31 Illustration of “simultaneous” collision, and definition of the d.sim parameter
If the distance of two or more projected target atoms to the undeflected trajectory of the
incident particle is less than d.sim (simultaneous collision distance), then the multibody
collision algorithm is invoked. For example, in Figure 31, 1 and 2 are considered to be
simultaneous collisions, but 3 is not. To change the default value of d.sim, use the command:
pdbSetDouble <material> d.sim <n>
The scattering events are computed for each target individually, and the final moment and
energy of the incident particle are computed by applying momentum and energy conservation
principles.
The simultaneous collisions are handled in the same way as for MARLOWE, except for the
location of the turning point. In MARLOWE, the turning point is assumed to be the average of
those of the simultaneous collision partners. In Sentaurus MC, the turning point is placed at a
point determined by the collision with the minimum impact parameter. Simulations have
indicated that such a scheme obtains better results for ultralow energy implantations, while it
has little impact on implantation energies above 5 keV. This allows Sentaurus MC to treat the
entire implantation energy range, including ultralow energy and very high energy, with the
same model, in exactly the same way.
Ion channeling, which is the preferential penetration of implanted ions along crystal axes or
planes, occurs naturally due to the inclusion of the crystal structure of the lattice. Both axial
and planar channeling show enhanced penetrations. The effect of the tilt and rotation
parameters is much more pronounced for implants into crystalline silicon than into amorphous
silicon.
13
2
d.sim
Lattice Atoms
Incident Particle
3: Ion Implantation
Monte Carlo Implantation
148 Sentaurus™ Process User Guide
H-2013.03
Electronic Stopping Model
A moving ion loses energy by inelastic electronic processes, which include both nonlocal and
local stopping power. Sentaurus MC uses the same electronic stopping model for both
amorphous and crystalline materials. For each collision, the energy loss due to electronic
stopping is:
(90)
(91)
(92)
where is the scaled dimensionless energy.
nloc.pre and nloc.exp are specified in the material parameter database and can be changed
by using:
pdbSet <material> <dopant> nloc.pre <n>
pdbSet <material> <dopant> nloc.exp <n>
Nonlocal Electronic Stopping
Nonlocal electronic stopping acts as the dragging (frictive) force on moving ions, which is
proportional to the ion velocity and is independent of the impact parameter:
(93)
(94)
where is the free flight path between collisions and is the ion energy at the stopping
power maximum.
The quantities and are given by [7][20]:
(95)
(96)
∆Eexnl ∆Enl
exloc ∆Eloc
⋅+⋅=
xnl xloc
+1=
xnl min nloc.pre εnloc.exp 1,⋅()=
ε
∆Enl
eLN
dens Se
⋅⋅=
SeLSS.pre Ses Emfes
1–
⋅⋅ ⋅=
L
Em
Ses
fes
Ses
1.212 Z1
76⁄ Z2
Z1
23⁄Z2
23⁄
+
32⁄
M1
12⁄
-------------------------------------------------------=
fes
E0Em
⁄
E0Em
⁄EmE0
⁄e2–++()ln
---------------------------------------------------------------------
δ2⁄Em
E0
-------
δ2⁄
+1δ⁄
=
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 149
H-2013.03
where:
■ is the ion atomic number.
■ is the composite target atomic number.
■ is the base of natural logarithm.
■ is a fitting parameter.
LSS.pre is specified in the material parameter database; to change it, use:
pdbSet <material> <dopant> LSS.pre <n>
Local Electronic Stopping
Local electronic loss is a result of the electron exchange between the moving ion and the target
atom, which is based on the Oen–Robinson model [21] and is dependent on the impact
parameter:
(97)
(98)
(99)
where scr.par is an adjustable screening length parameter that you can change by using:
pdbSet <material> <dopant> scr.par <n>
Damage Accumulation and Dynamic Annealing
As the ions travel through a crystalline target, they collide with the target atoms and displace
many of them from their lattice sites. In the binary collision approximation (BCA) code, it is
assumed that, if the transferred energy exceeds a certain threshold, the target atom is displaced
and, at this lattice site, a vacancy is generated. When the displaced atom comes to rest, it is
identified as an interstitial. This defect production rate can be evaluated either by the modified
Kinchin–Pease formula [22] or by simulating the full cascade. Sentaurus MC provides both
types of damage calculation.
Z1
Z2
e
δ1.425=
∆Ee
loc Se
2πa2
------------ b
a
---–
exp⋅=
af
au
0.3
-------
⋅=
fscr.par 1.45
Z1
25⁄
-------------
⋅=
3: Ion Implantation
Monte Carlo Implantation
150 Sentaurus™ Process User Guide
H-2013.03
Damage Accumulation Models
The default damage model calculates the deposit energy for each collision, which is
then converted to the number of point defects (Frenkel pairs) using the modified Kinchin–
Pease formula [22]:
(100)
where and disp.thr = 15 eV for silicon by default.
If the cascades switch is specified in the implant command, Sentaurus MC traces all of the
generated secondary recoils. After each collision, a calculation is performed to determine the
trajectories of the silicon lattice atoms that are knocked from their sites in the lattice by
collisions with implanted ions. A silicon atom is assumed to be knocked from its site when it
absorbs an energy greater than a damage threshold casc.dis from a collision.
The silicon atoms freed from the lattice can, in turn, knock other atoms from their sites so that
cascades of damage result. Sentaurus MC calculates the trajectories of these knock-ions with
the same detail as the implanted ions. A vacancy is assumed to have formed whenever a lattice
atom is knocked from its site. An interstitial is assumed to have formed whenever a silicon
lattice atom that has been knocked from its site comes to rest. This damage model can be used
to calculate the different profiles of interstitials and vacancies, that is, I–V separations.
disp.thr and casc.dis are specified in the material PDB and you can change them using:
pdbSet <material> <dopant> disp.thr <n>
pdbSet <material> <dopant> casc.dis <n>
Dynamic Annealing
Not all of the defects as calculated above will survive; some of the generated defects will
recombine within the cascade as well as with the preexisting defects. To achieve computational
efficiency, Sentaurus MC uses a statistical approach to account for the I–V recombination in
both intracascades and intercascades. The encounter probability of the projectile with
interstitials also is accounted for statistically. The net increase of the defects in a local region
with defect concentration is:
(101)
In the cascade damage model, surv.rat and sat.par are replaced with casc.sur and
casc.sat. To conserve particle numbers, interstitials and vacancies are recombined in pairs,
and the model distinguishes between recoiled interstitials and recoiled lattice atoms. When an
interstitial is recoiled, the local interstitial number decreases by one and no vacancy is
produced. On the other hand, when a lattice atom is recoiled, a vacancy is created. However,
defect recombination must be considered.
EDx()
nx() κED2disp.thr⋅()⁄()=
κ0.8=
Cx()
∆nx() surv.rat nx() 1sat.par Cx()⋅
Ndens
--------------------------------------–
⋅⋅=
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 151
H-2013.03
The intracascade recombination is accounted for by a factor casc.sur, while intercascade
recombination is accounted for by a probability , which describes that the
vacancy is not located within the capture radius of an interstitial.
When a recoil comes to rest, it is only allowed to recombine with vacancies from previous
cascades, which is described by a factor , but not with those of the same
cascade since this recombination has already been accounted for by vacancy intracascade
recombination in the previous step. surv.rat and casc.sur are specified in the material
PDB and you can change them using:
pdbSet <material> <dopant> surv.rat <n>
pdbSet <material> <dopant> casc.sur <n>
For light implant species, damage could saturate at certain concentrations due to the balance
between defect production and dynamic annealing. Damage saturation is controlled by the
parameters sat.par and casc.sat for the default damage model and cascade damage
model, respectively. The default value is 1 for all implant species. Therefore, with the default
parameter, the maximum damage is equal to the lattice density. If, for example, sat.par is set
to 4.35, damage saturates at 23% of the lattice density and cannot exceed the amorphization
threshold (1.15e22 cm–3 by default). Therefore, the crystal will never be amorphized in this
case. To change these parameters, use:
pdbSetDouble <material> <dopant> sat.par <n>
pdbSetDouble <material> <dopant> casc.sat <n>
NOTE For heavy species, a single cascade may amorphize the crystal.
Therefore, the intracascade parameter sat.par may not prevent the
amorphization even if it is set to a very large value.
Damage De-Channeling
The accumulated damage has a significant effect on the destination of the subsequent ions,
thereby altering the shape of the impurity profiles. This effect is known as damage de-
channeling. Sentaurus MC handles this problem by switching from the crystalline model to the
amorphous model based on the damage that has accumulated in the substrate. If the local defect
concentration is greater than the amorphization threshold, this local region is assumed to
be amorphized, and the amorphous collision model is used for this local region.
For the local regions with defect concentrations below the amorphization threshold, the
probability of selecting the amorphous model is proportional to the local defect concentration
and a random number call. The amorphous collision model is selected when:
(102)
1NIN⁄dens
()–
1NvN⁄dens
()–
Cx()
Cx()
Rrand amor.par Cx()
Ndens
------------⋅<
3: Ion Implantation
Monte Carlo Implantation
152 Sentaurus™ Process User Guide
H-2013.03
Increasing amor.par makes the profiles more like those implants into amorphous materials.
For the cascade damage model, amor.par is replaced with casc.amo. The parameters
amor.par and casc.amo are specified in the material PDB, and you can change them using:
pdbSet <material> <dopant> amor.par <n>
pdbSet <material> <dopant> casc.amo <n>
NOTE For low energy implants, due to very shallow projected ranges, the mesh
near the surface should be refined to account fully for the damage de-
channeling effect.
NOTE The amorphization process is not explicitly simulated by
Sentaurus MC. However, for the MC model, by common practice, when
a critical amount of damage is accumulated in a certain region, a crystal/
amorphous phase transition is assumed to occur in this region. For a
silicon target, this critical amorphization threshold is approximately
25% of the lattice density. Therefore, if the defect concentration reaches
more than for silicon, this region is considered to be
amorphized. Using this criterion, Sentaurus MC predicts the onset of
amorphization and the thicknesses of the amorphous layers for high-
dose implantations.
Improved Binary Collision Approximation Damage Model
During implantation, energetic ions penetrate into the target and lose their energy through
collisions with atoms and electrons. It is traditionally assumed that only energy deposited in
the form of nuclear collisions contributes to damage generation; whereas, energy transfers to
the electronic system are taken as inelastic losses. While energetic atoms are in the ballistic
regime (that is, they have energies well above the displacement threshold casc.dis), they can
be well simulated using binary collision approximation (BCA) algorithms. However, as their
energy decreases to the thermal regime (around and below the displacement threshold),
multiple interactions with target atoms become important. Molecular dynamics (MD)
simulations demonstrate that energy transfers among atoms at this low-energy regime can
generate amorphous pockets, thereby generating more damage than BCA models. The
improved BCA (iBCA) damage model is an attempt to simulate MD simulation results within
the framework of BCA.
The iBCA damage model implemented in Sentaurus MC implant is largely based on an the
published article [23] (for the detailed physical basis of the model, refer to this article).
This section briefly describes the model, its usage, and the parameters that are accessible to
users.
1.15 1022
× cm 3–
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 153
H-2013.03
The procedure followed in the iBCA model is:
1. The collision phase of the cascade is simulated with the conventional BCA model.
2. The BCA simulation provides the position of Frenkel pairs generated during the cascade,
the remaining energy of the recoils at the end of their trajectories, and the position and
energy of all the atoms that receive any amount of energy above the minimum energy
(MinHotEnergy). These particles are called hot particles.
3. At the end of the collision phase, there is a set of vacancies, interstitials, and hot particles.
Within BCA, the energy conservation principle applied to elastic binary collisions implies that
the energy of the incident particle must be equal to its energy after the collision plus the recoil
energy plus the energy required to take the recoil away from its lattice site.
A moving atom stops when its energy is insufficient to generate more subcascades. However,
the remaining energy of the generated interstitial at the end of its trajectory can still contribute
to generate more damage if low-energy interactions were modeled. To consider this effect
within the iBCA model, the residual energy of each generated interstitial is equally shared with
its neighboring atoms. Ballistic collision only considers the impinging atom and the closest
target atom (two-body interactions); however, as energy decreases, collisions with several
target atoms occur more often, and groups of energetic atoms are created as the cascade
develops (many body interactions).
After this energy rearrangement, you evaluate which atoms are disordered taking into account
their efficiencies:
(103)
where is the energy density, and and are the threshold energy density and damage
generation cost, respectively. If the calculated efficiency of a given atom is below zero, it is not
disordered.
If is between 0 and 1, the atom is disordered with a random probability given by its
efficiency. If is 1 or greater, the atom is disordered and a random neighbor is disordered
with the probability given by the remaining efficiency and so on.
To simulate the energy diffusion process: First, evaluate the efficiency of those atoms with the
highest amount of energy in their environment. Second, repeat the process until no further
energy remains to create more disordered atoms.
This scheme for damage generation can be regarded as a combination of the two traditional
BCA approaches for damage description. As in the full-cascade BCA, ion and recoil
trajectories are followed to generate damage at the atomic level and to provide the individual
positions of Frenkel pairs, but you also must consider the energy deposited in atoms not
eff ρET
–
DC
---------------=
ρ
ET
DC
eff
eff
eff 1–()
3: Ion Implantation
Monte Carlo Implantation
154 Sentaurus™ Process User Guide
H-2013.03
displaced by ballistic collisions. This energy is used to generate thermally disordered atoms
following a scheme similar to the modified Kinchin–Pease approach. Nevertheless, since the
residual deposited energies that are being considered to determine efficiencies are always at the
low-energy regime, the local character of damage generation is guaranteed. In addition, the
damage efficiency expression accounts for phase transformation (melting) and heat dissipation
through the dependency of the parameters and on the number of energetic neighbors.
This feature captures the nonlinear effects on damage generation due to the proximity of
several energetic atoms as it occurs in molecular implants.
To activate the iBCA damage model, specify iBCA in the implant command or switch on the
global switch:
pdbSet MCImplant iBCA 1
You can calibrate the iBCA damage model by changing the minimum energy for hot particles
(MinHotEnergy) and the maximum distance for the local neighbors
(DistLocalNeighbors):
pdbSet Silicon MinHotEnergy <n>
pdbSet Silicon DistLocalNeighbors <n>
By default, MinHotEnergy is 1 eV, and DistLocalNeighbors is 3.84e-4 .
In addition, you can calibrate the model by changing the formulas for the threshold energy
density (Et_iBCA) and the damage generation cost (Dc_iBCA) by modifying Tcl procedures.
As noted in the article [23], the default Tcl procedures for these quantities are defined as:
proc Et_iBCA { ln } {
set et [expr 11.348 * pow($ln+1, -0.837) + 0.931]
return $et;
}
proc Dc_iBCA { ln } {
set dc [expr 11.211*exp(-0.146*$ln + 0.00158*$ln*$ln)];
return $dc;
}
where ln is the number of local neighbors.
NOTE To avoid nonphysical results, only fine-tuning of these formulas is
recommended.
NOTE Because the iBCA damage model is substantially more CPU intensive
than the cascade damage model, only low-energy implant is practical
for this damage model.
ET
DC
µm

3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 155
H-2013.03
Crystal-TRIM Physical Models
Crystal-TRIM simulation is based on the binary collision approximation (BCA), which
represents the motion of ions in the target material as a set of binary collisions with the target
atoms [2].
BCA is valid in a wide range of projectile energies, from approximately 100 eV to many MeV.
It can, therefore, be employed over the whole range of energies of interest for ion implantation.
For energies below approximately 100 eV, collective interactions may play an increasingly
important role and BCA may become invalid. Nevertheless, the applications of Crystal-TRIM
to ultra low-energy implants lead to results that are still sufficiently good compared with
experimental data.
At each collision, the projectile loses a part of its energy due to elastic nuclear scattering at
target atoms and inelastic electronic iterations. The particles are assumed to come to rest if their
energy is in the order of 15 eV.
Single-Crystalline Materials
Nuclear Collisions and Collision Cascades
Nuclear scattering is treated by classical mechanics using a Coulomb-screened pair potential
(ZBL potential [18]). If the energy transfer to the target atoms exceeds the so-called
displacement threshold (approximately 15 eV for silicon), the target atom can leave its site and
become displaced (primary recoil).
By default, only the trajectories of implanted ions are simulated. The number of vacancies and
displaced target atoms produced at each collision is calculated approximately using the
modified Kinchin–Pease formula.
A full cascade–type of simulation is performed if the keyword cascades is used. The
trajectories of energetic recoils are calculated in the same way as for the original ions. A
primary recoil with sufficiently high initial energy can generate more recoils (collision
cascade). While both methods yield correct range profiles, only the full-cascade simulation
produces physically correct profiles of vacancies and displaced atoms. However, a full-cascade
simulation requires more computational time.
In single-crystalline silicon, vacancies and recoils are often identified with the vacancies and
interstitials responsible for transient-enhanced diffusion (TED) of dopants. The choice of a
diffusion model determines whether the full-cascade mode of Crystal-TRIM must be applied.
3: Ion Implantation
Monte Carlo Implantation
156 Sentaurus™ Process User Guide
H-2013.03
Electronic Stopping
Electronic energy loss of the projectile is treated using semiempirical models.
For crystalline target materials, the loss depends on the local electronic density in the
environment of the projectile. Therefore, the use of a local approach is particularly important
for investigations of channeling effects in single-crystalline substrates.
A simplified local approach, the so-called modified Oen–Robinson formula [21], is used. The
parameter CEX1 describes the variation of the electron density for a projectile moving in the
<110> direction of the crystal, while CEX2 does the same for any other direction. The value for
CEX1 and CEX2 are set in the parameter database and can be changed by using:
pdbSet <material> <dopant> CEX1 <n>
The value for CEX1 should be close to 1 or at least within the range of 0.5 and 3. The default
value for CEX2 is 2.
The automatic calibration of these parameters can be switched off individually using
AutoCEX1, AutoCEX2.
Amorphous Materials
Nuclear Collisions
In amorphous materials, nuclear collisions are described by assuming that consecutive binary
collisions are completely uncorrelated. The only structural parameter that influences nuclear
scattering is the average interatomic distance in the target material, which determines the
maximum free flight-path length to the next collision and the maximum impact parameter.
By default, the impact parameter is assumed to be distributed uniformly between zero and its
maximum value. The free flight-path length is constant and equal to the average interatomic
distance. Alternatively, a slightly different description of the structure of the amorphous
material is possible using the switch AdvancedAmorph:
pdbSet <material> <dopant> AdvancedAmorph 1
In this case, the free flight-path length is assumed to have a half-Gaussian distribution above
the interatomic distance scaled with a value of the parameter AMAV. The standard deviation is
controlled by the parameter AMDEV.
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 157
H-2013.03
The default values are AMAV=1 and AMDEV=0, which correspond to setting AdvancedAmorph
0. The values of AMAV and AMDEV can be set in the parameter database:
pdbSet <material> <dopant> AMAV { <n> <n> <n> <n> }
pdbSet <material> <dopant> AMDEV { <n> <n> <n> <n> <n> <n> }
The set of parameters is given as an array. A pair of entries always specifies the number and
the value of the parameter, that is:
pdbSet Silicon Boron AMAV {0 -1.25e-4 1 0.93}
sets the two parameters for the calculation of AMAV to –1.25 and 0.93, respectively.
AMAV and AMDEV are made dependent on the atomic number of an implanted ion and its energy.
For arsenic, boron, and phosphorus in silicon, calibrated values are available in the parameter
database, and the AdvancedAmorph flag is set to 1. The calibration can overwrite external
settings of these parameters performed with pdbSet. To switch off the calibration, use:
pdbSet Silicon Boron AutoAMAV 0
A similar parameter is available for AMDEV.
NOTE This model should not be applied to implantation energies below 10–
20 keV. For low-energy implants, especially of boron, the default values
lead to wrong results. You should select AMAV and AMDEV manually,
where AMAV should be close to 1 and AMDEV should be a positive
number.
A single-crystalline material can also be treated as amorphous by setting:
pdbSet <material> Amorphous 1
Table 8 Values for AMAV and AMDEV used in Crystal-TRIM AdvancedAmorph mode
Energy [keV] B/BF2 P As
AMAV AMDEV AMAV AMDEV AMAV AMDEV
10 0.9287 0.0275 0.8888 0.0700 0.9490 0.0400
30 0.9262 0.0141 0.8963 0.0700 0.9490 0.0400
50 0.9237 0.0113 0.9038 0.0700 0.9490 0.0400
100 0.9175 0.0088 0.9225 0.0700 0.9490 0.0400
200 0.9050 0.0000 0.9600 0.0700 0.9490 0.0400
400 0.8800 0.0000 1.0350 0.0700 0.9490 0.0400
3: Ion Implantation
Monte Carlo Implantation
158 Sentaurus™ Process User Guide
H-2013.03
Electronic Stopping
A nonlocal approach based on the ZBL formula [18] is used for amorphous materials. This
formula uses an average density of electrons and has only one fitting parameter, Lambda. This
factor is used for the scaling of the ion-screening length in the ZBL electronic-stopping cross
section. Default values of Lambda are also set in the parameter database. Values of Lambda
close to 1 (between 0.7 and 1.5) are recommended. The automatic calibration of this parameter
can be switched off by using AutoLambda.
Damage Buildup and Crystalline–Amorphous Transition
The damage accumulation leading to de-channeling of ions and recoils, and the subsequent
crystalline–amorphous transition is described by a phenomenological model [22]. It can be
completely switched off by using:
pdbSet Ctrim DamageAccumulation No
This switches off both the damage accumulation and de-channeling. During the current implant
step, no additional damage will be produced and the existing predamage will have no effect.
Two other model options are available.
Full Amorphization Above a Critical Value
This model leads to full amorphization in mesh elements if the damage probability reaches a
critical limit and is chosen by setting:
pdbSet Ctrim DamageAccumulation Full
Below a certain threshold described by the parameter DCrit, the damage probability PD is
assumed to depend linearly on the nuclear energy deposition per atom ( ). The
proportionality factor is DAcc. If PD is greater than this value, the volume element is
completely amorphized and PD = 1, that is:
(104)
In most cases, DCrit should be less than DAcc to allow amorphization for high implantation
doses.
En
PD
DAcc En, DAcc En
⋅DCrit≤⋅
1 , DAcc En
⋅DCrit>
=
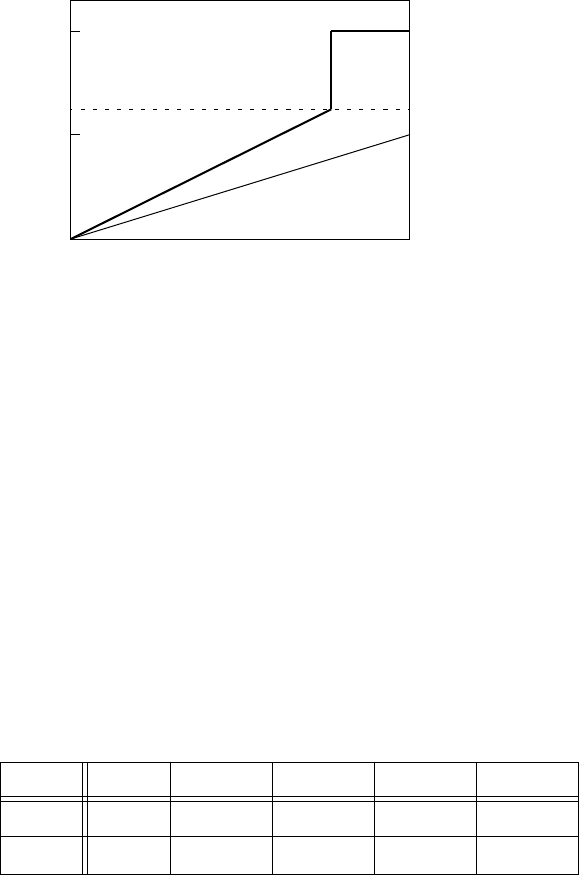
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 159
H-2013.03
Figure 32 Onset of amorphization after reaching DCrit for two values of DAcc
Saturation at Critical Value
pdbSet Ctrim DamageAccumulation Saturation
According to this model, below a threshold value DCrit, the damage probability PD has the
same linear behavior as in the ‘full’ model, but cannot grow above DCrit:
(105)
For both models, the values of the parameters DAcc and DCrit depend mainly on the atomic
number of the implanted ion.
Table 9 lists values for some species. The automatic calibration for these values can be
switched off using AutoDAcc and AutoDCrit.
Internal Storage Grid for Implantation Damage
By default, Crystal-TRIM stores the accumulated damage at the mesh. This makes the damage
accumulation dependent on the mesh and can lead to errors if the mesh is too coarse. Typically,
the amorphous boundary depends nonlocally on the mesh size closer to the surface of the
structure.
Ta bl e 9 Va l u es fo r DAc c an d D Cr it fo r m o s t i m po r t an t sp ec i es
BF2 As, Ga, Ge In, Sb, Sn B, C, N Al, P, Si
DAcc 0.15 0.3 0.3 0.1 0.2
DCrit 0.02 0.05 0.05 0.99 0.1
Vacancy Concentration
1.0
0.5
0.0
Damage Probability PD
DCrit
DAcc=0.8
DAcc=0.5
PD
DAcc En, DAcc En
⋅DCrit≤⋅
DCrit , DAcc En
⋅DCrit>
=
3: Ion Implantation
Monte Carlo Implantation
160 Sentaurus™ Process User Guide
H-2013.03
An internal grid can be used to accumulate and store as-implanted damage. This can be
switched on by using:
pdbSet Ctrim UseInternalGrid 1
This allows for a mesh-independent storage of the damage information. You can control the
grid spacing by using:
pdbSet Ctrim InternalGridSpacing <n>
In each grid cell, the accumulated as-implanted damage and the amorphization flag are stored.
During postprocessing, the accumulated as-implanted damage is transferred (interpolated)
from the internal grid to Sentaurus Process elements and then to Sentaurus Process nodes.
If two or more Crystal-TRIM steps directly follow each other, you can choose to leave the as-
implanted damage stored on the internal grid instead of transferring it to the mesh. The switch
keepdamage.igrid must be used within the implant command:
implant As crystaltrim keepdamage.igrid
The default is !keepdamage.igrid.
NOTE If keepdamage.igrid is used, the Sentaurus Process Damage dataset
will not be incremented in the postprocessing. If the subsequent
processing step is not a Crystal-TRIM implantation, the damage
information will be lost.
To access the damage information, which was stored on the internal grid during the previous
Crystal-TRIM implantation, the switch predamage.igrid (default is !predamage.igrid)
must be used within the implant command: An example is:
# first step
# no damage post-processing, keep damage on internal grid for the following
# steps
implant As crystaltrim keepdamage.igrid
# second step
# use pre-damage on igrid from the previous step
# no damage post-processing: one more step follows
implant B crystaltrim predamage.igrid keepdamage.igrid
# third step
# use pre-damage on igrid
implant P crystaltrim predamage.igrid
NOTE It is not possible to save the information from the internal grid to a file
using the struct command after the current Crystal-TRIM step.

3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 161
H-2013.03
Molecular Implantations
The MC method allows for the implantation of molecular ions or atomic cluster species such
as BF2. The assumption is that the molecule immediately breaks up into its constituents upon
impact on a solid surface.
Figure 33 Schematic of molecular implantation of BF2; one trajectory is calculated
for each atomic species, w is the weight assigned to the species
This is a valid approximation if the binding energy of the molecule is considerably smaller than
the implant energy (for example, for BF2: EB ~ 9 eV).
The implant energy is shared by several atoms according to:
(106)
where is the energy of the -th species, is the atomic mass, and is the statistical
weight according to the stoichiometry of the molecule.
The constituents move as separate particles. However, particles of different species are not
completely independent because of the interaction through the implantation damage.
Sentaurus Process supports several molecular species: BF2 (BF2), BCl2 (BCl2), B18H22
(B18H22), AsH2 (AsH2), and PH2 (PH2). An implantation of B18H22, for example, can be
performed with:
implant B18H22 energy=100 [ctrim | tmc]
BF2+
F (w=2)
B (w=1)
Ta r g et Surfa ce
Eienergy Mi
wjMj
j
-------------------⋅=
Ei
i
Mi
wj

3: Ion Implantation
Monte Carlo Implantation
162 Sentaurus™ Process User Guide
H-2013.03
The atomic masses, statistical weights, and the molecular composition are available in the
parameter database. A full molecular implantation is performed if the keyword
full.molecular is explicitly set, that is:
implant <dopant> [ctrim | tmc] full.molecular
In this case, the trajectories for all atomic species are calculated. At the end of the simulation,
datasets for each ballistic constituent of the original molecule are generated. This is the default.
Only the trajectory of the significant species (B in the case of BF2) is calculated
if !full.molecular is chosen.
NOTE This feature is available to both Sentaurus MC and Crystal-TRIM.
MC Implantation into Polysilicon
Polysilicon has three states of crystallinity:
■Amorphous
■Crystalline
■Polycrystalline
By default polysilicon is considered amorphous and can be treated as a single-crystalline or
polycrystalline material by using:
pdbSet PolySilicon Crystallinity Crystalline
pdbSet PolySilicon Crystallinity Polycrystalline
Crystal orientation (one of 100, 110, and 111) can be specified using the material-specific pdb
command:
pdbSet PolySilicon CrystalOrient 110
This command sets the crystal orientation for all polysilicon regions to <110>.
If the crystallinity is set to Polycrystalline, MC implantation checks for the existence of
the GSize dataset or GrainSize parameter. If neither is found, the grain model is disabled,
and the material is assumed to be single crystalline. The default grain size is [cm],
and can be changed with the command:
pdbSet PolySilicon GrainSize <n>
or by initializing the GSize dataset:
select z=<n> name=GSize
The units for both GrainSize and GSize are centimeters.
510
5–
×

3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 163
H-2013.03
The grain size also can be scaled with the GrainFactor parameter:
pdbSet PolySilicon GrainFactor <n>
The default GrainFactor is 1.
The polycrystalline model works by frequently switching between the crystal algorithm and
the amorphous algorithm. The probability of switching from the crystal model to the
amorphous model is determined by the accumulative path length in crystal (pathlength) and
polycrystalline grain size (GrainSize). It switches from the crystal model to the amorphous
model if:
(107)
where is a random number between 0 and 1. The polycrystalline model shares the same
random number sequence with other modules in MC implantation. Therefore, if the random
seed is reset, the random numbers used in the polycrystalline model are changed as well.
After an amorphous collision is processed, the pathlength is reset to zero, and the crystal
model is selected. The pathlength is accumulated again. The model used for the next
collision is again determined by the same rules. This process is repeated until the particle exits
the polycrystalline region.
NOTE This feature is available to both Sentaurus MC and Crystal-TRIM.
MC Implantation into Compound Materials with Molar
Fractions
A compound material with a spatially dependent molar fraction can be defined in the PDB. For
example, for single-crystalline silicon, the following PDB entry:
array set $Base {BinaryCompounds {String {
{ SiliconGermanium GeTotal "GeTotal/[pdbGetDouble Si LatticeDensity]" }
}}}
specifies a binary compound Si1–xGex with the mole fraction of Ge calculated from the
germanium concentration (GeTotal) divided by the silicon lattice density. This is the default
setting for SiliconGermanium in the PDB. MC models support implants into these
compound materials (binary, ternary, and quaternary).
Compound materials are detected automatically by using CompoundNumber,
BinaryCompound, TernaryCompound, QuaternaryCoumpound in the PDB. Due to more
computational demands, a minimum concentration is required to trigger MC implantation
pathlength Rrand GrainFactor GrainSize⋅()⋅>
Rrand
3: Ion Implantation
Monte Carlo Implantation
164 Sentaurus™ Process User Guide
H-2013.03
models to treat this material in a more sophisticated way. To specify this minimum
concentration, for example, use:
pdbSet Silicon SiliconGermanium.MCmin 1e20
If the concentration of GeTotal in any mesh node of silicon regions is greater than or equal to
, MC implantation model treats this material as a binary compound
SiliconGermanium. In this case, the average charge and mass of the material are calculated
individually for each mesh element. Lattice constant, nonlocal electron stopping power, and
Debye temperatures are interpolated linearly based on the mole fractions. The lattice is
constructed with the primary material, and each lattice site is assigned to a type of atom with
probability proportional to their mole fractions.
Sentaurus MC considers the fact that each specific lattice site will be occupied with certain
types of atoms only. Therefore, the substitution of the lattice atoms occurs only for those with
the same Group number. The default group number for each type of atom is the same as that
in the periodic table. To change the group number, for example, use:
pdbSet ImplantData Carbon Group 4
For example, in compound material Si1–x–yGexCy, Ge and C have the same group number (IV)
as Si, so both of them can substitute silicon atoms in its lattice sites.
Another example is InxGa1–xAs1-yPy; In and Ga belong to the same group (III), and As and P
belong to the same group (V). Suppose Ga occupies site 0, and As site 1 in zinc-blende
structures, then In can only occupy site 0, and P site can only occupy 1 with the occupation
probabilities proportional to their mole fractions.
Generally MC implant parameters, LatticeConstant, DebyeTemperature, and LSS.pre
vary with the mole fraction. In previous releases, the mole–fraction dependence of MC implant
parameters in compound material is assumed to be linear between base materials. In Version
H-2013.03, there are more options for II-V compounds:
■If a physical parameter is not specified in a ternary (or quaternary) material, the parameter
value is extracted by the linear interpolation with the parameter values of their base binary
materials. This is the default behavior.
■If the parameter is defined for the compound material, the given parameter value is used.
■If <parameter>.XTable is defined for compound material, the piecewise linear
interpolation specified with this table is used. For example:
pdbSetDoubleArray InGaAs LatticeConstant.XTable {<x1> <v1> ... <xn> <vn>}
■If <parameter>.XTable is undefined for compound material, <parameter>.X2 can be
used to define a quadratic interpolation. For example:
pdbSetDouble InGaAs LatticeConstant.X2 <n>
110
20
× cm 3–

3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 165
H-2013.03
To use these new parameter interpolation options, turn on the following switch:
pdbSet MCImplant Compound.Interpolation 1
By default, linear interpolation of physical parameters is used.
NOTE Although this feature is available to both Sentaurus MC and Crystal-
TRIM, you are strongly encouraged to use Sentaurus MC implant for
better results.
MC Implantation into Silicon Carbide
Sentaurus MC supports ion implantation into crystalline silicon carbide (SiliconCarbide)
with hexagonal lattice. The hexagonal system has four crystallographic axes: three -axes
(, , ) forming a plane, and a -axis that is normal to the plane. The crystallographic
planes and directions normally are described with four Miller indices (hkil). For the hexagonal
system, since the sum of the first three indices is zero, the third index sometimes can be
omitted.
Silicon carbide exists in many different crystal structures, called polytypes. All polytypes have
a hexagonal frame with a carbon atom situated above the center of a triangle of Si atoms and
underneath a Si atom belonging to the next layer. The difference among the polytypes is the
stacking sequence between the succeeding double layers of carbon and silicon atoms. For
example, 2H-SiC, 4H-SiC, and 6H-SiC have the AB, ABCB, and ABCACB stacking
sequences, respectively. 3C-SiC has an ABC stacking sequence and is the only form of SiC
with a zinc-blende crystal lattice structure. The default polytype for SiC is 4H. To change to a
different polytype, use the following command:
pdbSet SiliconCarbide Polytype {2H 3C 4H 6H}
NOTE The lattice constants may be different for different polytypes. For
convenience, Tcl procedures (set2H-SiC, set3C-SiC, set4H-SiC,
and set6H-SiC) are provided to set to different SiC polytypes.
Two silicon carbide wafer orientations (<0001> and <11-20>) are supported. To specify these
wafer orientations, use the pdb command:
pdbSet SiliconCarbide CrystalOrient {0001 1120}
The default wafer orientation is <0001>. For (0001) SiC wafer; the primary flat orientation is
<10-10>. For (11-20) SiC wafer, the primary flat orientation is <0001>.
For details of the model and comparison with experimental data for various implant conditions,
see [24].
a
a1
a2
a3
c

3: Ion Implantation
Monte Carlo Implantation
166 Sentaurus™ Process User Guide
H-2013.03
A miscut of – typically exists in SiC (1000) wafers. Sentaurus MC implantation takes
into account this wafer miscut by specifying caxis.tilt and caxis.rotation in the init
command. caxis.tilt is the angle by which the wafer normal is tilted with respect to the
a-axis in the crystal coordinate system. caxis.rotation is the angle that specifies the
direction into which the wafer normal is tilted. The default value of caxis.rotation is 0,
that is, the projection of the wafer normal to the crystal plane, formed by the b-axis and c-axis,
is coincidental to the <110> direction in silicon. If caxis.rotation=90, the wafer normal is
tilted by caxis.tilt towards the right with respect to the crystal coordinate system. (Or, in
terms of the simulation coordinate system, if caxis.rotation=90, the crystal coordinate
system is tilted towards the left with respect to the wafer normal.) By default, there is no wafer
miscut, that is, caxis.tilt=0.
Here is a simple example illustrating how to perform an MC implantation in SiC:
# Set up the structure
line x loc=0.0 tag=oxtop spac=0.001
line x loc=0.0015 tag=top spac=0.001
line x loc=0.5 spac=0.0025
line x loc=2.0 tag=bot spac=0.01
region Oxide xlo=oxtop xhi=top
region SiliconCarbide xlo=top xhi=bot
# Specify wafer miscut
init caxis.tilt=4 caxis.rotation=0
# Choose different polytype (default is 4H-SiC)
set6H-SiC
# Do the implantation
implant Aluminum energy=60 dose=1e13 tilt=0 rot=0 sentaurus.mc \
particles=10000 info=2
# Save the result
struct tdr=sic
NOTE This feature is available to Sentaurus MC only.
Recoil Implantation
Sentaurus MC implantation provides a general model for recoil implant, such as an oxygen
knock-on effect. Generally, recoil species are handled the same way as cascade atoms, except
that no vacancies are created at the displaced sites and the recoil species are not recorded as
interstitials when they stop. Instead, a separate dataset is created for each recoil species.
3.5°
8.5°

3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 167
H-2013.03
The recoil species is specified in the material composition. For example, to simulate the oxygen
knock-on effect, the following is defined in the parameter database:
Oxide -> Composition -> Component0 -> Name = Silicon
Oxide -> Composition -> Component0 -> StWeight = 1
Oxide -> Composition -> Component1 -> Name = Oxygen
Oxide -> Composition -> Component1 -> StWeight = 2
Oxide -> Composition -> Component1 -> Recoil = 1
To initiate oxygen recoil implant simulation, you must specify the keyword recoils in the
implant command:
implant <dopant> energy=<n> dose=<n> recoils
The datasets Oxygen_Implant and Oxygen are created, which contain displaced oxygen
distributions that can be used to analyze the oxygen knock-on effect.
NOTE This feature is available to Sentaurus MC implantation only.
Plasma Implantation
Three-dimensional tri-gate devices (FinFETs) have been employed at the 22 nm node and are
expected to continue at and beyond the 16 nm node. Doping of FinFETs must be 3D, and
conformal doping with plasma implantation (PLAD) is a promising approach. Likewise,
doping of planar devices is challenging, and PLAD offers capabilities not available in beamline
implantations.
To offer this simulation capability, Sentaurus Process provides a PLAD doping module that
accurately reflects both the hardware and process signatures as well as the physical properties
of the associated deposition, etching, sputtering, implantation, knock-on, defect creation, and
annihilation processes. This MC implantation module includes the following features:
■Perform alternating steps of deposition and MC implantation. The number of steps can be
specified by users.
■Deposition of material on the surface is performed isotropically (that is, constant growth
rate over the surface). The thickness is specified by users. A minimum thickness is imposed
by the program, which reduces the number of steps if necessary to prevent the deposition
of a layer that is too thin. The deposit material should be defined as usual, and material
composition of the layer must be specified by users.
■The MC implantation module allows the specification of multiple ions incidents on the
wafer. The ion species should be defined before implantation as usual, and some typical ion
species used in plasma implantation will be predefined. You can specify the dose, energy
distribution, and angular distribution of each ion species. The dose for each ion is applied
evenly for each step.
3: Ion Implantation
Monte Carlo Implantation
168 Sentaurus™ Process User Guide
H-2013.03
■An empirical model for conformal doping, in which the level of conformity can be
specified by users.
■In addition to computing the concentration of ions that penetrate through the deposited
overlayer, the MC implantation module allow for atoms to be knocked out of the overlayer
and into the wafer and tracks damage and amorphization as usual.
You mu st d efi ne the plasma so urc e bef or e impl an tatio n c an b e p erfo rm ed . To avo id ove rly
complex syntax in the implant command, Sentaurus Process provides two ways to specify the
plasma source: simple source and complex source.
Simple Source
Assuming that the multiple ion species in plasma have the same energy and angle distributions,
simply specify the multiple species as a list in the implant command (other parameters such
as dose, energy, tilt, and so on can be specified like a regular implantation):
plasma.source = {<species1>=<n> <species2>=<n> <species3>=<n> ...}
where:
■plasma.source specifies a list of ion species to be implanted. These species must be
predefined in ImplantData as usual.
■The number after each species is the fraction of the total dose (as specified by the dose
parameter) for the given species.
■All these species will have the same energy, tilt, en.stdev, and tilt.stdev as
specified.
Complex Source
In more complex cases, different species may have different energy and angle distributions. In
this case, each species can be specified with their own implantation parameters (energy,
tilt, en.stdev, and tilt.stdev). So for each species, define it with an implant
command:
implant species=<species1> energy=<n> tilt=<n> en.stdev=<n> tilt.stdev=<n>
implant species=<species2> energy=<n> tilt=<n> en.stdev=<n> tilt.stdev=<n>
implant species=<species3> energy=<n> tilt=<n> en.stdev=<n> tilt.stdev=<n>
...
where <species1>, <species2>, <species3> must be predefined in ImplantData as
usual. Only parameters that are different from the default values must be specified.
Then, you can perform the real implantation in the same way as in the simple source case:
implant plasma.source= {<species1>=<n> <species2>=<n> ...} dose=<n> energy=<n>
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 169
H-2013.03
tilt=<n> en.stdev=<n> tilt.stdev=<n> ...
where:
■plasma.source specifies a list of ion species to be implanted. These species must be the
same as those in previous implant commands.
■The regular implantation parameters (dose, energy, and so on) will be the default for
those species that are not specified. Essentially, this syntax is consistent with that for the
simple source and reduces to the simple source if no implantation parameters are specified
for each individual species.
Deposition of Material
To specify the deposition of the material during implantation, use the plasma.deposit
parameter in the implant command:
implant plasma.source= {<species1>=<n> <species2>=<n> ...}
plasma.deposit= {material=<c> thickness=<n> steps=<n>}
dose=<n> energy=<n> tilt=<n> ...
where:
■material is the name of the material to be deposited, which must be specified before the
implantation.
■thickness is the total thickness of the deposit material.
■steps is the number of steps of deposition.
The deposition and implantation are performed alternatively. If plasma.deposit is not
specified, or material is not specified, or thickness is not specified (or is equal to zero),
no deposition is performed.
Knock-on and Knock-off Effect
The MC implantation module simulates the dopant knock-on and knock-off effect by
specifying the recoils parameters in the implant command. In addition, you must specify
the recoil species to be simulated in material composition.
For example, assuming the deposit material is BHx, and Atom0 is Boron, the following
commands specify Boron as a recoil species:
pdbSetString BHx Composition Atom0 Name Boron
pdbSetDouble BHx Composition Atom0 StWeight 1
pdbSetBoolean BHx Composition Atom0 Recoil 1
pdbSetString BHx Composition Atom1 Name Hydrogen
pdbSetDouble BHx Composition Atom1 StWeight <x>
3: Ion Implantation
Monte Carlo Implantation
170 Sentaurus™ Process User Guide
H-2013.03
Conformal Doping
Conformal doping is an important characteristic in plasma implantation. However, due to the
complexity of plasma dynamics that involves manybody long-range interactions, the exact
mechanism for conformal doping in plasma implantation is still not well understood.
Physically, this may be possible if one of the following mechanisms or their combination
occurs, for example, in a trench:
■The ions become ionized anywhere in the ambient (including inside the trenches) and start
their acceleration towards the silicon surface.
■The ions scatter off other particles in the plasma/ambient (including inside the trenches)
and change their direction.
To account for such effects, an empirical model has been developed that is compatible with the
current plasma implantation. In this model, instead of launching all ions from above the device,
as in standard implantation, some ions are launched along the device surface (that is, the solid–
ambient interface). A fraction of ions launched along the surface can be specified by the
parameter conformity in the implant command:
implant <dopant> energy=<n> dose=<n> conformity=<n> sentaurus.mc
where conformity is a number between 0 and 1.0. For example, if conformity=0, you will
obtain standard plasma implantation results, and if conformity=1, you will obtain fully
conformal doping.
Other Plasma Implantation–related Parameters and Procedures
Sentaurus Process provides a simple model for taking into account the energy and tilt angle
distributions of the plasma source. Given the mean and standard deviation of the implantation
energy and tilt angles, Sentaurus MC implantation samples the given energy and tilt
distributions for each implantation particle. After the implantation energy and tilt angle are
determined, the particle tracing is computed using a standard procedure.
In addition to the normal implantation parameters, such as energy and tilt, you can specify the
standard deviation of implantation energy (en.stdev) or the standard deviation of the tilt
angle (tilt.stdev) or both. For example:
implant <dopant> plasma dose=<n> energy=<n> en.stdev=<n> tilt=<n>
tilt.stdev=<n> sentaurus.mc
where the implantation parameters energy and tilt are the mean energy and mean tilt,
respectively.
You a lso c an spe ci fy t he mi nimum e ne rg y th at is allowed for implantation using the command:
pdbSet MCImplant MinEnergyCutoff <n>
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 171
H-2013.03
Energy below the minimum energy will be truncated. The default minimum energy is zero. In
addition, you can specify the maximum energy that is allowed by using the command:
pdbSet MCImplant MaxNumStdevCutoff <n>
MaxNumStdevCutoff must be an integer (default is 5). The highest energy for a given implant
should not exceed the mean energy by the amount of en.stdev x MaxNumStdevCutoff.
Energy higher than this number will be truncated.
Energy Distributions
The energetic distribution of different molecular and atomic ions, after extraction from the
plasma, is known to cover the range from zero to the maximum energy , which is equal
to the product of the ion charge multiplied by the extraction voltage. Sentaurus Process allows
easy selection and addition of various energy distribution models. In addition to the default
Gaussian distribution, Sentaurus Process implements an alternative Burenkov model [25]. In
this model, the energy distribution, as presented by Tian et al. [26] as an integral number of
particles having their energy in a given interval, can be written in a differential form as
follows [25]:
(108)
The energy distribution presented in Eq. 108 is normalized, that is, the integral over all
possible energies of the extracted ions, ranging from 0 to , is equal to one. Burenkov et
al. have shown that by using the energy distribution given by Eq. 108, excellent agreement can
be obtained between simulations and experiments for BF3 plasma implantation [25].
Tilt Angle Distributions
Three different tilt angle distributions are offered in plasma implant. You can select a different
tilt angle distribution model by using:
pdbSet MCImplant PlasmaEnergyDistributionModel\
{Gaussian | Gaussian.Solid.Angle | Gaussian.3D}
The default Gaussian model:
(109)
This is a first order approximation in which the number of particles per solid angle has a sharp
maximum at tilt=0. This may not be ideal in certain situations.
Emax
fE() 5
6Emax
⋅
-------------------E
Emax
-----------
16/–
⋅=
fE()
Emax
ftilt() 1
2πσ
-------------- 1
2
---tilt
σ
------
2
–
exp=

3: Ion Implantation
Monte Carlo Implantation
172 Sentaurus™ Process User Guide
H-2013.03
The Gaussian.Solid.Angle is:
(110)
Here, the additional factor tilt considers that the solid angle which corresponds to a tilt angle
is proportional to the tilt angle. Note that the standard deviation ( ) has a slightly different
meaning from regular Gaussian distribution. In other words, only 1-1/e = 63.2% of ions will be
in the interval [0, ].
For Gaussian.3D (suggested by Burenkov et al.), you must add a random 3D vector, in which
each component has a random Gaussian distribution to the vertical unit vector, and calculate
the corresponding tilt angle. In this case, the knock-on process in the plasma may lead to
additional ion energy which has Gaussian distribution. Here again, has a different meaning
from standard Gaussian distribution; in other words, only ~40% of ions are within the interval
[0, ].
To select a different energy distribution model, use:
pdbSet MCImplant PlasmaEnergyDistributionModel {Gaussian | Burenkov}
To provide maximum flexibility for energy and tilt angle distributions, you also can provide
your own distributions by modifying the following Tcl procedures in Plasma.tcl:
Plasma::Energy_Distribution { energy stdev }
Plasma::Tilt_Distribution { mean stdev }
NOTE Sentaurus Process does not check the validity of these user-defined
distributions. If you change these distributions, you must provide the
correct distributions to ensure the correct implantation results.
NOTE This feature is available to Sentaurus MC implantation only.
MC Implantation Damage and Point-Defect Calculation
Sentaurus MC Damage Calculation
The damage in a Sentaurus MC implantation is computed either using the Kinchin–Pease
formula [22] (default) or with full cascades if the cascades parameter is specified in the
implant command. For details on damage calculations, see Damage Accumulation and
Dynamic Annealing on page 149. The calculation of the damage datasets for Sentaurus MC is
ftilt() tilt
2πσ
-------------- 1
2
---tilt
σ
------
2
–
exp=
σ
σ
σ
σ
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 173
H-2013.03
consistent with the analytic implantation model. At the end of an implantation step, the damage
for this step (Damage_LastImp) is added to the Damage profile using:
(111)
where + indicates the total damage as the sum of new damage and existing damage. The default
value for MCDFactor is 1 and can be changed at the implant command line or in the
parameter database:
implant <dopant> [tmc] [mc.dfactor=<n>]
pdbSetDouble <material> <dopant> MCDFactor <n>
The accumulated damage is taken into account automatically for subsequent MC
implantations, unless the Damage dataset is reset by the diffuse command.
Crystal-TRIM: Damage Probability
Defect accumulation in single-crystalline material and de-channeling due to the implantation
damage are treated dynamically using a phenomenological model [22]. The model is based on
the assumption of the formation of complex defects, such as amorphous pockets (APs) during
ion implantation.
The damage information calculated during Crystal-TRIM simulations is stored in the damage
probability dataset PD. This information is used in the model of APs to treat the nuclear
collisions in the partially damaged crystalline region. Within a certain volume element, PD
gives the probability that the collision can be treated as if the material were amorphous. The
material is locally considered to be completely amorphized if PD = 1.
The PD dataset is not deleted after an implant step and, consequently, can be reused in
subsequent Crystal-TRIM runs (damage history).
If the PD dataset has been deleted or has not yet been created, the Damage dataset is used to
initialize the damage history in all crystalline materials:
(112)
where Threshold is the amorphous threshold, which is the minimum of the lattice density and
amorphous density. Both parameters can be set in the parameter database:
pdbSet <material> LatticeDensity <n>
pdbSet <material> AmorpDensity <n>
At the end of the simulation, the Damage dataset is increased according to:
(113)
Damage += MCDFactor Damage_LastImp⋅
PD = Damage / Threshold, max(PD) = 1
Damage = Damage + PD_LastImp Threshold⋅
3: Ion Implantation
Monte Carlo Implantation
174 Sentaurus™ Process User Guide
H-2013.03
Point Defects
Elemental Material
Point-defect profiles after a MC run can be generated from the ballistic dopant profile using the
plus.one or effective.plus.n model, or from the ballistic vacancy (Frenkel pair) and
recoil profiles using the frenkel.pair model. The effective.plus.n model is the
default for all MC simulations. In this case, the ballistic dopant profile <dopant>_LastImp
is used according to Eq. 50, p. 118.
Interstitial and vacancy profiles can also be calculated using the ballistic vacancy dataset
Vac_LastImp generated during a MC run. The switch defect.model must be set to
frenkel.pair:
implant <dopant> [crystaltrim | sentaurus.mc] [defect.model=frenkel.pair]
Using the MC-specific factors MCIFactor and MCVFactor, the profiles are calculated
according to:
(114)
The default values for MCIFactor, MCVFactor, and IFactor are 1. These factors can be
changed in the parameter database:
pdbSet <material> <dopant> IFactor <n>
pdbSet <material> <dopant> MCIFactor <n>
pdbSet <material> <dopant> MCVFactor <n>
or on the command line:
implant <dopant> [crystaltrim | sentaurus.mc] [ifactor=<n>] [mc.ifactor=<n>]
[mc.vfactor=<n>]
Setting ifactor, mc.ifactor, and mc.vfactor in the implant command overwrites the
parameter database entries.
If cascades is enabled in MC implantation and the point-defect model is set to
frenkel.pair, the interstitial and vacancy profile is calculated using the concentration of the
recoil and vacancy as calculated based on the physics model:
implant <dopant> [crystaltrim | sentaurus.mc] [cascades] \
[defect.model=frenkel.pair]
Int_Implant += MCIFactor Vac_LastImp IFactor <dopant>_LastImp⋅+⋅
Vac_Implant += MCVFactor Vac_LastImp⋅
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 175
H-2013.03
In this case, the interstitial and vacancy densities increase according to the following:
(115)
Multicomponent Materials
In multicomponent materials, such as silicon carbide (SiC), the material is composed of
different types of atom. When an impurity is implanted into SiC, both silicon and carbon lattice
atoms can be displaced, thereby forming silicon interstitials or carbon interstitials, and leaving
behind silicon-site or carbon-site vacancies. Instead of classifying them together as interstitials
or vacancies, as in silicon, Sentaurus Process provides a mechanism to distinguish different
types of interstitial or vacancy.
To generate distinct types of point defect in multiple-component materials, you must switch on
the DistinctDefects flag, for example:
pdbSetBoolean SiliconCarbide DistinctDefects 1
By default, this flag is true for SiC but false for other materials. As a result, instead of
Int_Implant and Vac_Implant, the generated point-defect datasets in SiC are
IntSilicon_Implant, IntCarbon_Implant, VacSilicon_Implant, and
VacCarbon_Implant.
In this model, the total point-defect concentration is computed the same way as the elemental
material. The implantation parameters defect.model, ifactor, vfactor, mc.ifactor,
and mc.vfactor in the implant command still work. ifactor and vfactor are scaling
factors for interstitial profiles and vacancy profiles, respectively, in the plus.one defect
model; while mc.ifactor and mc.vfactor are scaling factors for interstitial profiles and
vacancy profiles, respectively, in the frenkel.pair defect model. The same Tcl procedure
CalcPlusNFactor calculates automatically the plus factors for the effective.plus.n
defect model.
Then, the individual point-defect concentration is computed by multiplying the total point-
defect concentration by the fraction of each component. The fraction of each component is, by
default, their stoichiometric weight, but it can be changed in the parameter database with the
parameters IFractor.Fraction and VFactor.Fraction.
For example, in SiC:
pdbSet SiC Composition Component0 IFactor.Fraction <n>
pdbSet SiC Composition Component1 IFactor.Fraction <n>
pdbSet SiC Composition Component0 VFactor.Fraction <n>
pdbSet SiC Composition Component1 VFactor.Fraction <n>
Int_Implant += <recoil>_LastImp <dopant>_LastImp+
Vac_Implant += <vacancy>_LastImp

3: Ion Implantation
Monte Carlo Implantation
176 Sentaurus™ Process User Guide
H-2013.03
If cascades is enabled in MC implantation and the point-defect model is set to
frenkel.pair, the interstitial and vacancy profile is calculated using the concentration of the
recoils and vacancies as calculated based on the physical model.
In this case, the interstitial and vacancy densities increase according to the following:
(116)
Statistical Enhancement
The energetic pseudoparticles in a MC simulation are statistical objects representing several
actual particles or only a fraction of an actual particle. Pseudoparticles start their motion at a
plane above the target parallel to the wafer surface. The starting surface is subdivided into
segments of equal size. The size of these segments can be controlled by setting dy and dz in
the MCImplant -> Intervals entry in the parameter database.
The number of pseudoparticles can be set by using:
pdbSet MCImplant Particles <n>
or:
implant <dopant> [crystaltrim | sentaurus.mc] [particles=<n>]
The default value for particles is 1000. The random number generator can be started with
a specified random seed. The integer value used can be set with the parameter RandomSeed:
pdbSet MCImplant RandomSeed <n>
The default is 1. Random seeds also can be chosen randomly by using the internal clock,
thereby giving different results for different runs. This feature is useful for statistical analysis
for MC implantations. To use this feature, use the following command:
pdbSet MCImplant Randomize 1
Trajector y Splittin g
Trajectory splitting artificially increases the number of trajectories calculated in regions with
low trajectory density. It can be switched on or off by using:
pdbSet MCImplant TrajectorySplitting 1
Int<component>_Implant += <component>_LastImp IFactor.Fraction <dopant>_LastImp⋅+
Vac<component>_Implant += <vacancy-component>_LastImp
3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 177
H-2013.03
If a projectile reaches an element with a small trajectory density, a split point is set, that is, the
particle is replaced by two daughter particles having half the statistical weight of the mother
projectile. Then, the trajectories of both daughter particles are simulated in the same manner
as for the original particle. Further splitting may occur that leads to a splitting tree related to
the mother projectile. At a split point, the two daughter projectiles start under identical
conditions.
However, the consideration of thermal vibrations of target atoms leads to a deviation of the
trajectories of the daughter projectiles after a few collisions. In this manner, a high number of
different particle trajectories with low statistical weight is obtained, which leads to an
important decrease of the statistical noise in the tail parts of the dopant distribution.
In Sentaurus MC, the maximum depth of the splitting tree is defined by a global parameter
MaxSplitLevels:
pdbSet MCImplant MaxSplitLevels <n>
In Crystal-TRIM, a similar parameter is defined for each species. For a given ion species, the
maximum depth of the splitting tree is defined by the parameter MaxSplits:
pdbSet <material> <dopant> MaxSplits <n>
pdbSet <material> <dopant> MaxSplitsPerElement <n>
The parameter MaxSplitsPerElement defines the maximum number of split events within
one element.
NOTE The trajectory splitting model is available to both Sentaurus MC and
Crystal-TRIM.
Dose Split
In the conventional pseudoparticle Monte Carlo approach, all particles have the same
weighting. In contrast, dose split algorithm uses smart particle weighting with first-coming
ions weighing less than later ions. This prevents crystalline from amorphizing too quickly,
thereby allowing more ions to enter the channeling regions. This model can drastically reduce
the noises of the channeling tails. By default, dose split is switched off. To activate the model,
use the command:
pdbSet MCImplant DoseSplit 1
The dose split model is especially effective for high-dose amorphizing implants, such as
arsenic implant with a dose of . For a typical run, the CPU time is about 2 to 3
times slower than that without dose split for the same number of particles. However, dose split
improves the statistics in the channeling tails by at least two orders of magnitude.
815
×10 cm 2–
3: Ion Implantation
Monte Carlo Implantation
178 Sentaurus™ Process User Guide
H-2013.03
To achieve the same statistical significance, the conventional approach requires at least 100
times more particles; this means that the effective speedup is about 30 to 50 times.
NOTE The dose split model is available to Sentaurus MC only.
Trajectory Replication
The trajectory replication algorithm uses the fact that in almost all 2D or 3D target structures,
several regions with 1D topology can be found. A particle trajectory going through such a part
can be copied many times by shifting its origin. Within the 1D region, each shifted trajectory
is a valid particle trajectory. Its reproduction by copying is much faster than its physical
calculation.
The subdivision into 1D parts or equivalence classes is performed automatically during the
implantation. A subdivision is performed using the segments of the start surface. At the
beginning, all the start segments are in the same equivalence class.
First, the whole trajectory tree is calculated including splits and recoils. The increments of all
concentration-type values between entering and leaving a grid element are stored for each
trajectory point (at least one per grid element). A start segment is chosen from the same
equivalence class. The starting point of the copy trajectory is set randomly within this start
segment. The point where the copied trajectory enters the material is found in the same way as
for the original trajectory. The vector between the first material point of the master and the
copied trajectory serves as a shift vector. All the increments of the master trajectory are
transferred point by point into grid elements that correspond to the shifted points.
If the materials are not identical within geometry tolerance ( ), or the initial
damage is different by more than 1% at the master and the replica points, replication fails and
the start segment is taken out of the present equivalence class and placed into a new equivalence
class. The generation of new equivalence classes stops after a certain number of particles has
been implanted. These initial particles are called probing ions.
Due to the random nature of ion trajectories, for the same structure, the equivalent classes as
discovered by the probing ions could be slightly different depending on the random seeds,
implanted species, or the number of probing ions.
The number of probing ions is empirically set to the total number of start segments. However,
depending on the situation, this number may be too small for 2D simulations; whereas, it may
be too large for 3D simulations. You can control this number by using the command:
pdbSet MCImplant ReplicationLearningFactor <n>
After this command, the new number of probing ions will be equal to the original number of
probing ions multiplied by ReplicationLearningFactor. Generally, the more probing
ions, the more equivalent classes will be created for a given structure. More equivalent classes
1.5 4–
×10 µm

3: Ion Implantation
Monte Carlo Implantation
Sentaurus™ Process User Guide 179
H-2013.03
will reduce the ratio of the replicated trajectories to the calculated trajectories, thereby
providing more accurate results at the expense of more CPU time.
The total number of implanted particles is given as the number of start segments multiplied by
the number of particles per segment, which can be set by using the parameter particles. Due
to the replication, the number of physically calculated trajectories is usually much smaller and
is given rather by the number of equivalence classes multiplied by particles.
The trajectory replication algorithm is based on the heuristic argument that the 1D part of the
structure should be equivalent. However, some parts of the 1D region may be close to the
sidewalls. Therefore, the dopant concentration is contributed to not only from the direct
exposure to the ion beam, but also from the particles scattered from the sidewalls and re-
entering the 1D region. In such situations (such as high-energy implant into a photoresist mask
or pocket implants), trajectory replication may not give accurate results near the sidewalls. In
addition, for high tilt pocket implants, saving CPU time by trajectory replication is limited.
Therefore, under such circumstances, you should switch off the trajectory replication.
NOTE Trajectory replication is switched on by default. To switch off trajectory
replication, use the global switch:
pdbSet MCImplant TrajectoryReplication 0
NOTE This feature is available in both Sentaurus MC and Crystal-TRIM.
Datasets
The datasets used in a MC run follow the same naming conventions as those used in analytic
implantation. Datasets unique to the MC implantation method are:
■The ballistic vacancy density Vac_LastImp.
■The damage probability PD, which is used to store and initialize damage history in
Crystal-TRIM.
■The nuclear energy deposition EnergyDeposition, which is created in Sentaurus MC
implantation.
Table 10 Datasets used in MC implantations
Dataset Description
EnergyDeposition
Accumulated energy deposition (in units of ) from nuclear collisions.
This dataset is created in Sentaurus MC implant only.
eV/cm3

3: Ion Implantation
Boundary Conditions and Domain Extension
180 Sentaurus™ Process User Guide
H-2013.03
Boundary Conditions and Domain Extension
Boundary conditions are needed in ion implantation simulations to account for the geometry
effects (such as shadowing) and lateral scattering of the implied structure. Both of these effects
require knowledge of the materials and damage concentration outside the simulation domain.
The required information is synthesized by the definition of the boundary conditions. The
following subsections describe how to specify these boundary conditions.
Damage
Accumulative damage (damage history). This dataset is deleted by the
diffuse command. For Sentaurus MC, at the end of an implant step, the
Damage_LastImp concentration is added to Damage, similar to analytic
implantation. For Crystal-TRIM, damage is generated using the PD dataset after
an implant step.
Damage_LastImp
Damage created during the last implant step. This dataset is used by
Sentaurus MC only.
<dopant>
Accumulative density of the dopant concentration. At the end of an implant
step, the <dopant>_LastImp concentration is added to <dopant>.
<dopant>_Implant
Accumulative density of the dopant concentration. At the end of an implant
step, the <dopant>_LastImp concentration is added to
<dopant>_Implant. This dataset is deleted by the diffuse command.
<dopant>_LastImp
Ballistic dopant concentration generated during the last implant step. It is
reset at the beginning of each implant step.
Int_Implant
Accumulative interstitial profile updated at the end of an implant step.
Int<component>_Implant
Accumulative interstitial profiles in multicomponent material with
DistinctDefects set to true, where <component> is the component
of the composition of the material. For example, in SiC, interstitial profiles
include IntSilicon_Implant and IntCarbon_Implant.
PD
Damage probability. This dataset is used by Crystal-TRIM only.
Vac_Implant
Accumulative vacancy profile updated at the end of an implant step.
Vac_LastImp
Ballistic vacancy density generated during the last implant step.
Vac<component>_Implant
Accumulative vacancy profiles in multicomponent material with
DistinctDefects set to true, where <component> is the component
of the composition of the material. For example, in SiC, interstitial profiles
include VacSilicon_Implant and VacCarbon_Implant.
Table 10 Datasets used in MC implantations
Dataset Description

3: Ion Implantation
Boundary Conditions and Domain Extension
Sentaurus™ Process User Guide 181
H-2013.03
Unified Implant Boundary Conditions
NOTE This is the preferred method for specifying implant boundary
conditions.
Sentaurus Process has two different sets of implant boundary conditions: one for analytic
implant and one for MC implant. To ensure consistent results between analytic and MC
implant, Sentaurus Process provides a unified method for specifying implant boundary
conditions. This method uses the implant command to specify the boundary conditions with
the following syntax:
implant boundary.conditions = {left=<c> right=<c> front=<c> back=<c>}
where the valid keywords are Periodic, Reflect, or Extend.
In contrast to the boundary conditions specified by PDB commands, if Periodic or Reflect
is specified in the implant command, TruePeriodic and TrueReflect will be used for
MC implant since these ensure the most consistent results between analytic and MC implants.
You do not n ee d to sp ec ify boundary conditions for all four sides. Extend boundary condition
is assumed for unspecified sides, except for periodic boundary conditions. Since periodic
boundary conditions must be paired, left and right or front and back, they must have the
same periodic boundary conditions. For simplicity, you need only specify periodic boundary
conditions on one side; the other side is automatically assumed to have the same periodic
boundary condition. If the other side is specified for a different type of boundary condition,
Sentaurus Process issues a warning and uses a periodic boundary condition.
Implant Boundary Conditions using PDB Commands
Although not recommended, these boundary conditions can be directly specified using PDB
commands. Advanced users can use these commands to adjust the implant boundary conditions
under certain circumstances.
NOTE Because this method for specifying implant boundary conditions is
obsoleted, you should use it only if it is absolutely necessary when
creating new input files.
3: Ion Implantation
Boundary Conditions and Domain Extension
182 Sentaurus™ Process User Guide
H-2013.03
Monte Carlo Implant
Boundary conditions determine how particles leaving the simulation domain at its outer
boundaries will be processed. It is assumed that the simulation domain is rectangular if viewed
from the top and is contained between:
LeftBoundary and RightBoundary (y-direction)
and:
BackBoundary and FrontBoundary (z-direction)
Legacy Periodic Boundary Conditions
In this boundary condition, boundaries exist only in solid regions. In solid regions, when a
particle reaches one side of the boundary, it is moved to the other side of the boundary.
However, in ambient (or gas), particles are free to enter or exit. Therefore, Periodic boundary
condition in MC implant only means translating the position of particles from one boundary to
the opposite boundary in solid regions. It takes the structure as it is, and does not extend to form
a true periodic structure.
To select these boundary conditions, use the following commands:
pdbSet MCImplant BoundaryPeriodicY 1
pdbSet MCImplant BoundaryPeriodicZ 1
This switches on Periodic boundary conditions in the left-right and front-back, respectively.
NOTE Because this boundary condition is obsoleted, you should not use it
when creating new input files.
TruePeriodic Boundary Conditions
This boundary condition considers the device as a true periodic structure. To select
TruePeriodic boundary conditions, use:
pdbSet MCImplant TruePeriodicY 1
pdbSet MCImplant TruePeriodicZ 1
TruePeriodicY and TruePeriodicZ are applied to left and right, and front and back,
respectively.
NOTE TruePeriodic has precedence over legacy Periodic boundary
condition and other types of boundary conditions. When Periodic is
specified in the implant command for unified boundary conditions,
3: Ion Implantation
Boundary Conditions and Domain Extension
Sentaurus™ Process User Guide 183
H-2013.03
TruePeriodic boundary condition is selected automatically in MC
implant.
Legacy Reflect Boundary Conditions
pdbSet MCImplant LeftBoundary Reflect
pdbSet MCImplant RightBoundary Reflect
pdbSet MCImplant FrontBoundary Reflect
pdbSet MCImplant BackBoundary Reflect
A particle hitting the boundary will have its position and direction of motion reflected with
respect to the boundary plane.
TrueReflect Boundary Conditions
Instead of reflecting the direction of the moving particles at the boundary, Sentaurus Process
provides a new boundary condition TrueReflect, which automatically reflects the structure,
performs the implantation, and then cuts the structure to its original domain. To specify the
TrueReflect boundary condition, use:
pdbSet MCImplant LeftBoundary TrueReflect
pdbSet MCImplant RightBoundary TrueReflect
pdbSet MCImplant FrontBoundary TrueReflect
pdbSet MCImplant BackBoundary TrueReflect
NOTE When Reflect is specified in the implant command for unified
boundary conditions, TrueReflect boundary condition is
automatically selected in MC implant.
NOTE Generally, reflective boundary conditions (including both Reflect and
TrueReflect) can be used only if there are the same reflect-symmetric
ion beams, such as tilt=0 (or projected tilt2D=0 in 2D), or
approximately, multiple rotation implantations. To improve
performance, averaging of the simulation results (including dopant and
damage fields) over the original and reflected domains can be
performed. If tilt (or tilt2D) < , Sentaurus Process automatically
averages the simulation results. In the case of mult.rot implantations
(or a sequence of multiple implant commands consisting of
mult.rot implantation), no automatically averaging is performed.
However, you can specify average in the implant command to do the
averaging. To overwrite such default behaviors, specify average or
!average in the implant command.
NOTE In cases where average is applied successfully, TrueReflect
generally achieves more accurate results than Reflect without the
significant performance penalty.
2°
3: Ion Implantation
Boundary Conditions and Domain Extension
184 Sentaurus™ Process User Guide
H-2013.03
Extending the Simulation Domain
pdbSet MCImplant LeftBoundary Extend
pdbSet MCImplant RightBoundary Extend
pdbSet MCImplant BackBoundary Extend
pdbSet MCImplant FrontBoundary Extend
The simulation domain is artificially extended in the corresponding direction. This
compensates for the decay of the profile near the boundaries of the simulation domain.
Sentaurus Process determines these extension lengths automatically by using tilt,
rotation, and slice.angle, and the user-defined parameters MinExtension and
ExtensionLength:
(117)
where the function is between 0 and 1. If tilt equals 0, equals 0. Therefore, for tilt
equals 0, extension length equals MinExtension. The default value for MinExtension is
0.1 , which is usually sufficient for low-energy implants. For high-energy implants, you
may need to increase MinExtension to avoid decaying concentration near the boundaries.
To control the size of the extension, use:
pdbSet MCImplant ExtensionLength <n>
pdbSet MCImplant MinExtension <n>
Transparent Boundary
pdbSet MCImplant LeftBoundary Transparent
pdbSet MCImplant RightBoundary Transparent
pdbSet MCImplant BackBoundary Transparent
pdbSet MCImplant FrontBoundary Transparent
All particles crossing the boundary leave the simulation domain and are lost.
NOTE For given boundary conditions other than Transparent, it is important
that the boundaries, which are lines in 2D and planes in 3D, are
continuous, that is, they should show no holes. The crossing of a particle
can only be registered if it happens within the material region. The
particle will finally leave the structure if it crosses the side while in a gas
region.
Ext Length MinExtension ExtensionLength ftilt rotation slice.angle,,()⋅+=
f
f
µm
3: Ion Implantation
Boundary Conditions and Domain Extension
Sentaurus™ Process User Guide 185
H-2013.03
Analytic Implant
Analytic implantation uses the same syntax as MC implantation for specifying boundary
conditions. It is assumed that the simulation domain is rectangular if viewed from the top and
is contained between:
LeftBoundary and RightBoundary (y-direction)
and:
BackBoundary and FrontBoundary (z-direction)
Extended Boundary Condition
pdbSet ImplantData LeftBoundary Extend
pdbSet ImplantData RightBoundary Extend
pdbSet ImplantData FrontBoundary Extend
pdbSet ImplantData BackBoundary Extend
The simulation domain is extended artificially in the corresponding direction. This
compensates for the decay of the profile near the boundaries of the simulation domain. The
extended structure is removed after the implant is completed. Extend is the default boundary
condition for ion implantation. To control the size of the lateral extension, use:
pdbSet ImplantData MaxLateralExtension <n>
Reflective Boundary Condition
pdbSet MCImplant LeftBoundary Reflect
pdbSet MCImplant RightBoundary Reflect
pdbSet MCImplant BackBoundary Reflect
pdbSet MCImplant FrontBoundary Reflect
In reflective boundary condition, a reflected image with respect to the domain boundary is first
constructed. Depending on the boundary condition specified on the other side, the composite
structure is either extended (if the other side is extended) or repeated (if the other side is also
reflective). The added structure including the reflected image is removed after the implant is
completed.
NOTE This boundary condition is equivalent to the TrueReflect boundary
condition in MC implant.
Periodic Boundary Condition
pdbSet ImplantData BoundaryPeriodicY 1
pdbSet ImplantData BoundaryPeriodicZ 1

3: Ion Implantation
Smoothing Implantation Profiles
186 Sentaurus™ Process User Guide
H-2013.03
These commands switch the periodicity in the left–right or front–back direction, respectively.
An array of periodic images is constructed outside the simulation domain before the implant is
performed. These added periodic images are removed after the implant is finished.
NOTE This boundary condition is equivalent to the TruePeriodic boundary
condition in MC implant.
Smoothing Implantation Profiles
The implantation profiles as produced by MC simulations are typically noisy, especially in low
concentration regions. This may sometimes cause converging problems or may require a very
small time step in diffusion. To overcome this problem, Sentaurus Process provides a facility
for smoothing the implant profiles.
The smoothing is enabled by using the following simple diffusion equation:
(118)
where:
■ is the concentration.
■ is the diffusion coefficient.
■ is the characteristic diffusing distance.
Smoothing All As-Implanted Profiles
To smooth all as-implanted profiles, specify the Boolean parameter smooth in the implant
command or, alternatively, use the global switch Smoothing, which can be specified as
follows:
pdbSet MCImplant Smoothing 1
In this case, all as-implanted fields are smoothed including dopant, damage, and point-defect
profiles.
Smoothing Dopant and Damage Fields
For flexibility, Sentaurus Process also provides facilities for smoothing selected fields by using
parameter smooth.field=<list of fields>. If this parameter is specified, only the
specified fields are smoothed. The valid fields are <dopant> or Damage. For example, for BF2
implantation, the valid fields are Boron, Fluorine, or Damage. Note that point defects
(interstitial and vacancy) are generally not independent and cannot be specified in
t∂
∂CDC∇⋅()–0=
C
D
Dt⋅

3: Ion Implantation
Automatic Extraction of Implant Moments
Sentaurus™ Process User Guide 187
H-2013.03
smooth.field. In addition, depending on the point-defect model used, the smoothing of
dopant or damage fields also may cause the point defects being smoothed.
You can control smoothing behavior by specifying the parameter
smooth.distance=<double array>. This list specifies the smoothing distances (diffusing
distance) for each of the fields as specified in smooth.field. If this list is missing, the
smoothing distances are retrieved from the PDB:
pdbSetDouble MCImplant Smooth <dopant> Smooth.Distance <n>
If no pdb parameter is available for a given species, the global default (2 nm) is used.
Smoothing Point Defects
If only point defects are smoothed, you must use the smooth command after the implant
command (instead of specifying smooth or smooth.field in the implant command). The
syntax is as follows:
smooth smooth.field=<list of fields> smooth.distance=<double array>
This is a general command that can be used to smooth any field. For example, to smooth point
defects after implantation, use the command:
smooth smooth.field= {Int_Implant Vac_Implant} smooth.distance= {1<nm> 5<nm>}
NOTE If using the smooth command to smooth a field, the pdb parameter for
smooth distance will not be read. Therefore, smooth.distance must
be specified in the smooth command if it is different from the default
2nm.
Automatic Extraction of Implant Moments
Implant moments are one of the most critical elements in analytic implantation. By default,
Sentaurus Process provides a large set of implant tables that cover many species and materials,
and a wide range of implantation conditions. However, occasionally, users want to explore new
species, new materials, or the implantation parameter space, which is outside of the supplied
implant tables. In this case, you need to do the experiments or to run the MC implantation
simulations to obtain accurate implantation profiles. Automatically extracting implant
moments bridges the gap of converting these raw profiles into the moments that can be used in
analytic implantation.
The critical part of automatic extraction of implant moments is the optimization (or least square
fit) algorithm, that is, given a profile or a set of pairs of data points , optimize the
m
xiyi
(,)

3: Ion Implantation
Automatic Extraction of Implant Moments
188 Sentaurus™ Process User Guide
H-2013.03
parameter set of the model function , so that the sum of the squares of the errors at
each point becomes minimal:
(119)
Sentaurus Process uses the same optimizer as TSUPREM-4, which implements the popular
Levenberg–Marquardt algorithm, also known as the damped least-squares method. Since this
algorithm only finds the local minimum, the initial parameter values may affect the extracted
results.
Required Parameters
To extract the moments, you must specify the parameters extract.moments and data.file
in the implant command. The parameter extract.moments simply indicates that instead of
performing an implantation or setting implantation parameters, the implant command is used
to extract implant moments. The parameter data.file specifies the ASCII data file from
where implant moments will be extracted.
Optional Parameters
To better control the extraction process, the following optional parameters are available in the
implant command:
■dualpearson (default), gaussian, and pearson specify the type of moments to be
extracted.
■rp, stdev, gamma, beta, rp2, stdev2, gamma2, beta2, and ratio specify the initial
values for optimization. If not specified, initial values will be guessed from the profile data.
■data.units, data.xco, data.col, data.xlo, data.xhi, data.min, and
data.max specify how the data in data.file will be interpreted and retrieved.
■max.iter specifies the maximum number of iterations allowed in the optimization loop.
Default is 500.
■tolerance specifies the tolerance of target errors. Default is 0.1.
β
fxβ,()
Sβ() yifx
iβ,()–[]
2
i1=
m
=

3: Ion Implantation
Automatic Extraction of Implant Moments
Sentaurus™ Process User Guide 189
H-2013.03
Output Format
Extracting implant moments provides two types of output, at the same time, to facilitate further
manipulation of the moments:
■Command line. This is useful for copying and inserting the output into the implant
command. The extracted moments are printed on the screen and in the log file in the format:
rp=<n> stdev=<n> gamma=<n> beta=<n> ...
■Tcl list. The output list of moments has the format:
{model dualpearson rp <n> stdev <n> gamma <n> beta <n> ...}
The output Tcl list can be converted into a Tcl array by using array set, which then can be
used to access the moments conveniently. For example:
set moms [implant extract.moments data.file=myfile]
array set m $moms
LogFile ''model = $m(model)''
LogFile ''rp = $m(rp)''
LogFile ''stdev = $m(stdev)''
Utilities
The Tcl script ImplantTableMaker can be used to guide users through selecting
implantation conditions to automatically create a Taurus format implant table from MC
implantations. This script must be run in interactive mode, and you must input various implant
parameters that are necessary to create an implant table. The resulting table is named
<species>_in_<material>_mystandard.
NOTE Since each implant profile is extracted independently, and the
Levenberg–Marquardt optimization algorithm can only find the local
minima, slightly different profiles may result in totally different implant
moments. Interpolation between these moments may not give optimal
results. Therefore, when using the Tcl script ImplantTableMaker, the
quality of implant tables cannot be guaranteed.

3: Ion Implantation
Loading External Profiles
190 Sentaurus™ Process User Guide
H-2013.03
Loading External Profiles
Loading Files Using load.mc
Precomputed profiles can be loaded to a given structure using the load.mc facility in
Sentaurus Process, that is:
implant <dopant> load.mc file=<name>
If the load.mc switch is set in the implant command, Sentaurus Process takes the TDR file
specified with the file selector and loads the datasets into the present structure. Interpolation
of the datasets is performed if the structure in the TDR file is different from the present
structure.
Sentaurus Process attempts to find the doping profiles required from the implant species and
the damage probability (for Crystal-TRIM) or damage dataset (for Sentaurus MC). For
example, in the following statement:
implant Boron load.mc file=my_data energy=10 dose=1e14
Sentaurus Process opens the files my_data{_fps}.tdr, and checks for the datasets
Boron_LastImp and PD_LastImp (for Crystal-TRIM) or Damage_LastImp (for
Sentaurus MC). If successful, these datasets are restored. If one or more of the required
datasets is missing, the respective fields remain empty. Then, during implantation
postprocessing, Boron_LastImp and Damage_LastImp are added to the Boron_Implant
and Damage datasets.
The following options are available in 2D structures:
■shift=<n>: Shifts the dataset along the y-axis.
■flip: Flips the dataset; the default is a flip to the left.
■left, right: Specifies the flipping direction.
■multiply=<n>: Multiplies the dopant data in the dataset by a factor; the damage remains
untouched.
NOTE The switch load.mc restores the datasets from the files without
checking the implant conditions specified in the implant command.
Therefore, load.mc by itself does not require the implant parameters
such as energy and dose be specified. However, Advanced Calibration
and CoImplant models may use these parameters (energy and dose)
for their calculations. Therefore, it is recommended that energy and
dose always be specified along with load.mc.

3: Ion Implantation
Multithreaded Parallelization of 3D Analytic Implantation
Sentaurus™ Process User Guide 191
H-2013.03
Automated Monte Carlo Run
If no TDR file with the specified name is found or the file selector is empty, a separate run
of Crystal-TRIM or Sentaurus MC is started to generate these files and the required datasets,
depending on the setting of the MC implantation model in the PDB (default is
sentaurus.mc):
pdbSet MCImplant model {crystaltrim | sentaurus.mc}
Sentaurus Process internally switches from the load.mc to crystaltrim or sentaurus.mc
mode. All implant parameters related to MC implantation (particles, cascades,
full.molecular) are used in this run.
NOTE The load.mc feature is designed to reuse precomputed MC results.
However, the profiles to load do not necessarily need to be generated
using the MC method.
Example
implant BF2 dose=1e14 energy=40 tilt=20 rotation=-90 load.mc \
file=bf2_1e14_40 particles=500 cascades
In the first run of this command, Sentaurus Process checks for the TDR file with the name
bf2_1e14_40. Since there is no file with this name, a full-cascade Crystal-TRIM or
Sentaurus MC run is started using the process parameters specified. At the end, the TDR file
bf2_1e14_40_fps.tdr is saved. The following are stored as well:
■All datasets related to the BF2 impurity profile (Boron_LastImp, Fluorine_LastImp).
■The damage probability (PD_LastImp) or damage (Damage_LastImp).
■The recoil profile (Silicon_LastImp) and vacancy profile (Vac_LastImp) because the
command is run in the full-cascade mode.
In a subsequent run of the same command, Sentaurus Process loads and restores these datasets
in a preprocessing step. The postprocessing is the same as after a MC run.
Multithreaded Parallelization of 3D Analytic Implantation
Parallel processing has become ubiquitous with the advent of multicore processors. The
performance of 3D analytic implantation can be improved dramatically by exploiting the
parallel processing power of multicore processors. In multithreaded mode, each thread works
on separate nodes, sharing the workload, thereby reducing the computation time.

3: Ion Implantation
Multithreaded Parallelization of Sentaurus MC Implantation
192 Sentaurus™ Process User Guide
H-2013.03
To engage the multithreaded parallelization of 3D analytic implantation, use the math
command:
math [ numThreads = <n> | numThreadsImp3d = <n> ]
where numThreads is a general keyword for MC implantation, 3D analytic implantation,
KMC, matrix assembly, and linear solver. However, numThreadsImp3d has a higher priority
over numThreads for 3D analytic implants and can be used to create the number of threads
specifically for 3D analytic implantation, which is different from that for other multithreaded
operations. The value of numThreads or numThreadsImp3d must be equal to the number of
cores in multicore processors.
You al so ca n mod ify t he stack s ize for each thread using the command:
math [ threadStackSize = <n> ]
The default stack size ( bytes) is usually sufficient for 3D analytic implantation.
Multithreaded Parallelization of Sentaurus MC
Implantation
NOTE This feature is available to Sentaurus MC only.
The performance of MC implantation (Sentaurus MC) also can be significantly improved by
using multithreaded parallelization. In this approach, a large job with many particles ( ) is
divided into multiple ( ) separate jobs with a smaller number of particles ( ). Sentaurus
Process then creates multiple threads and launches instances of Sentaurus MC implant.
Each instance of Sentaurus MC implant runs independently on its own thread. After these
threads are finished, the results are averaged, thereby improving the effective execution speed
for a large job.
To engage the multithreaded parallelization of Sentaurus MC implantation, use the math
command:
math [ numThreads = <n> | numThreadsMC = <n> ]
where numThreads is a general keyword for MC implantation, 3D analytic implantation,
KMC, matrix assembly, and linear solver. However, numThreadsMC has higher priority over
numThreads for MC implants and can be used to create the number of threads specifically for
Sentaurus MC implant, which is different from that for other multithreaded operations. The
value of numThreads or numThreadsMC should be equal to the number of cores in multicore
processors.
218 262144=
N
m
Nm⁄
m

3: Ion Implantation
References
Sentaurus™ Process User Guide 193
H-2013.03
You al so ca n mod ify t he stack s ize for each thread using the command:
math [ threadStackSize = <n> ]
The default stack size ( bytes) is usually sufficient for MC implantation.
References
[1] G. Hobler and S. Selberherr, “Two-Dimensional Modeling of Ion Implantation Induced
Point Defects,” IEEE Transactions on Computer-Aided Design, vol. 7, no. 2, pp. 174–
180, 1988.
[2] J. P. Biersack, “Basic Physical Aspects of High Energy Implantation,” Nuclear
Instruments and Methods in Physics Research, vol. B35, no. 2, pp. 205–214, 1988.
[3] J. F. Gibbons, W. S. Johnson, and S. W. Mylroie, Projected Range Statistics:
Semiconductors and Related Materials, Pennsylvania: Dowden, Hutchinson & Ross,
2nd ed., 1975.
[4] C. Zechner et al., “New Implantation Tables for B, BF2, P, As, In and Sb,” in 14th
International Conference on Ion Implantation Technology (IIT), Taos, NM, USA,
pp. 567–570, September 2002.
[5] S. Tian, V. Moroz, and N. Strecker, “Accurate Monte Carlo Simulation of Ion
Implantation into Arbitrary 1D/2D/3D Structures for Silicon Technology,” in MRS
Symposium Proceedings, Silicon Front-End Junction Formation—Physics and
Technology, vol. 810, San Francisco, CA, USA, pp. 287–292, April 2004.
[6] S. J. Morris et al., “An Accurate and Efficient Model for Boron Implants Through Thin
Oxide Layers into Single-Crystal Silicon,” IEEE Transactions on Semiconductor
Manufacturing, vol. 8, no. 4, pp. 408–413, 1995.
[7] S. Tian, “Predictive Monte Carlo ion implantation simulator from sub-keV to above
10 MeV,” Journal of Applied Physics, vol. 93, no. 10, pp. 5893–5904, 2003.
[8] M. Posselt, “Crystal-TRIM and Its Application to Investigations on Channeling Effects
During Ion Implantation,” Radiation Effects and Defects in Solids, vol. 130–131,
pp. 87–119, 1994.
218 262144=
3: Ion Implantation
References
194 Sentaurus™ Process User Guide
H-2013.03
[9] A. F. Tasch et al., “An Improved Approach to Accurately Model Shallow B and BF2
Implants in Silicon,” Journal of the Electrochemical Society, vol. 136, no. 3, pp. 810–
814, 1989.
[10] G. Hobler, E. Langer, and S. Selberherr, “Two-Dimensional Modeling of Ion
Implantation with Spatial Moments,” Solid-State Electronics, vol. 30, no. 4, pp. 445–
455, 1987.
[11] A. Stolmeijer et al., “General Expressions for the Impurity Distributions of B and P
Implanted in SiO2,” Journal of the Electrochemical Society, vol. 135, no. 9, pp. 2309–
2311, 1988.
[12] FLOOPS process and device simulator: http://www.flooxs.tec.ufl.edu/, October 2013.
[13] H. Ryssel, W. Krüger, and J. Lorenz, “Comparison of Monte Carlo Simulations and
Analytical Models for the Calculation of Implantation Profiles in Multilayer Targets,”
Nuclear Instruments and Methods in Physics Research, vol. B19/20, no. 20, pp. 40–44,
1987.
[14] G. Hobler and V. Moroz, “Initial Conditions for Transient Enhanced Diffusion: Beyond
the Plus-Factor Approach,” in International Conference on Simulation of
Semiconductor Processes and Devices (SISPAD), Athens, Greece, pp. 34–37, September
2001.
[15] S. Strauss et al., “Analytic model for ion channeling in successive implantations in
crystalline silicon,” Materials Science and Engineering B, vol. 124–125, pp. 376–378,
December 2005.
[16] S. Tian, “Accurate Monte Carlo simulation of fluorine and BF2 ion implantation into
crystalline silicon,” Nuclear Instruments and Methods in Physics Research B, vol. 215,
no. 3-4, pp. 403–412, 2004.
[17] H. Goldstein, Classical Mechanics, Cambridge, Massachusetts: Addison-Wesley Press,
1950.
[18] J. F. Ziegler, J. P. Biersack, and U. Littmark, “The Stopping and Range of Ions in
Solids,” The Stopping and Ranges of Ions in Matter, vol. 1, New York: Pergamon Press,
1985.
[19] M. T. Robinson and I. M. Torrens, “Computer simulation of atomic-displacement
cascades in solids in the binary-collision approximation,” Physical Review B, vol. 9,
no. 12, pp. 5008–5024, 1974.
[20] J. Lindhard and M. Scharff, “Energy Dissipation by Ions in the kev Region,” Physical
Review, vol. 124, no. 1, pp. 128–130, 1961.
[21] O. S. Oen and M. T. Robinson, “Computer Studies of the Reflection of Light Ions from
Solids,” Nuclear Instruments and Methods, vol. 132, pp. 647–653, 1976.
[22] M. Posselt et al., “Modeling of Damage Accumulation during Ion Implantation into
Single-Crystalline Silicon,” Journal of the Electrochemical Society, vol. 144, no. 4,
pp. 1495–1504, 1997.
3: Ion Implantation
References
Sentaurus™ Process User Guide 195
H-2013.03
[23] I. Santos et al., “Improved atomistic damage generation model for binary collision
simulations,” Journal of Applied Physics, vol. 105, p. 083530, April 2009.
[24] S. Tian, “Monte Carlo Simulation of Ion Implantation in Crystalline SiC With Arbitrary
Polytypes,” IEEE Transactions on Electron Devices, vol. 55, no. 8, pp. 1991–1996,
2008.
[25] A. Burenkov et al., “Simulation of BF3 Plasma Immersion Ion Implantation into
Silicon,” in 19th International Conference on Ion Implantation Technology (IIT),
Valladolid, Spain, pp. 233–236, June 2012.
[26] X. B. Tian, D. T. K. Kwok, and P. K. Chu, “Modeling of incident particle energy
distribution in plasma immersion ion implantation,” Journal of Applied Physics, vol. 88,
no. 9, pp. 4961–4966, 2000.
3: Ion Implantation
References
196 Sentaurus™ Process User Guide
H-2013.03

Sentaurus™ Process User Guide 197
I-2013.12
CHAPTER 4 Diffusion
This chapter provides information on the continuum models for
dopant and defect diffusion models and parameters. (For an
atomistic approach, see Chapter 5.)
Overview
During the fabrication process, dopants are introduced into the substrate with different
concentration profiles. As processing proceeds through various thermal annealing cycles, the
dopants diffuse and redistribute through the structure. The following effects contribute to
dopant redistribution and can be modeled by Sentaurus Process:
■Dopant (de)activation
■Dopant–defect interaction
■Chemical reactions at interfaces and in bulk materials
■Material flow
■Moving material interfaces
■Internal electric fields
Sentaurus Process is designed to address the challenges of integrated-circuit process modeling.
As technology development continues, the need for new process models increases. The
Alagator language is a versatile way to add and modify diffusion models quickly. This chapter
describes the diffusion models in Sentaurus Process. To modify or add new diffusion models,
see Modifying Diffusion Models on page 608.
The diffuse command represents the main simulation capabilities of Sentaurus Process. It
simulates:
■Thermal annealing of impurities.
■Material growth processes during annealing, for example, oxidation, silicidation, and
epitaxy (see Epitaxy on page 282 and Oxidation on page 615).
■Process-induced stress (see Chapter 9 on page 643).

4: Diffusion
Basic Diffusion
198 Sentaurus™ Process User Guide
I-2013.12
Basic Diffusion
The diffuse command is used to model the diffusion of impurities under oxidizing and non-
oxidizing conditions. The options of the diffuse command set diffusion conditions as well
as time-stepping options. (See diffuse on page 908 for all options.) For example, a command
for a simple non-oxidizing annealing at a temperature of for 10 s is:
diffuse temperature=900<C> time=10<s>
If you want to perform the same anneal with a wet (H2O) oxidizing ambient, execute the
following command:
diffuse temperature=900<C> time=10<s> H2O
A simple temperature ramp can be specified directly in the diffuse command by the keyword
ramprate. This keyword sets the change in the temperature over time:
diffuse temperature=900<C> time=10<min> O2 ramprate=10<C/min>
This example describes a dry oxidation of 10 minutes, starting at and ending at
. The same example can be repeated using the temp_ramp command as follows:
temp_ramp name=MyTempRamp temperature=900 time=10 O2 ramprate=10<C/min>
diffuse temp.ramp=MyTempRamp
The first line creates a temperature ramp with given conditions, and the second line specifies a
diffusion referring to this temperature ramp.
To describe more complex temperature cycles within one diffuse command, multiple
instances of the temp_ramp command can be used. A temperature ramp can consist of several
segments and, for each segment, one temp_ramp command is required. In addition, segments
can be grouped by using the same name for each segment. For example, a ramp-up, plateau,
and ramp-down can be specified as:
temp_ramp name=MyCycle temperature=500<C> time=5<min> H2O ramprate=100<C/min>
temp_ramp name=MyCycle temperature=1000<C> time=10<min> O2
temp_ramp name=MyCycle temperature=1000<C> time=10<min> ramprate=-50<C/min> \
last
diffuse temp.ramp=MyCycle
The keyword last in the third temp_ramp command declares the last segment of the
temperature ramp.
Sentaurus Process allows for thermal oxidation from O2 and H2O. The gas_flow command
is used to specify a mixed gas flow by specifying directly either the partial pressures of the gas
components or the flow [volume/time]. If the flows are defined, they are converted to partial
900°C
900°C
1000°C
4: Diffusion
Basic Diffusion
Sentaurus™ Process User Guide 199
I-2013.12
pressures by taking ratios. The use of the gas_flow command is similar to the temp_ramp
command; however, multiple gas flows using the same name must not be specified. When a
gas_flow is specified, it can be referred to from both the temp_ramp and diffuse
commands:
gas_flow name=MyGasFlow pH2O=0.5 pO2=0.5 pH2=0.1
To invoke the gas flow specification as given above, use:
temp_ramp name=MyTempRamp temperature=1000<C> time=10<min> gas.flow=MyGasFlow
diffuse temp.ramp=MyTempRamp
or:
diffuse temperature=1000<C> time=10<min> gas.flow=MyGasFlow
Sentaurus Process also allows you to select various diffusion models for point defects and
dopants (see Transport Models on page 205). Diffusion model setting and parameter setting are
performed with the pdbSet command. The basic settings are:
pdbSet <material> Dopant DiffModel <model>
where <model> can be any of Constant, Fermi, Pair, React, ChargedFermi,
ChargedPair, or ChargedReact.
Epitaxy can be simulated if either the Epi (also known as epi) or LTE ambient is specified in
either the temp_ramp or diffuse command. If Epi is specified, Silicon will grow on
Silicon and PolySilicon will grow on PolySilicon. If the LTE ambient is specified,
Silicon will again grow on Silicon, but PolySilicon will grow on Oxide, Nitride, and
PolySilicon.
pdbSet Silicon Dopant DiffModel Pair
diffuse temperature=800<C> time=60<min> Epi thick=0.01 \
epi.doping = {Germanium = 8e21}
This example sets the dopant diffusion model for all dopants in silicon to the Pair model and
grows a 0.01 thick epi layer with a Germanium concentration of .
It is also possible to set the initial diffusion time-step and the minimum annealing temperature
with the diffuse command.
diffuse temp.ramp=MyCycle minT=600<C> init=0.01<s>
This example uses the temp_ramp created in the earlier example. The initial time step is set to
0.01 s and the minimum annealing temperature is set to . The diffusion and reaction
equations will be switched off below but the mechanics will be solved.
µm
810
21
×
600°C
600°C

4: Diffusion
Basic Diffusion
200 Sentaurus™ Process User Guide
I-2013.12
If you want to set the minimum annealing temperature and initial time-step globally for all
diffusion commands, the following commands can be used:
pdbSet Diffuse minT {<n>}
pdbSet Diffuse InitTimeStep {<n>}
It is also possible to set minimum and maximum temperature limits for the annealing process
using the following commands:
pdbSet Diffuse minAnnealT <n>
pdbSet Diffuse maxAnnealT <n>
If the annealing temperature goes above or below these limits, Sentaurus Process will quit with
an error message.
See Viewing the Defaults: Parameter Database Browser on page 60 for other parameters
related to Diffuse.
Obtaining Active and Total Dopant Concentrations
By default, active and total dopant concentrations are only updated during diffusion steps. For
example, after an implant, the active concentrations (BoronActiveConcentration,
ArsenicActiveConcentration, and so on) and the total concentrations
(BoronConcentration, ArsenicConcentration, and so on) are not modified, which
makes them out of date.
Similarly for other commands that can change the dopant concentrations, the active and total
concentrations are not updated. These commands include, but are not limited to, select,
load, init, and profile.
After one of these commands is issued, the active and total dopant concentrations may not be
current. To update the active and total dopant concentrations use the diffuse time=0 ...
command.
NOTE To update the active and total concentrations of the dopants without
dopant redistribution, the diffuse command with zero time can be
used. For example, to calculate the active dopant concentration at
for the chosen diffusion model, use:
diffuse time=0.0 temperature=850
NOTE Since the diffuse command performs the recrystallization and the
initialization of clusters even with zero time, it must not be added
between consecutive implantation steps.
850°C

4: Diffusion
Basic Diffusion
Sentaurus™ Process User Guide 201
I-2013.12
See diffuse on page 908 for all options of the diffuse command.
Transport Models
Sentaurus Process has several basic transport models with varying levels of complexity for
computing flux, . This diversity of models is needed to balance accuracy with simulation
times, which vary widely depending on the model selection:
■The React (see React Diffusion Model on page 212) and ChargedReact (see
ChargedReact Diffusion Model on page 206) diffusion models, also known as five-stream
diffusion models, are the most advanced dopant diffusion models in Sentaurus Process.
They solve up to three separate equations per dopant – a substitutional dopant – and up to
two dopant–defect pairs and two defect equations. The ChargedReact model is the most
accurate model available in Sentaurus Process. but because of the large number of
equations required, it also is the most computationally expensive. The React model, which
is an uncharged version of the ChargedReact model, is provided for backward
compatibility.
■The Pair (see Pair Diffusion Model on page 216) and ChargedPair (see ChargedPair
Diffusion Model on page 214) diffusion models, also known as three-stream diffusion
models, assume that dopant–defect pairs are in local equilibrium but still solve for separate
point-defect equations. These models solve one equation per dopant and two defect
equations. The ChargedPair diffusion model allows the pairing coefficients to vary with
charge state. These models are the most commonly used for advanced CMOS processes as
they represent a balance between accuracy and computational expense. For extremely fast
ramp rates or for customized initial conditions, the ChargedReact model or React model
is a better choice. The Pair model, which is an uncharged version of the ChargedPair
model, is provided for backward compatibility.
■The Fermi (see Fermi Diffusion Model on page 219) and ChargedFermi (see
ChargedFermi Diffusion Model on page 217) diffusion models both assume that point
defects as well as dopant–defect pairs are in equilibrium. The ChargedFermi diffusion
model allows the diffusivity of each charge state to be set separately. An uncharged version
of the model is provided for backward compatibility. These models can be used for long-
term high-temperature anneals where the transient effect of annealing implant damage is
minimal.
■The Constant diffusion model (see Constant Diffusion Model on page 220), unlike all
other transport models, assumes a constant diffusivity and no electric-field effect, and is
used mainly for dopant diffusion in oxide.
The selection of transport model is specified as follows:
pdbSet <material> Dopant DiffModel <model>
J
4: Diffusion
Basic Diffusion
202 Sentaurus™ Process User Guide
I-2013.12
where <model> must have one of the valid diffusion model names – Constant, Fermi, Pair,
React, ChargedFermi, ChargedPair, or ChargedReact.
It is also possible to select a different diffusion model for each dopant in the same material. In
this case, use the command:
pdbSet <material> <dopant> DiffModel <model>
where <dopant> is a valid dopant name (for example, Boron).
The ChargedFermi, ChargedPair, and ChargedReact diffusion models take into account
each charged point defect individually. Otherwise, they are very similar to the Fermi and Pair
diffusion models.
NOTE Even though you can select any diffusion model individually for each
dopant, it is not recommended to mix the ChargedFermi,
ChargedPair, or ChargedReact models with the uncharged
versions.
Ta bl e 11 S um m ar y o f d o p a nt d i f fu s io n m o de l s an d p a r a me te rs
pdb command Diffusion model
Type
ChargedReact
React
ChargedPair
Pair
ChargedFermi
Fermi
Constant
pdbSet <material> <dopant> <defect> D Diffusivity
XXXX
pdbSet <material> <dopant> <defect> Dstar Diffusivity
XX
pdbSet <material> <dopant> Dstar Diffusivity
X
pdbSet <material> <defect> D Diffusivity
XXXXX
pdbSet <material> <dopant> <defect> ChargePair Pairing Coeff.
XX
pdbSet <material> <dopant> <defect> Binding Pairing Coeff.
XX
pdbSet <material> <dopant> <defect> kfFTM Rate
X
pdbSet <material> <dopant> <defect> kfKickOut Rate
X
pdbSet <material> <defect> ChargeStates Charging Coeff.
XXXXX
pdbSet <material> <defect> ChargeStatesScale Charging Coeff.
XXXXX
pdbSet <material> <defect> Cstar Concentration
XXXXXXX
pdbSet <material> <defect> KbulkChargeStates Charging Coeff.
XXXXX

4: Diffusion
Basic Diffusion
Sentaurus™ Process User Guide 203
I-2013.12
In Table 11, <material> is a valid material name, <dopant> is a valid dopant name, and
<defect> is either Int or Vac.
Recombination and Reaction Models
Many reactions and recombination models are available in Sentaurus Process. Different
diffusing species such as dopants, defects, and impurities will all have different recombination
and reaction terms. These terms come from the following models:
■Dopant clusters–solid solubility, transient, and dopant–defect cluster models (see Dopant
Activation and Clustering on page 292).
■Defect clusters–Equilibrium, {311}, Loop, LoopEvolution, 1Moment, 2Moment,
Full, and FRENDTECH models (see Defect Clusters on page 319).
■Impurity species:
• Carbon model, Nitrogen model (see NeutralReact Diffusion Model on page 220).
• Fluorine model (see Dopant Active Model: FVCluster on page 304).
Boundary Conditions
Sentaurus Process can simulate various boundary conditions for dopants and defects. You can
select eight different boundary conditions:
■HomNeumann can be applied to any boundary (see HomNeumann on page 357).
■Natural is for point defects (see Natural on page 358).
■Segregation is for dopants (see Segregation on page 361).
■Dirichlet is for dopants and defects (see Dirichlet on page 364).
■ThreePhaseSegregation is for dopants (see ThreePhaseSegregation on page 365).
■GrainBoundarySegregation is for dopants in polycrystalline materials (see Boundary
Conditions on page 258).
■GrainGrainBoundarySegregation is for dopants in polycrystalline materials (see
Boundary Conditions on page 258).
■Trap is for dopants such as fluorine and nitrogen in trap-dependent oxidation.
■TrapGen is for dopants such as nitrogen in N2O oxidation.
■Continuous is for dopants used only during epi growth (see Continuous on page 369).
The Natural and Dirichlet boundary conditions consider interstitial injection during
oxidation for oxidation-enhanced diffusion (OED) of dopants.

4: Diffusion
General Formulation
204 Sentaurus™ Process User Guide
I-2013.12
Other Materials and Effects
In addition to generic transport and recombination and reaction models, other effects can be
simulated:
■Polysilicon model (see Diffusion in Polysilicon on page 242).
■SiGe diffusion model (see Dopant Diffusion in SiGe on page 260).
■Epitaxy (see Epitaxy on page 282).
General Formulation
The general expression for the particle current of a diffusing species of charge is given by:
(120)
where is the concentration, is the diffusivity, is the electron concentration, and is
the intrinsic electron concentration.
The continuity equation for species of charge is given by:
(121)
where the recombination/reaction term is split into two parts: is a possible contribution
coming from the transport model selection (see Transport Models on page 205) and can
contain terms from other reactions, which are most often clustering reactions but could include
any type of reaction. Reactions that transform species into another species will introduce
positive terms into the expression for . Total dopant concentration of dopant will be
equal to the sum of all dopants, dopant–defect pairs, and any related clusters (for example,
).
For the models that do not consider different charge states, computation of the electron
concentration by default is given by the charge neutrality condition , where
is given by the active dopant concentrations (for example, where is the
active donor concentration and is the active acceptor concentration). For the charged
models, the charge states of the defects or defect pairs are considered individually.
A
c
JAcd–Ac
n
ni
----
c–∇Acn
ni
----
c
=
Ac
dAc
n
ni
A
c
t∂
∂Ac
∇J•Ac
–RAc
trans RAc
clus
–+=
RAc
trans
RAc
clus
Ac
RAc
clus
A
ATotal
cAcApair
cAclus
c
++=
n–pN∆++ 0=
N∆
∆NN
dNa
–=
Nd
Na

4: Diffusion
Transport Models
Sentaurus™ Process User Guide 205
I-2013.12
It is expected that the charge reactions are in equilibrium, so that the ratio in the various charged
states is set by the Fermi level:
(122)
where is a reference charge state, which is chosen as 0 for interstitials and vacancies, and is
chosen as the dopant charge for dopant–defect pairs. The are parameters that are set by
default to an Arrhenius expression. In addition, for the charged models, it is necessary to solve
a coupled equation for the electron concentration. The default equation is the same as for the
uncharged case, that is, the charge neutrality equation but, in this case,
is a function of because it contains contributions from charged defects or charged defect
pairs as well as dopants. It is also possible for both the charged and uncharged models to solve
the Poisson equation (see Electron Concentration on page 276).
Transport Models
Transport models compute the particle flux of dopants and are the core diffusion models solved
by Sentaurus Process. In addition to particle flux, pairing reactions can be computed depending
on the transport model selection. Transport models are usually used with one or more
clustering or activation models available. The reaction or clustering models will not modify the
dopant flux, but will compute terms to be added to from Eq. 121. The models are
described in detail here.
The selection of the transport model is made with the command:
pdbSet <material> <dopant> DiffModel <model>
where <material> is the material name; <dopant> can be either "Dopant" to apply to all
dopants or a named dopant such as boron, arsenic, phosphorus, antimony, and indium; and
<model> is one of the models ChargedReact, React, ChargedPair, Pair,
ChargedFermi, Fermi, or Constant.
Ta bl e 1 2 S o l ut io n na m es
Symbol Boron Arsenic Phosphorus Antimony Indium
Boron Arsenic Phosphorus Antimony Indium
BoronInt ArsenicInt PhosphorusInt AntimonyInt IndiumInt
BoronVac ArsenicVac PhosphorusVac AntimonyVac IndiumVac
BActive AsActive PActive SbActive InActive
Acr+kAcCAr
n
ni
----
c()–
=
r
kAc
n–pN∆++ 0=
N∆
n
Rclus
c
CA
CAI
CAV
CA
+

4: Diffusion
Transport Models
206 Sentaurus™ Process User Guide
I-2013.12
The transport for point defects is computed when Compute.Point.Defect is set to 1:
pdbSet <material> Compute.Point.Defect <0 | 1>
ChargedReact Diffusion Model
The ChargedReact diffusion model is the most general transport model in Sentaurus Process.
The model has an immobile substitutional dopant and up to two mobile charged dopant–defect
pair species. Mobile charged point defects are also included in the model.
The following reactions are considered:
(123)
(124)
(125)
(126)
(127)
The differential equations that are solved in this model are:
(128)
(129)
Ta bl e 1 3 Po i n t- d e fe c t n a m es
Symbol Interstitial Vacancy
Int Vac
EqInt EqVac
IntNeutralStar VacNeutralStar
IntNeutral VacNeutral
CX
CX
*
CX0
*
CX0
AzIi
+AI zj+()ij–()e–↔
AzVi
+AV zj+()ij–()e–↔
IiVj
+ij+()e–↔
AI zi+()
Vj
+Azij+()e–↔
AV zi+()
Ij
+Azij+()e–↔
t∂
∂CARAI
–RAV
–RAI V,RAV I,RA
clus
–++=
t∂
∂CAI ∇J•AI
–RAI RAI V,
–RAI
clus
–+=
4: Diffusion
Transport Models
Sentaurus™ Process User Guide 207
I-2013.12
(130)
(131)
(132)
where is the concentration of substitutional (and assumed to be immobile) dopants, is
the concentration of ‘free’ defects of type (either interstitials or vacancies), in other words,
those defects not in clusters or pairs. The reaction rates of the different species ( ) are defined
later in this section.
Next, the flux of the mobile defect pair is considered. Working with Eq. 120, p. 204 for the
charged pairs, the equation will be written in terms of the total concentration of pairs.
It is expected that the dopant-defect pairing reaction is in equilibrium, therefore, a set of
constants for this pairing is defined:
(133)
where is either I or V, is the charge of the dopant , and is the pairing coefficient for
the pair , and is given by:
(134)
To set , use:
pdbSet <material> <dopant> <defect> ChargePair <c> {<n>}
where <material> is a material name (see), <dopant> is one of the existing Sentaurus
Process dopants, <defect> is either Interstitial or Vacancy, <c> is the charge state, and
<n> is a Tcl expression that returns a number – it can be simply a number.
One commonly used Tcl procedure for setting parameters is Arrhenius. This procedure takes
a prefactor and an energy as arguments and returns .
t∂
∂CAV ∇J•AV
–RAV RAV I,
–RAV
clus
–+=
t∂
∂CIJI
∇•–RIV RAI
–– RAV I,
–RI
clus
–=
t∂
∂CVJV
∇•–RIV R–AV
–RAI V,
–RV
clus
–=
CA
CX
X
R
CAXzc+kAXcCAzCXc
=
X
z
A
kAXc
AX
kAXckAXc
0k–AXc
E
kBT
-------------
exp=
kAXc
prefactor energy
–
kBT
--------------------
exp⋅
4: Diffusion
Transport Models
208 Sentaurus™ Process User Guide
I-2013.12
The flux of the pairs is computed from Eq. 120, p. 204:
(135)
where is the total concentration of pairs that is the sum of the concentrations of pairs at
every charge state and is an effective diffusivity of dopant point-defect pairs at charge
state and is related to the self diffusivity by:
(136)
where is the equilibrium concentration of the neutral defects and is related to the total
equilibrium intrinsic concentration of defect by:
(137)
The quantities and , which by default follow an Arrhenius law, can be
changed by using the command:
pdbSet <material> <defect> Cstar {<n>}
To set , use:
pdbSet <material> <dopant> <defect> D <c> {<n>}
A set of equilibrium-charging constants, , for defect is defined:
(138)
where is either I or V, and is the charging coefficient for the defect and is given by:
(139)
JAX JAXc
c
–=
DAXc
n
ni
----
c–z–CAX
CX0
*kAXqkXq
n
ni
----
q–
q
-------------------------------------------------- n
ni
----
z
∇
c
–=
CAX
DAXc
c
dAXc
DAXcCX0
*kAXckXcd
AXc
=
DAXc
0D–AXc
E
kBT
---------------
exp=
CX0
*
X
CX0
*CXintrinsic()
*
kXc
c
-----------------------------=
CIintrinsic()
*
CVintrinsic()
*
DAXc
kXc
X
CXckXcCX0
n
ni
----
c–
=
X
kXc
X
kXckXc
0k–Xc
E
kBT
----------
exp=
4: Diffusion
Transport Models
Sentaurus™ Process User Guide 209
I-2013.12
To set , use:
pdbSet <material> <defect> ChargeStates <c> {<n>}
NOTE The neutral charge state must always be 1.0.
Similar to the pairs, the defect fluxes are computed from Eq. 120, p. 204:
(140)
where is the total concentration of defects that is the sum of the concentrations of defect
at every charge state and is the diffusivity of the defect of charge state and is given by:
(141)
To set , use:
pdbSet <material> <defect> D <c> {[Arrhenius <prefactor> <energy>]}
Now, the reaction rates can be written by considering Eq. 123, p. 206 to Eq. 127, p. 206 and
the general formula for the rate of all combinations of charge states:
(142)
Therefore, summing all possible charge states gives:
(143)
(144)
(145)
kXc
JXJXc
c
–=
kXcD
Xc
n
ni
----
c–CX
*
c
kXq
n
ni
----
q–
q
---------------------------------------------- CX
CX
*
-------
∇–=
CX
X
DXc
X
c
DXcdXc
=
DXc
0D–Xc
E
kBT
------------
exp=
DXc
AaBbABcca–b–()e+↔+
RAaBbc,, kAaBbc,,
fCAaCBbkAaBbc,,
rCABc
n
ni
----
ca–b–()
–
=
RAX KAX
fCACX
CAX
KAX
r
----------–
–≡
RAI V,KAI V,
fCAICVKAX
rCI
*CV
*CA
–()≡
RAV I,KAV I,
fC(AVCIKAX
rCI
*CV
*CA)–≡
4: Diffusion
Transport Models
210 Sentaurus™ Process User Guide
I-2013.12
where:
(146)
(147)
(148)
(149)
where is the forward reaction rate for the kick-out mechanism, and is either interstitial
or vacancy, and are forward reaction rates for the Frank–Turnbull mechanism and
is the equilibrium constant.
The forward ( ) kick-out reaction rates can be set by using the following commands:
pdbSet <material> <dopant> <defect> kfKickOut <c> {<n>}
where c is the charge state. By default, kfKickOut values for each charge state are given as:
(150)
where is the hopping length, which can be set using the command:
pdbSet <material> <dopant> <defect> lambdaK
Similarly, the forward ( , ) Frank–Turnbull reaction rates can be defined using the
commands:
pdbSet <material> <dopant> <defect> kfFTM <i,j> {<n>}
KAX
f
KfXKOikXi
n
ni
----
i–
i
kXc
n
ni
----
c–
c
---------------------------------------------
≡
KAX
r
kAXikXi
n
ni
----
i–
i
kXc
n
ni
----
c–
c
---------------------------------------
≡
KAI V,
f
Kf
AIiVj
,FTkAIikVj
n
ni
----
ij+()–
j
i
kAIc
n
ni
----
c–kVz
n
ni
----
z–
z
c
----------------------------------------------------------------------------
≡
KAV I,
f
Kf
AViIj
,FTkAVikIj
n
ni
----
ij+()–
j
i
kAVc
n
ni
----
c–kIz
n
ni
----
z–
z
c
----------------------------------------------------------------------------
≡
KAX
f
X
KAI V,
f
KAV I,
f
KAX
r
KfXKOi j,
KfXKOi
DAXi
CX
*kXi
i
kX0λ2
---------------------------------
≡
λ
KAI V,
f
KAV I,
f

4: Diffusion
Transport Models
Sentaurus™ Process User Guide 211
I-2013.12
NOTE The indices of the forward recombination rates have the form of .
Both and are integers and are separated by a comma; no space is
allowed between the indices.
The I–V recombination reaction is given as:
(151)
where:
(152)
The superscript ‘*’ refers to the equilibrium concentration, and the subscripts and are for
the interstitials and vacancies, respectively. The subscripts are the charge states of the
defects. is the bulk recombination rate for interstitials and vacancies at the charge stated
and , respectively. The bulk recombination rate for each charged point defect can be
set using the command:
pdbSet <mater> <defect> KbulkChargeStates <i,j> {<n>}
The equilibrium concentration of the unpaired point defect can be calculated by:
(153)
where is the scaled charging coefficient for the defect and can be set by using:
pdbSet <material> <defect> ChargeStatesScale <c> {<n>}
is set to as a default.
NOTE The indices for the parameter KbulkChargeStates have the form of
. Both and are integers and are separated by a comma; no space
is allowed between the indices.
ij,
i
j
RIV KIV CICVCI
*CV
*
–()=
KIV
CIintrinsic()
*CVintrinsic()
*
CI
*CV
*kIzkVz
z
z
----------------------------------------------------------KIiVjkIikVj
n
ni
----
ij+()–
j
i
=
I
V
zij,,
KIiVj
i
j
KIiVj
CX
*CXintrinsic()
*
kXc
n
ni
----
c–
c
ks
Xc
c
------------------------------=
ks
Xc
X
ks
Xc
kXc
ij,
i
j

4: Diffusion
Transport Models
212 Sentaurus™ Process User Guide
I-2013.12
React Diffusion Model
The React model is similar to the ChargedReact model, except that the reaction rates are
not charge state–dependent and the electron concentration is computed directly from the net
doping concentration. In addition, the Frank–Turnbull mechanism is not considered.
The reactions considered are:
(154)
(155)
where is the dopant, is the interstitial, and is the vacancy.
The following set of differential equations represents the model:
(156)
(157)
(158)
where is the concentration of substitutional (and assumed to be immobile) dopant and
is the concentration of ‘free’ defects of type (either interstitials or vacancies), that is, those
defects not in clusters or pairs.
Next, the flux of the mobile–defect pair is considered. In this model, the reaction rates are
assumed to be independent of the charge state, so the pair charging constants are only needed
for the flux of the pairs and are absorbed into the diffusivity of the pairs in this way:
(159)
AI+AI↔
AV+AV↔
A
I
V
t∂
∂CARAI
–RAV
–RA
clus
–=
t∂
∂CAX JAX
∇•–RAX RAX
clus
–+=
t∂
∂CXJX
∇•–RIV
–RAX
–RX
clus
–=
CA
CX
X
JAX
DAXc
n
ni
----
c–z–
c
–
BAX
------------------------------------------------CAX
CX
*
----------n
ni
----
z
∇=
4: Diffusion
Transport Models
Sentaurus™ Process User Guide 213
I-2013.12
where is the charge state of dopant , is either interstitial or vacancy, and is the
effective diffusivity of dopant point-defect pair at charge state and is related to the bare
diffusivity, by:
(160)
where is a set of equilibrium charging constants for defect defined by Eq. 138, p. 208
and Eq. 139, p. 208, and is the equilibrium concentration of the neutral defects defined by
Eq. 137, p. 208.
To set , use:
pdbSet <material> <dopant> <defect> D <c> {<n>}
where:
■<material> is a material name (see Material Specification on page 52).
■<dopant> is one of the existing Sentaurus Process dopants.
■<defect> is either Interstitial or Vacancy.
■<c> is the charge state.
■<n> is a Tcl expression that returns a number; it can be simply a number.
One commonly used Tcl procedure for setting parameters is Arrhenius. This procedure takes
a prefactor and an energy as arguments and returns .
You ca n mod if y t he enti re arr ay with t he co mmand (for example, arsenic–vacancy pairs):
pdbSet Si Arsenic Vac D {
0 {[Arrhenius 0.0 3.45]}
-1 {[Arrhenius 12.8 4.05]}
}
The defect flux is the same as the ChargedReact model and is given by Eq. 140, p. 209.
The reaction can be written as:
(161)
where is either interstitial or vacancy, is the binding coefficient of defect and dopant
, is the rate constant for the chemical reaction, and is the active portion of .
z
A
X
DAXc
c
dAXc
DAXcCX0
*BAXkXcd
AXc
=
DAXc
0D–AXc
E
kBT
---------------
exp=
kXc
X
CX0
*
DAXc
prefactor energy
–
kBT
--------------------
exp⋅
JX
RAX KAXr CA
+CX
CAX
BAX
----------–
≡
X
BAX
X
A
KAXr
CA
+
CA

4: Diffusion
Transport Models
214 Sentaurus™ Process User Guide
I-2013.12
The binding term between the defect and dopant also follows the Arrhenius law:
(162)
The term can be changed with the command:
pdbSet <material> <dopant> <defect> Binding {<n>}
The chemical reaction term is expressed with:
(163)
and can be modified by using the command:
pdbSet <material> <dopant> <defect> Krate {<n>}
The defect recombination rate is the same as in the ChargedReact model and is given by
Eq. 151, p. 211.
ChargedPair Diffusion Model
The ChargedPair diffusion model assumes that the dopant–defect pairs are in local
equilibrium with the dopant and defect concentration. Point defects themselves are not
assumed to be in equilibrium. The kick-out mechanism that describes the dopant–defect
pairing is given by:
(164)
(165)
and is assumed to be in equilibrium. In these two equations, is the dopant, is the interstitial,
is the vacancy, and is the charge state.
The differential equations solved with this model are:
(166)
(167)
BAX BAX0
BAXE
–
kT
----------------
exp=
KrKr0
K–rE
kT
------------
exp=
RIV
AzIc
+AI zc+()
↔
AzVc
+AV zc+()
↔
A
I
V
c
t∂
∂CAJA
∇•–RA
clus
–=
t∂
∂CXtotal
() JXJA
∇•–∇•–RIV
–RX
clus
–=
4: Diffusion
Transport Models
Sentaurus™ Process User Guide 215
I-2013.12
where is the total interstitial concentration including dopant–defect pairs
but excluding clusters, is the sum of AI and AV pair fluxes, is the defect flux, and
is the total dopant concentration excluding clusters.
To write an expression for the pair fluxes, it is necessary to first define the equilibrium
constants, , for the pairing reactions:
(168)
where is either interstitial or vacancy, is the charge state of the point defect, is the
active portion of , and is the concentration of the neutral point defect . The ionization
equilibrium constant is given by Eq. 138, p. 208 and Eq. 139, p. 208.
The pairing coefficients for the dopant–defect pairs with different charge states, , can be
modified with the command:
pdbSet <material> <dopant> <defect> ChargePair <c> {<n>}
where <material> is a material name (see Material Specification on page 52), <dopant> is
one of the existing Sentaurus Process dopants, <defect> is either Interstitial or
Vacancy, <c> is the charge state, and <n> is a Tcl expression that returns a number; it can be
simply a number.
The flux for the impurity is given by:
(169)
where is the effective diffusivity of dopant point-defect pair at charge state , is the
charge state of dopant , is the active portion of , is the concentration of the neutral
point defect , and is the equilibrium concentration of the same defect and is given by
Eq. 137, p. 208.
The effective diffusivity is related to the bare dopant–defect diffusivity, , by:
(170)
You ca n set by us in g:
pdbSet <material> <dopant> <defect> D <c> {<n>}
CXtotal CXCAX
+≡
X
JA
JX
CA
kAXc
CAX CA
+CX0kAXckXc
n
ni
----
c–
c
≡
X
c
CA
+
CA
CX0
X
kXc
kAXc
JADAXc
n
ni
----
c–z–CA
+CX0
CX0
*
-------- n
ni
----
z
∇
cX,
–=
DAXc
c
z
A
CA
+
CA
CX0
X
CX0
*
dAXc
DAXcCX0
*kXck
AXcdAXc
=
DAXc
0D–AXc
E
kBT
---------------
exp=
DAXc

4: Diffusion
Transport Models
216 Sentaurus™ Process User Guide
I-2013.12
NOTE Diffusion coefficients for the ChargedPair model and Dstar for
the ChargedFermi model include the interstitial efficiency factors.
Both the flux for the defects ( ) and the defect recombination rate ( ) are the same as the
ChargedReact model and are given by Eq. 140, p. 209 and Eq. 151, p. 211, respectively.
Pair Diffusion Model
The Pair diffusion model is similar to the ChargedPair model except that the reaction rates
are not charge state–dependent, and the electron concentration is computed directly from the
net doping concentration. In addition, the Frank–Turnbull mechanism is not considered. The
kick-out mechanism, which describes the dopant–defect pairing, is given by:
(171)
(172)
and is assumed to be in equilibrium. In these two equations, is the dopant, is the interstitial,
and is the vacancy.
As in the ChargedPair model, the following set of differential equations is solved:
(173)
(174)
where is the total interstitial concentration including dopant–defect pairs
but excluding clusters, is the sum of AI and AV pair fluxes, is the defect flux, and
is the total dopant concentration excluding clusters.
An equilibrium constant for the pairing reactions is defined and given by:
(175)
where is either interstitial or vacancy, is the binding coefficient of defect and dopant
, and is the active portion of .
The binding term between the defect and dopant also follows the Arrhenius law:
(176)
D
JX
RIV
AI+AI↔
AV+AV↔
A
I
V
t∂
∂CAJA
∇•–RA
clus
–=
t∂
∂CXtotal
() JXJA
∇•–∇•–RIV
–RX
clus
–=
CXtotal CXCAX
+≡
X
JA
JX
CA
CAX BAXCA
+CX
≡
X
BAX
X
A
CA
+
CA
BAX BAX0
BAXE
–
kT
----------------
exp=

4: Diffusion
Transport Models
Sentaurus™ Process User Guide 217
I-2013.12
The term can be changed with the command:
pdbSet <material> <dopant> <defect> Binding {<n>}
The flux for the impurity is given by:
(177)
where represents the diffusivity of dopant point-defect pairs at charge state , is the
charge state of dopant , is the active portion of , and is either interstitial or vacancy.
The effective diffusivity is related to the bare dopant–defect diffusivity, , by:
(178)
where is the equilibrium concentration of the neutral point defect and the ionization
equilibrium constant, , is defined by Eq. 138, p. 208 and Eq. 139, p. 208.
To modify diffusivity terms, use the command:
pdbSet <material> <dopant> <defect> D <charge> {<n>}
Both the flux for the defects ( ) and the defect recombination rate ( ) are the same as the
ChargedReact model and are given by Eq. 140, p. 209 and Eq. 151, p. 211, respectively.
ChargedFermi Diffusion Model
The ChargedFermi diffusion model is similar to the ChargedPair diffusion model, except
that charged point defects are considered to be in equilibrium; no point-defect equations are
solved for inert diffusion conditions if the point defect clusters are not turned on. If the point
defect clusters are turned on or the oxidation is on, the point defect equations will be turned on
automatically. The substitutional dopants are immobile and the total dopant flux is due to the
dopant–defect pairs. The following set of differential equations is solved along with the
potential equation Eq. 354, p. 276:
(179)
JADAXc
n
ni
----
c–z–CA
+CX
CX
*
-------n
ni
----
z
∇
cX,
–=
DAXc
c
z
A
CA
+
CA
X
dAXc
DAXcCX0
*kXcBAXdAXc
=
DAXc
0D–AXc
E
kBT
---------------
exp=
CX0
*
X
kXc
JX
RIV
t∂
∂CAJA
∇•–=
4: Diffusion
Transport Models
218 Sentaurus™ Process User Guide
I-2013.12
As in the ChargedPair model, a set of pairing constants ( ) that define the pair
concentration is defined:
(180)
where:
■ is either interstitial or vacancy.
■ is the charge state of the point defect.
■ is the charge state of dopant .
■ is the active portion of .
The ionization equilibrium constant is given by Eq. 138, p. 208 and Eq. 139, p. 208.
The dopant flux is given by:
(181)
where:
■ is the effective diffusivity of dopant point–defect pairs at charge state .
■ is the concentration of the neutral point defect .
■ is the equilibrium concentration of the same defect and is given by Eq. 137, p. 208.
■ will be equal to if the point-defect equations are switched off.
■ is related to the bare diffusivity by:
(182)
To set use:
pdbSet <material> <dopant> <defect> Dstar <c> {<n>}
The pairing coefficients for the dopant–defect pairs with different charge states, , can be
defined with the command:
pdbSet <material> <dopant> <defect> ChargePair <c> {<n>}
NOTE If the point-defect clusters are switched off and oxidation is switched
on, only the interstitial point-defect equation will be switched on.
kAXc
CAX CA
+CX0kAXckXc
n
ni
----
c–
c
≡
X
c
z
A
CA
+
CA
kXc
JA
JADAXc
n
ni
----
c–z–CA
+CX0
CX0
*
-------- n
ni
----
z
∇
cX,
–=
DAXC
c
CX0
X
CX0
*
CX0
CX0
*
DAXC
dAXc
DAXcCX0
*kAXckXcd
AXc
=
DAXc
0D–AXc
E
kBT
---------------
exp=
DAXc
kAXc

4: Diffusion
Transport Models
Sentaurus™ Process User Guide 219
I-2013.12
Fermi Diffusion Model
The Fermi diffusion model is more complex than the Constant diffusion model. It assumes
that point defects are in equilibrium and it includes electric-field effects. Therefore, the point-
defect equations are not solved.
As in the ChargedPair model, the following set of differential equations is solved:
(183)
where is the sum of AI and AV pair fluxes, and is the total dopant concentration
including clusters.
An equilibrium constant for the pairing reactions is defined and given by:
(184)
where is either interstitial or vacancy, is the binding coefficient of defect and dopant
, and is the active portion of . The dopant flux is given by:
(185)
where:
■ is the charge state of the point defect.
■ is the charge state of dopant .
■ is the active portion of .
■ is either interstitial or vacancy.
■ is the effective diffusivity of dopant point-defect pairs at charge state and is related
to the bare diffusivity by:
(186)
The pairing ratio only appears in the formula for and cannot be modified independently
in the Fermi model. You can set by using:
pdbSet <material> <dopant> <defect> Dstar <c> {<n>}
t∂
∂CAJA
∇•–=
JA
CA
CAX BAXCA
+CX
≡
X
BAX
X
A
CA
+
CA
JADAXc
n
ni
----
c–z–CA
+n
ni
----
z
∇
Xc,
–=
c
z
A
CA
+
CA
X
DAXC
c
dAXc
DAXcCX0
*BAXkXcd
AXc
=
DAXc
0D–AXc
E
kBT
---------------
exp=
BAX
D
DAXc

4: Diffusion
Transport Models
220 Sentaurus™ Process User Guide
I-2013.12
Constant Diffusion Model
The Constant diffusion model is the simplest diffusion model used in Sentaurus Process and
is mainly for dopant diffusion in oxide. It assumes that there is no interaction between dopants
and point defects, and that there are no electric-field effects on dopant diffusion. The point-
defect equations are also switched off. The impurity diffusion is given by:
(187)
where is the intrinsic diffusivity of the impurity and is the active portion of .
The diffusivity follows the Arrhenius law:
(188)
For example, the command:
pdbSet Silicon Arsenic Dstar {[Arrhenius 6.66e-2 3.44]})
sets the to and to 3.44 eV. The general format of the command
is:
pdbSet <material> <dopant> Dstar {<n>}
NOTE Unlike the ChargedFermi model, Dstar is not defined as an array for
the Constant model.
NeutralReact Diffusion Model
NeutralReact diffusion in silicon is close to the React model (see React Diffusion Model
on page 212) except that there are no charged atoms. The model can be switched on using the
command:
pdbSet <material> <dopant> DiffModel NeutralReact
NeutralReact diffusion in silicon is described by a kick-out mechanism [1]. Other
mechanisms such as dissociation and clustering can also be taken into account. In the
integration in Sentaurus Process, each of these mechanisms is described by one or more terms:
(189)
(190)
t∂
∂CADstar CA
+
∇()∇•=
Dstar
A
CA
+
CA
Dstar Dstar0
DstarE
–
kT
-------------------
exp=
Dstar0
6.66 10 2–
× cm2/s
DstarE
AI+AI↔
AI V+A↔

4: Diffusion
Transport Models
Sentaurus™ Process User Guide 221
I-2013.12
where is the substitutional dopant, is the mobile dopant–interstitial pair, is interstitial,
and is vacancy. The first reaction is the kick-out reaction and the second one is the
dissociation reaction. These reaction can be written as:
(191)
(192)
where:
■ is the concentration of substitutional dopant atoms.
■ is the concentration of mobile dopant atoms.
■ and are the forward reaction rates.
■ and are the binding coefficients.
To set, use:
pdbSet <material> <dopant> Interstitial Kf {<n>}
pdbSet <material> <dopant> Vacancy Kf {<n>}
pdbSet <material> <dopant> Interstitial Bind {<n>}
pdbSet <material> <dopant> Vacancy Bind {<n>}
The differential equations that describe the model are:
(193)
(194)
where is the diffusivity of mobile dopant–interstitial pairs and can be set using the
command:
pdbSet <material> <dopant> Interstitial D 0 {<n>}
Carbon Diffusion Model
Carbon diffusion is a typical example for the NeutralReact diffusion model. The kick-out
reaction rate is defined by:
pdbSet Silicon Carbon Interstitial Kf {\
[expr ([pdbGetElement Si Carbon D 0]/([pdbDelayDouble Si Carbon\
MigrationLength]* [pdbDelayDouble Si Carbon\
MigrationLength]*[pdbDelayDouble Si Int Cstar]))]\
}
A
AI
I
V
RKickOut KfI CACIBAICAI
–()≡
RDissociation KfV CAICVCI
*CV
*BAVCA
–()≡
CA
CAI
KfI
KfV
BAI
BAV
t∂
∂CARDissociation RKickOut
–=
t∂
∂CAI DAI0CAI
∇()RKickOut RDissociation
–+∇•=
DAI0

4: Diffusion
Transport Models
222 Sentaurus™ Process User Guide
I-2013.12
will set to .
is the migration length (cm) of carbon atoms and is the diffusivity of carbon, and these
parameters can be set using the commands:
pdbSet <material> Carbon MigrationLength {<n>}
pdbSet <material> Carbon D 0 {<n>}
For the details of the carbon-clustering model, see Carbon Cluster on page 303.
Nitrogen Diffusion Model
Nitrogen diffusion is defined according to the Constant diffusion model by default. However,
instead of the Constant model, the NeutralReact diffusion model can be used for nitrogen
diffusion. If the NeutralReact model is specified for nitrogen diffusion, the nitrogen dimer
forms and diffuses. The dimer is formed by the following reaction:
(195)
In the above reaction, is the monomer, in other words, nitrogen interstitial , and
denotes the dimer , which has the solution name NDimer. The nitrogen monomer and
dimer equations are formulated by:
(196)
(197)
(198)
(199)
(200)
The reaction for dimer formation is given by:
(201)
KfI
DC0
λ2CI
*intrinsic()
----------------------------------------
λ
DC0
NI NI+N2↔
NI
Ni
N2
Ni
()
2
t∂
∂CNRDissociation RKickOut
–RNV
–=
t∂
∂CNI DNI0CNI
∇()RKickOut RDissociation
–RN2
–+∇•=
t∂
∂CN2DN2CN2
∇()RN2RN2V
–+∇•=
t∂
∂CN2VRN2VRN2V2
–=
t∂
∂CN2V2RN2V2
=
RN2
RN2KfN2CNICNI BN2CN2
–()≡

4: Diffusion
Transport Models
Sentaurus™ Process User Guide 223
I-2013.12
where:
■ is the concentration of nitrogen monomers.
■ is the concentration of nitrogen dimers.
■ is the forward reaction rate.
■ is the binding coefficient.
To set, use:
pdbSet <material> NDimer Kf {<n>}
pdbSet <material> NDimer Bind {<n>}
NOTE NDimer is a cluster of nitrogen which can be diffused by the
NeutralReact diffusion model and initialized by nitrogen cluster
initialization. For details on the nitrogen clusters NV, N2V, a n d N 2V2,
see Nitrogen Cluster on page 304.
Mobile Impurities and Ion-Pairing
The ion-pairing model includes the pairing of positively and negatively charged dopant
ions [2][3][4]. Ion-pairing reduces the diffusivity of dopants where the concentration of
dopants of the opposite type is large. The ion-pairing model assumes that positively charged
donors can bind with negatively charged acceptors to form neutral pairs. The ion-pairing model
is significant because it allows the dependency of the impurity diffusivity to be modeled in both
n-type and p-type materials. In particular, it reduces the effective diffusivity of boron in n-type
materials without affecting its diffusivity at high p-type concentrations.
The model reduces the mobile concentration of dopant species by the following factors:
for donor species (202)
for acceptor species (203)
where:
■ and are the total concentrations of electrically active donors and acceptors,
respectively.
■ is the concentration of ion pairs.
■ and are the ion-pairing factors for donors and acceptors, respectively.
CNI
CN2
KfN2
BN2
fpd 1Np
Nd
------–
=
fpa 1Np
Na
------–
=
Nd
Na
Np
fpd
fpa

4: Diffusion
Solid Phase Epitaxial Regrowth Model
224 Sentaurus™ Process User Guide
I-2013.12
The concentration of ion pairs is given by:
(204)
The parameter is given by:
(205)
where Ion.Pair.Omega is a parameter for material; the default value for silicon and
polysilicon is 6.0 [3].
The ion-pairing model is enabled or disabled for each material by the Ion.Pair parameter.
By default, it is disabled for all materials.
Solid Phase Epitaxial Regrowth Model
The solid phase epitaxial regrowth (SPER) model simulates the movement of amorphous and
crystalline boundaries due to the recrystallization of the amorphous silicon and the dopant
dynamics during such process. The SPER model is switched on by:
pdbSet Diffuse SPER 1
The boundary movement is described with the specific solution fields, either the distance field
by the level-set method or the phase field by the phase-field method. You can select one of the
models by:
pdbSet Diffuse SPER.Model {LevelSet | PhaseField} ;# default=LevelSet
Level-Set Method
The level-set method solves the equation for the distance field , which is named with
AmorpDistance:
(206)
where:
■ is the recrystallization velocity perpendicular to a boundary surface.
■ is positive in an amorphous region, negative in a crystalline region, and zero at an
amorphous–crystalline boundary.
Np
Np
1
2
---NdNaΩ++()NdNaΩ++()
24NdNa
––()=
Ω
ΩIon.Pair.Omega ni
⋅=
ϕ
ϕ∂
t∂
------ v∇ϕ+0=
v
ϕ

4: Diffusion
Solid Phase Epitaxial Regrowth Model
Sentaurus™ Process User Guide 225
I-2013.12
The velocity is defined by:
(207)
where:
■ is the scaling factor.
■ is the orientation-dependent velocity.
■ is the scaling factor of velocity near surfaces.
■ is the shortest distance from the surface.
■ is the characteristic length for velocity reduction near surfaces.
pdbSet Silicon SPER V.Factor {<expression>} ;# f_v (unitless)
pdbSet Silicon SPER V100 {<n>} ;# cm/sec
pdbSet Silicon SPER V110 {<n>} ;# cm/sec
pdbSet Silicon SPER V111 {<n>} ;# cm/sec
pdbSet Silicon SPER VsurfScale {<n>} ;# s_v (unitless)
pdbSet Silicon SPER VsurfScaleLength {<n>} ;# L_vr (um)
The tensor mesh structure to solve the level-set equation is defined by:
pdbSet Grid SPER TensorMeshSpacing {X <n> Y <n> Z <n>} ;# (um)
The level-set algorithm used is the general time-stepping initial-value formulation as described
in MGOALS Interface on page 778.
It is assumed that all dopant atoms are mobile in an amorphous region. The diffusion
coefficient of the mobile species in the amorphous region is specified by:
pdbSet Silicon <dopant> DAmor {<n>} ;# cm2/sec
It has been experimentally observed that during regrowth of an amorphous layer, dopants can
be swept along by the amorphous–crystalline boundary. The physical mechanism for this
sweeping behavior is not well understood. To model this effect, a phenomenological model has
been introduced as follows:
(208)
where:
■ is the user-defined multiplication factor.
■ is the local speed of distance variation.
■ is the characteristic length of dopant drift.
■ is the drift probability near material interfaces.
v
vf
vvori V100 V110 V111,,()11sv
–() d
Lvr
-------–
exp–
⋅⋅=
fv
vori
sv
d
Lvr
t∂
∂X∇fDαvDLd11P–() d
Ldr
--------–
exp–
X∇αS
⋅=
fD
vD
Ld
P
4: Diffusion
Solid Phase Epitaxial Regrowth Model
226 Sentaurus™ Process User Guide
I-2013.12
■ is the shortest distance from material interfaces.
■ is the characteristic length for drift reduction near material interfaces.
■ is the amorphous state calculated by:
(209)
where is the phase transition width. is 1.0 and 0.0 in a completely amorphous and
crystalline region, respectively.
■ is the shifted amorphous state given by:
(210)
pdbSet Silicon SPER PhaseTransWidth {<n>} ;# w_T (um)
pdbSet Silicon SPER DriftWidth {<n>} ;# w_D (um)
pdbSet Silicon SPER DriftFactor {<expression>} ;# f_D (unitless)
pdbSet Silicon SPER DriftDistance {<n>} ;# d_D (um)
pdbSet Silicon SPER SurfaceDriftProbability {<n>} ;# P (unitless)
pdbSet Silicon SPER DriftReductionLength ;# L_dr (um)
pdbSetDouble Silicon <dopant> SPER.DriftLength {<n>} ;# L_d (um)
The parameters SurfaceDriftProbability and DriftReductionLength can be
specified for a specific dopant by:
pdbSetDouble Silicon <dopant> SPER.SurfaceDriftProbability {<n>}
pdbSetDouble Silicon <dopant> SPER.DriftReductionLength {<n>}
To control the clustering rate in the region between amorphous and crystalline regions, an
additional term can be defined by:
pdbSetString Si <cluster> SPERBoundaryTerm {<expression>}
pdbSetString Si <dopant> SPERBoundaryTerm {<expression>}
NOTE The term added by SPERBoundaryTerm of <cluster> must be
correctly subtracted by SPERBoundaryTerm of <dopant> so that the
total dose conservation is kept, for example:
pdbSetString Si As3 SPERBoundaryTerm "-1e1*(0.99*AsTotal-3.0*As3)"
pdbSetString Si Arsenic SPERBoundaryTerm "3e1*(0.99*AsTotal-3.0*As3)"
d
Ldr
α
α1
2
---erfc⋅ϕ
wT
------–()=
wT
α
αS
αS
1
2
---erfc⋅ϕdD
–
wD
----------------–()=

4: Diffusion
Solid Phase Epitaxial Regrowth Model
Sentaurus™ Process User Guide 227
I-2013.12
The full equation of a dopant is described by:
(211)
where is defined by SPERBoundaryTerm.
The maximum time step during SPER is set by:
(212)
Phase Field Method
Since the level-set method requires Cartesian grids to calculate the distance field, it may cause
instability from the difficult time-step control as well as the interpolation error due to the
decoupled method with the different mesh structure. The phase field method uses a consistent
mesh structure, so that the phase and the other solutions are coupled seamlessly into the
hydrodynamic Scharfetter–Gummel discretization scheme, which improves the convergence if
there is high drift due to an abrupt phase change.
The phase field method solves the equation for the phase field , which is named with
SPERPhase:
(213)
where:
■, , and are the relaxation time, the phase transition width, and the phase (–1 for
completely amorphous, 1 for completely crystalline), respectively.
■ and are given by the parameters PhaseTransWidth and Lambda.Fac, respectively.
The inverse of the relaxation time is calculated by:
(214)
where:
■ and are the isotropic and anisotropic multiplication factors, respectively.
■ is the relaxation rate.
t∂
∂X1α–()
t∂
∂X
crystal
α∇ Damor∇X
⋅∇++fDαvDLd11P–() d
Ldr
--------–
exp–
X∇αS
R9ϕ2
WT
2
---------–
exp⋅–⋅
=
R
tmax
∆min SPER.TimeStepScale WT
max v()
-----------------
⋅SPER.MaxTimeStep,()=
ϕ
τϕ∂
t∂
------ w2∇2ϕϕ
21–()ϕλϕ
21–()–()–=
τ
w
ϕ
w
λ
τ
τ1–fisofanisoREaniso ESv ESs
++
kT
---------------------------------------------–
exp=
fiso
faniso
R

4: Diffusion
Solid Phase Epitaxial Regrowth Model
228 Sentaurus™ Process User Guide
I-2013.12
■, and are the orientation-dependent, the hydrostatic stress–dependent, and
shear stress–dependent activation energies, respectively.
(215)
(216)
(217)
pdbSet Silicon SPER Relax.Rate {<n>} ;# R(1/sec)
pdbSet Silicon SPER R.Fac {<expression>} ;# f_iso (unitless)
pdbSet Silicon SPER R.Fac.Aniso {100 <n> 110 <n> 111 <n>} ;# f_aniso (unitless)
pdbSet Silicon SPER E.Aniso {100 <n> 110 <n> 111 <n>} ;# E_aniso (eV)
pdbSet Silicon SPER VFrecrys {<n>} ;# (cm-3)
pdbSet Silicon SPER Shear.Coupling {<n>} ;# (eV)
The phase field method assumes no diffusion in a crystalline region during SPER, so that the
diffusion equation of a dopant is formulated by:
(218)
where is the chemical potential energy difference to cause the dopant segregation at an
amorphous–crystalline boundary.
is given by the parameter SPER.Energy that you can define with a string expression:
pdbSet Silicon <dopant> SPER.Energy {<expression>} ;# E_seg (eV)
The maximum time step during SPER is set by:
(219)
As soon as regrowth is completed, the dopant activation in the regrowth region is performed
with the pdb parameter AmInit or the term ${Sol}AmInit.
NOTE Since the dopant active concentration is initialized after regrowth is
completed, the evaluated values of some terms, such as ${Sol}Total
and ${Sol}Active can be incorrect during SPER.
Eaniso
ESv
ESs
Eaniso E.Aniso ϕ∇
ϕ∇
------------
⋅
100 110 111,,
=
ESv PVFrecrys⋅=
ESs Shear.Coupling εxy εyz εzx
++()⋅=
X∂
t∂
------ 1ϕ–
2
------------
∇Damor∇XD
amorXEseg
kT
---------- ∇ϕ+
⋅=
Eseg
Eseg
tmax
∆min SPER.TimeStepScale τmin
4
----------
⋅SPER.MaxTimeStep,()=

4: Diffusion
Flash or Laser Anneal Model
Sentaurus™ Process User Guide 229
I-2013.12
Flash or Laser Anneal Model
The flash or laser anneal model becomes necessary for an advanced process that requires
diffusionless, but high activation. The model can simulate the inhomogeneous thermal
distribution, which results in better accuracy for stress calculation as well as heat transfer delay
to the region in which the devices form. The flash or laser model is not available for 3D
simulation yet.
The solution name of the local temperature (in kelvin) is Temperature. The model is
switched on by specifying the Boolean parameter laser in a diffusion statement. For example:
diffuse temperature=500 time=1<ms> laser
For a melting laser anneal, the phase field variable is introduced to describe whether the
material is liquid or solid. The solution name of the phase is HeatPhase. The melting laser
anneal model is invoked by switching on the Boolean parameter Use.Melting.Laser:
pdbSet Heat Use.Melting.Laser 1
The heat transfer equation is:
(220)
where:
■, , and are the conductivity, the mass density, and the specific heat capacity,
respectively. and can depend on temperature.
■ is the unit mass latent heat (Eq. 232). The phase-dependent term takes the heat
consumption (or generation) due to the solid-to-liquid (or liquid-to-solid) phase change
into account.
and vary with the liquid or solid phase as follows:
(221)
(222)
pdbSet <material> SpecificHeatCapacity {<expression>} ;# Cps (J/kg/K)
pdbSet <material> Liquid.SpecificHeatCapacity {<expression>} ;# Cpl (J/kg/K)
pdbSet <material> ThermalConductivity {<expression>} ;# ks (W/cm/K)
pdbSet <material> Liquid.ThermalConductivity {<expression>} ;# kl (W/cm/K)
T
ϕ
ϕ
ρcP
∂T
∂t
------ κT∇()∇•G30ρLϕ21ϕ–()
2
t∂
∂ϕ
++=
κ
ρ
cP
κ
cP
L
κ
cP
cPϕcPs 1ϕ–()cPl
+=
κϕκ
s1ϕ–()κ
l
+=
4: Diffusion
Flash or Laser Anneal Model
230 Sentaurus™ Process User Guide
I-2013.12
The heat generation rate is calculated by:
(223)
where , , and represent the intensity, absorptivity, and depth, respectively. The
absorptivity is given by the user-defined expression:
pdbSet <material> Absorptivity {<expression>} ;# cm-1
See Intensity Models for Flash Anneal on page 236 and Intensity Model for Scanning Laser on
page 238 for the intensity models.
The governing equation of the phase field is given by:
(224)
where:
■, , and are the melting interface mobility, the surface tension, and the interface
thickness, respectively. and depend on material crystallinity.
■ is the seed function to start melting.
■ is the interface response function to describe the front moving velocity of the flat
melting interface:
(225)
(226)
where the subscripts and of and indicate the amorphous and crystalline materials,
respectively. The is the degree of the structural disorder in a material that is calculated
by:
(227)
where is the Frenkel pair concentration by implantation damage, and is the
amorphous threshold to determine the amorphous and crystal transition.
pdbSet <material> Melting.Interface.Mobility {<n>} ;# uc (cm4/J/sec)
pdbSet <material> Amorphous.Melting.Interface.Mobility {<n>} ;# ua (cm4/J/sec)
pdbSet <material> Surface.Tension {<n>} ;# gammac (J/cm2)
pdbSet <material> Amorphous.Surface.Tension {<n>} ;# gammaa (J/cm2)
pdbSet <material> AmorpDensity {<n>} ;# Dmax (cm-3)
G
GIααld
l0=
ld=
–
exp⋅=
I
α
d
ϕ
∂ϕ
∂t
------ µγ ϕ∇2µγ
δ2
------ϕ1ϕ–()12ϕ–()5vint
δ
------- ϕ21ϕ–()
2sTϕ,()++–=
µ
γ
δ
µ
γ
sTϕ,()
νint
µαµ
a1α–()µ
c
+=
γαγ
a1α–()γ
c
+=
a
c
µ
γ
α
α1
2
---1 10 DFP
Dmax
------------
ln
tanh+
=
DFP
Dmax
4: Diffusion
Flash or Laser Anneal Model
Sentaurus™ Process User Guide 231
I-2013.12
The seed function is modeled by:
(228)
where is the melting point. and are the multiplier and the temperature to control
initial melting. The seed term in Eq. 224 is switched off when is reduced to less than the
SeedOffPhase value:
pdbSet Heat Seed.Factor {<expression>} ;# fs (unitless)
pdbSet Heat Seed.Temperature {<n>} ;# Ts (K)
pdbSet Heat SeedOffPhase {<n>} ;# unitless
It is known that the melting point varies with the dopant concentration, such as for germanium,
as well as material crystallinity. The melting point is calculated by:
(229)
where , , and are the degree of structural disorder, and the melting point of
amorphous material and crystalline material, respectively. is the concentration of the
dopant that affects the melting point. For example:
pdbSet Silicon Melting.Point 1690 ;# Tmc0
pdbSet Silicon Amorphous.Melting.Point 1420 ;# Tma0
pdbSet Silicon Dop.Dep.Melting.Point { Ge {960 1211} } ;# Tma1 and Tmc1
The interface response function is modeled by the Frenkel–Wilson law [5]:
(230)
where is the lattice density. The liquid-to-solid interface transfer rate is given by:
(231)
where the model can be selected by:
pdbSet <material> Melting.Velocity.Model {Arrhenius | FulcherVogel}
The latent heat and the liquid-to-solid interface transfer rate depend on crystallinity as follows:
(232)
sTϕ,() fs
δ2
µγ
------ϕ11
2
---erfc TT
m
–
Ts
----------------()–
–=
Tm
fs
Ts
ϕ
TmαTma0Tma1Tma0
–()
Cx
Cmax
------------+
1α–()Tmc0Tmc1Tma0
–()
Cx
Cmax
------------+
+=
α
Tma0
Tmc0
Cx
Vint
Vint v1ρLC
max
⁄
kTTm
-----------------------TT
m
–()
exp–
=
Cmax
ν
v
v0H
kT
------–
for Arrhenius modelexp
v0f
B
kT T
g
–()
-----------------------–
for Vogel–Fulcher modelexp
=
LαLa1α–()Lc
+=

4: Diffusion
Flash or Laser Anneal Model
232 Sentaurus™ Process User Guide
I-2013.12
(233)
(234)
(235)
(236)
(237)
where the subscripts and indicate the parameter for the amorphous and crystalline
materials, respectively.
pdbSet <material> Latent.Heat {<n>} ;# Lc (J/kg)
pdbSet <material> Amorphous.Latent.Heat {<n>} ;# La (J/kg)
pdbSet <material> Melting.Velocity.0 {<n>} ;# v0c (cm/sec)
pdbSet <material> Melting.Velocity.E {<n>} ;# Hc (eV)
pdbSet <material> Amorphous.Melting.Velocity.0 {<n>} ;# v0a (cm/sec)
pdbSet <material> Amorphous.Melting.Velocity.E {<n>} ;# Ha (eV)
pdbSet <material> FV.Melting.Velocity.0 {<n>} ;# v0c (cm/sec)
pdbSet <material> FV.Melting.Velocity.E {<n>} ;# Hc (eV)
pdbSet <material> FV.Melting.Velocity.T {<n>} ;# Tgc (K)
pdbSet <material> FV.Amorphous.Melting.Velocity.0 {<n>} ;# v0a (cm/sec)
pdbSet <material> FV.Amorphous.Melting.Velocity.E {<n>} ;# Ha (eV)
pdbSet <material> FV.Amorphous.Melting.Velocity.T {<n>} ;# Tga (K)
Dopant Diffusion in Melting Laser Anneal
Since a melting or solidification process occurs too quickly to observe the dopant diffusion in
a solid region, it is assumed that the dopant atoms diffuse only by entropic force and
temperature gradient. The different chemical potentials of dopants at liquid, solid, and
boundary regions induce the segregation. During melting laser anneal, the temperature varies
greatly depending on the location. Therefore, the dopant diffusion equation must be solved by
coupling it to the heat equation (Eq. 220) and the phase equation (Eq. 224):
(238)
v0αv0a1α–()v0c
+=
HαHa1α–()Hc
+=
v0fαv0fa 1α–()v0fc
+=
BαBa1α–()Bc
+=
Tg0αTga 1α–()Tgc
+=
a
c
t∂
∂CDCeq
Ceq0
----------- CCeq0
Ceq
-----------
∇
∇•=
4: Diffusion
Flash or Laser Anneal Model
Sentaurus™ Process User Guide 233
I-2013.12
(239)
(240)
where:
■, , and are the dopant diffusivities in a liquid, a liquid–solid interface, and
solid regions, respectively. is calculated by an Arrhenius formula with global
temperature.
■ and are the chemical potential energies in a solid state and an interface state
relative to that in a liquid state, respectively.
pdbSet Heat Max.Liquid.Phase {<n>} ;# phi_L (unitless)
pdbSet Heat Min.Solid.Phase {<n>} ;# phi_S (unitless)
pdbSet <material> <dopant> Dliquid.0 {<n>} ;# cm2/sec
pdbSet <material> <dopant> Dliquid.E {<n>} ;# eV
pdbSet <material> <dopant> Dils.0 {<n>} ;# cm2/sec
pdbSet <material> <dopant> Dils.E {<n>} ;# eV
pdbSet <material> <dopant> Dstar {<n>} ;# cm2/sec (Dsolid)
pdbSet <material> <dopant> Melting.Seg.E {<n>} ;# eV
pdbSet <material> <dopant> Melting.Intf.Seg.E {<n>} ;# eV
To solve the dopant diffusion equation by coupling it with the heat and phase equations, use:
solution name= <solution> Heat
By default, it is applied to boron, phosphorus, arsenic, antimony, and indium impurities.
The instant recrystallization of an amorphous region, that is, the initialization of cluster
solutions, is performed before diffusion. The cluster solutions are reset to zero in a melted
region during diffusion by multiplying by the cluster solutions, which implies that all
dopants in a liquid region are activated fully. Like the cluster solutions, the point-defect and
defect-cluster solutions are reset to zero in liquid regions.
Guideline for Parameter Setting
The default value ( cm) of the variable Heat.Phase.Width (interface thickness ) is
typically good for simulation of melting depths nm. For the larger melting depths (such
D
Dliquid Dils Dliquid
–()
ϕ
ϕL
------ for ϕϕ
L
≤()+
Dils for ϕLϕϕ
S
≤<()
Dsolid Dils Dsolid
–()
1ϕ–
1ϕ–S
--------------- for ϕSϕ≤()+
ϕDsolid 1ϕ–()Dliquid if ϕLϕS
>()+
=
Ceq
Ceq0
----------- ϕEseg 16ϕ21ϕ–()
2Eintf
+
kT
----------------------------------------------------------------–
exp=
Dliquid
Dils
Dsolid
Dsolid
Eseg
Eintf
ϕ
17–
×10
δ
~100

4: Diffusion
Flash or Laser Anneal Model
234 Sentaurus™ Process User Guide
I-2013.12
as ), a value of cm can reduce the simulation time and still maintain
reasonable accuracy:
pdbSet Si Heat.Phase.Width 1e-7 ;# (cm)
Mesh spacing in the molten region must always be smaller than Heat.Phase.Width ( ) to
obtain proper convergence. A larger mesh spacing results in a faster simulation and less noise.
Larger values of can speed up convergence even for a fixed mesh spacing. The simulated
dopant distribution depends on both mesh spacing and . For an equidistant 1D mesh, the
simulation results are almost the same for all mesh spacing < . For an inhomogeneous 1D
mesh, the simulated melting front speed changes when the solid–liquid interface reaches the
region of mesh inhomogeneity, unless the maximum mesh spacing is smaller than . A
mesh finer than may result in a larger CPU time. This basically means that mesh
refinements are allowed only if the background mesh is finer than . Therefore, an
equidistant mesh should be used in the melting region whenever possible.
If mesh spacing is not smaller than the inverse of absorptivity ( ), a numerical error appears
in the expression for heat generation. The total integrated dose of scaled HeatRate is a good
indicator for the presence of such an error which can be checked in the output file. For example,
for a 0.5-nm mesh and an cm-1, the scaled HeatRate dose is equal to
. The analytic total integrated HeatRate dose is equal to . A difference
of more than may cause pronounced increase of the melting depth. Two solutions are
possible:
■A finer mesh at the outer silicon interface. It may be limited by the increase of CPU time
related to the mesh inhomogeneity constraint previously described.
■The laser fluence can be multiplied by the factor TotalHeatRateDose. This is
performed automatically when pdbSet Heat Correct.Energy.Dose 1 is applied.
This option obtains good results at mesheswhen mesh spacing does not satisfy the criteria
mesh spacing << .
Saving a Thermal Profile
To save the thermal profile computed during this step to a file, use the write.temp.file
parameter of the diffuse command. In a subsequent simulation, you can use this file to create
a temperature ramp using the read.temp.file parameter of the temp_ramp command. For
more information, see diffuse on page 908 and term on page 1173.
1 µm
~2 7–
×10
δ
δ
δ
δ
δ8⁄
δ8⁄
δ8⁄
α
α1.46 6
×10=
1.0004 14
×10
1.0 14
×10
1.0 3–
×10
1.0 14
×10
1α⁄

4: Diffusion
Flash or Laser Anneal Model
Sentaurus™ Process User Guide 235
I-2013.12
Boundary Conditions
At the top surface, that is, the gas interface, the heat emission flux from the top material is given
by:
(241)
where is the environment temperature specified by temperature in the diffuse
command.
At the bottom, the boundary condition depends on whether the thermal resistor is attached. If
AttachThermalResistor is switched on, the emission flux at bottom is calculated by:
(242)
where and are the wafer thickness and the bottom coordinate of a simulation structure,
respectively. Otherwise:
(243)
The wafer thickness in micrometers is specified by:
pdbSet Heat WaferThickness <n>
At the sides, the flux is calculated by:
(244)
By default, SideHeatTransfer is set to zero for all materials.
Structure Extension
The heat transfer is much faster in comparison with an impurity or a point-defect diffusion. For
example, in silicon, the diffusion length of the heat temperature is 20–30 times longer than that
of interstitials at . Therefore, solving the heat equation requires a much larger structure
size than for diffusion equations. The model provides the method to temporarily extend the
current structure for solving the heat equation, and then recovers the original structure after
finishing the laser or flash anneal. The downward extension is controlled by the Boolean
parameter ExtendBottom.
F5.6703–10 12–Emissivity T4T0
4
–()⋅⋅ ⋅=
T0
Fκ
twxbot
–
--------------------
–TT
0
–()=
tw
xbot
FHeatSinkTransfer–TT
0
–()=
FSideHeatTransfer–TT
0
–()=
800°C

4: Diffusion
Flash or Laser Anneal Model
236 Sentaurus™ Process User Guide
I-2013.12
The location of the extended bottom is specified by WaferThickness, for example:
pdbSet Heat ExtendBottom 1
pdbSet Heat WaferThickness 700
which are defined by default.
Since the flash light source transfers heat to the whole wafer surface at the same time, no heat
flux is assumed at the structure sides so that you do not have to extend the structure along the
side directions. However, since the laser anneal scans a wafer by beaming a laser on a localized
spot, the structure must be extended to the side directions to correctly take into account the heat
transfer from the beamed spot. The extended distance in micrometers to the sides is defined
with:
pdbSet Heat SideExtension <n>
To reduce the computation time for the extension, one side among the left and right sides is
extended first, and then the extended structure is reflected on the side that is defined by:
pdbSet Heat ReflectSide <Left | Right | None>
For example, the following statements specify an extension of in the right direction
and a reflection of the extended structure on the left side:
pdbSet Heat SideExtension 200
pdbSet Heat ReflectSide Left
When ReflectSide is set to None, each side (that is, both the left and right sides) is extended.
The material of all the extended regions is set to HeatSubstrate. The thermal properties of
the HeatSubstrate material are defined internally to the same as the BulkMaterial
material (default value: Si). In the region of HeatSubstrate, only the heat equation is solved.
Intensity Models for Flash Anneal
Intensity can be specified by a Gaussian model, or a table lookup method, or a user-specified
model:
pdbSet Heat Intensity.Model { Gaussian | Table | User }
Gaussian Model
200 µm
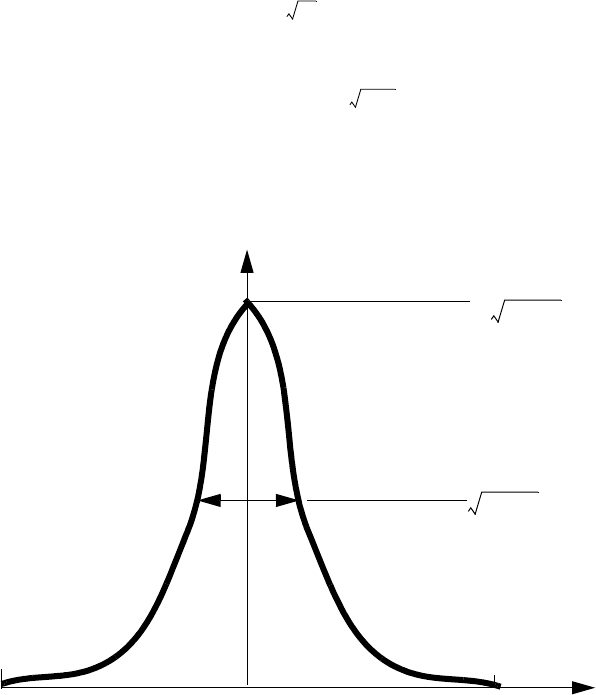
4: Diffusion
Flash or Laser Anneal Model
Sentaurus™ Process User Guide 237
I-2013.12
The intensity can be given by the Gaussian profile as follows:
(245)
(246)
where Pulse is the full width at half maximum (FWHM) time interval. The parameter
Fluence is the energy dose in .
Figure 34 Heat Intensity for flash anneal with respect to time
Table Lookup Method
The table of time versus intensity can be given by:
pdbSet Heat Intensity.Table { <t1> <I1> <t2> <I2> ... <tn> <In> }
The intensity values in the table can be scaled by:
pdbSet Heat Intensity.Table.Factor {<n>}
I
IFluence
2πts
-------------------------tt
0
–()
2
2ts
2
-------------------–
exp=
ts
Pulse
22 2ln
-------------------=
J/cm2
Time
Gaussian
PULSE
I
22π⁄ln Fluence/Pulse⋅
2π⁄ln Fluence/Pulse⋅
03 t
s6 ts

4: Diffusion
Flash or Laser Anneal Model
238 Sentaurus™ Process User Guide
I-2013.12
User-specified Model
You c an de fine t he h eat i nt ensit y profi le by us in g IntensityProfile (unit is ). The
heating time, that is, the light-sourcing time for the user-specified intensity is given by the
parameter HeatingTime. For example, for the sum of two different Gaussian intensities:
set ttime "\[simGetDouble Heat time\]"
set rt2pi [expr sqrt(2*3.141592)]
set tp1 3e-3 #from 3*sigma = 3*1e-3
set tp2 6e-3 #from 3*sigma = 3*2e-3
set ts1 2e-6 #from 2*sigma*sigma = 2*1e-3*1e-3
set ts2 8e-6 #from 2*sigma*sigma = 2*2e-3*2e-3
pdbSet Heat HeatingTime 12e-3
pdbSet Heat IntensityProfile "1e4/$rt2pi*exp(-((($ttime-$tp1)^2)/$ts1)) \
+2.5e3/$rt2pi*exp(-((($ttime-$tp2)^2)/$ts2))"
Here, [simGetDouble Heat time] returns the current time that is used to solve the heat
equation.
Intensity Model for Scanning Laser
The scanning laser beam is characterized with the scanning speed (cm/s), the beam width
(), and the beam fading distance () by diffraction:
pdbSet Heat ScanSpeed <n>
pdbSet Heat BeamWidth <n>
pdbSet Heat BeamFadeDistance <n>
The intensity specification for laser beam precedents that of a flash light source. When a
positive ScanSpeed is specified, the laser scanning model is assumed and the heat intensity is
calculated with the laser beam parameters.
J/cm2/s
µm
µm
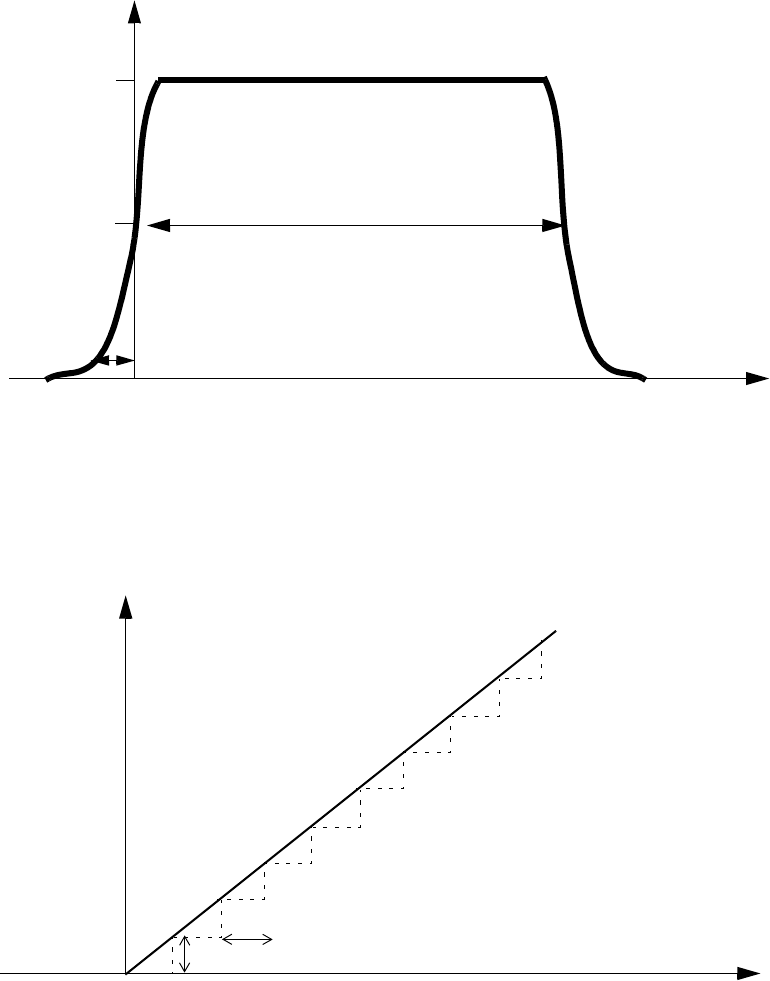
4: Diffusion
Flash or Laser Anneal Model
Sentaurus™ Process User Guide 239
I-2013.12
Two complementary error functions are multiplied to generate the laser beam intensity as
shown in Figure 35.
Figure 35 Heat intensity profile for laser beam
The laser beam moves by the distance (BeamWidth-BeamOverlap) after (BeamWidth-
BeamOverlap)/ScanSpeed anneal time step. As BeamOverlap approaches BeamWidth,
the simulation accuracy is improved.
Figure 36 Beam location along time
0Distance
BeamWidth
0.0786
0.5
1.0
I/BeamIntensity
BeamFadeDistance
1/4 erfc(-y/BeamFadeDistance) erfc((y-BeamWidth)/BeamFadeDistance)
0
Beam Location
Time
reality
model
(BeamWidth-BeamOverlap)/ScanSpeed (10-4s)
(BeamWidth-BeamOverlap)(um)
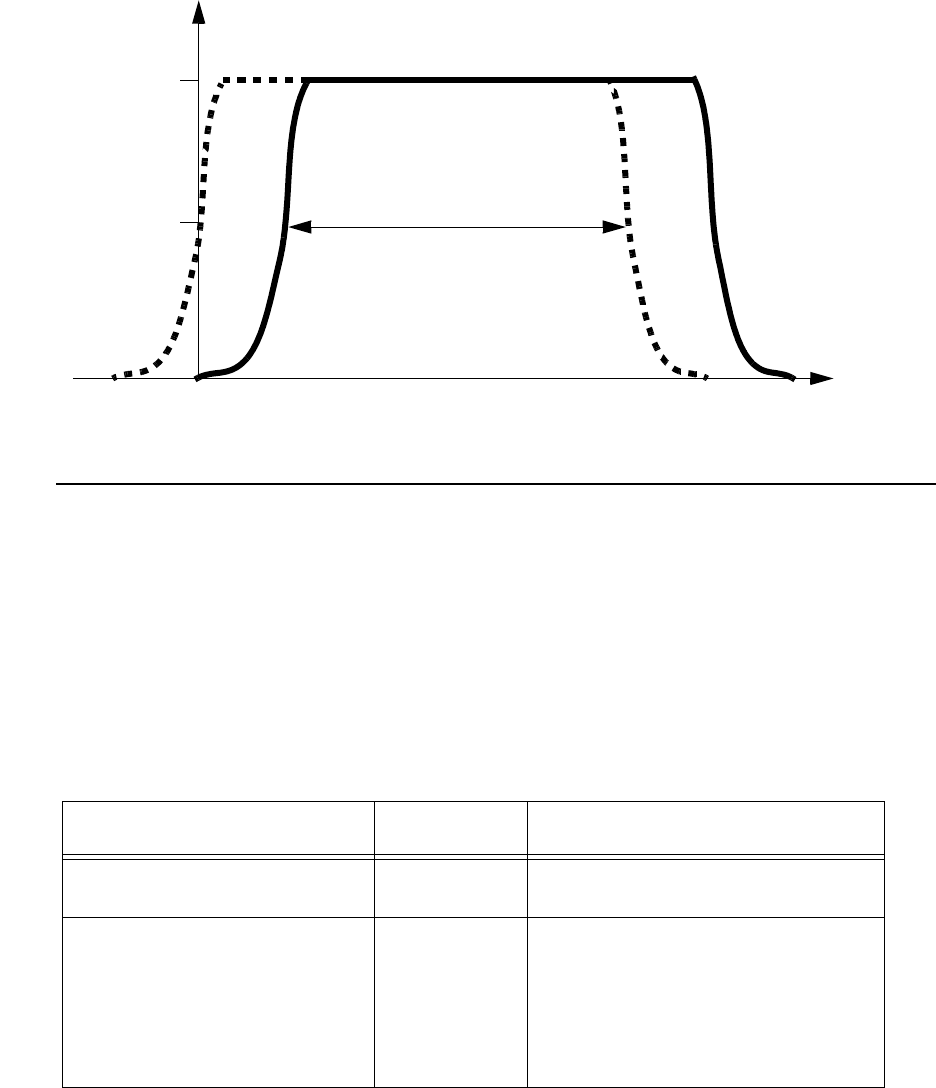
4: Diffusion
Flash or Laser Anneal Model
240 Sentaurus™ Process User Guide
I-2013.12
Figure 37 Laser beam displacement at each time step
Control Parameters
Table 14 lists the control parameters. These parameters must be used with:
pdbSet Heat
For example:
pdbSet Heat HeatingTime 12e-3
pdbSet Heat MaxTimeStep 500
Ta bl e 14 C on t ro l p a ra m e te rs
Parameter (with available options) Default value
Unit (if applicable)
Description
AttachThermalResistor <0|1> 0
Used to attach the thermal resistor to the
bottom. Options are 0 | 1.
BulkMaterial <material> Si
Indicates that the global temperature for solving
diffusion equations will be calculated by
averaging the local temperatures at interfaces of
the specified material. In addition, the material
HeatSubstrate of the extended region
will have the same thermal properties as
BulkMaterial ones.
0Distance
0.5
1.0
I/BeamIntensity
t = 0 s t=(BeamWidth-BeamOverlap)/ScanSpeed (10-4s)
BeamOverlap
4: Diffusion
Flash or Laser Anneal Model
Sentaurus™ Process User Guide 241
I-2013.12
ExtendBottom <0|1> 1
Extends bottom of the simulation structure to
the WaferThickness thickness. Options
are 0|1.
HeatingTime <n> 0.0
Defines the time the heating source is switched
on. Not applicable to the scanning laser model.
HeatSinkTransfer <n> 1e5
Defines the heat transfer rate coefficient at the
extended structure bottom. Only applicable
when AttachThermalResistor is set
to 0.
MaxTimeStep <n> 600.0
Defines the maximum time step for solving the
heat equation.
ReflectSide <Left|Right|None> Left
Specifies the side at which the structure will be
reflected after extending the structure to the
other side by SideExtension. Only
applicable for positive SideExtension.
Options are Left | Right | None.
SideExtension <n> 0.0
Defines the extended distance to side. The
extended region is set to the
HeatSubstrate material, which has the
same thermal properties as the
BulkMaterial ones. Note that only the
heat equation is solved in the region of
HeatSubstrate.
TempAverageBox
"<x1,y1,z1,x2,y2,z2>"
–Defines the box area to average the local
temperatures for calculating the global
temperature. It must be satisfied that x1<=x2
and y1<=y2 and z1<=z2.
pdbSet Heat TempAverageBox
"-0.1 0.1 0.0 1.0 2.0 0.0"
TimeSampleSize <n> 20.0
Specifies the number of time steps during the
sourcing of the heat energy. Not applicable to
the scanning laser model. The maximum time
step is given by the minimum time step among
MaxTimeStep and
for HeatingTime <= 0.0, or
HeatingTime/TimeSampleSize.
UpdateHeatRate <0|1> 0
Updates heat rate at each time step. Options are
0 | 1.
Ta bl e 14 C on t ro l p a ra m e te rs
Parameter (with available options) Default value
Unit (if applicable)
Description
ms
W/(cm2K)
s
µm
3Pulse ⋅
TimeSampleSize 22ln⋅
----------------------------------------------------------------------- (ms)

4: Diffusion
Diffusion in Polysilicon
242 Sentaurus™ Process User Guide
I-2013.12
Notes
■It takes three times the standard deviation time to reach the peak intensity. After six times
the standard deviation time, the heat source is switched off.
■The global temperature, which is calculated by averaging the local temperature
distribution, is used for solving the diffusion equations.
■When the Boolean parameter UseTemperatureField in mechanics is on, the local
temperature is used for solving the mechanics equations.
■The global temperature is calculated by:
(247)
where and are the volume and the local temperature at a node in BulkMaterial
material. By default, the nodes on the nonreflecting surfaces of BulkMaterial are taken.
When you set TempAverageBox, the nodes within the specified box are taken.
Diffusion in Polysilicon
Polysilicon has a microstructure composed of small monocrystalline grains of different
crystalline orientation. The grains are separated by 2D surfaces – the grain boundaries.
Sentaurus Process uses a two-stream model to simulate polycrystalline or granular materials.
Granular diffusion can be switched on with:
pdbSet PolySilicon Arsenic DiffModel Granular
WaferThickness <n> 700.0
Defines the wafer thickness to which the
simulation structure is to be extended if
ExtendBottom is set to 1.
Ta bl e 14 C on t ro l p a ra m e te rs
Parameter (with available options) Default value
Unit (if applicable)
Description
µm
Tglobal
ViTi
i1=
n
Vi
i1=
n
--------------------=
Vi
Ti

4: Diffusion
Diffusion in Polysilicon
Sentaurus™ Process User Guide 243
I-2013.12
Isotropic Diffusion Model
The dopant concentration for species is split into a fraction of dopants in the grain and a
fraction of dopants in the grain boundary, that is:
(248)
Here, denotes the total concentration inside the grain per grain volume and denotes the
concentration inside the grain boundaries per grain boundary volume. Both quantities are
defined in the entire polysilicon region representing average concentrations.
The ratio of grain volume to the polysilicon volume is known as the volume share. The volume
share of the grain regions depends on the shape and size of the grain. The volume share of
the grain boundary is defined as:
(249)
The grain volume share and grain boundary share are defined by the terms GVolShare and
GbVolShare, respectively.
The concentration of the grain boundary is assumed to be electrically inactive. The grain
density is identified with the active portion of the total concentration . The active
concentration is stored in the dataset <dopant>Active. The grain boundary concentration
is stored in the dataset <dopant>Gbc and is initialized with the portion of the
total dopant concentration in the grain boundary . In the absence of clusters, the total
dopant concentration is given as:
(250)
Grain Shape and the Grain Growth Equation
The microscopic shape and size of the grains is not described in the model. Instead the size,
orientation, and type of a prototype grain is used to compute all parameters that depend on the
grain size.
The growth model can be switched on by:
pdbSet PolySilicon Dopant DiffModel Granular
A
CAfgcA
gfgb cA
gb
⋅+⋅=
cA
g
cA
gb
fg
fgb 1fg
–=
CA
+
CA
gb fgb cA
gb
⋅=
fgb CA
⋅
CAfgCA
+CA
gb
+⋅=

4: Diffusion
Diffusion in Polysilicon
244 Sentaurus™ Process User Guide
I-2013.12
In Sentaurus Process, a columnar grain structure is assumed by default. The grains are assumed
to be columns that are oriented along the vertical axis, extending through the entire
polycrystalline layer.
Figure 38 Columnar (left) and cubic (right) grains: is the grain size, is the grain
boundary thickness, and is the layer thickness
The grain size defines the average edge length of the square cross section of the columns.
The grain size is stored in the dataset GSize. The volume share of the grain region is given as:
(251)
For cubic grains, the volume share is:
(252)
The grain shape and the initial values for the grain size (cm) and grain boundary thickness
(cm) can be set in the parameter database, that is:
pdbSet PolySilicon GrainShape <model>
pdbSet PolySilicon GrainSize 5.0e-6
pdbSet PolySilicon GrainBoundaryThickness 5.0e-8
where <model> is either Columnar or Cubic. It is assumed that the layer thickness is a
constant value set as:
pdbSet PolySilicon LayerThickness 1.0e-6
The grains grow during thermal processes. During the grain growth, the volume share of the
grains increases and the volume share of the grain boundary decreases. The grain growth is
modeled by:
(253)
LL
δδ
d
L
δ
d
L
fg
L
Lδ+
------------
2
=
fg
L
Lδ+
------------
3
=
L
δ
d
td
dLLτ2a0b0
2Dλ
kT
-------------------------=1
11
ar
-----+
-------------- 1cgb
CSi
---------------–
⋅⋅

4: Diffusion
Diffusion in Polysilicon
Sentaurus™ Process User Guide 245
I-2013.12
where denotes the ratio between the grain boundary volume inside the polycrystalline layer
and the grain boundary volume at the material interfaces of the polycrystalline layer bounding
other materials. The grain size, , is represented with the solution name GSize and can be
monitored like other solution fields.
The grain growth parameters can be specified in the parameter database in the material entry.
The following names are used: Tau, Lambda, and A0. The parameter is twice the
lattice spacing of silicon. The Arrhenius values for the various contributions to the silicon self-
diffusivity can be specified with the parameter Dself.
For columnar grains:
(254)
(255)
For cubic grains, this is:
(256)
(257)
The grain shape switches from Columnar to Cubic when the grain size reaches the layer
thickness and the grain shape is set to Columnar, that is:
pdbSet PolySilicon GrainShape Columnar
This is the default. No switching is performed if the grain shape is set to Cubic. The grain
growth equation is solved with the dopant diffusion equations.
Note that the GSize and the dopant distribution in the grain boundary <dopant>Gbc are not
reset automatically at the beginning of a new diffusion step. The pdb switch GbcNew can be
used to reset the grain size dataset GSize to the current value of GrainSize, that is:
pdbSet PolySilicon GrainSize 1e-6
pdbSet PolySilicon GbcNew 1
will reset the grain size in polysilicon to 10 nm. It also resets the <dopant>Gbc dataset to the
value calculated using the grain size and the grain-boundary volume share.
ar
L
τ
λ
a0
b0
D
ar
2Lδ+()d
2Lδ+()
2
------------------------=
td
dLL12Lδ+()
2
2Lδ+()d
------------------------+
τ2a0b0
2Dλ
kT
------------------------- 1cgb
CSi
---------------–
⋅=
ar
3L23Lδδ
2
++()d
2Lδ+()
3
--------------------------------------------- 1
2
---–=
td
dLLd3L23Lδδ
2
++()Lδ+()
3
+
d3L23Lδδ
2
++()Lδ+()
3
–
------------------------------------------------------------------------τ2a0b0
2Dλ
kT
------------------------- 1cgb
CSi
---------------–
⋅=
L
d

4: Diffusion
Diffusion in Polysilicon
246 Sentaurus™ Process User Guide
I-2013.12
The GSize and the initial <dopant>Gbc distribution in a newly deposited layer can be
specified in the fields and in the values list in the deposit command, that is:
deposit PolySilicon type=isotropic rate=1.0 time=0.1 fields= {GSize Arsenic} \
values= {4e-6 1e19}
This will initialize the GSize to 40 nm and the arsenic concentration to a constant value of
. The ArsenicGbc solution will be created and the value of the corresponding
dataset will be set automatically.
Diffusion Equations
The diffusion in polycrystalline materials is modeled with two separate diffusion fluxes for the
diffusion of inside the grains and the diffusion of along the grain boundaries. The
diffusion inside the grain regions is modeled as for crystalline silicon with the ChargedFermi
diffusion model. The diffusion fluxes are scaled with the ratio of the grain boundary volume to
the polysilicon volume, that is:
(258)
(259)
For details on the ChargedFermi model parameters, see ChargedFermi Diffusion Model on
page 217. The diffusivity for the grain interior is set as usual for the ChargedFermi
model, for example:
pdbSet PolySilicon Boron Int Dstar \
{ 0 {[Arr 0.743e2 3.56]} 1 {[Arr 0.617e2 3.56]} }
For the fluxes along the grain boundaries, the gradient of the concentration in the grain
boundary is multiplied by a constant diffusivity and the grain boundary volume share :
(260)
(261)
The grain boundary diffusivity can be set in the parameter database using:
pdbSet PolySilicon Arsenic Dgb {[Arrhenius 1100.0 3.53]}
1019 cm 3–
CA
+
cA
gb
JAf–gDAXc
n
ni
----
c–z–CA
+n
ni
----
z
∇
Xc,
=
t∂
∂CAJA
∇•–R–=
DAXc
fgb
t∂
∂CA
gb
JA
gb
∇•–R+=
JA
gb f–gbDA
gb 1cA
gb
CSi
--------–
cA
gb
()∇=
DA
gb
4: Diffusion
Diffusion in Polysilicon
Sentaurus™ Process User Guide 247
I-2013.12
In polycrystalline materials, a segregation reaction is assumed to occur at the surface of the
grains. The reaction describes the exchange of dopants between grain and grain boundary
regions. The reaction term is given as:
(262)
The segregation term depends on the transport coefficient , multiplied by the grain surface
area per unit volume of polysilicon .
The transport coefficient can be specified in the parameter database using:
pdbSet PolySilicon Arsenic Ksgb {[Arrhenius 1.630e4 3.586]}
The dopant segregation coefficient for the segregation between the grain and grain boundary
can be specified by using:
pdbSet PolySilicon Arsenic Sgb {[Arrhenius 2.75 -0.44]}
The grain surface area per unit volume depends on the grain structure. For columnar
grain structures, this is:
(263)
and, for cubic grains, this is:
(264)
Ta bl e 1 5 S o l ut io n na m es
Symbol Boron Arsenic Phosphorus Antimony Indium
Boron Arsenic Phosphorus Antimony Indium
BoronGbc ArsenicGbc PhosphorusGbc AntimonyGbc IndiumGbc
BActive AsActive PActive SbActive InActive
Ta bl e 1 6 S o l ut io n na m es fo r g ran ul ar m od e l
Symbol Solution name
GSize
R
RaL()KC
A
+1cA
gb
CSi
--------–
⋅cA
gb
sg
------- 1CA
+
CSi
--------–
⋅–⋅⋅=
K
aL()
K
sg
aL()
aL() 4L⋅
L
δ
+()
2
--------------------=
aL() 6L2
⋅
L
δ
+()
3
--------------------=
CA
cA
gb
CA
+
L

4: Diffusion
Diffusion in Polysilicon
248 Sentaurus™ Process User Guide
I-2013.12
Anisotropic Diffusion Model
The behavior of dopants in polycrystalline materials is strongly influenced by the boundaries
between crystalline grains. Dopant atoms tend to segregate from the interior of a grain to the
boundaries, which provide paths for rapid diffusion. The rate of segregation depends on the rate
of grain growth, while the number of diffusion paths along the boundaries depends on the grain
size. In addition, the boundaries of the polycrystalline material act like grain boundaries,
providing sites for electrically inactive dopant atoms and paths for diffusion. The diffusion
equations for the grain-interior and grain-boundary components of the doping profile are
solved separately. The equations are coupled by terms describing the segregation between the
grain interiors and grain boundaries. To determine the rate of segregation and the density of
grain-boundary diffusion paths, you also solve for the growth in grain size during high-
temperature processing. The boundaries of the polycrystalline region are included as explicit
grain boundaries in the diffusion and segregation equations. The model has been implemented
in Sentaurus Process [6][7][8][9].
The model is invoked by specifying the polycrystalline material, for example:
pdbSet PolySilicon PolyCrystalline 1
To set the model to the TSUPREM-4 compatible mode, use the command:
SetTS4PolyMode
Diffusion in Grain Interiors
Redistribution of dopants in polycrystalline materials occurs by the parallel diffusion of
dopants through the interiors of grains and along grain boundaries.
In the grain interiors, diffusion of the active dopant is given by:
(265)
where is the active concentration in the grain interior. The diffusivity and electric field
in the grain interior are calculated from the electron concentration , which is in turn
calculated from the doping concentrations . accounts for the segregation of dopant to
grain boundaries as described in Segregation Between Grain Interior and Boundaries on
page 251.
The parameter Grain.Crystallinity specifies the initial crystallinity of the grain interiors.
If Grain.Crystallinity is set to Crystalline, the initial active concentration is
determined by the pdb parameter AcInit or the term ${Sol}AcInit. If
∂cg
∂t
-------- ∇Dg
–∇cgzscg
qEg
kT
---------
–
G–⋅–=
cg
Dg
Eg
ng
cg
G

4: Diffusion
Diffusion in Polysilicon
Sentaurus™ Process User Guide 249
I-2013.12
Grain.Crystallinity is set to Amorphous, the initial active concentration is determined
by the pdb parameter AmInit or the term ${Sol}AmInit. The initialization is performed for
the remainder after some implantation atoms go to a grain boundary according to Eq. 274,
p. 251.
Grain Boundary Structure
Diffusion along grain boundaries is described in terms of the dopant concentration per unit
area of grain boundary , and the average area of grain boundaries per unit volume:
(266)
where is the average area of grain boundaries per unit volume in the bulk of the poly layer
and accounts for the dopant at interfaces between poly and other materials (or ambient).
is inversely proportional to the average grain size :
(267)
where GBGeomFactor is a geometric factor specified for the polycrystalline material, for
example:
pdbSet PolySilicon GBGeomFactor 2.0
is a function of position defined by the fact that its integral over any volume is equal to the
area of the polysilicon interface passing through that volume:
(268)
The concentration of dopants in the grain boundaries per unit volume of material is then given
by:
(269)
Diffusion along Grain Boundaries
The diffusion of dopant in the grain boundaries is given by:
(270)
The diffusivity and electric field along the grain boundaries are calculated from the
electron concentration ; is calculated by assuming that the net donor and acceptor
cgb
ρ′ ρ δif
+=
ρ
δif
ρ
Lg
ρGBGeomFactor
Lg
------------------------------------------------=
δif
Aif
δif Vd
Aif
=
cA
gb ρ′cgb
=
∂cA
gb
∂t
-----------∇FDgb
–∇cgb zscgb
qEgb
kT
------------
–
G+⋅–=
Dgb
Egb
ngb
ngb
4: Diffusion
Diffusion in Polysilicon
250 Sentaurus™ Process User Guide
I-2013.12
concentrations are calculated from , the equilibrium dopant concentrations in the grain
interior near the grain boundary, where is the segregation coefficient given by Eq. 276,
p. 251. accounts for the segregation of dopant to grain boundaries as described in
Segregation Between Grain Interior and Boundaries on page 251.
Diffusivity is given by:
(271)
Dgb and Dgb.Fermi are defined by:
pdbSet <material> <dopant> Dgb <value>
pdbSet <material> <dopant> Dgb.Fermi <array>
is a tensor that describes the diffusion paths available to dopant in the grain boundaries. It is
composed of two parts: . describes the available paths within the
bulk of the poly layer. For a horizontal poly layer, it is given by:
(272)
Because of the columnar grain structure, Dgb.F22 is larger than Dgb.F11, which implies that
diffusion through the layer is faster than diffusion parallel to the layer.
Dgb.F11 and Dgb.F22 are defined for the polycrystalline material, for example:
pdbSet PolySilicon Dgb.F11 1.0
pdbSet PolySilicon Dgb.F22 2.0
describes the available paths for diffusion along material interfaces. In the vicinity of a
horizontal interface, it has the value:
(273)
For the interface between polysilicon and silicon, the phenomenon of interfacial breakup
accompanied by epitaxial realignment can occur, as described in Interface Oxide Breakup and
Epitaxial Regrowth on page 255. is the fraction of the polysilicon–silicon interface that has
broken up. For layers or interfaces that are not horizontal, and are rotated by the angle
of the layer or interface, respectively, with respect to the horizontal axis.
cgb K⁄
K
G
Dgb
Dgb Dgb Dgb.Fermi ngb
ni
--------
c–
⋅
⋅=
F
FF
b1Fbu
–()Fif
+=
Fb
Fbdiag Dgb.F22
Lg
----------------------------Dgb.F11
Lg
----------------------------Dgb.F11
Lg
----------------------------
,,
=
Fif
Fif diag 0 δ,if δif
,()=
Fbu
Fb
Fif
4: Diffusion
Diffusion in Polysilicon
Sentaurus™ Process User Guide 251
I-2013.12
Segregation Between Grain Interior and Boundaries
When dopant is initially introduced into a polycrystalline material, some of the dopant
occupies sites in the interior of a grain and some occupies sites on a grain boundary. The initial
segregation of dopant is given by:
(274)
GBMaxDensity, GMaxConc, and GSegInit represent the density of available sites on grain
boundaries, and in the grain interiors and the initial segregation entropy, respectively. In the
case of ion implantation, and describe the additional dopant introduced by the
implantation; dopant that is present before the implantation is not redistributed.
Dopant atoms are free to move between sites in the interior of a grain and sites on the grain
boundary during high-temperature processing. The rate of segregation is given by:
(275)
The segregation coefficient is given by [10]:
(276)
GBMaxDensity, GMaxConc, GSegInit, and Sgb are defined for dopants, for example:
pdbSet PolySilicon Dopant GBMaxDensity 2.5e15
pdbSet PolySilicon Dopant GMaxConc 5e22
pdbSet PolySilicon Dopant GSegInit 1.0
pdbSet PolySilicon Boron Sgb {[Arrhenius 0.2 -0.38]}
The segregation velocities associated with the bulk of the poly region and the material
interfaces are given by:
(277)
GBVFactor is the parameter for the material. KsgbFactor and , which is defined by Vsgb,
are specified for dopants. For example:
pdbSet PolySilicon GBVFactor 1.33
pdbSet PolySilicon Dopant KsgbFactor {[Arr 4.0 0.0]}
pdbSet PolySilicon Dopant Vsgb {[Arr 1e7 3.0]}
cgb
GBMaxDensity
GMaxConc
------------------------------------------------GSegInit cg
=
cg
cgb
Gρqb1Fbu
–()δ
ifqif
+()f
gb cgf
g
cgb
K
-------
–
=
K
KGBMaxDensity
GMaxConc
------------------------------------------------Sgb=
qb
1
GBVFactor
------------------------------------∂Lg
∂t
---------KsgbFactorDg
Lg
------+=
qif
4: Diffusion
Diffusion in Polysilicon
252 Sentaurus™ Process User Guide
I-2013.12
and are the fractions of unfilled interior and boundary sites:
(278)
(279)
where the sum is taken over all the dopant species present in the structure. is the fraction
of the polysilicon–silicon interface that has broken up, as described in Interface Oxide Breakup
and Epitaxial Regrowth on page 255.
Grain Size Model
The grains in the polycrystalline material are assumed to be oriented as columns that extend
through the wafer. The structure is characterized by , the average grain size in the lateral
direction (in other words, in the plane of the layer), and a vector describing the orientation of
the columnar grains.
The initial grain size is determined by the temperature of the poly deposition process:
(280)
where:
■ is the deposition temperature (specified on the deposit command) in degree Celsius.
■ is the thickness of the amorphous silicon layer produced by low-temperature deposition.
■ is the distance from the bottom of the layer.
For high-temperature depositions, grain size depends on the thickness specified in the
deposit command. Dividing a deposition into multiple smaller depositions produces
different results for the grain size. For low-temperature depositions, the material is assumed to
be amorphous (a negative grain size is reported in printing or plotting). The initial grain size is
calculated from the thickness of the amorphous layer at the beginning of the next diffusion
step.
Frac.TA, GrainSize, GrainSizeFactor, and GrainSizeTempC are the material
parameters, for example:
pdbSet PolySilicon Frac.TA 0.5
pdbSet PolySilicon GrainSize 5e-6 ;# cm
pdbSet PolySilicon GrainSizeFactor {[Arr 0.1 0.0]}
pdbSet PolySilicon GrainSizeTempC 600.0
fg
fgb
f
g1cg
GMaxConc
--------------------------------–=
f
gb 1cgb
GBMaxDensity
------------------------------------------------
–=
Fbu
Lg
Lg
max Frac.TA taGrainSize,×() TcGrainSizeTempC≤
GrainSize 2GrainSizeFactor⋅+ z⋅ TcGrainSizeTempC>
=
Tc
ta
z
ta
4: Diffusion
Diffusion in Polysilicon
Sentaurus™ Process User Guide 253
I-2013.12
Surface Nucleation Model
An alternative model for the standard grain size model (see Grain Size Model on page 252) is
the surface nucleation model. In this model, grains are assumed to grow from small clusters
formed at the early stages of deposition. The kinetics of nucleation, under this assumption,
determines the average distance between clusters, thereby, the average starting grain size near
the surface upon which deposition occurs. The model for atomistic nucleation is derived from
rate equations for growth, surface diffusion, and desorption [11]. Several different regimes are
considered:
■Large deposition rates (compared to surface diffusion or evaporation) are labeled
“complete condensation”.
■Conditions where the deposition rate between surface diffusion and evaporation are labeled
“incomplete condensation”.
■Low deposition rates are labeled “extremely incomplete condensation”.
Besides the deposition rates, two types of nuclei are considered: 2D or 3D islands. The
following formulas are used to compute the nucleation density:
(281)
where and are given by Table 17 on page 254. The choice of regime is set with:
pdbSet SNG.Model <regime>
where <regime> is one of Complete, Initially.Incomplete, Extreme.Incomplete,
or None (default) meaning the model is switched off.
nn
0RpE
kT
------
exp=
p
E

4: Diffusion
Diffusion in Polysilicon
254 Sentaurus™ Process User Guide
I-2013.12
NOTE Default values for parameters of the surface nucleation model have not
been calibrated for any process. They have simply been set to give
approximately the same values as the grain size model in polysilicon
diffusion. This model is in an experimental state. The default values of
the model may change in the future if reasonable values can be found
for typical technology conditions.
Grain Growth
The growth of the grains during high-temperature processing is given by [12]:
(282)
Ta bl e 1 7 Fo r mu l as fo r s ur fa c e n u c l e a ti on m od e l
Regime 3D islands 2D islands
Extremely
incomplete
Incomplete
Complete
Table 18 Parameters of surface nucleation model set with
pdbSet PolySilicon <parameter> <value>
Symbol Parameter Name
Island dimension
SNG.Island.Dim
SNG.Growth.Flux
SNG.Prefactor
SNG.Critical.Island.Size
SNG.Adsorption.Energy
SNG.Diffusion.Energy
SNG.Critical.Island.Energy
p2
3
---i=
E2
3
---Eii1)EaEd)–+()+()=
pi=
EE
ii1)EaEd)–+()+=
p2
5
---i=
E2
5
---EiiEa
+()=
pi
2
---=
EE
iiEa
+=
pii2.5+()⁄=
EE
iiEd
+()i2.5+()⁄=
pii2+()⁄=
EE
iiEd
+()i2+()⁄=
R
n0
i
Ea
Ed
Ei
∂Lg
∂t
---------1
Lg
----- A0 DselfFactor Dself Egb
kT
-------- Fseg GEA
+××××=
4: Diffusion
Diffusion in Polysilicon
Sentaurus™ Process User Guide 255
I-2013.12
A0, DselfFactor, and Dself represent the empirical geometric factor, the enhancement
factor of silicon self-diffusivity at the grain boundary, and the silicon self-diffusivity in the
vicinity of a grain boundary, respectively:
pdbSet PolySilicon A0 6.0
pdbSet PolySilicon DselfFactor {[Arrhenius 5.6e-6 -1.73]}
pdbSet PolySilicon Dself { -2 {[Arrhenius 5.6e-6 2.86]}
-1 0.0
0 {[Arrhenius 4.29e-7 2.18]}
1 0.0
2 0.0 }
is the surface energy per atom associated with the grain boundary [12][13][14];
models the segregation drag effect; and models epitaxial regrowth of the poly layer (see
Interface Oxide Breakup and Epitaxial Regrowth on page 255).
(283)
(284)
is the thickness of the polycrystalline layer.
The segregation drag effect reduces the grain growth rate [15]:
(285)
SegDragExponent is defined for the material, for example:
pdbSet PolySilicon SegDragExponent 2.0
Interface Oxide Breakup and Epitaxial Regrowth
A thin interfacial oxide layer is typically present between a deposited polysilicon layer and any
underlying single-crystal silicon. This interfacial oxide presents a barrier to epitaxial
realignment of the poly layer. With sufficient high-temperature processing, the oxide layer
breaks up into a discrete set of small spheres, allowing epitaxial regrowth of the poly to
proceed.
Egb
Fseg
GEA
Egb 2LatticeSpacing Tu⋅⋅()
2Lambda 1
1Lambda.hfn
+
----------------------------------------------Lambda.1 Lg
tpoly
-----------
+
⋅⋅=
fn
Lg
2tpoly Lg
–
-------------------------- Lgtpoly
<
Lg
tpoly
----------- Lgtpoly
≥
=
tpoly
Fseg 1cgb
GBMaxDensity
------------------------------------------------
+
SegDragExponent–
=

4: Diffusion
Diffusion in Polysilicon
256 Sentaurus™ Process User Guide
I-2013.12
The oxide breakup is modelled by the formation of voids in the interfacial oxide layer
[16]–[19]. The radius of the voids increases as:
(286)
where:
■ is a constant.
■ is the initial oxide thickness.
■ is the activation energy of the breakup process.
■ is initialized to zero whenever poly is deposited on exposed silicon.
The fraction of the interface that is broken up is given by:
(287)
where is the areal density of the voids.
The parameters for the model are specified in terms of a characteristic breakup time for the
thinnest (5 Å) interfacial oxide layers:
(288)
is defined with the parameter PolyOxBreakTime, for example:
pdbSet PolySilicon PolyOxBreakTime {[Arrhenius 1.0 -5.0]} ;# seconds
(289)
In the present implementation, assume that all polycrystalline or single-crystalline interfaces
share a common oxide thickness given by:
(290)
PolyOxThickness is defined in the material:
pdbSet PolySilicon PolyOxThickness 5e-8 ;# cm
Rvoid
dRvoid
dt
----------------β
tox
3
------ exp Ebu
kT
--------
–
×=
β
tox
Ebu
Rvoid
Fbu 1exp πNEARvoid
2
–()–=
NEA
tbu
5A
·3
πNEAβ
--------------------- expEbu
kT
--------
×≡
tbu
dRvoid
dt
----------------1
tbu
------ 5A
·
tox
-------
3 1
πNEA
------------------=
tox PolyOxThickness=
4: Diffusion
Diffusion in Polysilicon
Sentaurus™ Process User Guide 257
I-2013.12
Epitaxial regrowth is modeled by increasing the poly grain size to a value larger than the
thickness of the poly layer. This grain growth is described by in Eq. 273, p. 250 for the
grain size:
(291)
It serves as a driving force for epitaxial regrowth from the interface at the silicon–polysilicon
interface. Parameters for this model are given by:
(292)
pdbSet PolySilicon EpiGrowthVelocity {[Arrhenius 100.0 3.0]} ;# cm2/sec
Dependence of Polysilicon Oxidation Rate on Grain Size
It has been observed experimentally that the oxidation rate for fine-grained polysilicon is faster
than for coarser-grained polycrystalline or single-crystalline silicon, presumably because of
enhanced oxidation at the grain boundaries.
This enhancement can be modeled by assuming a faster surface reaction rate where grain
boundaries intersect the oxide–poly interface:
(293)
where:
(294)
is the surface reaction rate in the absence of grain boundaries, is the reaction rate at a grain
boundary, and:
(295)
is the fraction of the surface within a distance of a grain boundary. The enhancement
factor at grain boundaries is specified as:
(296)
pdbSet Oxide_PolySilicon H2O GBFactor {[Arr 10.0 0.0]}
pdbSet Oxide_PolySilicon O2 GBFactor {[Arr 10.0 0.0]}
pdbSet Oxide_PolySilicon N2O GBFactor {[Arr 10.0 0.0]}
GEA
GEA FbuvEAδif
=
vEA EpiGrowthVelocity=
ks1f–()kgfkgb
+=
kg
B
A
--- Cox
C*
---------=
kgb
fmin Tδ
Lg
------1,
=
Tδ2⁄
kgb
kg
-------GBFactor=

4: Diffusion
Diffusion in Polysilicon
258 Sentaurus™ Process User Guide
I-2013.12
The effective thickness of the grain boundaries is given by , where
GBEffThick is specified for each material.
Boundary Conditions
Boundary Conditions for Grain Growth Equation
Several boundary conditions to control the grain size (GSize) in the grain growth equation are
available. The reflective (HomNeumann) boundary condition assumes that the interface value
grows like the bulk value. The minimum value (MinimumSize) boundary condition sets the
interface value at the minimum value for GSize. The minimum value is set with:
pdbSet <material> GSize minConc {<n>}
The initial size (InitialSize) boundary condition fixes the interface value at the initial value
of GSize.
The boundary conditions can be switched on using:
pdbSet Oxide_PolySilicon <dopant> BoundaryCondition [
{HomNeumann
MinimumSize InitialSize}]
The default setting for the grain growth boundary condition is HomNeumann.
Dopant Diffusion Boundary Conditions
There are three additional segregation-type boundary conditions available with the
polycrystalline diffusion model. The first can be selected with:
pdbSet Oxide_PolySilicon Arsenic BoundaryCondition GrainBoundarySegregation
The total dopant fluxes at the interfaces between the grain boundary and the neighboring layer
are balanced. The fluxes are given by:
(297)
where is the concentration of dopant on the other side of the interface, is the grain
boundary concentration, is the transfer rate, and is the segregation rate
of dopant in the grain boundary. To set these parameters, use:
pdbSet <interface material> <dopant> Transfer {<n>}
pdbSet <interface material> <dopant> SegregationGb {<n>}
TδGBEffThick=
jn•kTransfer CA
other CA
gb
fgbsgkgb
Segregation
----------------------------------------------–
=
CA
other
CA
gb
kTransfer
kgb
Segregation
A
4: Diffusion
Diffusion in Polysilicon
Sentaurus™ Process User Guide 259
I-2013.12
The coefficient is the grain–grain boundary segregation coefficient.
To select the second boundary condition, use:
pdbSet Oxide_PolySilicon Arsenic BoundaryCondition
GrainGrainBoundarySegregation
The total dopant fluxes at the interfaces between the grain and the neighboring layer and the
grain boundary and neighboring layer are balanced. The fluxes are given by:
(298)
(299)
and:
(300)
where is the active concentration of dopant in the grain and is the segregation
rate of dopant in the grain. To set this parameter, use:
pdbSet <interface material> <dopant> Segregation {<n>}
To select the third boundary condition, use:
pdbSet Oxide_PolySilicon Arsenic BoundaryCondition \
BulkGrainBoundarySegregation
The total dopant fluxes at the interfaces between the grain and the neighboring layer and
between the grain and the grain boundary are balanced. The fluxes are given by:
(301)
(302)
sg
jgn•kTransfer CA
other fgCA
+
kSegregation
-----------------------------–
=
jgb n•kTransfer CA
other CA
gb
sgkgb
Segregation
---------------------------------------–
=
jn•jgn•jgb n•+=
CA
+
kSegregation
A
jn•kTransfer CA
other fgCA
+
kSegregation
-----------------------------–
=
jgb n•kTransfer
fgCA
+
kSegregation
-----------------------------CA
gb
fgbsgkgb
Segregation
----------------------------------------------–
=

4: Diffusion
Dopant Diffusion in SiGe
260 Sentaurus™ Process User Guide
I-2013.12
NOTE For the anisotropic polycrystalline model, use the Segregation model
for the boundary condition:
pdbSet PolySilicon_Silicon Boron BoundaryCondition Segregation
pdbSet Oxide_PolySilicon Boron BoundaryCondition Segregation
pdbSet Gas_PolySilicon Boron BoundaryCondition Segregation
Dopant Diffusion in SiGe
The presence of germanium in silicon affects the dopant diffusion in various ways. First, the
band gap of silicon is lowered in the presence of germanium. Second, germanium affects the
point-defect equilibrium concentration. In addition, germanium can pair with boron forming
an immobile but electrically active species.
Bandgap Effect
The effect of the bandgap narrowing on the dopant diffusion arises from the change in the
intrinsic carrier concentration . This has been implemented in Sentaurus Process as
follows:
(303)
where is bandgap narrowing due to germanium content. It can be defined using the
command:
pdbSet <material> Germanium delEg {<n>}
The band gap, delEg ( ), is a function of germanium concentration and is given by:
(304)
(305)
(306)
(307)
where [20] is the bandgap narrowing in strained silicon, [21] is the bandgap
narrowing in the relaxed silicon, is a pseudomorphic factor that shows the degree of the
niSi()
niniSi()e
Eg∆–
2kT
--------------
=
Eg∆
Eg∆
Eg∆Egs∆Egr∆Egs∆–()1Fpm–()+≡
Egs
∆0.835x21.01x–=
Egr
∆0.33x20.55x–=
xCGe
5.0 22
×10
---------------------=
Egs∆
Egr∆
Fpm

4: Diffusion
Dopant Diffusion in SiGe
Sentaurus™ Process User Guide 261
I-2013.12
relaxation, and is the germanium fraction in silicon. is calculated with respect to lattice
mismatch in the substrate. For example:
(308)
where:
■ is the lattice-spacing of the silicon-germanium region.
■ is the lattice-spacing of silicon.
■ is the lattice-spacing calculated in the mechanics.
If the system is fully relaxed, is zero. If it is fully strained, is one. will be used
if the germanium percentage is greater than 0.1%, and the bandgap narrowing effects due to
other strain sources will be ignored (see Pressure-dependent Defect Diffusion on page 275).
Potential Equation
The permittivity of “Ge-doped” silicon can be calculated by the following formula, in which
is the germanium concentration in silicon:
(309)
where and are defined as PDB parameters:
pdbSet Ge Potential Permittivity 15.8
pdbSet Si Potential Permittivity 11.7
This calculation is included when setting:
pdbSetString Si CompSpecies "Germanium"
pdbSetBoolean Si Potential PermittivityContentEffect 1
The product of the density-of-states in the conduction and valence bands, , is
multiplied by the factor defined as:
pdbSetFunction Si Ge factorNcNv [GefactorNcNv]
when the following PDB value is set:
pdbSetBoolean Si Potential BandgapContentEffect 1
Such a factor can be defined as:
(310)
x
Fpm
Fpm aSiGe a–()
aSiGe aSi
–
---------------------------=
aSiGe
aSi
a
Fpm
Fpm
Eg∆
xGe
ε1xGe
–()ε
Si xGe
+×ε
Ge
×=
εSi
εGe
NcNv
×
1xGe
–0.47 0.81⁄×()
34⁄

4: Diffusion
Dopant Diffusion in SiGe
262 Sentaurus™ Process User Guide
I-2013.12
with the following line:
fproc GefactorNcNv { } { return "(1.0-(Germanium/5.0e22)*0.47/0.81)^0.75" }
Effects on Point-Defect Equilibrium Concentrations
The introduction of germanium acts thermodynamically on the equilibrium of the silicon
matrix. Compressive strain increases the equilibrium concentration of vacancies and decreases
the equilibrium concentration of interstitials, and the tensile strain has the opposite effect on
the point-defect equilibrium concentrations [22][23].
These effects are modeled in Sentaurus Process by modifying the equilibrium point-defect
concentrations:
(311)
(312)
where is the equilibrium concentration of point defects (interstitial or vacancy), and
is the activation volume change of equilibrium point defects due to the pressure .
The following set of commands can be used to modify :
pdbSet Silicon Interstitial Volume 8.59e-24
pdbSet Silicon Vacancy Volume -5.52e-24
To switch on strain effects on point defects, set the following switches:
pdbSet Silicon Interstitial CStarMod FermiPressureDependent
pdbSet Silicon Vacancy CStarMod FermiPressureDependent
is the total activation volume change of equilibrium point defects due to the presence
of germanium and is calculated from the activation volume change , the lattice
mismatch coefficient , and the germanium fraction in the structure (see Eq. 312). These
quantities can be modified using the following commands:
pdbSet Silicon Germanium Interstitial delVol 11.8
pdbSet Silicon Germanium Vacancy delVol 25.6
pdbSet Silicon Germanium LatticeMismatch 0.0425
NOTE delVol is given in units of eV and Volume is given in units of .
CX
*CX
*Si P 0≡,()e
VXPV
GeX
∆+∆()–
kT
--------------------------------------------
=
VGeX
∆VolGeX a∆SiGe
CGe
522
×10
-----------------∆=
CX
*
VX
∆
P
VX
∆
VGeX
∆
VolGeX
∆
a∆SiGe
cm3

4: Diffusion
Dopant Diffusion in SiGe
Sentaurus™ Process User Guide 263
I-2013.12
Effect of Ge on Point-Defect Parameters
In this version of Sentaurus Process, you can define the arbitrary Alagator expressions for the
point-defect basic parameter prefactors. The names of terms used for the vacancy and
interstitial equilibrium concentration are VacCStarFactor and IntCStarFactor,
respectively. Corresponding terms for the vacancy and interstitial diffusivity are
VacDiffFactor and IntDiffFactor. These expressions can be used to include the effect
of germanium on point-defect parameters. For example, the prefactor for the vacancy
equilibrium concentration in SiGe can be calculated as follows:
MultiplyTerm Si VacCStarFactor "exp((1.088*($x_Ge))*$Vt_i)"
where x_Ge is the germanium content, and
Vt_i
is in .
NOTE The MultiplyTerm command is not saved to the TDR files. If the input
file is split, the command must be included in the new input file.
Impact of Ge on Extended-Defect Parameters
The parameters IClusterDissIntFactor, C311DiffIntFactor, and CLoopTransfer
(used in the Full model for I-clusters) can be used to include the impact of germanium on
extended defects.
Impact of Dopant Diffusivities
The germanium chemical effect is simulated by the activation energy correction using
diffusivity prefactors. For example, in the case of boron, it is performed by the term
BoronIntDiffFactor, which can be defined in silicon:
MultiplyTerm Si BoronIntDiffFactor "exp(-0.227*($x_Ge)*$Vt_i)"
NOTE The MultiplyTerm command is not saved to the TDR files. If the input
file is split, the command must be included in the new input file.
During assembly of the diffusion equations, Sentaurus Process checks each dopant and
material for whether such diffusion factors exist. The diffusivity through dopant–interstitial or
dopant–vacancy pairs is then multiplied by the corresponding diffusion enhancement factors.
A separation between interstitial and vacancy effects is necessary because with increasing
germanium content of SiGe, the fractions of diffusion mediated by dopant–interstitial and
dopant–vacancy pairs change.
1(kT)⁄
eV 1–

4: Diffusion
Dopant Diffusion in SiGe
264 Sentaurus™ Process User Guide
I-2013.12
SiGe Strain and Dopant Activation
The solid solubility of dopants depends on the strain. In general, for compressive strain, the
solubility of atoms smaller than silicon increases; whereas, the solubility of larger atoms
decreases. In the Transient model, the stress effect is taken into account by introducing the
pressure-dependent parameters Solubility and TotalSolubility:
(313)
An example of the definition of boron pressure-dependent solid solubility is:
pdbSet Si B SS.Factor "exp(3.636e-24*Pressure*$kT_i)"
pdbSet Si B Total.SS.Factor "exp(3.636e-24*Pressure*$kT_i)"
Since the emission rate for the silicon side in the three-phase segregation model is proportional
to the solid solubility, a corresponding modification also must be included in the boundary
condition. For example, this can be achieved by the following line for boron:
pdbSetString Si B Side.SS.Factor "exp(3.636e-24*Pressure_Silicon*$kT_i)"
NOTE You can define the arbitrary Alagator expressions for the dopant solid
solubility prefactors in Sentaurus Process. The name of strings used for
the solid solubility, the total solid solubility, and the emission rate
correction are SS.Factor, Total.SS.Factor, and
Side.SS.Factor, respectively.
Germanium–Boron Pairing
Germanium can pair with boron and the pairs are known to be electrically active [24] but not
mobile:
(314)
In Sentaurus Process, this reaction is modeled with the following differential equation:
(315)
where is the concentration of germanium–boron pairs, is the concentration of
germanium, is the concentration of boron, and and are the forward reaction rate
and equilibrium constant, respectively.
SP() SP(0)P– VkT⁄()exp==
Ge B GeB↔+
t∂
∂CGeB Kf CGeCBKbCGeB
–()=
CGeB
CGe
CB
Kf
Kb
4: Diffusion
Diffusion in III–V Compounds
Sentaurus™ Process User Guide 265
I-2013.12
You ca n spec if y the m od el pa rameters with the commands:
pdbSet Silicon Germanium Boron Kf {<n>}
pdbSet Silicon Germanium Boron Kb {<n>}
Germanium diffusion is modeled by assuming a constant diffusion model:
(316)
where is the diffusivity of germanium and can be set using the command:
pdbSet Silicon Germanium Dstar {<n>}
NOTE The germanium–boron cluster model is switched off by default. To
switched it on, use:
solution add name=GeB ifpresent = "Germanium Boron" !negative
Of course, if boron is present in silicon, the reaction in Eq. 315 is automatically added to
Eq. 316.
Initializing Germanium–Boron Clusters
Initially, germanium–boron cluster concentrations are set to zero. If there is an existing cluster
concentration field, the field is used. To initialize the cluster concentration field, use the
select command in the input command file.
Diffusion in III–V Compounds
This section discusses diffusion in III–V compounds.
Material Conversion
At the beginning of a diffusion, the adjacent III–V materials or a III–V material doped with
other group III or V atoms can be merged into the proper ternary or quaternary compound
Ta bl e 1 9 S o l ut io n na m es fo r g er m an iu m m o d el
Symbol Solution name
Germanium
GeB
t∂
∂CGe DC
Ge
∇()∇•=
D
CGe
CGeB

4: Diffusion
Diffusion in III–V Compounds
266 Sentaurus™ Process User Guide
I-2013.12
materials. The conversion is performed when the Boolean parameter
Convert.IIIVMaterials is switched on:
pdbSet Diffuse Convert.IIIVMaterials <0|1> ;# default 1
When different III–V material regions are adjacent and there is a common material derived
from each III–V material, the regions merge into the common derived material region. For
example, if the neighbor region of a GaAs material region is InAs, two regions are merged and
converted into InGaAs material if InGaAs is the derived material from both GaAs and InAs.
The derived material is specified by:
pdbSet <material> Derived.Materials { <derivedmaterial list> }
For example:
pdbSet GaAs Derived.Materials { InGaAs AlGaAs GaPAs }
pdbSet InAs Derived.Materials { InPAs InGaAs }
When group III or group V atoms are doped into a III–V material, and the atoms are a different
species from the components of the material, the material is converted to the new III–V
material with the component list, including the doping species, if the doping concentration
exceeds the minimum concentration Min.Conv.Conc for conversion. For example, when
indium atoms are doped into a GaAs material region, GaAs is converted to InGaAs if InGaAs
is one of the derived materials of GaAs, and the maximum concentration of indium atoms in
the region exceeds the indium Min.Conv.Conc parameter value of InGaAs:
pdbSet InGaAs Indium Min.Conc {<n>}
The atoms of the material components are filled into the region before the material conversion
for the mole fraction calculation and the interdiffusion simulation.
Physical Parameter Interpolation
The parameter for the mole fraction of a ternary (or quaternary) material is specified by:
pdbSet <material> MoleFraction.Atoms { x <atom> } ;# ternary
pdbSet <material> MoleFraction.Atoms { x <atom1> y <atom2> } ;# quaternary
For example:
pdbSet InGaAs MoleFraction.Atoms { x Gallium } ;# In
(1-x)
Ga
(x)
As
pdbSet AlInGaAs MoleFraction.Atoms {x Aluminum y Indium} ;# Al
(x)
In
(y)
Ga
(1-x-y)
As

4: Diffusion
Diffusion in III–V Compounds
Sentaurus™ Process User Guide 267
I-2013.12
If a physical parameter is not specified on a ternary (or quaternary) material, the parameter
value is extracted by the linear interpolation with the parameter values of their base materials
that is, binary materials. For the value of the parameter of material :
(317)
(318)
(319)
NOTE For the energy bandgap and affinity, the second-order mole-fraction
dependency can be specified (see details in Table 22 on page 278).
Dopant Diffusion
To model dopant diffusion in a III–V material, the following assumptions are applied:
■Point defects diffuse by the second nearest neighbor hopping.
■Group II dopants react only with group III point defects.
■Group VI dopants react only with group V point defects.
■There are no antisite defects.
■The charging reaction is in equilibrium.
■There are two types of vacancy (that is, at group III and V sites): VacIII and VacV.
Since the substitutional concentrations of group IV dopants on group III sites and group V sites
are modeled and calculated separately, the autocompensation effect due to the amphoteric
behavior is implicitly taken into account.
ChargedReact Model
(320)
(321)
(322)
PM
M
PIIIAx()
IIIB1x–()
VCPIIIAVCxP
IIIBVC1x–()⋅+⋅=
PIIIAx()
IIIBy()
IIIC1x–y–()
VDPIIIAVDxP
IIIBVDyP
IIICVD
1x–y–()⋅+⋅+⋅=
PIIIAx()
IIIB1x–()
VCy()
VD1y–() PIIIAVCxy PIIIAVDx1y–()PIIIBVC
1x–()yP
IIIBVD
1x–()1y–()⋅+⋅+⋅+⋅=
∂AI
∂t
---------∇DAImjηj–
j
ηzAm
–AIm
αAIm
-----------ηzAm
∇
RAYm
ko()
Ym
+⋅RAIVm
ft()
–
m
=
∂AV
∂t
-----------∇DAVmjηj–
j
ηzAm
–AVm
αAVm
------------ ηzAm
∇
RAVm
ko()
+⋅RAVYm
ft()
Ym
–
m
=
∂Ams,
∂t
-------------- RAYm
ko()
Ym
–RAVm
ko()
–RAIVm
ft() RAVYm
ft()
Ym
++=
4: Diffusion
Diffusion in III–V Compounds
268 Sentaurus™ Process User Guide
I-2013.12
where:
(323)
(324)
(325)
(326)
and (327)
and (328)
(329)
(330)
(331)
(332)
RAYm
ko() kAYmj
ko()
Ymi,∗φYmjηj–
j
Ams,
Ym
Ym∗
--------- AIm
αAIm
-----------Yms,
Yms,∗
--------------
–
=
RAVm
ko() kAVmj
ko()
Vmi,∗φVmjηj–
j
Ams,
Vm
Vm∗
----------AVm
αAVm
------------–
=
RAIVm
ft() kAImjk
ft() αAImj,Vmi,∗φVmkηjk+()–
jk,
AIm
αAIm
-----------Vm
Vm∗
----------Ams,
–
=
RAVYm
ft() kAVmjk
ft() αAVmj,Ymi,∗φYmkηjk+()–
jk,
AVm
αAVm
------------ Ym
Ym∗
--------- Ams,
Yms,
Yms,∗
--------------
–
=
αAImαAImj,ηj–
j
=
αAImj,DAImjdAImj
⁄≡
αAVmαAVmj,ηj–
j
=
αAVmj,DAVmjdAVmj
⁄≡
AIIII
αAIIIIη1–
αAIIIIη1–αAIVη1rIIIV
⁄+
------------------------------------------------------------ AI=
AIV
αAIVη1
rIIIVαAIIII
η1–αAIVη1
+
---------------------------------------------------------AI=
AVIII
αAVIIIη1–
αAVIIIη1–αAVVη1rIIIV
⁄+
--------------------------------------------------------------- AV=
AVV
αAVVη1
rIIIVαAVIII
η1–αAVVη1
+
----------------------------------------------------------- AV=

4: Diffusion
Diffusion in III–V Compounds
Sentaurus™ Process User Guide 269
I-2013.12
Ta bl e 2 0 D e s c r i pt i o n o f s y m b ol s fo r t h e Ch ar g e d Re ac t m od e l
Symbol Description Unit
The Mendeleev group number of a constituent atom of III–V material, . Unitless
Substitutional dopant concentration at group lattice sites.
Dopant–interstitial pair concentration. For example, is SiInt for Silicon.
Dopant–group interstitial pair concentration.
Dopant–vacancy pair concentration. For example, is SiVac for Silicon.
Dopant–group vacancy pair concentration.
Self-interstitial concentration. For example, is GaInt or InInt, and is
AsInt in InGaAs.
Constituent atom concentration. For example, is Gallium or Indium, and
is Arsenic in InGaAs.
Self-interstitial concentration in equilibrium (Eq. 346, p. 272).
Self-interstitial concentration in intrinsic equilibrium:
pdbSet InGaAs GaInt Cstar {<n>}
Constituent atom concentration in equilibrium:
pdbSet InGaAs Gallium CsubStar {<n>}
Charge-state fractions of self-interstitials: . is specified,
for example, by:
pdbSet GaAs GaInt ChargeStates { -2 <n> ... 2 <n> }
Unitless
Va can cy conc entr atio n. i s VacIII and is VacV.
Va can cy conc entr atio n in equ ilib rium (Eq. 347, p. 272).
Va can cy conc entr atio n in int rins ic equi libr ium:
pdbSet GaAs VacIII Cstar {<n>}
Charge-state fractions of vacancies: . is specified, for
example, by:
pdbSet GaAs VacIII ChargeStates { -2 <n> ... 2 <n> }
Unitless
Effective diffusivity of dopant–group interstitial:
pdbSet <mat> <dopant> IntIII D { -2 <n> ... 2 <n> }
pdbSet <mat> <dopant> IntV D { -2 <n> ... 2 <n> }
Effective diffusivity of dopant–group vacancy:
pdbSet <mat> <dopant> VacIII D { -2 <n> ... 2 <n> }
pdbSet <mat> <dopant> VacV D { -2 <n> ... 2 <n> }
Self-diffusivity of dopant–group interstitial:
pdbSet <mat> <dopant> IntIII Dpair { -2 <n> ... 2 <n> }
pdbSet <mat> <dopant> IntV Dpair { -2 <n> ... 2 <n> }
m
mIIIV,{}∈
Ams,
m
cm 3–
AI
AI
cm 3–
AIm
m
cm 3–
AV
AV
cm 3–
AVm
m
cm 3–
Ym
YIII
YV
cm 3–
Yms,
YIII s,
YV
cm 3–
Ym
*
cm 3–
Ymi,
*
cm 3–
Yms,
*
cm 3–
φYm
φYmjφYmj′φ
Ymj′
j
⁄=
φYmj′
Vm
VIII
VV
cm 3–
Vm
*
cm 3–
Vmi,
*
cm 3–
φVm
φVmjφVmj′φ
Vmj′
j
⁄=
φVmj′
DAIm
m
cm2/s
DAVm
m
cm2/s
dAIm
m
cm2/s

4: Diffusion
Diffusion in III–V Compounds
270 Sentaurus™ Process User Guide
I-2013.12
Self-diffusivity of dopant–group vacancy:
pdbSet <mat> <dopant> VacIII Dpair { -2 <n> ... 2 <n> }
pdbSet <mat> <dopant> VacV Dpair { -2 <n> ... 2 <n> }
Ratio of dopant–group interstitial pair concentration to substitutional concentration
in equilibrium.
Unitless
Ratio of dopant–group vacancy pair concentration to substitutional concentration in
equilibrium.
Unitless
Charge of ionized substitutional atom at group lattice sites. Unitless
Ratio of electron concentration to intrinsic carrier concentration ( ). Unitless
Kick-out reaction rate at which kicks out , generates , and increases the
mole fraction of .
Kick-out reaction rate at which reacts with and generates a dopant–vacancy
pair .
Frank–Turnbull reaction rate at which reacts with and generates a
substitutional dopant .
Frank–Turnbull reaction rate at which reacts with , generates a substitutional
dopant , and increases the mole fraction of .
Reaction rate constant associated with , for example:
pdbSet GaAs Si Gallium kfKickOut { -2 <n> ... 2 <n> }
Reaction rate constant associated with , for example:
pdbSet GaAs Si VacV kfKickOut { -2 <n> ... 2 <n> }
Reaction rate constant associated with , for example:
pdbSet GaAs Si VacIII kfFTM { {-2,-2} <n> ... {2,2} <n> }
Reaction rate constant associated with , for example:
pdbSet GaAs Si Gallium kfFTM { {-2,-2} <n> ... {2,2} <n> }
Ratio of the substitutional concentration at group III lattice sites to group V sites in
intrinsic equilibrium. . Applies only to group IV dopants. For
example:
pdbSet GaAs Si Csub.Ratio {<n>}
Unitless
Ta bl e 2 0 D e s c r i pt i o n o f s y m b ol s fo r t h e Ch ar g e d Re ac t m od e l
Symbol Description Unit
dAvm
m
cm2/s
αAIm
m
αAVm
m
zAm
m
η
nn
i
⁄
RAYm
ko()
Ym
Ams,
AIm
Y
cm 3–/s
RAVm
ko()
Vm
Ams,
AVm
cm 3–/s
RAIVm
ft()
AIm
Vm
Ams,
cm 3–/s
RAVYm
ft()
AVm
Ym
Ams,
Y
cm 3–/s
kAYm
ko()
RAYm
ko()
cm 3–/s
kAVm
ko()
RAVm
ko()
cm 3–/s
kAIVm
ft()
RAIVm
ft()
cm 3–/s
kAVYm
ft()
RAVYm
ft()
cm 3–/s
rIIIV
rIIIV AIII s,AVs,
⁄()≡i∗

4: Diffusion
Diffusion in III–V Compounds
Sentaurus™ Process User Guide 271
I-2013.12
Fermi Model
The point-defect concentrations are assumed to be at thermal equilibrium:
(333)
where:
(334)
(335)
Constant Model
The point-defect concentrations are assumed to be at intrinsic equilibrium:
(336)
where:
(337)
(338)
Activation Model
The solid solubility model can be specified. For more information, see Dopant Active Model:
Solid on page 293.
For group IV dopants, the amount of substitutional concentration on group III and group V sites
is reduced by the ratio of a given parameter Csub.Clust.Ratio to the clustering
concentration, respectively:
(339)
∂A
∂t
------ ∇DImDVm
+()η
zm
–Ams,ηzm
()∇
⋅
m
=
AIII s,
η1–
η1–η1rIIIV
⁄+
------------------------------------As
=
AVs,
η1
rIIIVη1–η1
+
-------------------------------- As
=
∂A
∂t
------ ∇D∗Ams,
()∇()⋅
m
=
AIII s,
1
11rIIIV
⁄+
--------------------------- As
=
AVs,
1
rIIIV 1+
-------------------- As
=
AIII s,
+AIII s,
1
11Csub.Clust.Ratio⁄+
---------------------------------------------------------AsAs
+
–()–=

4: Diffusion
Diffusion in III–V Compounds
272 Sentaurus™ Process User Guide
I-2013.12
(340)
Point-Defect Diffusion
(341)
(342)
(343)
where:
(344)
(345)
(346)
(347)
Ta bl e 2 1 D e s c r i pt i o n o f s y m b ol s fo r t h e pa i r d i f f u si on m o de l
Symbol Description Unit
The Mendeleev group number of a constituent atom of III–V material, . Unitless
Self-interstitial concentration. For example, is GaInt or InInt, and is
AsInt in InGaAs.
Constituent atom concentration. For example, is Gallium or Indium, and
is Arsenic in InGaAs.
AVs,
+AVs,
1
Csub.Clust.Ratio 1+
--------------------------------------------------AsAs
+
–()–=
∂Ym
∂t
----------∇dYmj,Ymi,∗φYmjηj–
j
Ym
Ym∗
---------
∇
RYVm
–RYY′m
Y′m
–RAYm
ko() RAVYm
ft()
+()
A
–⋅=
∂Vm
∂t
---------- ∇dVmj,Vmi,∗φVmjηj–
j
Vm
Vm∗
----------
∇
RYVm
Ym
–RAVm
ko() RAIVm
ft()
+()
A
–⋅=
∂Yms,
∂t
--------------RYVmRYY′m
Y′m
RAYm
ko() RAVYm
ft()
+()
A
++=
RYVmkYVmjYmi,∗φYmj
()Vmi,∗φVmk
()η
jk+()–
jk,
Ym
Ym∗
--------- Vm
Vm∗
----------Yms,
Yms,∗
--------------–
=
RYY′mkYY′mjY′ms,∗Ymi,∗φYmj
()η
j–
j
Ym
Ym∗
--------- Y′ms,
Y′ms,∗
--------------- Y′m
Y′m∗
----------- Yms,
Yms,∗
--------------–
=
Ym∗Ymi,∗φYmjηj–
j
=
Vm∗Vmi,∗φVmjηj–
j
=
m
mIIIV,{}∈
Ym
YIII
YV
cm 3–
Yms,
YIII s,
YV
cm 3–

4: Diffusion
Diffusion in III–V Compounds
Sentaurus™ Process User Guide 273
I-2013.12
Self-interstitial concentration in equilibrium (Eq. 346, p. 272).
Self-interstitial concentration in intrinsic equilibrium:
pdbSet InGaAs GaInt Cstar {<n>}
Constituent atom concentration in equilibrium:
pdbSet InGaAs Gallium CsubStar {<n>}
Charge-state fractions of self-interstitials: . is specified,
for example, by:
pdbSet GaAs GaInt ChargeStates { -2 <n> ... 2 <n> }
Unitless
Va can cy conc entr atio n. i s VacIII and is VacV.
Va can cy conc entr atio n in equ ilib rium (Eq. 347, p. 272).
Va can cy conc entr atio n in int rins ic equi libr ium:
pdbSet GaAs VacIII Cstar {<n>}
Charge-state fractions of vacancies: . is specified, for
example, by:
pdbSet GaAs VacIII ChargeStates { -2 <n> ... 2 <n> }
Unitless
Self-interstitial diffusivity:
pdbSet InGaAs GaInt D { -2 <n> ... 2 <n> }
Vacancy diffusivity:
pdbSet GaAs VacIII D { -2 <n> ... 2 <n> }
Ratio of electron concentration to intrinsic carrier concentration ( ). Unitless
Kick-out reaction rate at which kicks out , generates , and increases the
mole fraction of . See Eq. 323, p. 268 for details.
Kick-out reaction rate at which reacts with and generates a dopant–vacancy
pair . See Eq. 324, p. 268 for details.
Frank–Turnbull reaction rate at which reacts with and generates a
substitutional dopant . See Eq. 325, p. 268 for details.
Frank–Turnbull reaction rate at which reacts with , generates a substitutional
dopant , and increases the mole fraction of . See Eq. 326, p. 268 for details.
Interstitial–vacancy bulk recombination rate for group .
Kick-out reaction rate at which kicks out and occupies the lattice site by
generating . The reaction increases the mole fraction of but decreases that of .
Reaction rate constant associated with , for example:
pdbSet GaAs GaInt KbulkChargeStates { -2 <n> ... 2 <n> }
Reaction rate constant associated with , for example:
pdbSet InGaAs GaInt Indium kfKickOut { -2 <n> ... 2 <n> }
Ta bl e 2 1 D e s c r i pt i o n o f s y m b ol s fo r t h e pa i r d i f f u si on m o de l
Symbol Description Unit
Ym
*
cm 3–
Ymi,
*
cm 3–
Yms,
*
cm 3–
φYm
φYmjφYmj′φ
Ymj′
j
⁄=
φYmj′
Vm
VIII
VV
cm 3–
Vm
*
cm 3–
Vmi,
*
cm 3–
φVm
φVmjφVmj′φ
Vmj′
j
⁄=
φVmj′
dYm
cm2/s
dVm
cm2/s
η
nn
i
⁄
RAYm
ko()
Ym
Ams,
AIm
Y
cm 3–/s
RAVm
ko()
Ym
Ams,
AVm
cm 3–/s
RAIVm
ft()
AIm
Vm
Ams,
cm 3–/s
RAVYm
ft()
AVm
Ym
Ams,
Y
cm 3–/s
RYVm
m
cm 3–/s
RYY′m
Ym
Y'ms,
Y'm
Y
Y'
cm 3–/s
kYVm
RYVm
cm 3–/s
kYY′m
RYY′m
cm 3–/s

4: Diffusion
Diffusion in III–V Compounds
274 Sentaurus™ Process User Guide
I-2013.12
Poisson Equation
(348)
The band structure parameter (Eq. 359, p. 278) relies on the mole-fraction dependent affinity
and energy bandgap. For details, see Poisson Equation for Hetero-junctions on page 278.
MoleFractionFields
The Tcl procedure MoleFractionFields returns the list of the constituent atom
concentrations for given mole-fractions by considering that the lattice density of an alloy varies
with mole-fractions. The usage is:
MoleFractionFields <alloy> <x-mole-fraction> <y-mole-fraction>
where y-mole-fraction is required only for quaternary materials.
For example:
sprocess> MoleFractionFields InGaAs 0.53
Indium = 9.514445e+21 Gallium = 1.072906e+22
sprocess> MoleFractionFields AlInGaAs 0.2 0.4
Aluminum = 4.122000e+21 Indium = 8.244000e+21 Gallium = 8.244000e+21
The proc MoleFractionFields is useful for adding the constituent atoms into a deposited
layer, for example:
deposit InGaAs thickness=0.1 fields.values= “[MoleFractionFields InGaAs 0.53]
Be=1e17”
εrε0ψθ–()∇()∇•
qp n–zAmAms,
()
m
A
zAm j+()α
AImj,ηj–
()
j
AIm
αAIm
-----------zAm j+()α
AVmj,ηj–
()
j
AVm
αAVm
------------
+
jYmi,∗φYmjηj–
j
Ym
Ym∗
--------- jVmi,∗φVmjηj–
j
Vm
Vm∗
----------+
Ym
m
+
m
A
++
–=

4: Diffusion
Pressure-dependent Defect Diffusion
Sentaurus™ Process User Guide 275
I-2013.12
Pressure-dependent Defect Diffusion
Eq. 153, p. 211 shows that depends on the Fermi level. However, you can select one of the
available models (Constant, FermiLevelDependent, FermiPressureDependent)
using the command:
pdbSet <material> <defect> CStarMod <model>
where defect is interstitial or vacancy, and model is one of the available models.
The Constant model simply sets to , the FermiLevelDependent model is
given in Eq. 153, and the FermiPressureDependent model includes both Fermi effects and
pressure-field effects.
The pressure effects are modeled in Sentaurus Process by modifying the equilibrium point-
defect concentrations:
(349)
where is the total equilibrium concentration of point defect (interstitial or vacancy).
is the activation volume change of equilibrium point defects due to the pressure and is given
by:
(350)
(351)
where is the dilatation, is the measure of the sphericity of the interstitial, is the radius
of the vacancy, is the Poisson ratio of silicon, is the surface tension of the vacancy, and
is the shear modulus of silicon. The following set of commands can be used to modify :
pdbSet Silicon Interstitial Volume 8.59e-24
pdbSet Silicon Vacancy Volume -5.52e-24
The unit of Volume is .
CX
*
CX
*
CXintrinsic()
*
CX
*CX
*Si P 0≡,()e
VXP∆()
kT
-------------------–
=
CX
*
X
VX
∆
P
VI
∆ε4πro
3
=
VV
∆-2πrs
231η–()
1η+
-----------------Γ
µ
---=
ε
ro
rs
η
Γ
µ
VX
∆
cm3

4: Diffusion
Electron Concentration
276 Sentaurus™ Process User Guide
I-2013.12
Electron Concentration
To calculate the electron concentration or, alternatively, the electron potential, Sentaurus
Process solves either the Poisson equation or charge balance equation. By default, the
ChargedReact, ChargedPair, and ChargedEquilibrium models all solve the charge
balance equation. The uncharged models do not require a separate equation because the
electron concentration can be computed directly from the net doping.
The Poisson equation is given by:
(352)
where is the permittivity, is the potential, and are the electron and hole
concentrations, and is the net charge.
Electrons and holes are always assumed to be in equilibrium, such that:
(353)
The charge balance equation is:
(354)
In Eq. 352 and Eq. 354, the must be calculated. The net charge is given by:
(355)
where:
■ is the charge state of the defect , interstitial, or vacancy.
■ is the concentration of the defect in the charge state .
■ is the charge state of dopant .
■ is the dopant and defect pair.
You ca n ex clud e or i nc lu de th e c ha rge d do pa nt –de fe ct pai rs or ch arg ed d ef ec ts in Eq. 355, for
example:
pdbSet Si Dopant ChargeModel DopantOnly
pdbSet Si Dopant ChargeModel DopantDefect
The first command, which is the default behavior for dopants, includes only the charged
dopants in silicon in Eq. 355. The second command includes the charged dopants as well as the
charged dopant–defect pairs in Eq. 355.
εψ∇()∇•qp n–N∆+()–=
ε
ψ
n
p
∆N
pn ni
2
=
n–pN∆++ 0=
∆N
N∆zjCAj
+
j
cCXc
c
zjc+()CAjX
cz
j
+()
Xcj,,
++=
c
X
CXc
X
c
zj
Aj
AjX
Aj
X

4: Diffusion
Electron Concentration
Sentaurus™ Process User Guide 277
I-2013.12
In a similar way:
pdbSet Si Defect ChargeModel None
pdbSet Si Defect ChargeModel Defect
The first command, which is the default behavior for defects, excludes the charged defects in
silicon in Eq. 355 and the second command includes them.
NOTE The diffusion models Constant, Fermi, Pair, and React always
exclude the charged dopant–defect pairs.
The Poisson equation is switched on or off with the command:
pdbSet Si Potential Poisson 1 | 0
NOTE The above switch is used to switch from or to the Poisson equation to or
from the charge balance equation.
If it is switched off, the charged defects and charged dopant–defect pairs are not included in
Eq. 355, and Eq. 356 is used to calculate the potential:
(356)
where is , and is the intrinsic concentration of electrons and can be set using the
command:
pdbSet <material> Potential ni {<n>}
To switch on or off the solution of the Poisson equation or the charge balance equation,
regardless of the diffusion model selected, use the commands:
pdbSetBoolean Potential ForcedTurnOff 0/1
pdbSetBoolean Potential ForcedTurnOn 1/0
NOTE If the potential equation is switched off, charge neutrality is assumed. If
the selected diffusion or cluster models use complex charges, this may
lead to instability in the code.
ψ1
Vti
------ 1
2ni
------- N∆N∆24ni
2
++()
log=
Vti
1kT⁄
ni

4: Diffusion
Electron Concentration
278 Sentaurus™ Process User Guide
I-2013.12
Poisson Equation for Hetero-junctions
(357)
where:
(358)
and (359)
(360)
(361)
(362)
(363)
Table 22 Description of symbols for Poisson equation
Symbol Description Unit
Intrinsic Fermi potential. V
Band structure parameter. V
Va cuu m pe rmitt iv it y ( ) . F /m
Relative permittivity:
pdbSet <material> Potential Permittivity {<n>}
Unitless
Ratio of electron concentration to intrinsic carrier concentration ( ). Unitless
Affinity that depends on the mole fraction . If Affinity is specified on the
material, then .
Otherwise, , where is
calculated by Physical Parameter Interpolation on page 266.
pdbSet <material> Potential Affinity {<n>}
pdbSetDouble <material> Potential Affinity.X1 {<n>}
pdbSetDouble <material> Potential Affinity.X2 {<n>}
eV
εrε0ψθ–()∇()∇•qp n–N∆+()–=
qθχ
Eg
2
------kT
2
------Nc
Nv
------
ln++=
nn
i
qψ
kT
-------
exp=
pn
i
qψ
kT
-------–
exp=
NcNc300 T
300
---------
32⁄
=
NvNv300 T
300
---------
32⁄
=
niNcNv
Eg
2kT
---------–
exp=
EgEg300 Egα
3002
300 β+
------------------ T2
Tβ+
-------------–
+=
ψ
θ
ε0
8.854 12–
×10 F/m
εr
η
nn
i
⁄
χ
x
χAffinity Affinity.X1 xAffinity.X2 x1x–()⋅+⋅+=
χχ
interpolated Affinity.X2 x1x–()⋅+=
χinterpolated

4: Diffusion
Electron Concentration
Sentaurus™ Process User Guide 279
I-2013.12
Turning on Use.DOS specifies to use the density-of-state to calculate the intrinsic carrier
concentration. If Use.DOS is turned off, is set to 0.0 and is given by the PDB parameter
ni:
pdbSet Silicon Potential Use.DOS 0
To solve Poisson’s equation in insulators or to apply the Dirichlet boundary condition at
conductor surfaces, use:
pdbSet <mat> Potential Poisson 1
For example:
pdbSet Oxide Potential Poisson 1
pdbSet Aluminum Potential Poisson 1
NOTE Dirichilet boundary condition is automatically applied at metal–
insulator or metal–semiconductor interfaces only when Poisson’s
equations are on in both neighboring materials.
NOTE Continuous boundary condition is automatically applied at insulator–
insulator, insulator–semiconductor, semiconductor–semiconductor
only when Poisson’s equations are on in both neighboring materials.
Band gap at 300 K, which depends on the mole fraction . If Eg300 is
specified on the material, then .
Otherwise, , where
is calculated by Physical Parameter Interpolation on page 266.
pdbSet <material> Potential Eg300 {<n>}
pdbSetDouble <material> Potential Eg300.X1 {<n>}
pdbSetDouble <material> Potential Eg300.X2 {<n>}
eV
Bandgap modification for temperature dependency:
pdbSet <material> Potential Eg.Alpha {<n>}
eV
Temperature constant for band gap depending on temperature:
pdbSet <material> Potential Eg.Beta {<n>}
K
Density-of-states of a conduction band.
Density-of-states of a conduction band at 300 K:
pdbSet <material> Potential Nc300 {<n>}
Density-of-states of a valence band.
Density-of-states of a valence band at 300 K:
pdbSet <material> Potential Nv300 {<n>}
Table 22 Description of symbols for Poisson equation
Symbol Description Unit
Eg300
x
Eg300 Eg300 Eg.X1 xEg.X2 x1x–()⋅+⋅+=
Eg300 Eg300interpolated Eg.X2 x1x–()⋅+=
Eg300interpolated
Egα
β
Nc
cm 3–
Nc300
cm 3–
Nv
cm 3–
Nv300
cm 3–
θ
ni

4: Diffusion
Electron Concentration
280 Sentaurus™ Process User Guide
I-2013.12
To define a conductor, set Conductor and WorkFunction parameters. For example:
pdbSet Aluminum Conductor 1
pdbSet Aluminum Potential WorkFunction 4.1
The default conductors are set as follows:
To specify to turn on Poisson for all material regions in a simulation structure, use:
pdbSet Compute All.Poisson 1
Bandgap Narrowing
If bandgap narrowing effects need to be considered, Sentaurus Process uses the effective
intrinsic electron density, , instead of . is given by:
(364)
where is the reduction in the bandgap energy of silicon and is defined as:
(365)
where is the user-defined bandgap narrowing and can be set using:
pdbSet <material> Potential delEg {<n>}
Ta bl e 23 D efa u l t c o nd u ct o r m at e r ia ls
Material Work Function (eV)
Aluminum
4.10
Colbalt
5.00
ColbaltSilicide
4.76
Copper
4.70
Nickel
5.20
NickelSilicide
4.84
Platium
5.50
Titanium
4.33
Tungsten
4.80
TungstenSilicide
4.76
TiSilicide
4.56
nie
ni
nie
nie nie
Eg∆
2kT
----------–
=
∆Eg
Eg∆Egu∆Egs∆+=
Egu∆

4: Diffusion
Electron Concentration
Sentaurus™ Process User Guide 281
I-2013.12
is the bandgap narrowing due to strain in the structure. To switch on this effect, the
intrinsic electron density model (niMod) must be set to StrainDependent. To select the
model, use:
pdbSet <material> Potential niMod <model>
niMod can have either the value Constant or StrainDependent. The Constant model
will ignore .
If the StrainDependent model is selected, will be calculated [25] using:
(366)
(367)
where is the strain in the respected direction, and are the dilatational deformation
potentials for the conduction and valence band valleys, respectively.
To set, use:
pdbSet Silicon Potential Ec Dilatational {
1 -8.6
2 -8.6
3 -8.6
}
pdbSet Silicon Potential Ev Dilatational {
1 -2.1
2 -2.1
}
and are the deviatoric deformation potential of conduction and
valence band valleys. They can be set using the commands:
pdbSet Si Potential Ec Deviatoric(1) {
1 9.5
2 0.0
3 0.0
}
pdbSet Si Potential Ec Deviatoric(2) {
1 0.0
2 9.5
3 0.0
}
pdbSet Si Potential Ec Deviatoric(3) {
1 0.0
2 0.0
Egs∆
Egs∆
Egs∆
Eci∆Dci εxx εyy εzz
++()Dcxiεxx Dcyiεyy Dcziεzz
+++=
Evi∆Dvi εxx εyy εzz
++()=
0.5Dvbi
2εxx εyy
–()
2εyy εzz
–()
2εzz εxx
–()
2
++()Dvdi
2εxy
2εxz
2εyz
2
++()+()±
ε
Dci Dvi
,
Dcxi Dcyi Dczi
,,
Dvbi Dvdi
,

4: Diffusion
Epitaxy
282 Sentaurus™ Process User Guide
I-2013.12
3 9.5
}
pdbSet Si Potential Ev Deviatoric(1) {
1 0.5
2 4.0
}
pdbSet Si Potential Ev Deviatoric(2) {
1 0.5
2 4.0
}
Sentaurus Process uses the averaged values of conduction and valence bands energies,
:
(368)
(369)
The bandgap narrowing becomes:
(370)
NOTE is ignored where (see Dopant Diffusion in
SiGe on page 260).
Epitaxy
Epitaxial growth is simulated when an Epi type ambient is specified on either the diffuse
command or in a temp_ramp ramp used by the diffuse command.
By default, two Epi type ambients are available: one is called Epi and the other is called LTE.
If Epi is specified, Silicon will grow on Silicon and PolySilicon will grow on
PolySilicon. If the LTE ambient is specified, Silicon will again grow on Silicon, but
PolySilicon will grow on Oxide, Nitride, and PolySilicon. The layer thickness is
specified with the thick parameter and doping is specified with the epi.doping parameter.
Epitaxy is solved using the Alagator general growth scheme (see Alagator for Generic Growth
on page 596). This allows the creation of new epi growth modes (that is, specifying which
materials grow) and material-dependent growth rates.
Eci,Evi∆∆
Ec∆kT 1
3
---e
Eci∆
kT
------------–
i1=
3
log–=
Ev∆kT 1
2
---e
Evi∆
kT
------------
i1=
2
log=
Egs∆Ec∆Ev∆–=
Egs∆
Germanium
5e22
------------------------------- 0.1%>

4: Diffusion
Epitaxy
Sentaurus™ Process User Guide 283
I-2013.12
An unlimited number of species can be incorporated into the epitaxial layer. Doping is
specified using the epi.doping and epi.doping.final parameters in either the diffuse
command or a temp_ramp included in a diffuse command. The parameters <material>
<solution> Cepi0 and <material> <solution> CepiE set the default value of fields
in the growing material. The defaults are overwritten by setting the epi.doping and
epi.doping.final parameters of the diffuse or temp_ramp commands. The same set of
equations as for the single-crystalline silicon is solved for the epitaxial silicon during the
diffusion step simulation. If the growth temperature goes below the minimum diffusion
temperature, the diffusion equations will be switched off, but the boundary conditions for
dopant incorporation will be applied. The Continuous boundary condition is applied to all
the mobile species at the interface between the epitaxial layer and single-crystalline silicon to
take into account the variable jump.
It is also possible to incorporate the auto-doping of dopants during the epitaxial growth using
the auto.doping parameter in either the diffuse command or a temp_ramp included in a
diffuse command. Auto-doping can be switched on only for dopants that are not listed in the
epi.doping or epi.doping.final parameters (see Epi Auto-Doping on page 285).
In certain examples, it is easier to specify resistivity to obtain the required doping concentration
in the epi layer. The resistivity can be specified using the epi.resist parameter in either the
diffuse command or a temp_ramp included in a diffuse command.
Two different methods can be selected to simulate the epitaxial growth. The epi.model
parameter of the diffuse command is used to switch between them:
■If epi.model=0 (default), a moving-boundary algorithm similar to the oxidation one is
applied.
■If epi.model=1, alternating doped deposition and inert annealing steps are used. Model 1
supports selective epitaxy, graded doping, and material-dependent growth rates, and can be
used with both the Sentaurus Structure Editor and MGOALS modes. Furthermore, for 3D
epitaxy, Model 1 is recommended because of the computational time and reliability issues
related to moving boundaries (Model 0) in 3D.
To set the grid spacing, use the epi.layers parameter. This sets the number of grid layers
that are deposited during the corresponding diffuse or temp_ramp steps.
NOTE Model 1 is recommended for 3D epitaxy and can be used with 2D.
Using LKMC for Deposition Shape
The shape of the growing epi layer can be controlled by lattice KMC (LKMC) to obtain more
realistic deposition shapes without having the performance penalty associated with pure

4: Diffusion
Epitaxy
284 Sentaurus™ Process User Guide
I-2013.12
atomistic mode. To use this mode, specify lkmc on the diffuse command and the pdb
parameter KMC Epitaxy, for example:
pdbSet KMC Epitaxy 1
diffuse time=1<s> temperature=550 Epi lkmc epi.thickness=0.02
In this example, the LKMC epi growth rate will be scaled such that the <100> direction (by
default the fastest direction) will grow 0.02 µm/s. For more information about controlling
LKMC epitaxy, see Epitaxial Deposition on page 519.
Epi Doping
Two parameters of the diffuse and temp_ramp commands are used to control doping:
epi.doping and epi.doping.final. Both parameters take a list of parameters, that is,
dopant and field names, as their arguments.
If a dopant or field name appears in only one of the lists or in both of the lists with the same
value, the value of the doping is constant throughout the step. If the dopant or field appears in
both lists with different values, a linear gradient of the doping is applied. For example:
temp_ramp name=t1 temperature=550 t.final=700 time=1<min>
temp_ramp name=t1 t.final=700 time=5<min> Epi thick=0.1<um> \
epi.doping = { Boron=1e18 Germanium=1e21 } \
epi.doping.final = { Germanium=5e21 }
diffuse temp.ramp=t1
In this example, epitaxy is simulated after an inert temperature ramp. During epitaxy, the boron
concentration is a constant , and germanium is ramped from to
. In addition, all these parameters can be set in the diffuse command, for
example:
diffuse temperature=700 time=5<min> LTE \
epi.doping.final = { Arsenic=1e18 } thick = 0.1<um>
In this case, a constant arsenic doping of is applied to an LTE epitaxial growth.
Initialization of Dopant Clusters in Epi
The dopant cluster concentration in an epitaxial layer is initialized by:
(371)
1018 cm 3–
1021 cm 3–
510
21
× cm 3–
1018 cm 3–
∆CAclust i,CAdoping clust i,, f+Aclust i,max CAdoping dopant,EpiIni–0.0,()⋅=

4: Diffusion
Epitaxy
Sentaurus™ Process User Guide 285
I-2013.12
where:
(372)
EpiInit and FractionEpi are specified by:
pdbSet <material> <dopant> EpiInit {<n>}
pdbSet <material> <cluster> FractionEpi {<n>}
The list of epi.doping can include both a dopant and its cluster solutions, for example:
epi.doping= { Boron= 1e20 B4= 1e19 }
With EpiInit= , the concentrations of active boron (Boron) and B4 (B4) are set to
and ( + ( – )/4); in other words, , respectively.
Epi Auto-Doping
The auto.doping parameter of the diffuse and temp_ramp commands controls doping.
The parameter takes a list of parameters, that is, dopant and field names, as its arguments. If a
dopant or field name appears in auto.doping and in either epi.doping or
epi.doping.final, auto-doping of this dopant is ignored. For example:
temp_ramp name=t1 temperature=550 t.final=700 time=1<min>
temp_ramp name=t1 t.final=700 time=5<min> Epi thick=0.1<um> \
auto.doping = { Boron Germanium } epi.doping = { Germanium=1e21 } \
diffuse temp.ramp=t1
In this example, only auto-doping of boron is simulated. The parameter auto.doping
switches on the following model automatically at Gas and epitaxially grown material interface:
(373)
where is the transfer rate, is the segregation rate, is the peak value of
the dopant concentration in the auto-doped region, is the minimum value of the dopant
concentration, is the decay rate of the auto-doping, and is the dopant concentration.
These parameters can be modified using the commands:
pdbSet <interface material> <dopant> TransferAutoDoping <n>
pdbSet <interface material> <dopant> SegregationAutoDoping <n>
pdbSet <interface material> <dopant> Cstar <n>
pdbSet <interface material> <dopant> minConc <n>
fAclust i,
FractionEpii
FractionEpii
i
-------------------------------------=
219
×10
219
×10
119
×10
120
×10
219
×10
319
×10
jn•kTransfer CA
*e
kDecay∆t
t
---------------------
–
CA
min
+
CA
kSegregation
-----------------------------–
=
kTransfer
kSegregation
CA
*
CA
min
kDecay
CA

4: Diffusion
Epitaxy
286 Sentaurus™ Process User Guide
I-2013.12
pdbSet <interface material> <dopant> DecayRate <n>
where <interface material> is the Gas and epitaxially grown material interface. In
Eq. 373, is the percentage of the simulation time since the diffusion started, and is the
total simulation time from the beginning to the end of diffusion. Eq. 373 is created
automatically and stored in a term called <dopant>AutoDoping. You can overwrite this by
defining your own reactions.
For example:
term name=BoronAutoDoping EpiOnSilicon /Gas add eqn = \
"1e-3*(1e16-Boron_EpiOnSilicon/0.1)"
NOTE Since the model does not solve equations in gas, the dose loss or gain of
the dopant is expected.
Epi Doping Using Resistivity
The epi.resist parameter of the diffuse and temp_ramp commands controls doping.
The parameter takes a list of parameters, that is, dopant name and resistivity, as its argument.
If more than one dopant name appears in the list, the doping concentration is calculated
individually for each dopant by ignoring the other ones. For example:
temp_ramp name=t1 temperature=550 t.final=700 time=1<min>
temp_ramp name=t1 t.final=700 time=5<min> Epi thick=0.1<um> \
epi.resist= { Arsenic=1e-2 Phosphorus=2e-3 }
diffuse temp.ramp=t1
In this example, epitaxy is simulated after an inert temperature ramp. During epitaxy, the
arsenic concentration is a constant and the phosphorus concentration is
. In addition, all of these parameters can be set in the diffuse command, for
example:
diffuse temperature=700 time=5<min> LTE \
epi.resist= { Arsenic=1e-2 Phosphorus=2e-3 } thick= 0.1<um>
The doping concentration calculations use the silicon-based mobility models (see Resistivity
on page 857).
Epi Growth Settings: Low-Temperature Epitaxy
Several parameters are available to allow for the simulation of effects seen in low-temperature
epitaxy (LTE). LTE growth can result in the growth of polysilicon on insulators such as oxide
∆t
t
4.3 1019
× cm 3–
9.7 1019
× cm 3–

4: Diffusion
Epitaxy
Sentaurus™ Process User Guide 287
I-2013.12
and nitride after a seed layer has nucleated. In addition, the growth rate may depend on the
starting material where the growth is occurring. To allow different growth rates and nucleation
times, Sentaurus Process uses temporary materials with distinct names that are converted back
to standard names at the end of the diffusion command. For example, during LTE,
LTEOnOxide is grown on oxide, and LTEOnSilicon is grown on silicon (there are also the
materials LTEOnPolySilicon and LTEOnNitride). After the diffusion step is complete,
LTEOnOxide is converted to PolySilicon, and LTEOnSilicon is converted to Silicon.
In addition, be aware that after material conversion, regions will merge if there are interfaces
with the same material on both sides.
To set the nucleation delay for LTE growth on oxide, use:
pdbSet Gas_<starting material> <ambient> NucleationDelay <n>
where <n> is in seconds. For the case of LTEOnOxide, <starting material> is Oxide and
<ambient> is LTE.
NOTE The exposure time is not saved, so nucleation must happen within one
diffuse command (use the temp_ramp command to create long
diffusion steps with optional ramp-up or ramp-down).
The growth rate for all materials is determined by default from the native layer thickness as
well as the thick and time parameters of the diffuse or temp_ramp commands. However,
the growth rate can be set manually using a callback procedure like this:
pdbSet <growing material>_Gas <ambient> GrowthRateProc <proc name>
Inside <proc name>, you should set the pdb parameter GrowthReaction. For example:
pdbSet Gas_LTEOnOxide LTE GrowthRateProc MyGRProc
proc MyGrProc { Mat Sol } {
set myGrowthRate 1.0e-7 ;# in cm/s
pdbSetString $Mat $Sol GrowthReaction "$myGrowthRate"
}
It is possible to set GrowthReaction to any Alagator expression not involving derivative
expressions or element values.
Simulating Facet Growth during Selective Epitaxy
There are two ways to switch on faceting:
■Using the angles.factors parameter of the temp_ramp (or diffuse) command.
■Using the PDB parameter <epimat gas interface> angles.factors along with
setting the parameter pdbSet Grid AnisotropicGrowth 1.
4: Diffusion
Epitaxy
288 Sentaurus™ Process User Guide
I-2013.12
The syntax for both angles.factors parameters is similar:
temp_ramp angles.factors= {
<interface material1>= { angle1(degrees) factor1(unitless) \
angle2 factor2 ...} \
<interface material2> = ... \
}
or:
pdbSet <interface material> angles.factors {
angle1(degrees) factor1(unitless) angle2 factor2 ...
}
where the interface material would be, for example, EpiOnSilicon_Gas for epi growth on
silicon and Gas_LTEOnSilicon for LTE on silicon. There are aliases for all materials, so the
order of the interface materials is not important.
To form facets, a large range of degrees near 0 that have a factor of 1.0 is needed. For larger
angles, the factor should monotonously decrease to 0 at the required facet angle. For example,
to form facets during epi on silicon, the following setting could be used:
temp_ramp thick=<thick> epi time=<time> temperature=<temp> epi.layers=<nlay> \
angles.factors = {
EpiOnSilicon_Gas = { 0.0 1.0 20.0 1.0 35.0 0.0 }
}
Controlling Where Facets Form
By default, facets form at all triple points. To switch off these facets, use:
pdbSet AnisoGrowthTriplePoints 0
By default, facets will not form on the outer boundaries. To switch on faceting on the outer
boundary, use:
pdbSet AnisoGrowOuterBoundaries 1
Time-stepping
This algorithm and anisotropic growth in general can be inherently unstable. If a ‘bump’
develops during growth, it may persist or perhaps even grow larger. To prevent bumps from
forming, it is necessary to take small time steps. The parameter dThicknessAnisoGrowth
can be used to control time-stepping during anisotropic growth. It sets a maximum thickness
per time step:
pdbSet Diffuse dThicknessAnisoGrowth <thickness in um> #; default 0.001um
35°

4: Diffusion
Other Effects on Dopant Diffusion
Sentaurus™ Process User Guide 289
I-2013.12
Other Effects on Dopant Diffusion
Pressure-dependent Dopant Diffusion
Dopant diffusivities can be enhanced or retarded due to stress or pressure. In addition to this,
shrinking device dimensions can cause significant stress or pressure gradients affecting dopant
diffusion further [25]. With this model, Sentaurus Process allows diffusivities and gradients to
be multiplied by user-defined factors as follows:
(374)
where is the concentration, is the diffusivity, and and are user-definable terms.
You can define both factors. To switch the model on or off, use the command:
pdbSet <material> <dopant> StressModel <model>
where model is None (off, default value) or PDependent (on).
For the definition of terms, see Chapter 6 on page 571.
For example, in the case of specified boron in silicon, this is given by:
term name=BoronIntSSFactor add Silicon eqn = {User defined equation}
term name=BoronIntSPFactor add Silicon eqn = {User defined equation}
To allow Sentaurus Process to use these terms, specify a term with the name
<dopant><defect>SSFactor or <dopant><defect>SPFactor.
If the model is switched on and you do not provide the terms, and are calculated as:
(375)
(376)
where and are activation volumes and can be set using the commands:
pdbSet <material> <dopant> <defect> delVolV {<n>}
pdbSet <material> <dopant> <defect> delVolS {<n>}
jDD
SS
C
DSP
----------
∇–=
C
D
DSS
DSP
DSS
DSP
DSS e
VVP∆()
kT
-------------------–
=
DSP e
VSP∆()
kT
------------------–
=
∆VV
∆VS

4: Diffusion
Other Effects on Dopant Diffusion
290 Sentaurus™ Process User Guide
I-2013.12
Diffusion Prefactors
Dopant diffusivities can be enhanced or retarded due to various new process conditions. If a
new model does not exist to simulate the observed behavior, you may want to multiply the
existing diffusivity with a prefactor. Sentaurus Process allows diffusivities to be multiplied by
user-defined factors as follows:
(377)
where is the concentration, is the diffusivity, and is the diffusion prefactor. For
example, in the case of specified boron in silicon, this is given by:
term name=BoronDiffFactor add Silicon eqn = "exp(0.042 * $Vti * 125 * \
Germanium / 5e22)"
The effective diffusivity of boron ( ) will be multiplied by
BoronDiffFactor. (For the definition of terms, see Using Terms on page 580.) Sentaurus
Process also allows each dopant–defect diffusivity to be multiplied by a different user-defined
factor:
(378)
where is the diffusivity of the dopant–interstitial pair, is the diffusivity of the dopant–
vacancy pair, and and are the diffusion prefactors for each dopant–defect pair. For
example:
term name=BoronIntDiffFactor add Silicon eqn = "exp(0.042 * $Vti * 125 * \
Germanium / 5e22)"
term name=BoronVacDiffFactor add Silicon eqn = "exp(0.042 * $Vti * 25 * \
Germanium / 5e22)"
In this example, diffusivity of boron–interstitial pairs ( ) and the diffusivity of boron–
vacancy pairs ( ) will be multiplied by BoronIntDiffFactor and
BoronVacDiffFactor, respectively.
To allow Sentaurus Process to use these terms, specify a term with the name
<dopant>DiffFactor or <dopant><defect>DiffFactor.
Anisotropic (optionally stress-dependent) diffusivities can be specified using the diffusion
prefactors as well. To do this, the diag operator must be used when specifying the diffusion
prefactor. For more information, see Special Functions on page 573.
jDD
FC∇–=
C
D
DF
DBDBI DBV
+=
jD
CIDIF DCVDVF
+()C∇–=
DCI
DCV
DIF
DVF
DBI
DBV

4: Diffusion
Other Effects on Dopant Diffusion
Sentaurus™ Process User Guide 291
I-2013.12
High-Concentration Effects on Dopant Diffusion
To model experimentally observed sharp increases in arsenic diffusion in silicon at high dopant
concentrations, diffusivity coefficients must be modified. Dunham and Wu [26] proposed that
interactions of vacancies with more than one dopant result in the enhancement of dopant–
vacancy pair diffusivity by a factor:
(379)
(380)
where is the active concentration of arsenic, and cm–3 and for
arsenic. The correction factor can be applied to all dopant–defect pairs as long as the
parameters are supplied. The model can be switched on with the command:
pdbSet <material> <dopant> <defect> HighConcDiffEffect 1
The default of the model is off (0). The model parameters are set using the command:
pdbSet <material> <dopant> <defect> Cref {<n>}
pdbSet <material> <dopant> <defect> Cpow {<n>}
For example:
pdbSet Silicon Arsenic Vacancy Cref 1.6e20
pdbSet Silicon Arsenic Vacancy Cpow 4.0
Hydrogen Effects on Dopant Diffusion
To model experimentally observed sharp increases in boron diffusion in oxide at the presence
of hydrogen, diffusivity coefficients of boron must be modified. Chakravarthi et al.[27]
proposed that the presence of hydrogen results in the enhancement of boron diffusivity by the
following factor:
(381)
(382)
where is the active concentration of hydrogen, and cm–3 and for
boron. If hydrogen is present in the structure, the enhancement factor for boron will be applied
automatically.
jDD
FC∇–=
DF1CA
Cref
----------
Cpow
+=
CAs
Cref 220
×10∼
Cpow 4∼
jDD
FC∇–=
DF1CH
Cref
----------
Cpow
+=
CH
Cref 115
×10∼
Cpow 1∼

4: Diffusion
Dopant Activation and Clustering
292 Sentaurus™ Process User Guide
I-2013.12
The model parameters are set using the command:
pdbSet <material> Hydrogen <dopant> Cref {<n>}
pdbSet <material> Hydrogen <dopant> Cpow {<n>}
For example:
pdbSet Oxide Hydrogen Boron Cref 1.6e20
pdbSet OxideHydrogen Boron Cpow 4.0
The correction factor can be applied to other dopants if the dopant is given in the dopant list
using the command:
pdbSet <material> Hydrogen Dopants <list>
where <list> is the list of dopants. For example:
pdbSet Oxide Hydrogen Dopants "Boron Arsenic"
The diffusion of hydrogen itself is modeled using the constant diffusion model (see Constant
Diffusion Model on page 220).
Dopant Activation and Clustering
It is possible to select a different clustering or activation model for each dopant in different
materials with the command:
pdbSet <material> <dopant> ActiveModel <model>
where <dopant> is a valid dopant name and <model> is one of the valid active models (None,
Solid, Transient, Cluster, ChargedCluster, BIC, FVCluster, or Equilibrium).
NOTE BIC is valid only for boron and is not recommended because the
ChargedCluster model is better suited for modeling boron–
interstitial clusters. FVCluster is valid only for fluorine.
Dopant Active Model: None
If ActiveModel is set to None, all dopants are assumed to be active. No solid solubility or
dopant clustering effects will be taken into account for dopant activation.

4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 293
I-2013.12
Dopant Active Model: Solid
If ActiveModel is set to Solid, a simple solid solubility for the dopant activation is
considered:
(383)
is calculated by:
(384)
where is the multiplication factor that is defined by:
pdbSet <material> <dopant> SS.Factor <expression>
and the solid solubility of the dopant that can be set with:
pdbSet <material> <dopant> Solubility {<n>}
when SS.Model is set to Analytic. However, when SS.Model is set to Table, the solid
solubility is taken from the temperature-versus-solid solubility table.
The SS.Model is defined by:
pdbSet <material> <dopant> SS.Model <Analytic or Table>
The temperature-versus-solid solubility table is defined by:
pdbSet <material> <dopant> SS.Table <temp1 ss1 temp2 ss2 ... temp# ss#>
With the table, the solid solubility for the given temperature is logarithmically interpolated or
extrapolated when the given temperature is out of range.
Dopant Active Model: Precipitation
Setting ActiveModel to Precipitation or setting ActiveModel to the other activation
model the list of More.Active.Model.List including Precipitation solves the
transient equation:
(385)
CA
+CA
ssCA
CA
ss CA
+()
--------------------------=
CA
SS
CA
SS fC
A
ss0
⋅=
f
CA
SS0
A
CAppt
∂
t∂
-----------------CAppt CAppt
*
–
τAppt
----------------------------------–=
4: Diffusion
Dopant Activation and Clustering
294 Sentaurus™ Process User Guide
I-2013.12
with the constraint:
(386)
is the precipitates concentration of the dopant and is the equilibrium precipitates
concentration given by:
(387)
where:
■ is the total chemical concentration.
■ represents the total dopant concentration in other clusters than precipitates.
■ is the equilibrium active concentration calculated by Dopant Active Model:
Equilibrium on page 306.
in Eq. 386 and Eq. 387 are included only when the other clustering model is invoked
with the list of More.Active.Model.List, for example:
pdbSet Si Boron More.Active.Model.List { Precipitation }
pdbSet Si Boron ActiveModel Transient
The time constant is given by:
for (388)
for (389)
The solution for is named with <species name>Ppts; for example, BPpts for boron
precipitates. The parameters ClusteringTime and DeclusteringTime are defined for the
precipitates in the material, for example:
pdbSet Silicon BPpts ClusteringTime {[Arr 8e-16 -4.2]}
pdbSet Silicon BPpts DeclusteringTime {[Arr 8e-16 -4.2]}
is the minimum active concentration defined by the parameter MinimumActive, for
example:
pdbSetString Silicon Boron MinimumActive "0.0"
Initializing Precipitation Model
The initialization of the precipitation concentration depends on the value of the parameters
AmInit and AcInit. The initial level of active concentration in amorphized and crystalline
0.0 CAppt max CAnCACl CAmin
+
–
–0.0,()≤≤
CAppt
CAppt
*
CAppt
*CAnCACl CAEq
–
–=
CA
nCACl
CAEq
nCACl
τAppt
τAppt ClusteringTime=
CAppt CAppt
*
≤
τAppt DeclusteringTime=
CAppt CAppt
*
>
CAppt
CAmin
+
4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 295
I-2013.12
regions can be specified per dopant as AmInit and AcInit, respectively. You can specify the
AmInit and AcInit parameters using:
pdbSet <material> <dopant> AcInit {<n>}
pdbSet <material> <dopant> AmInit {<n>}
If the AcInit parameter is not defined, the solid solubility of the dopant is used to calculate
the AcInit parameter. If you want AcInit and AmInit to be a function of other fields for a
specific dopant, define the terms <dopant>AcInit and <dopant>AmInit in your input files.
The precipitation model can be used with other activation models, for example:
pdbSet Si B ActiveModel BIC
pdbSet Si B More.Active.Model.List { Precipitation }
NOTE The items allowed in More.Active.Model.List are
Precipitation or ComplexCluster. The equilibrium activation
model cannot be used with the precipitation model.
When the precipitation model is invoked with other activation models, the initial
concentrations of the clusters and the precipitates are set as follows.
In the amorphous region:
(390)
(391)
where the fraction ratio for precipitation and for the cluster of other activation
model are written as:
(392)
(393)
In the crystalline region:
(394)
(395)
CAppt fAppt max CAAmInit–0.0,()⋅=
CAclust i,fAclust i,max CAAmInit–0.0,()⋅=
fAppt
fAclust i,
i
fAppt
FractionAmorppt
FractionAmorppt FractionAmordopant FractionAmori
i
++
-------------------------------------------------------------------------------------------------------------------------------------------------=
fAclust i,
FractionAmori
FractionAmorppt FractionAmordopant FractionAmori
i
++
-------------------------------------------------------------------------------------------------------------------------------------------------=
∆CAppt fAppt max CAimplant AcInit–0.0,()⋅=
∆CAclust i,fAclust i,max CAimplant AcInit–0.0,()⋅=

4: Diffusion
Dopant Activation and Clustering
296 Sentaurus™ Process User Guide
I-2013.12
where:
(396)
(397)
The parameters FractionAmor and FractionCryst for the precipitation are written as:
pdbSet <material> <precipitates> FractionAmor <number>
pdbSet <material> <precipitates> FractionCryst <number>
Dopant Active Model: Transient
If ActiveModel is set to Transient, a transient dopant-cluster model is used. The transient
dopant cluster is more complicated than the simple, solid solubility model. The following set
of equations is solved along with the appropriate diffusion model equations:
(398)
where:
■ is the forward-clustering reaction rate.
■ is the de-clustering rate.
■ is the active dopant concentration.
■ is the concentration of clusters.
■ is the number of substitutional dopants.
■ is the number of electrons participating in the reaction.
■ is the electron concentration.
■ is the intrinsic electron concentration.
■ is the electron concentration assuming that the dopant reached the limits of solid
solubility.
fAppt
FractionCrystppt
FractionCrystppt FractionCrystdopant FractionCrysti
i
++
----------------------------------------------------------------------------------------------------------------------------------------------=
fAclust i,
FractionCrysti
FractionCrystppt FractionCrystdopant FractionCrysti
i
++
----------------------------------------------------------------------------------------------------------------------------------------------=
t∂
∂CAC kf
n
ni
----
kc1
118
×10
-----------------
kc1–lc
+
ni
kcKFfwd CA
+
()
lcKFbwdkbnss
()
zlcni
zlc
–CAC
n
ni
----
zlc
–
–
=
kf
kb
CA
+
CAC
lc
kc
n
ni
nss
A

4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 297
I-2013.12
These quantities can be set using the commands:
pdbSet <material> <dopant> Kc {<n>}
pdbSet <material> <dopant> Lc {<n>}
pdbSet <material> <dopant> KcEqu {<n>}
pdbSet <material> <dopant> CluRate {<n>}
where KcEqu and CluRate correspond to and . Initialization of transient dopant clusters
is explained in Ion Implantation to Diffusion on page 353. The default value of KcEqu is
calculated by using:
(399)
where is the solid solubility of the unpaired dopant and is the solid solubility of total
concentration of the dopant. can be set directly using the command:
pdbSet Silicon Arsenic KcEqu 1e66
or can be set indirectly using the commands:
pdbSet Silicon Arsenic Solubility 1e20
pdbSet Silicon Arsenic TotSolubility 1e21
and its modifying factors:
pdbSet Silicon Arsenic SS.Factor 1
pdbSet Silicon Arsenic Total.SS.Factor 1
In addition, and are forward and backward reaction factors, respectively. They
can be defined as:
term name = <dopant>TClusterForwardFac <mater>
eqn = { User defined equation }
term name = <dopant>TClusterBackwardFac <mayer>
eqn = { User defined equation }
If they are not defined, their default value is 1.
Ta bl e 2 4 S o l ut io n na m es fo r t ra n s ie n t mo d el
Symbol Boron Arsenic Phosphorus Antimony Indium
B4 As3 P3 Sb3 In3
kb
kf
KcEqu lc
Css
lc
Csstot Css
–
----------------------------
=
Css
Csstot
KcEqu
KFfwd
KFbwd
CAC
4: Diffusion
Dopant Activation and Clustering
298 Sentaurus™ Process User Guide
I-2013.12
Initializing Transient Model
If you switch on the transient dopant cluster model or cluster model, initialization of the dopant
clusters is performed in the diffPreProcess procedure (see Ion Implantation to Diffusion
on page 353). The initialization of the dopant-cluster concentration depends on the value of the
parameters AmInit and AcInit. The initial level of active concentration in amorphized and
crystalline regions can be specified per dopant as AmInit and AcInit, respectively.
You ca n specif y the AmInit and AcInit parameters using:
pdbSet <material> <dopant> AcInit {<n>}
pdbSet <material> <dopant> AmInit {<n>}
If the AcInit parameter is not defined, the solid solubility of the dopant is used to calculate
the AcInit parameter. If you want AcInit and AmInit to be a function of other fields for a
specific dopant, define the terms <dopant>AcInit and <dopant>AmInit in your input files.
For example:
term name=ArsenicAcInit silicon add eqn = "Germanium/5e22 * [pdbDelayDouble
Silicon Arsenic Solubility]"
term name=ArsenicAmInit silicon add eqn = "Germanium/5e22*1e19"
In this case AcInit and AmInit for arsenic are replaced with ArsenicAcInit and
ArsenicAmInit.
If the dopant concentration is lower than AcInit in crystalline regions, dopants are considered
to be active. If the dopant concentration is higher than AcInit, the number of active dopants
is initially AcInit, and the concentration of clustered dopants is given by Dopant – AcInit.
The following outlines the initilization of dopant clusters.
In crystalline regions:
(400)
In amorphous regions:
(401)
where Dopant is the dopant name (for example, Boron, B4, and Boron_Implant).
Smoothing also can be applied to dopant profiles using the parameter AmorpGamma (see Ion
Implantation to Diffusion on page 353).
Dopant = Dopant + min(AcInit, Dopant_Implant)
DopantCluster = DopantCluster + Dopant_Implant - min(AcInit, Dopant_Implant)
Dopant = min(AmInit, Dopant+DopantCluster+Dopant_Implant)
DopantCluster = Dopant + DopantCluster + Dopant_Implant
min(AmInit, Dopant+DopantCluster+Dopant_Implant)–

4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 299
I-2013.12
Dopant Active Model: Cluster
If ActiveModel is set to Cluster, a dopant–defect cluster model is used. The model is
primarily implemented to simulate arsenic–vacancy clusters, but if the model parameters are
provided, it is possible to simulate other dopant–defect clusters.
The model assumes that arsenic–vacancy clusters are formed in silicon during arsenic
deactivation [28][29]. It is also assumed that neutral clusters ( ) are formed. Different
charge states are taken into account by:
(402)
(403)
(404)
where denotes interstitial charge, and are the electron and hole densities, and and
are the dopant and defect sizes, respectively.
The following commands can be used to set the reacting defect species, and , respectively:
pdbSet <material> <dopant> ClusterDefects <c>
pdbSet <material> <dopant> <defect> ClusterSizes {{<n> <n>}}
for example:
pdbSet Silicon Arsenic ClusterDefects Vac
pdbSet Silicon Arsenic Vacancy ClusterSizes {{4 1}}
where 4 is the number of arsenic atoms in the cluster and 1 is the number of vacancies in the
cluster. They form the As4Vacancy clusters.
NOTE Cluster sizes are defined as {i j} where i and j are integers and are
separated by a space.
The reaction rate for the cluster formation can be written as:
(405)
where is the active arsenic concentration, is the arsenic–vacancy cluster
concentration, is the neutral interstitial concentration, is the electron concentration,
AsmVk
mAs f AsmVkkI j[]
+↔+
fmkj–()e if kj m–()0<,=
fmkj–()h if kj m–()0≥,=
j
e
h
m
k
m
k
RjKfjCAs
mn
ni
----
mkj–KrjCAsmVkCI0
n
ni
----
kj
–≡
CAs
CAsmVk
CI0
n
ni
4: Diffusion
Dopant Activation and Clustering
300 Sentaurus™ Process User Guide
I-2013.12
is the intrinsic electron concentration, is the forward reaction rate, and is the backward
reaction rate. An additional assumption was made for all :
(406)
Then, the total rate is the sum of over all :
(407)
The forward reaction rate can be set using the command:
pdbSet <material> <dopant> <defect> CluRateChargeStates {<n>}
for example:
pdbSet Silicon Arsenic Vacancy CluRateChargeStates
{-2 {[Arrhenius 5.0e-42 7.8]}
-1 {[Arrhenius 5.0e-42 7.8]}
0 {[Arrhenius 5.0e-42 7.8]}
1 {[Arrhenius 5.0e-42 7.8]}
2 {[Arrhenius 5.0e-42 7.8]}}
The equilibrium reaction rate is calculated by:
(408)
where is the electron concentration assuming that arsenic reached the limits of solid
solubility and is the equilibrium concentration of interstitials. is given as:
(409)
where is the solid solubility of the unpaired arsenic dopants and is the solid
solubility of total arsenic concentration.
You c an either s et the so lid solu bi lity values or using the commands:
pdbSet Silicon Arsenic Solubility 1e20
pdbSet Silicon Arsenic TotSolubility 1e21
pdbSet Silicon Arsenic Vac KcEqu 1e66
Kfj
Krj
j
Krj
Kfj
------- Kequ
=
R
Rj
j
RK
fj
n
ni
----
kj
CAs
mn
ni
----
KequCAsmVkCI0
–
j
=
Kfj
Kequ
KcEqu
nss
ni
-------
m
CI
*
()
k
-------------------------------=
nss
CI
*
KcEqu
KcEqu mCss
m
Csstot Css
–
----------------------------
=
Css
Csstot
KcEqu

4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 301
I-2013.12
In addition, Solubility and TotSolubility can be changed through their modifying
factors:
pdbSet Silicon Arsenic SS.Factor 1
pdbSet Silicon Arsenic Total.SS.Factor 1
NOTE For high-concentration effects, see High-Concentration Effects on
Dopant Diffusion on page 291. For smoothing dopant profiles around
amorphous to crystalline regions, see Ion Implantation to Diffusion on
page 353.
Initializing Cluster Model
Initially, dopant–interstitial cluster concentrations (for example, As4Vac) are set to zero. If
there is an existing cluster concentration field, the field is used. If there are amorphized regions,
dopant–defect pairs and clusters are redistributed in these regions (see Dopant and Dopant-
Defect Cluster Initialization on page 315). In addition, you can initialize any of the cluster
concentration fields using the select command in the command file.
Dopant Active Model: NeutralCluster
The NeutralCluster model assumes that all reactants as well as clusters are neutral. If the
NeutralCluster model is selected, the following reactions are taken into account:
(410)
(411)
(412)
(413)
(414)
(415)
Ta bl e 2 5 S o l ut io n fo r cl us te r mo d e l
Symbol Arsenic
As4Vac
CAsmVk
AiIjI+AiIj1+
↔
AiIjV+AiIj1–
↔
AiIjAI+Ai1+Ij1+
↔
AiVjV+AiVj1+
↔
AiVjI+AiVj1–
↔
AiVjAV+Ai1+Vj1+
↔
4: Diffusion
Dopant Activation and Clustering
302 Sentaurus™ Process User Guide
I-2013.12
where are the number of dopant and defect atoms in the cluster, respectively. The clusters
can be specified using the command:
pdbSet <material> <dopant> <defect> ClusterSizes <list>
For example, the carbon clusters are defined by:
pdbSet Silicon Carbon Interstitial ClusterSizes {{1 0} {1 1} {2 0} {2 1} {3 1}}
NOTE This sets up the diffusion equations for , , , , and .
Cluster sizes given in the ClusterSizes list increase in size. For
example, the following is incorrect:
pdbSet Si Boron Int ClusterSizes { {1 0} {3 1} {1 1} }
NOTE If you add new cluster sizes, you must define the aliases for the new
clusters, for example:
alias C2I2 Carbon2Int2
The reactions are written as:
(416)
(417)
(418)
(419)
(420)
(421)
where:
■, , , , , and are the forward reaction rates.
■, , , , , and are the binding coefficients.
To set these parameters, use the following commands:
pdbSet <material> <dopant> Interstitial KfCluster <cluster> {<n>}
pdbSet <material> <dopant> Vacancy KfCluster <cluster> {<n>}
pdbSet <material> <dopant> <pair> KfCluster <cluster> {<n>}
pdbSet <material> <dopant> Interstitial BindCluster <cluster> {<n>}
ij,
C
CI
C2
C2I
C3I
RIAI KfIAiIjCAiIjCIBIAiIj
CAiIj1+
–()≡
RVAI KfVAiIjCAiIjCVBVAiIj
CAiIj1–
–()≡
RAI KfAIAiIjCAiIjCAI BAIAiIjCAi1+Ij1+
–()≡
RVAV KfVAiVjCAiVjCVBVAiVj
CAiVj1+
–()≡
RIAV KfIAiVjCAiVjCIBIAiVj
CAiVj1–
–()≡
RAV KfAVAiVjCAiVjCAV BAVAiVjCAi1+Vj1+
–()≡
KfIAiIj
KfVAiIj
KfAIAiIj
KfIVAiVj
KfIAiVj
KfAVAiVj
BIAiIj
BVAiIj
BAIAiIj
BVAiVj
BIAiVj
BAVAiVj

4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 303
I-2013.12
pdbSet <material> <dopant> Vacancy BindCluster <cluster> {<n>}
pdbSet <material> <dopant> <pair> BindCluster <cluster> {<n>}
where <cluster> is a valid cluster name (for example, C2, C2I, C3I, C3I2), and <pair> is
a dopant–interstitial pair (for example, CarbonInt) or a dopant–vacancy pair.
For example:
pdbSet Silicon Carbon CarbonInt KfCluster C3I2 {[Arrhenius 1e-10 0.3]}
sets the forward reaction rate of the CarbonInt and C3I2 reaction to {[Arrhenius 1e-10
0.3]}.
Initializing NeutralCluster Model
Initially, cluster concentrations are set to zero. If there is an existing cluster concentration field,
the field is used. If there are amorphized regions, dopant–defect pairs and clusters are
redistributed in these regions (see Dopant and Dopant-Defect Cluster Initialization on
page 315).
In addition, you can initialize any of the cluster concentration fields using the select
command in the command file.
Carbon Cluster
The carbon-clustering model uses the NeutralCluster model. The following solutions are
solved for the carbon model.
Ta bl e 2 6 S o l ut io n na m es fo r c ar bo n mo de l
Symbol Solution name
Carbon
CarbonInt
C2
C2I
C3I
C3I2
C4I2
C4I3
C
CI
C2
C2I
C3I
C3I2
C4I2
C4I3

4: Diffusion
Dopant Activation and Clustering
304 Sentaurus™ Process User Guide
I-2013.12
Nitrogen Cluster
The nitrogen-clustering model uses the NeutralCluster model. The following solutions are
solved for the nitrogen model.
NDimer is a cluster; therefore, it is initialized by FractionAmor and/or FractionCryst in
the same way as other cluster initializations.
NOTE If an activation model other than NeutralCluster is used for
nitrogen, the reactions between Nitrogen and NDimer are turned off.
NV is not a mobile pair but an immobile cluster. For example, the reaction is defined by:
pdbSet Si N Vac ClusterSizes { {1 0} {1 1} }
pdbSet Si N Vac KfCluster N {[expr [DiffLimit Silicon Vac 0.0]]}
pdbSet Si N Vac BindCluster N {[Arr 5e22 1.58]}
Dopant Active Model: FVCluster
If ActiveModel is set to FVCluster, the fluorine–vacancy cluster model is used. The model
based on fluorine–point defect interaction is implemented [30]. The primary reactions used in
the model are:
(422)
C5I3
C5I4
Ta bl e 2 7 S o l ut io n na m es fo r n it ro ge n m o de l
Symbol Solution name
Nitrogen
NitrogenInt
NV
NDimer
N2V
N2V2
Ta bl e 2 6 S o l ut io n na m es fo r c ar bo n mo de l
Symbol Solution name
C5I3
C5I4
N
NI
NV
N2
N2V
N2V2
F3VI+3F↔

4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 305
I-2013.12
(423)
where is mobile fluorine, is interstitial, is vacancy, and is an immobile cluster
(three fluorine atoms bound to a vacancy). The model assumes that is the dominant
cluster.
These reactions can be written as:
(424)
(425)
where and are the forward reaction rates. They can be defined using the commands:
pdbSet <material> Fluorine F3V KfI {<n>}
pdbSet <material> Fluorine F3V KfV {<n>}
is the diffusivity of neutral interstitials, is the concentration of fluorine–vacancy
clusters, is the concentration of fluorine, and and are the concentration of
interstitials and vacancies, respectively.
Quantities with a superscript (*) correspond to their equilibrium values. The equilibrium value
of can be set by:
pdbSet <material> Fluorine F3V Cstar {<n>}
The differential equations that describe the model are:
(426)
(427)
The fluorine diffusion coefficient, , can be set using the command:
pdbSet <material> Fluorine Dstar {<n>}
The quantity is subtracted from and the quantity is added to .
F3V3FV+↔
F
I
V
F3V
F3V
R1KI
≡DI0CF3VCI
CF
3
CF3V
*
------------–
R2KV
≡DI0CF3V
CF
3CV
CF3V
*CI
*CV
*
--------------------------–
KI
KV
DI0
CF3V
CF
CI
CV
CF3V
t∂
∂CFD0CF
∇()∇•3R13R2
++=
t∂
∂CF3VR1
–R2
–=
D0
R1
RI
clus
R2
RV
clus
4: Diffusion
Dopant Activation and Clustering
306 Sentaurus™ Process User Guide
I-2013.12
NOTE The fluorine model is switched off by default. To switch it on, use the
following commands:
solution add name = Fluorine ifpresent = "Fluorine" !negative
solution add name = F3V ifpresent = "F3V Fluorine" !negative
Initializing the FVCluster Model
You c an select a diff er ent ini ti alizat io n model for fluorine with the command:
pdbSet <material> Fluorine FVCluster.Init <model>
where <model> is either FV.Full or DAC.
If the FV.Full model is selected, it is assumed that the complete fluorine dose is implanted as
fluorine–vacancy clusters ( ). Since the ‘+1’ model is used to generate the excess
interstitials, this effectively introduces interstitials of a concentration equal to , except in
amorphous regions.
If the DAC model is selected, initially, the fluorine–vacancy cluster concentration is set to zero.
If there is an existing cluster concentration field, this field is used. If there are amorphized
regions, clusters are redistributed in these regions (see Dopant and Dopant-Defect Cluster
Initialization on page 315). In addition, you can initialize any of the cluster concentration fields
using the select command in the input command file.
NOTE The DAC model does not modify the vacancy field during initialization
of F3V.
Dopant Active Model: Equilibrium
If ActiveModel is set to Equilibrium, solid solubility or dopant clustering is considered.
In the clustering model, the active concentration of the dopant, , is obtained by solving:
(428)
where and are clustering parameters and can be set by:
pdbSet <material> <dopant> Kctn {<n>}
Ta bl e 2 8 S o l ut io n na m es fo r f lu or i n e mo de l
Symbol Solution name
Fluorine
F3V
CF
CF3V
CF3V
CF3V
CA
+
CAS CA
+KCTNCA
+
()+KCTN.F
=
KCTN
KCTN.F

4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 307
I-2013.12
pdbSet <material> <dopant> Kctn.F {<n>}
is also given by:
(429)
where is the total unpaired dopant concentration, and is calculated as described in
Dopant Active Model: Solid on page 293.
If the clustering parameters and are zero, the active concentration is determined
by Eq. 429; otherwise, Eq. 428 is solved numerically.
When the parameter Equil.Active.Conc is specified, the expression of
Equil.Active.Conc is evaluated for the active concentration in equilibrium, instead of
calculating Eq. 428 and Eq. 429. For example:
pdbSet Si B Equil.Active.Conc "(Boron>1E19)?(1E19):(Boron)"
Dopant Active Model: BIC
ActiveModel can be set to BIC for boron. The model is implemented to simulate boron–
interstitial clusters (BICs).
The model makes no assumptions regarding the diffusion model to generate the diffusion
equation for the substitutional and the mobile species. However, it should be used with either
the Pair or React model. The model does not take charge-state–dependent reaction rates into
account.
The following types of clustering reaction are taken into account:
(430)
(431)
Table 29 Solution names for equilibrium model
Symbol Boron Arsenic Phosphorus Antimony Indium
BoronEqu ArsenicEqu PhosphorusEqu AntimonyEqu IndiumEqu
CAS
CAS
CA
CA
SS CA1.1CA
SS
–()
2
0.4CA
SS
-------------------------------------–
CA
SS
=
CA0.9CA
SS
<
0.9CA
SS CACA
SS
≤≤
CA1.1CA
SS
>
CA
CA
SS
KCTN.F
KCTN
CAS
BiIjI+BiIj1+
↔
BiIjV+BiIj1–
↔
4: Diffusion
Dopant Activation and Clustering
308 Sentaurus™ Process User Guide
I-2013.12
(432)
where are the number of boron and interstitial atoms in the cluster , respectively.
The clusters can be specified using the command:
pdbSet Silicon Boron Interstitial ClusterSizes <list>
For example:
pdbSet Silicon Boron Interstitial ClusterSizes {{1 0} {1 1} {1 2} {2 1} \
{3 1} {3 2}}
will set up the diffusion equations for , , , and .
NOTE Cluster sizes given in the ClusterSizes list are increasing in size. For
example, the following command is incorrect:
pdbSet Si B Int ClusterSizes { {1 0} {3 1} {1 1} }
The reactions are written as:
(433)
(434)
(435)
where , , and are the forward reaction rates, and , , and are
the binding coefficients.
To set these parameters, the following commands can be used:
pdbSet <material> Boron Interstitial KfCluster <cluster> {<n>}
pdbSet <material> Boron Vacancy KfCluster <cluster> {<n>}
pdbSet <material> Boron BoronInt KfCluster <cluster> {<n>}
pdbSet <material> Boron Interstitial BindCluster <cluster> {<n>}
pdbSet <material> Boron Vacancy BindCluster <cluster> {<n>}
pdbSet <material> Boron BoronInt BindCluster <cluster> {<n>}
where <cluster> is a valid cluster name, for example, BI2, B2I, B3I, B3I2.
For example:
pdbSet Silicon Boron BoronInt KfCluster B3I2 {[Arrhenius 1e-10 0.3]}
BiIjBI+Bi1+Ij1+
↔
ij,
BiIj
BI2
B2I
B3I
B3I2
RIKfIBiIjCBiIjCIBIBiIj
CBiIj1+
–()≡
RVKfVBiIjCBiIjCVBVBiIj
CBiIj1–
–()≡
RCI KfBIBiIj
CBiIjCBI BBIBiIj
CBi1+Ij1+
–()≡
KfIBiIj
KfVBiIj
KfBIBiIj
BIBiIj
BVBiIj
BBIBiIj
4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 309
I-2013.12
sets the forward reaction rate of the BoronInt and B3I2 reaction to {[Arrhenius 1e-10
0.3]}.
The differential equations for the clusters are:
(436)
The reactions , , and are added to the appropriate point-defect equations, and
substitutional and mobile boron diffusion equations.
Initializing BIC Model
Initially, boron–interstitial cluster concentrations (for example, B3I) are set to zero. If there is
an existing cluster concentration field, the field is used. If there are amorphized regions, boron
defect pairs, and clusters are redistributed in these regions. For details, see Dopant and Dopant-
Defect Cluster Initialization on page 315. In addition, you can initialize any of the cluster
concentrations field using the select command in the input command file.
Dopant Active Model: ChargedCluster
ActiveModel can be set to ChargedCluster for dopant. The model is implemented to
simulate dopant-defect clusters including different charge states.
The model makes no assumptions regarding the diffusion model to generate the diffusion
equation for the substitutional and the mobile species. However, it should be used with either
the Pair or ChargedPair or React or ChargedReact model.
The following types of clustering reaction are taken into account:
(437)
Table 30 Solution names for BIC model
Symbol Solution name
Boron
BoronInt
BI2
B2I
B3I
B3I2
t∂
∂BiIjR–IR–VRBI
–=
RI
RV
RBI
CB
CBI
CBI2
CB2I
CB3I
CB3I2
CAmIn
cCI
z
+CAmIn1+
yyc–z–()e+↔
4: Diffusion
Dopant Activation and Clustering
310 Sentaurus™ Process User Guide
I-2013.12
(438)
(439)
(440)
where:
■ are the number of dopant and interstitial atoms in the cluster , respectively, and
they are the number of dopant and vacancy atoms in the cluster , respectively.
■ are the concentration of interstitials and vacancies.
■ are different charge states of clusters and point defects.
The clusters can be specified using the command:
pdbSet Silicon <dopant> <defect> ClusterSizes <list>
For example:
pdbSet Silicon Boron Interstitial ClusterSizes {{1 0} {1 1} {1 2} {2 1} \
{3 1} {3 2}}
sets up the diffusion equations for , , , and .
NOTE Cluster sizes given in the ClusterSizes list are increasing in size. For
example, the following command is incorrect:
pdbSet Si Boron Int ClusterSizes { {1 0} {3 1} {1 1} }
The reactions are written as:
(441)
(442)
(443)
(444)
CAmIn
cCAI
z
+CAm1+In1+
yyc–z–()e+↔
CAmIn
cCV
z
+CAmIn1–
yyc–z–()e+↔
CAmIn
cCAV
z
+CAm1+In1–
yyc–z–()e+↔
mn,
CAmIn
CAmVn
CICV
,
cyz,,
BI2
B2I
B3I
B3I2
RAmIn1+KfAmIn1+CAmIn
cCI
zKrAmIn1+CAmIn1+
yn
ni
----
yc–z–()
–
≡
RAm1+In1+KfAm1+In1+CAmIn
cCAI
zKrAm1+In1+CAm1+In1+
yn
ni
----
yc–z–()
–
≡
RAmIn1–KfAmIn1–CAmIn
cCV
zKrAmIn1–CAmIn1–
yn
ni
----
yc–z–()
–
≡
RAm1+In1–KfAm1+In1–CAmIn
cCAV
zKrAm1+In1–CAm1+In1–
yn
ni
----
yc–z–()
–
≡
4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 311
I-2013.12
where:
■, , , and are the forward reaction rates.
■, , , and are the equilibrium constants.
The forward reaction rates are a function of the lattice spacing ( ), the capture radius factor
( ), and the diffusion of point defect ( ) or dopant-defect pair ( ):
(445)
(446)
(447)
(448)
To set the capture radius factor parameter, use the following command:
pdbSet <material> <dopant> <defect|mobile> CaptureRadiusFactor <cluster> \
{<expression>}
where mobile is the mobile species (for example, BoronInt, BoronVac). For example:
pdbSet Silicon Boron BoronInt CaptureRadiusFactor B2 1.3
means that the capture radius for is 1.3 times the lattice spacing.
The equilibrium conditions are calculated internally using:
(449)
where is the cluster degeneracy, is the cluster charge, and is the formation energy
of the cluster.
To set these parameters, use the following commands:
pdbSet <material> <dopant> <defect> ClusterDegeneracy <cluster> {<n>}
pdbSet <material> <dopant> <defect> ClusterCharge <cluster> {<n>}
pdbSet <material> <dopant> <defect> ClusterFormE <cluster> {<expression>}
where <cluster> is a valid cluster name, for example, BI2, B2I, B3I, B3I2.
KfAmIn1+
KfAm1+In1+
KfAmIn1–
KfAm1+In1–
KrAmIn1+
KrAm1+In1+
KrAmIn1–
KrAm1+In1–
L
r
DI
zDV
z
,
DAI
zDAV
z
,
KfAmIn1+ α 4πLrAmIn1+DI
z
KfAm1+In1+ α 4πLrAm1+In1+DAI
z
KfAmIn1– α 4πLrAmIn1–DV
z
KfAm1+In1– α 4πLrAm1+In1–DAV
z
B2BoronInt+B3I↔
CAmIn
cNCA
m
522
×10()
m1–
-------------------------------- CI
CI
0
------ n
ni
----
m–c–e
Eform
kT
------------
=
N
c
Eform
4: Diffusion
Dopant Activation and Clustering
312 Sentaurus™ Process User Guide
I-2013.12
For example:
pdbSet Silicon Boron Int ClusterFormE B3I2 {0.3}
sets the formation energy of cluster B3I2 to 0.3 eV.
The differential equations for the clusters are:
(450)
The reactions are added to the appropriate point-defect equations, and substitutional and
mobile boron diffusion equations.
Initializing ChargedCluster Model
Initially, dopant–interstitial cluster concentrations (for example, B3I) are set to zero. If there is
an existing cluster concentration field, the field is used. If there are amorphized regions,
dopant-defect pairs and clusters are redistributed in these regions (see Dopant and Dopant-
Defect Cluster Initialization on page 315).
In addition, you can initialize any of the cluster concentration fields using the select
command in the command file.
Dopant Active Model: ComplexCluster
The reaction of the ComplexCluster model is as follows:
(451)
Table 31 Solution names for ChargedCluster model
assuming the base defect is boron
Symbol Solution name
Boron
BoronInt
BI2
B2I
B3I
B3I2
t∂
∂CAmInRAmIn1+
–RAm1+In1+
–RAmIn1–
–RAm1+In1–
–=
CB
CBI
CBI2
CB2I
CB3I
CB3I2
n1Xn
2Yn
3P1n4e++ + Xn1Yn2P2m1,m2P3m3e++↔
4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 313
I-2013.12
where:
■ and denote two different dopant species.
■ is a complex cluster.
■, , and denote point defects, that is, either a Si self-interstitial or vacancy.
■ indicates an electron.
The reaction is formulated by:
(452)
is the normalization factor which is specified by:
term name=${X
n1
Y
n2
P
2,m1
}NormValue <material> add eqn=<equation>
By default, is set to the intrinsic carrier concentration, .
The chemical elements of the complex cluster are defined by:
pdbSetDoubleArray <material> <X
n1
Y
n2
P
2,m1
> Component.List \
{<X> <n
1
> <Y> <n
2
> <P
2
> <m
1
>}
For example:
pdbSetDoubleArray Silicon BCI Component.List { Boron 1 Carbon 1 Int 1 }
When the charge state of the complex cluster, , , and are given, the other unknowns
, , and are determined automatically.
, , and are specified by:
pdbSetDoubleArray <material> <X
n1
Y
n2
P
2,m1
> Product.List \
{<P
3
> <m
2
> <Electron or Hole> <|m
3
|>}
For example:
pdbSetDoubleArray Silicon BCI Product.List { Electron 1 }
pdbSetDoubleArray Silicon BCI ChargeState { 0 1.0 }
With the two examples above, the reaction is defined as follows:
(453)
X
Y
Xn1Yn2P2m1,
P1
P2
P3
e
RK
fni
CA1
+
Cnorm
---------------
n1CA2
+
Cnorm
---------------
n2P1
P1
*
------
n3n
ni
----
n4KrCXn1Yn2P2m1,
P3
P3
*
------
m2n
ni
----
m3
–=
Cnorm
Cnorm
ni
Xn1Yn2P2m1,
P3
m2
m3
P1
n3
n4
P3
m2
m3
RK
fni
CB
+
ni
-------
CC
+
ni
-------
I
I*
----
KrCBCI
n
ni
----
–=
4: Diffusion
Dopant Activation and Clustering
314 Sentaurus™ Process User Guide
I-2013.12
The forward and reverse reaction rates, and , are calculated by:
(454)
(455)
where , , and are specified by:
pdbSetDouble <material> <X
n1
Y
n2
P
2,m1
> KF <number>
pdbSetDouble <material> <X
n1
Y
n2
P
2,m1
> KR <number>
term name=${X
n1
Y
n2
P
2,m1
}KFFactor <material> add eqn=<equation>
term name=${X
n1
Y
n2
P
2,m1
}KRFactor <material> add eqn=<equation>
By default, and are set to 1.0.
Initializing ComplexCluster Model
The initialization of the clusters in the ComplexCluster model is performed in a similar way
to that of the precipitation model (see Initializing Precipitation Model on page 294).
The ComplexCluster model can be used with other activation models, for example:
pdbSet Si B ActiveModel BIC
pdbSet Si B More.Active.Model.List { ComplexCluster }
NOTE The items allowed in More.Active.Model.List are
Precipitation or ComplexCluster.
For the initialization of the clusters with the other activation models, see Initializing
Precipitation Model on page 294.
When the multiple dopant species are involved in the reaction of the ComplexCluster model,
the initialization is more complicated because the dose conservations of both dopant species
must be satisfied. For example, the silicon substrate is doped with boron atoms
and carbon atoms, and the following specifications for their activation models
and initializations are given by:
pdbSet Si B ActiveModel ComplexCluster
pdbSet Si C ActiveModel ComplexCluster
pdbSet Si B AmInit 4E19
pdbSetDouble Si C AmInit 1E19
pdbSet Si BCI FractionAmor 1.0
In the amorphous region, the initial boron active concentration needs to be set to
. This means that the BCI concentration must be , while the initial
active concentration of carbon is set to . Therefore, the BCI concentration is
Kf
Kr
KffactorfKf0
×=
KrfactorrKr0
×=
Kf
Kr
factorf
factorr
factorf
factorr
110
20
× cm 3–
110
20
× cm 3–
410
19
× cm 3–
610
19
× cm 3–
110
19
× cm 3–

4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 315
I-2013.12
supposed to be set to . In this case, the smaller BCI concentration
is set for BCI, so that the active concentration of carbon is adjusted to ; although,
AmInit for carbon is given as .
Dopant and Dopant-Defect Cluster Initialization
If you switch on the BIC, ChargedCluster, CarbonCluster, React, ChargedReact, or
NeutralReact model, initialization of the dopant and dopant-defect clusters are performed
in the diffPreProcess procedure (see Ion Implantation to Diffusion on page 353). It is
possible to redistribute dopants and dopant-defect clusters in the amorphous and crystalline
regions after implantation.
The initialization of the dopant and dopant-defect clusters depends on the value of the
parameters AmInit, AcInit, FractionCryst, and FractionAmor. The initial level of
active concentration of dopants in amorphized and crystalline regions can be specified per
dopant as AmInit and AcInit, respectively. You can specify the AmInit and AcInit
parameters by using:
pdbSet <material> <dopant> AcInit {<n>}
pdbSet <material> <dopant> AmInit {<n>}
If the AcInit or AmInit parameter is not defined, the default value of is used. If you
want AcInit and AmInit to be a function of other fields for a specific dopant, define the terms
<dopant>AcInit and <dopant>AmInit in your input files. For example:
term name=ArsenicAcInit silicon add eqn = "Germanium/5e22 * [pdbDelayDouble
Silicon Arsenic Solubility]"
term name=ArsenicAmInit silicon add eqn = "Germanium/5e22*1e19"
In this case, AcInit and AmInit for arsenic are replaced with ArsenicAcInit and
ArsenicAmInit.
The FractionCryst and FractionAmor parameters are used to calculate the fraction of
dopants and dopant-defect clusters in crystalline and amorphized regions. To specify the
FractionCryst and FractionAmor parameters, use:
pdbSetDouble <material> <dopant | cluster> FractionCryst {<n>}
pdbSetDouble <material> <dopant | cluster> FractionAmor {<n>}
The following outlines the initilization of dopant clusters.
910
19
× cm 3–
610
19
× cm 3–
410
19
× cm 3–
110
19
× cm 3–
510
22
×

4: Diffusion
Dopant Activation and Clustering
316 Sentaurus™ Process User Guide
I-2013.12
In crystalline regions:
(456)
(457)
In amorphous regions:
(458)
(459)
where:
■Dopant is the dopant name (for example, Boron).
■DopantCluster is the cluster name (for example, B4I2).
■DopantTotal is the total dopant (for example, Boron+4*B4I2).
■Dopant_Implant is the implanted dopant (for example, Boron_Implant).
■DopantSize is the size of the dopant (for example, 4 for B4I2).
Dopant Trapping at EOR Defects
To simulate dopant trapping at EOR defects [31], interstitial clusters must be switched on (311
or 2Moment), loops must be switched on, and one of the following dopant diffusion models
also must be used:
■React (see React Diffusion Model on page 212)
■ChargedReact (see ChargedReact Diffusion Model on page 206)
■NeutralReact (see NeutralReact Diffusion Model on page 220)
To switch on the model, use:
pdbSet <material> <dopant> EORTrap { 1 | 0 }
Crytalline% FractionCryst
DopantSize FractionCryst
Dopant|Cluster
------------------------------------------------------------------------------------------=
MaxDopant = max(Dopant_Implant – AcInit,0)
DopantCluster = DopantCluster + Crystalline% MaxDopant×
Dopant = Dopant + Dopant_Implant – MaxDopant + Crystalline% MaxDopant×
Amorphous% FractionAmor
DopantSize FractionAmor
Dopant|DopantCluster
--------------------------------------------------------------------------------------------------------=
MaxDopant = max(DopantTotal + Dopant_Implant – AmInit,0)
DopantCluster = Amorphous% MaxDopant×
Dopant = DopantTotal + Dopant_Implant – MaxDopant Amorphous% MaxDopant×+
4: Diffusion
Dopant Activation and Clustering
Sentaurus™ Process User Guide 317
I-2013.12
The following type of clustering reactions are taken into account:
(460)
(461)
(462)
where is the mobile dopant, is the concentration of mobile dopants trapped at {311}
defects, and is the concentration of mobile dopants trapped at dislocation loops.
For these reactions:
■The first reaction describes the capture and release of mobile dopants at {311} defects.
■The second one describes the capture and release of mobile dopants at dislocation loops.
■The last reaction explains the transformation of mobile dopants trapped at a {311} defect
to mobile dopants trapped at a dislocation loop.
The capture rate for the mobile dopant and {311} defect reaction is written as:
(463)
where is the forward reaction rate, is the concentration of interstitials in {311}
defects and is the density of traps along the {311} defects for the mobile dopants.
To change , use:
pdbSet <material> <dopant> EORForwardReaction <trapped dopant> <n>
where <trapped dopant> is (for example, B311). For boron, it is currently set to:
(464)
where is the capture radius and is the diffusivity of neutral dopant defect pair, .
To change the capture radius, use:
pdbSet <material> <dopant> EORCaptureRadius <trapped dopant> <n>
is proportional to the length of the {311} defect. It is defined as a term and is user
definable:
term name = EORDopant311_Max <material> add eqn = {c}
CAI CA311 CA311 1+↔+
CAI CADL CADL 1+↔+
CA311 CADL
→
CAI
CA311
CADL
RA311
capture kf311 C311Cmax
311 CA311
–()≡
kf311
C311
Cmax
311
kf311
CA311
kf311 4πrA311CAIDAI
=
rA311
DAI
CAI
Cmax
311
4: Diffusion
Dopant Activation and Clustering
318 Sentaurus™ Process User Guide
I-2013.12
The release rate for the mobile dopant and {311} defect reaction is written as:
(465)
where is the emission rate of trapped mobile dopant from {311} defects. To change it,
use:
pdbSet <material> <dopant> EOREmissionRate <trapped dopant> <n>
The capture rate for the mobile dopant and dislocation loop reaction is written as:
(466)
where is the forward reaction rate and is the density of traps along the edge of
dislocation loops for the mobile dopants. To change , use:
pdbSet <material> <dopant> EORForwardReaction <trapped dopant> <n>
where <trapped dopant> is (for example, BDL). It is set to:
(467)
where is the capture radius. To change the capture radius, use:
pdbSet <material> <dopant> EORCaptureRadius <trapped dopant> <n>
is proportional to the density of dislocation loops. It is defined as a term and is user
definable:
term name = EORDopantDL_Max <material> add eqn = {c}
The release rate for the mobile dopant and dislocation loop reaction is written as:
(468)
where is the emission rate of trapped mobile dopant from a dislocation loop. To change
it, use:
pdbSet <material> <dopant> EOREmissionRate <trapped dopant> <n>
The unfaulting of {311} defects in the presence of trapped mobile dopants is given as:
(469)
RA311
release CA311EA311
≡
EA311
RADL
capture kfDL CDLCmax
DL CADL
–()≡
kfDL
Cmax
DL
kfDL
CADL
kfDL πrADLCAIDAI
=
rADL
Cmax
DL
RADL
release CADLEADL
≡
EADL
RA311 ADL→
Unfold KD311 DLoop→s311
CA311
C311
--------------≡
4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 319
I-2013.12
where is the unfaulting rate of {311} defects to dislocation loops and comes from
the Loop model (see Defect Cluster Model: Loop on page 328). is the size of {311} defect.
Initializing Dopant Trapping in EOR Model
Initially, trapped dopants at EOR are set to zero. If there is an existing cluster concentration
field, the field is used. If there are amorphized regions, dopant–defect pairs and clusters are
redistributed in these regions. For details, see Dopant and Dopant-Defect Cluster Initialization
on page 315. In addition, you can initialize any of the cluster concentration fields using the
select command in the command file.
Defect Clusters
The available cluster models are None, Equilibrium, 311, Loop, LoopEvolution,
FRENDTECH, 1Moment, 2Moment, Full, and the model is selected with the command:
pdbSet <material> <defect> ClusterModel <model>
where <defect> is interstitial or vacancy, and <model> is one of the valid model names.
These clusters play an important part in transient-enhanced diffusion (TED) of impurities
following ion implantation. The main effect of the models is to delay the onset of TED at low
temperatures and to distribute the diffusion enhancement over a longer period of time. This
eliminates the excessive diffusion at low temperatures that is predicted without any clustering.
In some cases, multiple cluster equations must be switched on. The following command can be
used:
pdbSet <material> <defect> MultiClusterModel < < cluster model> <list> >
For example:
pdbSet Silicon Int MultiClusterModel { Full {1Moment} }
Loop {311} }
Table 32 Solution names for EOR trap model
Symbol Solution name
BoronInt
B311
BDL
KD311 DLoop→
s311
CAI
CA311
CADL

4: Diffusion
Defect Clusters
320 Sentaurus™ Process User Guide
I-2013.12
switches on the 1Moment model if the interstitial cluster model Full is selected. In the same
way, it will switch on the 311 model if the interstitial cluster model Loop is selected.
For example:
pdbSet Si Int ClusterModel Full
pdbSet Si Int MultiClusterModel Full {2Moment Loop}
switches on the model given in [32].
In this model, seven equations are solved to describe the kinetics of self-interstitial clusters:
■Three data fields (I2, I3, I4) describe small interstitial clusters (SMICs).
■Two data fields (D311, density of {311} defects, and C311, density of interstitials bound
in {311} defects) describe the presence of {311} defects.
■Two data fields (DLoop, density of dislocation loops, and CLoop, density of interstitials
bound in dislocation loops) describe dislocation loops.
Defect Cluster Model: None
If ClusterModel is set to None, no point-defect clustering effects will be taken into account.
Defect Cluster Model: Equilibrium
If you set the model to Equilibrium, the following nonlinear algebraic equation along with
the related diffusion equations are solved:
(470)
where is the equilibrium constant, is either interstitial or vacancy, 4 is the size of the
cluster, and is the concentration of point-defect cluster. can be specified with:
pdbSet <material> <defect> KCluster {<n>}
NOTE The equilibrium cluster model is defined only for interstitials in silicon.
Defect Cluster Model: 311
If you set the defect cluster model to 311, the {311} point-defect model developed by Law and
Jones [33] is activated. It solves for the concentration of interstitials in the defects and the
concentration of defects , as well as for the concentration of interstitials in small
CX0
()
4kbCXC
–0=
kb
X
CXC
kb
C311
D311
4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 321
I-2013.12
interstitial clusters (SMICs). SMICs come in two types. The small type SmicS is assumed to
have a cluster size of 2. The larger type Smic is assumed to have a cluster size of = 4, by
default. This value is, however, user accessible:
pdbSet <material> C311 NSize {<n>}
Nucleation of defects occurs during the implantation process. Initial distribution of defects
comes from the implant code (see Ion Implantation to Diffusion on page 353), in particular, all
interstitials created during the implantation process are assumed to be in immobile SmicS.
Vacancies and interstitials recombine or may form di-interstitials and di-vacancies. Some
interstitials will also form small interstitial clusters (SmicS, Smic) or {311} defects. The SMICs
dissolve to the surface through the release of interstitials. The capture and release of interstitials
on the {311} defects occur only at the end of the defects and, therefore, are proportional to the
number of defects .
The reactions involved in the {311} model are given by:
(471)
(472)
(473)
(474)
where and are di-interstitials and di-vacancies, and is the binding coefficient
between the di-defect, for example, , and the base defect, (where refers to either I or V),
and is the forward reaction rate for the recombination of di-defects.
To change these variables, use the commands:
pdbSet <material> <di-defect> Kforward {<n>}
pdbSet <material> <di-defect> Bind {<n>}
(475)
(476)
(477)
(478)
Nsize
D311
II I
2
↔+
VV+V2
↔
RI2form kfCI
2BI2CI
–()=
RV2form kfCV
2BV2CV
–()=
I2
V2
BX2
I2
I
X
kf
I2VI↔+
V2IV↔+
V2I20↔+
RI2rec kfCI2CVCI2
*CV
*CI
CI
*
------–
=
4: Diffusion
Defect Clusters
322 Sentaurus™ Process User Guide
I-2013.12
(479)
(480)
where is the equilibrium concentration of the respective defect or di-defect, and is the
forward reaction rate for the respective recombination process.
To change these variables, use the commands:
pdbSet <material> <di-defect> KRecomb {<n>}
pdbSet <material> <di-defect> KBiMole {<n>}
pdbSet <material> <di-defect> Cstar {<n>}
Aggregation or emission of interstitials from SmicS and Smic are given by:
(481)
(482)
This process increases the concentration of interstitials in SmicS by .
(483)
(484)
This process increases the concentration of interstitials in Smic by . is the forward
reaction rate for SMIC–interstitial reactions, and and are the binding
coefficients between interstitials and SMICs.
To change these variables, use the commands:
pdbSet <material> SmicS KfI {<n>}
pdbSet <material> SmicS Bind {<n>}
pdbSet <material> Smic KfI {<n>}
pdbSet <material> Smic Bind {<n>}
The recombination of SmicS or Smic with a vacancy or bi-vacancies is given by:
(485)
(486)
This process decreases the concentration of interstitials in SmicS by .
RV2rec kfCV2CICV2
*CI
*CV
CV
*
-------
–
=
RI2V2rec kfCI2CV2CI2
*CV2
*
–()=
CX
*
kf
SmicS I+SmicS 1+()↔
RISmicSagg kfICSmicS CIBISmicS
–()=
1/cm3
Smic I+Smic 1+()↔
RISmicagg kfICSmic CIBISmic
–()=
1/cm3
kfI
BISmicS
BISmic
SmicS V+SmicS 1–()↔
RVSmicSrec kfVCVCSmicS
=
1/cm3
4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 323
I-2013.12
(487)
(488)
This process decreases the concentration of interstitials in Smic by .
(489)
(490)
This process decreases the concentration of interstitials in SmicS by .
(491)
(492)
This process decreases the concentration of interstitials in Smic by . and are the
diffusion-limited SMIC–(di-)vacancy capture rates and are defined as:
(493)
(494)
where and are the diffusivities of neutral vacancies and di-vacancies, and is the
lattice spacing of silicon.
To change these variables, use the commands:
pdbSet <material> Vacancy D 0 {<n>}
pdbSet <material> V2 D0 {<n>}
pdbSet <material> LatticeSpacing {<n>}
A Smic is assumed to contain interstitials, with a default value of 4. A SmicS contain two
interstitials less than a Smic. A SmicS can be converted to a Smic by combining with a di-
interstitial. Formation of a Smic from a SmicS and bi-interstitials is given by:
(495)
(496)
This process increases the concentration of interstitials in Smic by and decreases the
concentration of interstitials in SmicS by ( – 2) . is the forward reaction rate for
SMIC–di-interstitial reactions.
Smic V+Smic 1–()↔
RVSmicrec kfVCVCSmic
=
1/cm3
SmicS V2
+SmicS 2–()↔
RV2SmicSrec kfV2CV2CSmicS
=
2/cm3
Smic V2
+Smic 2–()↔
RV2Smicrec kfV2CV2CSmic
=
2/cm3
kfV
kfV2
kfV 4πaDV0
=
kfV 4πaDV2
0
=
DV0
DV2
0
a
Nsize
SmicS I2
+Smic→
RSmicform kfI2CSmicSCI2
=
Nsize
/cm3
Nsize
/cm3
kfI2
4: Diffusion
Defect Clusters
324 Sentaurus™ Process User Guide
I-2013.12
To change these variables, use:
pdbSet <material> SmicS KfI2 {<n>}
pdbSet <material> C311 NSize {<n>}
When a Smic combines with an additional di-interstitial, they form a {311} defect. Formation
of a new {311} defect from a Smic and bi-interstitials is given by:
(497)
(498)
This process increases the concentration of {311} defects by and the concentration of
interstitials in {311} defects by ( + 2) , and deceases the number of interstitials in a
Smic by . is the reaction rate.
To change these variables, use:
pdbSet <material> C311 KnI2 {<n>}
The emission of interstitials from {311} defects is given by:
(499)
(500)
This process decreases the concentration of interstitials in {311} defects by , but does
not change the number of {311} defects. The {311} defect simply became shorter. is the
concentration of interstitials in the {311} defects and is the decay rate.
To change these variables, use the commands:
pdbSet <material> C311 Decay {<n>}
The dissolution of a defect is given by:
(501)
(502)
This process changes the number of {311} defects, but does not affect the number of
interstitials in the {311} defects. The interstitials released in this process immediately
aggregate on other {311} defects. is the concentration of {311} defects and is the
defect decay rate.
I2Smic 311→+
R311nIform knI2CSmicCI2
=
1/cm3
Nsize
/cm3
Nsize
/cm3
knI2
C311 C311 1–()I+→
RC311Iem C311DRate
=
1/cm3
C311
DRate
D311 D311 1–→
RD311decay D311DRate
=
D311
DRate
4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 325
I-2013.12
A set of 14 interstitials can nucleate at the end of a {311} defect. The formation or dissolution
of a {311} defect from interstitials is given by:
(503)
(504)
This process increases the concentration of interstitials in {311} defects by 14 ; the
number of defects remains unchanged. is the forward reaction rate, is the capture
(reaction) range, and is the binding coefficient between the {311} defect and the
interstitial.
To change these variables, use:
pdbSet <material> C311 KfI {<n>}
pdbSet <material> C311 BindI {<n>}
pdbSet <material> C311 RangeI {<n>}
A set of 14 di-interstitials can nucleate at the end of a {311} defect. The formation of two {311}
defects from seven bi-interstitials is given by:
(505)
(506)
This process increases the concentration of interstitials in {311} defects by 28 ; the
number of defects remains unchanged. is the forward reaction rate, is the capture
(reaction) range, and is the binding coefficient between the {311} defect and the
interstitial.
To change these variables, use:
pdbSet <material> C311 BindI2 {<n>}
pdbSet <material> C311 KfI2 {<n>}
pdbSet <material> C311 RangeI2 {<n>}
A {311} defect can dissolve into interstitials or di-interstitials. The probability of this process
is proportional to the inverse length of the defect, which can be expressed as the ratio of the
concentration of defects and the concentration of interstitials in defects ( ). Then, the
dissolution of {311} defects is given by:
(507)
(508)
14I311↔
RC311Iform kfID31114RICIBD311I
–()=
/cm3
kfI
RI
BD311I
14I2311↔
RC311I2form D311kfI214RI2CI2BD311I2
–()=
/cm3
kfI2
RI2
BD311I2
D311 C311
⁄
RD311Iem D31114CRate
D311
C311
----------- kfIRIBD311I
=
RD311I2em D31114CRate
D311
C311
----------- kfI2RI2BD311I2
=

4: Diffusion
Defect Clusters
326 Sentaurus™ Process User Guide
I-2013.12
This process does not change the number of free interstitials or the number of interstitials in
{311} defects. It is assumed that all interstitials were released from the defect aggregate in
other {311} defects. is the spontaneous combustion rate and gives the percentage of
interstitials dissolved from {311} defects by dissolution of all defects.
To change these variables, use:
pdbSet <material> C311 CombRate {<n>}
The following set of differential equations is solved with the {311} model. The point-defect
equations are:
(509)
(510)
The di-defect equations are:
(511)
(512)
where is the diffusivities of di-defects (where refers to either I or V).
To change these variables, use:
pdbSet <material> <di-defect> D0 {<n>}
The equation for is given by:
(513)
The equation for is given by:
(514)
CRate
t∂
∂CIJIRIV
–∇•–2RI2form
–RI2rec RV2rec
–RC311Iform
–+=
+RC311Iem RISmicSagg
–RISmicagg
–
t∂
∂CVJVRIV
–∇•–2RV2form
–RV2rec RI2rec
–RVSmicSrec
–RVSmicrec
–+=
t∂
∂CI2DI2CI2
∇()∇•RI2form RI2rec
–RI2V2rec
–RSmicform R311nIform RC311I2form
–––+=
t∂
∂CV2DV2CV2
∇()∇•RV2form RV2rec
–RI2V2rec
–RV2SmicSrec RV2Smicrec
––+=
DX2
X
SmicS
t∂
∂CSmicS RVSmicSrec
–2RV2SmicSrec
–RISmicSagg RSmicform
–+=
Smic
t∂
∂CSmic RVSmicrec
–2RV2Smicrec
–RISmicagg RSmicformNSize R311nIformNSize
++ +=

4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 327
I-2013.12
The equation for the density of {311} defects is given by:
(515)
The equation for the concentration of interstitials in {311} defects is given by:
(516)
The initialization of {311} defect fields is given in Ion Implantation to Diffusion on page 353.
NOTE Even though the {311} model and the model parameters are given in
general format, they are defined only for silicon. If these models need to
be used in other materials, their parameters must be copied.
Initializing 311 Model
The defect-cluster concentrations I2, V2, C311, SmicS, Smic, and D311 are initialized in the
diffPreProcess procedure. The model assumes that all the free implant interstitials
(Int_Implant) are transferred to SmicS. Initially, other transient defect-cluster
concentrations are set to zero. If there is an existing cluster concentration field, the field is used.
By default, clusters are assumed to break apart in the amorphous regions. You can specify the
percentage of clusters retained in the amorphous region per cluster solution variable using the
parameter AmPercent:
pdbSet <mater> <cluster> AmPercent {<n>}
Ta bl e 3 3 S o l ut io n na m es fo r 3 11 m od e l
Symbol Solution name
C311
D311
Smic
SmicS
I2
V2
t∂
∂CD311 RD311 decay
–RD311Iem
–2RD311I2em
–R311nIform
+=
t∂
∂CC311 RC311Iem
–RC311Iform 2RC311I2fom Nsize 2+()R311nIform
++ +=
C311
D311
CSmic
CSmicS
CI2
CV2

4: Diffusion
Defect Clusters
328 Sentaurus™ Process User Guide
I-2013.12
For example:
(517)
(518)
The value of the AmPercent parameter must be between 0 and 1.
Defect Cluster Model: Loop
If you set the defect cluster model to Loop, the modified version of the dislocation loop
nucleation model [34] is used to solve for the concentration of interstitials in the defects
and the concentration of defects . The model assumes that the dislocation loops come
from unfaulted {311} defects. The unfaulting rate can be defect size–dependent or not.
The following switch can be used to change the unfaulting rate:
pdbSet <material> CLoop Unfault.Model <model>
where <model> is either Direct or SizeDependent.
Direct Model
If the Unfault.Model is set to Direct, the following reaction equations as well as the {311}
defect equations (see Defect Cluster Model: 311 on page 320) are solved:
(519)
(520)
(521)
where:
■ is the unfaulting rate of {311} defects to dislocation loops.
■ is the average radius of loops.
SmicS SmicS*AmPercent
SmicS+Int_Implant
=Amorphous regions
Crystalline regions
C311
C311*AmPercent
C311
=Amorphous regions
Crystalline regions
CLoop
DLoop
t∂
∂CLoop K311C311 2π2RLoopDLoopDI0KCLoop CICILoop
*
–()+=
CILoop
*CI
*e
γΩ
bkT
---------
e
µbΩ
4πRLoopkT 1υ–()
--------------------------------------------- 8RLoop
b
-----------------
ln
≡
t∂
∂DLoop K311D311 KDLoop
2DLoop
RLoop
2
------------------
–=
K311
RLoop

4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 329
I-2013.12
■ is the diffusivity of neutral interstitials.
■ is the shear modulus (dyn/ ).
■ is the stacking fault energy (dyn/cm).
■ is the atomic value of silicon ( ).
■ is the Poisson ratio.
■ is the magnitude of Burger’s vector.
■ and are fitting parameters for the model.
These parameters can be set using the following commands:
pdbSet <material> CLoop K311 {<n>}
pdbSet <material> Mechanics ShearModulus {<n>}
pdbSet <material> CLoop StackingFaultEng {<n>}
pdbSet <material> CLoop BurgersVec {<n>}
pdbSet <material> CLoop KCLoop {<n>}
pdbSet <material> DLoop KDLoop {<n>}
Size-dependent Model
If the Unfault.Model is set to SizeDependent, the following reaction equations and the
{311} defect equations (see Defect Cluster Model: 311 on page 320) are solved:
(522)
(523)
where is the scaling factor for the unfaulting rate and can be defined using the command:
pdbSet <material> CLoop KD311 {<n>}
is the user-defined term to further modify the unfaulting rate and can be defined using the
command:
term name=CLoopTransfer <material> eqn = {User defined Equation}
The model is used to simulate all three phases of dislocation nucleation and evolution:
nucleation, Ostwald ripening, and dissolution.
To modify the equilibrium concentration of interstitials at the loop boundaries ( ) by
complex prefactors, you can define the following terms in the command file:
term name=CLoopDissIntFactor silicon add eqn = { equation }
DI0
µ
cm2
γ
Ω
223–
×10
υ
b
KCLoop
KDLoop
t∂
∂CLoop K311C311
2CT2π2RLoopDLoopDI0KCLoop CICILoop
*
–()+=
t∂
∂DLoop K311D311C311CTKD311
2π2RLoopDLoop
2DI0KCLoopCILoop
*
CLoop
------------------------------------------------------------------------------------–=
KD311
CT
CILoop
*

4: Diffusion
Defect Clusters
330 Sentaurus™ Process User Guide
I-2013.12
increases dramatically as the loop radius becomes smaller. To avoid convergence
problems, is limited by a minimum loop radius ( ) and as follows:
(524)
The minimum radius and the damping factor are set with the commands:
pdbSet <material> CLoop RLoopMin {<n>}
pdbSet <material> CLoop DampFactor {<n>}
If the Loop model is used with the 2Moment model, to avoid convergence problems, the
interstitial evaporation terms in Eq. 562, Eq. 563, Eq. 564, and Eq. 565 are scaled by:
(525)
The damping factor for the {311} defects can be set:
pdbSet <material> C311 DampFactor {<n>}
Initializing Loop Model
Since the loop model relies on the {311} defect model, first, the {311} defect model is
initialized (see Initializing 311 Model on page 327). Then, the loop model fields CLoop and
DLoop are initialized. If there is no preexisting data field, the fields are set to and
, respectively. If there is an existing data field, the existing fields are used. By default,
the loops are assumed to break apart in the amorphous regions.
You c an s pe cif y the pe rc ent ag e of loops retained in the amorphous region per solution variable
using the parameter AmPercent:
pdbSet <mater> DLoop AmPercent {<n>}
pdbSet <mater> CLoop AmPercent {<n>}
For example:
(526)
Ta bl e 3 4 S o l ut io n na m es fo r l oo p mo de l
Symbol Solution name
CLoop
DLoop
CILoop
*
CILoop
*
RLoopMin
CLoop
dFactor
CILoop
*CILoop
*RLoopMin
()CILoop
*RLoop
()
CILoop
*RLoopMin
()CILoop
*RLoop
()+
---------------------------------------------------------------------------------------- 1CLoop
dFactor
CLoop CLoop
dFactor
+
-------------------------------------------–
=
Rlimit 1C311
dFactor
C311
dFactor C311
+
---------------------------------------–
=
CLoop
DLoop
110
×10
57
×10
DLoop
DLoop*AmPercent
DLoop
=Amorphous regions
Crystalline regions

4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 331
I-2013.12
(527)
The value of the AmPercent parameter must be between 0 and 1.
Defect Cluster Model: LoopEvolution
If you set the defect cluster model to LoopEvolution, the TS4 style loop evolution
model [35] is used. The rate of absorption of interstitials by dislocation loops is given by:
(528)
(529)
where:
■ is the average loop radius.
■ is the loop density.
■ is the diffusivity of interstitials.
■ is the fitting parameter.
■ is the shear modulus (dyn ).
■ is the stacking fault energy (dyn/cm).
■ is the atomic value of silicon ( ).
■ is the Poisson ratio.
■ is the magnitude of Burger’s vector.
These parameters can be set using the following commands:
pdbSet <material> CLoop ShearModulus {<n>}
pdbSet <material> CLoop StackingFaultEng {<n>}
pdbSet <material> CLoop BurgersVec {<n>}
pdbSet <material> CLoop KCLoop {<n>}
The evolution of the loop radius is given by:
(530)
where is the {111} planar density of silicon ( ).
CLoop
CLoop*AmPercent
CLoop
=Amorphous regions
Crystalline regions
RI2π2RLoopKCLoopDLoopDICICILoop
*
–()≡
CILoop
*CI
*e
γΩ
bkT
---------
e
µbΩ
4πRLoopkT 1υ–()
--------------------------------------------- 8RLoop
b
-----------------
ln
≡
RLoop
DLoop
DI
KCLoop
µ
/cm2
γ
Ω
223–
×10
υ
b
t∂
∂RLoop π
N0
------ KCLoopDICICILoop
*
–()=
N0
1.57 1015
× cm 2–

4: Diffusion
Defect Clusters
332 Sentaurus™ Process User Guide
I-2013.12
The equilibrium concentration of interstitials at the loop boundaries ( ) increases
dramatically as the loop radius becomes smaller. To avoid convergence problems, is limited
by a minimum loop radius ( ) as follows:
(531)
To set the minimum radius, use:
pdbSet <material> CLoop RLoopMin {<n>}
To set the density of loops to a fixed value, use:
pdbSet <mater> DLoop ConstantDensity {<n>}
Initializing LoopEvolution Model
To specify the initial loop radius, use:
pdbSet <mater> RLoop InitialRadius {<n>}
To set the density of loops to a fixed value, use:
pdbSet <mater> DLoop ConstantDensity {<n>}
or it will be calculated using the following:
(532)
where and can be set with:
pdbSet <material> CLoop Fraction {<n>}
pdbSet <material> CLoop Threshold {<n>}
NOTE If ConstantDensity is zero, Eq. 532 will be used.
Loops are produced in that portion of the structure where the interstitial concentration (due to
implant damage, before recombination) is in the range:
(533)
Ta bl e 3 5 S o l ut io n na m es fo r L oo pE vol ut io n m o d e l
Symbol Solution name
RLoop
CILoop
*
RI
RLoopMin
Damp0
RLoop
RLoopMin
----------------------- Damp Damp04.605()
Damp04.605+
--------------------------------------≡;≡
RIRIRLoop
()1e–Damp()–
()≡
RLoop
DLoop
Fraction CIThreshold–()
πN0RLoop
2
-------------------------------------------------------------------=
Fraction
Threshold
Dmax Damage Dmin>>

4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 333
I-2013.12
and can be set:
pdbSet <material> CLoop Dmin {<n>}
pdbSet <material> CLoop Dmax {<n>}
The concentration of interstitials corresponding to the edge of the amorphous region is from
the work of Fair and Pappas [2]. The concentration of interstitials is not reduced by the
formation of end-of-range loops if DLoop is set to a constant value. Pre-existing dislocation
loops in the region are presumed to be destroyed by the implant.
NOTE In this model, CLoop and DLoop are terms. Therefore, they should be
converted to data fields with the select command or added to the plx/
DFISE list to save in a file.
Defect Cluster Model: FRENDTECH
If you set the model to FRENDTECH, this activates the extended defect model developed by
FRENDTECH partners [36][37][38]. The model assumes the following reactions:
(534)
(535)
where and are the interstitial defects consisting of and silicon atoms. The
reactions can be written as:
(536)
(537)
and the model is given by:
(538)
where is the density of defects containing atoms.
Dmax
Dmin
In1–I+In
↔
In1+InI+↔
In
In1+
n
n1+
R1kfn1–CIn1–krnCIn
–≡
R2kfnCInkrn1+CIn1+
–()–≡
t∂
∂CInR1R2
+=
CIn
n

4: Diffusion
Defect Clusters
334 Sentaurus™ Process User Guide
I-2013.12
The emission rate is given by:
(539)
where:
■ is the diffusion coefficient of neutral interstitials.
■ is the equilibrium concentration of interstitials.
■ is the capture efficiency of the defect.
■ is its formation energy.
The capture rate is:
(540)
where is the concentration of interstitials. and can be defined using the command:
pdbSet <material> Interstitial BindCluster <cluster> {<n>}
where <cluster> is a valid cluster name (for example, I2, I3, I21) and <n> must be an
Arrhenius expression with a prefactor of and an activation energy of .
For example:
pdbSet Silicon Interstitial BindCluster I6 {[Arrhenius 3.82578e-07 -1.29]}
sets the to 3.82578 x 10–7 eV and to –1.29 eV for .
Since cluster sizes can change easily from a few atoms to a few thousand atoms, it is not
feasible to solve all cluster equations. Therefore, the number of equations to be solved is
reduced by using the method proposed by FRENDTECH partners [39]. The method allows for
the logarithmic discretization of clusters:
(541)
krn
krnDI0CI
*An1–
Reff
-------------e
E–fn
kT
----------
=
DI0
CI
*
An1–
Reff
-----------
Efn
kfn
kfnDI0CI
An
Reff
---------=
CI
An
Reff
--------
Efn
An
Reff
--------
Efn
An
Reff
--------
Efn
n6=
td
dρu() 1eu–
2
-------+
eu–
2α
------- kFuα+ρuα+kFuα–ρuα–
–()–=
+1 eu–
2
-------–
eu–
2α
------- kRuα+ρuα+kRuα–ρuα–
–()
+e2u–
2α2
----------kFuα+ρuα+2kFuρu
–kFuα–ρuα–
+()
+e2u–
2α2
----------kRuα+ρuα+2kRuρu
–kRuα–ρuα–
+()
4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 335
I-2013.12
where is the density function, and is the step of the logarithmic discretization. and
are related to the capture and emission rate and by the relations:
(542)
(543)
The discretization is regular on a regularities scale. For example, the step in the reduced region
is calculated by:
(544)
where is the biggest cluster size, is the number of small interstitial clusters in the
linear region, and is the number of steps in the reduced region.
You can specify , , and using the following commands:
pdbSet <material> Int BiggestClusterSize {<n>}
pdbSet <material> Int NumberofSmallClusters {<n>}
pdbSet <material> Int logSteps {<n>}
For example:
pdbSet Silicon Int BiggestClusterSize 10000
pdbSet Silicon Int NumberofSmallClusters 11
pdbSet Silicon Int logSteps 5
allow Sentaurus Process to solve the cluster equations for I2, I3, I4, I5, I6, I7, I8, I9, I10,
I11, I12, I46, I177, I679, I2605, I10000.
The capture and emission rate and are stored in the parameter database for cluster sizes
less than 10. If you do not specify the rates for bigger clusters, they will be calculated using the
following formulas:
(545)
(546)
ρu()
α
kFu
kRu
kfn
krn
kFukf
ne
u
=
=
kRukr
ne
u
=
=
αnmax 1–()()log nlastlin 1+()log–
nlogsteps
-------------------------------------------------------------------------------=
nmax
nlastlin
nlogsteps
nmax
nlastlin
nlogsteps
kfn
krn
An
Reff
---------
311{}
4πaL 4πaW 8πa2
++
11
2a
W
------
2
++
log 2a
W
------
log–11
2La+()
W
--------------------
2
++
log 2La+()
W
--------------------
log–+
1
a
---12a
W
------
2
+1
La+
------------ 12La+()
W
--------------------
2
++
----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------
-----------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------------=
Estrain
nµb2L
2π1v–()
----------------------- 2W
b
--------
log µb2W
2π
-------------- θcos()
2θsin()
2
1v–
------------------
+
2L
b
------
log+=

4: Diffusion
Defect Clusters
336 Sentaurus™ Process User Guide
I-2013.12
(547)
where:
■ and are the length and width of the {311} defect ( cm and
cm).
■ is the lattice spacing of silicon.
■ denotes the shear modulus of silicon ( dyn/cm).
■ is the Poisson ratio ( ).
■ is the length of Burger’s vector ( cm).
■ is the angle between Burger’s vector and the normal vector perpendicular to the plane of
the defect ( ).
■ is the stacking fault energy per atom ( eV).
Burger’s vector and the stacking fault energy can be set using the commands:
pdbSet <material> C311 BurgersVec {<n>}
pdbSet <material> Int StackingFauldEng {<n>}
NOTE Since FRENDTECH models solve for a range of cluster sizes,
simulations may be slower.
Initializing FRENDTECH Model
By default, the interstitial clusters are assumed to break apart in the amorphous regions. You
can specify the percentage of clusters retained in the amorphous region per solution variable
using the parameter AmPercent:
pdbSetDouble <mater> I200 AmPercent {<n>}
pdbSetDouble <mater> I100 AmPercent {<n>}
For example:
(548)
Table 36 Solution names for FRENDTECH model
Symbol Solution name
I2
I3
I10
Efn Estrain
n1+γn1+()⋅Estrain
nγn⋅+–+=
L
W
Ln510
9–
×⋅=
W410
7–
×=
a
µ
µ7.55 1011
×=
v
v0.3=
b
b1.1 10 8–
×=
θ
θ77.8°=
γ
γ0.38=
CI2
CI3
CI10
I200
I200*AmPercent
I299
=Amorphous regions
Crystalline regions

4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 337
I-2013.12
Since not all clusters have been incorporated into the parameter database, the pdbSetDouble
command must be used; the shorthand pdbSet command cannot be used to specify these
parameters. The value specified for the AmPercent parameter must be between 0 and 1.
Defect Cluster Model: 1Moment
Interstitial
If you set the model to 1Moment, the model for the formation and dissolution of interstitial
clusters ({311} or {113} defects) is included. The 1Moment model uses a single equation to
calculate the total number of interstitials bound in clusters. The following nonlinear algebraic
equation along with the related diffusion equations are solved:
(549)
where is the concentration of clustered interstitials, and and describe cluster
interaction with interstitials and vacancies, respectively.
The interstitial interaction includes two terms describing the clustering of interstitials and one
describing the de-clustering:
(550)
where is the concentration of unclustered interstitials, and is the equilibrium
concentration of interstitials. The first term models nucleation of the interstitial clusters at high
interstitial supersaturation. The second term models growth of the interstitial clusters by
consuming free interstitials. The third term models dissolution of the interstitial clusters by
emitting interstitials. If the 1Moment model is used with the Full cluster model, Eq. 550 is
modified as follows:
(551)
where is given in Defect Cluster Model: Full on page 346. The smallest large
cluster forms when the small cluster captures one free interstitial by the reaction rate
. The vacancy interaction includes one recombination and one generation term:
(552)
t∂
∂CICluster RCI RCV
–=
CICluster
RCI
RCV
RCI Ikfi
CI
()
Ifi
CI
*
()
Isfi
-----------------Ikfc
CI
()
Ifc
CI
*
()
Isfc
----------------- CICluster IkfciCI
+()Ikr CICluster
()
Icr
–+=
CI
CI
*
RCI max 1+()RsIcl max 1+()
Ikfc
CI
()
Ifc
CI
*
()
Isfc
----------------- CICluster IkfciCI
+()Ikr CICluster
()
Icr
–+=
RsIcl max 1+()
RsIcl max 1+()
RCV Vkrv
CV
()
Vrv
CV
*
()
Vsrv
--------------------CICluster
()
Vcrv Vkf CV
*
()
Vsfv CICluster
()
Vcfv
–=
4: Diffusion
Defect Clusters
338 Sentaurus™ Process User Guide
I-2013.12
where is the concentration of free vacancies and is the equilibrium concentration of
vacancies. The first term models dissolution of the interstitial clusters by consuming vacancies.
The second term models the emission of vacancies by the interstitial clusters.
To modify the dissolution rates and by complex prefactors, you can define the
following terms in the input file:
term name=IClusterDissIntFactor silicon add eqn = { equation }
term name=IClusterDissVacFactor silicon add eqn = { equation }
For extrinsic silicon, the prefactors can be made a function of to provide consistent TED
results.
If the 1Moment model is used with the Full cluster model, Eq. 552 is modified as follows:
(553)
where is given in Defect Cluster Model: Full on page 346. It is the reaction rate
of small interstitial clusters with vacancies. The reaction constants of the model can be
modified using the following commands:
pdbSet <material> ICluster Ikfi {<n>}
pdbSet <material> ICluster Ikfc {<n>}
pdbSet <material> ICluster Ikr {<n>}
pdbSet <material> ICluster Ifi {<n>}
pdbSet <material> ICluster Isfi {<n>}
pdbSet <material> ICluster Ifc {<n>}
pdbSet <material> ICluster Isfc {<n>}
pdbSet <material> ICluster Icf {<n>}
pdbSet <material> ICluster Icr {<n>}
pdbSet <material> ICluster Ikfci {<n>}
pdbSet <material> ICluster Vkrv {<n>}
pdbSet <material> ICluster Vrv {<n>}
pdbSet <material> ICluster Vsrv {<n>}
pdbSet <material> ICluster Vcrv {<n>}
pdbSet <material> ICluster Vkfv {<n>}
pdbSet <material> ICluster Vsfv {<n>}
pdbSet <material> ICluster Vcfv {<n>}
CV
CV
*
Ikr
Vkf
n
ni
----
RCV max 1+()RsIcV max 1+()
V+krv
CV
()
Vrv
CV
*
()
Vsrv
--------------------CICluster
()
Vcrv Vkf CV
*
()
Vsfv CICluster
()
Vcfv
–=
RsIcV max 1+()

4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 339
I-2013.12
The changes are accompanied by corresponding inverse changes in . Therefore, clustering
reduces the number of free interstitials, while the dissolution of clusters releases interstitials.
Vacancy
If you set the model to 1Moment, the model for the formation and dissolution of vacancy
clusters or voids is included. The 1Moment model uses a single equation to calculate the total
number of vacancies bound in clusters. The following nonlinear algebraic equation along with
the related diffusion equations are solved:
(554)
where is the concentration of clustered vacancies, and and describe cluster
interaction with vacancies and interstitials, respectively.
The vacancy interaction includes two terms describing the clustering of vacancies and one
describing the declustering:
(555)
where is the concentration of unclustered vacancies, and is the equilibrium
concentration of vacancies. The first term models nucleation of the vacancy clusters at high
vacancy supersaturation. The second term models growth of the vacancy clusters by
consuming free vacancies. The third term models dissolution of the vacancy clusters by
emitting vacancies. If the 1Moment model is used with the Full cluster model, Eq. 555 is
modified as follows:
(556)
where is given in Defect Cluster Model: Full on page 346. The smallest large-
cluster forms when the small cluster captures one free vacancy by the reaction rate
.
Ta bl e 3 7 S o l ut io n na m es fo r 1 Mo me nt m o de l of i nt e r s ti t i a l s
Symbol Solution name
ICluster
CI
CICluster
t∂
∂CVCluster RCV RCI
–=
CVCluster
RCV
RCI
RCV Vkfi
CV
()
Vfi
CV
*
()
Vsfi
-------------------Vkfc
CV
()
Vfc
CV
*
()
Vsfc
------------------- CVCluster VkfciCV
+()Vkr CVCluster
()
Vcr
–+=
CV
CV
*
RCV max 1+()RsVcl max 1+()
Vkfc
CV
()
Vfc
CV
*
()
Vsfc
------------------- CVCluster VkfciCV
+()Vkr CVCluster
()
Vcr
–+=
RsVcl max 1+()
RsVcl max 1+()
4: Diffusion
Defect Clusters
340 Sentaurus™ Process User Guide
I-2013.12
The interstitial interaction includes one recombination and one generation term:
(557)
where is the concentration of free interstitials, and is the equilibrium concentration of
interstitials. The first term models dissolution of the vacancy clusters by consuming
interstitials. The second term models the emission of interstitials by the vacancy clusters.
To modify the dissolution rates and by complex prefactors, you can define the
following terms in the command file:
term name=VClusterDissVacFactor silicon add eqn = { equation }
term name=VClusterDissIntFactor silicon add eqn = { equation }
If the 1Moment model is used with the Full cluster model, Eq. 557 is modified as follows:
(558)
where is given in Defect Cluster Model: Full on page 346. It is the reaction rate
of small vacancy clusters with interstitials. The reaction constants of the model can be modified
using the following commands:
pdbSet <material> VCluster Vkfi {<n>}
pdbSet <material> VCluster Vkfc {<n>}
pdbSet <material> VCluster Vkr {<n>}
pdbSet <material> VCluster Vfi {<n>}
pdbSet <material> VCluster Vsfi {<n>}
pdbSet <material> VCluster Vfc {<n>}
pdbSet <material> VCluster Vsfc {<n>}
pdbSet <material> VCluster Vcf {<n>}
pdbSet <material> VCluster Vcr {<n>}
pdbSet <material> VCluster Vkfci {<n>}
pdbSet <material> VCluster Ikrv {<n>}
pdbSet <material> VCluster Irv {<n>}
pdbSet <material> VCluster Isrv {<n>}
pdbSet <material> VCluster Icrv {<n>}
pdbSet <material> VCluster Ikfv {<n>}
pdbSet <material> VCluster Isfv {<n>}
pdbSet <material> VCluster Icfv {<n>}
RCI Ikrv
CI
()
Irv
CI
*
()
Isrv
------------------CVCluster
()
Icrv Ikf CI
*
()
Isfv CVCluster
()
Icfv
–=
CI
CI
*
Vkr
Ikf
RCI max 1+()RsVcI max 1+()
Ikrv
CI
()
Irv
CI
*
()
Isrv
------------------CVCluster
()
Icrv Ikf CI
*
()
Isfv CVCluster
()
Icfv
–+=
RsVcI max 1+()
4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 341
I-2013.12
The changes are accompanied by corresponding inverse changes in . Therefore, clustering
reduces the number of free vacancies; while the dissolution of clusters releases vacancies.
Initializing 1Moment Model
The initial concentration of interstitial clusters or vacancy clusters after implantation is set in
the diffPreProcess procedure (see Ion Implantation to Diffusion on page 353) and can be
changed using the parameter InitPercent as follows:
pdbSet <material> ICluster InitPercent {<n>}
pdbSet <material> VCluster InitPercent {<n>}
InitPercent is the percentage of free implant interstitials used to initialize the model, for
example:
(559)
(560)
The value of InitPercent must be between 0 and 1. The model assumes that existing
interstitial or vacancy clusters in the amorphized region break apart.
Defect Cluster Model: 2Moment
Interstitial
If you set the model to 2Moment, the model for the formation and dissolution of interstitial
clusters ({311} or {113} defects) and conversion of {311} clusters into dislocation loops are
included [39]. The model calculates the first two moments of the size distribution of interstitial
clusters, in other words, the number of clusters and the number of interstitials contained in the
clusters. It can be used with the existing model for dislocation loops (LoopEvolution),
although it is designed to include modeling of at least some dislocation loops.
Ta bl e 3 8 S o l ut io n na m es fo r 1 Mo me nt m o de l fo r vac an ci es
Symbol Solution name
VCluster
CV
CVCluster
ICluster Int_Implant*InitPercent
0
=Crystalline regions
Amorphous regions
VCluster Vac_Implant*InitPercent
0
=Crystalline regions
Amorphous regions
4: Diffusion
Defect Clusters
342 Sentaurus™ Process User Guide
I-2013.12
The 2Moment clustering model is an implementation of the Gencer analytic kinetic
precipitation model (AKPM). The density of clusters ( ) and concentration of interstitials
() contained in clusters are calculated as:
(561)
(562)
where and are the concentration and diffusivity of free interstitials. If the 2Moment
model is used with the Full cluster model, Eq. 561 and Eq. 562 are modified as follows:
(563)
(564)
, which gives the dependence of the dissolution rate on the average cluster size, is given
by:
(565)
where:
■ controls the dissolution of two atom clusters.
■ is the average number of interstitials in a cluster.
, which gives the dependence of the rate of interstitial release on the average cluster size,
is given by:
(566)
where and are chosen to make and continuous at . are the
solid solubility of smaller and larger clusters.
D311
C311
t∂
∂D311 DIλ0CI
2D311CIi
*Csγ0
–()=
t∂
∂C311 2t∂
∂D311 D+Iλ1D311 CICsCIi
*γ1
–()=
CI
DI
t∂
∂D311 RsIcl max 1+()
RsIcV max 1+()
DIλ0D311CIi
*Csγ0
––=
t∂
∂C311 max 1+()RsIcl max 1+()
RsIcV max 1+()
–()D+Iλ1D311 CICsCIi
*γ1
–()=
Csγ0
Csγ0
Css
K1
s1–
-----------
CslK31
s1–
-----------
α
=sn
crit
<
sn
crit
>
K1
sC
311 D311
⁄=
Csγ1
Csγ1
Css
s2–
sK
0
+
---------------1K02+()K2
sK
0
+
---------------------------+
Csl 1K+4
K02+
sK
0
+
---------------
α
=sn
crit
<
sn
crit
>
K3
K4
Csγ0
Csγ1
sn
crit
=
Css,Csl

4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 343
I-2013.12
To modify and by complex prefactors, you can define the following terms in the
command file:
term name=C311DissIntFactor silicon add eqn = { equation }
To set the model parameters, use the following commands:
pdbSet <material> C311 Lambda0 {<n>}
pdbSet <material> C311 Lambda1 {<n>}
pdbSet <material> C311 K0 {<n>}
pdbSet <material> C311 K1 {<n>}
pdbSet <material> C311 K2 {<n>}
pdbSet <material> C311 Alpha {<n>}
pdbSet <material> C311 NCritical {<n>}
pdbSet <material> Int SolubilitySmall {<n>}
pdbSet <material> Int SolubilityLarge {<n>}
Eq. 561 models the nucleation and dissolution of two-atom clusters. is the capture length
for these processes.
Eq. 562 models the absorption and release of interstitials by clusters. The three terms on the
right side model the absorption of interstitials during nucleation, the absorption of interstitials
by nucleated clusters, and the release of interstitials by nucleated clusters. is the capture
length for absorption and release of interstitials by nucleated clusters. is the
concentration of interstitials in equilibrium with a population of large {311} clusters.
For , some of the {311} defects unfault to form dislocation loops. These dislocation
loops are included in the 2Moment model by modifying the cluster dissolution rates.
controls the dissolution rate when loops are present, and is the concentration of
interstitials in equilibrium with a population of large dislocation loops, .
Vacancy
If you set the model to 2Moment – the model for the formation and dissolution of vacancy
clusters – the model calculates the first two moments of the size distribution of vacancy
clusters, that is, the number of clusters and the number of vacancies contained in the clusters.
Ta bl e 3 9 S o l ut io n na m es fo r 2 Mo me nt m o de l of i nt e r s ti t i a l s
Symbol Solution name
C311
D311
Csγ0
Csγ1
λ0
λ1
CIi
*Css
sn
crit
>
α
CIi
*Csl
CIiCsl
C311
D311
4: Diffusion
Defect Clusters
344 Sentaurus™ Process User Guide
I-2013.12
The 2Moment clustering model is an implementation of the Gencer analytic kinetic
precipitation model (AKPM). The density of clusters ( ) and concentration of vacancies
() contained in clusters are calculated as:
(567)
(568)
where and are the concentration and diffusivity of free vacancies.
If the 2Moment model is used with the Full cluster model, Eq. 567 and Eq. 568 are modified
as follows:
(569)
(570)
, which gives the dependence of the dissolution rate on the average cluster size, is given
by:
(571)
where:
■ controls the dissolution of two atom clusters.
■ is the average number of vacancies in a cluster.
, which gives the dependence of the rate of vacancy release on the average cluster size, is
given by:
(572)
are the solid solubility of clusters.
To modify and by complex prefactors, you can define the following terms in the
command file:
term name=CVoidDissVacFactor silicon add eqn = { equation }
Dvoid
Cvoid
t∂
∂Dvoid DVλ0CV
2DvoidCVi
*Csγ0
–()=
t∂
∂Cvoid 2t∂
∂Dvoid D+Vλ1Dvoid CVCsCVi
*γ1
–()=
CV
DV
t∂
∂Dvoid RsVcl max 1+()
RsVcI max 1+()
DVλ0DvoidCVi
*Csγ0
––=
t∂
∂Cvoid max 1+()RsVcl max 1+()
RsVcI max 1+()
–()D+Vλ1Dvoid CVCsCVi
*γ1
–()=
Csγ0
Csγ0Css
K1
s1–
-----------=
K1
sC
void Dvoid
⁄=
Csγ1
Csγ1Css
s2–
sK
0
+
---------------1K02+()K2
sK
0
+
---------------------------+=
Css
Csγ0
Csγ1

4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 345
I-2013.12
To set the model parameters, use the following commands:
pdbSet <material> CVoid Lambda0 {<n>}
pdbSet <material> CVoid Lambda1 {<n>}
pdbSet <material> CVoid K0 {<n>}
pdbSet <material> CVoid K1 {<n>}
pdbSet <material> CVoid K2 {<n>}
pdbSet <material> Vac SolubilitySmall {<n>}
Eq. 567 models the formation and dissolution of di-vacancy clusters. is the capture length
for these processes.
Eq. 568 models the absorption and release of vacancies by clusters. The three terms on the right
side model the absorption of vacancies during di-vacancy cluster formation, the absorption of
vacancies by clusters, and the release of vacancies by clusters. is the capture length for
absorption and release of vacancies by clusters. is the concentration of vacancies in
equilibrium with a population of large vacancy clusters.
Initializing 2Moment Model
The initial concentration of interstitial clusters after implants is set in the diffPreProcess
procedure (see Ion Implantation to Diffusion on page 353). By default, clusters are assumed to
break apart in the amorphous regions. You can specify the percentage of clusters retained in the
amorphous region per cluster solution variable using the parameter AmPercent:
pdbSet <material> C311 AmPercent {<n>}
pdbSet <material> D311 AmPercent {<n>}
pdbSet <material> CVoid AmPercent {<n>}
pdbSet <material> DVoid AmPercent {<n>}
For example:
(573)
The value of the AmPercent parameter must be between 0 and 1.
Ta bl e 4 0 S o l ut io n na m es fo r 2 Mo me nt m o de l of va ca n ci es
Symbol Solution name
CVoid
DVoid
λ0
λ1
CVi
*Css
Cvoid
Dvoid
D311
D311*AmPercent
D311
=Amorphous regions
Crystalline regions

4: Diffusion
Defect Clusters
346 Sentaurus™ Process User Guide
I-2013.12
Defect Cluster Model: Full
Interstitial
If you set the defect cluster model to Full, the TS4 style transient small interstitial cluster
model is used. The reactions associated with the size-n small interstitial cluster is as follows:
(574)
(575)
(576)
(577)
denote the n-size interstitial small cluster; are the interstitials and vacancies. The
transient equation for the n-size small interstitial cluster is:
(578)
can be set using the following command:
pdbSet Si Int CL.Size {<n>}
and and are described as follows:
(579)
(580)
(581)
(582)
(583)
In1–I+In
↔
InI+In1+
↔
InV+In1–
↔
In1+V+In
↔
In
IV,
t∂
∂CnRcI n() RcV n()
–= 2nn
max
<≤
nmax
RcI n()
RcV n()
RcI n() RcI n 1–()n()→()
RcI n() n1+()→()
–=
RcV n() RcV n() n1–()→()
RcV n 1+()n()→()
–=
RcI n 1–()n()→()
kfi
n()
CIn 1–()
CI
CI
*
------ kri
n()
CIn()
–=
RcI n() n1+()→()
kfi
n1+()
CIn()
CI
CI
*
------ kri
n1+()
CIn 1+()
–=
RcV n() n1–()→()
krv
n()
CIn()
CV
CV
*
-------kfv
n()
CIn 1–()
–=
4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 347
I-2013.12
(584)
here:
(585)
(586)
(587)
(588)
(589)
(590)
(591)
To set the reaction rate constants, , use:
pdbSet Si I2 kfI {<i,j>} {<n>}
pdbSet Si I2 krI {<i,j>} {<n>}
pdbSet Si I2 kfV {<i>} {<n>}
pdbSet Si I2 krV {<i>} {<n>}
pdbSet Si I3 kfI {<i>} {<n>}
pdbSet Si I3 krI {<i>} {<n>}
pdbSet Si I3 kfV {<i>} {<n>}
pdbSet Si I3 krV {<i>} {<n>}
pdbSet Si I4 kfI {<i>} {<n>}
pdbSet Si I4 krI {<i>} {<n>}
pdbSet Si I4 kfV {<i>} {<n>}
pdbSet Si I4 krV {<i>} {<n>}
pdbSet Si I5 kfI {<i>} {<n>}
RcV n 1+()n()→()
krv
n1+()
CIn 1+()
CV
CV
*
-------kfv
n1+()
CIn()
–=
kfi
n() CIi
*fnz
ikIz
n
ni
----
z–
z
=n2>
kfi
2() CIi
*2
CI
*
-------- f2qz
ikIzkIq
n
ni
----
zq+()–
zq,
=n2=
kri
n() CIi
*rnz
ikIz
n
ni
----
z–
z
=n2>
kri
2() CIi
*r2qz
ikIzkIq
n
ni
----
zq+()–
zq,
=n2=
kfv
n() CVi
*fnz
vkVz
n
ni
----
z–
z
=n2>
kfv
2() CIi
*CVi
*
CI
*
---------------- f2z
vkVz
n
ni
----
z–
z
=n2=
krv
n() CVi
*rnz
vkVz
n
ni
----
z–
z
=
fikfI fv
,kfV ri
,krI rv
,krV
== ==
4: Diffusion
Defect Clusters
348 Sentaurus™ Process User Guide
I-2013.12
The indices and are integers representing the charge state of interstitials and reacting
interstitials. The shorthand pdbSet command can be used for clusters up to size 5. For all other
clusters, the longhand pdbSetDoubleArray command must be used.
NOTE The indices for the parameters kfI and krI for I2 clusters have the form
. The indices are separated by a comma; no space is allowed between
the indices.
If you want to use same reaction rate constants for all charges, use:
pdbSet Si Int CL.All {1|0}
If CL.ALL is set to 1, the model uses the 0th indexed reaction rate constants (for example,
kfI(0,0) or kfI(0), krV(0) and so on) in the reaction calculation for all charged states.
To modify and by complex prefactors, you can define the following terms in the
command file, for example, for I2:
term name=I2DissIntFactor silicon add eqn = { equation }
term name=I2DissVacFactor silicon add eqn = { equation }
The net capture rate of free interstitials by the small interstitial clusters is given by:
(592)
The net capture rate of free vacancies by the small interstitial clusters is given by:
(593)
Ta bl e 41 S o lu t io n n am e s fo r f u ll m o d e l o f i n t e rs ti t i a l s
Symbol Solution name
I2
I3
I4
I5
i
j
ij,
kri
kfv
RsIcl Rcl 12→() Rcl n n 1+→()
n1=
max
+=
RsIcV RcV n n 1+→()
n1=
max
=
CI2
CI3
CI4
CI5

4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 349
I-2013.12
Vacancy
If you set the defect cluster model to Full, the TSUPREM-4-style transient small-vacancy
cluster model is used. The reactions associated with the -size small interstitial cluster are:
(594)
(595)
(596)
(597)
where denotes the -size small vacancy cluster, and are the interstitials and vacancies.
The transient equation for the -size small vacancy cluster is:
(598)
can be set using:
pdbSet Si Vac CL.Size {<n>}
and are described as follows:
(599)
(600)
(601)
(602)
(603)
(604)
n
Vn1–V+Vn
↔
VnV+Vn1+
↔
VnI+Vn1–
↔
Vn1+I+Vn
↔
Vn
n
IV,
n
t∂
∂CnRcV n() RcI n()
–= 2nn
max
<≤
nmax
RcV n()
RcI n()
RcV n() RcV n 1–()n()→()
RcV n() n1+()→()
–=
RcI n() RcI n() n1–()→()
RcI n 1+()n()→()
–=
RcV n 1–()n()→()
kfv
n()
CVn 1–()
CV
CV
*
-------krv
n()
Cvn()
–=
RcV n() n1+()→()
kfv
n1+()
CVn()
CV
CV
*
-------krv
n1+()
CVn 1+()
–=
RcI n() n1–()→()
kri
n()
CVn()
CI
CI
*
------ kfi
n()
CVn 1–()
–=
RcI n 1+()n()→()
kri
n1+()
CVn 1+()
CI
CI
*
------ kfi
n1+()
CVn()
–=
4: Diffusion
Defect Clusters
350 Sentaurus™ Process User Guide
I-2013.12
here:
(605)
(606)
(607)
(608)
(609)
(610)
(611)
To set the reaction rate constants, , use:
pdbSet Si V2 kfV {<i,j>} {<n>}
pdbSet Si V2 krV {<i,j>} {<n>}
pdbSet Si V2 kfI {<i>} {<n>}
pdbSet Si V2 krI {<i>} {<n>}
pdbSet Si V3 kfV {<i>} {<n>}
pdbSet Si V3 krV {<i>} {<n>}
pdbSet Si V3 kfI {<i>} {<n>}
pdbSet Si V3 krI {<i>} {<n>}
pdbSet Si V4 kfV {<i>} {<n>}
pdbSet Si V4 krV {<i>} {<n>}
pdbSet Si V4 kfI {<i>} {<n>}
pdbSet Si V4 krI {<i>} {<n>}
pdbSet Si V5 kfV {<i>} {<n>}
pdbSet Si V5 krV {<i>} {<n>}
pdbSet Si V5 kfI {<i>} {<n>}
pdbSet Si V5 krI {<i>} {<n>}
pdbSet Si V6 kfV {<i>} {<n>}
pdbSet Si V6 krV {<i>} {<n>}
kfv
n() CVi
*fnz
ikVz
n
ni
----
z–
z
=n2>
kfv
2() CVi
*2
CV
*
-------- f2qz
ikVzkVq
n
ni
----
zq+()–
zq,
=n2=
krv
n() CVi
*rnz
ikVz
n
ni
----
z–
z
=n2>
krv
2() CVi
*r2qz
ikVzkVq
n
ni
----
zq+()–
zq,
=n2=
kfi
n() CIi
*fnz
ikIz
n
ni
----
z–
z
=n2>
kfi
2() CIi
*CVi
*
CV
*
---------------- f2z
ikIz
n
ni
----
z–
z
=n2=
kri
n() CIi
*rnz
ikIz
n
ni
----
z–
z
=
fvkfV fi
,kfI rv
,krV ri
,krI
====
4: Diffusion
Defect Clusters
Sentaurus™ Process User Guide 351
I-2013.12
pdbSet Si V6 kfI {<i>} {<n>}
pdbSet Si V6 krI {<i>} {<n>}
pdbSet Si V7 kfV {<i>} {<n>}
pdbSet Si V7 krV {<i>} {<n>}
pdbSet Si V7 kfI {<i>} {<n>}
pdbSet Si V7 krI {<i>} {<n>}
pdbSet Si V8 kfV {<i>} {<n>}
The indices and are integers representing the charge state of vacancies and reacting
vacancies. The shorthand pdbSet command can be used for clusters up to size 8. For all other
clusters, the longhand pdbSetDoubleArray command must be used.
NOTE The indices for the parameters kfV and krV for V2 clusters have the
form of . The indices are separated by a comma; no space is allowed
between the indices.
If you want to use same reaction rate constants for all charges, use:
pdbSet Si Vac CL.All {1|0}
If CL.ALL is set to 1, the model uses the 0th indexed reaction rate constants (for example,
kfV(0,0) or kfV(0), krI(0) and so on) in the reaction calculation for all charged states.
To modify and by complex prefactors, you can define the following terms in the
command file, for example, for V2:
term name=V2DissVacFactor silicon add eqn = { equation }
term name=V2DissIntFactor silicon add eqn = { equation }
The net capture rate of free vacancies by the small vacancy clusters is given by:
(612)
The net capture rate of free interstitials by the small vacancy clusters is given by:
(613)
i
j
ij,
krv
kfi
RsVcl RcV 12→() RcV n n 1+→()
n1=
max
+=
RsVcI RcI n n 1+→()
n1=
max
=

4: Diffusion
Defect Clusters
352 Sentaurus™ Process User Guide
I-2013.12
Initializing Full Model
The initial concentration of interstitial or vacancy clusters after implantation is set in the
diffPreProcess procedure (see Ion Implantation to Diffusion on page 353). By default,
clusters are assumed to break apart in the amorphous regions. You can specify the percentage
of clusters retained in the amorphous region per cluster solution variable using the parameter
AmPercent:
pdbSet <material> <cluster> AmPercent {<n>}
For example:
(614)
The value of the AmPercent parameter must be between 0 and 1.
In addition, you can specify the initial concentration of interstitial or vacancy clusters after
implantations by using the parameter InitPercent as follows:
pdbSet <material> <cluster> InitPercent {<n>}
The parameter InitPercent is the percentage of free implant interstitials or vacancies used
to initialize the model, for example:
(615)
The value of InitPercent must be between 0 and 1.
Ta bl e 4 2 S o l ut io n na m es fo r f ul l mo de l o f v a c an ci e s
Symbol Solution name
V2
V3
V4
V5
V6
V7
V8
CV2
CV3
CV4
CV5
CV6
CV7
CV8
I4
I4*AmPercent
I4
=Amorphous regions
Crystalline regions
ICluster Int_Implant*InitPercent
0
=Crystalline regions
Amorphous regions

4: Diffusion
Ion Implantation to Diffusion
Sentaurus™ Process User Guide 353
I-2013.12
Ion Implantation to Diffusion
During the implantation, important data fields (see Chapter 3 on page 81) such as
Int_Implant, Vac_Implant, and Damage are created
Int_Implant and Vac_Implant represent the total number of interstitial and vacancy point-
defects coming from the ion implantation. Since it is possible that the point defects already
exist in the structure or amorphization occurred due to ion implantation, the point-defect fields
must be updated before any diffusion step.
Sentaurus Process calls a default procedure, diffPreProcess, to process these fields. The
main goal of the procedure is to process the point-defect fields and to store the processed fields
in the Interstitial and Vacancy data fields. These data fields represent the total number
of interstitials and vacancies that will be used to initialize the total number of unpaired
interstitials (Int) and vacancies (Vac) (see Initializing Solution Variables on page 355):
(616)
(617)
First, interstitials and vacancies from implants (Int_Implant, Vac_Implant) are added to
existing Interstitial and Vacancy fields in the crystalline regions. If the fields do not
exist, they are created and set to their equilibrium values.
The Damage field is used to determine whether the material is amorphized. The threshold value
for the amorphization can be set by:
pdbSet <material> AmorpDensity {<n>}
It is assumed that if a material amorphizes due to ion implantation, the amorphized portion of
the material will grow to a perfect crystalline material and point-defect densities in this region
will be equal to their thermal equilibrium values (see Eq. 617). If a material is an amorphized
material (that is, polysilicon), the point-defect densities in this material are set automatically to
their equilibrium values.
The abovementioned amorphization algorithm leads to very steep interstitial profiles at the
amorphous–crystalline boundary. This boundary can be softened using an error function. The
degree of smoothing can be controlled using the parameter AmorpGamma, that is:
pdbSet <material> AmorpGamma {<n>}
Interstitial Interstitial+Int_Implant
CI
*
=Crystalline regions
Amorphous regions
Vacancy Vacancy+Vacancy_Implant
CV
*
=Crystalline regions
Amorphous regions
4: Diffusion
Ion Implantation to Diffusion
354 Sentaurus™ Process User Guide
I-2013.12
The value of this parameter must be between 0 and 1, where 1 means a very steep transition.
Smoothing also will be applied to dopant profiles if the transient or cluster model is selected.
To find out whether a material is amorphous, use the commands:
pdbGet <material> Amorphous
pdbSet <material> Amorphous 1 or 0
When the point-defect concentrations are set to their equilibrium values in the amorphous
regions, their densities in non-amorphous regions are compared to the solid solubility values
of these defects in each material. If the solid solubility values are defined, the defect profiles
are cut off at the solid solubility values. To specify the solid solubility numbers for the
Interstitial and Vacancy fields, use:
pdbSet <material> Int TotSolubility {<n>}
pdbSet <material> Vac TotSolubility {<n>}
(618)
In addition to the processing of implant data fields, the diffPreProcess procedure
determines whether point-defect equations need to be solved. As a default behavior, Sentaurus
Process does not solve the defect equations if the dopant diffusion models are set to Fermi
(see Fermi Diffusion Model on page 219), Constant (see Constant Diffusion Model on
page 220), or ChargedFermi (see ChargedFermi Diffusion Model on page 217) in all
materials.
However, the interstitial point-defect equation will be solved for the same dopant diffusion
models if the oxidation is switched on and dopants are present in the structure. Both point-
defect equations are solved if the dopant diffusion models are set to Pair, React,
ChargedPair, or ChargedReact in any material.
Although it is not recommended, you may want to switch on or off the point-defect equations
for any chosen dopant diffusion model. In this case, use the commands:
pdbSetBoolean Defect Int ForcedTurnOff 1
pdbSetBoolean Defect Vac ForcedTurnOff 1
pdbSetBoolean Defect Int ForcedTurnOn 1
pdbSetBoolean Defect Vac ForcedTurnOn 1
NOTE These parameters are not in the parameter database and are provided for
advanced users.
To change the initialization of point defects in the amorphous regions, use:
pdbSet <material> Int Truncation.Model <model>
pdbSet <material> Vac Truncation.Model <model>
Interstitial min Interstitial, IntTotSolubility()=
Vacancy min Vacancy, VacTotSolubility()=

4: Diffusion
Initializing Solution Variables
Sentaurus™ Process User Guide 355
I-2013.12
where <model> is either None or Equilibrium. The default is None and follows the
initialization procedure previously explained in Initializing Solution Var ia ble s o n page 3 55.
The Equilibrium model sets the unpaired total interstitial (Int) and vacancy (Vac)
concentrations to the user-defined equilibrium values, (see Eq. 153, p. 211).
The diffPreProcess procedure also initializes the fluorine model (see Initializing the
FVCluster Model on page 306), the active dopant models Cluster and Transient (see
Initializing Transient Model on page 298), the {311} defect-clustering model (see Initializing
311 Model on page 327), and the 1Moment defect-clustering model (see Initializing 1Moment
Model on page 341).
When the preprocessing of the data fields is completed, most implant fields are deleted.
Sentaurus Process also calls the diffPostProcess procedure as soon as diffusion has
finished. In this procedure, remaining implant fields are cleared and total defect concentrations
are stored for use with the next diffusion command during initialization.
Initializing Solution Variables
Initialization of solution variables is typically a minor task in Sentaurus Process. You can
manipulate data and easily modify any data field using the select command (see select on
page 1117). Data fields can be added, subtracted, truncated, or manipulated in many ways.
You al so c an d efi ne sp ecial ca llback procedures to initialize solution variables in different
ways. This section covers the callback procedures and the keywords used by Sentaurus Process
to initialize solution variables.
No nonlinear or partial differential equations are solved to initialize dopant solutions. Dopant
data fields generated during implantation are simply added to existing ones. For example, if
you select the dopant diffusion model React (see React Diffusion Model on page 212), there
is no contribution to the dopant–defect pair fields from the implant. However, you can use the
select command to distribute dopants among the other fields as required.
Conversely, Sentaurus Process uses callback procedures (see Using Callback Procedures on
page 586) to initialize the total number of unpaired interstitials (Int) and vacancies (Vac),
which are used as solution names. Since extra dopant–defect equations are not solved for the
Pair, ChargedPair, or ChargedFermi dopant diffusion models, transferring all point
defects from implantation to their respective solution names may cause an artificial increase of
dopant–defect pairs in the structure. To prevent this artificial dopant–defect pair increase,
defects from implantation must be added to the total interstitials and vacancies.
There are two main callback procedures to initialize solution variables: InitSolve and
EquationInitProc. To initialize a solution variable, the keyword InitStep must be
CX
*
4: Diffusion
Initializing Solution Variables
356 Sentaurus™ Process User Guide
I-2013.12
defined with the solution variable. This is different from the typical use of callback procedures
(see Using Callback Procedures on page 586):
pdbSetString <material> <solution> InitSolve <callback procedure>
pdbSetString <material> <solution> EquationInitProc <callback procedure>
The procedures take three arguments: a material, a solution, and the name of the callback
procedure. For example, the command:
pdbSetString Si Int InitSolve ResetInt
‘informs’ the code to invoke the ResetInit procedure every time that solutions are checked.
This is usually performed at the very beginning of a diffusion step.
The ResetInit procedure could be defined as:
proc ResetInit { Mat Sol } {
pdbUnSetString $Mat $Sol Equation
}
When a solution variable requires initialization, Sentaurus Process searches for whether the
EquationInitProc callback procedure is used for the solution name. If it is used, Sentaurus
Process executes the procedure given with the command. Otherwise, you must provide the
initialization equation. The command:
pdbSetString Si Int EquationInitProc InitializeInt
‘informs’ Sentaurus Process to call the procedure InitializeInt before parsing the
initialization equation for the solution Int.
The InitializeInt procedure could be defined as:
proc InitializeInit { Mat Sol } {
pdbSetString $Mat $Sol Equation "$Sol - 1e17"
}
In this case, the initialization equation for the solution name Int will be set to Int-1e17=0.
When the initialization is completed, Int will have the value of in the specified
material. This is a trivial example, but you can define any valid equation in this procedure.
For example, the default initialization equation for Int in Sentaurus Process, which can change
depending on the dopants and diffusion models, can be:
Interstitial - (Int + (I0 * BActive * (( [expr [Arrhenius 5.68 0.48] * \
[pdbGetDouble Si Boron Int Binding] ] + [expr 0.0 * \
[pdbGetDouble Si Boron Int Binding] ] * Noni) * Noni + ( \
110
17
×

4: Diffusion
Boundary Conditions
Sentaurus™ Process User Guide 357
I-2013.12
[expr [Arrhenius 5.68 0.42] * [pdbGetDouble Si Boron Int Binding] ] + \
[expr 0.0 * [pdbGetDouble Si Boron Int Binding] ] * Poni) * Poni + \
([expr 1.0 * [pdbGetDouble Si Boron Int Binding] ])))) = 0
where I0, BActive, Noni, and Poni are a function of the solution Int.
NOTE InitPostProcess (see Figure 86 on page 601) can be used to save
and plot solution variables after the initialization is completed.
Boundary Conditions
Different boundary conditions can be selected in Sentaurus Process:
■HomNeumann
■Natural
■Segregation
■Dirichlet
■ThreePhaseSegregation
■Trap
■Trapgen
■Continuous
Even though you can select any boundary conditions, they should be used with appropriate
species. It is possible to set a general boundary condition for all dopants or a specific boundary
condition for a single species, for example:
pdbSet Oxide_Silicon Boundary BoundaryCondition HomNeumann
pdbSet Oxide_Silicon Int BoundaryCondition Dirichlet
The first line switches the boundary condition for dopants at an oxide–silicon interface from
its default Segregation boundary condition to the HomNeumann boundary condition. The
second line sets the boundary condition at the oxide–silicon interface for interstitials to the
Dirichlet boundary condition.
HomNeumann
It is assumed that there are no fluxes and transfers across the interface. This is chosen by default
at the left, right, and bottom boundaries, and can be applied to any boundary.

4: Diffusion
Boundary Conditions
358 Sentaurus™ Process User Guide
I-2013.12
Natural
This is the default boundary condition for point defects at gas–silicon and oxide–silicon
interfaces. The normal flux across an outer surface is given by:
(619)
where is the surface recombination rate, and and are the concentration of interstitials
or vacancies and equilibrium concentration of interstitials and vacancies, respectively. The
equilibrium concentration of point defects at the surface can be modified using user-defined
parameters (see Modifying Point-Defect Equilibrium Values at Surface on page 361). There
are four surface recombination velocity models:
■PDependent
■InitGrowth
■Simple
■Normalized
To set the models, use:
pdbSet <interface material> <defect> Surf.Recomb.Vel <model>
where:
■<interface material> is an interface material name (see Material Specification on
page 52).
■<defect> is either Interstitial or Vacancy.
■<model> is one of the model names.
In each case, the surface recombination rate depends on the motion of the interface due to
oxidation.
Surface Recombination Model: PDependent
The PDependent model is the pressure-dependent surface recombination model. The flux that
takes into account the interstitial injection during oxidation is given by:
(620)
jn•hC C
*
–()=
h
C
C*
jn•ks1kRat
Vox
VScale
---------------
kpow
Po
kppow
+
CX0CX0
*
–()Gox
–=

4: Diffusion
Boundary Conditions
Sentaurus™ Process User Guide 359
I-2013.12
where:
■ is the surface recombination rate.
■ is the generation rate.
■ is the oxygen partial pressure.
■ is the local oxidation rate (ReactionSpeed).
■ is the reference oxidation rate for bare, undoped silicon.
■, , and are model parameters.
To modify these parameters, use:
pdbSet <interface material> <defect> Ksurf {<n>}
pdbSet <interface material> <defect> Scale {<n>}
pdbSet <interface material> <defect> Krat {<n>}
pdbSet <interface material> <defect> Kpow {<n>}
pdbSet <interface material> <defect> Kppow {<n>}
The generation term, , is given by:
(621)
, , and are model parameters to adjust the interstitial injection during oxidation.
To modify these parameters, use:
pdbSet <interface material> <defect> Gpow {<n>}
pdbSet <interface material> <defect> Ggpow {<n>}
pdbSet <interface material> <defect> theta {<n>}
is the lattice density of silicon and can be set by:
pdbSet <material> LatticeDensity {<n>}
is the scaling factor for the generation rate and given by:
(622)
(623)
(624)
ks
Gox
Po
Vox
VScale
kRat
kpow
kppow
Gox
Gox θVox Lden
Vox
VScale
---------------
Gpow
Po
GgpowGScale
=
Gpow
Ggpow
υ
Lden
GScale
GScale
G0
G1Glow
=Glow 0 and Charged Model =
Glow 0≠
G0
CXi
*
CX
---------=
G1mm m 1ppp++++
mm n
ni
----
2PotOx mn
ni
----
PotOx 1pp
ni
----
PotOx pp p
ni
----
2PotOx
++++
---------------------------------------------------------------------------------------------------------------------------------------------------------=

4: Diffusion
Boundary Conditions
360 Sentaurus™ Process User Guide
I-2013.12
If the dopant diffusion model is set to ChargedFermi or ChargedPair or ChargedReact
and is zero, will be set to ; otherwise, it will be set to .
, , , , , and are model parameters that can be modified with the
commands:
pdbSet <interface material> <defect> PotOx {<n>}
pdbSet <interface material> <defect> mm {<n>}
pdbSet <interface material> <defect> m {<n>}
pdbSet <interface material> <defect> p {<n>}
pdbSet <interface material> <defect> pp {<n>}
pdbSet <interface material> <defect> Glow {<n>}
Surface Recombination Model: InitGrowth
The InitGrowth model is almost identical to PDependent (see Surface Recombination
Model: PDependent on page 358) surface recombination model except that is set
to 1 for nonoxidizing cases in Eq. 620.
Surface Recombination Model: Simple
The Simple model takes into account the interstitial injection through total free and
equilibrium point-defect concentrations during oxidation. The recombination flux is given by:
(625)
(626)
is given by Eq. 622.
Surface Recombination Model: Normalized
The Normalized model is a TSUPREM-4-type surface recombination model. This model
provides both the time dependence and the dependence on the oxidation conditions by using a
constant normalizing factor:
(627)
(628)
Glow
GScale
G0
G1
Glow
PotOx
mm
m
p
pp
Vox VScale
⁄
jn•ks1kRat
Vox
VScale
---------------
+
CXCX
*
–()Gox
–=
Gox θVox LdenGScale
=
GScale
jn•ksksvel
Vox
VScale
---------------kpow
CXCX
*
–()+
Gox
–=
Gox θLden Vox
Vox
VScale
---------------
Gpow
GScale
=

4: Diffusion
Boundary Conditions
Sentaurus™ Process User Guide 361
I-2013.12
To set the normalizing factor, use:
pdbSet <interface material> <defect> Ksvel {<n>}
is given by Eq. 622.
Modifying Point-Defect Equilibrium Values at Surface
The equilibrium value of point defects at the interface can be enhanced as follows:
(629)
is given by Eq. 624.
, , and are model parameters that can be modified with the commands:
pdbSet <interface material> <defect> VrefRate {<n>}
pdbSet <interface material> <defect> Fox {<n>}
pdbSet <interface material> <defect> Pox {<n>}
To switch on the enhancement, use the command:
pdbSet <interface material> <defect> HybridBC {1 | 0}
Segregation
This is the default boundary condition for dopants. The total dopant fluxes at the interfaces are
balanced. The fluxes are assumed to be proportional to the deviation from the segregation
equilibrium. The fluxes are given by:
(630)
where:
■is the concentration of dopant on one side of the interface.
■ is the concentration of dopant on the other side of the interface.
■ is the transfer rate.
■ is the segregation rate of dopant .
GScale
CX Enhanced
*CX
*1FoxG1
Vox
Vref
-------------
Pox
+
=
G1
Vref
Fox
Pox
jn•hC
A
aCA
b
s
-------–
=
CA
a
CA
b
h
s
A

4: Diffusion
Boundary Conditions
362 Sentaurus™ Process User Guide
I-2013.12
If the charge states of the dopants must be included or the boundary condition for dopant defect
pairs must be set, use the command:
pdbSet <interface material> <dopant> Surf.Recomb.Model <diffmodel> <model>
where:
■<interface material> is an interface material name (see Material Specification on
page 52).
■<dopant> is a valid dopant name.
■<diffmodel> is the Constant or Fermi or ChargedFermi or Pair or ChargedPair
or React or ChargedReact dopant diffusion model.
■<model> is either Default or PairSegregation.
Surface Recombination Model: Default
If the surface recombination model is set to Default for any dopant diffusion model, the
segregation fluxes are given by:
(631)
To set these parameters, use the commands:
pdbSet <interface material> <dopant> Transfer {<n>}
pdbSet <interface material> <dopant> Segregation {<n>}
NOTE If the dopant diffusion model is set to React or ChargedReact,
will be the substitutional dopant.
Surface Recombination Model: PairSegregation
If the surface recombination model is set to PairSegregation for the Constant, Fermi,
ChargedFermi, Pair, or ChargedPair diffusion models, the segregation fluxes are given
by:
(632)
(633)
jn•kTransfer CA
aCA
b
kSegregation
-----------------------------–
=
CA
jn•kTransferkrate
akrate
bCA
an
ni
----
b
zCA
an
ni
----
a
z
kSegregation
-----------------------------–
=
krate
ab⁄fI
CI0
*
CI0
*
-------
ab⁄
1fI
–()
CV0
*
CV0
*
--------
ab⁄
+
=

4: Diffusion
Boundary Conditions
Sentaurus™ Process User Guide 363
I-2013.12
where:
■ is the interstitial fraction of dopant trapping in equilibrium.
■ and are the parameters ( ) to control intersititial and vacancy injections
respectively.
To set, use:
pdbSet <interface material> <dopant> Trap.Fi {<n>}
pdbSet <interface material> <dopant> <defect> Scale.PairSegregation_${side}
{<n>}
To use the total unpaired interstitial concentration, instead of the neutral one, use:
pdbSet <interface material> <dopant> UseUnpairedTotalInt { 1|0 }
In this case, Eq. 633 will be:
(634)
If the dopant diffusion model is not Pair or ChargedPair model on interface side , is
set to 1. If the same is true for interface side , is set to 1.
If the surface recombination model is set to PairSegregation for React or ChargedReact
diffusion models, the segregation fluxes are given by:
(635)
(636)
(637)
(638)
fI
γI
γV
0γIγV
,1≤≤
krate
ab⁄fI
CI
*
CI
*
------
ab⁄
1fI
–()
CV
*
CV
*
-------
ab⁄
+
=
a
krate
a
b
krate
b
jn•kTransferkAIrate
aCI0
*
[]
bCA
an
ni
----
a
zCAI
bn
ni
----
b
z
kSegregation kAIz
bkIz
bn
ni
----
b
z–
z
-------------------------------------------------------------------–
=
jn•kTransferkAVrate
aCV0
*
[]
bCA
an
ni
----
a
zCAV
bn
ni
----
b
z
kSegregation kAVz
bkVz
bn
ni
----
b
z–
z
----------------------------------------------------------------------–
=
kAIrate
afI
CI0
*
[]
b
---------------krate
a
=
kAVrate
a1f–I
CV0
*
[]
b
----------------krate
a
=

4: Diffusion
Boundary Conditions
364 Sentaurus™ Process User Guide
I-2013.12
where and are the concentration of dopant-defect pairs. is the concentration of
the total unpaired dopant.
To use the total unpaired interstitial and vacancy concentrations, Eq. 637 and Eq. 638 will be:
(639)
(640)
Dirichlet
The Dirichlet boundary condition can be used with both point defects and dopants. However,
it can be set only at gas and any other material interfaces for dopants. In this way, ‘in-diffusion’
for a dopant can be simulated using the Dirichlet boundary condition. If the Dirichlet boundary
condition is selected, the defect or dopant concentration at the boundary is set to its equilibrium
value. The equilibrium value can be specified with:
pdbSet <material> <dopant|defect> Cstar <n>
where:
■<material> is a material name (see).
■<dopant> is a valid dopant name.
■<defect> is Interstitial or Vacancy.
■Cstar is the equilibrium value of the solution variable.
If the Dirichlet boundary condition is selected and the oxidation is switched on, the modified
Dirichlet boundary condition is used for interstitials. The equilibrium value of interstitial point-
defects at the interface is enhanced (see Modifying Point-Defect Equilibrium Values at Surface
on page 361) and the new equilibrium at the interface becomes:
(641)
CAI
CAV
CA
kAIrate
afI
CI0
*
[]
b
---------------
kIc
n
ni
----
c–
c
ks
Ic
c
---------------------------- krate
a
=
kAVrate
a1f–I
CV0
*
[]
b
----------------
kVc
n
ni
----
c–
c
ks
Vc
c
------------------------------krate
a
=
CI0ox
*CI0
*1FoxG1
Vox
Vref
-------------
Pox
+
=

4: Diffusion
Boundary Conditions
Sentaurus™ Process User Guide 365
I-2013.12
ThreePhaseSegregation
Dose loss during diffusion can be modeled with three-phase segregation in Sentaurus Process.
Dopants can segregate from both silicon and oxide to the silicon–oxide interface where they
are considered inactive. The model in Sentaurus Process is based on the original model by Lau
et al.[40], and Oh and Ward [41]. The diffusion equation at the interface is given by:
(642)
where is the diffusivity of the dopant at the interface, and and are the flux towards
the interface from material and material , respectively.
The diffusivity at the interface can be defined by:
pdbSet <interface material> <dopant> D <c> {<n>}
where:
■<interface material> is an interface material name (see Material Specification on
page 52).
■<dopant> is one of the existing Sentaurus Process dopants.
■<c> is the charge state.
■<n> is a Tcl expression that returns a number; it can be simply a number.
NOTE Only a neutral charge state is considered at the interface.
The fluxes and depend on the surface recombination model used. The surface
recombination models are Default or PairSegregation, and can be set for different
diffusion models using the command:
pdbSet <interface material> <dopant> Surf.Recomb.Model <diffmodel> <model>
where:
■<diffmodel> is the Constant, Fermi, ChargedFermi, Pair, ChargedPair, React,
or ChargedReact dopant diffusion model.
■<model> is either Default or PairSegregation.
t∂
∂CAD0CA
∇∇ FaFb
++=
D0
Fa
Fb
a
b
Fa
Fb
4: Diffusion
Boundary Conditions
366 Sentaurus™ Process User Guide
I-2013.12
Surface Recombination Model: Default
If the surface recombination model is set to Default for any dopant diffusion model, the
segregation fluxes are given by:
(643)
where:
■ is the trapping rate.
■ is the maximum number of sites in the adjacent bulk regions.
■ is the concentration of trapped dopant .
■ is the solid solubility of the dopant.
■ is the active concentration of dopant .
■ is the charge state of the dopant.
■ is the emission rate.
To set the model parameters, use:
pdbSet <interface material> <dopant> TrappingRate_<side> {<n>}
pdbSet <interface material> <dopant> EmissionRate_<side> {<n>}
pdbSet <interface material> CMax {<n>}
where <side> is one side of the interface and <interface material> is the interface
material. For example, the side would be either Oxide or Silicon for an oxide–silicon
interface.
Sentaurus Process allows the parameter to be multiplied by user-defined factors as
follows:
(644)
For example, in the case of oxide silicon interface, this is given by:
term name=CMaxFactor add Oxide /Silicon eqn = "exp(0.02)"
To allow Sentaurus Process to use this term, specify a term with the name CMaxFactor for the
interface material.
Similarly, you can modify and (the trapping and emission rates, respectively) using
user-defined factors such as:
and (645)
Fab⁄TRate
ab⁄CA
Tmax
[]
ab⁄CAi
i
–
CA
+
[]
ab⁄n
ni
----
ab⁄
z
ERate
ab⁄CACA
ss
[]
ab⁄CA
+
[]
ab⁄
–()–=
TRate
CA
Tmax
CAi
Ai
CA
SS
CA
+
A
z
ERate
CA
Tmax
CAFactor
Tmax CA
Tmax
×
TRate
ERate
TRate Factor–TRate
×
ERate Factor–ERate
×
4: Diffusion
Boundary Conditions
Sentaurus™ Process User Guide 367
I-2013.12
The rate factors are specified as follows:
term name=<dopant>EmissionRateFactor_<side> add <InterfaceMaterial1> /
<InterfaceMaterial2> eqn="<expr>"
term name=<dopant>TrappingRateFactor_<side> add <InterfaceMaterial1> /
<InterfaceMaterial2> eqn="<expr>"
The factors are specified for an interface, for a particular dopant and specific to the side from
which the interface is being approached. For example, given an oxide–silicon interface and an
arsenic dopant, the EmissionRateFactor from the oxide side can be specified as:
term name=ArsenicEmissionRateFactor_Oxide add Oxide /Silicon eqn="exp(2.0)"
You also can use the individual trap density by switching off the UseTotalInterfaceTrap
flag by:
pdbSet <interface material> <dopant> UseTotalInterfaceTrap 0
By default, the flag is switched on (1). If the flag is switched off, Eq. 643 becomes:
(646)
where is the maximum number of sites in the adjacent bulk region for this solution
variable.
To change this parameter, use:
pdbSet <interface material> <dopant> CMax {<n>}
NOTE If the dopant diffusion model is set to React or ChargedReact,
will be the substitutional dopant.
Sentaurus Process allows the parameter to be multiplied by user-defined factors as
follows:
(647)
For example, in the case of specified boron at the oxide–silicon interface, this is given by:
term name=BoronCMaxFactor add Oxide /Silicon eqn = "exp(0.02)"
To allow Sentaurus Process to use this term, specify a term with the name
<dopant>CMaxFactor for the interface material.
Fab⁄TRate
ab⁄CA
max
[]
ab⁄CA
–()CA
+
[]
ab⁄n
ni
----
ab⁄
zERate
ab⁄CACA
ss
[]
ab⁄CA
+
[]
ab⁄
–()–=
CA
max
CA
+
CA
max
CAFactor
max CA
max
×
4: Diffusion
Boundary Conditions
368 Sentaurus™ Process User Guide
I-2013.12
Sentaurus Process also allows the parameter to be multiplied by a user-defined factor
defined as Side.SS.Factor, for example:
pdbSetString Si B Side.SS.Factor "exp(3.636e-24*Pressure_Silicon/$kbT)"
Surface Recombination Model: PairSegregation
If the Surface Recombination model is set to PairSegregation for the Pair or
ChargedPair dopant diffusion model, the segregation fluxes are given by:
(648)
To use the total unpaired interstitial concentration, instead of the neutral one, use the command:
pdbSet <interface material> <dopant> UseUnpairedTotalInt { 1|0 }
In this case, in Eq. 648 will be replaced with .
If the surface recombination model is set to PairSegregation for the React or
ChargedReact diffusion model, the segregation fluxes are given by:
(649)
where is the interstitial fraction of dopant trapping in equilibrium and can be set using the
command:
pdbSet <interface material> <dopant> Trap.Fi {<n>}
CA
SS
Fab⁄
CI0
CI0
*
-------
ab⁄
fITRate
ab⁄CV0
CV0
*
--------
ab⁄
1f–I
()TRate
ab⁄
+
×=
CA
Tmax
[]
ab⁄CAi
i
–
CA
+
[]
ab⁄n
ni
----
ab⁄
z
CACA
ss
[]–
ab⁄ERate
ab⁄
TRate
ab⁄
-------------
CX0
CX0
*
--------
ab⁄
CX
CX
*
------ ab⁄
Fab⁄
fITRate
ab⁄
CI0
*
[]
ab⁄
-------------------- CA
Tmax
[]
ab⁄CAi
i
–
CAI
[]
ab⁄n
ni
----
ab⁄
z
kAIz
ab⁄kIz
ab⁄n
ni
----
b
z–
z
-------------------------------------------- CACA
ss
[]
ab⁄
–ERate
ab⁄
TRate
ab⁄
------------- CI0
[]
ab⁄
×=
1f–I
()TRate
ab⁄
CV0
*
[]
ab⁄
------------------------------ CA
Tmax
[]
ab⁄CAi
i
–
CAV
[]
ab⁄n
ni
----
ab⁄
z
kAVz
ab⁄kVz
ab⁄n
ni
----
b
z–
z
-------------------------------------------- CACA
ss
[]
ab⁄
–ERate
ab⁄
TRate
ab⁄
------------- CV0
[]
ab⁄
fI

4: Diffusion
Boundary Conditions
Sentaurus™ Process User Guide 369
I-2013.12
To use the total unpaired interstitial and vacancy concentration, in Eq. 649 will be scaled
with:
(650)
All other parameters have the usual meaning as explained above.
If the individual trap density is switched off, will be replaced with
.
and are the concentrations of dopant-defect pairs. is the concentration of the total
unpaired dopant. If the surface recombination model is set to PairSegregation for the
Constant, Fermi, or ChargedFermi diffusion model, the Default model will be used.
Trap
The Trap boundary condition is used to trap species at the interface. This boundary condition
is a combination of the Segregation model (see Segregation on page 361) and the
ThreePhaseSegregation model (see ThreePhaseSegregation on page 365).
The model is used mainly to trap nitrogen and fluorine during oxidation to reduce the oxidation
rate (see Trap-dependent Oxidation on page 626).
TrapGen
The TrapGen boundary condition defines not only dopant trapping, but also dopant generation
depending on the reaction velocity at a boundary.
Continuous
For all of the same material interfaces (for example, Silicon_Silicon), by default,
continuous flux and solution boundary conditions apply:
(651)
(652)
fI
kXc
n
ni
----
c–
c
ks
Xc
c
------------------------------
CA
Tmax
[]
ab⁄CAi
i
–
CA
max
[]
ab⁄CA
–()
CAI
CAV
CA
C1xi→()C2ix←()=
D1'C1n1
∇D2C2n2
∇=

4: Diffusion
Periodic Boundary Condition
370 Sentaurus™ Process User Guide
I-2013.12
where:
■Indices 1 and 2 indicate the two sides of the interface .
■ indicates the component of the dopant gradient normal to the interface.
■ is the diffusivity, and is the concentration of the solution variable.
If the boundary condition is not specified using the callback procedures for the solution
variable at the interface, the continuous boundary condition can be set using the command:
pdbSetBoolean <mater> <dopant> Continuous 1
where <mater> is the interface material, and <dopant> is the solution variable name. For
example:
pdbSetBoolean PolySilicon_Silicon Potential Continuous 1
will set the potential solution and its fluxes continuous at the polysilicon–silicon interface if
Potential is solved on both sides of the interface.
Periodic Boundary Condition
The periodic boundary condition can be applied when a device structure has a repetitive
pattern:
pdbSet Diffuse <Left | Right | Front | Back> Periodic <0 | 1>
The Front and Back definitions apply to 3D structures only.
Boundary Conditions at Moving Interfaces
Enhanced and Retarded Diffusion
During the growth of materials (for example, oxide and silicide), the reaction speed is
calculated at the moving interfaces. The data field is called ReactionSpeed. The reaction
speed can be used to simulate the enhanced dopant diffusion (for example, oxidation-enhanced
diffusion (OED)) or the retarded dopant diffusion (for example, oxidation-retarded diffusion
(ORD)) by allowing for the injection of interstitials and vacancies.
In Eq. 620, p. 358, the injection rate is given as a function of the reaction speed
(=
ReactionSpeed) and is used to simulate OED effects.
i
n
D
C
Vox
4: Diffusion
Common Dopant and Defect Dataset Names
Sentaurus™ Process User Guide 371
I-2013.12
Conserving Dose
The mesh of the simulated structure is modified during the growth of materials with each
diffusion step. Some elements of the mesh will become bigger and some will shrink during this
process. The change in the element size from one diffusion step to another will artificially
change the dopant doses in the structure.
This artificial effect has two components. One is due to the change of element sizes and the
other is due to the material consumption at the moving boundaries. The first effect is accounted
for internally by applying an up-wind term to the solution equations. The second effect is
accounted for using the Alagator scripting language. If the total dopant concentration on one
side of the interface is different from the other side, the total concentration of the consumed
material is used.
For example, if you assume that there are no dopant clusters and the React diffusion model
for boron is selected on the silicon side and the Constant diffusion model is selected on the
oxide side of an oxide–silicon interface, the total boron concentration would be
Boron+BoronInt+BoronVac on the silicon side and Boron on the oxide side. Since the
consumed material is silicon during the oxidation, the dopant consumption due to growth is
passed using the command:
pdbSetString Oxide_Silicon Boron Consumed_Silicon \
"Boron_Silicon+BoronInt_Silicon+BoronVac_Silicon"
This is performed automatically. If the React diffusion model for boron was also selected on
the oxide side, the total dopant concentration on the oxide side would be
Boron+BoronInt+BoronVac, and the following command would be used:
pdbSetString Oxide_Silicon Boron Consumed_Silicon "Boron_Silicon"
Common Dopant and Defect Dataset Names
Sentaurus Process does not solve the diffusion equations for the total dopant or defect
concentrations, but solves the equations for the total unpaired dopant and defect
concentrations. Sentaurus Process monitors the total dopant and defect concentration through
various terms. Depending on the diffusion models selected, Sentaurus Process will update
these terms. For example, the commands:
pdbSet Silicon Dopant DiffModel React
pdbSet Silicon B ActiveModel None
will set the dopant diffusion model in silicon to React and the active model to None for boron
in silicon. Assuming that there is only boron in the structure, various terms and data fields will
4: Diffusion
Common Dopant and Defect Dataset Names
372 Sentaurus™ Process User Guide
I-2013.12
be created after the diffusion. The most important ones are Boron, BActive, BTotal,
BoronInt, BoronVac, Int, ITotal, Interstitial, Vac, VacTotal, and Vacancy.
Boron, BoronInt, BoronVac, Int, and Vac are the solution names:
If the cluster models for both interstitial and boron are switched on, as follows:
pdbSet Si Dopant DiffModel React
pdbSet Si B ActiveModel Transient
pdbSet Si I ClusterModel 1Moment
some of the previous fields will be updated. Two additional solution variables, ICluster and
B4, will be solved. ICluster is the clustered interstitials used with the 1Moment model (see
Defect Cluster Model: 1Moment on page 337), and B4 is the clustered boron used with the
Transient model (see Dopant Active Model: Transient on page 296).
The changed fields will be:
BActive
Active boron concentration. (Since the active model is none, it will be equal
to Boron).
Boron
Total unpaired boron concentration (for example, no clusters, no boron–
defect pairs).
BoronInt
Concentration of boron–interstitial pairs.
BoronVac
Concentration of boron–vacancy pairs.
BTotal
Boron + BoronInt + BoronVac = Total boron concentration.
Int
Total unpaired interstitial concentration.
Interstitial
Total interstitial concentration used to initialize Int. (In this example, it will
be Int.)
ITotal
Int + BoronInt = Total interstitial concentration.
Vac
Total unpaired vacancy concentration.
Vacancy
Total vacancy concentration used to initialize Vac. (In this example, it will
be Vac.)
VTotal
Vac + BoronVac = Total vacancy concentration.
BTotal
Boron + 4*B4 + BoronInt + BoronVac = Total boron concentration.
ITotal
Int + BoronInt + ICluster = Total interstitial concentration.
4: Diffusion
Common Dopant and Defect Dataset Names
Sentaurus™ Process User Guide 373
I-2013.12
If the diffusion and cluster models are changed as follows:
pdbSet Si Dopant DiffModel ChargedPair
pdbSet Si B ActiveModel Transient
pdbSet Si I ClusterModel Equilibrium
Sentaurus Process uses the ChargedPair diffusion model (see ChargedPair Diffusion Model
on page 214) and the Transient active model (see Dopant Active Model: Transient on
page 296) for boron, and the Equilibrium cluster model (see Defect Cluster Model:
Equilibrium on page 320) for interstitials.
In this case, Boron, B4, Int, and Vac will be the solution names. BoronInt and BoronVac
will not be solved, but there will be BoronInt and BoronVac terms to calculate boron–
interstitial and boron–vacancy concentrations. In this case, total unpaired Boron also will
include BoronInt and BoronVac since they are not solved. The important fields are:
NOTE If the dopant, defect, or cluster fields are modified by other process steps
(for example, implant, deposition, and so on), terms that define active
dopant concentration and total dopant concentration may not be current.
They be updated with the next diffusion step.
BActive
Boron – BoronInt – BoronVac = Active boron concentration.
Boron
Total unpaired boron concentration.
BoronInt
Concentration of boron–interstitial pairs calculated using Eq. 168, p. 215.
BoronVac
Concentration of boron–vacancy pairs calculated using Eq. 168.
BTotal
Boron + 4*B4 = Total boron concentration.
ICluster
Equilibrium interstitial concentration calculated using Eq. 470, p. 320.
Int
Total unpaired interstitial concentration.
Interstitial
Int + 4*ICluster + BoronInt = Total interstitial concentration used
to initialize Int.
ITotal
Int + 4*ICluster + BoronInt = Total interstitial concentration.
Vac
Total unpaired vacancy concentration.
Vacancy
Vac + BoronVac = Total vacancy concentration used to initialize Vac.
VTotal
Vac + BoronVac = Total vacancy concentration.

4: Diffusion
Common Dopant and Defect Dataset Names
374 Sentaurus™ Process User Guide
I-2013.12
Table 43 Variable names used in diffusion and reaction solvers
Name Comment Solution (S), Term (T), Data field (F)
Antimony
Total unpaired antimony concentration S, F
AntimonyGbc
Antimony grain boundary concentration S, F
AntimonyInit
Antimony–interstitial pair concentration T or (S, F) (depends on the model)
AntimonyVac
Antimony–vacancy pair concentration T or (S, F) (depends on the model)
Arsenic
Total unpaired arsenic concentration S, F
ArsenicGbc
Arsenic grain boundary concentration S, F
ArsenicInt
Arsenic–interstitial pair concentration T or (S, F) (depends on the model)
ArsenicVac
Arsenic–vacancy pair concentration T or (S, F) (depends on the model)
As3
Three-arsenic cluster concentration (default size
is three, user-configurable)
S, F
As4Vac
Four-arsenic and a vacancy cluster concentration S, F
AsActive
Arsenic active concentration T
AsTotal
Total arsenic concentration T (for example,
Arsenic+4*As4Vac)
B2
Two-boron cluster concentration S, F
B2I
Two-boron and interstitial cluster concentration S, F
B2I2
Two-boron and two-interstitial cluster
concentration
S, F
B2I3
Two-boron and three-interstitial cluster
concentration
S, F
B3
Three-boron cluster concentration S, F
B3I
Three-boron and interstitial cluster concentration S, F
B3I2
Three-boron and two-interstitial cluster
concentration
S, F
B3I3
Three-boron and three–interstitial cluster
concentration
S, F
B3I4
Three-boron and four-interstitial cluster
concentration
S, F
B4
Four-boron cluster concentration (default size is
four, user-configurable)
S, F
BActive
Boron active concentration T
BI2
Boron and two-interstitial cluster concentration S, F
4: Diffusion
Common Dopant and Defect Dataset Names
Sentaurus™ Process User Guide 375
I-2013.12
Boron
Total unpaired boron concentration S, F
BoronGbc
Boron grain boundary concentration S, F
BoronInt
Boron–interstitial pair concentration T or (S, F) (depends on the model)
BoronVac
Boron–vacancy pair concentration T or (S, F) (depends on the model)
BTotal
Total boron concentration T (for example, Boron+2*B2I)
C2
Two-carbon cluster concentration S, F
C2I
Two-carbon and interstitial cluster concentration S, F
C311
Concentration of interstitials trapped in {311}
defects
S, F
C3I
Three-carbon and interstitial cluster
concentration
S, F
C3I2
Three-carbon and two-interstitial cluster
concentration
S, F
C4I2
Four-carbon and two-interstitial cluster
concentration
S, F
C4I3
Four-carbon and three-interstitial cluster
concentration
S, F
C5I3
Five-carbon and three-interstitial cluster
concentration
S, F
C5I4
Five-carbon and four-interstitial cluster
concentration
S, F
C6I5
Six-carbon and five-interstitial cluster
concentration
S, F
C6I6
Six-carbon and six-interstitial cluster
concentration
S, F
Carbon
Total unpaired carbon concentration S, F
CarbonInt
Carbon–interstitial pair concentration S, F
CLoop
Concentration of interstitials trapped in
dislocation loops
T or (S, F) (depends on the model)
CTotal
Total carbon concentration T
D311
Density of {311} defects S, F
DLoop
Density of dislocation loops T or (S, F) (depends on the model)
EqInt
Equilibrium interstitial concentration T
Table 43 Variable names used in diffusion and reaction solvers
Name Comment Solution (S), Term (T), Data field (F)

4: Diffusion
Common Dopant and Defect Dataset Names
376 Sentaurus™ Process User Guide
I-2013.12
EqVac
Equilibrium vacancy concentration T
F3V
Three-fluorine and vacancy cluster concentration S, F
Fluorine
Total unpaired fluorine concentration S, F
FluorineTotal
Total fluorine concentration T
GeB
Germanium–boron pair concentration S, F
Germanium
Total unpaired germanium concentration S,F
GermaniumTotal
Total germanium concentration T
H2O
Wet oxidant concentration S, F
I2
Two-interstitial cluster concentration S,F
ICluster
Interstitial cluster concentration T or (S, F) (depends on the model)
In3
Three-indium cluster concentration (default size
is three, user-configurable)
S, F
InActive
Indium active concentration T
Indium
Total unpaired indium concentration S, F
IndiumGbc
Indium grain boundary concentration S, F
IndiumInt
Indium–interstitial pair concentration T or (S, F) (depends on the model)
IndiumVac
Indium–vacancy pair concentration T or (S, F) (depends on the model)
Int
Total unpaired interstitial concentration S, F
Interstitial
Total interstitial concentration excluding cluster
solutions, used to initialize Int
F
IntNeutral
Neutral interstitial concentration (I0)T
InTotal
Total indium concentration T
IntTotal
Total interstitial concentration T
O2
Dry oxidant concentration S, F
P3
Three-phosphorus cluster concentration (default
size is three, user-configurable)
S, F
PActive
Phosphorus active concentration T
Phosphorus
Total unpaired phosphorus concentration S, F
PhosphorusGbc
Phosphorus grain boundary concentration S, F
PhosphorusInt
Phosphorus–interstitial pair concentration T or (S, F) (depends on the model)
Table 43 Variable names used in diffusion and reaction solvers
Name Comment Solution (S), Term (T), Data field (F)

4: Diffusion
References
Sentaurus™ Process User Guide 377
I-2013.12
References
[1] B. Colombeau and N. E. B. Cowern, “Modelling of the chemical-pump effect and C
clustering,” Semiconductor Science and Technology, vol. 19, no. 12, pp. 1339–1342,
2004.
[2] R. B. Fair and P. N. Pappas, “Diffusion of Ion-Implanted B in High Concentration P-
and As-Doped Silicon,” Journal of the Electrochemical Society, vol. 122, no. 9,
pp. 1241–1244, 1975.
[3] N. E. B. Cowern and D. J. Godfrey, “A Model for Coupled Dopant Diffusion in Silicon,”
in Fundamental Research on the Numerical Modelling of Semiconductor Devices and
Processes: Papers from NUMOS I, the First International Workshop on the Numerical
Modelling of Semiconductors, pp. 59–63, Dublin, Ireland: Boole Press, 1987.
[4] F. Wittel and S. T. Dunham, “Diffusion of phosphorus in arsenic and boron doped
silicon,” Applied Physics Letters, vol. 66, no. 11, pp. 1415–1417, 1995.
PhosphorusVac
Phosphorus–vacancy pair concentration T or (S, F) (depends on the model)
Potential
Electrostatic potential T or (S, F) (depends on the model)
PTotal
Total phosphorus concentration T
Sb3
Three-antimony cluster concentration (default
size is three, user-configurable)
S, F
SbActive
Antimony active concentration T
SbTotal
Total antimony concentration T
(for example,
Antimony+AntimonyVac)
Smic
Concentration of submicroscopic interstitial
clusters (default size 4)
S, F
SmicS
Concentration of smaller submicroscopic
interstitial clusters (default size 2)
S, F
V2
Two-vacancy cluster concentration S, F
Vac
Total unpaired vacancy concentration S, F
Vacancy
Total vacancy concentration excluding cluster
solutions, used to initialize Vac
F
VacNeutral
Neutral vacancy concentration (V0)T
VacTotal
Total vacancy concentration T
Table 43 Variable names used in diffusion and reaction solvers
Name Comment Solution (S), Term (T), Data field (F)
4: Diffusion
References
378 Sentaurus™ Process User Guide
I-2013.12
[5] A. Mittiga, L. Fornarini, and R. Carluccio, “Numerical modeling of laser induced phase
transitions in silicon,” Applied Surface Science, vol. 154–155, pp. 112–117, February
2000.
[6] S. K. Jones and A. Gérodolle, “2D Process Simulation of Dopant Diffusion in
Polysilicon,” COMPEL, vol. 10, no. 4, pp. 401–410, 1991.
[7] A. Gérodolle and S. K. Jones, “Integration in the 2D Multi-layer Simulator TITAN of
an Advanced Model for Dopant Diffusion in Polysilicon,” in Simulation of
Semiconductor Devices and Process (SISDEP), Zurich, Switzerland, vol. 4, pp. 381–
387, September 1991.
[8] S. K. Jones et al., “Complete Bipolar Simulation Using STORM,” in IEDM Technical
Digest, San Francisco, CA, USA, pp. 931–934, December 1992.
[9] A. G. O’Neill et al., “A new model for the diffusion of arsenic in polycrystalline
silicon,” Journal of Applied Physics, vol. 64, no. 1, pp. 167–174, 1988.
[10] M. M. Mandurah et al., “Dopant segregation in polysilicon silicon,” Journal of Applied
Physics, vol. 51, no. 11, pp. 5755–5763, 1980.
[11] J. A. Venables, Introduction to Surface and Thin Film Processes, Cambridge University
Press, 2000.
[12] L. Mei and R. W. Dutton. “A Process Simulation Model for Multilayer Structures
Involving Polycrystalline Silicon,” IEEE Transactions on Electron Devices,
vol. ED-29, no. 11, pp. 1726–1734, 1982.
[13] L. Mei et al., “Grain-Growth Mechanisms in Polysilicon,” Journal of the
Electrochemical Society, vol. 129, no. 8, pp. 1791–1795, 1982.
[14] C. V. Thompson, “Secondary grain growth in thin films of semiconductors: Theoretical
aspects,” Journal of Applied Physics, vol. 58, no. 2, pp. 763–772, 1985.
[15] D. Gupta, D. R. Campbell, and P. S. Ho, “Grain Boundary Diffusion,” in Thin Films–
Interdiffusion and Reactions, New York: John Wiley & Sons, pp. 161–242, 1978.
[16] C. Hill and S. K. Jones, “Modelling Diffusion in and from Polysilicon Layers,” in MRS
Proceedings, Polysilicon Thin Films and Interfaces, San Francisco, CA, USA, no. 182,
pp. 129–140, April 1990.
[17] S. A. Ajuria and R. Reif, “Early stage evolution kinetics of the polysilicon/single-crystal
silicon interfacial oxide upon annealing,” Journal of Applied Physics, vol. 69, no. 2,
pp. 662–667, 1991.
[18] J. D. Williams, Epitaxial Alignment of Polycrystalline Silicon and Its Implications for
Analogue Bipolar Circuits, Ph.D. thesis, University of Southampton, UK, 1992.
[19] F. Benyaïch et al., “Kinetic and structural study of the epitaxial realignment of
polycrystalline Si films,” Journal of Applied Physics, vol. 71, no. 2, pp. 638–647, 1992.
[20] D. Dutartre et al., “Excitonic photoluminescence from Si-capped strained Si1-xGex
layers,” Physical Review B, vol. 44, no. 20, pp. 11525–11527, 1991.
4: Diffusion
References
Sentaurus™ Process User Guide 379
I-2013.12
[21] R. Braunstein, A. R. Moore, and F. Herman, “Intrinsic Optical Absorption in
Germanium-Silicon Alloys,” Physical Review, vol. 109, no. 3, pp. 695–710, 1958.
[22] A. Pakfar, “Dopant diffusion in SiGe: modeling stress and Ge chemical effects,”
Materials Science and Engineering, vol. B89, pp. 225–228, February 2002.
[23] M. J. Aziz, “Thermodynamics of diffusion under pressure and stress: Relation to point
defect mechanisms,” Applied Physics Letters, vol. 70, no. 21, pp. 2810–2812, 1997.
[24] R. F. Lever, J. M. Bonar, and A. F. W. Willoughby, “Boron diffusion across silicon–
silicon germanium boundaries,” Journal of Applied Physics, vol. 83, no. 4, pp. 1988–
1994, 1998.
[25] G. E. Pikus and G. L. Bir, “Effect of Deformation on the Hole Energy Spectrum of
Germanium and Silicon,” Fizika Tverdogo Tela, vol. 1, no. 11, pp. 1642–1658, 1959.
[26] S. T. Dunham and C. D. Wu, “Atomistic models of vacancy-mediated diffusion in
silicon,” Journal of Applied Physics, vol. 78, no. 4, pp. 2362–2366, 1995.
[27] S. Chakravarthi et al., “Modeling the Effect of Source/Drain Sidewall Spacer Process on
Boron Ultra Shallow Junctions,” in International Conference on Simulation of
Semiconductor Processes and Devices (SISPAD), Boston, MA, USA, pp. 159–162,
September 2003.
[28] P. Fastenko, Modeling and Simulation of Arsenic Activation and Diffusion in Silicon,
Ph.D. thesis, University of Washington, Seattle, WA, USA, 2002.
[29] S. Chakravarthi et al., “Modeling of Diffusion and Activation of Low Energy Arsenic
Implants in Silicon,” in MRS Spring Meeting Proceedings, Symposium C, vol. 717, San
Francisco, CA, USA, pp. C3.7.1–C3.7.6, April 2002.
[30] M. Diebel et al., “Investigation and Modeling of Fluorine Co-Implantation Effects on
Dopant Redistribution,” in MRS 2003 Spring Meeting Proceedings, Symposium D,
vol. 765, San Francisco, CA, USA, p. D6.15, April 2003.
[31] E. M. Bazizi et al., “Modelling of Boron Trapping at End-of-Range defects in pre-
amorphized ultra-shallow junctions,” Materials Science and Engineering B, vol. 154–
155, pp. 275–278, December 2008.
[32] N. Zographos, C. Zechner, and I. Avci, “Efficient TCAD Model for the Evolution of
Interstitial Clusters, {311} Defects, and Dislocation Loops in Silicon,” in MRS
Symposium Proceedings, Semiconductor Defect Engineering—Materials, Synthetic
Structures and Devices II, vol. 994, San Francisco, CA, USA, p. 0994–F10–01, April
2007.
[33] M. E. Law and K. S. Jones, “A New Model for {311} Defects Based on In-Situ
Measurements,” in IEDM Technical Digest, San Francisco, CA, USA, pp. 511–514,
December 2000.
[34] I. Avci et al., “Modeling extended defect ({311} and dislocation) nucleation and
evolution in silicon,” Journal of Applied Physics, vol. 95, no. 5, pp. 2452–2460, 2004.
4: Diffusion
References
380 Sentaurus™ Process User Guide
I-2013.12
[35] R. Y. S. Huang and R. W. Dutton, “Experimental investigation and modeling of the role
of extended defects during thermal oxidation,” Journal of Applied Physics, vol. 74,
no. 9, pp. 5821–5827, 1993.
[36] C. J. Ortiz et al., “Modeling of extrinsic extended defect evolution in ion-implanted
silicon upon thermal annealing,” Materials Science and Engineering B, vol. 114–115,
pp. 184–192, December 2004.
[37] C. J. Ortiz et al., “A physically based model for the spatial and temporal evolution of
self-interstitial agglomerates in ion-implanted silicon,” Journal of Applied Physics,
vol. 96, no. 9, pp. 4866–4877, 2004.
[38] E. Lampin et al., “Prediction of boron transient enhanced diffusion through the atom-by-
atom modeling of extended defects,” Journal of Applied Physics, vol. 94, no. 12,
pp. 7520–7525, 2003.
[39] E. Lampin et al., “Combined master and Fokker–Planck equations for the modeling of
the kinetics of extended defects in Si,” Solid-State Electronics, vol. 49, no. 7, pp. 1168–
1171, 2005.
[40] F. Lau et al., “A Model for Phosphorus Segregation at the Silicon–Silicon Dioxide
Interface,” Applied Physics A, vol. 49, pp. 671–675, 1989.
[41] Y.-S. Oh and D. E. Ward “A Calibrated Model for Trapping of Implanted Dopants at
Material Interface During Thermal Annealing,” in IEDM Technical Digest, San
Francisco, CA, USA, pp. 509–512, December 1998.

Sentaurus™ Process User Guide 381
I-2013.12
CHAPTER 5 Atomistic Kinetic Monte Carlo
Diffusion
This chapter describes an alternative, atomistic simulation approach
to the diffusion and activation processes in Sentaurus Process. It
also provides alternatives for faceted solid phase epitaxial regrowth
and epitaxial deposition. For a continuum approach, see Chapter 4.
All diffusion models previously described are based on the conventional (continuum)
simulation approach. The atomistic approach described in this chapter is based partially on the
kinetic Monte Carlo (KMC) diffusion simulator DADOS [1][2][3], and is available with the
optional Sentaurus Process Kinetic Monte Carlo license.
Overview
The continuum approach to modeling dopant diffusion in process simulation tools is used to
solve a system of partial differential equations (PDEs) that describe transport of the dopants
and conservation of the dose. This approach has proven to be useful in designing
semiconductor devices in the past, but several trends in the manufacturing process of sub-100-
nm devices may make it difficult to maintain a high predictability in future devices.
The shrinking thermal budget significantly reduces diffusion and, therefore, reduces the need
to accurately model diffusion. On the other hand, dopant activation phenomena, including the
formation of a variety of dopant-defect pairs and extended defects of different configurations,
often do not reach thermodynamic equilibrium and necessitate transient rather than
equilibrium simulation. In the continuum diffusion model, this requires the use of one equation
per each dopant-defect configuration, which leads to a large number of equations to be solved.
The trend of reducing device sizes results in a small number of impurity atoms (as small as tens
or hundreds) that determine the threshold voltage of a transistor. It is likely that a limit soon
will be reached where small discretized distribution can no longer be accurately modeled with
a continuum description.
A Monte Carlo (MC)–based diffusion simulation provides a valuable alternative to the
continuum approach. Computational resources required for the MC diffusion simulation are
decreasing with device dimension because they are proportional to the number of dopants and
defects in the device. On the other hand, resources for continuum simulations increase as
modeling of ever more complex nonequilibrium phenomena are required. This trend has
5: Atomistic Kinetic Monte Carlo Diffusion
KMC Method
382 Sentaurus™ Process User Guide
I-2013.12
already gone a long way towards making the KMC diffusion method competitive with the most
detailed continuum diffusion methods today in terms of the required computational resources.
Unlike the continuum approach, the large number of different dopant-defect configurations
does not present a problem for the MC approach, which simply needs to introduce the
probabilities for the additional reactions. These probabilities are calculated based on the
binding energies that can be plugged in directly from experiments, molecular dynamics, or ab
initio calculations.
Besides, atomistic implantation and diffusion models provide a natural way of determining
statistical variations for a specific process flow/device geometry combination.
KMC Method
Sentaurus Process Kinetic Monte Carlo (Sentaurus Process KMC) considers only defects and
impurities, and ignores the lattice, (except for some SPER and epitaxial deposition models).
This drastically reduces memory requirements compared to molecular dynamics or lattice
KMC techniques and allows you to investigate simulation domains that are large enough to
contain deep-submicron devices. As Sentaurus Process KMC tracks the diffusion and
interaction of defects, the fastest process is the jumping of a point defect with a period of
approximately s. When there are no mobile point defects in the structure, the time step is
increased automatically to an emission of mobile particles from the surface or from an
extended defect, which has a period of approximately s.
Therefore, typically, Sentaurus Process KMC would begin with time steps of approximately
s. As the point defects are trapped by the clusters and extended defects, Sentaurus
Process KMC switches automatically to the larger time steps of the order of s that are
large enough to model typical process steps.
Operating Modes
Sentaurus Process KMC can operate in two different modes:
■The atomistic mode handles data atomistically throughout the simulation and is expected
to be the most accurate method.
■The nonatomistic mode allows Sentaurus Process KMC to be used for only part of a
simulation. Sentaurus Process KMC transfers data back and forth to the continuum solver
to allow you to take advantage of the efficiency of the continuum solver for steps closer to
equilibrium and to allow Sentaurus Process KMC to handle one or more steps that are far
from equilibrium (such as fast RTA/flash annealing) to take advantage of its accuracy.
10 9–
10 3–
10 9–
10 3–

5: Atomistic Kinetic Monte Carlo Diffusion
Operating Modes
Sentaurus™ Process User Guide 383
I-2013.12
Atomistic Mode
When Sentaurus Process KMC is in atomistic mode, the data fields are handled completely
atomistically. To select the atomistic mode at the very beginning of the simulation, use:
SetAtomistic
The SetAtomistic command sets the parameter AtomisticData to true, allowing the
Sentaurus Process commands diffuse, deposit, etch, implant, init, line, photo,
profile, region, select, strip, and struct to work in the atomistic mode when
possible, and to properly synchronize Sentaurus Process KMC when the structure changes.
SetAtomistic also sets the diffuse method as Sentaurus Process KMC, and the implant
mode as MC implantation. Finally, it calls PDE2KMC to atomize the available continuum fields
into atomistic ones. The atomistic mode can be finished using:
UnsetAtomistic
The UnsetAtomistic command calls the procedure KMC2PDE to translate the atomistic
quantities to fields and finishes the atomistic mode.
When AtomisticData is true, Sentaurus Process KMC does not populate continuum data
fields with its own results, unless instructed to do so (using kmc deatomize).
Restrictions
The following restrictions apply when using the atomistic mode:
■Do not use the command transform stretch.
■The load command only accepts the options tdr and replace.
The other commands work as expected, although the ones listed in Table 44 have been
especially adapted to operate in this mode.
Table 44 Commands adapted to work in atomistic mode
Command Extension
deposit
Synchronizes the Sentaurus Process KMC structure after deposition.
diffuse
If the parameter kmc is specified or Diffuse KMC is set to 1 in the parameter database,
diffuse calls Sentaurus Process KMC. If AtomisticData is not set to 1, a new
KMC object is created, and it will be removed at the end of the diffusion step. Oxidation,
nitridation, epitaxial deposition, and so on are accepted.
etch
Synchronizes the Sentaurus Process KMC structure removing the etched material, its
contained particles, and setting the material to gas.

5: Atomistic Kinetic Monte Carlo Diffusion
Operating Modes
384 Sentaurus™ Process User Guide
I-2013.12
Implant
Sentaurus Process KMC requires the damage morphology (coordinates of each point defect)
for its damage accumulation model; this information is not available in analytic implantations.
In atomistic mode, implant automatically uses the Sentaurus MC model as well as the
cascades option for storing full cascades. The implantation time also is needed because while
implanting, Sentaurus Process KMC automatically performs diffusion at the specified
temperature (default is ambient). Typically, the temperature and time during the implantation
affect only slightly the distribution of dopants, but they may affect damage accumulation,
amorphization, and subsequent recrystallization and impurity cluster formation. The
implantation time is returned by the function DoseRate defined in the file Implant.tcl.
This time is computed by default using a fixed dose rate equal to /cm2s. If a dose rate
is specified in an implant command by the dose.rate argument, it is used to compute the
implantation time for this particular implantation instead.
implant
Works in MC mode and sends the cascades directly to Sentaurus Process KMC. Dynamic
annealing also is simulated during the implant using Sentaurus Process KMC.
init
If a background concentration is specified, it is atomized and passed as particles to Sentaurus
Process KMC.
line
Adds a new line to the Sentaurus Process KMC internal mesh, when possible.
load
Loads a Sentaurus Process KMC distribution from a TDR file and replaces the current one.
math
Accepts numThreadsKMC.
photo
Creates photoresist mask and synchronizes the new Sentaurus Process KMC structure.
profile
Loads and atomizes a profile.
region
When region changes the material, Sentaurus Process KMC is synchronized.
select
(Only when select creates a new field or modifies an existing one). If this field is known
by Sentaurus Process KMC, the Sentaurus Process KMC concentration of particles is
synchronized with the value of the field, removing or creating extra particles.
strip
Sentaurus Process KMC is synchronized with the new material. If there are particles in the
stripped materials, they are removed.
struct
Automatically deatomizes some Sentaurus Process KMC data fields to make them accessible
when saving to a file. It also saves Sentaurus Process KMC restart information.
transform
The Sentaurus Process KMC structure is updated after the transformation. Particles are
removed or modified depending on the particular materials being created or removed. The
option stretch is not allowed.
Table 44 Commands adapted to work in atomistic mode
Command Extension
112
×10
5: Atomistic Kinetic Monte Carlo Diffusion
Operating Modes
Sentaurus™ Process User Guide 385
I-2013.12
For each implant dose and surface size, the number of implanted ions is computed. For
example, boron dose in a simulation cell with a surface of and
250-nm depth implants boron cascades (being a cascade
one ion and all its generated damage). The information is passed to Sentaurus Process KMC
for annealing. These diffusion steps occur internally and are not user-specified, but the total
“diffused” time can be controlled by the function DoseRate explained above. Afterwards, the
implant report issued by Sentaurus Process KMC names the backscattered particles as outside
particles.
Molecular implants are allowed. To perform a molecular implant, specify the name of the
molecule as the implant species (see Implant on page 384). The components of implanted
molecules are introduced as isolated species in Sentaurus Process KMC; in other words, an
implanted BF2 molecule will split into 2F and 1B inside Sentaurus Process KMC.
Diffuse
For the first diffuse after the implant, the use of a small temperature ramp-up is recommended.
The time for this ramp-up should be chosen as realistically as possible. At the end of an
implant, the simulation cell contains the implanted ions plus a large amount of damage (point
defects). During the ramp-up, this damage recombines and forms different types of clusters.
The use of a realistic ramp-up produces more accurate results.
You ca n set t he pdb parameters automaticRampUp and automaticRampDown to true to
automatically perform ramps whenever the requested diffuse temperature is different from the
current one. These ramps are performed with a ramp rate specified by rampUpRate and
rampDownRate in C/s:
sprocess> pdbGet KMC rampUpRate
100
sprocess> pdbGet KMC automaticRampUp
0
Sentaurus Process KMC performs different annealings at different temperatures during the
ramp-up. The objective is to perform few large annealings at low temperature and short ones
at high temperatures. This maintains a high accuracy without spending too much time at low
temperatures (changes in the temperature have a performance penalty).
The way these ramp-ups are performed can be configured using the following parameters of
the parameter database:
sprocess> pdbGet KMC dTBase
2.0
sprocess> pdbGet KMC nInit
1
sprocess> pdbGet KMC dTLimit
20.0
110
14
× cm 2–
40 40× nm2
40 10 7–
×40×10 7–
×1014
×1600=

5: Atomistic Kinetic Monte Carlo Diffusion
Operating Modes
386 Sentaurus™ Process User Guide
I-2013.12
The temperature steps for the highest temperature (the end of the ramp-up or the beginning of
the ramp-down) are computed as:
(653)
where is dTBase and is nInit.
For example, using the above parameters, in a ramp-down from to , the first
annealing is performed at – , the second one is performed at
, and so on. When is greater than dTLimit, the value
dTLimit is taken.
NOTE The above parameters are used only for temperature ramps induced by
automaticRampUp or automaticRampDown. For temperature ramps
specified with the command temp_ramp, the parameters to control the
ramp are specified in the commands temp_ramp or diffuse.
Oxidation options are allowed in Sentaurus Process KMC. For more information, see
Oxidation-enhanced Diffusion (OED) Model on page 512 and Oxidation on page 518.
Nonatomistic Mode
Sentaurus Process KMC also can be used only for one diffusion step, synchronizing the status
of the simulation before and after the diffuse step. This is performed with the kmc parameter
in the diffuse command as follows:
diffuse kmc temperature=<n> time=<n>
When Sentaurus Process KMC runs with AtomisticData set to false, a new Sentaurus
Process KMC simulation is launched at the beginning of the diffuse command:
■First, it receives the information (atomized from the data fields).
■Second, the diffusion is completed and, at the end of this command, Sentaurus
Process KMC transfers the information to Sentaurus Process as data fields.
■Third, the Sentaurus Process KMC information is removed from memory.
This is similar to the following commands:
SetAtomistic
diffuse temperature=<n> time=<n>
UnsetAtomistic
∆Tbase
()
ninit n+
∆Tbase
ninit
600°C
500°C
600°C
210+598°C=
598°C2
11+
–594°C=
∆Tbase
()
ninit n+

5: Atomistic Kinetic Monte Carlo Diffusion
Operating Modes
Sentaurus™ Process User Guide 387
I-2013.12
Atomistic/Nonatomistic Translation
These transformations of information back and forth from concentrations to particles are
performed by the Tcl procedures PDE2KMC and KMC2PDE. These transformations may degrade
the accuracy of the obtained results. By default, the transformation to continuum data is
mapped to the ChargedReact (five-stream) model.
If you need to add a customize transformation, you can rewrite the procedures PDE2KMCUser
and KMC2PDEUser with your own map. PDE2KMCUser (KMC2PDEUser) returns a string
mapping the array of transformation from continuum to atomistic (atomistic to continuum).
KMC2PDEUser cannot modify already existing fields, but it adds new ones. These maps contain
three columns: the name of the original field, the name of the translated field, and the factor to
be applied during the translation. For example, the following will transfer a new helium field
into Sentaurus Process KMC:
fproc PDE2KMCUser {} {
return "Helium He 1 \
HeInt Hei 1 \
HeVac HeV 1"
}
PDE2KMC uses the PDB parameter KMC Si Damage TrimField to trim the PDE fields
exceeding this maximum value. This is useful to trim the concentration of Is and Vs in
amorphized regions to more realistic values, avoiding the wasteful creation of excessive point
defects. A value being at least 20% higher than the Sentaurus Process KMC amorphization
threshold is suggested to properly amorphize the material.
In addition to the standard translations, there are special built-in translations for total
concentrations such as BTotal (BoronConcentration) and AsTotal
(ArsenicConcentration) and for the field NetActive (DopingConcentration). The
computation of the total concentration is performed for dopants and germanium and takes into
account all species. For example, Bi contributes 1B atom to BTotal, and B2I3 contributes 2B
atoms. The field NetActive is computed by summing all charged particles including
substitutional dopants, charged dopant-defect pairs, charged clusters, and so forth. The
NetActive field is only computed in materials in which the Boolean pdb parameter KMC
<material> Semiconductor is true.
For further customization, you can overload the Tcl procedures with your own. For more
information on how to create and manipulate continuum and atomistic data, see select on
page 1117 and kmc on page 995.
NOTE KMC2PDE and PDE2KMC may consume CPU time in large simulations.
To improve efficiency, KMC2PDE keeps track of a previous translation
and does not perform a new one if the previous one is still valid.
5: Atomistic Kinetic Monte Carlo Diffusion
Operating Modes
388 Sentaurus™ Process User Guide
I-2013.12
Sano Method
The Sano method for converting particles to continuum profiles can be performed inside
Sentaurus Process. The conversion is performed using the same module as the one available in
Sentaurus Mesh.
For more details about this method, see Mesh Generation Tools User Guide, Defining Particle
Profiles on page 35 and Mesh Generation Tools User Guide, Appendix B on page 179.
To apply the Sano method to all dopants and the computed quantity NetActive, select the
sano option of the UnsetAtomistic command. However, to take full advantage of having
the Sano method inside Sentaurus Process, do the following:
1. Generate a mesh tailored for device simulation, including the use of adaptive refinement
based on NetActive.
2. Add contacts using the contact command.
3. Use the tdr=<filename> and !Gas option of the struct command that will, by
default, create a mesh with contacts present and appropriate for device simulation.
As an example:
# Place mesh settings before UnsetAtomistic command in 3D, because
# a new mesh will be created during UnsetAtomistic using the
# current refinement settings
pdbSet Grid Adaptive 1
refinebox adaptive refine.fields= { BActive AsActive NetActive } \
rel.error= { BActive = 1.1 AsActive = 1.1 NetActive = 1e30 } \
max.asinhdiff= { NetActive = 5 } \
target.length = 1e5 refine.min.edge = 0.5<nm>
UnsetAtomistic sano
contact name = c1 box xlo = 0.0 ylo = 0.0025 xhi = 0.04 yhi = 0.0125 \
silicon adjacent.material=oxide
contact name = c2 box xlo = 0.025 ylo = -0.01 xhi = 0.075 yhi = 0.01 \
silicon
struct tdr= n10 !Gas
The following parameter is available to control the accuracy of the Sano smoothing
computation:
pdbSet KMC SanoMethod <species> ScreeningFactor <n>
The screening factor sets the inverse of the screening length of the Sano method. The smaller
the screening factor, the smoother the profile and the longer the computation time.

5: Atomistic Kinetic Monte Carlo Diffusion
Simulation Domain
Sentaurus™ Process User Guide 389
I-2013.12
For more options of the UnsetAtomistic command, see UnsetAtomistic on page 1187. In
addition, smoothing and remeshing based on Sano fields can be called directly using the grid
command (see grid on page 940).
Simulation Domain
All Sentaurus Process KMC simulations are performed internally in a 3D domain. If the
Sentaurus Process structure is 1D or 2D, the missing lateral dimensions are created
automatically to form a 3D simulation domain for Sentaurus Process KMC. For a 2D structure,
the extension in the z-direction is taken from MinZum and MaxZum. For a 1D structure, the
default extension is MinYum to MaxYum and MinZum to MaxZum.
To change the default values, use:
pdbSet KMC MinYum <n>
pdbSet KMC MaxYum <n>
The Sentaurus Process KMC simulation domain is the same as the Sentaurus Process
simulation domain, including the top of the simulation, and it cannot be changed. For Y and Z,
the values MinYum, MinZum and MaxYum, MaxZum are used only when they are not set up in
the input file (because the dimensionality of the simulation is smaller).
Consequently, the Sentaurus Process KMC dimensions fit Sentaurus Process dimensions. The
size of the Sentaurus Process KMC simulation domain is reported in the output, for example:
KMC domain (-0.1, 0, 0) to (0.02, 0.02, 0.025) um Sentaurus domain (-0.1, 0, 0)
to (0.02, 0.02, 0) um.
Recommended Domain Size
For 3D simulations of deep-submicron transistors with twofold symmetry, you should make
the simulation domain size one-quarter of the transistor. The Sentaurus Process KMC domain
is automatically the same.
For a 1D simulation (that can be compared to SIMS data), Sentaurus Process KMC uses as
small as possible lateral domain sizes to save CPU time. However, simulation domains with
lateral sizes smaller than 20 nm may be too small to represent extended defects. If you are
interested in a 1D profile with less statistical noise, you should increase the lateral size.
NOTE The minimum recommended size for accurate implant cascades and
damage accumulation is 40 nm x 40 nm.

5: Atomistic Kinetic Monte Carlo Diffusion
Internal Grid
390 Sentaurus™ Process User Guide
I-2013.12
The lateral domain area is multiplied by the implant dose to obtain the number of ions
implanted. For high impurity concentration levels, you may obtain enough particles in a
relatively small simulation domain. For example, the implant dose of creates 16000
ions for the 40-nm x 40-nm lateral domain side. This might be sufficient to obtain low
statistical noise, while any further increase in the 40-nm x 40-nm lateral domain size only slows
the simulation.
To obtain good statistics for lower concentrations or lower doses, you must increase the lateral
size of the simulation domain. For example, an implant dose of creates only 16 ions
for the 40-nm x 40-nm lateral domain.
One way to reduce the statistical noise without increasing the waiting time is to use the KMC
parallel features available for 1D and 2D simulations. For more information, see Parallelism on
page 393.
NOTE When using Sentaurus Process KMC, try to use the smallest (but
realistic) domain possible. If the simulation is too noisy or not
representative, increase the lateral size. CPU time typically is
proportional to the surface area. If a simulation with a
surface takes 5 minutes to finish, you can expect a
simulation to take four times longer.
Internal Grid
Sentaurus Process KMC uses an internal grid to:
■Store the geometry and material assignments of the structure being simulated.
■Accelerate the search for possible interaction partners for each defect in the simulation.
■Compute the electronic properties.
■Be the minimum volume of amorphized silicon.
■Be the base to compute the concentrations written in the TDR file (using the kmc extract
tdrWrite command).
The Sentaurus Process KMC grid is a tensor-product grid. This grid is different and isolated
from the regular Sentaurus Process grid. The minimum size for each rectangular grid box is set
to and a minimum value <0.8 nm in any axes will not be accepted. There
is no maximum size. The grid is built using Sentaurus Mesh and can be adjusted using the
following pdb parameters (default values in parenthesis):
XGrading
Grading in the x-direction. (1.05)
YMinCell
Minimum cell size in y-direction [ ]. (1.5 nm)
1015 cm 2–
1012 cm 2–
20 20× nm2
40 40× nm2
0.8 0.8×0.8× nm3
µm
5: Atomistic Kinetic Monte Carlo Diffusion
Internal Grid
Sentaurus™ Process User Guide 391
I-2013.12
There are no XMinCell and XMaxCell for x. The maximum size for X is fixed to 100 nm, but
is further controlled with refinements, as explained below. The final mesh is similar to the one
obtained by Sentaurus Mesh using the following script:
tensor {
mesh {
mincellsize = 8e-4
maxcellsize direction "x" 1e-1
maxcellsize direction "y" $YMaxCell
mincellsize direction "y" $YMinCell
maxcellsize direction "z" $ZMaxCell
mincellsize direction "z" $ZMinCell
minbndcellsize = 8e-4
maxbndcellsize = 1e-3
grading = {$XGrading $XGrading}
}
}
where $Name means the value of the parameter. The default parameters are set to try to
minimize the Manhattan geometry at the interfaces, while maximizing the performance of the
simulation. All of these parameters can be set using pdbSet KMC, for example:
pdbSet KMC ZMaxCell 3e-3
Further customization can be added in the form of refinements. To refine the KMC internal
grid, the command refineBox, with the parameter kmc, is used. By default, Sentaurus
Process KMC applies one refinement when using SetAtomistic. This refinement is defined
as:
refinebox kmc min = { 0 0 } max = { 0.1 2 } xrefine = { 0.0012 0.0015 0.0015 }
This default refinement can be changed overwriting the procedure kmcDefaultRefinement
with a user-defined refinement. For example, to remove the default refinement only:
fproc kmcDefaultRefinement { } {
LogFile "Removing the default refinement (by not defining it)..."
YMaxCell
Maximum cell size in y-direction [ ]. (2.5 nm)
ZMinCell
Minimum cell size in z-direction [ ]. (1.5 nm)
ZMaxCell
Maximum cell size in z-direction [ ]. (2.5 nm)
Always3DMeshing
Use Sentaurus Mesh to extrude 2D into 3D. By default, it uses an internal
algorithm. (false)
NonUniformTensor
Use a nonuniform tensor grid (true) or use the old uniform tensor method
(false) and the obsolete parameters BitsBoxes and MinXum. Using the
old uniform tensor method is strongly discouraged.
µm
µm
µm

5: Atomistic Kinetic Monte Carlo Diffusion
Randomization
392 Sentaurus™ Process User Guide
I-2013.12
}
Finally, the lines and spacing specified with the line command also will be included in the
simulation if possible.
For more information on the internal grid and how it affects the simulation, see Materials and
Space on page 398.
Randomization
You can investigate statistical variations of a process flow by selecting a different seed for the
random number generator used by Sentaurus Process KMC in each run. You can specify the
value of the seed changing the parameter randomSeed in the parameter database:
pdbSet KMC randomSeed <n>
The seed can be set to any value from 0 to 31328.
Boundary Conditions
By default Sentaurus Process KMC uses periodic boundary conditions at the left, right, front,
and back sides of the simulation domain. To change these conditions, use the parameters KMC
PeriodicBC_Y and KMC PeriodicBC_Z.
There also is an option to use only periodic boundary conditions for extended defects like
{311}s and dislocation loops. To define this option, set DebugFlag to 4 in the PDB. This
option applies periodic boundary conditions only to the extended defects and still uses
reflective boundary conditions for everything else. You can use this option to reduce the lateral
simulation domain for investigating 1D simulations. Even when the lateral simulation domain
is comparable or smaller than the typical length of the extended defect, the periodic boundary
conditions allow you to obtain meaningful results.
NOTE This option should not be used for 3D simulations if there are lateral
variations in geometry or profiles.
When Sentaurus Process KMC detects an improper choice of the periodic boundary
conditions, it changes the periodic conditions:
** Warning **
KMC. The material structure is not the same in the plane y=0 and y=ymax.
Periodic boundary conditions for defects have been disabled!

5: Atomistic Kinetic Monte Carlo Diffusion
Parallelism
Sentaurus™ Process User Guide 393
I-2013.12
The boundary conditions for the x-axis are relective. You can transform them into a sink using
the parameter sinkProbBottom. This parameter is defined for any material. For example, to
specify that 20% of the incoming positive interstitials should be sunk when reaching the
maximum coordinate in silicon, use:
pdbSet KMC Silicon I sinkProbBottom IP 0.2
It is also possible to define sink boundary conditions for the y- and z-axis, independently of the
general mirror or periodic conditions described below. Similarly to the sinks defined in the x-
axis, a probability for particles crossing the boundary to be annihilated will be defined. This
probability is applied before the general boundary conditions. For example, if a 50% sink is
defined for interstitials at the left boundary, 50% of them surviving the sink will be either
mirrored or moved to the opposite side to simulate periodicity.
The parameter names for the y-axis are sinkProbLeft and sinkProbRight and, for the
z-axis, they are sinkProbFront and sinkProbBack.
Parallelism
You can use several CPUs during a Sentaurus Process KMC simulation. This feature is
configured with the math command:
math numThreadsKMC=<n>
where <n> is the number of threads to launch. When also using MC implantation, the number
of threads used by Sentaurus Process KMC overwrites the number of threads used by MC
implantation.
The KMC to PDE "smooth" algorithm (see Smoothing Out Deatomized Concentrations on
page 537) also can work in parallel:
math numThreadsDeatomize=<n>
Sentaurus Process KMC uses the sparallel licenses in the same way as Sentaurus Process
does. In particular, if no licenses are available, the code will continue in serial or abort
depending on the go.serial or go.abort options specified by the user.
How Parallelism Works
Sentaurus Process KMC works in parallel by assuming that there is no space anisotropy in the
z-direction. This assumption is trivially true for 1D and 2D simulations, and generally false for
3D simulations. Consequently, parallelism is only allowed in simulations that have a 1D or 2D
domain in Sentaurus Process (even when internally all atomistic simulations are 3D).
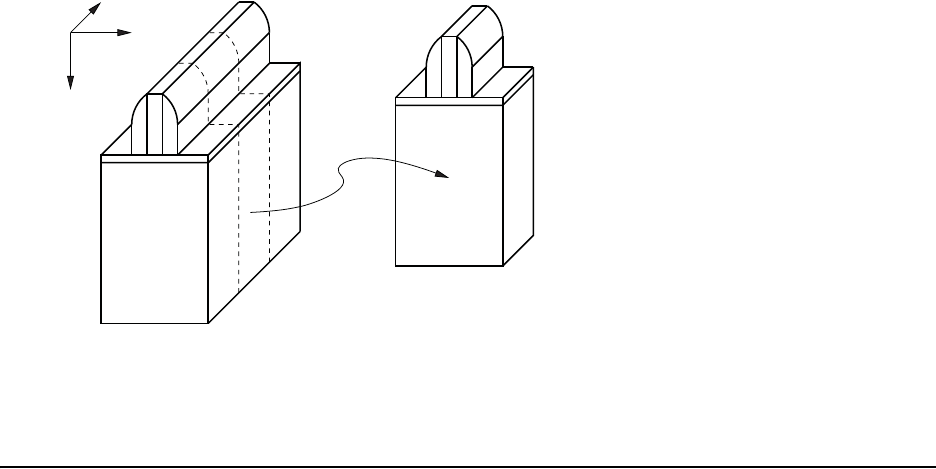
5: Atomistic Kinetic Monte Carlo Diffusion
Estimating CPU Time
394 Sentaurus™ Process User Guide
I-2013.12
The main simulation domain is divided into <n> subdomains; n is the number of threads to be
used. Each subdomain is then run as an independent simulation. Then, the boundary conditions
selected for the Z boundary are applied to the new Zi boundaries. At the end of the simulation
(implantation or diffusion), the main domain is recreated as the simple addition of all the
subdomains. All these “splits” and “forks” of the simulation domains are performed
automatically and are transparent to users.
When using parallelization, one big parallel simulation domain is run as several smaller
domains. At the end of the simulation, the third dimension will be collapsed and averaged to
produce a 2D result (third and second dimensions for 1D results). In any case, the subdomains
must be large enough to allow an accurate representation of the physics involved in the
simulation. In particular, since a minimum surface of 40 nm by 40 nm is recommended, the
minimum suggested size for parallel simulations is 40 nm in the y-axis and nm in the
z-axis, where is the number of threads.
When instructing Sentaurus ProcessKMC to work in parallel with
<n> threads, the domain is
divided into <n> subdomains in the z-direction, and each one is processed by a different CPU.
Later, the subdomains are appended together. These manipulations are transparent to users.
Figure 39 When Sentaurus Process KMC works in parallel with <n> threads, the domain is
divided into <n> subdomains in the z-direction, and each one is processed by a
different CPU. Later, the subdomains are appended together. Here, <n> = 3.
Estimating CPU Time
CPU time and memory required for KMC diffusion simulation are directly proportional to the
number of particles in the structure. For a typical 2-GHz machine, Sentaurus Process KMC
performs up to 1 million events (jumps) per second. In some 64-bit platforms, the number can
reach 2 million events per second.
n40×
n
Z
Y
X
CPU #1 #2 CPU #3
CPU #2
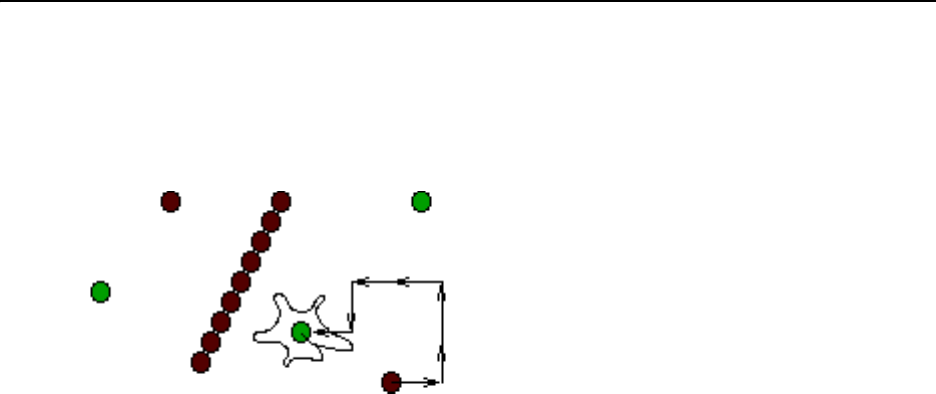
5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
Sentaurus™ Process User Guide 395
I-2013.12
In equilibrium conditions without implant damage, concentrations of mobile species are low
and events are rare. Therefore, the simulation requires few events to reach the required
diffusion time and proceeds quickly.
For transient-enhanced diffusion after an implant step, it takes some time to anneal the implant
damage. Depending on the implant conditions, each implanted ion generates up to
interstitials and vacancies. Each interstitial and vacancy makes up to jumps before
recombining at the surface. This means that it takes approximately 1 second of CPU time to
anneal one implanted ion.
NOTE The above numbers are only estimations. The CPU speed differs
depending on the machine, operative system, and other factors.
Clustering and emission processes take longer internally than diffusion (hops) processes in
Sentaurus Process KMC, and similar numbers of simulated clustering or declustering
processes may lead to a larger wall clock time.
NOTE The use of parallelization also changes the time estimation (see
Parallelism on page 393).
NOTE The use of different hopping modes (KMC HoppingMode) also changes
the time needed to run the simulation, with doublelong being the
fastest mode (see Hopping Mode on page 423).
Atomistic Diffusion Simulation with Sentaurus
Process KMC
The nonlattice KMC method tracks only atoms in defects, while lattice silicon atoms are not
included as shown in Figure 40.
Figure 40 The nonlattice KMC method tracks only atoms in defects; lattice silicon atoms are
not included
103
105
311
I
V

5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
396 Sentaurus™ Process User Guide
I-2013.12
In molecular dynamics, all lattice atoms and all defect atoms must be simulated, but in the
nonlattice KMC method, only defect atoms are considered. Lattice atoms vibrate with a high
frequency because of thermal energy and, occasionally, one of the point defects diffuses and
moves to a neighboring position. Since Sentaurus Process KMC uses the nonlattice KMC
method, it discards the lattice information and only follows the defect atoms. This greatly
affects CPU time, from approximately s for lattice vibrations to approximately s
for fast diffusing particles. During diffusion, moving particles can be captured by extended
defects that emit isolated particles with frequencies orders of magnitude smaller than
frequency of point defects jumps.
Sentaurus Process KMC takes the input parameters of migration, binding, emission energies,
and so on and simulates the frequencies at which these different events occur. Sentaurus
Process KMC starts with short time steps, but when the simulation evolves and the fast moving
point defects disappear, the average time step automatically changes to adapt to the new
situation.
Single particles can move alone or belong to an extended defect, like a {311}.
■For self-silicon point defects, in other words, interstitial and vacancy models, see Point
Defects, Impurities, Dopants, and Impurity-paired Point Defects on page 415.
■For diffusing dopants, see Impurities on page 418.
■For self-silicon extended defects, see Damage Accumulation Model: Amorphous Pockets
on page 432 and Extended Defects on page 442.
■For clusters involving dopants, see Impurity Clusters on page 472.
Units
The units used by Sentaurus Process KMC are:
■Micrometer ( ) for length
■Second (s) for time
■Electron volt (eV) for binding energies
■Atoms per for concentrations
■ for diffusivities
■ for stress activation volumes
10 13–
10 9–
µm
cm3
cm2s1–
nm3
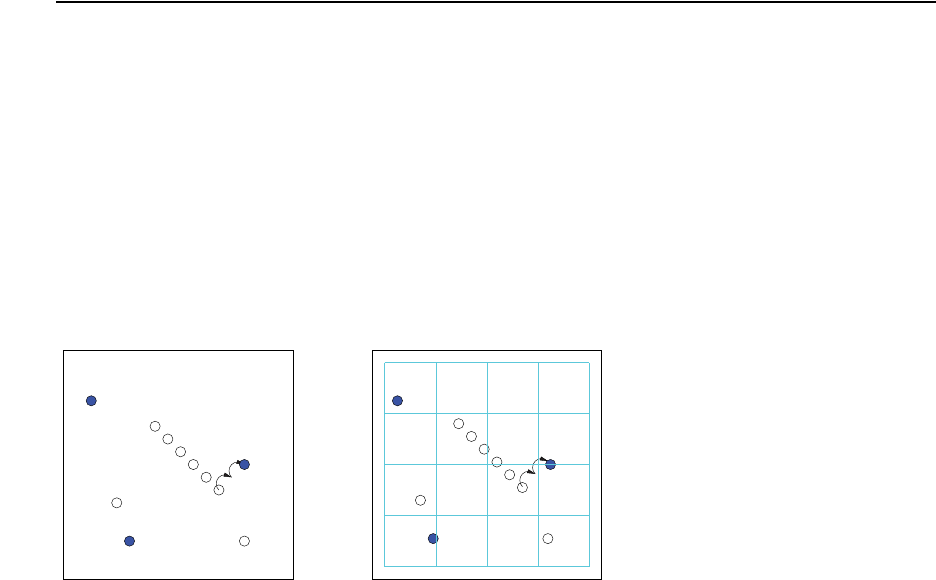
5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
Sentaurus™ Process User Guide 397
I-2013.12
NOTE These units are standard in atomistic and continuum simulators.
Nevertheless, Sentaurus Process KMC internally uses a
nonconventional unit to measure frequencies. For consistency with
migration prefactors, frequencies are measured in diffusivity units in the
input parameter files. However, even for migration prefactors, which are
in diffusivity units to easily compare them with experimental numbers,
Sentaurus Process KMC must transform them to frequency units in .
This is performed using the expression , where is the average
jump distance.
Space Management
Sentaurus Process KMC assumes an orthogonal simulation cell to manage space. The
minimum and maximum x-, y-, and z-dimensions (that is, the bounding box) are passed to
Sentaurus Process KMC as simulation parameters. Sentaurus Process KMC assumes that the
x-axis is the depth of the silicon wafer; whereas, yz is the wafer area.
When Sentaurus Process KMC has the simulation cell size, it splits the space (see Figure 41)
using Sentaurus Mesh. This creates an internal grid inside the rectangular simulation boundary
box. This grid is used only by Sentaurus Process KMC and is fully isolated from the Sentaurus
Process finite-element mesh. These rectangular elements cannot be smaller than twice the jump
distance ( ). To customize the internal grid, see Internal Grid on page 390.
Figure 41 Sentaurus Process KMC divides the space (left) into small rectangular elements
(right); these elements are used for neighbor search, amorphization, and charge
models
Sentaurus Process KMC shows the number of elements in its output:
KMC domain (-0.1, 0, 0) to (0.3, 0.16, 0.05) um
KMC NonUniformTensor. Boxes: 430125
X=155 (1 - 100)nm
Y=111 (0.903641 - 1.7048)nm
Z=25 (2 - 2)nm
s1–
υ6D
λ2
-------=
λ
0.8 nm
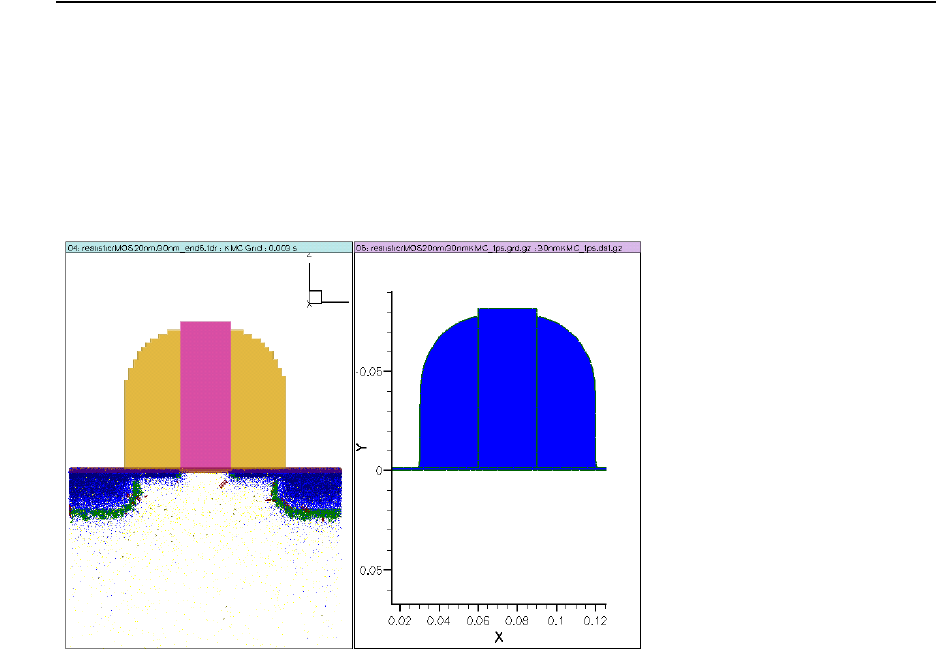
5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
398 Sentaurus™ Process User Guide
I-2013.12
In the above example, the x-axis minimum cell is 1 nm and the maximum one is 100 nm. For
y, these values are 0.9 and 1.7 nm, respectively. Finally, all of the cells have the same size in
the z-axis: 2 nm. There are 155 cells in the x-direction, and 11 and 25 for y and z, respectively.
The total number of elements is 430125.
NOTE Memory allocation depends on the number of internal elements and on
the number of particles. To modify the number of internal elements
using the parameter, see Internal Grid on page 390.
To add user lines to the simulation, use the line command. You also can specify the option
spacing in this command. In this case, and in contrast with the nonatomistic mode, there is a
minimum size of 0.8 nm between lines. If you specify a very thin spacing, and Sentaurus Mesh
tries to add some lines later to better refine a surface, these last lines could be discarded in the
KMC mesh only to keep the 0.8 nm limitation. Because of this, the use of spacing is not
suggested, and it is usually better to rely on the results of Sentaurus Mesh.
Materials and Space
The transfer of materials from Sentaurus Process to Sentaurus Process KMC is
straightforward: Each Sentaurus Process KMC element is assigned to a material type.
Sentaurus Process KMC creates interfaces whenever two elements are set to different
materials, except when instructed not to do so. Consequently, the shape and interfaces assigned
by Sentaurus Process KMC depend on how smooth the shapes are and how fine the internal
elements are (see Figure 42).
Figure 42 Interfaces between materials have a Manhattan structure in Sentaurus
Process KMC; each element is assigned to a material, and elements are always
rectangular
KMC Original

5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
Sentaurus™ Process User Guide 399
I-2013.12
The meshing algorithm included in Sentaurus Mesh tries to fit the interfaces using a
nonuniform tensor. This fit is perfect when the interfaces are flat, axis-aligned and there are no
limitations with the element size due to spacing or very thin features. Since the minimum
dimension for an element is 0.8 nm, interfaces thinner than that will not be accurately
represented.
After the material assignment, Sentaurus Process KMC checks that there are no mistakes in the
translation by reviewing the original interface elements and checking that there is a
corresponding KMC interface associated with them. Whenever this correspondence is not
satisfied, Sentaurus Process KMC issues a warning:
** Warning **
KMC. 1.79 percent of the Oxide/PolySilicon interface is lost when translating
to KMC. Please, review the results carefully.
... continuing execution
NOTE In some cases, these warnings may be produced by thin, but negligible,
structure shapes. In these cases, the percentage of interface lost is small.
When the percentage is significant, they point to important problems
that must be resolved before continuing the simulation.
Supported Materials
The materials already defined in Sentaurus Process KMC include:
■Silicon (crystalline silicon)
■Amorphous silicon
■Silicon oxide
■Polysilicon
■Nitride
■Gas
■Germanium
■Germanium oxide
■Amorphous germanium
Amorphous can be assigned by users, but it also is created automatically by changing the
crystalline regions during simulation when the damage reaches an amorphization level (see
Amorphization and Recrystallization on page 454).
5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
400 Sentaurus™ Process User Guide
I-2013.12
The materials supported by Sentaurus Process KMC are defined in the PDB:
It is possible to define or remove materials, already known by Sentaurus Process, into
Sentaurus Process KMC. They only need to be added as true or false to this list. For
materials not defined in Sentaurus Process, they must be introduced to Sentaurus Process first
with mater add. For example, to add a new material called AnotherSilicon and to remove
Nitride:
pdbSet KMC Materials AnotherSilicon true
pdbSet KMC Materials Nitride false
sprocess> pdbGet KMC Materials
Silicon true
AmorphousSilicon true
Oxide true
PolySilicon true
Germanium true
AmorphousGermanium true
GeOxide true
Nitride true
Cobalt true
CobaltSilicide true
Nickel true
NickelSilicide true
Platinum true
PlatinumSilicide true
Titanium true
TiSilicide true
Tungsten true
TungstenSilicide true
Gas true
Unknown true
5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
Sentaurus™ Process User Guide 401
I-2013.12
Every new material in Sentaurus Process KMC requires the following pdb parameters:
After the material is properly defined, the parameters for all the particles in this new material
must be defined as well. These parameters depend on the model defined for the new material
(for an overview of these parameters, see Including New Impurities on page 522). Finally,
parameters for all interfaces between the existing materials and the new material must also be
defined.
Model
The options are:
The discard model creates a material empty of particles (for example,
gas). All particles introduced in this material are discarded.
The simple model includes impurity clusters and simple diffusion of
dopants. It does not model Fermi-level dependencies, amorphization, solid
phase epitaxial regrowth (SPER), and extended defects. Oxide and nitride
are examples of ‘simple’ materials.
The full model allows all models: point defects (interstitials and
vacancies), extended defects, impurity clusters, damage accumulation,
amorphization, recrystallization, and the Fermi level–dependent diffusivity
models (for example, silicon).
ShortName
This is the short name of the material when reading parameters defined for
the interfaces. For example, the interface Oxide_Silicon contains the
parameters Eb_SurfOx and Eb_SurfSi, where Ox and Si are the short
names of silicon and oxide.
Semiconductor
A Boolean value. True for materials where DopingConcentration
(NetActive) should be computed and stored.
Crystalline
A Boolean value. True if the material is crystalline (silicon) and false if it is
amorphous (amorphous silicon).
Equivalent
Name of the amorphous/crystalline equivalent. For example, silicon will
have amorphous silicon as its equivalent, and the amorphous silicon
equivalent is silicon.
Oxide
Specify with true or false whether the material is an oxide or is not an
oxide, respectively. This is used for oxidation models. For example,
SiOxide and GeOxide have this field as true.
Alloy
Specify if this material can alloy with another material to form binary alloys
with corrections to activation energies. Write the alloy material here. For
example, for silicon alloying with germanium, write Germanium in the
Alloy field of the silicon material.

5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
402 Sentaurus™ Process User Guide
I-2013.12
NOTE To minimize the work of defining parameters for a new material, it is
advisable to disable the materials that will not be used, thereby avoiding
to define parameters for the interfaces between those materials and the
new one. For the dopant parameters in the new material, you can “copy”
the parameters from another material and redefine only some of them.
Any other material existing in Sentaurus Process, but not defined by Sentaurus Process KMC,
is mapped as ‘Unknown’. The model for this material is discard, and all particles inside these
materials are discarded and removed. They do not need parameters because they contain no
particles to simulate.
Material Alloying
Sentaurus Process KMC allows materials containing an alloying element to be treated in a
quasi-atomistic framework. Such an alloying element is specified using the Alloy parameter
of the material specification. In the following, as an example, it is assumed that the material is
silicon and the alloy is germanium (although, this can be reversed, or any pair of materials can
be used with "full" modeling).
The quasi-atomistic framework means that alloy particles (Ge) are not created as particles, but
they will be taken into account as a field, so as to produce a local concentration. This saves
memory and speeds up the concentration. Diffusion of the alloying element is possible using
the model specified in Alloy Diffusion on page 431.
The inclusion of an alloy changes the bandgap narrowing as specified in Narrowing due to
Presence of an Alloy on page 504. Such a model uses a quadratic interpolation to smooth from
the band gap of the pure material to the band gap of the pure alloy. Since the positions in the
band gap of all particles are scaled with the total band gap, the positions for charged defects
are scaled accordingly.
All activation energies for diffusion-, emission-, and activation-related processes are corrected
by a term linear on the alloy concentration. If a given mechanism is simulated by following an
Arrhenius expression similar to:
(654)
the mechanisms under an alloy concentration are corrected by a linear term in the activation
energy with the concentration of the alloy (Ge):
(655)
The same applies to the formation energies of point defects and the potential energies of
impurity clusters. Examples of these corrections are available in Alloy Effects on page 432.
νPE–kBT⁄()exp=
νPEαGe[]+()–kBT⁄()exp=
5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
Sentaurus™ Process User Guide 403
I-2013.12
Point Defects
Sentaurus Process KMC can distinguish between different interstitials, depending on the
material, using different syntax. The syntax "I" refers to an interstitial in the particular
material where it is positioned; otherwise, a more concrete notation must be used. For example,
in a structure where the first half is Ge and the second half I, "I" refers to Gei in the first half
and Sii in the second half. While Sii, for example, would have the same meaning as in the
second half, but it would produce Si in the first half.
Ambiguous Alloying
Sentaurus Process KMC allows the use of a material (for example, Si) with an alloy (for
example, Ge) without having to define the material alloyed (Ge). Nevertheless, it is also
possible to define the alloyed material as the main material (Ge) with the other one as the
alloyed material (Si). However, having all alloys defined twice is ambiguous. For example,
Si0.2Ge0.8 can be defined as main Si with 80% Ge, or as main Ge with 20% Si.
The main material is the one specified in the input file as material. In this case, it can be
silicon, and then you can use the select command or similar to include 80% Ge (or
germanium) and 20% silicon. Unfortunately, the complexity of the models and their calibration
produce different results when these two ways to have the same alloy are used. To solve this
issue, using Si as the main material up to 80% Ge concentration and Ge up to 20% silicon is
suggested.
Time Management
The main component in Sentaurus Process KMC is an algorithm that sequentially selects the
possible random events (migration of point defects, emission of extended defects, and so on)
according to their corresponding frequencies, similar to the Bortz–Kalos–Liebowitz (BKL)
algorithm so widely used in KMC methods. Figure 43 illustrates the selection procedure for
the atomistic configuration shown in Figure 40 on page 395, consisting of three vacancies (V),
two interstitials (I), and one {311} defect.
Figure 43 Events are selected according to their rates, which in turn, depend on the current
atomistic configuration
rand
3000 3200 32100
V jump
I jump
{311} emission

5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
404 Sentaurus™ Process User Guide
I-2013.12
Assuming the vacancy and interstitial migration frequencies are and ,
respectively, and the {311} emission rate is , to simulate 1 s, you have to simulate a total
of 3210 events. Consequently, simulating one event corresponds to simulating 1/3210 s. This
implies that the simulated time step is not fixed, but depends on the particular simulation
configuration. In addition, you must choose a V with a probability of 3000/3210, and Is and
{311} defects with probabilities of 200/3210 and 10/3210. A random number between 1 and
3210 (or 0 and 3209) is generated. For example, in Figure 43, the number 3147 selects an I
migration event.
After several migration events, when one interstitial reaches and interacts with a vacancy, the
simulator generates an IV pair called an amorphous pocket (AP) (see Damage Accumulation
Model: Amorphous Pockets on page 432).
The simulation contains two Vs, one Is, one IV, and one {311}. You can assume the IV pair will
recombine with a frequency of . The new random number will be between 1 and
2000 + 100 + 500 + 10 = 2610. Consequently, the time step will be 1/2610 s. If the IV
recombination event is chosen and this IV pair is annihilated, the new simulation contains only
two Vs, one I, and one {311}, and the following time step is 1/2110 s.
NOTE The time step is not a fixed quantity in Sentaurus Process KMC, but it
depends on the state of the simulation.
The current time step depends on two factors:
■Number of particles in the simulator
■Frequency of the events associated with the particles or defects or both
And these frequencies depend on three factors:
■Type of event
■Arrhenius plot associated with the event
■Temperature
Simulation and CPU Times
The time needed to complete a simulation depends on several factors:
■How fast the computer simulates one event
■How many events need to be simulated
■The hopping mode chosen
■The presence of lattice KMC models (for SPER and epitaxial deposition)
■Whether you use parallel capabilities
1000 s 1–
100 s 1–
10 s 1–
500 s 1–
5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
Sentaurus™ Process User Guide 405
I-2013.12
The number of events to be simulated is inverse to the average time step (which changes during
the simulation, as explained above). The speed at which the simulator processes events depends
on the type of simulated events. Migration events usually are simulated rapidly. Simulations
involving changes in the electronic concentration or temperature or both are much slower
because updating the dependencies with the temperature and the Fermi level takes extra time.
Generally, the smaller the simulation, the shorter the time.
In simulations with implants, the implant MC module also adds time to the simulation while
computing the cascades. In amorphizing conditions, the Sentaurus Process KMC amorphous
model requires extra time to smooth out the damage and create amorphous layers. Finally,
simulations with strong gradients in the electronic concentration need more charge updates,
which take extra time.
During oxidation, several remeshings must be performed to update the Sentaurus
Process KMC structure to the new Sentaurus Process oxide thickness, consuming extra time.
Finally, the hopping mode allows you to chose whether long hops or double hops are allowed,
thereby speeding up the simulation. Both are switched on by default (see Hopping Mode on
page 423).
NOTE Yo u ca n es ti mate t he t im e n ee ded for simulation by running a small
simulation and assuming the CPU time is proportional to the number of
particles which is proportional to the surface area.
Sentaurus Process KMC shows the status of the current simulation, printing log messages each
time the temperature changes during a temperature ramp, or whenever there is a new snapshot
(see Snapshots on page 407). These log messages are as following:
Reaction : 31s to 63s step : 32s temp: 950.0C
Mechanics: 31s to 63s step : 32s temp: 950.0C
Diffusion: 31s to 63s step (d): 32s temp: 950.0C
KMC: Time(s) Temp(C) Events Events/s Average step(s) %Done
46.416 950.01 3600728007 1877077 4.749174e-08 7.74% (36% V)
Reaction : 1.05min to 2.117min step : 1.067min temp: 950.0C
Mechanics: 1.05min to 2.117min step : 1.067min temp: 950.0C
Diffusion: 1.05min to 2.117min step (d): 1.067min temp: 950.0C
KMC: Time(s) Temp(C) Events Events/s Average step(s) %Done
100.000 950.01 4742533558 1865695 4.692958e-08 16.67% (33% V)
Reaction : 2.117min to 4.25min step : 2.133min temp: 950.0C
Mechanics: 2.117min to 4.25min step : 2.133min temp: 950.0C
Diffusion: 2.117min to 4.25min step (d): 2.133min temp: 950.0C
KMC: Time(s) Temp(C) Events Events/s Average step(s) %Done
215.444 950.01 6881748852 1868310 5.396525e-08 35.91% (34% I)
Reaction : 4.25min to 7.125min step : 2.875min temp: 950.0C
Mechanics: 4.25min to 7.125min step : 2.875min temp: 950.0C
Diffusion: 4.25min to 7.125min step (d): 2.875min temp: 950.0C
Reaction : 7.125min to 10min step : 2.875min temp: 950.0C
5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
406 Sentaurus™ Process User Guide
I-2013.12
Mechanics: 7.125min to 10min step : 2.875min temp: 950.0C
Diffusion: 7.125min to 10min step (d): 2.875min temp: 950.0C
KMC: Time(s) Temp(C) Events Events/s Average step(s) %Done
464.159 950.01 11927967061 1864825 4.928753e-08 77.36% (35% I)
Elapsed time for diffuse 7.8672e+03s
600.000 950.01 14721335445 1882323 4.862968e-08 100.00% (35% I)
It is easy to identify the log immediately after the Diffusion statement because it is preceded
by a “KMC:” header. In the above example, the total simulated (annealed) time is 600 s. The
(current) temperature is . A total of events has been simulated so far, and the
simulator processed 1.9 million events each CPU second. The averaged time step is
. Finally, Sentaurus Process KMC writes the particle with the biggest percentage
of diffusion jumps. In the example, the diffusion of the neutral interstitial (I) has taken 33 to
36% of the total diffused particles.
This information may change from one simulation to another, and also at different times during
the same simulation. The events per second and average step statistics are recomputed between
sentences.
Parallelism and CPU Time
Table 45 lists the CPU times that can be expected when running on different numbers of
threads. However, the exact time depends on the particular simulation.
The total simulation time is superlinear for and starts saturating for . The reason
for being superlinear for a small number of threads is that the CPU time depends superlinearly
on size. Consequently, simulating a size XYZ/n takes less CPU time than S/n. Nevertheless, as
increases, different mechanisms (such as the waiting time of threads to be synchronized)
conspire to degrade the total simulation time.
Ta bl e 4 5 A p p rox im at e C P U t im e s w he n u s i ng mu lt ip le t hr e ad s
Number of independent
threads
Approximate time Improvement
1 S 1x
2S/2.32.3x
4S/4.84.8x
8 S/8 8x
12 S/9.5 9.5x
16 S/10.5 10.5x
950°C
1.4 1010
×
4.9 10 8–
× s
n8<
n12≥
n
5: Atomistic Kinetic Monte Carlo Diffusion
Atomistic Diffusion Simulation with Sentaurus Process KMC
Sentaurus™ Process User Guide 407
I-2013.12
Snapshots
A snapshot is an interruption of the normal Sentaurus Process KMC simulation flow to print
the status of the simulation and to allow you to run a customizable Tcl command (see Movie
on page 407). To control these interruptions, set the pdb parameters listed in Table 46.
For example:
pdbSet KMC Decade 2
produces two snapshots per decade, as in:
Time(s) Temp(C) Events Events/s Average step(s) %Done
0.000 26.85 0
548.138 700.00 1 5.481385e+02 0.05% (35% I)
1492.073 700.00 5 2.359835e+02 0.15% (33% I)
3639.863 700.00 7867 2.731862e-01 0.36% (35% I)
10130.431 700.00 14553 9.707701e-01 1.01% (35% I)
31902.727 700.00 35862 1.021742e+00 3.19% (36% I)
100030.228 700.00 205821 169959 4.008467e-01 10.00% (35% I)
317268.817 700.00 668173 4.698554e-01 31.73% (34% I)
1000000.000 700.00 1754838 1086665 6.282812e-01 100.00% (35% I)
Movie
The Sentaurus Process KMC Movie command is similar to the Sentaurus Process Movie
command and executes the contents of the parameter KMC Movie any time a new snapshot is
generated. You can use this command interactively to obtain information about the simulation,
to add data to the TDR file of Sentaurus Process KMC, and so on.
For example, it can be used to add concentration information and the positions of particles to
the TDR file during the simulation:
pdbSet KMC Movie {kmc extract tdrAdd concentrations defects}
or to plot the evolution of damage while the simulation is still running:
pdbSet KMC Movie {
Ta bl e 46 S na p sh ot p ara me te rs
Parameter Description
Decade <n>
Sets how many snapshots will be generated per decade. 0 disables it.
InitOutputTime <n>
No snapshots per decade are generated when the simulated time is smaller than n.
maxSnapshots <n>
Maximum number of snapshots to be stored. After this limit is reached, the oldest
ones are erased to make space for the new ones.
5: Atomistic Kinetic Monte Carlo Diffusion
Particles
408 Sentaurus™ Process User Guide
I-2013.12
kmc deatomize name=ITotal; sel z=log(ITotal+1); plot.1d label=I clear;
kmc deatomize name=VTotal; sel z=log(VTotal+1); plot.1d label=V !clear
}
Time Internal Representation and Limitations
There are no internal limits for the frequencies used in Sentaurus Process KMC. Nevertheless,
very high frequencies (typically produced by small migration energies) can lead to slow
simulations.
Particles
Particles are represented in Sentaurus Process KMC with three spatial coordinates (x, y, z) and
two labels:
■The particle type label identifies the species, charge state, and role of the particle in the
simulation.
■The defect type indicates when the particles are agglomerated with others or when they
stand alone.
Particle Types
To obtain a list of the standard particles currently defined for Sentaurus Process KMC, use the
command:
sprocess> kmc particletypes
I V B As C F In O P Sb N H IMM IM IP IPP VMMM VMM VM VP VPP VPPP Asi AsiP AsV
AsVP AsVM Bi BiP BiM Ci FV FI Ini IniM InV InVM Pi PiP PV PVM PVP Sbi SbiP SbV
SbVP SbVM Ge Gei Si Sii
Dopants are user defined in Sentaurus Process KMC, while interstitials and vacancies are fixed
and cannot be customized. The standard list of interstitials and vacancies defined in Sentaurus
Process KMC is:
I Silicon self interstitial–neutral
IMM Silicon self interstitial-double negative
IM Silicon self interstitial–negative
IP Silicon self interstitial–positive
IPP Silicon self interstitial-double positive

5: Atomistic Kinetic Monte Carlo Diffusion
Particles
Sentaurus™ Process User Guide 409
I-2013.12
The dopants are defined in the parameter database. By default, particles for As, B, P, In, C, F,
N, Nn (N2), H, and Sb are defined. For example, the default particles for As, B, and P are:
NOTE Since P at the end of the name means positive, to specify a cluster
containing phosphorus, the P cannot be at the end in any case. This
means that AsiP is an arsenic–interstitial positive, while PAsi is a
phosphorus arsenic–interstitial cluster. Similarly, AsP will be
interpreted as arsenic positive, while PAs is phosphorus–arsenic.
VVacancy–neutral
VMMM Vacancy-triple negative
VMM Vacancy–double negative
VM Vacancy–negative
VP Vacancy–positive
VPP Vacancy–double positive
VPPP Vacancy-triple positive
As Substitutional arsenic–positive
Asi Interstitial arsenic–neutral
AsiP Interstitial arsenic–positive
AsV Vacancy arsenic–neutral
AsVM Vacancy arsenic–negative
AsVP Vacancy arsenic–positive
BSubstitutional boron–negative
Bi Interstitial boron–neutral
BiM Interstitial boron–negative
BiP Interstitial boron–positive
PSubstitutional phosphorus–positive
Pi Interstitial phosphorus–neutral
PiP Interstitial phosphorus–positive
PV Vacancy phosphorus–neutral
PVM Vacancy phosphorus–negative
PVP Vacancy phosphorus–positive
5: Atomistic Kinetic Monte Carlo Diffusion
Particles
410 Sentaurus™ Process User Guide
I-2013.12
NOTE These lists show the most commonly used particles, but the list is not
exhaustive. An exhaustive list contains all point defects (Is and Vs) with
charges from –3 to +3, and all impurity pairs with charges from –2 to +2.
Particles in Models
The dopants allowed in the simulation are defined in the parameter database under the label
KMC Impurities. The database lists the impurity name, the charge, and a Boolean parameter
indicating whether the particle is allowed in the Sentaurus Process KMC simulation.
The particle name and the charge must be delimited by a comma without spaces. For example,
in the case of arsenic and boron:
pdbSet KMC Impurities As,1 true
pdbSet KMC Impurities B,-1 true
When the dopant type has been defined, the paired particles(particles with I or V) can be
defined in KMC Pairs. The definitions are a string containing the name of the pair, the name
of the dopant, the type of pair (I or V) and the charge. These fields are separated with commas.
Finally, a Boolean parameter instructs Sentaurus Process KMC to take the particle into
account.
For arsenic and boron:
pdbSet KMC Pairs Asi,As,I,0 true
pdbSet KMC Pairs AsiP,As,I,1 true
pdbSet KMC Pairs AsV,As,V,0 true
pdbSet KMC Pairs AsVP,As,V,1 true
pdbSet KMC Pairs AsVM,As,V,-1 true
pdbSet KMC Pairs BV,B,V,0 false
pdbSet KMC Pairs Bi,B,I,0 true
pdbSet KMC Pairs Bi,B,I,0 true
pdbSet KMC Pairs BiP,B,I,0 true
pdbSet KMC Pairs BiM,B,I,-1 true
In the previous example, the particle boron vacancy is not defined (set to false). A particle is
not defined when it does not appear in the Pairs list, or when its Boolean variable is false.
The following rules must be followed when defining particles:
■The dopants used in the definitions of Pairs must exist in the list of impurities
■A charge n can only be defined when a charge n – 1 or n + 1 is already defined, except for
pairs with same charge states as their dopants.
■Charge states without possible pairing reactions cannot be defined.
■The charge must be –1, 0, or +1.
5: Atomistic Kinetic Monte Carlo Diffusion
Particles
Sentaurus™ Process User Guide 411
I-2013.12
■Pairs with neutral state; and with the charge of its dopant, always must be defined.
For example, when As+ is defined as an impurity, AsV+ can be defined. The existence of AsV+
allows you to define AsV0 and AsV-. AsV-- cannot be defined (no charge –2). For B-, you can
define Bi-, Bi0, and Bi+.
Alias
Aliases of particle names are defined in KMC aliases, which is a list of particle names and
alternative names separated by commas. These aliases are used only when Sentaurus
Process KMC tries to map a name as particles or defects. For example, if there is an alias such
as:
Bi BoronInt,BI
the commands:
kmc present defectname=Bi
kmc present defectname=BI
are the same, and:
sel z=1e19 name=Bi
sel z=1e19 name=BI
sel z=1e19 name=BoronInt
create fields with different names, but the atomized particle is the same (Bi).
Colors
You can cha ng e t he defa ult v isu alization color for the atomistic representation of particles and
defects in Tecplot SV, or add new colors to existing particles and defects. The list of colors is
KMC Colors, and it is an array of particle names and colors in #rrggbb format (red, green,
blue).
Particles and Parameters
New particles need new parameters. For every impurity specified in Impurities, a new file
must be created for each material folder and surface. The name of these files is obtained using
the command alias with the name of the Sentaurus Process KMC impurity as a parameter:
sprocess> alias B
Boron

5: Atomistic Kinetic Monte Carlo Diffusion
Particles
412 Sentaurus™ Process User Guide
I-2013.12
Table 47 lists the parameters required for materials defined to use the simple model.
When amorphous materials (where the best example is amorphous silicon) use the dangling
bond model (see Indirect Diffusion on page 457), the parameters listed in the previous table
plus those in Table 48 are required.
Table 49 lists the parameters used on interfaces.
Table 47 Nonsilicon material parameters
Parameter Description
Dm, Em
Diffusion parameters.
VD, VF
Activation volumes for stress.
sinkProbTop, sinkProbBottom, sinkProbLeft,
sinkProbRight, sinkProbFront, sinkProbBack
Boundary conditions.
Implement_Complex
Parameters for clustering.
ReactionsPointDefect, ReactionsCluster
Binary interactions.
Ta bl e 4 8 E x t ra p a ra me te rs fo r a mo r p h ou s ma te r ia ls
Parameter Description
EmGe, EfGe
Ge correction to migration energies.
gamma
Coefficient for dangling bond creation.
Db, Eb
Binding prefactor and energy.
Table 49 Parameters used on interfaces
Parameter Description
EBarrier_Surf?,
Eb_Surf?
Interface binding energies for dopants. ? denotes the material. Gas for gas, Si
for silicon, Ox for oxide, Ni for nitride, Po for polysilicon, and Unknown for
the rest.
Db_Surf
Surface emission prefactor for dopants.
EMax_Surf, C0Max_Surf
Maximum number of particles trapped at the surface.
Evaporation_Surf
Probability to evaporate (annihilate).
VF_Surf?
Activation volume for stress. ? denotes the material.
5: Atomistic Kinetic Monte Carlo Diffusion
Particles
Sentaurus™ Process User Guide 413
I-2013.12
Table 50 lists the parameters required for materials using full modeling.
Finally, amorphous, simple and full materials allow the definition of impurity clusters. These
clusters are defined using the parameters listed in Table 51.
For further explanations on these parameters, see the comments in the parameter database and
the model descriptions.
Ta bl e 5 0 F ul l ma te r i al p ar a m e t e rs
Parameter Description
Dm, Em
Diffusivities.
Db, Eb
Binding prefactors and energies.
VD, VF
Activation volumes for stress.
e0
Electronic levels.
EfGe, EmGe
Corrections for germanium.
P_recrysDeposit, E_recrysDeposit,
recrysDepositThreshold, C0_recrysMaxActive,
E_recrysMaxActive, recrysMaxTotal,
recrysMaxSize, recrysDeposit_Complex, E_recrys,
E_recrys_exponent, recrysDeposit_Active,
e0_Complex
SPER model (recrystallization).
sinkProbTop, sinkProbBottom, sinkProbLeft,
sinkProbRight, sinkProbFront, sinkProbBack
Boundary conditions.
ReactionsPointDefect, ReactionsCluster,
ReactionsClusterI, ReactionsClusterV,
ReactionsClusterIV, ReactionsLoop,
ReactionsVoid, Reactions311
Binary interactions.
Table 51 Parameters used for impurity clusters
Parameter Description
Implement_Complex
Whether a cluster exists or not.
Etotal_Complex
Cluster potential energy.
e0_Complex
Cluster charge.
VF_Complex
Stress dependency.
CaptVol_Complex
Cluster capture volume used for emission.
D0_Cluster
Cluster emission prefactors.
EbarrierDopant_Complex, EbarrierIV_Complex
Cluster emission barriers.
Dm_Cluster, Em_Cluster
Cluster migration parameters

5: Atomistic Kinetic Monte Carlo Diffusion
Particles
414 Sentaurus™ Process User Guide
I-2013.12
Undefining Particles
Particles can be undefined erasing their definition in the parameter database or setting its
Boolean to false. If an impurity is undefined or erased, Sentaurus Process KMC also
undefines all its pairs.
For example, to undefine indium in a simulation, use:
pdbSet KMC Impurities In,-1 false
To undefine only boron interstitials with a positive charge, use:
pdbSet KMC Pairs BiP,B,I,1 false
NOTE Undefining particles that will not be used in the simulation saves some
small memory and CPU time. If an undefined dopant is used (for
example, it is implanted or introduced with select), it causes an error.
Defect Types
For a list of the defects implemented in Sentaurus Process KMC, use the command:
sprocess> kmc defecttypes
PointDefect AmorphousPocket Void ThreeOneOne Loop ImpurityCluster Interface
TwinDefect Amorphous LatticeAtom Rejected PD
Defects implemented in Sentaurus Process KMC include those listed in Table 52.
Table 52 Defects implemented in Sentaurus Process KMC
Defect Description
Amorphous
Amorphous region inside the crystalline silicon. Only I, V, impurity clusters, and dopants are
allowed. See Amorphization and Recrystallization on page 454.
AmorphousPocket
Disordered agglomeration of Is and Vs (damage). Only I and V particle types are allowed.
See Damage Accumulation Model: Amorphous Pockets on page 432.
ImpurityCluster
Impurity clusters. Agglomeration of dopants with Is or Vs. See Impurity Clusters on
page 472.
Interface
Dopants trapped in the interfaces (for example, Si/SiO2).
See Interfaces and Surfaces on page 507.
LatticeAtom
Atom in the silicon lattice used for SPER or epitaxial deposition. See LKMC: Fully
Atomistic Modeling of Solid Phase Epitaxial Regrowth on page 463
Loop
Dislocation loops. Extended Is defect. Only Is are allowed.
See Dislocation Loops on page 447.

5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
Sentaurus™ Process User Guide 415
I-2013.12
NOTE Not all possibilities of particle and defect types are allowed. Some
particle types, like the paired ones (Asi, BiM…), are only allowed as
PointDefect. Others, like As or B, can stand alone (PointDefect),
can be trapped in interfaces (Interface), or can belong to an impurity
clusters (ImpurityCluster). Neutral interstitials, for example, can
stand alone (PointDefect), can be in damaged clusters
(AmorphousPocket), {311} defects (ThreeOneOne), or dislocation
loops (Loop). Single particles can be mobile point defects (in other
words, interstitials and vacancies), immobile impurity atoms (like
substitutional boron and arsenic), and also mobile impurity-defect pairs
such as Bi or AsV. All are considered PointDefect.
Point Defects, Impurities, Dopants, and Impurity-paired
Point Defects
Interstitials and Vacancies
Interstitials and vacancies in Sentaurus Process KMC perform a diffusion event in each axis
(x, y, and z) at a frequency given by the expression:
(656)
Defect Description
PointDefect, PD
Single particles (IMM, IM, I, IP, IPP, VMM, VM, V, VP, VPP, As, and B) or paired ones
(Asi, AsiP, AsVM, AsV, AsVP, BiM, Bi, and BiP) that do not belong to any extended
defect or particle agglomeration; in other words, impurities, dopants, and impurity-paired
point defects.
Rejected
A particle coming from MC implant which is determined to be in a material which does not
support the particle. For example, a point defect in a simple material such as oxide.
ThreeOneOne
{311} rod-like extended defects. Only Is. are allowed.
See {311} Defects (ThreeOneOne) on page 442.
TwinDefect
Can be formed during SPER if the twin defect model is turned on. See Defect Generation
during SPER on page 467.
Void
Vacancy clusters with spherical shape. Only Vs are allowed. See Voids on page 451.
Table 52 Defects implemented in Sentaurus Process KMC
j
νjm
ν0m,
3
-----------Emσ'j∆Vpar σ'i∆Vort
()∆EmGe()+
ij≠
++
kBT()⁄–
exp⋅=
5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
416 Sentaurus™ Process User Guide
I-2013.12
where:
■ is the jump frequency for the axis .
■ is the prefactor.
■ is the migration energy.
■ are the principal stresses (the stresses in the coordinate system where all the stress
tensor nondiagonal components are null).
■ is the activation volume for stress-parallel diffusion.
■ is the activation volume for stress-perpendicular diffusion.
■ is the correction due to germanium concentration.
These parameters are called Dm (prefactor), Em (energy), VD (activation volumes), and EmGe
(for ) in the parameter database. They are defined only in the full model materials:
sprocess> pdbGet KMC Si I Dm
IMM 5e-2
IM 5e-2
I 5e-2
IP 5e-2
IPP 5e-2
sprocess> pdbGet KMC Si I Em
IMM 0.8
IM 0.8
I 0.8
IP 0.8
IPP 0.8
sprocess> pdbGet KMC Si I VD
IMM 0,0
IM 0,0
I 0,0
IP 0,0
IPP 0,0
sprocess> pdbGet KMC Si I EmGe
0
The activation volumes for parallel and perpendicular diffusion, respectively, are separated by
a comma (no spaces) in VD.
The stresses are produced by Sentaurus Process and imported by Sentaurus Process KMC.
This stress tensor is diagonalized to obtain the principal stresses . The directions x, y, and z
used in the equation refer to the system in which the stress tensor is diagonal.
NOTE Yo u ca n c alibr at e thes e p aram et ers if necessary to change the point-
defect diffusivity and DC product.
νjm
j
ν0m,
Em
σ'i
∆Vpar
∆Vort
∆EmGe() α
mGe[]=
αm
σij
σ'i
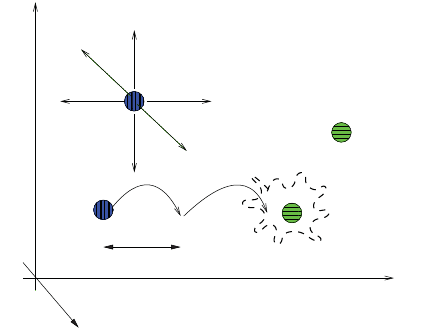
5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
Sentaurus™ Process User Guide 417
I-2013.12
Sentaurus Process KMC simulates point-defect migration, modifying the particle coordinates
in the orthogonal directions a fixed distance, called , which corresponds to the second
neighbor’s distance in the silicon lattice (0.384 nm). The value of can vary spacially (as
explained in Hopping Mode on page 423).
After each diffusion event, the charge state of the point defect is updated according to the new
local Fermi level (see Fermi-Level Effects: Charge Model on page 490 and Updating Charged
States on page 497). Whenever a jumping point defect encounters another particle, defect, or
interface, the jumping point defect interacts according to the specific situation. These
interactions are allowed depending on the following:
■Incoming species—for example, substitutional boron plus interstitial (B + I) is allowed,
and the incoming species form a boron interstitial. Boron plus vacancy is not allowed. This
interactions can be enabled or disabled in the parameter database.
■Energetics—Sentaurus Process KMC allows interactions for {311} defects, dislocation
loops, and pairing because the binding energies are greater than 0. For impurity clusters, if
the reaction is unfavourable, the newly formed defect breaks up and dissolves in the
original components or is rejected before reacting.
■Charge states—interactions between repulsive species are forbidden, except for the
‘percolation’ model (see Percolation on page 477).
Mobile particles can interact with other mobile particles or with the particles belonging to
extended defects, whenever they enter in the capture radius of the other particle or defect (see
Figure 44). The capture radius for a mobile particle is , assumed to be the same as the
jumping distance. For extended defects, the capture volume is the sum of the capture volumes
of its constituent particles. Mobile particles can interact with surfaces/interfaces as explained
in Interfaces and Surfaces on page 507.
Figure 44 Point defects diffuse by jumping a distance in any orthogonal direction and can
interact with neighbor particles
λ
λ
λ
Jump
I+V InteractionLambda
Simulation Cell x
y
z
Particle Type: Interstitial
Defect: PointDefect
Coords: x1, y1, z1 Particle Type: Vacancy
Defect: PointDefect
Coords: x2, y2, z2
Possible
Migrations
λ
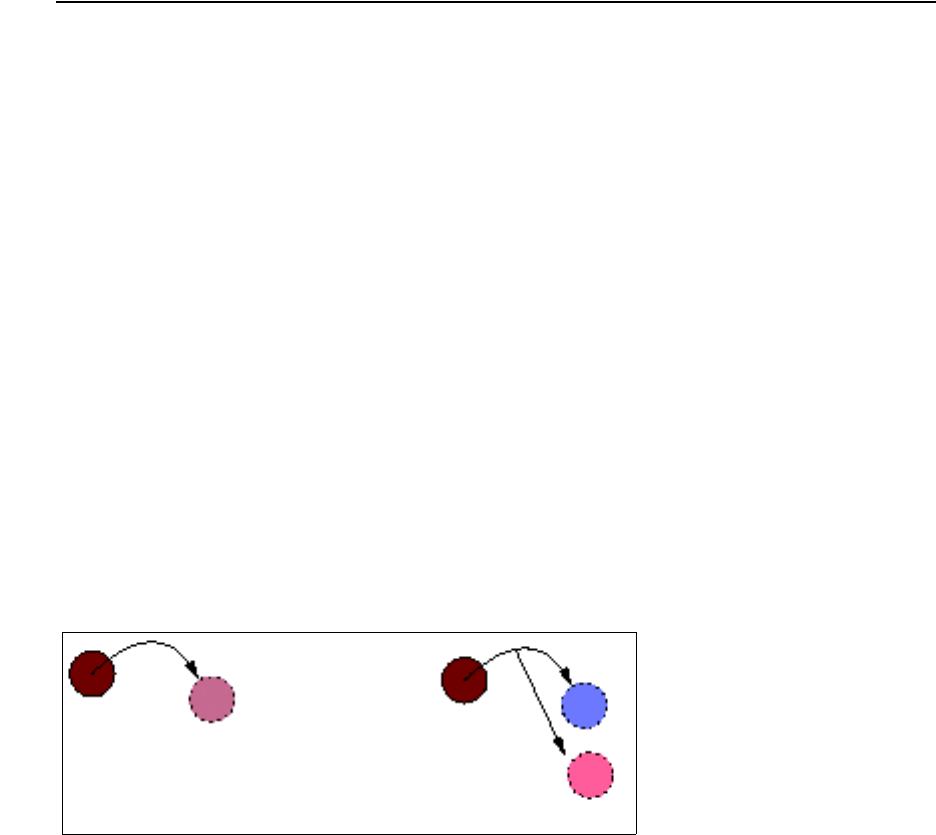
5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
418 Sentaurus™ Process User Guide
I-2013.12
Impurities
Isolated impurities in Sentaurus Process KMC can be in a substitutional state or can be paired
with interstitials or vacancies. Substitutional impurities are electrically active and typically
immobile. The acceptor and donor impurities (Groups III and V of the periodic table,
respectively) can move in silicon only by pairing with an interstitial or a vacancy, as shown in
the literature [4][5][6][7][8]. Other impurities, such as fluorine, may diffuse without the aid of
an extra I or V (see Impurities Diffusing without Pairing on page 525).
Impurity atoms are modeled like interstitials or vacancies. They have a position and a defect
type and particle type. The defect type is PointDefect, and the particle type characterizes the
species, charge state, and the presence of a paired I or V. For example, BiM indicates a
negatively charged boron paired with an interstitial.
NOTE Sentaurus Process KMC assumes interstitial particles and substitutional
particles paired with an interstitial as the same configuration; in other
words, Bi is the same as IB or BI, and there is only one position (three
coordinates) for it.
Paired impurities can perform two possible types of events (see Figure 45):
■Diffusion jump
■Breakup of the impurity–defect pair
Figure 45 Impurity pairs can diffuse or break up into a substitutional
plus an interstitial or a vacancy
NOTE Fluorine is modeled as an interstitial particle (in the default parameters,
not in Advanced Calibration). Consequently, fluorine diffuses without
pairing. See Impurities Diffusing without Pairing on page 525.
Migration (Diffusion)
The diffusion event is defined as for point defects (see Eq. 656, p. 415). Nevertheless, the
equation defines an instant diffusivity that is different from the effective diffusivity. Effective
I
B
Migration Breakup
Bi
Bi
5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
Sentaurus™ Process User Guide 419
I-2013.12
diffusivity measured in experiments involves a large number of microscopic migration steps
and long times. Microscopically, dopants diffuse using the kick-out mechanism. For example,
when an interstitial reacts with a substitutional boron, a boron–interstitial pair is generated:
. In contrast with the boron in substitutional position, the generated pair is mobile.
Then, begins to diffuse, using the diffusivity parameters specified in Eq. 656. After some
time, the interstitial boron breaks up, releasing the interstitial. This boron will not move until
a new incoming I reacts with it. Consequently, the macroscopic diffusivity is related not only
with the boron interstitial diffusivity, but also with its breakup frequency as:
(657)
depends on the Bi and I migration prefactors and on the Bi breakup prefactor.
is related with the Bi microscopic migration energy , the formation energy
of an interstitial and the Bi binding energy (assuming there are no stress or
SiGe corrections):
(658)
Finally, the total boron diffusivity is given as the sum of the contribution of all mobile species.
For boron interstitial, and assuming there are three mobile species, negative, neutral, and
positive:
(659)
where represents the microscopic pair diffusivity of charge .
Breakup
The breakup event for an interstitial-impurity pair can be described as:
(660)
where is an interstitial-impurity pair, is a substitutional impurity atom, and is an
interstitial silicon atom. This breakup event happens with a frequency given by:
(661)
where is the prefactor and is the activation energy, defined as the binding energy plus
the migration energy of the emitted species and the SiGe and stress corrections:
BI+Bi
→
Bi
Deff B() D0
eff B() Ediff B()
kBT
--------------------–
exp=
D0
eff B()
Ediff B()
EmBi
()()
EfI()()
EbBi
()()
Ediff B() EmBi
()EfI() EbBi
()–+=
DB() DB
i
-
()
Bi
-
[]
B-
[]
---------- DB
i
0
()
Bi
0
[]
B-
[]
-----------DB
i
+
()
Bi
+
[]
B-
[]
-----------++=
DB
i
x
()
x
AiAsI+↔
Ai
As
I
νbk ν0bk,Ebk kBT()⁄–()
1
3
---∆–Em
ikBT()⁄()exp
i
xyz,,
exp⋅=
ν0bk,
Ebk
5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
420 Sentaurus™ Process User Guide
I-2013.12
(662)
where:
■ is the hydrostatic pressure, computed as the mean value of the
principal stresses.
■ are the activation volumes for the formation energies.
■ is the germanium concentration.
■ accounts for the variation of the formation energy with the germanium concentration.
The corrections to the migration energies induced by the stress and SiGe are (as previously
explained in Interstitials and Vacancies on page 415):
(663)
Sentaurus Process KMC assumes that the activation volume and the SiGe variation for the
formation energy do not depend on the charge. All the charge states of the same species share
the same activation volume and SiGe dependencies for the formation energy.
Figure 46 shows the energies for boron involved in this mechanism. It is easy to deduce that a
change in the formation energies due to stress and SiGe will change the binding energy as:
(664)
Figure 46 Energies involved in the kick-out mechanism for B–+ I0 = Bi–. Migration energy of
the interstitials and boron interstitial are specified in the parameter database as
Em. The binding energies are Eb, and the I formation energy is specified as
Eform. The formation energy for dopants in pure silicon is assumed to be 0
because the dopants are already in the simulation; the dopants are not created
by the system.
Ebk Ai
() EbAi
()EmI() P∆VfI() ∆VfA() ∆VfAi
()–+()Ge[]α(fI() α
fA() α
fAi
())–+++ +=
P1
3
---–
σ'xσ'yσ'z
++()=
∆Vf
Ge[]
αf
∆Em
iσ'j∆Vpar αmGe[] σ'i∆Vort
()
ij≠
++=
∆EbBi
() ∆EfI() ∆EfB() ∆EfBi
()–+=
Em (Bi)
Em (I)
Eb (Bi)
Ef (Bi)
Em (I)
Ef (I)
Ef (B)
5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
Sentaurus™ Process User Guide 421
I-2013.12
Percolation
In a percolation event, an impurity can react with any other defect in its neighborhood without
need for diffusion. In this aspect, it can simulate the reactions that occur through distortions in
the lattice but without the need for the migration of particles. The neighborhood of the particle
is defined in the same way as for diffusing point defects.
The percolation rate, that is, the frequency at which the particle attempts to interact with any
valid defect in its neighborhood, is defined as:
(665)
where and are the prefactors for percolation, specified as input parameters. Percolation
only applies to substitutional dopants or impurities. It can provide an extra mechanism for
dopant deactivation at very high concentrations.
Parameters
The dopant diffusion parameters are stored in the parameter database for each material and
dopant, under the names Dm, Em for diffusivities, and Db, Eb for binding energies. Dm and Db
are prefactors, Em and Eb energies. The activation volumes for the formation energies have the
name VF and is called EfGe. The prefactor and the activation energy for percolation are
called D0_Percolation and E0_Percolation, respectively.
The following parameters can be changed:
pdbSet KMC <material> <Dopant> Dm <particle type> <value>
pdbSet KMC <material> <Dopant> Em <particle type> <value>
pdbSet KMC <material> <Dopant> Db <particle type> <value>
pdbSet KMC <material> <Dopant> Eb <particle type> <value>
pdbSet KMC <material> <Dopant> VF <particle type> <value>
pdbSet KMC <material> <Dopant> VD <particle type> <value>,<value>
pdbSet KMC <material> <Dopant> EfGe <particle type> <value>
pdbSet KMC <material> <Dopant> D0_Percolation <value>
pdbSet KMC <material> <Dopant> E0_Percolation <value>
For the migration energy, the prefactor, the SiGe dependency, and the activation volumes for
stresses, the specified material must be modeled as full or simple (in other words, any
material that does not discard particles). For binding energies, only the full materials are
valid. Percolation parameters are applied to simple and full materials.
Immobile species (substitutional dopants) have the migration prefactor set to 0, and the
migration energy high, to clarify that the species will not perform diffusion steps. Finally, since
Sentaurus Process KMC assumes substitutional atoms to be ionized (in other words, B– and
νper Pper Eper
kBT
---------–
exp=
P
E
αf
5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
422 Sentaurus™ Process User Guide
I-2013.12
As+), the binding parameters (both the prefactor and the binding energy) are only defined for
pairing reactions with a neutral I or V, like or . The binding
energies for the other breakup reactions are computed automatically using these parameters.
Parameter Examples
Silicon migration energies of boron particles:
sprocess> pdbGet KMC Si B Em
B 5.
BiM 0.5
Bi 0.25
BiP 1.1
Prefactors of the above energies:
sprocess> pdbGet KMC Si B Dm
B 0.
BiM 1.e-3
Bi 1.e-3
BiP 1.e-3
Migration energies for boron in oxide. The only allowed boron particle is B.
sprocess> pdbGet KMC Oxide B Em
B 3.53
Binding energy of boron in silicon:
sprocess> pdbGet KMC Si B Eb
BiM 0.3
and prefactor:
sprocess> pdbGet KMC Si B Db
BiM .37
Activation volumes for the formation energies of boron interstitial:
sprocess> pdbGet KMC Si B VF
BiM -0.0044
Variation of formation energy with Ge concentration:
sprocess> pdbGet KMC Si B EfGe
0
B–I0
+Bi
–
→
As+V0
+AsV+
→

5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
Sentaurus™ Process User Guide 423
I-2013.12
Finally, the binding energy cannot be defined for any material but full model ones, the result
should be blank:
sprocess> pdbGet KMC PolySilicon B Eb
NOTE Yo u c an c hange the se pa ra met er s whenever necessary to calibrate
intrinsic and extrinsic dopant diffusivity under equilibrium conditions.
For nonequilibrium conditions, you also can change the extended
defects if necessary.
Hopping Mode
The parameter KMC HoppingMode controls the way Sentaurus Process KMC performs
diffusion events. This mode accepts the modes short, long, double, and longdouble, and
it is set by default to longdouble. Changing the hopping mode only changes the results
statistically (in other words, it is similar to changing the random seed); although, it may change
the CPU time significantly. The default hopping mode, longdouble, is the fastest one.
The short Mode
The short mode implies that the jumping distance for all the diffusion events is the same
and is equal to the second neighbor distance. In addition, only one diffusion event is performed
at a time.
The long Mode
The long mode implies that the code increases the hopping distance to , being an integer
number, in regions where there are no particles with which to interact. This increase improves
the performance of the code in this area (in theory, by a factor of ). In practice, the overall
speed improvement is a factor of 2 or smaller, mainly because the empty regions where there
are no particles to interact with are limited. The overhead is caused by the long mode
implementation.
Figure 47 Even if the long hop model is available, it is used only for particles diffusing
on empty volumes
λ
nλ
n
n2
Standard ‘short hop’
model
Volume suitable for using
the ‘long hop’ model

5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
424 Sentaurus™ Process User Guide
I-2013.12
The double Mode
Setting HoppingMode to double allows Sentaurus Process KMC to perform two diffusion
events in one. Nevertheless, to properly account for interactions, the intermediate diffusion
event is still simulated by Sentaurus Process KMC. Using this hopping mode saves 10% or less
of CPU time.
The longdouble Mode
The longdouble mode is the default mode and enables both long and double hops.
NOTE For more information on these models, refer to the literature [9].
Enabling and Disabling Interactions
Interactions between particles are important in Sentaurus Process KMC. Whenever one mobile
particle jumps into another particle, Sentaurus Process KMC tries to make both particles
interact. These interactions may or may not be possible depending on whether the interaction
is allowed and if it is energetically possible.
The interactions allowed between one mobile particle and other particles are specified in the
parameter database as the parameter ReactionsPointDefect. The interactions between this
type of defect are assumed to be always energetically favorable.
Consequently, changing the parameter ReactionsPointDefect is the only way to establish
whether a moving particle will interact with other (mobile or immobile) particles. To change
this parameter, use:
pdbSet KMC <material> <dopant> ReactionsPointDefect <string> <true/false>
This parameter needs a string and Boolean value. The Boolean value specifies if the interaction
is allowed (true) or not (false). The string contains the name of the two interacting particles,
separated by a comma. For example:
sprocess> pdbGet KMC Si C ReactionsPointDefect
C,I true
C,V false
C,Ci true
Ci,I true
Ci,V true
Ci,Ci true
5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
Sentaurus™ Process User Guide 425
I-2013.12
Therefore, in this example, the interaction between C and V is disabled. To enable it, use the
command:
pdbSet KMC Si C ReactionsPointDefect C,V true
When enabling an interaction, the result does not have to be specified because Sentaurus
Process KMC already knows it. The possible interaction results are:
The reactions for each single charged state must be introduced, so the charged I also interacts
with V and with other charged states of V:
sprocess> pdbGet KMC Si I ReactionsPointDefect
I,V true
I,VM true
I,VMM true
I,VP true
I,VPP true
IP,V true
IM,V true
IM,IP true
(...)
All interactions are listed in the parameter database. With that list of interactions, you can
understand which reactions are considered and how they work.
Interaction Rules
Sentaurus Process KMC does not accept all possible interactions within every two particles,
but only interactions with a physical meaning, or with an available model. Consequently, the
following rules apply:
■Reactions must include existing particles.
■Some reactions are only allowed in materials with full modeling.
PointDefect
Pairing reactions, in other words, dopants pairing with interstitials or
vacancies to generate dopant-interstitial or dopant-vacancy particles.
For example, B + I, or As + IM.
AmorphousPocket
Reaction of interstitials or vacancies between them.
For example, I+ IM, I+ V, or V + V. They involve both damage
formation (mixed I and V) and small cluster creation (only I or V).
ImpurityCluster
Reactions involving the formation of impurity clusters, in other words,
when the result has dopants and interstitials or vacancies with two or
more of each. For example, Bi + I or AsV + AsV.
5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
426 Sentaurus™ Process User Guide
I-2013.12
■Reactions for a pair must be defined in the file of the involved dopant (for example, a
reaction with Bi must be in the boron file, not in interstitial).
■If the result of a reaction does not exist, the reaction is discarded (in other words, the
reaction C + V is specified, but the particle CV is not defined).
■Repulsive reactions are not allowed (for example, Bi– + Bi
–) except for ‘percolation’
models such as As + As or B + B (see Percolation on page 477).
■Reactions must satisfy microscopic reversibility. For example, if the reverse reaction is not
possible, the reaction is discarded.
■Reactions creating impurity clusters must give a defined cluster. For example, Bi + Bi is
allowed as long as there is a B2I2 cluster defined; in this case, Bi + BiM would also be
allowed.
■Only reactions producing defined PointDefect, ImpurityCluster, or
AmorphousPocket are allowed. For example, Bi + C will produce an error message if
there is no BCI cluster defined.
Examples
The interactions for boron are:
sprocess> pdbGet KMC Si B ReactionsPointDefect
B,I true
Bi,I true
Bi,V true
BiM,V true
BiP,V true
B,Bi false
B,BiP true
B,IP true
Bi,Bi true
Bi,VM true
Bi,VMM true
Bi,VP true
Bi,VPP true
BiM,VP true
BiM,VPP true
BiP,VM true
BiP,VMM true
B and I can react, giving a mobile Bi particle. B and IP also give a Bi particle. The charge state
of the resulting Bi particle is computed automatically by Sentaurus Process KMC depending
on the Fermi level, temperature, and Bi levels in the band gap. B– + IM is an electrostatically
repulsive reaction, and is not allowed.

5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
Sentaurus™ Process User Guide 427
I-2013.12
Bi possible charge states are neutral, positive, and negative. The reactions for these also states
should be specified. Bi and its different charges can react with I, V, and Bi. Bi + I produces an
impurity cluster. Only reactions microscopically reversible are allowed. Because a BI2 cluster
breaks up as Bi + I, any nonrepulsive reaction involving Bi and I is allowed. Bi + V recombines
the IV pair, depositing substitutional boron. All nonrepulsive reactions between Bi and V are
allowed, and all are specified in this example. Finally, there are more ways to produce impurity
clusters including BiP + B, producing B2I, and , as long as giving
B2I2.
The reaction B + V is not specified here. Typing B,V false produces the same effect. Setting
this reaction to true implies defining a BV particle (and its parameters) and specifying
reactions for this BV particle, such as .
NOTE Only advanced users should change the default interaction list because
improper modifications can drastically change the diffusion models.
Defining Nonstandard Interactions
Sentaurus Process KMC allows you to define a nonstandard interaction. These interactions are
intended to provide a mechanism for exceptional models that are not possible to be
implemented using the standard models and interactions. These reactions are of the type:
(666)
A, B, and C must be single particles (point defects or dopants). They are defined as
SpecialReaction in the folder including the first species:
pdbSet KMC Si A SpecialReaction A,B,C true
The reactions defined with this mechanism are not reversible: C will not break into A and B
back.
Interaction Rules
The difference between a regular interaction and a nonstandard one is that the set of rules the
latter one obeys is a very reduced subset of the rules for the regular one. In particular, a
nonstandard interaction must follow only these rules:
■Reactions must include existing particles. The result is always a point defect, not a cluster
or another defect type.
■Reactions are only allowed in materials with full modeling.
■Reactions must be defined in the files of the involved dopant (for example, a reaction with
Bi must be in the boron file).
Bi
aBi
b
+B2I2
↔
ab⋅0≤
BVI+B→
AB+C→

5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
428 Sentaurus™ Process User Guide
I-2013.12
■If the result of a reaction does not exist, the reaction is discarded (that is, the reaction C +
V is specified, but the particle CV is not defined).
In particular, these reactions can be nonconservative. For example, you can define a carbon–
interstitial interaction giving arsenic ( ). These reactions are nonstandard because
they lack a physical sense, but they are allowed because they offer extra flexibility to define
new models.
Example
A model for nitrogen diffusion can be defined using a nonstandard interaction, in particular,
when you want to model the following:
(667)
(668)
where is mobile but is immobile. The second reaction is not a problem. You can
define a dopant called Nn to be , and make it mobile, and you can define an NnV as the
result of Nn + V. These reactions are standard. The problem is that it is impossible to have a N
+ N reaction giving as Nn using the standard mechanisms. For this exception, you define as
N and use the special reaction:
pdbSet KMC Si N SpecialReaction N,N,Nn true
NOTE Special reactions are printed in the log file:
KMC. Using special non-reversible reaction N + N -> Nn
Stress Effects on Point Defects, Impurities, Dopants, and
Impurity-Paired Point Defects
The stress model for Sentaurus Process KMC is disabled by default, but can be enabled by
setting the variable KMC Stress to 1:
pdbSet KMC Stress 1
or by adding the parameter kmc.stress in the command line of diffuse:
diffuse kmc.stress time=...
Sentaurus Process KMC uses the stress provided by Sentaurus Process, but Sentaurus
Process KMC does not compute it. The stress fields are updated from Sentaurus Process for
each diffuse step.
CI+As→
NiNi
+N2i,
→
N2i,VN
2V↔+
N2i,
N2V
N2i,
Ni
5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
Sentaurus™ Process User Guide 429
I-2013.12
Stress local dependency is introduced into Sentaurus Process KMC using the correction of the
migration and binding energies of point defects and impurity-paired point defects.
Stress also affects the bandgap narrowing, as explained in Bandgap Narrowing on page 500.
Migration Energy
An anisotropic correction to the migration energy is introduced as:
(669)
where are the corrections to the migration energy when diffusing in the axis; are
the principal stresses; and and are the activation volumes for diffusion parallel
and perpendicular to stress, respectively. They are included in the PDB as VD.
The relation between the axes and the standard ones is established by a rotation R tensor. This
tensor diagonalizes the stresses tensor:
(670)
The default setting of the parameter ChangeAxis is false which disables this rotation, using
the standard xyz axis instead of the ones.
NOTE Setting ChangeAxis to true changes the direction of hopping
depending on the local stresses. This, in turn, dramatically impacts
{311} dissolution because the structure is sensitive to the direction of
the migrating incoming particles.
For more information, see Interstitials and Vacancies on page 415.
Binding Energy
The binding energy of an impurity-paired point defect Ai is corrected by:
(671)
where is the activation volume for the formation energy. The activation energy for an
impurity-paired point-defect breakup is defined (without stress) as the sum of the binding
∆Exm
∆Eym
∆Ezm
∆Vpar ∆Vort ∆Vort
∆Vort ∆Vpar ∆Vort
∆Vort ∆Vort ∆Vpar
σ'x
σ'y
σ'z
=
∆Eim
i'
σ'i
∆Vpar
∆Vort
i'
R[]
σxx σxy σxz
σxy σyy σyz
σxz σyz σzz
R[]
T
σ'x00
0σ'y0
00σ'z
=
i'
∆EbAi
() 1
3
---σxx σyy σzz
++()∆VfI() ∆VfAi
()–()=
∆Vf

5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
430 Sentaurus™ Process User Guide
I-2013.12
energy plus the migration energy of the emitted species (I for Ai). Then, an extra correction of
the migration energy of the emitted species under stress is needed.
Since the migration energy corrections depend on the axis, but the breakup of an impurity pair
in Sentaurus Process KMC does not, an average of the corrections for all the axes is performed,
and the frequency is computed as:
(672)
where is the breakup frequency when there is no stress.
For more information, see Impurities on page 418.
Alloys
Alloys are included in Sentaurus Process KMC simulations as a field instead of as a particle.
Using Ge as an example of an alloy in Si, it means that Sentaurus Process KMC discards the
particular position of Ge (the xyz coordinates) and only keeps track of how many Ge atoms
were introduced in each internal element. Doing this saves a huge amount of memory, while
allowing Sentaurus Process KMC to account for SiGe effects. Ge is handled as usual, except
for the following limitations:
■There are no Ge particles in the atomistic 3D plot.
■Ge (including implanted Ge) is shown as a field in the atomistic 3D plot.
■There are no events or reports associated with Ge because there are no Ge particles.
■There are no Ge models (for example, no Ge clusters) except for Ge diffusion.
This model can be used to:
■Simulate Ge diffusion.
■Include corrections to the migration and formation energies of point defects and impurity-
paired defects when diffusing in SiGe materials.
■Include bandgap corrections due to SiGe.
NOTE Sentaurus Process KMC considers the effect of germanium whenever
germanium is present. Continuum parameters (such as Silicon
SiliconGermanium.ConversionConc) do not affect Sentaurus
Process KMC simulations.
νstressbk νbk
0∆EbkBT()⁄–()
1
3
---∆Em
ikBT()⁄–()exp
i
xy z,,
exp=
ν0bk
5: Atomistic Kinetic Monte Carlo Diffusion
Point Defects, Impurities, Dopants, and Impurity-paired Point Defects
Sentaurus™ Process User Guide 431
I-2013.12
Alloy Diffusion
Ge will be used as an example of an alloy in Si material. Here, the Ge model diffusion
implemented has been based partially on [10]. This model defines the diffusion of Ge in an
indirect way through the use of of Is and Vs for a Si1–xGex material as:
(673)
where and are the transport capacity associated with Ge and Si interstitials in
Si1–xGex, respectively.
It can be assumed that these s follow the equation:
(674)
where , , , and are input parameters specified in the PDB. Both interstitials
and vacancies have a different .
NOTE The Ge diffusion model is switched on by default. To switched it off, set
and to null values.
Parameters and Parameter Examples
The parameters , , , and are specified in the PDB with the names
D0alphaSi, D0alphaGe, EalphaSi, and EalphaGe. In particular:
sprocess> pdbGet KMC Si I EalphaSi
0.4
sprocess> pdbGet KMC Si I EalphaGe
0
sprocess> pdbGet KMC Si I D0alphaSi
35
sprocess> pdbGet KMC Si I D0alphaGe
2.2
sprocess> pdbGet KMC Si V EalphaSi
0.25
sprocess> pdbGet KMC Si V EalphaGe
0
sprocess> pdbGet KMC Si V D0alphaSi
30
sprocess> pdbGet KMC Si V D0alphaGe
2.2
α
αI
DI
Ge x()
DI
Si x()
------------------=
DI
Ge x()
DI
Si x()
α
αx() α
0Si,
1x–α0Ge,
x1x–()ESi xEGe
+
kBT
-------------------------------------------–
exp=
α0Si,
α0Ge,
ESi
EGe
α
α0Si,
α0Ge,
α0Si,
α0Ge,
ESi
EGe

5: Atomistic Kinetic Monte Carlo Diffusion
Damage Accumulation Model: Amorphous Pockets
432 Sentaurus™ Process User Guide
I-2013.12
Alloy Effects
The following sections discuss alloy effects on point defects, impurities, dopants, and impurity-
paired point defects. In the next sections, Ge in silicon is used as an example for these models.
Migration and Formation Energies
The corrections and are added to the migration and
formation energies, respectively. is the germanium concentration, and are the
dependencies of energies with Ge concentration for migration and formation.
Binding Energy
The binding energy of an impurity-paired point defect is corrected by:
(675)
For more information, see Impurities on page 418.
Bandgap Narrowing
The Ge inclusion changes the band gap as explained in Bandgap Narrowing on page 500.
Introducing Alloys in the Simulation
Alloys can be introduced in the simulation by:
■Using implantation.
■Using the select command.
■Atomizing a previous continuum structure with the alloy.
■Using kmc add to explicitly add it.
■Depositing a layer doped with the alloy.
Damage Accumulation Model: Amorphous Pockets
Damage accumulation evolution, that is, the evolution of small interstitial and vacancy clusters
after ion implantations, is a crucial step that affects the subsequent formation of extended
defects and impurity clusters. This accumulation generates the transient-enhanced diffusion
(TED) of commonly used dopants, such as boron.
Experimentally, electron irradiation and light-ion implantation create isolated point defects
inside the silicon. In contrast, heavy ions generate highly disordered regions called amorphous
∆EmαmGe[]=
∆EfαfGe[]=
Ge[]
αmαf
,
Ai
∆EbAi
() Ge[]α
fI() α
fA() α
fAi
())–+()=
5: Atomistic Kinetic Monte Carlo Diffusion
Damage Accumulation Model: Amorphous Pockets
Sentaurus™ Process User Guide 433
I-2013.12
pockets (APs) as a consequence of the implanted cascades. Depending strongly on the
temperature, ion mass, and dose rate, this disordered region can dissolve as a result of internal
recombination or can grow until an amorphous layer is created. The activation energy for
annealing this damage varies in the literature, 0.9 eV [11] at room temperature, 1.2 eV for
to , but it is much less than the 2.7 eV reported for truly amorphized amorphous
layers. This means the damage accumulation depends on the dynamic annealing, ripening, and
dissolution history of the APs during the implant process. This annealing can have a quasi-
continuum range of activation energies.
There is much discussion on how this damage is annealed. Some papers point to an annealing
of the disordered region [12]: APs using an internal recombination of IV pairs rather than
through the emission of point defects. Only when the AP does not contain further IV pairs does
it begin to emit its remaining Is or Vs, behaving as a small I or V cluster.
Sentaurus Process KMC simulates the damage accumulation using APs, disordered collections
of point defects (Is and Vs) stable at low temperatures. APs dissolve fast at higher temperatures,
leaving only clusters with the net excess of Is or Vs. APs can contain IV pairs or only Is and Vs.
In the first case, APs try to recombine the pairs; in the second, APs behave as small clusters
and can emit their constituent particles. Whenever an AP containing only Is or Vs (but not IV
pairs) reaches a threshold size, the AP transforms into an extended defect ({311}s for Is, voids
for Vs).
APs can grow capturing new incoming point defects, and they can dissolve by internal
recombination of IV pairs or by particle emission when there are no more IV pairs (see
Figure 48).
Figure 48 Growth of APs showing their internal recombination
400°C
550°C
Growth
Growth
2D Projection
Internal Recombination
Amorphous Pocket
5: Atomistic Kinetic Monte Carlo Diffusion
Damage Accumulation Model: Amorphous Pockets
434 Sentaurus™ Process User Guide
I-2013.12
Shape
APs have an irregular shape. Sentaurus Process KMC does not reshape the defect as new;
incoming particles join the AP: particles are left in their incoming positions. Figure 49 shows
some APs resulting from an implanted cascade.
Figure 49 APs formed in Sentaurus Process KMC after some implanted as cascades:
interstitials are red, vacancies are green, arsenic is yellow
Growth
APs capture any incoming point defect (I or V) within their capture radius. The capture radius
of APs is the sum of all their constituent particles. Point defects with any charge state are
captured by APs containing both Is and Vs. Only neutral Is or Vs are captured when APs
contain only Is or only Vs.
Recombination
APs containing IV pairs (that is, APs with both interstitials and vacancies) can recombine pairs
using a recombination event, which recombines one I with one V at a time. This event is
performed with a frequency given by:
(676)
where the prefactor is called D0_AmorphousPocket in the parameter database, and
size is the size of the cluster.
νdiss ν0diss,sizeβEdiss size()P∆Vdiss αIV Ge[]++()–/kBT()()exp⋅=
ν0diss,
5: Atomistic Kinetic Monte Carlo Diffusion
Damage Accumulation Model: Amorphous Pockets
Sentaurus™ Process User Guide 435
I-2013.12
The size of a cluster InVm is a Tcl procedure of n and m specified in the file KMC.tcl under the
name getAmorphousPocketSize:
fproc getAmorphousPocketSize { sizeI sizeV } {
return [expr ($sizeI + $sizeV)/2]
}
It can be modified by users. is an exponent called ExponentAmorphousPocket.
depends on the size of the AP and is specified as a list of energies for each size
(Eb_AmorphousPocket). The different sizes are specified as IxeVx. If some energy is not
specified for a size, Sentaurus Process KMC takes the linear interpolation between the last two
specified values. For sizes higher than the last specified size, the last specified energy is
assigned automatically. is the Germanium correction to recombination
(Eb_AmorphousPocketGe) and finally, is the hydrostatic pressure and is the
activation volume for the AP dissolution (VFAmorphousPocket).
Parameters
The parameters needed by the damage accumulation model are specified using:
pdbSet KMC Si Damage <Parameter> <value>
An example of these parameters is:
sprocess> pdbGet KMC Si Damage D0_AmorphousPocket
0.0005
sprocess> pdbGet KMC Si Damage ExponentAmorphousPocket
0.66
sprocess> pdbGet KMC Si Damage VFAmorphousPocket
0
sprocess> pdbGet KMC Si Damage Eb_AmorphousPocket
IV 0.65
I199V199 2.4
sprocess> pdbGet KMC Si Damage Eb_AmorphousPocketGe
0
For the above parameters, the Eb_AmorphousPocket values for IxVx, with x > 1 and x < 199
will be generated by Sentaurus Process KMC as a linear interpolation between the points (1,
0.65) and (199, 2.4):
Silicon/Damage Eb_AmorphousPocket(IV) = 0.65
Silicon/Damage Eb_AmorphousPocket Interpolated (2) = 0.658838
Silicon/Damage Eb_AmorphousPocket Interpolated (3) = 0.667677
...
NOTE Yo u c an ch an ge the se pa ram et ers to calibrate the damage accumulation
model.
β
Ediss
αIV
P
∆Vdiss
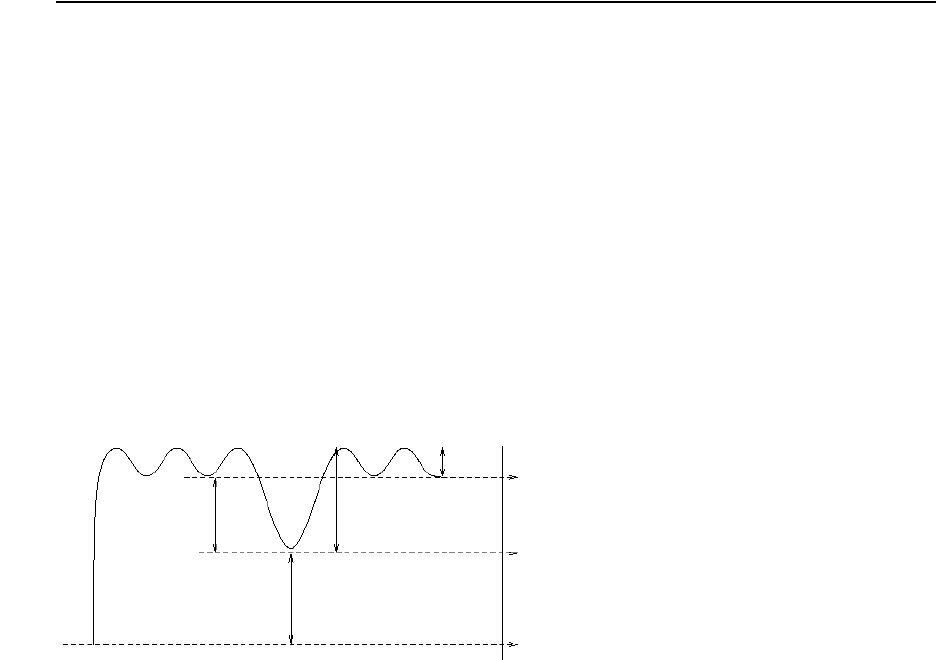
5: Atomistic Kinetic Monte Carlo Diffusion
Damage Accumulation Model: Amorphous Pockets
436 Sentaurus™ Process User Guide
I-2013.12
NOTE The maximum size allowed for IV clusters is I249V249.
Emission
When all IV pairs have been recombined, APs behave as small I or V clusters, allowing the
emission of their extra constituent particles. These defects emit neutral Is or Vs particles with
a frequency given by:
(677)
The prefactor is proportional to the input parameter D0_Cluster, but also includes a
dependency on the size of the cluster. The activation energy for emission of an X (in other
words, either I or V) is:
(678)
the sum of the corrected binding energy (that depends on the cluster size) and the migration
energy. The cluster size is defined as the number of contained Is or Vs (see Figure 50).
Figure 50 Energies involved in emission of an interstitial from an n-size cluster
D0_Cluster is the constant proportional to the emission prefactor, and Eb_Cluster is the
cluster binding energy, where dependency with size is explicitly assigned. For sizes bigger than
the last specified cluster, the binding energies are computed using:
(679)
where:
■ (Eb_LargeCluster) is the binding energy for the largest cluster.
■ (Eb_SmallestCluster) is the binding energy for the smallest cluster (size 1).
■ (exponent_Cluster) is the exponent, usually 2/3 or 3/4.
νemit ν0emit,E–emit size()/kBT()()exp⋅=
Eemit EbX() EmX() ∆EbX() ∆EmX()++ +=
Em
E(n–1) + E(1)
E(n)
E(n–1)
Eb E diss
Ef
Ebsize()EbL,EbL,EbS,
–()
sizeasize 1–()
a
–
2a1–
-------------------------------------------
–=
EbL,
EbS,
a
5: Atomistic Kinetic Monte Carlo Diffusion
Damage Accumulation Model: Amorphous Pockets
Sentaurus™ Process User Guide 437
I-2013.12
Figure 51 shows some binding energy values and compares them with the numbers obtained
using Eq. 679.
Figure 51 Interstitial cluster (left) and vacancy cluster (right) binding energies; discrete
values are assigned in the parameter database and the continuum energies are
computed using Eq. 679
Finally, the correction applied for the migration is the normal one:
(680)
and for the binding of the particle to the cluster is:
(681)
where is called VFCluster in the PDB and is the Germanium correction to
binding (Eb_ClusterGe).
Parameters
The parameters for Is and Vs emission are specified only for the silicon material. They can be
found in the interstitial and vacancy files included in the parameter database.
Prefactors
sprocess> pdbGet KMC Si I D0_Cluster
150.0
sprocess> pdbGet KMC Si V D0_Cluster
10
2.1
2.2
2.3
2.4
2.5
2.6
2.7
2.8
2.9
3
0 20 40 60 80 100 120
Binding Energy Eb [eV]
I Cluster Size
Interstitial Cluster Binding Energies
Discrete
Continuum energies
-0.5
0
0.5
1
1.5
2
2.5
3
3.5
0 5 10 15 20 25 30
Binding Energy E
b
[eV]
V Cluster Size
Vacancy Cluster Binding Energies
Discrete
Continuum energies
∆EmX() α
mGe[]
1
3
---∆Em
ikBT()⁄–()exp
i
xy z,,
+=
∆EbX() P∆Vb
extended X() α
Cl Ge[]+=
∆Vb
extended
αCl
5: Atomistic Kinetic Monte Carlo Diffusion
Damage Accumulation Model: Amorphous Pockets
438 Sentaurus™ Process User Guide
I-2013.12
Energies
sprocess> pdbGet KMC Si I Eb_Cluster
I2 2.45
I3 2.45
(...)
I13 2.853
I103 2.889
sprocess> pdbGet KMC Si V Eb_Cluster
V2 1.4
V3 1.4
V4 2.4
(...)
For sizes between specified sizes (for example, I14 to I102), the parameters are computed as
linear interpolations of the specified values:
Silicon/I Eb_Cluster(I13) = 2.853
Silicon/I Eb_Cluster Interpolated (14) = 2.8534
(...)
Silicon/I Eb_Cluster Interpolated (102) = 2.8886
Silicon/I Eb_Cluster(I103) = 2.889
and parameters for Eq. 678, p. 436:
sprocess> pdbGet KMC Si I Eb_SmallestCluster
2.51
sprocess> pdbGet KMC Si I Eb_LargeCluster
3.09
sprocess> pdbGet KMC Si I exponent_Cluster
0.75
sprocess> pdbGet KMC Si V Eb_SmallestCluster
1.5
sprocess> pdbGet KMC Si V Eb_LargeCluster
3.7
sprocess> pdbGet KMC Si V exponent_Cluster
0.6667
NOTE When changing these parameters, their values affect not only the
damage accumulation model, but also the interstitial and vacancy
supersaturation and, consequently, the transient-enhanced diffusion
(TED). Because the damage accumulation model is the seed for
subsequent extended defects or recrystallization, these values also affect
the formation of extended defects.

5: Atomistic Kinetic Monte Carlo Diffusion
Damage Accumulation Model: Amorphous Pockets
Sentaurus™ Process User Guide 439
I-2013.12
Amorphous Pockets Life Cycle
Whenever two point defects (I + I, V + V or I + V) interact with each other, an AP is generated.
When the AP is formed, subsequent incoming Is or Vs are captured and added to the AP. If the
AP contains at least one IV pair, the AP recombines the IV pairs. The IV pair frequency depends
on the number of IV pairs present in the AP. If there is only Is or Vs in the AP, it emits Is or Vs.
The evolution of APs can follow three paths:
■APs can dissolve recombining internal IV pairs, emitting point defects, or both.
■AP containing only Is or Vs may be transformed into extended defects: {311} defects for
Is and voids for Vs.
■If the concentration of some of the boxes (the internal Sentaurus Process KMC grid
elements) containing the AP reaches a concentration threshold, the element is considered
amorphous and its particles are removed from the AP. Consequently, APs only exist in
crystalline silicon.
For example, assuming there is an AP with two Vs and seven Is (I7V2), since the AP contains
both Is and Vs, the only possible event is the recombination of IV pairs. The first IV pair
recombines with the recombination energy assigned to size 2, leaving an I6V1. The second
recombination energy, with a recombination energy assigned to size 1, leaves an I5 AP. This
AP begins to emit Is, with a frequency associated to its size (5). However, if it captures a V, it
becomes an I5V1 and must recombine the IV pair with an associated recombination size of 1.
An AP must satisfy the following conditions before being transformed into a {311} or void:
■It can contain only Is or only Vs, but not both.
■It must be bigger than or equal to a threshold size.
■Transition must be enabled.
The threshold size is specified with the parameters Min311Size and MinVoidSize for Is and
Vs, respectively. The transition is enabled by a value between 0 and 1. This value is computed
as:
(682)
The prefactor is specified as D0_APto311 for {311}s and D0_APtoVoid for voids, and the
energies as E_APto311 and E_APtoVoid. For {311}s are the corrections for pressure and
Ge; for voids :
.(683)
The volume correction is called VF311toLoop, and the Germanium one VF311toLoop.
PE
0E∆E+()–()kBT()⁄()exp×=
E0
∆E
∆E0=
∆EPV
311toLoop α311toLoop Ge[]+=

5: Atomistic Kinetic Monte Carlo Diffusion
Damage Accumulation Model: Amorphous Pockets
440 Sentaurus™ Process User Guide
I-2013.12
For , the transition is disabled; for , it is enabled. For , the value is rounded
to 1. Values between 0 and 1 establish a probability for the transition.
Parameters
Minimum sizes for the transitions:
sprocess> pdbGet KMC Si I Min311Size
33
sprocess> pdbGet KMC Si V MinVoidSize
27
Transition probabilities:
sprocess> pdbGet KMC Si Damage D0_APto311
200000000.0
sprocess> pdbGet KMC Si Damage E_APto311
1.3
sprocess> pdbGet KMC Si Damage D0_APtoVoid
200000000.0
sprocess> pdbGet KMC Si Damage E_APtoVoid
1.3
Interactions of Amorphous Pockets
To change the default AP interactions, use the parameters ReactionsClusterI,
ReactionsClusterV, and ReactionsClusterIV. These parameters control the reactions
between APs containing only interstitials, only vacancies, or both. APs can react not only with
Is and Vs, but also with dopants. In this latter case, the result of the reaction must be specified.
Interaction with Point Defects: I and V
To customize AP reactions, change the parameters defined for I, V, and IV clusters using the
command:
pdbSet KMC Si Damage <ReactionsClusterType> <species> <true/false>
For example, the command:
pdbSet KMC Si Damage ReactionsClusterI I false
disables the reaction for small clusters.
P0=
P1=
P1>
InI+In1+
→
5: Atomistic Kinetic Monte Carlo Diffusion
Damage Accumulation Model: Amorphous Pockets
Sentaurus™ Process User Guide 441
I-2013.12
Consequently, it disables the ripening of these clusters. The line:
pdbSet KMC Si Damage ReactionsClusterV I false
disables the recombination of I with small vacancy clusters.
Parameters
Small interstitial and vacancy clusters may react with neutral interstitials and vacancies.
Charged interstitials or vacancies are not allowed due to microscopic reversibility reasons:
sprocess> pdbGet KMC Si Damage ReactionsClusterI
I true
V true
sprocess> pdbGet KMC Si Damage ReactionsClusterV
I true
V true
APs with both Is and Vs accept interstitials or vacancies with any charge. In this case, because
they do not emit particles, there are no microscopic reversibility restrictions:
sprocess> pdbGet KMC Si Damage ReactionsClusterIV
I true
IP true
IM true
V true
VP true
VPP true
VM true
VMM true
Interaction with Impurities
APs do not trap impurities, but can interact with them. In this interaction, impurities can lose a
point defect, becoming substitutional (for example, ) or can gain some
of them being transformed into an impurity cluster (for example, ).
Consequently, the interaction within impurities and APs plays a crucial role in deactivating
dopants, typically during implantation and low-temperature anneals.
To control these interactions, use:
pdbSet KMC Si <impurity> <ReactionsClusterType> <species,result> <true/false>
ReactionsClusterType can be ReactionsClusterI for small I clusters,
ReactionsClusterV for small V clusters and ReactionsClusterIV for mixed clusters.
BiI2V3
+BI3V3
+→
BiI2V3
+BI2IV3
+→

5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
442 Sentaurus™ Process User Guide
I-2013.12
For example, the reaction can be disabled for mixed clusters with:
pdbSet KMC Si B ReactionsClusterIV BiM,BI2 false
NOTE A comma must separate the incoming particle from the result, without
any space in between.
Parameters
The reactions between boron (for example) and mixed clusters can be displayed with:
sprocess> pdbGet KMC Si B ReactionsClusterIV
BiM,BI2 true
Bi,BI2 true
BiP,BI2 true
B,BI2 true
There are no reactions between boron and vacancy clusters:
sprocess> pdbGet KMC Si B ReactionsClusterV
The reactions between boron and small I clusters are disabled:
sprocess> pdbGet KMC Si B ReactionsClusterI
Bi,BI2 false
BiM,BI2 false
Extended Defects
Small clusters are defined as immobile agglomerations of interstitials or vacancies, and are
modeled using the AP defects previously explained. When the number of Is or Vs in these
clusters grows above a specified threshold, the small clusters are converted into extended
defects ({311} or void types). Finally, when the ripening of {311}s overcomes some limit, the
{311}s are transformed into dislocation loops.
{311} Defects (ThreeOneOne)
The {311} rod-like defects are associated with TED [13]. Consequently, they need a realistic
simulation, in both shape and energetic values. Its shape is like rectangular stripes of
interstitials lying on a {311} plane along a <110> direction. The paper [14] gives an atomic
model for its structure, whose stability has been confirmed by theoretical studies [15][16][17].
Bi
–InVm
+BI2In1–Vm
+→
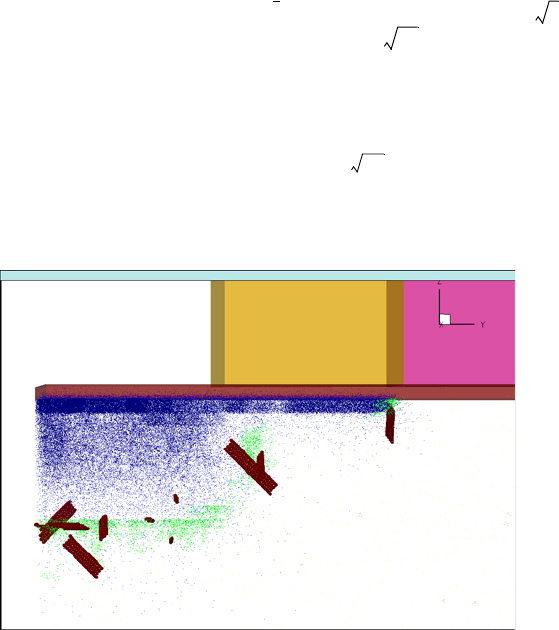
5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
Sentaurus™ Process User Guide 443
I-2013.12
Shape
Sentaurus Process KMC models {311} defects as parallel stripes (rows) of I particles lying in
one of the twelve orientations, randomly chosen, of a {311} plane. The {311} shape is modeled
as Nr rows of Is lying on a line with a distance of between Is in the same line,
and as Nc columns keeping a distance of between them, with , the
silicon lattice constant.
The ratio between length ( ) and width ( ) is given by:
(684)
being . This ratio is maintained reshaping the {311} defect (that is, changing the
number of row and columns) when necessary (see Figure 52).
Figure 52 {311} defects are simulated by Sentaurus Process KMC as parallel stripes (rows)
of I particles lying in a {311} plane: red is silicon interstitials in {311}; green is I
and V in APs; and blue is arsenic
The {311} defects only exist above a size threshold. Smaller defects are assumed to be APs,
and they have an irregular shape (see Amorphous Pockets Life Cycle on page 439).
When {311} defects grow enough, they are transformed into dislocation loop defects (see
Dislocation Loops on page 447). The threshold size (number of interstitials) between {311}
defects and dislocation loops is assumed to follow an Arrhenius plot:
(685)
The formation energy of the dislocation loop must be smaller than the {311} formation energy
at the threshold size; otherwise, the threshold is taken as the size where both energies are equal.
<011>
a2⁄
a22 4⁄
a0.543 nm=
L
W
WCL≈
C0.5 nm=
size prefactor Ek
BT()⁄()exp×=
5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
444 Sentaurus™ Process User Guide
I-2013.12
Both prefactor and E are parameters available in the database with names D0_311toLoop
and E_311toLoop, respectively.
Parameters
The parameters to control the transformation between {311}s and loops are specified for
interstitials in silicon:
sprocess> pdbGet KMC Si I D0_311toLoop
1.6
sprocess> pdbGet KMC Si I E_311toLoop
0.68
NOTE These parameters can be changed to fit the {311} to dislocation loop
transition size.
Capture
Each time a neutral I point defect interacts with an I belonging to a {311} defect, the {311}
captures the point defect. Since {311} defects grow and shrink at their ends, the new particle
is attached at the nearest end of the defect. When the end cannot grow because it is too close
to a interface or a boundary, the other end is used.
When 311DopantModel is set to 1, impurities also can be trapped. These trapped impurities
will remain in the captured location until they are re-emitted. Only neutral impurities (or
neutral impurity pairs) are captured and re-emitted.
Emission
To preserve microscopic reversibility between the capture and the emission processes, emitted
particles (neutral interstitials) are taken randomly from one of the two ends and released from
a random point at the {311} surface (see Figure 53 on page 445).
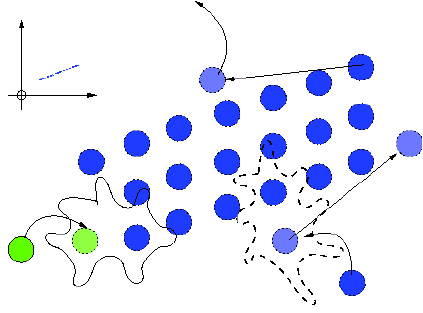
5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
Sentaurus™ Process User Guide 445
I-2013.12
Figure 53 Recombination of defects in a {311} defect
The emission frequency is computed as in APs by:
(686)
where the considerations for the AP emission, including all the corrections, apply. The binding
energies are taken from the list supplied with the Eb_Cluster parameter. These energies are
shared with the APs. As explained above, for sizes less than a threshold value, defects are
considered APs. Otherwise, they are rearranged as {311} defects. Consequently, only binding
energies for sizes equal or bigger than APs-{311} threshold applies for {311} defects.
Corrections are applied for both pressure and Germanium content. These corrections are
specified as VF311 and Eb_311Ge, respectively.
{311}s may also emit captured dopants if the 311DopantModel is set to 1. The emission
frequency for them is:
(687)
being the prefactor and activation energy called D0_311 and Eb_311, respectively, in the PDB.
Parameters
For impurity re-emission, the parameters are:
sprocess> pdbGet KMC Si In D0_311
200.0
sprocess> pdbGet KMC Si In Eb_311
2.0
2D Projection
Emission
Recombination Growth
νemit ν0emit,E–emit size()P∆V311 α311 Ge[]++()/kBT)()()exp×=
νemit Ai
() ν
0emit,Ai
() Eemit Ai
()kBT()⁄()–()exp×=
5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
446 Sentaurus™ Process User Guide
I-2013.12
Recombination
The {311} defects recombine incoming Vs with any charge by annihilation of the Is at the
nearest {311} defect end. When {311} defects dissolve, they do not become APs when the
threshold AP-{311} size is reached. The emission frequency depends on the binding energy,
and the binding energy only depends on the size of the defect. Since a interstitial cluster and a
small {311} defect have the same binding energy when they have the same size, the defect
shape affects only the capture volume, but not the emission frequency. Consequently,
rearranging {311} as small defects and vice versa only changes the capture volume, and these
changes are negligible for small clusters. Nevertheless, the capture volume differences between
small {311} defects and irregular clusters are negligible, and there is no information about the
shape of dissolving {311} defects.
Finally, when a {311} reaches size 2, it releases the particles as two interstitials and the {311}
disappears.
Interactions
Interactions between {311} defects and mobile particles can be modified with:
pdbSet KMC Si <I/V/Impurities> Reactions311 <species> <true/false>
Growth reactions ( ) are controlled with I, and recombination reactions ( ) with V.
{311} defects can break up paired dopants capturing the interstitial or recombining the vacancy
(for example, ). The remaining dopant will be immediately released or
captured (and re-emitted later) by the defect depending on the value of the parameter
311DopantModel (0 releases dopants, 1 traps it). A captured dopant can be re-emitted. These
reactions enable the {311} to decrease the impurity diffusion.
Only neutral Is react with {311} defects and, consequently, only paired dopants with the same
charge as the substitutional dopant react with {311} defects. Any charge state is allowed for
the recombination of vacancies.
Parameters
Use the following for growth and recombination:
sprocess> pdbGet KMC Si I Reactions311
I true
sprocess> pdbGet KMC Si V Reactions311
V true
InI+
InV+
BiIn
+BIn1+
+→

5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
Sentaurus™ Process User Guide 447
I-2013.12
For paired impurity breakup (boron, for example), use:
sprocess> pdbGet KMC Si B Reactions311
BiM false
sprocess> pdbGet KMC Si I 311DopantModel
0
You can define the parameter 311DopantModel globally as a default for all dopants, but
define it locally with a different value that overwrites the global value for a particular dopant.
For example:
pdbSet KMC Si I 311DopantModel 0
pdbSetDouble KMC Si B 311DopantModel 1
sets the model for all the impurities as ‘release dopant,’ except for boron.
Dislocation Loops
Dislocation loops are planar defects lying on {111} planes [18]. A dislocation loop can be
either a faulted dislocation loop (FDL) or perfect dislocation loop (PDL). FDLs are circular
stacking faults surrounded by a dislocation line. PDLs are not implemented in Sentaurus
Process KMC.
{311} defects are the precursors of dislocation loops. When the implant conditions (available
concentration of I, distance to the free surface) are appropriate, {311} defects grow until they
reach the threshold size and transform into dislocation loops. Dislocation loops are more stable
than {311} defects; consequently, the supersaturation created by dislocation loops is lower.
Shape
The shape of dislocation loops is computed as a filled circle in a {111} orientation (see
Figure 54). All {111} orientations are allowed, and one is randomly chosen.
Figure 54 A dislocation loop taken from a Sentaurus Process KMC simulation

5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
448 Sentaurus™ Process User Guide
I-2013.12
Capture
Dislocation loops capture any incoming neutral interstitial. The original position is lost, and
the particle is moved to the proper position in the disk. The capture radius is the sum of the
capture radius of the constituent particles.
When the ReactionsLoop is set and LoopDopantModel is true, dislocation loops capture
incoming impurities. When LoopDopantModel is false, the impurity is not captured.
However, when it carries a point defect (in other words, is an impurity pair), the pair is broken;
the impurity is deposited as a substitutional impurity; and the point defect reacts with the loop.
Emission
Dislocation loops emit neutral interstitials with a frequency given by:
(688)
includes both a prefactor and a linear dependency with the dislocation loop size, and
is the binding energy of the dislocation loop, which only depends on the size.
Sentaurus Process KMC computes the binding energies as:
(689)
The dislocation loop formation energies are taken from [19] as:
(690)
where:
■ is the loop radius.
■ is the stacking fault energy per unit area.
■ is the shear modulus.
■ is the Poisson ratio.
■ is Burger’s vector modulus.
■ is the silicon lattice parameter.
■ is the atomic density in a {111} plane, in .
The above parameters are specified in the parameter database. is called gamma, is mu,
is nu, and is named burgVectMod. The emission prefactor is called D0_Loop.
νemiss ν0emiss,
Ebloop,size()EmI() ∆EmI() ∆Ebloop,
++ +
kBT
----------------------------------------------------------------------------------------------------------–
exp×=
ν0emiss,
Ebloop,size()
Ebloop,size()EfI() Efloop,size 1–()Efloop,size()–+=
Efloop,size()πγR2a2µ
61 ν–()
--------------------R8R
b
-------
log⋅⋅ nEfI()–+=
Rsize πd111
()⁄=
γ
µ
ν
b
a
d111
nm 2–
γ
µ
ν
b
5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
Sentaurus™ Process User Guide 449
I-2013.12
The corrections applied to the migration energy of interstitials are the usual ones:
(691)
and for the binding of the particle to the loop is:
(692)
where is called VFLoop, and is called Eb_LoopGe in the PDB.
Captured impurities (when LoopDopantModel is true) re-emit impurities into the bulk with a
frequency given by:
(693)
being the prefactor and activation energy called D0_Loop and Eb_Loop in the PDB,
respectively.
Parameters
The parameters needed for the simulation of dislocation loops are defined for interstitials in
silicon:
sprocess> pdbGet KMC Si I D0_311toLoop
1.6
sprocess> pdbGet KMC Si I E_311toLoop
0.68
sprocess> pdbGet KMC Si I D0_Loop
1000000.0
sprocess> pdbGet KMC Si I gamma
0.4375
sprocess> pdbGet KMC Si I mu
472
sprocess> pdbGet KMC Si I nu
0.3
sprocess> pdbGet KMC Si I burgVectMod
0.3135
sprocess> pdbGet KMC Si I VFLoop
0
sprocess> pdbGet KMC Si Eb_LoopGe
0
∆EmI() α
mGe[]
1
3
---∆Em
ikBT()⁄–()exp
i
xy z,,
+=
∆Ebloop,P∆Vb
loop αloop Ge[]+=
∆Vb
loop
αloop
νemit Ai
() ν
0emit,Ai
() Eemit Ai
()kBT()⁄()–()exp×=
5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
450 Sentaurus™ Process User Guide
I-2013.12
For impurity re-emission, the parameters are:
sprocess> pdbGet KMC Si In D0_Loop
200.0
sprocess> pdbGet KMC Si In Eb_Loop
2.0
NOTE These parameters can be changed to fit the dislocation loop formation
and dissolution.
Figure 55 shows how a dislocation loop grows capturing interstitials, and how it shrinks
recombining incoming vacancies or emitting interstitials.
Figure 55 Emission, capture, and recombination of point defects in a dislocation loop
Interactions
The interactions between dislocation loops and mobile particles are:
■Growth reactions. Only with neutral Is:
pdbSet KMC Si I ReactionsLoop I <true/false>
■Recombination reactions. Any vacancy:
pdbSet KMC Si I ReactionsLoop <V/VM/VP/VMM/VPP> <true/false>
■Impurity pairs break up and interact with the associated point defect (I or V). Only with
pairs with the substitutional charge state the same as the substitutional dopant (for example,
Bi– for B–). The interstitial or vacancy is trapped or recombined, and the dopant is released
(0) or trapped (1) depending on the model used. The model is specified for all dopants
(default value) using the parameter LoopDopantModel for interstitials.
Dislocation Loop
Recombination
Emission
Growth
2D Projection

5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
Sentaurus™ Process User Guide 451
I-2013.12
This particular default can be overwritten for one particular dopant.
pdbSet KMC Si I LoopDopantModel <true/false>
pdbSetDouble KMC Si <dopant> LoopDopantModel <true/false>
Parameters
Loops trap interstitials, but the recombination of vacancies is disabled:
sprocess> pdbGet KMC Si I ReactionsLoop
I true
sprocess> pdbGet KMC Si V ReactionsLoop
V false
Loops can break up some paired dopants, for example, boron:
sprocess> pdbGet KMC Si B ReactionsLoop
BiM true
sprocess> pdbGet KMC Si I LoopDopantModel
0
Voids
Small vacancy defects have been reported (using paramagnetic resonance and
photoluminescence) [20][21][22][23]. Theoretical studies [24][25] indicate that some of these
small clusters can be particularly stable. Sentaurus Process KMC models these small clusters
as APs and, consequently, they have irregular shapes. Nevertheless, size-dependent binding
energies are considered for their V emission (see Damage Accumulation Model: Amorphous
Pockets on page 432).
Vacancy clusters appear as spheroidal voids when they are big enough to be seen by TEM [26].
Tight-binding molecular dynamics studies show that the binding energies are a function of the
cluster size [27] (see Figure 56 on page 452).
5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
452 Sentaurus™ Process User Guide
I-2013.12
Figure 56 Voids are simulated with a spherical shape; this one contains 654 vacancies
Shape
The threshold size between irregular small vacancy clusters (APs) and voids is specified with
the parameter MINVoidSIZE. Another parameter, MAXVOIDSDIAM, is used to set up the
maximum-allowed diameter (in nanometers) for these defects.
Reshaping the small clusters into voids above the mentioned limit is necessary to maintain the
right volume/surface ratio, as the V cluster grows. A large cluster of n vacancies is reshaped to
be spheroidal, occupying the volume corresponding to the same number of silicon lattice sites.
Sentaurus Process KMC manages the void shape to assert that its density is correct.
Parameters
The parameters for voids are specified for silicon material and vacancy as species:
sprocess> pdbGet KMC Si V MinVoidSize
27
sprocess> pdbGet KMC Si V MaxVoidDiam
5.0
5: Atomistic Kinetic Monte Carlo Diffusion
Extended Defects
Sentaurus™ Process User Guide 453
I-2013.12
Capture
Voids capture neutral vacancies, rearranging them to have a spheroidal shape. Figure 57 shows
the possible interactions between voids and point defects.
Figure 57 Voids are big cluster vacancies with a spherical shape that grow trapping neutral
vacancies and shrink by recombination and vacancy emission
Emission
Vo i d s e m i t n e u t r a l va c a n c i e s w i t h a f r e q u e n c y :
(694)
is a prefactor which includes a constant and a dependency with the surface of the void, and
is the binding energy of a void. These binding energies are assigned in the
parameter database together with the small vacancy cluster binding energies. For information
on how to locate and modify them, see Amorphous Pockets Life Cycle on page 439. For voids,
only the values for sizes bigger than the AP-Void threshold apply.
Finally, corrections to the migration energy of vacancies and the binding of them to the void
are applied. The migration correction is the usual one:
(695)
and for the binding energy, it is corrected using the parameter for small vacancy clusters:
(696)
where is Eb_VoidGe.
Void
Recombination
Emission
Growth
2D Projection
νν
0Ebvoid,size()∆Ebvoid,EmV() ∆EmV()+++()kBT()⁄–()exp×=
ν0
Ebvoid,size()
∆EmV() α
mGe[]
1
3
---∆Em
ikBT()⁄–()exp
i
xy z,,
+=
∆Ebvoid,P∆Vb
cluster V() α
void Ge[]+=
αvoid

5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
454 Sentaurus™ Process User Guide
I-2013.12
Recombination
Voids recombine incoming interstitials with any charge.
Interactions
Interactions between void defects and other particles fall into these categories:
■Trapping of neutral vacancies (growth):
pdbSet KMC Si V ReactionsVoid V <true/false>
■Recombination of interstitials:
pdbSet KMC Si V ReactionsVoid <I/IM/IP> <true/false>
■Impurity pair breakup. Voids do not trap impurities, but they can trap or recombine the
interstitial or vacancy associated with a paired impurity. (For example,
). The pair must have the same charge as the substitutional dopant (in
other words, Bi– for B–, AsV+ for As+).
pdbSet KMC Si <impurity> ReactionsVoid <species> <true/false>
Parameters
Voids trap vacancies and recombine interstitials:
sprocess> pdbGet KMC Si V ReactionsVoid
V true
sprocess> pdbGet KMC Si I ReactionsVoid
I true
Voids may break up some pairs. For example, Bi– is disabled:
sprocess> pdbGet KMC Si B ReactionsVoid
BiM false
Amorphization and Recrystallization
A predictive atomistic process simulator must include an amorphization model. Nevertheless,
accounting for each particle and position during the amorphization, although possible [28], is
not convenient for the sizes, times, and computer resources involved in process modeling.
Despite this, amorphization involves the destruction of the lattice structure. Without a lattice,
the KMC method, which discards the lattice and tracks only defects, is opened to a
quasiatomistic approach, as explained in this section.
BiVn
+B–Vn1–
+→
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
Sentaurus™ Process User Guide 455
I-2013.12
Figure 58 shows a generic damage concentration profile after an implant.
Figure 58 Damage concentration profile after an implant
There are two different concentrations in Figure 58 (A and C) and one concentration threshold
called Amorphization. This threshold is stored in the parameter database in the damage
section as AmorphizationThreshold:
sprocess> pdbGet KMC Si Damage AmorphizationThreshold
1.5e+22
NOTE Yo u c an c ha nge t his l im it if n ec essa ry. T he d am age a cc umul at ion m od el
is dependent on the AmorphizationThreshold.
The behavior of the simulator while adding new point defects (damage) differs depending on
the local concentration of the internal grid elements. A new point defect is inserted into a box
depending on the concentration of that box. If the concentration is smaller than the
Amorphization threshold (C, crystalline region), the point defect is inserted as it is. In other
words, a particle is placed inside the simulator with its three coordinates, the defect, and
particle type. Finally, the damage concentration can be higher than the amorphization threshold
(region A (amorphous) in Figure 58). In this case, if a particular crystalline volume (specified
by the parameter minAmorphousVol) has an averaged damage concentration larger than the
threshold, the entire internal volume is assumed to be amorphous. The atomistic 3D
coordinates for Is and Vs are discarded for amorphous boxes because the definition of a point
defect is now meaningless in an amorphous region, and only their concentration is stored.
Finally, the material of the internal box changes from crystalline (such as silicon) to amorphous
(such as amorphous silicon) and an interface, which is capable of simulating a three-phase
segregation model, is created between them.
NOTE For amorphous regions, the atomistic 3D information is discarded, and
only the number of particles is stored. When asking Sentaurus
Process KMC for the atomistic information (the 3D coordinates for
each particle), you should not expect to obtain Is and Vs in amorphous
regions.
A
C
Amorphization
a–c

5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
456 Sentaurus™ Process User Guide
I-2013.12
NOTE To obtain the amorphous–crystalline interface, use the command:
kmc extract acinterface
Amorphous Defects
An amorphous defect is a special defect assigned to each grid element of Sentaurus
Process KMC with a damage level above the amorphization threshold.
Material
Amorphous defects are always associated with amorphous materials. Each amorphous internal
element is paired with an amorphous defect.
Shape
The shape of an amorphous defect coincides with the element containing it. Amorphous layers
are created as a set of several amorphous defects. Consequently, amorphous layers can follow
any complicated amorphous geometry, but always as a set of Sentaurus Process KMC
elements.
Growth
Amorphous defects do not grow because they are limited to the size of the element. Amorphous
layers grow when new elements are amorphized and become amorphous. These amorphous
elements capture any incoming particle.
Recombination
These defects can recombine their damage and become crystalline silicon. Amorphous defects
do not emit particles; recrystallization is the only event they can perform.
Diffusion in Amorphous Materials
Two models are available for diffusion in amorphous materials:
■A simpler, direct diffusion model
■An indirect diffusion model that uses dangling bonds as an intermediate species
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
Sentaurus™ Process User Guide 457
I-2013.12
To select the model to use, set KMC <material> Damage amorphous.bonds to true,
where <material> is the crystalline material.
Direct diffusion
Dopants can diffuse in amorphous materials using direct diffusion. The implemented
diffusivity is:
(697)
where the parameters and are input parameters.
Parameters
The parameters and needed for diffusion in amorphous materials are specified in the
PDB as Dm and Em, respectively, under the amorphized material:
pdbSet KMC <amorphous material> <dopant> Em <dopant> <value>
For example:
pdbSet KMC AmorphousSilicon B Em B 0.8
pdbSet KMC AmorphousSilicon B Dm B 1e-3
NOTE The alias aSi can be used for AmorphousSilicon.
Indirect Diffusion
The observed boron diffusion in amorphous silicon does not seem to obey a standard Fick’s
law with constant diffusivity prefactors and activation energy, thereby making the direct
diffusion model in amorphous silicon inaccurate. A different model has been proposed
[29][30] that relies on the presence and distribution of dangling bonds and floating bonds and
that interacts with the boron atoms. In this model, an initial number of dangling bonds
(threefold-coordinated atoms) and floating bonds (fivefold-coordinated atoms) is created
during amorphization.
These dangling bonds and floating bonds are allowed to evolve using a simple direct diffusion
for dangling bonds and for floating bonds. Dangling bonds and floating bonds can
interact with them, annihilating each other. Dangling bonds also can interact with boron (or any
other user-defined impurity) with a proportionality constant .
In this model, boron in amorphous silicon can exist in two different states: an immobile
fourfold-coordinated state and a highly mobile threefold-coordinated state. Boron
changes between these two states by capturing and releasing a dangling bond. The threefold
DmX() Dm0,X() EmX() kBT()⁄()–()exp=
Dm0,
Em
Dm0,
Em
n0
Dd
Df
α
B4
B3
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
458 Sentaurus™ Process User Guide
I-2013.12
mobile boron is allowed to diffuse with a simple Arrhenius plot. Boundary conditions can be
set at the AmorphousSilicon_Silicon and AmorphousSilicon_Oxide interfaces for
dangling bond (DB) and floating bond (DB) recombination. Finally, despite the initial
concentration of dangling bonds produced by amorphization, an extra contribution of
is added to produce a total DB concentration of:
(698)
where is a coefficient relating to the presence of boron atoms in amorphous silicon with an
excess of dangling bonds, and is the concentration of boron in amorphous silicon.
Consequently, the following reactions are allowed:
(699)
(700)
Implementation
To minimize the number of species and physical mechanisms, the implementation of indirect
diffusion through dangling bonds and FBs has been done by renaming:
■Dangling bonds as interstitials in amorphous silicon
■Floating bonds as vacancies in amorphous silicon
■ as substitutional boron in amorphous silicon
■ as interstitial boron in amorphous silicon
In this way, all that is needed is to allow I and V inclusion, and the following reactions in
amorphous silicon:
(701)
(702)
The B interaction with and further emission by are modeled as a regular kickout
mechanism. Consequently, the parameter is modeled indirectly through the binding energy
and prefactor of the pair.
When amorphizing an element with volume , an initial number of Is and Vs will be
created inside, where is the silicon density. If there are boron atoms inside or boron atoms
are introduced through implantation or any other means (for example, using the select or
profile commands), an extra number of Is will be introduced per boron atom.
n0
γB[]
nBn0γB[]+=
γ
B[]
DB FB+∅→
B4DB+B3
↔
B4
B3
IV+∅→
BI+Bi
↔
I
Bi
α
Bi
∆V
n0∆Vδ
δ
γ
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
Sentaurus™ Process User Guide 459
I-2013.12
Parameters
The parameters needed for this model are introduced in different places. If you want to model
the indirect diffusion of boron in amorphous silicon (other impurities or amorphous materials
are accepted also), you can use aSi as an alias for AmorphousSilicon.
Impurity Clusters in Amorphous Materials
Impurities diffusing in amorphous materials can interact with each other and form impurity
clusters. In contrast with impurity clusters in crystalline volumes, the amorphous impurity
clusters do not contain interstitials or vacancies, and are only an agglomeration of impurities.
Consequently, they can only re-emit the trapped impurities. With this exception, they behave
as regular impurity clusters. For further information, see Impurity Clusters on page 472.
Table 53 Parameters used for indirect diffusion in amorphous silicon
Parameter Description Symbol
KMC aSi amorphous.bond true
Model activation. None
KMC aSi I Dm I <value>
KMC aSi I Em I <value>
Dangling bond diffusion.
KMC aSi V Dm V <value>
KMC aSi V Em V <value>
Floating bond diffusion.
KMC aSi B Dm Bi <value>
KMC aSi B Em Bi <value>
B3 diffusion. None
KMC aSi B Eb Bi <value>
KMC aSi B Eb Bi <value>
B3–B4 reaction rate.
KMC aSi B ReactionsPointDefect B,I true
.None
KMC Si Damage amorphous.n0 <value>
Initial dangling bond and floating bond
percentage (versus silicon density).
KMC aSi B gamma <value>
Number of dangling bonds created per
boron atom.
Dd
Df
α
B4DB+B3
↔
n0
γ

5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
460 Sentaurus™ Process User Guide
I-2013.12
Recrystallization
Two recrystallization models are implemented:
■The simple KMC quasiatomistic model assigns a recrystallization rate to each amorphous
defect for recrystallization simulations. No orientation dependencies are allowed.
■The fully atomistic model uses a lattice kinetic Monte Carlo (LKMC) method to simulate
the evolution of the amorphous–crystalline interface. This model includes orientation-
dependent solid phase epitaxial regrowth (SPER) and facet formation.
The recrystallization model is set up with:
pdbSet KMC <material> Damage Model.SPER <model>
where <material> is the crystalline material (typically, silicon), and <model> is one of the
following:
■LKMC model
■Simple KMC quasiatomistic model
KMC: Quasiatomistic Solid Phase Epitaxial Regrowth
Recrystallization is implemented as a special event performed by the amorphous defects. At a
given temperature, every amorphous defect can recombine all its internal damage, in other
words, recrystallize, with a frequency . The recrystallization of several amorphous
defects with different recrystallization frequencies, depending on their recrystallization axis
and the number of amorphous neighbors, generates an advancing recrystallization front, as can
be seen in Figure 59 on page 461.
νrecryst
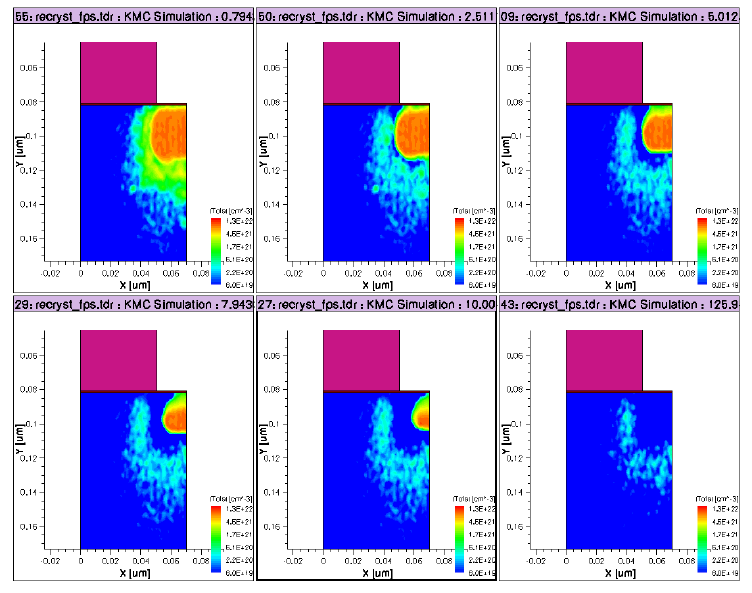
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
Sentaurus™ Process User Guide 461
I-2013.12
Figure 59 Source and gate of a transistor during SPER, as simulated by Sentaurus
Process KMC. The total concentration of interstitials is represented. The time
evolution is shown while the recrystallization front (a consequence of the
recrystallization of isolated amorphous defects) is moving. At the end, there is
only damage near the a-c interface. This remaining damage may form end-of-
range (EOR) defects.
Therefore, if w is the length of an amorphous defect in the recrystallization direction, the
frequency associated with the recrystallization is v/w. This recrystallization velocity v is
computed as an Arrhenius function that includes dependencies on both the local Fermi level
and the presence of impurities [31]:
(703)
parameters are specified as E_recryst. is the activation volume for SPER
(dependency on hydrostatic pressure) called VFRecryst, and n is the percentage of
amorphous material around a given element. The time it takes to recrystallize an amorphous
cell depends on the number of amorphous neighbors; the more neighbors that are amorphous,
the longer it takes. The longer recrystallization takes, the more stable the amorphous defect, so
Vn() V0
Fermi n() Erecryst n() P∆VSPER c++()–kBT()⁄()exp×=
Erecrys n()
∆VSPER

5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
462 Sentaurus™ Process User Guide
I-2013.12
its activation energy is bigger. accounts for the prefactor, including dependency on the
Fermi level. This dependency is introduced as:
(704)
being and the input parameter called V0_recryst. Doping is the local amorphous
element doping, and is a calibration parameter (different for n-type and p-type materials)
called V0_recrys_ntype and V0_recrys_ptype, respectively.
Finally, takes into account the changes in SPER regrowth due to local impurity
concentration. This correction term is modeled as:
(705)
is the parameter controlling how much each impurity changes the planar
recrystallization activation energy (assumed to have 50% amorphous neighboring elements).
is an exponent to control how this correction depends on the dopant concentration. A null
impurity concentration gives a zero correction, while an impurity concentration of
produces . Consequently, represents the planar
recrystallization activation energy if the sample contains only the impurity, while allows it
to control the transition between these two opposite situations. These parameters are called
E_recrys and E_recrys_exponent, respectively in the PDB.
This model, in which elements with fewer amorphous neighbors recrystallize faster, extends
the ideas described in the literature [28] for amorphous elements. This simple method can
simulate the faster recrystallization of amorphous corners or thin amorphous panhandlers
embedded in crystalline silicon.
Finally, if a recrystallization event that will break the compactness of the amorphous layer is
detected, its recrystallization will be retarded by the parameter CompactFactor. This
prevents the formation of amorphous isolated islands and ensures a better compactness of the
amorphous material.
Parameters
The parameters for the recrystallization model are:
sprocess> pdbGet KMC Si Damage VFRecrys
0
sprocess> pdbGet KMC Si Damage V0_recrys
0 1.7e8
99 1.7e8
100 0
sprocess> pdbGet KMC Si Damage E_recrys
0 1.72
V0
Fermi
V0
Fermi n() V0n()1KDoping×+()=
V0n()
K
c
c1Impurity[]
5 1022
×
----------------------------
x
–
Erecrys 50() Impurity[]
5 1022
×
----------------------------
x
EfImpurity()+
Impurities
=
EfImpurity()
x
510
22
×
cE
fImpurity()
Erecrys 50()–=
EfImpurity()
x
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
Sentaurus™ Process User Guide 463
I-2013.12
15 1.72
40 2.7
70 2.7
95 3.3
99 5
NOTE To produce consistent notation, the suffix has changed from recryst
to recrys.
The unspecified values between two specified ones are computed by linear interpolation.
The parameters controlling the Fermi level and impurity concentration dependencies are
specified for each dopant (and material). For example, for boron, they are:
sprocess> pdbGet KMC Si B E_recrys
2.7
sprocess> pdbGet KMC Si B E_recrys_exponent
1
LKMC: Fully Atomistic Modeling of Solid Phase Epitaxial Regrowth
It is well known that the SPER velocity depends on the substrate orientation with approximate
ratios of 20:10:1 for orientations (100), (110), and (111), respectively. In addition, the
recrystallization of thin layers in fin transistors is shown as an ‘arrow tip’ shape formed by two
(111) planes that slow down the SPER, leading to the formation of polycrystalline silicon in
regions still not recrystallized.
This model, based on the literature [32], introduces the lattice in the amorphous–crystalline
interface and assigns a recrystallization event to each of the atoms there. When an internal
mesh element is detected to be amorphous as explained in Amorphization and
Recrystallization on page 454, the silicon lattice is recreated around it. This lattice takes into
account the wafer orientation specified in the init command. Those lattice atoms belonging
to crystalline elements are assigned a “crystalline” flag, while those belonging to the
amorphous element are assigned an “amorphous” flag. This produces the initial amorphous–
crystalline interface. At this point, even when the amorphous–crystalline interface still follows
the contour of the internal mesh, it is formed by a set of individual lattice atoms.
From this point, different recrystallization rates are assigned to each atom at the interface. The
interface is defined as the set of lattice atoms that, having an amorphous state, has at least one
first neighbor with a crystalline state. Any other lattice atom that does not belong to this
interface has a recrystallization rate of 0. This means that crystalline-lattice atoms have a zero
probability of recrystallizing (because they are already crystalline).

5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
464 Sentaurus™ Process User Guide
I-2013.12
In some cases where regular SPER is very slow, random nucleation and growth can produce
polysilicon material [33] not simulated here. Inclusion of defective silicon created during
SPER also is not simulated.
For amorphous lattice atoms belonging to the interface (in other words, surrounded by at least
one crystalline lattice atom), a SPER rate is assigned. The model assumes that an atom in the
amorphous phase must form two undistorted bonds with its first neighbors in the silicon phase
to become crystalline. For amorphous atoms close to a (001) surface, this happens naturally.
For (011) surfaces, two adjacent amorphous atoms have to cluster together so that each atom
has two undistorted bonds. Finally, for (111) orientations, three atoms are needed to cluster
together.
Consequently, there will be three different recrystallization prefactors – K(1), K(2), and K(3)
– depending on the number of amorphous atoms needed to complete two undistorted bonds.
These K(1), K(2), and K(3) prefactors will be related but not proportional to the different (001),
(011), and (111) SPER velocities. In particular, K(2) and K(3) are probabilities for two and
three atoms, respectively, to come together in an amorphous phase and form spontaneously
undistorted crystalline bonds between them. Consequently, K(2) is expected to be smaller than
K(1), and K(3) is expected to be smaller than K(2), by several orders of magnitude.
Each of these lattice atoms is given a recrystallization frequency of:
(706)
are the K(1), K(2), and K(3) prefactors explained above where:
■ is a Fermi-level correction similar to Eq. 704.
■ is taken as in Eq. 703.
■, , and are the absolute value of the shear stresses.
■ is a parameter coupling the shear stresses.
■ and are the same terms as those defined in Eq. 703.
Figure 60 on page 465 shows the evolution of an amorphized fin during SPER when this model
is used, after 2-, 4-, and 6-minute annealing at . When the arrow tip is formed by the
two lateral 111 planes, the recrystallization is almost stopped (middle and right images). The
planes are formed by the presence of the oxide–silicon interface. Since the oxide does not
provide the needed undistorted bonds for silicon recrystallization, it is used as a starting point
for the (111) plane formation.
A similar model using LKMC for epitaxial regrowth can be read in Epitaxial Deposition on
page 519.
νLKMC ν0
Fermi Kn() Erecryst
LKMC εxy εxz εyz
++()λP∆VSPER c+++()
kBT
-----------------------------------------------------------------------------------------------------------------------–
exp××=
Kn()
ν0
Fermi 1KDoping×+=
Erecryst
LKMC
Erecrys 50()
εxy
εxz
εyz
λ
P∆VSPER
c
550°C
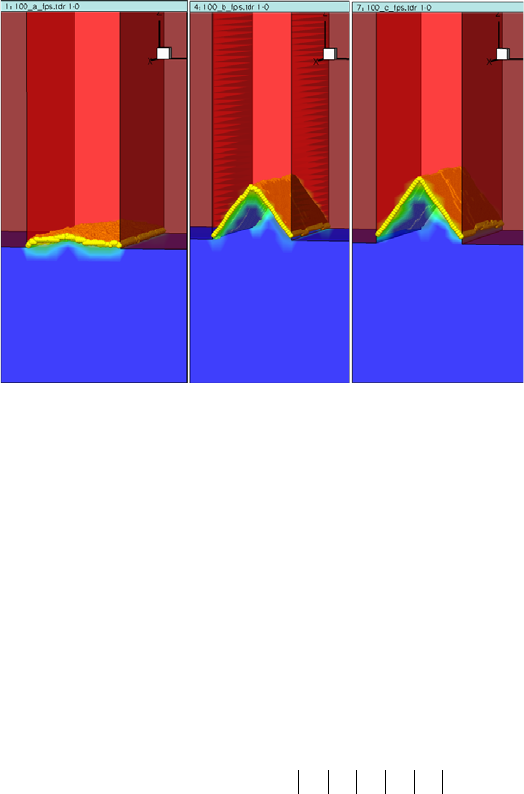
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
Sentaurus™ Process User Guide 465
I-2013.12
Figure 60 SPER evolution (blue is crystalline silicon; red is amorphous one) with time (left to
right) 2, 4, and 6 minutes at ) of a thin (20 nm) silicon fin. The oxide (brown
material) does not provide the correct template for the lattice atoms to form
undistorted bonds, stopping the recrystallization and leading the way to the (111)
planes. When the two (111) planes are formed, no further fast (100) SPER is
possible, and the SPER occurs through the very slow and defect-prone (111)
recrystallization.
Several corrections are applied to the recrystallization rate of a lattice atom. Three of them –
the pressure correction ( ), the impurity correction (through the term ), and the
Fermi-level correction ( ) – are the same in both this model and the simple KMC model
(see KMC: Quasiatomistic Solid Phase Epitaxial Regrowth on page 460).
Shear-Strain Correction
The correction for shear strain, , is unique to this model. Its inclusion
allows the LKMC model to successfully simulate the evolution of line-shaped amorphized
regions. The experimental rate at the corners of line-shaped amorphized regions is very small,
producing a pinching of the SPER interface at the corners [34]. This can be simulated with the
inclusion of this shear stress term [32]. The shear strain is generated during amorphization due
to the different density of the amorphous phase. The expansion of the amorphous phase is not
possible in embedded amorphous regions. The compression of the amorphous phase leads to a
sharp gradient of shear stress at the corners. The model uses the shear strain to simulate the
anomalous regrowth patterns and facet formation experimentally seen in rectangular-shaped
amorphized regions, as shown in Figure 61 on page 466.
550°C
P∆VSPER
c
ν0
Fermi
εxy εxz εyz
++()λ
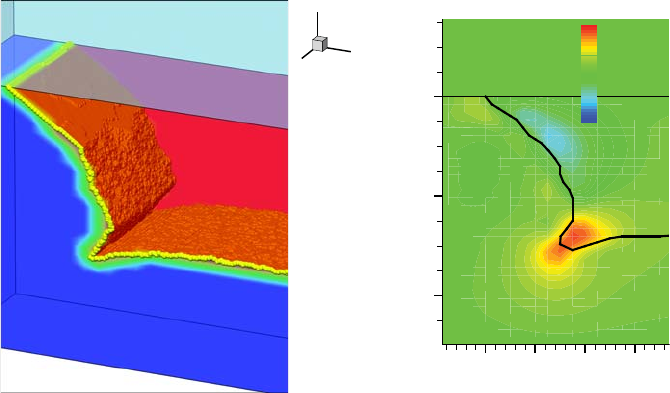
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
466 Sentaurus™ Process User Guide
I-2013.12
Figure 61 Recrystallization of a rectangular-shaped amorphous region using the LKMC
model in Sentaurus Process KMC
Figure 61 (left) shows the distribution of lattice atoms at the amorphous interface side. A (111)
plane, featuring a small nano-island, can be observed close to the interface. The trench formed
at the corner is due to the perturbation introduced by shear strain. Figure 61 (right) shows the
xy shear strain distribution; its maximum intensity occurs at the corner.
Since this model relies on the strain created by the different density of the amorphous material
versus the silicon one, some extra commands must be introduced in the script to account for it.
First, a new material to account for amorphous silicon in the mechanics simulator must be
introduced:
mater add name = Amorph
Mechanics properties for this new material must be defined:
pdbSetDoubleArray Silicon Amorph Conc.Strain {0 0 1 0.02}
pdbSetBoolean Silicon Mechanics UpdateStrain 1
and Sentaurus Process KMC must be instructed that stress is being taken into account:
pdbSet KMC Stress 1
Finally, the synchronization between the atomistic and the mechanics simulator is automatic.
After every mechanics step, the KMC Stress 1 parameter instructs Sentaurus Process KMC
to update the stress and strain fields. After each diffusion (atomistic diffusion) step, Sentaurus
Process KMC updates the “Amorph” distribution by automatically calling the procedure
KMCSync written in KMC.tcl. This procedure, which can be modified by the user but, in
X [µm]
Y [µm]
0.01 0.02 0.03 0.04
0
0.02
0.04
ElasticStrainELXY [1]
1.1e-02
6.1e-03
1.2e-03
-3.6e-03
-8.5e-03
-1.3e-02
X
Z
Y

5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
Sentaurus™ Process User Guide 467
I-2013.12
principle, does not need to be, contains the lines responsible for updating the amorphous region
in mechanics to properly account for the strain and stress:
LogFile IL2 "A/C synchronization: KMC -> PDE"
kmc deatomize name=AC
sel Silicon z=1e22*AC name=Amorph store
Parameters
Table 54 lists the parameters used in this model. It is assumed that silicon (Si) is the crystalline
material and amorphous silicon (aSi) is the amorphous material.
Defect Generation during SPER
It is known that when (111) planes have formed in a simulation, the recrystallization beyond
these planes is defective, and silicon of low quality, or even polysilicon, is formed. A simple
predictive LKMC model for defect formation during SPER based on [35] and [36] is included.
Such modeling is performed by assigning two tags after every recrystallization event in the
lattice: a normal tag for sites sharing the substrate configuration, and a defective tag for sites
assumed not to bond to their neighbors and that form twin defects. Although this modeling
does not physically set the atoms in twin positions, but only assigns them a tag while remaining
Table 54 Parameters for LKMC model
Parameter name as typed in parameter database Description Symbol
KMC Si Damage Model.SPER <model>
Use LKMC to set the model, KMC to unset. None
KMC Si Damage prefactor.SPER.100 <value>
Value for the prefactor associated with 100 SPER. K(1)
KMC Si Damage prefactor.SPER.110 <value>
Value for the prefactor associated with 110 SPER. K(2)
KMC Si Damage prefactor.SPER.111 <value>
Value for the prefactor associated with 111 SPER. K(3)
KMC Si Damage Shear.Coupling <value>
Shear-strain coupling parameter.
KMC Si Damage VFRecrys <value>
SPER pressure correction (same as the KMC
model).
KMC Si Damage E_recrys 50 <value>
Activation energy for recrystallization (same as the
KMC model).
KMC Si Damage V0_recrys_ntype <value>
KMC Si Damage V0_recrys_ptype <value>
Fermi-level corrections (same as the KMC model).
KMC Si Damage E_recrys <value>
KMC Si Damage E_recrys_exponent <value>
Impurity corrections (same as the KMC model).
KMC Si Damage Lattice.Constant <value>
Lattice constant. None
λ
∆VSPER
Erecryst
LKMC
ν0
Fermi
c
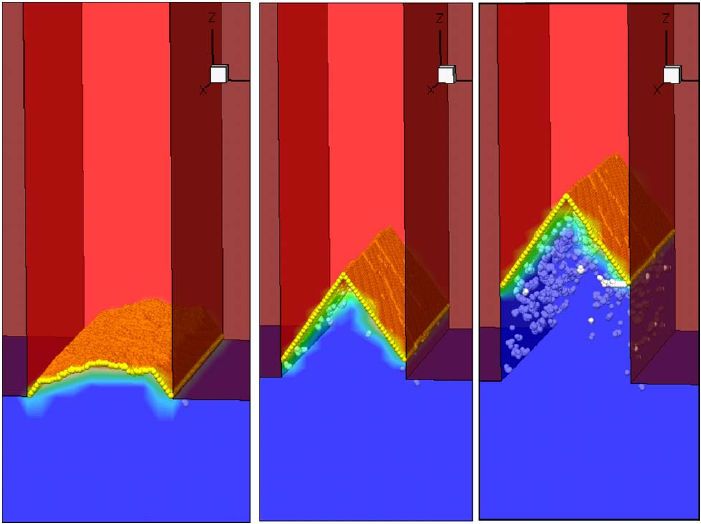
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
468 Sentaurus™ Process User Guide
I-2013.12
in a perfect crystalline position, it is sufficient to predict the defective regions in silicon and to
slow down SPER in a similar way to experiments [35][36].
The new defective sites are produced by two mechanisms:
■Recrystallization of (111) sites, having a probability Pdef of becoming defective.
■Recrystallization of atoms in the neighborhood of defective sites, inheriting such tags and
becoming also defective.
The definition of a coordination number, a keystone in this model to identify the microscopic
configurations, also is modified to distinguish between normal and defective sites. In this way,
the formation of defects slows down the recrystallization of neighboring sites.
The formed defects are represented in the non-LKMC module as an IV twin defect in the TDR
file. No actions are associated with them in the regular KMC simulator. Consequently, when
they are formed, twin defects do not disappear and do not interact with other particles. They
are created for users to identify the regions predicted to have highly defective silicon.
The only new parameter needed for the model is the probability of (111) recrystallizations to
produce twin defects. This parameter is specified in:
pdbSet KMC Si Damage probability.SPER.defect <0-1>
Figure 62 Evolution of a thin (20 nm width) amorphized silicon fin (amorphous is red,
crystalline is blue) annealed at 600oC; arrow-shaped a/c interface is represented
by yellow atoms and formation of defects (twins) is shown as white spheres
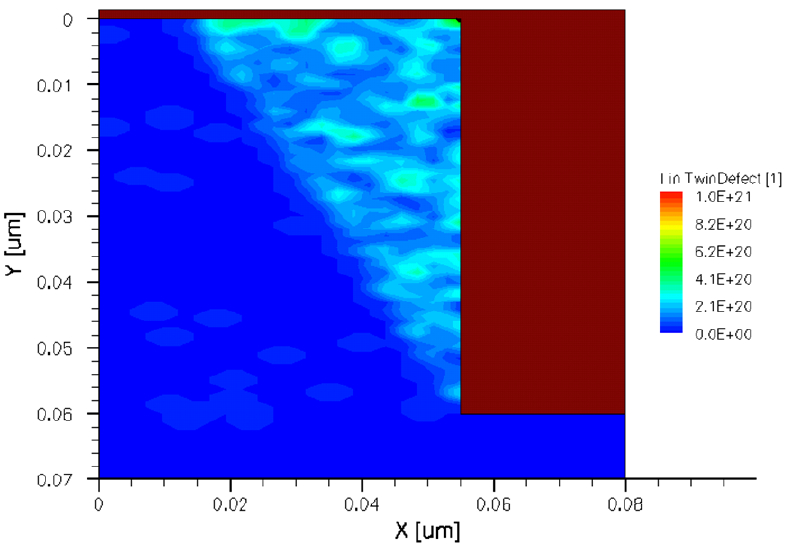
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
Sentaurus™ Process User Guide 469
I-2013.12
Figure 63 Concentration of twin defects representing defective silicon formation when
SPER is in an amorphous region close to SiO2 (silicon is blue, SiO2 is brown)
Figure 62 on page 468 and Figure 63 show two examples where twin-defect formation is
involved. Figure 62 represents the formation of defects during the SPER of a thin silicon fin.
Figure 63 shows the defective triangular-shaped region, bounded by a (111) plane, typical of
SPER close to SiO2-filled trenches.
Redistributing Damage
The recrystallization event forces all IV pairs inside an amorphous defect to recombine. The I
or V excess is redistributed to the neighboring amorphous boxes if any. Otherwise, the excess
is recombined at the surface. If there is no free surface/interface neighboring amorphous boxes,
it is left as point defects. If the recrystallization front has crossed several elements, the amount
of excess point defects can be high. When the defects are deposited in the crystalline silicon,
they grow and ripen into extended defects depending on the annealing conditions.
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
470 Sentaurus™ Process User Guide
I-2013.12
Parameters
The parameter depositExcessDamage controls whether to redistribute the excess or to
discard it. In simulations with buried amorphous layers, setting this parameter to true is
suggested:
sprocess> pdbGet KMC Si Damage depositExcessDamage
0
Impurity Sweep/Deposit
The recrystallization process may affect the impurity concentration. The recrystallization front
moves indium and other dopants away, changing the concentration profiles [37][38]. To model
this effect, the amorphous defects transfer impurities during recrystallization:
■Dopants usually (recrysDeposit) remain in the box or move away with the
recrystallization front (see Figure 64 on page 471). The two available models for this
movement are Elements and Hops, chosen by the RedistributionModel parameter:
•The
Elements model takes all the particles in one internal element and moves
to the adjacent one. If moving the dopant with the
recrystallization front increases the concentration of the neighboring element more
than a limit (recrysMaxTotal), it will be deposited in the current element, no matter
what its moving probability.
•The
Hops model goes particle-by-particle inside the affected element and decides
whether the particle should be displaced a second neighbor distance, depending on
recrysDeposit. If the particle is not displaced, it remains where it was. The
algorithm continues with the next particle (which may still be the same one, pushing it
again through the adjacent element little by little) until no more particles remain. To
prevent the concentration of displaced particles being too high, the algorithm forces the
deposit probability to be 1 when a particle has 25 or more dopant neighbors. The
algorithm corrects this probability by linear interpolation starting when the number of
neighbors is a given a percentage of 25. This percentage is controlled by the parameter
recrysDepositThreshold.
■When the box is recrystallized, if the remaining dopant concentration is bigger than the
solubility limit (C0_recrysMaxActive, E_recrysMaxActive) after SPER, the extra
dopants are deposited as impurity clusters. These clusters have a limited size, and there are
two different models to deposit these clusters depending on whether recrysMaxSize is
defined.
n
nrecrysDeposit×
5: Atomistic Kinetic Monte Carlo Diffusion
Amorphization and Recrystallization
Sentaurus™ Process User Guide 471
I-2013.12
Figure 64 Impurity sweep example showing that arsenic has been pushed through the
surface during recrystallization or SPER
The parameters for the recrystallization model are defined only for impurities in silicon (or
other full material). P_recrysDeposit and E_recrysDeposit define (prefactor and
energy) the probability for a dopant remaining in the same box after the recrystallization front
passes. Setting this value to 1 disables the swept of impurities. recrysMaxTotal establishes
the maximum concentration piled up during SPER. recrysMaxActive is the maximum
allowed concentration of an active dopant in the recrystallized areas. Finally, if
recrysMaxSize is defined (and it is by default), the old model to limit the maximum size of
the deposited impurity clusters will be used. To undefine this parameter, use:
sprocess> pdbUnSetDouble KMC Si B recrysMaxSize
This instructs Sentaurus Process KMC to use the new model to deposit impurity clusters after
SPER. This model deposits the clusters specified in recrysDeposit_Complex with the
probabilities defined there.
Finally, the active dopants after SPER are deposited as substitutional impurities, but you can
change this default using recrysDeposit_Active. This parameter accepts a list of
impurities and impurity pairs with the probability to be deposited. For example:
sprocess> pdbSet KMC Si F recrysDeposit_Active F .1
sprocess> pdbSet KMC Si F recrysDeposit_Active Fi .9
This deposits 90% of the ‘active’ fluorine as Fi, and 10% as F.
NOTE Specifying a high probability for a cluster with a number of dopants
greater than 1 does not necessarily means that you will obtain only that
cluster. For example, if you specify that B2I should be 100% of the
deposited clusters, but Sentaurus Process KMC finds only one boron
particle in an element, Sentaurus Process KMC will not form a B2I.
1022
1021
1020
1019
05
Total A s
Swept As
Amorphous layer
before recrystallization
Depth [nm]
SiO2
Concentration [cm–3]

5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
472 Sentaurus™ Process User Guide
I-2013.12
Parameters
The recrystallization parameters for dopants can be obtained as:
sprocess> pdbGet KMC Si As P_recrysDeposit
0.3
sprocess> pdbGet KMC Si As E_recrysDeposit
0.0
sprocess> pdbGet KMC Si As recrysMaxActive
1e+21
sprocess> pdbGet KMC Si As recrysMaxSize
4
sprocess> pdbGet KMC Si B recrysDeposit_Complex
B3I3 .40 B2I3 .30 BI2 .30
sprocess> pdbGet KMC Si B recrysDeposit_Active
B 1.0
To see which model is being used, use:
sprocess> pdbGet KMC Si Damage RedistributionModel
Elements
The concentration thresholds associated with each model are:
sprocess> pdbGet KMC Si B recrysMaxTotal
2e+22
sprocess> pdbGet KMC Si B recrysDepositThreshold
75
Impurity Clusters
At certain concentrations, dopants are electrically inactive in crystalline silicon [7]. At the
same time, high I concentration can make a fraction of boron electrically inactive even when
its concentration is below its solubility [39]. This phenomena can be explained by a BmIn
clustering mechanism [15][40] or dopant precipitation [7]. Sentaurus Process KMC considers
these mechanisms, implementing the impurity clusters.
Recent studies [41] show that boron precipitation in amorphous silicon occurs through
formation of a boron complex, thereby making the inclusion of impurity clusters in amorphous
materials necessary. Consequently, pure dopant clusters, , are allowed in amorphous
materials and other materials modeled as simple.
The AnBo...Xm impurity clusters allow powerful modeling of the interaction of several
impurities between them. For example, fluorine–boron clusters (FnBoIm and FnBoVm) can be
tried to explain the effects of boron coimplanted with fluorine, or AsnPoVm clusters to allow a
satisfactory explanation, as seen in [52]. Nevertheless, the most common use of impurity
Bn

5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
Sentaurus™ Process User Guide 473
I-2013.12
clusters is the traditional one where only one dopant is present. Consequently, except where
indicated, the description of impurity clusters that follows assumes that the clusters are in the
more common form AnXm with only one impurity involved. Starting with Version I-2013.12,
impurity clusters have a simple model for diffusion, that is, they can migrate. For more
information, see Diffusion on page 474.
Finally, impurity clusters do not need to be neutral. Consequently, the charge state of each
impurity cluster can be defined by using the parameter e0_Complex. When needed, an
impurity cluster with a particular charge as AnXmcharge will be denoted. In addition, clusters
can react with charged particles, as long as the reaction is not between a cluster and a particle
with the same sign, in other words, it is not an electrostatic repulsive reaction.
To simplify the following descriptions, this section describes, the AnXm clusters with only one
impurity, and neutral reactions (in other words, reactions similar to
, ). To see how the model works when this is not the case,
see Charge Dependency on page 485.
NOTE Since P is used both for positive and phosphorus, clusters containing
phosphorus cannot have the P at the end of the cluster name. For
example, AsIP will be interpreted as a positive interstitial arsenic, while
AsPI or PAsI will be a phosphorus–arsenic–interstitial cluster; the same
is true for AsP, PAs, and so on.
Shape
An impurity cluster is an irregular agglomeration of impurities (A, B, ...) with or without
interstitials and vacancies (X) that can be written as AnBo...Xm, with n impurity atoms of type
A, o of type B, and so on, and m Is or Vs. If m = 0, it is a pure impurity cluster (the only ones
allowed in simple materials).
For Sentaurus Process KMC, the notation AnBo...Xm means any possible configurations with
n impurities of type A, o of type B, and so on, and m interstitial (vacancy) atoms. The
interstitial (vacancy) atoms can be both silicon self-interstitials or dopant atoms in an
interstitial position. Since Sentaurus Process KMC assumes all the AnBo...Xm configurations
to be the same with only one effective formation energy, A,B, ... are represented always as a
substitutional but inactive dopant or impurity, and X as a silicon interstitial or vacancy.
AnXm
aAXb
+An1+Xm1+
c
↔
ab+c=

5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
474 Sentaurus™ Process User Guide
I-2013.12
Figure 65 AsV impurity clusters simulated with Sentaurus Process KMC; blue is arsenic
and green is vacancy
Diffusion
A diffusion mechanism has been added to impurity clusters. This means that impurity clusters
can perform migration hops similar to the ones performed by point-defects, impurities, and
dopants. An impurity cluster migration event involves all its constituent particles: The whole
cluster is displaced. The particle coordinates are modified isotropically at a fixed distance of
= 0.384 nm in the orthogonal direction (parallel to the x-axis, y-axis, or z-axis).
The migration rate for impurity clusters is assumed to be:
(707)
where is the prefactor for each cluster, called Dm_Cluster in the parameter database,
and is the migration energy for each cluster, specified as Em_Cluster in the PDB.
Parameters
As previously stated, the names of the diffusion parameters for impurity clusters are
Dm_Cluster and Em_Cluster:
sprocess> pdbGet KMC Si As Dm_Complex As2V
1e-3
sprocess> pdbGet KMC Si As Em_Complex As2V
1.5
λ
νmν0m,EmkBT⁄–()exp×=
ν0m,
Em
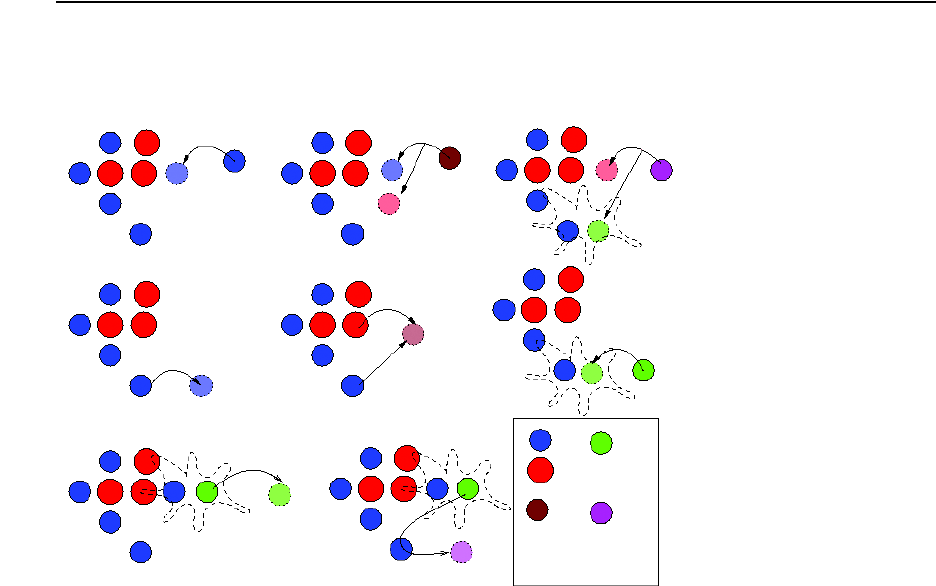
5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
Sentaurus™ Process User Guide 475
I-2013.12
Limitations
The migration events for impurity clusters, although similar to the ones for single particles, do
not share all their features. In particular, the following limitation applies:
■The diffusivity for impurity defects is global, isotropic, and constant. There are no SiGe,
stress, strain, or charge dependencies on diffusivity.
■Impurity defects do not interact with interfaces at all (Si–SiO2 and so on). All interfaces
are considered mirrors.
■Periodic boundary conditions or mirror conditions are correctly applied to the limits of the
simulation box.
■There is no recombination probability at the boundaries. This means that the parameters
sinkProbTop, sinkProbBottom, sinkProbBack, sinkProbFront, sinkProbLeft,
and sinkProbRight do not apply to diffusing impurity clusters.
■Speedup migration does not apply to impurity cluster diffusion: no long hops or double
hops.
Growth
Impurity clusters grow trapping neutral mobile particles (see Figure 66).
Figure 66 Impurity clusters are disordered agglomerations of dopants and silicon point
defects that trap and emit particles. FT mechanisms and IV recombinations also
are possible.
(a) (b) (c)
(d) (e) (f)
(g) (h) I
A
Ai
V
Av
Growth Growth Growth
Emission
Emission
Recombination
Frank–Turnbull
Frank–Turnbull and
Emission
Impurity Clusters
5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
476 Sentaurus™ Process User Guide
I-2013.12
For an AnIm cluster, both I and Ai particles can be trapped:
(708)
The trapping is not automatic as it was for extended defects. In extended defects, the binding
energy is always positive, so the trapping is always allowed. For impurity clusters, the cluster
can grow in both Is (Vs) or dopants. The energy between the initial and final states is not always
favorable. Sentaurus Process KMC computes the probability for an impurity cluster AnIm to
trap an I or Ai as:
(709)
where:
(710)
and is an optional energy barrier.
The binding energies are computed using the potential impurity cluster energies:
(711)
(712)
The potential energy for the neutral , assuming that the substitutional is negative, is given
by , where the binding energy includes the pressure and Ge corrections.
The minus sign accounts for the fact that the binding must have a sign that is opposite that of
the potentials.
These potentials energies are computed as:
(713)
where is specified in the parameter database for each impurity in silicon as
Etotal_Complex. is the activation volume to take into account the hydrostatic
pressure dependency, also defined for each impurity cluster size as VF_Complex. The energy
barriers are called EbarrierIV_Complex and EbarrierDopant_Complex. The
EbarrierIV is defined for emission and capture of interstitials and vacancies, and
EbarrierDopant for dopants or paired dopants. For example, to set the potential energy of a
BIC, such as BI2 to some value:
pdbGet KMC Si B Etotal_Complex BI2 <n>
AnIm
aIb
+AnIm1+
c
↔
AnIm
aAi
b
+An1+Im1+
c
↔
Pcapture
Ecapture
kBT
--------------------–
exp=
EAnImcapture Ebarrier AnIm
()max 0 EAnImb
–,()+=
Ebarrier AnIm
()
EAnImb
EAnImbI() Epot AnIm1+
()Epot AnIm
()–=
EAnImbAi
() Epot An1+Im1+
()Epot AnIm
()–Epot Ai
()+=
Ai
A
E–bAi
–
()e–0,()Ai
()–
Epot AnIm
()Epot
0AnIm
()P∆Vpot AnIm
()+=
Epot
0AnIm
()
∆Vpot AnIm
()
5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
Sentaurus™ Process User Guide 477
I-2013.12
and, for As4V:
pdbGet KMC Si As Etotal_Complex As4V <n>
Initial Seeds
The clusters , , , and are the initial seeds for the impurity cluster ripening. The
formation of is discussed in Percolation on page 477. The others are formed by the
reactions:
■
■
■
■
where and are the charge states for the reactants, and for the result. All these reactions
provide a cluster starting with impurities or impurity pairs. In the cases where for the
first reactions or for the last one, the reaction is not neutral, and the special
considerations of Charge Dependency on page 485 should be taken.
These initials seeds can be enabled and disabled independently by the parameter
ReactionsPointDefect, as explained in Enabling and Disabling Interactions on page 424.
Percolation
Some dopants deactivate without visible diffusion when they are in high concentrations [42].
They also can form impurity clusters [43]. Sentaurus Process KMC models this deactivation
allowing the substitutional dopants to interact with impurity clusters or with other dopants right
after its inclusion in the simulation (for example, after being implanted or selected).
As can react with As giving As2. Substitutional As does not migrate, so this reaction is only
possible when two arsenic are close enough to each other. The higher the arsenic concentration,
the higher this possibility. An As + As2 reaction and As3 + As reaction also are possible. These
species also are immobile. They react only when they are close enough. Consequently, the
probability of forming an As4 cluster using this mechanism is low because it needs four As
atoms close enough to each other. This probability increases with the concentration. With high
concentration, the probability is not negligible, and the substitutional As react with each other
forming As clusters and becoming inactive.
NOTE The reaction between two substitutional dopants to give an impurity
cluster is the only exception to the rule that two particles with the same
charge will not interact.
A2I
AI2
A2I2
A2
A2
AaAi
b
+A2Ic
↔
Ai
aIb
+AI2
c
↔
Ai
aAi
b
+A2I2
c
↔
AaAa
+A2
c
↔
a
b
c
ab+c≠
2ac≠
5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
478 Sentaurus™ Process User Guide
I-2013.12
In simple materials, such as amorphous silicon, this ‘percolation’ model is the only one
allowing impurity clusters to be formed. Since all particles are neutral in simple materials, the
percolation reaction does not constitute an exception there.
Parameters
The potential and binding energies for impurity clusters are defined only in materials with
full and simple modeling, including amorphous. They are defined in arrays whose index is
the cluster name. For example, for F clusters:
sprocess> pdbGet KMC Si F Etotal_Complex
FV2 -4.20
F2 -0.5 F2V -4.63 F2V2 -7.07
F3 -1 F3V -7.08 F3V2 -9.04
F4 -1.5 F4V -7.12 F4V2 -11.47
F5 -3 F5V -8.5 F5V2 -13.29
F6 -4.5 F6V -9.7 F6V2 -16.09
F7V 3
FI2 -4.20
F2I -4.63 F2I2 -7.07
F3I -7.08 F3I2 -9.04
F4I -7.12 F4I2 -11.47
F5I -8.5 F5I2 -13.29
F6I -9.7 F6I2 -16.09
F7I 3
A particular value for only one element also can be obtained. The current potential energy for
As4V is:
sprocess> pdbGet KMC Si As Etotal_Complex As4V
-5.4
The barriers are, by default, not defined:
sprocess> pdbGet KMC Si F EbarrierIV_Complex
sprocess> pdbGet KMC Si F EbarrierDopant_Complex
The charge value for the clusters is retrieved with:
sprocess> pdbGet KMC Si F e0_Complex
sprocess> pdbGet KMC Si B e0_Complex
B3 0 B3I 0 B3I2 0 B3I3 0
B2 0 B2I 0 B2I2 0 B2I3 0
BI2 0
No value means that they are neutral.

5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
Sentaurus™ Process User Guide 479
I-2013.12
NOTE The impurity cluster model and the activation or deactivation of clusters
can be calibrated further fitting the potential energies. For further
accuracy, Advanced Calibration also can be used.
When a particular energy for a particular configuration is not specified (in other words, when
input in the parameter file exists for an AnIm or AnVm impurity cluster), Sentaurus
Process KMC assumes this configuration to be unstable. When a barrier energy is not
specified, a value of 0 eV (no barrier) is assigned.
All impurities are allowed to form impurity clusters with I, V, or both. If an impurity does not
form impurity clusters, the default can be changed, modifying the parameters and the
interactions (see Interactions on page 487).
Emission
Impurity clusters can emit both neutral interstitials (vacancies) or mobile dopants:
(714)
(715)
Sentaurus Process KMC computes the emission frequencies as:
(716)
The emission energies are:
(717)
(718)
is the migration energy of the emitted species, and both and have
been shown above. The emission prefactors for dopant and I or V emission depend on the
model used.
When UseCaptVol_Complex is set to true, the emission prefactors are proportional to the
capture volumes of the impurity clusters:
■
■
AnImAnIm1–I+↔
AnImAn1–Im1–Ai
+↔
νemission ν0emission,
Eemission
kBT
----------------------–
exp×=
Eemission I() EmI() Ebarrier AnIm
()max 0 EAnImbI(),()++=
Eemission Ai
() EmAi
()Ebarrier AnIm
()max 0 EAnImbAi
(),()++=
Em
EAnImbI()
EAnImbAi
()
ν0emission,Ai
() KA
i
()Vcapt AnIm
()=
ν0emission,I() KI()Vcapt AnIm
()=
5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
480 Sentaurus™ Process User Guide
I-2013.12
is the capture volume for each impurity cluster, defined in the PDB as
CaptVol_Complex, and the constant is a parameter named D0_Cluster in the PDB.
NOTE The unit of the capture volumes is the capture volume of one single
point defect.
If UseCaptVol_Complex is set to false, Sentaurus Process KMC uses the default old
model, in which the capture volumes are internally fixed to be for emission and
for emission. The use of this default model is not suggested, since it does not lead to
microscopic reversibility.
When a particle is emitted, the impurity cluster tests if the number of remaining particles is
enough to maintain the cluster. If there is only an interstitial (vacancy) or an interstitial and a
dopant, the cluster dissolves leaving an interstitial or a mobile, paired dopant, respectively.
Parameters
The prefactor constants are:
sprocess> pdbGet KMC Si As D0_Cluster
As,AsV 2.1
As,V 10
sprocess> pdbGet KMC Si B D0_Cluster
B,Bi 3
B,I 150
The notation for these prefactors is as follows: two strings are needed, separated by a comma.
The second string represents the emitted particle for which the parameter is being defined. The
first string represents the type of cluster. This first string is needed to define a different prefactor
for emitting a Bi from a B cluster rather than from a hypothetical BF cluster. For example, the
emission of Bi from a B2I2 will use B,Bi, while from a B2F3I2 will use BF,Bi. This last one
can be defined in KMC Si B and KMC Si F, but if defined in both of them with different values,
it will produce an error.
The capture volume parameters are:
sprocess> pdbGet KMC UseCaptVol_Complex
0
sprocess> pdbGet KMC Si B CaptVol_Complex
B3 1 B3I 1 B3I2 2 B3I3 3
B2 1 B2I 1 B2I2 2 B2I3 2
BI2 1
The notation for capture volumes is as following: if only the cluster is specified, that applies to
all emissions for that particular size, but if a cluster size and a particular particle are specified,
Vcapt AnIm
()
K
m
I
min n m,()
Ai

5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
Sentaurus™ Process User Guide 481
I-2013.12
separated by a comma, that applies to that cluster emitting only that particle. For example, the
following applies only to emission if I by B2I2:
pdbSet KMC Si B CaptVol_Complex B2I2,I 6
The rest of the parameters needed for emission are the same as in Growth on page 475.
Recombination
Impurity clusters can trap incoming neutral Vs (Is) and recombine them with internal Is (Vs):
(719)
The capture probability is:
(720)
The associated energies are:
(721)
and:
(722)
After the IV pair recombination, the cluster size is tested and, if necessary, dissolved, as
previously explained.
Parameters
The parameters used for recombination of point defects are the same as in Growth on page 475.
Frank–Turnbull Mechanism
A generalized Frank–Turnbull (FT) mechanism is the emission of a neutral V (I) from an AnIm
(AnVm) impurity cluster by the formation of a Frenkel pair (IV):
(723)
AnImV+AnIm1–
↔
Pcapture
Ecapture
kBT
--------------------–
exp=
EAnImcapture EAnImbEpot V()–Epot I()–=
EAnImbEA
nIm1–
()EA
nIm
()–=
AnImIVA
nIm
++ AnIm1+V+→→
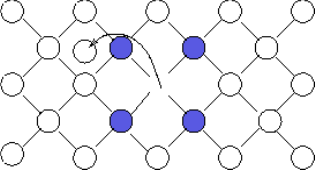
5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
482 Sentaurus™ Process User Guide
I-2013.12
or:
(724)
Sentaurus Process KMC includes the FT mechanism to maintain microscopic reversibility.
Since impurity clusters recombine incoming Is or Vs, the opposite mechanism (FT) also is
needed. Usually, this mechanism is unfavorable, but for some particular configurations, the
energetic differences between them can enable the formation of IV pairs and, therefore, the
emission of particles using the FT mechanism (see Figure 67).
Figure 67 Example of FT mechanism: an As4 cluster can emit an interstitial to become an
As4V impurity cluster: blue is arsenic and white is silicon
The vacancy (interstitial) emission frequency is computed as:
(725)
being:
(726)
and:
(727)
where the potential energies for the clusters and the point defects include pressure and Ge
corrections.
Parameters
The parameters used are the same as in Growth on page 475. The potential energies for
interstitial and vacancies are specified for the material as Ef:
sprocess> pdbGet KMC Si I Ef
4.0
sprocess> pdbGet KMC Si V Ef
3.8
AnVmIVA
nVm
++ AnVm1+I+→→
νemission ν0emission,
Eemission
kBT
----------------------–
exp×=
Eemission V() EmV() max 0 EAnImbV(),()+=
EAnImbV() Epot I() Epot V() Epot AnIm1+
()Epot AnIm
()–++=

5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
Sentaurus™ Process User Guide 483
I-2013.12
The corrections to the potential energies for I and V are VF and EfGe for pressure and Ge,
respectively:
sprocess> pdbGet KMC Si I EfGe
I 0.0
sprocess> pdbGet KMC Si I VF
I 0.0
The prefactor for I and V emission is computed automatically for impurity clusters with only
one dopant (for example, BnIm or AsnVm clusters) and must be specified for other cases. For
example, in a case with AsPV clusters, the prefactors for Frank–Turnbull emission are
specified as follows:
pdbSet KMC Si As D0_Cluster AsP,V 50
pdbSet KMC Si As D0_Cluster AsP,I 50
The following is also valid:
pdbSet KMC Si P D0_Cluster AsP,V 50
pdbSet KMC Si P D0_Cluster AsP,I 50
Complementary Recombination
Some impurities diffuse using both the interstitial and vacancy mechanisms. For these cases,
the impurity clusters can react with both of them. For example, an AnVm impurity cluster can
grow trapping AsV, as previously explained, and can interact with an incoming Asi, trapping
the As and recombining the I with one internal vacancy. This implies to take into account the
reaction:
(728)
These complementary recombinations of neutral particles are allowed with a probability of:
(729)
The capture energies are computed as:
(730)
AsnVmAsi
+Asn1+Vm1–
↔
Pcapture
Ecapture kBT()⁄–()exp Ecapture 0>
1Ecapture 0≤
=
Ecapture
Epot An1+Vm1–
()Epot AnVm
()–EfV()–EfI()–Epot Ai
()–m0>
Epot An1+Vm
()Epot AnVm
()–Epot Ai
()–EmI() EmAi
()–+ m0n1>,≡
EA
iA→() m0n1≡,≡
=
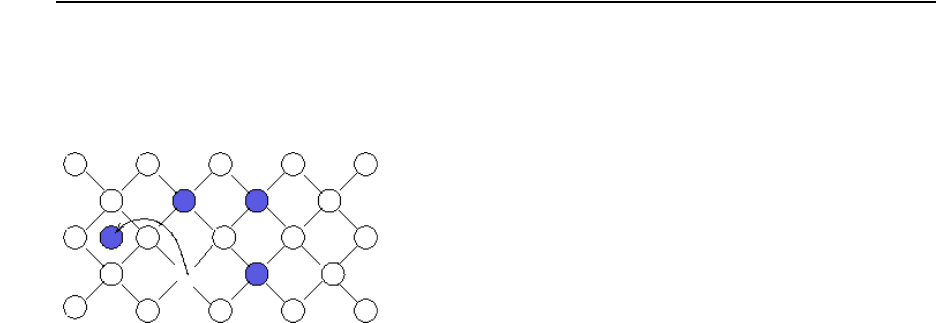
5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
484 Sentaurus™ Process User Guide
I-2013.12
where is an internal parameter that cannot be changed.
Parameters
The parameters used are the same as in Frank–Turnbull Mechanism on page 481. The potential
energy for the paired dopant is the binding energy of the pair corrected with the Fermi-level
dependency.
Complementary Emission
To maintain microscopic reversibility, the reaction reverse to the complementary
recombination must be defined (see Figure 68).
Figure 68 Example of complementary emission: the cluster emits an interstitial that takes
an impurity and generates a vacancy; blue is arsenic and white is silicon
The equation for this process is:
(731)
Its emission frequency is computed using the emission frequency equation:
(732)
where:
(733)
EA
iA→()
AnImAnImIV++ AVAn1–Im1+
+→→
Eemission EmAV
()max 0 EAnImbAV
(),()+=
EAnImbAV
() EfI() EfV() Epot An1–Vm1+
()Epot AnIm
()–++=

5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
Sentaurus™ Process User Guide 485
I-2013.12
Parameters
The parameters used are the same as in Recombination on page 481.
For simple impurity clusters, those with only one impurity (BnIm, AsnVm, and so on), the
prefactor for emission is calculated automatically. For complex impurity clusters, (AsnPoVm
and so on), the prefactor must be written explicitly:
pdbSet KMC Si As D0_Cluster AsP,Asi 50
Charge Dependency
Neutral Reactions
In the previous discussions, all the impurity clusters are assumed to be neutral and,
consequently, there are no explicit charge Fermi-level dependencies. Nevertheless, there are
nonexplicit dependencies. In particular, for clusters emitting impurity-paired dopants, the
emission energy depends on the binding of the paired dopants, which, in turn, contains a Fermi-
level dependency.
The Fermi-level dependency of the binding energy is related to the level of the neutral-paired
dopant in the band gap. This level also depends on the temperature and the bandgap narrowing.
All the previous dependencies are included by default, except the indirect dependency on the
bandgap narrowing, which can be switched off and on using:
pdbSet KMC <material> BandGap Correct_Complex <true/false>
Nonneutral Reactions
Assume the following reaction:
(734)
The potential energy for is defined with respect to a ground state that produces the
impurity cluster in a neutral reaction. This means that:
(735)
BnIm
aBoIp
bXc
+→
BnIm
a
nB-mV0an+()–e-an+0<
an+()h+an+0>
++
5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
486 Sentaurus™ Process User Guide
I-2013.12
is the ground state for . Consequently, an account of holes and electrons must be
followed during the reaction. In particular, calling the initial cluster and the final one , these
accounts are:
■,
■,
The final state must account for the charge in the emitted particle. Calling the charge of the
substitutional dopant of the emitted species (in other words, if or
if ), the final accounts for holes and electrons are:
■
■
This allows writing the first energetic term for the binding energy as:
(736)
The second term is the obvious difference in potential energies:
(737)
The binding energy of the emitted particle is also needed, including the transition from the
neutral state to the current emitted state:
(738)
In addition, only in cases where a Frank–Turnbull emission is involved ( or
), the pair recombination energy is:
(739)
This gives a binding energy of:
(740)
That finally allows computing the emission energy:
(741)
AsnVm
a
i
f
hi
+an+an+0>
0an+0<
=
ei
-0an+0>
a–n–an+0<
=
hf1
+bo+no+0<
0ba+o<
=
ef1
-0bo+0>
b–o–bo+0<
=
d
X
d1–=
XcBi
c
=
d0=
XcIc
=
hf
+hf1
+cd–cd–0>
0cd–0<
+=
ef
-ef1
-0cd–0>
dc–cd–0<
+=
Ech esarg ei
-ef
-
–()EgeF
–()hi
+hf
+
–()eF
+=
Eclusters Epot BnIm
()Epot BoIp
()–=
d
EbXc
() EbXd
()EX
ddc–()e-dc–0>
cd–()h+cd–0>
+Xc
→
+=
XcVc
=
XcBVc
=
Erecom EfI() EfV()+=
EBnImbXc
() Ech esarg Eclusters
–– EbXc
()–Erecom
+=
Eemission Xc
() EmXc
()Ebarrier BnIm
()max 0 EBnImbXc
(),()++=

5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
Sentaurus™ Process User Guide 487
I-2013.12
NOTE All the previous energies (potential, binding, migration, and so on) are
computed including hydrostatic pressure, SiGe, and bandgap narrowing
local corrections.
Interactions
You can modify all the interactions involved in the impurity cluster model. The impurity
clusters can be enabled or disabled with the Boolean parameter Implement_Complex. For
example, to disable the FnVm impurity clusters, use:
pdbSet KMC Si F Implement_Complex 0
When the impurity clusters are enabled, you can set and unset the particular reactions using the
ReactionsCluster parameter:
pdbSet KMC Si <dopant> ReactionsCluster <reaction> <true/false>
where reaction is a string with two fields, separated by a comma. The first field is the name
of the impurity cluster, and the second is the name of the reacting particle. Spaces are not
allowed between these fields. The setting or unsetting of these reactions enables or disables the
specified reactions and its reverse ones. This is performed to maintain the microscopic
reversibility. For example, to disable the capture of a vacancy by As2 to growth to As2V:
pdbSet KMC Si As ReactionsCluster As2,V false
This also disables the inverse reaction, in other words, the emission of V by As2V. To e n a b l e
the recombination of I by an As4V cluster:
pdbSet KMC Si As ReactionsCluster As4V,I true
This reaction also enables the FT emission of I by As4, that is, .
Enabling a reaction does not mean that the reaction will happen; it depends on the energetics.
If the reaction is unfavorable, it will not occur (but the inverse will). Disabling a reaction will
forbid the reaction to occur, even if it is described by the parameters as favorable. Any reaction
not listed in ReactionsClusters is disabled.
Complex Impurity Clusters
To enable impurity clusters with more than one dopant (for example, an As2PV), the switches
for both the As and P clusters should be on:
pdbSetBoolean KMC Si As Implement_Complex 1
pdbSetBoolean KMC Si P Implement_Complex 1
As4As4VI+→
5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
488 Sentaurus™ Process User Guide
I-2013.12
To enable the cluster reaction (assuming As2 is predefined), use the As or
the P to define the reaction. If both are used with different values, an error message will be
displayed. In this example, they are defined in As:
pdbSet KMC Si As ReactionsCluster As2,PV true
The energy and capture volume of this new cluster must be defined as usual:
pdbSet KMC Si P Etotal_Complex PAs2V -3.0
pdbSet KMC Si P CaptVol_Complex PAs2V 1.3
The I, V, and PV emission prefactors are:
pdbSet KMC Si P D0_Cluster PAs,I 0.5
pdbSet KMC Si P D0_Cluster PAs,V 0.5
pdbSet KMC Si P D0_Cluster PAs,PV 0.1
Finally, for AsV emission from the cluster As2PV, allow the reaction by
defining:
pdbSet KMC Si P ReactionsClusters PAs,AsV true
This also enables the formation of the clusters through the reaction of these particles.
Parameters
To show the parameters involved in the impurity cluster reactions, arsenic is used as an
example. AsV clusters are allowed:
sprocess> pdbGet KMC Si As Implement_Complex
1
Since AsV clusters are allowed in the Sentaurus Process KMC simulation, they require some
enabled reactions. The reactions are explained in Percolation on page 477 and allow
deactivation without arsenic diffusion:
sprocess> pdbGet KMC Si As ReactionsCluster
As2,As true
As3,As true
Reactions to grow capturing V and to shrink emitting them are:
As2,V true
As3,V true
As4,V true
As2PV+As2PV↔
AsP AsV+As2PV↔

5: Atomistic Kinetic Monte Carlo Diffusion
Impurity Clusters
Sentaurus™ Process User Guide 489
I-2013.12
Capture or emission of AsV is:
As2,AsV true
As3,AsV true
Recombination of interstitials and Frank–Turnbull emission of interstitials are:
As2V,I true
As3V,I true
As4V,I true
Recombination of interstitials, capture of arsenic, and emission of Asi are:
As2V,Asi true
As3V,Asi true
The following rules must be satisfied to allow a reaction between a particle and an impurity
cluster:
■The first field must be a correct impurity cluster, and the second must be a defined particle.
■The particle must be an interstitial or a vacancy of a paired dopant. The resulting cluster
must be defined (in Etotal_Complex and CaptVol_Complex).
■Only nonrepulsive interactions are allowed, except for percolation. The reactions do not
need to conserve the charge.
Impurity clusters require an initial impurity cluster or ‘seed’ to begin the ripening. This initial
cluster is formed with the reactions of two particles. These reactions are explained in Enabling
and Disabling Interactions on page 424.
Setting Up Impurity Clusters in a Material
To set up an impurity cluster in a material (for example, in amorphous silicon), the
following PDB parameters must be created:
■First, the cluster must be allowed:
pdbSetBoolean KMC aSi B Implement_Complex true
■Second, an emission prefactor for the cluster, cluster potential energies, barriers, capture
volumes, and stress corrections must be defined:
pdbSetDoubleArray KMC aSi B D0_Cluster { B 3.0 }
pdbSetDoubleArray KMC aSi B Etotal_Complex { B2 -0.45 B3 -2.5 }
pdbSetDoubleArray KMC aSi B EbarrierDopant_Complex { }
pdbSetDoubleArray KMC aSi B CaptVol_Complex { B2 1.5 B3 2.0 }
pdbSetDoubleArray KMC aSi B VF_Complex { }
Bn

5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
490 Sentaurus™ Process User Guide
I-2013.12
■Third, for these clusters to form, you must introduce a reaction path:
pdbSetArray KMC aSi B ReactionsPointDefect { B,B true }
pdbSetArray KMC aSi B ReactionsCluster { B2,B true }
■None of this will happen without a mobile particle allowing for growing and emission:
pdbSet KMC aSi B Dm B 3.0e-3
pdbSet KMC aSi B Em B 2.1
NOTE The impurity cluster parameters (energies, barriers, capture volumes,
stress corrections) must be specified in the PDB only when the cluster
is implemented (with Implement_Complex true). This saves many
parameters in the description of impurities without impurity clusters.
Fermi-Level Effects: Charge Model
Point defects (I, V) and impurity atoms (B, As) can appear in different charge states in silicon,
while extended defects and impurity clusters have a fixed charge state in Sentaurus
Process KMC. Impurity atoms are neutral in materials using the simple model.
For example, interstitials and vacancies can be triple negative, double negative, double positive,
triple positive, neutral, positive, or negative. Some species and charged states are listed in
Table 55. You can customize these definitions. The maximum charge state for point defects is
and for impurity paired defects .
Ta bl e 55 S p ec i e s an d c h a r ge d s t at es o f Se nt au r us P ro c es s K M C
II
+++, I++, I+, I0, I-, I--, I---
IPPP, IPP, IP, I, IM, IMM, IMMM
VV
+++, V++, V+, V0, V-, V--, V---
VPPP, VPP, VP, V, VM, VMM, VMMM
As As+, Asi+, Asi0, AsV+, AsV0, AsV-
As, AsiP, Asi, AsVP, AsV, AsVM
BB
-, Bi+, Bi0, Bi-
B, BiP, Bi, BiM
CC
0,Ci0
C, Ci
FF
0, FI0, FV0
F, FI, FV
Sb Sb+,Sbi0, Sbi+, SbV-, SbV0, SbV+
Sb, Sbi, SbiP, SbVM, SbV, SbVP
3±
2±

5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
Sentaurus™ Process User Guide 491
I-2013.12
Charge states can be modeled using different approaches. The most intuitive approach is
adding a charge ‘label’ to each particle. Nevertheless, because the migration energy (and
maybe some other parameters) change with the charged state, each of these states requires a
full set of parameters.
Sentaurus Process KMC Approach
No charge label is defined for the particles. The charge is implicitly assumed in each particular
particle, and there are different particles for each charged state. This implies the necessity of
defining the interactions one by one, according not only with the particle type, but also with its
charge state.
The charge is represented in a quasiatomistic approach to account for the fact that the electron
transport is several orders of magnitude faster than the atomic transport. The charge
magnitudes (for example, Fermi level and bandgap width) are associated with each internal box
in the simulation. Consequently, there can be local changes between different boxes, but the
charge magnitudes are considered to be homogenous in each Sentaurus Process KMC internal
element.
Assumptions
Sentaurus Process KMC takes the energy reference in the valence band. The following
assumptions also are taken:
■Charge reactions are faster than structural reactions [44]. Consequently, the charges are
updated instantaneously.
■Formation energy for neutral species (for example, Ef(I0)) are not dependent on the Fermi
level. Sentaurus Process KMC takes the formation energies for neutral species as
parameters using them to compute the energies for the nonneutral species.
■Potential energies for impurity clusters are not dependent on the Fermi level. For example,
Sentaurus Process KMC defines the potential energy for as the energy returned by
the system in the reaction (assuming that ).
■The electronic level dependency with temperature is proportional to the bandgap
temperature dependency. The same applies for the bandgap narrowing. This assumption
allows Sentaurus Process KMC to establish proportionality relations to compute the
In In-, Ini0, Ini-, InV0, InV-
In, Ini, IniM, InV, InVM
PP
+, Pi0, Pi+, PV0, PV-, PV+
P, P i, PiP, PV, PVM, PVP
Ta bl e 55 S p ec i e s an d c h a r ge d s t at es o f Se nt au r us P ro c es s K M C
AsnVm
a
nAs+mV0na–()e-
++ AsnVm
a
→
na–0>

5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
492 Sentaurus™ Process User Guide
I-2013.12
electronic levels and bandgap narrowing at different temperatures using a known value for
one particular temperature.
■Substitutional dopants are always ionized; that is, substitutional boron is always B– and
substitutional arsenic As+.
■The properties inside each Sentaurus Process KMC element are constant. Properties can
change between internal elements.
For further references on similar KMC charge models, see [1][45][46].
Formation Energies for Charged Species
Taking I+ as an example, in the reaction:
(742)
the energy needed to take an electron from an I0 and obtain I++ e– is denoted as e(+,0), and is
measured from the valence band. The formation energy for a positive interstitial is:
(743)
where is the Fermi level. Consequently, the concentration between different interstitial
charge species using as a reference the neutral concentration is:
(744)
(745)
The electronic levels (for T = 0 K) are specified in the parameter database as e0. They are
defined only for silicon. They can be changed with:
pdbSet KMC Si <I/V/impurity> e0 <species> <n>
for example:
pdbSet KMC Si I e0 IP 0.35
I0I+e-
+→
EfI+
() EfI0
() eFe+,0()–+=
eF
I0
[]
I+
[]
--------- eFe+,0()–
kBT
---------------------------
exp=
I-
[]
I0
[]
---------eFe0-,()–
kBT
---------------------------
exp=

5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
Sentaurus™ Process User Guide 493
I-2013.12
Parameters
The bandgap levels for interstitials and vacancies can be retrieved with:
sprocess> pdbGet KMC Si I e0
IM 1.0
IP 0.35
sprocess> pdbGet KMC Si V e0
VMM 1.06
VM 0.6
VP 0.03
VPP 0.13
They also are specified for dopants, like As:
sprocess> pdbGet KMC Si As e0
AsVM 0.77
AsVP 0.3
AsiP 0.1
NOTE The modification of these parameters affects both extrinsic and intrinsic
diffusion.
Binding Energies for Particles
The binding energy needed for pairing and breakup reactions is only specified for the reaction
with the neutral interstitial or vacancy. For example, for boron, the binding energy is specified
for the reaction B– + I0 = Bi–. The other binding energies (for example, B– + I+ = Bi0) are
computed using the binding energy for the above reaction and the energy levels associated to
the charge transitions [46]:
(746)
The activation energy for the breakup is . Because electronic levels scale
with (as shown below), a slight dependency with T is introduced in these calculated binding
energies.
Binding Energies for Impurity Clusters
For an example of how to compute the binding energy for an impurity cluster, see Nonneutral
Reactions on page 485.
EbBi
0
() EbBi
-
()eB
i
()0-,()eI()+0,()–+=
Bi
0
EbBi
0
()EmI+
()+
Eg

5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
494 Sentaurus™ Process User Guide
I-2013.12
Temperature Dependency
The bandgap width used in Sentaurus Process KMC is given by the expression [47]:
(747)
Using the assumption of proportionality with the band gap, Sentaurus Process KMC assumes
that the electronic levels at different temperatures can be computed as:
(748)
Effective state density of conduction and valence bands follows similar expressions:
(749)
(750)
Finally, Sentaurus Process KMC uses the values to compute the intrinsic levels and intrinsic
carrier densities:
(751)
(752)
Parameters
The needed parameters are specified in the parameter database under the folder BandGap:
Eg0
Bandgap width at 0 K ( ).
Agap
Bandgap width temperature dependency parameter ( ).
Bgap
Bandgap width temperature dependency parameter ( ).
Nc300
Effective state density for the conduction band, at 300 K ( ).
Nv300
Effective state density for the valence band, at 300 K ( ).
EgT() EgT=0()
AT2
BT+
-------------–=
ej 1j,+()T() ej 1j,+()0() EgT()
Eg0()
--------------×=
NcT() Nc300() T
300
---------
×expNc
=
NvT() Nv300() T
300
---------
×expNv
=
eiT() EgT()
2
-------------- kBT
2
---------
Nv
Nc
------
ln+=
niT() NcNv
EgT()
2kBT
--------------–
exp×=
Eg0()
A
B
Nc300()
Nv300()

5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
Sentaurus™ Process User Guide 495
I-2013.12
They can be changed using pdbSet. For example, to set the bandgap width at 0 K, use:
pdbSet KMC Si BandGap Eg0 1.17
The parameters for the bandgap temperature dependency are defined for silicon in the
BandGap folder:
sprocess> pdbGet KMC Si BandGap Eg0
1.17
sprocess> pdbGet KMC Si BandGap Agap
0.000473
sprocess> pdbGet KMC Si BandGap Bgap
636.0
sprocess> pdbGet KMC Si BandGap Nc300
3.2e+19
sprocess> pdbGet KMC Si BandGap Nv300
1.8e+19
sprocess> pdbGet KMC Si BandGap expNc
1.5
sprocess> pdbGet KMC Si BandGap expNv
1.5
Charge Attractions and Repulsions
The short-range repulsions between charged particles have been implemented forbidding the
interactions between particles in the same charge state (except for the percolation reactions; see
Percolation on page 477). Long-range forces are considered automatically due to the bias
induced in the particle migration by the local Fermi level.
Fermi-Level Computation
Sentaurus Process KMC computes the Fermi level assuming charge neutrality and Fermi–
Dirac statistics. It simply makes the number of charges in each cell element equal to the
concentration of substitutional dopants and charged impurity clusters in the box. The presence
of mobile charged particles is neglected.
The charge concentration for each element is an average of the charge concentration in the
neighborhood. The averaging radius is taken as the parameter smoothRadius. The power of
expNc
Effective state density temperature dependency parameter (conduction).
expNv
Effective state density temperature dependency parameter (valence).
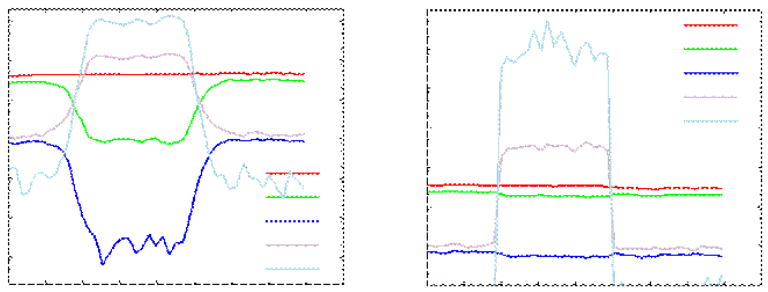
5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
496 Sentaurus™ Process User Guide
I-2013.12
this average is controlled with the parameter smoothPower. This average is important because
of the atomistic nature of the simulation.
Without this averaging, a medium-dose doped sample, with some elements filled up with
particles and some empty ones, could be considered as a set of intrinsic (empty) boxes and a
few boxes with a very high concentration.
For example, a dopant concentration of corresponds to one particle in .
The volume of an internal element may be as small as . This means one particle per
10 boxes. Without any charge averaging, a moving interstitial would diffuse intrinsically in
nine empty boxes and extrinsically in one box. With the average, the interstitial ‘sees’ the right
concentration of and diffuses according to this concentration (see Figure 69).
Figure 69 Simulated vacancy profiles for a p-sample (from 30 to 60 nm) for different
vacancy charged states: (left) smoothing out the charge concentration and (right)
incorrect results without smoothing
Parameters
This smoothing is a default Sentaurus Process KMC feature performed by an ultrafast
algorithm and is controlled only by the cutoff radius (in nanometers) specified in the parameter
database:
sprocess> pdbGet KMC Si BandGap smoothRadius
7
110
20
× cm 3–
10 nm3
1 nm3
110
20
× cm3
1011
Concentration [cm–3]
Depth [nm]
10 20 30 40 50 60 70 80 90 100
1010
108
107
106
105
V0
V–
V––
V+
V++
Depth [nm]
10 20 30 40 50 60 70 80 90 100
1014
Concentration [cm–3]
1013
108
107
1012
109
1010
1011
V0
V–
V––
V+
V++

5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
Sentaurus™ Process User Guide 497
I-2013.12
NOTE To switch off this local computation and to set up a constant, user-
defined dopant concentration, experienced users may want to use:
pdbSetDouble KMC setDopantConc <dopant concentration>
where dopant concentration is a positive quantity for n-type devices and
a negative one for p-type devices.
Updating Charged States
The charge model of Sentaurus Process KMC assumes that the electronic transport and
reactions are faster than the atomic transport and reactions. Therefore, it is necessary to
implement mechanisms to update the charge distribution (and the local Fermi level) that
follows the structural changes. Since the equilibrium ratios depend only on the Fermi level, it
is necessary to update them each time the Fermi level varies.
There are two reasons for local changes in the Fermi level:
■Mobile particles diffusing between elements with different Fermi levels
■Change of the electronic concentration in one element
Besides, each time a new particle appears or disappears because of pairing or breakup
reactions, it is necessary to ensure that the charge state of the new particle is consistent with its
local Fermi level.
Different mechanisms are implemented to maintain the right charge ratios. All are performed
at the same time, but they apply to different scenarios.
Electronic Concentrations and Charge-State Ratios
An update algorithm periodically reviews all the particles and updates the Fermi level and the
proportions of charged particles in each element. The algorithm:
■Smooths the charge distribution.
■Computes the Fermi level for each box using the charge neutrality assumption.
■Establishes the appropriate charge ratios.
NOTE This update algorithm slows down the simulation. It is crucial to follow
the changes in the Fermi level, but without spending too much CPU
time.
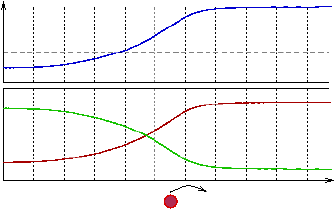
5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
498 Sentaurus™ Process User Guide
I-2013.12
Mobile Particles
Mobile particles see different Fermi levels when they move from one element to another.
Therefore, it is needed to update its charge each time it crosses the boundaries between boxes.
At the same time, particles change their charge state to maintain the proper charge distribution;
consequently, they need extra updates. This is implemented with an algorithm that updates the
charge of mobile particles each time they perform a migration jump. This algorithm also
considers the migration frequency of each particle, as explained in [46], to avoid artificial
concentration increases in the slow diffusing species concentration.
Figure 70 A mobile particle (I–) sees different electronic properties when jumping from one
element to a different one. Its charge state must be updated to reproduce the
expected macroscopic concentration.
Pairing and Breakup Reactions
After pairing or breakup reactions, some species appear and disappear in the Sentaurus
Process KMC elements. To ensure that the concentration of these species maintains the correct
proportions, a breakup, pairing charge update mechanism is implemented. It computes the
probability of the new particles to be in a particular charge state.
Parameters
The charge update algorithm only uses one external parameter in the database called
ChargeVarPercent and accounts for the maximum relative error allowed for the Fermi-level
updates. This parameter is a compromise between accuracy and efficiency.
Decreasing its value leads to more accurate but slower simulations, and the charge model can
overload your computer resources.
Increasing its value speeds up the simulation at the cost of accuracy:
sprocess> pdbGet KMC ChargeVarPercent
0.25
NOTE Only small modifications to this parameter are recommended.
n(x) ni
1+(x)
x
1–7
1–(x)
Concentration

5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
Sentaurus™ Process User Guide 499
I-2013.12
Electric Drift
The charge model of Sentaurus Process KMC considers the:
■Introduction of an electric field, related to the local charge variations.
■Existence of forces acting over the charged species; these forces generate a bias in the
diffusion – the electric drift.
Sentaurus Process KMC models electric bias modifying the jump probabilities to account for
the space anisotropy produced when an electric field is present.
A particle inside the electric field can jump in both directions, but the probability of jumping
following the electric field is higher. Consequently, a ‘migration barrier’ is implemented. The
barriers are related to the relative concentration of each species. For example, for an I+ jumping
from a position in a box to a position in a different box, if , where is
the probability of an interstitial having a positive charge, the jump is always possible.
Otherwise, there is a probability of of being rejected.
For an I+ particle, the jump is accepted with a probability of:
(753)
where typically, if there is no bandgap narrowing effects, and:
(754)
The subscripts 1 and 2 refer to magnitudes in different elements. Figure 71 shows an energy
diagram of this process. The number of rejected jumps for each axis is shown in a report at the
end of the annealing.
Figure 71 Energy diagram showing the jump process
x2
x1
Px
2
()Px
1
()>
P
1Px
2
()[]–Px
1
()[]⁄
Px
2
()
Px
1
()
--------------eFe+,0()–[]
2eFe+,0()–[]
1
–
kBT
---------------------------------------------------------------------------
exp=
e+0,()
2e+0,()
1
=
Px
2
()
Px
1
()
--------------eF2eF1
–
kBT
----------------------
exp=
Jump Prob. = 1
Jump Prob. < 1
eF2 – eF1
x1x2
5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
500 Sentaurus™ Process User Guide
I-2013.12
Bandgap Narrowing
Narrowing due to Dopant Concentration
Sentaurus Process KMC includes doping-induced energy shifts of the conductions band
minimum and the valence band maximum. The narrowing of the fundamental band gap is
presented as the function [48] for n-type semiconductors:
(755)
(756)
and for p-type semiconductors:
(757)
(758)
The total bandgap narrowing is:
(759)
Since the distance between bands shrinks, Eq. 759 gives negative values.
Figure 72 Bandgap narrowing; Sentaurus Process KMC assumes the valence band has
zero energy
∆Egdc Acn1/4 N+
1018
----------
1/4
Acn1/3 N+
1018
----------
1/3
Acn1/2 N+
1018
----------
1/2
++=
∆EgdvAvn1/4 N+
1018
----------
1/4
Avn1/3 N+
1018
----------
1/3
Avn1/2 N+
1018
----------
1/2
++=
∆Egdc Acp1/4 N-
1018
----------
1/4
Acp1/3 N-
1018
----------
1/3
Acp1/2 N-
1018
----------
1/2
++=
∆EgdvAvp1/4 N-
1018
----------
1/4
Avp1/3 N-
1018
----------
1/3
Avp1/2 N-
1018
----------
1/2
++=
∆Egd EgdcEgdv
–=
eF
Ev
Ec
DEv
DEc
Ev=0
eF
Ec
5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
Sentaurus™ Process User Guide 501
I-2013.12
Parameters
The parameters and so on are extracted from [47] and are listed in the parameter
database for BandGap in silicon. For the conduction band:
pdbGet KMC Si BandGap Acn1_4
0
sprocess> pdbGet KMC Si BandGap Acn1_3
-0.01484
sprocess> pdbGet KMC Si BandGap Acn1_2
0.00078
sprocess> pdbGet KMC Si BandGap Acp1_4
-0.01627
sprocess> pdbGet KMC Si BandGap Acp1_3
0
sprocess> pdbGet KMC Si BandGap Acp1_2
-0.00018
For the valence band:
sprocess> pdbGet KMC Si BandGap Avn1_4
0.01508
sprocess> pdbGet KMC Si BandGap Avn1_3
0
sprocess> pdbGet KMC Si BandGap Avn1_2
0.00074
sprocess> pdbGet KMC Si BandGap Avp1_4
0
sprocess> pdbGet KMC Si BandGap Avp1_3
0.01846
sprocess> pdbGet KMC Si BandGap Avp1_2
-0.00263
Narrowing due to Strain
There are two models available for modeling the narrowing due to stress. A simple one and a
full narrowing model.
The full narrowing model is chosen setting the pdb parameter FullNarrowing to true. This
model is the same as in Bandgap Narrowing on page 280:
(760)
(761)
Acn1/4
Eci∆Dci εxx εyy εzz
++()Dcxiεxx Dcyiεyy Dcziεzz
+++=
Evi∆Dvi εxx εyy εzz
++()0.5Dvbi εxx εyy
–()
2εyy εzz
–()
2εzz εxx
–()
2
++()±=
+Dvdi εxy
2εxz
2εyz
2
++()

5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
502 Sentaurus™ Process User Guide
I-2013.12
Sentaurus Process KMC also uses the averaged values of conduction and valence bands
energies:
(762)
(763)
defining the narrowing due to strain effects as:
(764)
The simple model computes the narrowing as:
(765)
where the parameter is called DiScalar.
When Ge is present, the narrowing is computed as a linear interpolation between the narrowing
produced by strain for pure Si ( ), and the one for pure Ge ( ). In this way, the total
narrowing for Si1–xGex is:
(766)
where is the relative Ge concentration specified in Si1–xGex.
Parameters
The parameters used for the full model for pure Si are defined in the Sentaurus Process KMC
dataset as:
sprocess> pdbGet KMC Si BandGap EcDilatational
1 -8.6
2 -8.6
3 -8.6
sprocess> pdbGet KMC Si BandGap EvDilatational
1 -2.1
2 -2.1
sprocess> pdbGet KMC Si BandGap EcDeviatoric(1)
1 9.5
2 0.0
3 0.0
Ecs
∆kT 1
3
---e
Eci∆
kT
------------–
i1=
3
log–=
Evs
∆kT 1
2
---e
Evi∆
kT
------------
i1=
2
log=
∆Egs ∆Ecs ∆Evs
–=
∆Egs Kεxεyεz
++=
K
∆Egs
Si
∆Egs
Ge
∆Egs ∆Egs
Si x∆Egs
Ge ∆Egs
Si
–()+=
x
5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
Sentaurus™ Process User Guide 503
I-2013.12
sprocess> pdbGet KMC Si BandGap EcDeviatoric(2)
1 0.0
2 9.5
3 0.0
sprocess> pdbGet KMC Si BandGap EcDeviatoric(3)
1 0.0
2 0.0
3 9.5
sprocess> pdbGet KMC Si BandGap EvDeviatoric(1)
1 0.5
2 4.0
sprocess> pdbGet KMC Si BandGap EvDeviatoric(2)
1 0.5
2 4.0
The parameter for the simple model for pure Si is:
sprocess> pdbGet KMC Si BandGap DiScalar
1.75
The parameters used for pure Ge are similar to the ones for pure Si, but with the Ge. prefix:
sprocess> pdbGet KMC Si BandGap Ge.EcDilatational
1 0.59 2 0.59 3 0.59
sprocess> pdbGet KMC Si BandGap Ge.EvDilatational
1 -1.24 2 -1.24
sprocess> pdbGet KMC Si BandGap Ge.EcDeviatoric(1)
1 -9.42 2 0.0 3 0.0
sprocess> pdbGet KMC Si BandGap Ge.EcDeviatoric(2)
1 0.0 2 -9.42 3 0.0
sprocess> pdbGet KMC Si BandGap Ge.EcDeviatoric(3)
1 0.0 2 0.0 3 -9.42
sprocess> pdbGet KMC Si BandGap Ge.EvDeviatoric(1)
1 2.55 2 5.50
sprocess> pdbGet KMC Si BandGap Ge.EvDeviatoric(2)
1 2.55 2 5.50
sprocess> pdbGet KMC Si BandGap Ge.DiScalar
1.75
Finally, the simple (0) or full (1) narrowing models are selected:
sprocess> pdbGet KMC Si BandGap FullNarrowing
0
5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
504 Sentaurus™ Process User Guide
I-2013.12
Narrowing due to Presence of an Alloy
The narrowing due to an alloy concentration is computed as (assuming, in this example, that
Ge is the alloy in Si material):
(767)
where is the germanium concentration, and are the parameters needed for the
quadratic interpolation between the silicon gap (1.12 eV) and the Ge gap (0.78 eV). They are
respectively called GeNarrowing and GeNarrowing2.
Parameters
sprocess> pdbGet KMC Si BandGap GeNarrowing
6.8e-24
sprocess> pdbGet KMC Si BandGap GeNarrowing2
0
This is simply (1.12-0.78)/5e22.
Bandgap Narrowing Use
The value of , computed as:
(768)
is used to correct , , , and the dopant levels in the gap, . For these last
ones, they are assumed to be proportional to the band gap. This means that these new values,
after applying the bandgap narrowing correction, are:
(769)
It is interesting to note that .
Whenever a Sentaurus Process KMC model needs a bandgap level, the bandgap narrowing–
corrected value is used. The only exception is the activation energy for the impurity pair
emission from impurity clusters where the narrowing correction can be controlled by:
pdbSet KMC <material> BandGap Correct_Complex <false/true>
NOTE The bandgap narrowing due to doping, stress, and SiGe is always
switched on by default. To disable it, set the proper parameters to zero.
∆EgGe Ge[]β
1β2Ge[]+()–=
Ge[]
β1β2
,
∆Eg
∆Eg∆Egdop ∆Egs ∆EgGe
++=
ei
ni
eF
ejj 1+,()A()
ejj 1+,()A()
corrected ejj 1+,()A()1∆Eg
Eg
----------+
=
∆Eg0<
5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
Sentaurus™ Process User Guide 505
I-2013.12
Charge Model and Boron Diffusion Example
The known charged states of Bi are Bi–, Bi0, and Bi+ [21][22]. The three states are included in
Sentaurus Process KMC, although the inclusion of Bi+ is only important for systems far from
equilibrium. The pairing, breakup, and charge reactions related to Bi are represented in the
current Sentaurus Process KMC model by the reactions:
(770)
(771)
(772)
(773)
Direct breakup of Bi+ is not included because I++ is not implemented. Boron effective
diffusivity is given by the sum of the contribution of all mobile species:
(774)
Using the Maxwell–Boltzmann approximation, the previous equation is usually written as:
(775)
where is the interstitial supersaturation, and and are the hole concentration and the
intrinsic concentration, respectively.
The relations between the above diffusivity components and the microscopic parameters are
[46]:
(776)
(777)
I0B-
+Bi
-
↔
I+B-
+Bi
0
↔
Bi
-Bi
0Bi
+
↔↔
I0I+
↔
DB()
DB() DB
i
-
()
Bi
-
[]
B-
[]
----------DB
i
0
()
Bi
0
[]
B-
[]
-----------DB
i
+
()
Bi
+
[]
B-
[]
-----------++=
DB() SIDXB() DPB()
p
ni
---- DPP B()p
ni
----
2
++=
SI
p
ni
DXB() vcaptDI
0
()I0
[]
*νmBi
-
()
νbreak Bi
-
()
--------------------------
=
DPB() vcaptDI
0
()I0
[]
*νmBi
0
()
νbreak Bi
0
()
-------------------------- eB
i
()0-,()ei
–
kBT
------------------------------------
exp=

5: Atomistic Kinetic Monte Carlo Diffusion
Fermi-Level Effects: Charge Model
506 Sentaurus™ Process User Guide
I-2013.12
(778)
is the intrinsic level.
These expressions can be used as a bridge between the parameters of continuum simulators and
those used by Sentaurus Process KMC. The above continuum expression assumes Maxwell–
Boltzmann and quasi-equilibrium conditions, which are not needed in Sentaurus
Process KMC.
Charge Model and Arsenic Diffusion Example
A similar analysis can be performed for arsenic, which has both vacancy and interstitial
contributions, related to AsV and Asi defects. The arsenic reactions are:
(779)
(780)
(781)
(782)
and:
(783)
(784)
(785)
(786)
(787)
DPP B() vcaptDI
0
()I0
[]
*νmBi
-
()
νbreak Bi
-
()
--------------------------eB
i
()0-,()eB
i
()+,0()2ei
–+
kBT
------------------------------------------------------------------------
exp=
ei
I0As+
+Asi
+
↔
I-As+
+Asi
0
↔
I0I-
↔
Asi
+Asi
0
↔
V0As+AsV+
↔+
V-As+AsV0
↔+
V-- As+AsV-
↔+
V0V-V--
↔↔
AsV+AsV0AsV-
↔↔

5: Atomistic Kinetic Monte Carlo Diffusion
Interfaces and Surfaces
Sentaurus™ Process User Guide 507
I-2013.12
All the previously mentioned contributions are included in KMC. Consequently:
(788)
which in continuum models is usually reduced to:
(789)
is the fraction of interstitial-assisted diffusion. Note, however, that this last continuum
description conveys several simplifying assumptions compared with the model included in
Sentaurus Process KMC. The common assumption that and fit an Arrhenius plot is
only true if the contributions of AsV+ and Asi+ have the same activation energy. The same
applies for the AsV0 and Asi0 contributions. The continuum equation also assumes that the
interstitial fraction, , is independent of the Fermi level (the same for the three charged states)
and is independent of the temperature.
Interfaces and Surfaces
An interface is the extension between two regions with different materials. The most common
interface is the silicon–oxide interface. Sentaurus Process KMC allows for modeling all
interfaces between two different materials.
As explained in Materials and Space on page 398, Sentaurus Process KMC divides the space
in small rectangular elements and assigns to each of them a material. The interfaces are the set
of element faces between different materials.
The element faces are independent. The interface behaves as the sum of all of its faces, but such
an ‘interface’ does not really exist. What exists are the element faces, all of them emitting and
trapping with different rates depending on its area, local stress, and so on. In the following
sections, these element faces are called interface.
Interfaces set the equilibrium concentration for self-silicon point defects and the solubility
concentration for impurities. Sentaurus Process KMC models the interfaces differently for
silicon point defects than for impurities.
DAs() DAsV
+
()
AsV+
[]
As+
[]
------------------DAs
i
+
()
Asi
+
[]
As+
[]
--------------DAsV
0
()
AsV0
[]
As+
[]
----------------- DAs
i
0
()
Asi
0
[]
As+
[]
--------------DAsV
-
()
AsV-
As+
[]
--------------
++ ++=
DAs() fISI1fI
–()SV
–[]DXAs()DMAs()
n
ni
---- DMM As()
n
ni
----
2
++×=
fI
DX
DM
fI
5: Atomistic Kinetic Monte Carlo Diffusion
Interfaces and Surfaces
508 Sentaurus™ Process User Guide
I-2013.12
Figure 73 Sentaurus Process KMC interfaces are the set of element faces between
different materials
Different Interface Models
The parameter Model specifies the behavior of an interface:
pdbSet KMC Oxide_Silicon Model Allcharges
The different models are:
Allcharges
Three-phase segregation model for dopants. Emission and capture of all the
charge states of point defects on materials with full modeling. Capture of
all the charge states of impurity-paired defects on materials with full
modeling.
Amorphous
When one material is full and the other is simple, this interface acts as an
asymmetric mirror. Particles going from the simple to the full material
are reflected, while particles going from the full to the simple material
are allowed to pass. No trapping or emission of particles on either side.
Interface
Three-phase segregation model for dopants. Emission and capture of neutral
point defects on materials with full modeling.
none
No interface between materials. This model is only possible when the model
of the materials involved in the interface is the same; that is, both are
simple or full.
Reflective
The interface acts as a mirror. Particles are not trapped. No emission of
particles on either side.
Boxes
SiO2
Si
Interface
Small Box Face
5: Atomistic Kinetic Monte Carlo Diffusion
Interfaces and Surfaces
Sentaurus™ Process User Guide 509
I-2013.12
Interfaces for Self-Silicon Point Defects
It is common to define a recombination length as the distance from the surface needed to
obtain the equilibrium concentration (see Figure 74).
Figure 74 Recombination length is the distance between the interface and the point in which
the prolongation of the point-defect concentration joins its equilibrium
concentration
The microscopic meaning of can be associated with the probability of a point defect being
trapped at the surface:
(790)
where is the point-defect jumping distance. The smaller , the better sink is the surface.
For interstitials in the silicon–oxide interface, it is close to a perfect sink with < 5 nm
[49][50].
Capture
Interfaces capture neutral interstitials and vacancies with the probability set in Eq. 790. When
is set to zero, the probability is set to 1, a perfect sink.
Emission
In [1], the point-defect (for example, interstitials) equilibrium concentration is related to the
interface frequency emission prefactor and energy as:
(791)
Lr
Extended defects ({311})
creating I supersaturation
I concentration
I equilibrium concentration
Silicon bar
Surface
Prolongation
x = 0
Recombination length
Lr
Ptrap
λ
Lr
-----=
λ
Lr
Lr
Lr
I[]
*2
λ
---D0FS
a2
--------------EfI()
kBT
------------–
exp×=
5: Atomistic Kinetic Monte Carlo Diffusion
Interfaces and Surfaces
510 Sentaurus™ Process User Guide
I-2013.12
where the surface frequency emission is:
(792)
where:
■sites is the number of capture sites in the interface (proportional to its surface and equal
to ).
■ is the surface emission prefactor.
■ and are the migration and formation energies of the point defects, respectively.
■ are the regular corrections to migration and formation due to stress.
■ are the corrections to migration and formation due to Ge concentration,
explained below.
The point defects are emitted from a randomly chosen position at the surface. Only neutral Is
or Vs are emitted when the interface model is Interface. All charge states are emitted and
captured when using Allcharges. In equilibrium, these two models give the same results.
Stress
The presence of stress changes the migration and formation energy of interstitial and vacancies
and, consequently, the emission frequency. Each interface (where, as previously stated,
interface was called to the independent element faces) is oriented in a unique axis ‘j’, and the
projections of the principal axes into ‘j’ should be accounted. Then, the total emission
frequency is:
(793)
where are the projections of the principal axis into the surface axis. Finally, for each axis,
the migration and formation energies including stress effects are computed as:
(794)
where , , and are the perpendicular and parallel activation volumes for
diffusion and the activation volume for formation, respectively.
For more information on these parameters and the stress models, see Stress Effects on Point
Defects, Impurities, Dopants, and Impurity-Paired Point Defects on page 428.
νsites Ptrap
×D0FS 6
λ2
----- D0m,
×Em∆Em
stress ∆Em
Ge Ef∆Ef
stress ∆Ef
Ge
+++++
kBT
-----------------------------------------------------------------------------------------------------------------–
exp×=
2
a2
----- YZ
D0FS
Em
Ef
∆Em
stress ∆Ef
stress
,
∆Em
Ge ∆Ef
Ge
,
νPjaxisνaxis
axis
x'y'z',,
=
Pjaxis
Em
stress Ef
stress
+EmEfσ'axis∆Vpar σ'i∆Vort
1
3
---∆Vfσ'i
i
+
iaxis≠
++ +=
∆Vort
∆Vpar
∆Vf
5: Atomistic Kinetic Monte Carlo Diffusion
Interfaces and Surfaces
Sentaurus™ Process User Guide 511
I-2013.12
Alloys
The presence of an alloy (assumed to be Ge in this example) changes the migration and
formation energy of point defects in the following way:
(795)
(796)
where is the germanium concentration, and are the dependencies of migration and
formation with germanium, specified as EmGe and EfGe in the PDB.
Parameters
The parameters that control the point-defect interface model can be found in the PDB by
looking in the Oxide_Silicon folder. By default, interfaces, other than the oxide–silicon
interface, have their point-defect interface model set to None and do not require any
parameters. The formation energies are listed for the material, not for the interface.
For example, the formation energy of interstitials in silicon is under Silicon, not in
Oxide_Silicon or any other interface.
The migration energies are displayed in the point defect section of the file (see Point Defects,
Impurities, Dopants, and Impurity-paired Point Defects on page 415). The surface values can
be easily obtained using the command line.
For example, for interstitials and vacancies in the silicon–oxide interface:
sprocess> pdbGet KMC Oxide_Silicon I D0FS_Si
5000.0
sprocess> pdbGet KMC Silicon I Ef
4.0
sprocess> pdbGet KMC Oxide_Silicon I RecLnm_Si
0
sprocess> pdbGet KMC Oxide_Silicon V D0FS_Si
800.0
sprocess> pdbGet KMC Silicon V Ef
3.8
D0FS_Mat Surface emission prefactor, . Mat is the short name of the
material.
Ef Formation energy, .
RecLnm_Mat Recombination length, . Mat is the short name of the material.
∆Em
Ge αGe[]=
∆Ef
Ge βGe[]=
Ge[]
αβ,
D0FS I()
EfI()
Lr

5: Atomistic Kinetic Monte Carlo Diffusion
Interfaces and Surfaces
512 Sentaurus™ Process User Guide
I-2013.12
sprocess> pdbGet KMC Oxide_Silicon V RecLnm_Si
0
and for vacancies in the silicon–gas interface:
sprocess> pdbGet KMC Gas_Silicon V D0FS_Si
800.0
sprocess> pdbGet KMC Gas_Silicon V RecLnm_Si
0
sprocess> pdbGet KMC Si V Ef
3.8
NOTE Yo u ca n mo dify t hese va lue s. Cha nges in the formation energy or
surface emission prefactor will modify the DC equilibrium product of
point defects and the diffusivity of all the species. A change, both in the
formation and migration parameters, that maintains the DC product
constant will not produce this undesirable effect, but may change the
extended defects dissolution times.
Oxidation-enhanced Diffusion (OED) Model
The current flux of I (V) across an outer surface in Sentaurus Process KMC is described in the
previous model as:
(797)
being:
(798)
and the jumping distance. An extra term is included to account for oxidation:
(799)
This term tries to combine the Sentaurus Process continuum model (see Surface
Recombination Model: PDependent on page 358) with an atomistic implementation. In
particular, its definition is:
(800)
where is a vectorial prefactor and is the normal to the interface, so that gives the
proper component for a planar, axis-oriented, interface in an internal element.
jn⋅KsI[] I*
[]–()=
Ks
1
6
---νmI()λ
2Lr
⁄=
λ
jn⋅KsI[] I*
[]–()Gox
–=
Gox
Gox θn⋅()Vox Gscale
Vox
Vscale
--------------
Gpow EθP∆Vθ
+
kBT
---------------------------–
exp=
θ
n
θn⋅()

5: Atomistic Kinetic Monte Carlo Diffusion
Interfaces and Surfaces
Sentaurus™ Process User Guide 513
I-2013.12
is the ReactionSpeed computed by the PDE solver in Sentaurus Process and used here
by Sentaurus Process KMC. and are additional model parameters to adjust the
interstitial injection. is a term defined to account for Fermi-level effects, and defined
similarly to the continuum one as:
(801)
is the activation energy for point-defect injection and, finally, is a parameter to include
a hydrostatic dependency for OED.
Consequently, this is a hybrid model in which the continuum solver computes and generates a
ReactionSpeed value to be used by Sentaurus Process KMC to compute the point-defect
injection prefactor.
NOTE Boundary movement is allowed in this model and is switched on by
default. This implies that the model serves to generate a more adequate
point-defect injection from the interface during oxidation processes,
while a remeshing mechanism changes the oxide thickness. If you do
not want the oxide thickness to change, set Grid
Reaction.Modify.Mesh to 0.
Parameters
Table 56 lists the parameters defined for the oxide–silicon interface only.
To use this model, call diffuse with any oxidation parameter (for a list of oxidation
parameters for diffuse, see diffuse on page 908).
Ta bl e 56 Par am et e rs us ed i n OE D m od e l
Parameter name Description
ox_Etheta
Activation energy .
ox_xtheta, ox_ytheta, ox_ztheta
Components for the vector used as a prefactor.
ox_VFtheta
Pressure correction .
ox_Vscale, ox_Gpow
ReactionSpeed control: and .
ox_pp, ox_p, ox_Ep, ox_m, ox_mm,
ox_Em, ox_pot
Parameters used to compute the Fermi-level dependencies introduced
by . In particular, prefactor and activation energies for pp, p,
m, and mm terms, and exponent.
Vox
Vscale
Gpow
Gscale
Gscale
mm m 1ppp++++
mm n
ni
----
2PotOx mn
ni
----
PotOx 1pn
ni
----
PotOx–pp n
ni
----
2PotOx–
+++ +
--------------------------------------------------------------------------------------------------------------------------------------------------------------=
Eθ
∆Vθ
Eθ
θ
∆Vθ
Vscale
Gpow
Gscale
5: Atomistic Kinetic Monte Carlo Diffusion
Interfaces and Surfaces
514 Sentaurus™ Process User Guide
I-2013.12
Interfaces for Impurities
The interface model of impurities in Sentaurus Process KMC follows a three-phase segregation
model. Particles can be emitted to both sides of the interface or can stay trapped at the interface.
Figure 75 shows the atomistic mechanisms and energies for trapping and detrapping
impurities.
Figure 75 Dopants reaching the interface may be trapped by it with a different binding
energy for each interface side. Energy barriers for capture and emission also can
be present.
These interfaces are modeled between any two materials; however, depending on the material
model, the interface will behave differently.
Simple Material Side
The simple material side faces a material using the simple model. In these materials, only
direct diffusion of dopants is allowed. Since there are no paired dopant impurity point defects,
the model is as follows: Dopants arriving at the nonsilicon side may be captured with certain
probability, and they can be remitted later.
Capture
The capture probability is:
(802)
Oxide/Silicon Interface
Barrier (Si)
SiliconOxide
Barrier (Ox)
Binding (Ox) Binding (Si)
Pnon-Sicap A() Barrier A() kBT()⁄–()1Trapped A()
MaxTrapped A()
-------------------------------------------–
exp=

5: Atomistic Kinetic Monte Carlo Diffusion
Interfaces and Surfaces
Sentaurus™ Process User Guide 515
I-2013.12
where is the dopant being trapped, Barrier is the barrier energy, Trapped is the number of
particles trapped at the interface, and MaxTrapped is the maximum number of them that can be
trapped.
If the particle is trapped, there is a probability to evaporate (annihilate) the just-trapped dopant.
Emission
Interfaces emit particles to the nonsilicon side with a frequency given by:
(803)
The emission is proportional to the number of trapped dopants and to a parameter Pref that
acts as a prefactor. The emission energy is:
(804)
The migration energy contains stress and Ge corrections. The binding energy contains a
pressure correction:
(805)
Parameters
The energy barrier to a nonsilicon interface is introduced as EBarrier_Surf<mat>, where
<mat> is Si, aSi, Ox, Ni, Po Gas, and Unknown, or any other user-defined material short name:
The values are always specified in the interface parameter file:
sprocess> pdbGet KMC Oxide_Silicon B EBarrier_SurfOx
B 0.1
Si Silicon
aSi Amorphous silicon
Ox Oxide
Ni Nitride
Po PolySilicon
Gas Gas
Unknown Unknown
A
νnon-Siemiss A() Trapped A()Pref Ener kBT()⁄–()exp=
Ener A() Barrier A() EmA() Binding A()++=
∆Eb
surface A() P∆Vb
surface A()=
5: Atomistic Kinetic Monte Carlo Diffusion
Interfaces and Surfaces
516 Sentaurus™ Process User Guide
I-2013.12
The probability to evaporate a trapped particle is called Evaporation_Surf:
sprocess> pdbGet KMC Oxide_Silicon B Evaporation_Surf
B 0 Bi 0
The pressure correction to the binding energy of the dopants to the surface is given by the
parameter VF_Surf<mat>:
sprocess> pdbGet KMC Oxide_Silicon B VF_SurfOx
The maximum number of trapped particles per cubic centimeter follows an Arrhenius plot with
prefactor C0Max_Surf:
sprocess> pdbGet KMC Oxide_Silicon B C0Max_Surf
2e+14
and energy EMax_Surf:
sprocess> pdbGet KMC Oxide_Silicon B EMax_Surf
0
The prefactor for emission is called Db_Surf:
sprocess> pdbGet KMC Oxide_Silicon B Db_Surf
B 1e-3
Finally, the binding energy of dopants is Eb_Surf<mat>:
sprocess> pdbGet KMC Oxide_Silicon B Eb_SurfOx
B 0.28
Full Material Side
The particles transporting dopants (or impurities) in materials with full modeling are not
typically the dopants themselves, but impurity-paired point defects. In other words, an impurity
plus an interstitial or a vacancy. When these pairs reach the interface, if they are trapped, the
accompanying interstitial or vacancy is recombined, and the dopant itself is piled at the surface.
Consequently, the dopant cannot be emitted unless an incoming interstitial or vacancy reacts
with it, carrying it away from the interface.
Capture
Neutral (or charged, if the model Allcharges is selected) impurity-paired point defects are
trapped at the surface with a probability given by:
(806)
PSicap Ai
() Barrier Ai
()kBT()⁄–()1Trapped A()
MaxTrapped A()
-------------------------------------------–
exp=
5: Atomistic Kinetic Monte Carlo Diffusion
Interfaces and Surfaces
Sentaurus™ Process User Guide 517
I-2013.12
Different barriers can be assigned to andand, consequently, different recombination
probabilities. The number of trapped particles and maximum trapped particles is assigned to
the interface and is shared between both sides.
Emission
Particles are not emitted by themselves, but the interface allows particles to be moved to the
material bulk. Point defects (interstitials and vacancies) can react with the dopants trapped at
the surface, forming mobile impurity-paired point defects. The probability of these reactions
being successful depends on the binding of the dopant to the surface and the barrier energy:
(807)
where:
(808)
The binding is corrected with a pressure-dependent term:
(809)
Parameters
The parameters that control the maximum number of trapped particles have already been
discussed in Simple Material Side on page 514. The barrier energy is called
EBarrier_SurfSi:
sprocess> pdbGet KMC Oxide_Silicon As EBarrier_SurfSi
Asi 0.0
AsV 0.0
and the binding energy is Eb_SurfSi:
sprocess> pdbGet KMC Oxide_Silicon As Eb_SurfSi
Asi 0.1
AsV 0.1
The stress correction is given by:
sprocess> pdbGet KMC Oxide_Silicon B VF_SurfSi
Ai
AV
Pemiss
Si Ai
() Ener Ai
()kBT()⁄–()exp=
Ener Ai
() Binding Ai
()Barrier Ai
()+=
∆Eb
surface Ai
() P∆Vb
surface Ai
()=
5: Atomistic Kinetic Monte Carlo Diffusion
Oxidation
518 Sentaurus™ Process User Guide
I-2013.12
Oxidation
Sentaurus Process KMC is fully coupled with oxidation. Consequently, any oxidation
conditions issued in the diffuse command of Sentaurus Process are transferred to Sentaurus
Process KMC. Setting Grid DoNotMove.Reaction to 1 (it is 0 by default) disables
boundary movement at the oxide–silicon interface. Otherwise, the Sentaurus Process oxidation
algorithm is allowed to work during the reaction step, and the new structure (with expanded
oxide) is imported into Sentaurus Process KMC immediately before the atomistic diffusion
step. The velocities at which the interfaces and the oxide move are used to compute the
displacement of the particles.
Sentaurus Process KMC uses the displacement to relocate the displaced particles and finishes
the remeshing. After this, regular atomistic diffusion occurs. Since there are several
interpolations performed in this process, minor inaccuracies in the final position of particles
can be introduced during remeshing, especially during large oxidations. Regular diffusion
occurring at the same time as oxidation should make these interpolation inaccuracies
negligible.
In principle, Sentaurus Process KMC can be used successfully for 1D, 2D, and 3D oxidation.
In particular, since the precision of the Sentaurus Process KMC solution does not depend on a
fine continuum mesh, a coarse Sentaurus Process mesh can be specified, increasing the
stability of oxidation, while the Sentaurus Process KMC part takes care of the position of
particles. The example in Figure 76 shows the results of such an approach.
Sentaurus Process KMC also allows OED (see Oxidation-enhanced Diffusion (OED) Model
on page 512).
Figure 76 Example of Sentaurus Process KMC coupled with an oxidation in 3D: (left) KMC
simulation in which the internal mesh is coupled to (right) continuum oxidation
simulation
Y
Z
X
Y
Z
X

5: Atomistic Kinetic Monte Carlo Diffusion
Epitaxial Deposition
Sentaurus™ Process User Guide 519
I-2013.12
Epitaxial Deposition
Epitaxial deposition can be performed by using one of the following:
■Regular Sentaurus Process epitaxy
■Native epitaxial deposition using an LKMC model based on [51]
This section describes the native epitaxy model only. The model is switched off by default and
must be switched on for the native epitaxy:
pdbSet KMC Epitaxy true
The model shares many features with LKMC (to fully understand this model, see LKMC: Fully
Atomistic Modeling of Solid Phase Epitaxial Regrowth on page 463).
NOTE The native epitaxy model can be used in both atomistic and
nonatomistic modes. When used in the nonatomistic mode with the
parameter lkmc specified on the diffuse command, doping and
diffusion are controlled by the continuum solver,. The deposition shape
is controlled by KMC. For more information, see Using LKMC for
Deposition Shape on page 283 .
The model introduces the silicon lattice and assigns a flag to each lattice position. This flag is
switched on for lattice positions that match the silicon material and is switched off for positions
lying in the gas. The simulator assigns a frequency to all the off positions to become on, that
is, to accept a silicon atom (coming from the gas). Only off lattice positions that have an on
lattice in the neighborhood have a frequency different from zero. Consequently, the silicon
grows slowly, simulating an epitaxial deposition.
The frequency for an off position at the silicon–gas interface to accept a silicon atom and
become on is:
(810)
where is the frequency for selective epitaxial growth (SEG), very similar to the one
written in Eq. 706, p. 464:
(811)
is a prefactor that accounts for the local microscopic growth for each
configuration. This prefactor depends on two variables: n and m. n can be 100, 110, or 111
defined very similarly to K(1), K(2), and K(3) in LKMC: Fully Atomistic Modeling of Solid
Phase Epitaxial Regrowth on page 463. m is used to distinguish between configurations with
νEpi K'νSEG
LKMC
×=
νSEG
LKMC
νSEG
LKMC KSEG
LKMC site() ESEG
LKMC ∆Esite()+
kBT
---------------------------------------------–
exp×=
KSEG
LKMC site()
5: Atomistic Kinetic Monte Carlo Diffusion
Epitaxial Deposition
520 Sentaurus™ Process User Guide
I-2013.12
the same n but different second neighbor coordination numbers. In this model, also published
in [51], 100 configurations only are split into three different ones: 100, 100.7, and 100.8 for
100 configurations with six or fewer, seven, and 8 or more second neighbor coordination
numbers.
is a correction energy applied to special sites. It is used to simulate the formation of
{311} facets during SEG. As such, only 1 site is defined to have a non null correction: the
{311} local configuration. This configuration is in two different situations a) and b). a) is a
(100) generic site that lacks half of its third neighbors, and b) is a (110) where the second atom
in the chain needed to define the place as 110 would have first coordination number equal to 2.
Assigning a slower rate to configuration a) prevents the {311} facet becoming a {111} facet.
The slower rate for configuration b) assures that the local {311} configuration is not broken by
a lateral (110) regrowth.
is an empirical factor to fit the epitaxial deposition thickness to the specified thickness. This
empirical factor is computed automatically by Sentaurus Process KMC trying to match the
thickness specified in the processing conditions. In particular:
(812)
where:
■thickness is the specified thickness.
■time is the annealing time.
■ is the length of a nanolayer of recrystallized silicon. A nanolayer is assumed to be
half the lattice constant.
■ is the frequency for recrystallization in a
pure (100) substrate.
■ is an empirical constant, available in the PDB as Damage
prefactor.thickness, that relates the microscopic growth of a lattice atom in a (100)
local neighborhood with the macroscopic growth of a (100) substrate.
This epitaxial deposition can create {111} facets and maintain the same (100):(110):(111)
growth rate as SPER. Under regular selective epitaxial conditions, the shapes generated agree
with experimental ones.
At the end of the LKMC epitaxial deposition, the simulator smooths the generated atomistic
interface and reinserts it into Sentaurus Process. The algorithm used to mesh the atomistic
shape uses the parameter KMC Simplify.Geometry. A bigger value provides a faster and
more stable insertion, but with a loss of surface details.
The inclusion of doping is possible during LKMC epitaxy. If a doping profile with a linear
change is indicated, the included doping will be linear with time, and not with thickness.
∆Esite()
K'
K'Kthickness thickness νSEG
LKMC 100()time Knanolayer
××()⁄×=
Knanolayer
νSEG
LKMC 100()K100() ESEG
LKMC kBT()⁄–()exp×=
Kthickness

5: Atomistic Kinetic Monte Carlo Diffusion
Epitaxial Deposition
Sentaurus™ Process User Guide 521
I-2013.12
Finally, the generation of an LKMC starting surface from Sentaurus Process and the reinsertion
after LKMC epitaxy are delicate and time-consuming operations if they are performed only
once at the start of the diffuse command and at the end. This means that the state of the
simulator at intermediate steps during the diffuse command may not be synchronized with
the KMC simulator.
NOTE To avoid problems, the KMC Movie command is disabled during LKMC
epitaxy. In addition, the diffuse movie command must not be used
during LKMC epitaxial deposition.
Parameters
The parameters needed for this model are defined in the parameter database under KMC Si
Epitaxy. Table 57 lists the parameters for the site prefactors.
Table 58 lists the parameters for the activation energies.
Table 59 lists the parameters for the thickness.
As an example, some of these parameters can be obtained as:
sprocess> pdbGet KMC Si Epitaxy prefactor.SEG.100
3.3e+15
Table 57 Parameters for site prefactors
Parameter Description
prefactor.SEG.100.8
For (100) sites with 8 or more second neighbor coordination number
prefactor.SEG.100.7
For (100) sites with 7 second neighbor coordination number
prefactor.SEG.100
For (100) sites with 6 or fewer second neighbor coordination number
prefactor.SEG.110
For (110) sites
prefactor.SEG.111
For (111) sites
Table 58 Parameters for activation energies
Parameter Description
energy.SEG
Overall activation energy for SEG
energy.SEG.311
Correction for 311 planar SEG
Table 59 Parameters for the thickness
Parameter Description
prefactor.thickness
Empirical factor to fit the thickness in epitaxial growth
5: Atomistic Kinetic Monte Carlo Diffusion
Including New Impurities
522 Sentaurus™ Process User Guide
I-2013.12
sprocess> pdbGet KMC Si Epitaxy prefactor.SEG.100.8
1.8e+18
sprocess> pdbGet KMC Si Epitaxy prefactor.SEG.111
3.5e+14
sprocess> pdbGet KMC Si Epitaxy energy.SEG
2.7
Figure 77 shows an example of epitaxial deposition using LKMC.
Figure 77 (Left) Initial shape and (right) final shape after LKMC epitaxial deposition with
thickness of 175 nm
Including New Impurities
You can customize Sentaurus Process KMC to include other impurities not supported by
default. The modifications affect the parameter database and the file KMC.tcl. Nevertheless,
it is not necessary or recommended to modify the files included in the Sentaurus Process
distribution. All the modifications can be included in the input file.
For the parameter database, the pdbSet family of commands allows overwriting previous
values or defining new ones. For the procedures written in the file KMC.tcl, defining the new
procedure in the input file is enough; the new one will be executed instead of the old one.
The modifications include the following:
1. Include the new impurity X in Sentaurus Process KMC under the label named
KMC Impurities.
5: Atomistic Kinetic Monte Carlo Diffusion
Including New Impurities
Sentaurus™ Process User Guide 523
I-2013.12
2. Include the impurity-related particle pairs (Xi or XV or both) in KMC Pairs (see Particles
in Models on page 410). If your model does not have impurity pairs (in other words,
simple material), you do not need to specify them, including Dm and Em.
3. Be careful about which charge states you include because not all are allowed; you must
specify parameters for all those included.
4. Include possible aliases for the particle in KMC Aliases. If the particle already exists for
Sentaurus Process, include this name as an alias in Sentaurus Process KMC (see Alias on
page 411).
5. You can customize the colors for this particle (although it is optional) in Tecplot SV in
KMC Colors (see Colors on page 411).
6. To create parameters for the new particle, define the parameters explained in Particles and
Parameters on page 411 under KMC <mat> X, where <mat> is every material defined in
your simulation, and X is the name of the new defined dopant. Be sure to include all of
them. Parameters for the impurity cluster model are not needed if Implement_Complex
is set to false. All others require values since they specify how the surface and amorphous
regions interact with the new dopant X.
7. Specify the reactions for KMC <mat> X in ReactionsPointDefect. Typical reactions
here include the pair formation (such as X,I true) and impurity cluster formation (such
as Xi,X true). These reactions only need to be defined in materials with the full model.
8. Specify also the reactions with damage and extended defects if there are any. You can leave
these fields empty. See Interaction with Impurities on page 441, Interactions on page 446,
Interactions on page 450, and Interactions on page 454.
If you include impurity clusters, ensure the following:
1. Define reactions that create impurity clusters from two isolated particles.
2. Enable the right impurity clusters for your simulation with Implement_Complex (see
Impurity Clusters on page 472).
3. Fill the parameters for the impurity clusters. You can leave the energy barriers empty, but
you must specify Etotal_Complex energies and CaptVol_Complex capture volumes.
4. Write the reactions for your impurity clusters. If you have specified an energy for a
particular impurity cluster, then that impurity cluster should be reachable through some
reactions. Include the reactions with dopants and point defects in ReactionsCluster
(see Interactions on page 487).
5. If you need dopant deactivation without diffusion for high concentrations, see Percolation
on page 477.
5: Atomistic Kinetic Monte Carlo Diffusion
Including New Impurities
524 Sentaurus™ Process User Guide
I-2013.12
Finally, you must define some variables set in the KMC.tcl file placed in TclLib. This can
(and should) be performed locally in your input file:
1. Add the names of your new impurities and pairs to the nameOf array.
2. Complete the map of MC implantation to Sentaurus Process KMC with MCnameOf.
If you need to transfer information back and forth from continuum to KMC, you also must
modify the procedures PDE2KMCUser and KMC2PDEUser:
■Add the new particles and clusters to the lists in PDE2KMCUser. The first field is the field
name in Sentaurus Process (continuum models), the second is the name in Sentaurus
Process KMC, and the third is the conversion factor. For example:
fproc PDE2KMCUser {} {
return "Dopant X 1 \
DopantInt Xi 1 \
DopantVac XV 1 \
DopantCluster X2 .5 \
DopantCluster X3 .3333"
}
■Add the new particles in KMC2PDEUser. The first name is the name in Sentaurus
Process KMC, the second is the Sentaurus Process field, and the third is the factor. For
example:
fproc KMC2PDEUser { } {
return "X Dopant 1\
Xi DopantInt 1\
XV DopantVac 1\
X2 DopantCluster 1\
X3 DopantCluster 1.5\
X4 DopantCluster 2"
}
NOTE MCnameOf can be used to manipulate some KMC species without
actually implementing them. For example, an unknown implant can be
performed to simulate preamorphization. If Sentaurus Process KMC
does not support the unknown dopant, use
set MCnameOf(dopant) "I" to instruct the MC implantation code
to pass the dopant atoms as interstitials to Sentaurus Process KMC. This
way, all of the damage generated by an unknown dopant implant will be
correctly calculated and passed, and the dopant ions will be considered
to be silicon interstitials by Sentaurus Process KMC.

5: Atomistic Kinetic Monte Carlo Diffusion
Including New Impurities
Sentaurus™ Process User Guide 525
I-2013.12
Impurities Diffusing without Pairing
Sentaurus Process KMC allows impurities to diffuse using two different mechanisms:
■Normal diffusion
■Diffusion without pairing
Normal Diffusion
For impurities with +1 or –1 charge, in other words, dopants, the substitutional dopant is active,
but it does not diffuse. The substitutional dopant reacts with interstitials or vacancies, forming
a pair that diffuses. These pairs break up with a given frequency, releasing the dopants back
into the substitutional positions.
Diffusion without Pairing
For neutral impurities, the normal diffusion is still available. An alternative diffusion
mechanism is migration without pairing. In these cases, the impurity diffuses as it is, that is,
the substitutional impurity has a nonzero diffusivity and continues forming pairs with point
defects.
Diffusion without pairing has the following characteristics:
■The impurity has nonzero diffusivity. The pairs can exist, but they do not have to diffuse:
sprocess> pdbGet KMC Silicon F Dm
F 5e-3
FV 0
FI 0
sprocess> pdbGet KMC Silicon F Em
F .8
FV 5
FI 5
■When a pair (for example, FI) breaks up, Eq. 661 and Eq. 662, p. 420 still apply. The
migration energy of the impurity is not accounted for.
■The impurities cannot interact with extended defects, but their pairs can, as explained in
Extended Defects on page 442.
■Impurities interact with interfaces. Interfaces re-emit impurities.
■Impurity clusters are possible with some variations:
• Reactions within the impurities (for example, F + F) apply to the moving particles and
not only the percolation model.

5: Atomistic Kinetic Monte Carlo Diffusion
Reports
526 Sentaurus™ Process User Guide
I-2013.12
• Impurity clusters emit point defects and impurities, as explained in Emission on
page 436, but the capture and emission reactions for impurities are:
(813)
• Consequently, the binding energies involved in the capture and emission of impurities
will be:
(814)
• Recombination, FT, and complementary FT mechanism are implemented as well. The
binding energies for them are not changed since the emission recombination of Ai (for
AnIm clusters) or AV (for AnVm clusters) is not involved.
Reports
Sentaurus Process KMC prints several reports in the log file including:
■Models used
■Particle distribution
■Cluster distribution
■Defect activity
■Interactions
■Event
Models Used Report
Sentaurus Process KMC reports the models used immediately after being initialized. A
summary is printed for each impurity, dopant, and point defect:
+----------------+-------------------------------------------------+
| KMC models | Silicon |
+----------------+-------------------------------------------------+
|Interstitial | |
| DiffModel |Direct(I) |
| ChargeModel |I( -1 0 1 ) |
| ClusterModel |I+I AmorphousPocket Void ThreeOneOne Loop |
| SPERModel |Clean |
|Vacancy | |
| DiffModel |Direct(V) |
| ChargeModel |V( -2 -1 0 1 2 ) |
| ClusterModel |V+V AmorphousPocket Void ThreeOneOne |
AnImAA
n1+Im
↔+
EAnImbA() Epot An1+Im
()Epot AnIm
()–=

5: Atomistic Kinetic Monte Carlo Diffusion
Reports
Sentaurus™ Process User Guide 527
I-2013.12
| SPERModel |Clean |
|Arsenic | |
| DiffModel |Kick-out(Asi) Kick-out(AsV) |
| ChargeModel |Asi( 0 1 ) AsV( -1 0 1 ) |
| ClusterModel |As+As AsnVm |
| SPERModel |As4Vm 30% deposited 70% moved |
|Boron | |
| DiffModel |Kick-out(Bi) |
| ChargeModel |Bi( -1 0 1 ) |
| ClusterModel |BnIm AmorphousPocket Loop |
| SPERModel |B3Im 100% deposited 0% moved |
|Fluorine | |
| DiffModel |Direct(F) |
| ChargeModel |FI( 0 ) FV( 0 ) |
| ClusterModel |F+F FnIm FnVm |
| SPERModel |F2Im F2Vm 30% deposited 70% moved |
+----------------+-------------------------------------------------+
| Stress model | Disable |
+----------------+-------------------------------------------------+
| SPER model |Non-Lattice KMC |
+----------------+-------------------------------------------------+
This summary is printed for any particle allowed in the simulation, even if this particle is not
going to be used.
Particle Distribution Report
The activation report lists how many dopants exist per material and the state of the material:
-- KMC Particle distribution report
Material Dopant Total State
Ta bl e 60 S e nt au r us P ro ce ss K M C mo de ls
Model Description
ChargeModel
The particles and their allowed charged states are displayed.
ClusterModel
The interactions between the impurity or point defect and extended defects and clusters are
displayed.
DiffModel
The diffusion model can be direct or kick-out. Kick-out means that the particle does not
diffuse unless paired with an interstitial or vacancy.
SPERModel
Recrystallization model shows the percentage of dopant being deposited, and the bigger
deposited cluster, if any. Point defects are just cleaned during the recrystallization.
Stress model
Disabled or enabled.
SPER model
The algorithm for SPER can be non-lattice KMC (isotropic) or lattice KMC (anisotropic).

5: Atomistic Kinetic Monte Carlo Diffusion
Reports
528 Sentaurus™ Process User Guide
I-2013.12
Nitride As 28604 100.00% mobile
Oxide As 759 100.00% mobile
Oxide_Silicon As 1777 100.00% trapped
PolySilicon As 21986 100.00% mobile
Silicon As 58394 41.55% active
Oxide B 25 100.00% mobile
PolySilicon B 283 100.00% mobile
Silicon B 828 99.52% active
The different states depend on the material:
Cluster Distribution Report
This report shows the distribution of clusters versus size for each material and reports how
many clusters are in the simulation and their types:
-- KMC impurity cluster distribution report --
Name #number
--- Silicon ---
As4I 4889 As2 4062 As4 1110 As2V 571
As3I 121 As2I 95 As3 56 As3V 19
As4V 9 As4I2 6 B2I2 2
For example, in the above report, all the BICs are B2I2. The As–vacancy clusters are distributed
between different types, but the most common one is As4I.
Defect Activity Report
Sentaurus Process KMC displays the point defects, impurities, dopants, extended defects,
clusters, amorphous areas, recrystallizations, and surface emission accounted for during the
simulation:
-- KMC defect activity report --
First: Time Events Temp | Last: Time Events Temp Label
0.000 0.00e+00 27 | 3e-01 2.86e+09 672 PointDefect (I)
0.000 0.00e+00 27 | 2e-01 2.86e+09 772 PointDefect (V)
2.285897e-02 1.04e+06 27 | 75610 here PointDefect (As)
full material Particles can be active (substitutional dopant) or inactive (anything else).
simple material Particles can be mobile (single impurity) or immobile (impurity in a
cluster).
Interface Number of particles trapped at the interface.
5: Atomistic Kinetic Monte Carlo Diffusion
Reports
Sentaurus™ Process User Guide 529
I-2013.12
0.000 0.00e+00 27 | 1132 here PointDefect (B)
9.639 1.47e+06 27 | 4e-01 2.86e+09 572 PointDefect (IM)
1.657462e-01 1.08e+03 27 | 3e-01 2.86e+09 672 PointDefect (IP)
11.025 1.52e+06 27 | 2e-01 2.86e+09 722 PointDefect (VMM)
2.849592e-02 3.60e+01 27 | 2e-01 2.86e+09 722 PointDefect (VM)
103.606 2.99e+06 27 | 2e-01 2.86e+09 772 PointDefect (VP)
2.352941e-04 1.19e+07 650 | 1e-03 1.85e+09 1230 PointDefect (VPP)
6.114947e-01 1.08e+06 27 | 2e-01 2.86e+09 722 PointDefect (AsV)
13.397 1.60e+06 27 | 2e-01 2.86e+09 722 PointDefect (AsVM)
6.057627e-01 1.08e+06 27 | 2e-01 2.86e+09 772 PointDefect (AsVP)
0.000 1.04e+06 27 | 8e-01 2.86e+09 222 PointDefect (Asi)
8.988 1.45e+06 27 | 33 here PointDefect (AsiM)
9.790420e-01 1.10e+06 27 | 4e-01 2.86e+09 622 PointDefect (AsiP)
9.143608e-01 2.14e+04 27 | 6e-04 2.55e+07 1000 PointDefect (BV)
1.154401e-01 5.63e+02 27 | 1e-03 1.68e+09 1230 PointDefect (BVM)
1.501662e-01 9.14e+02 27 | 7e-04 3.78e+07 1040 PointDefect (BVP)
0.000 0.00e+00 27 | 3e-01 2.86e+09 672 PointDefect (Bi)
7.337895e-03 2.00e+00 27 | 3e-01 2.86e+09 672 PointDefect (BiM)
1.501662e-01 9.14e+02 27 | 1e-03 7.36e+08 1300 PointDefect (BiP)
0.000 0.00e+00 27 | 3e-02 2.85e+09 862 AmorphousPocket (I)
5.772006e-03 1.00e+00 27 | 1e-03 2.07e+09 1180 AmorphousPocket (V)
0.000 0.00e+00 27 | 2e-03 2.43e+09 1030 AmorphousPocket (IV)
3.254430e-04 1.29e+07 720 | 8e-04 1.36e+08 1130 Void
2.241841e-03 1.04e+07 950 | 1 here ThreeOneOne
0.000 1.04e+06 27 | 5111 here ImpurityCluster (AsI)
7.615 1.40e+06 27 | 599 here ImpurityCluster (AsV)
7.573 1.40e+06 27 | 5228 here ImpurityCluster (As)
1.154401e-02 6.00e+00 27 | 2 here ImpurityCluster (BI)
17.278 4.97e+06 27 | 7e-04 5.66e+07 1070 ImpurityCluster (B)
0.000 0.00e+00 27 | 1464 here Elements emitting I
0.000 0.00e+00 27 | 1464 here Elements emitting V
3.300695e-04 1.29e+07 730 | 936 here Elements emitting As
7.478722e-04 6.83e+07 1080 | 1e-03 2.15e+09 1130 Elements emitting Asi
47.284 2.31e+06 27 | 7e-04 6.21e+07 1110 amorphous (Recryst.)
0.000 0.00e+00 27 | 23025 here LatticeAtom
The report contains two columns with three subcolumns each. The first report shows when the
model was first used; the last report shows when the model was last used. If the model is still
being used, the number of particles or defects using it is displayed followed by “here.” The
three subcolumns report the time, number of simulated events, and temperature.
For example, the previous report shows the first {311} defect (ThreeOneOne) was formed at
, with a temperature of , and with one {311} still in the simulation. There
was silicon amorphous, from 47 s, to at . Since any anneal resets
the time to zero, the first time applies to a previous anneal or implant (since there is damage
accumulation, in other words, room temperature annealing, during implants).
2.2 10 3–
× s
950°C
27°C
6.7 10 4–
× s
1110°C

5: Atomistic Kinetic Monte Carlo Diffusion
Reports
530 Sentaurus™ Process User Guide
I-2013.12
For the interface models, the report shows how many interfaces are in the simulation (I and V),
and how many of them contain trapped dopants (936 for As). It also lists the first and last time
the interfaces let As go in the form of Asi.
This information shows how the different models were used during the simulation and when
the damage was annealed.
Interactions Report
This reports shows, for each material and interface, all the reactions between a mobile particle
(point defect or impurity-pair point defect) and the number of times they happened.
The first column lists the name of the interacting defect, the second the interaction itself, and
the third the number of times it happened from the beginning of the simulation. Columns 4, 5,
6, and 7 are the same as 2 and 3. This report explains which reactions may be important and
which are not. For example, in the report below, the reaction I+VP (31 times) is negligible in
comparison with I+V (111848 times) and does not play a significant role in this simulation for
the formation of AP.
Finally, depending on the defect reported, the output can be slightly different.
PointDefect
-- KMC interactions report --
Reaction #Times Reaction #Times Reaction #Times---
Silicon ---
PointDefect I+I 278778 I+V 111848 I+As 1435719
PointDefect I+B 644803 I+IM 862 I+IP 7856
PointDefect I+VMM 83 I+VM 3316 I+VP 31
PointDefect I+AsV 273 I+AsVP 2480 I+AsVM 35
PointDefect I+Bi 2809
PointDefect V+V 56907 V+As 25324 V+IM 103
PointDefect V+IP 3541 V+VMM 288 V+VM 4248
PointDefect V+VP 77 V+Asi 12480 V+AsiP 3405
PointDefect V+Bi 433 V+BiP 1924 V+BiM 44
PointDefect As+As 243 As+IM 1313602 As+VMM 38966
PointDefect As+VM 30332 As+AsVM 2666
PointDefect B+IP 285066 B+BiP 203
It includes the reaction between two mobile particles.
5: Atomistic Kinetic Monte Carlo Diffusion
Reports
Sentaurus™ Process User Guide 531
I-2013.12
Indirect Diffusion
When using the indirect diffusion model for amorphous materials, the results are similar to
crystalline ones, but I and V mean dangling bond and floating bond, respectively.
Reaction #Times Reaction #Times Reaction #Times
--- AmorphousSilicon ---
PointDefect I+V 40475 I+B 30707
AmorphousPocket
AmorphousPocket Ix+I 713391 Ix+V 210933 Ix+VMM 305
AmorphousPocket Ix+VM 4575 Ix+VP 25
AmorphousPocket Vx+I 31202 Vx+V 43040 Vx+IM 126
AmorphousPocket Vx+IP 11372 Vx+Bi 1281 Vx+BiM 47
AmorphousPocket IxVy+I 312602 IxVy+V 910338 IxVy+B 204
AmorphousPocket IxVy+IM 154 IxVy+IP 15555 IxVy+VMM 110
AmorphousPocket IxVy+VM 4930 IxVy+VP 5 IxVy+Bi 4683
AmorphousPocket IxVy+BiP 618 IxVy+BiM 106
It includes the reaction between small interstitial clusters (Ix), small vacancy clusters (Vx), and
APs including both Is and Vs (IxVy). To keep the report small, all the sizes are condensed into
only one Ix, Vx, or IxVy.
ThreeOneOne
ThreeOneOne Ix+I 1035217
All the {311} sizes are condensed under the term Ix.
Loop
Loop Ix+I 177885 Ix+BiM 10
All the dislocation loop sizes are written under the term Ix.
ImpurityCluster
ImpurityCluster B2+I 31 B2+Bi 6
ImpurityCluster B3+I 3
ImpurityCluster B2I+I 463 B2I+V 25 B2I+Bi 21
ImpurityCluster B3I+I 27591 B3I+V 1148
ImpurityCluster BI2+V 3835 BI2+Bi 278
ImpurityCluster B2I2+I 1518 B2I2+V 86 B2I2+Bi 1314
ImpurityCluster B3I2+I 26
ImpurityCluster B2I3+V 26

5: Atomistic Kinetic Monte Carlo Diffusion
Reports
532 Sentaurus™ Process User Guide
I-2013.12
ImpurityCluster As2+V 1728 As2+As 5 As2+AsV 1251
ImpurityCluster As3+V 100 As3+AsV 69
ImpurityCluster As4+V 3
ImpurityCluster As2V+I 56605 As2V+Asi 20411
ImpurityCluster As3V+I 87723 As3V+Asi 18437
ImpurityCluster As4V+I 1973488
Since impurity clusters are important for the correct activation and deactivation of dopants, and
their sizes are small numbers, all are written in the report.
Interface
--- Oxide_Silicon ---
Interface I 11364 I+As 73 V 1141
Interface AsV 410 Asi 1440
The name of each particle interacting with any interface, and the number of times it happened,
is reported last.
Event Report
The event report is the reverse of the reaction report. The reaction report shows the forward
reactions; the events report shows the reverse ones. Since the reactions and other events depend
strongly on the defects, this report changes from defect to defect.
PointDefect
-- KMC event report --
Name Jump X Jump Y Jump Z Break-up
PointDefect I 2921360317 2921305233 2921406601
PointDefect V 1285293026 1285290404 1285265687
PointDefect As 1120 1071 1163
PointDefect B 7 23 15
PointDefect IM 228653836 228646801 228625103
PointDefect IP 1074734274 1074760443 1074719693
PointDefect VMM 86102860 86115025 86090069
PointDefect VM 846251937 846307626 846269323
PointDefect VP 7881794 7875672 7878359
PointDefect Asi 1144728 1144688 1142864 832840
PointDefect AsiP 51512 52143 51812 1894402
PointDefect AsV 18255 17863 17967 30572
PointDefect AsVP 3544 3436 3438 22783
PointDefect AsVM 3585 3586 3460 37804
PointDefect Bi 13459181 13453297 13458878 257929
PointDefect BiP 94930 95115 95524
5: Atomistic Kinetic Monte Carlo Diffusion
Reports
Sentaurus™ Process User Guide 533
I-2013.12
PointDefect BiM 124899 123825 124860 670411
The second column shows the name of the mobile particle. The 3rd, 4th, and 5th columns show
how many diffusion events (hops or jumps) have every particle perform in the x-, y-, and z-axis,
respectively. In the absence of anisotropies, these three numbers must be approximately the
same. Finally, the last column shows the number of breakups. Since not all the mobile particles
can break up (for example, Bi will break in B + I, but I cannot break up), some of the particles
will have an empty column there. The relative number between the number of diffusion steps
and the number of breakups gives an estimation of the stability of the particle. The more stable
the particle (more diffusion events and less breakups), the larger its long-hop distance.
When using the double-hop model, the report looks like:
Name Jump >< Jump >> Jump >^ Break-up
PointDefect I 288583696 288599278 1154233402
PointDefect V 524892 527369 2103188
PointDefect IM 32935856 32946513 131772254
PointDefect IP 153307399 153285675 613169175
PointDefect VMM 373744 374963 1498954
PointDefect VM 256003 255776 1022437
PointDefect VP 309 260 1111
PointDefect VPP 4 4 10
PointDefect AsV 14570 14473 58109 47289
PointDefect AsVM 223313
PointDefect AsVP 348 371 1464 3547
PointDefect Asi 102330 102738 411863 839856
PointDefect AsiP 8695 8669 34730 843920
PointDefect BV 41 30 157 3
PointDefect BVM 166 181 682 280
PointDefect BVP 1
PointDefect Bi 26231 25952 104499 21468
PointDefect BiM 9798 9735 39016 42747
In this report, the third column reports the number of jumps in opposite directions. The fourth
column reports the number of jumps in the same direction, and the fifth column lists jumps in
orthogonal directions. For further information, see Hopping Mode on page 423.
AmorphousPocket
Name IV Recom I Emis V Emis
AmorphousPocket Ix 300905
AmorphousPocket Vx 59627
AmorphousPocket IxVy 802007
Ix are small interstitial clusters. They can only emit interstitials. Vx are small vacancy clusters
that can only emit vacancies. Finally, IxVy are APs. They can recombine (destroy) an internal
IV pair.
5: Atomistic Kinetic Monte Carlo Diffusion
Reports
534 Sentaurus™ Process User Guide
I-2013.12
ThreeOneOne
Name I Emis
ThreeOneOne Ix 1038899
{311}s can only emit neutral interstitials.
Loop
Name I Emis
Loop Ix 181927
Dislocation loops, like the {311}s, can only emit neutral interstitials.
ImpurityCluster
Name Emis
ImpurityCluster B3 10 V
ImpurityCluster B3I3 913 I 14203 Bi
ImpurityCluster B2I3 4710 I
ImpurityCluster B3I2 13570 I
ImpurityCluster B2I2 656 I
ImpurityCluster BI2 1 I 32 Bi
ImpurityCluster B2I 578 BiP
An impurity cluster (for example, a BIC) emits Bi and I. B2I also can emit BiP particles.
Finally, an internal Frenkel pair can be created, trapping the I and emitting the V. This has been
the case in this simulation for 10 B3 ( ). Since BV is not defined by default, it
cannot be emitted.
Name Emis
ImpurityCluster B3 1 V
ImpurityCluster B2 10 V
ImpurityCluster B3I2 6 I
ImpurityCluster C2I 3 I 1 V 32 Ci
ImpurityCluster B3I 1 V 6 Bi
ImpurityCluster B2I 107 Bi
ImpurityCluster CB2I 84 Bi 2 Ci
ImpurityCluster CBI 42 I 1 Bi
In this case, apart from more or less standard boron and carbon clusters, there is a hypothetical
carbon–boron–interstitial (CBI) cluster.
Two members of this CBI cluster are present here,
CB2I, emitting Bi and Ci, and CBI, emitting I and Bi.
B3B3IV+→
5: Atomistic Kinetic Monte Carlo Diffusion
Reports
Sentaurus™ Process User Guide 535
I-2013.12
Amorphous Defects
Name Recryst.
Amorphous Ele. 4932 B3I3 4598 B2I3 7087 BI2
This is an example of recrystallization depositing inactive boron in different cluster
configurations. In this case, the simulator tries to deposit the impurity clusters with a proportion
of 30%, 30%, and 40% for B3I3, B2I3, and BI2, respectively.
Lattice Atoms
Name SPER
LatticeAtom I 2070434
Example of output related with epitaxial growth, showing the number of atoms that were
incorporated into crystalline silicon.
Simple Materials
An event report is written for simple materials as well:
--- AmorphousSilicon ---
Name Jump >< Jump >> Jump >^ Break-up
PointDefect B 4 2 16
Name Recryst.
amorphous I 7575
Name Jump >< Jump >> Jump >^
Rejected PD As 9 42
Indirect Diffusion
The report for amorphous materials with indirect diffusion is similar to the one of crystalline
materials, but the I and V mean dangling bond and floating bond, respectively.
--- AmorphousSilicon ---
Name Jump >< Jump >> Jump >^ Break-up
PointDefect I 408765 409824 1637334
PointDefect V 244177 244724 975635
PointDefect Bi 456685 457074 1824373 30707
Name Jump >< Jump >> Jump >^
Rejected PD I 141130
Rejected PD V 85422
Rejected PD Bi 155922

5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
536 Sentaurus™ Process User Guide
I-2013.12
Extracting KMC-related Information
You c an r equest Se nta uru s Process K MC info rm ation in one of t he foll owi ng ways:
■Using the Sentaurus Process interface (in some cases, the information must be translated
to Sentaurus Process fields using kmc deatomize before calling the Sentaurus Process
commands):
•struct command
• With the
select, print, WritePlx, and plot commands
■Calling directly the Sentaurus Process KMC kernel:
• Writing Sentaurus Process KMC TDR files
• Extracting atomistic information with the
kmc extract command (see kmc on
page 995)
Calling Sentaurus Process KMC directly has the following advantages:
■More information can be obtained than using the regular interface.
■The atomistic continuum conversions needed to compute the concentrations are more
accurate.
■The atomistic information (in other words, 3D coordinates and shape of defects) can be
displayed.
■Simulations can be saved and loaded.
Transferring Fields from KMC to Continuum Information:
deatomize
Sentaurus Process KMC is independent of the mesh and fields of Sentaurus Process.
Consequently, after a diffusion in atomistic mode (see Atomistic Mode on page 383), there are
no Sentaurus Process fields to visualize. You can instruct Sentaurus Process KMC to create
fields with KMC information. For example, to deatomize the simulation and convert the 3D
positions into concentrations, use:
kmc deatomize name=<field>
When the field is created, Sentaurus Process KMC will not modify it unless there is a new
deatomize command. This means that the field is synchronized with the Sentaurus
Process KMC simulation when it is created. However, after that, if the simulation changes (for
example, performing another diffusion), the field will conserve the initial values.
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 537
I-2013.12
The fields created by kmc deatomize are:
■Concentration of particles (number of particles per volume unit). It could be substitutional
(B, As, ...), paired (AsV, Bi), and the charge state is included (AsV is neutral; AsVM is
negative). If these particles are mapped as mobile in the KMC.tcl file (see Including New
Impurities on page 522), the field will be computed as an average of time. Otherwise, the
field will contain the instantaneous concentration.
■Total concentration of impurities (number of particles in any defect per volume unit):
Itotal, BTotal,…
■Concentration in the interface (number of particles in the interface per volume unit):
BInterface, AsInterface…
■Concentration in amorphous material (number of particles in amorphous layers per volume
unit): AsAmorphous, BAmorphous…
■Concentration of a particular extended defect (number of defects, where one defect
contains more than one particle, per volume unit): I54, V23, I1026…
■Concentration of a particular AP (number of APs, where an AP contains more than one
particle): IV, I3V4…
■Concentration of a particular impurity cluster (number of impurity clusters, where an
impurity cluster contains more than one particle): B2I3, As4V…
■Active concentrations (active number of dopants per volume unit, net, p-type, n-type, and
total): NetActive, pNetActive, nNetActive, and tNetAtive.
■Germanium concentration: Ge.
Some fields compute the defect concentration (concentration of APs, impurity clusters, and
extended defects). You can transform them into particle concentrations multiplying by the size
of the defect.
For example, you can obtain the concentration of boron particles in B2, B3, BI2, and B2I2 in the
field BICs with the following set of commands:
kmc deatomize name=B2; kmc deatomize name=B3
kmc deatomize name=BI2; kmc deatomize name=B2I2
select z="2*B2 + 3*B3 + BI2 + 2*B2I2" name=BICs
Smoothing Out Deatomized Concentrations
The direct deatomization of Sentaurus Process KMC quantities into continuum mesh elements
produces values with strong gradients between neighboring elements. This is especially true
for small concentrations, where Sentaurus Process KMC contains a few particles that are
deatomized as an “all or nothing” distribution; that is, some cells may contain one particle, and
this is a concentration of , while others contain no particles, thereby having a zero
concentration.
1∆V()⁄
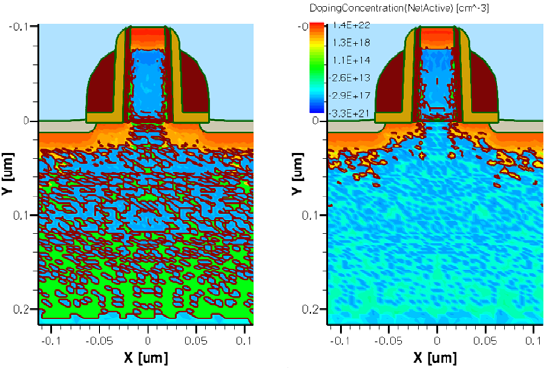
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
538 Sentaurus™ Process User Guide
I-2013.12
In some cases, especially when the Sentaurus Process KMC output is used as a device
simulation input, a smoother concentration is desirable, as seen in Figure 78. This can be
performed by setting the PDB parameter:
KMC Smooth.Field <field> <number>
where:
■<field> is the field name to be smoothed (for example, NetActive).
■<number> is an integer.
Figure 78 Comparison of NetActive concentration in simulations performed with (left)
Smooth.Field equal to 0 (disabled) and (right) equal to 1
A value of 0 produces no smoothing; larger numbers produce more smoothed profiles. The
smoothing algorithm works as follows:
■For each node in the standard Sentaurus Process mesh, the number of particles
associated with the node is counted.
■The concentration set to that node is, in principle, , where is the Voronoï
volume associated with the node.
■ is compared to , where is the number specified in Smooth.Field for this field.
■If the field name does not exist in Smooth.Field, 0 is assumed.
■If , the smoothing algorithm applies. Starting at the node, the algorithm looks for
particles not associated with the Voronoï volume of the node, with an increasing radius.
■When particles are found in a radius , it stops searching. At this point, there are
particles inside the radius, outside the Voronoï volume, and inside the
Vo r o n o ï v o l u m e .
N
NV
Voronoi
⁄
VVoronoi
N
M
M
NM<
MN–
R
M
MN–
N

5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 539
I-2013.12
■Then, the concentration of the node will be:
(815)
where is the volume associated with a segment (1D), circle (2D), or circumference (3D)
of radius , and weight is the PDB value Smooth.Weight:
pdbSet KMC Smooth.Weight 0.01
This technique is not intended to perfectly conserve the total dose, but to fill the nodes with low
concentrations with values depending on the distance to the nearest particles. The factor
weight is included to limit the extra dose introduced in those nodes.
The Smooth.Field parameter is used whenever a deatomization is performed. This includes
calling directly kmc deatomize from the command line or indirectly through KMC2PDE or
another procedure.
NOTE The algorithm to smooth atomistic concentrations can be extremely
slow in simulations with a large number of nodes.
The smooth algorithm can be relatively slow for simulations with a large number of nodes or
a large number of particles. This can be resolved by calling it in parallel mode with the
following option:
math numThreadsDeatomize=<n>
This option is independent from the KMC parallel mode and does not interfere with it. It
applies to the smooth algorithm only, not to the whole deatomization.
Adding and Obtaining Defects in Simulations: add,
defects.add, and defects.write
Sentaurus Process KMC allows you to add defects to the simulation using the commands:
kmc add queue name=<defect name> coordx=<x> coordy=<y> coordz=<z> [amorphous]
[crystalline]
kmc add
First, the defects are queued in the creation queue with the command:
kmc add queue
You can queue as many defects as you want. Queueing a defect does not actually introduce it
in the simulation. You must use the command kmc add alone to empty all of the queue by
generating defects in the simulation.
NV
Voronoi
⁄weight NM–()()VR
⁄+
VR
R
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
540 Sentaurus™ Process User Guide
I-2013.12
The option name specifies the defect to be created (examples are B, B2I4, I54, and
BInterface). The options coordx, coordy, and coordz provide the place where the defect
(or the center of mass of the defect) will be created. Finally, the optional arguments
amorphous and crystalline provide a way to tell the simulator whether this defect should
be created in an amorphous or a crystalline material. For example, if the current material is
crystalline, but the option amorphous is specified, the simulator not only creates a defect, but
also changes the material from crystalline to amorphous phase.
An alternative way to add defects to the simulation is using the defects.read command,
which requires the name of a text file to be specified with defects.read, for example:
kmc defects.read=my_filename
This command takes all the defects specified in the file and adds them consecutively in a very
similar way to the add command. Similar to the add command, only the center of mass is
specified for defects having more than one particle.
One line specifies one defect. The format of each line is:
defect_type defect_name coord_x coord_y coord_z
where:
■defect_type is the generic name of the defect, for example, PointDefect, Loop,
ThreeOneOne, Interface, ImpurityCluster.
■defect_name is the particular name of the defect, for example, B, P, I50, BiM, B3I2.
■coord_x, coord_y, coord_z are the coordinates (in nanometers) for the center of mass
of the defect.
For example:
Interface P 1.000000 11.250000 13.000000
PointDefect As 5.032000 5.320000 1.032000
ThreeOneOne I50 10.000000 10.222565 9.777436
PointDefect B 10.000000 5.000000 6.000000
PointDefect BiM 30.000002 5.000000 14.000000
ThreeOneOne I100 19.554871 20.445129 20.000000
Loop PI59 13.000000 13.000000 13.000000
PointDefect Bi 26.000000 26.000000 26.000000
ImpurityCluster B2I 6.235641 7.115777 7.077633
ImpurityCluster B3I2 4.287220 7.497602 4.604665
PointDefect Ge 6.696747 7.492962 0.834044
Similarly, the command kmc defects.write writes all the defects currently in the
simulation into a file with the above format. The name of the file must be specified as:
kmc defects.write=my_filename
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 541
I-2013.12
Files written with defects.write can be read later with defects.read. Since only the
center of mass of the particle is written, this is an inaccurate way to save a simulation. To save
a simulation, use instead struct or kmc extract tdrWrite.
Using the Sentaurus Process Interface
The select, print, WritePlx, and plot Commands
The commands select, print, WritePlx, and plot work as expected. If you need a field
for them, create it using kmc deatomize (see Transferring Fields from KMC to Continuum
Information: deatomize on page 536).
In particular, select creates particles inside Sentaurus Process KMC whenever the name of
the field is recognized as a particle or defect. These names are:
Example
For a typical situation with a 1D SIMS-like simulation (implant and anneal), the 1D profiles
can be extracted in a .plx file using WritePlx:
SetPlxList BTotal B
WritePlx file
Calling WritePlx without selecting the list with SetPlxList also works. It generates a list
of the most common fields:
WritePlx file
Dopants As, B, …
Impurities C,F, …
Paired particles Bi, AsV, Ci, …
Point defects I,V
Any of the above particles with a different charge IM, VPP, BiM, AsVP, …
Amorphous BAmorphous, AsAmorphous, …
Amorphous pockets IV, I4V5, …
Dopants or impurities in an interface BInterface, AsInterface, …
Extended defects I43, V21, …
Impurity clusters AsV4, BI2, …
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
542 Sentaurus™ Process User Guide
I-2013.12
In nonatomistic mode, the fields must be deatomized first:
kmc deatomize name=BTotal
kmc deatomize name=B
The init Command
The init command works as expected. Background dopings can be assigned. A TDR file to
be loaded can also be assigned with:
init tdr=filename
and, if the file has been saved with Sentaurus Process KMC and contains atomistic information
(see the option defects for tdrAdd, kmc on page 995), Sentaurus Process will load it, and
the simulation can be continued.
NOTE Loading a file and continuing a simulation with init will give results
similar, but not identical, to performing the simulation without the save/
load process. The differences between the results are only statistical; in
other words, both represent possible solutions.
The struct Command
The struct command works as expected, except that by default it creates some extra fields to
be saved. It generates these extra fields by calling the function preKMC with the argument
struct. You can modify or customize this function in the KMC.tcl file. By default, preKMC
tries to generate and store fields similar to a five-stream model from the KMC information.
The struct command also saves restart information, allowing the Sentaurus Workbench
#split command to work properly with Sentaurus Process KMC.
The load Command
The load command accepts the options kmc and replace only. It is intended to load a TDR
Sentaurus Process KMC simulation to replace the existing one. It performs the necessary
conversions between the existing internal Sentaurus Process KMC and the one read from the
TDR file, conserving the existing one.
The deposit Command
The deposit command works as expected, including depositing doped layers.

5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 543
I-2013.12
The diffuse Command
The diffuse command works as expected with Sentaurus Process KMC, except that the
diffusion is done with the atomistic solver. In particular, the options for oxidation and
silicidation are supported (see Oxidation on page 518). Epitaxial options also are supported.
For lattice LKMC epitaxy, see Epitaxial Deposition on page 519.
Nonatomistic Mode
When Sentaurus Process KMC operates in the nonatomistic mode (see Nonatomistic Mode on
page 386), it transforms the five-stream model fields into atomistic information before every
annealing and converts the atomistic information to five-stream model information after any
annealing. Sentaurus Process KMC is disabled between annealings; consequently, all
information should be accessed using the standard Sentaurus Process interface. The only way
to access atomistic information is by using the Movie option during the Sentaurus
Process KMC annealings.
The transformation from five-stream to atomistic information is performed in the function
preKMC with the argument diffuse in the KMC.tcl file. The function preKMC is called
automatically before a Sentaurus Process KMC diffusion in nonatomistic mode. After the
diffusion, the function postKMC, with argument diffuse, is invoked to convert the atomistic
information into five-stream fields. You can modify and customize both functions.
Atomistic Mode
When operating in atomistic mode, Sentaurus Process KMC does not automatically perform
any transformation from atomistic to continuum, or vice versa, except if the command struct
is called (see Atomistic Mode on page 383).
Calling Directly the Sentaurus Process KMC Kernel
The best way to access the atomistic information is by calling directly the Sentaurus
Process KMC kernel using the kmc command. The option extract of this command accesses
the KMC raw information directly and returns it in different formats, or creates a TDR file to
be opened with Sentaurus Workbench Visualization.
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
544 Sentaurus™ Process User Guide
I-2013.12
Writing and Displaying TDR Files with KMC Information
The command:
kmc extract tdrWrite filename=<filename>
creates files to be displayed by Sentaurus Workbench Visualization. This file contains a
collection of states or snapshots. Each snapshots is taken at a particular time during the
simulation; the collection of snapshots gives information about the time evolution during the
simulation. Each snapshot can contain concentrations and histograms, atomistic information,
or both. These snapshots are created with the command:
kmc extract tdrAdd [concentrations] [defects] [stress] [histogram]
[visual={<fields>} [list={<fields>}]
The parameter concentrations includes a list of standard fields. These fields are the total
concentration for each particle, the time-averaged concentrations of mobile particles,
NetActive, the electronic concentration, the concentration of impurities in the surface,
amorphous and impurity clusters, and the concentration of point defects in APs, amorphous
layers, impurity clusters, and extended defects. These concentrations are displayed with the
same dimension as the simulation.
Since Sentaurus Process KMC always works in 3D, for 1D simulations, the displayed
concentrations are averaged for yz planes and, for 2D simulations, they are averaged in z-lines.
An extra A/C field also is stored to let you know whether the material is amorphous or
crystalline.
The histogram option includes a 2D graph representing the APs and impurity cluster
histograms. These histograms give the number of defects existing in the simulation for each
different size (I and V for APs, impurities, and point defects for impurity clusters). One-
dimensional histograms, giving the number of extended defects versus its size, are included as
well when using the histogram option. Stress and strain information can be added to these
concentrations and histograms with the stress option.
Figure 79 on page 545 shows one snapshot saved with the concentrations and histogram
options for a 2D simulation.
Regarding NetActive, the parameter KMC tdr smoothDopingConcentration controls
whether this concentration is smoothed. By default, it is not smoothed because smoothing it
artificially decreases the channel length. On the other hand, the smooth algorithm partially
removes irregularities and other atomistic features that may appear on the p-n junctions. This
smoothing is different than the one controlled by Smooth.Field and is explained in
Smoothing Out Deatomized Concentrations on page 537.
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 545
I-2013.12
While smoothDopingConcentration controls the smoothing of only NetActive in the
Sentaurus Process KMC TDR file, Smooth.Field controls the smoothing of any field when
translating the atomistic information into the continuum mesh concentration.
The parameter defects includes atomistic information about the defects in the simulation.
This atomistic information can be used to obtain an atomistic 3D plot that is independent of the
simulation dimensions. It offers the most realistic representation of the simulation.
Figure 79 Example of a TDR file of Sentaurus Process KMC displayed with Sentaurus
Workbench Visualization. The concentrations and histogram options have been
used; 1D histograms for clusters and 2D histograms for APs and impurity clusters
are included. The concentration (in this case, for a 2D simulation) for several
fields is included with this option.
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
546 Sentaurus™ Process User Guide
I-2013.12
Figure 80 shows an example of the information saved with the defects option.
Figure 80 Example of TDR file of Sentaurus Process KMC including atomistic information;
the defects option has been used
The defects option also includes extra information necessary to restart or load the simulation.
A file saved with this parameter can be reloaded into Sentaurus Process KMC, and the
simulation can be continued. To load the simulation, use the command init.
Using the KMC Movie option, you can include the command kmc extract tdrAdd to view
the evolution of the simulation with time.
Finally, the option visual stores atomistic 3D information in a way similar to defects. The
differences between using visual and defects are:
■Files saved with visual cannot be restarted.
■The visual option produces smaller file sizes than defects. In particular, visual stores
the defects, but it does not store the information needed to restart. The visual files are
intended for visualization purposes only.
■The visual option requires a list of defects, separated by commas, to be saved. The all
option saves all the defects.

5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 547
I-2013.12
■Other valid defects include defect names (such as BiM or B2I3), general defect names
(such as ThreeOneOne), and material names (Silicon). This lets you control which
defects will be saved and visualized later, making it easier to visualize information without
saving a large file with all the 3D information.
Plotting Only Some Particles
Sentaurus Process visualization does not represent all the particles inside APs. Since the
number of particles (interstitials and vacancies) in APs can be large after an implant, only 1 in
50 particles is visualized by default. This default behavior is written in the TDR file as set by
the parameter VisualizeDamage:
sprocess> pdbGet KMC VisualizeDamage
50
Sentaurus Process visualization can overwrite the default set for each file using the option:
-s:psf n
where n is the new value of VisualizeDamage:
NOTE Setting VisualizeDamage to 1 causes Sentaurus Workbench
Visualization to plot all the particles present in the simulation. This
number can be very large immediately after an implant.
Time-averaged Concentration Name
The default field name for time-averaged concentrations in the TDR file is ‘mobile’, but it can
be defined with the parameter KMC tdr averageTag.
Inquiring about KMC Profiles, Histograms, and Defects
Besides the select command and the TDR files, you can access Sentaurus Process KMC
information using the kmc extract command (see kmc on page 995):
■kmc extract histogram—extracts histograms for extended defects, impurity defects,
and APs.
■kmc extract profile—extracts concentrations and stresses in 1D, 2D, and 3D.
■kmc extract supersaturation—extracts the concentration relative to the
equilibrium concentration for point defects.
■kmc extract defects—obtains the atomistic information of the defects.
■kmc extract dose—extracts doses, that is, concentrations in .
cm 2–
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
548 Sentaurus™ Process User Guide
I-2013.12
■kmc extract materials—obtains information about the different materials in the
simulation.
■kmc extract acinterface—obtains the position of the amorphous–crystalline
interfaces in the simulation.
The histogram Option
kmc extract histogram name=[meansize [minsize=<n>]]
[materialname=<material>]
Sentaurus Process KMC includes several models where the defects are not isolated, but
agglomerated in extended defects that can contain many particles. The histogram option
allows you to extract information about the sizes (number of particles) of these extended
defects.
The histogram option needs a valid name to compute the following available histograms:
The optional parameter meansize displays the average size of clusters instead of displaying
the whole list of clusters when using this parameter. Without specifying minsize, the average
size begins with size 0.
Finally, the parameter materialname restricts the output to the material specified instead of
the whole simulation.
Interstitial-extended
defects
Set name=I to extract information about the small I clusters,
{311} defects, and dislocation loops.
Va ca nc y- ex te nd e d de fe ct s Se t name=V to extract information about the small V clusters
and voids.
B interstitial clusters or
other clusters with I
Set name=XI to extract information about the dopant named
‘X’. For example, for boron impurity clusters (BICs),
name=BI.
As vacancy clusters or
other clusters with V
Set name=XV. For example, set name=AsV to extract
information about the arsenic–vacancy clusters.
Cluster with multiple
impurities
For example, set name=PAs for clusters with both P and As.
Amorphous pockets Set name=IV.
IV
BI
I
…
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 549
I-2013.12
Interstitial Histograms
These histograms extract the number of defects in the simulation for each size. The histograms
contain information about the small, irregular clusters (see Amorphous Pockets Life Cycle on
page 439), {311} defects (for size bigger than the established limit), and dislocation loops. For
example:
LogFile [kmc extract histogram name=I]
gives a list of extended defects with I. In this example:
sprocess> LogFile [kmc extract histogram name=I]
I2 302
I3 104
I4 42
I5 12
I6 4
I72 1
I677 1
sprocess> LogFile [kmc extract histogram name=I meansize]
4.11373
sprocess> LogFile [kmc extract histogram name=I meansize minsize=10]
374.5
Vacancy-extended Defects Histogram
These histograms are similar to the interstitial-extended defects histogram, except that the
extracted number of particles versus size is for vacancies:
sprocess> LogFile [kmc extract histogram name=V]
V5 1
V7 2
V8 5
V9 2
V10 5
(...)
sprocess> LogFile [kmc extract histogram name=V meansize]
12.9143
sprocess> LogFile [kmc extract histogram name=V meansize minsize=10]
15
Amorphous Pockets Histogram
The AP histograms contain the number of cluster versus I and V size. APs with null Is or Vs
can be considered as APs or small I or V clusters.
sprocess> LogFile [kmc extract histogram name=IV]
I2 367
I3 69
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
550 Sentaurus™ Process User Guide
I-2013.12
I4 22
I5 1
IV 823
I2V 249
I3V 61
I4V 23
I5V 2
V2 408
IV2 251
I2V2 111
(...)
The average size can be requested for these clusters. It will return values for both I and V:
sprocess> LogFile [kmc extract histogram name=IV meansize]
I2.3501V0.796781
The parameter meansize applies here and specifies the minimum size to begin the average for
both species.
Boron–Interstitial Clusters
The boron–interstitial cluster histogram offers information about the number of BICs for each
BIC configuration (BnIm).
sprocess> LogFile [kmc extract histogram name=BI]
B2I 16
B3I 347
sprocess> LogFile [kmc extract histogram name=BI meansize]
B2.95592I1
Arsenic–Vacancy Clusters
The arsenic–vacancy cluster histogram offers information about the number of arsenic and
vacancies in impurity clusters for each configuration (AsnIm):
sprocess> LogFile [kmc extract histogram name=AsV]
As2 277
As3 109
As4 3
As2V 752
As3V 281
As4V 178
sprocess> LogFile [kmc extract histogram name=AsV meansize]
As2.47V0.756875

5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 551
I-2013.12
The profile Option
Sentaurus Process KMC computes the profiles concentration versus size as a convenient way
to directly obtain useful data.
kmc extract profile [timeaveraged] name=
[materialname=] [coordx=] [coordy=] [coordz=]
For example, 1D profiles can be compared with SIMS experiments. The profiles are an average
of the concentration of particles. For 3D, Sentaurus Process KMC takes a volume to be
averaged equal to an element defined by the parameters extractDeltaX, extractDeltaY,
and extractDeltaZ. For 2D and 1D, this volume includes all the elements in y and z,
respectively.
The parameter name chooses the profiles to be obtained. holes and electrons return the
concentration of holes and electrons, respectively. GapNarrowing returns the bandgap
narrowing, in eV. Stress can be stressXX, stressYY, and stressZZ, and strain is one of
strainXX, strainXY, strainXZ, strainXY, strainXZ, or strainYZ. A particle name
(like Bi, IM, or AsVP) returns the concentration of all the particles in the simulation matching
the given one. A cluster name (for example, As4V) will return the concentration of that cluster
in the simulation. For particles, the concentration of particles is returned; for clusters, the
concentration of clusters is returned. For example, an As4 cluster is considered to be four As
particles when you request the concentration of As, but only one defect when you request the
concentration of As4 clusters. An impurity name followed by the word “Total”, like BTotal,
will return the total profile of that impurity (active, inactive, in pairs, clusters and so on) in the
simulation.
When a particle profile is specified, the optional parameter defectname can be used to further
specify the kind of particle. For example, the command:
kmc extract profile name=I
computes the concentration of interstitials in any kind of defect, in other words, the total
interstitial concentration.
holes
electrons
particles BAs
i…,,()
clusters InVmBnIm…,,()
stress
strain
GapNarrowing
dopants
totals BTotal,AsTotal()
defectname=
ThreeOneOne
Interface
Loop
…
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
552 Sentaurus™ Process User Guide
I-2013.12
The command:
kmc extract profile name=I defectname=ImpurityCluster
computes the concentration of interstitials in impurity clusters.
The parameter defectname should not be specified with cluster concentration, electrons,
holes, GapNarrowing, stresses or strains.
Finally, the parameter materialname restricts the output to the material specified instead of
the whole simulation.
The returned concentration has the same dimensionality as the performed simulation. Use the
optional parameters coordx, coordy, and coordz to change this default. These parameters
specify cutlines. For example, in a 3D simulation the command:
kmc extract profiles name=I
returns the concentration for all the volume elements in the simulation. The command:
kmc extract profiles name=I coordx=20<nm> coordz=10<nm>
returns a 1D profile with concentrations in the plane x = 20 <nm> and z = 10 <nm>.
Finally:
kmc extract profiles name=I coordx=20<nm> coordy=15<nm> coordz=10<nm>
returns only one value, the concentration at the specified point.
The use of the parameters coordx, coordy, and coordz depends on the simulation
dimensions. As previously explained, specifying coordx for 1D, 2D, or 3D simulations,
coordy for 2D or 3D, or coordz for 3D, returns the concentrations only on elements
including the specified cutlines. On the other hand, the use of coordy or coordz in 1D
simulations, or coordz in 2D, is quite different. In this case, the result returned is not averaged
for the whole remaining dimensions (y and z for 1D, z for 2D), but only calculated in the
specified cutlines. In other words, specifying coordy in a 3D simulation returns all the
concentrations in the x, z volumes for the y specified in coordy; while specifying coordy in
a 1D simulation returns concentrations versus the x-axis, but instead of being averaged for
every y and z, they will be averaged only for every z in the plane marked by coordy. In 3D, it
will reduce the size of the output (since only the output for the specified plane y is written). In
1D, the output has the same number of lines (one for each x position), but the concentration
displayed is different because it is averaged into z only, and not into y and z.
Sentaurus Process KMC returns the instantaneous concentration by default. For mobile
particles the instantaneous concentration does not usually contain any information rather than
noise. The parameter timeaveraged instructs Sentaurus Process KMC to return the average
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 553
I-2013.12
concentration of mobile particles between the current time and the last time Sentaurus
Process KMC created a snapshot (see Snapshots on page 407).
For example, Figure 81 has been produced with the input script:
pdbSet KMC MaxYum 40e-3
pdbSet KMC MaxZum 40e-3
SetAtomistic
pdbSet KMC Movie ""
pdbSet IncrementalHops 0
line x loc=0.0 tag=xleft spacing = 0.002
line x loc=1.5e-3 tag=xmed spacing = 0.002
line x loc=5e-3 tag=xright spacing = 0.002
region oxide xlo=xleft xhi=xmed
region silicon xlo=xmed xhi=xright
init
diffuse time=1e7<s> temp=700 info=1
kmc extract tdrClear
kmc extract tdrAdd concentrations
kmc extract tdrWrite filename=equil
The concentrations of neutral, positive, and negative interstitials also can be obtained with:
sprocess> kmc extract profile timeaveraged name=IP coordx=2.5<nm>
162487
sprocess> kmc extract profile timeaveraged name=I coordx=2.5<nm>
1.0276e+06
sprocess> kmc extract profile timeaveraged name=IM coordx=2.5<nm>
18170.5
Figure 81 Equilibrium concentrations of neutral (green), positive (red), and negative (blue)
interstitials at 700oC
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
554 Sentaurus™ Process User Guide
I-2013.12
The supersaturation Option
kmc extract supersaturation name=
Supersaturation is the current concentration relative to the equilibrium concentration:
(816)
Sentaurus Process KMC computes the supersaturation for Is and Vs. The current global
concentration is calculated involving a time average between the current time and the last
snapshot:
sprocess> LogFile [kmc extract supersaturation name=I]
1.00555
To obtain the supersaturation evolution with time, use the KMC Movie parameter as shown in
the following example:
SetAtomistic
set kmcSupersat ""
pdbSet KMC MaxYum 30e-3
pdbSet KMC MaxZum 30e-3
pdbSet KMC Movie {lappend kmcSupersat $time [kmc extract \
supersaturation name=I]}
pdbSet KMC automaticRampUp 1
line x loc=0.0 tag=xleft spacing = 0.002
line x loc=1.5e-3 tag=xmed spacing = 0.002
line x loc=350e-3 tag=xright spacing = 0.002
region oxide xlo=xleft xhi=xmed
region silicon xlo=xmed xhi=xright
init
implant Silicon energy=40 dose=2e13 tilt=7
diffuse time=100000<s> temp=600
LogFile $kmcSupersat
produces the results:
10.0007 4.29101e+07
21.5462 2.51728e+07
46.4218 1.58636e+07
100.004 1.41889e+07
215.493 7.67109e+06
464.238 3.44625e+06
1000.26 1.86513e+06
2156.46 477255
4645.69 322049
I
V
supersaturation X() X[]
X[]
*
-----------=
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 555
I-2013.12
10002.8 113849
21551.6 132116
46419.1 110329
100006 81506.4
Figure 82 shows a comparison with experimental results.
Figure 82 Supersaturation evolution with time, after a silicon implant (2 x 1013 dose, 40 keV
energy) annealed 1 x 105s at 600oC. (Experimental points modified from [50] to
use the same B diffusivity.) Points are experiments, lines are KMC.
The defects Option
The defects option allows access to the raw atomistic information of any simulation. The
obtained information must match the parameters name and defectname when they are
specified.
kmc extract defects
[countparticles] [countdefects] [materialname=<material>]
[acinterface]
When there are no restrictions, all defects are accessed. For example, for the following added
defects:
kmc add queue name=BI2 coordx=2<nm> coordy=2<nm> coordz=3<nm>
kmc add queue name=I2 coordx=3<nm> coordy=2e-3 coordz=4e-3
kmc add queue name=I3 coordx=4<nm> coordy=3<nm> coordz=1<nm>
kmc add
108
107
106
105
104
103
102
105
104
103
102
101
100
Time [s]
Interstitial Supersaturation
600oC
name particles BA
i…,,()
clusters InVmBnIm…,,()
=
defectname=
ThreeOneOne
Interface
Loop
…
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
556 Sentaurus™ Process User Guide
I-2013.12
The obtained defects are:
sprocess> kmc extract defects
0.00201273 0.00238963 0.00298179 I ImpurityCluster 0
0.00196125 0.00204962 0.00249501 I ImpurityCluster 0
0.00177392 0.00225923 0.00247647 B ImpurityCluster 0
0.00320253 0.00162722 0.00466375 I AmorphousPocket 1
0.00222551 0.00139856 0.004411 I AmorphousPocket 1
0.0044946 0.00317389 0.00130069 I AmorphousPocket 2
0.00441116 0.00353071 0.000842549 I AmorphousPocket 2
0.00403856 0.00351353 0.00084327 I AmorphousPocket 2
sprocess> kmc extract defects name=I
0.00201273 0.00238963 0.00298179 I ImpurityCluster 0
0.00196125 0.00204962 0.00249501 I ImpurityCluster 0
0.00320253 0.00162722 0.00466375 I AmorphousPocket 1
0.00222551 0.00139856 0.004411 I AmorphousPocket 1
0.0044946 0.00317389 0.00130069 I AmorphousPocket 2
0.00441116 0.00353071 0.000842549 I AmorphousPocket 2
0.00403856 0.00351353 0.00084327 I AmorphousPocket 2
sprocess> kmc extract defects name=I defectname=ImpurityCluster
0.00201273 0.00238963 0.00298179 I ImpurityCluster 0
0.00196125 0.00204962 0.00249501 I ImpurityCluster 0
sprocess> kmc extract defects name=BI2
0.00201273 0.00238963 0.00298179 I ImpurityCluster 0
0.00196125 0.00204962 0.00249501 I ImpurityCluster 0
0.00177392 0.00225923 0.00247647 B ImpurityCluster 0
The six columns present in the output are:
■X-coordinate of the defect
■Y-coordinate of the defect
■Z-coordinate of the defect
■Particle name
■Defect name.
■Number of defect. Particles with the same number belong to the same defect.
NOTE The command kmc extract defects can produce large outputs.
When there are amorphous defects in the simulation, the result may not be the expected.
Amorphous defects do not store the damage, but only its concentration (see Amorphous
Defects on page 456). Consequently, amorphous defects will not report any interstitial or
vacancy inside them. Impurities are stored and displayed. Nevertheless, if the indirect diffusion
model is used in amorphous silicon, the dangling bonds, floating bonds, and mobile and
immobile impurities will be obtained as I, V, Bi and B (for boron) (see Indirect Diffusion on
page 457).
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 557
I-2013.12
The parameter materialname restricts the output to the material specified instead of the
whole simulation.
The options countparticles and countdefects do not display the atomistic information,
but they count the number of particles and defects, respectively, for the given conditions. For
example, this example shows how to count the particles and defects listed in the last example:
sprocess> kmc extract defects countparticles
8
sprocess> kmc extract defects countdefects
3
sprocess> kmc extract defects name=I countparticles
7
sprocess> kmc extract defects name=I defectname=AmorphousPocket countdefects
2
The parameter acinterface displays the lattice atoms belonging to the amorphous–
crystalline interface. All the lattice atoms can be obtained by using acinterface detailed.
This parameter generates an output only when the LKMC model is used for recrystallization
(see LKMC: Fully Atomistic Modeling of Solid Phase Epitaxial Regrowth on page 463).
The dose Option
The dose option extracts the concentration per surface unit (in other words, cm–2) for the
whole simulation cell. dose can be used to look at the evolution of the species with the time.
kmc extract dose
[countdefects] [materialname=<material>]
The parameters name and defectname restrict the species to compute the dose. name can be
any particle or defect. defectname can be specified only when name is a particle, and it
restricts the particles to be of the specified type. A list of defect types is obtained with kmc
defecttypes. A “total” name: the name of an impurity followed by the work “Total”, like
BTotal, applies to all the circumstances where the specified impurity is present, that is, in
clusters, substitutional, pairs, and so on.
This command counts the number of particles (such as kmc extract defects with the
same restrictions and the countparticles option) and it divides this number by the surface
area. Finally, the parameter materialname restricts the output to the material specified
name
Particles I AsV Bi
-…,,,()
Defects InVnInVmBnIm…,, , ,()
Totals AsTotal BTotal ...,,()
=
defectname
ImpurityCluster
ThreeOneOne
…
=
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
558 Sentaurus™ Process User Guide
I-2013.12
instead of the whole simulation. For interfaces, instead of specifying a material name, specify
a particle name such as AsInterface, BInterface.
As an example, the following input file implants silicon into silicon and extracts the dissolution
of {311} during annealing:
set silicon_depth 350e-3
set size 50e-3
set SiO2gate 1.5e-3
SetAtomistic
set sol ""
pdbSet KMC Movie {lappend sol $time [kmc extract dose name=I \
defectname=ThreeOneOne]}
pdbSet KMC MaxZum $size
pdbSet KMC MaxYum $size
pdbSet KMC GasUm $SiO2gate
pdbSet KMC automaticRampUp true
line x loc=0.0 tag=xtop spacing = 0.002
line x loc=$silicon_depth tag=xbottom spacing = 0.002
region silicon xlo=xtop xhi=xbottom
init
deposit oxide fill coord=[expr -$SiO2gate]
implant silicon energy=40 dose=5e13 tilt=7
diffuse time=3100 temp=670
LogFile $sol
This example produces the results:
46.4176 2.88e+12
100.01 9.56e+12
215.464 1.104e+13
464.175 1.596e+13
1000 2.172e+13
2154.45 3.144e+13
4641.63 3.736e+13
10000 3.044e+13
21544.5 1.388e+13
46417.4 9.6e+11
100233 0
5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 559
I-2013.12
Figure 83 shows the comparison with experimental data [8].
Figure 83 Dissolution of {311} extended defects at 670oC after a silicon into silicon implant
of 40 keV, 5 x 1013 cm–2
The materials Option
kmc extract materials [detailed] [coordx=] [coordy=] [coordz=]
The command kmc extract materials produces the list of materials currently in the
simulation:
sprocess> kmc extract materials
Silicon Oxide Gas
The option detailed produces a list of coordinates and materials. This list contains the same
number of dimensions as the simulation (except if you use coordx, coordy, or coordz). For
example, in a 1D simulation:
sprocess> kmc extract materials detailed
-0.002 Gas
-0.000625 Oxide
0.00075 Oxide
0.002125 Oxide
0.0035 Oxide
0.004875 Oxide
0.00625 Oxide
0.007625 Oxide
0.009 Oxide
0.010375 Silicon
0.01175 Silicon
0.013125 Silicon
0.0145 Silicon
Simulation
Experiment
1000 10000 100000
1014
1013
1012
1011
Interstitials in Extended Defects [cm–2]
Time [s]

5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
560 Sentaurus™ Process User Guide
I-2013.12
0.015875 Silicon
0.01725 Silicon
The parameters coordx, coordy, and coordz affect the output of detailed by changing its
dimensionality. They work exactly the same as in kmc extract profile (see The profile
Option on page 551).
The acinterface Option
kmc extract acinterface [coordx=] [coordy=] [coordz=]
The command kmc extract acinterface produces the list of amorphous–crystalline
interfaces found in the simulation given a 1D cutline. No extra options are needed in 1D. In 2D,
you must specify coordx or coordy. In 3D, you must specify two of three parameters
coordx, coordy, and coordz. For example, in 1D:
sprocess> kmc extract acinterface
Silicon Amorphous/Crystalline 0.0165
When the KMC model for SPER is detected, the option acinterface displays all the places
where the amorphous–crystalline field crosses the threshold specified in
KMC ACInterfaceAt. A value of 1 means perfectly amorphous and 0 means perfectly
crystalline. The default value is 0.9. The output also displays the materials at both sides of the
interface, displaying first the material with a smaller coordinate value. In this case, the interface
is at , and the transition is from Amorphous to Crystalline as the x-coordinate
increases. When the LKMC model is used for SPER, the parameter ACInterfaceAt is
ignored, and a more precise interface is extracted directly from the LKMC atomistic
information.
Common Dopant and Point-Defect Names
Several KMC commands contain a name=<particlename> parameter. Table 61 on page 561
lists several of these names and the commands where they are applicable. Since dopants can be
defined as needed, the names depend on the simulation parameters. In particular, they contain
names defined when using Advanced Calibration. A list of the names can be obtained using
kmc particletypes. Clusters also are included in the list. Since the number of different
clusters is large, only a partial cluster list is included as an example (see Table 62 on page 564).
The commands considered for the referenced tables, and the symbols used to represent them,
are:
■kmc add queue (Add)
■kmc extract profile (Pro)
■kmc extract histograms (His)
0.0165 µm

5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 561
I-2013.12
■kmc extract dose (Dos)
■kmc extract defects (Def)
■kmc extract supersaturation (Sup)
■kmc deatomize (Dea)
■kmc present (Pre)
The defects used in the descriptions are:
■Point defects, self-silicon point defects like interstitials and vacancies.
■Extended defects, agglomeration of self-silicon point defects.
■Clusters, agglomeration of impurities or dopants, with or without point defects.
■Substitutional dopants or impurities.
■Paired dopants or impurities, paired with point defects.
■Amorphous pockets, agglomeration of point defects (also sometimes called damage).
■Defects attached to interfaces.
Ta bl e 61 N a me s us ed i n S e n ta u ru s Pr oc es s K M C
Name Description Used in
I
Neutral interstitial in point defects, clusters, and
extended defects
Sup, His, Add, Pro, Dos, Def, Dea, Pre
IMM
I--, point defect Add, Pro, Dos, Def, Dea, Pre
IM
I-, point defect Add, Pro, Dos, Def, Dea, Pre
IP
I+, point defect Add, Pro, Dos, Def, Dea, Pre
IPP
I++, point defect Add, Pro, Dos, Def, Dea, Pre
ITotal
Total interstitial concentration; similar to I + IMM +
IM + IP + IPP
Dea, Pre, Pro, Dos
V
Neutral vacancy in point defects, clusters, and
extended defects
Sup, His, Add, Pro, Dos, Def, Dea, Pre
VMMM
V---, point defect Add, Pro, Dos, Def, Dea, Pre
VMM
V--, point defect Add, Pro, Dos, Def, Dea, Pre
VM
V-, point defect Add, Pro, Dos, Def, Dea, Pre
VP
V+, point defect Add, Pro, Dos, Def, Dea, Pre
VPP
V++, point defect Add, Pro, Dos, Def, Dea, Pre
VPPP
V+++, point defect Add, Pro, Dos, Def, Dea, Pre
VTotal
Total vacancy concentration; similar to V +
VMMM + VMM + VM + VP + VPP + VPPP
Dea, Pre, Pro, Dos

5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
562 Sentaurus™ Process User Guide
I-2013.12
B
Boron, substitutional, at interfaces or in clusters Add, Pro, Dos, Def, Dea, Pre
BV
Neutral-paired defect, boron vacancy Add, Pro, Dos, Def, Dea, Pre, His
BVM
BV-, paired defect Add, Pro, Dos, Def, Dea, Pre
BVP
BV+, paired defect Add, Pro, Dos, Def, Dea, Pre
Bi
Neutral-paired defect, boron interstitial Add, Pro, Dos, Def, Dea, Pre, His
BiM
Bi-, paired defect Add, Pro, Dos, Def, Dea, Pre
BiP
Bi+, paired defect Add, Pro, Dos, Def, Dea, Pre
BTotal
Total boron concentration; similar to B + BV +
BVM + BVP + Bi + BiM + BiP
Dea, Pre, Pro, Dos
BInterface
Boron at interfaces Add, Pro, Dos, Def, Dea, Pre
As
Arsenic, substitutional at interfaces or in clusters Add, Pro, Dos, Def, Dea, Pre
AsV
Neutral-paired defect of arsenic and a vacancy Add, Pro, Dos, Def, Dea, Pre, His
AsVM
AsV-, paired defect Add, Pro, Dos, Def, Dea, Pre
AsVP
AsV+, paired defect Add, Pro, Dos, Def, Dea, Pre
Asi
Neutral-paired defect of arsenic and an interstitial Add, Pro, Dos, Def, Dea, Pre, His
AsiM
Asi-, paired defect Add, Pro, Dos, Def, Dea, Pre
AsiP
Asi+, paired defect Add, Pro, Dos, Def, Dea, Pre
AsTotal
Total arsenic; similar to As + AsV + AsVM + AsVP
+ Asi + AsiM + AsiP
Dea, Pre, Pro, Dos
AsInterface
Arsenic attached at interfaces Add, Pro, Dos, Def, Dea, Pre
C
Carbon, substitutional, at interfaces or in clusters Add, Pro, Dos, Def, Dea, Pre
CV
Neutral-paired defect of carbon and a vacancy Add, Pro, Dos, Def, Dea, Pre, His
Ci
Neutral-paired defect of carbon and an interstitial Add, Pro, Dos, Def, Dea, Pre, His
CInterface
Carbon attached at interfaces Add, Pro, Dos, Def, Dea, Pre
CTotal
Total carbon; similar to C + CV + Ci Dea, Pre, Pro, Dos
F
Fluorine, substitutional at interfaces or in clusters Add, Pro, Dos, Def, Dea, Pre
FI
Neutral-paired defect of fluorine with an interstitial Add, Pro, Dos, Def, Dea, Pre, His
FV
Neutral-paired defect of fluorine with a vacancy Add, Pro, Dos, Def, Dea, Pre, His
FTotal
Total fluorine; similar to F + FI + FV Dea, Pre, Pro, Dos
Ta bl e 61 N a me s us ed i n S e n ta u ru s Pr oc es s K M C
Name Description Used in

5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
Sentaurus™ Process User Guide 563
I-2013.12
FInterface
Fluorine attached at interfaces Add, Pro, Dos, Def, Dea, Pre
In
Indium, substitutional, at interfaces or in clusters Add, Pro, Dos, Def, Dea, Pre
InV
Neutral-paired defect of indium and a vacancy Add, Pro, Dos, Def, Dea, Pre, His
InVM
InV-, paired defect Add, Pro, Dos, Def, Dea, Pre
InVP
InV+, paired defect Add, Pro, Dos, Def, Dea, Pre
Ini
Neutral-paired defect of indium and an interstitial Add, Pro, Dos, Def, Dea, Pre, His
IniM
Ini-, paired defect Add, Pro, Dos, Def, Dea, Pre
IniP
Ini+, paired defect Add, Pro, Dos, Def, Dea, Pre
InTotal
Total indium; similar to In +InV + InVM + InVP +
Ini + IniM + IniP
Dea, Pre, Pro, Dos
InInterface
Indium attached at interfaces Add, Pro, Dos, Def, Dea, Pre
P
Phosphorus, substitutional, at interfaces or in
clusters
Add, Pro, Dos, Def, Dea, Pre
PV
Neutral-paired defect of phosphorus and a vacancy Add, Pro, Dos, Def, Dea, Pre, His
PVM
PV-, paired defect Add, Pro, Dos, Def, Dea, Pre
PVP
PV+, paired defect Add, Pro, Dos, Def, Dea, Pre
Pi
Neutral-paired defect of phosphorus and an
interstitial
Add, Pro, Dos, Def, Dea, Pre, His
PiM
Pi-, paired defect Add, Pro, Dos, Def, Dea, Pre
PiP
Pi+, paired defect Add, Pro, Dos, Def, Dea, Pre
PTotal
Total phosphorus; similar to P + PV + PVM + PVP
+ Pi + PiM + PiP
Dea, Pre, Pro, Dos
PInterface
Phosphorus attached at interfaces Add, Pro, Dos, Def, Dea, Pre
Sb
Antimony, substitutional, at interfaces or in clusters Add, Pro, Dos, Def, Dea, Pre
SbV
Neutral-paired defect of antimony and a vacancy Add, Pro, Dos, Def, Dea, Pre, His
SbVM
SbV-, paired defect Add, Pro, Dos, Def, Dea, Pre
SbVP
SbV+, paired defect Add, Pro, Dos, Def, Dea, Pre
Sbi
Neutral-paired defect of antimony and an interstitial Add, Pro, Dos, Def, Dea, Pre, His
SbiP
Sbi+, paired defect Add, Pro, Dos, Def, Dea, Pre
SbTotal
Total antimony; similar to Sb + SbV + SbVM +
SbVP + Sbi + SbiP
Dea, Pre, Pro, Dos
Ta bl e 61 N a me s us ed i n S e n ta u ru s Pr oc es s K M C
Name Description Used in

5: Atomistic Kinetic Monte Carlo Diffusion
Extracting KMC-related Information
564 Sentaurus™ Process User Guide
I-2013.12
SbInterface
Antimony at interfaces Add, Pro, Dos, Def, Dea, Pre
N
Nitrogen, substitutional, at interfaces or in clusters Add, Pro, Dos, Def, Dea, Pre
NTotal
Total nitrogen, same as N Dea, Pre, Pro, Dos
NInterface
Nitrogen at interfaces Add, Pro, Dos, Def, Dea, Pre
Nn
N2, substitutional, at interfaces or in clusters Add, Pro, Dos, Def, Dea, Pre
NnV
Moving N2V particle Add, Pro, Dos, Def, Dea, Pre, His
NnTotal
Similar to Nn + NnV Dea, Pre, Pro, Dos
NnInterface
N2 stored at the interface Add, Pro, Dos, Def, Dea, Pre
H
Hydrogen, substitutional, at interfaces or in clusters Add, Pro, Dos, Def, Dea, Pre
HTotal
Total hydrogen, only H Dea, Pre, Pro, Dos
HInterface
Hydrogen at interfaces Add, Pro, Dos, Def, Dea, Pre
Ge
Germanium (stored as a field, not as a particle) Add, Pro, Dos, Def, Dea, Pre
Table 62 Some cluster names used in Sentaurus Process KMC
Name Comment Used in
IV
Interstitial–vacancy amorphous pocket. His, Add, Pro, Dos, Def, Dea, Pre
I3V2
Amorphous pocket. Any other IxVy with x and y
integers is also valid.
Add, Pro, Dos, Def, Dea, Pre
I8
Extended defect formed by eight interstitials. Any
other Ix, where x is an integer, is also valid.
Add, Pro, Dos, Def, Dea, Pre
V4
Extended defect formed by four vacancies. Any
other Vx, with x an integer, is valid.
Add, Pro, Dos, Def, Dea, Pre
B2I3
Boron–interstitial cluster. Other integers are also
valid.
Add, Pro, Dos, Def, Dea, Pre
As4V
Arsenic–vacancy cluster. Other integers are also
valid.
Add, Pro, Dos, Def, Dea, Pre
B2IC3
Dopant cluster. Any combination of dopants with
(or without) interstitials or vacancies is valid.
Add, Pro, Dos, Def, Dea, Pre
P2As2I
Another example for dopant cluster Add, Prod, Dos, Def, Dea, Pre
Ta bl e 61 N a me s us ed i n S e n ta u ru s Pr oc es s K M C
Name Description Used in

5: Atomistic Kinetic Monte Carlo Diffusion
Advanced Calibration for Sentaurus Process KMC
Sentaurus™ Process User Guide 565
I-2013.12
Advanced Calibration for Sentaurus Process KMC
The default parameters used in Sentaurus Process KMC are inherited from previous versions
of Sentaurus Process KMC and may not be accurate for modern processing conditions. A more
accurate calibration for Sentaurus ProcessKMC has been performed by the Advanced
Calibration team and is available using the command:
AdvancedCalibration
This command includes the calibration of point-defect diffusivity, extended defects formation
and dissolution, boron diffusivity, boron–interstitial clustering process (activation and
deactivation of boron), surface trapping and re-emission of boron, and so on.
This command must be written after SetAtomistic since it detects the presence of an
atomistic simulation to load the Advanced Calibration parameters related to Sentaurus
Process KMC:
SetAtomistic
...
AdvancedCalibration
In cases where Advanced Calibration for Sentaurus Process KMC must be loaded, but it is not
possible to call it after SetAtomistic, the following workaround can be used. In particular,
this is the preferred mode to call Advanced Calibration for Sentaurus Process KMC in hybrid
simulations, and the only way to do it when using the kmc option in the diffuse command:
pdbSet AtomisticData 1
AdvancedCalibration
pdbSet AtomisticData 0
NOTE For more information on the Advanced Calibration parameters and
methodology, refer to the Advanced Calibration for Process Simulation
User Guide.
NOTE The use of Advanced Calibration is strongly recommended.

5: Atomistic Kinetic Monte Carlo Diffusion
References
566 Sentaurus™ Process User Guide
I-2013.12
References
[1] I. Martin-Bragado, Simulación atomística de procesos para Microelectrónica, Ph.D.
thesis, Universidad de Valladolid, Valladolid, Spain, 2004.
[2] M. Jaraiz et al., “Atomistic Front-End Process Modelling: A Powerful Tool for Deep-
Submicron Device Fabrication,” in International Conference on Simulation of
Semiconductor Processes and Devices (SISPAD), Athens, Greece, pp. 10–17, September
2001.
[3] N. Strecker, V. Moroz, and M. Jaraiz, “Introducing Monte Carlo Diffusion Simulation
into TCAD tools,” in Technical Proceedings of the International Conference on
Modeling and Simulation of Microsystems (Nanotech 2002), vol. 1, San Juan, Puerto
Rico, USA, pp. 462–465, April 2002.
[4] R. A. Casali, H. Rücker, and M. Methfessel, “Interaction of vacancies with interstitial
oxygen in silicon,” Applied Physics Letters, vol. 78, no. 7, pp. 913–915, 2001.
[5] N. Cowern and C. Rafferty, “Enhanced Diffusion in Silicon Processing,” MRS Bulletin,
vol. 25, no. 6, pp. 39–44, 2000.
[6] N. E. B. Cowern et al., “Impurity Diffusion via an Intermediate Species: The B-Si
System,” Physical Review Letters, vol. 65, no. 19, pp. 2434–2437, 1990.
[7] P. M. Fahey, P. B. Griffin, and J. D. Plummer, “Point defects and dopant diffusion in
silicon,” Reviews of Modern Physics, vol. 61, no. 2, pp. 289–388, 1989.
[8] P. A. Stolk et al., “Physical mechanisms of transient enhanced dopant diffusion in ion-
implanted silicon,” Journal of Applied Physics, vol. 81, no. 9, pp. 6031–6050, 1997.
[9] I. Martin-Bragado, N. Zographos, and M. Jaraiz, “Long and double hop kinetic Monte
Carlo: Techniques to speed up atomistic modeling without losing accuracy,” Materials
Science and Engineering B, vol. 154–155, pp. 202–206, December 2008.
[10] P. Castrillo et al., “Atomistic Modeling of Defect Diffusion in SiGe,” in International
Conference on Simulation of Semiconductor Processes and Devices (SISPAD), Vienna,
Austria, pp. 9–12, September 2007.
[11] P. J. Schultz et al., “Crystalline-to-amorphous transition for Si-ion irradiation of
Si(100),” Physical Review B, vol. 44, no. 16, pp. 9118–9121, 1991.
[12] T. Diaz de la Rubia and G. H. Gilmer, “Structural Transformations and Defect
Production in Ion Implanted Silicon: A Molecular Dynamics Simulation Study,”
Physical Review Letters, vol. 74, no. 13, pp. 2507–2510, 1995.
[13] D. J. Eaglesham et al., “Implantation and transient B diffusion in Si: The source of the
interstitials,” Applied Physics Letters, vol. 65, no. l8, pp. 2305–2307, 1994.
[14] S. Takeda, “An Atomic Model of Electron-Irradiation-Induced Defects on {113} in Si,”
Japanese Journal of Applied Physics, vol. 30, no. 4A, pp. L639–L642, 1991.
5: Atomistic Kinetic Monte Carlo Diffusion
References
Sentaurus™ Process User Guide 567
I-2013.12
[15] S. M. Hu, “Diffusion in Silicon and Germanium,” Atomic Diffusion in Semiconductors,
London: Plenum Press, pp. 217–350, 1973.
[16] J. Kim et al., “Extended Si {311} defects,” Physical Review B, vol. 55, no. 24,
pp. 16186–16197, 1997.
[17] M. Kohyama and S. Takeda, “Atomic structure and energy of the {113} planar
interstitial defects in Si,” Physical Review B, vol. 46, no. 19, pp. 12305–12315, 1992.
[18] B. de Mauduit et al., “Identification of EOR defects due to the regrowth of amorphous
layers created by ion bombardment,” Nuclear Instruments and Methods in Physics
Research B, vol. 84, no. 2, pp. 190–194, 1994.
[19] F. Cristiano et al., “Formation energies and relative stability of perfect and faulted
dislocation loops in silicon,” Journal of Applied Physics, vol. 87, no. 12, pp. 8420–8428,
2000.
[20] G. D. Watkins and J. W. Corbett, “Defects in Irradiated Silicon: Electron Paramagnetic
Resonance of the Divacancy,” Physical Review, vol. 138, no. 2A, pp. A543–A555,
1965.
[21] G. D. Watkins, “Defects in irradiated silicon: EPR and electron-nuclear double
resonance of interstitial boron,” Physical Review B, vol. 12, no. 12, pp. 5824–5839,
1975.
[22] G. D. Watkins, “Erratum: Defects in irradiated silicon: EPR and electron-nuclear double
resonance of interstitial boron,” Physical Review B, vol. 13, no. 10, p. 4644, 1976.
[23] B. Hourahine et al., “Identification of the hexavacancy in silicon with the optical
center,” Physical Review B, vol. 61, no. 19, pp. 12594–12597, 2000.
[24] D. J. Chadi and K. J. Chang, “Magic numbers for vacancy aggregation in crystalline Si,”
Physical Review B, vol. 38, no. 2, pp. 1523–1525, 1988.
[25] S. K. Estreicher, J. L. Hastings, and P. A. Fedders, “The ring-hexavacancy in silicon: A
stable and inactive defect,” Applied Physics Letters, vol. 70, no. 4, pp. 432–434, 1997.
[26] O. W. Holland and C. W. White, “Ion-induced damage and amorphization in Si,”
Nuclear Instruments and Methods in Physics Research B, vol. 59/60, pp. 353–362, July
1991.
[27] A. Bongiorno and L. Colombo, “Interaction between a monovacancy and a vacancy
cluster in silicon,” Physical Review B, vol. 57, no. 15, pp. 8767–8769, 1998.
[28] L. Pelaz et al., “Atomistic modeling of amorphization and recrystallization in silicon,”
Applied Physics Letters, vol. 82, no. 13, pp. 2038–2040, 2003.
[29] S. Mirabella et al., “Mechanism of Boron Diffusion in Amorphous Silicon,” Physical
Review Letters, vol. 100, p. 155901, 2008.
[30] I. Martin-Bragado and N. Zographos, “Indirect boron diffusion in amorphous silicon
modeled by kinetic Monte Carlo,” Solid-State Electronics, vol. 55, no. 1, pp. 25–28,
2011.
B80
4
5: Atomistic Kinetic Monte Carlo Diffusion
References
568 Sentaurus™ Process User Guide
I-2013.12
[31] N. Zographos and I. Martin-Bragado, “A Comprehensive Atomistic Kinetic Monte
Carlo Model for Amorphization/Recrystallization and its Effects on Dopants,” in MRS
Symposium Proceedings, Doping Engineering for Front-End Processing, vol. 1070,
p. 1070-E03-01, March 2008.
[32] I. Martin-Bragado and V. Moroz, “Facet formation during solid phase epitaxy regrowth:
A lattice kinetic Monte Carlo model,” Applied Physics Letters, vol. 95, p. 123123, 2009.
[33] R. Duffy et al., “Solid phase epitaxy versus random nucleation and growth in sub-20 nm
wide fin field-effect transistors,” Applied Physics Letters, vol. 90, no. 24, p. 241912,
2007.
[34] K. L. Saenger et al., “An examination of facet formation during solid phase epitaxy of
line-shaped amorphized regions in (001) and (011) Si,” Journal of Applied Physics,
vol. 101, no. 10, p. 104908, 2007.
[35] I. Martin-Bragado, “{111} local configurations: The main source of silicon defects
during solid phase epitaxial regrowth modeled by lattice kinetic Monte Carlo,” Applied
Physics Letters, vol. 98, no. 23, p. 233109, 2011.
[36] I. Martin-Bragado, “Importance of twin defect formation created by solid-phase
epitaxial growth: An atomistic study,” Scripta Materialia, vol. 66, no. 3–4, pp. 186–189,
2012.
[37] V. C. Venezia et al., “Dopant redistribution effects in preamorphized silicon during low
temperature annealing,” in IEDM Technical Digest, Washington, DC, USA, pp. 489–
492, December 2003.
[38] O. Dokumaci et al., “Transient Enhanced Diffusion and Dose Loss of Indium in
Silicon,” in MRS Symposium Proceedings, Si Front-End Processing—Physics and
Technology of Dopant-Defect Interactions, vol. 568, San Francisco, CA, USA, pp. 205–
210, April 1999.
[39] P. A. Stolk et al., “Trap-limited interstitial diffusion and enhanced boron clustering in
silicon,” Applied Physics Letters, vol. 66, no. 5, pp. 568–570, 1995.
[40] L. Pelaz et al., “B diffusion and clustering in ion implanted Si: The role of B cluster
precursors,” Applied Physics Letters, vol. 70, no. 17, pp. 2285–2287, 1997.
[41] S. Mirabella et al., “Mechanism of Boron Diffusion in Amorphous Silicon,” Physical
Review Letters, vol. 100, p. 155901, April 2008.
[42] D. C. Müller, Deactivation and Activation of Donors in Silicon, Series in
Microelectronics, vol. 151, Konstanz, Germany: Hartung-Gorre, 2004.
[43] P. M. Rousseau et al., “Arsenic deactivation enhanced diffusion: A time, temperature,
and concentration study,” Journal of Applied Physics, vol. 84, no. 7, pp. 3593–3601,
1998.
[44] C. Rafferty, “Progress in Predicting Transient Diffusion,” in International Conference
on Simulation of Semiconductor Processes and Devices (SISPAD), Cambridge, MA,
USA, pp. 1–4, September 1997.
5: Atomistic Kinetic Monte Carlo Diffusion
References
Sentaurus™ Process User Guide 569
I-2013.12
[45] I. Martin-Bragado et al., “Physical modeling of Fermi-level effects for decanano device
process simulations,” Materials Science and Engineering B, vol. 114–115, pp. 284–289,
December 2004.
[46] I. Martin-Bragado et al., “Fermi-level effects in semiconductor processing: A modeling
scheme for atomistic kinetic Monte Carlo simulators,” Journal of Applied Physics,
vol. 98, p. 053709, September 2005.
[47] C. Persson, U. Lindefelt, and B. E. Sernelius, “Band gap narrowing in n-type and p-type
3C-, 2H-, 4H-, 6H-SiC, and Si,” Journal of Applied Physics, vol. 86, no. 8, pp. 4419–
4427, 1999.
[48] S. C. Jain and D. J. Roulston, “A Simple Expression for Band Gap Narrowing (BGN) in
Heavily Doped Si, Ge, GaAs and GexSi1–x Strained Layers,” Solid-State Electronics,
vol. 34, no. 5, pp. 453–465, 1991.
[49] D. R. Lim, C. S. Rafferty, and F. P. Klemens, “The role of the surface in transient
enhanced diffusion,” Applied Physics Letters, vol. 67, no. 16, pp. 2302–2304, 1995.
[50] N. E. B. Cowern et al., “Transient enhanced diffusion in preamorphized silicon: the role
of the surface,” Nuclear Instruments and Methods in Physics Research B, vol. 148,
no. 1–4, pp. 257–261, 1999.
[51] I. Martin-Bragado and V. Moroz, “Modeling of {311} facets using a lattice kinetic
Monte Carlo three-dimensional model for selective epitaxial growth of silicon,” Applied
Physics Letters, vol. 98, no. 15, p. 153111, 2011.
[52] B. Sahli et al., “Ab initio calculations of phosphorus and arsenic clustering parameters
for the improvement of process simulation models,” Material Science and
Engineering B, vol. 154–155, pp. 193–197, December 2008.
5: Atomistic Kinetic Monte Carlo Diffusion
References
570 Sentaurus™ Process User Guide
I-2013.12

Sentaurus™ Process User Guide 571
H-2013.03
CHAPTER 6 Alagator Scripting Language
This chapter discusses the Alagator scripting language, which is
used to specify partial differential equations and boundary
conditions for use with diffusion simulations.
The equations are expressed in a Newton iteration–ready form. They are specified as text
strings that are assumed to be equal to zero. Most mathematical operators are supported to
specify equations, and various operators for differential terms are available. For terms that
include the gradient (grad) operator, Sentaurus Process automatically calculates the
divergence. It is not necessary to specify the divergence in the equations.
Binary operators, functions, constants, and parameters are supported. Care must be exercised
with Tcl expansion of variables and strings, as usually users want variables to be evaluated at
run-time, not when they are read.
Operators
The Alagator language operators and variables consist of binary operators, simple functions,
differential functions, string names, solution names, subexpressions, constants, and
parameters.
Binary and Unary Operators
Most common binary algebraic operators are supported. Addition (+), subtraction (–),
multiplication (*), and division (/) are included. Unary negation is also supported with the usual
mathematical rules applying. Typical precedence rules apply (see Table 63 on page 572).
Parentheses are supported for grouping operations. In addition to the basic four mathematical
operators, power (^) is also supported; for example, a^b raises a to the b power.
NOTE The Tcl command expr pow(a,b) is not supported in the Alagator
language when a and b do not evaluate to an integer or double value.
Many comparison operators are implemented. These do not support derivative operations, so
they cannot be used in the gradient expression. However, they can be used in the select
command (see select on page 1117). The operators >, <, >=, <=, ==, and != are implemented

6: Alagator Scripting Language
Operators
572 Sentaurus™ Process User Guide
H-2013.03
with their usual meanings. Care must be used with equals and not equals, since a comparison
of floating point values in this way can be problematic.
Logical operators and (&&) and or (||) are also provided for use with callbacks and
initialization. When these operators are used as part of a differential equation, care must be
taken as the Newton method does not ensure convergence for problems that are not first-order
continuous.
In addition to these, a conditional operator (?:) is provided, which takes three operands. The
first operand is a condition, the second operand is the value of the entire conditional expression
if the condition is true, and the third operand is the value of the entire conditional expression if
the expression is false. For example, the command:
sel z = "(Vac>1e15) ? (1e15) : (Vac)" name = Vac
sets the value of
Vac
to on mesh points where
Vac
is greater than and does
not change
Vac
on mesh points where
Vac
is smaller than . Since the select
command works on mesh nodes, the conditional operator is very useful for truncating profiles.
Simple Functions
All simple functions take one argument that must be enclosed in parentheses. The argument
can be any expression. Most common functions are available, including ‘exp’ natural
exponentiation, ‘log’ natural log, ‘log10’ log base 10, and ‘sqrt’ square root. Additionally, the
complementary error function ‘erfc’ and error function ‘erf’ are provided to help build initial
doping profiles. All of these functions have supported derivatives and can be used in the
specification of partial differential equations (PDE).
‘abs’ and ‘sign’ provide an absolute value and sign operation. The sign operation is positive if
the argument is greater than zero and minus one for less than zero. These functions do not
provide derivatives and cannot be used as part of a differential equation.
Ta bl e 63 O pe ra to r pr ec ed e nc e
Operator Description Operator Description
^Power&&Logical and
-Unary minus||Logical or
*, / Multiplication, division ?: Conditional operator
+, – Addition, subtraction , Comma operator for lists
<, <=, >=, >, ==, != Equality, inequality
110
15
×
110
15
×
110
15
×

6: Alagator Scripting Language
Operators
Sentaurus™ Process User Guide 573
H-2013.03
Differential Functions
The differential functions are used in partial differential equations only and are not evaluated
with the select command. There are two differential operators: ddt and grad. Time
derivatives are supported with the ddt operator. It takes a single argument and computes the
first-time derivative of the argument for use in a partial differential equation. Time-step
integration is provided automatically using the Bank–Rose TRBDF method [1].
Spatial derivatives are supported in two ways. A simple gradient is supported with grad.
Implied is the evaluation in a discrete sense and the integral around a control volume. For this
reason, div is not required. For example, A*B*grad(C) is treated as div(A*B*grad(C)).
Special Functions
The diag Operator
The special operator diag provides the modeling of anisotropic diffusion. The special operator
(anisotropic diffusion matrix) has the form:
(817)
where a, b, and c are any valid Alagator expression. It also can include diffusion solution
variables. For discretization, the diag operator projects the vector to the edge direction. For
1D structures, a must be specified. For 2D structures, a and b must be specified. For 3D
structures, a, b, and c must be defined.
The following expressions are valid:
ddt(C
1
)-D
1
*diag(a,b,c)*grad(C
1
)
ddt(C
1
)-D
1
*diag(a,b,c)*grad(C
1
)-D
2
*diag(g,h,i)*grad(C
2
)
ddt(C
1
)-D
1
*diag(a,b,c)*grad(C
1
)-D
2
*grad(C
2
)
# Addition of two diag operators (diag(...)+diag(...))
ddt(C
1
)-(D
1
*diag(a,b,c)+D
2
*diag(g,h,i))*grad(C1)
The following expressions are not valid:
# Multiplication of two diag operators (diag(...)*diag(...))
ddt(C1)-(D1*diag(a,b,c)*diag(g,h,i))*grad(C1)
# Division of two diag operators (diag(...)/(diag(...))
ddt(C1)-(D1*diag(a,b,c)/diag(g,h,i))*grad(C1)
diag(a,b,c)
a00
0b0
00c
≡

6: Alagator Scripting Language
Operators
574 Sentaurus™ Process User Guide
H-2013.03
# Addition of a diag operator with a constans (diag(...)+C)
ddt(C1)-(D1*diag(a,b,c,d,e,f)+D2)*grad(C1)
You c an use bui lt -in f un ction s to define a nis ot ro py. For ex amp le :
(818)
term name=BoronDiffFactor Silicon add eqn = "diag(exp(3.0*ElasticStrainKK_x \
* [simDelayDouble Diffuse Vti]),exp( -2.0 * ElasticStrainKK_y * \
[simDelayDouble Diffuse Vti]),1.0)"
The above command makes the effective diffusivity of boron anisotropic. If
<dopant>DiffFactor (see Alagator for Diffusion on page 575) is already defined (that is,
AdvancedCalibration), perform the following:
set locterm [term name=BoronDiffFactor Silicon print]
term name=BoronDiffFactor Silicon add eqn = "($locterm) * \
(diag(exp(3.0*ElasticStrainKK_x * [simDelayDouble Diffuse Vti]), \
exp( -2.0 * ElasticStrainKK_y * [simDelayDouble Diffuse Vti]),1.0))"
String Names
Strings that are not recognized as real numbers, operators, or functions are handed to the
resolution phase of the parser. These strings are compared to four sets of possible matches. The
first set is valid solution names created with the solution command (see solution on
page 1145). The second set is the data field name. The third set is named subexpressions
created with the term command (see term on page 1173). Finally, any remaining strings are
passed to the Tcl expression function to see if they can be parsed to a real number constant.
This allows parameters from the parameter database to be used in differential equations.
Solution Names and Subexpressions: Terms
Solution names (for example, Boron, 311) must match the string exactly as specified in the
solution command (see solution on page 1145). These are the most important resolutions
since they allow linking of the equations to the variables to be solved. Derivatives are
automatically taken of all equations with respect to each solution name found in the equation.
Terms are useful for common subexpressions. The string used in the equations must match
exactly the name given in the term command (see term on page 1173).
diag(e
3σx
kT
---------
e
2σy
kT
---------
–
1),,
e
3σx
kT
---------
00
0e
2σy
kT
---------
–
0
00 1
≡

6: Alagator Scripting Language
Alagator for Diffusion
Sentaurus™ Process User Guide 575
H-2013.03
NOTE Terms and data fields with the same name can coexist in the structure.
During equation parsing, the term name will have higher priority than
the data field name for a region.
Constants and Parameters
Anything that does not match a term or solution is handed to the Tcl expression parser to see
if it evaluates as a valid expression. The result is treated as a constant. The evaluation of the
expression is performed again in the code if the temperature or time changes, so that parameters
can have explicit dependencies on these values.
When defining parameters, care must be given to the nested declaration. Especially when
parameters are derived using the pdbDelayDouble command from other parameters, the Tcl
expression parser may be unable to expand the whole expression and evaluate it correctly. For
example:
pdbSetDouble Si Test Param1 {[Arr 1 2]} (1)
pdbSetDouble Si Test Param2 {2.0*[pdbDelayDouble Si Test Param1]}(2)
pdbSetDouble Si Test Param3 {2.0*[pdbGetDouble Si Test Param1]}(3)
pdbGet Si Test Param2 (4)
pdbGet Si Test Param3 (5)
The first three lines set Param1, Param2, and Param3. Param2 and Param3 are derived
parameters from Param1. While Param2 uses pdbDelayDouble to obtain the value of
Param1, Param3 uses pdbGetDouble. When retrieving data, line 4 will return an error
message and line 5 will return a valid double number without an error message. The error
message is issued because pdbDelayDouble returns an expression of Param1, which is
treated as a string by the Tcl parser during the evaluation of Param2. To prevent such errors,
Param2 can be encapsulated with the expr command:
pdbSetDouble Si Test Param2 {[expr 2.0*[pdbDelayDouble Si Test Param1]]}
NOTE If Sentaurus Process cannot evaluate an expression correctly, it assumes
that it is zero.
Alagator for Diffusion
In this section, an example is used to illustrate how to specify equations using the Alagator
scripting language. The general expression for diffusion of species is given by:
(819)
CX
t∂
∂CXD∇CX
∇•=

6: Alagator Scripting Language
Alagator for Diffusion
576 Sentaurus™ Process User Guide
H-2013.03
and will be translated to Alagator language as:
ddt(CX)-D*grad(CX) = 0
where CX is the solution variable and D is the diffusivity term (see Solution Names and
Subexpressions: Terms on page 574).
NOTE Do not specify the div operator in front of D*grad(CX); div is
implied.
NOTE The grad operator must work on function of solution variable.
NOTE Since the examples given in this chapter use parameters that are not in
the PDB, long-hand pdbSet commands are used (see pdbSet and
Related Commands on page 1056).
Basics
The simplest diffusion equation uses a constant diffusivity and can be described by Fick’s first
law and second law. Two main steps are required to initialize and solve this equation. First, a
solution must be defined (see solution on page 1145). Second, the equation must be entered
into the parameter database.
A minimum of two commands can be used to accomplish this, for example:
solution name=CX add !negative !damp solve
pdbSetString Silicon CX Equation "ddt(CX) - 1e-15*grad(CX)"
The first line creates a new solution named CX and adds it to the solution list. The solution
cannot take negative values and numeric damping is not applied to the updates of the Newton
iteration. The solution is always to be solved.
NOTE Aliases are defined only for pdb commands. In the above example, the
solution name used in the definition of partial differential equations
must match the one defined with the solution command.
The second line makes an entry into the parameter database. This is created for the material
Silicon and the solution variable CX. An entry is made for an Equation, which is the
predefined entry that Alagator looks for to find a differential equation. The string value set is
the differential equation that will be solved for this variable in this material. The equation uses
a time operator (ddt) and gradient operator (grad) to implement a simple diffusion equation.
The divergence operator is implied and computed as part of the discretization of the equations.
Whenever a gradient operator grad is in an Alagator script, it is assumed that the divergence
will be taken of that term during assembly.
6: Alagator Scripting Language
Alagator for Diffusion
Sentaurus™ Process User Guide 577
H-2013.03
In this example, the equation to be solved is d(CX)/dt-div(1.0e-15*grad(CX))=0. The
solution name in the equation (
CX) must also be identical to the name in the solution
command. The diffusivity in this example is hard wired to be .
In this first example, the diffusivity does not depend on the temperature. To use a temperature-
dependent diffusivity, use the following command lines:
set diff {[Arrhenius 0.138 1.37]}
pdbSetString Silicon CX Equation "ddt(CX) - $diff * grad(CX)"
In the first line, a local Tcl variable is created to hold a string representing the diffusivity. The
braces are necessary to prevent immediate evaluation of the Arrhenius function (see Arrhenius
on page 884). The Arrhenius function is a predefined helper function that allows for the simple
creation of Arrhenius expressions. It uses the temperature set by the diffusion command or
the SetTemp command (see SetTemp on page 1130). The presence of braces means that the
Arrhenius function is inserted directly into the parameter database equation and is evaluated
during the diffusion. For each diffusion time-step, the Arrhenius function will then be
evaluated at the current temperature.
A further enhancement can be made by adding the diffusivity to the parameter database. This
allows other users to change the value in the equation by accessing the properties directly. The
following changes make the equation dependent on the stored value in the database:
pdbSetDouble Silicon CX D {[Arrhenius 0.1 3.62]}
set diff [pdbDelayDouble Silicon CX D]
pdbSetString Silicon CX Equation "ddt(CX) - $diff * grad(CX)"
The first line sets the diffusivity in the database. This can be made permanent by directly
editing the hierarchy files. The second line uses pdbDelayDouble to return the expression
stored in the database. This is necessary so that the evaluation of the expression does not occur
until the diffuse command is executed. Now, the equation depends on the database entry.
(You can change this entry to observe the effect of different diffusivities on the final profile.)
In addition, it is possible to solve for CX only after it is introduced into the structure or
otherwise present in the material. This is performed by modifying the solution command:
solution name=CX !negative !damp ifpresent=CX add
The ifpresent option enables the solution as a variable for the diffusion equation only if a
real data field exists with that name. This means that only structures already having CX defined
(with the select command, for example) will solve the differential equation. This is useful
for controlling CPU time and matrix size by not requiring solutions of systems that do not have
that species present.
10 15– cm2/s

6: Alagator Scripting Language
Alagator for Diffusion
578 Sentaurus™ Process User Guide
H-2013.03
Setting Boundary Conditions
Dirichlet Boundary Condition
The previous example can be enhanced by adding a boundary condition to allow in-diffusion
of this species from a gas source. For simplicity, it is assumed that the gas source fixes the
surface concentration of species CX at .
The following two commands create this boundary condition:
pdbSetBoolean Gas_Silicon CX Fixed_Silicon 1
pdbSetString Gas_Silicon CX Equation_Silicon "CX_Silicon - 5.0e19"
Both commands work on the gas–silicon interface for the CX variable. The first command states
that the value is to be fixed on the silicon side, that is, a Dirichlet boundary condition is to be
applied. The keyword Fixed is used only with the Dirichlet boundary condition.
Fluxes will be ignored at this node and the boundary condition will control the concentration.
The _Silicon option on Fixed indicates the value is to be set on the silicon side. This is
critical because there can be three components on any interface, one for each material and one
for the interface. The second command sets the boundary condition equation on the silicon side
to be the concentration of .
NOTE Equations are set to zero by definition. The CX variable also has
_Silicon appended to indicate that the concentration is set on the
silicon side.
NOTE The interface names are lexically ordered. Most interface names are set
to the right order using the alias command (for example,
Silicon_Gas will be interpreted as Gas_Silicon). If a new interface
name is introduced, the order must be followed.
Segregation Boundary Condition
If a segregation-type boundary condition is needed, for example, at the oxide–silicon interface,
the following two commands will create this boundary condition:
pdbSetString Oxide_Silicon CX Equation_Oxide "(1.6e-7*(CX_Oxide - \
CX_Silicon/0.28))"
pdbSetString Oxide_Silicon CX Equation_Silicon "-(1.6e-7*(CX_Oxide - \
CX_Silicon/0.28))"
Of course, this boundary condition assumes that the diffusion equation for CX is solved in the
oxide region as well. Otherwise, the diffusion equations would be unbalanced at this interface.
510
19
× cm 3–
510
19
× cm 3–
6: Alagator Scripting Language
Alagator for Diffusion
Sentaurus™ Process User Guide 579
H-2013.03
Both of the commands work on the oxide–silicon interface. The _Silicon and _Oxide
options on Equation indicate the side of the interface to which the given flux will be applied.
The same options on the solution variable CX indicate whether the solution variable value at
this interface is taken from the oxide side or the silicon side. It should also be noted that the
fluxes have opposite signs. The first number ( ) in the flux equation is the transfer
coefficient and the second number (0.28) is the segregation coefficient.
Natural Boundary Condition
If a natural boundary condition is needed, the following three commands will create this
boundary condition:
set Ksurf {[Arrhenius 1.17e6 1.37]}
set CXStar {[Arrhenius 3.6e27 3.7]}
pdbSetString Gas_Silicon CX Equation_Silicon "- $Ksurf * (CX_Silicon - \
$CXStar)"
The first two lines set the surface recombination rate and the equilibrium concentration, and
the third line sets the equation. In this case, a flux is added to the equation on the silicon side,
so there is no need for the Fixed flag. Recombination at an interface obtains a negative sign
and generation obtains a positive sign. The variable again needs to have _Silicon appended
to indicate the value on the silicon side of the interface.
Interface Traps
Interfaces can act like traps for diffusing species. It is also possible that the trapped species may
diffuse along the interface and segregate into neighboring materials.
The following commands create this boundary condition:
set diff {[Arrhenius 5.e-14 1.2]}
set Ktrap {[Arrhenius 1.17e-2 1.37]}
set CXStar {[Arrhenius 3.6e27 3.7]}
pdbSetString Gas_Silicon CX Equation "ddt(CX) -$diff * grad(CX) - $Ktrap * \
(CX_Silicon - $CXStar)"
pdbSetString Gas_Silicon CX Equation_Silicon "-$Ktrap * (CX_Silicon - \
$CXStar)"
The first three lines set the diffusivity, trapping rate, and equilibrium concentration of CX at the
gas–silicon interface, respectively. The fourth line sets the trapping equation at the gas–silicon
interface. The solution variable name (CX) without the suffix _Silicon indicates the value at
this interface. The variable with _Silicon appended indicates the value on the silicon side of
the interface. The grad() operator has the usual meaning as previously explained (see Basics
1.6 10 7–
×

6: Alagator Scripting Language
Alagator for Diffusion
580 Sentaurus™ Process User Guide
H-2013.03
on page 576). The last term in the equation is the flux, which depends on the trapping rate. The
last line adds the corresponding flux to the equation on the silicon side.
External Boundary Condition
To set a boundary condition at the outer sides, use the command:
pdbSetString <material> <solution> Equation_<boundary> <string>
where:
■<material> is the material name.
■<solution> is the solution name.
■<boundary> is specified with one of the following: LeftSide, RightSide,
FrontSide, BackSide, or Bottom.
The specified equation is added to the bulk equation of the solution at the nodes on the specified
side.
Using Terms
It is possible to extend the previous example by introducing a new solution variable, CY, and
the following recombination reaction between CX and CY:
(820)
(821)
The reaction states that two species (CX and CY) annihilate each other when they react. is
the forward reaction rate, and and are the equilibrium values of the solution
variables. The new solution variable is assumed to diffuse according to Fick’s law of diffusion.
Building on the previous example, the above equation can be implemented by the following
command lines:
solution add name=CX !damp !negative solve
solution add name=CY !damp !negative solve
set Kf {[Arrhenius 4.2e-11 0.1]}
set CXStar {[Arrhenius 3.6e27 3.7]}
set CYStar {[Arrhenius 4.0e26 3.97]}
set RCXCY "$Kf * (CX * CY- $CXStar * $CYStar)"
set diff {[Arrhenius 0.138 1.37]}
XY+0↔
RCXCY KfCXCY CX*CY*
–()≡
Kf
CX∗
CY∗
6: Alagator Scripting Language
Alagator for Diffusion
Sentaurus™ Process User Guide 581
H-2013.03
pdbSetString Silicon CX Equation "ddt(CX) - $diff * grad(CX) + $RCXCY"
set diff {[Arrhenius 0.02 0.3]}
pdbSetString Silicon CY Equation "ddt(CY) - $diff * grad(CY) + $RCXCY"
The first two lines create solutions for CX and CY. The third line is the forward reaction rate.
The next two lines set the equilibrium concentrations of CX and CY. The sixth line sets the
RCXCY variable to be a subexpression for the recombination reaction. Any excess of CX and CY
is annihilated until the concentrations are at the equilibrium product. Finally, the diffusivity is
obtained and the equation is set, similar to the previous example. The recombination reaction
is added to both solution variable equations.
A common error is not to add reaction terms to all affected equations. When the recombination
is positive, it forces the time derivative to become negative to make the equation equal to zero.
The implementation cited above is difficult to read. The RCXCY variable is used more than once
and may need to be used in other equations. To reduce the maintenance of the code, a term can
be created and used everywhere, not only in the local scope where the term is defined (see term
on page 1173).
A term is a common subexpression that can be used in multiple instances. When the term
appears in multiple equations, the values are easily retrieved from memory and accumulated,
for example:
solution add name=CX !damp !negative solve
solution add name=CY !damp !negative solve
set Kf {[Arrhenius 4.2e-11 0.1]}
set CXStar {[Arrhenius 3.6e27 3.7]}
set CYStar {[Arrhenius 4.0e26 3.97]}
term name=RCXCY Silicon eqn = "$Kf * (CX * CY- $CXStar * $CYStar)"
set diff {[Arrhenius 0.138 1.37]}
pdbSetString Silicon CX Equation "ddt(CX) - $diff * grad(CX) + RCXCY"
set diff {[Arrhenius 0.02 0.3]}
pdbSetString Silicon CY Equation "ddt(CY) - $diff * grad(CY) + RCXCY"
This is almost identical to the previous example, except that a term was created, not a local
variable. Due to this change, the dollar sign (indicative of a Tcl variable) is no longer needed
in the equation. It has become a simple text string, which will be resolved to the term. The terms
are kept until you exit the simulator, so it can be used in other equations or in a select
command (if you want to monitor the recombination rates).

6: Alagator Scripting Language
Alagator for Diffusion
582 Sentaurus™ Process User Guide
H-2013.03
A further enhancement can be made by adding the diffusivity, equilibrium concentrations, and
reaction rate to the parameter database as previously. The following changes make the equation
dependent on the stored values in the database:
pdbSetDouble Silicon CX D {[Arrhenius 0.138 1.37]}
pdbSetDouble Silicon CY D {[Arrhenius 0.02 0.3]}
pdbSetDouble Silicon CX Kf {[Arrhenius 4.2e-11 0.1]}
pdbSetDouble Silicon CX Cstar {[Arrhenius 3.6e27 3.7]}
pdbSetDouble Silicon CY Cstar {[Arrhenius 4.0e26 3.97]}
solution add name=CX !damp !negative solve
solution add name=CY !damp !negative solve
set Kf [pdbDelayDouble Silicon CX Kf]
set CXStar [pdbDelayDouble Silicon CX Cstar]
set CYStar [pdbDelayDouble Silicon CY Cstar]
term name = RCXCY Silicon eqn = "$Kf * (CX * CY- $CXStar * $CYStar)"
set diff [pdbDelayDouble Silicon CX D]
pdbSetString Silicon CX Equation "ddt(CX) - $diff * grad(CX) + RCXCY"
set diff [pdbDelayDouble Silicon CY D]
pdbSetString Silicon CY Equation "ddt(CY) - $diff * grad(CY) + RCXCY"
As stated in the previous section, the first five lines use pdbDelayDouble to return the
expression stored in the database. This is necessary so that the evaluation of the expression does
not occur until the diffuse command is executed.
Callback Procedures
Callbacks allow additional ‘intelligence’ to be built into the equations by allowing procedures
to be called at run-time. These procedures build the Alagator equation strings according to
user-specified options. By selecting model switches, you can choose between different
physical models to be represented in the equation strings. By having callback procedures that
use a material name, a dopant name, or a defect name as arguments, the same type of equation
can be built for several materials, dopants, and defect species. In Sentaurus Process, all
frequently used equations are built-in callback procedures.
The callback procedure–related keywords in Alagator are:
■InitGrowth
■InitSolve
■InitProc
■EquationGrowthProc
6: Alagator Scripting Language
Alagator for Diffusion
Sentaurus™ Process User Guide 583
H-2013.03
■EquationInitProc
■EquationProc
■GrowthRateProc
InitGrowth and EquationGrowthProc are used to define generic growth equations (see
Alagator for Generic Growth on page 596), and GrowthRateProc is used with the epi model
(see Epitaxy on page 282). Using the pdbSet command, you can point Alagator to use various
Tcl callback procedures.
This section focuses on the remaining keywords. All of these keywords provide a Tcl callback
procedure name to Sentaurus Process:
Callbacks during Execution of diffuse Command
The Tcl callback procedures are called at various stages during the execution of a diffuse
command. In addition to the callback procedures, Sentaurus Process calls the
diffPreProcess Tcl procedure before executing the diffuse command and the
diffPostProcess Tcl procedure after executing the diffuse command. The default
behavior is described in Ion Implantation to Diffusion on page 353.
EquationInitProc
Provides the Tcl callback procedure name that sets up the
initialization equations.
EquationProc
Provides the Tcl callback procedure name that sets up diffusion
equations.
InitProc
Provides the Tcl callback procedure name that is usually used to reset
or delete existing parameter database equations or terms at the
beginning of a diffusion simulation.
InitSolve
Provides the Tcl callback procedure name that is used to reset or
delete existing parameter database equations or terms.
6: Alagator Scripting Language
Alagator for Diffusion
584 Sentaurus™ Process User Guide
H-2013.03
Figure 84 shows the flowchart of this process. Sections relating to generic growth are omitted.
They are explained in Alagator for Generic Growth on page 596.
Figure 84 Flowchart with calls to the callback procedures during the execution of a diffuse
command by Sentaurus Process; callbacks related to material growths are
omitted
diffuse
diffPreProcess
For each: solution and material
InitSolve callback procedure:
Next
For each: marked solution and material
EquationInitProc callback procedure
Solve initialization
InitPostProcess
For each: solution and material
EquationProc callback procedure
Solve diffusion
diffPostProcess
InitProc callback procedure
For each: solution and material
Next
equations
Next
Next
equations
6: Alagator Scripting Language
Alagator for Diffusion
Sentaurus™ Process User Guide 585
H-2013.03
The diffPreProcess Procedure
The execution of every diffuse command starts with a call to the diffPreProcess Tcl
procedure. The diffPreProcess Tcl procedure is used to initialize various data fields or to
preprocess the existing data fields. For example, the truncation of interstitial and vacancy
profiles in the amorphous regions is performed in this procedure. In diffPreProcess, the
point-defect equations are switched on or off according to the diffusion models selected.
Initialization
After the execution of diffPreProcess, Sentaurus Process checks for all materials and
solution names, and whether a procedure name is specified for the keyword InitSolve for
the material and solution names. If a procedure name is defined, the procedure will be called,
using the material name and solution name as arguments.
Subsequently, Sentaurus Process checks for all marked materials and solutions to see if a
procedure name is specified with the keyword EquationInitProc for the solution variable
that needs to be initialized. If this is the case, the procedure will be executed with the material
name and solution name as arguments. Typically, in this procedure, the equation string is built,
which is to be solved at time=0 in the diffusion solver. Alternatively, if no callback procedure
name is defined for the keyword EquationInitProc for a material and solution, the equation
string to be used for the initialization can be specified directly on the command line.
After that, Sentaurus Process will solve the initialization equations for all materials and
solutions that need to be initialized. The initialization equations are solved for the initial
temperature of the temperature ramp specified in the diffuse statement. Such an initialization
is usually not required for all solutions. It is typically necessary for solutions whose initial
value depends in a complex way on the data fields (see Complex Initialization Procedures:
InitSolve and EquationInitProc on page 592).
If initialization is not required at all, it can be omitted using the !isolve option with the
diffuse command.
A Tcl procedure called InitPostProcess is provided for convenience. It is called after the
initialization is completed. It can be used to plot or save the solution variable profiles after the
initialization. By default, InitPostProcess is an empty procedure.
Building and Solving Diffusion Equations
After the initialization, Sentaurus Process checks for all materials and solution names to see
whether a procedure name is specified for the keyword InitProc. If the procedure is defined,
it will be called for the specified material name and solution name. These procedures are
usually used to set the equation strings to empty strings and to remove terms defined in
previous diffusion steps. By having empty equation strings, the equations and terms can be
built up piecewise, by adding expressions for each selected model that contributes to an

6: Alagator Scripting Language
Alagator for Diffusion
586 Sentaurus™ Process User Guide
H-2013.03
equation or a term. This is necessary because different diffusion models may be used for
different diffusion steps, and because additional species may be added between diffusion steps,
which may require terms to be added to the equations for existing species.
In the next step, Sentaurus Process checks for all materials and solution names to see whether
a procedure name is specified for the keyword EquationProc. If the procedure is defined, it
will be called with the material name and the solution name as parameters. These procedures
are used to set the diffusion equations for the solution variable. Alternatively, if no callback
procedure is defined for a material and a solution, the equation string can be set in a command
line without specifying any callback procedures.
NOTE If the callback procedure EquationProc is provided for a material
name and solution name, it will typically overwrite any equation
specified on the command line for this material and solution.
After the diffusion equations are set, Sentaurus Process solves the equations for the whole
temperature cycle specified in the diffuse statement.
The diffPostProcess Procedure
Finally, the procedure diffPostProcess will be called. The main purpose of the procedure
is to delete the data fields that are no longer needed and to store the total concentration of point
defects. The procedures diffPreProcess, diffPostProcess, and InitPostProcess
can be found in the Tcl library (see Ion Implantation to Diffusion on page 353) in the file
DiffProcess.tcl.
Using Callback Procedures
In this section, the previous examples will be implemented using callback procedures. First,
the callback procedure keywords InitProc and EquationProc will be explained since they
are the most widely used. Then, the examples will be expanded to include the use of other
keywords and procedures.
6: Alagator Scripting Language
Alagator for Diffusion
Sentaurus™ Process User Guide 587
H-2013.03
Setup Procedure: InitProc
The keyword InitProc is used to clean up equation strings. It specifies the name of the
callback procedure to be called by Sentaurus Process. For example, the first command below:
defines the ResetEquations procedure as the callback procedure of the solution variable CX
in silicon. The callback procedure itself takes two arguments: a material name and a solution
name. In this example, Sentaurus Process will call the ResetEquations procedure with two
arguments. The first argument Mat will be Silicon and the second argument Sol will be CX.
The argument names Sol and Mat are arbitrary, and they can be any valid Tcl variable but the
first argument is always the material name and the second argument is always the solution
name.
The procedure is called every time the solutions are checked during the diffusion. The
procedure prints the message 'This callback procedure unsets CX equation in
Silicon' and removes the pdb equation if it was defined.
Note that neither the solution name CX nor the material name Silicon is used in the
implementation of the ResetEquations callback procedure. Therefore, the callback
procedure is a generic procedure and can be used for several materials and solutions. This
example can be extended with the following commands:
pdbSetString Silicon CX InitProc ResetEquations
pdbSetString Silicon CY InitProc ResetEquations
pdbSetString Oxide CX InitProc ResetEquations
pdbSetString Oxide CY InitProc ResetEquations
In this case, the same callback procedure, ResetEquations, is used for the solution variables
CX and CY in the materials oxide and silicon. Sentaurus Process will print the following
messages:
This callback procedure unsets CX equation in Oxide.
This callback procedure unsets CX equation in Silicon.
This callback procedure unsets CY equation in Oxide.
This callback procedure unsets CY equation in Silicon.
The advantage of using callback procedures is clear. With four new command lines, the
equations for CX and CY in both oxide and silicon can be unset. At the same time, there is only
pdbSetString Silicon CX InitProc ResetEquations
proc ResetEquations { Mat Sol } {
LogFile "This callback procedure unsets $Sol equation in $Mat."
pdbUnSetString $Mat $Sol Equation
}
6: Alagator Scripting Language
Alagator for Diffusion
588 Sentaurus™ Process User Guide
H-2013.03
one callback procedure to maintain. If you change the callback procedure, the changes will
apply to all four settings.
Equation Procedure: EquationProc
The primary responsibility of the equation procedure is to construct the equation string. It uses
the keyword EquationProc in the parameter database. The keyword determines which
callback procedure name will be called by Sentaurus Process. For example, the first command
below:
defines the SetEquations procedure as the callback procedure of the solution variable CX in
silicon. The callback procedure itself takes two arguments: a material name and a solution
name. In this example, Sentaurus Process will call the SetEquations procedure with two
arguments. The first argument Mat will be Silicon and the second argument Sol will be CX.
The argument names Sol and Mat are arbitrary, and they can be any valid Tcl variable, but the
first argument is always the material name and the second argument is always the solution
name.
The SetEquations procedure is called every time the solutions are checked during the
simulation. In the above example, the procedure will print the message ‘This callback
procedure sets CX equation in Silicon.’ and will set the pdb equation for CX in
silicon.
NOTE Neither the solution name CX nor the material name Silicon is used in
the implementation of the SetEquations callback procedure.
Therefore, the callback procedure is a generic procedure. The equation
setting is similar to the one explained in Basics on page 576. The only
difference is that instead of using a solution name and material name,
only Tcl variables are used.
This example can be extended with the following commands:
pdbSetString Silicon CX EquationProc SetEquations
pdbSetString Silicon CY EquationProc SetEquations
pdbSetString Silicon CX EquationProc SetEquations
proc SetEquations { Mat Sol } {
LogFile "This callback procedure sets $Sol equation in $Mat."
pdbSetString $Mat $Sol Equation "ddt($Sol) -[Arrhenius 0.138 1.37]*grad($Sol)"
}
6: Alagator Scripting Language
Alagator for Diffusion
Sentaurus™ Process User Guide 589
H-2013.03
In this case, the same callback procedure, SetEquations, is used for the solution variables
CX and CY in silicon. Sentaurus Process will print the following messages:
This callback procedure sets CX equation in Silicon.
This callback procedure sets CY equation in Silicon.
The advantage of using callback procedures is clear. With two new command lines, you can set
the diffusion equations for CX and CY in silicon. At the same time, there is only one callback
procedure to maintain. If you change the callback procedure, the changes will apply to both
settings.
The above implementation uses the same diffusivity for both CX and CY. To use different
diffusivities for each solution variable, the callback procedure SetEquations must be
modified and diffusivities for each solution variable should be set as follows:
pdbSetString Silicon CX EquationProc SetEquations
pdbSetString Silicon CY EquationProc SetEquations
pdbSetDouble Silicon CX D {[Arrhenius 0.138 1.37]}
pdbSetDouble Silicon CY D {[Arrhenius 0.02 0.3]}
proc SetEquations { Mat Sol } {
LogFile "This callback procedure sets $Sol equation in $Mat."
set diff [pdbDelayDouble $Mat $Sol D]
pdbSetString $Mat $Sol Equation "ddt($Sol) - $diff * grad($Sol)"
}
The first change above is the setting of CX and CY diffusivities in the database. The second
change is in the SetEquations callback procedure. Instead of having a hard-wired diffusivity
number, pdbDelayDouble is used to obtain the expression stored in the database. Now, the
diffusivities depend on the database entry. You can change these entries to observe the effect
of different diffusivities on the final profile.
The example given in Using Terms on page 580 can be enhanced further by using both terms
and callback procedures as follows:
pdbSetDouble Silicon CX D {[Arrhenius 0.138 1.37]}
pdbSetDouble Silicon CY D {[Arrhenius 0.02 0.3]}
pdbSetDouble Silicon CX Kf {[Arrhenius 4.2e-11 0.1]}
pdbSetDouble Silicon CY Kf {[Arrhenius 4.2e-11 0.1]}
pdbSetDouble Silicon CX Cstar {[Arrhenius 3.6e27 3.7]}
pdbSetDouble Silicon CY Cstar {[Arrhenius 4.0e26 3.97]}
pdbSetString Silicon CX Recomb “CY”
pdbSetString Silicon CY Recomb “CX”
solution add name=CX !damp !negative solve
solution add name=CY !damp !negative solve
6: Alagator Scripting Language
Alagator for Diffusion
590 Sentaurus™ Process User Guide
H-2013.03
pdbSetString Silicon CX EquationProc SetEquations
pdbSetString Silicon CY EquationProc SetEquations
proc SetEquations { Mat Sol } {
LogFile "This callback procedure sets $Sol equation in $Mat."
set diff [pdbDelayDouble $Mat $Sol D]
set Kf [pdbDelayDouble $Mat $Sol Kf]
set Recomb [pdbGetString $Mat $Sol Recomb]
set CXStar [pdbDelayDouble $Mat $Sol Cstar]
set CYStar [pdbDelayDouble $Mat $Recomb Cstar]
term name = RCXCY $Mat eqn = "$Kf * ($Sol * $Recomb- $CXStar * $CYStar)"
pdbSetString $Mat $Sol Equation "ddt($Sol) - $diff * grad($Sol) + RCXCY”
}
First, the diffusivity, equilibrium concentration, and forward reaction rate for the
recombination of CX and CY are stored in the parameter database. Since CX and CY recombine
with each other, this information (Recomb) is also stored in the database. Then, the callback
procedure is modified to read these database entries. The Tcl variable Recomb in the callback
procedure will have the value of CY for CX and CX for CY. The forward recombination rate
RCXCY is the same for both solution variables. The callback procedure will be called once for
CX and once for CY. During each call, the term RCXCY will be created. Since the term name does
not depend on the solution name, the first term created during the CX equation setup will be
deleted during the CY equation setup. This is performed intentionally for this example since
both equations use exactly the same term. If you want to create a unique term for each call, the
callback procedure must be modified as follows:
proc SetEquations { Mat Sol } {
LogFile "This callback procedure sets $Sol equation in $Mat."
set diff [pdbDelayDouble $Mat $Sol D]
set Kf [pdbDelayDouble $Mat $Sol Kf]
set Recomb [pdbGetString $Mat $Sol Recomb]
set CXStar [pdbDelayDouble $Mat $Sol Cstar]
set CYStar [pdbDelayDouble $Mat $Recomb Cstar]
term name = R${Sol}${Recomb} $Mat eqn = "$Kf * ($Sol * $Recomb- $CXStar * \
$CYStar)"
pdbSetString $Mat $Sol Equation "ddt($Sol) - $diff * grad($Sol) + \
R${Sol}${Recomb}"
}
In this case, two terms will be created: RCXCY and RCYCX.
6: Alagator Scripting Language
Alagator for Diffusion
Sentaurus™ Process User Guide 591
H-2013.03
Preprocessing and Postprocessing Data: diffPreProcess,
UserDiffPreProcess, diffPostProcess, UserDiffPostProcess
Sentaurus Process can initialize solution variable fields on the command line using various
commands, such as select (see select on page 1117) and profile (see profile on
page 1093). If the initialization can be standardized, it is better to use the diffPreProcess
callback procedure. By default, diffPreProcess is used to initialize the data fields for
interstitials, vacancies, dopants, dopant clusters, dopant-defect clusters, and defect clusters (see
Ion Implantation to Diffusion on page 353). The procedure also switches on and off point-
defect solutions and various cluster solutions.
You ca n over writ e the pr oc ed ure diffPreProcess. For example:
proc diffPreProcess { } {
LogFile "This procedure is used to initialize data fields CX and CY"
sel z = "CX + 2.0e18 * exp( -(x-0.5)*(x-0.5) / (0.01 * 0.01) ) + 1.0" \
name=CX store
sel z = "CY + CX * 0.1" name=CY store
}
This procedure will create a Gaussian profile for the solution variable CX with a peak at the
depth x = 0.5 and a maximum concentration of . Ten percent of the CX profile will
be added to the existing data field CY.
More complex examples can be created by combining pdb commands and the Tcl callback
procedures, for example:
pdbSetDouble Silicon CY minDose 1e10
proc diffPreProcess { } {
LogFile "This procedure is used to initialize data fields CX and CY"
sel z = "CX + 2.0e18 * exp( -(x-0.5)*(x-0.5) / (0.01 * 0.01) ) + 1.0" \
name=CX store
sel z = CY
set dose [FindDose]
if { $dose > [pdbGetDouble Silicon CY minDose] } {
solution add name=CY !damp !negative solve
} else {
solution add name=CY !damp !negative nosolve
}
}
In this example, a parameter called minDose is created for CY in the database to set the
minimum allowed dose for diffusion to occur. In the callback procedure diffPreProcess,
218
×10 cm 3–
6: Alagator Scripting Language
Alagator for Diffusion
592 Sentaurus™ Process User Guide
H-2013.03
the CX profile is set as previously explained. The second and fourth lines calculate the dose of
CY in silicon. The ‘if-else’ statement retrieves the minimum dose value from the database. If
the existing dose of CY is below the minimum dose, the solution for CY is switched off;
otherwise, it is switched on.
To enforce additional actions to be performed upon diffusion preprocessing, it is not necessary
to overwrite the default implementation of the procedure diffPreProcess. Instead, to
preserve the default initialization of data fields, it is recommended to redefine the procedure
UserDiffPreProcess. By default, UserDiffPreProcess is an empty procedure and is
called from the procedure diffPreProcess as one of the last commands.
The callback procedure diffPostProcess is called at the end of diffusion. By default, it is
used to store the total concentrations of point defects and to delete some temporary data fields
such as Int_Implant and Vac_Implant. In the last command line of diffPostProcess,
the procedure UserDiffPostProcess is called, which is empty by default. If you want to
add commands to be executed after diffusion, it is recommended to redefine the procedure
UserDiffPostProcess.
Complex Initialization Procedures: InitSolve and EquationInitProc
In some cases, the initialization of solution variables can be very complex and cannot be
accomplished by using the select command (see select on page 1117). In these cases,
Sentaurus Process defines the initialization equations using the Alagator language and callback
procedures.
Assume that the CX solution variable is initialized by solving the following equation:
(822)
where is the total concentration of CX, and and are user-defined initialization
parameters. Depending on the value of and , you need to solve Eq. 822.
To initiate the initialization setup procedure, the solution name must be defined as:
solution add name=CX !damp !negative solve InitStep
The keyword InitStep allows this solution variable to be initialized.
CXTotal CX αCX()
β
+=
CXTotal
α
β
α
β

6: Alagator Scripting Language
Alagator for Diffusion
Sentaurus™ Process User Guide 593
H-2013.03
The keyword InitSolve determines which callback procedure name will be called by
Sentaurus Process. For example, the first command below:
defines the ResetInitEquations procedure as the callback procedure of the solution
variable CX in silicon. The callback procedure itself takes two arguments: a material name and
a solution name. In this example, Sentaurus Process will call the ResetInitEquations
procedure with two arguments. The first argument Mat will be Silicon and the second
argument Sol will be CX. In this example, the solution variable CX is marked to be initialized
in silicon. If there is no such setting for the solution variable CY, CY will not be initialized.
After calling the callback procedures defined by InitSolve, Sentaurus Process will look for
an equation string for the solution variable. This is performed by defining a callback procedure
using the keyword EquationInitProc.
In this procedure, the equation string for initialization is constructed. For example, the first
command below:
defines the SetInitEquations procedure as the callback procedure of the solution variable
CX in silicon. The callback procedure itself takes two arguments: a material name and a
solution name. In this example, Sentaurus Process will call the SetInitEquations
procedure with two arguments. The first argument Mat will be Silicon and the second
argument Sol will be CX. The argument names Sol and Mat are arbitrary, and they can be any
valid Tcl variable, but the first argument is always the material name and the second argument
is always the solution name.
The EquationInitProc and EquationProc keywords work in the same way. The callback
procedure defined with the keyword EquationInitProc is called only during initialization.
pdbSetString Silicon CX
InitSolve ResetInitEquations
proc ResetInitEquations { Mat Sol } {
LogFile "This callback procedure unsets $Sol equation in $Mat during initialization."
pdbUnSetString $Mat $Sol Equation
}
pdbSetString Silicon CX
EquationInitProc SetInitEquations
proc SetInitEquations { Mat Sol } {
LogFile "This callback procedure sets $Sol equation in $Mat during initialization."
}
pdbSetString $Mat $Sol Equation "${Sol}_Implant - $Sol - ($alpha * $Sol)^$beta"
set alpha [pdbDelayDouble $Mat $Sol Alpha]
set beta [pdbDelayDouble $Mat $Sol Beta]

6: Alagator Scripting Language
Alagator for Diffusion
594 Sentaurus™ Process User Guide
H-2013.03
The callback procedure defined with the keyword EquationProc is called only during
diffusion. It is assumed that Alpha and Beta are already entered into the database and
CXTotal is defined.
Diffusion Summary: pdb, TclLib, SPROCESS.models
So far, it has been shown how to set up diffusion equations and boundary equations, and how
to initialize solution variables. The complete example can be divided into three major parts.
The first part is the pdb entries as follows:
pdbSetDouble Silicon CX D {[Arrhenius 0.138 1.37]}
pdbSetDouble Silicon CY D {[Arrhenius 0.02 0.3]}
pdbSetDouble Silicon CX Kf {[Arrhenius 4.2e-11 0.1]}
pdbSetDouble Silicon CY Kf {[Arrhenius 4.2e-11 0.1]}
pdbSetDouble Silicon CX Cstar {[Arrhenius 3.6e27 3.7]}
pdbSetDouble Silicon CY Cstar {[Arrhenius 4.0e26 3.97]}
pdbSetString Silicon CX Recomb "CY"
pdbSetString Silicon CY Recomb "CX"
pdbSetDouble Silicon CX Alpha {[Arrhenius 1.03103e-17 -0.4]}
pdbSetDouble Silicon CX Beta {4.0}
This part corresponds to the parameter database (see Parameter Database on page 55). All
default Sentaurus Process model parameters are stored in the parameter database. The second
part is the definition of the names of the solution variables and the names of the callback
procedures:
solution add name=CX !damp !negative solve
solution add name=CY !damp !negative solve
pdbSetString Silicon CX EquationProc SetEquations
pdbSetString Silicon CY EquationProc SetEquations
pdbSetString Silicon CX InitProc ResetEquations
pdbSetString Silicon CY InitProc ResetEquations
pdbSetString Gas_Silicon CY InitProc ResetEquations
pdbSetString Gas_Oxide CX InitProc ResetEquations
pdbSetString Silicon CX InitSolve ResetInitEquations
pdbSetString Silicon CX EquationInitProc SetInitEquations
The information for the default Sentaurus Process models are stored in the TclLib directory
(see Environment Variables on page 47) in the file SPROCESS.models, which is read by
Sentaurus Process as soon as the simulation starts. The file also contains information with
regard to solver types, implant directories, and material names.
6: Alagator Scripting Language
Alagator for Diffusion
Sentaurus™ Process User Guide 595
H-2013.03
The third part is the definition of diffusion and initialization models. The models are stored in
the TclLib directory (see Environment Variables on page 47):
proc UserDiffPreProcess { } {
LogFile "This procedure is used to initialize data fields CX and CY"
sel z = "CX + 2.0e18 * exp( -(x-0.5)*(x-0.5) / (0.01 * 0.01) ) + 1.0" \
name=CXTotal store
sel z = "CY + CXTotal * 0.1" name = CY store
}
proc ResetEquations { Mat Sol } {
LogFile "This callback procedure resets $Sol equation in $Mat."
pdbUnSetString $Mat $Sol Equation
}
proc SetEquations { Mat Sol } {
LogFile "This callback procedure sets $Sol equation in $Mat."
set diff [pdbDelayDouble $Mat $Sol D]
set Kf [pdbDelayDouble $Mat $Sol Kf]
set Recomb [pdbGetString $Mat $Sol Recomb]
set CXStar [pdbDelayDouble $Mat $Sol Cstar]
set CYStar [pdbDelayDouble $Mat $Recomb Cstar]
term name = RCXCY $Mat eqn = "$Kf * ($Sol * $Recomb- $CXStar * $CYStar)"
pdbSetString $Mat $Sol Equation "ddt($Sol) - $diff * grad($Sol) + RCXCY"
}
proc ResetInitEquations { Mat Sol } {
LogFile "This callback procedure unsets $Sol equation in $Mat during \
initialization."
pdbUnSetString $Mat $Sol Equation
}
proc SetInitEquations { Mat Sol } {
LogFile "This callback procedure sets $Sol equation in $Mat during \
initialization."
set alpha [pdbDelayDouble $Mat $Sol Alpha]
set beta [pdbDelayDouble $Mat $Sol Beta]
pdbSetString $Mat $Sol Equation "${Sol}_Implant - $Sol
- (($alpha*$Sol)^$beta)"
}
6: Alagator Scripting Language
Alagator for Generic Growth
596 Sentaurus™ Process User Guide
H-2013.03
Alagator for Generic Growth
The generic growth scheme uses most of the standard definitions used in diffusion. Read
Alagator for Diffusion on page 575 before continuing with this section.
Basics
In this section, examples of varying complexity are used to illustrate how to specify growth
equations using the Alagator scripting language. Most of the definitions are identical to those
of the diffusion equation. However, the keywords used to define the callback procedures and
the definition of solution variables differ. In addition, the reaction equations interact with the
reaction command.
Figure 85 Flux, ambient concentration, and motion of the growth front during the growth
process
Consider a reaction where the ambient GX reacts with silicon and forms a new material called
TEOS. The schematic in Figure 85 shows the ambient concentration at each interface and the
motion of the growth during the process. J represents the fluxes towards the Gas_TEOS and
Silicon_TEOS interfaces, and the flux of GX inside TEOS.
Since TEOS is a new material, first, it must be entered into the existing material list using the
command:
mater add name=TEOS
Then, the reacting materials must be defined using the reaction command (see reaction on
page 1099).
It is possible to create a reaction based on existing ambients, but in this case, a new react type
(named GX) ambient is created:
ambient name=GX react add
reaction name=TEOSreaction mat.l=Silicon mat.r=Gas mat.new=TEOS \
new.like=oxide ambient.name=GX diffusing.species=GX
n
nTEOS
Silicon
Gas
JGas_TEOS JTEOS JSilicon_TEOS
GXGas_TEOS
GXSilicon_TEOS
6: Alagator Scripting Language
Alagator for Generic Growth
Sentaurus™ Process User Guide 597
H-2013.03
Silicon (mat.l) is on the left side of the reacting interface and Gas (mat.r) is the material
on the right side of the reacting interface. The newly formed material (mat.new) at the reacting
interface is TEOS. The new material and its interfaces with other materials also are defined to
be like Oxide and Oxide interfaces (for example, TEOS = Oxide, PolySilicon_TEOS =
Oxide_PolySilicon). For the GX reaction, it requires an ambient, and the ambient name is
GX. The reaction will not occur unless the ambient GX is present in a gas flow or directly in the
diffuse command. (For more information about how to specify ambients, see Ambients and
Gas Flows on page 617.) Reactions that require a react-type ambient cannot have more than
one diffusing species name. If the reaction does not require an ambient, it can have multiple
names of diffusing species. In this case, the reaction occurs if the reacting interfaces exist in
the structure.
The reaction command automatically adds the diffusing species GX to the general solution
list. This is performed internally by using the command:
solution name=GX add !negative GrowthStep solve
where GrowthStep identifies this solution name as a reaction solution name. When the
solution name and the new material are defined, the reaction and diffusion equations are written
as follows:
pdbSetString TEOS GX Equation "ddt(GX)- \[Arrhenius 0.2 1.86\]*grad(GX)"
pdbSetString Gas_TEOS GX Equation_TEOS "-(GX_TEOS - 1e17)"
pdbSetString Silicon_TEOS GX Equation_TEOS "-5e-2*(GX_TEOS)"
pdbSetString Silicon_TEOS GX GrowthReaction " 5e-2*(GX_TEOS)"
The first line sets the diffusion equation of GX in TEOS. The next two lines set the boundary
fluxes at the Gas_TEOS and Silicon_TEOS interfaces. One of the commands works on the
Gas_TEOS interface and the other one works on the Silicon_TEOS interface. The _TEOS
option on Equation indicates the side of the interface to which the given flux will be applied.
The same option on the solution variable GX indicates that the solution variable value at this
interface is taken from the TEOS side. These settings are identical to the ones described in
Setting Boundary Conditions on page 578.
The last line is unique to the generic growth equations. The keyword GrowthReaction is
used to define the growth reaction flux at the reacting interface. In this example, the growth
reaction flux is identical to the diffusion flux at the reacting interface. In addition, the sign of
the growth reaction flux is the opposite of the sign of the diffusion flux.
Finally, it is necessary to specify the ambient in a gas flow (see gas_flow on page 935) and use
it with the diffusion command or specify it directly in the diffusion command in order
for the reaction to occur. For example:
gas_flow name=gxflw partial_pressure = { GX = 1.0 }
diffuse time=100 temp=1000 gas_flow = gxflw
6: Alagator Scripting Language
Alagator for Generic Growth
598 Sentaurus™ Process User Guide
H-2013.03
or:
diffuse time=100 temp=1000 GX
will both switch on reactions involving the ambient GX and will set the partial pressure of GX
to 1. Using the gas_flow command is more flexible in that the partial pressure can be set to
any value (not just 1.0) or the partial pressure can be computed from gas flows and gas
reactions.
Epi Reactions
This is an example of using the reaction command to create a new epitaxial growth mode:
ambient name=MyEpi epi add
# Now, create the new temporary material to be used during epi growth
# the name <Ambient>On<Material> is not necessary, it is just
# the same convention as used internally
mater name=MyEpiOnNitride add
reaction name= MyEpiOnNiReact mat.l=Nitride mat.r=Gas mat.new=MyEpiOnNitride \
ambient.name=MyEpi new.like=PolySilicon mat.final=PolySilicon
In this example, there is an additional parameter mat.final, which is the final name of the
epi material. There is a conversion from mat.new to mat.final at the end of the diffuse
command. For details on how to set up epi reactions and growth rates, see Epitaxy Growth
Rate: GrowthRateProc on page 605.
The model can be enhanced by adding the model parameters to the PDB. This allows other
users to change the values in the equations by accessing the properties directly. The following
changes make the equations dependent on the stored value in the database:
pdbSetDouble TEOS GX Dstar "\[Arrhenius 0.2 1.86\]"
pdbSetDouble Gas_TEOS GX Cstar "1e17"
pdbSetDouble Silicon_TEOS GX Kfd "5e-2"
pdbSetDouble Silicon_TEOS GX Kfg "5e-2"
set diff [pdbDelayDouble TEOS GX Dstar]
pdbSetString TEOS GX Equation "ddt(GX)- $diff * grad(GX)"
set GXStar [pdbDelayDouble Gas_TEOS GX Cstar]
pdbSetString Gas_TEOS GX Equation_TEOS "-(GX_TEOS - $GXStar)"
set GKfd [pdbDelayDouble Silicon_TEOS GX Kfd]
pdbSetString Silicon_TEOS GX Equation_TEOS "-$GKfd*(GX_TEOS)"
set GKfg [pdbDelayDouble Silicon_TEOS GX Kfg]
pdbSetString Silicon_TEOS GX GrowthReaction " $GKfg*(GX_TEOS)"
6: Alagator Scripting Language
Alagator for Generic Growth
Sentaurus™ Process User Guide 599
H-2013.03
The first lines set the diffusivity, equilibrium value of GX, and forward reaction rates for the
diffusion and growth fluxes in the database. This can be made permanent by directly editing
the hierarchy files. The pdbDelayDouble command is used to return the expression stored in
the database. This is necessary so that the evaluation of the expression does not occur until the
diffuse command is executed. Now, the equation depends on the database entry.
It is possible to use terms with the generic growth equations. For example:
pdbSetDouble TEOS GX Dstar "\[Arrhenius 0.2 1.86\]"
pdbSetDouble Gas_TEOS GX Cstar "1e17"
pdbSetDouble Silicon_TEOS GX Kfd "5e-2"
pdbSetDouble Silicon_TEOS GX Kfg "5e-2"
set diff [pdbDelayDouble TEOS GX Dstar]
pdbSetString TEOS GX Equation "ddt(GX)- $diff * grad(GX)"
set GXStar [pdbDelayDouble Gas_TEOS GX Cstar]
pdbSetString Gas_TEOS GX Equation_TEOS "-(GX_TEOS - $GXStar)"
term name = Reaction TEOS eqn = "GX"
set GKfd [pdbDelayDouble Silicon_TEOS GX Kfd]
pdbSetString Silicon_TEOS GX Equation_TEOS "-$GKfd*(Reaction_TEOS)"
set GKfg [pdbDelayDouble Silicon_TEOS GX Kfg]
pdbSetString Silicon_TEOS GX GrowthReaction "$GKfg*(Reaction_TEOS)"
a term called Reaction is created in TEOS. Note that the term also takes the _TEOS option to
indicate that the value of Reaction will be taken from the TEOS side.
The velocities regarding the growth reaction flux are calculated internally as follows:
(823)
where is the growth reaction flux defined using the pdbSetString command and
GrowthReaction keyword as previously explained. Beta is the stoichiometry of the growing
material, Expansion.Ratio is the conversion ratio from consumed material to the growing
material, and Density.Grow is the density of the growing material. The default values for
Beta, Expansion.Ratio, and Density.Grow are 1, 2.2, and , respectively. They
can be changed by using the following commands:
pdbSetDouble <interface material> <ambient> Beta <n>
pdbSetDouble <interface material> <ambient> Expansion.Ratio <n>
pdbSetDouble <interface material> <ambient> Density.Grow <n>
υGrowth
Beta
Expansion.Ratio * Density.Grow
---------------------------------------------------------------------------------FGrowth
=
FGrowth
2.2 22
×10

6: Alagator Scripting Language
Alagator for Generic Growth
600 Sentaurus™ Process User Guide
H-2013.03
For example, for the above example, you can change these values with the following
commands:
pdbSetDouble Silicon_TEOS GX Beta 1.1
pdbSetDouble Silicon_TEOS GX Expansion.Ratio 2.0
pdbSetDouble Silicon_TEOS GX Density.Grow 3e22
If Expansion.Ratio is set to 0, the material will dissolve but the new material will not form.
(For example, silicon will dissolve but no TEOS will form. This is useful for silicidation.)
Callback Procedures
Callbacks allow additional ‘intelligence’ to be built into the equations by allowing procedures
to be called at run-time. These procedures build the equation strings according to user-specified
options. By selecting model switches, the user can choose between different physical models
to be represented in the equation strings. Having callback procedures that use a material name
and a solution name as arguments, the same type of equation can be built for several materials,
dopants, and defect species. In Sentaurus Process, all frequently used equations are built-in
callback procedures.
There are six callback procedure–related keywords in Alagator: InitProc, InitSolve,
EquationInitProc, EquationProc, InitGrowth, and EquationGrowthProc.
InitProc, EquationInitProc, InitSolve, and EquationProc are used to define
generic diffusion equations (see Alagator for Diffusion on page 575). In this section, the
procedures specified by the keywords InitGrowth and EquationGrowthProc are
explained:
■InitGrowth provides the Tcl callback procedure name that is usually used to reset or
delete existing pdb equations or terms at the beginning of a diffusion simulation.
■EquationGrowthProc provides the Tcl callback procedure name that sets up equations
for material growth.
The Tcl callback procedures are called at various stages during the execution of the diffuse
command. In addition to the callback procedures, Sentaurus Process calls the
diffPreProcess Tcl procedure before executing the diffuse command, and the
diffPostProcess Tcl procedure after executing the diffuse command (see Ion
Implantation to Diffusion on page 353).
Figure 86 on page 601 shows the flowchart of the execution of a diffuse statement by
Sentaurus Process, including generic material growth. The sections regarding diffusion are
represented on a smaller scale and are shown in Figure 84 on page 584.
6: Alagator Scripting Language
Alagator for Generic Growth
Sentaurus™ Process User Guide 601
H-2013.03
Figure 86 Flowchart with calls to the callback procedures during execution of diffuse
command
growthPreProcess
diffuse
For each: reactant solution and material
InitGrowth callback procedure
Next
For each: reactant solution and material
EquationGrowthProc callback procedure
Solve generic
diffPostProcess
Next
growth equations
For each: solution and material
InitSolve callback procedure:
Next
For each: marked solution and material
EquationInitProc callback procedure
Solve initialization
InitPostProcess
For each: solution and material
EquationProc callback procedure
Solve diffusion
InitProc callback procedure
For each: solution and material
Next
equations
Next
Next
equations
Diffusion process
diffPreProcess
6: Alagator Scripting Language
Alagator for Generic Growth
602 Sentaurus™ Process User Guide
H-2013.03
Setup Procedure: InitGrowth
The procedures specified by the keyword InitGrowth are used to clean up the equation
strings and terms. The keyword determines the callback procedure name to be called by
Sentaurus Process. For example, the first command below:
defines the ResetEquations procedure as the callback procedure of the solution variable GX
in silicon. The callback procedure itself takes two arguments: a material name and a solution
name. In this example, Sentaurus Process will call the ResetEquations procedure with two
arguments. The first argument Mat will be TEOS and the second argument Sol will be GX. The
argument names Sol and Mat are arbitrary, and they can be any valid Tcl variable, but the first
argument is always the material name and the second argument is always the solution name.
The above procedure ResetEquations will print the message ‘This callback
procedure unsets GX equation in TEOS.’ and will remove the pdb equation for GX
in TEOS if it was defined. Note that neither the solution name GX nor the material name TEOS
is used in the ResetEquations callback procedure. Therefore, the callback procedure is a
generic procedure. The example can be extended with the following commands:
pdbSetString TEOS GX InitGrowth ResetEquations
pdbSetString Gas_TEOS GX InitGrowth ResetEquations
pdbSetString Silicon_TEOS GX InitGrowth ResetEquations
In this case, the same callback procedure, ResetEquations, is used for the solution variables
GX inside TEOS, and at the Gas_TEOS and Silicon_TEOS interfaces. Sentaurus Process will
print the following messages:
This callback procedure resets GX equation in TEOS.
This callback procedure resets GX equation in Gas_TEOS.
This callback procedure resets GX equation in Silicon_TEOS.
Since the interface equations can be written for the interface material itself or the neighboring
materials, all of them must be unset including the growth reaction equation. In this case, you
can write a special Tcl procedure for the interfaces as follows:
proc ResetInterfaceEquations {Mat Sol} {
set mater1 [FirstMat $Mat]
set mater2 [SecondMat $Mat]
pdbSetString TEOS GX InitGrowth ResetEquations
proc ResetEquations { Mat Sol } {
LogFile "This callback procedure unsets $Sol equation in $Mat."
}
pdbUnSetString $Mat $Sol Equation
6: Alagator Scripting Language
Alagator for Generic Growth
Sentaurus™ Process User Guide 603
H-2013.03
pdbUnSetString $Mat $Sol Equation
LogFile "This callback procedure resets $Sol equation in $Mat."
pdbUnSetString $Mat $Sol Equation_$mater1
LogFile "This callback procedure resets $Sol equation in $Mat on $mater1 \
side."
pdbUnSetString $Mat $Sol Equation_$mater2
LogFile "This callback procedure resets $Sol equation in $Mat on $mater2 \
side."
pdbUnSetString $Mat $Sol GrowthReaction
LogFile "This callback procedure resets $Sol growth reaction equation in \
$Mat."
}
pdbSetString Gas_TEOS GX InitGrowth ResetInterfaceEquations
pdbSetString Silicon_TEOS GX InitGrowth ResetInterfaceEquations
The callback procedure, ResetInterfaceEquations, is similar to the ResetEquations
procedure. The commands FirstMat and SecondMat used in the procedure return the names
of the neighboring materials (for example, Gas and TEOS for Gas_TEOS). Then, the equations
set for either side of the interface or the interface are unset including the generic growth
reaction equation. This special procedure is called for GX at the Gas_TEOS and
Silicon_TEOS interfaces. Sentaurus Process will print the following messages:
This callback procedure resets GX equation in TEOS.
This callback procedure resets GX equation in Gas_TEOS.
This callback procedure resets GX equation in Gas_TEOS on Gas side.
This callback procedure resets GX equation in Gas_TEOS on TEOS side.
This callback procedure resets GX growth reaction equation in Gas_TEOS.
This callback procedure resets GX equation in Silicon_TEOS.
This callback procedure resets GX equation in Silicon_TEOS on Silicon side.
This callback procedure resets GX equation in Silicon_TEOS on TEOS side.
This callback procedure resets GX growth reaction equation in Silicon_TEOS.
Equation Procedure: EquationGrowthProc
The primary responsibility of the equation procedure is to construct the equation string for
material growth reaction. It uses the keyword EquationGrowthProc in the parameter
database. The keyword specifies the name of the callback procedure to be called by Sentaurus
Process.
6: Alagator Scripting Language
Alagator for Generic Growth
604 Sentaurus™ Process User Guide
H-2013.03
For example, the first command below:
has the effect that, according to the flowchart presented in Figure 86 on page 601, Sentaurus
Process calls the procedure SetEquations with the arguments TEOS and GX.
The SetEquations procedure is called every time the solutions are checked during the
reaction. The procedure above will print the message ‘This callback procedure sets
GX equation in TEOS.’ and will set the pdb equation for GX in TEOS. Note that neither the
solution name GX nor the material name TEOS is used in the SetEquations callback
procedure. Therefore, the callback procedure is a generic procedure. The equation setting is
similar to the one explained in Basics on page 576. The only difference is that instead of using
a solution name and a material name, only Tcl variables are used. Since the interface equations
settings are different, this example can be extended with the following commands:
pdbSetString Gas_TEOS GX EquationProc SetInterfaceEquations
pdbSetString Silicon_TEOS GX EquationProc SetInterfaceEquations
proc SetInterfaceEquations { Mat Sol } {
set mater1 [FirstMat $Mat]
set mater2 [SecondMat $Mat]
if { [pdbIsAvailable $Mat $Sol Cstar] } {
set GXStar [pdbDelayDouble $Mat $Sol Cstar]
pdbSetString $Mat $Sol Equation_$mater2 "-(${Sol}_$mater2 - $GXStar)"
LogFile "This callback procedure sets $Sol equation in $Mat on $mater2 \
side."
} else {
set GKfd [pdbDelayDouble $Mat $Sol Kfd]
pdbSetString $Mat $Sol Equation_$mater2 "-$GKfd*(${Sol}_$mater2)"
LogFile "This callback procedure sets $Sol equation in $Mat on $mater2 \
side."
set GKfg [pdbDelayDouble $Mat $Sol Kfg]
pdbSetString $Mat $Sol GrowthReaction " $GKfg*(${Sol}_$mater2)"
LogFile "This callback procedure sets $Sol growth reaction equation in \
$Mat."
}
}
pdbSetString TEOS GX EquationGrowthProc SetEquations
proc SetEquations { Mat Sol } {
LogFile "This callback procedure sets $Sol equation in $Mat."
}
pdbSetString $Mat $Sol Equation "ddt($Sol) -$diff * grad($Sol)"
set diff [pdbDelayDouble $Mat $Sol Dstar]
6: Alagator Scripting Language
Alagator for Generic Growth
Sentaurus™ Process User Guide 605
H-2013.03
Here, the same callback procedure, SetInterfaceEquations, is used to set up the interface
equations on both the Gas_TEOS and Silicon_TEOS interfaces. The commands FirstMat
and SecondMat used in the procedure return the names of the neighboring materials (for
example, Gas and TEOS for Gas_TEOS). The command pdbIsAvailable returns true (1) if
the Cstar value is entered to the parameter database for the given solution name and material.
In this example, it will return true for the Gas_TEOS interface and false for the Silicon_TEOS
interface. Using this information and the ‘if-else’ statement, the equations can be set for the
Gas_TEOS and Silicon_TEOS interfaces. Now, the diffusivities, reaction rates, and
equilibrium values depend on the database entries.
Epitaxy Growth Rate: GrowthRateProc
The GrowthRateProc callback procedure can be used to set the GrowthReaction pdb
variable during epitaxial growth. The requirements are similar to EquationGrowthProc,
except that there can be a different GrowthRateProc for each epitaxial ambient. For example:
proc mygrproc { Mat Amb } {
pdbSetString $Mat $Amb GrowthReaction \
"([simDelayDouble Diffuse EpiThick]- \
[pdbDelayDouble $Mat $Amb NativeOffset])/ \
[simDelayDouble Diffuse AnnealStepTime]"
}
pdbSet Gas_LTEOnOxide LTE GrowthRateProc mygrproc
This example demonstrates a number of simulation status values that are available to the
GrowthRateProc implementer. The quantity simDelayDouble Diffuse EpiThick is
the value of the thick parameter set in the diffuse or temp_ramp commands. The quantity
pdbDelayDouble $Mat $Amb NativeOffset is set to the native layer thickness if a native
layer was deposited; otherwise, it is 0. Finally, simDelayDouble Diffuse
AnnealStepTime is the total time of the current temp_ramp segment or diffuse time.
If a new material is being deposited that is not a standard Sentaurus Process epi material, the
following parameters should be set:
pdbSetDouble <mat.new>_Gas <ambient.name> Expansion.Ratio 1.0
pdbSetDouble <mat.new>_Gas <ambient.name> Density.Grow 1.0
where <mat.new> is the name of the new material being grown and <ambient.name> is the
name of the ambient triggering the growth of <mat.new>.
The growthPreProcess Procedure
The execution of every diffuse command starts with a call to the growthPreProcess Tcl
procedure. The growthPreProcess Tcl procedure is used to initialize various reaction-
related data fields or to preprocess the existing data fields.

6: Alagator Scripting Language
Alagator for Generic Growth
606 Sentaurus™ Process User Guide
H-2013.03
To enforce additional actions to be performed upon generic growth preprocessing, it is not
necessary to overwrite the default implementation of the procedure growthPreProcess.
Instead, to preserve the default initialization of data fields, it is recommended to redefine the
procedure UserGrowthPreProcess. By default, UserGrowthPreProcess is an empty
procedure and is called from the procedure growthPreProcess as one of the last commands.
Generic Growth Summary: pdb, TclLib,
SPROCESS.models
So far, it has been shown how to set up reaction equations and fluxes. The complete example
can be divided into three major parts. The first part consists of pdb entries as follows:
pdbSetDouble TEOS GX Dstar "\[Arrhenius 0.2 1.86\]"
pdbSetDouble Gas_TEOS GX Cstar "1e17"
pdbSetDouble Silicon_TEOS GX Kfd "5e-2"
pdbSetDouble Silicon_TEOS GX Kfg "5e-2"
This part corresponds to the parameter database (see Parameter Database on page 55). All
default Sentaurus Process model parameters are stored in the parameter database. The second
part is the definition of reaction variable names, material names, and callback procedure
names:
mater add name=TEOS
reaction name=TEOSreaction mat.l=Silicon mat.r=Gas mat.new=TEOS \
new.like=oxide diffusing.species=GX ambient
pdbSetString TEOS GX InitGrowth ResetEquations
pdbSetString Gas_TEOS GX InitGrowth ResetInterfaceEquations
pdbSetString Silicon_TEOS GX InitGrowth ResetInterfaceEquations
pdbSetString Gas_TEOS GX EquationGrowthProc SetInterfaceEquations
pdbSetString Silicon_TEOS GX EquationGrowthProc SetInterfaceEquations
pdbSetString TEOS GX EquationGrowthProc SetEquations
The information for the default Sentaurus Process models is stored in the TclLib directory
(see Environment Variables on page 47) in the SPROCESS.models file, which is read by
Sentaurus Process as soon as the simulation starts. The file also contains information regarding
solver types, implant directories, and material names.
The third part is the definition of diffusion/reaction models. The models are stored in the
TclLib directory (see Environment Variables on page 47):
proc ResetEquations {Mat Sol} {
LogFile "This callback procedure resets $Sol equation in $Mat."
pdbUnSetString $Mat $Sol Equation
}
6: Alagator Scripting Language
Alagator for Generic Growth
Sentaurus™ Process User Guide 607
H-2013.03
proc ResetInterfaceEquations { Mat Sol } {
set mater1 [FirstMat $Mat]
set mater2 [SecondMat $Mat]
pdbUnSetString $Mat $Sol Equation
LogFile "This callback procedure resets $Sol equation in $Mat."
pdbUnSetString $Mat $Sol Equation_$mater1
LogFile "This callback procedure resets $Sol equation in $Mat on $mater1 \
side."
pdbUnSetString $Mat $Sol Equation_$mater2
LogFile "This callback procedure resets $Sol equation in $Mat on $mater2 \
side."
pdbUnSetString $Mat $Sol GrowthReaction
LogFile "This callback procedure resets $Sol growth reaction equation in \
$Mat."
}
proc SetEquations { Mat Sol } {
LogFile "This callback procedure sets $Sol equation in $Mat."
set diff [pdbDelayDouble $Mat $Sol Dstar]
pdbSetString $Mat $Sol Equation "ddt($Sol) - $diff * grad($Sol)"
}
proc SetInterfaceEquations { Mat Sol } {
set mater1 [FirstMat $Mat]
set mater2 [SecondMat $Mat]
if { [pdbIsAvailable $Mat $Sol Cstar] } {
set GXStar [pdbDelayDouble $Mat GX Cstar]
pdbSetString $Mat $Sol Equation_$mater2 "-(${Sol}_$mater2 - $GXStar)"
LogFile "This callback procedure sets $Sol equation in $Mat on $mater2 \
side."
} else {
set GKfd [pdbDelayDouble $Mat $Sol Kfd]
pdbSetString $Mat $Sol Equation_$mater2 "-$GKfd*(${Sol}_$mater2)"
LogFile "This callback procedure sets $Sol equation in $Mat on $mater2 \
side."
set GKfg [pdbDelayDouble $Mat $Sol Kfg]
pdbSetString $Mat $Sol GrowthReaction " $GKfg*(${Sol}_$mater2)"
LogFile "This callback procedure sets $Sol growth reaction equation in \
$Mat."
}
}

6: Alagator Scripting Language
Modifying Diffusion Models
608 Sentaurus™ Process User Guide
H-2013.03
Modifying Diffusion Models
In Sentaurus Process, all equations of dopant diffusion models are constructed using callback
procedures as previously explained. If you want to add a new expression to an existing equation
or term, or to subtract a new expression from an existing equation or term, one of the following
can be used:
■UserAddEqnTerm
■UserSubEqnTerm
■UserAddToTerm
■UserSubFromTerm
■MultiplyTerm
NOTE The UserAddEqnTerm, UserSubEqnTerm, UserAddToTerm,
UserSubFromTerm, and MultiplyTerm commands should not be
used within the callback procedures. They are designed to change
diffusion equations without using the callback procedures.
NOTE The UserAddEqnTerm, UserSubEqnTerm, UserAddToTerm,
UserSubFromTerm, and MultiplyTerm are saved to the TDR files.
If the input file is split, the commands must not be included in the new
input file. However a user defined variable or term used with these
command, the variable or term must be included in the new input file or
the variable must be saved with define command (see define on
page 897) and the term must be stored with the store parameter (see
term on page 1173).
UserAddEqnTerm and UserSubEqnTerm
The commands UserAddEqnTerm and UserSubEqnTerm allow you to add a new expression
to an existing solution variable equation or to subtract the new expression from an existing
solution variable equation. The commands have the format:
UserAddEqnTerm <material> <solution> <expression> <side>
UserSubEqnTerm <material> <solution> <expression> <side>
where:
■<material> is any valid material name.
■<solution> is any valid solution variable name.
■<expression> is the new expression to be added to or subtracted from the solution
variable.

6: Alagator Scripting Language
Modifying Diffusion Models
Sentaurus™ Process User Guide 609
H-2013.03
■<side> is the side of the interface material where the new expression will be added or
subtracted.
For example, the command:
UserAddEqnTerm Silicon CY "{2e-15*(CY*CY-1e-16*CX)}"
adds the expression "{2e-15*(CY*CY-1e-16*CX)}" to the Vac equation in silicon during
the PDE solve. Since the equations can be set in three different ways for interfaces, you have
the option to specify the side to which the expression will be added or subtracted. For example,
the commands:
UserAddEqnTerm Oxide_Silicon CX "(CX_Oxide - CX_Silicon)" Silicon
UserSubEqnTerm Oxide_Silicon CX "(CX_Oxide - CX_Silicon)" Oxide
add the expression "(CX_Oxide - CX_Silicon)" to the Oxide_Silicon interface
equation for CX on the Silicon side, and subtract the same expression from the
Oxide_Silicon interface equation for CX on the Oxide side, respectively. If no side
information is given, the expression will be added to the Oxide_Silicon interface equation
for CX.
UserAddToTerm and UserSubFromTerm
The commands UserAddToTerm and UserSubFromTerm are used to add a new expression
to an existing term or to subtract the new expression from an existing term. The commands
have the format:
UserAddToTerm <material> <term> <expression>
UserSubFromTerm <material> <term> <expression>
where:
■<material> is any valid material name.
■<term> is an existing term name.
■<expression> is the new expression to be added to or subtracted from the existing term.
For example the command:
UserAddToTerm Silicon
VTotal
"2*CX"
adds the expression "2*CX" to the term VTotal in silicon.
In the same way, the command:
UserSubFromTerm Silicon
VTotal
"2*CX"

6: Alagator Scripting Language
References
610 Sentaurus™ Process User Guide
H-2013.03
subtracts the expression "2*CX" from the term VTotal in silicon.
References
[1] R. E. Bank et al., “Transient Simulation of Silicon Devices and Circuits,” IEEE
Transactions on Electron Devices, vol. ED-32, no. 10, pp. 1992–2007, 1985.

Sentaurus™ Process User Guide 611
H-2013.03
CHAPTER 7 Advanced Calibration
This chapter provides details about the use of Advanced Calibration
in Sentaurus Process.
Overview
Synopsys’ Consulting and Engineering is working continually on improving the simulation
models and optimizing the model parameters for the latest technology. This effort is based on
long-standing experience of model calibration for customers and a comprehensive, growing
database of state-of-the-art secondary ion mass spectrometry (SIMS) profiles.
With Advanced Calibration in Sentaurus Process, you have a set of models and parameters that
have been calibrated to a broad range of technologies, from power devices to advanced CMOS.
With these parameters, you can obtain accurate results for many processes in device fabrication
such as ion implantation, dopant diffusion and activation, ultrashallow junction formation, and
surface dose loss.
The Advanced Calibration set of models and parameters is located in a single file. For the
current Sentaurus Process, it has the file name AdvCal_2013.12.fps and is located in the
directory $STROOT/tcad/$STRELEASE/lib/sprocess/TclLib/AdvCal.
The models and parameters of AdvCal_2013.12.fps are recommended for all technologies
for silicon, SiGe, and germanium substrates. The models have been tested extensively in 1D
and 2D simulations, and have proven to be accurate and robust.
Advanced Calibration also is available for Sentaurus Process Kinetic Monte Carlo (see
Advanced Calibration for Sentaurus Process KMC on page 565).
Using Advanced Calibration
Advanced Calibration is the recommended starting point for process simulation of all silicon-
based and germanium-based devices.To use Advanced Calibration in Sentaurus Process, at the
beginning of the input file, insert the line:
AdvancedCalibration

7: Advanced Calibration
Additional Calibration by Users
612 Sentaurus™ Process User Guide
H-2013.03
or:
AdvancedCalibration 2013.12
This will source the file $STROOT/tcad/$STRELEASE/lib/sprocess/TclLib/AdvCal/
AdvCal_2013.12.fps.
You have t he o pt ion t o use A dva nce d Ca lib ra tio n pa ram et ers a nd mo de ls f rom p revious
releases, for example:
AdvancedCalibration 2013.03
This will source the file $STROOT/tcad/$STRELEASE/lib/sprocess/TclLib/AdvCal/
AdvCal_2013.03.fps.
Additional Calibration by Users
Advanced Calibration is based on the assumption that all parameters that are not changed in
the parameter files are the Sentaurus Process default parameters. To use the Advanced
Calibration file AdvCal_2013.12.fps, it must be sourced before the real process
description.
You can f ur ther i nc rease th e a ccurac y of a c ertain technology by additional fine-tuning of
physical parameters. This should be performed by experienced users with a good
understanding of the diffusion models and callback procedures of Sentaurus Process.The best
way to perform this is to put all additional calibration in a user calibration file, for example,
user_calibration.fps. This file includes all project-specific changes of physical
parameters or callback procedures with respect to Advanced Calibration.
In the process simulation file, at the beginning of the process simulation, insert the lines:
AdvancedCalibration 2013.12
source ./user_calibration.fps
This method has two distinct advantages:
■There is a clear separation between the process flow, which is contained in the Sentaurus
Process input file, and the selection of physical models and parameters. During calibration
of Sentaurus Process for a specific technology, you can first set up the process flow in the
input file of Sentaurus Process and then improve the accuracy of the process simulation by
making changes only in their parameter file. Conversely, if you want to apply the same
models and parameters to a different process, it is only necessary to change the file
containing the process flow.
7: Advanced Calibration
Additional Calibration by Users
Sentaurus™ Process User Guide 613
H-2013.03
■The Advanced Calibration file is used as a starting point. The user calibration file (for
example, user_calibration.fps) is usually short and clear. You can see all parameter
changes with respect to the original Advanced Calibration at a glance.
NOTE For detailed documentation of the contents and physical models
included in Advanced Calibration as well as a discussion of its accuracy
and limitations, refer to the Advanced Calibration for Process
Simulation User Guide.
To accelerate process simulation for power technologies, use the procedures
AdvancedPowerDeviceMode and AdvancedPowerDeviceModeReset. For details, refer
to the Advanced Calibration for Process Simulation User Guide.
7: Advanced Calibration
Additional Calibration by Users
614 Sentaurus™ Process User Guide
H-2013.03

Sentaurus™ Process User Guide 615
I-2013.12
CHAPTER 8 Oxidation and Silicidation
This chapter describes the oxidation models available in Sentaurus
Process.
Oxidation
Sentaurus Process can simulate the thermal oxidation of silicon. Due to the conversion ratio
from Si to SiO2 being greater than one, new ‘volume’ is generated, which, in turn, leads to the
motion of materials and mechanical stress in the structure. The oxidation process has three
steps:
■Diffusion of oxidants (H2O, O2) from the gas–oxide interface through the existing oxide to
the silicon–oxide interface.
■Reaction of the oxidant with silicon to form new oxide1.
■Motion of materials due to the volume expansion, which is caused by the reaction between
silicon and oxide.
The oxidant diffusion equation is solved using the generic partial differential equation (PDE)
solver of Sentaurus Process. For the simulation of thermal oxidation, there are two
requirements:
■The silicon or polysilicon region is in contact with gas or an oxide region, which, in turn,
is in contact with gas.
■The diffuse command specifies a reactive atmosphere.
If silicon or polysilicon is in contact with gas at the beginning of a thermal oxidation, an initial
oxide layer is created automatically. The default thickness of this layer is 1.5 nm. The value of
the initial oxide thickness is specified in the parameter database by:
pdbSet Grid NativeLayerThickness 1.5e-7
which controls the native layer thickness for oxidation and silicidation. There are several ways
to specify a reactive atmosphere. Furthermore, temperature can vary during oxidation, and the
ambient can contain contributions from different oxidants. The following sections describe
how to handle these cases using Sentaurus Process.
It is important to note that oxidation occurs in conjunction with mechanics. The details of the
mechanical equations, boundary conditions, and material models are given in the next chapter.
1. In this chapter, oxide refers to SiO2.

8: Oxidation and Silicidation
Oxidation
616 Sentaurus™ Process User Guide
I-2013.12
Basic Oxidation
The diffuse command is used to specify two reactive ambients for oxidation, either H2O or
O2. The oxidation temperature and time must be given. For example, a command for a simple
oxidation using wet ambient temperature at for 10 minutes is:
diffuse temperature=1000<C> time=10<min> H2O
A simple temperature ramp can be specified directly in the diffuse command by the keyword
ramprate. This keyword sets the change in the temperature over time:
diffuse temperature=1000<C> time=10<min> O2 ramprate=10<C/min>
This example describes a dry oxidation of 10 minutes, starting at and ending at
.
NOTE The value of ramprate can be negative if the temperature is required
to decrease.
Temperature Cycles
The second example given in Basic Oxidation also can be specified by using the temp_ramp
command, for example:
temp_ramp name=MyTempRamp temperature=1000 time=10 O2 ramprate=10<C/min>
diffuse temp_ramp=MyTempRamp
The first line creates a temperature ramp with given conditions, and the second line specifies a
diffusion referring to this temperature ramp.
To describe more complex temperature cycles within one diffuse command, multiple
instances of the temp_ramp command can be used. A temperature ramp can consist of several
segments and, for each segment, one temp_ramp command is required. In addition, segments
can be grouped by using the same name for each segment. For example, a ramp-up, plateau,
and ramp-down can be specified as:
temp_ramp name=MyCycle temperature=1000<C> time=5<min> H2O ramprate=20<C/min>
temp_ramp name=MyCycle temperature=1100<C> time=10<min> O2
temp_ramp name=MyCycle temperature=1100<C> time=10<min> ramprate=-10<C/min>
diffuse temp_ramp=MyCycle
1000°C
1000°C
1100°C

8: Oxidation and Silicidation
Oxidation
Sentaurus™ Process User Guide 617
I-2013.12
If you want to set the minimum and maximum reaction/oxidation time steps in minutes
globally, for all diffusion commands, the following commands can be used:
pdbSet Diffuse MinGrowthStep <n>
pdbSet Diffuse MaxGrowthStep <n>
See Parameter Database on page 55 for other diffusion-related parameters.
Ambients and Gas Flows
Sentaurus Process has a flexible scheme for dealing with gas flows. By default, several
ambients are available, and you can also create additional ones for new reactions (see reaction
on page 1099). Table 64 lists the ambients that are available by default.
The react and inert ambients can be specified in any combination using the gas_flow
command. The inert ambients are inert in the sense that they do not switch on material
reactions. However, inert ambients can be used in gas flows to change the partial pressure of
react ambients through gas reactions or just taking part of the total pressure as is the case with
N2, for example. As the name implies, react ambients cause material reactions to occur, such
as oxidation. The epi-type ambients trigger epitaxial growth and should not be used with any
other ambient.
To specify an ambient is present and to set the partial pressure to 1.0 * total pressure, use the
shorthand parameter name <ambient name> in the diffuse or gas_flow command. The
parameter pressure sets the total pressure and also is available on the diffuse or gas_flow
command line. The default for total pressure is 1 atm. Only one ambient should be specified
using the shorthand parameter <ambient name>. For epitaxy, specify the appropriate ambient
by name.
Ta bl e 64 Avai l a bl e a mb i en t s
Ambient name Ambient type Reactions
O2 react
Oxidation
H2O react
Oxidation
HCl inert
Gas reactions only
N2 inert
None
H2 inert
Gas reactions only
N2O react
Oxynitridation
Epi epi
Standard epitaxy
LTE epi
Low-temperature epitaxy
8: Oxidation and Silicidation
Oxidation
618 Sentaurus™ Process User Guide
I-2013.12
Specifying Gas Flows
The gas_flow command is used to specify a mixed gas flow by specifying directly either the
partial pressures of the gas components or the flow <volume/time>. When a gas_flow is
specified, it can be referred to from the temp_ramp and diffuse commands.
The gases present during diffusion can either be specified as partial pressures or using gas
flows. When using flow specifications, the partial pressure is computed from gas reactions, the
presence of inert gases, and the total pressure. Alternatively, the partial pressure can be
specified directly. The partial pressure can be set with either the p<ambient name>
parameters or the partial.pressure parameter of the gas_flow command:
gas_flow name=MyGasFlow pH2O=0.5 pO2=0.5
or:
gas_flow name=MyGasFlow partial.pressure = {H2O=0.5 O2=0.5}
Specifying directly in the diffuse command is also possible, for example:
diffuse pH2O=0.5 pO2=0.5 temperature=1000 time=10<min>
Instead of specifying partial pressures directly, the gas components can be given in terms of
flows using flow<ambient name> or the flows parameter, for example:
gas_flow name=MyGasFlow flowH2O=0.5 flowO2=0.5 flowH2=0.2 flowN2=1.0
or:
gas_flow name=MyGasFlow flows = {H2O=0.5 O2=0.5 H2=0.2 N2=1.0}
If flows are specified, Sentaurus Process calculates the partial pressures of the components
assuming a complete reaction of the gases. Because the only effect of inert ambients in
Sentaurus Process is to change the partial pressure of reacting ambients, inert ambients should
only be set using flows in the gas_flow command.
To invoke the gas flow specification as given above, use:
temp_ramp name=MyTempRamp temperature=1000<C> time=10<min> gas_flow=MyGasFlow
diffuse temp_ramp=MyTempRamp
or:
diffuse temperature=1000<C> time=10<min> gas_flow=MyGasFlow

8: Oxidation and Silicidation
Oxidation
Sentaurus™ Process User Guide 619
I-2013.12
Computing Partial Pressures
Given a flow of O2 in addition to a flow of H2 or HCl for example, a chemical reaction between
the components is taken into account: O2 is reduced and H
2O increases. A complete
stoichiometric reaction is assumed. The final flows1 after the reaction are computed in the
AmbientReactions procedure as shown in Eq. 824.
If :
(824)
else:
(825)
In the case where not all of the H2 is consumed by the reaction, a warning is displayed. If a
contribution of HCl is given, the equations read as follows.
If :
(826)
else:
(827)
1. The index init refers to the initial flows specified by users in the gas_flow command, and final describes the
flow after the chemical reaction.
flowO2init 0.5 flowH2init
⋅>
flowO2final flowO2init 0.5 flowH2init
⋅–=
flowH2Ofinal flowH2Oinit flowH2init
+=
flowH2final 0=
flowO2final 0=
flowH2Ofinal flowH2Oinit 2flowO2⋅init
+=
flowH2final flowH2init 2flowO2init
⋅–=
flowO2init flowHClinit
>
flowO2final flowO2init 0.5 flowHCl⋅init
–=
flowH2Ofinal flowH2Oinit flowHClinit
+=
flowH2final 0=
flowO2final 0=
flowH2Ofinal flowH2Oinit 2flowO2⋅init
+=
flowHClfinal flowHClinit flowO2init
–=

8: Oxidation and Silicidation
Oxidation
620 Sentaurus™ Process User Guide
I-2013.12
The final flows are used internally to compute the partial pressure of each component. Partial
pressures are the relevant quantity for the subsequent simulation. These are computed as:
(828)
where holds for a certain component of the gas mixture and pressure is the total
pressure.
In Situ Steam-generated Oxidation
Switching on the in situ steam-generated (ISSG) Boolean parameter in the gas_flow
command specifies that the gas flow condition is to be recognized for the ISSG oxidation (see
In Situ Steam-generated Oxidation on page 632). For example:
gas_flow name=ISSGflow pressure=12<torr> flowH2=6 flowO2=12 ISSG
Oxidant Diffusion and Reaction
For the rigorous simulation of the oxidation process, the dissolution of the oxidant species at
the gas–oxide interface, the transport through the existing or already grown oxide, and the
consumption at the oxide–silicon interface have to be simulated. The dissolution and
consumption are modeled by boundary conditions; for the oxidant transport, a diffusion
equation is solved in the oxide layer.
The oxidant species H2O, O2, and N2O are defined in the SPROCESS.models file (see Default
Simulator Settings: SPROCESS.models File on page 53) using the reaction command (see
reaction on page 1099):
reaction name=dryoxSi mat.l=Silicon mat.r=Gas mat.new=oxide \
diffusing.species=O2 ambient.name=O2
reaction name=wetoxSi mat.l=Silicon mat.r=Gas mat.new=oxide \
diffusing.species=H2O ambient.name=H2O
reaction name=n2ooxSi mat.l=Silicon mat.r=Gas mat.new=oxide
diffusing.species=N2O ambient.name=N2O
For mixed oxidant flows, for each species, one diffusion–reaction system is solved. For each
oxidant, one dataset is allocated: H2O or O2 or N2O [ ].
Growth reaction fluxes at the reacting interfaces are defined using the Alagator scripting
language (see Alagator for Generic Growth on page 596). These fluxes are divided internally
by the particle density of oxide in order to obtain the growth velocities. Manipulation of these
pComp pressure
flowCompfinal
flowCompfinal
Comp
---------------------------------------------
⋅=
Comp
1/cm3

8: Oxidation and Silicidation
Oxidation
Sentaurus™ Process User Guide 621
I-2013.12
fluxes is essential for the implementation of empirical growth models, such as the Massoud
model, which is not yet covered by a diffusion equation.
In the case of a mixed gas flow, the contributions of both fluxes are summed. At the reaction
front, the following reactions are assumed:
(829)
The conversion from Si to SiO2 leads to a volume increase of 125%, which leads to motion and
mechanical stresses in the compound.
The oxidant diffusion described by Fick’s law leads to the diffusion equation:
(830)
where is the diffusivity of the oxidant and is the particle flux. The flux of oxidants in the
normal direction to the surface, going from the gas region to the oxide, is given by:
(831)
where is the mass transfer coefficient and is the solid solubility of the oxidant. If is
sufficiently large, the concentration of oxidant at the gas–oxide interface is approximately
equal to the solid solubility.
The coefficient is defined in the parameter database as MassTransfer and can be set using
the commands:
pdbSet Gas_Oxide O2 MassTransfer <n>
pdbSet Gas_Oxide H2O MassTransfer <n>
pdbSet Gas_Oxide N2O MassTransfer <n>
The solid solubility is a function of the pressure:
(832)
where is the reference solid solubility.
Its value can be set using the following commands:
pdbSet Oxide O2|H2O|N2O CL0 <n>
pdbSet Oxide O2|H2O|N2O CLW <n>
where the symbol | stands for the logical or.
Si O2
+ SiO2
→
Si 2H2O+ SiO22H2
+→
t∂
∂cj∇+0 where jDc∇–=,=
D
j
jhc
*c–()⋅=
h
c*
h
h
c*
c*pComp cref
*
⋅=
cref
*cL0e
cLw
kT
--------
–
=
8: Oxidation and Silicidation
Oxidation
622 Sentaurus™ Process User Guide
I-2013.12
The flux caused by the chemical reaction at the oxidation front is described by:
(833)
The stoichiometry coefficient is 1 for O2 and 2 for H2O, is the chemical reaction rate, and
is the particle density at the oxide–silicon interface. The reaction rate and diffusivity are
computed from the linear and parabolic rate constants used in the Deal–Grove model.
Transition to Linear and Parabolic Rate Constants
Assuming the stationary state in Eq. 830, the growth rate in the 1D case can be described by
the Deal–Grove model:
(834)
where describes the thickness of the 1D oxide layer. This equation can be solved
analytically. The parabolic rate constant is given by and the linear rate constant is given by
B/A. A deeper analysis reveals relations between the parabolic rate and diffusivity, and the
linear rate and reaction rate. Assuming :
(835)
where is the equivalent oxygen concentration in oxide, for example, it is equal to the
concentration of H2O and one half (1/2) the concentration of O2. Both the parabolic rate and
linear rate are functions of pressure and temperature. For the temperature dependency, two
Arrhenius functions, for a low-temperature and high-temperature regime, are available:
(836)
Taking pressure dependence into account, reads:
(837)
The parameters B0.h, BW.h, B0.l, BW.l, Bp.dep, and BT.break can be found in the
parameter database in Oxide H2O |O2 |N2O.
jβkc
si
=
β
k
csi
dxox
dt
-----------B
2xox A+
---------------------=
xox
B
hk»
DβBcox
2c*
---------------=
kcox
c*
------- B
A
---
≈
Cox
BT()
B0.h BW.h
kBT
------------–
, if TBT.break>exp⋅
B0.l BW.l
kBT
------------–
, elseexp⋅
=
B
BBpT,()BT()pComp
Bp.dep/[bar]
==
8: Oxidation and Silicidation
Oxidation
Sentaurus™ Process User Guide 623
I-2013.12
An equivalent set of equations is solved for the linear rate B/A:
(838)
with the corresponding set of parameters, BA0.h, BAW.h, BA0.l, BAW.l, BAp.dep, and
BAT.break. These can be found in the parameter database:
Oxide_Silicon H2O | O2 |N2O 100 | 110 | 100
Oxide_PolySilicon H2O | O2 |N2O 100 | 110 | 100
and can be set using the pdbSet command (for example, pdbSet O2 110 BA0.h <n> or
pdbSet H2O 100 BAT.break <n>).
Parameters defining the diffusivity and parabolic rate constant are bulk properties and,
therefore, are defined in oxide. Parameters defining the reaction rate and linear rate constants
are interface properties and, therefore, are defined on interfaces. This data can also depend on
the crystal orientation when crystalline materials are involved.
Massoud Model
The Massoud model is an empirical model that describes an enhanced growth rate in the initial
regime of the oxidation. The model can be seen as an extension of the Deal–Grove model and
is in good agreement with measurement. Sentaurus Process uses a slightly different form of the
originally suggested model:
(839)
is the one-dimensional unmasked oxide thickness. To account for the enhanced growth in
the initial regime, the second term of Eq. 839 contributes to the flux (compare with Eq. 834).
Both the parameters and depend on the crystal orientation and temperature:
(840)
B
A
--- T()
BA0.h BAW.h
kBT
----------------–
, if TBAT.break>exp⋅
BA0.l BAW.l
kBT
----------------–
, elseexp⋅
=
dxox
dt
-----------B
2xox A+
---------------------Cxox
L
-------–
exp+=
xox
L
C
CT()
C0.h CW.h
kBT
------------–
, if TMBAT.break>exp⋅
C0.l CW.l
kBT
------------–
, elseexp⋅
=
8: Oxidation and Silicidation
Oxidation
624 Sentaurus™ Process User Guide
I-2013.12
(841)
The parameters L0.h, LW.h, L0.l, LW.l, C0.h, CW.h, C0.l, CW.1, and MBAT.break can be
found in the parameter database:
Oxide_Silicon O2 | H2O | N2O 100 | 110 | 111
Oxide_PolySilicon O2 | H2O | N2O 100 | 110 | 111
Orientation-dependent Oxidation
For different crystal orientations, different reaction rates can be applied. Internally, Sentaurus
Process computes the data fields Ori100, Ori110, and Ori111. If the normal vector on an
interface coincides with a certain crystal orientation, the value for this orientation is one; if it
is orthogonal, the value equals zero. Interpolation is used to compute the rates on orientations
not coinciding with the crystallographic directions.
When saving results in TDR format, these data fields are not stored; however, they can be
accessed by using the Alagator scripting language. The Tcl procedure proc
OxidantReaction creates the terms ReactionRateO2 and ReactionRateH2O:
(842)
The reaction rates , , and are computed from the linear rates B/A given for
different orientations. The parameters L0 and C used in the Massoud model depend on the
crystal orientation as well.
For information about the TDR format, refer to the Sentaurus™ Data Explorer User Guide.
Stress-dependent Oxidation
Stress-dependent oxidation (SDO) usually refers to the coupling of the oxidant diffusivity and
reaction rate to the local stress field. To handle the stress-dependent oxidant diffusion and
stress-dependent reaction rate, two data fields are created internally. The data field Pressure
is stored by default, while NStress is not; however, both can be accessed by using the
Alagator scripting language.
The data fields Pressure and NStress are defined as:
(843)
LT()
L0.h LW.h
kBT
-------------–
, if TMBAT.break>exp⋅
L0.l LW.l
kBT
-------------–
, elseexp⋅
=
kk
<100>Ori100 k<110>Ori110 k<111>Ori111++=
k<100>
k<110>
k<111>
Pressure 1
3
---– σxx σyy σzz
++()=
8: Oxidation and Silicidation
Oxidation
Sentaurus™ Process User Guide 625
I-2013.12
and:
(844)
The components of the stress tensor are given by and the normal vector at the reaction front
is given by . The definition of NStress is only meaningful at an interface. If:
pdbSetBoolean Oxide Oxidant SDO 1
or:
pdbSet Oxide_PolySilicon H2O | O2 | N2O SDO 1
pdbSet Oxide_Silicon H2O | O2 | N2O SDO 1
is selected, the reaction rate and diffusivity are modified in the following way:
(845)
and:
(846)
The activation volume VD, being a bulk property, is defined in Oxide O2 |H2O |N2O. The
activation volume Vk controls the impact of the normal stress at the reaction front and,
therefore, is defined on interfaces:
Oxide_Silicon | Oxide_PolySilicon O2 | H2O | N2O
For example:
pdbSet Oxide_Silicon O2 Vk <n>
To fully enable stress-dependent reaction rate, use the command:
pdbSet Diffuse SDReactionRate 1
is the maximum stress factor and is used to cap the exponential parts. is defined in
Oxide O2 |H2O |N2O as MaxStressFactor.
For example:
pdbSet Oxide O2 MaxStressFactor <n>
NStress σjknjnk
k
j
–=
σjk
nj
kNStress T,()kT()min Smax e
NStress Vk⋅
kBT
-----------------------------------------–
,
⋅=
DPressure T,()DT()min Smax e
Pressure VD⋅
kBT
---------------------------------------------–
,
⋅=
Smax
Smax

8: Oxidation and Silicidation
Oxidation
626 Sentaurus™ Process User Guide
I-2013.12
For improved numerical stability, the exponential part can be approximated by a reciprocal
function for a small exponent and a linear function for a large exponent. This option is switched
off by default and can be switched on with the following command:
pdbSet Mechanics TS4CappedExp 1
and replaces the maximum stress factor used to cap the exponential part.
Trap-dependent Oxidation
Impurities such as nitrogen and fluorine can be trapped at Oxide_Silicon interfaces during
oxidation. This will reduce the number of oxidizing sites; therefore, the oxidation rate is
reduced. To switch on the model, use the command:
pdbSet <interface material> O2 | H2O | N2O TrapDependent 1 | 0
The list of trapped impurities is given with the command:
pdbSet <interface material> O2 | H2O | N2O TrapList { Trapped impurity list }
For example, the following command switches on the trapping flux for nitrogen and fluorine:
pdbSet Oxide_Silicon O2 TrapList {Nitrogen Fluorine}
Two models are available for the trapping flux of impurities: Trap and TrapGen.
Trap Model
The trapping flux of impurities is described with the interface Trap model by ignoring the
detrapping flux. The total impurity flux at interfaces is the sum of the trapping flux into
interfaces and the two-phase segregation. This can be achieved by setting the boundary
condition to Trap (see Boundary Conditions on page 357). For example:
pdbSet Oxide_Silicon Nitrogen BoundaryCondition Trap
Since the surface reaction rate is proportional to the number of available oxidizing sites, the
rate of oxidant consumption at the oxidizing interface is given by:
(847)
where:
■ is the surface recombination rate.
■ is the oxidant concentration at the interface.
■ and are the impurity trapped density and the maximum trap density,
respectively.
Fk
sCoi 1σC
σTCMax
-------------------–
ni
=
ks
Coi
σC
σTCMax

8: Oxidation and Silicidation
Oxidation
Sentaurus™ Process User Guide 627
I-2013.12
The maximum trap density is orientation dependent and can be specified using the following
commands:
pdbSet <interface material> <trapped impurity> 100 CMax {<n>}
pdbSet <interface material> <trapped impurity> 110 CMax {<n>}
pdbSet <interface material> <trapped impurity> 111 CMax {<n>}
TrapGen Model
The interface TrapGen model calculates not only the trapping flux, but also the generation flux
of impurities. The generation flux by reaction due to the Gen.Ambient gas is added to the
Gen.Material side. For example:
pdbSet Oxide_Silicon Nitrogen BoundaryCondition TrapGen
pdbSet Oxide_Silicon Nitrogen Gen.Ambient N2O
pdbSet Oxide_Silicon Nitrogen Gen.Material Oxide
The generation flux in the interface TrapGen model is calculated by:
(848)
where:
■ is the generation density.
■ is the reaction velocity.
■ is the normalization velocity.
■ is the power of normalized velocity.
, , and are specified with the parameters Gen.Density, Gen.Vnorm, and
Gen.Power, respectively.
Dopant-dependent Oxidation
A dopant-dependent oxidation rate is incorporated through the electron concentration
dependence as:
(849)
where:
(850)
Fρvv
vnorm
-------------
αni
=
ρ
v
vnorm
α
ρ
vnorm
α
kT n
ni
-----,
kT()lc⋅=
lc 1γVCV1–()+=
8: Oxidation and Silicidation
Oxidation
628 Sentaurus™ Process User Guide
I-2013.12
(851)
and:
(852)
The quantities in Eq. 852 are given by the following formulas:
(853)
(854)
(855)
(856)
(857)
(858)
(859)
(860)
The dependence on carrier concentration is a function of the location along the oxidizing
interface.
Dopant-dependent oxidation is switched off by default and can be switched on for O2 and H2O,
respectively, with:
pdbSetBoolean Oxide_Silicon O2 DopantDependentReaction 1
pdbSetBoolean Oxide_Silicon H2O DopantDependentReaction 1
γVGAMMA0GAMMAW–
kT
-------------------------------
exp×=
CV
1C+ni
n
-----
C_n
ni
-----
C=n
ni
-----
2
+++
1C+C_C=
+++
------------------------------------------------------------------------------=
C+E+Ei
–
kT
-----------------
exp=
C_EiE_
–
kT
-----------------
exp=
C=2EiE_
–E=
–
kT
---------------------------------
exp=
E+0.35eV=
E_Eg0.57eV–=
E=Eg0.12eV–=
Ei
Eg
2
------0.75 0.719()kTln+=
Eg1.17 4.73 4–
×10()T2
T636+
------------------------------------eV–=
8: Oxidation and Silicidation
Oxidation
Sentaurus™ Process User Guide 629
I-2013.12
In Eq. 851, the quantities GAMMA0 and GAMMAW can be set for O
2 and H
2O ambients,
respectively, as follows:
pdbSetDouble Oxide_Silicon O2 Gamma0 2360
pdbSetDouble Oxide_Silicon O2 GammaW 1.1
and:
pdbSetDouble Oxide_Silicon H2O Gamma0 2360
pdbSetDouble Oxide_Silicon H2O GammaW 1.1
The quantities and are defined as procedures called DFactorEg and DFactorEi, each
taking a single argument, which is temperature. If you want to overwrite them, use:
proc DFactorEg { temp } {
# enter the function here
}
Finally, Eq. 852 is implemented using the expressions for , , , and , where is
identically equal to 1. To overwrite them for O2 and H2O ambients, respectively, use:
pdbSetDoubleArray Oxide_Silicon O2 \
DopantReactFactor {1 <expr 1> 0 <expr 2> -1 <expr 3> -2 <expr4>}
and:
pdbSetDoubleArray Oxide_Silicon H2O \
DopantReactFactor {1 <expr 1> 0 <expr 2> -1 <expr 3> -2 <expr4>}
Diffusion Prefactors
The reactant diffusivities can be enhanced or retarded due to various new process conditions.
If a new model does not exist to simulate the observed behavior, you may want to multiply the
existing diffusivity with a prefactor. Sentaurus Process allows diffusivities to be multiplied by
user-defined factors. For example, in the case of specified O2 and H2O, these are given by:
term name=O2DiffFactor add Oxide eqn=1.0e18/(1.0*N2ox+1.0e18)
term name=H2ODiffFactor add Oxide eqn=1.0e18/(1.0*N2ox+1.0e18)
The effective diffusivity of O2 and H
2O will be multiplied by O2DiffFactor and
H2ODiffFactor, respectively. In this example, the diffusivity of both reactants will be a
function of the dataset N2ox. See Chapter 6 on page 571 for the definition of terms.
Eg
Ei
C+
C0
C–
C=
C0

8: Oxidation and Silicidation
Oxidation
630 Sentaurus™ Process User Guide
I-2013.12
Oxidation with Dielectric on Top
Thermal oxidation of silicon with a dielectric on top can be simulated in Sentaurus Process
using an Alagator generic growth script. Besides the three oxidation steps outlined at the
beginning of this chapter, there are two additional ones:
■Diffusion of oxidants (H2O, O2) from the gas–dielectric interface through the dielectric to
the dielectric–oxide interface;
■Diffusion of oxidants (H2O, O2) from the dielectric to oxide.
The first step involves the dissolution of the oxidant species at the gas–dielectric interface and
the oxidant transport in the bulk dielectric. The second step is modeled by the boundary
condition between the dielectric and oxide.
This oxidation mode can be enabled or disabled with the commands
SetDielectricOxidationMode on page 1124 and UnsetDielectricOxidationMode on page 1189.
N2O Oxidation
In N2O oxidation or oxynitridation, nitrogen is trapped at Si–SiO2 interfaces so that the number
of oxidizing sites and, in turn, the oxidation rate are reduced. N2O oxidation is performed by
specifying the N2O parameter in the diffuse statement. For the thick oxidation regime (in
other words, the Deal–Grove model), the parameters for N2O oxidation are specified similar to
O2 or H2O. However, for thin oxidation, the Massoud model is modified by multiplying the
nitrogen effect as follows:
(861)
can be defined for each of the three available silicon orientations and for polysilicon by
specifying the N.Thin.Max values for the N2O ambient. For example:
pdbSet Oxide_Silicon N2O 100 N.Thin.Max {Double {[Arrhenius 1.55e14 0.0]}}
pdbSet Oxide_PolySilicon N2O 100 N.Thin.Max {Double {[Arrhenius 1.55e14 0.0]}}
SiC Oxidation
Carbon atoms are generated during SiC oxidation. Some carbon atoms diffuse into oxide and
react with diffusing oxidants. The reaction of carbon atoms with oxygen atoms generates
carbon oxide (CO). CO2 formation requires higher energy than CO formation, so that only CO,
which is assumed to evaporate instantly, is accounted for. Some carbon atoms are captured at
oxidizing interfaces and reduce the oxidant reaction rate. Although silicon atoms also are
rthin Cxox
L
-------–
exp 1 σN
σmax
------------–
=
σmax

8: Oxidation and Silicidation
Oxidation
Sentaurus™ Process User Guide 631
I-2013.12
generated and diffuse into oxide, it is assumed that the Si–O reaction mainly occurs at SiC–
oxide interfaces because Si diffusivity in oxide is very low. Therefore, two-stream, that is,
carbon and oxidant, diffusion in oxide is taken into account. The default parameter set is
provided for Si-face, that is, (0001) and C-face, that is, (0001) in both O2 and H2O ambients
and for (1120) in O2:
(862)
(863)
(864)
where:
■ is the carbon concentration in oxide, which is named CarbonReact.
■ is the reaction rate of a carbon atom and an oxidant.
■ is the diffusivity of a carbon atom in oxide.
■ is the oxidant concentration (O2 or H2O).
■ is the oxidant diffusivity that is calculated from the parabolic parameter B.
■ is the concentration for normalization (the Cox parameter of the oxidant is used).
■ is the captured carbon density at interfaces.
■ is the trapping rate of carbon atoms to the interface (cm3/s).
■ is the emission rate of carbon atoms from the interface (cm3/s).
■ is the maximum carbon concentration in oxide.
■ is the maximum carbon trap density at the interface (cm–2).
, , , , , and are given by, for example:
pdbSetDouble Oxide CarbonReact O2 Reaction.Rate <n>
pdbSetDouble Oxide CarbonReact Dstar <n>
pdbSetDouble Oxide CarbonReact CMax <n>
pdbSetDouble Ox_SiC CarbonReact 0001 O2 Trapping.Rate <n>
pdbSetDouble Ox_SiC CarbonReact 0001 O2 Emission.Rate <n>
pdbSetDouble Ox_SiC CarbonReact 0001 CMax <n>
At the oxide surface, the incoming fluxes of oxidants and carbon atoms into oxide are
expressed by:
(865)
∂COx
∂t
-------------DOx COx
∇()∇kbCC
COx
COx
o
---------
–=
∂CC
∂t
---------- DCCC
∇()∇kbCC
COx
COx
o
---------–=
∂σc
∂t
---------tσcmax,σc
–()CCeCCmax,σc
–=
Cc
kb
DC
COx
DOx
COx
o
σc
t
e
CCmax,
σcmax,
kb
DC
CCmax,
t
e
σcmax,
FOx hC
∗COx
–()=

8: Oxidation and Silicidation
Oxidation
632 Sentaurus™ Process User Guide
I-2013.12
(866)
where is the mass transfer rate of carbon atoms:
pdbSet Gas_Oxide CarbonReact MassTransfer <n>
At the SiC–oxide interface, the incoming fluxes of oxidants and carbon atoms into oxide are
modeled by:
(867)
(868)
where:
■ is the oxidant reaction rate at the interface, which is calculated from the B/A value.
■ is the ratio of the carbon generation rate to the oxidant reaction rate. is given by:
pdbSetDoubleArray Oxide_SiliconCarbide CarbonReact 0001 O2 Reaction.Factor <n>
NOTE The parameter interpolation of the arbitrary crystal orientation of
polytype crystalline materials is not supported yet. Therefore,
regardless of the surface geometry, uniform crystal orientation is
assumed, which is given by:
pdbSet SiliconCarbide Crystal.Orient <0001 | 000-1 | 11-20>
In Situ Steam-generated Oxidation
The low-pressure combustion of hydrogen–oxygen mixtures is effective in producing high-
quality oxides. Combustion-like chemical reactions are initiated over the heated wafer,
producing a high density of gas-phase radicals (O– and OH–) that react rapidly with silicon.
The model for such ISSG empirically describes the oxidation by the radical O–, which
dominates the typical ISSG oxidation. When the pressure is too low, which means the
hydrogen–oxygen mixtures flow too fast, the reactant residence time is too short for chemical
activity to occur. On the other hand, when the pressure increases over some extent, the oxygen-
atom density is localized and falls off rapidly downstream of the flame so that the narrow
reaction zone prevents oxygen atoms from reaching the wafer surface. The oxygen-atom
concentration at the wafer surface is modeled by [1]:
(869)
FCksCC
–=
ks
FOx ki1σc
σcmax,
----------------–
COx
–=
FCrCFOx
∂σc
∂t
---------–=
ki
rC
rC
C∗Cmax pH2flow(, )BPD αβPmax P;,,()RRZ Plim P;()⋅⋅=
8: Oxidation and Silicidation
Oxidation
Sentaurus™ Process User Guide 633
I-2013.12
calculates the maximum oxygen-atom concentration depending on the partial
pressure of hydrogen and the total flow of the hydrogen–oxygen mixtures:
(870)
(871)
(872)
(873)
determines the profile of the oxygen-atom concentration with a given
pressure. The dependence on the pressure is modeled by the beta prime distribution (BPD) as
follows:
(874)
(875)
where and are specified by the parameters Alpha and Beta, respectively. The pressure
at the peak oxygen-atom concentration, , is modeled as follows:
(876)
defines the rapid reaction zone where the oxygen atoms do not reach the silicon
surface:
(877)
(878)
The values of the parameters from Eq. 872 through Eq. 878 can be modified by:
pdbSet Oxide ISSG <parameter> <value>
Cmax pH2flow(, )
flow flowH2flowO2
+=
pH2flowH2flow⁄=
Cmax pH2flow(, )Cmax pH2
()flow 1slm⁄()
C.FLOW.W
⋅=
Cmax pH2
()C.H2.L.0 min C.H2.Break pH2
,()
C.H2.L.W C.H2.H.S max pH2C.H2.Break–0,()⋅+⋅=
BPD αβPmax P;,,()
BPD αβPmax P;,,()
Pn
α1–1Pn
+()
α–β–
α1–
β1+
------------
α1–αβ+
β1+
-------------
α–β–
--------------------------------------------------------------=
Pn
α1–
β1+
------------ P
Pmax
------------⋅=
α
β
Pmax
Pmax P.Max.H2.0 pH2
P.Max.H2.W P.Max.Flow.0 flow 1slm⁄()
P.Max.Flow.W
⋅+⋅=
RRZ Plim P;()
RRZ Plim P;()
1
2
---erfc P.Limit.Smooth PP
lim
–()1torr⁄⋅()=
Plim P.Limit.0 P.Limit.A P.Limit.B eP.Limit.W pH2
⋅–
⋅+
flow⋅+=
8: Oxidation and Silicidation
Silicide Models
634 Sentaurus™ Process User Guide
I-2013.12
The diffusivity and reaction rates of the oxygen atoms can be modified respectively by:
pdbSet Oxide ISSG D <value>
pdbSet Oxide_Silicon ISSG Ks <value>
The process conditions to invoke the ISSG oxidation are defined in the diffuse or gas_flow
commands, for example:
diffuse temp=1000 time=1 pressure=12<torr> flowH2=6 flowO2=12 ISSG
Silicide Models
Sentaurus Process allows you to define models for new materials and reactions. This ability has
been used to define models for the growth of titanium, tungsten, cobalt, and nickel silicides.
The following sections describe the kinetics of TiSi2 growth, the specification of the model and
parameters, and suggestions for modeling other silicides.
TiSi2 Growth Kinetics
Figure 87 Velocities in 1D silicidation process
Titanium silicide is assumed to form when silicon atoms react in the silicide with titanium at
the titanium silicide–titanium (TiSi2–Ti) interface. The dissolution of silicon and the
consumption of titanium lead to the deformation of the material layers in the structure.
NOTE While the discussion that follows describes the growth of TiSi2 on
silicon, it also applies to growth of TiSi2 on polycrystalline silicon.
The silicidation process has three main steps:
■Dissolution of silicon and diffusion of silicon atoms from the TiSi2–silicon interface
through the existing TiSi2 to the Ti–TiSi2 interface.
■Reaction of silicon with titanium to form new TiSi2.
n
n
υTiSi υ
Gas Ti TiSi2Si
CSi
υgrow
Si
dissolve
2

8: Oxidation and Silicidation
Silicide Models
Sentaurus™ Process User Guide 635
I-2013.12
■Motion of materials due to the volume expansion, which is caused by the reaction between
diffused silicon and titanium, and also by the dissolution of silicon at the TiSi2–silicon
interface.
NOTE The name of the silicide reactant field (which represents the
concentration of silicon atoms in the silicide) is SiliconReact.
If silicon or polysilicon is in contact with titanium at the beginning of a thermal silicidation, an
initial TiSi2 layer is created automatically as in the case of oxidation. The thickness of this layer
is, by default, 1.5 nm.
The value of the initial TiSi2 thickness is specified in the parameter database by:
pdbSet Grid NativeLayerThickness 1.5e-7
which also controls the native layer thickness for oxidation.
Sentaurus Process automatically recognizes the silicidizing interfaces and switches on the
reaction equations.
TiSi2 Formation Reactions
At the TiSi2–silicon interface, there is the reaction:
(879)
where is the silicon as a diffusing species on the silicon material side and is the
silicon as a diffusing species on the TiSi2 material side. Therefore, silicon (on the Si side of the
interface) reacts to form silicon atoms (on the TiSi2 side of the interface). The reaction is
reversible, allowing for the reformation of silicon (if silicon is released by nitridation of TiSi2,
for example):
(880)
and are the diffusion flux and growth reaction flux, respectively, at the TiSi2–silicon
interface. The forward rate of this reaction depends only on temperature, while the reverse rate
is also proportional to the concentration of diffusing silicon atoms in TiSi2. is the
concentration of silicon in TiSi2 and is the equilibrium concentration of silicon at the
TiSi2–silicon interface. Beta is the stoichiometry of the growing material whose default is 1.0.
is the mass transfer coefficient.
SiSi SiTiSi2
↔
SiSi
SiTiSi2
RfKfCSi Cstar
–()≡
RgBeta Rf
≡
Rf
Rg
CSi
CStar
Kf

8: Oxidation and Silicidation
Silicide Models
636 Sentaurus™ Process User Guide
I-2013.12
To change them, use:
pdbSet Silicon_TiSilicide SiliconReact Kf <n>
pdbSet Silicon_TiSilicide SiliconReact Cstar <n>
pdbSet Silicon_TiSilicide SiliconReact Beta <n>
For each silicon atom removed from the silicon side of the interface, the volume of silicon is
reduced by:
(881)
where Density.Grow is the density of the growing material whose default value is .
To change it, use:
pdbSet Silicon_TiSilicide SiliconReact Density.Grow <n>
There is no new material formation at the TiSi2–silicon interface. Silicon dissolves at this
interface and is transported across the TiSi2 layer by simple diffusion:
(882)
where is the concentration of silicon in TiSi2 and is the diffusivity of silicon in TiSi2.
The following command changes the diffusivity:
pdbSet TiSilicide SiliconReact Dstar {<n>}
At the TiSi2–titanium interface, there is the reaction:
(883)
This reaction is assumed to be irreversible:
(884)
and are the diffusion flux and growth reaction flux, respectively, at the TiSi2–titanium
interface. The reaction rate is proportional to the concentration of diffusing silicon at the TiSi2
side of the interface. is the concentration of silicon in TiSi2 and is the equilibrium
concentration of silicon at the titanium–TiSi2 interface. Beta is the stoichiometry of the
growing material whose default is 0.5. is the mass transfer coefficient.
∆VBeta
Density.Grow
-----------------------------------=
522
×10
t∂
∂CSi Dstar CSi
∇()∇•=
CSi
Dstar
SiTiSi20.5Ti+0.5TiSi2TiSi2
→
RfKfCSi Cstar
–()≡
RgBeta Rf
≡
Rf
Rg
CSi
Cstar
Kf

8: Oxidation and Silicidation
Silicide Models
Sentaurus™ Process User Guide 637
I-2013.12
To change them, use:
pdbSet TiSilicide_Titanium SiliconReact Kf <n>
pdbSet TiSilicide_Titanium SiliconReact Cstar <n>
pdbSet TiSilicide_Titanium SiliconReact Beta <n>
The volumes of titanium and TiSi2 change according to:
(885)
where Expansion.Ratio is the conversion ratio from consumed material to the growing
material, and Density.Grow is the density of the growing material. The default values for
Expansion.Ratio and Density.Grow are 2.42 and , respectively. They can be
changed by using the commands:
pdbSet TiSilicide_Titanium SiliconReact Expansion.Ratio <n>
pdbSet TiSilicide_Titanium SiliconReact Density.Grow <n>
Tungsten-, Cobalt-, and Nickel-Silicide Models
The tungsten-, cobalt-, and nickel-silicide models are identical in form to the titanium-silicide
model. The parameters of the models are different, however, reflecting the differences between
the materials (see [2][3] for the tungsten-silicide model and [4][5] for the cobalt-silicide
model). The names of the relevant materials are Tungsten and TungstenSilicide (WSi2),
Cobalt and CobaltSilicide (CoSi2), and Nickel and NickelSilicide (NiSi).
Stress-dependent Silicidation
The stress-dependent silicidation model is experimental, and can become unstable and produce
irregular shapes. Fundamental changes to the model are possible in future releases.
Similar to oxidation, the silicide reaction rate and the reactant diffusivity can be affected by
local stress. For the silicide reaction, the speed of the reaction is assumed to be affected by the
total stress energy, so that the stress effect is incorporated symmetrically with respect to tension
versus compression.
To switch on stress-dependent diffusion, use the command:
StressDependentSilicidation <silicide>
where <silicide> can be set only to NickelSilicide.
V∆Beta
Expansion.Ratio * Density.Grow
---------------------------------------------------------------------------------=
2.34 22
×10

8: Oxidation and Silicidation
Silicide Models
638 Sentaurus™ Process User Guide
I-2013.12
When the stress-dependent silicidation model is switched on, the reaction rate given in Eq. 880
and Eq. 884 (that is, at both metal–silicide and silicon–silicide interfaces) is suppressed by the
normal stress:
(886)
Similarly, the diffusivity of the reactant SiliconReact becomes pressure dependent:
(887)
The activation volume VD is a bulk property and is defined in the silicide. The activation volume
Vk controls the impact of the normal stress at the reaction front and is defined on interfaces:
Nickel_NickelSilicide | NickelSilicide_Silicon
For example:
pdbSet Nickel_NickelSilicide SiliconReact Vk <n>
pdbSet NickelSilicide SiliconReact VD <n>
In addition to switching on stress dependency of the silicide reaction, the command
StressDependentSilicidation reduces the viscosity of the silicide to a point where
viscous relaxation occurs at typical silicidation temperatures (see StressDependentSilicidation
on page 1156). Similar mass relaxation effects have been proposed in the literature [6][7]. To
modify the relaxation of the silicide, use one or both of the following commands:
pdbSet NickelSilicide Mechanics Viscosity0 <n>
pdbSet NickelSilicide Mechanics ViscosityW <n>
Oxygen-retarded Silicidation
The silicidation process may be influenced by the presence of oxygen in the silicide. This
oxygen is assumed to enter the silicide at interfaces with silicon dioxide and to diffuse in the
silicide according to Fick’s law. The oxygen retards the reaction of silicon atoms at the silicide–
silicon and silicide–metal interfaces and the diffusion of silicon in the silicide; this is called
oxygen-retarded silicidation (ORS). The retardation factor is assumed to be in the form of:
(888)
where ORSOxygen is the retardant solution name. The model can be switched on and off by
using the command:
pdbSet TiSi2 Silicon ORS {0 | 1}
KfNStress T,()KfT() e
abs NStress()Vk⋅
kBT
--------------------------------------------------------
–
=
DPressure T,()DT() e
abs Pressure()VD⋅
kBT
------------------------------------------------------------
–
=
Rfactor 1ORSOxygen
122
×10
--------------------------------–≡

8: Oxidation and Silicidation
Silicide Models
Sentaurus™ Process User Guide 639
I-2013.12
If the model is on, is multiplied by of Eq. 880, p. 635, of Eq. 882, p. 636, and
of Eq. 884. You can define the retardation factors using the term command, for example:
term name=SiliconReactFactor add TiSilicide /Titanium \
eqn = "((1-ORSoxygen_TiSilicide/1e22)>0)?(1-ORSoxygen_TiSilicide/1e22):(0.0)"
term name=SiliconDiffFactor add TiSilicide \
eqn = "((1-ORSoxygen/1e22)>0)?(1-ORSoxygen/1e22):(0.0)"
term name=SiliconReactFactor add Silicon /TiSilicide \
eqn = "((1-ORSoxygen_TiSilicide/1e22)>0)?(1-ORSoxygen_TiSilicide/1e22):(0.0)"
NOTE The oxygen-retarded silicidation model can cause instabilities (such as
a zigzag shape of the silicide boundary) because of a high concentration
of silicon near the oxide. Solving the silicon diffusion equation in steady
state reduces such instability. It can be switched on using the command:
pdbSet NickelSilicide Silicon Steady 1
Triple-Point Control
During silicidation, triple points where more than two materials come together (such as oxide,
silicon, silicide node) may move inadvertently due to material consumption around the node.
To control the movement, a retardation factor around the triple point is applied to the velocities.
The retardation factor is assumed to be:
(889)
Factor is the suppression value at the triple point, and Distance is the rolloff length for
silicidation triple-point suppression. The distance determines how far the suppression factor
will be effective from the triple point. SDistance is the distance to the nearest node from the
triple point and is calculated internally. The other parameters can be changed using the
following commands:
pdbSet <mater> SilicidationTripleDistance {<n>}
pdbSet <mater> SilicidationTripleFactor {<n>}
where <mater> is the interface material (for example, Silicon_TiSilicide). If you want
to switch the triple-point control on or off, use the command:
pdbSet Mechanics SilicidationCorrection {1 | 0}
Rfactor
Rf
DStar
Rf
RFactor
2 1 2Factor–()
π
----------------------------------- SDistance
Distance
------------------------
atan+=
8: Oxidation and Silicidation
Dopants and Defects in Oxides and Silicides
640 Sentaurus™ Process User Guide
I-2013.12
Dopants and Defects in Oxides and Silicides
Dopants in oxides and silicides are modeled in the same way as in other nonsemiconductor
materials. Transport within a silicide or an oxide is governed by simple diffusion (in other
words, no electric field effects). For details on segregation at material interfaces, see Boundary
Conditions on page 357. Point defects can participate in reactions at interfaces with silicon.
While the current oxidation model specifies the generation of interstitials by the consumption
of silicon, the current titanium silicide model specifies the generation of vacancies by the same
mechanism.
Numerics
During oxidation or silicidation, one material grows at the expense of another material. To
handle the growth of materials, Sentaurus Process uses two different time loops – inner and
outer – as shown in Figure 88 for the case of oxidation.
Figure 88 Flowchart for simulation of material growth
Outer Time Loop
The diffusion equation for the oxidants is solved using the general PDE solver in Sentaurus
Process. In addition, a predictor for the next time step (oxidation time step) is computed. When
the concentrations of oxidants at the oxide–silicon interface are known, the corresponding
growth velocities can be computed. These velocities serve as a boundary condition for the
mechanics problem. After solving the mechanics problem, the velocity field in the entire
structure is known. At this point, the program enters the inner time loop.
Outer Time Loop
Inner Time Loop
Solve Oxidant
Diffusion
Solve Mechanics
Solve Dopant
Diffusion
Velocity at the
Reaction Front
Velocity Field
Remove Small
Elements
Move Mesh
Points
Compute Maximum
∆t on Given Mesh

8: Oxidation and Silicidation
Numerics
Sentaurus™ Process User Guide 641
I-2013.12
Inner Time Loop
Given a mesh and the velocity field, a time step (grid time step) can be computed so that
elements do not collapse when applying the velocities to the nodes of the mesh (moving mesh).
In the next time step, the dopant diffusion is solved using the general PDE solver and a
predictor for the next time step (diffusion time step) is computed. Then, mesh points are moved
according to the velocity field and, subsequently, small mesh elements are removed.
After removing small elements, the next grid time step is computed. The smaller of the two
time steps (grid time step and diffusion time step) is applied in the next time cycle. The inner
time loop runs as long as the time step of the outer loop (oxidation time step) is fulfilled. Then,
the code goes into its second time step of the outer loop. An example of typical output during
oxidation is:
...
Reaction Solve from 14.86min to 15.11min. Time step: 15.32s.
Mechanics Solve from 14.86min to 15.11min. Time step: 15.32s.
Diffusion Solve from 14.86min to 14.99min. Time step: 8.144s.
Diffusion Solve from 14.99min to 15.11min. Time step: 7.176s.
Reaction Solve from 15.11min to 15.37min. Time step: 15.4s.
Mechanics Solve from 15.11min to 15.37min. Time step: 15.4s.
Diffusion Solve from 15.11min to 15.25min. Time step: 8.361s.
Diffusion Solve from 15.25min to 15.29min. Time step: 2.077s.
Diffusion Solve from 15.29min to 15.37min. Time step: 4.967s.
...
This output reproduces the time-stepping scheme: Reaction Solve and Mechanics Solve
occur in the outer time loop; whereas, Diffusion Solve occurs in the inner time loop.
As previously mentioned, after solving the mechanics problem, velocities are given on all mesh
points. Mesh points are moved according to these velocities. This leads to a change in the
geometry and, in some cases, also to a change in the topology of the structure at each time step.
At a reactive interface, for example at the oxidation front, two velocities apply: one describes
the growth of a material and one describes the consumption of another material.
The velocity describing the growth of the material is used to solve the mechanics problem, and
the velocities describing the consumption of a material are used to update the structure or mesh.
Therefore, mesh elements on the growing side of the oxidation front are stretched, and
elements on the shrinking side are compressed. Edges on the growing side, which become too
8: Oxidation and Silicidation
References
642 Sentaurus™ Process User Guide
I-2013.12
long with time, are split. Edges and elements on the shrinking side of the interface which
become too small are removed. This is demonstrated in Figure 89.
Figure 89 Meshing strategy during thermal oxidation
References
[1] N. Sullivan et al., “Exploring ISSG Process Space,” in 9th International Conference on
Advanced Thermal Processing of Semiconductors (RTP), Anchorage, AK, USA, pp. 95–
110, September 2001.
[2] S-L. Zhang, R. Buchta, and M. Östling, “A study of silicide formation from LPCVD-
tungsten films: Film texture and growth kinetics,” Journal of Materials Research, vol. 6,
no. 9, pp. 1886–1891, 1991.
[3] G. Giroult, A. Nouailhat, and M. Gauneau, “Study of a WSi2/polycrystalline silicon/
monocrystalline silicon structure for a complementary metal-oxide-semiconductor for a
compatible self-aligned bipolar transistor emitter,” Journal of Applied Physics, vol. 67,
no. 1, pp. 515–523, 1990.
[4] C. M. Comrie and R. T. Newman, “Dominant diffusing species during cobalt silicide
formation,” Journal of Applied Physics, vol. 79, no. 1, pp. 153–156, 1996.
[5] R. Stadler et al., “Ab initio calculations of the cohesive, elastic, and dynamical properties
of CoSi2 by pseudopotential and all-electron techniques,” Physical Review B, vol. 54,
no. 3, pp. 1729–1734, 1996.
[6] S.-L. Zhang and F. M. d’Heurle, “Stresses from solid state reactions: a simple model,
silicides,” Thin Solid Films, vol. 213, no. 1, pp. 34–39, 1992.
[7] F. Cacho et al., “Numerical modeling of stress build up during nickel silicidation under
anisothermal annealing,” Materials Science and Engineering B, vol. 135, no. 2, pp. 95–
102, 2006.
Silicon Oxide

Sentaurus™ Process User Guide 643
H-2013.03
CHAPTER 9 Computing Mechanical Stress
This chapter discusses the computation of mechanical stress in
Sentaurus Process.
Overview
Mechanical stress plays an important role in process modeling. It controls the structural
integrity of the device, the yield from the process depends on stresses, the mobility of charged
carriers is changed by stresses, and leakage currents also are a function of the stress in the
system.
On a finer scale, stresses can affect dopant diffusion rates by modifying the band gap. They can
affect oxidation rates and, therefore, can alter the shapes of thermally grown oxide layers.
In modern process flows, accurate computation of stress is important. However, there is a
continual trend toward designing process flows that produce the right types of stress in the
device. With appropriate stresses, device performance can be enhanced significantly.
Stress computation simulations are performed in four distinct steps:
■First, the equations for mechanics are defined. The equations used in Sentaurus Process
define force equilibrium in the quasistatic regime.
■Second, the boundary conditions for these equations are defined. For the elliptic equations
that arise from the equations of force equilibrium, boundary conditions are needed on all
boundaries. Sentaurus Process allows Dirichlet or Neumann boundary conditions provided
that certain minimum criteria are met. The minimum criterion is to constrain the structure
sufficiently so that it has no rigid body modes.
■Third, material properties are defined. This is the part where the relationship between
stresses and strains is defined. Some materials may hold stresses for a given strain without
relaxing; these are elastic materials. Others may relax the stresses away; these are viscous
or viscoelastic materials. Sentaurus Process provides viscoelastic constitutive equations
for the computation of mechanical stresses. By setting parameters appropriately, the border
cases of a purely viscous and a purely elastic material can be simulated as well. The
viscoelastic models used in Sentaurus Process provide a choice between the Maxwell
model and the Standard Linear Solid model. The viscosity can depend on the local shear
stresses, which make the viscosity a locally varying quantity and can lead to nonlinear
mechanical behavior. In addition to elastic and viscoelastic materials, there are other
materials to model irreversible deformation and temperature-dependent volume change.

9: Computing Mechanical Stress
Material Models
644 Sentaurus™ Process User Guide
H-2013.03
Sentaurus Process provides nonlinear material models for incremental plasticity,
deformation plasticity, viscoplasticity and creep, and swelling.
■Fourth, the mechanisms that drive the stresses are defined. In Sentaurus Process, this is
performed through intrinsic stresses, thermal mismatch, material growth, lattice mismatch
(silicon germanium), and densification. All these processes are additive in the linear elastic
regime. In the nonlinear regime, they must be updated from the available stress history.
Stress is solved in all materials. However, during an inert diffusion, the stress computation can
be switched off. Parameters describing material behavior, which will be introduced in this
chapter, can be found in the parameter database:
<material> Mechanics
Some examples are Viscosity0, ViscosityW.
In the following sections, the constitutive equations are discussed in detail. These tensor
equations can be split into two parts:
■The dilatational part, which corresponds to the trace of the tensor, describes the material
behavior in the case of a pure volume change.
■The deviatoric part describes an arbitrary deformation but without changing the volume.
For example, the strain tensor can be decomposed as follows:
(890)
This decomposition will be used in subsequent equations to discuss the constitutive equation
for the dilatational and deviatoric parts independently.
Material Models
Sentaurus Process implements the viscous, viscoelastic, and elastic models in a general
manner, where the viscous model and elastic model can be derived from the viscoelastic model.
The viscous and viscoelastic models use shear stress–dependent viscosity. The elastic model
also has the anisotropic elasticity where the elastic coefficients are dependent on the crystal
orientation. The plasticity models describe the material behavior beyond yield, independent of
the rate of loading. The viscoplasticity and creep models provide rate-dependent plastic
behavior, while the swelling material models provide temperature-dependent volume change.
εij ε'
ij
deviatoric
1
3
---εkk
k
δij
dilatational
+=

9: Computing Mechanical Stress
Material Models
Sentaurus™ Process User Guide 645
H-2013.03
Viscoelastic Materials
The viscoelastic material response is characterized by elastic and viscous components. The
combined response depends on how elastic and viscous stresses or strains are coupled.
Sentaurus Process provides two commonly used combinations:
■Maxwell model
■Standard Linear Solid model
Maxwell Model
The viscoelastic behavior for the Maxwell model is obtained by combining elastic and viscous
responses in series. The stress–strain equations are written in terms of dilatational and shear
components. The equations for the volumetric part of the stress tensor1 take the form:
(891)
and:
(892)
where is the bulk viscosity. In addition, the relation of the stress and strain tensor to the
hydrostatic pressure is shown. The bulk modulus can be computed from the Poisson ratio
PoissRatio and Young’s modulus YoungsMod as:
(893)
The deviatoric part of the stress tensor is described by:
(894)
where is the shear viscosity. The shear modulus can be computed from the Poisson ratio
and Young’s modulus as:
(895)
1. The subscripts of vectors and tensors hold for the Cartesian coordinates x, y, and z.
σ
·v
K
----- σv
ηvTσ,s
()
-----------------------+ 3ε
·v
=
σkk
k
3p–3σv
==
ηv
p
K
KYoungsMod
31 2 PoissRatio⋅–()
--------------------------------------------------------=
σ'
·ij
G
------- σ'
ij
η'Tσ,s
()
---------------------+2ε'
·ij
=
η'
G
GYoungsMod
21 PoissRatio+()
-------------------------------------------------=
9: Computing Mechanical Stress
Material Models
646 Sentaurus™ Process User Guide
H-2013.03
By default, the viscoelastic response is applied to the deviatoric parts. The linear elastic model
is used for the pressure-volume response, that is:
(896)
To apply the viscoelastic response to both the deviatoric parts and the volumetric part, use:
pdbSet Mechanics NoBulkRelax 0
The shear viscosity is a function of the shear stress and the temperature , where:
(897)
Usually, the value of ViscosityW is negative and, therefore, the shear viscosity decreases
with increasing temperature. The bulk viscosity has a similar Arrhenius expression defined by
the parameters Viscosity0.K and ViscosityW.K. The dependency on the shear stress
is discussed in Shear Stress–dependent Viscosity on page 648.
Standard Linear Solid Model
In the Standard Linear Solid model, the material behavior is modeled by combining elastic
response in parallel with the Maxwell model-based viscoelastic response:
(898)
where is the elastic stress and is the viscoelastic stress. The difference compared to the
Maxwell model allows the total stress to be nonzero even after the viscoelastic stress has
relaxed away:
(899)
The dilatational and deviatoric components of the elastic and viscoelastic stresses are written
in the usual form:
(900)
(901)
(902)
σvKεkk
k
=
η'
T
η'T() Viscosity0 ViscosityW
kBT
--------------------------------–
exp⋅=
η'
σs
σij σij
el σij
ve
+=
σij
el
σij
ve
σij t∞→()σ
ij
el
=
σv
el 3Kbaseεv
=
σ'ij
el 2Gbaseε'ij
=
σ
·v
ve
K
--------σv
ve
ηvTσs
ve
(, )
-----------------------+ 3ε
·v
=
9: Computing Mechanical Stress
Material Models
Sentaurus™ Process User Guide 647
H-2013.03
and:
(903)
where:
■ and are the bulk and shear moduli for the elastic response, respectively.
■ and are the bulk and shear moduli for the elastic part of the viscoelastic response,
respectively.
■ and are the bulk and shear viscosities, respectively.
■ and are the dilatational and deviatoric components of mechanical strain,
respectively.
To enable the Standard Linear Solid model, use:
pdbSetSwitch <material> Mechanics ViscoElasticity.Model SLS-Maxwell
The default value for the above parameter is Maxwell for the Maxwell model.
The elastic response for the Standard Linear Solid model is inactive, by default, so that the
material behavior is similar to that of the Maxwell model. The elastic response can be activated
by providing nonzero values for bulk and shear moduli:
pdbSetDouble <material> Mechanics BaseBulkModulus <n>
pdbSetDouble <material> Mechanics BaseShearModulus <n>
The material parameters for viscoelastic response are specified in the same manner as for the
Maxwell model. By default, the dilatational component of viscoelastic stress is assumed to be
purely elastic:
(904)
To activate viscoelastic response for the dilatation component, use:
pdbSet Mechanics NoBulkRelax 0
To visualize elastic and viscoelastic responses, this model provides additional output fields.
Stresses and strains for the elastic response can be viewed with the BaseStressEL and
BaseElasticStrainEL fields, respectively. Creep strains for the viscoelastic response
can be viewed with the CreepStrainEL field:
(905)
σ'
·ij
ve
G
---------σ'
ij
ve
η'Tσs
ve
(, )
----------------------+ 2ε'
·ij
=
Kbase
Gbase
K
G
ηv
η'
εv
ε'ij
σv
ve 3Kεv
=
εij
cr
εij
cr σv
ve
3ηv
---------σ'ij
ve
2η'
---------+
td
0
t
=

9: Computing Mechanical Stress
Material Models
648 Sentaurus™ Process User Guide
H-2013.03
The solution for viscoelastic stress is time dependent. It also becomes nonlinear when viscosity
is a function of viscoelastic shear stress. Therefore, the Newton method is used to solve for
stresses. At the end of each Newton iteration, a check is made on whether the convergence
criteria have been satisfied. More iterations are performed until all the criteria are satisfied
within the specified tolerance or until the maximum number of iterations is reached. For details
on convergence criteria and time-stepping for mechanics, see Time-Step Control for
Mechanics on page 871.
Purely Viscous Materials
Oxide and nitride, by default, are treated as viscoelastic materials. However, the viscosity is a
function of the temperature (see Eq. 897). With increasing temperature, the viscosity
decreases, that is, the material becomes increasingly more liquid. When the viscosity reaches
a very low value, the first term in Eq. 894 can be neglected:
(906)
Eq. 906 describes the deviatoric part of a purely viscous material. The relaxation time
typically gives a good estimate of the behavior of a viscoelastic material. If is
much greater than the process time, the material is in the elastic regime. The material behaves
viscoelastically if is in the range of the process time. If is very small, the material is in the
viscous regime.
Shear Stress–dependent Viscosity
For viscous and viscoelastic materials, the viscosity may depend on the temperature and the
shear stress . The temperature dependency is described by Eq. 897. The dependency on the
shear stress is given by:
(907)
The shear stress is computed from the local stress distribution based on the second invariant
of the deviatoric part of the stress tensor:
(908)
The viscosity breakdown value can be determined by:
(909)
σ'
jk
η'Tσ,s
()
--------------------- 2ε'
·jk
=σ'
jk 2η'ε'
·jk
=⇔
τη'G⁄=
τ
τ
τ
σs
ησ
sT,()ηT() σsσcrit
⁄
σsσcrit
⁄()sinh
-----------------------------------
⋅=
σs
σs
3
2
---σ
k
j
'
jk
σ'
kj
=
σcrit
σcrit
2kBT
vcrit
-------------=

9: Computing Mechanical Stress
Material Models
Sentaurus™ Process User Guide 649
H-2013.03
where:
(910)
By default, oxide and nitride are treated as viscoelastic materials with shear stress–dependent
viscosity. The values for Vcrit0 and VcritW also are set in the PDB:
pdbSetDouble <material> Mechanics Vcrit0
pdbSetDouble <material> Mechanics VcritW
Purely Elastic Materials
If the viscosity in Eq. 894 is chosen high enough, the second term on the left can be neglected
and the equation reads:
(911)
This equation describes the deviatoric part of a purely elastic material. By default, silicon and
polycrystalline silicon are treated as purely elastic materials. To achieve this, the viscosity of
these materials is set to .
NOTE and are the primary parameters describing elastic materials, and
not Young’s modulus and the Poisson ratio. When changing material
properties with the pdb command, only a change of the primary
parameters has an effect on the simulation. To obtain Young’s modulus
and the Poisson ratio, use the following commands, respectively (see
KG2E on page 993 and KG2nu on page 994):
KG2E <BulkModulus> <ShearModulus>
KG2nu <BulkModulus> <ShearModulus>
NOTE When material data is given in terms of Young’s modulus and the
Poisson ratio, use the following commands to convert them to the shear
modulus and the bulk modulus, respectively (see Enu2G on page 920
and Enu2K on page 921):
Enu2G <YoungsModulus> <PoissonRatio>
Enu2K <YoungsModulus> <PoissonRatio>
vcrit T() Vcrit0 VcritW
kBT
-------------------–
exp⋅=
σ'
·jk
G
-------- 2ε'
·jk
=σ'
jk 2Gε'
jk
=⇔
140
×10 poise
K
G

9: Computing Mechanical Stress
Material Models
650 Sentaurus™ Process User Guide
H-2013.03
Anisotropic Elastic Materials
The stress and strain relations for anisotropic elastic materials can be described using:
(912)
where and are the components of the engineering stress and strain, respectively, and
is the component of the stiffness matrix. The engineering stress (i=1, …, 6) corresponds to
the stress-tensor components σxx, σyy, σzz, σxy, σyz, σxz, and the engineering strain
(j=1, …, 6) corresponds to the strain-tensor components εxx, εyy, εzz, 2εxy, 2εyz, 2εxz.
NOTE The engineering shear-strain components differ from the shear-strain
tensor components by a factor of 2.
Cubic Crystal Anisotropy
The mechanical responses of a crystalline solid vary along various crystal orientations. For a
cubic crystal, the axes of reference are chosen to be parallel to the crystal axes. In a coordinate
system with axes aligned along the crystal axes, the symmetric stiffness matrix C has the
following nonzero components:
C11 = C22 = C33, C12 = C23 = C13
C44 = C55 = C66
All other components are zeros. The anisotropic stress and strain relation is completely defined
when three independent modulus parameters C11, C12, and C44 are specified.
The degree of anisotropy for a given material can be measured by the departure from unity of
the ratio A = 2 C44/(C11 – C12). The anisotropic model reduces to the isotropic model if
the ratio A is equal to 1. When the simulation coordinate axes do not coincide with the crystal
axes, the stiffness matrix C must be transformed accordingly. For this, note that C is actually a
rank-4 tensor.
By default, the anisotropic elasticity model is switched off. The following command is required
to switch on the model:
pdbSet Silicon Mechanics Anisotropic 1
The values of these three modulus parameters with respect to the cubic crystal axis can be
defined using the following commands, which also show the default values for the crystalline
silicon:
pdbSet Silicon Mechanics C11 16.57E11
pdbSet Silicon Mechanics C12 6.39E11
pdbSet Silicon Mechanics C44 7.96E11
σiCijεj
=
σi
εj
Cij
σi
εj
9: Computing Mechanical Stress
Material Models
Sentaurus™ Process User Guide 651
H-2013.03
The unit for these default values is dyn/cm2.
This model depends on the wafer.orient and slice.angle parameters specified in the
init command.
Orthotropic Model
Orthotropic materials have three planes of symmetry. In a coordinate system with axes aligned
along the symmetry planes, the symmetric stiffness matrix C has the following nonzero
components:
C11, C22, C33, C44=C55=C66, C12=C21, C13=C31, C23=C32
Orientation
The symmetry planes of the model, by default, coincide with the simulation axes. You can
choose the symmetry plane directions by switching off this default:
pdbSetBoolean <mat> Mechanics Ortho.Axis.Sim.Aligned 0
and specifying the axes in the wafer coordinate system (see Wafer Coordinate System on
page 66). Then, these axes will depend on the slice.angle, wafer.orient, and
flat.orient arguments specified in the init command. If these arguments are not given
(and the above pdb flag is switched off), the default values of slice.angle, wafer.orient,
and flat.orient will be used. The default wafer in-plane symmetry plane directions are
given by wafer.orient= {0 0 1}, flat.orient= {1 1 0}, that is, at a angle to
the xy plane of the wafer coordinate system)
NOTE When the symmetry plane directions are specified, they remain the
same for the entire structure and cannot be set regionwise.
Orthotropic material properties can be described by specifying nine independent parameters,
namely, the Young’s moduli in the symmetry planes (E1, E2, E3), the directional shear moduli
(G12, G23, G13), and the directional Poisson ratios (ν12, ν13, ν23). The other directional Poisson
ratio are calculated from:
(913)
The stiffness matrix components are calculated from the specified material properties:
(914)
(915)
45°
υij
Ei
------ υji
Ej
------=
Ckk Ek1νijυji
–()=
Cij Eiυji υjkυki
+()=
9: Computing Mechanical Stress
Material Models
652 Sentaurus™ Process User Guide
H-2013.03
and .
By default, the orthotropic model is switched off, and it is switched on using the command:
pdbSet <material> Mechanics Orthotropic 1
The material properties can be specified as:
pdbSetDouble <material> Mechanics <material parameter> <n>
specifically:
pdbSet Silicon Mechanics YoungsModulus1 162E10
pdbSet Silicon Mechanics YoungsModulus2 162E10
pdbSet Silicon Mechanics YoungsModulus3 162E10
pdbSet Silicon Mechanics PoissonRatio12 0.28
pdbSet Silicon Mechanics PoissonRatio13 0.28
pdbSet Silicon Mechanics PoissonRatio23 0.28
pdbSet Silicon Mechanics ShearModulus12 63.28E10
pdbSet Silicon Mechanics ShearModulus13 63.28E10
pdbSet Silicon Mechanics ShearModulus23 63.28E10
The values given also are the default values used. The units for the Young’s modulus and shear
modulus are dyn/cm2.
Orthotropic thermal expansion also is considered in this material model, and different
coefficients of thermal expansion can be specified along the three symmetry planes:
pdbSet Silicon Mechanics ThExpCoeff1 3E-06
pdbSet Silicon Mechanics ThExpCoeff2 3E-06
pdbSet Silicon Mechanics ThExpCoeff3 3E-06
The specified values are the default values.
Temperature-dependent material properties can be specified for all the material parameters
specified above. The variation of a property can be specified as:
(916)
where the reference value is the material parameter value specified in the input deck.
The values can be specified as:
pdbSet <material> Mechanics <material parameter>Rate <n>
For example:
pdbSet Silicon Mechanics ThExpCoeff1Rate 0
ij k≠,
ξ
ξT() ξ
ref ξ
·TT
ref
–()+=

9: Computing Mechanical Stress
Material Models
Sentaurus™ Process User Guide 653
H-2013.03
Plastic Materials
Materials such as metals show linear elastic behavior at lower stresses but undergo permanent
deformation at higher stresses. At low temperatures, permanent deformation in these materials
is not sensitive to the rate of loading. Such material behavior is defined as plastic or elastic-
plastic. Depending on the type of loading, plastic deformations may be computed using
incremental plasticity or deformation plasticity.
To switch on the plastic material model, use the command:
pdbSet <material> Mechanics IsPlastic <n>
Incremental Plasticity
Plastic material behavior under nonmonotonic loading is modeled using incremental
formulation.
Incremental plasticity uses the von Mises yield criterion with associative flow and bilinear
hardening. The von Mises yield criterion for isotropic solid materials takes the form:
(917)
where is the back stress, is an isotropic hardening variable, and:
(918)
is a function describing the change of yield surface with progressive yielding. The Einstein
summation convention is used to define the tensor product in the above equation. is the
yield stress in uniaxial tension. is the isotropic hardening modulus, which is constant for
bilinear isotropic hardening. To set these two parameters, use the commands:
pdbSet <material> Mechanics FirstYield <n>
pdbSet <material> Mechanics Hardening.Modulus.Isotropic <n>
Under a small strain assumption, the strains (and strain rates) are decomposed additively:
(919)
where are the elastic strains, and are the plastic strains.
For incremental plasticity, the plastic strains are determined by the plastic flow rule:
(920)
Fσ'ij qij α,,()σ'ij qij
–()σ'ij qij
–()Yα()–0==
qij
α
Yα() 2
3
---σyHisoα+()=
σy
Hiso
ε
ij εij
eεij
p
+=
εij
e
εij
p
ε
·ij
pγ
·∂Q
∂σij
----------
=
9: Computing Mechanical Stress
Material Models
654 Sentaurus™ Process User Guide
H-2013.03
where is the slip rate, and is the plastic potential. Plastic flow is assumed to be volume
preserving, so that plastic strain is purely deviatoric:
(921)
For associative plastic flow, the plastic potential is set equal to the yield function . The
evolution of the isotropic hardening variable and the back-stress variable are given by:
(922)
and:
(923)
where is the kinematic hardening modulus, and is the equivalent plastic strain rate. To
set the kinematic hardening modulus, use the command:
pdbSet <material> Mechanics Hardening.Modulus.Kinematic <n>
For linear isotropic hardening, the hardening modulus is interpreted as the slope of the stress
versus the plastic strain curve (as obtained from uniaxial tension test) . It differs from
the elastic-plastic tangent modulus, which is defined as the slope of the stress versus total strain
curve .
For combined isotropic and kinematic hardening, a common choice for hardening moduli is:
; ; (924)
where is a constant.
To switch on the incremental plasticity model, use:
pdbSet <material> Mechanics Plasticity.Model Incremental
The rate equations are discretized using backward Euler scheme and then solved using a radial
return mapping algorithm (see [1] for more details).
The nonlinear nature of the plasticity model requires Newton iterations to achieve the
equilibrium state for each loading step. At the end of each iteration, a check on the satisfaction
of convergence criteria is made. More Newton iterations are performed until all the
convergence criteria are satisfied within the specified tolerance or until the maximum number
of iterations is reached. See Time-Step Control for Mechanics on page 871 for details on
convergence criteria and time-stepping for mechanics.
γ
·0≥
Q
εij
pδij 0ε'ij
p
εij
p
==
Q
F
α
·e
·p2
3
---γ
·
==
q
·ij
2
3
---γ
·Hkin
σ'ij qij
–()
σ'kl qkl
–()σ'kl qkl
–()
---------------------------------------------------------=
Hkin
e
·p
Hiso
dσ
dεp
--------=
Eep dσ
dε
------=
Hkin 1β–()H=
Hiso βH=
β01,[]∈
H

9: Computing Mechanical Stress
Material Models
Sentaurus™ Process User Guide 655
H-2013.03
NOTE To define the plastic model, use nonzero values for the isotropic or the
kinematic hardening modulus along with yield stress. In the absence of
hardening, the numeric simulation of plastic deformation may become
unstable.
Deformation Plasticity
Plastic materials that do not have well-defined yield stress can be modeled using deformation
plasticity. This model is based on the Ramberg–Osgood formula [2][3], which is only valid for
monotonic loading. It is used mostly for plastic deformation around crack tips since it is well
suited to the J-integral calculation.
For one dimension, an additive decomposition of strains under a small strain assumption is
given as:
(925)
where and are material parameters, is the stress in one dimension, is the total strain
in one dimension, and is Young’s modulus.
Extending the formula to three dimensions, the strain components can be expressed as:
(926)
where is the equivalent stress.
The plastic flow is assumed to be associative and is governed by the von Mises yield criterion.
Under monotonic loading, the total plastic strain can be written as:
(927)
where is the total equivalent plastic strain.
Inverting the plastic strain expression gives:
(928)
with defining the work hardening exponent.
εε
eεp
+σ
E
--- ασy
E
-----
σ
σy
-----
n
+==
α
n
σ
ε
E
elastic plastic
εij εij
eεij
p
+σ'ij
2G
------- σkkδij
9K
--------------+3
2
---ασy
E
-----
σeq
σy
--------
nσ'ij
σeq
--------
+==
σeq 3
2
---σ'ijσ'ij
=
εij
p3
2
---epσ'ij
σeq
--------=
epασy
E
-----
σeq
σy
-------
n
=
σeq σy
E
ασy
----------
m
ep
()
m
=
m1
n
---=

9: Computing Mechanical Stress
Material Models
656 Sentaurus™ Process User Guide
H-2013.03
To switch on the deformation plasticity model, use:
pdbSet <material> Mechanics Plasticity.Model Deformation
The deformation plasticity equations do not require any integration due to total stresses and
strains. However, the nonlinear expressions require Newton iterations to achieve the
equilibrium state for each loading step. At the end of each iteration, a check on the satisfaction
of convergence criteria is made. More Newton iterations are performed until all the
convergence criteria are satisfied within the specified tolerance or until the maximum number
of iterations is reached. See Time-Step Control for Mechanics on page 871 for details on
convergence criteria and time-stepping for mechanics.
NOTE Deformation plasticity must be used only with monotonic loading since
the equations are not valid for unloading. This model must be used if the
J-integral must be calculated around a crack tip with plastic strains.
Viscoplastic Materials
Materials, such as metals at high temperatures, exhibit rate-dependent plasticity also known as
viscoplasticity or creep. There are different ways to model such behavior:
■Anand model
■Power law creep
Anand Model
The Anand model [4][5] is used for rate-dependent plasticity that combines creep and plastic
deformation.
Assuming small strains, the strain rates and strains can be decomposed into elastic and
viscoplastic parts in an additive manner:
(929)
(930)
The elastic strains are evaluated using Hooke’s law, while the Anand model is used to evaluate
the viscoplastic part. The Anand model assumes that plastic deformation occurs at all values
of strain, so instead of a yield function, a constitutive equation is used to relate stresses to
viscoplastic strains.
ε
·ij ε
·ij
eε
·ij
vp
+=
εij εij
eεij
vp
+=
9: Computing Mechanical Stress
Material Models
Sentaurus™ Process User Guide 657
H-2013.03
The flow rule for evolution of viscoplastic strains (volume preserving) is assumed to be of the
familiar form:
(931)
The equivalent viscoplastic strain rate at constant temperature is given by a constitutive
equation:
(932)
where is deformation resistance. It is defined in terms of an isotropic hardening function as:
(933)
The saturation value of deformation resistance at a given temperature and strain rate is
expressed as:
(934)
In the above formulation:
■ is a pre-exponential factor.
■ is the activation energy.
■ is the universal gas constant.
■ is absolute temperature in kelvin.
■ is the stress multiplier.
■ is the strain rate sensitivity.
■ is the constant of athermal hardening or softening.
■ is the exponent of athermal hardening or softening.
■ is the coefficient for the saturation value of deformation resistance.
■ is the exponent for the saturation value of deformation resistance.
Values for the material parameters and , and the initial value for
deformation resistance are obtained by fitting experimental data for stress–strain (obtained
from tension or compression tests conducted at various temperatures and strain rates) to the
above equations. For details on how to obtain such data, refer to the literature [4][6][7][8].
ε'
·ij
vp 3
2
---e
·vp σ'ij
σ'
---------σ'; σ'ijσ'ij
==
e
·vp fσ's,()AQ
RT
-------–
ξσ'
s
---------
sinh 1m⁄
exp==
s
s
·hs()e
·vp h01s
s∗
-----–
a1s
s∗
-----–
e
·vp a1≥;sgn==
s∗s
˜e
·vp
A
------- Q
RT
-------
exp
n
=
A
Q
R
T
ξ
m
h0
a
s
˜
n
AQξmh
0as
˜
,,,, ,,
n
s0

9: Computing Mechanical Stress
Material Models
658 Sentaurus™ Process User Guide
H-2013.03
To set these parameters, use the commands:
pdbSetDouble <material> Mechanics Viscoplasticity.A <n>
pdbSetDouble <material> Mechanics Viscoplasticity.Q <n>
pdbSetDouble <material> Mechanics Viscoplasticity.Xi <n>
pdbSetDouble <material> Mechanics Viscoplasticity.m <n>
pdbSetDouble <material> Mechanics Viscoplasticity.h0 <n>
pdbSetDouble <material> Mechanics Viscoplasticity.a <n>
pdbSetDouble <material> Mechanics Viscoplasticity.stilde <n>
pdbSetDouble <material> Mechanics Viscoplasticity.n <n>
pdbSetDouble <material> Mechanics Viscoplasticity.s0 <n>
NOTE For other materials, use the long form of the pdb commands to set
parameter values.
A new material named Solder has been added to the PDB to model viscoplastic behavior. The
default values for the above parameters for Solder material are based on 96.5Sn3.5Ag solder
alloy as reported in [6].
To solve the above nonlinear equations, the rate terms are discretized using the backward Euler
method, and the resulting algebraic equations are evaluated locally at every integration point
using the Newton–Raphson iterative scheme.
To switch on the viscoplastic material model, use the command:
pdbSet <material> Mechanics IsViscoPlastic <n>
This flag must be switched on during the simulation if viscoplastic deformation exists. The
nonlinear nature of the viscoplasticity model also requires Newton iterations to achieve
equilibrium of mechanics equations at each loading step. At the end of each iteration,
convergence criteria are checked. More iterations are performed until all the convergence
criteria are satisfied within the specified tolerance or until the maximum number of iterations
is reached. See Time-Step Control for Mechanics on page 871 for details on convergence
criteria and time-stepping for mechanics.
NOTE To avoid convergence problems, use small time steps at the beginning
of the analysis. You can increase the number of time steps later, during
the analysis, if it does not adversely affect the solution.
Power Law Creep
The power law creep [9], also known as the Bailey–Norton creep, assumes creep strain to be
of the following form:
(935)
ε
·cr AQ
RT
-------–
exp σnmt
˜m1–()
=

9: Computing Mechanical Stress
Material Models
Sentaurus™ Process User Guide 659
H-2013.03
where, for multiaxial loading:
■ is the equivalent creep strain rate.
■ is a pre-exponential factor.
■ is the activation energy.
■ is the universal gas constant.
■ is the absolute temperature in kelvin.
■ is the equivalent stress or the von Mises stress.
■ is the time (different from physical time).
■ and are exponents.
The above form is referred to as a time hardening form. A more commonly used form called
the strain hardening form is obtained by eliminating the time variable:
(936)
Under a small strain assumption, strains (and strain rates) can be decomposed additively as:
(937)
with creep strains being distinct from plastic strains.
Creep flow is assumed to be volume preserving ( ) and is governed by:
(938)
When incremental plasticity is also active, the creep flow rule is modified to account for
hardening:
(939)
where:
■ is the equivalent stress.
■ is the back stress for kinematic hardening.
■Plastic flow equations are solved simultaneously with creep flow.
ε
·cr 2
3
---ε
·ij
crε
·ij
cr
=
A
Q
R
T
σ3
2
---σ'ijσ'ij
=
t
˜
n
m
ε
·cr mA Q
RT
-------–
exp σnεcr
()
m1–()
1
m
----
=
εij εij
eεij
pεij
cr
++=
εij
crδij 0=
ε
·ij
cr 3
2
---ε
·cr σ'ij
σ
-------
=
ε
·ij
cr 3
2
---ε
·cr σ'ij qij
–
σ
--------------------
=
σ3
2
---σ'ij qij
–()σ'ij qij
–()=
qij

9: Computing Mechanical Stress
Material Models
660 Sentaurus™ Process User Guide
H-2013.03
The material parameters , , , and are obtained by fitting experimental data. To set
these parameters, use the commands:
pdbSet <material> Mechanics Creep.A <n>
pdbSet <material> Mechanics Creep.Q <n>
pdbSet <material> Mechanics Creep.n <n>
pdbSet <material> Mechanics Creep.m <n>
NOTE For materials other than Solder, use the long form of these commands
to set parameter values.
The default values for the above parameters have been added to the PDB to the Solder
material based on the 96.5Sn3.5Ag solder alloy as reported in [10].
To solve the creep equations, the rate terms are discretized using the backward Euler method,
and the resulting algebraic equations are evaluated locally at every integration point using the
Newton–Raphson iterative scheme.
To switch on the creep material model, use the command:
pdbSet <material> Mechanics IsCreep <n>
This flag must be switched on during the simulation if creep deformation exists. The nonlinear
nature of the creep model also requires global Newton iterations to achieve equilibrium of
mechanics equations at each loading step. At the end of each iteration, convergence criteria are
checked. More iterations are performed until all the convergence criteria are satisfied within
the specified tolerance or until the maximum number of iterations is reached. See Time-Step
Control for Mechanics on page 871 for details on convergence criteria and time-stepping for
mechanics.
NOTE To avoid convergence problems, use small time steps at the beginning
of the analysis. You can increase the number of time steps later, during
the analysis, if it does not adversely affect the solution.
Swelling
Swelling refers to volumetric expansion of material. Swelling material behavior is defined by
specifying strain rates at various temperatures that are interpolated linearly. To switch on the
model, use the command:
pdbSetBoolean <material> Mechanics IsSwelling <n>
A
Q
n
m

9: Computing Mechanical Stress
Material Models
Sentaurus™ Process User Guide 661
H-2013.03
The swelling strain rate data is specified with a double array:
pdbSetArray <material> Mechanics SwellingStrainRate
Temperature { <temp> {<SSR1> <SSR2> <SSR3>}
<temp> {<SSR1> <SSR2> <SSR3>} ... }
where <temp> is the temperature in degree Celsius, and <SSR1>, <SSR2>, and <SSR3> are
strain rates in the x-, y-, and z-direction, respectively, in .
Strain rates can be the same (isotropic) or different (anisotropic) in each of the three directions.
For cyclic temperature loading, strain rate data must be given for loading (temperature
increment) as well as unloading (temperature decrement). For example:
pdbSetDouble Mold Mechanics SwellingStrainRate Temperature {
27 {0.0 0.0 0.0}
77 {0.001 0.001 0.001}
127 {0.002 0.002 0.002}
80 {0.0012 0.0012 0.0012}
25 {0.0 0.0 0.0}
}
If strain rate data is not given for unloading, loading data is used for increasing as well as
decreasing temperatures.
Since strains are assumed to be small, swelling strain rates are added to other strain rates:
(940)
and are integrated over time to give total strains:
(941)
where for .
For a given material, only one set of strain rate data can be specified for a solve step. If
necessary, different strain rate data may be specified for the same material in a subsequent solve
step.
Mole Fraction–dependent Mechanical Properties
The mechanical properties of compound materials change with the ratio of substance
concentration, that is, mole fraction. The mole fraction is calculated with the interpolated
compound lattice density. Mole fraction dependency can be switched on with:
pdbSet Mechanics Compound.Interpolation 1
s1–
ε
·ij ε
·ij
eε
·ij
sw
+=
εij εij
eεij
sw
+=
εij
sw ε
·ij
sw 0==
ij≠
9: Computing Mechanical Stress
Material Models
662 Sentaurus™ Process User Guide
H-2013.03
and is applied to:
■Bulk modulus and shear modulus for isotropic materials
■C11, C12, and C44 for anisotropic elastic materials
■Thermal expansion coefficient
The PDB parameters Derived.Materials and MoleFraction.Atoms are used to define
the compound material. The following commands define SiGe as a compound material:
pdbSetString Si Derived.Materials { SiGe }
pdbSetString Ge Derived.Materials { SiGe }
pdbSetArray SiGe MoleFraction.Atoms { x Ge }
The material SiGe is set to be a compound by default. More information about compound and
alloy materials can be found in Diffusion in III–V Compounds on page 265.
The mole fraction dependency for the above-listed mechanical properties can be defined
separately. By default, a linear interpolation is used. If the parameter is defined for the
compound material, the given parameter value is used.
SiGe is treated as material silicon with germanium concentration without material conversion
of the binary compound. Therefore, the current approach cannot be directly applied to material
SiGe.
The mole fraction of Si1–xGex is solved from the following equation:
(942)
The mole fraction definition here is different from the formulation used in older SiGe–related
models:
(943)
NOTE Do not mix the new syntax with the old syntax (see Deprecated Syntax
for Mole Fraction–dependent Mechanical Properties of Binary
Compounds on page 663). Some of the old syntax may exist in the
Advanced Calibration script.
xCGe
LDGexLD
Si 1x–()+
--------------------------------------------------=
xCGe
LDSi
-----------=
9: Computing Mechanical Stress
Material Models
Sentaurus™ Process User Guide 663
H-2013.03
Deprecated Syntax for Mole Fraction–dependent Mechanical
Properties of Binary Compounds
The mole fraction–dependent model for binary compounds can be applied to the following
elastic moduli:
■Bulk modulus and shear modulus for isotropic materials
■C11, C12, and C44 for anisotropic elastic materials
The parameter interpolation is computed by:
(944)
where:
■ denotes the elastic moduli of binary compound .
■ is the mole fraction.
■ is an interpolation function with and .
The mole fraction–dependent moduli for Si1–xGex are linear combinations of the elastic moduli
of each material. The binary compound Si1–xGex is treated as silicon regions with germanium.
The mole fraction of Si1–xGex is calculated as germanium concentration divided by the silicon
lattice density. The mole fraction–dependent model can be switched on for silicon with:
pdbSetBoolean Si IsCompound 1
Next, a list of binary compound materials with mole fraction–dependent elastic moduli is
created by:
pdbSetString Mechanics BCompoundList {Silicon Germanium}
Finally, the interpolation function is defined as a double array, in other words,
{x1 f1 x2 f2 x3 f3 ... }. For example, a linear interpolation function can be specified with:
pdbSetDoubleArray SiliconGermanium CompoundInterp {0 0 1 1}
For nonlinear relations, a piecewise linear interpolation profile is used.
The mole fraction dependency can be applied to thermal expansion coefficients of materials
using the command:
pdbSet Mechanics Compound.ThExpCoeff 1
when the binary compound model is switched on.
CAB CA1fx()–()CBfx()+=
CAB
A1x–Bx
x
f
f0() 0=
f1() 1=

9: Computing Mechanical Stress
Material Models
664 Sentaurus™ Process User Guide
H-2013.03
Temperature-dependent Mechanical Properties
The mechanical properties of materials are different at high temperature from those at room
temperature. The elastic modulus of typical materials decreases as temperature rises. Some
materials show non-negligible changes of mechanical properties at different temperatures.
The temperature dependency and mole fraction dependency are handled under the same PDB
switch. The temperature dependency can be switched on with:
pdbSet Mechanics Compound.Interpolation 1
and is applied to:
■Bulk modulus and shear modulus for isotropic materials
■C11, C12, and C44 for anisotropic elastic materials
■Thermal expansion coefficient
The temperature dependency for the above-listed mechanical properties can be defined
separately. The available options are:
■The linear dependency is defined with the PDB parameter <parameter>.T1. For
example:
pdbSet <mat> Mechanics BulkModulus.T1 <n>
■The parameter is then calculated with the formula:
where the unit of temperature is degree Celcius.
■The piecewise linear dependency can be specified with:
pdbSetDoubleArray SiliconGermanium Mechanics ShearModulus.TTable
{ <T1> <v1> ... <Tn> <vn> }
The temperature unit is degree Celcius.
The linear dependency defined by <parameter>.T1 is ignored.
NOTE Do not mix the new syntax with the old syntax (see Deprecated Syntax
for Temperature-dependent Mechanical Properties). Some of the old
syntax may exist in the Advanced Calibration script.
PT() PP.T1T26.85–()+=

9: Computing Mechanical Stress
Plane Stress Analysis
Sentaurus™ Process User Guide 665
H-2013.03
Deprecated Syntax for Temperature-dependent Mechanical Properties
The linear temperature dependency of the elastic moduli can be defined by a rate coefficient.
For isotropic elasticity, this parameter is specified with:
pdbSet <material> Mechanics YoungsModulusRate <n>
For anisotropic elasticity, the coefficients are defined with:
pdbSet <material> Mechanics C11Rate <n>
pdbSet <material> Mechanics C12Rate <n>
pdbSet <material> Mechanics C44Rate <n>
The linear temperature dependency can be applied to the thermal expansion coefficient using
the command:
pdbSet <material> Mechanics ThExpCoeffRate <n>
to specify the rate coefficient. For the nonlinear temperature-dependent thermal expansion
coefficient, a piecewise linear interpolation function is used. To switch on the model, use the
command:
pdbSet Mechanics Interp.ThExpCoeff 1
The piecewise linear function is specified with:
pdbSet <material> Mechanics ThExpCoeff.Interp {<temp> <LCTE> <temp>
<LCTE> ... }
For the first yield stress of plastic materials, an Arrhenius expression is used for the
temperature-dependent effect. The prefactor and exponent of the Arrhenius expression are
specified respectively with the following commands:
pdbSet <material> Mechanics FirstYield <n>
pdbSet <material> Mechanics FirstYieldW <n>
Plane Stress Analysis
In two-dimensional problems, the elastic models implemented above follow the plane strain
formulation by default. Under the plane strain assumption:
(945)
While this is good for structures where the strain in the third direction is very small compared
to the cross section, it would give inaccurate results for thin structures. Thin plate-like
εzz 0 σzz 0≠;=

9: Computing Mechanical Stress
Equations: Global Equilibrium Condition
666 Sentaurus™ Process User Guide
H-2013.03
structures where one dimension is very small compared to the other two can be modeled under
the plane stress assumption:
(946)
The strain is obtained as a function of other strains, for example, for purely elastic
structures:
(947)
The plane stress model can be switched on for a particular region using:
pdbSetBoolean <material> Mechanics PlaneStress 1
NOTE Yo u c an c ombine pla ne st ress a nd plane strain formulations within a
structure by switching on plane stress in only a few regions. However,
such a simulation is not advisable.
If both plane stress and plane strain regions are present in a structure, the material thickness of
regions can be specified by:
pdbSetDouble <material> Mechanics Thickness <d>
where <d> is specified in micrometers.
Equations: Global Equilibrium Condition
The equations for mechanics in Sentaurus Process are the quasistatic equations of force
equilibrium.
The strain rate tensor is related to the symmetric part of the velocity gradient and is given by:
(948)
Strain is then related to stresses through any of the material models defined in Material Models
on page 644. For all models, the global equilibrium condition is given by:
(949)
The above equations are solved using the finite-element method. The solution is a vector
representing the velocity components at each node. These velocities are used to compute the
εzz 0 σzz
;≠0=
εzz
εzz νε
xx εyy
+()–=
ε
·jk
1
2
---vj
∂
xk
∂
------- vk
∂
xj
∂
-------+
=
∂σjk v()
xk
∂
-------------------
k
0=

9: Computing Mechanical Stress
Boundary Conditions
Sentaurus™ Process User Guide 667
H-2013.03
strain and stresses. The stresses and the boundary conditions determine the mechanical state of
the system.
NOTE The stress and strain are derivatives of the velocity. They are, therefore,
computed at one order of accuracy lower than the solution variable. This
also means that they are discontinuous across the elements. When
visualized, the stress values may appear badly converged even if the
linear solver has converged.
In addition, the quasistatic mechanics equations are elliptic in nature and, therefore, are prone
to high levels of shape dependence. This is most frequently seen at gate corners during
polysilicon reoxidation steps or at the corners of the STI trench during liner oxidation. These
equations also exhibit a high sensitivity to the mesh modification algorithms at these corners.
NOTE At sharp corners, the mechanics equations have a singularity. Therefore,
it is not possible to discretize at a corner correctly using regular types of
element.
Boundary Conditions
Equations for stress equilibrium require boundary conditions to define the system completely.
Figure 90 Default mechanics boundary conditions in Sentaurus Process axis orientation
In Sentaurus Process, various boundary conditions can be selected using:
pdbSet Mechanics <side> BoundaryCondition <model>
where:
■<side> is Left, Right, Front, or Back.
■<model> is HomNeumann or Dirichlet.
Vx=0
Vy=0 Vy=0
9: Computing Mechanical Stress
Boundary Conditions
668 Sentaurus™ Process User Guide
H-2013.03
The default boundary conditions are zero velocities in the direction perpendicular to the
boundary planes. Since velocities are set to fixed values along the boundaries, these boundary
conditions are referred to as Dirichlet boundary conditions in directions perpendicular to
boundary planes. The HomNeumann boundary condition is used when the plane must be free.
For example, if you want to set the ‘right’ plane to be free, use the command:
pdbSet Mechanics Right BoundaryCondition HomNeumann
The HomNeumann boundary condition implies a zero normal stress (shown in Figure 91).
Figure 91 HomNeumann boundary condition on ‘right’ boundary plane
Dirichlet boundary conditions are imposed using the penalty method, by default. To adjust the
penalty factor, use the command:
pdbSet Mechanics Boundary.Penalty.Factor {<n>}
The default penalty factor is 1.0e12. The larger this factor, the more accurate the enforcement
of Dirichlet boundary conditions. However, using an extremely large penalty factor could lead
to an ill-conditioned matrix and, therefore, could slow down the linear equation solver.
Alternatively, you can use the matrix reduction method to impose Dirichlet boundary
conditions. To choose the penalty method or matrix reduction method, use the command:
pdbSet Mechanics Boundary.Method.Type {<model>}
where {<model>} is either Penalty or MatrixReduction.
NOTE To ensure the structure is bounded by a perfect rectangle, the
displacements computed by these general boundary conditions are not
applied to the structure. However, they evaluate the stresses correctly.
This assumption is consistent with the small deformation assumption
within each mechanics time step.
Vx=0
Vy=0 sy=0

9: Computing Mechanical Stress
Boundary Conditions
Sentaurus™ Process User Guide 669
H-2013.03
Example: Applying Boundary Conditions
line x loc=-0.02 tag=e spacing=0.005
line x loc=0 tag=a spacing=0.005
line x loc=0.2 tag=b spacing=0.05
line y loc=0 tag=c spacing=0.05
line y loc=2 tag=d spacing=0.05
region silicon xlo=a xhi=b ylo=c yhi=d
region oxide xlo=e xhi=a ylo=c yhi=d
init !DelayFullD
pdbSetDouble Mechanics RefThExpCoeff 0
pdbSet Mechanics Right BoundaryCondition HomNeumann
pdbSet Oxide Mechanics Viscosity0 1e40
pdbSet Oxide Mechanics ViscosityW 0
temp_ramp name=tr1 temperature=600 ramprate=30<K/min> time=10<min>
diffuse temp_ramp=tr1
struct tdr=rampup
diffuse time=10 temp=900 wet
struct tdr=postout
Sentaurus Process also provides a general way to specify boundary conditions for stress
analysis through the stressdata command:
stressdata bc.location=<c> bc.value= { dx=<n> | dy=<n> | dz=<n> }
where:
■bc.location can be Left|Right|Front|Back|Bottom.
■dx, dy, and dz are used to specify displacement rates (default unit: cm/s).
The displacement rates are applied to the area defined through bc.location, where
Left|Right|Front|Back|Bottom refer to the outer boundary surfaces of the simulation
domain. At least at one node, the displacement along any coordinate system direction must be
fixed to remove the rigid body motion.

9: Computing Mechanical Stress
Boundary Conditions
670 Sentaurus™ Process User Guide
H-2013.03
Pressure Boundary Condition
The pressure boundary condition is used to apply uniform pressure on the exterior boundary.
The direction of the loading depends on the normal of the exterior surface. To apply the
pressure boundary condition, use the stressdata command, for example:
stressdata bc.location = <c> bc.value = {pressure=<n>}
where bc.location can be left, right, front, back, bottom.
Advanced Dirichlet Boundary Condition
A more advanced Dirichlet-type boundary condition also can be defined, which specifies both
the translational and rotational velocities on the boundaries. It is defined using the command:
stressdata bc.location=<c> bc.rotation.axis= {xa=<c> | ya=<c> | za=<c>} \
bc.value= {dx=<n> | dy=<n> | dz=<n> | rx=<n> | ry=<n> | rz=<n>}
where:
■bc.location can be Left|Right|Front|Back|Bottom.
■dx, dy, and dz specify displacement rates (default unit: cm/s).
■rx, ry, and rz specify rotational velocities (default unit: rad/s).
■xa, ya, and za specify the coordinates of the point around which the rotation occurs
(default unit: cm).
Periodic Boundary Condition
The periodic boundary condition is used for structures with a periodically repeating pattern.
This condition is used on periodic structures with assigned master and slave boundaries. The
9: Computing Mechanical Stress
Boundary Conditions
Sentaurus™ Process User Guide 671
H-2013.03
slave boundary has the same deformation profile as the master boundary. In Figure 92, the left
and right boundaries are bound together by the periodic boundary condition.
Figure 92 Periodic boundary condition
To apply the periodic boundary condition to the outer bounding surfaces, use the command:
pdbSet Mechanics <Left | Right | Front | Back> Periodic 1
If this command is specified on a sidewall, the opposite sidewall is defined automatically as a
periodic boundary. Conflict of boundary condition definitions are checked on all sidewalls. For
example, to apply a periodic boundary condition on the left and right sidewalls, use one of the
following commands:
pdbSet Mechanics Left Periodic 1
pdbSet Mechanics Right Periodic 1
Both the periodic and coupling boundary conditions are implemented using the penalty
method. To adjust the penalty factor, use the command:
pdbSet Mechanics Constraint.Penalty.Factor <n>
The default penalty factor is . The larger this factor, the more accurately the periodic
or coupling boundary conditions will be enforced. Using an extremely large penalty factor
could lead to an ill-conditioned matrix and, therefore, slow down or even fail the linear
equation solver.
NOTE If you choose to apply periodic boundary conditions, all other boundary
conditions defined through the old pdbSet method will be ignored and
must be redefined using the stressdata command.
UU
UU
U = U ; U = U ...
1L
1R
1R
2R2L
1L2R2L
1.0 13
×10

9: Computing Mechanical Stress
Time Step Control
672 Sentaurus™ Process User Guide
H-2013.03
Time Step Control
It may be necessary to use time step control when viscous or viscoelastic materials are present
in the structure. Usually, when other dopants also are present, the time steps will be sufficiently
limited by the diffusion solver. If dopants are not present or for materials with a low viscosity,
the time step control for the Maxwell model can be switched on with:
pdbSet Mechanics Visc.Step.Control 1
Time-stepping can be controlled with the displacement increment and with the relative
relaxation time. To switch on these two options, use:
pdbSet Mechanics Visc.Step Limit.Disp 1
and:
pdbSet Mechanics Visc.Step Limit.ScaleT 1
respectively. For more control parameters, see “Mechanics Visc.Step” in the Parameter
Database Browser.
For the standard linear solid model for viscoelasticity, time step control is activated
automatically (see Time-Step Control for Mechanics on page 871 for details).
Stress-causing Mechanisms
Every mechanical system needs a set of stress-driving mechanisms to reach a stressed state.
The stress-inducing mechanisms in Sentaurus Process are listed here.
Stress Induced by Growth of Material
During the oxidation process, volume is produced. Consuming silicon of volume 1 during
thermal oxidation produces oxide of volume 2.25. This process introduces velocities at a
growing interface: a velocity vector pointing into the silicon describes the proceeding of the
oxidation front, and a velocity vector pointing into the oxide accounts for the volume expansion
as described above. The latter is responsible for the generation of mechanical stresses and,
therefore, is used as a boundary condition for the mechanical problem.

9: Computing Mechanical Stress
Stress-causing Mechanisms
Sentaurus™ Process User Guide 673
H-2013.03
Densification-induced Stress
A typical densification process uses thermal heating to increase the density of a porous
material. As the material density increases, its volume shrinks and the volume shrinkage
generates stresses.
The densification-induced stress computation is switched on using the density.increase
parameter in the diffuse command or the temp_ramp command, such as:
diffuse temperature=1000<C> time=30<min> \
density.increase= {[<regionName> = value] [<material> = value]}
temp_ramp name=dens time=1 temp=1000 density.increase= { oxide=0.02 }
diffuse temp_ramp=dens
The total amount of density increase can be specified per material or per region for a given
diffuse (or temp_ramp) step as shown above. A proportional amount of density increase is
applied during each time step of the densification process.
The densification operation can be performed for all existing materials, as well as new
materials defined using the mater command:
mater add name=TEOS new.like=oxide
diffuse time=1 temp=1000 density.increase = { TEOS = 0.03 }
For densification processes involving large amounts of volume shrinkage, the material
boundaries and meshes can be updated using the following settings:
pdbSet Grid Inert.Modify.Mesh 1
pdbSetDouble TEOS Grid MinimumVelocity 0
For a complete densification process that has distinguished density changes, multiple diffuse
steps can be used with different density increases for each segment of the process.
Selectively Switching Off Grid Movement
The parameter MinimumVelocity can be used to selectively switch off point or interface
movement. This can be useful, for example, when a mechanics simulation computes a small
amount of boundary movement that is either unwanted or could cause element quality to suffer
in the vicinity, and the approximation of no movement is acceptable. In general, the command
is:
pdbSet <material> Grid MinimumVelocity <speed>
If <material> is a bulk material (no underscore), the parameter applies to bulk points. If the
speed of the bulk points is less than <speed> (in cm/s), Sentaurus Process truncates the speed

9: Computing Mechanical Stress
Stress-causing Mechanisms
674 Sentaurus™ Process User Guide
H-2013.03
to zero. This truncation is applied to material silicon by default. On the other hand, if
<material> is an interface material (having an underscore such as
PolySilicon_Silicon), the parameter only applies to points on that interface. This
truncation is applied to material Silicon by default.
NOTE The moving mesh operations can become unstable for values of
MinimumVelocity that are neither very large nor zero. Very large
values stop all motion, and 0 allows all motion.
Stress Caused by Thermal Mismatch
Temperature changes during the process described by the temp_ramp command or the
keyword ramprate in the diffuse command lead to stress in the structure caused by the
different thermal expansion coefficients of the relevant materials. When necessary, the stress
computation can be switched off by using the stress.relax flag:
diffuse temperature=1000<C> time=30<min> !stress.relax
NOTE If viscous or viscoelastic materials are present in the structure, the stress
distribution may change even without a change in the temperature due
to viscoelastic relaxation.
By default, stresses are computed during inert annealing for 2D simulations. To
apply !stress.relax to all inert annealing steps, use the command:
pdbSet Compute NoStressRelax 1
The thermal expansion coefficient for certain materials can be found in the parameter database
as follows:
<material> Mechanics ThExpCoeff
Thermal expansion only affects the dilatational part of the constitutive equation:
(950)
The change in the temperature is described by and is the relative
thermal expansion coefficient of a certain material with respect to the thermal expansion
coefficient of the substrate.
In certain examples, like bending, you may want to use absolute expansion coefficients instead
of relative. This can be achieved by setting a parameter called RefThExpCoeff as follows:
pdbSetDouble Mechanics RefThExpCoeff 0.
σkk 3Kεkk αrel∆T–()=
∆T
αrel αmat αsubs
–=

9: Computing Mechanical Stress
Stress-causing Mechanisms
Sentaurus™ Process User Guide 675
H-2013.03
All the thermal expansion coefficients are computed with respect to the substrate. This
reference value is changed by setting a certain region as substrate and resetting the thermal
expansion coefficient. A region can be tagged as the substrate in several ways:
■Use the substrate keyword when defining regions with the region command before the
init command.
■If a saved structure is being loaded into Sentaurus Process, a region is tagged as the
substrate with the command:
region name=<region_name> substrate
The reference thermal expansion coefficient can be directly set with:
pdbSetDouble Mechanics RefThExpCoeff <n>
This command overwrites the reference thermal expansion coefficient setting from the
substrate.
Materials expand differently in different temperature ranges. The linear dependency of the
thermal expansion coefficient on temperature can be specified by:
pdbSet Silicon Mechanics ThExpCoeffRate 4e-9
So the total thermal expansion coefficient at the elevated temperature can be expressed as:
(951)
where the room temperature is set to 300 K.
Lattice Mismatch
The presence of impurities, such as germanium and carbon, can change the lattice parameters
of crystalline silicon. This effect has been exploited in two ways technologically:
■Introducing an impurity during epitaxy to form a strained layer.
■Growing a substrate (typically, a very thick layer grown on a standard substrate) to produce
a customized lattice constant.
However, most technological applications are based on the first use, for example, when SiGe
source/drain pockets are grown on silicon substrates. For strained SiGe epitaxy, Sentaurus
Process automatically computes and applies the strain, and no user input is necessary.
T
aThExpCoeff ThExpCoeffRate+TRoomTemperature–()×=
9: Computing Mechanical Stress
Stress-causing Mechanisms
676 Sentaurus™ Process User Guide
H-2013.03
For customized lattice-spacing substrates or other material systems, more setup of the tool is
required. This section explains the theory and implementation of this model and gives an
example. Figure 93 shows a simple SiGe wafer.
Figure 93 Simple SiGe wafer for customizing lattice-spacing
There are four main regions of the manufactured substrate. The silicon region has the graded
buffer layer where the Ge concentration increases linearly from zero to the required
concentration. The manufacturing process of this layer is designed such that all the dislocations
are forced energetically to nucleate here, and the wafer is completely relaxed. The relaxed layer
that is grown on top of the graded layer has no dislocations and no strain. The lattice-spacing
of this layer is determined by the Ge mole fraction. The lattice-spacing of this layer controls
the strains obtained in the top strained layer. The top strained layer is grown depending on the
kind of strain required. If this layer is to be in a tensile state, the Ge concentration here must be
less than that of the relaxed layer. In the case of a compressive state, the Ge concentration must
be greater than that of the relaxed layer. This layer has a thermodynamic limit on its thickness
since the strain energy it contains should be less than the dislocation nucleation energy. The
strain energy is directly proportional to the volume that is under the strain. The strain profile of
germanium in silicon is given approximately by:
(952)
where is the Ge mole fraction calculated as the germanium concentration divided by the
silicon lattice density.
In the relaxed region, Sentaurus Process modifies the lattice-spacing. This results in no stresses
due to the presence of germanium. In the strained region, the lattice-spacing is fixed by the
lattice-spacing in the relaxed region. Now, using the Ge mole fraction in the strained region,
Ge Concentration
Relaxed Layer
Graded Layer
Pure Silicon
Strained Layer
0.01
0.5
0.7
X µm
ε0.0425x=
x
9: Computing Mechanical Stress
Stress-causing Mechanisms
Sentaurus™ Process User Guide 677
H-2013.03
the effective unstrained lattice-spacing is computed, and the stresses are based on the difference
of the effective lattice-spacing and the lattice-spacing of the relaxed region.
For example, assume the strained layer has no Ge: The effective lattice-spacing is that of silicon
given by . The lattice-spacing of the relaxed SiGe part is, for example, . The strain in
the strained region is:
(953)
The strain computed using Eq. 953 is applied as a biaxial strain in the y- and z-directions.
Using the Lattice Mismatch Model
For the most common case of SiGe layers grown on silicon substrates, the model is switched
on by default, and strain is computed and updated as necessary. For simulating other material
systems, a few settings are required to instruct Sentaurus Process how the strain should be
computed.
If the substrate is not silicon or it is not the lowest most region in the substrate (that is, the
largest x-coordinate), you must identify the substrate region in the wafer. Use the region
command and include the keyword substrate for the appropriate region. If there is no
substrate defined in the loaded structure, use the following command to tag a region as a
substrate:
region name=<region_name> substrate
Regions isolated from the substrate by nonsubstrate materials must be tagged as substrate to
account for the lattice mismatch effect.
For systems other than SiGe, Sentaurus Process must know the strain profile of the field in the
substrate. The strain_profile command is used to specify this. The strain is specified as a
piecewise linear function of the mole fraction. For Ge in silicon, it is:
strain_profile Silicon species=Germanium strain= {0 0.0425} ratio= {0 1}
or:
pdbSet Silicon Germanium Conc.Strain {0 0 1 0.0425}
The lattice mismatch model for SiGe is switched on by default. Next, for customized lattice-
spacing substrates, the substrate must be given a strain profile. The strain profile can be
specified with the substrate_profile command or the profile command as a piecewise
linear function of the x-coordinate:
profile region=<region_name> name=Germanium \
concentration= {1e10 1e10 2e22 2e22 1e10 1e10} \
xcoord= {0 0.01 0.011 0.5 0.7 10} linear
LSi
LSiGe
εLSiGe LSi
–()LSi
⁄=
9: Computing Mechanical Stress
Stress-causing Mechanisms
678 Sentaurus™ Process User Guide
H-2013.03
The location of the top of the relaxed region must be specified in Sentaurus Process. Generally,
this should not be at the top of the relaxed layer (see Figure 93 on page 676) because
germanium diffusion during any anneal step can cause unrealistic stress values to appear in this
area. The best location for the top of the relaxed region is approximately two-thirds of the
relaxed layer thickness from the top of the relaxed layer. In this example, it is approximately
. This reference position can be set with the command:
pdbSet Silicon Mechanics TopRelaxedNodeCoord 0.35e-4
NOTE In most cases, when the simulation does not require any SiGe substrate
(for example, when SiGe source/drain pockets are grown on silicon
substrates), this parameter is not needed. The reference lattice-spacing
is the one of the substrate; Sentaurus Process detects automatically the
adjacent silicon-like regions and applies to them the lattice mismatch
model. For this reason, the value of this parameter defaults to the bottom
coordinate of the structure.
Finally, for these concentrations to take effect and all mechanics computations to occur, you
must add a short diffusion step if there is none.
The update_substrate command is deprecated. If used, the update_substrate
command should be called only once for initialization assuming all strain profiles have not
been accounted for lattice-mismatch strains.
During dopant redistribution, the lattice-spacing and lattice mismatch strains are updated, and
the doping concentration at the top of the relaxed layer may change. To disable automatic
updating of lattice-mismatch strains, use:
pdbSet Silicon Mechanics UpdateStrain 0
To switch off the lattice-spacing tracking at the top of the relaxed layer, use:
pdbSet Mechanics LatticeHistory 0
Total Concentration Model
The total concentration model computes the total contribution of lattice mismatch stress with
the current impurity concentration and the elastic moduli at the current temperature. For binary
compound materials, the elastic moduli are computed with the current mole fraction. With this
approach, the lattice mismatch stress is history independent and can change even with an
unchanged doping profile.
0.35 µm

9: Computing Mechanical Stress
Stress-causing Mechanisms
Sentaurus™ Process User Guide 679
H-2013.03
This is the default lattice mismatch model. To switch off this model by computing the lattice
mismatch stress increment with the elastic moduli during doping profile change, use the
command:
pdbSet Mechanics Total.Concentration.Model 0
Reference Concentration Model
The reference concentration model is a simplified lattice mismatch model, which does not
distinguish the relaxed region and strained region by specifying the location of the top of the
relaxed region. Only relative concentration accounts for the lattice-spacing and strain changes.
For example, the strain in SiGe is:
(954)
and the lattice-spacing is computed by , where is the lattice density
of silicon and is the reference Ge concentration in SiGe defined by:
pdbSet Silicon Germanium Ref.Concentration 1e22
The lattice-spacing and strain from this model may not be physical in the relaxed region.
The reference concentration model is used when the structure is flipped for backside
processing. To switch on this model, use the command:
pdbSet Mechanics Reference.Concentration.Model 1
Strained Deposition
Impurity-induced stress can be introduced locally during deposition to account for a lattice-
spacing change due to stress rebalancing. For example, the SiGe lattice-spacing during
unconstrained growth gradually returns to the unconstrained SiGe lattice-spacing. The lattice
mismatch effect should diminish during the SiGe growth.
ε0.0425 CGe Cref
–()CSi
⁄⋅=
LSiGe LSi 1ε+()⋅=
CSi
Cref

9: Computing Mechanical Stress
Stress-causing Mechanisms
680 Sentaurus™ Process User Guide
H-2013.03
The following steps are performed when strained deposition is enabled:
1. Deposit a new layer.
2. Apply a doping profile, and compute the lattice spacing of the newly deposited layer.
3. Set the lattice spacing of the deposited layer to that of the underlayer, and compute the
mismatch strain.
4. Perform stress relaxation to establish stress equilibrium, and update the lattice spacing.
5. Merge layers if needed.
To correctly catch the relaxation effect, the thickness of the deposited layer must be chosen
properly; a fine mesh is required. Multiple deposition can be particularly useful in such cases.
To switch on this model, use the command:
pdbSet Mechanics StrainedDeposition 1
and set the option Strained.Lattice in the deposit command.
The total concentration model is disabled during strained deposition. The reference
concentration model should not be used with strained deposition.
Edge Dislocation
The existence of crystal lattice defects, such as dislocation, affects the channel stress state. The
impact of edge dislocation is included by superposing the dislocation-induced stress field from
elasticity theory. Each edge dislocation can be defined with:
stressdata apply.dislocation dislocation.origin= {<n> <n> <n>}
para.orient= {<n> <n> <n>} perp.orient= {<n> <n> <n>} region=<c> <material>
where:
■dislocation.origin is the location of the dislocation core.
■para.orient specifies the direction of the edge dislocation or the direction of the half
plane.
■perp.orient is Burger’s vector in the perpendicular direction to the half plane.
Here, the magnitude of perp.orient is the slip distance. You must supply either a region
name or a material name. If region is specified, the stress field is superposed to this region.
If material is specified, the stress field is applied to all regions of crystalline material. To save
the edge dislocation geometry information to a TDR file for visualization, specify the
command option saveTDR with the edge dislocation definition.
9: Computing Mechanical Stress
Stress-causing Mechanisms
Sentaurus™ Process User Guide 681
H-2013.03
Figure 94 Edge dislocation located at the origin O; n1 is Burger’s vector and n2 is the
direction of the half plane
Singularity exists in the analytic solution at the dislocation core. Without using a nonlinear
atomistic theory, the stresses in the core region within a few magnitudes of Burger’s vector to
the dislocation core are smoothed away. The factor for this core radius can be defined with:
pdbSet Mechanics Dislocation.Coresize.Factor 2.0
A prototype model for positioning the edge dislocations is available by minimizing the elastic
strain energy [11]. The stress field from each edge dislocation is superposed. The elastic strain
energy is determined after force equilibrium with edge dislocations at their initial locations.
The initial location of edge dislocation serves as the initial guess and can be defined by:
stressdata !apply.dislocation dislocation.origin= {<n> <n> <n>} \
para.orient= {<n> <n> <n>} perp.orient= {<n> <n> <n>} region=<c>
where:
■dislocation.origin is the initial location of the edge dislocation.
■!apply.dislocation is specified to delay applying the dislocation-induced stress field.
Multiple edge dislocations with different Burger’s vectors can be defined separately with this
syntax.
When all the edge dislocations for minimizing the elastic strain energy are specified, you can
start the optimization with the command:
stressdata origin.max= {<n> <n> <n>} origin.min= {<n> <n> <n>} \
optimize.dislocation
where origin.max and origin.min define the range of dislocation positions in the specified
region. Some additional parameters for optimization convergence control also can be defined
in this command (see stressdata on page 1151).
n2
n1
O

9: Computing Mechanical Stress
Stress-causing Mechanisms
682 Sentaurus™ Process User Guide
H-2013.03
The movement of edge dislocations depends on the gradient of the total elastic strain energy
computed from a discrete integral over all elements. The target of the optimization is set to -5
multiplied by the absolute value of the starting elastic strain energy. This factor can be changed
with:
pdbSetDouble Mechanics Energy.Optimization.Factor <n>
The coordinates of the edge dislocations after optimization are returned in a Tcl list formatted
as <x1> <y1> <x2> <y2> ... for two dimensions, and <x1> <y1> <z1> <x2> <y2>
<z2> ... for three dimensions. The final stress state remains the same as before the edge
dislocations are introduced. The edge dislocations may stop at the local minimum where the
elastic strain energy has not reached the global minimum. In such a case, a new optimization
step must be started with the initial guess of the edge dislocation positions adjusted based on
the previous optimization result. It is also helpful to refine the mesh.
Intrinsic Stress
Certain process steps require the deposition of materials with intrinsic stresses. Sentaurus
Process can be used to model these process steps. The intrinsic stresses (StressELXX,
StressELYY, StressELZZ, StressELXY, StressELYZ, StressELZX) can be prescribed in
the deposit command (see deposit on page 901). After stress relaxation, the resulting stresses
will be less than the prescribed ones by default. You can scale the prescribed stresses so that
for a flat surface, the relaxed stress will be the same as the prescribed stress. To scale the
stresses, use the command:
pdbSet Mechanics StressRelaxFactor 1
For deposition in 3D, you can specify stresses in specific layers using the stressdata
command (see stressdata on page 1151). For example:
stressdata nitride syyi=1.4e10
sets the yy component of the intrinsic stress in the nitride to .
1.4 10
×10 dyn/cm2

9: Computing Mechanical Stress
Stress-causing Mechanisms
Sentaurus™ Process User Guide 683
H-2013.03
For interconnect simulations, intrinsic stresses in metal lines can be modeled as width
dependent [12] with either a linear relation or a logarithmic relation, using the parameters
defined through the stressdata command:
■If modeled as a linear relation, the total intrinsic stresses are given by:
■If modeled as a natural logarithmic relation, the total intrinsic stresses are given by:
where are defined through the parameters sxxi, sxx1, sxx2, and base of
the stressdata command. The other two components ( and ) are defined in the same
way, and is calculated internally with respect to the region (not material) boundaries.
Stress Rebalancing after Etching and Deposition
When materials are removed from or added to a given structure, physical stress distributions
generally change with the corresponding geometry and boundary changes. In simulations, a
stress-rebalancing step is required to re-establish the stress equilibrium in the structure and to
conform the stress distributions to the new boundaries. By default, a stress-rebalancing
operation is called after etching or deposition is performed. To omit the stress-rebalancing step,
use:
pdbSet Mechanics EtchDepoRelax 0
Automated Tracing of Stress History
Thermal residual stress in a given device structure is a function of its fabrication history, which
consists of process steps at various temperatures and temperature ramps in between. To model
stress evolution accurately, all temperature ramps should be traced. When the pdb parameter
StressHistory is switched on, for example:
pdbSet Mechanics StressHistory 1
the temperature gaps between process steps such as diffusion, deposition, and etching are
detected and filled with instant stress-rebalancing, solving for thermal mismatch strains and
stresses.
σxx σxxi σxx1
wx
wb
------+=
σyy σyyi σyy1
wy
wb
------+=
σzz σzzi σzz1
wz
wb
------+=
σxx σxxi σxx2
wx
wb
------
ln+=
σyy σyyi σyy2
wy
wb
------
ln+=
σzz σzzi σzz2
wz
wb
------
ln+=
σxxi σxx1σxx2wb
,,,
yy
zz
w

9: Computing Mechanical Stress
Saving Stress and Strain Components
684 Sentaurus™ Process User Guide
H-2013.03
Saving Stress and Strain Components
By default, stress-tensor components are saved on both elements and nodes. The elastic
portions of the strain-tensor components also are saved on both elements and nodes by default.
The elastic strains are computed from stresses using isotropic elasticity by default. The
anisotropic elasticity also can be used for a given crystalline material when the corresponding
pdb parameter Anisotropic is set. The elastic strain-field computing and saving operation
can be omitted by using the following command:
pdbSet Mechanics saveElasticStrain 0
The stress tensor can be decomposed and the resulting dilatational and deviatoric stress
components can be saved on nodes when the following pdb parameter is switched on:
pdbSet Mechanics decomposeStress 1
Description of Output Variables
The mechanics module in Sentaurus Process assumes that stresses and strains are defined on
elements. However, not all tools can read or visualize element values. For this reason,
Sentaurus Process performs an element-to-node interpolation of stresses as a postprocessing
step and writes both forms of stresses to output.
The element stresses are prefixed by StressEL and the nodal stresses are prefixed by Stress.
The tensor components are given by the post-fix (XX, YY, ZZ, XY, YZ, ZX).
In history-dependent materials, you cannot create a simple, closed-form relation between
stresses and strains. It is useful, however, to compute the elastic part. The elastic component of
the strain is an indicator of the stored strain energy in the system. In addition, the elastic
component of the strain is the total strain in elastic materials such as silicon and polysilicon.
Pressure is one-third of the negative of the trace of the stress tensor:
(955)
The field LatticeSpacing represents the lattice-spacing of the crystal at the location of the
node. This is controlled by the presence of lattice-altering species such as germanium or carbon
in the structure. In addition, the strain_profile command must be specified.
In Sentaurus Process, the select command is used to perform Tcl-level and Alagator-level
operations. To access the stress components, use the select command.
P1
3
---σii
i
–=
9: Computing Mechanical Stress
Saving Stress and Strain Components
Sentaurus™ Process User Guide 685
H-2013.03
The stresses and strains are represented as symmetric tensors. To access the xx, yy, and zz
components of nodal stress values, the variable references for the select command are
Stress_xx, Stress_yy, and Stress_zz, respectively. To access the xy, yz, and zx
components, use Stress_xy, Stress_yz, and Stress_zx, respectively.
For element values, the Boolean keyword element of the select command must be set to
true. To access the xx, yy, and zz components of the element stress values, the variable
references for the select command are StressEL_xx, StressEL_yy, and StressEL_zz,
respectively. To access the xy, yz, and zx components, use StressEL_xy, StressEL_yz, and
StressEL_zx, respectively.
The old variable names for accessing the components of stress and strain tensors are supported
as well. A comparison of the new names and the corresponding deprecated names is included
in Table 66 on page 688.
Table 65 presents descriptions of the mechanics-related output data and whether the variables
apply to elements or nodes.
Table 65 Variable names in Sentaurus Process output files
Variable name Element/Node Description Unit
Displacement_x
Node X component of displacement cm
Displacement_y
Node Y component of displacement cm
Displacement_z
Node Z component of displacement cm
ElasticStrainXX
Node XX component of elastic strain Unitless
ElasticStrainXY
Node XY component of elastic strain Unitless
ElasticStrainYY
Node YY component of elastic strain Unitless
ElasticStrainYZ
Node YZ component of elastic strain Unitless
ElasticStrainZX
Node ZX component of elastic strain Unitless
ElasticStrainZZ
Node ZZ component of elastic strain Unitless
PlasticStrainXX
Node XX component of plastic strain Unitless
PlasticStrainXY
Node XY component of plastic strain Unitless
PlasticStrainYY
Node YY component of plastic strain Unitless
PlasticStrainYZ
Node YZ component of plastic strain Unitless
PlasticStrainZX
Node ZX component of plastic strain Unitless
PlasticStrainZZ
Node ZZ component of plastic strain Unitless
PlasticStrainEQV
Node Equivalent plastic strain Unitless
9: Computing Mechanical Stress
Saving Stress and Strain Components
686 Sentaurus™ Process User Guide
H-2013.03
ViscoPlasticStrainXX
Node XX component of viscoplastic strain Unitless
ViscoPlasticStrainXY
Node XY component of viscoplastic strain Unitless
ViscoPlasticStrainYY
Node YY component of viscoplastic strain Unitless
ViscoPlasticStrainYZ
Node YZ component of viscoplastic strain Unitless
ViscoPlasticStrainZX
Node ZX component of viscoplastic strain Unitless
ViscoPlasticStrainZZ
Node ZZ component of viscoplastic strain Unitless
ViscoPlasticStrainEQV
Node Equivalent viscoplastic strain Unitless
CreepStrainELXX
Element XX component of creep strain Unitless
CreepStrainELXY
Element XY component of creep strain Unitless
CreepStrainELYY
Element YY component of creep strain Unitless
CreepStrainELYZ
Element YZ component of creep strain Unitless
CreepStrainELZX
Element ZX component of creep strain Unitless
CreepStrainELZZ
Element ZZ component of creep strain Unitless
CreepStrainELEQV
Element Equivalent creep strain Unitless
SwellingStrainELXX
Element XX component of swelling strain Unitless
SwellingStrainELXY
Element XY component of swelling strain Unitless
SwellingStrainELYY
Element YY component of swelling strain Unitless
SwellingStrainELYZ
Element YZ component of swelling strain Unitless
SwellingStrainELZX
Element ZX component of swelling strain Unitless
SwellingStrainELZZ
Element ZZ component of swelling strain Unitless
LatticeSpacing
Node Lattice-spacing cm
Pressure
Node Pressure Pa
StressELXX
Element XX component of element stress Pa
StressELXY
Element XY component of element stress Pa
StressELYY
Element YY component of element stress Pa
StressELYZ
Element YZ component of element stress Pa
StressELZX
Element ZX component of element stress Pa
StressELZZ
Element ZZ component of element stress Pa
StressXX
Node XX component of node stress Pa
Table 65 Variable names in Sentaurus Process output files
Variable name Element/Node Description Unit
9: Computing Mechanical Stress
Saving Stress and Strain Components
Sentaurus™ Process User Guide 687
H-2013.03
StressXY
Node XY component of node stress Pa
StressYY
Node YY component of node stress Pa
StressYZ
Node YZ component of node stress Pa
StressZX
Node ZX component of node stress Pa
StressZZ
Node ZZ component of node stress Pa
MisesStress
Node von Mises stress Pa
DeformationResistance
Node Deformation resistance Pa
ElasticEnergyDens
Node Elastic strain energy density J/m3
PlasticEnergyDens
Node Plastic strain energy density J/m3
ViscoPlasticEnergyDens
Node Viscoplastic strain energy density J/m3
CreepEnergyDensEL
Element Creep strain energy density J/m3
BaseStressELXX
Element XX component of elastic element stress for
Standard Linear Solid viscoelasticity model
Pa
BaseStressELXY
Element XY component of elastic element stress for
Standard Linear Solid viscoelasticity model
Pa
BaseStressELYY
Element YY component of elastic element stress for
Standard Linear Solid viscoelasticity model
Pa
BaseStressELYZ
Element YZ component of elastic element stress for
Standard Linear Solid viscoelasticity model
Pa
BaseStressELZX
Element ZX component of elastic element stress for
Standard Linear Solid viscoelasticity model
Pa
BaseStressELZZ
Element ZZ component of elastic element stress for
Standard Linear Solid viscoelasticity model
Pa
BaseElasticStrainELXX
Element XX component of elastic element strain for
Standard Linear Solid viscoelasticity model
Unitless
BaseElasticStrainELXY
Element XY component of elastic element strain for
Standard Linear Solid viscoelasticity model
Unitless
BaseElasticStrainELYY
Element YY component of elastic element strain for
Standard Linear Solid viscoelasticity model
Unitless
BaseElasticStrainELYZ
Element YZ component of elastic element strain for
Standard Linear Solid viscoelasticity model
Unitless
BaseElasticStrainELZX
Element ZX component of elastic element strain for
Standard Linear Solid viscoelasticity model
Unitless
Table 65 Variable names in Sentaurus Process output files
Variable name Element/Node Description Unit
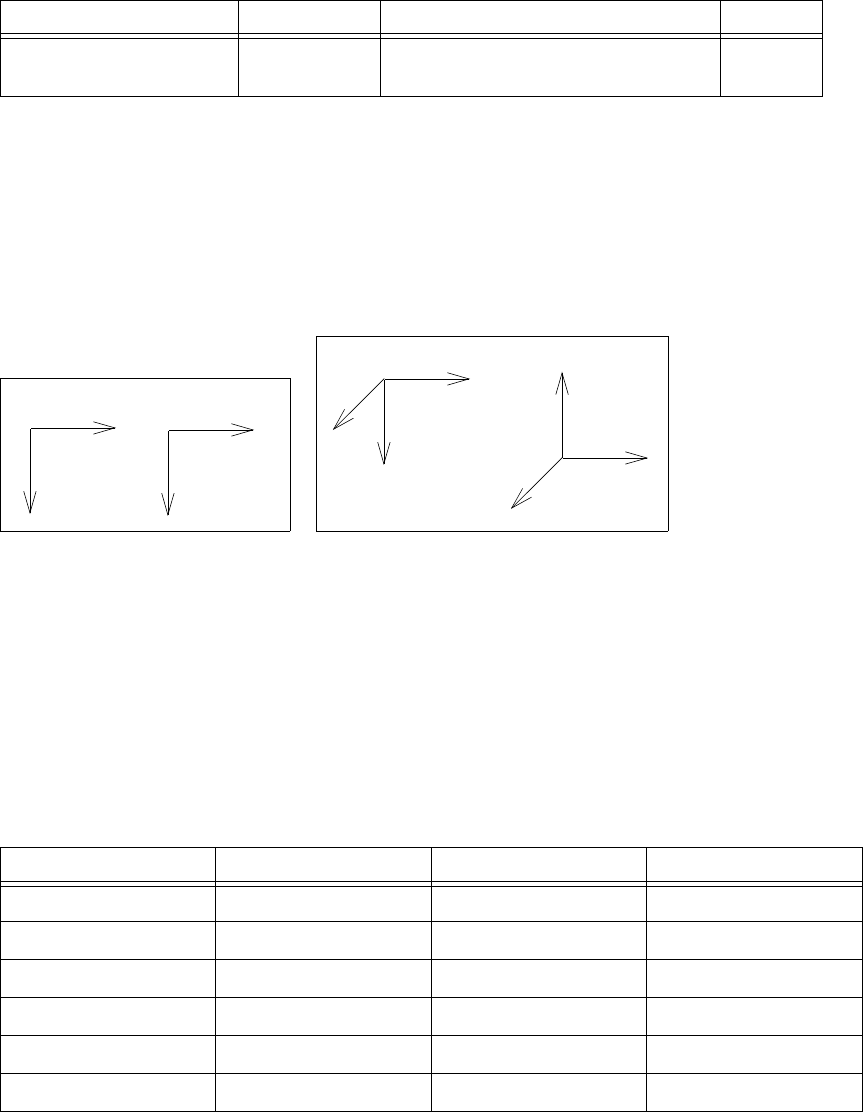
9: Computing Mechanical Stress
Saving Stress and Strain Components
688 Sentaurus™ Process User Guide
H-2013.03
NOTE The stresses and strains in the output file are according to the UCS,
unless you explicitly request to save in the DF–ISE coordinate system
by using the math coord.dfise command. The UCS is the same as
the Sentaurus internal coordinate system, but differs from the DF–ISE/
TDR coordinates. Therefore, it is important to note the directions of the
axes in 2D and 3D (see Figure 95).
Figure 95 (Left) Axis orientation in 2D and (right) axis orientation in 3D
The axis directions in DF–ISE/TDR coordinates are different in 2D and 3D in the UCS.
Figure 95 shows the axis orientation in 2D and in 3D. Consequently, the values of the stress
and strain components change.
For information about the TDR format, refer to the Sentaurus™ Data Explorer User Guide.
Table 66 maps the fields from the select command to the fields in the output files of
Sentaurus Process. The axis directions in Sentaurus Process are the same in 2D and 3D.
BaseElasticStrainELZZ
Element ZZ component of elastic element strain for
Standard Linear Solid viscoelasticity model
Unitless
Ta bl e 66 Va ri abl e na me s in o u t p ut f il es
Field name Field name (deprecated) 2D 3D
StressEL_xx StressELKK_x StressEL-YY StressEL-ZZ
StressEL_yy StressELKK_y StressEL-XX StressEL-YY
StressEL_zz StressELKK_z StressEL-ZZ StressEL-XX
StressEL_xy StressELIJ_x StressEL-XY -StressEL-YZ
StressEL_yz StressELIJ_y
Not applicable
StressEL-XY
StressEL_zx StressELIJ_z
Not applicable
-StressEL-ZX
Table 65 Variable names in Sentaurus Process output files
Variable name Element/Node Description Unit
X
X
Y
Y
UCS DF–ISE/TDR
UCS DF–ISE/TDR
Z
Z
X
X
Y
Y

9: Computing Mechanical Stress
Saving Stress and Strain Components
Sentaurus™ Process User Guide 689
H-2013.03
Stress_xx StressKK_x Stress-YY Stress-ZZ
Stress_yy StressKK_y Stress-XX Stress-YY
Stress_zz StressKK_z Stress-ZZ Stress-XX
Stress_xy StressIJ_x Stress-XY -Stress-YZ
Stress_yz StressIJ_y
Not applicable
Stress-XY
Stress_zx StressIJ_z
Not applicable
-Stress-ZX
Pressure
Not applicable
Pressure Pressure
MisesStress
Not applicable
MisesStress MisesStress
PrincipalStress1
Not applicable
PrincipalStress1 PrincipalStress1
PrincipalStress2
Not applicable
PrincipalStress2 PrincipalStress2
PrincipalStress3
Not applicable
PrincipalStress3 PrincipalStress3
ElasticStrain_xx ElasticStrainKK_x ElasticStrain-YY ElasticStrain-ZZ
ElasticStrain_yy ElasticStrainKK_y ElasticStrain-XX ElasticStrain-YY
ElasticStrain_zz ElasticStrainKK_z ElasticStrain-ZZ ElasticStrain-XX
ElasticStrain_xy ElasticStrainIJ_x ElasticStrain-XY -ElasticStrain-YZ
ElasticStrain_yz ElasticStrainIJ_y
Not applicable
ElasticStrain-XY
ElasticStrain_zx ElasticStrainIJ_z
Not applicable
-ElasticStrain-ZX
PlasticStrain_xx PlasticStrainKK_x PlasticStrain-YY PlasticStrain-ZZ
PlasticStrain_yy PlasticStrainKK_y PlasticStrain-XX PlasticStrain-YY
PlasticStrain_zz PlasticStrainKK_z PlasticStrain-ZZ PlasticStrain-XX
PlasticStrain_xy PlasticStrainIJ_x PlasticStrain-XY -PlasticStrain-YZ
PlasticStrain_yz PlasticStrainIJ_y
Not applicable
PlasticStrain-XY
PlasticStrain_zx PlasticStrainIJ_z
Not applicable
-PlasticStrain-ZX
PlasticStrainEQV
Not applicable
PlasticStrainEQV PlasticStrainEQV
ViscoPlasticStrain_xx ViscoPlasticStrainKK_x ViscoPlasticStrain-YY ViscoPlasticStrain-ZZ
ViscoPlasticStrain_yy ViscoPlasticStrainKK_y ViscoPlasticStrain-XX ViscoPlasticStrain-YY
ViscoPlasticStrain_zz ViscoPlasticStrainKK_z ViscoPlasticStrain-ZZ ViscoPlasticStrain-XX
ViscoPlasticStrain_xy ViscoPlasticStrainIJ_x ViscoPlasticStrain-XY -ViscoPlasticStrain-YZ
ViscoPlasticStrain_yz ViscoPlasticStrainIJ_y
Not applicable
ViscoPlasticStrain-XY
Ta bl e 66 Va ri abl e na me s in o u t p ut f il es
Field name Field name (deprecated) 2D 3D
9: Computing Mechanical Stress
References
690 Sentaurus™ Process User Guide
H-2013.03
The directionality is the same for other tensor fields such as StressEL and ElasticStrain.
Tracking Maximum Stresses
During a typical process flow, the maximum stresses may be reached in a process step and,
subsequently, the stresses may fall. If the material is prone to failure through delamination or
nucleation of dislocations, the failure may occur when the maximum stress is reached. To
always track the maximum stresses, set the following parameter:
pdbSet Mechanics SaveMaxStress 1
The StressMaxEL field is updated when the current stress is greater than the stored stress. In
this way, the maximum is maintained throughout the process flow. The maximum element
stresses and the von Mises stress are computed and stored.
By using the stressdata command (see stressdata on page 1151), a list of maximum stresses
(hot spots) and their locations can be obtained. The hot spots can be evaluated by one of the six
stress components (sxx, syy, szz, sxy, syz, and szx), the von Mises stress, the principal stress,
or the hydrostatic stress (negative pressure value or the pressure). The command returns a list
of maximum stress values (largest magnitude, largest tensile, largest compressive) and the
corresponding location coordinates.
References
[1] J. C. Simo and T. J. R. Hughes, Computational Inelasticity, vol. 7, New York: Springer,
1998.
[2] W. Ramberg and W. R. Osgood, Description of Stress-Strain Curves by Three
Parameters, National Advisory Committee for Aeronautics, Technical Note No. 902,
Washington, DC, USA, July 1943.
[3] J. Lubliner, Plasticity Theory, Macmillan: New York, 1990.
ViscoPlasticStrain_zx ViscoPlasticStrainIJ_z
Not applicable
-ViscoPlasticStrain-ZX
ViscoPlasticStrainEQV
Not applicable
ViscoPlasticStrainEQV ViscoPlasticStrainEQV
Displacement_x
Not applicable
Displacement-Y -Displacement-Z
Displacement_y
Not applicable
Displacement-X Displacement-Y
Displacement_z
Not applicable Not applicable
Displacement-X
Ta bl e 66 Va ri abl e na me s in o u t p ut f il es
Field name Field name (deprecated) 2D 3D
9: Computing Mechanical Stress
References
Sentaurus™ Process User Guide 691
H-2013.03
[4] S. B. Brown, K. H. Kim, and L. Anand, “An Internal Variable Constitutive Model for
Hot Working of Metals,” International Journal of Plasticity, vol. 5, no. 2 pp. 95–130,
1989.
[5] G. G. Weber et al., “An Objective Time-Integration Procedure for Isotropic Rate-
Independent and Rate-Dependent Elastic-Plastic Constitutive Equations,” International
Journal of Plasticity, vol. 6, no. 6, pp. 701–744, 1990.
[6] G. Z. Wang et al., “Applying Anand Model to Represent the Viscoplastic Deformation
Behavior of Solder Alloys,” Journal of Electronic Packaging, vol. 123, no. 3, pp. 247–
253, 2001.
[7] Q. Wang et al., “Anand Parameter Test for Pb-Free Material SnAgCu and Life
Prediction for a CSP,” in 8th International Conference on Electronic Packaging
Technology (ICEPT), Shanghai, China, pp. 1–9, August 2007.
[8] J. Wilde et al., “Rate Dependent Constitutive Relations Based on Anand Model for
92.5Pb5Sn2.5Ag Solder,” IEEE Transactions on Advanced Packaging, vol. 23, no. 3,
pp. 408–414, 2000.
[9] H. J. Frost and M. F. Ashby, Deformation-Mechanism Maps, Pergamon Press: Oxford,
1982.
[10] S. Wiese, F. Feustel, and E. Meusel, “Characterisation of constitutive behaviour of
SnAg, SnAgCu and SnPb solder in flip chip joints,” Sensors and Actuators A, vol. 99,
no. 1–2, pp. 188–193, 2002.
[11] R. Gatti et al., “Dislocation engineering in SiGe heteroepitaxial films on patterned Si
(001) substrates,” Applied Physics Letters, vol. 98, no. 12, p. 121908, 2011.
[12] Y.-C. Joo, J.-M. Paik, and J.-K. Jung, “Effect of Microstructure and Dielectric Materials
on Stress-Induced Damages in Damascene Cu/Low-k Interconnects,” in MRS
Symposium Proceedings, Materials, Technology and Reliability of Advanced
Interconnects, vol. 863, San Francisco, CA, USA, p. B7.6/O11.6, March 2005.
9: Computing Mechanical Stress
References
692 Sentaurus™ Process User Guide
H-2013.03

Sentaurus™ Process User Guide 693
H-2013.03
CHAPTER 10 Mesh Generation
This chapter describes the mesh algorithms and meshing parameters
available in Sentaurus Process.
Overview
Sentaurus Process automatically generates meshes as they are needed. The behavior of the
automatic-meshing scheme is different in 3D than in 1D and 2D because of the time required
to generate 3D meshes. In 1D and 2D, meshes are generated after every geometry operation
such as etch, deposit, and transform. In 3D, meshes are only generated immediately before
steps that require a bulk mesh, such as a diffuse or an implant command, and structure
saving. This scheme can reduce the time spent when there are multiple geometry-changing
steps without a diffuse or an implant command (or any other step requiring a mesh) in
between.
Sentaurus Process uses Sentaurus Mesh as its mesh generation engine. Details of the meshing
algorithms are provided, but for simplification, Sentaurus Mesh is used throughout.
The mesh generation process starts with a bisection algorithm, which places mesh points as
instructed by the user. Afterwards, the mesh elements are created using a modified Delaunay-
meshing algorithm. Refer to the Mesh Generation Tools User Guide for details about Sentaurus
Mesh.
The meshes generated within Sentaurus Process can be refined adaptively, statically, or as a
combination of adaptive and static refinements. The refinement can be specified using one of
the major types of refinement box:
■Field based (adaptive meshing)
■Mask based
■Uniform (standard)
■Interface axis-aligned
■Interface offsetting (offset normal to the interface)
All these refinement types are user controllable. In addition, Sentaurus Mesh enforces mesh
smoothing to limit the changes in element size from one element to the next. This smoothing
is important for mechanics accuracy and convergence behavior (see Mesh Refinement on
page 694).

10: Mesh Generation
Mesh Refinement
694 Sentaurus™ Process User Guide
H-2013.03
One important algorithm affecting refinement behavior is the UseLines algorithm. This
algorithm inserts lines created using the line command into the internal bisection algorithm
before any other lines are introduced. Further mesh refinement proceeds by bisecting the boxes
created by the UseLines lines. This has the effect of isolating static regions of a structure from
regions where the boundaries are moving due to geometric operations. Geometry movement
naturally causes perturbations to the mesh lines. The UseLines lines compartmentalize this
mesh movement to minimize solution degradation from interpolation. For more information,
see UseLines: Keeping User-defined Mesh Lines on page 722.
NOTE Because this internal bisection algorithm in Sentaurus Process is
different than the one used to create mesh refinement in the stand-alone
Sentaurus Mesh tool, it is not possible to create meshes identical to
those created with Sentaurus Mesh. However, element quality, stability,
and the Delaunay properties should be qualitatively the same.
Mesh Refinement
Mesh refinement is a two-step process:
■First, you define the refinement box.
■Second, the mesh is refined when the next remesh occurs either with an explicit
grid remesh call or during standard geometry modifications such as etch, deposit, clip,
or native layer formation.
The refinement boxes remain valid unless the list of refinement boxes is cleared with the
refinebox clear command.
All refinement boxes have refinement criteria that add mesh and constraints that can be used to
limit where the mesh refinement occurs. One type of refinement criteria is available for each
type of box, and it essentially defines the box type. The refinement criteria and, therefore, the
refinement box type can be either static or adaptive. All types of refinement box can be mixed
as required. The refinement box constraints are specified along with the refinement criteria in
the refinebox command and can be used in combination within one command.
The constraints available include:
■A material constraint using the materials parameter that takes a list of materials.
■Region constraints using the regions parameter that takes a list of region names.
■The min and max parameters that limit the size of the refinement box (which by default
applies to all of the space).

10: Mesh Generation
Mesh Refinement
Sentaurus™ Process User Guide 695
H-2013.03
Refinement information also can be extracted and written to a file readable by Sentaurus Mesh
using the mshcmd flag in conjunction with the tdr parameter of the struct command (see
struct on page 1158).
Viewing Mesh Refinement
To aid in setting mesh refinement, you can store the current minimum edge length in each
direction as a field using the command:
pdbSet Grid Set.Min.Edge 1
When specified, Sentaurus Process computes the smallest edge length in each direction and
saves it in three fields:
■MinXEdgeLength
■MinYEdgeLength (for 2D or 3D structures)
■MinZEdgeLength (for 3D structures)
In addition, it prints the average edge length to the screen.
Static Refinement
Standard Refinement Boxes
The standard refinement box allows you to specify a smoothly varying mesh density inside the
refinement box at three locations in the x-, y- and z-directions using the xrefine and
yrefine, and zrefine parameter lists, respectively. If all three xrefine, yrefine, and
zrefine values are specified, the mesh density varies quadratically in that direction. If two
are specified, the variation is linear from top to bottom. If only one value is specified, a constant
mesh density is assumed.
Refinement boxes also can be limited to refine only in one specific material or region using the
regions or materials parameter.
Examples
This is an example of specifying two refinement boxes and calling remesh:
refinebox min= {-0.25 0.4 0.0} max= {0.4 0.6 1.0} xrefine= {0.1 0.06 0.1} \
yrefine= {0.1 0.01 0.1} zrefine = {0.01} oxide
refinebox min= {0.6 0.6} max= {0.8 0.8} xrefine= 0.1 silicon
grid remesh
10: Mesh Generation
Mesh Refinement
696 Sentaurus™ Process User Guide
H-2013.03
NOTE Calculating the linear or quadratic variation of the mesh density when
two or three x-, y-, or z-direction values are given requires the
specification of min and max. If min and max are not specified and at
least one region is specified, the minimum and maximum values of the
bounding box for that region serve as min and max for the calculation.
If more than one region is specified, only the bounding box of the first
region is used for the calculation, although all regions are used as
constraints to the refinement.
Interface Refinement Boxes
Refinement near interfaces can be specified using the refinebox command. So it is possible
to have a large global default minimum interface mesh-spacing, for example, and a smaller
localized value inside a box. The parameters affecting interface refinement are demonstrated
in the following examples:
■Set the mesh criteria near the interface. This is the maximum size the first normal edge can
be, and it is possible for the edge to be 0.5 min.normal.size:
pdbSet Grid SnMesh min.normal.size <n>
■Set the growth rate of the edge size away from the interface:
pdbSet Grid SnMesh normal.growth.ratio.2d <n>
pdbSet Grid SnMesh normal.growth.ratio.3d <n>
■Set min.normal.size or normal.growth.ratio or both locally within a refinement
box:
refinebox min.normal.size = <n> normal.growth.ratio = <n> \
[interface.materials=<list> | interface.mat.pairs = <list of pairs>]
Interface Offsetting Refinement Boxes
In addition, the Sentaurus Mesh offsetting algorithm can be used to create offsetting layers at
interfaces by giving the offsetting keyword, which also permits regionwise interface
specification in addition to the materialwise possibility:
refinebox offsetting min.normal.size = <n> normal.growth.ratio = <n> \
[interface.materials=<list> | interface.mat.pairs = <list of pairs>] \
[interface.regions=<list> | interface.region.pairs = <list of pairs>]
For Sentaurus Mesh offsetting, an additional keyword offsetting.maxlevel defines the
number of layers to be generated at the interface.
10: Mesh Generation
Mesh Refinement
Sentaurus™ Process User Guide 697
H-2013.03
The offsetting.maxlevel can be defined globally using the mgoals command, or on a
materialwise or regionwise basis using the refinebox command as shown in the following
three possibilities:
mgoals offsetting.maxlevel = <i>
refinebox offsetting.maxlevel = <i> interface.materials= { <string list> }
refinebox offsetting.maxlevel = <i> interface.regions= { <string list> }
NOTE For Sentaurus Mesh offsetting, offsetting.maxlevel can only be
defined on a material or region basis with interface.materials or
interface.regions or globally, not with interface.mat.pairs
or interface.region.pairs.
NOTE For Sentaurus Mesh offsetting, min.normal.size and
normal.growth.ratio can only be defined by material pair or region
pair with interface.mat.pairs or interface.region.pairs or
globally, not with interface.materials or interface.regions.
Offset-meshing parameters defined at interfaces using interface.mat.pairs or
interface.region.pairs are interpreted in a symmetric way by default. This means that,
given the specification of a material or region pair , the parameters are defined for both
at the interface and for at the interface. If the !double.side keyword is given,
Sentaurus Mesh interprets in a nonsymmetric way, that is, only for at the
interface.
Refinement Inside a Mask
Mask-based refinements are similar to standard refinements (see Standard Refinement Boxes
on page 695), except that they have an additional constraint that is defined by a volume
specified by a previously existing mask. This constraint is applied in addition to the normal box
constraint defined by the min and max parameters. Mask-based refinements are a way to have
layout driven refinements.
For example, if you specify min and max, the refinement area will be the intersection of the
specified rectangle and the mask. If you specify a material name, the final refinement will be
the intersection of the regions with such a material and the mask.
These constraints are specified using the refinebox command with the following options:
■A mask name (mask).
■Minimum and maximum coordinates in x where the refinement will be applied
(extrusion.min and extrusion.max).
■An optional parameter to see if the refinement should extend some distance apart from the
mask (extend).
x1x2
⁄
x1
x2
x2
x1
x1x2
⁄
x1
x2
10: Mesh Generation
Mesh Refinement
698 Sentaurus™ Process User Guide
H-2013.03
Negative masks are also allowed. Mask boundaries are never interpreted as being infinite in any
direction, even if they extend far from the simulation boundary. Consequently, shrinking a
refinement by specifying a negative extension parameter might leave a region uncovered, even
if the mask originally extended past the boundary. For example, if a mask from (–0.010 to 1)
covers a domain from (0 to 2), applying an extend parameter of –0.02 will produce a
refinement extending from (0.010 to 0.98), thereby leaving the region from 0 to 0.010
unrefined.
Example
First, create a mask, and then a refinement box can be issued:
polygon name=pol segments= { -0.5 -0.5 -.25 -.5 -.25 -.05 .25 -.05 .25 -.5 \
.5 -.5 .5 0 -.5 0 }
mask name = "Mask" polygons= { pol }
#now that there is a mask it can be used to produce a refinement.
refinebox name = "refi_mask" mask = "Mask" xrefine= { .075 .075 .075 } \
yrefine= { .075 .075 .075 } extrusion.min = 0 extrusion.max = 0.05 \
extend = -0.1
Refinement Near Mask Edges
Refinement also can be constrained to be near mask edges. This mask edge–based refinement
has three parameters available in the refinebox command:
■mask.edge.refine.extent
■mask.edge.mns
■mask.edge.ngr
The parameter mask.edge.refine.extent must be specified to switch on mask edge–
based refinement and to set the lateral extent of the refinement from the mask edge. Vertically,
the mask edge–based refinement can be controlled with the x-coordinate of the min and max
parameters. The minimum mesh spacing near the mask edge is set with mask.edge.mns (the
default is taken from the pdb parameter Grid SnMesh min.normal.size), and the growth
of the edge length away from the mask edge is specified with mask.edge.ngr (default is 1.0,
meaning the constant edges of lengths mask.edge.mns in the normal direction).
NOTE Similar to the pdb parameter Grid SnMesh min.normal.size,
actual edge lengths may be up to two times smaller than
mask.edge.mns at the mask edge because of the binary-tree
refinement algorithm.
An example of using mask edge–based refinement is:
polygon name=p1 segments= { 1.0 1.0 1.0 5.0 3.0 5.0 3.0 2.5 2.0 2.5 2.0 1.0 }
mask name=m1 polygons = p1
10: Mesh Generation
Mesh Refinement
Sentaurus™ Process User Guide 699
H-2013.03
refinebox clear
# Prevent mesh propagation by defining regular coarse mesh
refinebox yrefine = 0.5 zrefine = 0.5
# Add edge-based refinement
refinebox mask = m1 mask.edge.mns = 0.08 mask.edge.refine.extent = 0.25
grid remesh
The resist layer was created later using the command;
photo mask = m1 thickness = 0.05
Figure 96 shows the result.
Figure 96 Mask edge–based refinement shown on the mask and in silicon
Adaptive Refinement
Tailoring a mesh to a specific problem with static refinement boxes can be tedious and time-
consuming. In addition, for some applications, dopant profiles evolve so much during the
process that the areas where a finer mesh was needed at the beginning are very different from
the areas where a finer mesh is needed at the end.
To accurately capture the entire evolution with a static mesh, it is necessary to put a fine mesh
over large areas of the structure leading to long simulation times and large memory use.
Adaptive meshing in Sentaurus Process addresses these issues.
10: Mesh Generation
Mesh Refinement
700 Sentaurus™ Process User Guide
H-2013.03
The adaptive-meshing feature has a major component: field-based refinement.
For details, see Tips for Adaptive Meshing on page 709.
The refinement parameters and criteria are the same for adaptive implantation as for field-
based. When adaptive meshing is switched on, field-based refinement is performed during
every remesh step and for any dimension (in 1D, 2D, or 3D). This happens for all etch, deposit,
implant, native layer, regrid, and transform operations. In addition, during solve at a specified
step interval, a check of the current mesh is made to determine whether a remesh is required;
then the remesh is performed if necessary. For details, see Adaptive Meshing during Diffusion
on page 707. Finally, when adaptive meshing is used during implantation, in addition to
adaptively refining the newly implanted species and damage, adaptive refinement (also based
on existing fields) is applied simultaneously.
Adaptive Refinement Criteria
Numerous refinement functions are available to deal with differing fields and situations. All
functions involve some comparison between values on neighboring nodes and possible values
between neighboring nodes. In some cases, the same refinement function is available in
Sentaurus Mesh, and similar results to Sentaurus Mesh refinement will be obtained. The
following refinement criteria are available:
■Relative difference (default)
■Absolute difference
■Logarithmic difference
■Inverse hyperbolic sine (asinh) difference
■Gradient
■Local dose error
■Interval refinement
These refinements can be applied globally (default) or they can be limited as follows:
■Boxwise
■Materialwise
■Regionwise
Detailed descriptions of the refinement types, their respective control parameters, and
instructions for applying refinement constraints are given in subsequent sections. The default
adaptive meshing parameters have been set to apply only relative difference criteria to the
whole structure, and they typically produce a fairly coarse mesh. It is necessary to set one
criterion or more to produce a mesh sufficiently fine to reach a required accuracy.

10: Mesh Generation
Mesh Refinement
Sentaurus™ Process User Guide 701
H-2013.03
Adaptive meshing is switched off by default. To switch on adaptive meshing, use:
pdbSet Grid Adaptive 1
pdbSet Grid SnMesh UseLines 1 ;# Recommended with adaptive meshing
Relative Difference Criteria
The relative difference between two neighboring nodes is computed as follows:
(956)
where is the field value on node , and is the field-specific refinement parameters set
with:
pdbSet Grid <Field> Refine.Abs.Error <n>
or def.abs.error and abs.error, which are parameters of the refinebox command.
If the value of the expression in Eq. 956 is greater than the maximum relative difference, the
edge between node 1 and node 2 is split. To set the maximum relative difference, use:
pdbSet Grid <Field> Refine.Rel.Error <n>
or def.rel.error and rel.error, which are parameters of the refinebox command.
The quantity <Field> is the name of the field, and <n> is a unitless number for
Refine.Rel.Error and Refine.Abs.Error; the units are the same as the units of the field.
The default values for Refine.Abs.Error and Refine.Rel.Error are set from <Field>
= AdaptiveField, except for the standard dopants, point defects, and Damage that have
entries in the PDB.
The density of the mesh is sensitive to Refine.Rel.Error because it represents the target
relative change of the field across an edge. For many standard situations, a number of the order
of 1.25 gives a coarse mesh, and a number of approximately 0.5 often gives a fine mesh. The
parameter sets a smooth cutoff such that values of the field below result in no refinement.
NOTE The relative difference criteria should only be used with fields that are
always positive.
Absolute Difference Criteria
The absolute difference between two neighboring nodes is computed simply:
(957)
2C1C2
–
C1C2α++()
----------------------------------
Ci
i
α
α
α
C1C2
–

10: Mesh Generation
Mesh Refinement
702 Sentaurus™ Process User Guide
H-2013.03
where is the field value on node . If the value of the expression in Eq. 957 is greater than
the maximum absolute difference, the edge between nodes 1 and 2 is split. The maximum
allowable absolute difference can be set with:
pdbSet Grid <Field> Refine.Max.Difference <n>
Logarithmic Difference Criteria
The logarithmic (base 10) difference between two neighboring nodes is computed as follows:
(958)
where is the field value on node , and is the low value cutoff that can be set with:
pdbSet Grid <Field> Refine.Abs.Error <n>
or def.abs.error and abs.error, which are parameters of the refinebox command.
If the value of the expression in Eq. 958 is greater than the maximum logarithmic difference,
the edge between nodes 1 and 2 is split. To set the maximum logarithmic difference, use:
pdbSet Grid <Field> Refine.Max.LogDiff <n>
or def.max.logdiff and max.logdiff, which are parameters of the refinebox
command.
NOTE The logarithmic difference criteria should only be used with fields that
are always positive. Use the asinh criteria for fields that can have
negative values such as stresses.
Inverse Hyperbolic Sine (asinh) Difference Criteria
The asinh difference between two neighboring nodes is computed as follows:
(959)
where is the field value on node . If the value of the expression in Eq. 959 is greater than
the maximum asinh difference, the edge between nodes 1 and 2 is split. To set the maximum
asinh difference, use:
pdbSet Grid <Field> Refine.Max.AsinhDiff <n>
or def.max.asinhdiff and max.asinhdiff, which are parameters of the refinebox
command.
Ci
i
C1α+()log C2α+()log–
Ci
i
α
C1
()asinh C2
()asinh–
Ci
i

10: Mesh Generation
Mesh Refinement
Sentaurus™ Process User Guide 703
H-2013.03
Gradient Criteria
The gradient between two neighboring nodes is computed as follows:
(960)
where is the field value on node , and is the length of the edge between nodes and .
If the value of the expression in Eq. 960 is greater than the maximum gradient, the edge
between the two nodes is split. To set the maximum gradient, use:
pdbSet Grid <Field> Refine.Max.Gradient <n>
or def.max.gradient and max.gradient, which are parameters of the refinebox
command.
Local Dose Error Criteria
If an edge between two neighboring nodes is not split, the local dose error is computed as
follows:
(961)
where:
■ is the field value on node .
■ is the concentration at the midpoint between nodes and .
■ is the length of the edge between nodes and .
■ is the box size perpendicular to the edge between nodes and (see Figure 97).
Figure 97 (Left) One-dimensional and (right) 2D representation of dose loss criteria
The function in Figure 97 (left) is taken from the previous mesh (or from an analytic
implantation). The box with four points in Figure 97 (right) represents one cell of the mesh
C1C2
–
l12
-------------------
Ci
i
lij
i
j
0.5C12 0.25C–10.25C2
–l12s12
Ci
i
Cij
i
j
lij
i
j
sij
i
j
Ci
Cij
Cj
lij
sij
ij
lij
10: Mesh Generation
Mesh Refinement
704 Sentaurus™ Process User Guide
H-2013.03
refinement tree. The shaded area is the part of the 2D field under consideration. The dose in the
shaded area is computed in two ways:
■As is
■If the edge between and is split
If the difference between these two ways is greater than max.dose.error, the edge is split.
The box size is 1.0 (unitless) in 1D; it is the box width (in cm) in 2D; and it is the box area
( ) perpendicular to the edge –
in 3D. If the value of the expression in Eq. 961 is greater
than the normalized maximum local dose error, the edge between the two nodes is split. The
local dose error can be set with:
pdbSet Grid <Field> Refine.Max.DoseError <n>
where <n> has units of , or def.max.dose.error and max.dose.error, which are
parameters of the refinebox command.
The local dose error is first multiplied by the simulation size before comparing it to the
expression in Eq. 961. The simulation size is 1.0 (unitless) in 1D, the simulation width (in cm)
in 2D, and the simulation lateral area in 3D (in ).
To estimate the total dose loss, you must estimate how many nodes carry a significant
concentration of the field in question and then multiply that number by the local dose error to
obtain approximately the maximum total dose error expected. (In practice, the dose error is
often considerably less than this.) This quantity is relatively easy to understand and is less
sensitive than some other parameters to process conditions.
Interval Refinement
Interval refinement provides a way to refine the mesh such that field values within a certain
interval are well resolved. Interval refinement produces mesh edges of a specified length
wherever the field values are within a specified interval. Four parameters are required to define
an interval refinement:
■A minimum and maximum value
■ and
■A target length,
■A target length scaling,
To preserve the anisotropy of the mesh, interval refinement examines each edge of a refinement
cell and calculates an effective edge length defined by:
(962)
i
j
cm 2–
i
j
cm 2–
cm 2–
Cmin
Cmax
lt
s
leff
leff abs r1r2
–()C∇⋅()=
10: Mesh Generation
Mesh Refinement
Sentaurus™ Process User Guide 705
H-2013.03
where and are the endpoints of the edge, and is the average gradient of the field in
the refinement cell. Edges that are nearly parallel to the contours of the field have effective edge
lengths near zero. Edges that are nearly perpendicular to the contours have effective edge
lengths near their actual edge length. Since edges are split only when they are longer than a
given target length, edges that are parallel to the field contours are allowed to be longer than
those that are perpendicular.
Interval refinement will split any edge whose effective edge length exceeds the effective target
length. The effective target length is calculated differently depending on whether the field
values on the edge overlap the interval specified by (refinebox min.value) and
(refinebox max.value).
Let and be the values of the field on the endpoints of the edge. If the relation
is satisfied for any value of between and , the edge overlaps the
interval.
For edges that overlap the interval, the effective target length is exactly the target length that
you specify (refinebox target.length), that is:
(963)
For edges that do not overlap:
(964)
where is either or , is either or , and the values of and are
chosen to minimize the difference.
The formula for outside the interval produces a graded mesh with an edge length that falls
off parabolically with distance from the interval. Default values for the parameters of the
interval refinements are defined in the PDB.
Table 67 lists refinebox parameters in the left column that can be used to specify boxwise
refinement. The right column lists the corresponding PDB parameters that can be used to
specify refinement criteria globally.
Ta bl e 67 S um m ar y o f r e f i n e me n t p a ram et er s
refinebox parameter Corresponding entry in parameter database
def.rel.error, rel.error Grid <Field> Refine.Rel.Error
def.abs.error, abs.error Grid <Field> Refine.Abs.Error
def.max.difference, max.difference Grid <Field> Refine.Max.Difference
def.max.logdiff, max.logdiff Grid <Field> Refine.Max.LogDiff
r1
r2
C∇
Cmin
Cmax
C1
C2
Cmax CC
min
>>
C
C1
C2
lteff lt=
lteff lt 1Ca
log Cb
log–+()
2s=
Ca
Cmin
Cmax
Cb
C1
C2
Ca
Cb
lteff
10: Mesh Generation
Mesh Refinement
706 Sentaurus™ Process User Guide
H-2013.03
Localizing Adaptive Meshing using refinebox Command
Adaptive meshing has been implemented through generalized refinement boxes. As such,
adaptive refinement and the refinement parameters themselves can be set in a boxwise manner.
The default adaptive refinement box covers the entire structure and relies on global parameters
and field-based parameters for its default values. If you specify an adaptive refinement box, the
default box is not created.
NOTE For field-based refinement, any adaptive refinebox that is manually
created overrides the default adaptive refinebox. The default adaptive
refinebox (that covers the entire structure) can be created explicitly
with the command refinebox adaptive.
You ca n c re at e o ne or m ore a da ptive re fin ement boxes with different parameters. The most
commonly used parameters control the size of the box (min and max), and the minimum and
maximum edge lengths (refine.min.edge and refine.max.edge).
The default list of fields upon which to refine includes all dopants, point defects, and clusters
in the structure. This list can be modified in several ways. For example, the following command
overrides the default list:
pdbSet Grid <field> DoNotAdapt 1
The next example adds Field1 and Field2 to the default list for this particular box:
refinebox refine.add.fields = { Field1 Field2 ... }
The following command redefines the list of fields to be used as the basis for refinement; if set,
this command overrides any add or subtract settings:
refinebox refine.fields = { Field1 Field2 ... }
def.max.asinhdiff, max.asinhdiff Grid <Field> Refine.Max.AsinhDiff
def.max.gradient, max.gradient Grid <Field> Refine.Max.Gradient
def.max.dose.error, max.dose.error Grid <Field> Refine.Max.DoseError
min.value Grid <Field> Refine.Min.Value
max.value Grid <Field> Refine.Max.Value
target.length Grid <Field> Target.Length (um)
target.length.scaling Grid <Field> Target.Length.Scaling
Ta bl e 67 S um m ar y o f r e f i n e me n t p a ram et er s
refinebox parameter Corresponding entry in parameter database
10: Mesh Generation
Mesh Refinement
Sentaurus™ Process User Guide 707
H-2013.03
Examples
To switch on adaptive meshing, use:
pdbSet Grid Adaptive 1
To apply adaptive meshing only inside a box and to set the anisotropic edge minimum in the
same box, use:
refinebox min= {0.0 0.0} max= {0.01 0.5} refine.min.edge= {0.001 0.25} adaptive
To refine only considering arsenic and boron, use:
refinebox refine.fields = {Arsenic Boron} adaptive
To create a default box and, in addition, to create a refinement box where is modified locally
for all species and is modified for only boron, use:
refinebox adaptive
refinebox min= {0.0 0.0} max= {0.01 0.5} def.rel.error= 0.9 \
abs.error= {Boron = 1.0e14} adaptive
Adaptive Meshing during Diffusion
Adaptive meshing during diffusion is switched on by default when adaptive meshing is
switched on (in other words, pdbGet Grid Adaptive returns 1). There is an additional
control that allows for the prevention of adaptive meshing at low temperatures, which is
specified like this:
pdbSet Grid Min.Adaptive.Temp <Temp C>
In any case, by default adaptive meshing is not performed during oxidation or silicidation. You
can switch on adaptive meshing during these steps by setting:
pdbSet Diffuse Compute.Regrid.Steps 10 ;# during inert annealings
pdbSet Diffuse Growth.Regrid.Steps -1 ;# during oxidation and silicidation
pdbSet Diffuse Epi.Regrid.Steps -1 ;# during epitaxy
where <number> is the fixed interval of time steps. For the first parameter
Compute.Regrid.Steps, the default is 10 steps and, for the other two, the default is –1,
meaning it is off by default. After the specified number of steps is taken, the mesh is checked
to see if the refinement criteria are satisfied (within some tolerance); a remesh is performed if
necessary. Currently, the use of adaptive meshing during oxidation and epitaxy is possible.
rF
αF

10: Mesh Generation
Mesh Refinement
708 Sentaurus™ Process User Guide
H-2013.03
The refinement criteria check is performed as follows: Axis-aligned edges are checked to see
if they satisfy:
actual < Refine.Factor * error / maxerror (965)
where:
■Refine.Factor is a direction-dependent parameter of the PDB under Grid.
■error is the error functions given in Eq. 956–Eq. 961.
■maxerror is the maximum error parameter associated with each refinement type.
■actual is the ‘actual’ edge length.
There is a cutoff percentage PDB parameter Grid Refine.Percent that limits the percent
of edges that fail (Eq. 965) before a remesh is called. This check procedure is performed for
every Diffuse Compute.Regrid.Steps whether a remesh is called or not. You can omit
the refinement criteria check (which can be time-consuming for large meshes) and force a
remesh by setting:
pdbSet Grid Refine.Check 0
Table 68 summarizes the parameters available for adaptive meshing for diffusion.
Adaptive Meshing during Implantation
Adaptive meshing during implantation is active whenever adaptive meshing is switched on (in
other words, pdbGet Grid Adaptive returns 1). It also may be enabled or disabled for each
implant step using the adaptive parameter of the implant command.
Table 68 Adaptive meshing parameters
Parameter Comment
Compute Compute.Regrid.Steps
Number of diffusion steps before refinement criteria are checked to
decide if remeshing is required.
Compute Pre.Regrid.Save
To help with tailoring the mesh, files can be saved immediately before
adaptive remeshing occurs during diffusion. The files are named
<input_file_stub>_preregrid_###_fps.tdr
where <input_file_stub> would be, for example n1, if the
input file was n1_fps.cmd and ### is an increasing index starting
with 001.
Grid Refine.Check
If Grid Refine.Check is set to 1 (true), then refinement criteria
are checked and remeshing occurs if necessary.
If it is set to 0 (false), remeshing occurs without a check.
Grid Refine.Factor
Tolerance factor for marking an edge too long.
Grid Refine.Percent
Allowed percentage of edges too long.
10: Mesh Generation
Mesh Refinement
Sentaurus™ Process User Guide 709
H-2013.03
Adaptive meshing during implantation differs from adaptive meshing for other process steps in
one key respect: When performing an implantation step, the implanted concentrations are
defined by analytic expressions instead of discretized field values. Therefore, the final values
for the implanted fields are not known before the remeshing step begins so they must be
computed as the mesh is refined.
By default, refinement on damage is handled differently from refinement on dopants. For the
analysis of damage, the gradient is usually uninteresting, but the location of the crystal–
amorphous interface is often critical. Therefore, refinement should be added to the mesh, not
according to the damage gradient, but rather according to whether the damage is near the
crystal–amorphous threshold. This is accomplished using an interval refinement (see Interval
Refinement on page 704). By default, the minimum and maximum values of the interval are set
to the value of the crystal–amorphous threshold ( ). The target length is
, and the target length scaling is 1.0.
NOTE The default target-length setting of 2 nm can produce many mesh points
for amorphizing implants in 3D. You should first try using a larger
setting and then reduce it if necessary.
As the mesh is constructed, each cell of the refinement tree is evaluated to determine whether
the refinement criteria are satisfied. The criteria for as-implanted fields are computed for each
edge of the cell. If any criterion is not satisfied, the cell is split and the as-implanted
concentrations are computed at the newly introduced points. This process continues until all
refinement criteria are satisfied (or the minimum edge length is reached) for all cells on the
refinement tree. Therefore, the constructed mesh satisfies the refinement criteria for all fields
present in the structure, not solely the implanted fields.
For adaptive meshing during MC implantation, the analytic module is used to compute
refinement, the mesh is formed, and afterwards, the implant profiles are computed with the MC
module.
Tips for Adaptive Meshing
The following list gives useful suggestions when using adaptive meshing:
■When setting boxwise meshing criteria, remember that any global criteria you have
specified still apply inside the box. This means you cannot use boxwise meshing criteria to
establish less stringent meshing criteria (such as a larger relative error) inside a box because
the more stringent global criteria still apply. If you want to use different criteria for different
parts of the structure, set the global criteria to the least stringent criteria and use boxes for
more stringent criteria.
■To switch on adaptive meshing and use all the defaults, all that is needed is pdbSet Grid
Adaptive 1. The main parameter for adjusting the amount of refinement is pdbSet
Grid AdaptiveField Refine.Rel.Error, which defaults to 1.5. In many cases, this
1.15 1022
× cm 3–
0.002 µm

10: Mesh Generation
Mesh Refinement
710 Sentaurus™ Process User Guide
H-2013.03
does not refine sufficiently. Decreasing the value causes more refinement. The number of
mesh points is sensitive to this value, and it is not generally recommended to use a value
less than 0.25. This parameter generally meshes doping gradients well, but may leave the
peaks too coarse. To refine the peaks, the best criterion to use is maximum dose error (Grid
AdaptiveField Max.Dose.Error).
■To override the default refinement box used for field-based refinement (which covers the
whole structure and applies to all solution variables), you need only to create an adaptive
refinement box. To add criteria in addition to the default criteria, for example, to add finer
criteria under the gate while preserving standard parameters elsewhere, you can create your
own default refinement box. For example:
refinebox adaptive
refinebox min= {-0.01 -0.01} max= {0.15 0.05} adaptive def.rel.error=0.75
■The default refinement setting for implant damage can give too fine a mesh. Increase
Grid AdaptiveField Refine.Target.Length from the default value of 0.002 to
reduce refinement.
Default Refinement
In two dimensions, by default, interface refinement is applied to any interface in which one of
the neighboring bulk regions is of material Silicon, Polysilicon, or Oxide. In three
dimensions, by default, interface refinement is applied only to interfaces where one of the
neighboring bulk regions is Silicon. For other interfaces, the min.normal.size criterion
is not applied. To view currently defined refinement boxes (including default refinement
boxes), use:
refinebox print
Additional interface refinement can be specified with the command:
refinebox interface.materials= {<material1> <material2> ...}
This command specifies refinement at all interfaces to both <material1> and <material2>.
refinebox interface.mat.pairs= {<material1> <material2>}
This command specifies interface refinement at all interfaces where one side of the interface is
<material1> and the other side is <material2>.
The interfaces that are refined are the union of interface.materials (all interfaces
touching materials in the list) and interface.mat.pairs (only refined on material pairs
found in the list first and second, third and fourth, and so on).
The default min.normal.size for all interface refinement boxes including the default ones
is taken from the pdb parameter Grid SnMesh min.normal.size. Similarly, the default

10: Mesh Generation
Mesh Refinement
Sentaurus™ Process User Guide 711
H-2013.03
value of normal.growth.ratio for all interface refinement boxes is taken from the pdb
parameter Grid SnMesh normal.growth.ratio.2d in two dimensions and from Grid
SnMesh normal.growth.ratio.3d in three dimensions.
To add an interface refinement, use the refinebox command.
To remove an existing interface refinement, first do refinebox clear, and then start again.
Examples
The interfaces to be refined are defined as follows:
# Change the default min.normal.size (in micrometers)
pdbSet Grid SnMesh min.normal.size 2.0e-3
# Now modify which materials to apply interface refinement
# refine at all interfaces to silicon and poly (use the global min.normal.size
# and normal.growth.ratio)
refinebox clear
refinebox interface.materials= {silicon poly}
The next example shows refinement only at the silicon–oxide and polysilicon–oxide interfaces,
and specifies a local value for interface refinement parameters:
refinebox clear
refinebox min.normal.size = 0.005 normal.growth.ratio = 3 \
interface.mat.pairs= { Silicon Oxide PolySilicon Oxide }
Refinement Box Manipulations: Using
transform.refinement
Several transformation can be performed on refinement boxes using the
transform.refinement command. This command works like the transform command
(see Stress Handling on page 757), except for refinement boxes. This command accepts the
following options:
■Transformation – either translate, stretch, cut, rotate, flip, or reflect.
■Transformation options – depend on the type of transformation. A displacement is required
for translate, axis and angle for rotate, a box for cut, reflecting plane for reflect,
the length and direction for stretch, and so on.
■name – applies the transformation to a particular refinement box if specified or to all of
them otherwise.
■name.new – specifies the name of the transformed refinement.
10: Mesh Generation
Mesh Settings
712 Sentaurus™ Process User Guide
H-2013.03
■keep.original – specifies whether to keep the original. Specifying !keep.original
transforms the specified refinement box, while setting it to keep.original preserves the
original refinement box and creates a new transformed one. This option is useful when you
want to “copy and paste” refinements by, for example, translating them to a different
position while keeping the original in place.
For example:
transform.refinement name="refbox" name.new="newRefBox" \
translate= { 0.1 0 0 } keep.original
creates a new refinement called newRefBox identical to refbox but displaced in x.
Mesh Settings
The following tables list the parameters available for Sentaurus Mesh. To set the parameters in
Table 69, use:
pdbSet Grid SnMesh <Parameter name> <value>
Table 69 Parameters available for Sentaurus Mesh
Parameter Default Description
CoplanarityAngle 175
Any pair of faces with an angle of CoplanarityAngle
or more will be considered coplanar.
CoplanarityDistance 1.0e-6
Maximum deformation caused to the boundary when
swapping the edge shared by a pair of adjacent faces.
DecimateBeforeImprint true
Decimates the boundary before imprinting it with the axis-
aligned mesh.
DelaunayTolerance 1.0e-4
Specifies how close the ridges and boundary faces conform to
the Delaunay criterion.
DelaunayToleranceMat
Specifies an array pair of materials and tolerances to be used
in those materials. For example:
pdbSet Grid SnMesh DelaunayToleranceMat
{Silicon 0.01 Oxide 1.0}
DelaunayToleranceReg
Specifies an array pair of regions and tolerances to be used in
those regions.
DelaunayType constrained
Types of mesh generated by Sentaurus Mesh. Available types
are box method, conformal, or constrained.
DelPsc false
Indicates whether the Delaunay refinement for piecewise
smooth complex (DelPSC) algorithm is applied to the
boundary at the beginning of a mesh generation step.
0.1 µm
degrees
µm

10: Mesh Generation
Mesh Settings
Sentaurus™ Process User Guide 713
H-2013.03
DelPscAccuracy 1e-4
Specifies the accuracy used by the DelPSC algorithm when
approximating high-curvature areas. This parameter is used
during standard mesh generation (as opposed to using DelPSC
during oxidation).
EdgeProximity 0.05
Specifies the minimum ratio of the edges generated when an
edge is split.
FaceProximity 0.05
Specifies the minimum ratio of the faces generated when a
face is split.
ImprintCoplanarFacesOnly true
Imprints the binary tree on the coplanar sets of faces. This is
useful to avoid over-refinement in curved areas.
ImprintCoplanarityAngle 179
Angle used to decide when two faces are coplanar. If two
adjacent faces have an angle greater than this value, they will
be added to the set of faces to be imprinted with the binary
refinement tree cells.
max.box.angle.2d 120
Maximum angle in binary tree (2D only).
max.box.angle.3d 150
Maximum angle in binary tree (3D only).
MaxAspectRatio 1e6
Specifies the maximum-allowed aspect ratio of an element in
the binary tree.
MaxBoundaryCutRatio.2d 0.01
Specifies the maximum-allowed ratio between the lengths of
adjacent axis-aligned edges cutting material boundaries (2D
only).
MaxBoundaryCutRatio.3d 0.01
Specifies the maximum-allowed ratio between the lengths of
adjacent axis-aligned edges cutting material boundaries (3D
only).
MaxConnectivity 1e37
Specifies the maximum number of elements connected to a
point in the final mesh.
MaxNeighborRatio 3.0
Specifies the maximum-allowed ratio between the
circumscribed spheres of neighboring elements.
MaxPoints 100000
Maximum number of points allowed by the Sentaurus Mesh
delaunization module.
MaxSolidAngle 360
Specifies the maximum solid angle allowed in the elements of
the mesh (3D only).
MaxTetQuality 1e37
Specifies the minimum shortestEdge/
circumradius ratio allowed in the mesh (3D only).
MinAngle 0
Specifies the minimum angle allowed in the elements of the
mesh (2D only).
minedge 2.0e-6
Minimum edge length request.
Table 69 Parameters available for Sentaurus Mesh
Parameter Default Description
µm
degrees
degrees
degrees
degrees
µm

10: Mesh Generation
Controlling Mesh during Oxidation
714 Sentaurus™ Process User Guide
H-2013.03
Controlling Mesh during Oxidation
Oxidation creates new regions and dramatically alters the shape of existing ones. Controlling
the mesh is important. This section covers some mesh control methods.
TS4 Mesh Library
The TSUPREM-4 moving-boundary meshing library is available from within Sentaurus
Process (hereafter, referred to as the TS4 mesh library). By default, the TS4 mesh library
performs the 2D mesh update.
The following statement switches off the TS4 mesh library and specifies the use of the old
mesher:
pdbSet Grid UseTS4Mesh 0
2D.MeshLib is an alias of UseTS4Mesh.
The TS4 mesh library deposits the native oxide layer before oxidation by default. Since it
removes all grids inside a gas region, the simulation performance is improved without any loss
of accuracy. To use the MGOALS native layer in two dimensions, instead of the TS4 mesh
library, use the command:
pdbSet Grid UseTS4Native 0
SliverAngle 175
Limits the maximum dihedral angle on one element when the
delaunizer performs the sliver removal step.
SliverDistance 1e-2
Limits the amount of “damage” done to the standard Voronoï
diagram by the sliver removal algorithm. Note that the grid
produced by the sliver removal algorithm is weighted
Delaunay, so the standard Voronoï diagram is “damaged”
unless the Voronoï weights are stored (see the
StoreDelaunayWeight parameter). When the box
method library reads those weights, it calculates the correct
Voronoï diagram and coefficients to solve the PDEs.
StoreDelaunayWeight 0
When set to 1, stores the Delaunay–Voronoï weight
(DelVorWeight) for the box method library.
UseLines true
Specify 1 or 0. UseLines is specified in the line
command in the mesh generated by Sentaurus Mesh.
Table 69 Parameters available for Sentaurus Mesh
Parameter Default Description
degrees
µm
10: Mesh Generation
Controlling Mesh during Oxidation
Sentaurus™ Process User Guide 715
H-2013.03
2D.MeshLib.Native is an alias of UseTS4Native.
For silicidation, a gas mesh is used by default. The following statement forces the use of the
TS4-style gas mesh instead of the default gas mesh:
pdbSet Grid UseTS4GasMesh 1
Control Parameters in TS4Mesh
The control parameters are specified with:
pdbSet Grid TS4Mesh <control parameter> <value>
The available control parameters are:
■MergeSubAndAdd <0|1> (default: 1)
It optimizes the speed performance by merging the grid subtraction and addition
procedures.
■DoSubAfterStep <0|1> (default: 0)
Grid subtraction is performed after each diffusion step, while grid addition is performed
after each mechanics step followed by the diffusion step. Switching on this flag forces only
one diffusion step per each mechanics step. When this flag is switched on,
MergeSubAndAdd is ignored.
■SubTimeFactor <double> (default: 1.5)
The time step given by mechanics for grid removal is scaled by SubTimeFactor.
NOTE Do not change the default.
■MinSpaceOnInterface <double> (default: 2e-6 [ ])
The nodes on an interface mesh must be rebuilt after meshing on the moving boundary
since the bulk meshes along the interface can be added or removed. Instead of destroying
and rebuilding the interface mesh, the TS4 mesh library tries to reuse the original node data
on the interface mesh to minimize the interpolation error. The original nodes are detected
when the location difference is less than MinSpaceOnInterface.
NOTE Do not change the default.
■ExactGridSpace <0|1> (default: 1)
On the growing material side of the interface, the triangular mesh elements expand. To
maintain solution accuracy in the material (for example, calculating the diffusion of
oxidant in the oxide), you must add nodes to the growing material. The addition of nodes
µm
10: Mesh Generation
Controlling Mesh during Oxidation
716 Sentaurus™ Process User Guide
H-2013.03
to the growing material is controlled by perp.add.dist, ExactGridSpace, and
LocalGridSpace.
NOTE perp.add.dist is the grid control parameter of each material, for
example: pdbSet Oxide Grid perp.add.dist 0.01e-4
Precise grid spacing is obtained by adding new nodes in a growing layer at the distance
specified by perp.add.dist from the existing node in the layer. Because only one node
can be added at each point on an interface during a simulation time step, the size of the time
step may need to be reduced to achieve the required spacing. This reduction in the time step
can be disabled by specifying:
pdbSet Grid TS4Mesh ExactGridSpace 0
By default, ExactGridSpace is set to 1 to allow reducing the number of time steps to
control the grid spacing. The algorithm does not allow grid points to be added at spacings
less than 1 A, and control of the spacing may not be precise for spacings less than 2 A.
■LocalGridSpace <0|1> (default: 1)
The grid control algorithms and parameters apply to the entire structure. To avoid adding a
very fine grid in field regions when growing gate oxides, an option allows the grid spacing
to vary with the oxide growth rate. When LocalGridSpace is switched on, the grid
spacing to be used at each point in the growing material is:
h = (vmax/v) perp.add.dist
where v is the growth rate at a point in the structure, and vmax is the maximum growth rate
at all interfaces of the same type in the structure. LocalGridSpace is switched on by
default.
■OrderFlatTri <0|1> (default: 1)
When the area of a shrinking triangle becomes less than 1e-15 (cm2) after a time step, the
triangle is removed. When the shrinking triangle to be removed is located at a material
interface and the removal of the triangle will result in a bad mesh, the material type of the
shrinking triangle is replaced with the type of the growing neighbor material, instead of
removing it. When those triangles are adjacent to each other, the reordering algorithm for
the replacements smooths the interface shape after conversion.
■MinAreaRemovalRatio <double> (default: 10.0)
When a region has only one triangle surrounded by neighbors of different materials and its
area is less than MinAreaRemovalRatio multiplied by 1e-15 (cm2), the material type of
the triangle is replaced with the neighbor material that shares the longest edge with the
triangle.
■Min.Split.Distance <double> (default: 1e-8 [cm])
When multiple regions with the same material meet at one point, the point is split by
inserting new elements. The parameter determines the minimum split distance.

10: Mesh Generation
Controlling Mesh during Oxidation
Sentaurus™ Process User Guide 717
H-2013.03
Moving Mesh and Mechanics Displacements
The displacements computed by the mechanics solution during oxidation are applied to the
nodes after checking against the MinimumVelocity criterion defined for each region.
Velocity is the computed solution variable and is multiplied by the time step to compute
displacements. The nodes are moved by this amount.
The computed velocities are compared against MinimumVelocity and, if the computed
velocity is greater than MinimumVelocity, the displacements are computed and applied. The
MinimumVelocity is set with the command:
pdbSetDouble Silicon Grid MinimumVelocity <n>
Grid Spacing
Grid spacing in the growing region is controlled by perp.add.dist. The value is in units of
centimeter, and the edges in growing regions are checked to see whether they are nearly
perpendicular to the interface.
If they are perpendicular, they are split if their length exceeds the perp.add.dist number.
This value is set with the command:
pdbSet Oxide Grid perp.add.dist 2e-7 ;# unit is cm
Grid Cleanup
During oxidation or silicidation, the growing region increases at the expense of a shrinking
region. The shrinking regions then have a problem of small edges. Below a certain value, these
edges must be removed entirely, and the mesh around them must be adjusted.
The short edge criterion is specified by the Remove.Dist parameter, which is specified in
centimeters and is set as follows:
pdbSet Silicon Grid Remove.Dist 3e-8
NOTE Due to mesh quality constraints, this number must be kept above a value
of .
210
8–
× cm

10: Mesh Generation
Controlling Mesh during Oxidation
718 Sentaurus™ Process User Guide
H-2013.03
Maximum-allowed Rate of Growth
For a minimal simulation time, it would be best if the entire thickness growth were simulated
in one step. However, this is not possible. The reasons for this include:
■Nonconvergence of diffusion equations.
■Inability to track material interfaces if they grow more than one edge length of a triangle
or a tetrahedron, and so on.
In the new growing region, new nodes are introduced and the data is interpolated from the
nearby nodes; if growth is too fast, significant interpolation errors could occur.
The rate of growth can be controlled by the parameters dThickness and IncreaseRatio.
The dThickness parameter (specified in micrometers) defines the maximum-allowed
oxidation front displacement per time step and is set as follows:
pdbSet Diffuse dThickness 0.001
The IncreaseRatio parameter is the factor by which the time integration step is allowed to
grow.
Miscellaneous Tricks
Since Sentaurus Process oxidation does not allow the interface to traverse more than one
element thickness at a time, speed can be achieved by having elements with longer edge lengths
near the interface. This can be controlled by refinement boxes or the pdb parameter
Grid SnMesh min.normal.size. Large structures, like those used in power devices, may
need min.normal.size of , while submicron CMOS devices need .
The mesh away from the interface is unrefined based on the pdb parameter
Grid SnMesh normal.growth.ratio.3d. If the mesh is not unrefining fast enough, this
number can be increased.
In large structures, the interface fidelity may not need to be as tight as that of 45-nm or 32-nm
gate transistors. The MGOALS accuracy parameter can be increased to , which will
cause MGOALS to clean up interfaces of small (sub– ) features and ensure smooth long
edges that speed up oxidation.
These are options available to the process engineer; however, care must be exercised in varying
these parameters since they may affect the final structure significantly.
0.01 µm
8 Å
1 Å
1 Å

10: Mesh Generation
Controlling Mesh during Oxidation
Sentaurus™ Process User Guide 719
H-2013.03
Meshing for 3D Oxidation
Maintaining a conformal high-quality mesh during the simulation of 3D oxidation is very
difficult because of the following requirements: moving boundaries, accurate dopant profiles,
dose conservation, minimization of the number of mesh points, and maintaining high-quality
mesh elements. In particular, handling the frequent collision of the oxidation front with points
inside the silicon, polysilicon, or gas regions can cause intractable problems for the local mesh
operations needed for maintaining dose conservation.
Before each diffusion time-step, the mesh is checked for the maximum possible time step until
the first tetrahedron element collapses (becomes flat). If necessary, the time step is reduced. In
the diffusion simulation, all mesh points are moved using the velocity and the time step. The
mesh topology is not changed during the diffusion time step. At the end of the diffusion time
step, the mesh quality is improved flat elements are removed allowing for the next time step to
be sufficiently large.
MovingMesh
This section describes an experimental feature that can be used for 3D oxidation. This feature
called MovingMesh is activated with the following command before the diffuse command:
pdbSet Grid Use.MovingMesh 1 ;# switched on by default
Three important parameters control MovingMesh:
pdbSet Oxide Grid perp.add.dist 0.005e-4 ;# centimeter
pdbSet Grid Remove.Dist 0.001e-4 ;# centimeter
pdbSet Grid MovingMesh Remove.Dist.On.Interface 0.0001e-4 ;# centimeter
The perp.add.dist parameter specifies the distance that the oxide interface can move
before new mesh points are inserted in the oxide. The unit is in centimeter.
The Remove.Dist parameter specifies the shortest distance the mesh vertices are allowed
from the interface into the bulk. Shorter than this distance, the vertices will be removed. Do not
specify a distance larger than the minimum material thickness. For a typical example with
1.5 nm native layer, Remove.Dist of 1.0 nm or less is appropriate. The unit is in centimeter.
The Remove.Dist.On.Interface parameter controls small triangles on material interfaces.
Triangles with an edge shorter than this distance will be removed. A smaller number makes the
interfaces smoother, but results in a larger number of triangles.
10: Mesh Generation
Controlling Mesh during Oxidation
720 Sentaurus™ Process User Guide
H-2013.03
The oxide interfaces can develop problematic geometric features like knife edges, noisy
surfaces, or extremely thin gaps. You can enable geometry repair and surface remeshing by
using:
pdbSet Grid MovingMesh Repair.Geometry 1 ;# switched on by default
The criteria to trigger geometry repair are based on the minimum dihedral angle and the
maximum face angle:
pdbSet Grid MovingMesh Repair.Geometry.Min.Dihedral.Angle 5 ;# degree
pdbSet Grid MovingMesh Repair.Geometry.Max.Face.Angle 175 ;# degree
If the minimum dihedral angle between two triangles is below the threshold or the maximum
face angle of a triangle is above the threshold, the geometry repair procedure starts.
The geometry repair procedure involves a multimaterial level-set (MLS) formulation. The
resolution of the level-set cell size is controlled by:
pdbSet Grid MovingMesh Repair.Geometry.Resolution 0.001 ;# micrometer
NOTE The parameter Repair.Geometry.Resolution should be, at most,
one-third the thickness of the thinnest region. Otherwise, the thin region
may be considered noise, and it disappears.
The boundary representation (brep) of the new geometry must go through a meshing algorithm
for curved surfaces called the Delaunay refinement for piecewise smooth complex (DelPSC)
that improves the quality of triangles on brep surfaces. This algorithm is enabled by:
pdbSet Grid MovingMesh Apply.Brep.DelPSC 1 ;# switched on by default
DelPSC performs adaptive sampling on ridges (1D geometric feature) according to the
refinement fields, the curvatures of the ridges, and the proximity among the ridges. On each
surface patch (2D geometric feature), DelPSC performs adaptive sampling according to the
refinement fields and the curvatures of the surface.
Ridge sampling also is controlled by:
pdbSet Grid MovingMesh Apply.Brep.DelPSC.Resolution 0.005 ;# micrometer
The above parameter ensures no ridge edge will be longer than the specification. It is useful,
for example, when you have a straight line (no curvature) next to curved surfaces. You want the
sampling points on the straight line to be fine enough to support the adjacent curved surfaces.
Note that you no longer require Apply.Brep.DelPSC.Resolution to be as small as the thin
native layer thickness, because of the adaptive sampling based on proximity between nearby
ridges.
10: Mesh Generation
Controlling Mesh during Oxidation
Sentaurus™ Process User Guide 721
H-2013.03
To control accuracy in high curvature areas, you can specify the acceptable distance between
the old and new curved surfaces using:
pdbSet Grid MovingMesh Apply.Brep.DelPSC.Accuracy 0.0001 ;# micrometer
These are typical settings for a small transistor structure:
pdbSet Oxide Grid perp.add.dist 2e-7 ;# cm 2nm
pdbSet Silicon Grid perp.add.dist 1e-6 ;# cm 10nm
pdbSet Grid Remove.Dist 9e-8 ;# cm 9A
pdbSet Grid MovingMesh Remove.Dist.On.Interface 3e-8 ;# cm 3A
pdbSet Grid MovingMesh Repair.Geometry.Resolution 3e-4 ;# um 3A
pdbSet Grid MovingMesh Apply.Brep.DelPSC.Resolution 3e-3 ;# um 3nm
pdbSet Grid MovingMesh Apply.Brep.DelPSC.Accuracy 1e-4 ;# um 1A
These are typical settings for a large power structure.
pdbSet Oxide Grid perp.add.dist 2e-6 ;# cm 20nm
pdbSet Silicon Grid perp.add.dist 1e-5 ;# cm 100nm
pdbSet Grid Remove.Dist 9e-7 ;# cm 9nm
pdbSet Grid MovingMesh Remove.Dist.On.Interface 3e-7 ;# cm 3nm
pdbSet Grid MovingMesh Repair.Geometry.Resolution 3e-3 ;# um 3nm
pdbSet Grid MovingMesh Apply.Brep.DelPSC.Resolution 3e-2 ;# um 30nm
pdbSet Grid MovingMesh Apply.Brep.DelPSC.Accuracy 1e-3 ;# um 1nm
MovingMesh has facilities for troubleshooting run-time failures. A typical setting would be:
# Switch on level-1 diagnostics
pdbSetDouble debugLevel MovingMesh 1 ;# switched off by default
# Save intermediate result every 100 time steps
pdbSetDouble Grid MovingMesh Save.Interval 100 ;# switched off by default
# Save diagnostic files for Repair.Geometry and DelPSC
pdbSetDouble Grid MovingMesh Repair.Geometry.Monitor 1
;# switched off by default
In level-1 diagnostics, the intermediate result will be saved after a certain number of time steps
in the files:
<NodeName>_MovingMeshGridTimeStep<xxxx>.tdr
The frequency of saving is specified by the Save.Interval parameter. The files are written
after mechanics and before diffusion to analyze the grid-limited time step.
In the event of failure in Repair.Geometry, the level-1 diagnostics will save files with names
such as:
<NodeName>_remeshBrep{In,MLS,PSC,Out}.tdr
<NodeName>_applyBrepDelPSC{In,Out}.tdr

10: Mesh Generation
UseLines: Keeping User-defined Mesh Lines
722 Sentaurus™ Process User Guide
H-2013.03
They are useful for checking whether the resolution parameters are adequate. The most likely
cause of failure is a too coarse resolution to capture thin oxide layers and other small geometric
features.
If the parameter Repair.Geometry.Monitor
is set to 1, the intermediate files with names
such as:
<NodeName>_remeshBrep_<xxxx>_{In,MLS,PSC,Out}.tdr
<NodeName>_applyBrepDelPSC_<xxxx>_{In,Out}.tdr
will be saved every time the repair geometry operation is triggered. These files are useful to
monitor how the MLS and DelPSC algorithms perform at various points in a simulation.
UseLines: Keeping User-defined Mesh Lines
During the init command, the line location and spacing specifications given by line
commands are expanded into ticks and stored in the PDB and in TDR files. This is performed
by default.
By carefully placing lines, you can isolate areas of the structure that changed (because of
etching, deposition, and so on) from those that do not (such as bulk silicon). In this way, the
mesh in areas that do not change will have the least amount of change, the least interpolation,
and the most accurate results. Even the mesh in regions that do change will have a similar
starting point and should also have minimal mesh-point movement coming from remeshing.
Lines for Sentaurus Process Kinetic Monte Carlo are stored separately from the lines used with
continuum solvers (in other words, Sentaurus Mesh meshes). By default, line commands are
applied to both KMC and continuum meshes. Use the parameter !kmc or!mgoals to not
apply a particular line command. For example, for a line command to apply only to
continuum, use line !kmc.
In the simplest case, all the line commands are specified before the init command, and they
are saved and reused every time a remesh is performed. However, there are other cases
described in the following sections that allow this feature to be more powerful.
All the line commands are specified before the init command, and they are saved and
reused every time a remesh is performed. However, there are other cases described in the
following sections that allow this feature to be more powerful.

10: Mesh Generation
UseLines: Keeping User-defined Mesh Lines
Sentaurus™ Process User Guide 723
H-2013.03
Using line Commands after init Command
The expansion of lines from line commands into ticks (in other words, all starting mesh line
locations) is performed only at the point that the lines in that direction are needed. For example,
x-lines are always expanded in the init command, but y-lines are only expanded when the
first etch with a mask is given. Therefore, it is possible to load a 1D structure, give y-lines, and
then expand to 2D, or give both y- and z-lines and expand to 3D.
After a particular direction or dimension is expanded, it is only possible to insert one tick at a
time in that direction using the line command (in other words, the spacing parameter is
thereafter ignored). For example, this could be because you identified the amorphous–
crystalline interface in silicon.
This is handled by specifying one of the following:
■The line command in a dimension greater than the current dimension. For example, a y-
line specification when the simulation is in 1D.
■The line command in a dimension at or less than the current dimension. For example,
either an x-line or a y-line specified when the simulation is in 2D.
For more information about the operating dimension, see Automatic Dimension Control on
page 74.
Dimension within Current Spatial Dimension
This is encountered if the user-specified x-lines and the current spatial dimension of analyses
is 1D. Or, it could happen if you specify x- or y-lines in 2D, or x-, y-, or z- lines in 3D.
In this case, the line command ignores the spacing parameter and tries to insert only one
tick as long as that tick (line) is not too close to an existing tick.
Inside the init command, the line commands are expanded into ticks using the spacing
specifications for the dimension as they are needed. When additional line commands are
given for dimensions where the ticks have already been expanded, the spacing parameter is
ignored and one additional tick is added as long as it is not too close to an existing tick.
Dimension Greater Than Current Spatial Dimension
This is encountered if you specified y- or z-lines, and the current spatial dimension of analyses
is 1D. Or, it could happen if you specify z-lines and the current spatial dimension of analyses
is 2D.
10: Mesh Generation
UseLines: Keeping User-defined Mesh Lines
724 Sentaurus™ Process User Guide
H-2013.03
In this case, the line command is considered in its entirety, and the spacing parameter is
used. All the intermediate lines are included in the list of ticks kept.
Creating More Than One Structure
You c an cr ea te mo re tha n o ne s tr uct ur e us in g line, region and init commands in one
command file when using the UseLines feature. To ensure lines from the first structure are
not inserted into subsequent structures, it is important to issue line clear before starting the
definition of a new structure. For example:
#####################################################
line x loc=0 spacing=0.001
line x loc=1 spacing=0.1
line y loc=0
line y loc=1
region silicon
init !DelayFullD
# initial structure gives 96 nodes
LogFile [grid qual nodes]
grid remesh
# after remesh, 112 nodes
# because Sentaurus Process adds interface refinement
LogFile [grid qual nodes]
####################################################
# the lines from above are removed
# to start a new structure
line clear
line x loc=0
line x loc=1
line y loc=0
line y loc=1
region silicon
init !DelayFullD
# this tiny structure has only have 8 nodes
LogFile [grid qual nodes]
grid remesh
# grid remesh gives 44 nodes
LogFile [grid qual nodes]

10: Mesh Generation
UseLines: Keeping User-defined Mesh Lines
Sentaurus™ Process User Guide 725
H-2013.03
The UseLines and transform Commands
The ticks must be handled in a special manner with the transform reflect, transform
stretch, transform rotate, transform translate and transform cut commands.
The reflect Command
In the reflected region, the ticks are created after applying lateral inversion along the
appropriate plane.
The stretch Command
On applying stretch at a given coordinate in a given direction, the existing ticks in the
stretched area are translated by the amount of the stretch. You must insert lines in the stretched
area appropriately.
The rotate Command
When applying rotate, the ticks also are rotated and properly transferred between x-ticks, y-
ticks and z-ticks.
The translate Command
The translate command shifts the ticks by the specified amount.
The cut Command
The lines in the part of the structure that is cut are deleted.
Examples
Testing line Commands
Use the following example to test line commands:
line x loc=0 tag=a spacing=0.05
line x loc=0.1 spacing=0.05
line x loc=1 tag=b spacing=0.05
line y loc=0 tag=c spacing=0.01
10: Mesh Generation
UseLines: Keeping User-defined Mesh Lines
726 Sentaurus™ Process User Guide
H-2013.03
line y loc=0.6 tag=d spacing=0.1
region silicon xlo=a xhi=b ylo=c yhi=d
init !DelayFullD
deposit oxide thickness=0.002 iso
grid remesh info=2
line y loc=0.026
line y loc=0.027
line y loc=0.028
line y loc=0.029
line y loc=0.025
line y loc=0.024
line y loc=0.023
grid remesh
deposit poly thickness=0.18 iso
mask name=m1 left=-0.1 right=0.025
etch aniso thickness=0.2 poly mask=m1
struct tdr=linetest
Showing Clearing Lines for a New Structure
Use the following example to show clearing lines and to prepare for another structure definition
within the same command file:
line x loc=0 tag=a spa=0.125
line x loc=1 tag=b spa=0.125
line y loc=0 tag=c spa=0.125
line y loc=1 tag=d spa=0.125
region silicon xlo=a xhi=b ylo=c yhi=d
init
grid FullD
line clear
line x loc=0 tag=a spa=0.125
line x loc=1 tag=b spa=0.125
line y loc=0 tag=c spa=0.125
line y loc=1 tag=d spa=0.125
region silicon xlo=a xhi=b ylo=c yhi=d
init
line y loc=0.3 spa=0.01
grid FullD

10: Mesh Generation
Data Interpolation
Sentaurus™ Process User Guide 727
H-2013.03
Data Interpolation
Sentaurus Process stores a copy of the mesh with all its data before performing any geometry-
changing operation. This is the reference mesh used to interpolate data onto the new mesh. In
3D, a mesh is generated only when it is necessary, so you can have multiple etch, deposit,
photo, and strip commands without the need to remesh in between. When a new mesh is
required, data is interpolated from the stored mesh and data.
Data interpolation is performed material-wise. This is important because some nodal data can
be discontinuous at material interfaces; for example, segregation causes a jump in
concentration at the silicon–oxide interface. In addition, the precise location of an interface can
change slightly due to numeric noise in geometry-moving algorithms. Therefore, it is
necessary to allow the data to be interpolated from points in the old mesh nearby, but only from
the same material.
Data also can be interpolated from materials that are Like materials (that is, the material in the
old mesh is Like the material in the new mesh, or the material in the new mesh is Like the
material in the old mesh). When interpolating data at an interface, the preference is to use data
from the same region, then data from the same material, and finally data from Like materials.
If no match is found, then 0 is set for all data at that point.
For data defined on elements, the overlap of elements from the old mesh to the new mesh is
used for weighting. Similar to nodal data, interpolation of elements near interfaces uses the
region, material, and Like material preference order.
Multithreaded interpolation can be used to speed up interpolation in large 3D structures.
Because of the memory-intensive nature of interpolation, typically, the performance benefit of
multithreading saturates at two threads and can even decrease when using more than four
threads. Therefore, for interpolation, the suggested maximum is to use two threads using the
command:
math numThreadsInterp=2
Troubleshooting
Sometimes, the mesh generation step fails and it is not clear what the problem may be. The
following are recommendations of where to look when problems arise during meshing:
■Set InfoDefault to 2 or higher, for example:
pdbSet InfoDefault 2
10: Mesh Generation
Troubleshooting
728 Sentaurus™ Process User Guide
H-2013.03
■When Sentaurus Mesh prints the message:
"Short edge 1e-8 around points (x1, y1, z1) (x2, y2, z2)"
look at the input structure around the coordinates (x1, y1, z1) or (x2, y2, z2), and
check whether there is a singularity in that area (a crack, fold, surface overlap, and so on).
Sometimes, these singularities are the product of an etching or a deposition step, and action
can be taken to improve the quality of the structure.
■Check the quality of the boundary printed for the steps preceding the mesh generation
process. In particular, the following line provides an indication of quality (this is output if
InfoDefault is 2 or higher):
minDihedralAngle: <angle> [near (x1, y1, z1),(x2, y2, z2)] at
region=Nitride_1.
If you see an angle of less than in the geometry, this may indicate a problem in the
structure at the given coordinates. The recommendation is then to look at the preceding
process steps in Tecplot SV, and to see whether they can be modified to avoid creating the
problem.
To visualize the problem area in Tecplot SV, you can create a rectangular zone that can be
used as a marker to identify the problem. Select Data > Create Zone > Rectangular, and
input a box where one of the corners is a coordinate reported by Sentaurus Mesh as
problematic. The second corner of the box must be calculated manually to give a box large
enough to be seen in the visualization window. Then, hide all materials except the box that
you created. Magnify the rectangular zone, and display the material that was reported in
the minDihedralAngle message. Now, you should see the artifact. Sometimes, you need
to rotate the structure around the rectangular box to see what is happening to the geometry.
■In addition to this, you may need to add !repair to the etch or deposit command. This
prevents the structure from being repaired and makes it easier to spot the problem in
Tecplot SV.
■It is recommended to frequently save snapshots of the boundary file of the structure,
especially before all mesh generation operations. This will help you to investigate possible
problems in the input to the mesh generator. To accomplish this, use the command:
struct tdr.bnd=fileName
3°
10: Mesh Generation
Troubleshooting
Sentaurus™ Process User Guide 729
H-2013.03
Figure 98 (Left) Example of artifact in geometry and (right) magnification of artifact
10: Mesh Generation
Troubleshooting
730 Sentaurus™ Process User Guide
H-2013.03

Sentaurus™ Process User Guide 731
H-2013.03
CHAPTER 11 Structure Generation
This chapter describes the etching, deposition, and other geometry
manipulations available in Sentaurus Process.
Overview
Sentaurus Process offers the following interfaces to perform etching and deposition:
■MGOALS (1D, 2D, and 3D)
■Sentaurus Structure Editor (3D only)
■Sentaurus Topography and Sentaurus Topography 3D (limited availability)
By default, Sentaurus Process uses an internal module, MGOAL, which is based on polygonal
boundary representations. This is the preferred module for performing geometric etching and
deposition because of its robust and flexible algorithms. The etching and deposition operations
are always simulated using geometric shapes or simple mathematical formulations; no physical
processes are simulated. For a description of the MGOALS interface, see MGOALS Interface
on page 778.
In 3D, Sentaurus Process offers and interfaces with the geometric library in Sentaurus Structure
Editor to perform basic and most common geometry-modeling process steps. For a description
of the Sentaurus Structure Editor interface, see Sentaurus Structure Editor Interface on
page 791.
For physical etch and deposition, Sentaurus Process provides an interface to Sentaurus
Topography and Sentaurus Topography 3D. For a description of the interfaces, see Sentaurus
Topography Interface on page 794.
Functionality
Sentaurus Process provides a number of etching and deposition operations, in addition to
purely geometric operations to help shape the geometry of the devices.
11: Structure Generation
Overview
732 Sentaurus™ Process User Guide
H-2013.03
Etching
The following types of etching are supported (see Etching on page 733 for more details):
Deposition
The following types of deposition are supported (see Deposition on page 751 for more details):
Masks and Photoresist
Masks offer an effect (similar to a masking layer) to limit the etch or deposition process to a
certain window or to provide a convenient way to mimic lithographic patterning (see The mask
and photo Commands on page 764).
Geometry Creation and Transformations
You als o c an create an d i nse rt polygo ns an d polyh ed ra , or re ad an ex isting 3D s tr uctur e f ro m a
file (see Inserting Polygons in Two Dimensions on page 785 and Inserting Polyhedra in Three
Dimensions on page 786).
The shape library provides commands for generating some special shapes in Sentaurus Process
(see Shape Library on page 757).
In addition, several geometric transformations are available including reflection, translation,
rotation, flipping, cutting, and stretching (see Geometry Transformations on page 772).
■Isotropic
■Anisotropic
■Directional
■Polygonal
■Trapezoidal
■Fourier
■Crystallographic
■Chemical-mechanical polishing (CMP)
■Piecewise linear
■Isotropic
■Anisotropic
■Directional
■Polygonal
■Fill
■Fourier
■Crystallographic
■Trapezoidal (3D only)

11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 733
H-2013.03
Etching and Deposition Types and Options
Three main specifications are required for all etching and deposition steps:
■Etch or deposit type
■Material or materials to be etched, or material to be deposited
■Amount of material to be removed or deposited
Etching
The types of available etching are:
NOTE To remove materials exposed to the top gas, use the strip command
instead of the etch command. The strip command is used
specifically for this purpose. It is more straightforward, less prone to
user error, and more robust in delivering the expected results:
strip Photoresist
type=angles.rates
Etch according to a definition of a piecewise linear etch rate.
type=anisotropic
Etch in the vertical direction only.
type=cmp
Perform CMP. The coordinate of the new surface must be specified as
coord.
type=crystal
Angle-dependent etching where etch rate is dependent on the
crystallographic direction.
type=directional
Etch in one specific direction only.
type=fourier
Angle-dependent etching where etch rate is a cosine expansion of the
etching angle.
type=isotropic
Etch rate is uniform in all directions.
type=polygon
Etch according to a user-supplied polygon (2D only).
type=trapezoidal
Etching allowing undercut and taper angle specifications (2D only),
or taper and bottom angle specifications (3D).

11: Structure Generation
Etching and Deposition Types and Options
734 Sentaurus™ Process User Guide
H-2013.03
Each etch type requires the setting of parameters particular to that etch type. Many options are
available and certain options are available only with certain etch types. Table 70 summarizes
the syntax options for each etch type.
Table 70 Options for etch and deposit command syntax (E=etching, D=deposition)
Area Parameter name
isotropic
anisotropic
directional
cmp
polygon
fourier
crystal
trapezoidal
angles.rates
Rate
rate
ED ED ED E
angles.rates
E
coeffs
ED
mat.coeffs
E
crystal.rate
ED
Stop criteria
time
ED ED ED ED ED E E
thickness
ED ED ED ED
etchstop
EE E EE E
coord
E
etchstop.overetch
EE E EE E
isotropic.overetch
EE
Shape
polygon
ED
angle
ED
undercut
Ea
a. In 2D when not using force.full.levelset.
bottom.angle
Eb
b. In 3D when not using force.full.levelset.
bottom.thickness
Eb
direction
ED
ambient.rate
Ec
c. When used in conjunction with force.full.levelset.
Beam
sources
ED E
shadowing
EcDED ED E
shadowing.nonisotropic
ED
Mesh
ast
ED ED ED E ED ED ED E
Adaptive
ED ED ED E ED ED ED E
Mode
force.analytic
EEE
force.full.levelset
ED E ED EdDd
d. Full level-set is the default scheme for Fourier and crystallographic.
EdDd E
1D
ED
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 735
H-2013.03
To specify the etch type, the parameter type is used. For some etch types, one of the following
keywords can be used instead as a shorthand for specifying type: isotropic,
anisotropic, trapezoidal, or cmp. The amount to be etched is specified as either
thickness (by specifying rate and time) or an etchstop material with etchstop.
Etch types have been implemented in Sentaurus Process using three different methods
(analytic, fast level-set, and general time-stepping level-set), which are described in MGOALS
Interface on page 778.
These methods may require different inputs to perform the steps and may take different effects
into account. The method is selected depending on the specified parameters and the structure
to be etched.
NOTE The simplest and fastest algorithm possible is chosen by default.
If simple isotropic, anisotropic, directional, or CMP etching of a single material is requested
and for polygonal etching, an analytic algorithm is tried first. The analytic algorithm is the
fastest and most accurate. However, in some cases, the resulting etching front might intersect
itself. Because the analytic algorithm cannot handle this situation, the fast level-set method is
used.
NOTE Although these methods are fast and can handle most simple etching
tasks, they do not consider shadowing or visibility effects, and they
cannot etch more than one material at a time.
The general time-stepping level-set method is chosen if you specify any rate versus angle-type
etching (Fourier or crystallographic), or if you choose to etch different materials at different
rates, or if the parameter force.full.levelset is specified. In addition, the general time-
stepping level-set method can handle multiple etching beams and, optionally, shadowing.
The general time-stepping level-set scheme used in Sentaurus Process has the same limitations
as all level-set methods:
■Sharp corners in the evolving front are rounded.
■Small front movement requires a fine level-set mesh, resulting in large memory use and
long simulation times.
■The accuracy is limited by the size of the level-set mesh.
Besides the etching type, the materials to be etched and the amount of material to etch must be
specified. The amount of material to be etched can be specified in three ways:
■Thickness
■Rate and time
■Using an etch stop
11: Structure Generation
Etching and Deposition Types and Options
736 Sentaurus™ Process User Guide
H-2013.03
The etch rate may be specified using etch beams that are created in the beam command. Beams
can be used only with the Fourier etch type. If an etch stop is specified, the etching stops as
soon as the specified material is exposed to gas.
In addition, a mask specification can be given for all etch types, except CMP and polygonal to
limit the areas where material is removed.
NOTE The trapezoidal etch type supports mask specification in 3D only.
Etching Tips
Some tips for etching are:
■If the total etch thickness is exactly the same thickness as the layer to be etched, numeric
roundoff errors can cause thin pieces of the material to be retained. You should etch a little
more (for example, 0.1%) than the thickness of the layer.
■Etching small thicknesses using a small isotropic.overetch or etching large
structures can cause MGOALS to allocate a large amount of memory and increase
simulation time to solve the level-set equation. The MGOALS parameter resolution can
be increased for the simplified boundary movement mode, and the parameters dx and dy
can be increased in the general boundary movement mode to reduce memory consumption.
However, this may impact the accuracy.
Etching Type: Isotropic
Isotropic etching removes material at the same rate in all directions. You can specify more than
one material to be etched isotropically, in which case, the generalized level-set boundary
movement module is invoked.
When isotropic etching uses the level-set method (either fast or general time-stepping), the
final surface is obtained by solving a differential equation on a discrete mesh.
NOTE To control errors in the fast level-set method, use the resolution
parameter in the mgoals command. For the general etch method, use
either resolution or the dx and dy parameters of the mgoals
command.
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 737
H-2013.03
An example of a single-material isotropic etch is (see Figure 99):
etch silicon thickness = 0.05 type=isotropic
Figure 99 Single-material isotropic etch
An example of a multiple-material isotropic etch is (see Figure 100):
mgoals resolution=0.04
etch material = {Silicon Oxide Poly} rate = {1.0 1.5 1.0} time = 0.05 \
type=isotropic info=2
Figure 100 Multiple-material isotropic etch
Etching Types: Anisotropic and Directional
Anisotropic etching is designed primarily to work with masks or masking layers. It also is
frequently used to create spacers. Anisotropic etching etches material away in a direction that
11: Structure Generation
Etching and Deposition Types and Options
738 Sentaurus™ Process User Guide
H-2013.03
is purely vertically downwards. It works well with structures such as those shown in
Figure 101.
Figure 101 Anisotropic etching
Anisotropic etching can take more than one material if the same rate is specified for all etched
materials. This can be useful to create multiple spacers since it does not produce small gaps
which are hard to avoid when etching one spacer at a time. If you want to use different rates for
each material then a better alternative can be Fourier etching.
If anisotropic etching is performed to etch the shaded region in the structure shown in
Figure 102, instabilities can arise. The resulting structure can be very different depending on
the numeric roundoff errors.
Figure 102 For this structure, anisotropic etching would not be stable
Anisotropic operations are sensitive to numeric noise at vertical or nearly vertical walls.
If the etch command is supposed to remove the entire layer, care must be taken to overetch by
a small amount to prevent thin regions remaining due to numeric roundoff error.
NOTE It is more robust and better to use the strip command to remove all
exposed layers of a certain material.
The directional etching method is similar to the anisotropic method. In this case, the specified
etching rate is applied in the direction of the etching beam. Visibility effects are not considered.
The etching window is determined from user-defined masks and from the exposed areas of the
etched material (see Figure 103 on page 739).
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 739
H-2013.03
For example:
etch silicon thickness = 0.05 type=anisotropic
Figure 103 Anisotropic etching
In the next example, directional etching is demonstrated. The parameter direction sets the
direction of the etching beam by setting values for {x y z}. This direction vector is normalized
to 1.0 before being used by the etching module.
etch material=silicon rate = 0.05 time=1.0 type=directional direction = {1 1}
Figure 104 Directional etching (angled wall)
etch material=silicon rate = 0.05 time=1.0 type=directional direction = {1 -1}
Figure 105 Directional etch undercutting
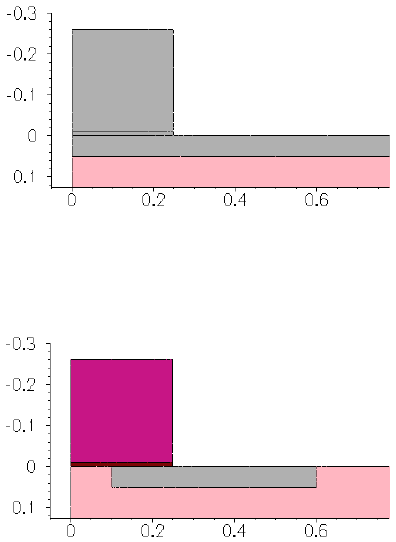
11: Structure Generation
Etching and Deposition Types and Options
740 Sentaurus™ Process User Guide
H-2013.03
Etching Types: Polygonal and CMP
Polygonal etching provides a way of modifying a region without having to define etching rates
or the direction of the etch. The specified polygon is used to intersect the mesh elements. The
elements inside the polygon are replaced by gas.
CMP is handled as a special case of polygonal etching. Mesh elements are intersected at the
specified coordinate. All elements of the specified material above the coordinate are reassigned
to gas.
etch type=cmp coord = 0.05 material=all
Figure 106 CMP
# Etch with polygon given as x1 y1 x2 y2 ... xn yn
etch type=polygon material=silicon polygon= {-0.1 0.1 0.1 0.1 0.1 0.6 -0.1 0.6}
Figure 107 Polygonal etch
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 741
H-2013.03
Etching Type: Fourier
In Fourier etching, the etching rate is a function of the angle between the incident etching beam
and the normal vector of the surface being etched. This allows for reasonably directional
etching with control of the slope of sidewalls. The coefficients are defined using coeffs
(for a single-material etch) or mat.coeffs (for a multimaterial etch), and the etching rate is
computed according to:
(966)
where:
■ is the angle between the incident beam and the normal to the surface being etched.
■ is the factor given in the beam command for beam .
Any number of coefficients can be given for each material. If the parameters are chosen
such that negative etch rates would result in some slope angles, no etching will occur on the
parts of the surface that have that slope. It is common to set the parameters such that the
etch rate for angles less than a certain angle are positive and drop below zero (resulting in no
etching) above that angle. This produces a trench with a rounded bottom and a sidewall given
by the angle where the etch rate drops to zero.
Fourier etching uses the full level-set model formulated after Lax–Friedrichs. This formulation
shows good stability, leading to good accuracy of etching wall-angle control. The Lax–
Friedrichs formulation results in slightly less corner sharpness.
Etching Beams
The beam command is used to define the direction and relative strength of etching beams to be
used with Fourier etching. The syntax is:
beam name=<beam_name> incidence=<angle> | direction= {<x> <y> <z>}
factor=<relative_strength>
The angle of incidence of each beam is specified either by the incidence parameter in the
beam command (incidence=0 defines a vertical beam), or by a direction vector, which is
normalized automatically to unit length. To be clear, the angle in Eq. 966 is measured from
the surface perpendicular to the angle of incidence for beam . The relative strength factor
is used to mix the strength of different beams. Each etching beam must be given a unique name.
Etching beams are assumed to be collimated, that is, a slight angular spread of beam direction
is not considered.
An
etch rate factoriAjθi
cosj
j0=
n
i0=
m
=
θi
i
factori
i
Aj
Aj
Aj
θi
i
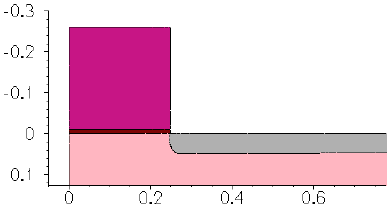
11: Structure Generation
Etching and Deposition Types and Options
742 Sentaurus™ Process User Guide
H-2013.03
The parameter sources of the etch command specifies the list of names of etching beams to
be used in an etch operation.
A Fourier etching example follows (see Figure 108):
mgoals resolution=0.02
beam name=src1 direction = {1 0 0} factor=1
etch material=silicon type=fourier sources = src1 coeffs = {0 0 1.0} time=0.05
Figure 108 Fourier etching
Through the selection of Fourier coefficients, the angle of the etching wall can be controlled to
a large degree. In particular, the first coefficient in the list, , corresponds to the equivalent
of the rate of isotropic etching. The second coefficient in the list, , corresponds
approximately to the equivalent of the rate of anisotropic or directional etching. The
approximate formula for determining the etch wall angle is given as:
(967)
where is the angle of the etch wall measured from the horizontal plane. For example, the
choice of and results in an etch wall at an angle of approximately
from the horizontal as shown in Figure 109 on page 743:
beam name=src1 direction= {0.1 0 0} factor=1
mgoals full.resolution= 0.05 resolution= 0.05
etch material= {silicon} type=fourier sources= {src1} coeffs= {-0.5 0.7071} \
time=1.0.
A0
A1
ϕA–0
A1
---------
cos 1–
≈
ϕ
A00.5–=
A10.7071=
45°
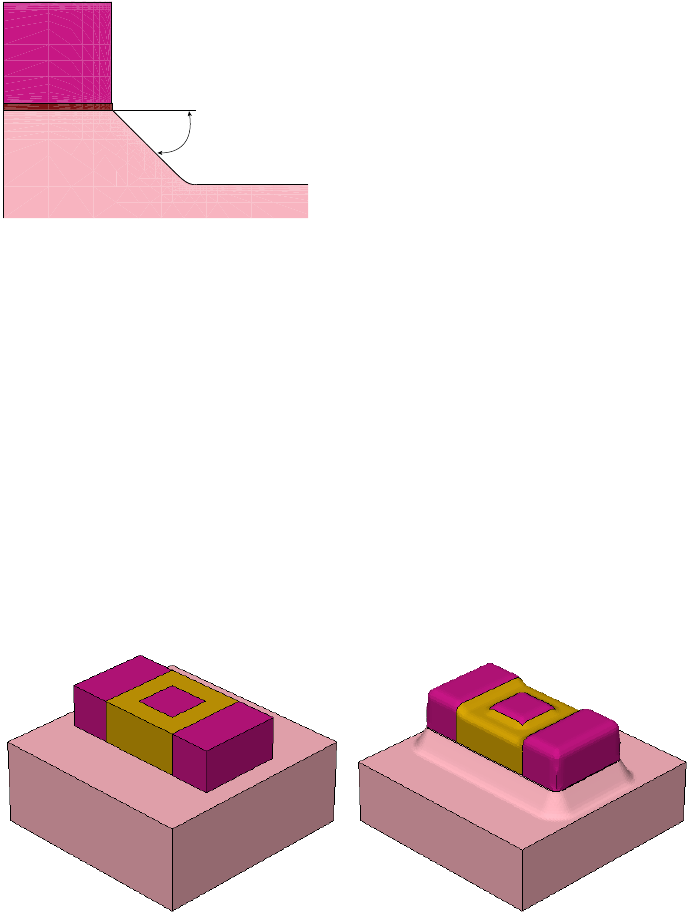
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 743
H-2013.03
Figure 109 Example of Fourier etch
Another Fourier etching example shows the functionality in 3D and how multiple rates for
multiple materials are specified using mat.coeffs. For 3D Fourier etching, also use the
command sde off.
Etching coefficients chosen for this example are illustrative and may not be physically
meaningful:
beam name=src1 direction= {1 0 0} factor=1
mgoals dx=0.1 dy=0.1 dz=0.1
sde off
etch info=10 sources= {src1} mat.coeffs= { Silicon= {-1 2} \
Nitride= {-0.7 1.2} Oxide= {0.01} PolySilicon= {-0.05 0.2} } type=fourier \
remesh=false time=1
Figure 110 Three-dimensional multimaterial Fourier etching example before etching (left)
before etching and (right) after etching
Currently, in 3D Fourier etching, shadowing is not implemented as it is in two dimensions.
ϕ
11: Structure Generation
Etching and Deposition Types and Options
744 Sentaurus™ Process User Guide
H-2013.03
Keywords shadowing and shadowing.nonisotropic
The keyword shadowing.nonisotropic is used instead of shadowing when you want to
allow only the 0th-order Fourier coefficient to etch areas where the beam is shadowed. The
keyword shadowing prevents all Fourier etching in areas shadowed from the beam; while the
keyword shadowing.nonisotropic prevents only the Fourier coefficients of order one and
higher from etching in areas where the beam is shadowed.
Be aware that even when this parameter is specified, the 0th-order Fourier coefficient
should continue to etch areas where the beam is shadowed. This permits a pseudo-isotropic
etching that is independent of shadowing, while at the same time the full Fourier etching occurs
only in areas where the beam is not shadowed.
Etching Type: Crystallographic
The parameter crystal_rate defines etching rates for different crystallographic
orientations. These rates are used for type=crystal. For details on wafer orientation, see
Defining the Crystal Orientation on page 73. Crystallographic etching rates are specified in
crystal_rate as a list of Miller indices and corresponding etching rates. The currently
accepted indices are <100>, <110>, and <111>, as in the following example:
crystal_rate= {"<100>" =0.8 "<110>" =0.35 "<111>" =0.003}
Interpolation of the rate at a given point along the etch front is calculated as a linear
combination of the <100>, <110>, and <111> rates weighted by the component of the etch
front normal vector along the corresponding crystallographic direction.
NOTE You must add a space between the double quotation mark (") after the
orientation and the equal sign.
A crystallographic etching example follows (see Figure 111 on page 745):
etch material=silicon type=crystal crystal_rate= {"<100>" =1.0 "<110>" =0.5 \
"<111>" =0.001} time=0.25
A0
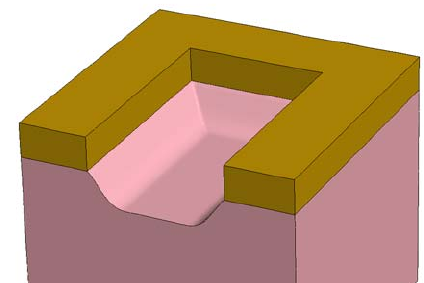
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 745
H-2013.03
Figure 111 Crystallographic etching
Etching Type: Trapezoidal
The trapezoidal etch model provides a simple but flexible approximation to a number of real
etching processes. The location of the etch is determined by masking layers (that is, layers of
nonetchable material that, if nonexistent, can be easily created with the photo command). In
3D, the etch location can be specified through the definition of a mask.
The trapezoidal model uses the following parameters to specify the shape of the region to be
removed:
■thickness specifies the vertical depth (or a combination of rate and time).
■angle specifies the angle (in degrees) of the resulting sidewalls.
■undercut specifies the horizontal penetration of the etch under the edges of the masking
layer. It only works in two dimensions.
■bottom.angle and bottom.thickness specify the angle and thickness of the sidewalls
for a second etching after thickness and angle are already etched (3D only).
These parameters can be used to approximate a number of real etching processes including:
■Combinations of vertical and isotropic etches.
■V-groove etches.
■Etches that produce retrograde sidewall profiles.
NOTE Trapezoidal etching is not supported by Sentaurus Structure Editor.
Consequently, the command sde off must be issued.
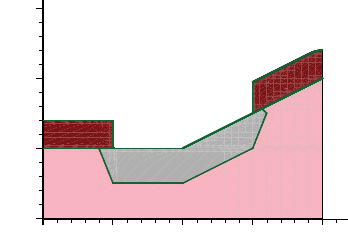
11: Structure Generation
Etching and Deposition Types and Options
746 Sentaurus™ Process User Guide
H-2013.03
Trapezoidal 2D Etching
An etch with the trapezoidal model is performed in three steps:
1. A vertical etch to depth thickness is performed. This etch does not apply to portions of
the surface that are masked by nonetchable materials or shadowed by etchable or
nonetchable materials; nor is it used on segments of the surface that form an angle greater
than angle to the horizontal.
2. A horizontal etch is performed. Surfaces that were exposed at the start of Step 1 are etched
horizontally by the distance undercut. Surfaces that were exposed during Step 1 are
etched by a distance proportional to the length of time between when they first became
exposed and the end of Step 1. Therefore, a sidewall exposed three-fourths of the way into
Step 1 is etched horizontally by one-fourth of undercut. (An exception is made when an
angle greater than is specified; this case is described below.)
3. Where overhangs of etchable material are present at the end of Step 2, a vertical upwards
etch (that is, in the direction) is performed. On surfaces that were exposed at the start of
Step 2, this etch is to a distance undercut. On surfaces that were first exposed during the
course of Step 2, the distance of this etch is reduced in proportion to the time from the start
of Step 2. This step approximates the undercutting of the mask due to the isotropic
component of the etch.
NOTE Trapezoidal etching using a mask definition is not supported in 2D.
When the thickness, angle, and undercut parameters satisfy the relationship:
(968)
the etch approximates a vertical etch with an isotropic component. This is the case whenever
two or fewer of the parameters thickness, angle, and undercut are specified with the
option Trapezoidal.Etch.Undercut set to 1 (default is 0):
pdbSet Grid Trapezoidal.Etch.Undercut 1
etch material=silicon type=trapezoidal thickness=0.25 undercut=0.1
Figure 112 Trapezoidal etching example 1
90°
thickness undercut angle()tan⋅=
X [µm]
Y [µm]
0 0.5 1 1.5 2
-0.5
0
0.5
1
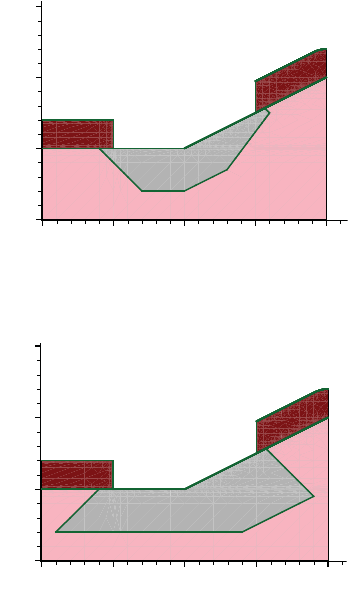
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 747
H-2013.03
The left half of Figure 112 on page 746 shows the result when etching a planar substrate. The
etch region is a trapezoid of depth thickness, extending a distance undercut beneath the
mask edge, and with a sidewall slope of angle degrees. The right half of Figure 112 shows the
result when etching a nonplanar surface.
Step 1 of the sequence etches the exposed surface vertically to a depth of thickness
micrometers. Step 2 etches the resulting sidewall in the horizontal direction, producing an
undercutting of the mask and the sloped sidewall. In this case, Step 3 also has an effect, etching
upwards from the undercut region. Therefore, the hook in the final silicon profile is the result
of approximating the isotropic component of the etch. In every case, the intersection between
the bottom of the etch region and the sidewall occurs directly under the edge of the mask.
Figure 113 and Figure 114 show what happens when Eq. 968 is not satisfied.
etch material=silicon type=trapezoidal thickness=0.3 undercut=0.1 angle=45
Figure 113 Trapezoidal etching example 2
etch material=silicon type=trapezoidal thickness=0.3 undercut=0.1 angle=135
Figure 114 Trapezoidal etching example 3
In Figure 113, you have thickness/undercut < tan(angle). In this case, the sloped sidewall
of the etch extends out under the opening in the mask. The intersection between the bottom of
the etch region and the sidewall is no longer directly beneath the edge of the mask. If the mask
opening is narrow enough, the bottom of the etch region disappears entirely, resulting in a V-
groove etch. To produce this etch shape, Step 1 of the etch process is modified to reduce the
X [µm]
Y [µm]
0 0.5 1 1.5
-0.5
0
0.5
1
2
X [µm]
Y [µm]
0 0.5 1 1.5 2
-0.5
0
0.5
1
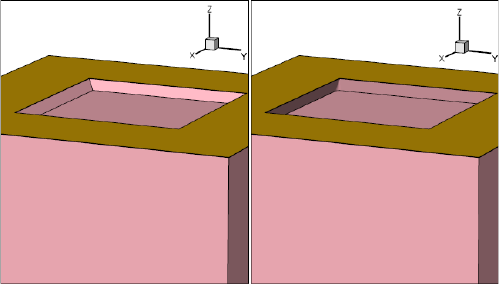
11: Structure Generation
Etching and Deposition Types and Options
748 Sentaurus™ Process User Guide
H-2013.03
depth of the vertical etch near the edges of the mask opening. Note that, in this situation, even
the smallest amount of nonetchable material can produce a triangular mound of unetched
material in the final structure.
Figure 114 on page 747 shows the case with an angle greater than . In this case, the bottom
of the etched region is wider than the opening in the masking layer, producing overhanging
sidewalls. This etch is accomplished by modifying Step 2 of the procedure to etch further
horizontally at the bottom of the sidewalls formed by Step 1 than at the top. The apparent etch
depth of at the right side of the mask opening is the result of a vertical etch
of the original sloped surface (Step 1) followed by a horizontal etch of the sloped
“bottom wall” that results from Step 1.
Trapezoidal 3D Etching
There are two possible cases for trapezoidal 3D etching:
■A thickness (or rate and time) and an angle (optional) are specified. If angle is not
specified, it is considered to be (vertical). In contrast with case 2, angle allows any
value between and . Figure 115 contains examples for both cases.
■A thickness (or rate and time) and an angle (optional) plus bottom.thickness and
bottom.angle (optional) are specified. If angle is not specified, it defaults to
(vertical). Here, a special condition applies: angle must be greater than or equal to ,
while bottom.angle must be smaller than or equal to . In other words, the first
etching penetrates behind the mask, while the second one does the opposite. Figure 116 on
page 749 shows an example of this type.
NOTE For 3D trapezoidal etching to succeed, the initial etching surface must
be more or less flat.
Figure 115 Trapezoidal 3D etching: (left) angle=45 thickness=0.3 and (right) angle = 110,
thickness=0.44
90°
0.5 µm
0.3 µm
0.4 µm
90°
0°
180°
90°
90°
90°
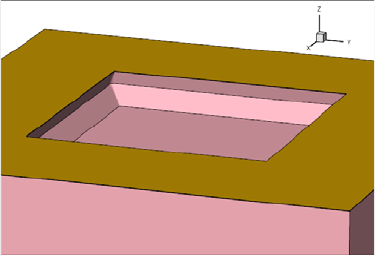
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 749
H-2013.03
Figure 116 Trapezoidal 3D etching produced with the options angle=110 thickness=0.3
bottom.angle=45 bottom.thickness=0.400
Trapezoidal Etching Using force.full.levelset
When the keyword force.full.levelset is specified in conjunction with trapezoidal
etching:
■Etch depth is controlled by rate and time, not by thickness.
■The parameter ambient.rate is used to approximate the underetch effect otherwise
available in the non-force.full.levelset case controlled in 2D by undercut. If
undercut is specified, ambient.rate will be approximated by , and
a warning message will be issued. The parameter ambient.rate also approximates
underetching, controlled in three dimensions in the non-force.full.levelset case by
the combination of angle, bottom.angle, and bottom.thickness.
■The parameters supported by the level-set solver such as shadowing become available.
■An additional parameter roundness (default value 1.0) can be used to increase the
curvature of etching sidewalls.
Etching Type: Piecewise Linear
In piecewise linear etching, the etch rate is a user-defined piecewise linear function of the angle
between the incident etching beam and the normal vector of the surface being etched. You
define the points of angle versus rate on a material-by-material basis, as per the following
syntax:
angles.rates= { \
materialA = { angleA
0
rateA
0
angleA
1
rateA
1
... angleA
n
rateA
n
} \
materialB = { angleB
0
rateB
0
angleB
1
rateB
1
... angleB
n
rateB
n
} \
...
}
where .
undercut time⁄
anglen1–anglen
≤
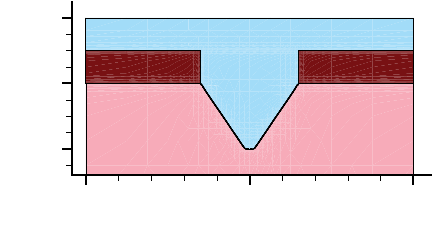
11: Structure Generation
Etching and Deposition Types and Options
750 Sentaurus™ Process User Guide
H-2013.03
The rates and angles are interpreted as follows:
■Angles are given in degrees in the range [ , ].
■The rate for angle < angle0 will be rate0, and the rate for angle > anglen will be 0.
■The rate is calculated as the linear interpolation of the nearest two angle/rate pairs within
which the angle lies.
While Fourier etching and trapezoidal etching also define the etch rate according to the angle
between the beam direction and the surface normal, a piecewise linear function is a more
general parameterization of etch rate versus angle that users control directly.
Etching Beams
The beam command defines the direction and relative strength of etching beams to be used with
piecewise linear etching (see Etching Beams on page 741).
The parameter sources of the etch command specifies the list of names of etching beams to
be used in an etching operation.
Example of Piecewise Linear Etching
This example is a piecewise linear etching (see Figure 117):
beam name=src1 direction= {1 0 0} factor=1
etch type=angles.rates sources=src1 time=0.2 \
angles.rates= { Silicon= { 25 1.0 45 0.3 } }
Figure 117 Piecewise linear etching
The piecewise linear function must be defined by users as smoothly as possible, avoiding
discontinuous changes, to ensure well-defined level-set results.
0°
180°
X [µm]
Y [µm]
0 0.5 1
-0.2
0
0.2

11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 751
H-2013.03
Deposition
To specify the deposition type, use either the parameter type or one of the parameters
anisotropic, isotropic, fill, or fourier. To specify the thickness of the deposited
layer for isotropic and anisotropic deposition, use either the parameter thickness, or the
rate and time parameters. Besides the deposition type and thickness, you must specify the
material to be deposited (only one material is allowed per deposit command). To do this,
specify the material name in the command or the parameter material=<material_name>.
The following deposition types are supported:
The number of steps for a deposition is specified as steps=<n>. The specified time or
thickness is subdivided accordingly. Subdividing a deposition into several steps might be
useful if stresses are initialized in the deposited layer. A stressed film of a given thickness can
be deposited at the same time or in several steps. Sentaurus Process simulates stress
rebalancing after each deposition step. Multistep deposition is known to generate more realistic
stress profiles compared to depositing the entire layer and then performing one stress
rebalancing calculation.
By default, the material is deposited on the surface exposed to the upper gas region. If the
structure has buried gas bubbles, they will be left untouched. To deposit inside those gas
bubbles specify the fill.buried parameter in the deposit command.
type=anisotropic
Performs anisotropic deposition.
type=isotropic
Performs isotropic deposition.
type=fill
Performs a fill of the structure with the specified material up to the
coordinate specified with the parameter coord.
type=fourier
Performs Fourier deposition.
type=polygon
Performs a polygonal deposition which requires the polygon
argument. The specified polygon is used to intersect all mesh
elements of material gas. Then, elements inside the polygon are
assigned to the specified material. (Not available in 3D.)
type=trapezoidal
Performs a trapezoidal deposition which requires the angle and
thickness arguments. (3D only)
anisotropic
Equivalent to type=anisotropic.
isotropic
Equivalent to type=isotropic.
fill
Equivalent to type=fill.
fourier
Equivalent to type=fourier.
11: Structure Generation
Etching and Deposition Types and Options
752 Sentaurus™ Process User Guide
H-2013.03
Mask Naming
The name of a mask also can be specified in the deposit command. In this case, the material
is deposited outside the specified mask. Deposition inside a mask requires the mask to be
inverted by specifying the parameter negative in the mask command defining the mask (see
Photoresist Masks on page 767).
For deposition, the analytic method, the fast level-set method, and the full level-set method are
available. In 2D, the analytic method is the preferred method for performing deposition, and
the level-set method is used when the analytic method is not possible because a front collision
is detected. In 3D, the analytic method is used for anisotropic deposition, the fast level-set
method is used for isotropic deposition, and the full level-set method is used for Fourier
deposition.
In the newly deposited region, constant field values can be initialized. For isotropic deposition,
you can define piecewise linear solution fields as a function of the distance from the original
surface.
Deposition Type: Isotropic
For simple conforming deposition, the boundary is offset an equal distance in all directions.
An isotropic deposition example follows (see Figure 118):
deposit nitride thickness = 0.05 type=isotropic
Figure 118 Isotropic deposition
Deposition Types: Fill and Polygonal
Fill is a special case of polygonal deposition. The mesh elements of material gas are intersected
at the specified coordinate. Gas elements below the coordinate are reassigned to the specified
material.
NOTE Polygon deposition is not available in 3D.
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 753
H-2013.03
Figure 119 is a polygonal deposition example:
deposit type=polygon material=nitride polygon= {0.1 0.1 0.1 0.6 -0.31 \
0.6 -0.31 0.1}
Figure 119 Polygonal deposition
Figure 120 shows a fill example:
deposit material=nitride type=fill coord= -0.31
Figure 120 Fill example
Deposition Type: Crystallographic
Crystallographic deposition takes advantage of the full level-set method to grow single
materials whose rate of growth is determined by the crystallographic directions. The crystal is
assumed to be cubic regardless of the material being deposited. Deposition rates can be set for
the <100>, <110>, and <111> directions. These rates will be applied to their respective
equivalent directions based on cubic symmetry, for example, the <100> rate will apply to the
<010>, <001>, <-100>, <0-10>, and <00-1> directions.
Figure 121 on page 754 is a crystallographic deposition example:
deposit type=crystal material=nitride time = 0.05 \
crystal.rate = { <100> = 1.0 <110> = 0.1 <111> = 0.05 }
11: Structure Generation
Etching and Deposition Types and Options
754 Sentaurus™ Process User Guide
H-2013.03
Figure 121 Crystallographic deposition (vertical direction is <100>, lateral direction is <011>)
Crystallographic deposition also can be simulated using an atomistic technique (see Epitaxial
Deposition on page 519).
Deposition Type: Fourier
Fourier deposition takes advantage of the full level-set method to grow single materials whose
rate of growth is defined by a function of the angle between the surface normal of the material
boundary and the deposit beams. The definition and calculation of the deposit shape are exactly
analogous to the case of Fourier etching as described in Etching Type: Fourier on page 741.
For Fourier deposition, first, define the deposition beam in the same way as in Fourier etching:
beam name=<beam_name> incidence=<angle> | direction= {<x> <y> <z>}
factor=<relative_strength>
Use the sources and coeffs parameters in the same way as in Fourier etching, shown in the
following Fourier deposition example:
beam name=src1 direction= {1 0 0} factor=1
deposit nitride time=0.2 fourier sources= {src1} coeffs= { -0.3 0.7 }
The coeffs parameter has units of µm/minute and, by default, time is given in units of
minutes. The Fourier deposition in 3D is only available in the default sde off mode:
NOTE The relationship between the A0 and A1 parameters in Fourier
deposition can be used to create a good reproduction of the
TSUPREM-4 deposit parameter ANISOTRO by using the formula
A1/A0 = ANISOTRO – 1.
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 755
H-2013.03
Figure 122 Two-dimensional Fourier deposition: for purposes of illustration, nitride is
deposited selectively on silicon using Fourier deposition and, then, an additional
Fourier deposition step adding polysilicon is performed using nitride as the
selective deposit material
Deposition Type: Trapezoidal
Trapezoidal deposition creates a shape in 3D with sidewalls of a defined height and angle.
Height is defined by thickness. Angle is defined by angle, measured in degrees from horizontal,
where angle=90 is vertical; angle > spreads outward as height increases, and angle
< closes inward with increasing height.
NOTE Trapezoidal deposition is available only in 3D.
Separate outward and inward deposition steps can be used to create diamond-like shapes.
Trapezoidal deposition requires a flat starting surface, for which the fill command can be
used.
The syntax is demonstrated with the following example, whose results are shown in
Figure 124:
# first step deposition
deposit material= PolySilicon type=trapezoidal \
selective.material= Silicon thickness= 0.25 angle= 120
# provide flat surface for second step
deposit type=fill coord= -0.25 Resist
# second step deposition
deposit material= PolySilicon type=trapezoidal \
selective.material= PolySilicon thickness= 0.25 angle= 60
X [µm]
Y [µm]
0 1 2
0
0.5
1
1.50.5
90°
90°
11: Structure Generation
Etching and Deposition Types and Options
756 Sentaurus™ Process User Guide
H-2013.03
Figure 123 Trapezoidal deposition. Left: Prior to deposition. Center: First step, deposition
with angle=120. Right: Second step, deposition with angle=60.
Selective Deposition
Selective deposition is optionally available. Using the selective.materials parameter,
you can select one or more materials to seed growth of the overlayer. When using 2D or 3D
MGOALS, multiple selective.materials can be specified. In 3D SDE mode, only one
material can be specified in the selective.materials list. Selective deposition can be used
with isotropic, anisotropic, or Fourier deposition types.
Fields in Deposited Layers
For isotropic deposition, piecewise linear fields can be specified in the deposited layer. A
doping command must be used for each field; each doping command must assign a unique
name. A list of names of the doping is then specified in the parameter doping of the deposit
command as a string array:
doping name=strainGe field=Germanium depths= {0 0.1} values= {1e22 1e22}
deposit material= oxide doping= {strainGe} type= isotropic \
thickness=0.1
These commands create a linear germanium field in the newly deposited oxide layer. Depth 0
corresponds to the initial surface (the bottom of the new layer).
Constant field values can be defined for all types of deposition as follows:
deposit material= oxide type= isotropic thickness=0.1 \
fields.values= {Vacancy=1e10 Germanium=2e22}
NOTE To create layers with intrinsic stress, use the field names StressELXX,
StressELXY, StressELYY, StressELZZ, StressELYZ, and

11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 757
H-2013.03
StressELXZ. It is not necessary to specify all components of stress.
Those that are not specified are assumed to be initially at zero. If stresses
are added in this way, they will be rebalanced after the deposition is
completed. The actual value of the stress may differ from the value that
was deposited.
Constant concentrations can be defined for known solution fields (known solution fields must
have been defined before the deposit command, either in the SPROCESS.models file or the
command file of the user) as:
deposit material= {oxide} type= isotropic thickness= 0.1 Vacancy \
concentration=1e10
Stress Handling
In addition to optionally including an automatic ramp-up or ramp-down before etching and
deposition, by default, Sentaurus Process automatically rebalances the stresses after etching
and deposition. This updates the stress fields at the temperature of the etch or deposit step based
on the new geometry.
For the best stress results, it is necessary to control the temperature history. This includes
thermal ramp-up to process temperature, back to room temperature, and similarly ramp-up and
ramp-down for etch, and also for deposition. However, as a minimum, the elastic stress
rebalancing can be handled automatically by switching on the stress history (see Chapter 9 on
page 643 and Automated Tracing of Stress History on page 683).
It is sometimes useful to switch off this stress rebalancing step in 3D because the rebalance
triggers a new mesh to be created and, in 3D, meshing is delayed until it is needed. Therefore,
if you are more concerned about simulation time than stress accuracy, you should specify:
pdbSet Mechanics EtchDepoRelax 0
to allow multiple etch and deposit steps to be performed without a mesh being generated in
between.
Shape Library
The shape library provides commands for generating some special-shaped polyhedra in
Sentaurus Process. These shapes are created using Sentaurus Structure Editor. The shape
library is an interface to use those shapes in Sentaurus Process.
There are two ways to use the shapes from the shape library: MGOALS mode or SDE mode.
In MGOALS mode, polyhedra are created using Sentaurus Structure Editor. The generated
11: Structure Generation
Etching and Deposition Types and Options
758 Sentaurus™ Process User Guide
H-2013.03
polyhedra then can be inserted into a Sentaurus Process structure using the insert command.
The MGOALS mode is activated with the command sde off.
In SDE mode, polyhedra are not created directly. Instead, the Sentaurus Structure Editor
structure itself is modified by inserting the shapes (replacing other materials). To activate the
SDE mode, use the command sde on.
In both modes, the correct coordinate transformation for the UCS (specified using math
coord.ucs) and for the DF–ISE coordinate system (default, or specified using math
coord.dfise) is applied.
The commands available in the shape library are:
■PolyHedronSTI creates a shallow trench isolation (STI)–shaped polyhedron.
■PolyHedronSTIaccc creates an STI concave active corner-shaped polyhedron.
■PolyHedronSTIaccv creates an STI convex active corner-shaped polyhedron.
■PolyHedronCylinder creates a cylinder-shaped polyhedron.
■PolygonWaferMask creates a wafer mask polygon.
■PolyHedronEpiDiamond creates an epitaxial diamond-shaped polyhedron.
Additional commands that create parameterized custom shapes can be defined by users using
the scripting capabilities of Sentaurus Process and Sentaurus Structure Editor.
PolyHedronSTI
The syntax of the PolyHedronSTI command is:
PolyHedronSTI name direction X0 Y0 Depth Zmin Zmax Tsti Asti Hsti Rd Rb Ru
[material]
where:
■The name parameter is set to the name for the polyhedron.
■The direction parameter can be set to left, right, front, or back, which tells the facing
direction of the STI polyhedron.
■The optional material parameter is used to specific the material of the inserted shape in
the SDE mode. In the MGOALS mode, the material of the inserted shape can be specified
in the insert command.
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 759
H-2013.03
■For other parameters, see Figure 124.
Figure 124 Parameters for generating STI-shaped polyhedron
Figure 125 shows some generated STI shapes in different directions. Figure 126 on page 760
shows STI shapes with different Tsti and Rb values.
Figure 125 STI-shaped polyhedra in different directions
R
d
(X0,Y0)
A
sti
R
b
Depth
H
sti
T
sti
R
u
Y
X
RightLeft
Front Back
Zmin
Zmax

11: Structure Generation
Etching and Deposition Types and Options
760 Sentaurus™ Process User Guide
H-2013.03
Figure 126 STI-shaped polyhedra with different Tsti and Rb
PolyHedronSTIaccc
The syntax of the PolyHedronSTIaccc command is:
PolyHedronSTIaccc name direction X0 Y0 Z0 Tsti Asti Hsti Rd Rb Ru Rac
[material]
where:
■The name parameter is set to the name for the polyhedron.
■The direction parameter can be set to rb (right back), lb (left back), lf (left front), or
rf (right front).
■The Rac parameter is the radius of the STI concave corner.
■For other parameters, see Figure 124 on page 759.
Figure 127 shows STI concave corner-shaped polyhedra in different directions.
Figure 127 STI concave corner-shaped polyhedra in different directions: (from left to right)
left back, right back, left front, and right front
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 761
H-2013.03
PolyHedronSTIaccv
The syntax of the PolyHedronSTIaccv command is:
PolyHedronSTIaccv name direction X0 Y0 Z0 Depth Tsti Asti Hsti Rd Rb Ru Rac
[material]
where:
■The name parameter is set to the name for the polyhedron.
■Same as the PolyHedronSTIaccc command, the direction parameter can be set to rb,
lb, lf, or rf.
■The Rac parameter is the radius of the convex corner.
■For other parameters, see Figure 124 on page 759.
Figure 128 shows a generated STI convex corner-shaped polyhedron.
Figure 128 STI convex corner-shaped polyhedron
Figure 129 (left) shows a structure generated by combining the above three STI commands.
Figure 129 (right) illustrates the directions of the STI shapes.
Figure 129 (Left) STI structures and (right) polyhedron directions
Back
lb rb
Left Right
lf rf
Front
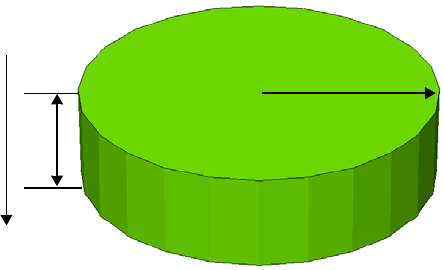
11: Structure Generation
Etching and Deposition Types and Options
762 Sentaurus™ Process User Guide
H-2013.03
PolyHedronCylinder
The syntax of the PolyHedronCylinder command is:
PolyHedronCylinder name X0 Y0 Z0 Rc Hc [material]
where:
■The name parameter is set to the name for the polyhedron.
■Other parameters give the center coordination, the radius, and the height for the cylinder
(see Figure 130).
Figure 130 Cylinder-shaped polyhedron
PolygonWaferMask
The syntax of the PolygonWaferMask command is:
PolygonWaferMask name Y0 Z0 Rw Lf
where:
■The name parameter is set to the name for the polyhedron.
■Other parameters give the location and size for the mask (see Figure 131 on page 763).
NOTE This command only works in MGOALS mode.
Hc
X0
(Y0,Z0)Rc
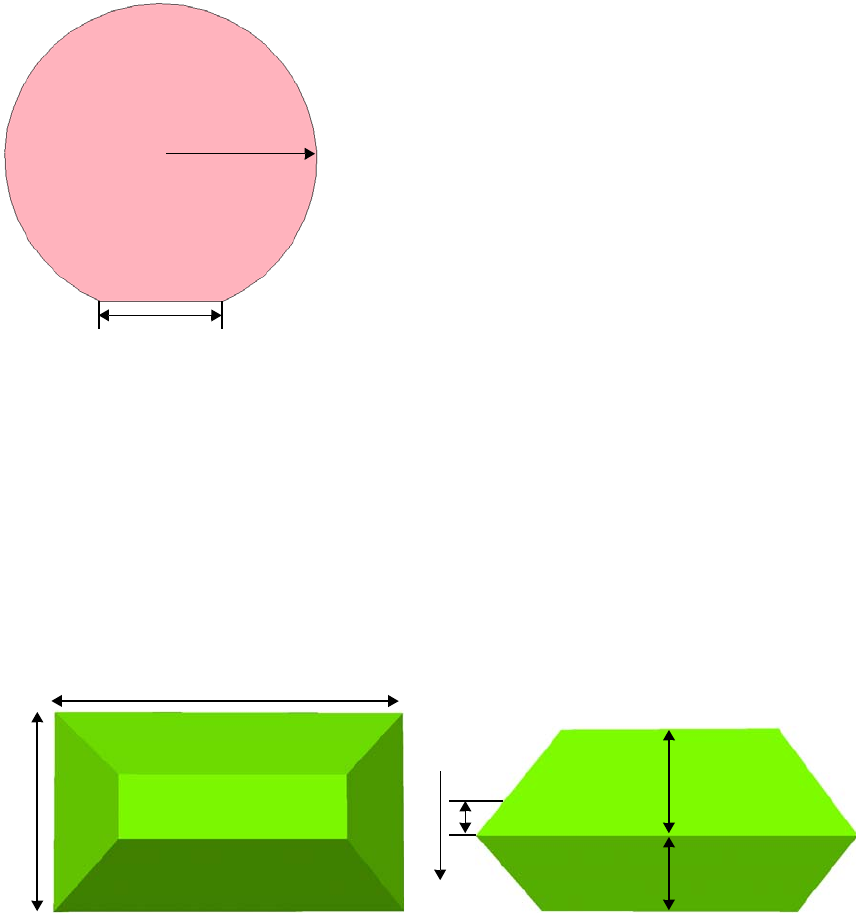
11: Structure Generation
Etching and Deposition Types and Options
Sentaurus™ Process User Guide 763
H-2013.03
Figure 131 Wafer mask-shaped polygon
PolyHedronEpiDiamond
The syntax of the PolyHedronEpiDiamond command is:
PolyHedronEpiDiamond name X0 Y0 Z0 Wepi Lepi Hup Hdown Drecess [material]
where:
■The name parameter is set to the name for the polyhedron.
■For other parameters, see Figure 132.
Figure 132 Epitaxial diamond-shaped polyhedron
(Y0,Z0)
Rw
Lf
Wepi
Lepi
(Y0,Z0)
X0
Drecess
Hup
Hdown

11: Structure Generation
The mask and photo Commands
764 Sentaurus™ Process User Guide
H-2013.03
The mask and photo Commands
A quick way to mimic lithographic patterning is using the photo command. The photo
command takes a mask and effectively does a resist spin-on step followed by an exposure bake
and etch. The resist layer produced has vertical walls and is by default a negative of the mask,
but the positive sense can optionally be created as well.
In addition to the photo command, the etch and deposit commands allow you to specify a
mask directly. The parameter mask in the etch, deposit, and photo commands specifies the
name of one mask that has been previously defined using a mask command or by reading in
masks from a layout using the IC WorkBench EV Plus interface (see Chapter 12 on page 817).
The mask will have an effect similar to a masking layer; it limits the etch or deposition process
to a certain window. By default etching is not performed for points inside the mask, unless the
parameter negative is used in the mask definition. Similarly, deposition of a new layer in the
deposit command and of the photoresist layer in the photo command is performed outside
the mask unless the negative parameter is specified in the mask command in which case
deposition happens inside the mask only.
NOTE Always specify the masks and the simulation domain such that masks
do not end exactly on the boundary of the simulation domain, but end
inside or extend safely beyond the boundary of the simulation domain.
The mask command creates a mask. You can define the geometry of the mask directly in the
command file or can read masks from a layout file. Masks defined in the command file must
be given a name; otherwise, the names are read from the layout file.
If the parameter list is specified in a mask command, information about the existing masks
is printed. If name is specified as well, information about the specified mask is printed.
If the parameter clear is specified in a mask command, all previous mask definitions are
removed. If a name is specified as well, only the specified mask is removed.
A mask can be defined directly in the command file by using three different types of geometry
object:
■Segments
■Rectangles
■Polygons
Each mask may be composed of an arbitrary number of such objects.
11: Structure Generation
The mask and photo Commands
Sentaurus™ Process User Guide 765
H-2013.03
Segments are defined as:
mask name= pmask segments= { -0.1 0.6 0.7 1.1 }
Pairs of subsequent values define the y-coordinates of the beginning and end of one mask
segment. Therefore, an even number of coordinates must be specified in the segments
parameter. The pairs may be defined in arbitrary order, and the segments defined by pairs of
coordinates may touch or overlap each other. In 3D, masks defined by segments are extended
over the entire range of z-coordinates.
A rectangular mask is defined as:
mask name=nimask left=0.2 right=1 front=0.2 back=1 negative
The left and right parameters define the minimum and maximum extensions of the mask
along the y-axis; the front and back parameters define the minimum and maximum
extensions of the mask along the z-axis.
NOTE Only one rectangle can be specified per mask command. The front
and back parameters may be omitted; in this case, the mask is
equivalent to a mask with one segment. Additional mask commands
with the same name can be used to add rectangles. The rectangles
defined for a mask may arbitrarily intersect or touch each other.
Masks also can be defined by a list of names of polygons. These named polygons must have
been defined before the mask command using one polygon command for each named
polygon:
polygon name=LShape2 segments= {0.0 -1.5 0.0 -0.5 0.5 -0.5 0.5 1.5 1.5 \
1.5 1.5 -1.5}
mask name=Mask2 polygons= {LShape2} negative
NOTE The segments in the polygon command are defined as a sequence of y-
and z-coordinates. The polygon is closed implicitly by connecting the
last point to the first. Each polygon for a mask must not touch or
intersect itself. The polygon may be specified with arbitrary orientation
(clockwise or counterclockwise in the yz plane). It does not matter if
different polygons for a mask touch or intersect each other.
Masks also can be defined by a combination of all three types of geometry object: segments,
rectangles, and polygons. Different objects may touch or overlap each other.
In 1D, the entire simulation domain is masked if the coordinate origin is masked. Any point
along the y-axis in a 2D simulation and any point of the yz rectangle of the simulation domain
in a 3D simulation are inside the mask if they are contained in any one of the geometry objects
11: Structure Generation
The mask and photo Commands
766 Sentaurus™ Process User Guide
H-2013.03
defined for the mask. Specifying the parameter negative inverts the mask. In other words,
any point outside all the geometry objects defined for the mask is masked.
These commands can be used to invert a mask at any time after it has been defined:
mask name=aaa negative
mask name=aaa !negative
NOTE In the etch command, the masked area is not etched. While in the
photo and deposit commands, the photoresist or the specified
material is deposited in the unmasked area.
Masks can also be combined using a set of Boolean operations. The operations are specified
using the bool parameter (see Boolean Masks on page 767).
Layouts that have been defined in the GDSII format can be read into Sentaurus Process using
the ICWBEV Plus interface (see Chapter 12 on page 817). Alternatively, masks can also be
read from a layout file in DF–ISE format (default file extension .lyt). To read the layout
directly into Sentaurus Process, the name of the layout file must be specified as
layoutfile=aaa.lyt in a mask command.
All mask rectangles and polygons read from a .lyt file are converted into named polygons.
These polygons then are collected into masks according to the names contained in the .lyt file.
When using a layout file, the relation between the layout coordinate system and the Sentaurus
Process coordinate system may need to be defined. By default, the layout-x axis corresponds
to the Sentaurus Process z-axis. The layout-y axis corresponds to the Sentaurus Process y-axis.
This definition matches the default definition of the parameter slice.angle in the init
command and the coordinate x- and y-axes when displaying the Sentaurus Process simulation
results.
The coordinate transformation between the Sentaurus Process coordinate system and the
layout coordinate system can be defined in two ways:
■In the mask command that specifies the layout file, the name of one mask may be specified.
If a mask with the specified name is contained in the layout file, it is used to position and
orientate the simulation domain in the layout.
■Otherwise, a mask with the specified name must have been defined before using a mask
command. The specified mask is defined in layout coordinates. It may be defined as a
rectangle or a polygon, containing at least two points.
In the case of a rectangle defined in a mask command, the point with the minimum layout-x
and layout-y coordinate is used as the origin of the Sentaurus Process coordinate system. The
direction from (min.layout-x, min.layout-y) to (min.layout-x, max.layout-y) is
used as the Sentaurus Process y-axis.

11: Structure Generation
The mask and photo Commands
Sentaurus™ Process User Guide 767
H-2013.03
If a polygon of at least two points is used (for example, a mask defined as a polygon), the first
point defined is used to place the origin of the Sentaurus Process coordinate system. The
direction from the first to the second point of the mask is used as the orientation of the
Sentaurus Process y-axis.
The local coordinates of the specified mask with respect to the selected Sentaurus Process y-
axis and origin are used as default extensions of the simulation domain. If a polygon mask with
only two points is used, the default extension in the z-direction is 0. The default extension in
the y-direction is defined by the distance between the two points. The default extensions in the
y- and z-directions as defined by the mask are reported. If no extensions have been defined
using the line y command or the line z command or both commands, the default extensions
are defined for the simulation when the layout file is read.
If a layout file is loaded, but no mask name is specified, the Cutline2D command that may
have been specified in the init command to define the parameter slice.angle will be used
to orientate the coordinate systems. The first point specified in the Cutline2D command is
used as the origin of the Sentaurus Process coordinate system. The direction from the first to
the second point is chosen as the direction of the Sentaurus Process y-axis. If Cutline2D is
used, no default extensions of the simulation domain are defined.
Photoresist Masks
To define photoresist layers, use the photo command and specify a mask. Sentaurus Process
defines photoresist layers by specifying the minimum thickness of the resist and selecting the
name of a mask that has been defined by the mask command. By default, the photoresist will
be deposited outside the specified mask and will have a flat top similar to spin-on resist. If the
parameter negative has been specified when defining the mask, a photoresist is created
inside the mask.
Boolean Masks
Two masks can be combined using the bool parameter of the mask command. The Boolean
operations include: +, ^, * and –. In addition, masks can be transformed using the following
operations: rotate, scale, mirror, array, bias, over_under, under_over, and
offset.
The bool option only accepts simple expressions as described in the examples in Table 71.
Complex nested expressions (for example, bool= "(M1 + M2) – bias(–50, M3 + M4)")
are not possible in this implementation and, therefore, must be reduced to simple operations.

11: Structure Generation
The mask and photo Commands
768 Sentaurus™ Process User Guide
H-2013.03
In addition, the bool option cannot be used together with the layoutfile, polygon, and
negative options.
Ta bl e 71 B oo l e an o pe ra ti on s
Operation Example Description
+ bool= "mask1+mask2"
Unites (merges) mask1 and mask2.
– bool= "mask1-mask2"
Subtracts mask2 from mask1.
Note that this is a geometric subtraction. The
program takes mask1 and removes from it the
portion of mask2 which overlaps mask1.
In particular, the operation mask1 - mask2 is
not equivalent to mask1 + (-mask2)
* bool= "mask1*mask2"
Produces the intersection of mask1 and mask2.
^ bool= "mask1^mask2"
Produces a mask that contains the nonoverlapping
portions of mask1 and mask2 (XOR operation).
– bool= "-mask"
Produces a mask that is the complement of the
input mask.
rotate bool= "rotate(direction,mask)"
Produces a mask that is rotated with respect to the
input mask. The direction parameter can be
either left-90 or right-90.
scale bool= "scale(factor,mask)"
Produces a mask that is scaled with respect to the
input mask using the floating-point value of
factor.
mirror bool= "mirror(axis,mask)"
Mirrors a mask with respect to a local axis
specified by x or y.
array bool= "array(n
x
, n
y
, d
x
, d
y
, mask)"
Produces an array of masks separated by a
distance specified with dx and dy.
bias bool= "bias(delta,mask)"
All mask edges on the input mask are offset in the
normal direction by the specified amount. A
positive delta value expands the mask, while a
negative delta shrinks it. Zero or negative area
sections of the mask are eliminated from the output
mask. Overlapping sections of the mask are
merged.
over_under bool= "over_under(delta, mask)"
Expands and then shrinks the input mask by delta.
This effectively merges areas in close proximity
and is equivalent to bias(delta, bias
(-delta, mask)).
under_over bool= "under_over(delta, mask)"
Shrinks and then expands the input mask by delta.
This eliminates small areas and is equivalent to
bias(delta, bias(-delta, mask)).
nxny
×

11: Structure Generation
The mask and photo Commands
Sentaurus™ Process User Guide 769
H-2013.03
Line Edge Roughness Effect
Line edge roughness (LER) is the deviation of feature edges from ideal straight lines due to
statistical fluctuations in photolithographic processes. Sentaurus Process uses the
line_edge_roughness command to apply randomized deviations to straight mask edges,
for example:
line_edge_roughness normal= "Z" masks= {mask1} correlation.length= 25.00<nm> \
standard.deviation= 5.00<nm> max.segment.length=5.00<nm>
The random noise function applied to mask edges by the line_edge_roughness
command is generated from the power spectrum of a Gaussian autocorrelation function. The
Gaussian autocorrelation shape is characterized by the standard deviation distance specified
by the standard.deviation parameter and the correlation length , specified by the
correlation.length parameter:
(969)
is obtained by Fourier synthesis, applying the inverse Fourier transform to Eq. 969, after
adding random phases. In this way, random deviations of the mask edges can be obtained from
run to run, which correspond to LER profiles having the same standard deviation and
correlation length .
These random deviations are added in discrete form to the mask edges in question. First, the
mask edge is subdivided into discrete segments complying with the user parameter
max.segment.length. Second, the deviation at each segment endpoint is added in the
direction normal to the initial mask edge orientation.
LER is applied by default to all edges of the mask. You can limit which edges in a named mask
are to receive LER by the parameter normal, which specifies either the y-axis (Y) or the z-axis
(Z). If normal is specified, only those edges in the named masks normal to the given axis are
chosen for LER to be applied. LER is applied only once per mask. Mask segments along the
device bounding box do not receive LER.
The parameter !random.reseed bypasses the reseeding of the random number generator
before the random phases are added. By using this parameter, the shape of the noise function
and, therefore, the LER result, can be reproduced from one run to the next if needed for
comparison.
offset bool= "offset(dy, dz, mask)"
Translates the mask by the specified amount. The
dz parameter is ignored in 2D.
Ta bl e 71 B oo l e an o pe ra ti on s
Operation Example Description
frandom
∆
Λ
Autocorrelation frandom
()∆
2πΛex2Λ2
⁄()–
=
frandom
∆
Λ
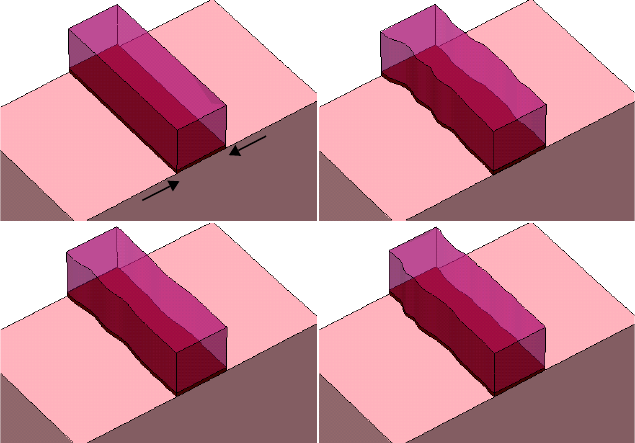
11: Structure Generation
The mask and photo Commands
770 Sentaurus™ Process User Guide
H-2013.03
The parameter random.seed can reproduce specific LER calculations from one run to the
next by setting the same random seed in both runs. When stored in a TDR file in split
simulations, the parameter random.seed is included when saving line_edge_roughness
to the TDR file, even if it is not specified by users, to ensure proper reproduction of the same
LER in a subsequent reload of the TDR file.
NOTE The structure is extruded automatically to three dimensions if it is less
than three dimensions and the line_edge_roughness command is
used.
Figure 133 Example of 3D structure with LER applied using different values of standard
deviation and correlation length : (A) no LER applied, (B) LER applied with
= 4 nm and = 20 nm, (C) LER applied with = 2 nm and = 20 nm, and
(D) LER applied with = 2 nm and = 12 nm
The following strategy is used to address the problem of nearly collinear LER mask points that
may trigger removal by decimation during meshing. Avoiding the decimation of nearly
collinear LER mask points is desirable because removing such points may perturb the power
spectrum of the Gaussian autocorrelation function represented by the mask shape and may also
result in meshing difficulties.
If the parameter max.tries is set to a nonzero value, LER masks are checked for nearly
collinear points, which would result in decimation by the mesher:
line_edge_roughness max.tries=30
If any points in the LER mask are decimated by the mesher, based on the current
mgoals accuracy setting, the LER mask is rejected and the LER generation process is
AB
CD
50 nm
∆
Λ
∆
Λ
∆
Λ
∆
Λ
11: Structure Generation
The mask and photo Commands
Sentaurus™ Process User Guide 771
H-2013.03
restarted. After each restart, the detection and restart process is repeated until an acceptable
LER mask is generated or until the number of attempts exceeds max.tries.
When max.tries is exceeded, Sentaurus Process stops with an error message that suggests
using a smaller value of mgoals accuracy or a larger value of max.segment.length in
the line_edge_roughness command.
The default value of max.tries is 0, meaning no decimation check is performed, no retries
are attempted, and the initial mask LER is accepted as it is, even with nearly collinear points.
Mirrored Boundary Conditions
The addition of line edge roughness (LER) is inherently 3D and nonsymmetric, thus not easily
compatible as a simulation problem with the assumption of symmetry reduction. LER added
to a structure reduced by symmetry may produce rough geometrical transitions at joined
geometrical boundaries when pieces are reunited.
To accommodate the possibility of joining symmetry-reduced structures, an additional
keyword smooth.points defines smoothing of LER at structure boundaries. The default
value of smooth.points is 0 (zero), which turns off smoothing.
The use of nonzero smooth.points alters the LER function that creates mirrored boundary
conditions at the structure boundaries. The smoothing occurs at the intersection of masks with
the structure boundary sides. The parameter smooth.points defines the number of segments
in the discrete LER function, counting from the mask’s boundary edge, that should be adjusted
or smoothed. The length of segments in the discrete LER function is determined using
max.segment.length.
Smoothing is calculated by generating the LER function, then creating a virtual mirroring at
the structure boundary edge where a cubic spline interpolation is applied with a mirror
boundary condition. The spline fit is made with the LER at smooth.points from the
boundary. The smoothed LER function is then applied to the mask.
Applying nonzero values of smooth.points affects somewhat the randomness and power
spectrum characteristics of the LER offset function at the boundaries as a tradeoff for
permitting the smooth joining of symmetry-reduced structures.

11: Structure Generation
Geometry Transformations
772 Sentaurus™ Process User Guide
H-2013.03
Geometry Transformations
Transformations supported in Sentaurus Process are reflection, stretch, cut, rotation,
translation, and flip:
■The transform reflect command is used with left, right, front, or back to
perform the reflection centered on the outer boundary of the simulation domain. At the
reflection side, regions are not merged immediately to allow a clean transform cut
afterward if required. The grid can be merged with grid merge command manually, but
if not, the structure will naturally be merged for any geometry changing operation later
(except transform reflect). It is also possible to discard the original structure when
reflecting by specifying the optional parameter !keep.original.
■The transform stretch command is used to to extend the mesh. The algorithm cuts the
mesh in two pieces using a cut plane defined by the location parameter and the left,
right, front, or back parameters. The resulting pieces of mesh are translated
perpendicular to the cut plane to create a gap of size length in the direction given by the
left, right, front, or back parameters. After this translation, a set of new elements is
inserted to connect both sections of the mesh.
■The transform cut command is used to crop the structure. The algorithm cuts the mesh
in two pieces using a cut plane defined by the location parameter and the left, right,
front, or back parameters. If the location parameter is omitted, the algorithm will cut
the structure in the middle. The left, right, front, or back parameters tell the
algorithm to remove the portion of the structure along the given direction. For a more
general crop operation, the cut parameter can be used with the min and max parameters
that specify the cropping box.
■The transform rotate command is used to rotate a structure. An axis and angle
should be specified. Only , , and angles are allowed for the x-axis, and
for the y- and z-axis. In the case of 2D simulations, rotations will produce an
extruded 3D simulation.
■The transform translate command shifts the structure by the specified quantity.
In 2D, all transformations are performed internally by Sentaurus Process. In 3D, if the SDE
mode is on, the appropriate Scheme commands are dispatched to Sentaurus Structure Editor to
transform the structure. If the SDE mode is off, Sentaurus Process performs the reflect,
rotate, translate. stretch and cut operations internally.
NOTE The transformations stretch and flip are not supported by Sentaurus
Process KMC.
90°
180°
270°
180°
11: Structure Generation
Geometry Transformations
Sentaurus™ Process User Guide 773
H-2013.03
Refinement Handling during Transformation
All transformations apply to the existing refinements created with either the refinebox or the
line command by default.
To disable this feature, use:
Grid Transform.Updates.Refinement
to
false
Detailed descriptions of refinement handling are provided with each operation here.
Contact Handling during Transformation
The only special contact handling occurs during transform reflect. In this case, contacts
that straddle or touch the reflecting plane are enlarged to the reflected area (only one contact
remains). The remainder of the contacts are duplicated and are renamed by appending a suffix
as follows:
■For right or left reflection, the contact on the left after reflection will be named <original
contact name>.1 (where <original contact name> was the name of the contact
before the reflection operation), and the contact on the right after reflection will be named
<original contact name>.2.
■Similarly, for front or back reflection, the front contact (which has a larger z-coordinate)
will be named <original contact name>.2, and the back contact will be named
<original contact name>.1.
■For up or down reflection, the upper contact will be named <original contact
name>.1, and the lower contact will be named <original contact name>.2.
You ca n ren am e cont ac ts af te r th e reflect command, or at any time, using the command:
contact name= <old contact name> new.name= <new contact name>
For example, after reflection, you can use the command:
contact name= SourceDrainContact.1 new.name= Source
where the original contact name before reflection was SourceDrainContact.
NOTE It is recommended to specify all contacts after all transform operations
other than transform reflect to avoid problems during contact
creation.

11: Structure Generation
Geometry Transformations
774 Sentaurus™ Process User Guide
H-2013.03
The transform reflect Command
The transform reflect command is used to reflect the structure about the left, right, front,
or back boundary (at minimum y, maximum y, maximum z, or minimum z). If any remeshing
or other mesh modification operations are performed after a transform reflect command,
the symmetry will be lost. An extra !keep.original parameter discards the original
structure leaving only the reflected one.
Examples:
transform reflect left
transform reflect ymin
transform reflect front
transform reflect left !keep.original
NOTE The option remesh is disabled in reflect because it may disrupt the
symmetry of the reflected structure. However, the command grid
merge can be used afterward to remove same-material interfaces at the
reflecting plane.
Refinement Handling during Reflection
In the case of !keep.original, lines coming from both the line command and refinement
boxes are reflected along with the structure. However, when the original structure is kept, some
special handling is required.
Typically, during any geometry operation, lines created with the line command that have been
defined outside the bounding box will be removed. Therefore, there is no danger of the
reflected lines conflicting with the original lines.
For refinements created with refinement boxes, if the refinement box is constrained spatially
(that is, the min or max parameter has been used in the definition), then the box will be
duplicated, and the name of the new box will be reflected_<name> where <name> is the
name of the original refinement box. If the original and reflected refinement boxes overlap,
there is no problem since the refinement criteria are the same.
The transform stretch Command
The transform stretch command stretches the structure in the left, right, down, up, front,
or back directions at a given coordinate location by offsetting one side of the structure by the
specified length. If there is no vertical line of edges at the specified location, MGOALS creates

11: Structure Generation
Geometry Transformations
Sentaurus™ Process User Guide 775
H-2013.03
such a line and then stretches the structure. The data at the two ends of the stretched region is
exactly the same as that of the unstretched mesh.
Examples:
transform stretch location=0.001 length=5 right
transform stretch down loc=0.5 length=200 info=2
NOTE Do not use together with the atomistic mode (KMC).
Refinement Handling during Stretch
During the stretch operation, mesh lines that were created with the line command in the part
of the structure being stretched are translated with the structure. No new lines are introduced
into the expanded region. The refinement boxes that straddle the stretch location are increased
in size by the stretch distance to follow the structure.
The transform cut Command
The transform cut command cuts at or near the requested coordinate location. The location
defines a line in 2D or a plane in 3D that divides the structure into the left and right, or front
and back, or up and down parts. You can select the left/right, or front/back, or up/down region
to be removed. If a line of element edges in 2D or a plane of element faces in 3D can be
identified by MGOALS, the operation eliminates only the elements in the removed region. This
works well if a structure had been reflected and needs to be cut back to the original
(unreflected) structure. If a line of element edges in 2D or a plane of element faces in 3D cannot
be found, a mesh-cutting operation is performed. By default, MGOALS tries to find a mesh
line or plane near the specified coordinate. Then, MGOALS removes entire mesh elements
rather than cutting mesh elements to avoid arbitrarily small edges and poor element quality. To
disable the search feature and perform the operation exactly where specified, use
the !mesh.align option of the transform command, which will invoke a remesh
unless !remesh also is specified.
Examples:
transform cut location=0.5 right
transform cut left loc = 0.0
You also ca n use the cut command to crop the mesh by specifying a rectangle/brick defined
by the upper-left-front and lower-right-back corners, specified with the min and max
parameters. The cut operation retains the region enclosed by the rectangle/brick. By default,
MGOALS tries to find a mesh line or plane near the specified coordinate and removes the
whole mesh elements instead of cutting the mesh (which could lead to arbitrarily small edges
and poor element quality). To suppress searching of a nearby mesh line or plane and to perform

11: Structure Generation
Geometry Transformations
776 Sentaurus™ Process User Guide
H-2013.03
the operation exactly at the specified location, specify !mesh.align. This automatically
invokes a remeshing unless !remesh is specified.
Examples:
transform cut min= {-2 -1} max= {11 0}
transform cut min= {-10 1.35 0.15} max= {10 1.65 0.4}
Refinement Handling during Cut
During the cut operation, lines created with the line command that are outside of the
simulation domain after the cut are removed and, similarly, any refinement box that lies
completely outside the simulation domain after the cut is removed as well.
The transform flip Command and Backside Processing
The transform flip command was introduced specifically to allow a convenient way to
perform process steps on the back of a wafer. During the transform flip command, the
structure is rotated about a line by default in the center of the structure parallel to the y-
axis. Therefore, the structure is upside down after the flip and in the same location. Because
most operations in Sentaurus Process require a Gas region on top, a gas region is added
automatically. In addition, many meshing operations require a solid material at the bottom of
the structure, so the Gas region that was previously on top of the structure is converted to an
auxiliary material called BackMat. If the structure is flipped again, the reverse happens,
namely, BackMat is converted to Gas, and Gas is converted to BackMat. Any operation is
allowed on a structure that has been flipped one or more times; however, the current bottom of
the structure is never an active surface for any operation such as oxidation, epi, etching, and
deposition.
There is great flexibility in the handling of the auxiliary material at the back of the structure.
The material itself defaults to BackMat as mentioned, but you can choose another material
using the command pdbSet Grid Back.Material <material>.
The material BackMat inherits its parameters from (is Like) Gas so that it behaves like gas
for dopant diffusion simulation. For implantation, the material is converted to Photoresist
so no implant tables are required for this material. Similarly, for mechanics, the only way to
obtain Gas-like mechanics boundary conditions at interfaces to the back material is to use an
actual Gas region. Therefore, the back material is converted automatically to Gas before each
mechanics call and is converted back directly afterwards. Finally, when a region of material
BackMat is saved in a structure, it is first converted to Gas, so that other tools reading the
structure will have the proper material. However, it is also given a Sentaurus Process–specific
tag, so that Sentaurus Process knows the region should actually be BackMat.
180°

11: Structure Generation
Geometry Transformations
Sentaurus™ Process User Guide 777
H-2013.03
Another important point regarding mechanics simulations on flipped structures is that only a
modified version of the lattice mismatch model is available. With this model, the command
substrate top.relaxed.coord is no longer available for modification. Instead, the
lattice-mismatch strain is added using the reference concentration model. This model uses a
reference concentration and bases all strains directly on the difference between the strain field
and that reference (see Reference Concentration Model on page 679).
NOTE To avoid switching from the standard lattice mismatch model to the
reference concentration lattice mismatch model after transform
flip is used, switch on the reference concentration model. This should
be performed before initial structure creation using:
pdbSet Mechanics Reference.Concentration.Model 1
for those simulations using transform flip.
NOTE When performing laser annealing on a structure that has been flipped,
the following settings are recommended (see also Flash or Laser Anneal
Model on page 229):
pdbSet Grid Zero.Back.Material 0
mater add name= MyBackMat new.like=Silicon add
pdbSet Grid Back.Material MyBackMat
pdbSet ImplantData Back.Material MyBackMat
NOTE Do not use together with the atomistic mode (KMC).
Refinement Handling during Flip
Refinements during a flip operation are handled in the same way as refinements during
reflection in the case of !keep.original. Lines coming from both the line command and
refinement boxes are reflected along with the structure.
The transform rotate Command
The transform rotate command rotates the structure the specified angle in the specified
axis using (0,0,0) as the rotation center. It accepts two parameters axis and angle to specify
the rotation axis and angle, respectively. For Y and Z, 180 degrees can be specified. For X, 90,
180, and 270 degrees are allowed.
It might happen that during the rotation the existing initial gas has to be moved to a side or the
bottom of the structure instead of being at the top. In these cases, new gas will be added to the
top.

11: Structure Generation
MGOALS Interface
778 Sentaurus™ Process User Guide
H-2013.03
The transform rotate command applies to 3D and 2D simulations. For 2D simulations, a
rotation in the z-axis will produce another 2D simulation, but rotations in the x-axis and y-axis
will produce a structure equivalent to extruding the z-axis and then performing the rotation.
Refinement Handling during Rotation
During a rotation operation, lines coming from both the line command and refinement boxes
are rotated along with the structure.
The transform translate Command
The transform translate command does not change the aspect of the structure. It only
adds the coordinate specified in the translate parameter to all the nodes, that is, it displaces
the structure or shifts it in space. It is equivalent to changing the origin of coordinates by a fixed
quantity.
Similarly, mesh lines created with the line command and the bounding box of refinement
boxes (which are specified with the min and max parameters of the refinebox command) are
translated with the structure.
MGOALS Interface
By default, etching and deposition operations are performed using the MGOALS library in 1D,
2D, and 3D. The MGOALS library operates as follows:
■The starting structure is analyzed for the interfaces that will change during the operation.
■The geometry-changing operations are performed.
■In 2D, the entire structure is remeshed. During remeshing, nodes in the silicon region are
retained as much as possible in their original locations. In most cases, a high percentage of
the nodes are retained after remeshing. This minimizes interpolation errors. In 3D, the
structure is remeshed only if the next step requires an up-to-date mesh.
MGOALS Boundary-moving Algorithms
MGOALS uses either an analytic method or a fast level-set method to perform boundary-
modifying operations. In general, the analytic method is faster, less memory intensive, and
more accurate. However, it cannot handle deposition in concave regions or etching of convex
areas when there are boundary collisions and self-intersections.
11: Structure Generation
MGOALS Interface
Sentaurus™ Process User Guide 779
H-2013.03
The analytic method is fast, accurate, and uses a simplified string algorithm. Due to speed and
accuracy advantages, MGOALS always tries to perform an analytic operation. If self-
intersections are detected in the new boundary, MGOALS automatically switches from the
analytic method to the fast level-set method. Both the analytic and the fast level-set methods
can handle simple etch and deposition processes.
Besides the analytic method and the fast level-set method, a general time-stepping level-set
method is available to handle more complex etch types such as Fourier, crystallographic, and
multimaterial etching, and to include shadowing effects.
Both level-set methods use an approach similar to that described in [1]:
■First, the level-set method identifies the interface or the part of an interface to be moved.
This computation is based on nonetched overlayers, masks, and if necessary, visibility due
to directional constraints specified by the user.
■Second, evolution of the moving interface is performed using either the fast-marching
scheme, which solves the time-independent boundary-value formulation of the Hamilton–
Jacobi equation (or Eikonal equation), or “full levelset,” a time-dependent, initial-value
formulation of the same equation. The fast-marching scheme computes the new boundary
location for all times in a single step. The nature of the equation is such that it captures and
handles collisions. However, the equation cannot identify when the collision actually
occurred. The “full levelset” formulation which is a time-dependent, initial-value
formulation is used for multimaterial, Fourier, and crystallographic etching, and for
handling shadowing effects. Its time-stepping algorithm allows for recalculating the front
velocity at every time step.
In MGOALS, the fast-marching and level-set equations are solved on a separate Cartesian
mesh that is independent of the simulation grid. For a description of the parameters that control
the Cartesian mesh, see MGOALS Boundary-moving Parameters on page 780.
After solving the level-set equations, the newly created boundary is extracted from the level-
set function on the Cartesian mesh and then incorporated into the simulation mesh. The exact
replication of the extracted boundary in the mesh can be expensive and can transfer unwanted
noise from the level-set solution into the structure. To resolve these issues, MGOALS allows a
certain smoothing to be performed on the extracted boundary.
In 2D, to incorporate the new boundary into the simulation grid, a simplified meshing step is
performed. A simple mesh is created for the modified regions and connected to the mesh in
unchanged regions. Since this mesh is not suitable for process simulation, by default, a full
remesh is performed after each etching and deposition step.
In 3D, almost all boundary-modification operations performed by MGOALS use the analytic
method. The only exceptions are isotropic deposition and etching, which are performed using
the fast level-set method. The new material boundary is integrated into the structure using a set
of polyhedral Boolean operations.
11: Structure Generation
MGOALS Interface
780 Sentaurus™ Process User Guide
H-2013.03
MGOALS Boundary-moving Parameters
Parameters to specify the resolution of the Cartesian mesh and the interface/boundary fidelity
are defined in an mgoals command before the etch or deposit command. These parameters
are applied to the entire structure. The interface quality and resolution are controlled by
accuracy, resolution, and full.resolution. The actual size and placement of the
Cartesian mesh bounding box is calculated starting with the initial interface being etched,
extended based on the time and rates given by the user, or in the case of etchstop materials,
extended based on the distance from the initial front to the etchstop materials.
The accuracy Parameter
The accuracy parameter is used to control the noise and features at an interface. A small value
of accuracy allows only small deviations between the boundary extracted from the level-set
function and the piecewise linear segments incorporated into the simulation mesh. As a result,
a large number of small segments may be created. In addition, a value of accuracy that is too
small may interpret numeric noise as surface features, which MGOALS requires to reproduce
in the simulation mesh. The default value for accuracy is .
Figure 134 The curved surface represents the extracted new boundary and the piecewise
linear segments represent the simplified boundary incorporated into the
simulation mesh. The accuracy parameter ensures that u accuracy.
The resolution Parameter
The value of the resolution parameter controls the element size in the Cartesian mesh used
to perform level-set based etching and deposition. Since the thickness of the layer to be
deposited or etched is user specified, the grid size is defined as a fraction of the thickness of
the modified layer. The size of each grid element of the level-set mesh is given by the product
of the value of the resolution and the etch or deposit thickness.
The resolution is specified in the mgoals command and the etch or deposit thickness is
specified in the etch or deposit command, respectively. This scheme usually provides a
good approximation of the required level-set resolution and is computationally efficient. The
default value for resolution is 0.1.
NOTE Providing a small resolution parameter for thin layers may lead to
excessive time and memory consumption. For example, if an isotropic
deposit of 1 nm thickness is performed with resolution=0.1, a level-
set grid size of 1Å will result.
1.0 5–
×10 µm
u
≤
11: Structure Generation
MGOALS Interface
Sentaurus™ Process User Guide 781
H-2013.03
NOTE For a thick etching or deposition, it may be necessary to reduce the
resolution. For example, a deposition with
resolution=0.1 leads to a level-set grid size of 100 nm that may lead
to a poor approximation of sharp corners and rounded areas in the new
boundary.
General Time-Stepping Level-Set Parameters
The general time-stepping level-set method has a few additional parameters to control and
balance accuracy, simulation time, and memory use. Usually, the full time-stepping level-set
method is used in situations where more intricate boundaries will be generated. The full time-
stepping level-set method is needed for Fourier, crystallographic, and multimaterial etching
types, and for etching with shadowing on. It is also used if the force.full.levelset
parameter is given.
The full.resolution parameter can be used for the time-stepping level-set algorithm in the
same way resolution is used for the fast level-set algorithm. You also can specify the actual
spacing of the Cartesian mesh in the x-direction or y-direction or both directions, with the dx
and dy parameters (and the z-direction in 3D with dz). Reducing the mesh size causes the time-
stepping method to allocate more memory, to take smaller time steps, and to increase the solve
time for each time step, thereby increasing the overall simulation time.
NOTE In previous releases, the Upwind formulation of the time-stepping level-
set method provided users with the mgoals parameters
reinitfrequency and reinititerations to control the frequency
and quality of level-set reinitialization. The currently implemented
Lax–Friedrichs formulation does not provide these parameters to users,
since reinitialization is performed at every time step.
Level-Set Cartesian Mesh and Resolution: Internal Calculations
The Cartesian mesh extent or bounding box, the resolution and full.resolution
criteria, and the grid spacing criteria dx, dy, and dz interplay in the following ways.
The Cartesian mesh encompasses the initial interface between the Gas and all the materials the
user has defined to be etched. It also encompasses the entire movement of the etching front
expected throughout the entire etch process. In the case of time-based etching with the time
keyword, an etching distance is computed based on the requested etch time multiplied by the
maximum expected etch rate. The resolution or full.resolution keyword is then used
as the approximate mesh spacing unless overridden by dx, dy, or dz. The number of resulting
Cartesian mesh lines follows as required to achieve the desired mesh spacing within the
Cartesian mesh bounding box.
In the case of material etchstop, the bounding box of the Cartesian mesh is calculated based on
the initial Gas/etch-material interface, and its extent is determined by the position of etchstop
1 µm

11: Structure Generation
MGOALS Interface
782 Sentaurus™ Process User Guide
H-2013.03
materials in the simulation domain—the estimated maximum etching distance. The target
mesh spacing in 2D is the resolution or full.resolution multiplied by the estimated
maximum etching distance. In 3D, the target mesh spacing is set to the min.levelset.size,
as the estimated maximum etching distance is not calculated in 3D for this purpose. From the
Cartesian mesh bounding box size and the target mesh spacing, possibly overridden by dx, dy,
and dz, the number of Cartesian mesh lines is determined.
Limitations of Level Set
As a general approach, while level set is especially useful for shadowing, multimaterial,
sophisticated etch velocity functions (for example, surface normal dependent or crystal
direction dependent), and complex evolution of etch surfaces (that is, complicated structures),
it is generally not a good choice when sharp or exact corners, and straight or exact etch walls,
are required, such as in anisotropic etching.
This limitation is due to the implicit representation of the structure as a rectilinear grid of
distance functions used to calculate the evolution of the moving surfaces.
MGOALS 3D Boundary-moving Algorithms
In 3D, a combination of level set, fast marching, and analytic techniques are used to perform
geometric operations similar to the 2D mode. In 3D, MGOALS can reliably handle
complicated polyhedral boundaries. This mode can perform any of the geometric operations
contained in the deposit, etch, photo, polyhedron, and transform commands.
NOTE After any one of these process steps is performed in 3D, all subsequent
geometry operations should be performed using MGOALS.
The use of the level-set method for thin etches or deposits can be prohibitively CPU and
memory intensive, especially for large structures or for very thin etch or deposit steps. To
address this issue, etches and deposits less than 1 nm use the analytic method by default. The
thickness of this cutoff can be modified using the analytic.thickness parameter of the
mgoals command.
To produce meshes with the highest quality elements and the fewest points, you should reduce
the number of interfaces in the structure. This is especially true for 3D. However, because
region-merging is inconsistent with the paint-by-numbers mode, the default behavior in 3D is
not to merge regions. When the MGOALS mode is switched on, it is assumed that you are not
running the paint-by-numbers mode, so region-merging is switched on.
NOTE When the command sde off is specified, region-merging is switched
on regardless of the previous setting.
11: Structure Generation
MGOALS Interface
Sentaurus™ Process User Guide 783
H-2013.03
Summary of MGOALS Etching and Deposition Algorithms
Table 72 on page 783 summarizes the algorithms used internally to implement etching and
deposition:
■Level set (LF) – General time-stepping level-set algorithm with Lax–Friedrichs (LF)
formulation.
This is the most general algorithm. All level-set algorithms have the disadvantage of a
certain amount of rounding at corners and edges. The LF formulation has added stability,
which can result in slightly more rounding at corners and edges.
■Level set (UW) – General time-stepping level-set algorithm with Upwind (UW)
formulation.
The UW formulation is less stable and less reliable. This algorithm is used only for 2D
isotropic single-material etching with shadowing or for 2D isotropic multimaterial etching.
■Fastmarch (fast level-set method) – This is used in simple 1D or 2D directional and
anisotropic etching.
■Geometric – Three-dimensional etching algorithm creates a shaped boundary
representation tool, which is applied through Boolean operations to the existing structure
“blank” to obtain the final etching shape.
■Analytic – One-dimensional or 2D etching algorithm calculates and inserts an analytically
calculated etching shape into the device structure.
Table 72 Summary of etching algorithms used for different etching input parameters
Material Shadowing Etching type Structure
dimension
Etchstop mechanism
Time and rate Material Etchstop Thickness
Single material No shadowing Isotropic 1D/2D Analytic Level set (UW) Analytic
3D Geometric Level set (LF) Geometric
Fourier 1D/2D/3D Level set (LF) Level set (LF) Not supported
Directional 1D/2D Fastmarch Fastmarch Fastmarch
3D Geometric Level set (LF) Geometric
Anisotropic 1D/2D Fastmarch Fastmarch Fastmarch
3D Geometric Level set (LF) Geometric
11: Structure Generation
MGOALS Interface
784 Sentaurus™ Process User Guide
H-2013.03
MGOALS Backward Compatibility
Default parameters and algorithm settings used by the MGOALS library may change from
release to release in the pursuit of more accurate, more realistic, and more stable structure
generation results. To use default parameters and settings from a previous release, enter the
required release as a string parameter in the mgoals command, for example:
mgoals "G-2012.06"
Partial support for this backward compatibility is available starting with Version D-2010.03
with more complete support starting with Version E-2010.12.
Shadowing Isotropic 2D Level set (UW) Level set (UW) Level set (UW)
3D Level set (LF) Level set (LF) Level set (LF)
Fourier 2D/3D Level set (LF) Level set (LF) Not supported
Directional 2D/3D Level set (LF) Level set (LF) Level set (LF)
Anisotropic 2D/3D Level set (LF) Level set (LF) Level set (LF)
Multimaterial No shadowing Isotropic 2D Level set (UW) Level set (UW) Level set (UW)
3D Level set (LF) Level set (LF) Geometric
Fourier 2D/3D Level set (LF) Level set (LF) Not supported
Directional 2D Level set (LF) Level set (LF) Level set (LF)
3D Level set (LF) Level set (LF) Geometric
Anisotropic 2D Level set (LF) Level set (LF) Level set (LF)
3D Level set (LF) Level set (LF) Geometric
Shadowing Isotropic 2D Level set (UW) Level set (UW) Level set (UW)
3D Level set (LF) Level set (LF) Geometric
Fourier 2D/3D Level set (LF) Level set (LF) Not supported
Directional 2D Level set (LF) Level set (LF) Level set (LF)
3D Level set (LF) Level set (LF) Geometric
Anisotropic 2D Level set (LF) Level set (LF) Level set (LF)
3D Level set (LF) Level set (LF) Geometric
Table 72 Summary of etching algorithms used for different etching input parameters
Material Shadowing Etching type Structure
dimension
Etchstop mechanism
Time and rate Material Etchstop Thickness

11: Structure Generation
MGOALS Interface
Sentaurus™ Process User Guide 785
H-2013.03
Boundary Repair Algorithm
Anisotropic or directional operations can produce residual material when the walls of the
etched material are not perfectly vertical or aligned to the etching beam. These residual
materials usually cause problems to the mesh generator since they contain sharp angles and
small features that cannot be meshed. To correct this problem, MGOALS has implemented a
repair algorithm that analyzes the structure and eliminates small, unwanted features.
The repair algorithm can be used with the etch, deposit, and photo commands. Those
commands include a Boolean parameter named repair that controls the repair algorithm. The
repair algorithm is enabled by default in 3D and disabled in 2D. To activate or deactivate the
repair algorithm, include repair or !repair in the command specification. For example:
etch material= {Silicon} type=anisotropic rate = 0.001 time=1.0 !repair
Inserting Segments in One Dimension
The insert command defines and inserts regions defined by segments in one dimension. You
can choose which materials or regions are replaced, and the name and material of the new
region.
Multiple regions can be inserted in one step. However, to insert multiple regions, the name
cannot be specified. If multiple regions are inserted, machine-generated names are used. For
more information, see insert on page 982.
Inserting Polygons in Two Dimensions
Two-dimensional regions defined by polygons can be created and inserted directly into a 2D
simulation.
Polygons are created with the polygon command. This command accepts several options to
specify how to create the polygon:
■points is followed by a list of points defining the polygon.
■rectangle, with min and max, specifies the rectangle limits.
■segments is followed by pairs of numbers specifying the coordinate where each segment
starts and the previous one finishes.
■xy specifies the polygon will be created in the xy plane.
Since the standard use of the polygon command (see The mask and photo Commands on
page 764 and Using Polygon and Rectangle Mask in 2D Simulation on page 797) is to
create masks for etch, deposit and photo, the default coordinates are y and z for the

11: Structure Generation
MGOALS Interface
786 Sentaurus™ Process User Guide
H-2013.03
segments and min and max options. Consequently, the option xy must be specified in
order for the polygon to be created in the xy plane instead of the yz plane.
■tdr is followed by the name of a TDR file to import the polygon from, and the parameter
materials instructs the reader which polygon to read if there are many.
For examples of polygon creation, see Polygon Creation and Insertion in MGOALS2D on
page 809.
The insert command takes a mandatory polygon parameter containing the name of the
polygon and inserts it into the structure. It allows specifying the parameters
replace.materials, new.material, replace.regions, and new.region in a very
similar way to the insertion in 3D (see Inserting Polyhedra on page 789).
For an example of polygon insertion, see Polygon Creation and Insertion in MGOALS2D on
page 809.
Inserting Polyhedra in Three Dimensions
Regions defined by polyhedra can be inserted into an existing structure in three dimensions.
The polyhedron command creates a polyhedron <phname> and adds it to the internal
polyhedron list:
polyhedron name=<phname>
(tdr=<filename> [!rotate] [materials = {mat1 mat2}] [regions = { r1 r2 }])||
(brick = { <minx> <miny> <minz> <maxx> <maxy> <maxz> })||
(polygons = { <polname> } min=<value> max=<value> })||
(polygons = { <polname_1> ... <polname_n> })
You can build a polyhedron in four different ways; however, only one of them can be used at a
time in one polyhedron command:
■Reading it from a TDR boundary file.
■Creating a rectangular prism (brick) polyhedron.
■Extruding a 2D <polname> polygon in the x-dimension.
■Creating a polyhedron from the beginning using its constituent polygonal faces
<polname_1> to <polname_n>.
Reading Polyhedra from a TDR Boundary File
The polyhedron option:
tdr=<filename> [!rotate] [materials = {mat1 mat2}] [regions = { r1 r2 }]
11: Structure Generation
MGOALS Interface
Sentaurus™ Process User Guide 787
H-2013.03
reads all the polyhedra included in a TDR boundary file called <filename>.
The parameter materials is optional and is used to choose which materials are included. In
addition to explicit material names, the keyword bulk.materials is available to specify all
nongaseous materials.
The parameter regions is optional and is used to choose which regions of the boundary are
included.
If neither regions nor materials is specified, all regions are assumed to be included. If both
regions and materials are specified, the union of the two is assumed.
The extra option !rotate is used to avoid the automatic rotation that Sentaurus Process
performs when reading polyhedra to transfer them from the TDR boundary file (assumed to be
in DF–ISE coordinates) to the Sentaurus Process structure in Sentaurus Process coordinates.
Several polyhedra can be included in the TDR file. Any valid TDR boundary file is allowed,
regardless of the tool used to create it.
For an example, see Reading a TDR file on page 812.
Creating a Rectangular Prism
The polyhedron option:
brick = { <minx> <miny> <minz> <maxx> <maxy> <maxz> }
creates a rectangular prism given its two corners in Sentaurus Process coordinates.
For an example, see Defining a Brick on page 815.
Extruding a 2D Polygon
The polyhedron option:
polygons = { <polname> } min=<value> max=<value> }
takes an existing 2D polygon (created with the polygon command) and extrudes it in the
x-direction from min to max to build a 3D polyhedron. The command expects the polygon to
be planar. Only one polygon name is expected in the polygons list.
For an example, see Extruding a 2D Polygon on page 813.
11: Structure Generation
MGOALS Interface
788 Sentaurus™ Process User Guide
H-2013.03
Creating a Polyhedron from Its Constituent Polygonal Faces
The polyhedron option:
polygons = { <polname_1> ... <polname_n> }
builds a polyhedron given its definition as a set of polygons. The polygons are <polname_1>
to <polname_n>. Obviously, the command expects the polygon list to form a valid
polyhedron, that is, a compact, enclosed, nonintersecting 3D space. The polygons can be
created with the polygon command.
For an example, see Creating a Polyhedron using Polygons on page 814.
Sentaurus Structure Editor Interface: External Mode
This mode differs from the standard sde mode in that a structure can be created inside
Sentaurus Structure Editor independent of the existing Sentaurus Process structure. The
minimum syntax needed for creating an external structure is:
sde external { <sde commands> }
Where <sde commands> are scheme commands sent directly to Sentaurus Structure Editor.
As an option, a polyhedron can be specified to initialize the structure, and after sde
external, further geometric commands such as etch, deposit, and transform operate on
the external structure until the command sde off is specified. For more options for the sde
command, see sde on page 1114. To create a polyhedron from the external structure, the
parameter external.sde of the polyhedron command must be given. In the following
example, an aluminum sphere is inserted into the Sentaurus Process structure:
math coord.ucs
sde external {
(sdegeo:create-sphere (position 0.4 0.0 0.0) 0.9 "Aluminum" "Aluminum_1")
}
polyhedron name= sphere external.sde
sde off
insert polyhedron= sphere
NOTE Commands sent directly to Sentaurus Structure Editor through the sde
command need to consider the Sentaurus Process coordinate system. In
the previous example, the UCS coordinate system (same as Sentaurus
Process coordinate system) is used, so the x coordinate is vertical (–x is
up), and y and z are lateral directions. If math coord.ucs is not
specified, the z-axis is vertical (+z is up) and x and y are lateral.
11: Structure Generation
MGOALS Interface
Sentaurus™ Process User Guide 789
H-2013.03
Inserting Polyhedra
The insert command is:
insert polyhedron=<phname> [replace.materials= { mat1 mat2 }]
[replace.regions= {r1 r2 }] [new.material=<matname>] [new.region=<regname>]
NOTE The parameter polyhedron is mandatory and specifies the name of the
polyhedron with which to operate. This polyhedron must be previously
defined with the polyhedron command (see Inserting Polyhedra in
Three Dimensions on page 786).
The parameter replace.materials is optional and specifies a list that indicates the
materials to be replaced by the polyhedron. In addition to explicit material names, the keyword
bulk.materials is allowed. If bulk.materials is specified, it means that all materials in
the structure, except gas, will be replaced.
The parameter replace.regions is optional and specifies a list that indicates the regions to
be replaced by the polyhedron. If neither replace.regions nor replace.materials is
specified, it means that all materials are replaced. If both replace.regions and
replace.materials are specified, the union of the two is assumed.
The parameter new.material is optional. If set, all the regions in the polyhedron will change
to the specified material. This option does not change the polyhedron information except
temporarily during the duration of the insert command. The material name in the inserted
polyhedron is inserted, but not in the original polyhedron.
The parameter new.region is optional and valid only when there is one region. When set, the
region name is set to the specified one after insertion. The region name in the inserted
polyhedron is affected, but not the original polyhedron.
The insert command can be used to perform polyhedron etch and polyhedron deposit as well
as the more general polyhedron insert functionality. Polyhedron etch is performed by
specifying new.material=gas in the insert command or by creating a gas polyhedron.
Polyhedron deposit is performed by specifying replace.materials=gas in the insert
command as well as choosing one or more bulk regions or materials in the polyhedron
command, such as materials=bulk.materials or new.material=Silicon.
NOTE The boundaries of the polyhedra to be inserted must not overlap any
interfaces or outer boundaries of the structure. Otherwise, it is likely the
operation will fail.
For examples of polyhedron insertions, see Reading a TDR file on page 812, Extruding a 2D
Polygon on page 813, Creating a Polyhedron using Polygons on page 814, and Defining a
Brick on page 815.

11: Structure Generation
MGOALS Interface
790 Sentaurus™ Process User Guide
H-2013.03
Structure Assembly in MGOALS Mode
Sentaurus Process can read an existing 2D or 3D structure from a file and paste it into the
current 2D or 3D simulation, respectively.
To perform structure assembly, use:
paste direction = [back | front | left | right] tdr= <filename>
where:
■2D direction can be left, or right.
■3D direction can be back, front, left, or right.
■filename is the file to paste, in TDR format, which must be specified.
Sentaurus Process automatically shifts the structure read from the file to the appropriate
quantity in x, y, and z to fit to the current structure. Nevertheless, Sentaurus Process will not
automatically stretch the incoming structure. Consequently, for the operation to succeed, the
sizes of the pasting planes of the incoming structure and the existing one must be the same.
The values of the fields are conserved for each structure and are interpolated at the interface
between the structures.
NOTE Structure assembly requires that the structure read from the file must
have the same dimensionality, 2D or 3D, as the existing structure.
NOTE In 3D, structure assembly must be done in 3D mode. That is, structure
assembly is not available when sde on is specified.
Multithreading
Some of the more sophisticated etching and deposition types require the use of the level-set
method (such as multimaterial etching, crystal etching and deposition, Fourier etching and
deposition, use of etch stops or shadowing). This can be time-consuming, especially for 3D
summations. To minimize simulation time, the MGOALS library allows for a multithreaded
solution of the level-set equations.
The multithreaded operation can be invoked using:
math numThreads = <n>
or:
math numThreadsMGoals = <n>

11: Structure Generation
Sentaurus Structure Editor Interface
Sentaurus™ Process User Guide 791
H-2013.03
where <n> is an integer. It is suggested to keep <n> at or below 4 to obtain reliable speed
improvement.
Sentaurus Structure Editor Interface
By default, etching and deposition operations are performed using the MGOALS library in 1D,
2D and 3D. However, the Sentaurus Structure Editor can also be used to perform 3D etch,
deposit, and geometry transformation operations.
Sentaurus Structure Editor is a 3D geometry editor that uses the ACIS solid geometry modeling
kernel and the Scheme scripting language. Structures are created using CAD operations and
process emulation operations. All 3D etch, deposit, strip, photo, mask, and transform
commands are translated into appropriate Scheme commands that are then dispatched to
Sentaurus Structure Editor.
Sentaurus Structure Editor is integrated as a library in Sentaurus Process. The command
controlling Sentaurus Structure Editor from within Sentaurus Process is the sde command.
Sentaurus Structure Editor also can be used as a stand-alone tool to build the final structure by
using both its GUI and scripting capability. Then, the final structure can be used in Sentaurus
Process either as a boundary file or after remeshing the structure with one of the available
stand-alone mesh generation tools. The mesh or the boundary for the final structure is loaded
and before each implant or diffuse step, the material of all regions, not yet present in the
structure for the process step, is changed to gas.
Finally, there is an external mode. The Sentaurus Structure Editor external mode allows
independent (in other words, external) structures built in Sentaurus Structure Editor to be
inserted into structures created with MGOALS. This mode was designed to take advantage of
the best of MGOALS (advanced geometry-moving algorithms) and Sentaurus Structure Editor
(solid modeling capabilities). For more information on this mode, see Sentaurus Structure
Editor Interface: External Mode on page 788.
Hereafter, standard Sentaurus Structure Editor interface mode (sde on) will be referred to as
the SDE mode.
As usual, simulations may start in one or two dimensions. If a 3D mask is encountered and if
z-lines have been defined, the structure will be extruded to three dimensions, and if the SDE
mode is switched on, the Sentaurus Structure Editor interface will be initialized. All subsequent
structure-modifying steps in the etch, deposit, strip, photo, and transform commands
are dispatched to Sentaurus Structure Editor.
NOTE Reading a discretized 3D structure in Sentaurus Structure Editor can be
unstable; most isotropic operations (deposit or etch) will fail if this
11: Structure Generation
Sentaurus Structure Editor Interface
792 Sentaurus™ Process User Guide
H-2013.03
method to initialize Sentaurus Structure Editor had been used.
Therefore, when initializing a 3D simulation, you should store and
load .sat files, rather than simply loading a 3D TDR boundary or grid
file.
When the 3D structure has been initialized in Sentaurus Structure Editor, structure generation
commands (mask, etch, deposit, photo, strip, and transform) are translated by
Sentaurus Process into appropriate Scheme commands and then dispatched to Sentaurus
Structure Editor.
NOTE Currently, a few options of the etch command cannot be translated into
appropriate Scheme constructs: Fourier etching, trapezoidal etching,
crystallographic etching, and shadowing effects are not supported in
3D. The parameter etchstop only works with cmp but not with other
etch types.
The modified structure will be retrieved from Sentaurus Structure Editor and remeshed when
a command that requires the geometry and the mesh to be synchronized (for example,
implant, diffuse, and struct commands that write the mesh to a file) is found in the
Sentaurus Process command file.
This ‘lazy’ remeshing (only when needed) minimizes the number of 3D remeshing operations
and, therefore, increases both the robustness and speed of the 3D structure generation and
remeshing.
There is a separate command to configure and control the interface to Sentaurus Structure
Editor and to specify Scheme commands directly. The syntax of this Sentaurus Structure Editor
command is:
sde {<scheme command>} [on] [off] [remesh] [logfile=<c>] [SdeCheck]
The command:
sde on
must be specified in each 3D simulation so that the simulation is performed using the Sentaurus
Structure Editor interface.
By default MGOALS is used in 3D, but if sde mode has been switched on, it can be turned off
again using::
sde off
11: Structure Generation
Sentaurus Structure Editor Interface
Sentaurus™ Process User Guide 793
H-2013.03
The parameter logfile provides a file name to record all Scheme commands that are
dispatched to Sentaurus Structure Editor. At the end of the simulation, a complete Scheme
script is generated that can be used in a stand-alone run, for example:
sde -l logfile.scm
for debugging, testing different algorithms, or fine-tuning a few command parameters for
Sentaurus Structure Editor without rerunning the Sentaurus Process simulation. These
modified parameters and algorithm selections can later be incorporated into the etch,
deposit, and other commands by specifying the parameter sde in these commands:
deposit oxide thickness=5<nm> isotropic sde= {"algorithm" "lopx" "adaptive" \
#t "radius" 0.075}
etch silicon thickness=0.2<um> isotropic sde= {"algorithm" "lopx" "radius" \
0.07 "vexity" "convex" "blend-global" "steps" 1 "overetch" 0.2}
deposit oxide thickness=5<nm> isotropic sde= {"algorithm" "lopx"}
NOTE The Scheme language is incompatible with the Tcl used by Sentaurus
Process. Therefore, all Scheme commands and parameter settings must
be enclosed by a pair of braces. The opening brace must be on the same
line as the sde parameter.
In the sde command (not the parameter), the braces may contain any number of Scheme
commands, each of which starts on a new line.
Since the braces protect the Scheme commands and parameter settings from being parsed by
Tcl, they must not contain any calls to Tcl procedures in Tcl expressions. The Scheme language
provides its own set of expressions, parameter definitions, and other language constructs.
You s ho uld i nc rease t he defa ul t v erb osity level when working with the sde command:
pdbSet InfoDefault 1
The Sentaurus Structure Editor library does not provide any error-processing facility for errors
that have occurred during the solid modeling operations. This can become time consuming if
a structure generation step fails and a long diffuse or implant simulation is performed for
an incorrect structure. To avoid this, use a few runs with the -f command-line option to adjust
the commands and to verify that the proper structure is created. In addition, by default, all
boundary files that are written in struct commands in the fast mode and before remeshing
are read and checked for any geometric inconsistencies. If any defects are observed, the
simulation is stopped with an error. To prevent this checking, specify the
parameter !SdeCheck.
NOTE By default, Sentaurus Process performs stress relaxation at the end of
each etching and deposition step. This requires that a boundary-fitted

11: Structure Generation
Sentaurus Topography Interface
794 Sentaurus™ Process User Guide
H-2013.03
mesh be constructed at the end of each step. If you do not want to track
the stress through all the process steps, use the pdbSet Mechanics
EtchDepoRelax 0 command before starting 3D structure generation.
NOTE To prevent adjacent regions of the same material (for example gas
regions) from merging, switch off region-merging using the command
pdbSet Grid No3DMerge 1. During the process, as more regions
need to be considered (for example, nitride spacer), appropriate
materials must be reverted from gas to the required materials.
Finally, the option Grid Auto3DMergeAndSeparate (off by default) adds the following
commands at the end of photo and depo when switched on:
(sdegeo:bool-unite (find-material-id 'depositedMaterial'))
(sde:separate-lumps)
Only the last one is added after etching.
Sentaurus Topography Interface
Sentaurus Process provides an interface to both Sentaurus Topography, the 2D physical etch
and deposit simulator, and Sentaurus Topography 3D, the 3D physical etch and deposit
simulator.
Sentaurus Topography
Each sptopo command first transfers the current 2D geometry from Sentaurus Process to
Sentaurus Topography. Then, it dispatches the command to Sentaurus Topography. Finally, it
retrieves the modified 2D geometry from Sentaurus Topography and remeshes it using the
MGOALS mesher in 2D.
Sending a new geometry from Sentaurus Process to Sentaurus Topography has been restricted
to cases where the geometry has actually been modified in Sentaurus Process after last
retrieving the structure from Sentaurus Topography, for example, when using Sentaurus
Process etch or deposit command. Provisions also are made to detect whether Sentaurus
Topography has actually modified the structure or simply a definition of it; for example, a new
machine has been added to Sentaurus Topography. Remeshing is restricted to the commands
that actually have changed the structure.
11: Structure Generation
Sentaurus Topography Interface
Sentaurus™ Process User Guide 795
H-2013.03
During the syntax check, Sentaurus Topography commands are dispatched to Sentaurus
Topography and checked for syntactical correctness. The supported syntax of the sptopo
command is:
sptopo <sptopo command>
or:
sptopo {
<sptopo command>
<sptopo command>
...
}
The first form of the sptopo command allows use of all the usual Sentaurus Process Tcl
constructions in the parameter specifications of <sptopo_command>. This form of the
command is parsed through the Tcl interpreter. Otherwise, the syntax used for the
<sptopo_command> is the same as in each of the commands for a stand-alone Sentaurus
Topography run. In Sentaurus Process command files, each sptopo command must start with
the sptopo string.
The pair of braces in the second form of the command prevents this sptopo command from
being parsed by the Tcl interpreter. No Tcl expressions must be used in the second form of the
sptopo command. On the other hand, any number of Sentaurus Topography commands can
be provided in the second form of the command, each on a separate command line. If necessary,
the structure will be sent from Sentaurus Process to Sentaurus Topography once at the
beginning, and retrieved and remeshed once at the end of the entire command sequence.
Examples:
sptopo {
deposit material=Oxide thickness = 0.005
deposit material=PolySilicon thickness = 0.180
}
Two planar deposition steps are performed in Sentaurus Topography:
■The first fills the structure with oxide up to 5 nm above the top material position.
■The second adds a planar layer of 180 nm polysilicon.
The structure is sent to Sentaurus Topography once, retrieved, and remeshed once at the end of
both deposition steps.
If masks are required in a Sentaurus Topography simulation, segments can be specified in the
sptopo command as shown below. Alternatively, the Sentaurus Process photo command can
be used with a mask to define a photoresist layer that will protect certain areas from being
etched in a sptopo etch command.

11: Structure Generation
Sentaurus Topography Interface
796 Sentaurus™ Process User Guide
H-2013.03
The Sentaurus Process strip command or the command:
sptopo etch material=Photoresist complete
can be used later to remove the entire photoresist layer.
sptopo {
mask name=m1 s0=-1.1 e0=-0.3 s1=0.3 e1=1.1
etch material=PolySilicon depth=0.185 mask=m1
machetch name=oxe1 material=Oxide anisotropy=1 rate=1
etch machname=oxe1 time = 0.02 dx=0.03 dy=0.03 mask=m1
}
The preceding example defines a mask in Sentaurus Topography including:
■Geometric etching of polysilicon which is strictly vertical and restricted to the outside of
the specified mask segments
■An anisotropic etching machine
■Execution of an anisotropic oxide etching in Sentaurus Topography
To increase the default verbosity level when working with the sptopo command, use:
pdbSet InfoDefault 1
For a complete list of commands, parameters, and syntax rules of the Sentaurus Topography
simulator, refer to the Sentaurus™ Topography User Guide.
Sentaurus Topography 3D
Sentaurus Process provides an interface to Sentaurus Topography 3D. This interface makes
advanced etching and deposition models of Sentaurus Topography 3D available from within
Sentaurus Process.
The subset of 3D commands that are needed for etching and deposition is available through the
interface. One single command, topo, in Sentaurus Process enables all the interface
functionality. The topo command is followed by the respective Sentaurus Topography 3D
commands:
topo <Sentaurus Topography 3D command>
For a list of the supported Sentaurus Topography 3D commands, refer to the Sentaurus™
Topography 3D User Guide.
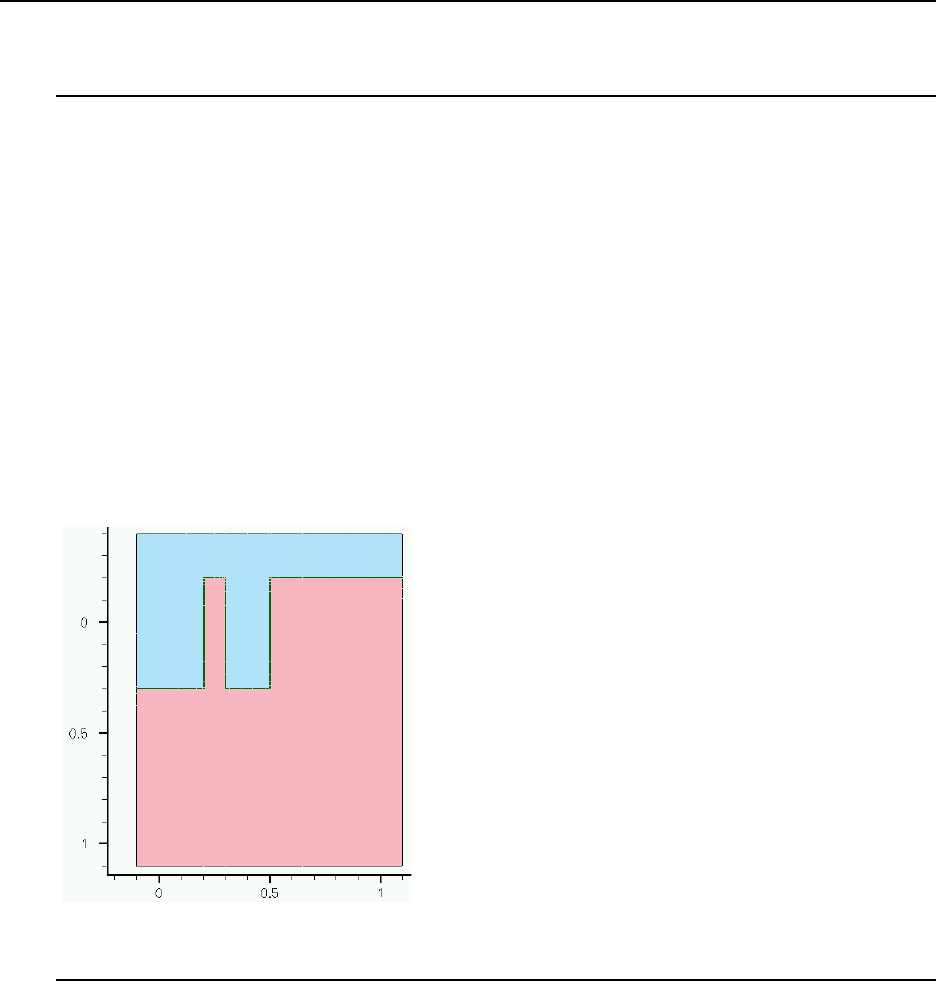
11: Structure Generation
Examples
Sentaurus™ Process User Guide 797
H-2013.03
Examples
Using Polygon and Rectangle Mask in 2D Simulation
line x loc=-0.3 tag=ox
line x loc=-0.2 tag=top
line x loc=1.1 tag=bot
line y loc=-0.1
line y loc=1.1
region silicon xlo=top xhi=bot
init
polygon name=LShape2 segments= {-0.1 -1.5 -0.1 -0.5 0.5 -0.5 0.5 1.5 1.5 \
1.5 1.5 -1.5}
mask name=Mask2 polygons= {LShape2} negative left=0.2 right=0.3 front=-0.1 \
back=0.2
etch silicon anisotropic thickness=0.5 mask=Mask2
struct tdr=final
Figure 135 Final result of 2D anisotropic etching with rectangle and polygon mask
3D Etching after 2D LOCOS Simulation (Sentaurus
Structure Editor Interface)
A 2D simulation result is loaded (LOCOS with nitride layer), modified in 2D, extruded to 3D,
and in 3D the polysilicon and oxide are etched.
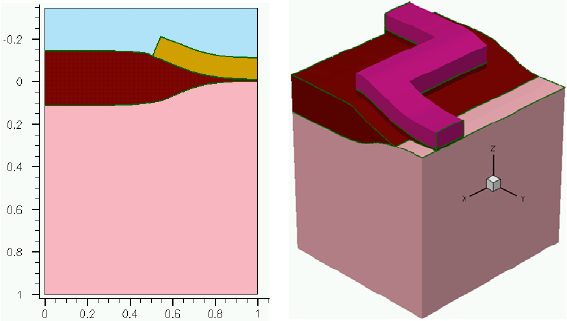
11: Structure Generation
Examples
798 Sentaurus™ Process User Guide
H-2013.03
# load a 2D locos structure
init tdr=initial
mgoals min.normal.size=0.02 accuracy=1e-4 max.box.angle=165 \
normal.growth.ratio=4
refinebox min= {-10 -10 -10} max= {10 10 10} xrefine=0.4 yrefine=0.4 \
zrefine=0.4
# --- allow expanding structure to 3D ---
line z loc=-0.6 spacing=0.1
line z loc=0.5 spacing=0.1
# still in 2D
strip nitride
deposit PolySilicon thickness=100<nm> isotropic
struct tdr=locos1
sde logfile=2d3d.scm on
pdbSet InfoDefault 1
polygon name=LShape2 segments= {-0.1 -0.4 0.6 -0.4 0.6 0.2 1.1 0.2 \
1.1 0.4 0.4 0.4 0.4 -0.2 -0.1 -0.2}
mask name=Mask2 polygons= {LShape2}
etch PolySilicon anisotropic mask=Mask2 thickness=0.41
etch Oxide thickness=30<nm> anisotropic
struct tdr=locos2
Figure 136 (Left) Initial 2D structure after LOCOS formation and (right) final result after
extruding to 3D and etching of poly and oxide
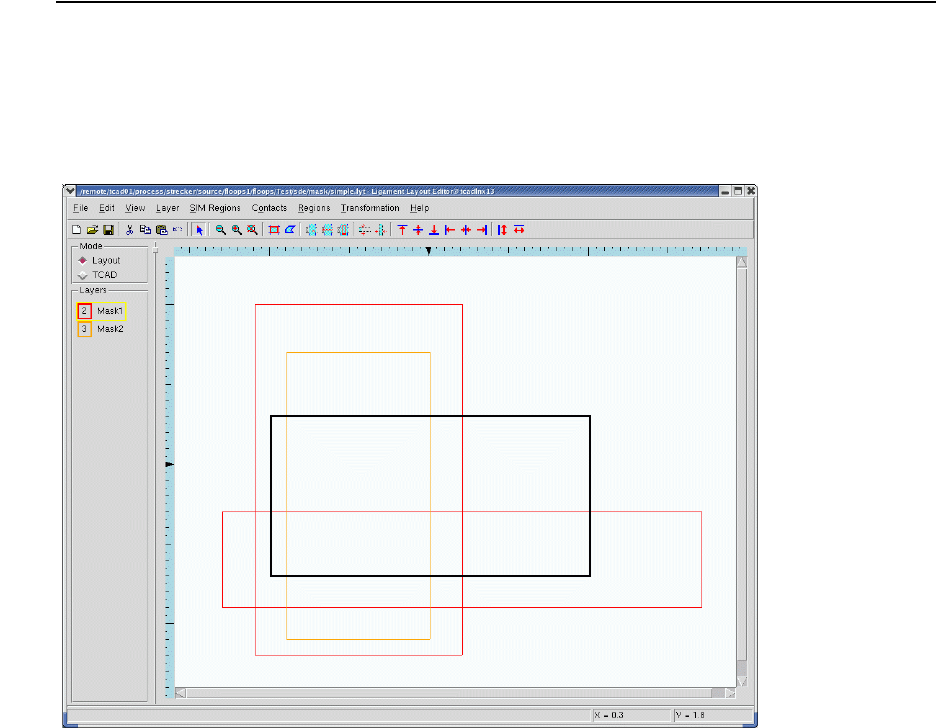
11: Structure Generation
Examples
Sentaurus™ Process User Guide 799
H-2013.03
Using Layout File for 3D Etching (Sentaurus Structure
Editor Interface)
This series of examples demonstrates how to use the layout file simple.lyt to define masks
for 3D etching.
Figure 137 View of simple.lyt file in Ligament Layout Editor
Input file mask0_fps.cmd: The origin of the Sentaurus Process coordinate system coincides
with the origin in simple.lyt; the y-axis of Sentaurus Process is aligned to the vertical axis
in simple.lyt:
line x loc=-0.25 tag=gastop spac=0.05
line x loc=0.0 tag=substop spac=0.01
line x loc=1.5 tag=subsbottom spac=0.2
line y loc=1.65 spac=0.1
line y loc=1.95 spac=0.1
line z loc=0.15 spac=0.1
line z loc=0.6 spac=0.1
region silicon xlo=substop xhi=subsbottom
mgoals min.normal.size=0.02 accuracy=1e-4 max.box.angle=165 \
normal.growth.ratio=4
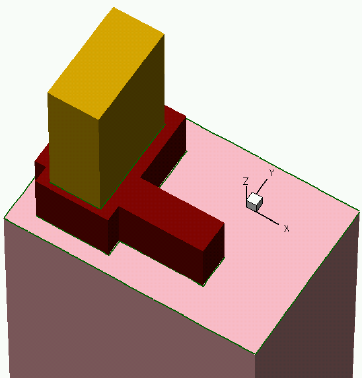
11: Structure Generation
Examples
800 Sentaurus™ Process User Guide
H-2013.03
refinebox min= {-10 -10 -10} max= {10 10 10} xrefine=0.2 yrefine=0.2 \
zrefine=0.2
refinebox interface.materials = {Silicon Oxide}
init
deposit oxide thickness=100<nm> isotropic
sde logfile=mask0.scm on
pdbSet InfoDefault 1
mask layoutfile=simple.lyt
# deposition with masks deposits where there is no mask
# so invert the mask used for deposition
mask name=Mask2 negative
deposit nitride thickness=0.25<um> anisotropic info=1 mask=Mask2
struct bndfile=mask0_0.bnd
etch oxide thickness=120<nm> type=anisotropic mask=Mask1
struct bndfile=mask0_1.bnd
struct tdr=mask0
Figure 138 Final structure after simulation of mask0_fps.cmd
11: Structure Generation
Examples
Sentaurus™ Process User Guide 801
H-2013.03
Input file mask1_fps.cmd: A Cutline2D is used to place the simulation domain in the layout
file. The extensions of the 3D simulation domain in the y- and z-directions must be specified
using line commands. The cutline is defined as the diagonal through the structure used in
mask1_fps.cmd, such that the Sentaurus Process origin is shifted and the coordinate system
is rotated compared to mask0_fps.cmd:
line x loc=-0.25 tag=gastop spac=0.05
line x loc=0.0 tag=substop spac=0.01
line x loc=1.5 tag=subsbottom spac=0.2
line y loc=0. spac=0.1
line y loc=0.4 spac=0.1
line z loc=-0.2 spac=0.1
line z loc=0.15 spac=0.1
region silicon xlo=substop xhi=subsbottom
mgoals min.normal.size=0.02 accuracy=1e-4 max.box.angle=165 \
normal.growth.ratio=4
refinebox min= {-10 -10 -10} max= {10 10 10} xrefine=0.2 yrefine=0.2 \
zrefine=0.2
refinebox interface.materials = {Silicon Oxide}
# define a coordinate transformation for a placement and rotation
# of a layout file
init slice.angle=[CutLine2D 1.65 0.15 1.95 0.6]
deposit oxide thickness=100<nm> isotropic
sde logfile=mask1.scm on
pdbSet InfoDefault 1
# do not specify any name ==> use the cutline from the init command
# to place the Sentaurus Process coordinate system in the layout.
# the first specified point becomes the origin of the Sentaurus Process
# coordinate system and the direction of the cutline becomes the direction
# of the Sentaurus Process z-axis.
mask layoutfile=simple.lyt
# deposition with masks deposits where there is no mask
# so invert the mask used for deposition
mask name=Mask2 negative
deposit nitride thickness=0.25<um> anisotropic info=1 mask=Mask2
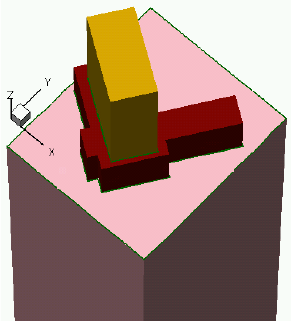
11: Structure Generation
Examples
802 Sentaurus™ Process User Guide
H-2013.03
struct bndfile=mask1_0.bnd
etch oxide thickness=120<nm> type=anisotropic mask=Mask1
struct bndfile=mask1_1.bnd
struct tdr=mask1
Figure 139 Final result of mask1_fps.cmd
Input file mask2_fps.cmd: A SIM3D mask is used, defined in the layout file:
# Use a layout file and place it according to the mask SIM3D, defined in the
# layout file itself.
# SIM3D defines an axis aligned rectangle in the layout plane. The point
# with the smallest layoutX and layoutY coordinates defines the
# origin of the Sentaurus Process coordinate system. The direction of the
# layoutX axis is used for the Sentaurus Process z-axis
# and the direction of the layoutY axis is used for the Sentaurus Process
# y-axis.
# The width of the rectangle (max(layoutX)-min(layoutX)) defines the
# default extension in Sentaurus Process z-direction.
# The height of the rectangle (max(layoutY)-min(layoutY)) defines the
# default extension in Sentaurus Process y-direction.
# If you specify line y and/or line z, your definitions are used.
# Otherwise the default extensions are used to define line y and line z
# when reading the layout file.
line x loc=-0.25 tag=gastop spac=0.05
line x loc=0.0 tag=substop spac=0.01
line x loc=1.5 tag=subsbottom spac=0.2
region silicon xlo=substop xhi=subsbottom
mgoals min.normal.size=0.02 accuracy=1e-4 max.box.angle=165 \
normal.growth.ratio=4
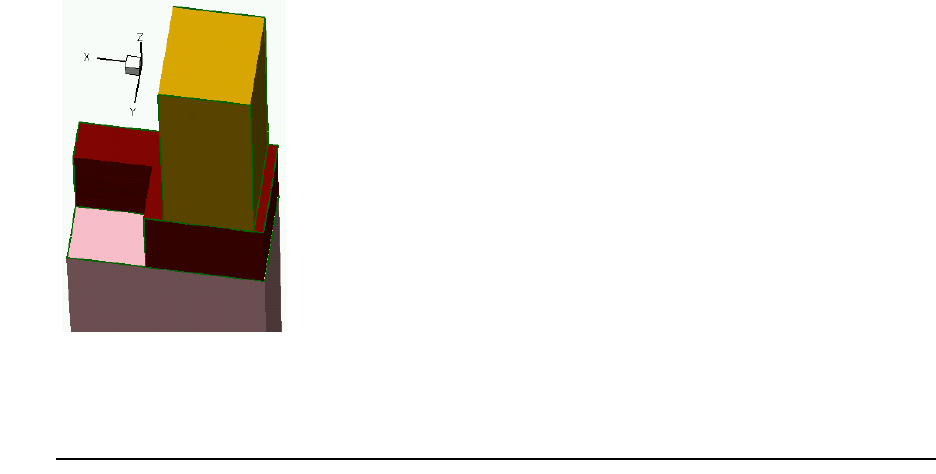
11: Structure Generation
Examples
Sentaurus™ Process User Guide 803
H-2013.03
refinebox min= {-10 -10 -10} max= {10 10 10} xrefine=0.2 yrefine=0.2 \
zrefine=0.2
refinebox interface.materials = {Silicon Oxide}
init
deposit oxide thickness=100<nm> isotropic
sde logfile=mask2.scm on
pdbSet InfoDefault 1
mask layoutfile=simple.lyt name=SIM3D
# In Sentaurus Process, deposition with masks deposits where
# there is no mask so invert the mask used for deposition
mask name=Mask2 negative
deposit nitride thickness=0.25<um> anisotropic info=1 mask=Mask2
struct bndfile=mask2_0.bnd
etch oxide thickness=120<nm> type=anisotropic mask=Mask1
struct bndfile=mask2_1.bnd
struct tdr=mask2
Figure 140 Final simulation result for mask2_fps.cmd; the y- and z-extensions are not
specified in the command file but are taken from the SIM3D mask in simple.lyt
(black line in Figure 137 on page 799)
3D Trench Etching, Sloped Sidewall with Predefined Angle
(Sentaurus Structure Editor Interface)
line x loc=-0.25 tag=gastop spac=0.05
line x loc=0.0 tag=substop spac=0.01
line x loc=0.5 tag=subsbottom spac=0.2
line y loc=0.0 spac=0.1
line y loc=0.4 spac=0.1
11: Structure Generation
Examples
804 Sentaurus™ Process User Guide
H-2013.03
line z loc=0 spac=0.1
line z loc=0.6 spac=0.1
region silicon xlo=substop xhi=subsbottom
mgoals min.normal.size=0.02 accuracy=1e-4 max.box.angle=165 \
normal.growth.ratio=4
refinebox min= {-10 -10 -10} max= {10 10 10} xrefine=0.2 yrefine=0.2 \
zrefine=0.2
refinebox interface.materials = {Silicon Oxide}
init concentration=1.4e+15<cm-3> field=boron wafer.orient=100
deposit oxide thickness=0.01 type=isotropic
fset NitrideThick 0.1
deposit nitride thickness=0.1 type=isotropic
mask name=STI left=-1 right=0.2 front=-1 back=0.4
deposit Photoresist isotropic thickness=0.5
sde logfile=sti3d.scm on
pdbSet InfoDefault 1
etch Photoresist anisotropic thickness=0.5*1.5 mask=STI
struct bndfile=photo1.bnd
fproc etchAngle { Angle Material Depth } {
set alpha [expr ${Angle}*atan(1.0)/45.0] ; #Degree to radiant conv.
set x1 [expr sin($alpha)] ; #x-component of etch directional vector
set x2 [expr cos($alpha)] ; #y-component of etch directional vector
set x3 [expr cos($alpha)] ; #z-component of etch directional vector
set etchRate [expr 1.0/sin($alpha)]
etch material=$Material time=$Depth type=directional \
direction= { $x1 $x2 $x3 } rate=$etchRate
}
fset NitrideAngle 89.0
etchAngle $NitrideAngle Nitride $NitrideThick*1.5
etch Oxide anisotropic thickness=0.02
strip Photoresist
fset TrenchAngle 84.0
fset TrenchDepth 0.2
etchAngle $TrenchAngle Silicon $TrenchDepth
struct bndfile=final.bnd
struct tdr=final
11: Structure Generation
Examples
Sentaurus™ Process User Guide 805
H-2013.03
Figure 141 Final result of simulation of STI etching with predefined sidewall angles
3D Etching after 2D LOCOS Simulation using MGOALS
An example, similar to 3D Etching after 2D LOCOS Simulation (Sentaurus Structure Editor
Interface) on page 797, but with some extra processing steps, can be performed using
MGOALS. The script is as follows:
# Switch off stress relaxation after depo/etch
pdbSet Mechanics EtchDepoRelax 0
# Load a 2D locos structure
init tdr=initial
mgoals resolution=0.2 min.normal.size=0.02 accuracy=1e-4 max.box.angle=165 \
norm
refinebox min= {-10 -10 -10} max= {10 10 10} xrefine=0.4 yrefine=0.4 zrefine=0.
# --- allow expanding structure to 3D ---
line z loc=-0.6 spacing=0.1
line z loc=0.5 spacing=0.1
# Still in 2D
strip nitride
deposit PolySilicon thickness=100<nm> isotropic
struct tdr=locos1
#sde logfile=2d3d.scm on
11: Structure Generation
Examples
806 Sentaurus™ Process User Guide
H-2013.03
sde off
pdbSet InfoDefault 2
pdbSet Grid sMesh 1
polygon name=LShape2 segments= {-0.1 -0.4 0.6 -0.4 0.6 0.2 1.1 0.2 1.1 0.4}
mask name=Mask2 polygons= {LShape2}
etch PolySilicon anisotropic mask=Mask2 thickness=0.41
struct tdr=locos2
etch Oxide thickness=30<nm> anisotropic
struct tdr=locos3
mgoals resolution=0.3
deposit oxide thickness=10<nm> isotropic
struct tdr=locos4
mgoals resolution=0.2
deposit nitride thickness=100<nm> isotropic
struct tdr=locos5
etch nitride thickness=120<nm> anisotropic
struct tdr=locos6 bnd
The initial 2D LOCOS structure (initial.tdr) is the left one represented in Figure 136 on
page 798. The sde off command is used to overwrite the standard setting and MGOALS is
used.
Figure 142 Script result simulated with MGOALS; polysilicon and silicon
are not shown to better reveal the structure features

11: Structure Generation
Examples
Sentaurus™ Process User Guide 807
H-2013.03
Structure Assembly in MGOALS
The following scripts creates a tdr file to be “pasted”
set gate 30e-3
set sti 55e-3
set sti_thick 100e-3
set gas_top 86e-3
set silicon_depth 300e-3
set zsize 640e-3
set SiO2gate 1.5e-3
set spacer 30e-3
set spacer_thick 30e-3
set extra 60e-3
set poly 80e-3
set factor 1.1
sde off
line x loc=0.0 tag=xtop
line x loc=$silicon_depth tag=xbottom
line y loc=0.0 tag=yleft
line y loc=[expr 2*$extra + 2*$spacer + 2*$sti + $gate] tag=yright spacing =
0.002
line z loc=0
line z loc=0.05
line z loc=0.1
region silicon xlo=xtop xhi=xbottom
init
#STI
set ll1 [expr 0]
set rr1 [expr $sti]
set ll2 [expr $sti + 2*$extra + 2*$spacer + $gate]
set rr2 [expr $ll2 + $sti]
mask negative name=sti_mask left=$ll1<um> right=$rr1<um>
mask negative name=sti_mask left=$ll2<um> right=$rr2<um>
etch silicon thickness=$sti_thick mask=sti_mask anisotropic
deposit oxide fill coord=[expr -$SiO2gate]
#poly gate
deposit polysilicon thickness=$poly isotropic
set ll3 [expr $sti + $extra+$spacer]
set rr3 [expr $ll3 + $gate]
mask name=gate_mask left=$ll3 right=$rr3
etch polysilicon mask=gate_mask anisotropic thickness=$poly
#spacer
deposit nitride thickness=$spacer_thick isotropic
etch nitride thickness=[expr $spacer_thick * $factor] anisotropic
11: Structure Generation
Examples
808 Sentaurus™ Process User Guide
H-2013.03
#rotate and transform into a 3D structure
transform rotate angle=90 axis = {X}
struct tdr=rotateX
Figure 143 on page 809 (upper right) shows the created structure.
Now, a simple structure is created with the same dimensions in the plane to be pasted and the
structures are put together:
set xsize 0.3
set ysize 0.1
set zsize 0.32
line x loc=0.0 tag=xleft
line x loc=$xsize/2 tag=xmed
line x loc=$xsize tag=xright
line y loc=0.0 tag=ybottom
line y loc=$ysize/2 tag=ymed
line y loc=$ysize tag=ytop
line z loc=0 tag=zinit
line z loc=$zsize/2 tag=zmed
line z loc=$zsize tag=zend
region oxide xlo=xleft xhi=xmed ylo=ymed yhi=ytop zlo=zinit zhi=zmed
region silicon xlo=xleft xhi=xmed ylo=ymed yhi=ytop zlo=zmed zhi=zend
region silicon xlo=xleft xhi=xmed ylo=ybottom yhi=ymed zlo=zinit zhi=zend
region silicon xlo=xmed xhi=xright ylo=ybottom yhi=ytop zlo=zinit zhi=zend
init
sde off
struct tdr = orig
paste direction = "right" tdr = "rotateX"
struct tdr = pasted_right
Figure 143 (upper left) shows the structure created before the paste operation, and the final
result is shown in the lower-left figure.
11: Structure Generation
Examples
Sentaurus™ Process User Guide 809
H-2013.03
Figure 143 (Upper left) The initial structure, (upper right) the structure read from the file, and
(lower left) the final structure after paste command
Polygon Creation and Insertion in MGOALS2D
The following script creates a 2D structure and saves it into a file:
set gate 30e-3
set sti 55e-3
set sti_thick 100e-3
set gas_top 86e-3
set silicon_depth 300e-3
set zsize 640e-3
set SiO2gate 1.5e-3
set spacer 30e-3
set spacer_thick 30e-3
set extra 60e-3
set poly 80e-3
set factor 1.1
refinebox min = { 0 0 } max = { 0.1 2 } xrefine = { 0.0012 0.0015 0.0015 }
line x loc=0.0 tag=xtop
line x loc=$silicon_depth tag=xbottom
line y loc=0.0 tag=yleft
line y loc=[expr 2*$extra + 2*$spacer + 2*$sti + $gate] tag=yright spacing =
11: Structure Generation
Examples
810 Sentaurus™ Process User Guide
H-2013.03
0.002
region silicon xlo=xtop xhi=xbottom
init
#STI
set ll1 [expr 0]
set rr1 [expr $sti]
set ll2 [expr $sti + 2*$extra + 2*$spacer + $gate]
set rr2 [expr $ll2 + $sti]
mask negative name=sti_mask left=$ll1<um> right=$rr1<um>
mask negative name=sti_mask left=$ll2<um> right=$rr2<um>
etch silicon thickness=$sti_thick mask=sti_mask anisotropic
deposit oxide fill coord=[expr -$SiO2gate]
#poly gate
deposit polysilicon thickness=$poly isotropic
set ll3 [expr $sti + $extra+$spacer]
set rr3 [expr $ll3 + $gate]
mask name=gate_mask left=$ll3 right=$rr3
etch polysilicon mask=gate_mask anisotropic thickness=$poly
#spacer
deposit nitride thickness=$spacer_thick isotropic
etch nitride thickness=[expr $spacer_thick * $factor] anisotropic
struct tdr=orig
Now, a triangular polygon is created in the xy plane and is called box:
point name=p1 coord = { -0.2 0.2 }
point name=p2 coord = { 0.2 0.25 }
point name=p3 coord = { 0.1 0.1 }
polygon name=box xy points = { p1 p2 p3 }
Finally, the triangular polygon is inserted into the original structure as Aluminum, but only in
the nitride and silicon materials:
insert polygon = "box" replace.materials = { "Silicon" "Nitride" } \
new.material = "Aluminum"
struct tdr=points
Figure 144 on page 811 (upper left) shows the initial structure, and the lower-left figure shows
the results after the insertion.
You a lso c an re ad th e po ly gon fro m a TD R fil e an d insert it later. The following script reuses
the files from the previous example by doing that. It reads the Aluminum material as a polygon,
and inserts it in the original script as oxide in silicon material only:
init tdr=orig
polygon name=box xy tdr = "points_bnd.tdr" materials = "Aluminum"
insert polygon = "box" replace.materials = "Silicon" new.material = "Oxide" \
11: Structure Generation
Examples
Sentaurus™ Process User Guide 811
H-2013.03
info=4
struct tdr=tdr
Figure 144 (lower left) shows the results after the insertion.
Figure 144 Use of insert command in a 2D simulation: (upper left) initial 2D structure, (lower
left) triangular polygon being inserted in the initial structure for silicon and nitride
only, and (upper right) the results of reading the triangle of the lower-left figure
and inserting it as oxide in the silicon of the initial structure
11: Structure Generation
Examples
812 Sentaurus™ Process User Guide
H-2013.03
Polyhedron Creation and Insertion in MGOALS
All the following examples use the structure in Figure 145 as a starting point to be modified by
polyhedron creation and insertion.
Figure 145 Initial 3D structure used for polyhedron creation and insertion examples
Reading a TDR file
This example uses two spheres created with Sentaurus Structure Editor using the following
script:
(sde:clear)
(sdegeo:create-sphere (position 3.0 3.0 4.7) 0.9 "Silicon" "Silicon_1")
(sdegeo:create-sphere (position 0.7 0.7 4.7) 0.9 "Gas" "Gas_2")
(sdeio:save-tdr-bnd "all" "sphere.tdr")
The spheres are inserted into Sentaurus Process. The gas sphere etches the material, while the
silicon one is deposited:
init tdr=initial
refinebox clear
sde off
polyhedron name=sphere tdr=sphere.tdr materials = { Silicon Gas }
insert polyhedron=sphere
struct bndfile=result
11: Structure Generation
Examples
Sentaurus™ Process User Guide 813
H-2013.03
Figure 146 shows the result of the above script.
Figure 146 (Left) Polyhedra included in a TDR boundary file and (right) effect of inserting
them in initial structure
Extruding a 2D Polygon
This example creates a simple 2D polygon – a triangle:
init tdr=initial
math coord.ucs
refinebox clear
sde off
polygon name=triangle segments = { -4.2 -3.0 3.2 0.5 -4.7 4.2 }
The polyhedron command uses the triangle to extrude in the x-direction:
polyhedron name=prism polygons = { triangle } min=-6 max=2
Finally, to insert it, specify new.material as gas to etch the extruded polygon:
insert polyhedron=prism new.material=Gas
struct bndfile=result
11: Structure Generation
Examples
814 Sentaurus™ Process User Guide
H-2013.03
Figure 147 shows the result.
Figure 147 Etching an extruded polygon
Creating a Polyhedron using Polygons
This example defines the polyhedra from the beginning using polygons:
init tdr=initial
refinebox clear
sde off
point name=p1 coord = { -6.5 2.0 2.0 }
point name=p2 coord = { -2.0 4.0 1.5 }
point name=p3 coord = { -2.0 2.0 3.0 }
point name=p4 coord = { -2.0 1.5 1.0 }
polygon name=face1 points = { p1 p2 p3 }
polygon name=face2 points = { p1 p3 p4 }
polygon name=face3 points = { p1 p2 p4 }
polygon name=face4 points = { p2 p3 p4 }
polyhedron name=tetrahedron polygons = { face1 face2 face3 face4 }
Now, the initial structure is etched using the new polyhedron:
insert polyhedron=tetrahedron new.material=Gas
struct bndfile=result
11: Structure Generation
Examples
Sentaurus™ Process User Guide 815
H-2013.03
Figure 148 shows the simulation results.
Figure 148 Etching of a polyhedron defined by using polygons: (left) 3D general view and
(right) y-plane cut view
Defining a Brick
The brick option provides a convenient way to define a rectangular prism by defining the
lower and upper corners. The script is as follows:
init tdr=initial
refinebox clear
sde off
polyhedron name=smallCube brick = { -6 -4 -2 -1 4.5 1 }
Now, you can use the polyhedron to insert an oxide brick into the simulation:
insert polyhedron=smallCube new.material=Oxide
struct bndfile=result
Figure 149 shows the result.
Figure 149 Oxide brick inserted in initial structure

11: Structure Generation
References
816 Sentaurus™ Process User Guide
H-2013.03
References
[1] J. A. Sethian, Level Set Methods and Fast Marching Methods: Evolving interfaces in
computational geometry, fluid mechanics, computer vision, and materials science,
Cambridge: Cambridge University Press, 1999.

Sentaurus™ Process User Guide 817
H-2013.03
CHAPTER 12 ICWBEV Plus Interface for
Layout-driven Simulations
This chapter presents strategies for using the IC WorkBench
EV Plus–TCAD Sentaurus interface.
Overview
The IC WorkBench EV Plus (ICWBEV Plus)–TCAD Sentaurus interface drives the TCAD
simulations from the GDSII or OASIS layout file provided by designers, which could be at any
level of integration in the hierarchy: full chip, test chip, or a single cell.
The TCAD simulation domain can be conveniently chosen using specific markups in the layout
file. A single process flow can be defined for all devices in the layout and can be applied easily
with minimal adjustments for 1D, 2D, and 3D simulation domains. For meshing, it provides
the unique feature of layout-driven meshing. Electrical contacts can be defined easily using
auxiliary masks.
This chapter includes the following sections:
■ICWBEV Plus Introduction for TCAD Users on page 818 provides basic ICWBEV Plus
training, especially with relevance to TCAD.
■Files Relevant to ICWBEV Plus–TCAD Sentaurus on page 828 introduces the relevant
files and file formats used in the ICWBEV Plus–TCAD Sentaurus interface. Specifically,
the Sentaurus markup file (*_mkp.mac) and TCAD layout file (*_lyt.mac).
■ICWBEV Plus Batch Mode and Macros on page 834 introduces working with macros and
running ICWBEV Plus in batch mode.
■TCAD Layout Reader of Sentaurus Process on page 835 presents the TCAD layout reader
of Sentaurus Process that provides a file-based interface between ICWBEV Plus and
Sentaurus Process.

12: ICWBEV Plus Interface for Layout-driven Simulations
ICWBEV Plus Introduction for TCAD Users
818 Sentaurus™ Process User Guide
H-2013.03
ICWBEV Plus Introduction for TCAD Users
Before discussing the ICWBEV Plus–TCAD Sentaurus interface, it is important to have an
understanding of ICWBEV Plus itself.
The general ICWBEV Plus training is a good starting point. Here, the focus is mainly on
ICWBEV Plus operations that are most relevant to TCAD Sentaurus users.
For details, refer to the ICWBEV Plus tutorials and manuals, which are accessed through the
main window of ICWBEV Plus (Help > Topics). The homepage of the online documentation
is displayed (see Figure 150).
Figure 150 Homepage of ICWBEV Plus online documentation
The first step consists of opening a layout file, which is typically in GDSII format.
Opening GDSII Layout Files
To open a GDSII or an OASIS layout file:
1. Set the environment variable ICWBEV_USER to activate the ICWBEV Plus Sentaurus User
Mode:
setenv ICWBEV_USER SENTAURUS
2. Launch ICWBEV Plus by typing:
icwbev
3. Select the file to be opened: File > Open > Browse for GDSII > Select GDSII > Open.
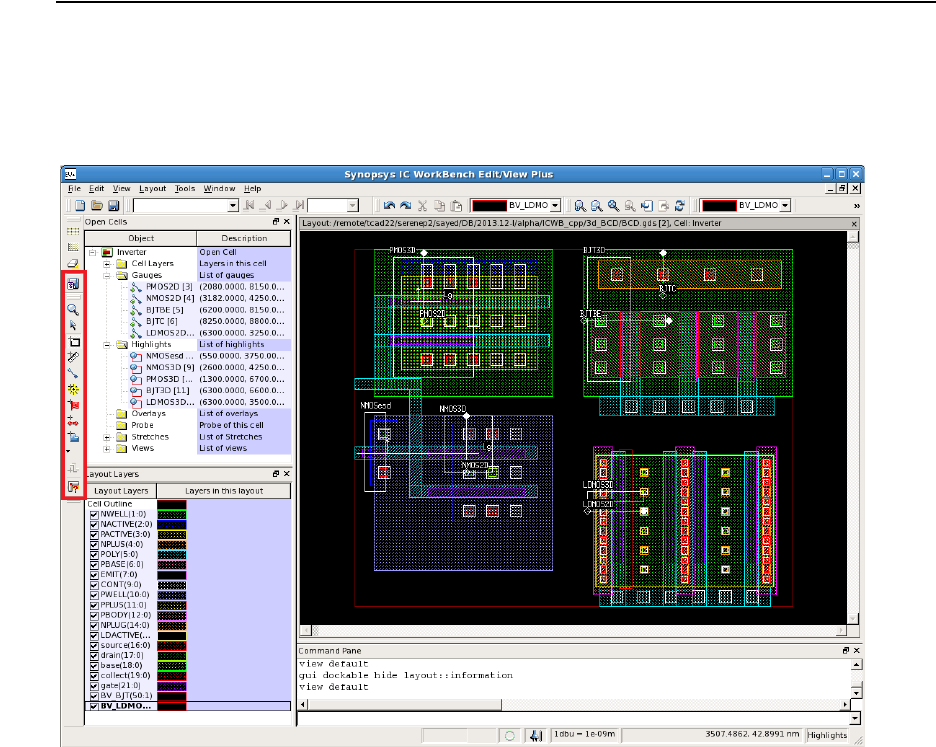
12: ICWBEV Plus Interface for Layout-driven Simulations
ICWBEV Plus Introduction for TCAD Users
Sentaurus™ Process User Guide 819
H-2013.03
NOTE Use ICWBEV Plus Version I-2013.12 or later. Earlier versions feature
a slightly different user interface. Refer to the Sentaurus™ Process
User Guide Version H-2013.03 for an introduction to the user interface
for earlier versions of ICWBEV Plus. For G-2012.06-SP5 versions of
ICWBEV Plus, use setenv ICWBEV_USER SENTAURUS_BETA to
activate the user interface described here.
Graphical User Interface of ICWBEV Plus
Figure 151 shows the graphical user interface (GUI) of ICWBEV Plus and illustrates the
typical layout of work, panes, and toolbars. The panes can be moved and reconfigured as
needed.
Figure 151 ICWBEV Plus main window, with TCAD Sentaurus–specific buttons (shown in
red box at left)
Figure 151 includes the following TCAD-relevant items in the GUI:
■The Open Cells pane contains details about layers and markups including the TCAD-
relevant markups.
■The Layout Layers pane shows the list of layers.
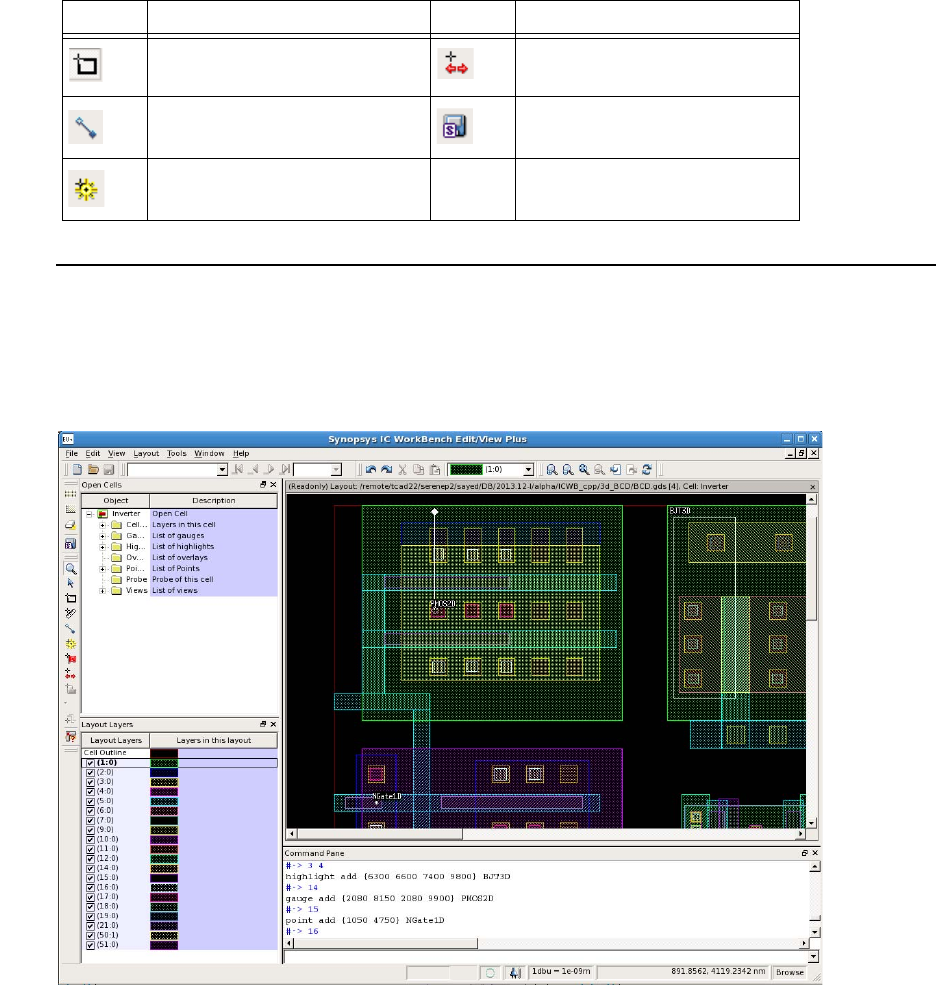
12: ICWBEV Plus Interface for Layout-driven Simulations
ICWBEV Plus Introduction for TCAD Users
820 Sentaurus™ Process User Guide
H-2013.03
■The Command Pane shows commands after GUI operations. Commands also can be
entered directly in this pane.
Table 73 describes the relevant toolbar buttons of the TCAD Sentaurus toolbar.
Sentaurus Markups
Sentaurus markups are used to add the simulation domain in 1D, 2D, and 3D domains as
needed. The Command Pane in Figure 152 shows the commands after adding Sentaurus
markups in the layout using GUI actions.
Figure 152 Adding Sentaurus markups to a layout
Table 73 TCAD Sentaurus–specific toolbar buttons
Button Description Button Description
3D simulation domain (highlight) Stretch utility
2D simulation domain (gauge) Save markups or save TCAD layout
1D simulation domain (point)
12: ICWBEV Plus Interface for Layout-driven Simulations
ICWBEV Plus Introduction for TCAD Users
Sentaurus™ Process User Guide 821
H-2013.03
To add a 3D simulation domain (highlight), a 2D simulation domain (gauge), or a 1D
simulation domain (point), click the respective toolbar button and draw a rectangle, a line, or a
point on the layout, respectively.
NOTE A 2D simulation domain (gauge) has a direction. The starting point is
given as an open diamond, and the endpoint is given as a filled diamond.
A gauge that runs parallel to an edge of a layer must have a finite
orthogonal distance to that edge. If a gauge is collinear with the edges
of a layer, this edge might not be included in the 2D mask.
The default naming convention is:
■For highlights, SIM3D<n>.
■For gauges, SIM2D<n>.
■For a point, SIM1D<n>.
where <n> is an automatically incremented number.
For 2D TCAD simulations, it can be useful to work with composite simulation domains, for
example, when the different contacts in a device layout cannot be connected by a single straight
line. In this case, it is not possible to perform a 2D device simulation after a 2D process step
using a single 2D simulation domain. However, a 2D TCAD simulation using a composite 2D
simulation domain is feasible. In this case, the various 2D cuts in the layout are daisy-chained
to form a composite 2D simulation domain.
An example of a composite simulation domain is shown in Figure 153, which shows a close-
up of the layout of a bipolar transistor with two 2D TCAD simulation domains. The simulation
domain labeled BJTBE cuts through two base–contact fingers and one emitter finger. The
simulation domain labeled BJTC cuts through the collector contact.
NOTE The two simulation domains are orthogonal and not contiguous.
Figure 153 Layout of a bipolar transistor with two 2D simulation domains: BJTBE cuts
through two base–contact fingers and one emitter finger, and BJTC cuts through
the collector contact
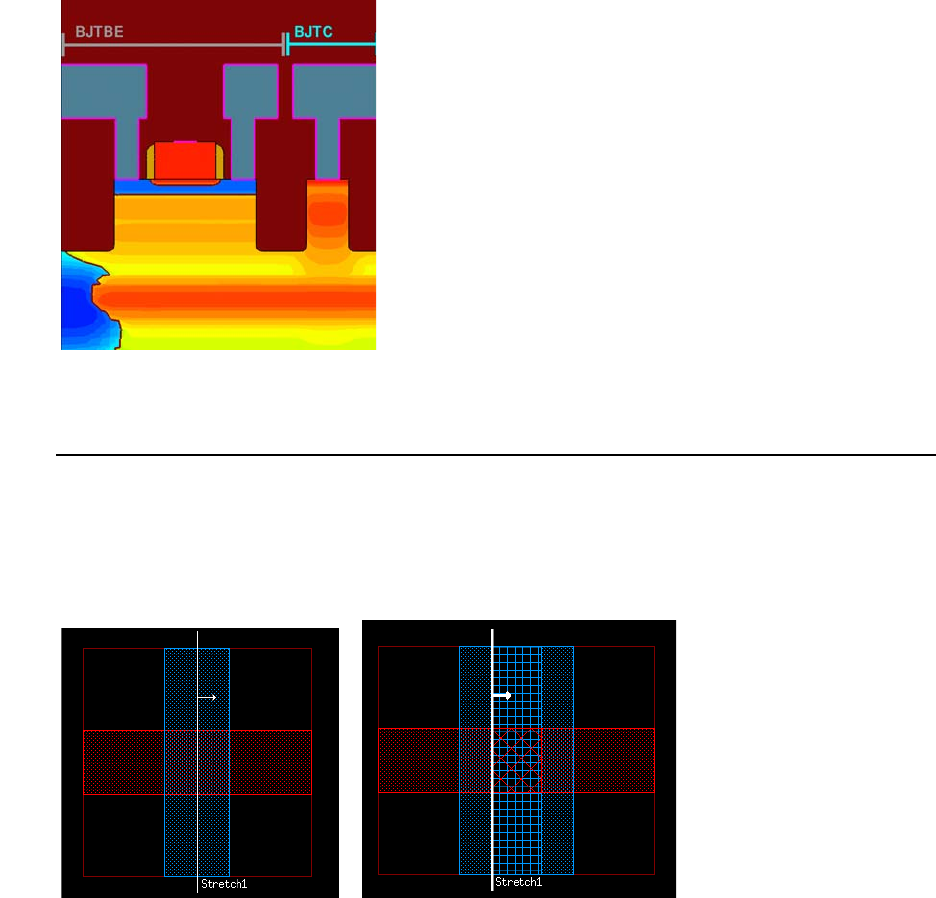
12: ICWBEV Plus Interface for Layout-driven Simulations
ICWBEV Plus Introduction for TCAD Users
822 Sentaurus™ Process User Guide
H-2013.03
Figure 154 shows the 2D TCAD simulation results obtained with a composite simulation
domain consisting of both the BJTBE and BJTC domains. Using a composite simulation
domain allows simulating a functional bipolar junction transistor (BJT) even for a 2D TCAD
simulation.
Figure 154 Two-dimensional TCAD simulation results using composite simulation domain
consisting of the 2D domains BJTBE and BJTC
Stretch Utility
The stretch utility provides a convenient way to parameterize a layout by inserting a uniformly
stretched segment into the layout. For example, this feature can be used to generate a set of
transistors that have different gate lengths but are otherwise identical.
Figure 155 (Left) Snapshot of sample ICWBEV Plus layout with stretch utility line and (right)
effective layout seen by Sentaurus Process when the layout is loaded with a
positive stretch amount
12: ICWBEV Plus Interface for Layout-driven Simulations
ICWBEV Plus Introduction for TCAD Users
Sentaurus™ Process User Guide 823
H-2013.03
A stretch line must be defined in ICWBEV Plus first. The stretch amount is set after loading
the TCAD layout with the Sentaurus Process command:
icwb stretch name= "<stretch-name>" value= <stretch-amount>
This feature can be used for simple parameterization of layouts for quantities such as threshold
rolloff. Figure 156 shows a close-up of the layout containing an NMOS transistor. In addition,
two stretch lines are shown. The stretch line labeled NMOS_W is used to vary gate width, and the
one labeled NMOS_L is used to vary the gate length in an NMOS.
To add a stretch line:
■Click the stretch utility toolbar button, and draw a line across the required region in the
layout.
NOTE The stretch line must cross the entire simulation domain to which it
should be applied. Stretch lines can be used for 2D and 3D simulation
domains.
Figure 156 Adding stretch lines in a layout to vary gate width and gate length
Figure 157 on page 824 shows the resulting changes in the NMOS gate width and gate length.
The default naming scheme for a stretch line is Stretch<n>.
For example, to apply a stretch at run-time in Sentaurus Process, use a command such as:
icwb stretch name= "NMOS_W" value=@Stretch@
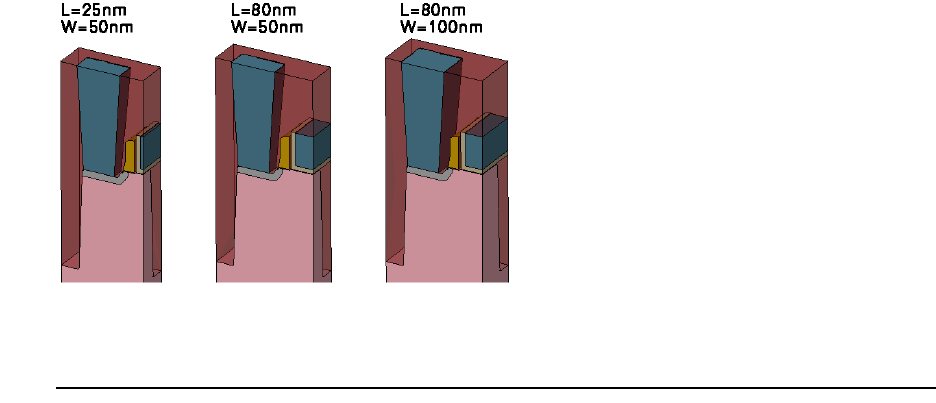
12: ICWBEV Plus Interface for Layout-driven Simulations
ICWBEV Plus Introduction for TCAD Users
824 Sentaurus™ Process User Guide
H-2013.03
where NMOS_W is the name of the stretch variable. Here, the amount of stretching is defined
using the Sentaurus Workbench variable @Stretch@. A positive stretch value is used for
expansion; a negative value leads to shrinkage.
Figure 157 Effect of stretch utility on 3D NMOS structure showing variation in width and
length
Renaming Markups
Markups can be renamed and edited.
To rename markups:
1. View > Views > Open Cells.
2. Expand the markup type, for example, Highlights.
3. Click the respective Sentaurus Process markup to edit the name.
4. Click the coordinates to edit the coordinate values.
Figure 158 on page 825 shows the Open Cells pane, displaying the list of Sentaurus Process
markups and their coordinates that can be edited as required.
12: ICWBEV Plus Interface for Layout-driven Simulations
ICWBEV Plus Introduction for TCAD Users
Sentaurus™ Process User Guide 825
H-2013.03
Figure 158 Open Cells pane showing list of objects with descriptions
Auxiliary Layers
Auxiliary layers are used, for example, to denote the position of electrical contacts in a layout.
To add auxiliary layers, first a layer must be declared and attributes must be defined. Figure 159
illustrates how to define a layer and its attributes.
Figure 159 Defining a layer and its attributes

12: ICWBEV Plus Interface for Layout-driven Simulations
ICWBEV Plus Introduction for TCAD Users
826 Sentaurus™ Process User Guide
H-2013.03
To add auxiliary layers, draw a polygon defining the region of the layer:
1. Open the layout for editing: Edit > Cell.
2. Select the active layer.
3. Select the shape tool.
4. Draw a polygon.
Editing Polygons
If required, polygons can be edited. You can edit polygons by either:
■Resizing a rectangle.
■Converting a rectangle to a polygon.
Resizing a Rectangle
To resize a rectangle:
1. If not already open, open the layout for editing: Edit > Cell.
2. Activate the selector tool.
3. Click the polygon edge to select it.
4. Move the edge as needed.
Figure 160 shows a rectangle highlighted for editing.
Figure 160 Moving the edge of a rectangle: select the rectangle and drag an edge

12: ICWBEV Plus Interface for Layout-driven Simulations
ICWBEV Plus Introduction for TCAD Users
Sentaurus™ Process User Guide 827
H-2013.03
Converting a Rectangle to a Polygon
To convert a rectangle to a polygon:
1. Click the polygon edge to select it.
2. Right-click and select Split Edge.
3. Move the edge as needed.
Figure 161 illustrates the procedure.
Figure 161 Converting a rectangle to a polygon
Nonaxis-aligned Simulation Domains
The ICWBEV Plus–TCAD Sentaurus interface supports nonaxis-aligned domains. To realize
nonaxis-aligned simulation domains, the GDSII layout is rotated by a given angle, and the
TCAD simulation domain is added as discussed in Sentaurus Markups on page 820.
To rotate a GDSII layout:
1. Layout > Transform.
2. In the Cell Transformation dialog box, enter the values of the fields as required.
Figure 162 on page 828 shows the transformation of a GDSII layout and the transformation
parameters.
12: ICWBEV Plus Interface for Layout-driven Simulations
Files Relevant to ICWBEV Plus–TCAD Sentaurus
828 Sentaurus™ Process User Guide
H-2013.03
Figure 162 Rotation of layout with transformation parameters
Files Relevant to ICWBEV Plus–TCAD Sentaurus
After adding Sentaurus Process markups in ICWBEV Plus, the markup information is saved in
two different files:
■Sentaurus markup file: This file format is based on the standard ICWBEV Plus macro
language. It can be used to reload and re-edit Sentaurus Process markup. It also contains a
reference to the original, potentially large, GDSII file.
■TCAD layout file: This file format is used as an internal file format for the exchange of
layout information between ICWBEV Plus and TCAD Sentaurus. It is based on the
standard ICWBEV Plus macro language. This file is flat and does not contain a reference
to the GDSII file. It is a small file because it contains only the parts of the layout needed
for TCAD Sentaurus.
After performing the necessary operations on the layout file, save the resulting Sentaurus
markup file.
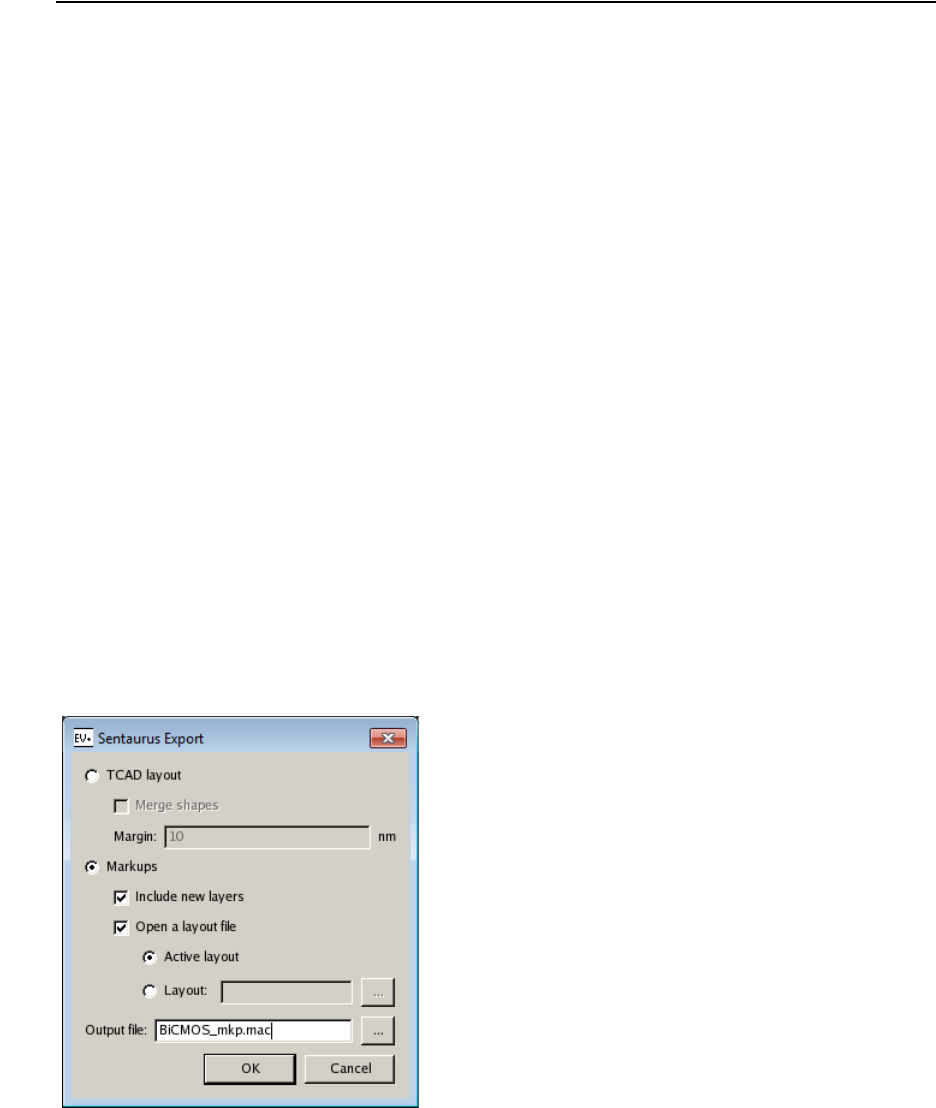
12: ICWBEV Plus Interface for Layout-driven Simulations
Files Relevant to ICWBEV Plus–TCAD Sentaurus
Sentaurus™ Process User Guide 829
H-2013.03
Saving the Sentaurus Markup File
To save a Sentaurus markup file:
1. Click the Save markups button (see Table 73 on page 820).
The Sentaurus Export dialog box is displayed (see Figure 163).
2. Select the Markups option.
3. If you have added auxiliary layers, which you do not want to store in the GDSII file, select
the Include new layers option.
NOTE Including new layers in the markup file keeps the original GDSII file
intact.
4. To open a specific layout file when reloading the markup file, select the Open a layout file
option:
a) Active layout: Select this option when using a centrally located GDSII layout. This
option is particularly useful when working with a very large full-chip layout.
b) Layout: Select this option when working with an edited or a local version of the GDSII
layout. Specify the name of the layout.
5. Type the file name in the Output file field.
The recommended extension for saving the file is _mkp.mac, for example,
BiCMOS_mkp.mac.
6. Click OK.
Figure 163 Sentaurus Export dialog box

12: ICWBEV Plus Interface for Layout-driven Simulations
Files Relevant to ICWBEV Plus–TCAD Sentaurus
830 Sentaurus™ Process User Guide
H-2013.03
The corresponding script command is:
sentaurus export markups <name>_lyt.mac [-newLayers]
[-active | -reference <gdsfilename>]
Contents of Sentaurus Markup File
This section describes a typical markup file with a brief explanation. For a description of
keywords, refer to the IC WorkBench EV Plus User Guide.
Version information:
# Sentaurus markups information - Wed Sep 11 15:11:17 2013
# Version - G-2012.06-SP5.355225 (Production)
Setting for treating self-intersection: By default, all layers are ORed. This convention is
expected by all subsequent tools and, therefore, this setting should not be altered.
default winding 1
Pointer to layout file:
layout open [<path>]/BICMOSinverter.gds Inverter
Global transformations:
cell transform 1 0 0 0 0
Layer declarations and display settings:
layer configure 1:0 -name NWELL -fill #00ff00 -pattern fill12-a
-outline #00ff00 -lineStyle solid -lineWidth 1
Open cell for editing (here, for adding new polygons):
cell edit_state 1
Auxiliary layers:
polygon -layer 20:0 {950 5150 950 5350 1150 5350 1150 5150}
...
Simulation domains:
point add {8900 8100} Emit1D
gauge add {2100 7900 2100 10000} PMOS
highlight add {7900 7550 9900 9900} BJT3D
...
12: ICWBEV Plus Interface for Layout-driven Simulations
Files Relevant to ICWBEV Plus–TCAD Sentaurus
Sentaurus™ Process User Guide 831
H-2013.03
File end:
select clear
catch {view default}
Reloading the Markup File
To edit a markup file, you must reload it.
To reload a markup file:
1. File > Open.
The Open File dialog box is displayed (see Figure 164).
2. In the Files of type field, select Flag Files (*.mac) or Macro Files (*.mac).
Figure 164 Open File dialog box for reloading the markup file
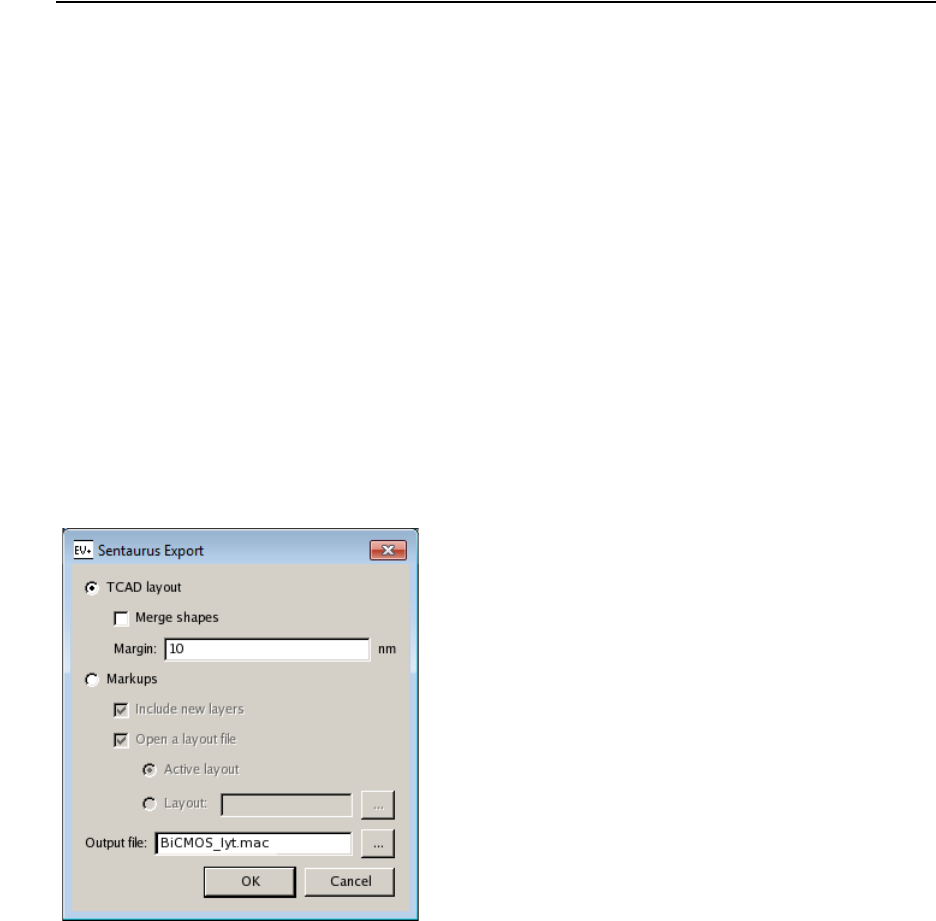
12: ICWBEV Plus Interface for Layout-driven Simulations
Files Relevant to ICWBEV Plus–TCAD Sentaurus
832 Sentaurus™ Process User Guide
H-2013.03
Saving the TCAD Layout File
To save the TCAD layout file:
1. Click the Save TCAD layout button (see Table 73 on page 820).
The Sentaurus Export dialog box is displayed (see Figure 165).
2. Select the TCAD layout option.
3. A layer in a layout can be defined by a large number of touching or overlapping polygons.
To merge all these polygons into a smaller number of possibly more complex polygons,
select the Merge shapes option.
4. For better viewing, layers are padded 10 units (nm). To change the padding value, edit the
Margin field.
5. Type the file name in the Output file field.
The recommended extension for the TCAD layout file is _lyt.mac, for example,
BiCMOS_lyt.mac.
6. Click OK.
Figure 165 Sentaurus Export dialog box, saving the TCAD layout file
The corresponding script command is:
sentaurus export layout <name>_lyt.mac [-merge | -noMerge] [-margin <number>]

12: ICWBEV Plus Interface for Layout-driven Simulations
Files Relevant to ICWBEV Plus–TCAD Sentaurus
Sentaurus™ Process User Guide 833
H-2013.03
Contents of TCAD Layout File
This section describes the contents of the TCAD layout file and the differences between the
contents of the Sentaurus markup file and TCAD layout file.
Version information: Same as Contents of Sentaurus Markup File on page 830.
Setting for treating self-intersection: Same as Contents of Sentaurus Markup File.
Pointer to layout file: Commented out.
Global transformations: Commented out.
Initialization of this self-contained layout:
layout new <cell name> -dbu 1e-09
Layer declarations and display settings: Same as Contents of Sentaurus Markup File.
File end: same as Contents of Sentaurus Markup File.
Simulation domains in a layout file are described below. Point, gauge, and highlight
coordinates are mentioned first, and all polygons associated with the given simulation domains
are listed:
■1D simulation domains:
point add {8900 8100} Emit1D
polygon -layer 1:0 {8890 8090 8910 8090 8910 8110 8890 8110}
...
■2D simulation domains:
gauge add {2100 7900 2100 10000} PMOS
rectangle -layer 1:0 {2090 10010 2110 7890}
...
■3D simulation domains:
highlight add {7900 7550 9900 9900} BJT3D
polygon -layer 1:0 {7890 7540 9910 7540 9910 9910 7890 9910}
...

12: ICWBEV Plus Interface for Layout-driven Simulations
ICWBEV Plus Batch Mode and Macros
834 Sentaurus™ Process User Guide
H-2013.03
Reloading the TCAD Layout File
For debugging purposes, reload the TCAD layout file.
To reload the file:
■File > Open > Macro File (*.mac).
NOTE Do not extract a TCAD layout from a reloaded TCAD layout. TCAD
layout files should always be extracted from the Sentaurus markup file.
ICWBEV Plus Batch Mode and Macros
Starting ICWBEV Plus in Batch Mode
To extract the TCAD layout file in batch mode, add the following command to the end of the
markup file:
sentaurus export layout <name>_lyt.mac [-merge | -noMerge] [-margin <number>]
exit
To start ICWBEV Plus in batch mode run the following command from the shell prompt:
> icwbev -nodisplay -run <name>_mkp.mac
ICWBEV Plus Macros
ICWBEV Plus macros can be used to create simple layouts. An example of a macro is:
default winding 1
layout new a -dbu 1e-09
cell transform 1.0 0.0 0 0.0 0.0
layer add 0:0
layer configure 0:0 -name {} -fill #ff0000
layer add 1:0
layer configure 1:0 -name {} -fill #ff0000
cell edit_state 1
polygon -layer 0:0 {0 0 0 100 50 100 50 0}
polygon -layer 1:0 {-25 25 75 25 75 75 -25 75}
select clear
catch {view default}
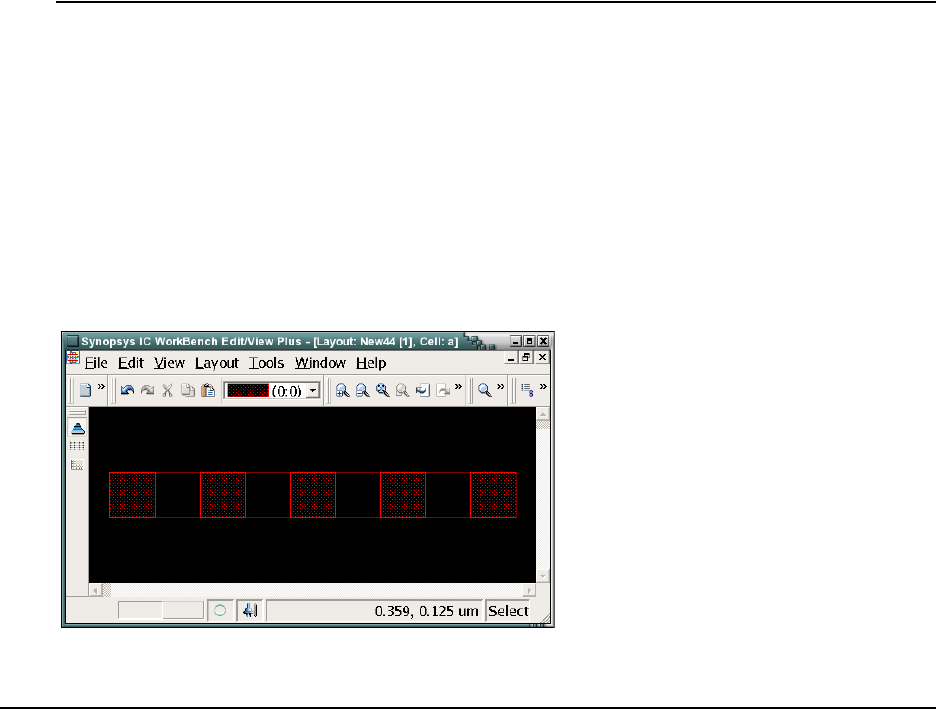
12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
Sentaurus™ Process User Guide 835
H-2013.03
Tcl-based Macros for Layout Parameterization
The macro language of ICWBEV Plus is Tcl based. Figure 166 shows a rectangle that has been
replicated four times. The following Tcl command performs the replication:
layout new a -dbu 1e-09
layer configure 0:0 -name {}
cell edit_state 1
set SHIFTS [list 0 100 200 300 400]
foreach SHIFT $SHIFTS {
eval rectangle -layer 0:0 { $SHIFT 0 [expr 50+$SHIFT] 50 }
}
Figure 166 Shift operation using macros
TCAD Layout Reader of Sentaurus Process
The TCAD layout reader of Sentaurus Process provides a file-based interface between
ICWBEV Plus and Sentaurus Process. Some of its key features include:
■Loading the TCAD layout (optional rescaling)
■Layout query functions
■Selecting a simulation domain
■Applying stretches
■Creating masks
■Mask-driven meshing
■Mask-driven contact assignment
The following sections discuss these features in detail.

12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
836 Sentaurus™ Process User Guide
H-2013.03
Loading the TCAD Layout
To load a TCAD layout in Sentaurus Process, use the following command:
icwb filename= "<filename.mac>" [scale=<scale>]
Coordinates found in the TCAD layout file are multiplied by the value of the optional
parameter scale as the file is read.
For example, to load the TCAD layout file BiCMOS_lyt.mac and apply a rescaling factor of
to convert the ICWBEVPlus default unit of nanometer to the Sentaurus Process
default unit of micrometer:
icwb filename= "BiCMOS_lyt.mac" scale=1e-3
Finding Simulation Domains
To generate a list of the simulation domains:
icwb list domains
For example:
set Domains [icwb list domains]
-> icwb: Domains -> Emit1D NBODY NMOS BJT3D PMOS3D
Finding Layer Names and Layer IDs
Each layer in the TCAD layout file has a unique ID of the form <int>:<int>, for example
3:0. A layer also can have an optional explicit layer name such as NWELL. If no explicit layer
name has been set in ICWBEV Plus, the TCAD layout reader uses the layer ID as the default
layer name. The TCAD layout reader refers to layers always by the layer name.
To find the layer names:
icwb list layerNames
For example:
set LNames [icwb list layerNames]
-> icwb: LNames -> NWELL NPDIFF POLY EMIT METAL CONT ndrain ngate nsource base
emitter collect
11000⁄

12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
Sentaurus™ Process User Guide 837
H-2013.03
To find the layer IDs:
icwb list layerIDs
For example:
set LIDs [icwb list layerIDs]
-> icwb: LIDs -> 1:0 2:0 3:0 4:0 5:0 6:0 7:0 8:0
Selecting the Simulation Domain
To select a single or a composite simulation domain:
icwb domain = <domain-name> | <list-of-2d-domain-names>
For example, to select a single simulation domain (which can be 1D, 2D, or 3D):
icwb domain = { PMOS }
To define a composite simulation in 2D:
icwb domain = { NBODY NMOS PMOS BJTBE BJTC }
Loading a GDSII Layout
To load a GDSII layout directly in Sentaurus Process, use the command:
icwb gds.file=<c> cell=<c> layer.names= {<list>} layer.numbers= {<list>}
sim2d | sim3d= {<n>} [domain.name=<c>] [scale= <n>]
[stretches= {<c>= {<n>}}]
For example:
icwb gds.file= BCD.gds cell= Inverter \
layer.numbers= "1:0 2:0 3:0 4:0 5:0 6:0 7:0 8:0" \
layer.names= "NWELL NACTIVE PACTIVE NPLUS POLY PBASE EMIT CONT" \
sim3d = "6300 3500 7750 3750" \
stretches= {lgate= {7025 3400 7025 3850}} scale= 1e-3
The domain.name argument defines the name of the simulation domain. If no name is
specified, SIM3D is used for a 3D domain, and SIM2D is used for a 2D domain.
The domain will be set to be the current domain automatically, so you do not need to call
icwb domain=<c> before using other icwb commands.

12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
838 Sentaurus™ Process User Guide
H-2013.03
NOTE The simulation domain and the stretches are defined using layout
coordinates.
This option does not support composite simulation domains.
This option does not require access to ICWBEV Plus.
Finding Domain Dimensions
To find the domain dimensions:
icwb dimension
This command returns 3 for 3D simulation domains (highlight), 2 for 2D simulation domains
(gauge), and 1 for 1D simulation domains (point).
For example:
set DIM [icwb dimension]
-> icwb: dimension -> 3
Finding Bounding Box of Domain
To find the coordinates of the bounding box of the simulation domain in the global layout
coordinates:
icwb bbox xmin | xmax | ymin | ymax
For example:
set LXmin [icwb bbox xmin] ; set LXmax [icwb bbox xmax]
set LYmin [icwb bbox ymin] ; set LYmax [icwb bbox ymax]
-> icwb: Layout Bounding Box -> 7.9 9.9 7.55 9.9
To find the coordinates of the bounding box that automatically recenters the simulation domain
to start at the origin:
icwb bbox left | right | back | front
For example:
set Ymin [icwb bbox left] ; set Ymax [icwb bbox right]
set Zmin [icwb bbox back] ; set Zmax [icwb bbox front]
-
> icwb: Centered Bounding Box -> 0 2.35 0 2
NOTE Sentaurus Process works with the centered coordinates.

12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
Sentaurus™ Process User Guide 839
H-2013.03
Interface with line Commands
After storing the bounding box of the simulation domain in the Tcl variables such as Ymin,
Ymax, Zmin, and Zmax, these variables can be used in line commands to define the initial
substrate and mesh in Sentaurus Process. For example:
if { $DIM == 3 } {
line y location= $Ymin spacing=100.0 tag=left
line y location= $Ymax spacing=100.0 tag=right
line z location= $Zmin spacing=100.0 tag=back
line z location= $Zmax spacing=100.0 tag=front
set Ydim "ylo=left yhi=right"
set Zdim "zlo=back zhi=front"
} elseif { $DIM == 2 } {
line y location= $Ymin spacing=100.0 tag=left
line y location= $Ymax spacing=100.0 tag=right
set Ydim "ylo=left yhi=right"
set Zdim ""
} else {
line y location=-0.5 spacing=100.0 tag=left
line y location= 0.5 spacing=100.0 tag=right
set Ydim "ylo=left yhi=right"
set Zdim ""
}
eval region silicon xlo=top xhi=bot $Ydim $Zdim
Creating Masks
To create a mask from a layer:
icwb.create.mask layer.name= (<string>|<string list>)
[name= <string>] [polarity= (positive|negative)] [info=<n>]
[shift= {dy dz}] [stretchypos= {yo dy}] [stretchyneg= {yo dy}]
[stretchzpos= {zo dz}] [stretchzneg= {zo dz}]
Masks can be created in the following ways:
■Mask name defaults to the layer name:
icwb.create.mask layer.name= POLY
■Give an explicit name to the mask. For example, to distinguish between the positive mask
and negative counterparts:
icwb.create.mask layer.name= NWELL name= NWELL polarity= positive
icwb.create.mask layer.name= NWELL name= NOTNWELL polarity= negative
12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
840 Sentaurus™ Process User Guide
H-2013.03
Several layers can be ORed to create a single mask. The following command illustrates the OR
procedure:
icwb.create.mask layer.name= "NPDIFF PPDIFF NPLUG PBASE" name= STI info=1
The info flag directs more detailed information about the mask creation process to the log file.
To automatically create mask layers with both polarities, use the following macro:
icwb.create.all.masks
The resulting mask names are <layername>_p for the positive version and <layername>_n
for the negative version.
The optional keywords shift, stretchypos, stretchyneg, stretchzpos, and
stretchzneg allow you to modify individual layers during the mask generation with
icwb.create.mask. For example, to generate a mask that corresponds to layer 1:0 shifted
by along the y-direction and by along the z-direction, use:
icwb.create.mask layer.name= 1:0 shift= {0.25 -0.1}
For 2D simulation domains, the z-shift can be omitted.
The keywords starting with the word stretch allow you to stretch individual layers in a
manner similar to the icwb stretch command. (The latter, however, is applied to all layers
and takes the location of the stretch from the TCAD layout file.) The remaining part of the
keyword determines if the stretch is applied along the y- or z-direction, and if the layer is
stretched to the positive or negative side of the stretch position. For example, to move the
vertices of layer 1:0, which have a y-coordinate less than 1.2 by –0.25 µm, use:
icwb.create.mask layer.name= 1:0 stretchyneg= {1.2 -0.25}
This command operates on layer vertex coordinates and does not check if the resulting polygon
is valid. When using these commands to shrink layers, you must ensure that the resulting
polygons are still well defined, for example, not self intersecting.
More than one shift and stretch keyword can be used in an icwb.create.mask
command. As these operations may not be commutative, you must note the order in which
these operations are applied if more than one is used. First, the shift is applied, and then
stretchypos, stretchyneg, stretchzpos, and finally stretchzneg are applied.
NOTE This order is hard coded and not influenced by the order the keywords
appear on the command line.
0.25 µm
0.1–µm
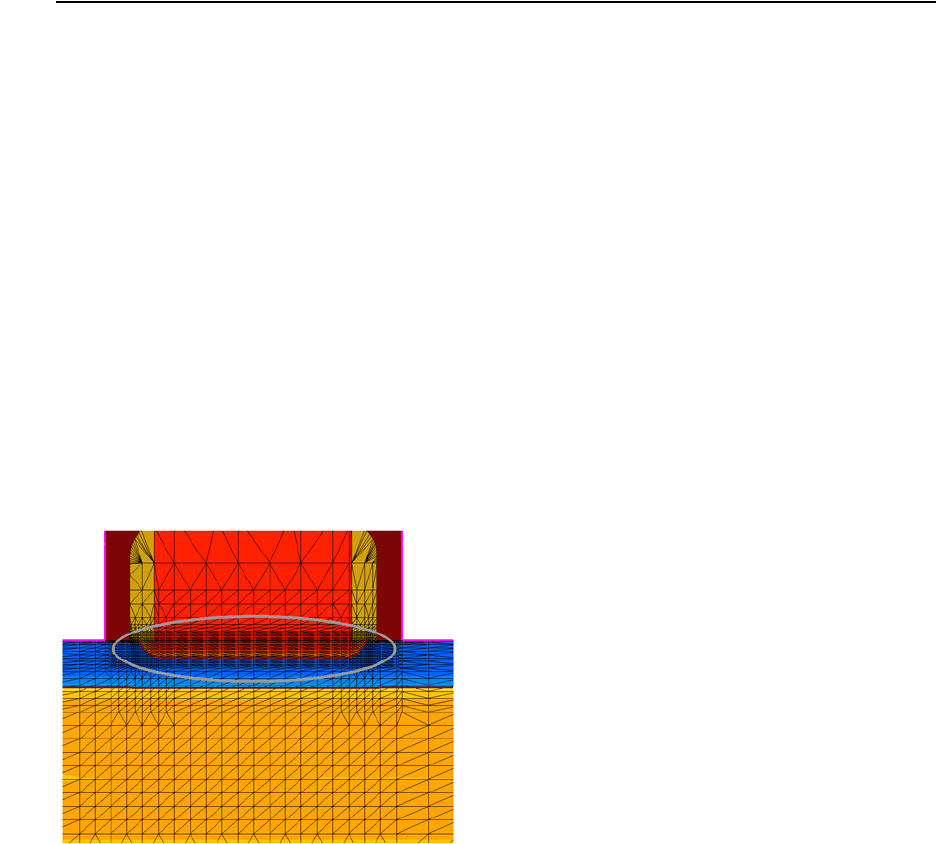
12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
Sentaurus™ Process User Guide 841
H-2013.03
NOTE If you have a large layout with masks containing many polygons, it can
take some time until the Tcl function icwb.create.all.masks
parses and creates the masks. In this situation, you can use the command
icwb create.all.masks to create all the masks (see icwb on
page 951). This command works like the Tcl version but creates the
masks much faster.
Layout-driven Meshing
To create a refinement box that is tied to layers in the ICWB TCAD layout file:
icwb.refine.mask layer.name= (<string> | <string list>) [name= <string>]
[oversize=<n>] xtop=<n> xbot=<n> <other options> [info=<n>]
Layout-driven meshing can be particularly useful when meshing in critical regions, such as the
channel and emitter areas of BiCMOS devices. The following example illustrates the use of the
POLY layer for meshing placement:
icwb.refine.mask name= UnderPoly layer.name= POLY oversize= 0.1 \
xtop= -1.51 xbot= -1.35 info= xrefine= 0.02 yrefine= 0.02
icwb.refine.mask name= SiOxPo layer.name= POLY oversize= 0.1 \
xtop= -1.51 xbot= -1.35 min.normal.size= 0.005 \
interface.mat.pairs= {Silicon Oxide Silicon Polysilicon}
The oversize parameter gives the option to mesh in areas wider than the layer. Figure 167
and Figure 168 on page 842 demonstrate the use of the oversize parameter in the emitter
region of a BJT and the channel region of an NMOS, respectively.
Figure 167 Meshing in emitter region of BJT; the oversize parameter is set to 0.1 µm
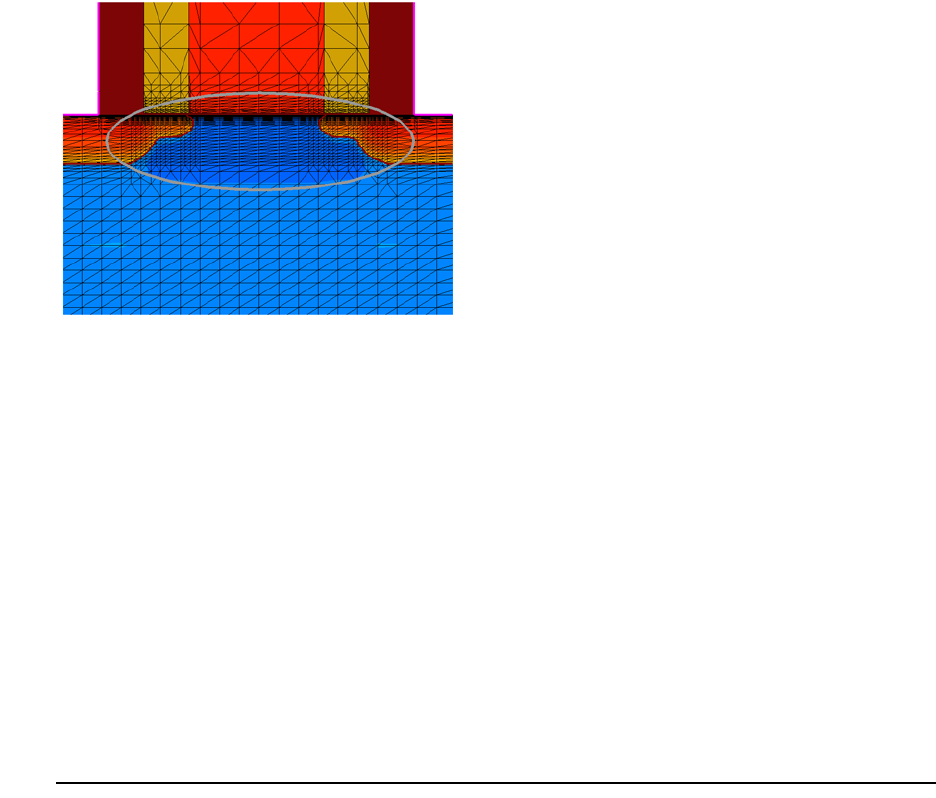
12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
842 Sentaurus™ Process User Guide
H-2013.03
Figure 168 Meshing in channel region of NMOS; the oversize parameter is set to 0.1 µm
The utility command icwb.refine.mask interfaces with the standard refinebox command
by providing information about the refinement extent (minimum and maximum) based on the
selected layer.
NOTE The extent of the refinement box in the primary direction must be given
explicitly with the keywords xtop and xbot. Any other commands are
passed on.
Layout-driven mesh refinements are applied under the layer itself. They
cannot be applied under the inverse of a layer. Consider defining
auxiliary layers in ICWBEV Plus to facilitate layout-driven meshing in
areas that do not coincide with an existing layer.
This feature supports only layers with axis-aligned edges. Slanted edges
may result in a large number of refinement boxes, which may not
appropriately represent the original shape.
Layout-driven Contact Assignment
The icwb.contact.mask command creates contacts for subsequent device simulations that
are tied to a layer in the TCAD layout file. The command serves as an interface between the
TCAD layout and the Sentaurus Process contact command by automatically obtaining the
lateral placement of the contact from the specified layout layer, taking the vertical placement
from the argument list and passing all other options directly to the contact command. The
syntax of the command is:
icwb.contact.mask layer.name= (<string> | <string list>)
[name=<string>] <other options> [info=<n>]
12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
Sentaurus™ Process User Guide 843
H-2013.03
The command supports both box and point contact types:
■A box-type contact consists of elements at the surface of one region or material inside the
box. The lateral extent of the box is determined automatically from the layer segment (2D)
or the polygon bounding box (3D), while the vertical extent is taken from the contact
command keywords xlo and xhi.
■A point-type contact contains all the boundary elements of one region. The lateral position
of the point is determined automatically as the midpoint of the layer segment (2D) or the
polygon bounding box (3D), while the vertical position is taken from the contact
command keyword x.
Often, there is no layer in the layout provided by designers that can be used readily for the
creation of contacts. In this case, add auxiliary layers in ICWBEV Plus to be used as markups
for device contacts.
The following example demonstrates the assignment of gate and drain contacts using layout-
driven contact assignment:
icwb.contact.mask layer.name= ndrain name= drain point aluminum \
replace x= -2.0
icwb.contact.mask layer.name= ngate name= gate box polysilicon \
adjacent.material=oxide xlo= -2.05 xhi= -1.95
The location in the primary direction must be given explicitly with either the keyword x for a
point-type contact, or with the keywords xlo and xhi for a box-type contact. Any other
command is passed on to the contact command. The keyword name is optional. If no name
is given, the name of the layer is used as the contact name.
Figure 169 on page 844 shows a layout on which auxiliary layers have been added for layout-
driven contact assignment. Figure 170 on page 844 shows the 2D boundary after the process
simulation with Sentaurus Process depicting the gate, drain, and source contacts.
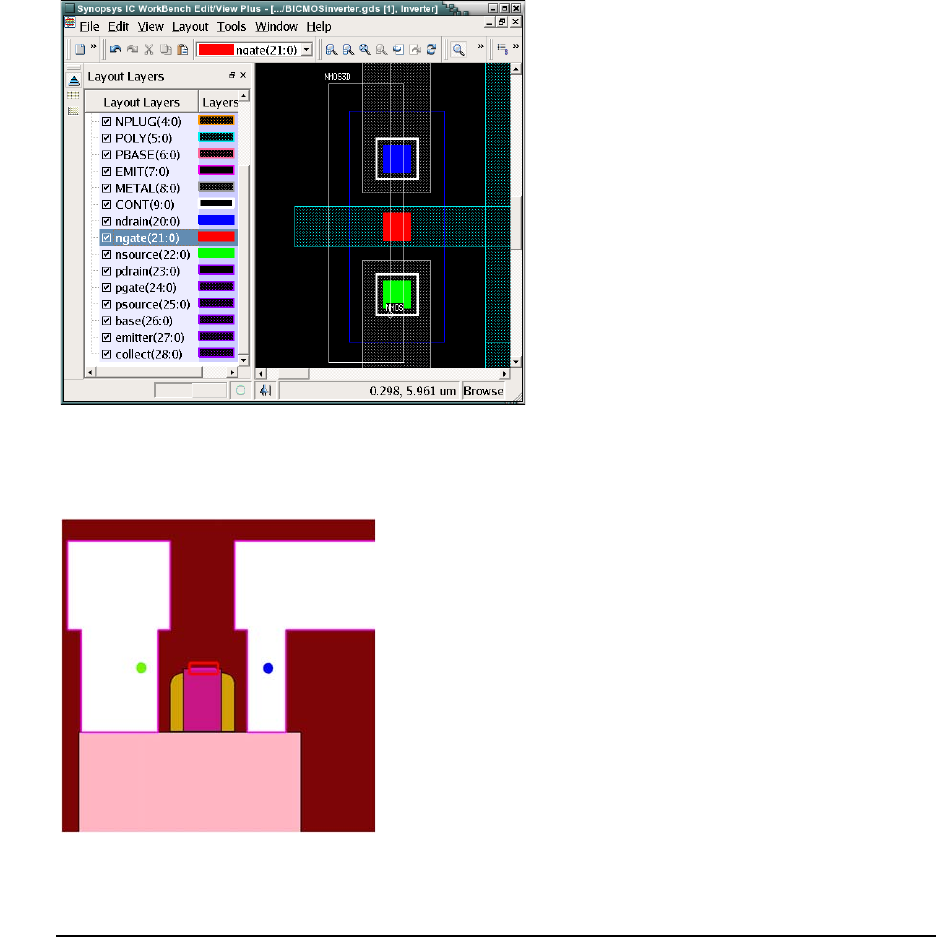
12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
844 Sentaurus™ Process User Guide
H-2013.03
Figure 169 Auxiliary layers added for gate, source, and drain contacts are represented by
rectangles of solid color in the layout
Figure 170 Final boundary after TCAD simulation showing gate (red), drain (blue), and
source (green)
Aligning Wafer and Simulation Domain
To correctly support tilted process steps for 2D and 3D simulation domains, the alignment
between the wafer and the simulation domain must be declared using the slice.angle
keyword in the init command.
12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
Sentaurus™ Process User Guide 845
H-2013.03
The TCAD layout reader command icwb slice.angle.offset returns the relative angle
of the active simulation domain so that the slice angle can be adjusted as needed.
Table 74 lists the returned slice.angle offset values for:
■A 3D simulation domain (SIM3D1).
■A 2D domain along the layout x-axis extending from left to right (SIM2DXLR), right to left
(SIM2DXRL), along the layout y-axis from top to bottom (SIM2DYTD) and bottom to top
(SIM2DYDT).
The following commands realize a tilted process:
set SliceAngle -90
set SliceOffset [icwb slice.angle.offset]
init silicon field=Boron concentration=1e13 \
slice.angle= [expr $SliceAngle+$SliceOffset]
implant phosphorus dose=4e12<cm-2> energy=100<keV> tilt=30 rot=0
Figure 171 shows a sample layout with a 3D simulation domain and the four 2D simulation
domains previously discussed. Figure 172 on page 846 and Figure 173 on page 846 show the
dopant profiles after the tilted implants for the different simulation domains.
Figure 171 Structure layout where implant is performed at highlighted L-shaped red region
Ta bl e 7 4 S l i c e a ng le o ff s et va lu es fo r di ffe r e n t d om ai ns
Domain SliceOffset value
SIM3D1 90
SIM2DXLR 0
SIM2DXRL 180
SIM2DYDT 90
SIM2DYTD –90
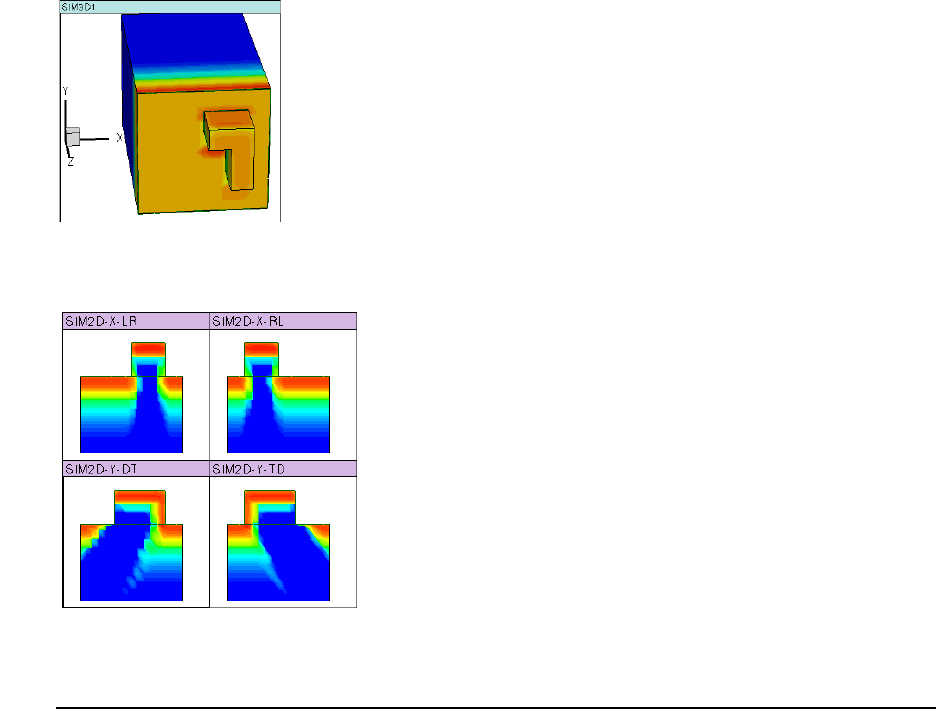
12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
846 Sentaurus™ Process User Guide
H-2013.03
Figure 172 Three-dimensional implanted profile
Figure 173 Two-dimensional implanted profile for selected slice angles as mentioned in
Tab l e 74 o n pa g e 84 5
Additional Query Functions
The TCAD layout reader of Sentaurus Process provides additional layout query functions. For
example, the following command returns a list of segments in the given layer for a 2D
simulation domain:
set Segments [icwb list.segments layer.name= "<layer-name>"]
For a 3D simulation domain, the following command returns a list containing the bounding
boxes for all polygons in the given layer (this command also can be used for 2D):
set PolyBBoxes [icwb list polygon.bounding.boxes layer.name= "<layer-name>"]
For a 3D simulation domain, the following command returns a list containing a tessellated
representation of polygons in the given layer (this command also can be used for 2D):
set PolyTessel [icwb list polygon.tessellations layer.name= "<layer-name>"]
12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
Sentaurus™ Process User Guide 847
H-2013.03
Figure 174 Sample layout containing two polygons
For example, Figure 174 shows a simple layout containing two polygons in layer 0:0. The
following commands:
■Load the TCAD layout file (here, called ORG_lyt.mac).
■Select the 2D simulation domain SIM2D1.
■Query the segment, the bounding boxes, and the tessellations:
icwb filename= "ORG_lyt.mac"
icwb domain= "SIM2D1"
set Segments [icwb list.segments layer.name= "0:0"]
LogFile "Segments $Segments"
# -> Segments 100 200 300 400 500 800
set BBoxes [icwb list polygon.bounding.boxes layer.name= "0:0"]
LogFile "BBoxes: $BBoxes"
# -> BBoxes: {100 0 200 0} {300 0 400 0} {500 0 800 0}
set Tessellations [icwb list polygon.tessellations layer.name= "0:0"]
LogFile "Tessellations: $Tessellations"
# -> Tessellations: {100 0 200 0} {300 0 400 0} {500 0 800 0}
NOTE The bounding box and tessellation queries are supported for 2D, and
they return flat rectangles. The returned y-values are the same as for the
segment query; however, zeros are padded for the z-direction.
When loading the 3D simulation domain SIM3D2, the set of rectangles returned by the
polygon.bounding.boxes query and the polygon.tessellations query are different:
icwb filename= "ORG_lyt.mac"
icwb domain= "SIM3D2"
set BBoxes [icwb list polygon.bounding.boxes layer.name= "0:0"]
LogFile "BBoxes: $BBoxes"
# -> BBoxes: {100 100 300 400} {100 500 300 800}
set Tessellations [icwb list polygon.tessellations layer.name= "0:0"]
LogFile "Tessellations: $Tessellations"
# -> Tessellations: {100 100 150 200} {100 200 150 300} {100 300 150 400}
12: ICWBEV Plus Interface for Layout-driven Simulations
TCAD Layout Reader of Sentaurus Process
848 Sentaurus™ Process User Guide
H-2013.03
# {100 500 150 600} {100 700 150 800} {150 100 300 200} {150 300 300 400}
# {150 500 300 600} {150 600 300 700} {150 700 300 800}
The polygon.bounding.boxes query returns the bounding box rectangle for each polygon
in the layer, while the polygon.tessellations query breaks each polygon into a set of
rectangles and then returns these rectangles. The set of rectangles covers the same area as the
original polygon, while the bounding box rectangles may cover a larger area.
Figure 175 shows the rectangles returned by the two query functions as an ‘effective/
equivalent’ layout for better comparison with the original layout shown in Figure 174 on
page 847.
NOTE The tessellation procedure supports only polygons with axis-aligned
edges.
Figure 175 Set of rectangles returned by (left) polygon.bounding.boxes query and (right)
polygon.tessellations query for polygons shown in Figure 174

Sentaurus™ Process User Guide 849
H-2013.03
CHAPTER 13 Extracting Results
This chapter presents strategies for analyzing simulation results.
Overview
This chapter covers basic tasks such as obtaining a list of materials currently in the structure,
and obtaining 1D dopant profiles from 2D or 3D structures to more complex ones, such as
looping through all materials and extracting Pearson parameters for each material. The
following commands perform these tasks: select, slice, layers, interface,
interpolate, print.1d, FitPearson, mater, FitPearsonFloor, FitArrhenius, and
FitLine.
All these Sentaurus Process commands are built-in procedures designed to work with the tool
command language (Tcl). These commands allow you to take full advantage of the
programmability of the Sentaurus Process input language and provide a powerful framework
for performing complex customized tasks.
These commands return or accept a Tcl list to perform their respective functions. The Tcl list
can be viewed and processed by the user, passed to another function, written to a file, or read
from a file. For example, the slice command returns a Tcl list of pairs where the value
gives the depth and the value gives the value chosen with the select command. This
list can be viewed with the Tcl puts command, written to a file with the Tcl open and puts
commands, or processed with another command such as fitting a Pearson function to the profile
with the FitPearson command.
An understanding of basic Tcl commands and Tcl lists is helpful to utilize fully the flexibility
of these commands. For convenience, some basic aspects of Tcl are described to enable you to
work efficiently with these commands, and examples of basic results analysis are provided.
Saving Data Fields
Sentaurus Process automatically saves all solutions, active dopant concentrations, total dopant
concentrations, electrostatic potential, net active concentrations, point-defect concentrations,
and total point-defect concentrations along with mechanical stress results in a TDR file. The
total and active concentrations of dopants and the total concentration of defects are created only
after a diffusion step. Therefore, if there is no diffusion, some of these fields may be missing
xy
x
µm[]
y

13: Extracting Results
Selecting Fields for Viewing or Analysis
850 Sentaurus™ Process User Guide
H-2013.03
in the TDR file. You can control the fields that will be saved in the TDR file using the
SetDFISEList command (see SetDFISEList on page 1122).
Since the active and total dopant concentrations are defined as terms in Sentaurus Process, they
are converted automatically to data fields with the same name when saved to TDR files.
Because the conversion is handled internally, the data fields are not kept in the memory.You
can create new data fields and store them in a TDR file. For example:
sel z= "BActive/BTotal" name=BActiveRatio store
will divide the active boron concentration by the total boron concentration and save the results
in a data field called BActiveRatio. The parameter store ensures that the newly created
field will be saved in a TDR file. To see whether a field will be saved in a TDR file, the select
command with the option permanent can be used (see select on page 1117), for example:
sel name= BActiveRatio permanent
will return 1 if the field will be saved in a TDR file. Otherwise, it will return 0.
Selecting Fields for Viewing or Analysis
Most analytic tasks begin with the select command, which is used to select a data field to be
viewed or operated on (see select on page 1117). A data field in Sentaurus Process is a quantity
that varies over the simulation domain, such as dopant concentration or electrostatic potential
distribution. The value of the data field is defined with an Alagator expression and is set with
z parameter of the select command.
The expression can be simply the name of a solution variable (such as Boron, H2O, or
Stress_x) or it can be a complex expression depending on what is required. If the expression
is the simple name of an existing data field, the select command selects this data field.
If it is more complex expression, the select command creates a corresponding data field and
then selects it, for example:
select z= BTotal ;# Select the total boron concentration
# term
select z= "Arsenic+Phosphorus-Boron" ;# Create and select a data field using
# solution variables
select z= log10(BActive) ;# Create and select a data field from
# the active boron concentration
The list of available data fields can be retrieved by using select list. The name that can
appear in the expression of the z parameter can be either a data field or a term.

13: Extracting Results
Obtaining 1D Data Cuts
Sentaurus™ Process User Guide 851
H-2013.03
A term is defined with the term command and is also an expression containing solution
variables, data fields, constants, and so on (see term on page 1173). Numerous terms are
created automatically in the diffuse command (see diffuse on page 908) and any of these
terms can be selected.
When a data field is selected (or created and selected) with the select command, the data field
can be viewed or operated on. The following commands can operate on the selected field:
slice, layers, interpolate, print.data, print.1d, and plot.1d.
Obtaining 1D Data Cuts
After a select command has been issued, you can obtain 1D cuts through the data along one
of the principal axes using the slice or print.1d command. The slice command returns
a list of coordinate data pairs. To make a cut perpendicular to x, specify the x parameter,
similarly for y and z.
The print.1d command returns a list of data-point lists. Each data-point list contains the
coordinate, data value, and the material name at that coordinate. Again, a cut perpendicular to
x is made by specifying the xparameter and, similarly, for the y and z cuts.
The plot.1d command can be used to view profiles with a temporary X11 graphics tool.
Examples
Sentaurus Process can run in interactive mode if there is no command file given on the
command line. In this case, you are prompted with the sprocess> prompt for commands. If
a command file is given, commands are read from this file. In interactive mode, the return value
of the commands is always displayed. You can set variables to the return value of a command
by using the syntax:
set var [command]
In this case, command is executed and a Tcl variable var is created if it does not already exist,
and the value of var is set to the return value of command. In addition, the return value of
command is displayed. It is also possible to write the return value to a user-defined file. The
following examples demonstrate the differences and functionality of the slice and print.1d
commands:
sprocess> select z=Vacancy
sprocess> slice y=0.6
{-1.000000e-02 4.804720e+16
-9.340278e-03 5.869015e+16
13: Extracting Results
Examples
852 Sentaurus™ Process User Guide
H-2013.03
...
0.000000e+00 5.969905e+17
0.000000e+00 7.075867e+17
7.421875e-04 7.618894e+17
...
sprocess> print.1d y=0.6
{ Distance Value Material }
{ -1.00000e-02 4.80472e+16 Oxide }
{ -9.34028e-03 5.86902e+16 Oxide }
...
{ 0.00000e+00 5.96991e+17 Oxide }
{ 0.00000e+00 7.07587e+17 Silicon }
{ 7.42188e-04 7.61889e+17 Silicon }
...
Here, the slice command returns raw coordinate data pairs, whereas print.1d returns a
header and coordinate data–material triplets. In both cases, the coordinates are given in
micrometers and the concentration is in .
To illustrate how data from these functions can be manipulated with Tcl, suppose you require
a 1D profile of Vacancy, which starts with 0.0 as the first coordinate, and the Vacancy
concentration to be in .
First, create a Tcl list from the data returned by the slice, and convert data in that list to a new
list, such as:
set myList[lindex [slice y=0.6] 0] ;# Create a new list from slice
# command called myList
set offset [lindex myList 0] ;# Grab the offset, that is, the
# first coordinate
list modList ;# Create new Tcl list where modified
# data will reside
foreach { coord data } $myList {\
lappend modList [expr $coord-$offset] ;#Convert coordinate by subtracting
# the offset and append to modlist
lappend modList [expr $data*1.0e-12] ;# Convert data to um^-3 units and
# append to modlist
}
The above example uses the following Tcl commands;
■lindex retrieves a given element of a list.
■list creates a list.
■lappend appends an element to the end of a list.
■expr evaluates a math expression.
■foreach is used for looping.
cm 3–
µm3–

13: Extracting Results
Determining the Dose: Layers
Sentaurus™ Process User Guide 853
H-2013.03
For example, to write modList to a file called xy.dat:
set fileID [open xy.dat w] ;# Use the Tcl open command
# to open a file for writing
foreach { x y } $modList { puts $fileID "$x $y" } ;# Write modList line by line
close $fileID
Determining the Dose: Layers
The layers command (see layers on page 1008) computes the dose of the selected data field
along one of the principal axes. The syntax to specify the cut is the same as the slice
command (see slice on page 1141). As with the other commands, the information is returned
as a list of lists:
sprocess> sel z=Vacancy
sprocess> set layerInfo [layers y=0.5]; # For a 2d structure,
either x or y must be specified
{ Top Bottom Integral Material}
{-2.06000e-01 -6.00000e-03 9.98843e+14 Silicon}
{-6.00000e-03 0.00000e+00 3.97970e+09e Oxide)
{ 0.00000e+00 1.00000e+00 2.81858e+05 PolySilicon}
The top and bottom coordinates are in micrometers. To obtain the total integrated dose along
y=0.5, use:
sprocess> set total 0
0
# Loop over layerInfo list of lists skipping header list,
# and retrieve the 3rd element of each list (first element has 0 index)
# which corresponds to the Integral for that layer.
sprocess> for { set i 1 } { $i < [llength $layerInfo] } { incr i } {
> set total [expr $total + [lindex [lindex $layerInfo $i] 2]]
>}
sprocess> puts $total
9.991288377e+14
sprocess>
In addition to the Tcl commands used in the previous section, this example uses the following:
■llength returns the size of a given list.
■incr increases an integer by 1.
For more information about the layers command, see layers on page 1008.
13: Extracting Results
Extracting Values and Level Crossings: interpolate
854 Sentaurus™ Process User Guide
H-2013.03
Extracting Values and Level Crossings: interpolate
The interpolate command has two purposes: to obtain the position at which a profile
crosses a particular value and to retrieve a value at a particular location in space. Interpolation
is used to accomplish both tasks.
The four main parameters of this command are x, y, z, and val. The command operates on a
selected data field. In 1D, you must supply either x or val. If x is supplied, Sentaurus Process
returns the value at x. If val is supplied, Sentaurus Process returns the locations at which the
selected profile crosses val.
In 2D, two of the four parameters must be given (not z) and, in 3D, three of the four parameters
must be given. For example, in 2D, if x and val are given, the locations along x where val is
crossed are returned. If x and y are given, the value at this location is returned. For more
information, see interpolate on page 991.
Extracting Values during diffuse Step: extract
The extract command is used to extract historical data during diffuse steps. This
command allows you to define the data extraction script with the command parameter. The
extraction script is composed typically of the select command for choosing the data field for
extraction and the interpolate command for retrieving the value at a specified location.
Only values returned by the interpolate command, at each time step, are stored in the
historical data values.
For example, to extract the boron concentration at position in the silicon and the YY
component of the element stress at position in the oxide for each diffuse
substep:
extract name=etest command= {
sel z=Boron
interpolate Silicon x=0.04
sel z=StressEL_yy element
interpolate Oxide x=-0.001
}
This command must be defined before the diffuse step. After the diffuse steps of interest,
the following command retrieves the extracted data values for the defined extraction etest:
extract name=etest print
0.04 µm
0.001– µm

13: Extracting Results
Extracting Values during diffuse Step: extract
Sentaurus™ Process User Guide 855
H-2013.03
The values are returned as a Tcl list with the format:
<time1> <Boron1> <Syy1> <time2> <Boron2> <Syy2> ...
The following script demonstrates how to manipulate this list for more formatted output:
set extdata [extract name=etest print]
foreach { time bval sval } $extdata {
puts "$time $bval $sval"
}
Output from the above script is:
0.000000e+00 4.738727e+15 6.000000e+09
1.000000e-04 4.738727e+15 4.793201e+09
2.503231e-04 4.738727e+15 4.793201e+09
5.509694e-04 4.738727e+15 4.793201e+09
1.152262e-03 4.738727e+15 4.793201e+09
...
For more information, see extract on page 931.
Optimizing Parameters Automatically
Previously, users had to rely on the optimizer in Sentaurus Workbench to perform parameter
optimization. Sentaurus Process provides a built-in capability for parameter automatic
optimization with the new optimize command. To use this feature, a Tcl procedure must be
created which takes as input the current values of the parameters to be optimized, and returns
a corresponding result. Although writing the procedure is a small extra task for the user, this
design allows greater flexibility in the types of optimization that can be performed. The
procedure can specify anything from a simple analytic function to a complete process
simulation flow. The form of the result is a vector of values from which an error is computed
based on user-defined target data. This generic flow allows for a variety of applications (such
as dopant profiles resulting from multiple process steps or the thickness of an oxide layer).
To use the automatic parameter optimization feature, the Tcl procedure is introduced into an
input file or sourced from an external file before calling the new command optimize.
Table 75 lists some of the parameters available in the optimize command:
Table 75 Parameters for optimize
Parameter Name Description
model.function
Name of the user defined Tcl procedure
model.parameters
Names of the parameters to be optimized
param.init
Starting values of parameters

13: Extracting Results
Fitting Routines: FitLine, FitArrhenius, FitPearson, and FitPearsonFloor
856 Sentaurus™ Process User Guide
H-2013.03
The optimize command also allows you to weight target values, log the history of
optimization steps, and specify the maximum number of iterations and other convergence
criteria. For more details, see optimize on page 1042.
Fitting Routines: FitLine, FitArrhenius, FitPearson, and
FitPearsonFloor
The following commands provide fitting capabilities in Sentaurus Process:
■The FitLine command is used to find the best offset and slope for a given set of data, for
example:
sprocess> foreach temp { 700 800 900 1000 } {
> lappend dat $temp
> lappend dat [expr 110 + 10*$temp]
>}
sprocess> FitLine $dat ;# Get the slope, offset, and correlation factor
10.0 110.0 1.0
■The FitArrhenius command is used to find the best prefactor and energy for an
Arrhenius fit of a given profile, for example:
sprocess> list dat ;# This is the list to be passed to FitArrhenius
sprocess> foreach temp { 700 800 900 1000 } {
> SetTemp $temp
> lappend dat $temp ;# dat will contain "temp" - "Arrhenius val" pairs
> lappend dat [Arrhenius 0.1 1.0]; ;# Arrhenius takes prefactor and
# activation energy
>}
sprocess> FitArrhenius $dat ;# Send the list to FitArrhenius
0.0999999308634 1.00030363776 -0.999999999866 ;# Return prefactor, energy
# and corr factor
sprocess>
The FitArrhenius command takes a list of temperature–function pairs. The unit of
temperature is degree Celsius. The return value is a list where the first member is the
param.lower
Lower limits of parameters
param.upper
Upper limits of parameters
param.log
Parameters that vary logarithmically during optimization
target | target.file
Options used to specify the target values, either via a Tcl array or from a file
Table 75 Parameters for optimize
Parameter Name Description

13: Extracting Results
Resistivity
Sentaurus™ Process User Guide 857
H-2013.03
prefactor, the second member is the activation energy [eV], and the third member is the
correlation factor. Absolute values of the correlation factor close to one are desirable.
■The FitPearson command is used to extract the best Pearson parameters of a profile.
■The FitPearsonFloor command is a modification of FitPearson and is used to set a
floor for the data value so that only data points with values greater than the given floor are
used for the Pearson fit.
An example of using this command is one that contains the command PearsonProfile,
which can be called to create a Pearson–IV, Pearson–V, or Pearson–VI profile depending
on the parameters sent. It takes as its arguments the name of the data field to be created and
a list of parameters in this order: dose [cm–2], projected range, standard deviation,
skewness, and kurtosis. FitPearsonFloor takes the minimum value and a list of x y
values, which are to be fit to a Pearson. It returns parameters in the same order as the list
of parameters for PearsonProfile:
sprocess> PearsonProfile Arsenic {1e14 0.0650 0.0228 0.577 3.4390}
;# Corresponds to 100keV As implant
sprocess> select z= Arsenic
sprocess> FitPearsonFloor 1.0e10 [lindex [slice silicon] 0]
9.99999659675e+13 0.0649998507454 0.0227994405041 0.576718866676
3.4368839917
sprocess>
Resistivity
The background concentration of the wafer can be defined using the resistivity of the wafer.
You can define th e r es ist ivit y o f t he wa fer with the init command, which requires a field name
to calculate the background concentration. For example:
line x location = 0 tag=top
line x location = 10 tag=bot
region silicon xlo = top xhi = bot
init field=boron silicon resistivity=1.4
sets the boron concentration of the wafer to in silicon.
The resistivity is given in , and the resistivity is calculated by:
(970)
where:
■ is the electron density.
■ is the background concentration.
■ is the mobility.
1.08 16
×10 cm 3–
Ωcm
RHO qNµ()
1–
=
q
N
µ
13: Extracting Results
Resistivity
858 Sentaurus™ Process User Guide
H-2013.03
Three mobility models can be selected using the command:
pdbSet <material> <dopant> Mobility.Model <model>
where
<model>
can be Model1 [1], or Model2
[2], or Model3 [3]. They are given as:
■Model1:
(971)
■Model2:
(972)
■Model3:
(973)
(974)
where , and are fitting
parameters for the empirical formulas.
The fitting parameters can be set using the following commands:
pdbSet <material> <dopant> uMin {<n>}
pdbSet <material> <dopant> uMin2{<n>}
pdbSet <material> <dopant> uMax {<n>}
pdbSet <material> <dopant> uM {<n>}
pdbSet <material> <dopant> uNr {<n>}
pdbSet <material> <dopant> uNs {<n>}
pdbSet <material> <dopant> uPC {<n>}
pdbSet <material> <dopant> uBeta{<n>}
pdbSet <material> <dopant> uAlpha {<n>}
pdbSet <material> <dopant> A0 {<n>}
pdbSet <material> <dopant> A1 {<n>}
pdbSet <material> <dopant> A2 {<n>}
pdbSet <material> <dopant> A3 {<n>}
µµ
min
µmax µmin
–
1N
Nr
------
α
+
----------------------------+=
µµ
mine
PC
N
------
–µmax µmin2
–
1N
Nr
------
α
+
-------------------------------µm
1Ns
N
------
β
+
-----------------------–+=
µpµmine
PC
N
------
–µmax
1N
Nr
------
α
+
------------------------+=
µn10
A0A1XA
2X2A3X3
++ +
1B1XB
2X2B3X3
++ +
------------------------------------------------------------
=
XN
Nr
------
log=
Nr, µmin, µmin2, µmax, µm, PC, A0, A1, A2, A3, B1, B2, B3β,
α

13: Extracting Results
Sheet Resistance
Sentaurus™ Process User Guide 859
H-2013.03
pdbSet <material> <dopant> B1 {<n>}
pdbSet <material> <dopant> B2 {<n>}
pdbSet <material> <dopant> B3 {<n>}
In addition to these models, a user-defined mobility model can be set using the command:
pdbSet <material> <dopant> Mobility.Equation <String Expression>
For example, the Mobility1 model for boron in silicon can be set using the command:
pdbSet Silicon Boron Mobility.Equation \
"(49.705+(467.729-49.705)/(1+abs(tNetActive/1.606e+17)^0.7))"
tNetActive
is the internal name for .
NOTE To change the model and default model paramters, use the
SetMobilityModel command (see setMobilityModel on page 1127).
Sheet Resistance
The sheet resistance and p-n junction depth of a semiconductor layer in the vertical direction
are calculated using the command:
SheetResistance <args>
where <args> must be the y–cross section in 2D, and the y– and z–cross sections in 3D. For
example, in 3D:
SheetResistance y=0.4 z=-0.1
The sheet resistance formula is given by:
(975)
where is mobility of the holes ( ) or electrons ( ) given in Eq. 971 or Eq. 972 or Eq. 973.
The active concentration of dopants is calculated at the last diffusion temperature. The electron,
, and hole, , concentrations are calculated assuming charge neutrality at a temperature of
300 K.
Sheet resistance can be calculated only after a diffusion statement or activation step. For
example:
diffuse time=0.0 temperatur=1000
N
Rs
1
qµnnµpp+()xd
pni
pni1+
---------------------------------------------------=
µX
p
n
n
p

13: Extracting Results
References
860 Sentaurus™ Process User Guide
H-2013.03
SheetResistance y=9.4 z=-0.1
Since not all data fields are stored in the TDR file, the sheet resistance may not be calculated
after loading the TDR file even though the last command was a diffusion command.
References
[1] D. A. Antoniadis, A. G. Gonzalez, and R. W. Dutton, “Boron in Near-Intrinsic <100>
and <111> Silicon under Inert and Oxidizing Ambients—Diffusion and Segregation,”
Journal of the Electrochemical Society, vol. 125, no. 5, pp. 813–819, 1978.
[2] G. Masetti, M. Severi, and S. Solmi, “Modeling of Carrier Mobility Against Carrier
Concentration in Arsenic-, Phosphorus-, and Boron-Doped Silicon,” IEEE Transactions
on Electron Devices, vol. ED-30, no. 7, pp. 764–769, 1983.
[3] W. R. Thurber et al., The Relationship Between Resistivity and Dopant Density for
Phosphorus- and Boron-Doped Silicon, National Bureau of Standards Special
Publication 400-64, Washington, DC, USA, May 1981.

Sentaurus™ Process User Guide 861
H-2013.03
CHAPTER 14 Numerics
This chapter discusses numerics-related issues, time integration
methods, and the linear solvers used in Sentaurus Process.
Overview
In Sentaurus Process, during the simulation of diffusion steps, three different sets of nonlinear
partial differential equations must be solved:
■Oxidant diffusion and reaction
■Dopant diffusion and reaction
■Stress equations
In the case of silicidation, the transport and reactions of dissolved silicon or dissolved metal is
handled similar to the oxidant diffusion and reaction.
The oxidant, dopant, and point-defect equations are solved on the simulation mesh using a
trapezoidal rule/backward differentiation formula (TRBDF) time discretization, a finite
volume (box) method for the spatial integration, and a Newton method to solve the nonlinear
equations.
For the discretization of the nonlinear stress equations, piecewise linear finite elements are
used. If stress history is tracked, the stress equations are solved, not only during the simulation
of diffusion steps, but also at the end of etch and deposit steps.
Various direct and iterative solvers are integrated in Sentaurus Process to solve the large
systems of linear equations in each Newton iteration. By default, for all equations in 1D
simulations and for mechanics equations in 2D simulations, the parallel direct solver
PARDISO is used. For diffusion equations in two dimensions and for all equations in three
dimensions, the iterative solver ILS is used. The solver can be selected using the math
command:
math ils
This command selects the solver ILS for all types of equation in 1D, 2D, and 3D. Separate
selections can be made for the various spatial dimensions and for the solution of mechanics
equations and diffusion equations (the same settings are used for both oxidant and
dopant–point defect equations).

14: Numerics
Setting Parameters of the Iterative Solver ILS
862 Sentaurus™ Process User Guide
H-2013.03
The parameters Flow and diffuse select the type of equation, and the parameter dim
specifies the spatial dimension:
math Flow dim=2 ils
math diffuse dim=2 pardiso
math Flow dim=3 pardiso
If a direct solver is used, a modified Newton method is used by default; Sentaurus Process tries
to avoid the recomputation and factorization of a new matrix and will reuse the last factorized
matrix, as long as the convergence rate remains sufficiently high. For the iterative solvers, by
default, a modified Newton scheme is used as well.
The math command is used to specify various parameters for the Newton iterations and to
define resources and specifications for the linear solvers (see math on page 1027).
For the default settings for ILS, refer to the parameter database browser (PDB). More detailed
settings for ILS can be made using pdbSet commands as described in the next section.
Setting Parameters of the Iterative Solver ILS
The iterative solver ILS is used by default to solve the linear systems for diffuse in 2D
simulations and for both Flow and diffuse in 3D simulations. Default parameters for ILS
have been added to the parameter database. To specify modified parameters for ILS, such as
the type of iterative scheme, the number of iterations, the output verbosity, or the memory
resources, use the pdbSet commands.
NOTE In the pdbSet commands, parameters must be specified separately for
each type of problem (Flow or diffuse) and for each dimension (1D,
2D, or 3D).
NOTE ILS is not recommended for use in 1D simulations because of the simple
structure of matrices arising in 1D cases. The default direct solver
PARDISO is the correct choice for 1D simulations.
Different ILS parameters can be specified for diffuse and Flow, both in 2D or 3D. In general,
the pdbSet command for the ILS parameters has the form:
pdbSet Math [diffuse | Flow] [2D | 3D] ILS.[command] [value]
The following ILS commands are available:
ILS.compact Boolean
ILS.fgmres.restart Double
ILS.gmres.restart Double
ILS.fit Double
14: Numerics
Setting Parameters of the Iterative Solver ILS
Sentaurus™ Process User Guide 863
H-2013.03
ILS.ilut.tau Double
ILS.leftPreconditioner Boolean
ILS.maxit Double
ILS.method String
ILS.nonsymmOrdering String
ILS.okayForModNewton Boolean
ILS.preconditioner String
ILS.recompute.ordering Double
ILS.refine.residual Double
ILS.refine.iterate Double
ILS.scaling String
ILS.symmOrdering String
ILS.tolabs Double
ILS.tolrel Double
ILS.tolunprec Double
ILS.useILSRCFile Boolean
ILS.verbose Double
To select the GMRES method, for example, gmres(60), use:
pdbSet Math diffuse 3D ILS.method gmres
pdbSet Math diffuse 3D ILS.gmres.restart 60
To specify the FlexibleGMRES method, for example, fgmres(40), to solve the stress
equations, use:
pdbSet Math Flow 3D ILS.method fgmres
pdbSet Math Flow 3D ILS.fgmres.restart 40
pdbSet Math Flow 3D ILS.fit 5
To select the efficient reuse mode (on each 3D diffuse time step, the costly reordering is applied
only once to a first Jacobian system), specify:
pdbSet Math diffuse 3D ILS.recompute.ordering 2
and to return to reordering for every system, use:
pdbSet Math diffuse 3D ILS.recompute.ordering 1
To improve the accuracy and convergence of iterative linear solvers, use an enhanced option
available in Version H-2013.03 by specifying (default value is 0):
pdbSet Math [diffuse | Flow] [2D | 3D] ILS.refine.iterate 1
To use the 3D diffuse gmres solver in the advanced parallel implementation, specify:
pdbSet Math diffuse 3D ILS.hpc.mode 3

14: Numerics
Partitioning and Parallel Matrix Assembly
864 Sentaurus™ Process User Guide
H-2013.03
In this command, the value 3 activates algorithmic improvements made in Versions H-2013.03
and H-2013.03, while the value 2 corresponds to Versions G-2012.06 and F-2011.09, and the
value 1 corresponds to Version E-2010.12.
To select the solvers for mechanics, STS2 or STCG2 for 2D, and STS3 or STCG3 for 3D, for
example, sts3, use:
pdbSet Math Flow 3D ILS.method sts3
pdbSet Math Flow 3D ILS.tolrel 1e-10
pdbSet Math Flow 3D ILS.ilut.tau 5e-4
pdbSet Math Flow 3D ILS.scaling diagsym
pdbSet Math Flow 3D ILS.nonsymmOrdering none
For the mechanics solvers STS2, STS3, STCG2, and STCG3, it is mandatory to specify
ILS.scaling
as
diagsym, and ILS.nonsymmOrdering as none. It is also recommended
to specify the value for the parameter ILS.ilut.tau
in the range of – .
To improve convergence of the mechanics solver STS2 or STS3, use an enhanced version of
the solver by specifying, respectively (default value is 0):
pdbSet Math Flow 2D ILS.refine.sts 1
or:
pdbSet Math Flow 3D ILS.refine.sts 1
The enhanced version takes advantage of results from previous solve steps, so the actual
performance gain can vary depending on the simulation setup, and it performs best when there
is a sequence of mechanical solve steps, as in temperature ramps.
To active the STS solver which improves robustness and speed, use:
pdbSet Math Flow 3D ILS.refine.sts 2
Partitioning and Parallel Matrix Assembly
Sentaurus Process can assemble the diffusion and mechanics matrices in parallel on multicore
machines. To switch on the parallel assembly, use:
math { numThreads = <n> | numThreadsAssembly = <n> }
where numThreads is the number of threads that would be used during the matrix assembly.
numThreads is a general keyword (see math on page 1027) used by both implant and linear
solvers. If you want to use a different number of threads for diffusion matrix assembly, use the
keyword numThreadsAssembly. If the number of threads is greater than 1, Sentaurus Process
first creates the threads.
54–
×10
55–
×10
14: Numerics
Partitioning and Parallel Matrix Assembly
Sentaurus™ Process User Guide 865
H-2013.03
To modify the thread stack size, use:
math threadStackSize = <n>
NOTE Parallel assembly of the matrix is performed only for inert anneals. It is
recommended that numThreadsAssembly does not exceed the
number of actual cores of the computer. Parallel assembly of the matrix
is not available for moving-boundary problems such as oxidation and
silicidation.
Sentaurus Process then partitions the mesh structure into levels, and each level is divided into
different domains at the beginning of the diffusion step. For example, Figure 176 shows a
structure with three levels. The first level (blue) (L0) has four domains: D0, D1, D2, and D3.
Elements belonging to each domain on the same level do not cross over to the other domains.
The second level (orange) (L1) also has four domains: D0, D1, D2, and D3. Again, the
elements on the same level do not cross over to the other domains. The third level (green) (L2)
has only one domain: D0. This is the last level and contains all the elements not included in the
previous levels.
To control the partitioning, use:
math { maxNumberOfDomains=<n> | NumberOfElementsPerDomain=<n> }
14: Numerics
Partitioning and Parallel Matrix Assembly
866 Sentaurus™ Process User Guide
H-2013.03
Figure 176 Partitioned mesh structure
NOTE maxNumberOfDomains is the maximum number of domains that each
level of partition can have. It is recommended that
maxNumberOfDomains equals or is greater than the number of threads
used. NumberOfElementsPerDomain is the number of elements that
should go to each domain.
The final number of domains at each level is determined by:
(976)
To partition the mesh, based on material type, give weight to each material using the command:
pdbSet <material> PartitionWeight <n>
For example:
pdbSet Silicon PartitionWeight 10
pdbSet Gas PartitionWeight 0
domains min maxNumberOfDomains Number of Edges
NumberOfElementsPerDomain
-----------------------------------------------------------------------------------(, )=
14: Numerics
Matrix Size Manipulation
Sentaurus™ Process User Guide 867
H-2013.03
gives more weight to silicon mesh elements than the gas mesh elements during partitioning.
This allows Sentaurus Process to distribute the work among the threads more evenly since there
is no matrix assembly for the gas mesh.
Partition weights for mechanics assembly can be specified separately with:
pdbSet <material> Mechanics PartitionWeight <n>
This allows balancing the workload among threads according to the stress analysis methods for
different material behaviors. If the partition weights for mechanics assembly are not defined,
the partition weights for diffusion assembly are used by default.
Matrix Size Manipulation
The size of the matrix used during diffusion assembly is automatically determined based on the
number of solution variables and nodes in the structure. In most cases the allocated matrix size
is more than sufficient. If the matrix size becomes insufficient during the assembly, the matrix
size will be increased automatically by 10%. You can change the default 10% value by using
the command:
pdbSet Math Matrix.Size.Scale <n>
where the value <n> should be greater than one.
You al so can in cr eas e t he au to mat ic ally de termined matrix size using the command:
pdbSet Math Assembly.Matrix.Size.Scale <n>
where the value <n> should be greater than one.
NOTE Be careful when choosing the matrix scaling values because it can
exhaust the computer memory for large scaling values.
Node and Equation Ordering
Because the order of nodes in meshes does not follow a specific order by default, adjacent
nodes may be far from each other in the internal node list. The order may not have much effect
on simulation time for small examples (such as 2D), but it may degrade 3D results. The nodes
in the structure can be ordered meshwise or globally using the command:
pdbSet Math < 1D | 2D | 3D > Reorder.Nodes <model>
where <model> is None (default), Mesh, or Global.

14: Numerics
Time Integration
868 Sentaurus™ Process User Guide
H-2013.03
The default order of equation numbering in the structure is based on the meshes. Each node in
the mesh receives an equation number from a solution variable and the same is repeated for the
next solution. This may create many distributed entries in the assembly matrix. Again, the order
may not greatly affect the simulation time for small examples (such as 2D), but it may degrade
3D results. It is possible to number equations based on solutions by taking a node in the mesh,
numbering it for each solution variable, and moving to the next node in the mesh. This creates
better-distributed entries in the assembly matrix. The order can be changed using the
command:
pdbSet Math < 1D | 2D | 3D > Reorder.Equations <model>
where <model> is None (default) or Solution.
Time Integration
The TRBDF method [1] is used for time integration by default for time-dependent problems.
It also is possible to choose the backward Euler method for the time integration. The following
command can be used to switch between methods:
math {tr_bdf | euler}
A TRBDF integration step consists of a trapezoidal step followed by a backward difference
step. A second trapezoidal solution is used to estimate the local truncation error and to
determine the size of the next time step.
The local truncation error can be estimated by either a Milne’s device (the default method) or
the divided difference method. The following command switches between methods:
math {milne | difference}
The local truncation error for the next time-step estimation can be modified using the
command:
pdbSet Math Time.Step.Function {<model>}
where <model> is Damped, UnDamped, or Linear. The Damped model applies a logarithmic
damping function to the truncation error if the error is greater than 1.0. The UnDamped model
does not modify the error. The Linear model applies a linear damping function to the
truncation error if the error is greater than 1.0. The Linear model matches that of
TSUPREM-4.
When the geometry of a simulation structure evolves, one cycle of the TRBDF time integration
requires the geometric coefficients at three incidents, that is, . To
reduce the computational time to calculate the geometric coefficients, especially in three
dimensions, the geometric coefficients at can be set to the interpolated values by
tt
0t0∆tTR
+t0∆t+,,=
tt
0∆tTR
+=

14: Numerics
Time-Step Control
Sentaurus™ Process User Guide 869
H-2013.03
assuming that the coefficients change linearly during , which reduces the number of the box
method calls by one third:
pdbSet Math 3D Use.Interpolated.Geom.Coeff 1
or:
math dimension=3 use.interpolated.geom.coeff
Time-Step Control
This section discussed different time-step controls.
Time-Step Control for PDEs
Sentaurus Process provides automatic time-step control. You can modify some of the control
parameters.
The first time step of the diffuse command uses the initial time given with the diffuse
command (see diffuse on page 908).
In an ideal situation, oxidation, mechanics, and diffusion time steps are equal to each other, and
the next time step is increased by the IncreaseRatio:
(977)
where is the next diffusion time step, is the current time step, and is the
IncreaseRatio. The default for IncreaseRatio is 2.
Use the following command to change IncreaseRatio:
pdbSet Diffuse IncreaseRatio {<n>}
In some cases, the ideal time step can be solution limited , grid limited, or reduced:
■Solution limited is the case when the time step is shortened to decrease the local truncation
error; in a log file, such steps are marked by (s).
■Grid limited is the case when the time step is reduced because of a grid motion; in a log
file, such steps are marked by (g).
■Reduced is the case when the time step is reduced to prevent overstepping of oxidation or
mechanics steps; in log files, such steps are marked by (r).
∆t
tn1+Iratiotn
=
tn1+
tn
Iratio
14: Numerics
Time-Step Control
870 Sentaurus™ Process User Guide
H-2013.03
If convergence is not achieved, the next time step is reduced by the ReduceRatio:
(978)
The following command can be used to change ReduceRatio:
pdbSet Diffuse ReduceRatio {<n>}
For more information about the convergence during diffusion, use the command:
pdbSet Diffuse Convergence.Info {1 | 0}
Typical output with information level=2 will look like:
Iter Potential Boron Arsenic Int Vac B4
1 4.345e-02 2.244e+01 2.512e+00 5.096e+03 1.835e+02 2.599e-06
-- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- -- --
Mesh: bulk Mater: Silicon
Org. Val. Org. Updt. Org.-Updt Apld. Updt. Error Location
Boron 5.7e+18 1.8e+17 5.5e+18 5.5e+18 3.1e+02 ( 1.7e-5, 0.0, 0.0 )
Arsenic 7.2e+16 3.1e+14 7.2e+16 7.2e+16 4.3e+01 ( 1.7e-5, 0.0, 0.0 )
Int 2.8e+12 -1.3e+13 1.5e+13 1.5e+13 4.5e+04 ( 1.5e-5, 0.0, 0.0 )
Vac 3.5e+14 5.5e+13 2.9e+14 2.9e+14 1.5e+03 ( 1.7e-5, 0.0, 0.0 )
Largest update: Int in Silicon @ ( 1.5e-5, 0.0, 0.0 )
where:
■Org.Val is the original value at the node.
■Org.Updt is the original update at the node.
■Org.-Updt is the Original Value – Original Update.
■Apld. Updt. is the applied update.
Sentaurus Process provides three time-step control models (TSCMs):
■A local model (LTS) based on the current time step and described above.
■Two history-based models involving all previous time steps within the diffuse command:
BPTS and NGLTS.
BPTS uses the biggest previous time step from the history, such that Eq. 977 is modified as:
(979)
NGLTS uses the latest nongrid limited time step, and Eq. 977 is modified as:
(980)
t
˜n1+Rratiotn1+
=
tn1+IratiotBPTS n,
=
tn1+IratiotNGLTS n,
=

14: Numerics
Time-Step Control
Sentaurus™ Process User Guide 871
H-2013.03
Use the following command to choose the TSCM:
pdbSet Diffuse TSCM <model>
where <model> can be LTS, BPTS, or NGLTS.
Error Control for PDEs
To control errors during transient simulation, Sentaurus Process uses the following to calculate
the error:
(981)
where the sum is taken over all solution variables, and is the update for solution .
TransRelErr and AbsErr are the transient relative error and absolute error for the solution
variables, respectively. They can be set using the commands:
pdbSet <mater> <solution> Transient.Rel.Error <n>
pdbSet <mater> <solution> Abs.Error <n>
where <mater> is the material name.
To control errors during nonlinear Newton iterations, Sentaurus Process uses the following to
calculate the error:
(982)
where RelErr is the relative error for the solution variables. It can be set using the command:
pdbSet <mater> <solution> Rel.Error <n>
NOTE If the error control parameter is not defined in the PDB for a material or
a solution, the long-hand command pdbSetDouble must be used.
Time-Step Control for Mechanics
Automatic time-step control for mechanics is activated only if the structure contains certain
nonlinear features that require Newton iterations, such as plasticity, viscoplasticity, and creep.
The size of the time step is adjusted based on satisfaction of certain convergence criteria.
e1
N
----ui
soliTransRelErr AbsErr+×
-----------------------------------------------------------------------
2
i1=
N
12⁄
=
ui
soli
e1
N
----ui
soliRelErr AbsErr+×
---------------------------------------------------------
2
i1=
N
12⁄
=
14: Numerics
Time-Step Control
872 Sentaurus™ Process User Guide
H-2013.03
Convergence Criteria
To check the convergence of Newton iterations for mechanics equations [2][3], the criteria are:
■Force residual
■Energy
■Displacement
The force residual criterion checks the satisfaction of force equilibrium by comparing the
maximum norm of the residual ( ) against a reference value:
(983)
The reference value of the force residual is computed automatically by taking a norm of the
element force residual vector at the first Newton iteration in a time step.
The energy criterion checks the satisfaction of the minimization of energy at equilibrium by
comparing the change in energy against a reference value:
(984)
The reference value of energy is computed automatically by taking a dot product of the force
residual and the displacement increment ( ) vector at the first Newton iteration in a
time step.
The displacement criterion checks the satisfaction of the solution accuracy by comparing the
maximum norm of the displacement increment against a reference value:
(985)
The reference value of displacement is computed automatically by taking a norm of the
displacement increment vector at the start of the first Newton iteration in a time step.
The force residual and the energy criteria are checked by default. Optionally, the energy
criterion may be replaced by the displacement criterion. Use the following parameters to
activate or deactivate any of the convergence criteria:
pdbSet Mechanics Convergence.Force.Check <n>
pdbSet Mechanics Convergence.Energy.Check <n>
pdbSet Mechanics Convergence.Displacement.Check <n>
where <n> is either 0 (deactivate) or 1 (activate).
RF
external F internal
–=
Rn1+
i
∞εRRn1+
1
≤
∆un1+
iRn1+
i
⋅ε
E∆unRn1+
1
⋅≤
∆uv∆t=
∆un1+
i
∞εu∆un
≤
14: Numerics
Time-Step Control
Sentaurus™ Process User Guide 873
H-2013.03
The choices for the force residual and the displacement reference value norms that can be set
using the following command are:
pdbSet Mechanics Convergence.Check.Norm [RMS | ABS]
where RMS refers to the root mean square value (default) and ABS refers to the mean absolute
value.
The reference values for any of the convergence criteria can be changed by using the
commands:
pdbSet Mechanics Convergence.Force.RefVal <n>
pdbSet Mechanics Convergence.Energy.RefVal <n>
pdbSet Mechanics Convergence.Displacement.RefVal <n>
where <n> is a suitable positive value.
By default, the tolerance for each of the convergence criteria is set to 0.001 and can be changed
by using the commands:
pdbSet Mechanics Convergence.Force.Tolerance <n>
pdbSet Mechanics Convergence.Energy.Tolerance <n>
pdbSet Mechanics Convergence.Displacement.Tolerance <n>
where <n> is a value between 0.0 and 1.0.
Convergence criteria are checked in every iteration until either they are satisfied or the
maximum number of Newton iterations is reached. The default value for the maximum number
of Newton iteration is 8, and this can be changed using the command:
pdbSet Mechanics MaxIterations <n>
where <n> is a value greater than zero.
Time-Step Adjustment
The first time step for solving mechanics equations is set to the initial time given with the
solve command. The size of subsequent time steps is decided based on the convergence
history of the preceding time step. The time-step size is extended if the preceding time step
converges quickly. The maximum value is limited to the maximum time given with the solve
command. The time-step size is reduced if the preceding time step takes too many iterations to
converge; it remains unchanged if the preceding time step takes a moderate number of
iterations to converge.
The time-step size also is adjusted to keep the viscoplastic or creep strain error or value within
tolerance when using such material models. The viscoplastic strain error or value is checked
14: Numerics
Time-Step Control
874 Sentaurus™ Process User Guide
H-2013.03
for Anand model (see Anand Model on page 656) while creep strain error or value is checked
for Power Law creep and Standard Linear Solid models (see Power Law Creep on page 658
and Standard Linear Solid Model on page 646). Use the following commands to change the
default settings for the viscoplastic or creep strain error or value:
pdbSet Mechanics StrainVP.Error.Tolerance <n>
pdbSet Mechanics StrainVP.Value.Tolerance <n>
pdbSet Mechanics StrainCr.Error.Tolerance <n>
pdbSet Mechanics StrainCr.Value.Tolerance <n>
where <n> is a value between 0.0 and 1.0 for both. By default, only the viscoplastic or creep
strain error criterion is used with a tolerance of 0.02.
Time-Step Cutback
An automatic time-step cutback procedure interrupts the Newton iteration loop and restarts the
time step with a smaller size when any of the following issues is encountered:
■Convergence criteria are not satisfied within the maximum number of Newton iterations.
■The solution converges very slowly over several iterations.
■The solution diverges over several iterations.
■Convergence criteria are satisfied, but viscoplasticity equations fail to converge.
■Convergence criteria are satisfied, but the viscoplastic or creep strain error or value is
greater than the tolerance.
To check convergence details during solve steps, specify info=1 in the solve command.

14: Numerics
References
Sentaurus™ Process User Guide 875
H-2013.03
References
[1] R. E. Bank et al., “Transient Simulation of Silicon Devices and Circuits,” IEEE
Transactions on Electron Devices, vol. ED-32, no. 10, pp. 1992–2007, 1985.
[2] O. C. Zienkiewicz and R. L. Taylor, The Finite Element Method, Butterworth-
Heinemann: Oxford, 5th ed., 2000.
[3] K. J. Bathe and A. P. Cimento, “Some Practical Procedures for the Solution of Nonlinear
Finite Element Equations,” Computer Methods in Applied Mechanics and Engineering,
vol. 22, no. 1, pp. 59–85, 1980.
14: Numerics
References
876 Sentaurus™ Process User Guide
H-2013.03

Sentaurus™ Process User Guide 877
I-2013.12
APPENDIX A Commands
This appendix describes the commands of Sentaurus Process.
Syntax Conventions
The commands are designed to optimize the use of the tool command language (Tcl).
The following conventions are used for the command syntax:
■Each command starts on a new line.
■A backslash (\) is used to extend a command on to multiple lines if it appears as the last
character on the line. It is not included on the last line of a command. For example:
icwb.refine.mask name= SiOxPo layer.name= POLY \
oversize= 0.1 xtop= -1.51 xbot= -1.35 min.normal.size= 0.005 \
interface.mat.pairs= {Silicon Oxide Silicon Polysilicon}
■Braces – { } – indicate a list. The braces are part of the syntax.
■Brackets – [ ] – indicate an optional argument. The brackets are not part of the syntax.
■Parentheses – ( ) – indicate grouping of arguments. The parentheses are not part of the
syntax.
■A vertical bar – | – indicates options, only one of which can be specified. Vertical bars are
not part of the syntax.
■Angle brackets – < > – indicate specific type of input. The angle brackets are not part of
the syntax.
NOTE An exception to this is that angle brackets must be included in the syntax
if units are specified in a command. For example:
line x location= 0<um> spacing= 0.02<um> tag= surf
■The following type identifiers are used:
•<c>: Replace with a string. In general, strings are enclosed in double quotation marks
(" "), for example:
line_edge_roughness normal= "Z" masks= {mask1} \
correlation.length= 25.00<nm> standard.deviation= 5.00<nm>
•<i>: Replace with an integer.
•<n>: Replace with a floating-point number.

A: Commands
Syntax Conventions
878 Sentaurus™ Process User Guide
I-2013.12
•<list>: Replace with a Tcl list of values. The list must be enclosed in braces, for
example, min= {-5.0 0.0 0.0}.
•<field>: Replace with the name of a field.
•<material>: Replace with the name of a material.
•<solution>: Replace with the name of a solution.
■Boolean arguments are true (or on) if they appear on the command line (for example,
negative).
■Boolean arguments are false (or off) if they appear on the command line preceded by an
exclamation mark (for example, !negative).
Example of Command Syntax
An example of command syntax is:
command
argument1=<n>[<unit1>]
argument2=<i>
argument3=<i>
argument4
...
There are two types of argument:
■Named arguments must be specified with a value, for example, name=Germanium.
■Unnamed arguments supply only the value, for example, <material> would be replaced
by Oxide.
If you specify units for an argument:
■Omit spaces between the value and unit.
■Include the angle brackets with the unit, for example, xlo=5<um>.
If you specify lists:
■Lists must be enclosed in braces.
■Elements in the list must be separated by space.
■You m ust in se rt s pa ce between t he e qua l sign a nd the o pe ning br ac e. Fo r ex amp le :
transform translate= {-1 0 0}

A: Commands
Syntax Conventions
Sentaurus™ Process User Guide 879
I-2013.12
Common Arguments
Nearly all Sentaurus Process commands (with the exception of those implemented as Tcl
procedures as well as a few others) support two common optional arguments:
■info=<i>
This argument sets the amount of information to be printed to the screen and the log file. It
can take the value 0, 1, or 2. The default is 0, which is the minimum amount. Higher values
give more details about the status of the simulation as well as model and parameter
selection information.
This argument can be used with any other argument combination for nearly all commands
that are not Tcl procedures.
For example, to specify that more information should be printed to the screen and the log
file for the etch command, use:
etch info= 2
■parameters
This argument prints all the available arguments for a command.
For example, to print the arguments for the extract command, use:
extract parameters
NOTE When you use the parameters argument, the complete list of
arguments for that command is generated. However, some of the
arguments in this list are not documented. These parameters are used for
debugging purposes only.

A: Commands
alias
880 Sentaurus™ Process User Guide
I-2013.12
alias
Sets and prints aliases.
Syntax
alias <c> [<c>] [-list]
Arguments
<c>
If one argument is specified, only one alias for this value is printed.
If two arguments are specified, a new alias is set.
-list
Prints a list of allowed aliases.
Description
This interactive mode command sets and prints aliases.
If the first argument is -list, a list of allowed aliases is printed. Otherwise, only one alias
corresponding to the first argument is printed.
Examples
Set a new alias of Temperature:
alias Temp
Temperature
Print the list of allowed aliases:
alias -list
Print an alias of Temp:
alias Temp

A: Commands
ambient
Sentaurus™ Process User Guide 881
I-2013.12
ambient
Creates new ambients for material growth reactions, such as oxidation or silicidation, or creates
a new epi growth mode.
Syntax
ambient
name=<c>
(add | clear | delete | list | print)
[epi | inert | react]
Arguments
add
Creates a new ambient.
clear
Clears all known ambients (only useful in very special situations).
delete
Deletes the named ambient.
epi, inert, react
When creating a new ambient, you can set the ambient type:
•epi ambients are used to create new epi growth modes or models.
•inert ambients are used in gas_flow commands to create gas reactions or to dilute
active ambients.
•react creates an active ambient and is used to define material growth reactions such
as oxidation.
list
Returns a Tcl list of all available ambients.
name
Defines the name to be used in the diffuse, gas_flow, reaction, and temp_ramp
commands to identify this ambient.
A: Commands
ambient
882 Sentaurus™ Process User Guide
I-2013.12
print
Prints all available ambients.
Description
The ambient names can be used in the diffuse, gas_flow, reaction, and temp_ramp
commands. Table 64 on page 617 lists the available ambients.
Examples
Create a new ambient for a new oxidation model:
ambient name= MyO2 add react
Create a new ambient for a new epi growth mode or model:
ambient name= MyEpi add epi
List the available ambients:
ambient list
See Also
diffuse on page 908
gas_flow on page 935
reaction on page 1099
temp_ramp on page 1166

A: Commands
ArrBreak
Sentaurus™ Process User Guide 883
I-2013.12
ArrBreak
Creates two Arrhenius expressions that depend on a break temperature.
Syntax
ArrBreak <n> <n> <n> <n> <n>
Arguments
<n>
The first argument is the prefactor and the second argument is the activation energy [eV]
of the first Arrhenius expression.
The third argument is the prefactor and the fourth argument is the activation energy [eV]
of the second Arrhenius expression.
The final argument is the break temperature [ ].
Description
This command creates two Arrhenius expressions and switches from the first expression to the
second one at the given break temperature.
The first Arrhenius expression is computed when the temperature is below the break
temperature. The second Arrhenius expression is computed when the temperature is equal to
or greater than the break temperature.
Examples
Create two Arrhenius expressions – and – with a break temperature
of . The first Arrhenius expression is computed when , and the second
Arrhenius expression is computed when :
ArrBreak 3.0 0.5 2.0 0.4 825.0
°C
3.0 e0.5–kBT⁄
2.0 e0.4–kBT⁄
825°C
T825°C<
T825°C≥

A: Commands
Arrhenius
884 Sentaurus™ Process User Guide
I-2013.12
Arrhenius
Creates an Arrhenius expression.
Syntax
Arrhenius <n> <n>
Arguments
<n>
The first argument is the prefactor and the second argument is the activation energy [eV]
of the Arrhenius expression.
Description
This command creates an Arrhenius expression.
Examples
Create the Arrhenius expression :
Arrhenius 4.0 0.5
4.0 e0.5–kBT⁄

A: Commands
beam
Sentaurus™ Process User Guide 885
I-2013.12
beam
Creates a beam for multiple-beam etching.
Syntax
beam
name=<c>
(direction= {<x> <y>} | incidence=<n>)
factor=<n>
[list]
Arguments
direction
Defines the angle of incidence of the beam using a direction vector. The specified direction
vector is normalized automatically to unit length.
factor
If multiple beams are defined, factor defines the relative strength of each beam.
incidence
Defines the angle of incidence of the beam. An angle of 0 is vertical. The angle is measured
counterclockwise, that is, a positive angle implies a beam ray entering from the upper left
towards the lower right. A negative angle implies a beam ray entering from the upper right
towards the lower left.
list
Returns a Tcl list of known beams.
name
Specifies the beam name to be referenced using sources of the etch command.
Description
This command defines the direction and relative strength of etchant beams. The beam name is
referenced in the etch command. The angle of incidence of the beam can be given using
direction or incidence. The relative strength factor is used to mix the strength of
different beams. Etchant beams are assumed to be collimated, that is, a slight angular spread of
beam direction is not taken into account.
A: Commands
beam
886 Sentaurus™ Process User Guide
I-2013.12
Examples
Define a vertical beam called source1 and a beam called source2 at half the strength of
source1 at an angle of (positive angle implies that the beam travels from the upper left
to the lower right). A third beam called source3 at one-tenth the strength of source1 enters
from the upper right slightly towards the lower left:
beam name= source1 incidence= 0 factor= 1
beam name= source2 incidence= 10 factor= 0.5
beam name= source3 direction= {1 -0.1} factor= 0.1
See Also
etch on page 923
10°

A: Commands
bound
Sentaurus™ Process User Guide 887
I-2013.12
bound
Extracts the boundary of a material or region, and returns the outline as a list of coordinates.
Syntax
bound
<material> | region=<c>
Arguments
<material>
Specifying a material extracts the boundary of all regions of the specified material. For
information about specifying materials, see Material Specification on page 52.
region
Specifying a region extracts the boundary of that region.
Description
This command is used to plot the limits of the regions for further processing. It returns a list of
lists of coordinates of the boundary. The outer lists are distinct parts of the regions. Each outer
list comprises a complete circle around that part. Each inner list contains coordinate pairs in
order around the regions. The coordinate pairs are written in xy order around the material.
NOTE This command is not available for 3D simulations.
Examples
Return the boundary of oxide material:
bound oxide
Return the boundary of the region named Silicon_1:
bound region= Silicon_1

A: Commands
Compatibility
888 Sentaurus™ Process User Guide
I-2013.12
Compatibility
Applies parameters consistent with the default values of a previous release.
Syntax
Compatibility <c>
Arguments
<c>
Specifies the release from which to apply parameters. Aliases are available for the release,
so it is not necessary to know the release foundation letter. For example, 2012.06 can be
used instead of G-2012.06.
Description
If used, this command must be the first command in the command file, so that all subsequent
commands that depend on the default values take into account the compatibility setting (see
Compatibility With Previous Releases on page 54.
Examples
Apply parameters consistent with Version G-2012.06:
Compatibility 2012.06

A: Commands
contact
Sentaurus™ Process User Guide 889
I-2013.12
contact
Defines a contact for subsequent device simulation. Intended only for adding contacts to
structures created for device simulation.
Syntax
contact
[add] [clear]
[depth=<n>] [list] [list.existing]
[left] [right] [back] [front] [top] [bottom]
[name=<c>] [new.name=<c>]
[print]
[region=<c>]
[SearchRadius=<n>][<m>|<cm>|<um>|<nm>]
[sidewall]
[width=<n>]
(
[box] [<material>] [adjacent.material=<c>] [cut.mesh]
[xlo=<n>][<m>|<cm>|<um>|<nm>]
[xhi=<n>][<m>|<cm>|<um>|<nm>]
[ylo=<n>][<m>|<cm>|<um>|<nm>]
[yhi=<n>][<m>|<cm>|<um>|<nm>]
[zlo=<n>][<m>|<cm>|<um>|<nm>]
[zhi=<n>][<m>|<cm>|<um>|<nm>] |
[point]
[x=<n>][<m>|<cm>|<um>|<nm>]
[y=<n>][<m>|<cm>|<um>|<nm>]
[z=<n>][<m>|<cm>|<um>|<nm>]
[replace]
)
Arguments
add
If the contact command is called multiple times with the same name, it overwrites the
previous definition by default. If add is specified, the contact command will instead add
to the existing contact indicated by name or create a new contact if it does not already exist.
adjacent.material
Specifies a second material for the contact. Only elements at the interface between the two
materials are allowed for the contact.
A: Commands
contact
890 Sentaurus™ Process User Guide
I-2013.12
box, point
Selects one of the supported contact types:
•A box-type contact consists of elements at the surface of one region or material inside
the box, defined for the contact. When choosing a box-type contact, the mesh is cut
where the box intersects the chosen region to give an accurate size for the contact (see
cut.mesh). Occasionally, this cutting produces poor quality mesh elements. In such
cases, cutting can be switched off with pdbSet Grid Cut.At.Contacts 0. Use
the line command to insert lines in the mesh to retain contact size accuracy if
required.
• A point-type contact contains all the boundary elements of one region. The region can
be specified or the material and the x-, y-, and z-coordinates of one point can be
specified to select the region.
clear
Clears the list of all contacts. If name is specified, clear removes only the specified
contact.
cut.mesh
By default, when a box contact is created, the mesh is cut at the contact borders to ensure
accurate contact dimensions. Specifying !cut.mesh switches off mesh cutting, providing
better element quality at the contact borders, but possibly sacrificing accuracy of the
contact borders. The contact will only include nodes of the existing mesh within the contact
borders.
depth
Depth of the contact in micrometers.
left, right, back, front, top, bottom
These arguments selectively switch on certain outer boundaries, which are the simulation
outer domain and not the bulk nongas outer boundary. Nongas outer boundaries are treated
like interfaces and can be specified using adjacent.material=gas.
By default, only top and bottom are switched on, and the rest are switched off. If any of
these arguments are specified, internal interfaces are switched off. In addition, sidewall
is equivalent to specifying all these arguments.
list
Prints a list of currently defined contacts.
A: Commands
contact
Sentaurus™ Process User Guide 891
I-2013.12
list.existing
Returns all contacts currently existing in the bulk structure if this Boolean parameter is
specified. Adding contacts to the mesh is delayed by default. However, additional contacts
that do not appear in the list returned with this parameter may be read directly from a file,
and thus exist in the structure.
<material>
Specifies the material for the contact. Contacts in DF–ISE and TDR files are always
defined as a set of surface elements. Only elements at the surface of volume regions of the
specified material are selected. For information about specifying materials, see Material
Specification on page 52.
name
Name of the contact.
new.name
Used with name to change the name of a contact from that specified by name to that
specified by new.name.
print
Prints the contact information.
region
Name of the volume region to be used for the contact. Only surface elements of that region
are selected for the contact.
replace
If specified, in the DF–ISE or TDR file, the material of the region of a contact is replaced
by gas for point contacts.
NOTE Sometimes, regions of material gas are not saved. The material of the
region in the subsequent simulation is not affected.
SearchRadius
If contacts have been read from a DF–ISE or TDR file, they are added to the current list of
contacts defined in Sentaurus Process. When saving the current structure to a DF–ISE or
TDR file, surface elements from the current simulation mesh are selected for the contacts
if all their points are in the vicinity of the original contact elements. Default unit: .
µm
A: Commands
contact
892 Sentaurus™ Process User Guide
I-2013.12
sidewall
Allows only surface elements on the external boundary of the simulation domain (left,
right, front, back) to be selected for a contact. By default, only surface elements at material
interfaces and surface elements at the top and bottom of the simulation domain are selected
for contacts. Default: false.
width
Width of the contact in micrometers.
x, y, z
Define the coordinates of a point for a point-type contact. If some coordinates of the point
are omitted, the region is selected using the specified coordinates only. Default unit: .
xlo, xhi, ylo, yhi, zlo, zhi
Define the low and high values in each of the coordinate directions for a box-type contact.
If some coordinates are omitted, the current bounds of the simulation domain are used.
Default unit: .
Description
This command defines new contacts, deletes contacts, and prints contact information. The
contact command can be called multiple times with the same name if the add argument is
specified. In this case, the contact will have multiple parts. Contacts are written to TDR files in
the struct command. They are not otherwise used in Sentaurus Process.
NOTE Contacts are only intended for structures written for device simulation.
They should be specified immediately before the final struct
command used to write a structure for device simulation.
NOTE Contacts are not transformed using the transform command.
NOTE If a TDR file containing a boundary or mesh is read into Sentaurus
Process (during the init command), contacts defined in this file are
added to the list of contacts.
Examples
List all available contacts:
contact list
Define a contact named gate, which will consist of boundary elements of the region
containing the point (-0.05,0.0). The region material will be replaced by gas:
µm
µm
A: Commands
contact
Sentaurus™ Process User Guide 893
I-2013.12
contact name= gate x= -0.05 y= 0.0 replace point
Define a box-type contact containing the surface elements of an aluminum region inside the
specified box:
contact box Aluminum xlo= -0.01 ylo= -0.46 xhi= 0.1 yhi= -0.16 name= source
Define the substrate contact at the bottom of the simulation domain, and switch off interior
interfaces:
contact bottom name= substrate
Define a contact named lfcontact on the left side (minimum y-coordinate) and the front
(maximum z-coordinate) of the simulation domain for that part of the simulation domain inside
the box (0,0,0) -> (1,1,1) and not on any interior interfaces:
contact left front name= lfcontact xlo= 0 ylo= 0 zlo= 0 xhi= 1 yhi= 1 zhi= 1
See Also
integrate on page 985
line on page 1010
struct on page 1158

A: Commands
contour
894 Sentaurus™ Process User Guide
I-2013.12
contour
Plots a contour of the selected variable or named data field at the value specified on a 2D plot.
Syntax
contour
[color=<c>] [name=<c>] [print] [value=<n>]
[x=<n>][<m>|<cm>|<um>|<nm>]
[y=<n>][<m>|<cm>|<um>|<nm>]
[z=<n>][<m>|<cm>|<um>|<nm>]
Arguments
color
Specifies the line color of the contour. It can be any color supported by X11 hardware and
named in the color database.
name
Name of the data field. It allows plots to be created without using the select command.
Default: Z_Plot_Var.
print
Specifies that the contour values must be printed not plotted. The output is compatible with
xgraph. In addition, a set of Tcl lists is returned.
value
Specifies the value at which the contour line should be plotted. If boron has been selected,
a value of 1.0e16 would produce a line of constant boron concentration at that
concentration.
x, y, z
Specify the plane on which contouring is performed. In two dimensions, they need not be
specified. In three dimensions, two arguments must be specified to indicate the plane of
calculation of the contour. Default unit: .
Description
The value must be specified in the range of the computed variable. For example, if plotting log
boron, the value must be in the range 10 to 20 not 1e10 to 1e20.
µm
A: Commands
contour
Sentaurus™ Process User Guide 895
I-2013.12
The contour command assumes that the plot.2d command has been specified and that the
screen is configured to plot a 2D graphic. If this has not been set, the routine most likely will
produce unhelpful results.
Examples
Draw a line at an isoconcentration of :
contour value= 1e10
See Also
Compatibility on page 888
plot.2d on page 1066
slice on page 1141
1010

A: Commands
CutLine2D
896 Sentaurus™ Process User Guide
I-2013.12
CutLine2D
Computes the slice angle when given a cut in wafer coordinates.
Syntax
CutLine2D <x1> <y1> <x2> <y2>
Arguments
<x1> <y1> <x2> <y2>
Endpoints of the simulation cutline in wafer coordinates.
Description
Given a cut in wafer coordinates defined by the endpoints (<x1>,<y1>) and (<x2>,<y2>), the
CutLine2D command computes the slice angle.
Examples
Set the cutline for the simulation from (0,0) to (1,0):
init slice.angle= [CutLine2D 0 0 1.0 0]
See Also
Understanding Coordinate Systems on page 66
init on page 978

A: Commands
define
Sentaurus™ Process User Guide 897
I-2013.12
define
Defines a Tcl variable.
Syntax
define <name> <value>
Arguments
<name>
Any user-defined parameter name.
<value>
Any number or string value.
Description
The define command is equivalent to the Tcl command set, except that variables defined
with set are not saved or re-stored in TDR files. Variables defined using the define command
are saved or re-stored.
The define and fset commands are equivalent.
Examples
Define the Tcl variable LG, which is stored in and loaded from a TDR file. It can be used in any
Tcl expression:
define LG 0.02
See Also
Tcl documentation for description of set syntax

A: Commands
defineproc
898 Sentaurus™ Process User Guide
I-2013.12
defineproc
Defines a Tcl procedure.
Syntax
defineproc <name> { <procedure_arguments> } {
<body_of_procedure>
}
Arguments
<name>
Name of the Tcl procedure.
<procedure_arguments>
Lists the arguments of the named Tcl procedure.
<body_of_procedure>
Describes the Tcl procedure.
Description
The defineproc command is equivalent to the Tcl command proc, except that procedures
defined with proc are not saved or re-stored in TDR files. Procedures defined using
defineproc are saved or re-stored.
The defineproc and fproc commands are equivalent.
Examples
Define the Tcl procedure relerr, which is stored in and loaded from a TDR file:
defineproc relerr { newVal RefVal name my_err } {
upvar my_err fl
set denom [ expr abs($newVal)+abs($RefVal)+1e-20 ]
set deviation [expr 100*abs(($RefVal - $newVal)/$denom)]
if { $deviation > 0.5 } {
LogFile IL0 "Compare: $name= $newVal, ref= $RefVal, relerr= $deviation
\n --> failed\n"
set fl [ expr $fl+1 ]
} else {
LogFile IL0 "Compare: $name= $newVal, ref= $RefVal, relerr= $deviation
ok\n"
A: Commands
defineproc
Sentaurus™ Process User Guide 899
I-2013.12
}
}
See Also
Tcl documentation for description of proc syntax

A: Commands
DeleteRefinementboxes
900 Sentaurus™ Process User Guide
I-2013.12
DeleteRefinementboxes
Deletes a set of refinement boxes based on a pattern.
Syntax
DeleteRefinementboxes pattern=<c>
Arguments
pattern
Specifies the pattern to use.
Description
This command finds all the refinement boxes with names that match the defined pattern and
deletes them. The pattern is expanded according to standard Tcl rules.
Examples
Delete all refinement boxes that have names such as root_1, root_2, and root_3:
DeleteRefinementboxes pattern= "root*"

A: Commands
deposit
Sentaurus™ Process User Guide 901
I-2013.12
deposit
Deposits a new layer.
Syntax
deposit
( [<material>] [anisotropic | crystal | fill | fourier | isotropic]
[coord=<n>][<m>|<cm>|<um>|<nm>]
[thickness=<n>][<m>|<cm>|<um>|<nm>] )
[1D] [Adaptive][angle=<n>]
[coeffs= {<A0> <A1> <A2> ... <An>}]
[crystal.rate= {"<100>"=<n> "<110>"=<n> "<111>"=<n>}]
[direction= <list>]
[doping= <list>]
([<fieldname>] | [species=<c>]
[concentration=<n>][<m-3>|<cm-3>|<um-3>|<nm-3>])
[fields.values= <list>]
[fill.buried]
[force.full.levelset]
[mask=<c>]
[mat.coeffs= {
<mat1>= {<A0> <A1> <A2> ... <An>}
<mat2>= {<A0> <A1> <A2> ... <An>}
...
<matn>= {<A0> <A1> <A2> ... <An>} }]
[material=<c>]
[polygon= <list>]
[rate=<n>]
[region.name=<c>]
[remesh] [repair]
[sde=<c>]
[selective.materials= <list>]
[shadowing] [shadowing.nonisotropic]
[sources= {<beam1> <beam2> ... <beamn>}]
[steps=<n>] [Strained.Lattice]
[temperature=<n>][<C>|<K>]
[time=<n>][<hr>|<min>|<s>]
[type= anisotropic | crystal | directional | fill | fourier | isotropic |
polygon]|trapezoidal
Arguments
1D
Usually, a polygon deposition automatically increases the dimension to two dimensions
before performing the operation. Specify 1D to prevent this behavior.
A: Commands
deposit
902 Sentaurus™ Process User Guide
I-2013.12
Adaptive
If specified, Adaptive switches on adaptive meshing for deposition. Parameters for
adaptive meshing are described in Adaptive Refinement on page 699. The default is the
return value of pdbGet Grid Adaptive.
angle
Specifies the side wall angle in degrees when using type=trapezoidal. An angle of
is vertical; > spreads outward with increasing deposit thickness; < closes inward
with increasing deposit thickness.
anisotropic, crystal, fill, fourier, isotropic
Specify the type of deposition:
•anisotropic defaults to vertically downward deposition.
•crystal specifies crystal deposition.
•fill is used to fill the structure with the specified material up to the specified
coordinate.
•fourier specifies Fourier-type deposition. When using Fourier deposition, the
coefficients must be specified using coeffs or mat.coeffs.
•isotropic implies the same rate in all directions.
These deposition types must be used with thickness or coord, not with rate and time.
coeffs
List of single-material coefficients used in Fourier deposition.
coord
Specifies the x-coordinate for type=fill. Default unit: .
crystal.rate
List of deposition rates defined per crystallographic direction in the format:
{"<100>"=<depo_rate> "<110>"=<depo_rate> "<111>"=<depo_rate>}
direction
Specifies the direction for directional deposition as a list of x- and y-coordinates of the
deposition vector. The x-coordinate must be positive. Positive-y indicates a right-pointing
deposition beam, and negative-y indicates a left-pointing beam.
90°
90°
90°
µm
A: Commands
deposit
Sentaurus™ Process User Guide 903
I-2013.12
doping
List of names of doping profiles that have been previously defined with the doping
command.
<fieldname>, species, concentration
These arguments allow a doped layer to be deposited. species specifies the name of the
data field to be incorporated (you can add a new user species this way). Instead of
specifying species, you can specify a field name (for example, boron, arsenic,
phosphorus, and indium). The default value and unit for concentration is .
fields.values
List of parameters where the parameter name is the name of the field to be introduced in
the deposited layer, and the value is the initial value. A list of fields of any name can be
initialized with this argument and, for solution variables or stress components, units are
accepted. For example:
fields.values= {boron= 1e18<cm-3>}
fill.buried
By default, the material is deposited on the surface exposed to the upper gas region. If the
structure has buried gas bubbles, they are untouched. Use fill.buried to deposit inside
those gas bubbles.
force.full.levelset
By default, the simplest algorithm is chosen to perform the deposition. However,
sometimes the algorithm chosen generates incorrect results if the topology of the structure
is complicated. Specifying force.full.levelset switches on the general level-set
time-stepping algorithm that correctly handles these structures.
mask
Name of a mask to be used for the deposition.
NOTE The material is deposited outside of the mask. If deposition inside the
mask is required, negative must be specified in the mask command.
mat.coeffs
List of multimaterial coefficients used in Fourier deposition with a different
set of coefficients defined for each material.
1010 cm 3–
A0A1…An
,,,
A: Commands
deposit
904 Sentaurus™ Process User Guide
I-2013.12
<material>
Allows the specification of the deposited material. For information about specifying
materials, see Material Specification on page 52.
material
Specifies the material to be deposited. Overrides the <material> specification.
polygon
Specifies a list of x- and y-coordinates for the deposition. This is used only for 2D
deposition with type=polygon. The list of coordinates must define a single polygon with
no self-intersections. The first and last points are connected implicitly to close the polygon.
The specified material is deposited inside the polygon. The default unit for the coordinates
is .
rate
Deposition rate. Default unit: /minute.
region.name
Name of the region created by the deposit command. The name must not contain an
underscore (_) or a period (.) because these characters have special meaning.
remesh
Performs a remeshing after the deposition.
repair
In 3D MGOALS mode, small regions are removed automatically by default. Sometimes,
this causes small gas bubbles in the structure or other problems. Use !repair to switch
off the small region removal.
sde
Specifies the arguments and algorithms for 3D Sentaurus Structure Editor. By default,
arguments such as rate, thickness, time, and type are translated into appropriate
Sentaurus Structure Editor commands.
If an algorithm is specified, it overwrites the algorithm used by default for isotropic or
anisotropic deposition, for example:
sde= {"algorithm" "lopx"}
sde= {"algorithm" "lopx" "radius" 0.07}
µm
µm
A: Commands
deposit
Sentaurus™ Process User Guide 905
I-2013.12
selective.materials
Specifies that deposition will occur only on a list of selected materials. For 2D simulations,
MGOALS is used and multiple materials can be selected. For 3D simulations, the
Sentaurus Structure Editor interface is called and only one material can be selected. In
either case, only one material can be deposited.
shadowing
In two dimensions, switches on the inclusion of shadowing effects if
force.full.levelset is specified or for Fourier deposition. The visibility of each
surface area to each beam is calculated at every level-set time step. In 3D MGOALS mode,
this argument enables shadowing effects on both directional and anisotropic deposition.
The interface to Sentaurus Structure Editor ignores shadowing.
shadowing.nonisotropic
Use instead of shadowing to allow the 0th-order Fourier coefficient to deposit in areas
where the beam is shadowed.
sources
Defines the source beams for level-set deposition.
steps
Subdivides a deposition into more than one step. If necessary, stress relaxation is calculated
at the end of each step. Default: 1.
Strained.Lattice
Specifies strained deposition.
temperature
Deposition temperature used for stress relaxation only. Default value and unit: .
thickness
Thickness of the deposited layers. Default unit: .
time
Deposition time. It must be specified if the rate argument is used. Default unit: minute.
26.85°C
µm
A: Commands
deposit
906 Sentaurus™ Process User Guide
I-2013.12
type
Explicitly specifies the type of deposition to be performed:
•type=anisotropic performs deposition in the vertical direction only, which must be
used with the rate and time arguments.
•type=crystal performs crystallographic deposition. The crystal.rate argument
also must be specified.
•type=directional performs anisotropic etching using a specified direction.
•type=fill performs a fill of a specified material up to the coordinate specified with
the coord argument.
•type=fourier performs Fourier deposition, which requires the coefficients to be
specified with either coeffs or mat.coeffs.
•type=isotropic performs isotropic deposition, which must be used with the rate
and time arguments.
•type=polygon performs a polygonal deposition (in two dimensions), which requires
the polygon argument.
•type=trapezoidal performs a trapezoidal deposition, which requires the angle
and thickness arguments, and recommended selective.materials. This is
supported in 3D only.
Arguments: Deprecated
fields
List of fields to be introduced in the deposited layer. The data values for these fields are
specified with the values argument. For stresses, use the field names StressELXX,
StressELXY, StressELYY, StressELZZ, StressELXZ, StressELYZ. It is not
necessary to specify all stress components. Those not specified are assumed to be zero
initially and are updated during the stress rebalance at the end of the deposition.
values
Data values of the fields introduced in the deposited layer by the fields argument.
Description
This command simulates a deposition step.
A: Commands
deposit
Sentaurus™ Process User Guide 907
I-2013.12
Examples
Isotropic deposition of a oxide layer:
deposit thickness= 0.2 oxide isotropic
Same as above; thickness is defined by rate and time:
deposit rate= {0.2} material= {oxide} type= isotropic time= 1
Add an intrinsic isotropic stress of to the deposited nitride layer before the post-
deposition mechanics rebalancing step:
deposit thickness= 0.1 nitride \
fields.values= {StressELXX= 1e9<Pa> StressELYY= 1e9<Pa> \
StressELZZ= 1e9<Pa>}
See Also
doping on page 917
mask on page 1020
mgoals on page 1037
0.2 µm
109Pa

A: Commands
diffuse
908 Sentaurus™ Process User Guide
I-2013.12
diffuse
Simulates thermal annealing, densification, and any material growth process during annealing
– oxidation, silicidation, and epitaxy.
Syntax
diffuse
(temp.ramp=<c> |
time=<n>[<hr>|<min>|<s>] temperature=<n>[<C>|<K>])
[Adaptive]
[<ambient> O2 | H2O | N2O | N2 | H2 | HCl | Epi | LTE]
[angles.factors= {
[<interface_mat1>= <list>]
[<interface_mat2>= <list>] }]
[auto.doping= <list>]
[coeffs= {<A0> <A1> <A2> ... <An>}]
[crystal.rate= {"<100>"=<n> "<110>"=<n> "<111>"=<n>}]
[delNT=<n>][<C>|<K>]
[delT=<n>][<C>|<K>]
[delTox=<n>][<C>|<K>]
[density.increase= <regionName>=<n> | <material>=<n>]
[deposit.type=<c>]
[epi.doping= <list>]
[epi.doping.final= <list>]
[epi.layers=<i>]
[epi.model=<i>]
[epi.resist= {[<dopant1=<n>[<ohm-cm>]] [dopant2=<n>[<ohm-cm>]] ...}]
[epi.thickness=<n>][<m>|<cm>|<um>|<nm>]
[eqnInfo]
[flow<ambient>=<n>][<l/min>]
[flows= {
[<ambient1>=<n>][<l/min>]
[<ambient2>=<n>][<l/min>]
...}]
[gas.flow=<c>]
[iiiv.epi=<c>]
[init=<n>][<hr>|<min>|<s>]
[isolve] [ISSG]
[kmc] [kmc.stress] [laser] [lkmc]
[mat.coeffs= {
<mat1>= {<A0> <A1> <A2> ... <An>}
<mat2>= {<A0> <A1> <A2> ... <An>}
...
<matn>= {<A0> <A1> <A2> ... <An>} }]
[maxstep=<n>][<hr>|<min>|<s>]
[mgoals.native]
A: Commands
diffuse
Sentaurus™ Process User Guide 909
I-2013.12
[minT=<n>][<C>|<K>]
[movie=<c>]
[p<ambient>=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[partial.pressure= {
[<ambient1>=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[<ambient2>=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
...}]
[pressure=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[ramprate=<n>][<C/s>|<K/s>|<C/min>|<K/min>]
[reload] [reloadHeat] [reloadReact]
[repair]
[sources= {<beam1> <beam2> ... <beamn>}]
[stress.relax]
[t.final=<n>][<C>|<K>]
[write.temp.file=<c>]
Arguments
Adaptive
If specified, Adaptive switches on adaptive meshing for this diffusion step. Parameters
for adaptive meshing are described in Adaptive Meshing during Diffusion on page 707.
The default is the return value of pdbGet Grid Adaptive.
<ambient>
Shorthand specification to set the ambient partial pressure the same as the total pressure. If
an ambient is specified this way, it must be the only ambient set in the diffuse command.
In addition to the oxidation-type ambients (O2, H2O, N2O, ISSG), the epitaxial ambients
Epi and LTE are available.
angles.factors
Specifies interface-specific anisotropic epi growth rate factors as a numeric list. This
argument specifies a piecewise linear growth rate factor versus angle for each growing
interface (the factors must be between 0 and 1). For example, to create a silicon facet
and a polysilicon facet, use:
angles.factors= {
EpiOnSilicon_Gas= {0.0 1.0 25.0 1.0 30 0.0}
EpiOnPolySilicon_Gas= {0.0 1.0 35.0 1.0 40 0.0}
}
auto.doping
List of species for which the auto-doping model will be switched on during epitaxial
growth.
30°
40°
A: Commands
diffuse
910 Sentaurus™ Process User Guide
I-2013.12
coeffs
List of single-material coefficients used in Fourier deposition when
epi.model=1 and deposit.type=fourier.
crystal.rate
List of etching rates defined per crystallographic direction in the format:
{"<100>"=<dep_rate> "<110>"=<dep_rate> "<111>"=<dep_rate>}
used for crystallographic deposition when epi.model=1 and deposit.type=crystal.
delNT
Defines the maximum temperature step during a temperature ramp-down if specified.
Default unit: degree Celsius. It also can be defined globally with the command:
pdbSet Diffuse delNT {<n>}
delT
Defines the maximum temperature step during a temperature ramp-up if specified. Default
unit: degree Celsius. It also can be defined globally with the command:
pdbSet Diffuse delT {<n>}
delTox
Defines the maximum temperature step during a temperature ramp for oxidation or growth
if specified. Default unit: degree Celsius. It also can be defined globally with the command:
pdbSet Diffuse delTox {<n>}
density.increase
Density increase. The value can be specified per region <regionName>=<n> or per
material <material>=<n>.
deposit.type
When epi.model=1, epitaxy is solved as a series of alternating deposition and diffuse
steps. This argument specifies the deposition type. The allowed values are:
•isotropic (default)
•crystal (in which case, crystal.rate must be specified)
•fourier (in which case, either coeffs or mat.coeffs must be specified)
A0A1…An
,,,
A: Commands
diffuse
Sentaurus™ Process User Guide 911
I-2013.12
epi.doping
List of parameters where the parameter name is the name of the species to be initialized
and the value is the initial value. A list of fields of any name can be initialized with this
argument and, for solution variables, units are accepted. For example:
epi.doping= {boron= 1e18<cm-3> GSize= 1<nm> myfield= 1}
epi.doping.final
List of parameters where the parameter name is the name of the species to be initialized
and the value is the final value. A list of fields of any name can be initialized with this
argument and, for solution variables, units are accepted. For example:
epi.doping.final= {boron= 1e18<cm-3> GSize= 1<nm> myfield= 1}
epi.layers
Number of layers of mesh lines required during epitaxial growth (for both epi.model=0
and epi.model=1). The default is –1, which indicates that 10 layers should be used if
epi.model=1, and 40 layers should be used if epi.model=0. However, if the global
parameter given by:
pdbSet Silicon Grid epi.perp.add.dist <n>
is set to a positive integer, epi.layers is ignored (whether set or not), and
epi.perp.add.dist is used to determine the distance between mesh lines.
For epi.model=1, the number of layers is adjusted if the deposited layer thickness is less
than the parameter Grid MinEpiDepositThickness in SDE mode or
Grid MinEpiDepositThicknessMGoals3D in 3d MGOALS mode.
epi.model
Specifies the epitaxial method to use:
•epi.model=0 (default) applies a moving-boundary algorithm similar to oxidation.
•epi.model=1 uses alternating doped deposition and inert annealing steps.
In three dimensions, epi.model=0 is not as stable or robust as epi.model=1, so
epi.model=1 is recommended.
epi.resist
List of parameters with dopant name and resistivity to calculate the background dopant
concentration. If more than one dopant name appears in the list, the doping concentration
is calculated individually for each dopant by ignoring the other ones.
epi.thickness
Sets the epitaxial layer thickness to be deposited. Default unit: .
µm
A: Commands
diffuse
912 Sentaurus™ Process User Guide
I-2013.12
eqnInfo
Prints the equation updates to the log file during the Newton iteration.
flow<ambient>, flows
List of gas flows in the reaction chamber. The gas flows are used to computed the partial
pressures of the active ambients (those causing material growth). Flows can be specified
using either a parameter name composed of flow + <ambient> (for example, flowO2 and
flowHCl where O2 and HCl are ambient names) or flows that takes a list of parameters
with the names of the ambients, for example:
flows= {O2= 1.0<l/min> HCl= 1.0<l/min>}
Table 64 on page 617 lists the available ambients, but this list can be extended by using the
ambient command. When a gas flow is specified as a combination of flows (and not when
using partial pressures), a complete reaction of the ambients is assumed to occur, for
example, O2 + 2H2 -> 2H2O. Besides gas reactions, the addition of inert gases changes the
partial pressure of the material-growing ambients. For example, if the flows of only N2 and
O2 are specified and are equal, the partial pressure of O2 will be <total pressure>/2.0
where <total pressure> is given by the pressure argument.
NOTE Flows and partial pressures must not be specified together in the same
gas_flow command.
gas.flow
Specifies a gas flow to be used for this diffusion step.
NOTE Do not use with other gas_flow command arguments or with the
temp_ramp command.
iiiv.epi
Specifies the name of an III-V material for epitaxial growth. An ambient should not be
specified if this parameter is specified.
init
First time step. The default is 0.0001 s, which is sometimes inappropriate for defect
simulations, particularly in cases of damage. Default unit: minute.
isolve
Switches off the initial solve for models that must have an equation solved to set the initial
conditions. In these cases, you can set an initial condition and switch off the default
initialization.
A: Commands
diffuse
Sentaurus™ Process User Guide 913
I-2013.12
ISSG
Switches on in situ steam-generated (ISSG) oxidation.
kmc
Allows the diffuse command to use Sentaurus Process Kinetic Monte Carlo (Sentaurus
Process KMC).
kmc.stress
Switches on the stress effect for Sentaurus Process KMC.
laser
Switches on laser annealing.
lkmc
Invokes the lattice kinetic Monte Carlo (LKMC) model during epitaxial growth without the
need to use a SetAtomistic simulation. The generation of the grown epitaxial surface is
performed by atomistic LKMC in a way that is transparent to users. Use lkmc for standard
continuum simulations with epitaxial LKMC growth.
mat.coeffs
List of multimaterial coefficients used in Fourier deposition when
epi.model=1 and deposit.type=fourier.
maxstep
Maximum time step. Default unit: minute.
mgoals.native
Use the MGOALS library to deposit a native oxide. This is often helpful with complex
structures that have several triple points.
minT
Minimum annealing temperature. If the diffusion temperature falls below this value, the
diffusion solver is switched off. If it occurs during a ramp, the time-stepping is altered such
that diffusion switches on or off exactly at this temperature. Default value and unit: .
movie
Allows you to specify actions that occur during the annealing step. For every time step of
the diffusion, the string value of the movie argument is executed.
A0A1…An
,,,
450°C
A: Commands
diffuse
914 Sentaurus™ Process User Guide
I-2013.12
p<ambient>, partial.pressure
List of the partial pressures of active ambients. Partial pressure specifications must not be
used with flow, flow<ambient>, or pressure specifications. Specify partial pressures
using either a parameter name composed of p + <ambient> (for example, pO2 and pN2O
where O2 and N2O are active ambient names) or partial.pressure that takes a list of
parameters with the names of the ambients, for example:
partial.pressure= {O2= 1.0<atm> N2O= 1.0<atm>}
Table 64 on page 617 lists the available ambients, but this list can be extended by using the
ambient command. These partial pressures are assumed to contribute to the oxidation or
user-defined reaction processes. No reaction between the species is assumed. Default unit:
atm.
NOTE Only the partial pressures of the active ambients are used directly in the
oxidation reaction equations, so setting the partial pressure of inactive
(in the sense that they cause the material growth reaction) ambients such
as N2 or HCl has no effect.
pressure
The (total) pressure of the ambient gas. This setting takes effect only if flows or
flow<ambient> is defined explicitly. If gas.flow is specified, the pressure is set in the
corresponding gas_flow command. Default value and unit: 1.0 atm.
ramprate
Temperature change during anneal. Default value and unit: .
reload, reloadHeat, reloadReact
Allows the diffusion, laser annealing, or reaction equation to be parsed at each time step.
repair
In 3D MGOALS mode, small regions are removed automatically by default. Sometimes,
this causes small gas bubbles in the structure or other problems. Use !repair to switch
off the removal of small regions.
sources
Defines deposition sources used in Fourier deposition when epi.model=1 and
deposit.type=fourier are specified.
stress.relax
Switches off relaxation of stresses during diffusion with an inert ambient. The default is
true for two dimensions and false for three dimensions.
0.0°C/s
A: Commands
diffuse
Sentaurus™ Process User Guide 915
I-2013.12
t.final
Final temperature for a temperature ramp-up or ramp-down. It is used if ramprate is not
given. The ramp time is calculated automatically. Default unit: degree Celsius.
temp.ramp
Name of a temperature ramp created with the temp_ramp command.
temperature
Annealing temperature. Default unit: degree Celsius.
time
Annealing time. Default unit: minute.
write.temp.file
Stores the thermal profile created during laser annealing. The format of the file is two
columns: time (in seconds) and temperature (in degree Celsius). This file can be used to
create a temp_ramp to allow subsequent simulations to use the computed temperature
profile without the need to resimulate laser annealing.
Arguments: Deprecated
epi.dopants
List of the data fields to be incorporated into the epitaxial layer. Deprecated in favor of
epi.doping.
Description
This command performs annealing (either continuum or KMC) or, if the diffusion time is set
to 0, it activates dopants and performs a stress update. The command arguments set diffusion
conditions as well as time-stepping options.
Diffusion model and parameter setting are performed with the pdbSet command. The basic
settings are:
pdbSet <material> Dopant DiffModel <model>
where <model> can be one of Constant, Fermi, Pair, React, ChargedFermi,
ChargedPair, or ChargedReact.
Temperature ramps are specified by first creating a list of ramping steps using the temp_ramp
command. Then, the ramp is applied using temp.ramp. All temp_ramp command arguments
can be specified with the diffuse command.
A: Commands
diffuse
916 Sentaurus™ Process User Guide
I-2013.12
To specify an oxidizing ambient, there are different methods:
■Use the shorthand <ambient> flags.
■Use flow<ambient> or p<ambient>.
■Use the gas_flow command and set the gas.flow argument to the name set in the
gas_flow command.
■Use gas_flow inside the temp_ramp command, and use temp.ramp to set the name of
the temp_ramp to be used.
Table 64 on page 617 lists the available ambients and includes O2, H2O, HCl, N2, H2, and N2O,
which can be used in oxidation specification, as well as two epi ambients Epi and LTE for
specifying epitaxial growth. For more information on Epi and LTE, see Epitaxy on page 282.
Examples
A simple low temperature anneal for 30 s:
diffuse time= 30<s> temp= 900
An oxidation step for a thick isolation oxide:
diffuse time= 90 temperature= 1000 H2O
A diffusion step using the temperature ramp named spike:
diffuse temp.ramp= spike
See Also
ambient on page 881
gas_flow on page 935
temp_ramp on page 1166
term on page 1173
900°C

A: Commands
doping
Sentaurus™ Process User Guide 917
I-2013.12
doping
Defines a named piecewise linear doping profile that can be used with the deposit command.
Syntax
doping
field=<c> name=<c>
depths= <list>
values= <list>
stress.values= <list>
[clear] [list] [location= vertex | element] [log.grad]
Arguments
clear
Clears all doping specifications.
depths
Numeric list of the depths at which the values are applied.
field
Name of the field; it can be the name of the dopant. For stresses, use the field names:
•StressELXX
•StressELXY
•StressELYY
•StressELZZ
•StressELXZ
•StressELYZ
It is not necessary to specify all stress components. Those not specified are assumed to be
zero initially and are updated during the stress rebalance at the end of deposition.
list
Returns the names of all doping specifications.
location
Location where the field is to be applied. Default: vertex.
A: Commands
doping
918 Sentaurus™ Process User Guide
I-2013.12
log.grad
Specifies a piecewise logarithmic doping profile.
name
Name of the profile to be used in the deposit command.
stress.values
Numeric list of the values of the stress field. The default unit is .
values
Numeric list of the values of the dopant field. The default unit is .
Description
This command allows a doping profile specification that can be used inside the deposit
command to add doping and other fields to the newly deposited layer (on either vertices or
elements).
Examples
Create a doping profile definition with the name init_boron that consists of a boron profile
linearly increasing from at the starting surface to at and beyond in the
deposited layer. This doping profile definition can be used with the deposit command to
create the specified profile:
doping name= init_boron field= Boron values= {1e10 1e20} depths= {0 0.1}
Add an intrinsic isotropic stress of to the deposited layer before the post-
deposition mechanics rebalancing step:
doping name= film_Sxx field= StressELXX stress.values= {1e9 1e9} depths= {0 1}
doping name= film_Syy field= StressELYY stress.values= {1e9 1e9} depths= {0 1}
doping name= film_Szz field= StressELZZ stress.values= {1e9 1e9} depths= {0 1}
See Also
deposit on page 901
dyn/cm2
cm 3–
110
×10
120
×10
0.1 µm
109dyn/cm2

A: Commands
element
Sentaurus™ Process User Guide 919
I-2013.12
element
Extracts the grid for a specified material and returns the grid as a list of coordinates.
Syntax
element <material> [region]
Arguments
<material>
Name of the material. For information about specifying materials, see Material
Specification on page 52.
region
Limits output to only one region if specified.
Description
This command can be used to plot the grid. It returns a list of coordinates that define the grid.
Each of the outer lists makes up a continuous line through the grid. Each inner list contains
coordinate pairs in order for that line.
NOTE This command is not available for 3D simulations.
Examples
Return the grid of oxide material:
element oxide

A: Commands
Enu2G
920 Sentaurus™ Process User Guide
I-2013.12
Enu2G
Computes the shear modulus from Young’s modulus and the Poisson ratio.
Syntax
Enu2G <n> <n>
Arguments
<n>
The first value is Young’s modulus.
The second value is the Poisson ratio.
Description
The same units are assumed for all moduli.
Examples
Compute the shear modulus from Young’s modulus (1.620e12 dyn/cm2) and the Poisson ratio
(0.28):
Enu2G 1.620e12 0.28

A: Commands
Enu2K
Sentaurus™ Process User Guide 921
I-2013.12
Enu2K
Computes the bulk modulus from Young’s modulus and the Poisson ratio.
Syntax
Enu2K <n> <n>
Arguments
<n>
The first value is Young’s modulus.
The second value is the Poisson ratio.
Description
The same units are assumed for all moduli.
Examples
Compute the bulk modulus from Young’s modulus (1.620e12 dyn/cm2) and the Poisson ratio
(0.28):
Enu2K 1.620e12 0.28

A: Commands
equation
922 Sentaurus™ Process User Guide
I-2013.12
equation
Allows test parsing and resolution of an equation string.
Syntax
equation eqn=<c> [nodal]
Arguments
eqn
String to be checked.
nodal
If specified, nodal returns the nodal part of the string specified by eqn.
Description
The equation string is parsed, broken into pieces, and derivatives are taken and printed. This
command is useful for debugging problems with the resolver and parsing, as equation strings
can be tried before being run.
Examples
Parse and resolve the string exp(Potential*$Vti):
equation eqn= "exp(Potential*$Vti)"
See Also
solution on page 1145

A: Commands
etch
Sentaurus™ Process User Guide 923
I-2013.12
etch
Removes part or all of an exposed layer.
Syntax
etch
[1D] [Adaptive] [ambient.rate=<n>] [angle=<n>]
[angles.rates= {
matA= {angleA0 rateA0 angleA1 rateA1 ... angleAn rateAn}
matB= {angleB0 rateB0 angleB1 rateB1 ... angleBn rateBn}
... }]
[anisotropic | cmp | isotropic | trapezoidal]
[bottom.angle=<n>] [bottom.thickness=<n>]
[coeffs= {<A0> <A1> <A2> ... <An>}]
[coord=<n>][<m>|<cm>|<um>|<nm>]
[crystal.rate= {"<100>"=<n> "<110>"=<n> "<111>"=<n>}]
[direction= <list>]
[etchstop= {<mat1> <mat2> ...} [etchstop.overetch=<n>]]
[force.full.levelset]
[isotropic.overetch=<n>]
[levelset.upwind]
[mask=<c>]
[mat.coeffs= {
<mat1>= {<A0> <A1> <A2> ... <An>}
<mat2>= {<A0> <A1> <A2> ... <An>}
...
<matn>= {<A0> <A1> <A2> ... <An>} }]
[<material>]
[material= <list>]
[polygon= <list>]
[rate= <list>]
[remesh] [repair] [roundness=<n>]
[sde=<c>]
[shadowing] [shadowing.nonisotropic]
[sources= {<beam1> <beam2> ... <beamn>}]
[temperature=<n>][<C>|<K>]
[thickness=<n>][<m>|<cm>|<um>|<nm>]
[time=<n>][<hr>|<min>|<s>]
[type= anisotropic | cmp | crystal | directional | fourier | isotropic |
polygon | trapezoidal]
[undercut=<n>]
A: Commands
etch
924 Sentaurus™ Process User Guide
I-2013.12
Arguments
1D
Usually, a polygon etching automatically increases the dimension to two dimensions
before performing the operation. Set 1D to prevent this behavior.
Adaptive
If specified, Adaptive switches on adaptive meshing for this etching step. Parameters for
adaptive meshing are described in Adaptive Refinement on page 699. The default is the
return value of pdbGet Grid Adaptive.
ambient.rate
Used with type=trapezoidal and force.full.levelset to approximate the
underetch effect.
angle
Specifies the etching angle for type=trapezoidal.
angles.rates
Specifies a set of angle rate pairs that gives a piecewise linear etch rate versus angle. To be
stable, smaller angles must have a higher etch rate. Specify the etching rate to go to zero
above some angle produces facets.
anisotropic, cmp, isotropic, trapezoidal
Specifies the type of etching:
•anisotropic: Anisotropic etching.
•cmp: Chemical-mechanical polishing (CMP).
•isotropic: Isotropic etching.
•trapezoidal: Trapezoidal etching.
bottom.angle
Specifies the angle for the bottom of a trapezoidal etching in three dimensions.
bottom.thickness
Specifies the thickness for the bottom of a trapezoidal etching in three dimensions.
coeffs
List of single-material coefficients used in Fourier etching.
A0A1…An
,,,
A: Commands
etch
Sentaurus™ Process User Guide 925
I-2013.12
coord
The x-coordinate to work with type=cmp. Default unit: .
crystal.rate
List of etching rates defined per crystallographic direction in the format:
{"<100>"=<etch_rate> "<110>"=<etch_rate> "<111>"=<etch_rate>}
direction
Numeric list of x-, y-, and z-values specifying the etching direction for
type=directional.
NOTE All three values must always be specified. In two dimensions, the z-
value must always be zero.
etchstop
Materials, instead of time, can be given as etch-stopping criteria. In this case, etching
continues until any of the given etchstop materials is exposed. An additional overetch is
performed, with a time equal to etchstop.overetch (default: 10%) multiplied by the
accumulated time required to expose the first etchstop material.
In three dimensions, etchstop is ignored when Sentaurus Structure Editor is used.
NOTE Materials, in addition to time, can be given as etch-stopping criteria for
Fourier etching. If both time and etchstop are given, Fourier etching
stops when either of the two criteria is first met.
etchstop.overetch
When etchstop is defined, an additional overetch is performed when the first etchstop
material becomes exposed to gas. The duration of this overetch step is the already
performed etching time multiplied by etchstop.overetch. Default: 0.1 (10%).
force.full.levelset
By default, the simplest algorithm is chosen to perform the etching. However, sometimes,
the algorithm chosen generates incorrect results if the topology of the structure is
complicated. Specifying force.full.levelset switches on the general level-set time-
stepping algorithm, which correctly handles these structures.
isotropic.overetch
Specifies a required amount of isotropic etching following anisotropic etching. The
thickness is specified as a fraction of the anisotropic component. This argument is not
implemented in MGOALS3D.
µm
A: Commands
etch
926 Sentaurus™ Process User Guide
I-2013.12
levelset.upwind
Used with force.full.levelset to choose the Upwind formulation of the full level-
set algorithm. The Upwind algorithm is less stable and less robust than the Lax–Friedrichs
algorithm, which is the default.
mask
Name of the mask to be used for the etching.
mat.coeffs
List of multimaterial coefficients used in Fourier etching with a different set
of coefficients defined for each material.
<material>
Specifies the material to be etched. For information about specifying materials, see
Material Specification on page 52.
material
String list of materials for multimaterial etching.
polygon
Numeric list of x- and y-coordinates for type=polygon. The list of coordinates {x0 y0
x1 y1 x2 y2 ... xn yn} defines a single polygon that must not be self-intersecting.
The first and last points are connected implicitly to close the polygon. The material inside
the polygon is etched. The default unit of the coordinates is .
rate
Numeric list of etching rates. Default unit: /minute.
remesh
Performs remeshing after the etching.
repair
In MGOALS3D mode, small regions are removed automatically by default. Sometimes,
this causes small gas bubbles in the structure or other problems. Use !repair to switch
off the small region removal.
roundness
Tuning argument for the curvature of the etch sidewalls in the case of trapezoidal etching
when force.full.levelset is used. The default value is 1.0. Increased values up to 2.0
or 3.0 increase the curvature of the etch sidewall calculated by the level-set solver.
A0A1…An
,,,
µm
µm
A: Commands
etch
Sentaurus™ Process User Guide 927
I-2013.12
sde
String used to specify parameters and algorithms for 3D Sentaurus Structure Editor. By
default, arguments such as rate, thickness, time, and type are translated into
appropriate Sentaurus Structure Editor commands. If an algorithm is specified using sde,
it overwrites the algorithm used by default for isotropic or anisotropic etching, for example:
sde= {"algorithm" "lopx"}
sde= {"algorithm" "lopx" "radius" 0.07}
shadowing
In two dimensions, shadowing switches on the inclusion of shadowing effects if
force.full.levelset is specified or for Fourier etching. The visibility of each surface
area to each beam is calculated at every level-set time step.
In MGOALS3D, shadowing enables shadowing effects on both directional and
anisotropic etching.
The Sentaurus Structure Editor interface ignores shadowing.
shadowing.nonisotropic
Used instead of shadowing to allow the 0th-order Fourier coefficient to etch areas where
the beam is shadowed.
sources
Defines the etching source beams for level-set etching.
temperature
Etching temperature. Default value and unit: .
thickness
Thickness that is removed in the etching. Default unit: .
time
Refers to the etching time. It must be specified if rate is used. Default value and unit:
1.0 minute.
NOTE If both time and etchstop are given for Fourier etching, the Fourier
etching stops when either of these two criteria is met.
26.85°C
µm
A: Commands
etch
928 Sentaurus™ Process User Guide
I-2013.12
type
Specifies the type of etching to be performed:
•type=anisotropic performs etching in the vertical direction only and must be used
with the rate and time arguments.
•type=cmp performs CMP and is used with the argument coord.
•type=crystal performs etching in two dimensions or three dimensions using etching
rates dependent on the crystallographic direction defined by the crystal.rate
argument.
•type=directional performs anisotropic etching in other directions and must be
used with the direction, rate, and time arguments.
•type=fourier performs angle-dependent etching in two dimensions or three
dimensions where the rate-versus-angle functions are defined by a cosine series using
coeffs or mat.coeffs.
•type=isotropic performs isotropic etching, which must be used with the rate and
time arguments.
•type=polygon performs polygonal etching in two dimensions and is used with the
polygon argument.
•type=trapezoidal performs TSUPREM-4-like trapezoidal etching in two
dimensions defined by the thickness, undercut, and angle arguments, and in
three dimensions defined by the thickness, angle, bottom.thickness, and
bottom.angle arguments.
undercut
Distance to etch below the nonetchable material in 2D trapezoidal etching.
Description
This command etches a layer exposed to the top gas. Several materials can be etched at the
same time. There are different modes to perform etching:
■The MGOALS mode uses either an analytic or a level-set method performed by the
MGOALS library.
■A general level-set time-stepping mode can handle more sophisticated etching capabilities
such as multimaterial etching, Fourier etching, multiple beam, and shadowing.
A: Commands
etch
Sentaurus™ Process User Guide 929
I-2013.12
Examples
Etch a silicon layer anisotropically in the direction indicated by direction. A mask
called m1 is used during etching:
etch time= 2.0 rate= {0.1} material= {silicon} type= directional \
direction= {1.0 1.0 0.0} mask= m1
Etch silicon for 0.1 minute at a rate of per minute, using source1 as the etching beam,
including shadowing effects, and with a Fourier response to the etchant defined by
and :
beam name= source1 factor= 1.0 incidence= -30
etch material= {silicon} shadowing sources= {source1} type= fourier \
coeffs= {0.1 0 0 1} time= 0.1
Define multimaterial Fourier etching for 0.2 minutes. The Fourier coefficients for each material
are given separately within mat.coeffs:
beam name= source_beam factor= 1.0 incidence= -30
etch material= {Silicon Nitride Oxide PolySi} sources= {source_beam} \
mat.coeffs= { Silicon= {0 0.5} Nitride= {0 1} Oxide= {0 0.75} \
PolySilicon= {0.5} } type= fourier time= 0.2
See Also
deposit on page 901
mask on page 1020
mgoals on page 1037
0.2 µm
1µm
A00.1=
A31=


A: Commands
extract
Sentaurus™ Process User Guide 931
I-2013.12
extract
Extracts historical data during a diffuse step.
Syntax
extract
[clear]
[command= {<c> <c> ...}]
[name=<c>] [print]
[syntax.check.value=<c>]
Arguments
clear
Clears the stored historical data.
command
List of commands for data interpolation.
name
Name of data extraction.
print
Returns the extracted data values as a Tcl list with all interpolated variables.
syntax.check.value
Sets a value to be returned only during syntax-checking mode. Sometimes, the value
returned by a command can cause a false syntax-check error because the value returned by
the command would not be the value during the normal run mode. Setting this value avoids
such problems.
Description
Specifies the commands for data extraction during a diffuse step.
The extracted historical data can be returned as a Tcl list. Typically, the extracted data is in
internal units. Internal units are CGS; for example, the unit for stress is dyn/cm2.
A: Commands
extract
932 Sentaurus™ Process User Guide
I-2013.12
Examples
Extract and store the boron concentration at the position in the silicon for each diffuse
step:
extract name= etest command= {select z= Boron interpolate Silicon x= 0.04}
Return the extracted data values with the extraction name etest in a Tcl list (for example, if
two values are extracted in etest, the Tcl list takes the format <time1> <value1_1>
<value2_1> <time2> <value1_2> <value2_2> ...):
extract print name= etest
Clear all stored historical data:
extract clear
See Also
Extracting Values during diffuse Step: extract on page 854
interpolate on page 991
0.04 µm
A: Commands
fbreak
Sentaurus™ Process User Guide 933
I-2013.12
fbreak
Starts interactive mode.
Syntax
fbreak
Description
This command interrupts the execution of command files and starts the interactive mode.
See Also
exit on page 930
fcontinue
fcontinue
Resumes execution of command files.
Syntax
fcontinue
Description
This interactive mode command resumes the execution of command files in batch mode.
See Also
exit on page 930
fbreak

A: Commands
fexec
934 Sentaurus™ Process User Guide
I-2013.12
fexec
Executes system commands.
Syntax
fexec
Description
Executes system calls through the Tcl command exec (with exactly the same syntax). Using
fexec, the system calls are not executed during syntax-checking as they would be if the plain
exec command were used.
Examples
List the contents of the current directory:
fexec ls
See Also
Tcl documentation for description of exec syntax
fproc
The defineproc and fproc commands are equivalent.
See defineproc on page 898.
fset
The define and fset commands are equivalent.
See define on page 897.

A: Commands
gas_flow
Sentaurus™ Process User Guide 935
I-2013.12
gas_flow
Specifies a gas mixture for use with the diffuse or temp_ramp command.
Syntax
gas_flow
(clear | list | name=<c> | print)
[<ambient>]
[flow<ambient>=<n>][<l/min>]
[flows= {
[<ambient1>=<n>][<l/min>]
[<ambient2>=<n>][<l/min>]
...}]
[ISSG]
[p<ambient>=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[partial.pressure= {
[<ambient1>=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[<ambient2>=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
...}]
[pressure=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
Arguments
<ambient>
Shorthand specification to set the ambient partial pressure the same as the total pressure.
Only active ambients can be specified in this way, and only one ambient can be specified.
The active ambients are O2, H2O, and N2O.
clear
Clears the global list of gas mixtures.
flow<ambient>, flows
List of gas flows in the reaction chamber. The gas flows are used to compute the partial
pressures of the active ambients (those causing material growth). You can specify flows
using either a parameter name composed of flow + <ambient> (for example, flowO2 and
flowHCl where O2 and HCl are ambient names) or flows that takes a list of parameters
with the names of the ambients, for example:
flows= {O2= 1.0<l/min> HCl= 1.0<l/min>}
Table 64 on page 617 lists the available ambients, but this list can be extended by using the
ambient command. When a gas flow is specified as a combination of flows (and not when
using partial pressures), a complete reaction of the ambients is assumed to occur, for
example, O2 + 2H2 -> 2H2O.
A: Commands
gas_flow
936 Sentaurus™ Process User Guide
I-2013.12
Besides gas reactions, the addition of inert gases changes the partial pressure of the
material-growing ambients. For example, if the flows of only N2 and O2 are specified and
are equal, the partial pressure of O2 will be <total pressure>/2.0 where <total
pressure> is given by pressure.
NOTE Flows and partial pressures must not be specified together in the same
gas_flow command.
ISSG
Switches on in situ steam-generated oxidation.
list
Generates a list of gas mixtures and returns a Tcl list that can be operated on as such. The
default action for commands is to print the return, so if no handling is required, this
argument prints a list of names of defined gas mixtures. If a name is specified, that gas
mixture only is listed with details.
name
Identifies the gas mixture description and specifies it in a diffuse or temp_ramp
command.
p<ambient>, partial.pressure
List of the partial pressures of active ambients. Partial pressure specifications must not be
used with flows, flow<ambient>, or pressure specifications. You can specify partial
pressures using either a parameter name composed of p + <ambient> (for example, pO2
and pN2O where O2 and N2O are active ambient names) or partial.pressure that takes
a list of parameters with the names of the ambients, for example:
partial.pressure= {O2= 1.0<atm> N2O= 1.0<atm>}
Table 64 on page 617 lists the available ambients, but this list can be extended by using the
ambient command. These partial pressures are assumed to contribute to the oxidation or
user-defined reaction processes. No reaction between the species is assumed. Default unit:
atm.
NOTE Only the partial pressures of the active ambients are used directly in the
oxidation reaction equations, so setting the partial pressure of inactive
(in the sense that they cause the material growth reaction) ambients such
as N2 or HCl has no effect.
pressure
The (total) pressure of the ambient gas. Default value and unit: 1.0 atm.
A: Commands
gas_flow
Sentaurus™ Process User Guide 937
I-2013.12
print
Prints the gas flow information.
Description
This command specifies a gas mixture for thermal oxidation or user-defined gas material
reactions, and can be set in the diffuse command or temp_ramp command. Specification in
multiple temp_ramp commands allows changing the gas flow during a temperature ramp. If
gas flows are specified by flows, a complete gas reaction between the contributing gas types
is assumed. The partial pressure of the active ambients (for example, material-growing
ambients O2, H2O, and N2O) are the quantities directly needed to compute oxidation rates.
If flows is specified, the partial pressures are computed from gas reactions, the mix of
remaining gases after the reaction, and the total pressure. If partial pressures of the active
ambients are specified, they are used directly. The default value is 0 for all arguments, except
pressure. Table 64 on page 617 lists the available ambients and includes O2, H2O, HCl, N2,
H2, and N2O as well as two epi ambients Epi and LTE that do not apply to this command.
Examples
These are three equivalent flow specifications for the gas mixture myflow:
gas_flow name= myflow pressure= 0.8 flows= {O2= 3.1 H2O= 1.2 H2= 0.8}
gas_flow name= myflow pressure= 0.8 flowO2= 3.1 flowH2O= 1.2 flowH2= 0.8
gas_flow name= myflow pressure= 0.8 flowO2= 3.1 flows= {H2O= 1.2 H2= 0.8}
Similar syntax for partial pressures: Three equivalent specifications for the gas mixture mypp.
When setting partial pressures, pressure, flow<ambient>, and flows must not be used:
gas_flow name= mypp partial.pressure= {O2= 3.1 H2O= 1.2}
gas_flow name= mypp pO2= 3.1 pH2O= 1.2
gas_flow name= mypp pO2= 3.1 partial.pressure= {H2O= 1.2}
See Also
diffuse on page 908
term on page 1173

A: Commands
graphics
938 Sentaurus™ Process User Guide
I-2013.12
graphics
Updates or initiates graphics specified by the option command.
Syntax
graphics
[cmd=<c>] [configure=<c>] [connect] [display=<c>]
[host=<c>] [maxdepth] [on | off] [update]
Arguments
cmd
Specifies the update command. Default: plot.tec grd data.
configure
Runs a plot.tec configuration command. Any valid plot.tec argument can be
specified.
connect
Allows connection to the Tecplot SV interface.
display
Specifies the display for Tecplot SV. The default is the value of the DISPLAY environment
variable.
host
Specifies the host on which Tecplot SV will run. The default behavior is explained in
Tecplot SV User Guide, Launching or Connecting to Tecplot SV on page 13.
maxdepth
Specifies the command depth limit.
off
Disables automatic updating of graphics.
on
Enables automatic updating of graphics using the command given by cmd.
A: Commands
graphics
Sentaurus™ Process User Guide 939
I-2013.12
update
Executes the update command.
Description
This command controls automatic graphics updating or specifies a command sequence that
will be called automatically after structure- or data-changing steps. The default display uses an
interface to Tecplot SV. To use the interface, you must open an interprocess
communication–enabled Tecplot SV first from the command line, for example:
unix:> tecplot_sv -s:ipc
After the main window has opened, subsequent runs of Sentaurus Process connect to
Tecplot SV and open a new frame if the command graphics on is issued.
Examples
Specify the datasets to display:
graphics configure= "xyshow=Boron* Stress*"
Switch graphics on and use the Tecplot SV interface:
graphics on
Automatically call the old 2D graphics display for data- or structure-changing steps:
graphics on cmd= "plot.2d grid gas"
See Also
plot.tec on page 1070

A: Commands
grid
940 Sentaurus™ Process User Guide
I-2013.12
grid
Performs grid operations and computes statistics about the mesh.
Syntax
grid
[bbox | bbox.cm | bbox.um | bulk.nodes | bulk.regions | dimension |
elements | interface.nodes | interface.regions | max.angle |
max.connectivity | max.edge | max.volume | max.volume.ratio |
max.volume.ratio.location | min.angle | min.edge | min.edge.vertices |
min.volume | min.volume.location | nodes | obtuse | total.nodes |
total.volume | vertices]
[brep.faces | brep.min.angle | brep.min.angle.location | brep.min.edge |
brep.min.edge.location | brep.stats | brep.vertices]
[Gas] [interface.area] [interpolate]
[<material>]
[merge] [mesh.stats]
[mingrid=<n>][<m>|<cm>|<um>|<nm>]
[remesh [Adaptive]] | (2D | 3D | FullD) | refine.check]
[rename [print.names]]
[sano.list] [sano.materials] [sano.remesh] [sano.smooth]
[smooth.brep [
repair.resolution=<n>[<m>|<cm>|<um>|<nm>]
delpsc.resolution=<n>[<m>|<cm>|<um>|<nm>]
delpsc.accuracy=<n>[<m>|<cm>|<um>|<nm>]]][set.min.edge] [set.volume]
[syntax.check.value=<c>]
Arguments: Regridding, Renaming, Refinement
2D, 3D, FullD
Extrudes grid to higher dimension. The line commands must be issued before extruding
to a higher dimension. For two dimensions, at least two y-lines must have been specified.
For three dimensions, two y-lines and two z-lines must have been specified. FullD
extrudes to the highest possible dimension.
Adaptive
If specified, Adaptive switches on adaptive meshing if remesh is specified. Parameters
for adaptive meshing are described in Adaptive Refinement on page 699. The default is the
return value of pdbGet Grid Adaptive.
interpolate
Performs interpolation if remesh is specified.
A: Commands
grid
Sentaurus™ Process User Guide 941
I-2013.12
merge
Merges adjacent regions of the same material into one region.
NOTE Do not use in combination with other arguments.
mingrid
Specifies the minimum-allowed grid spacing. Default unit: .
print.names
Prints the region names if rename is specified.
refine.check
Returns 1 if remeshing is needed based on refinement criteria; otherwise, it returns 0.
remesh
If specified, the mesh is re-created using the active mesh generator.
rename
Renames all regions of the structure according to the material they contain and the smallest
y-coordinate point of the region, that is, from the bottom of the structure upwards. Multiple
regions of the same material with the smallest y-coordinate within the given coordinate
interval will increase the associated index towards the positive x-axis, from left to right (see
print.names).
set.min.edge
Must be used by itself. When specified, Sentaurus Process computes the smallest edge
length in each direction and saves it in three fields: MinXEdgeLength, MinYEdgeLength
(for 2D or 3D structures), and MinZEdgeLength (for 3D structures). In addition, this
argument stores the element volumes in the field ElementVolume. When set.min.edge
is specified, the average edge length in each direction is returned and can be used to set a
Tcl variable, for example:
set aveEdgeLength [grid set.min.edge]
set.volume
Sets element volumes as element values over the mesh. This field is not updated
automatically.
syntax.check.value
Sets a value to be returned only during syntax-checking mode. Sometimes, the value
returned by a command can cause a false syntax-check error because the value returned by
µm
A: Commands
grid
942 Sentaurus™ Process User Guide
I-2013.12
the command would not be the value during the normal run mode. Setting this value avoids
such problems.
Arguments: Grid Statistics
Gas
By default, quality (except volume) and bbox measures include the gas mesh. To exclude
gas in the quality or bbox measure, use !Gas.
<material>
If specified, limits the measured grid statistics to the specified material.
Arguments: Reporting Grid Statistics
bbox
Prints the bounding box of the structure (coordinates in ).
bbox.cm
Prints the bounding box of the structure (coordinates in cm).
bbox.um
Prints the bounding box of the structure (coordinates in ).
bulk.nodes
Returns the number of bulk nodes.
bulk.regions
Returns the number of bulk regions.
dimension
Returns the current simulation dimension.
elements
Returns the number of elements.
interface.area
Computes the area of interfaces. It can be limited to one particular interface by specifying
a material interface such as "Silicon /Oxide" ( in two dimensions, in three
dimensions).
µm
µm
µm
µm2
A: Commands
grid
Sentaurus™ Process User Guide 943
I-2013.12
interface.nodes
Returns the number of interface nodes.
interface.regions
Returns the number of interface regions.
max.angle
Returns the maximum angle in degrees between edges (two dimensions) or faces (three
dimensions).
max.connectivity
Returns the maximum number of edges connected to a single node.
max.edge
Returns the maximum edge length in .
max.volume
Returns the maximum element volume ( in two dimensions, in three
dimensions).
max.volume.ratio
Returns the maximum ratio of volumes of two elements that share the same face (three
dimensions only).
max.volume.ratio.location
Returns the location where the maximum volume ratio occurs (coordinates in ).
mesh.stats
Indicates that all bulk mesh statistics must be printed. Here, all mesh statistics are listed as
command arguments if they can be individually queried. They also are printed with
mesh.stats or are listed below if they are available only by using mesh.stats:
•bbox: Bounding box (minimum and maximum extents) of the mesh.
•bulk.nodes: Number of nodes in mesh excluding those in the interface meshes (this
gives two nodes for each vertex on an interface and one node for each bulk vertex).
•bulk.regions: Number of regions in the mesh.
•dimension: Simulation dimension.
•elements: Number of elements in the mesh.
µm
µm2
µm3
µm
A: Commands
grid
944 Sentaurus™ Process User Guide
I-2013.12
•interface.nodes: Number of interface nodes in mesh. This will be the same as the
number of interface vertices.
•interface.regions: Number of interface regions in the mesh.
•max.angle: Maximum of all angles of all elements in the mesh.
•max.connectivity: Maximum number of edges sharing one vertex in the mesh.
•max.edge: Length of maximum edge in the mesh (in ).
•max.volume: Maximum element volume (in where D is the dimension).
•max.volume.ratio: Maximum ratio of volumes (larger volume to smaller volume)
of neighboring elements.
•max.volume.ratio.location: Location where maximum ratio of volumes occurs.
•min.angle: Minimum of all angles of all elements in the mesh.
•min.edge: Length of minimum edge in the mesh (in ).
•min.edge.vertices: Endpoints of minimum edge.
•min.volume: Minimum element volume (in where D is the dimension).
•min.volume.location: Location of the center of the element with the minimum
volume.
•nodes: Total number of nodes in mesh. At interfaces, there are three nodes for each
vertex. In the bulk, there is one node for each vertex.
•obtuse: Percentage of triangles or tetrahedra that have obtuse angles.
•total.nodes: Same as nodes, that is, the total number of nodes in mesh.
•total.volume: Volume of mesh (in where D is the dimension).
•vertices: Number of vertices in mesh.
min.angle
Returns the minimum angle in degrees between edges (two dimensions) or faces (three
dimensions).
min.edge
Returns the minimum edge length in .
min.edge.vertices
Returns the vertices of the minimum edge.
µm
µmD
µm
µmD
µmD
µm
A: Commands
grid
Sentaurus™ Process User Guide 945
I-2013.12
min.volume
Returns the element with the smallest area in two dimensions (in ) or the smallest
volume in three dimensions (in ).
min.volume.location
Prints the location of the smallest element (coordinates in ).
nodes
Same as bulk.nodes.
obtuse
Returns the percentage of triangles or tetrahedra that have obtuse angles.
total.nodes
Returns the total number of nodes in the mesh. Note the difference between points and
nodes: There is a node for each region sharing an interface point in addition to each point
not on an interface.
total.volume
Returns the total volume of the bounding box (cm in one dimension, in two
dimensions, and in three dimensions). The unit of the angles reported is degree. If
none of these is chosen, all values are reported.
vertices
Returns the total number of vertices in the mesh.
Arguments: Boundary Representation (Brep) Statistics Reporting
brep.faces
Returns the number of faces in the brep.
brep.min.angle
Returns the brep minimum angle in degrees.
brep.min.angle.location
Returns the coordinates of the minimum angle (coordinates in ).
brep.min.edge
Returns the minimum edge length (in ) (three dimensions only).
cm2
µm3
µm
cm2
cm3
µm
µm
A: Commands
grid
946 Sentaurus™ Process User Guide
I-2013.12
brep.min.edge.location
Returns the coordinates of the minimum edge length (three dimensions only) (coordinates
in ).
brep.stats
Indicates that all brep statistics must be printed. Here, all brep statistics are listed as
command arguments if they can be individually queried. They also are printed with
brep.stats or are listed below if they are available only by using brep.stats:
•brep.are.all.boundaries.on.bbox: Returns true if all outer boundaries are on
a flat bounding box; otherwise, false.
•brep.bbox: Returns the bounding box of the brep.
•brep.conformal: Returns true if the brep is conformal; otherwise, false.
•brep.dimension: Returns the dimension of the brep.
•brep.max.angle: Returns the brep maximum angle in degrees.
•brep.max.angle.from.flat: Returns -brep maximum angle (three
dimensions only).
•brep.max.angle.location: Returns the coordinates (in ) of the maximum
angle (three dimensions only).
•brep.min.dihedral.angle: Returns the minimum dihedral angle in degrees in the
brep (three dimensions only).
•brep.min.dihedral.angle.location: Returns the coordinates (in ) of the
minimum dihedral angle (three dimensions only).
•brep.min.dihedral.angle.material: Returns the material where the minimum
dihedral angle is located (three dimensions only).
•brep.regions: Returns the number of regions in the brep.
•brep.total.area: Returns the total brep interface area in three dimensions or the
bulk area in two dimensions (in ).
•brep.total.volume: Returns the brep volume (three dimensions only) (in ).
brep.vertices
Returns the number of vertices in the brep.
µm
180°
µm
µm
µm2
µm3
A: Commands
grid
Sentaurus™ Process User Guide 947
I-2013.12
Arguments: Sano Smoothing and Remeshing
sano.list
Sets the list of fields for sano.remesh or sano.smooth operations. By default, the list
contains the active dopant concentrations. The field NetActive
(DopingConcentration) is updated automatically during Sano operations and does not
need to be explicitly included here.
sano.materials
Sets the list of materials where the Sano method is applied. The default is Silicon. It is
not recommended to include other materials unless special care is taken to configure
Sentaurus Process KMC for those materials because, by default, the KMC models are
simplistic in materials other than silicon.
sano.remesh
Switches on a special Sano remesh mode. Usually, remeshing based on Sano fields and
Sano field creation are performed with the UnsetAtomistic command (see
UnsetAtomistic on page 1187), which calls grid sano.remesh and grid
sano.smooth. More detail is provided here if nonstandard behavior is required. During
grid sano.remesh, certain fields – Sano fields – can be the target of adaptive
refinement. By default, the list of Sano fields contain active dopants.
NOTE With regard to the Sano remesh mode:
•NetActive (DopingConcentration) is updated automatically
using active Sano fields during sano.remesh.
•This mode does not create any new fields in the structure. Sano
fields can be created using sano.smooth in a separate grid
command.
• This mode does not automatically switch on adaptive meshing.
sano.smooth
Converts KMC particle distributions to finite-element fields using the Sano method.
Usually, this conversion is performed with the UnsetAtomistic command (see
UnsetAtomistic on page 1187), which calls grid sano.remesh and grid
sano.smooth. More detail is provided here if nonstandard behavior is required. The list
of fields that are converted by default contains the active dopants that are present. To
change this list, use sano.list.
NOTE With regard to sano.smooth:
•NetActive (DopingConcentration) is updated automatically
using Sano fields during sano.smooth.
A: Commands
grid
948 Sentaurus™ Process User Guide
I-2013.12
•It specifies that only KMC particles are converted to new finite-
element fields on the existing mesh; no remeshing occurs.
Arguments: Smoothing Boundary Representation (Brep)
smooth.brep
Removes noise and sharp features with Smooth boundary representation (brep). First a
multimaterial level-set algorithm removes any noise that may be present in the Brep. Then,
surface remeshing creates good-quality triangles on uneven surfaces using DelPSC
algorithm (Delaunay refinement for Piecewise Smooth Complex).
repair.resolution
Specifies the level-set cell size to remove noise and sharp features. It should be, at most,
one-third the thickness of the thinnest region. Otherwise, the thin region may be considered
noise and it disappears. Uses the default value from pdbGet Grid
Repair.Geometry.Resolution.
delpsc.resolution
Controls the size of small triangles in DelPSC algorithm. Uses the default value from
pdbGet Grid Apply.Brep.DelPSC.Resolution.
delpsc.accuracy
Controls the deviation between the new curved surfaces from DelPSC algorithm and the
surfaces from multimaterial level-set algorithm. Uses the default value from pdbGet Grid
Apply.Brep.DelPSC.Accuracy.
Description
This command allows you to:
■Remesh.
■Merge regions.
■Extrude.
■Rename regions.
■Measure and report on various mesh statistics.
■Measure and report on various boundary representation (referred to as brep) statistics.
■Convert KMC particles into finite-element fields using the Sano method.
■Remesh while adaptively refining Sano fields.
A: Commands
grid
Sentaurus™ Process User Guide 949
I-2013.12
To retrieve mesh or brep statistics, you can either:
■In a single pass, all statistics can be computed and returned in a Tcl array that can be
accessed by the name of the measured value, for example:
array set returnArray [grid mesh.stats]
set numNodes $returnArray(total.nodes)
set numElements $returnArray(elements)
■To retrieve only the required parameter, use:
set numNodes [grid nodes]
NOTE If you are interested in several mesh statistics, it is more efficient to
retrieve all statistics in one pass and read them from a Tcl array.
To limit grid or brep statistics to a particular material or interface, specify the material on the
command line. For example:
■To limit brep statistics to silicon, use:
grid brep.stats Silicon
■To limit brep statistics to the oxide–silicon interface, use:
grid brep.stats Silicon /Oxide
Several mesh statistics parameters compute a measure of element quality including:
■avg.element.quality (average element quality)
■best.element.quality
■worst.element.quality
■lt3.element.quality (percentage of elements whose quality is less than 0.3)
■gt6.element.quality (percentage of elements whose quality is greater than 0.6)
These arguments are computed and returned as part of mesh.stats, but they are not
separately available.
NOTE For the purpose of this command, quality is defined as:
•Triangles: 4.0 * sqrt(3.0) * area (sum of side lengths)
2.
• Tetrahedra: The ratio of the radius of the inscribed sphere to the
radius of the circumsphere.
Examples
Recreate the mesh using currently specified refinements:
grid remesh

A: Commands
help
950 Sentaurus™ Process User Guide
I-2013.12
Report the percentage of obtuse elements in silicon:
grid obtuse silicon
Compute all mesh statistics, and then read the number of nodes and the number of elements
from the statistics array into numNodes and numElements, respectively:
array set returnArray [grid mesh.stats]
set numNodes $returnArray(total.nodes)
set numElements $returnArray(elements)
To have different Sano lists for remesh versus smoothing, use the following commands:
KMC2PDE
grid sano.remesh sano.list= BActive
grid sano.smooth sano.list= {AsActive BActive}
pdbSet Diffuse KMC 0
pdbSet Implant MC 0
pdbSet AtomisticData 0
kmc off
To remove noise and sharp features, use the brep command:
grid smooth.brep delpsc.resolution=0.02 delpsc.accuracy=0.0002 \
repair.resolution = 0.003
See Also
line on page 1010
help
Prints a list of all commands available in Sentaurus Process.
Syntax
help
Description
This command can be used in interactive mode as well as in command files.

A: Commands
icwb
Sentaurus™ Process User Guide 951
I-2013.12
icwb
IC WorkBench (ICWB)–related functions.
Syntax
icwb bbox (xmin | xmax | ymin | ymax | left | right | front | back)
icwb create.all.masks
icwb dimension
icwb domain=<c>
icwb domain= [list "<domain_name1>" "<domain_name2>" ... "<domain_namen>"]
icwb filename=<c> [scale=<n>]
icwb gds.file=<c> cell=<c> layer.names= {<list>} layer.numbers= {<list>}
sim2d | sim3d= {<n>} [domain.name=<c>] [scale=<n>] [stretches= {<c>= {<n>}}]
icwb layer.name=<c> list polygon.names
icwb list domains
icwb list (layerIDs | layerNames)
icwb list polygon.bounding.boxes layer.name=<c>
icwb list polygon.tessellations layer.name=<c>
icwb polygon.name=<c> list.segments
icwb slice.angle.offset
icwb stretch name=<c> value=<n>
Description
The keyword icwb introduces commands used to operate with ICWB TCAD layout files. The
different uses of the keyword icwb are given here, along with their syntax and corresponding
descriptions:
icwb bbox (xmin | xmax | ymin | ymax | left | right | front | back)
Returns the corresponding coordinate, which can be one of the following in either ICWB
coordinates (xmin, xmax, ymin, ymax) or Sentaurus Process coordinates (left, right,
front, back).
icwb create.all.masks
Faster version of the icwb.create.all.masks command; intended to create large
masks from complex layouts.
A: Commands
icwb
952 Sentaurus™ Process User Guide
I-2013.12
icwb dimension
Returns the dimension of the current domain. For the following domain types, the
corresponding value for dimension is returned:
• Point: 1
•Gauge: 2
• Highlight: 3
icwb domain=<c>
icwb domain= [list "<domain_name1>" "<domain_name2>" ... "<domain_namen>"]
Defines the current domain. Setting the name of the current domain is a prerequisite for
other ICWB commands that implicitly depend on the current domain being defined.
The second variation allows for the concatenation of multiple gauge domains, reorientated
into one linear simulation domain.
icwb filename=<c> [scale=<n>]
Reads an ICWB TCAD layout file with the extension .mac. Coordinates in the ICWB file
are multiplied by the optional scale argument as the file is read. The ICWB TCAD layout
file must be read as a prerequisite to other ICWB commands that act on the domains and
masks defined in that file.
icwb gds.file=<c> cell=<c> layer.names= {<list>} layer.numbers= {<list>}
sim2d | sim3d= {<n>} [domain.name=<c>] [scale=<n>]
[stretches= {<c>= {<n>}}]
Reads a GDSII layout file:
The gds.file argument specifies the input GDSII file name, and cell specifies the cell
name. The layer.numbers is a list of selected layers from the GDSII file, and
layer.names is a list of names for those layers.
The domain.name argument defines the name of the simulation domain. If no name is
specified, SIM3D is used for a 3D domain, and SIM2D is used for a 2D domain. The domain
will be set to be the current domain automatically, so you do not need to call icwb
domain=<c> before using other ICWB commands. However, you can call icwb
domain=<c> to set another preferred current domain.
sim2d | sim3d indicates whether it is a 2D or 3D simulation domain. The simulation
domain is defined by two points, with each point defined by x- and y-coordinates in the
GDSII coordinate system. For two dimensions, the two points are endpoints of a segment.
The segment must be horizontal or vertical in the GDSII coordinate system. For three
dimensions, the two points are the two opposite corners of the simulation domain. The first
point is the left-back corner and the second point is the right-front corner in the internal
coordinate system.
A: Commands
icwb
Sentaurus™ Process User Guide 953
I-2013.12
The stretches argument is a list of stretches, with each stretch having a name and being
defined by a segment with two points. For a 3D domain, the segment must cross the
bounding box of the domain. For a 2D domain, the segment must intersect with the 2D
domain.
The scale argument is the same as that in the icwb filename=<c> [scale=<n>]
command. Coordinates in the GDSII file are multiplied by scale as the file is read in to
Sentaurus Process.
This command can be called multiple times to set multiple simulation domains. The GDSII
layout file or the ICWB TCAD layout file must be read as a prerequisite to other ICWB
commands that act on the domains and masks defined in that file.
icwb layer.name=<c> list polygon.names
Lists polygon names given a layer name.
icwb list domains
Queries the names of the current domains.
icwb list (layerIDs | layerNames)
Returns a list of layer IDs or layer names of the ICWB TCAD layout file.
icwb list polygon.bounding.boxes layer.name=<c>
Returns the bounding box rectangle for each polygon in the layer.
icwb list polygon.tessellations layer.name=<c>
Breaks each polygon in the layer into a set of rectangles, and then returns these rectangles,
icwb polygon.name=<c> list.segments
Lists polygon segments given a polygon name.
icwb slice.angle.offset
Returns the relative angle of the active simulation domain.
icwb stretch name=<c> value=<n>
Applies the given stretch by the given amount to the current domains. The order of applied
stretches is important since the location of other stretches can change given the application
of one stretch.

A: Commands
icwb.contact.mask
954 Sentaurus™ Process User Guide
I-2013.12
icwb.contact.mask
Creates contacts for subsequent device simulations that are tied to a layer in the ICWB TCAD
layout file.
Syntax
icwb.contact.mask
layer.name= <c> | <list>
[name=<c>] [<other_arguments>]
Arguments
layer.name
Name of a layer in the ICWB TCAD layout file.
name
Name of the contact. The name defaults to the layer name.
<other_arguments>
Any other arguments supported by the contact command.
Description
This command serves as an interface between the ICWB TCAD layout and the contact
command by automatically obtaining the lateral placement of the contact from the specified
ICWB layer, taking the vertical placement from the argument list and passing all other options
directly to the contact command.
The icwb.contact.mask command supports both box-type and point-type contacts:
■A box-type contact consists of elements at the surface of one region or material inside the
box. The lateral extent of the box is determined automatically from the layer segment (two
dimensions) or the polygon bounding box (three dimensions), while the vertical extent is
taken from the contact command arguments xlo and xhi.
■A point-type contact contains all the boundary elements of one region. The lateral position
of the point is determined automatically as the midpoint of the layer segment (two
dimensions) or the polygon bounding box (three dimensions), while the vertical position is
taken from the contact command argument x.
For details on how to define contacts, see contact on page 889.
A: Commands
icwb.contact.mask
Sentaurus™ Process User Guide 955
I-2013.12
NOTE A layer used for layout-driven contact placements can consist of only a
single segment or polygon in each ICWB simulation domain. It may be
necessary to create auxiliary layers in IC WorkBench EV Plus for the
placement of contacts.
NOTE For 3D simulations, sometimes the placement of contacts in Sentaurus
Process causes meshing problems. In this case, use the similar layout-
driven contact placement feature in Sentaurus Structure Editor.
Examples
The following icwb.contact.mask commands create contacts for subsequent device
simulations tied to the layers named emitter and pdrain, respectively, in the example ICWB
TCAD layout file:
icwb.contact.mask layer.name= emitter box polysilicon \
adjacent.material= oxide xlo= -2.05 xhi= -1.95
icwb.contact.mask layer.name= pdrain name= drain point aluminum replace \
x= -2.0

A: Commands
icwb.create.all.masks
956 Sentaurus™ Process User Guide
I-2013.12
icwb.create.all.masks
Creates positive and negative mask versions for all layers found in the currently active ICWB
simulation domain.
Syntax
icwb.create.all.masks
Description
The names of the masks are given by the layer names and the suffix _p for the positive and _n
for the negative version of the mask.
For example, if the TCAD layout contains a layer with the name TRENCH, the corresponding
mask names are TRENCH_p and TRENCH_n.
NOTE Use the command-line option -n to suppress automatic syntax-
checking in Sentaurus Process when using this feature. The syntax-
checker cannot determine which masks are available. Therefore, it may
incorrectly flag the use of an unknown mask.

A: Commands
icwb.create.mask
Sentaurus™ Process User Guide 957
I-2013.12
icwb.create.mask
Creates a mask for subsequent use in etch, deposit, or photo commands from one or more
ICWB layers.
Syntax
icwb.create.mask
layer.name= <c> | <list>
[name=<c>]
[polarity= positive | negative]
[shift= {<dy> <dz>}]
[stretchyneg= {<yo> <dy>}] [stretchypos= {<yo> <dy>}]
[stretchzneg= {<zo> <dz>}] [stretchzpos= {<zo> <dz>}]
Arguments
layer.name
Name of one or more layers in the ICWB TCAD layout file. If more than one layer name
is given, the resulting mask is the union of the polygons (3D) or segments (2D) from all the
layers listed. (Use the icwb list layerNames command to obtain a list of all layer
names.)
name
Name of the mask. The mask name defaults to the layer name. If the layer name list
contains more than one entry, the first layer name is used.
polarity
Sets the polarity. Select negative to invert a mask. The polarity is assumed to be
positive by default, that is, points inside the mask are considered masked.
shift
Shifts the layers by the specified amount before creating the mask.
stretchyneg, stretchypos, stretchzneg, stretchzpos
Stretches the layer before creating the mask. The last four characters of the arguments
determine whether the stretch is applied along the y- or z-direction and whether the layer
is stretched to the positive or negative side of the stretch position.
More than one shift and stretch* argument can be used in the icwb.create.mask
command. As these operations may not commute, it is important to note the order in which
these operations are applied if more than one is used. First, shift is applied, and then
stretchypos, stretchyneg, stretchzpos, and finally stretchzneg are applied.
A: Commands
icwb.create.mask
958 Sentaurus™ Process User Guide
I-2013.12
Description
This command serves as an interface between the ICWB TCAD layout and the mask and
polygon commands, and provides a convenient way to generate 1D, 2D, and 3D masks
consisting of the points, segments, or polygons from one or more ICWB layers based on a
dimension-independent syntax. The command automatically determines the dimension of the
currently active ICWB simulation domain.
Examples
The following icwb.create.mask commands create masks from the corresponding layers
named by layer.name:
icwb.create.mask layer.name= NWELL polarity= negative
icwb.create.mask layer.name= NWELL name= NOTNWELL
icwb.create.mask layer.name= "NPDIFF PPDIFF NPLUG PBASE" name=STI info=1
icwb.create.mask layer.name= 1:0 stretchyneg= {1.2 -0.25}
See Also
deposit on page 901
etch on page 923
mask on page 1020
photo on page 1061
polygon on page 1082

A: Commands
icwb.refine.mask
Sentaurus™ Process User Guide 959
I-2013.12
icwb.refine.mask
Creates refinement boxes that are tied to layers in the ICWB TCAD layout file.
Syntax
icwb.refine.mask
layer.name= <c> | <list>
xbot=<n> xtop=<n>
[name=<c>] [oversize=<n>] [<other_arguments>]
Arguments
layer.name
Name of one or more layers in the ICWB TCAD layout file. If more than one layer name
is given, refinement boxes are created for the polygons (three dimensions) or segments
(two dimensions) from all mentioned layers.
name
Name of the refinement box. The name defaults to the layer name. If the layer name list
contains more than one entry, the first layer name is used. If more than one polygon (three
dimensions) or segment (two dimensions) is found, an index is appended to the refinement
box name.
<other_arguments>
Any other arguments supported by the refinebox command.
oversize
To refine an area wider than the polygon bounding box (three dimensions) or the segment
(two dimensions), specify a nonzero value (unit is ). This value is used to increase the
refinement boxes beyond the extent given by the polygon bounding boxes or segments.
xbot
Bottom or maximum x-coordinate of the refinement box extent.
xtop
Top or minimum x-coordinate of the refinement box extent.
µm
A: Commands
icwb.refine.mask
960 Sentaurus™ Process User Guide
I-2013.12
Description
This command serves as an interface between the ICWB TCAD layout and the refinebox
command by automatically obtaining the lateral dimension of the refinement box from the
specified ICWB layers, taking the vertical refinement box dimensions from the argument list,
and passing all other options directly to the refinebox command.
For a 2D or 3D ICWB simulation domain, a refinement box is created for each segment or
polygon found in the specified layers. For three dimensions, the lateral extent of respective
refinement boxes is given by an axis-aligned tessellation of the polygon.
Using oversize increases the area of refinement beyond the extent of the actual segments or
polygon bounding boxes. The nonzero value of oversize is subtracted from or added to the
minimum and maximum segment or polygon bounding box coordinates, respectively.
NOTE Layout-driven refinement is available only for the area under the given
layer itself, not for the inverse of a layer. If refinement is needed in an
area not covered by the layer, you must create the inverse of the layer as
an auxiliary layer explicitly in IC WorkBench EV Plus.
Examples
The following icwb.refine.mask commands create refinement boxes tied to the layer in the
ICWB TCAD layout file named POLY:
icwb.refine.mask name= UnderPoly layer.name= POLY oversize =0.1 \
xtop= -1.51 xbot= -1.35 xrefine= 0.02 yrefine= 0.02
icwb.refine.mask name= SiOxPo layer.name= POLY oversize= 0.1 \
xtop= -1.51 xbot= -1.35 min.normal.size= 0.005 \
interface.mat.pairs= {Silicon Oxide Silicon Polysilicon}
See Also
refinebox on page 1101

A: Commands
implant
Sentaurus™ Process User Guide 961
I-2013.12
implant
Specifies implantation model parameters and implants an ion species into a wafer.
Syntax
implant
[angle.dependent]
[beta=<n>] [beta2=<n>]
[boundary.conditions= {left=<c> right=<c> front=<c> back=<c>}
[cap.dependent]
[dam.suf=<c>] [dam.table=<c>] [damage] [data.suf=<c>] [dataset=<c>]
[depth.dependent]
[dualpearson | gaussian | pearson | pearson.s | point.response]
[eff.caplayer.thick=<n>] [eff.channeling.suppress]
[en.stdev=<n>] [energy=<n>]
[file=<c>]
[frenkel.pair.offset= {<n> <n> <n>}]
[gamma=<n>] [gamma2=<n>]
[ge.effect]
[i.plus.offset= {<n> <n> <n>}]
[imp.table=<c>]
[lat.scale=<n>][<m>|<cm>|<um>|<nm>]
[lat.scale2=<n>][<m>|<cm>|<um>|<nm>]
[lat.stdev=<n>][<m>|<cm>|<um>|<nm>]
[lat.stdev2=<n>][<m>|<cm>|<um>|<nm>]
[lexp=<n>] [lexp2=<n>]
[<material>]
[min.conc=<n>][<m-3>|<cm-3>|<um-3>|<nm-3>])
[ratio=<n>]
[rp=<n>][<m>|<cm>|<um>|<nm>]
[rp2=<n>][<m>|<cm>|<um>|<nm>]
[species=<c>]
[stdev=<n>][<m>|<cm>|<um>|<nm>]
[stdev2=<n>][<m>|<cm>|<um>|<nm>]
[tables= Default | AdvCal | Dios | Tasch | Taurus | TSuprem4]
[tilt=<n>] [tilt.stdev=<n>]
[ts4.material=<c>] [ts4.prefix=<c>] [ts4.species=<c>]
[v.plus.offset= {<n> <n> <n>}]
[y.position=<n>] [z.position=<n>]
[Adaptive] [average]
[backscattering] [beam.dose]
[cascades]
[conformity=<n>]
[contamination= {energy=<n> dose.fraction=<n>}]
[crit.dose=<n>][<cm-2>]
A: Commands
implant
962 Sentaurus™ Process User Guide
I-2013.12
[crystaltrim | sentaurus.mc]
[current=<n>]
[data.col=<n>] [data.file=<c>]
[data.max=<n>] [data.min=<n>] [data.units=<c>]
[data.xcol=<n>] [data.xhi=<n>] [data.xlo=<n>]
[defect.model= effective.plus.n | frenkel.pair | plus.one | user.defined]
[dfactor=<n>]
[dose=<n>][<cm-2>]
[dose.rate=<n>][<cm-2/s>]
[en.stdev=<n>]
[energy=<n>][<eV>|<keV>|<MeV>]
[extract.moments] [extrude]
[flip ([left] | [right])]
[fp.ifactor=<n>] [fp.vfactor=<n>]
[full.molecular] [get.moments]
[iBCA] [ifactor=<n>] [ion.movie] [KMC] [load.mc]
[match= range | dose | no]
[material=<c>]
[max.iter=<n>]
[mc.dfactor=<n>] [mc.ifactor=<n>] [mc.vfactor=<n>]
[mult.rot=<i>] [multiply=<n>]
[oxide.thickness=<n>][<m>|<cm>|<um>|<nm>]
[pai] [particles=<n>]
[plasma]
[plasma.deposit= {material=<c> thickness=<n> steps=<n>}]
[plasma.source= {<species1>=<n> <species2>=<n> ...}]
[point.implant]
[postprocess] [postprocessonly] [preprocess]
[primary= beam | wafer]
[profile.reshaping]
[randomize] [range.sh] [recoils]
[rotation=<n>][<degree>]
[rp.offset]
[save1d] [save1d.file=<c>] [save1d.unit=<c>]
[secondary.ions]
[shift=<n>]
[smooth]
[smooth.distance= {<double array>}]
[smooth.field= {<list>}]
[<species>]
[temperature=<n>]
[tilt=<n>][<degree>]
[tilt.stdev=<n>]
[tolerance=<n>]
[ts4.backscattering]
[vfactor=<n>]
A: Commands
implant
Sentaurus™ Process User Guide 963
I-2013.12
Arguments: Specifying Parameters
angle.dependent
Declares the tilt- and rotation-dependent range parameters in the implant table used. By
default, the Dios tables are all set to angle independent. For all other tables, angle-
dependent moments are assumed by default.
beta
Overwrites the kurtosis found in the specified implant table.
beta2
Overwrites the second kurtosis found in the specified implant table for the dual Pearson
model.
boundary.conditions= {left=<c> right=<c> front=<c> back=<c>}
Specifies the unified implant boundary conditions which will be used by both analytic and
MC implant. The valid keywords are Periodic, Reflect, or Extend.
cap.dependent
Specifies that the implant moments are cap (screening) layer dependent in the dual Pearson
model.
dam.suf
Specifies the file name suffix for Taurus™ tables that contain the required implant damage
data in the format <ion>_damage_in_<material>_<suffix>.
dam.table
Defines the implant table containing moments for the primary and lateral damage
distributions.
damage
Switches on or off the damage calculation based on the Hobler model.
data.suf
Specifies the file name suffix for Taurus tables that contain the required implant data in the
format <ion>_in_<material>_<suffix>.
dataset
Used for the data name that is created when an implantation is performed.
A: Commands
implant
964 Sentaurus™ Process User Guide
I-2013.12
depth.dependent
Switches on or off the lateral standard-deviation depth dependency.
dualpearson, gaussian, pearson, pearson.s, point.response
Specifies which type of distribution to use.
eff.caplayer.thick
Efficiency factor for the summation of layer thicknesses to calculate the total screening
(cap) layer thickness. Default: 1.
eff.channeling.suppress
Switches on the effective channeling suppression model. The default is 1 for the Taurus/
TSUPREM-4 mode, and 0 otherwise.
energy
Specifies the plasma implantation energy for the species. If energy is specified for a given
ion species, this energy will be used for this species instead of the common energy as
specified in the performing branch of the implant command. Used for plasma
implantation only. Default unit: keV.
en.stdev
Specifies the standard deviation of plasma implantation energy for the species. If
en.stdev is specified for a given ion species, its value will be used for this species instead
of the common energy as specified in the performing branch of the implant command.
Used for plasma implantation only. Default unit: keV.
file
Name of the file used in the point.response implantation model and the load.mc
mode.
frenkel.pair.offset
Specifies the amount of spatial shift for frenkel.pair. The actual shift occurs for
interstitials. This argument takes a list of numeric values. The first, second, and third values
in the list are taken as the x-, y-, and z-value, respectively. The missing value is treated as
zero.
gamma
Overwrites the skewness found in the specified implant table.
A: Commands
implant
Sentaurus™ Process User Guide 965
I-2013.12
gamma2
Overwrites the second skewness found in the specified implant table for the dual Pearson
model.
ge.effect
Specifies that the effect of Ge must be taken into account if its concentration is sufficiently
large. Default: false.
i.plus.offset
Specifies the amount of spatial shift for plus interstitials as a list of numeric values. The
first, second, and third values in the list are taken as the x-, y-, and z-value, respectively.
The missing value is treated as zero.
imp.table
Defines the implant table containing moments for the primary and lateral dopant
distributions.
lat.scale
Scaling factor for the lateral standard deviation. Default: 1. Default unit: unitless.
lat.scale2
Scaling factor for the second lateral standard deviation. Default: 1. Default unit: unitless.
lat.stdev
Overwrites the lateral standard deviation found in the specified implant table. Default unit:
.
lat.stdev2
Overwrites the second lateral standard deviation found in the specified implant table for the
dual Pearson model. Default unit: .
lexp
Overwrites the linear exponential tail length found in the specified implant table.
lexp2
Overwrites the second linear exponential tail length found in the specified implant table.
<material>
Specifies a material for which specification of model parameters is performed. For
information about specifying materials, see Material Specification on page 52.
µm
µm
A: Commands
implant
966 Sentaurus™ Process User Guide
I-2013.12
min.conc
Minimum concentration of the implanted species. Default unit: .
ratio
Ratio between the amorphous part of the dose and the total dose for the dual Pearson
model.
rp
Overwrites the projected range found in the specified implant table. Default unit: .
rp2
Overwrites the second projected range found in the specified implant table for the dual
Pearson model. Default unit: .
species
Name of one of the solution variables of the simulation, for example, Boron.
stdev
Overwrites the standard deviation found in the specified implant table. Default unit: .
stdev2
Overwrites the second standard deviation found in the specified implant table for the dual
Pearson model. Default unit: .
tables
Changes the implant tables and model switches in all materials. The settings are
overwritten for one particular species if species is specified. Otherwise, the implant
tables and model switches are overwritten for all species in all materials. The options that
correspond to different available tables are:
•Default (tables extracted from Monte Carlo simulations with Crystal-TRIM).
•AdvCal (makes the Default table data available in the Taurus Process table format).
•Dios (tables used by default in Dios).
•Tasch (University of Texas implant tables).
•Taurus (the Taurus Process table set).
•TSuprem4 (TSUPREM-4 native implant tables).
cm 3–
µm
µm
µm
µm
A: Commands
implant
Sentaurus™ Process User Guide 967
I-2013.12
tilt
Specifies the tilt angle for the species. If tilt is specified for a given ion species, the tilt
angle as specified is used for this species instead of the common tilt as specified in the
performing branch of the implant command. Used for plasma implantation only.
tilt.stdev
Specifies the standard deviation of the tilt angle for the species. If tilt.stdev is specified
for a given ion species, its value is used for this species instead of the common value of
tilt.stdev as specified in the performing branch of the implant command. Used for
plasma implantation only.
ts4.material
Specifies the name of the material as used in TSUPREM-4. This is used for TS4-style
tables only.
ts4.prefix
Specifies the prefix used in TSUPREM-4 native implant tables. Valid prefixes include
default, none, ch, dual, le, tr, ut, and scr.
ts4.species
Specifies the TS4 implant table name for the dopant, for example, chboron, tr.arsenic.
This is used for TS4-style tables only.
v.plus.offset
Specifies the amount of spatial shift for plus vacancies as a list of numeric values. The first,
second, and third values in the list are taken as the x-, y-, and z-value, respectively. The
missing value is treated as zero.
y.position
Point of reference in the y-direction for the automated 1D Monte Carlo run.
z.position
Point of reference in the z-direction for the automated 1D Monte Carlo run.
Arguments: Performing Implantation
Adaptive
If specified, Adaptive switches on adaptive meshing for both analytic and Monte Carlo
implantation. Parameters for adaptive meshing are described in Adaptive Meshing during
Implantation on page 708. The default is the return value of pdbGet Grid Adaptive.
A: Commands
implant
968 Sentaurus™ Process User Guide
I-2013.12
average
Specifies whether to average the as-implanted profiles over the reflected domains in the
case of TrueReflect boundary conditions. The default is true if the tilt angle (or tilt2D
in the case of a 2D structure) is less than , and false otherwise.
backscattering
Switches on or off the integration algorithm that accounts for particles backscattered from
the surface. Default: true.
beam.dose
Switches to beam dose control. The default is 1 for the Taurus/TSUPREM-4 mode, and 0
otherwise.
cascades
Runs Monte Carlo simulations in a full-cascade mode. Default: false.
conformity
Specifies the conformal doping fraction between 0 and 1:
•conformity=0 is standard implantation.
•conformity=1 is fully conformal doping.
contamination= {energy=<n> dose.fraction=<n>}
Specifies the energy contamination implantation parameters, where energy is the
contaminated energy, and dose.fraction is the fraction of the specified dose. The
contaminated dose is dose.fraction x dose, while the main implantation dose is then
(1 – dose.fraction) x dose.
crit.dose
Defines a parameter used for the point-defect profile calculation in the plus.one and
frenkel.pair models in the case of analytic implantation.
crystaltrim, sentaurus.mc
Selects the simulation of ion implantation using a Monte Carlo simulator, either
Crystal-TRIM or Sentaurus MC.
current
Beam line current. Default value and unit: 0 A.
2°
A: Commands
implant
Sentaurus™ Process User Guide 969
I-2013.12
data.col
Specifies the column number of concentration data in data.file used by
extract.moments. Default: 2.
data.file
Specifies the file name of the data for extract.moments.
data.max
Specifies the maximum value of the concentration data to be loaded from data.file.
Concentration data above data.max is ignored by extract.moments. Default value and
unit: .
data.min
Specifies the minimum value of the concentration data to be loaded from data.file.
Concentration data smaller than data.min is ignored by extract.moments. Default
value and unit: .
data.units
Specifies the units of the depth (x-)coordinate in data.file. Valid values are um, nm, and
cm. Default: um.
data.xcol
Specifies the column number of the depth (x-)coordinate in data.file used by
extract.moments. Default: 1.
data.xhi
Specifies the maximum value of the depth (x-)coordinate to be loaded from data.file.
Depths greater than data.xhi are ignored by extract.moments. Default: .
data.xlo
Specifies the minimum value of the depth (x-)coordinate to be loaded from data.file.
Depths smaller than data.xlo are ignored by extract.moments. The default value is
the first depth data in data.file.
defect.model
Selects the model used to calculate point defects:
•The
effective.plus.n model dynamically calculates an NFactor using an energy-
dependent and a dose-dependent fitting formula.
130
×10 cm 3–
114
×10 cm 3–
110
×10
A: Commands
implant
970 Sentaurus™ Process User Guide
I-2013.12
•For frenkel.pair, interstitial and vacancy profiles are calculated from the damage
profile resulting from the last implantation.
•plus.one selects the +1 model.
•The
user.defined model allows you to define your own models.
dfactor
Scaling factor for the damage profile calculation in analytic implantation. Default: 1.
dose
Dose of the implantation. Default value and unit: .
dose.rate
Dose rate of the implantation. If dose.rate is specified in the implant command, its
value is used with the assumption of a uniform dose rate. If it is not specified, the dose rate
is calculated from the DoseRate Tcl procedure in Implant.tcl. This argument is useful
for KMC simulations only.
en.stdev
Standard deviation of implantation energy for plasma implantation. Default: 0.0. Used for
plasma implantation only.
energy
Implantation energy. Default value and unit: 250 keV.
extract.moments
Specifies that the implant command will extract the implant moments from the ASCII
data file as specified by data.file.
extrude
Extrudes the 1D or 2D structure into a pseudo-3D structure before analytic implantation.
This makes 1D or 2D simulation results nearly identical to those in three dimensions.
Default: false.
flip ([left] | [right])
Flips the profile loaded with load.mc to the left. Optionally, left and right are
direction specifiers for the flip argument.
114
×10 cm 2–
A: Commands
implant
Sentaurus™ Process User Guide 971
I-2013.12
fp.ifactor
Scaling factor for the interstitial profile calculation in the frenkel.pair model. Used for
analytic implantation only.
fp.vfactor
Scaling factor for the vacancy profile calculation in the frenkel.pair model. Used for
analytic implantation only.
full.molecular
Follows all particles in a Crystal-TRIM or SentaurusMC run for a molecular species.
Default: true.
get.moments
Returns the implant moments instead of performing the actual implantation. The return
value of the implant command is a list of key–value pairs. For example, for the Gaussian
model, the return list would be "model gaussian rp <n> stdev <n>". For the dual
Pearson model, the return list would be:
model dualpearson rp <n> stdev <n> beta <n> gamma <n> rp2 <n> stdev2 <n>
beta2 <n> gamma2 <n> ratio <n>
Default: false.
iBCA
Switches on the improved BCA (iBCA) damage model. Default: false. Used in
Sentaurus MC implantations only.
ifactor
Scaling factor for the interstitial profile calculation in the plus.one and frenkel.pair
models. It is used for the frenkel.pair model in the case of analytic implantation only.
Default: 1.
ion.movie
Plots the positions of implanted ions during a Monte Carlo run.
KMC
Switches on the KMC mode for Monte Carlo implantation (both Crystal-TRIM and
Sentaurus MC). In this mode, dynamic annealing is performed using Sentaurus Process
KMC.
A: Commands
implant
972 Sentaurus™ Process User Guide
I-2013.12
load.mc
Loads an external profile specified with file. You can select either Crystal-TRIM or
Sentaurus MC for an automated Monte Carlo run by setting the model parameter in the
parameter database, for example:
pdbSet MCImplant model crystaltrim
Default: sentaurus.mc.
match
Selects the algorithm of the simulation in multilayer structures:
•range (default) selects the numeric range scaling algorithm.
•dose selects the dose-matching algorithm.
•no switches off both algorithms.
material
Specifies the material for get.moments from which implant moments are extracted.
Default: silicon.
max.iter
Maximum number of iterations to extract the dual-Pearson implant moments from
data.file. Default: 500.
mc.dfactor
Scaling factor for the damage profile calculated by Sentaurus MC. Default: 1.
mc.ifactor
Scaling factor for the interstitial profile calculation in the frenkel.pair model. Used for
Monte Carlo simulations only.
mc.vfactor
Scaling factor for the vacancy profile calculation in the frenkel.pair model. Used for
Monte Carlo simulations only.
mult.rot
Specifies multiple stages of implantations at different rotation angles. For example,
mult.rot=4 means one-quarter of the dose is implanted at the first rotation angle, the next
quarter at the original rotation angle plus , the next quarter at the original rotation angle
plus , and the last quarter at the original rotation plus . Default: 1.
90°
180°
270°
A: Commands
implant
Sentaurus™ Process User Guide 973
I-2013.12
multiply
Profile loaded with load.mc is multiplied by this value.
oxide.thickness
Specifies the oxide thickness for get.moments. For oxide thickness–dependent implant
tables, the implant moments are interpolated with respect to the specified oxide thickness.
Default: 0.0. Default unit: .
pai
Switches on the preamorphization implant (PAI) mode. The PAI model takes
preamorphization into account by converting the damage into effective screening layer
thicknesses used for the moment lookup in screening (cap) layer–dependent tables. The
default is 1 for the Taurus/TSUPREM-4 mode, and 0 otherwise.
particles
Number of pseudoparticles that will be started per surface segment during Monte Carlo
simulations.
plasma
Switches on plasma implantation. Valid for Sentaurus MC implantations only.
plasma.deposit= {material=<c> thickness=<n> steps=<n>}
Specifies the parameters for deposition during plasma implantation, where material
specifies the deposit material, thickness specifies the total thickness of the deposited
material, and steps specifies the number of steps Sentaurus Process will perform
alternatively between Monte Carlo implantation and material deposition. The default
thickness is 0.0, and the default number of steps is 1.
plasma.source= {<species1>=<n> <species2>=<n> ...}
Specifies a list of plasma ion species where <species1>, <species2>, and so on can be
any predefined species, and the number after each species specifies the fraction of the total
dose as specified by dose.
point.implant
Switches on the point implant mode, in which all particles are implanted into a central
location of the implant surface. Valid for Sentaurus MC implantations only.
postprocess
Switches on postprocessing of *_LastImp datasets. Default: true.
µm
A: Commands
implant
974 Sentaurus™ Process User Guide
I-2013.12
postprocessonly
Postprocesses existing *_LastImp datasets only (implantation itself is omitted).
preprocess
Switches on preprocessing. Default: true.
primary
Defines the interpretation of the range and lateral range parameters:
•beam (default) switches to the beam projection mode. In this case, the primary
moments are applied along the projection of the ion beam onto the simulation plane,
and the lateral integration is performed perpendicular to the projection of the ion beam.
•wafer switches to the wafer normal mode. Here, the primary distribution function and
the moments are interpreted orthogonally to the wafer surface.
profile.reshaping
Switches on the profile reshaping model. The default is 1 for the Taurus/TSUPREM-4
mode, and 0 otherwise.
randomize
If specified, randomizes the random seed (by using an internal clock) each time the
command file is run. Therefore, each run will produce different results. Used for Monte
Carlo implantations only. Default: false.
range.sh
Switches on the proportional range shift mode. The channeling part of the profile is shifted
proportionally to the ratio of the amorphous and the channeling range. The shift is the same
for both contributions if the model is switched off. The default is 1 for the Taurus/
TSUPREM-4 mode, and 0 otherwise.
recoils
Switches to the recoil implantation mode, such as simulating the oxygen knock-on effect.
Used for Sentaurus MC implantations only. Default: false.
rotation
Rotation angle of the wafer in the implanter. Default value and unit: .
90–°
A: Commands
implant
Sentaurus™ Process User Guide 975
I-2013.12
rp.offset
Specifies the offset for the projected ranges of the first and second Pearson moments
extracted by extract.moments. The extracted projected ranges are shifted by
rp.offset. Default value and unit: .
save1d
Specifies that the 1D profiles as calculated by Sentaurus MC implantation will be saved in
(x,y) format. These saved files have the particle names as the file name extension. Valid for
1D or quasi-1D structures only.
save1d.file
Specifies the file name for the 1D profiles as calculated by Sentaurus MC implantation.
These saved files have the particle names as the file name extension. Valid for 1D or quasi-
1D structures only.
save1d.unit
Specifies the unit of the x-axis for the 1D profiles as calculated by Sentaurus MC
implantation. Valid units are A, nm, and .
secondary.ions
When this argument is switched off, secondary ion fields (for example, fluorine in a BF2
implantation) are not created for Monte Carlo implantation. Default: true.
shift
Shifts the profile loaded with load.mc by a certain amount along the y-axis.
smooth
Switches on smoothing of the as-implanted profiles after Monte Carlo implantation. If
smooth.field is not specified, all the as-implanted profiles are smoothed. Default: false.
smooth.distance
Specifies the smoothing distance for each field as specified in smooth.field. Default
value and unit: 2.0 nm.
smooth.field
Specifies a list of fields to be smoothed. The valid fields are dopant names or Damage. For
example, for BF2 implantation, Boron, Fluorine, or Damage are valid names.
<species>
Any of the previously defined species names can be specified.
0.0 µm
µm
A: Commands
implant
976 Sentaurus™ Process User Guide
I-2013.12
temperature
Implantation temperature (wafer temperature). Default value and unit: .
tilt
Angle normal to the substrate at which the impurity was implanted. Default value and unit:
.
tilt.stdev
Standard deviation of the tilt angle for plasma implantation. Default: 0.
tolerance
Error tolerance for convergence to extract the dual-Pearson implant moments from
data.file. Default value and unit: 0.1%.
ts4.backscattering
Switches the TS4 backscattering model on or off. In this model, the portion of the profile
distribution that sticks out of the solid structure is assumed to be lost, resulting in slightly
less dose than the nominal dose. Default: false.
vfactor
Scaling factor for the vacancy profile calculation in the plus.one and frenkel.pair
models. It is used for the frenkel.pair model in the case of analytic implantation only.
Default: 1.
Arguments: Deprecated
igrid.file
Specifies the file name for storing the damage information in the internal grid of Crystal-
TRIM. This is not used in Sentaurus MC.
keepdamage.igrid
Keeps the damage information stored at the internal grid between two runs of Crystal-
TRIM. This is not used in Sentaurus MC.
mpp
Switches on or off the multiprocess parallelization (MPP) feature for Monte Carlo
implantations. However, if the pdb parameter ParallelJobs is equal to 1, no parallel
jobs will be run even if mpp is on. Default: true.
26.84°C
7°
A: Commands
implant
Sentaurus™ Process User Guide 977
I-2013.12
predamage.igrid
Uses the damage from the internal grid for a Crystal-TRIM run. This is not used in
Sentaurus MC.
Description
There are two main branches to this command. The first allows you to specify parameters for
the analytic model. It can be performed by specifying tables or species. The second
performs an implantation into the current structure. Either analytic functions or Monte Carlo
simulations (Crystal-TRIM or Sentaurus MC) can be used.
Examples
Change all implant specifications for the species boron from the default to the Dios
implantation tables and models:
implant species= Boron tables= Dios
Change the default implant table for boron in silicon to my_table.tab and the implantation
model to pearson. In addition, switch off the damage calculation for boron in silicon:
implant species= Boron Silicon imp.table= my_table.tab pearson !damage
Specify a 100 keV implantation of phosphorus with a dose of . The previously
assigned data files and models are used to obtain range statistics:
implant Phosphorus dose= 1e14 energy= 100
Specify an implantation of arsenic with a dose of and energy of 60 keV.
Sentaurus MC simulation will be used. Point-defect profiles will be calculated from the
damage profile:
implant Arsenic dose= 1e15 energy= 60 sentaurus.mc defect.model= frenkel.pair
114
×10 cm 2–
115
×10 cm 2–

A: Commands
init
978 Sentaurus™ Process User Guide
I-2013.12
init
Sets up the mesh and background doping levels.
Syntax
init
[Adaptive] [bnd=<c>]
[caxis.rotation=<n>] [caxis.tilt=<n>]
[clear]
[concentration=<n>][<m-3>|<cm-3>|<um-3>|<nm-3>]
[DelayFullD] [dfise=<c>] [done]
[field=<c>]
[flat.orient= <list>]
[load.commands] [<material>]
[pdb] [pdb.only] [resistivity]
[sat=<c>] [scale=<n>] [sigmac=<c>]
[slice.angle=<n>][<degree>]
[tdr=<c>] [top]
[wafer.orient= <list>]
Arguments
Adaptive
When loading a TDR file containing geometry but no mesh, a mesh is generated
automatically. This argument determines whether adaptive meshing is used. The default is
the return value of pdbGet Grid Adaptive.
bnd
Selects the .bnd format file for reading. The command reads the (2D or 3D) boundary file
and meshes it with MGOALS using the arguments and refinement boxes previously
defined.
caxis.rotation
Specifies the orientation of the wafer miscut, that is, the rotation angle of the wafer normal
with respect to the crystal coordinate system. This argument is used in both analytic and
Sentaurus MC implantations. The default value and unit is , that is, the projection of the
wafer normal to the crystal plane formed by the b-axis and c-axis is coincidental to the
<110> direction in silicon.
0°
A: Commands
init
Sentaurus™ Process User Guide 979
I-2013.12
caxis.tilt
Specifies the magnitude of the wafer miscut, that is, the tilt angle of the wafer normal from
the crystal c-axis. This argument is used in both analytic and Sentaurus MC implantations.
The default value and unit is , that is, there is no wafer miscut.
clear
Clears all the current structure data in memory. Default: true.
concentration
Concentration of the incorporated data field. The only available unit is , but any nodal
quantity (with any internal unit) can be initialized with this argument if no unit is specified.
Default: 0.0.
DelayFullD
By default, Sentaurus Process generates a minimum-dimensional structure, which will be
extruded to higher dimensions when Sentaurus Process encounters a mask. To generate a
full-dimensional structure, use !DelayFullD.
dfise
Selects a pair of DF–ISE format files for reading. The .grd,.dat or.grd.gz,.dat.gz
extensions are searched for automatically. For example, if dfise=filename, Sentaurus
Process looks for filename.grd, filename.dat, or filename.grd.gz,
filename.dat.gz. The DF–ISE format has a different default orientation from the
internal format of Sentaurus Process. A rotation is applied to the structure.
done
Returns 1 if the initialization is performed; otherwise, returns 0.
field
Name of the data field to be initialized everywhere in the structure.
flat.orient
Crystal orientation of the wafer flat or notch specified as a numeric list. Default:
flat.orient= {1 1 0}.
load.commands
Loads the commands in the TDR format file. Default: true.
<material>
Specifies a material for doping. It must be used with the field argument.
0°
cm 3–
A: Commands
init
980 Sentaurus™ Process User Guide
I-2013.12
pdb
Loads pdb parameters along with geometry and data in the TDR file. Default: true.
pdb.only
Loads only pdb parameters without geometry and data in the TDR file. Default: false.
resistivity
Sets the value of the field by requesting a resistivity. This argument only works for fields
that have the resistivity pdb parameters set (which, by default, are only As, B, P, Sb, and
In in silicon).
sat
Specifies to read the structure file in the Sentaurus Structure Editor format.
scale
Coordinates of the input structure are divided by the specified value. The default is 1.0e4,
which converts the units of the DF–ISE coordinate system (micrometer) to the units used
by Sentaurus Process (centimeter).
sigmac
Specifies to read the structure file in Sigma-C 3D format.
slice.angle
Angle of the simulation domain with respect to the wafer coordinate system. Default value
and unit: .
The slice.angle can be specified using a CutLine2D command:
init slice.angle= [CutLine2D 1.65 0.15 1.95 0.6]
tdr
Specifies the TDR format file to read. The _fps.tdr extension is appended to the
specified file name automatically if one is not supplied.
The TDR file can contain a variety of information depending on which tool was used to
write the file. By default, Sentaurus Process writes files with sufficient information to
restart a simulation. This includes current parameter settings, stored commands (such as
polygon, mask, contact), bulk mesh and data, and, in three dimensions, a boundary (see
Saving a Structure for Restarting the Simulation on page 76). If such a file is specified, all
this data is read and used to restart the simulation. It is also possible to read TDR files that
include only bulk mesh and data, or only a boundary. If only a boundary is available,
Sentaurus Process creates a mesh using current refinement criteria. Finally, a TDR file can
contain information for restarting a Sentaurus Process KMC simulation with a KMC
90–°
A: Commands
init
Sentaurus™ Process User Guide 981
I-2013.12
structure and other restart information. This type of file is saved by the kmc extract
command if the atomistic mode is switched on (see Atomistic Mode on page 383 and Using
the Sentaurus Process Interface on page 541).
If the --fastMode option is on and init does not find the specified file, it looks for
a.
bnd file instead.
For information about the TDR format, refer to the Sentaurus™ Data Explorer User
Guide.
top
Specifies that the gas is found at the smallest x-value (at the top of the structure). If !top
is specified, the gas is added at the highest x-value (at the bottom). Default: true.
wafer.orient
Wafer orientation specified as a numeric list. Default: wafer.orient= {0 0 1}.
Description
Sets up the mesh from either a rectangular specification or a file. The command also allows
initialization of the background doping concentration and type.
Examples
Read in a structure previously saved in tmp.grd.gz and tmp.dat.gz files:
init dfise= tmp
Finish a rectangular mesh and set up a boron doping of :
init field= Boron concentration= 1e15
See Also
CutLine2D on page 896
line on page 1010
region on page 1110
struct on page 1158
115
×10

A: Commands
insert
982 Sentaurus™ Process User Guide
I-2013.12
insert
Inserts segments into 1D structures, polygons into 2D structures, and polyhedra into 3D
structures.
Syntax
insert
polygon=<c> | polyhedron=<c> | segments= {<n1> <n2> ...}
[Adaptive]
[new.material=<c>] [new.region=<c>]
[replace.materials= {<mat1> ... <matn>}]
[replace.regions= {<reg1> ... <regn>}]
Arguments
Adaptive
If specified, Adaptive switches on adaptive meshing if remeshing. Parameters for
adaptive meshing are described in Adaptive Refinement on page 699. The default is the
return value of pdbGet Grid Adaptive.
new.material
Sets the material for the inserted segment, polyhedron, or polygon. This argument is
mandatory for all but TDR polyhedra or polygons.
new.region
Sets the name of the inserted region. It works with one segment or polygon only, that is, it
does not work if the segment, polygon, or polyhedron contains more than one region.
NOTE This argument is not allowed for TDR polyhedra.
polygon
Specifies the polygon to insert. Only polygons created with the xy argument of the
polygon command are allowed. The polygon argument fails if the simulation is three
dimensional, or if it is one dimensional and cannot be extruded to two dimensions because
there are no y-lines available.
polyhedron
Specifies the polyhedron to insert. It needs a 3D simulation or a simulation that can be
extruded to three dimensions.
A: Commands
insert
Sentaurus™ Process User Guide 983
I-2013.12
replace.materials
Specifies a list that indicates the materials to be replaced by the polyhedron. In addition to
explicit materials, the argument bulk.materials is allowed. If bulk.materials is
used, it means that all materials in the structure, except gas, will be replaced.
replace.regions
Specifies a list of regions to be replaced by the polyhedron.
segments
Segments are defined as a list of an even number of coordinates (in ). If more than two
coordinates are specified, unique region names are generated for each segment or region.
Description
Segments are defined using the segments argument, but polygons and polyhedra must be
defined using the polygon and polyhedron commands, respectively. You must specified one
of the segments, polyhedron, or polygon arguments.
When specified, replace.materials and replace.regions provide a list of materials
and regions to be replaced. If neither is specified, all materials will be replaced. If both are
specified, the union of them will be replaced. The new.material argument changes the
polyhedron material temporarily.
The insert command can be used to perform polyhedron or polygon etching and deposition
as well as the more general polyhedron or polygon insert functionality. Polyhedron or polygon
etching is performed by specifying new.material=gas or by creating a gas polyhedron. You
also can do the same in one dimension with segments, but this is the same as CMP or fill.
Polyhedron or polygon deposition is performed by specifying replace.materials=gas as
well as having one or more bulk materials in the polyhedron or polygon, or defining them
temporarily with new.material.
The insert command operates only in the MGOALS3D mode for polyhedra. If the SDE
mode is switched on, calling this command will set sde off.
Examples
Etch the structure using a polyhedron called prism:
insert polyhedron= prism new.material= Gas
µm
A: Commands
insert
984 Sentaurus™ Process User Guide
I-2013.12
Replace all materials in the structure with a polyhedron called smallCube, and fill the
polyhedron with oxide:
insert polyhedron= smallCube new.material= Oxide
Replace the nitride, and only the nitride, in the simulation with oxide inside the polyhedron
called smallCube:
insert polyhedron= smallCube replace.materials= {Nitride} new.material= Oxide
Replace all materials in the structure with a polygon called Channel, and fill the polygon with
material Silicon2, and the region is named ChannelRegion. This polygon can be inserted
without merging with neighboring silicon regions:
mater add name= Silicon2 new.like= Silicon alt.matername= Silicon
insert polygon= Channel new.material= Silicon2 new.region= ChannelRegion
See Also
Inserting Polyhedra in Three Dimensions on page 786
polygon on page 1082
polyhedron on page 1086

A: Commands
integrate
Sentaurus™ Process User Guide 985
I-2013.12
integrate
Returns the volume integration of the named quantity.
Syntax
integrate
[absolute] [average] [element] [interfaces]
[<material>]
[max= {<n> <n> <n>}]
[min= {<n> <n> <n>}]
[mode= mesh | boundary | jagged]
[name=<c>] [region=<c>]
[skipgas] [syntax.check.value=<c>]
Arguments
absolute
Specifies that integration is performed with the absolute values of the named quantity.
average
Specifies that the average value of the named quantity is computed and added to the
returned Tcl list.
element
Specifies that integration is performed elementwise. In this case, it is expected that the
quantity specified by name is an elemental quantity.
interfaces
Specifies that integration is performed on interface meshes.
<material>
Used to limit integration to regions of the specified material. For information about
specifying materials, see .
max
List of numbers defining the x-, y-, and z-coordinates of the lower-right front corner of the
cutting box in the internal coordinate system. For 1D, 2D, and 3D structures, a list of one,
two, or three numbers is required, respectively. The possible maximum number is used for
missing numbers.
A: Commands
integrate
986 Sentaurus™ Process User Guide
I-2013.12
min
List of numbers defining the x-, y-, and z-coordinates of the upper-left back corner of the
cutting box in the internal coordinate system. For 1D, 2D, and 3D structures, a list of one,
two, or three numbers is required, respectively. The possible minimum number is used for
missing numbers.
mode
Specifies the integration mode within a given box:
•The
mesh mode (default) uses a mesh-cutting algorithm.
•The
boundary mode for three dimensions cuts the boundary to a cuboid and remeshes
the cuboid using the given mesh refinements. The boundary mode is for 3D
simulations only.
•The
jagged mode includes all nodes contained entirely within the given box for
integration.
name
Quantity to be integrated. Default: Z_Plot_Var.
region
Limits integration to only the region specified.
skipgas
Specifies that integration is omitted on invisible meshes.
syntax.check.value
Sets a value to be returned only during syntax-checking mode. Sometimes, the value
returned by a command can cause a false syntax-check error because the value returned by
the command would not be the value during the normal run mode. Setting this value avoids
such problems.
Description
This command integrates the field specified with the name argument (by default, the last
unnamed select command field) over the entire structure or within a given box specified by
min and max. If material is specified, the integration is limited to regions of the given
material. If a region is specified, the integration is limited to only the named region.
The command by default expects the quantity to be nodal and the integration is performed
nodewise, but if element is given, an elemental quantity is expected and the integration
proceeds elementwise.
A: Commands
integrate
Sentaurus™ Process User Guide 987
I-2013.12
A Tcl list is returned where the first value is the integrated value.
The second value is the volume of the computed regions:
■In <value_unit>*cm for one dimension
■In <value_unit>* for two dimensions
■In <value_unit>* for three dimensions
where <value_unit> is the unit of the named quantity.
The third value is the dose (the integrated value divided by the simulated area in in all
dimensions). The fourth and fifth values are the minimum and maximum of the named
quantity, respectively. If average is specified, the averaged result for the named quantity is
appended to the returned Tcl list.
Examples
Return the integral boron in all silicon regions in the structure, the volume of all silicon regions,
and the combined boron dose in all silicon regions:
integrate silicon name= Boron
Return the integrated pressure elementwise in the entire structure (excluding gas) and the
volume of the structure (excluding gas):
select z= "1.0/3*(StressELXX+StressELYY+StressELZZ)"
integrate element
Return a list of integral, volume, dose, minimum, maximum, and average values of boron
within the box defined by the upper-left corner (0.0, 0.0) and the lower-right corner (10.0, 0.2):
integrate name= Boron average min= {0. 0.} max= {10. 0.2}
Return the integrated term BActive, the volume, and the combined dose in all silicon regions
in the structure. The term BActive is first converted to a temporary data field before
integration:
select z= BActive
integrate silicon
Return a list of integral, volume, dose, minimum, and maximum of boron within the cuboid
defined by the upper-left back corner (0.0, 0.0, 0.0) and the lower-right front corner (0.4, 0.4,
0.4):
integrate name= Boron mode= jagged min= {0. 0. 0.} max= {0.4 0.4 0.4}
cm2
cm3
cm 2–

A: Commands
interface
988 Sentaurus™ Process User Guide
I-2013.12
interface
Returns the location or the value of the selected data field at a material interface.
Syntax
interface
[All] [data] [<material>] [name=<c>]
[p1= {<n> <n> <n>} p2= {<n> <n> <n>}]
[precision=<n>]
[side=<c>]
[syntax.check.value=<c>]
[x=<n>][<m>|<cm>|<um>|<nm>]
[y=<n>][<m>|<cm>|<um>|<nm>]
[z=<n>][<m>|<cm>|<um>|<nm>]
Arguments
All
If specified, all interface locations are returned. Otherwise, only the first value is returned.
data
If specified, the value of the selected data field at the interface is returned.
<material>
Usually works with an interface description and returns the location or value of the selected
quantity at the interface. For information about specifying materials, see .
NOTE If an interface is not specified, an error occurs. If the specified interface
does not exist in the current structure, an error is reported.
name
Name of the data field to be returned when data is specified. Default: Z_Plot_Var.
p1, p2
Specify the two endpoints of a cutline. Both p1 and p2 must be specified together as a list
of numeric values. Only the first <dim> numbers from each list is read, where <dim> is the
spatial dimension of the simulation. Specifying the endpoints with p1 and p2 allows for
nonaxis-aligned cuts. Endpoints also can be used to limit axis-aligned cuts instead of
cutting through the entire structure.
A: Commands
interface
Sentaurus™ Process User Guide 989
I-2013.12
precision
Controls the number of precision digits of floating values (in scientific notation).
Default: 6.
side
Takes its value from one of the two bulk materials consisting of the interface or the
‘interface’ (literally) itself. If side is not specified, the ‘interface’ itself is assumed. If
side is specified as one of the bulk materials, the value of the selected data field for the
bulk material is returned. This argument is effective only if data is specified.
syntax.check.value
Sets a value to be returned only during syntax-checking mode. Sometimes, the value
returned by a command can cause a false syntax-check error because the value returned by
the command would not be the value during the normal run mode. Setting this value avoids
such problems.
x, y, z
Provide the description of a line to look for the interface. These arguments are unnecessary
in 1D simulations. In 2D simulations, one of x and y must be specified. In 3D simulations,
two of these must be specified. Default value and unit: .
Description
This command returns the position of an interface or returns the value of the selected data field
if data is specified. Therefore, the command can be used to prepare plots of material
thickness, silicon consumption, or material growth. It also is used to provide an argument to
the interpolate command, which returns a list if there is more than one interface. The list-
processing commands of Tcl, particularly lindex, are very helpful.
Examples
Return the silicon–oxide interface at a lateral position of :
interface y= 1.0 silicon /oxide
Return the top position of the oxide:
interface oxide /gas
Return any oxide–nitride interfaces between (1.0, 1.0) and (1.1, 1.1). This specification is valid
for one or two dimensions, but not three dimensions. In one dimension, it returns the interfaces
between 1.0 and 1.1:
interface p1= {1.0 1.0} p2= {1.1 1.1} Nitride /Oxide
0.0 µm
1.0 µm

A: Commands
interpolate
Sentaurus™ Process User Guide 991
I-2013.12
interpolate
Returns the requested position or value at a specified location.
Syntax
interpolate
<material> [name=<c>]
[syntax.check.value=<c>]
[value=<n>]
[x=<n>][<m>|<cm>|<um>|<nm>]
[y=<n>][<m>|<cm>|<um>|<nm>]
[z=<n>][<m>|<cm>|<um>|<nm>]
Arguments
<material>
Limits the search to a single material. For information about specifying materials, see
Material Specification on page 52.
name
Name of a data field. This allows printing without using the select or tclsel
commands. Default: Z_Plot_Var.
syntax.check.value
Sets a value to be returned only during syntax-checking mode. Sometimes, the value
returned by a command can cause a false syntax-check error because the value returned by
the command would not be the value during the normal run mode. Setting this value avoids
such problems.
value, x, y, z
The combination of these arguments determines how the command operates:
• In 1D simulations, you must supply either
x or value. If x is supplied, Sentaurus
Process returns the value at x. If value is supplied, Sentaurus Process returns the
locations at which the selected profile crosses value.
• In 2D simulations, two of the four arguments must be given (not
z).
•In 3D simulations, three of the four arguments must be given.
For example, in 2D, if x and value are given, the locations along x where value is crossed
are returned. If x and y are given, the value at the location (x,y) is returned. The default
unit of x, y, and z is .
µm
A: Commands
interpolate
992 Sentaurus™ Process User Guide
I-2013.12
Description
This powerful command analyzes simulation results. It returns the interpolated value of one
coordinate given the other two coordinates in three dimensions defined by x- and y-variables,
and the data field. It also works for 1D simulations by returning values as a function of one
coordinate.
This command returns a Tcl list of values if more than one is found. For example, there may
be several junctions found along a given line. All of these are returned and can be processed by
standard Tcl list operations. In most cases, this command returns a single value. The returned
value is in internal units. Internal units are CGS; for example, the unit for stress is dyn/cm2.
In addition, this command can return the data value at a specified position in the structure or
return the position at which a specified data value occurs.
Examples
Return the value of the data field at the position ( , ) in the oxide:
interpolate oxide x= 1.0 y= 1.0
Return a list of zero crossings in silicon of the data field along the vertical line y = :
interpolate y= 0.0 silicon value= 0.0
Return a list of zero crossings in silicon along a horizontal line at a depth of :
interpolate silicon x= 2.0 value= 0.0
Return the value of the data field at :
interpolate x= 1.0 silicon
Return the the yy component of the element stress field at :
interpolate x= 1.0 oxide name=StressEL_yy
Return a list of zero crossings in silicon material:
interpolate silicon value= 0.0
See Also
interface on page 988
plot.xy on page 1076
point.xy on page 1080
1.0 µm
1.0 µm
0.0 µm
2.0 µm
1.0 µm
1.0 µm

A: Commands
KG2E
Sentaurus™ Process User Guide 993
I-2013.12
KG2E
Computes Young’s modulus from the bulk modulus and the shear modulus.
Syntax
KG2E <n> <n>
Arguments
<n>
The first value is the bulk modulus.
The second value is the shear modulus.
Description
The same units are assumed for all moduli.
Examples
Compute Young’s modulus from the bulk modulus 1.2272e12 dyn/cm2 and the shear modulus
6.328e11 dyn/cm2:
KG2E 1.2272e12 6.328e11

A: Commands
KG2nu
994 Sentaurus™ Process User Guide
I-2013.12
KG2nu
Computes the Poisson ratio from the bulk modulus and the shear modulus.
Syntax
KG2nu <n> <n>
Arguments
<n>
The first value is the bulk modulus.
The second value is the shear modulus.
Description
The same units are assumed for all moduli.
Examples
Compute the Poisson ratio from the bulk modulus 1.2272e12 dyn/cm2 and the shear modulus
6.328e11 dyn/cm2:
KG2nu 1.2272e12 6.328e11

A: Commands
kmc
Sentaurus™ Process User Guide 995
I-2013.12
kmc
Specifies options for the atomistic kinetic Monte Carlo (KMC) mode.
Syntax
kmc
add |
add queue name=<c> [amorphous | crystalline] [clustertype name=<c>]
[coordx=<n>][<m>|<cm>|<um>|<nm>]
[coordy=<n>][<m>|<cm>|<um>|<nm>]
[coordz=<n>][<m>|<cm>|<um>|<nm>] |
deatomize name=<c> [active] [<material>] |
defects.read=<c> |
defects.write=<c> [defectname=<c>] [materialname=<c>] |
defecttypes [<material>] |
(extract
acinterface
[coordx=<n>][<m>|<cm>|<um>|<nm>]
[coordy=<n>][<m>|<cm>|<um>|<nm>]
[coordz=<n>][<m>|<cm>|<um>|<nm>] |
defects
[name=<c>] ([acinterface] [detailed])
[countdefects] [countparticles]
[defectname=<c>] [materialname=<c>] |
dose [countdefects] [defectname=<c>] [materialname=<c>] [name=<c>] |
histogram name=<c> [materialname=<c>] [meansize [minsize=<n>]] |
materials
[coordx=<n>][<m>|<cm>|<um>|<nm>]
[coordy=<n>][<m>|<cm>|<um>|<nm>]
[coordz=<n>][<m>|<cm>|<um>|<nm>] [detailed] |
profile
name=<c>
[coordx=<n>][<m>|<cm>|<um>|<nm>]
[coordy=<n>][<m>|<cm>|<um>|<nm>]
[coordz=<n>][<m>|<cm>|<um>|<nm>]
[defectname=<c>] [materialname=<c>] [timeaveraged] |
supersaturation [name=<c>] |
tdrAdd [concentrations] ([defects] | [visual= <list>]) [histogram]
[list= <list>] [Stress] |
tdrClear |
tdrWrite [filename=<c>]) |
materialtypes | off | particletypes | PDEupdated | present [name=<c>] |
report
A: Commands
kmc
996 Sentaurus™ Process User Guide
I-2013.12
Arguments
acinterface
The kmc extract acinterface command returns the 1D coordinates of the
amorphous–crystalline transitions present in the simulation. If the simulation has
dimension, it needs cutlines, specified by coordx, coordy, or coordz.
The kmc extract defects acinterface command returns the atomistic position of
lattice atoms in the amorphous–crystalline interface, when using the LKMC
recrystallization mode.
active
Used by kmc deatomize to deatomize only the active part of a dopant.
add
Instructs Sentaurus Process KMC to add a new defect into the simulation cell. The defect
to be included must be first sent to the queue using kmc add queue. Here, the defect is
specified with name, and the coordinates with coordx, coordy, and coordz. When all
defects are in the queue, the command kmc add, without any arguments, passes the defects
from the queue to Sentaurus Process KMC, erasing the queue.
amorphous, crystalline
When added to kmc add, amorphous creates the defect and changes the material to
amorphous.
When used with kmc add, crystalline creates the defect in a crystalline phase of the
material, locally recrystallizing the area where the defect will be added when necessary.
clustertype
Defines the type of defect specified in name. For example, kmc add queue
clustertype name=I56 returns if this cluster is considered an amorphous pocket (AP),
a {311}, or a loop.
concentrations
Instructs Sentaurus Process KMC to generate concentration information to be included in
the TDR file. It is used with kmc extract tdrAdd.
coordx, coordy, coordz
Specify the x-, y-, and z-coordinates needed for the kmc add queue command. They also
are used to specify the cutlines in the kmc extract profile and kmc extract
materials commands. Default unit: .
N
N1–
µm
A: Commands
kmc
Sentaurus™ Process User Guide 997
I-2013.12
countdefects
Used with kmc extract defects to instruct Sentaurus Process KMC to count the
number of defects instead of listing the particles in the defects.
Used with kmc extract dose to compute the dose of defects, not particles. For example,
the dose of loops is different from the dose of particles in loops.
countparticles
Used with kmc extract defects to instruct Sentaurus Process KMC to count and return
the number of particles, instead of listing them.
deatomize
Instructs Sentaurus Process KMC to build a new data field and fill it with the
concentrations taken from the KMC simulation. name is the field to create. For
deatomize, the argument name also can be XTotal or NetActive, where X means any
dopant. It accepts the active argument to account for the active part of the dopant only.
defectname
Specifies an optional name of a defect (for example, ThreeOneOne, ImpurityCluster)
for:
•kmc defects.write
•kmc extract defects
•kmc extract dose
•kmc extract profile
This argument is used to further refine name. For example, if name=I, using defectname
refines these interstitials to interstitials as point defects, or in impurity clusters, and so on.
defects
kmc extract defects returns the defects currently present in the simulation.
kmc extract tdrAdd defects appends to the TDR file an atomistic 3D view of the
defects currently contained in the simulation, allowing for visualization, and loading and
continuing the simulation.
defects.read
Specifies the name of a text file from which to read its defects and to insert them in the
current simulation.
A: Commands
kmc
998 Sentaurus™ Process User Guide
I-2013.12
defects.write
Specifies the name of a text file into which to write all the current defects in the simulation.
Use defectname and materialname to filter the defects written.
defecttypes
Using kmc defecttypes returns the name of the defects that can be used by
defectname.
detailed
Produces different results depending on the context:
•kmc materials detailed returns the current list of materials for each internal
KMC element, including the element coordinates.
•kmc extract defects acinterface detailed works as kmc extract
defects acinterface, but returns all the lattice atoms in the LKMC model, and not
solely those belonging to the amorphous–crystalline interface.
dose
Using kmc extract dose returns doses (concentration in ). The argument
countdefects returns the dose of defects, instead of the dose of particles (which is the
default). The defects and particles included in this dose are refined with name and
defectname.
extract
kmc extract retrieves information about the current status of the KMC simulation. This
information is mainly concentrations, histograms, the atomistic positions of defects, and
the types of defect. kmc extract also is used to control the information written to the
TDR file.
It is mandatory to use another argument with kmc extract:
•acinterface returns the 1D coordinates of the amorphous–crystalline transitions
present in the simulation.
•defects returns the current list of particles in the simulation.
•dose computes and returns the dose of given particles or defects or both.
•histogram retrieves the number of defects depending on their size.
•materials retrieves the materials currently in the simulation.
•profile returns the concentration of particles, defects, electrons, and holes in the
simulation or any cutline of the simulation.
cm 2–
A: Commands
kmc
Sentaurus™ Process User Guide 999
I-2013.12
•supersaturation returns the concentration relative to equilibrium for interstitials
and vacancies.
•tdrAdd, tdrClear, and tdrWrite control the information to be written to the TDR
file.
filename
Specifies the name of the TDR file to be written by kmc extract tdrWrite.
histogram
kmc extract histogram returns the number of defects for each defect size. The
argument name determines the returned histogram. If name is XI or XV (X being a dopant
or impurity), it returns the histogram of impurity clusters for that dopant with Is or Vs. If
the name is I or V, it produces the histogram of I or V extended defects. Finally, if name=IV,
it returns the AP histograms.
If meansize is included, the returned value is not a list of defects and sizes, but the average
size for these defects. The minimum size for computing this average is zero by default, but
it can be changed using minsize.
kmc extract tdrAdd histogram includes histogram information in the TDR file.
list
Adds a list of fields to be included in the TDR file. This argument is used in kmc extract
tdrAdd. Any specific defect name is allowed, and generic defect names (as obtained by
kmc defecttypes) also are allowed. For example, I8 adds this cluster to the TDR file,
but AmorphousPockets adds any AP existing in the simulation.
<material>
Specifies the material name for kmc deatomize or kmc defecttypes. For information
about specifying materials, see .
materialname
Restricts the output to the material specified. materialname adds a condition to the
output of kmc extract defects, profile, dose, and histogram.
materials
kmc extract materials returns a list of materials currently present in the simulation.
kmc extract materials detailed returns the coordinates and the materials of the
KMC elements.
A: Commands
kmc
1000 Sentaurus™ Process User Guide
I-2013.12
materialtypes
Returns the subset of materials allowed in the Sentaurus Process KMC simulation. Any
material not listed here is assigned as ‘unknown’.
meansize
Used only with kmc extract histogram. It instructs Sentaurus Process KMC to
compute the average size for the specified defect type. The minimum size needed to take
the defect into account is 0, unless minsize is specified.
minsize
Used only with kmc extract histogram meansize. It instructs Sentaurus Process
KMC to use the specified value as the minimum size to take any cluster into consideration
when computing the average cluster size.
name
Specifies the name of the field, particle, or defect for the following arguments. In the
following, is the name of a valid dopant (such as B or As). Any defect means very
detailed defects such as B2I3, I8, I4V5, and AsV4. Any particle means point defects, dopants,
impurities, or impurity- and dopant-paired point defects in any charge state, that is, any
name obtained with kmc particletypes (for example, I, VMM, BiP, or F):
•add is the name of the defect or particle to be added. XAmorphous and XInterface also
are acceptable.
•clustertype is any defect.
•deatomize is any defect or any particle. XAmorphous, XInterface, XTotal,
NetActive, pNetActive, nNetAtive, and tNetActive also are valid.
•defects is any defect or any particle.
•dose is any defect or any particle.
•histogram: XI, XV, I, V, and IV are the only valid names.
•present is any defect or any particle. XAmorphous, XInterface, and XTotal also are
valid.
•profile is any defect or any particle. Holes, electrons, XAmorphous, and XInterface
also are valid.
•supersaturation allows only I or V.
off
Use kmc off to delete the Sentaurus Process KMC information and to remove the current
KMC object from memory.
X
A: Commands
kmc
Sentaurus™ Process User Guide 1001
I-2013.12
NOTE Use with caution.
particletypes
Returns a list of valid particle names. This list may change between simulations, depending
on the dopants specified in the parameter database.
PDEupdated
Returns true if the state of Sentaurus Process KMC did not change since the last time the
PDEs were synchronized (by using KMC2PDE).
present
Returns true when the species specified in name is in the KMC simulation.
profile
Using kmc extract profile returns concentrations. These concentrations contain 1D,
2D, or 3D information depending on the simulation dimension. Cutlines can be specified
with coordx, coordy, and coordz. Each of these arguments reduces the dimensions of
the output by 1. The concentrations in 1D simulations can be directly compared with SIMS
profiles.
The argument name specifies the particle or defect from which the concentration is
obtained. If name is a valid particle name (see particletypes), defectname can be
used with a valid defect name (see defecttypes) to further refine name. When a particle
is specified using name, profile returns the concentration of particles; otherwise, it
returns the concentration of defects.
Some particular names that can be used are:
•holes, electrons, GapNarrowing, and gap for electronic concentrations.
•stressXX, stressXY, stressZZ, strainXX, strainYY, strainZZ, strainXY,
strainXZ, and strainYZ for mechanical properties.
•dopants for the net active concentration.
•Ge for the germanium concentration.
queue
Adds the new defect, specified by name, into the simulation at the coordinates coordx,
coordy, and coordz. Adding defects to the queue will not put them in the KMC
simulation. To transfer the defects from the queue to the simulation, use kmc add without
any other arguments.
A: Commands
kmc
1002 Sentaurus™ Process User Guide
I-2013.12
report
Instructs Sentaurus Process KMC to generate a list of defects created during the simulation.
This list includes the first and last time the defect was seen and the temperature. If the
defect is still in the simulation, the report also gives the number of defects. In addition, a
report is printed automatically at the end of the diffuse and implant commands.
Stress
Using kmc extract tdrAdd stress adds the stress and strain distribution as imported
by Sentaurus Process KMC.
supersaturation
Using kmc extract supersaturation returns the value of the global supersaturation
(concentration over equilibrium concentration). The argument name must be specified; it
can only be or .
tdrAdd
Using kmc extract tdrAdd instructs Sentaurus Process KMC to add a new snapshot of
information ready to be written into a TDR file. This information is stored in memory and
is written using kmc extract tdrWrite. In addition, kmc extract tdrAdd without
arguments adds an empty snapshot. The tdrAdd options are:
•concentrations computes and adds 1D, 2D, or 3D concentrations for each particle
and defect.
•defects adds atomistic 3D information. It allows you to see the shape and position of
the defect, and to load and continue the simulation.
•histogram adds histograms for extended defects and impurity clusters.
•list adds user-specified defects. For example, concentrations only adds ‘I in
ThreeOneOne’, but list can be used to add I45, I65, and so on. Specifying the name of
a defect (as obtained in kmc defecttypes) adds all the clusters in this particular
defect for each existing size in the simulation.
•stress includes stress fields in the file.
•visual includes atomistic information for visualization purposes in the file.
tdrClear
kmc extract tdrClear removes all snapshots previously added using kmc extract
tdrAdd from memory.
I
V
A: Commands
kmc
Sentaurus™ Process User Guide 1003
I-2013.12
tdrWrite
kmc extract tdrWrite instructs Sentaurus Process KMC to write all snapshots
(previously added using tdrAdd) to a file. The name of the file is specified using
filename.
timeaveraged
Must be used with kmc extract profile. It generates time-averaged concentrations of
particles, instead of instantaneous ones. The averaging is performed between two
snapshots. Since timeaveraged only makes sense for mobile particles, the argument
name must be a valid particle not a defect.
visual
When added to kmc extract tdrAdd, it includes atomistic information that can be used
for visualization purposes only, and not for restarting (in contrast with kmc extract
defects).
visual attempts to produce a file as small as possible without losing atomistic
information. You must specify a list of the type of defects to be saved. Generic defects
(such as ThreeOneOne, Void), specific ones (such as B2I3), or materials (Silicon)
specified act as filters for the defects to be save.
To save all of them, use visual=all.
Description
This command sends direct instructions to Sentaurus Process Kinetic Monte Carlo (Sentaurus
Process KMC), which is used for diffusion. The main options are:
■add creates defects in the simulation.
■deatomize creates fields according to the atomistic concentrations.
■extract returns physical information (mainly concentrations) about the simulation. It has
different arguments including defects, dose, histogram, profile, tdrAdd,
tdrClear, and tdrWrite.
Examples
Create an I8 cluster, a mobile B in interstitial position, and the BIC B2I2 in the queue, and add
the queue to the KMC simulation:
kmc add queue coordx= 10<nm> coordy= 15<nm> coordz= 6<nm> name= I8
kmc add queue coordx= 3<nm> coordy= 8<nm> coordz= 7<nm> name= B2I2
kmc add queue coordx= 5<nm> coordy= 5<nm> coordz= 5<nm> name= Bi
kmc add
A: Commands
kmc
1004 Sentaurus™ Process User Guide
I-2013.12
Return whether an I543 is a ThreeOneOne or a loop in this simulation:
kmc clustertype name= I543
Create a BTotal data field, which will be computed by Sentaurus Process KMC including all
the boron in any defect in the simulation:
kmc deatomize name= BTotal
Create an As4V data field, filled with the As4V information taken from the KMC simulation:
kmc deatomize name= As4V
Return a list of the different defect types modeled by Sentaurus Process KMC:
kmc defecttypes
Return a list containing the particles currently in the KMC simulation, including its defect type,
defect number, and coordinates. Particles with the same defect number belong to the same
defect:
kmc extract defects
Return a list of each I particle currently in the KMC simulation:
kmc extract defects name= I
Return the B particles in impurity clusters currently in the simulation:
kmc extract defects name= B defectname= ImpurityCluster
Return the number of particles in B2I3 defects:
kmc extract defects name= B2I3 countparticles
Return the number of B2I3 defects in the simulation:
kmc extract defects name= B2I3 countdefects
Return a list of particles currently in the KMC simulation. It also returns the ‘hidden’ particles,
which are regenerated by Sentaurus Process KMC using internal information (see Damage
Accumulation Model: Amorphous Pockets on page 432):
kmc extract defects detailed
Return the As4V defects present in the simulation:
kmc extract defects name= As4V
A: Commands
kmc
Sentaurus™ Process User Guide 1005
I-2013.12
Return the electron concentration computed by Sentaurus Process KMC:
kmc extract profile name= electrons
Return the BIC histograms:
kmc extract histogram name= BI
Return the average size of I in extended defects. The minimum size to be included in this
average is set to 40:
kmc extract histogram name= I minsize= 40
Return the concentration of I2V3 defects:
kmc extract profile name= I2V3
Return the concentration of holes in the KMC simulation:
kmc extract profile name= holes
Return the concentration of boron in any defect:
kmc extract profile name= B
Return the concentration of substitutional boron:
kmc extract profile name= B defectname= PointDefect
Return the dose (concentration in ) of interstitials in {311}s:
kmc extract dose name= I defectname= ThreeOneOne
Return the list of materials currently present in the simulation:
kmc extract materials
Return the coordinates of each KMC element and its material:
kmc extract materials detailed
Return the concentration of mobile interstitials in the simulation:
kmc extract profile timeaveraged name= I
Return the interstitial supersaturation:
kmc extract supersaturation name= I
cm 2–
A: Commands
kmc
1006 Sentaurus™ Process User Guide
I-2013.12
Clear all the previous stored information, add a new snapshot with the concentration of
particles and the atomistic information for any defect, and write the information in a TDR file
called example.tdr. Since defects is included, this file also can be used to load and
continue the simulation:
kmc extract tdrClear
kmc extract tdrAdd concentrations defects
kmc extract tdrWrite filename= example.tdr
Return the list of materials supported by Sentaurus Process KMC:
kmc materialtypes
Return the list of particles supported by Sentaurus Process KMC. This list can be changed
using pdb commands:
kmc particletypes
Return true (1) or false (0) depending on the presence of BI2 in the simulation:
kmc present name= BI2
Exit Sentaurus Process KMC and remove all its associated information from memory:
kmc off
Print a list of the simulated defects with the first and last time they were seen and the
temperature in the simulation:
kmc report
In one dimension, return the concentration of B2I2 at x = 10 nm. In two dimensions, return the
concentration of B2I2 in the line x = 10 nm. In three dimensions, return the concentration of
B2I2 in the plane x = 10 nm:
kmc extract profile name= B2I2 coordx= 10<nm>
Return the concentration in the line y with x = 10 nm and z = 15 nm:
kmc extract profile name= B2I2 coordx= 10<nm> coordz= 15<nm>
Create a TDR snapshot with any {311} defect and the BI2 and B2I2 defects:
kmc extract tdrAdd list= {ThreeOneOne BI2 B2I2}
See Also
Chapter 5 on page 381
deposit on page 901
diffuse on page 908

A: Commands
KMC2PDE
Sentaurus™ Process User Guide 1007
I-2013.12
etch on page 923
implant on page 961
integrate on page 985
line on page 1010
photo on page 1061
profile on page 1093
region on page 1110
select on page 1117
stressdata on page 1151
struct on page 1158
KMC2PDE
Translates atomistic KMC information to Sentaurus Process.
Syntax
KMC2PDE
Description
This command translates the atomistic information stored in the KMC diffusion into
continuum five-stream quantities and transfers it into the standard Sentaurus Process mesh.
Consequently, there are two transformation involved here:
■Deatomization of particles into concentrations
■Translation of Sentaurus Process KMC field names into Sentaurus Process field names
The deatomization is performed in a standard way by calling kmc deatomize for each
existing Sentaurus Process KMC field. If the Sentaurus Process mesh is too coarse, the
continuum fields will look smooth, but some information may be lost. On the other hand, if the
Sentaurus Process mesh is too fine, isolated islands of concentration may form following its
corresponding atomistic concentrations.
The translation is made as accurately as possible by mapping as many KMC species into
similar five-stream fields. When this one-to-one mapping is not possible or not unique,
acceptable approximations can be taken. For example, Bi will be translated into BoronInt,
but B2I2, BI2, and so on will be translated into only BCluster. A complete list of these
translations is available in the file KMC.tcl.
See Also
UnsetAtomistic on page 1187

A: Commands
layers
1008 Sentaurus™ Process User Guide
I-2013.12
layers
Prints material interfaces and integrated data field values.
Syntax
layers
[<material>] [merge] [name]
[precision=<n>] [print.logfile]
[region.names] [syntax.check.value=<c>]
[x=<n>][<m>|<cm>|<um>|<nm>]
[y=<n>][<m>|<cm>|<um>|<nm>]
[z=<n>][<m>|<cm>|<um>|<nm>]
Arguments
<material>
Used to limit the reporting of layers to regions of the specified material. For information
about specifying materials, see Material Specification on page 52.
merge
Specifies that the adjacent regions with the same material should be merged. Default: false.
name
Name of a data field. This allows printing without using the select or tclsel
commands. Default: Z_Plot_Var.
precision
Controls the number of precision digits of floating values (in scientific notation).
Default: 12.
print.logfile
Allows output to be written to the log file.
region.names
Specifies that region names must be printed in addition to the material names for each
region in the structure.
syntax.check.value
Sets a value to be returned only during syntax-checking mode. Sometimes, the value
returned by a command can cause a false syntax-check error because the value returned by
A: Commands
layers
Sentaurus™ Process User Guide 1009
I-2013.12
the command would not be the value during the normal run mode. Setting this value avoids
such problems.
x, y, z
Specify the constant values of a line along which sectioning will be performed. In one
dimension, these arguments are not necessary. In two dimensions, only one of x or y can
be specified for a given device. Specifying x produces a horizontal slice through the device,
and y specifies a vertical slice. An easy way to remember this is that the cross section is
taken at the constant value specified. For a 3D simulation, two of these three arguments
must be specified. Default unit: .
Description
This command prints the material interfaces and integrates the selected data field in each
region. It is most useful for examining doping because it gives the integrated doping in each
layer. This command can be simulated with the interpolate and interface commands,
and it returns a Tcl list of each material.
Examples
In a 1D simulation, list all material interfaces:
layers
In a 2D simulation, list all material interfaces at a lateral position of and integrate the
data field named Boron:
layers y= 0.0 name= Boron
See Also
interface on page 988
interpolate on page 991
select on page 1117
tclsel on page 1164
µm
0.0 µm

A: Commands
line
1010 Sentaurus™ Process User Guide
I-2013.12
line
Specifies the position and spacing of mesh lines.
Syntax
line
location=<n>[<m>|<cm>|<um>|<nm>] (x | y | z)
[clear] [kmc] [mgoals]
[spacing=<n>][<m>|<cm>|<um>|<nm>]
[tag=<c>]
Arguments
clear
Clears lines in preparation for a new structure definition, or removes all ticks stored for the
UseLines method (see UseLines: Keeping User-defined Mesh Lines on page 722).
kmc, mgoals
Lines for KMC and MGOALS (continuum) meshes are stored separately. By default, line
commands are applied to both KMC and continuum meshes. Use negative values for these
arguments to not apply mesh lines to one or the other. For example, for a line command
to apply only to a continuum mesh, use !kmc.
location
Location along the chosen axis. Default unit: .
spacing
Local grid spacing. Each mesh line has a characteristic required spacing. Lines are graded
from one spacing to the next over the interval. The default is a spacing equal to the largest
interval between the neighboring lines. Default unit: .
tag
Lines can be labeled for later reference by region commands. The label can be any word.
x, y, z
Orientation of the mesh line. Specifying x places a mesh line at a constant x-value. A series
of line x commands would specify the horizontal grid locations during the simulation.
µm
µm
A: Commands
line
Sentaurus™ Process User Guide 1011
I-2013.12
Description
All line commands must precede region commands, which in turn must be followed by the
init command. Lines must be given in increasing order.
When used to create the initial mesh, only rectilinear structures can be specified with the line
and region commands, that is, rectangular regions in two dimensions and cuboid-shaped
regions in three dimensions.
Sentaurus Process uses the unified coordinate system (UCS):
■x is the direction normal to the wafer with positive-x oriented into the bulk of wafer.
■y is perpendicular to the x-direction and lies along the wafer surface; y is in the lateral
direction.
■The z-direction is used for three dimensions, and the direction is given by .
By default, Sentaurus Process delays promoting a structure until it is necessary (by use of a
higher dimensional mask). The lines specifying the higher dimensions are stored until they are
needed. During the init command, the line and spacing information is expanded into mesh
‘ticks’ that are stored in the PDB. These ticks are used every time a mesh is created if
UseLines is switched on (see UseLines: Keeping User-defined Mesh Lines on page 722).
After an init command, if new lines are specified and UseLines is switched on, the
spacing argument is ignored, and only one tick or mesh line at a time may be added. To create
an entirely new structure, the command line clear must first be issued to clear old lines and
mesh ticks before issuing new line, region, and init commands.
Examples
There are three user-specified y-lines and two user-specified x-lines. Taking the y-lines as an
example, there is a finer spacing in the center than at the edges. After processing, Sentaurus
Process produces a mesh with x-lines at 0.0, 0.42, 0.69, 0.88, 1.0, 1.12, 1.31, 1.58, and 2.0.
Around the center, the spacing is 0.12, approximately what will be requested. At the edge, the
spacing is 0.42 because that is as coarse as it could become without having an interval ratio
greater than 1.5 (a fixed quantity). If the interval ratio was allowed to be 9, for example, there
would be one interval of 0.9 and one interval of 0.1 on each side. In this example, specifying a
spacing of 1 at the edges is redundant because that is what the spacing of the user-specified
lines was already:
line x location= 0 spacing= 0.02 tag= surf
line x location= 3 spacing= 0.5 tag= back
line y location= 0 spacing= 1 tag= left
line y location= 1 spacing= 0.1
line y location= 2 spacing= 1 tag= right
XY×

A: Commands
line_edge_roughness
Sentaurus™ Process User Guide 1013
I-2013.12
line_edge_roughness
Adds line edge roughness (LER) to named masks.
Syntax
line_edge_roughness
correlation.length=<n>[<m>|<cm>|<um>|<nm>]
masks= <list>
max.segment.length=<n>[<m>|<cm>|<um>|<nm>]
normal= "Y" | "Z"
standard.deviation=<n>[<m>|<cm>|<um>|<nm>]
[max.tries=<n>] [min.radius=<n>] [smooth.points=<n>]
[random.reseed] [random.seed=<n>]
Arguments
correlation.length
Specifies the correlation length of the randomized LER. Corresponds approximately to the
concept of wavelength. Default value and unit: 20 nm.
masks
Specifies as a list the names of masks to receive LER defined by the other arguments in the
line_edge_roughness command.
max.segment.length
Specifies the maximum segment length. Mask edges are subdivided into segments that are
approximately this size or smaller before LER deviation is added to each. Default value and
unit: 1 nm.
max.tries
Specifies the maximum number of LER mask generation attempts. For nonzero values,
detection of nearly collinear points is performed after LER generation, and the LER
process is restarted if decimation occurs based on the mgoals accuracy value. The
default value is 0, meaning that the LER mask is accepted as it is, with no decimation
detection.
min.radius
When normal is not specified, where two mask edges receiving LER meet, the corner is
first rounded before LER is applied. This allows a well-defined application of LER and
avoids discontinuous jumps in the resulting mask shape. The rounding radius is the greater
of min.radius and twice the correlation.length.
A: Commands
line_edge_roughness
1014 Sentaurus™ Process User Guide
I-2013.12
normal
Defines the normal axis. Only mask segments normal to this axis receive LER. This axis is
also the reference axis along which the LER deviation is added to the given mask segment.
The default is to add LER to all edges of the mask.
random.reseed
Before the calculation of LER, the random number generator is reseeded to ensure each call
of line_edge_roughness results in randomized noise that is uncorrelated with other
calls of line_edge_roughness.
To switch off random reseeding, use !random.reseed to reproduce the same LER from
call to call. Default: true.
random.seed
Used to reproduce specific LER calculations from one run to the next by setting the same
random seed in both runs. Ignored when !random.reseed is used.
smooth.points
Alters the LER function at the structure boundaries allowing for better mirrored boundary
conditions when uniting symmetry-reduced structure parts at those boundaries. Defines the
number of segments in the discrete LER function, counting from the mask’s boundary
edge, which should be adjusted or smoothed. Default: 0, implying no smoothing.
standard.deviation
Specifies the standard deviation of the randomized LER. Corresponds approximately to the
concept of added noise amplitude. Default value and unit: 2 nm.
Description
This command adds LER to the named masks, along the mask edges normal to the given
normal axis ("Y" or "Z"). LER can be added to a mask only once. See Boolean Masks on
page 767.
Examples
Add LER to the mask named mask1 along mask segments normal to the z-axis. These
segments are subdivided into smaller segments of length smaller than or equal to 5 nm. LER is
characterized by a correlation.length of 25 nm and standard.deviation of 5 nm.
The random number generator is reseeded automatically before LER is calculated:
line_edge_roughness normal= "Z" masks= {mask1} \
correlation.length= 25.00<nm> standard.deviation= 5.00<nm> \
max.segment.length= 5.00<nm>

A: Commands
load
1016 Sentaurus™ Process User Guide
I-2013.12
load
Loads data from a file and interpolates it onto the current mesh.
Syntax
load
(dfise=<c> | grdfile=<c> datfile=<c> | tdr=<c>)
(merge | rename | replace | sum)
[actions= <list>] [fast.tdr.ave= <list>]
[flip (left | right | front | back | up | down)]
[offset= {<n> <n>}]
[shift=<n>] [species= <list>]
[transform= {<n> <n> <n> <n> <n> <n>}]
Arguments
actions, species
These lists specify species-by-species actions. The species name must be one of those in
the loaded .dat file. Each action in the actions list must be one of merge, rename,
replace, or sum.
dfise
Specifies the input file name. Sentaurus Process checks for all standard suffixes for both:
• Grid files (.
grd,_fps.grd,.grd.gz,_fps.grd.gz)
• Data files (.
dat,_fps.dat,.dat.gz,_fps.dat.gz)
fast.tdr.ave
Averages the data from a list of TDR files (all with identical meshes to the current mesh)
and replaces the current data with the averaged data from the files.
NOTE Do not use fast.tdr.ave with any other argument. To use it, specify
a list of files, for example:
fast.tdr.ave= {mydata1.tdr mydata2.tdr mydata3.tdr}
flip (left | right | front | back | up | down)
For 2D structures only. Performs a flip of the data in the indicated direction about the outer
boundary before interpolation. This argument must be used with a direction, for example,
flip front.
A: Commands
load
Sentaurus™ Process User Guide 1017
I-2013.12
grdfile, datfile
Specifies the exact names of the DF–ISE files (must not be used with dfise).
merge
Adds only new datasets that do not currently exist in the structure.
offset
For 2D structures only. Offsets the data by a vector before loading it.
rename
Adds new datasets and renames them by adding the suffix __load.
replace
Adds new datasets and replaces existing datasets with new datasets of the same name.
shift
Shifts the data laterally before loading it.
sum
Adds new datasets and sums matching datasets.
tdr
Specifies the input file name with TDR format. Sentaurus Process checks for standard file
names with the .tdr extension.
transform
Provides a general interface for translating or rotating the structure to be loaded before
interpolation.
In one dimension, one value must be specified: the shift in the x-coordinate.
In two dimensions, six values must be specified: rxx, ryx, rxy, ryy, offsetx, offsety.
In three dimensions, 12 values must be specified: rxx, ryx, rzx, rxy, ryy, rzy, rxz, ryz,
rzz, offsetx, offsety, offsetz. First, the offset is applied, and then the rotation
matrix is applied (it does not have to be an orthogonal matrix).
Description
This command interpolates data from TDR or DF–ISE grid and data files onto the current
mesh. The file to be loaded must have the same dimension as the existing structure. There are
several options for handling the new and old datasets. First, the actions can be applied
A: Commands
load
1018 Sentaurus™ Process User Guide
I-2013.12
individually to selected datasets using species and actions. If the species list appears,
the actions list must be specified and must have the same number of members as the
species list. If this is the case, only the species in the species list are taken from the external
datasets. If the species list does not appear, one of the global actions is used. The default
behavior is a global sum where new datasets are added and, if there is an existing dataset with
the same name, the external data is added (summed) with the existing dataset.
The other actions that can be performed are:
■merge takes only the new datasets that do not currently exist in the structure.
■rename renames new datasets by appending __load to the name, which can be
manipulated with the select command as required.
■replace replaces current datasets with new datasets of the same name.
■In atomistic mode, only tdr and replace can be used to replace the current simulation
contents with the particles stored in a TDR file.
Examples
Replace all existing datasets with those in the file in_fps.tdr:
load tdr= in replace
Load the TDR file in_fps.tdr, sum Arsenic_Implant with the existing
Arsenic_Implant (if available), and replace the existing Damage_Implant data field by the
one in the in_fps.tdr file:
load tdr= in species= {Arsenic_Implant Damage_Implant} actions= {sum replace}
See Also
select on page 1117

A: Commands
LogFile
Sentaurus™ Process User Guide 1019
I-2013.12
LogFile
Prints a message to the screen and to the log file.
Syntax
LogFile
<c>
[IL0 | IL1 | IL2 | IL3]
Arguments
<c>
Specifies the message to be printed to the screen and to the log file.
IL0, IL1, IL2, IL3
Specifies the information level.
Description
This command prints messages to the terminal window in which Sentaurus Process is running
and to the log file. If IL0, IL1, IL2, or IL3 is given and this command is called from within a
Sentaurus Process command, the message is printed only if the information level is equal to or
greater than the one specified.
Examples
Print the string "Step 25" to the log file and the screen:
LogFile "Step 25"
Print the contents of the Tcl variable DebugInfo only if info=1 or higher has been specified
in the calling command:
LogFile IL1 "$DebugInfo"

A: Commands
mask
1020 Sentaurus™ Process User Guide
I-2013.12
mask
Creates a mask for subsequent use in etch, deposit, or photo commands.
Syntax
mask
[bbox] [bool=<c>] [clear] [covered.status]
[cut.x=<n> (materials= <list> | regions= <list>)]
[get.segments] [get.segments.z]
[list] [name=<c>] [negative]
(
[layoutfile=<c>] [polygons= <list>] [segments= <list>] |
[left=<n>][<m>|<cm>|<um>|<nm>]
[right=<n>][<m>|<cm>|<um>|<nm>]
[front=<n>][<m>|<cm>|<um>|<nm>]
[back=<n>][<m>|<cm>|<um>|<nm>]
)
Arguments
bbox
Returns the mask bounding box. The command returns a list of lists where the values are
in centimeters: {ymin zmin} {ymax zmax}.
bool
Performs Boolean operations between masks. It cannot be used with layoutfile,
polygons, and negative. See Boolean Masks on page 767
clear
Clears the list of all masks. If name is specified, it clears only that mask.
covered.status
Used to obtain information about the coverage of the simulation domain. The following
strings may be returned:
•covered: The mask completely covers the simulation domain.
•uncovered: The mask does not cover the simulation domain at all.
A: Commands
mask
Sentaurus™ Process User Guide 1021
I-2013.12
•partial.2d: The mask partially covers the domain, but in a way that the mask does
not promote the simulation dimension (that is, the mask does not vary in the z-direction
over the simulation domain).
•partial: The mask partially covers the simulation domain, and its use in etching or
deposition forces the simulation to three dimensions.
cut.x, materials, regions
The cut.x argument is only available in two dimensions. It must be used with either
regions or materials to create a mask. The mask is created by taking a cut through the
set of regions created by a union of regions listed in regions and regions of one of the
materials listed in materials. The cut is taken at the x-coordinate specified by cut.x,
and the resulting outline is used to create the mask.
get.segments, get.segments.z
Retrieves segments that result from cutting the mask at z=get.segments.z. The default
value of get.segments.z is the midpoint of the simulation domain in the z-direction.
layoutfile
Name of a layout file in DF–ISE layout format. All masks defined in the layout file will be
read. By default, the origin of the layout file and the internal coordinate system coincide.
The lateral coordinate of the layout file will be used as the Sentaurus Process y-coordinate,
and the vertical coordinate of the layout file will be used as the Sentaurus Process
‘-z’-coordinate.
You ca n pla ce th e Senta ur us P roc es s s imulat io n domain anywhere in the layout file by
specifying either name together with layoutfile or a CutLine2D in the init
command. If name is specified, it must refer either to a mask that has been previously
defined or to one of the masks in the layout file (SIM3D or SIM2D).
If a rectangle mask is used, defined as SIM3D in the layout file or by referring to a
previously defined mask, the minimum coordinates of the rectangle define the origin of the
internal coordinate system. The layout –x-axis and –y-axis define the Sentaurus Process z-
axis and y-axis, respectively.
If CutLine2D or SIM2D or a polygon mask is used, the origin of the UCS is defined by
the first point. The direction of the Sentaurus Process y-axis is aligned to the direction of
the specified line. If a polygon mask is used, only the first two points are required for the
placement of the simulation domain in the layout file.
Default extensions of the 2D or 3D simulation domain in the y- and z-directions are defined
from the specified mask. For SIM2D or a two-point polygon, the default extension along
the y-axis is defined for a rectangle, polygon, or SIM3D mask; default extensions are
defined in both the y-direction and z-direction. If you did not specify line y and line z
commands, the default extensions are used.
A: Commands
mask
1022 Sentaurus™ Process User Guide
I-2013.12
left, right, front, back
Specify the corners of one rectangle. The rectangle is added to the current list for the mask.
If several rectangles must be specified for a mask, several mask commands must be used
with the same name. Default unit: .
list
Prints a list of all currently defined masks. If name is specified, it prints information for that
mask only.
name
Name of a mask. If used with clear or list, only the specified mask will be reported or
removed. If defining a new mask, name must be specified.
negative
Inverts the type of mask. By default, points inside the mask are considered masked. For
example, the commands mask name=xyz negative and mask name=zyx !negative
invert an existing mask xyz.
polygons
Specifies a mask as a list of named polygons. The named polygons must have been defined
using polygon commands.
segments
Specifies a list of coordinates of mask segments. Several mask segments can be specified
at the same time. The first coordinate defines the beginning of a segment, the second
coordinate defines the end of the segment, the third defines the beginning of the second
segment, and so on. In 3D simulations, mask segments are extended across the entire
structure in the z-direction. Default unit: .
Description
This command manages and creates masks for use with subsequent etch, deposit, or photo
commands. Mask definitions are stored in TDR files and re-stored when loading a TDR file in
the init command. Masks can be defined by rectangles, polygons, and segments, or they can
be read using the ICWBEV Plus interface (see Chapter 12 on page 817), or they can be read
from a DF–ISE layout file.
Masks are created additively. If more than one mask command is issued with the same name,
the union of the specified masks is assumed. To change a mask, first clear it and then assign a
new specification (in two separate calls to the mask command).
µm
µm
A: Commands
mask
Sentaurus™ Process User Guide 1023
I-2013.12
Examples
Define a mask named field:
mask name= field left= 0.0 right= 10.0
The position of this mask is the same as in the previous example:
mask name= mgoals segments= {0.0 10.0}
Read the layout file and define the position and default extensions of the Sentaurus Process
simulation domain by one of the masks in the layout file. The default extensions of the
simulation domain are y: 0...length of SIM3D z: 0...width of SIM3D. They are ignored if you
defined line y and line z commands:
mask layoutfile= simple.lyt name= SIM3D
Define a mask as a two-point polygon, load the layout file, and align the origin and y-axis of
the default coordinate system with the specified mask. The default extension of the simulation
domain along the y-axis is 0...length of the segment:
polygon name= cutline segments= {1.65 0.15 1.95 0.6}
mask name= cutline polygons= {cutline}
mask layoutfile= simple.lyt name= cutline
Load a layout file, and align the origin and the y-axis of the default coordinate system with the
specified CutLine2D:
init slice.angle= [CutLine2D 1.65 0.15 1.95 0.6]
mask layoutfile= simple.lyt
NOTE There are no default extensions defined in this case. You must specify
line y explicitly.
# mask bbox example - return value in cm
mask name= m1 left= 0.1<um> right= 0.2<um> front= 0.3<um> back= 0.4<um>
set m1bbox [mask name= "m1" bbox]
# $m1bbox == {1.0e-05 3.0e-05} {2.0e-05 4.0e-05}
Return information about all masks in array format:
mask list
Return a list of mask names:
array set maskinfo [mask list]
LogFile "Mask names: [array names maskInfo]"
A: Commands
mask
1024 Sentaurus™ Process User Guide
I-2013.12
Print all information about the mask named PolygonMask:
array set polyInfo $maskInfo(PolygonMask)
LogFile "Contents of PolygonMask: [array names polyInfo]"
Print the number of polygons in the mask named PolygonMask:
LogFile "Number of polygons in PolygonMask: [llength $polyInfo(polygons)]"
Print the first polygon in the mask named PolygonMask:
LogFile "polygon 0 in PolygonMask: [lindex $polyInfo(polygons) 0]"
See Also
deposit on page 901
etch on page 923
photo on page 1061
point on page 1078
polygon on page 1082

A: Commands
mater
Sentaurus™ Process User Guide 1025
I-2013.12
mater
Returns a list of all materials in the current structure, and adds new materials to a global list.
Syntax
mater
[add] [alt.matername=<c>]
[bbox | bbox.cm | bbox.um] [cropped.bbox]
[Interface] [like.interpolate] [list.all]
[max= {<n> <n> <n>}] [min= {<n> <n> <n>}]
[name=<c>] [new.like=<c>]
[syntax.check.value=<c>]
Arguments
add
Adds a new material specified by name to the global material list.
alt.matername
Defines the alternative material to be used when saving a structure. When using the TDR
format, regions that are converted using alt.matername are converted back properly to
the simulation material when the TDR file is read in from the init command. Although
common materials such as SiGe and III–Vs are by default handled this way, special
situations may require additional conversions when transferring to device simulation.
bbox, bbox.cm, bbox.um
If specified, the mater command returns the maximum extents of the material in two
points in centimeter or micrometer.
cropped.bbox
Returns the cropped bounding box of a material within a user-defined bounding box.
Interface
Returns a list of interface materials in the current structure.
like.interpolate
Usually, the interpolation code interpolates data from and to materials that are like each
other (see Like Materials: Material Parameter Inheritance on page 57). Use
like.interpolate to prevent such interpolation.
A: Commands
mater
1026 Sentaurus™ Process User Guide
I-2013.12
list.all
Lists all the materials defined.
max, min
Specify the bounding box of a material. These arguments compute the cropped bounding
box of a material.
name
Name of the material.
new.like
Name of the existing material from which all default values are inherited. For newly created
materials, pdb parameters for this material are checked first and, if not found, the ‘Like’
material parameters are used (see Like Materials: Material Parameter Inheritance on
page 57).
syntax.check.value
Sets a value to be returned only during syntax-checking mode. Sometimes, the value
returned by a command can cause a false syntax-check error because the value returned by
the command would not be the value during the normal run mode. Setting this value avoids
such problems.
Description
This command returns a list of all materials in the current structure. The format of the list is
compatible with the material specification for the program. Bulk material names are returned
if no arguments are given. Interface materials can be obtained by using Interface.
This command also computes the cropped bounding box of a material that lies within a user-
specified bounding box defined by max and min. The name argument is given as input.
Examples
Add germanium material to the global material list and make it inherit the default values from
silicon material:
mater add name= Germanium new.like= Silicon

A: Commands
math
Sentaurus™ Process User Guide 1027
I-2013.12
math
Sets the numeric and matrix parameters. Parameters set with the math command are stored in
TDR files by default.
Syntax
math
[AMS.NegErrCntrl] [FTS.NegErrCntrl] [LocTrnErrCntrl] [NegErrCntrl]
[diffuse | flow]
[dimension= 1 | 2 | 3]
[pardiso | ils] [scale]
[fullNewton | modNewton]
[newtonDeriv] [newtonRate1=<n>] [newtonRate2=<n>]
[newtonStats=<i>] [newtonSteps1=<i>] [newtonTries1=<i>]
[coord.dfise]
[coord.transform coord.translate (coord.read | coord.write)]
[coord.ucs] [coord.xyz] [coord.yxz] [coord.-zyx]
[maxNumberOfDomains=<i>]
[NumberOfElementsPerDomain=<i>]
[numThreads=<i>]
[numThreadsAssembly=<i>]
[numThreadsBoxMethod=<i>]
[numThreadsDeatomize=<i>]
[numThreadsILS=<i>]
[numThreadsImp3d=<i>]
[numThreadsInterp=<i>]
[numThreadsKMC=<i>]
[numThreadsMC=<i>]
[numThreadsMGoals=<i>]
[numThreadsPardiso=<i>]
[numThreadsSano=<i>]
[numThreadsSnMesh=<i>]
[numThreadsTopo=<i>]
[parallel.license= go.serial | go.wait | go.abort | go.recheck]
[threadStackSize=<i>]
[limit.precision] [milne | difference] [tr_bdf | euler]
[use.interpolated.geom.coeff] [voronoitriangle]
A: Commands
math
1028 Sentaurus™ Process User Guide
I-2013.12
Arguments: Solver Selection
AMS.NegErrCntrl
Allows stricter error control for each solve time step after an adaptive meshing step by
calculating the error from negative updates instead of damped results.
diffuse, flow
Specifies the type of equation to which the command specification applies. If omitted, it
applies to all equation types.
dimension
Specifies the dimensionality to which the command specification applies. If omitted, it
applies to all dimensions.
FTS.NegErrCntrl
Allows stricter error control for the first solve time step by calculating the error from
negative updates instead of damped results.
LocTrnErrCntrl
Allows stricter error control for each solve time step by modifying the handling of negative
updates:
•1 (
|upd|/org*lte+abs)
•0 (
|upd|/org+lte*abs)
LocTrnErrCntrl can be switched on for individual solution variables in specific
materials using:
pdbSetBoolean <mater> <solution> LocTrnErrCntrl 1
NegErrCntrl
Allows stricter error control at each Newton iteration step by calculating the error from
negative updates instead of damped results. NegErrCntrl can be switched on for
individual solution variables in specific materials using:
pdbSetBoolean <mater> <solution> NegErrCntrl 1
pardiso, ils
Specifies the type of linear solver to apply to the system:
•pardiso selects the parallel direct solver PARDISO, which is based on the LU
factorization with pivoting of the matrix. PARDISO decomposes the matrix.
A: Commands
math
Sentaurus™ Process User Guide 1029
I-2013.12
•ils selects the iterative linear solver ILS, including preconditioners, iterative methods,
scaling, and convergence criteria. (You can change the default settings of ILS
parameters by specifying pdbSet Math commands.) To set ILS parameters in the
parameter database, see Setting Parameters of the Iterative Solver ILS on page 862.
scale
Applies row/column scaling to the matrix in an attempt to make it better conditioned. This
is a recommended argument. No scaling is performed if the modified Newton scheme
(modNewton) is used.
Arguments: Newton Method
fullNewton, modNewton
Specifies the type of nonlinear equation solver to use:
•fullNewton performs a matrix factorization at each step.
•modNewton tries to reuse one matrix factorization for several solve steps.
The full Newton method may be more robust, but it can use more solution time than the
modified Newton method:
•modNewton is the default in two dimensions for both PARDISO and ILS.
•modNewton is the default for ILS in three dimensions.
newtonDeriv
Allows the Jacobian computation to be switched on during the modified Newton step.
Default: false.
newtonRate1
For the modified Newton method, if the solution for any of the Newton steps 1 through
newtonSteps1 is newtonRate1 or more times better than the previous step, the next step
can be a solve-only step. Otherwise, the next step will perform a matrix factorization.
Default: 4.0.
newtonRate2
For the modified Newton method, if the solution for any of the Newton steps
newtonSteps1+1 onwards is newtonRate2 or more times better than the previous step,
the next step can be a solve-only step. Otherwise, the next step will perform a matrix
factorization. Default: 32.0.
newtonStats
Prints information on Newton iterations.
A: Commands
math
1030 Sentaurus™ Process User Guide
I-2013.12
newtonSteps1
For the modified Newton method, any of the Newton steps 1 through newtonSteps1 must
improve the solution by the factor newtonRate1 over the previous step. Otherwise, the
next step will be a full Newton step. For Newton steps newtonSteps1+1 onwards, the
solution at each step must improve by the factor newtonRate2. Otherwise, the next step
will be a full Newton step. Default: 12.
newtonTries1
Number of first modified Newton step breakdowns allowed before switching to the full
Newton method. Default: 2.
Arguments: Time Discretization
milne, difference
Controls whether the next time step is estimated using the Milne’s device or the divided
difference method. Default: milne.
tr_bdf, euler
Specifies the type of time discretization scheme to use. The options are TR-BDF(2) or the
backward Euler method. Default: tr_bdf.
Arguments: Parallel Processing
Sentaurus Process provides parallel processing for Monte Carlo implantation, interpolation,
3D analytic implantation, the KMC charge model, the matrix assembly, the box method, and
the linear solvers by generating multiple threads to accelerate simulations on multicore shared-
memory computers. By default, only one processor (thread) is used.
NOTE The number of threads must not exceed the number of actual CPUs
(cores) of the computer.
NOTE Observe the following general recommendations to obtain the best
results from a parallel run: Speedup is only obtained for sufficiently
large problems. In general, the mesh should have at least 10000 nodes.
Three-dimensional problems are good candidates for parallelization.
NOTE You must run a parallel job on an unloaded computer. As soon as
multiple jobs compete for processors, performance decreases
significantly (a parallel job could run even longer than a serial one).
NOTE The parallel execution of the matrix assembly on the solvers PARDISO
and ILS produces different rounding errors. Therefore, the number of
Newton iterations in particular may change.
A: Commands
math
Sentaurus™ Process User Guide 1031
I-2013.12
NOTE Parallel performance scalability of the different modules (such as
implant, assembly, and linear solver) can vary dramatically.
NOTE Yo u do n ot need to se t th e OpenMP environment variable
OMP_NUM_THREADS. You need only specify the number of threads
required in the math command.
To use more than one thread, specify the following arguments of the math command in the
command file:
maxNumberOfDomains
Modifies the maximum number of domains each level of partition can have (see
Partitioning and Parallel Matrix Assembly on page 864).
NumberOfElementsPerDomain
Modifies the number of elements that must go to each domain (see Partitioning and Parallel
Matrix Assembly on page 864).
numThreads
Specifies the number of parallel threads for Sentaurus Process. Applies to Sentaurus MC
implantation, interpolation, 3D analytic implantation, Sentaurus Process KMC, matrix
assembly, the box method, and the solvers PARDISO and ILS.
To run MGOALS, Sentaurus Process KMC, matrix assembly, the box method, or the
solvers with a different number of threads, specify the following arguments:
•numThreadsAssembly
•numThreadsBoxMethod
•numThreadsDeatomize
•numThreadsILS
•numThreadsImp3d
•numThreadsInterp
•numThreadsKMC
•numThreadsMC
•numThreadsMGoals
•numThreadsPardiso
• numThreadsSano
•numThreadsSnMesh
A: Commands
math
1032 Sentaurus™ Process User Guide
I-2013.12
•numThreadsTopo
Separately, these arguments have priority over numThreads.
numThreadsAssembly
Number of threads used for matrix assembly. Parallel assembly of the matrix applies only
to inert anneals.
numThreadsBoxMethod
Number of threads used for the box method.
numThreadsDeatomize
Number of threads used when deatomizing KMC particles into continuum finite-element
fields.
numThreadsILS
Number of threads for the ILS solver.
Some parallel implementations of a default diffusion iterative solver gmres can be
activated by the command:
pdbSet Math diffuse <2D | 3D> ILS.hpc.mode <0 | 1 | 2 | 3>
For the high-performance computing mode, the options are 0 (default), and the algorithmic
parallel enhancements are activated by:
•1 for Version E-2010.12
•2 for Versions F-2011.09, G-2012.06, and H-2013.03
•3 for Version I-2013.12
numThreadsImp3d
Number of threads used for 3D analytic implantation.
numThreadsInterp
Number of threads used for interpolation.
numThreadsKMC
Number of threads used for Sentaurus Process KMC diffusion.
NOTE Parallelism for Sentaurus Process KMC only works for 1D or 2D
Sentaurus Process simulations.
A: Commands
math
Sentaurus™ Process User Guide 1033
I-2013.12
numThreadsMC
Number of threads used for Sentaurus MC implantation. This value is over written by
numThreadsKMC when this last one is present.
numThreadsMGoals
Number of threads used for MGOALS-relatedoperations.
numThreadsPardiso
Number of threads when running PARDISO.
numThreadsSano
Number of threads used for the Sano method for KMC particle to finite-element field
smoothing computation.
numThreadsSnMesh
Number of threads when running the meshing engine.
numThreadsTopo
Number of threads used when calling Sentaurus Topography to perform etching and
deposition steps.
parallel.license
If you run a simulation in parallel mode but the number of parallel licenses is insufficient,
Sentaurus Process proceeds in serial mode (default behavior or if
parallel.license=go.serial is specified), or waits for parallel licenses
(parallel.license=go.wait), or aborts (parallel.license=go.abort). The
option parallel.license=go.recheck checks for parallel licenses at each parallel
step, regardless of whether or not the licenses were available at the previous step.
threadStackSize
Stack size for each thread. Default stack size is bytes (see Partitioning and
Parallel Matrix Assembly on page 864).
Arguments: Coordinate System Input and Output Selection
When saving structures and meshes, a coordinate transformation is applied. By default, the
transformation is to the TDR coordinate system (which is the same as the DF–ISE and
visualization coordinate systems discussed in Understanding Coordinate Systems on page 66).
NOTE This coordinate system is different for 1D, 2D, and 3D structures.
Arguments in this section can be used to change how files are written
218 262144=
A: Commands
math
1034 Sentaurus™ Process User Guide
I-2013.12
and read. Files written in alternative coordinate systems will be rotated
when read by other tools. Nevertheless, it can be useful to write files in
the Sentaurus Process native coordinate system to assist in writing
command files. Even though the structure will appear rotated when it is
displayed in Tecplot SV, the coordinates will match those in the
Sentaurus Process command file, which can be helpful when setting up
refinement boxes, masks, and so on.
coord.dfise
Reads or writes files in the DF–ISE coordinate system. This must be used only to revert
coordinate systems. If the dimension of the structure changes, math coord.dfise must
be recalled.
coord.transform, coord.translate, coord.read, coord.write
Both coord.transform and coord.translate allow a general coordinate
transformation through specification of a rotation matrix defined as follows:
coord.transform= {a11 a12 a13 a21 a22 a23 a31 a32 a33}
where aij (i=row, j=column) are the members of the rotation matrix, and:
coord.translate= {x y z}
specifies a translation vector (in the internal coordinate system).
Both coord.transform and coord.translate must be used with either coord.read
or coord.write to indicate the transformation is specifying the transformation for
reading or writing, respectively. When specifying coord.write, the inverse of the
specified transformation is applied when reading. When coord.read is specified, the
inverse transformation is applied when writing.
coord.ucs, coord.xyz
Reads and writes files in the unified coordinate system (UCS).
coord.yxz
Same as coord.dfise in two dimensions.
coord.-zyx
Same as coord.dfise in three dimensions.
A: Commands
math
Sentaurus™ Process User Guide 1035
I-2013.12
Arguments: General
limit.precision
For Intel x86 architecture, this argument limits floating-point operations to 64 bits instead
of the default 80 bits. This setting is applied by default to improve reproducibility among
different platforms.
use.interpolated.geom.coeff
Switches on the method to be used for interpolating geometric coefficients for TRBDF,
which reduces the number of box method calls by one third.
voronoitriangle
Switches on the internal box method calculation.
Description
This command is used to specify the:
■Different coordinate systems.
■Number of threads and the arguments used for parallel processing on shared-memory
computers.
■Default options on the matrix packages to be used for different equations.
■Arguments for the Newton method.
■Different time discretization schemes.
Examples
Use the ILS solver for mechanics in the 2D case:
math flow dim= 2 ils
Use the PARDISO solver with two threads for the PDE system in the 2D case and specify
nested dissection (ND) ordering for PARDISO:
math diffuse dim= 2 pardiso numThreadsPardiso= 2 scale
pdbSetDouble Pardiso.Ordering 2
NOTE To run in parallel mode, the solvers PARDISO and ILS must be used
with ND ordering for both the 2D and 3D cases. For example, to specify
ND ordering, use:
pdbSetDouble Pardiso.Ordering 2
pdbSet Math diffuse 2D ILS.symmOrdering nd
A: Commands
math
1036 Sentaurus™ Process User Guide
I-2013.12
Select the solver ILS for all equations in two dimensions. Print Newton statistics at the end of
each diffuse command. In the first case, the modified Newton method and the TR-BFDF(2)
methods are used. In the second case, the Euler and the full Newton methods are specified:
math dimension= 2 ils newtonStats= 1
math dimension= 2 ils newtonStats= 1 euler fullNewton

A: Commands
mgoals
Sentaurus™ Process User Guide 1037
I-2013.12
mgoals
Modifies the default parameters for geometric operations available in the MGOALS library.
Syntax
mgoals
[accuracy=<n>][<m>|<cm>|<um>|<nm>]
[analytic.thickness]
[aniso.etching.fragment.tol=<n>]
[aniso.etching.protect.materials]
[dx=<n> dy=<n> dz=<n>]
[fill.buried]
[force.analytic | force.full.levelset | force.full.levelset.depo |
force.full.levelset.etch]
[fourier.local.diffusivity]
[full.resolution=<n>][min.gas.thickness=<n>][<m>|<cm>|<um>|<nm>]
[max.number.levelset.cells=<n>]
[min.levelset.size=<n>][<m>|<cm>|<um>|<nm>]
[offsetting.maxlevel=<i>]
[print.params]
[reinitfrequency=<n>] [reinititerations=<n>]
[remove.floating.regions]
[repair.2d] [repair.angle=<n>]
[resolution=<n>]
[use.brep.2d]
H-2013.03-SP2 | H-2013.03-SP1 |H-2013.03 |G-2012.06-SP2 | G-2012.06 |
F-2011.09-SP1 | F-2011.09 | E-2010.12 | D-2010.03]
Arguments
accuracy
Specifies the error that can be tolerated in transferring the new interface definition from the
level-set grid to the simulation grid. There is a compromise between smoothness and the
number of grid points. Smoother grids need more points on curved regions. Default value
and unit: .
analytic.thickness
For etching and deposition steps of layers of thickness of 1 nm or less, an analytic method
performs the etching because thin etches using the level-set method can be prohibitively
CPU and memory intensive. For very large or very small structures, a 1-nm cutoff may be
inappropriate, so this argument can be used to modify the thickness where the analytic
method is used.
1.0 5–
×10 µm
A: Commands
mgoals
1038 Sentaurus™ Process User Guide
I-2013.12
aniso.etching.fragment.tol
Removes fragments remaining from 3D anisotropic etching. The tolerance measures the
ratio of the volume and the surface of a region. Default: 1.0e-6.
aniso.etching.protect.materials
When switched on, the 3D anisotropic algorithm attempts to protect areas shadowed by
buried materials. Default: false.
dx, dy, dz
Explicitly set the level-set grid spacing in each direction. If set, these arguments override
the automatic setting of dx, dy, and dz, which uses resolution.
fill.buried
For deposition, material is deposited on the surface exposed to the upper gas region. With
fill.buried specified, deposition also occurs inside the buried gas bubbles that may
exist.
force.analytic
When performing isotropic etching or deposition, this argument forces the use of an
analytic algorithm instead of a level-set algorithm even when a boundary collision will
occur. For very large structures or very small etching or deposition thicknesses, the level-
set algorithm may consume too much memory and time.
force.full.levelset, force.full.levelset.depo, force.full.levelset.etch
Defines the general level-set time-stepping algorithm as the default algorithm for both
etching and deposition, or etching only, or deposition only, respectively.
fourier.local.diffusivity
Controls the artificial diffusion parameter. If fourier.local.diffusivity is
specified, the solution is more accurate but the corners are less sharp. For complex Fourier
rates, switch off fourier.local.diffusivity to enhance stability.
full.resolution
Usually, the full-time stepping level-set method is used in situations where more intricate
boundaries will be generated. The full-time stepping level-set method is needed for Fourier,
crystallographic, and multimaterial etching types, and for etching with shadowing on. This
argument allows a separate resolution setting for these cases. The default value is 0.025
compared to 0.1 for resolution.
A: Commands
mgoals
Sentaurus™ Process User Guide 1039
I-2013.12
max.box.angle
Specifies the maximum angle in the interior of any region where MGOALS can put locally
an exact Cartesian grid. Default value and unit: .
max.number.levelset.cells.
Issues a warning when the maximum number of cells used by the level-set mesh exceeds a
limit. The default value is cells (or 1000x1000x1000 cells).
min.gas.thickness
Minimum thickness of the gas layer at the top of the simulation structure. Default value and
unit: .
min.levelset.size
Specifies the minimum size for the level-set mesh. Usually, the level-set mesh size scales
with the operation according to the resolution factor and the etching or deposition
thickness. However, for thin etching or deposition steps, this may lead to a small level-set
mesh causing excessive memory use and simulation time. Often, it is not necessary (for
thin layers, a mesh size between thickness/2.0 and thickness/3.0 is usually sufficient). This
argument limits the mesh size and, therefore, limits computational expense. Default value
and unit: .
offsetting.maxlevel
Specifies the number of offsetting layers at an interface when Sentaurus Mesh offsetting is
used at an interface.
print.params
Prints the current MGOALS parameters.
reinitfrequency
Level-set reinitialization is performed every reinitfrequency time step in level-set
operations. A reinitialization algorithm is run to condition the level-set distance function to
reduce the effect of contour bunching, which can cause etching distances to be less than
expected. The default value is 0, which means that no reinitialization is performed.
reinititerations
The internal reinitialization algorithm reinitializes first the 0 level set and works outwards
from the front with higher numbers of iterations. Default: 1. This argument only comes into
operation if reinitfrequency is nonzero.
120°
1.0 7–
×10
0.1 µm
1.0 4–
×10 µm
A: Commands
mgoals
1040 Sentaurus™ Process User Guide
I-2013.12
remove.floating.regions
Determines whether MGOALS automatically removes regions that are not attached to the
bottom of the structure. Default: true.
repair.2d
Controls the default behavior of the boundary repair operation in two dimensions. By
default, boundary repairs are disabled in two dimensions.
repair.angle
Controls the dihedral angle at which repairs are performed. The algorithm attempts to
repair any surface section with a dihedral angle less than repair.angle. Default: .
resolution
Specifies the minimum number of level-set cells across the thickness of a deposited or
etched layer. For example, resolution=0.2 implies five cells. Default: 0.1.
use.brep.2d
Switches on the brep structure mode for two dimensions when handling structural changes
such as 2D etching and 2D deposition. Default: false.
The boundary representation (brep) structure mode in two dimensions handles structural
changes similarly to how structural changes are handled by default in three dimensions. A
brep of the structure is used and modified rather than the volume mesh.
Using brep reduces run-times by avoiding unnecessary meshing operations and increases
stability and accuracy by eliminating both boundary simplification and variable
interpolation associated with remeshing between structural modification operations.
H-2013.03-SP2, H-2013.03-SP1, H-2013.03, G-2012.06-SP2, G-2012.06,
F-2011.09-SP1, F-2011.09, E-2010.12, D-2010.03
Sets the backward compatibility of parameters and algorithms to the specified release.
Support is available for Versions H-2013.03-SP2, H-2013.03-SP1, H-2013.03, G-2012.06-
SP2, G-2012.06, F-2011.09-SP1, F-2011.09, E-2010.12, and D-2010.03 (partial
support).Description
Description
This command allows you to define parameters for MGOALS-related operations.
1°
A: Commands
mgoals
Sentaurus™ Process User Guide 1041
I-2013.12
Examples
Explicitly set the vertical level-set mesh spacing to and the horizontal level-set mesh
spacing to . Reinitialization of the level-set distance function is performed every five
time steps, and every reinitialization is performed to an internal iteration accuracy of four
iterations:
mgoals dx= 0.01 dy= 0.02 reinitfrequency= 5 reinititerations= 4
0.01 µm
0.02 µm

A: Commands
optimize
1042 Sentaurus™ Process User Guide
I-2013.12
optimize
Performs optimization of specified parameters to achieve desired target values.
Syntax
optimize
model.function = <c>
model.parameters = {c1 c2 c3 ...}
[param.init = {c1=<n> c2=<n> c3=<n> ...}]
[param.lower = {c1=<n> c2=<n> c3=<n> ...}]
[param.upper = {c1=<n> c2=<n> c3=<n> ...}]
[param.log = {c1 ...}]
target = {n} | target.file = <c>
[max.iter = <n>]
[tolerance = <n>]
[weight = {n}]
[min.abs = {n}]
[min.rel = {n}]
[history = <c>]
Arguments
history
Outputs the history of optimization to the file provided by the option. The history of
parameters, corresponding target results, and RMS errors are displayed in columns with
each row corresponding to each loop.
max.iter
Specifies the maximum number of iterations allowed in the optimization loop. Default is
500.
min.abs
Lists the minimum absolute error for each target. If specified, the list must be the same
length as the number of targets. It is the minimum target value for which absolute error is
used to calculate the target error during optimization. This value is compared with the
absolute target value. Default is given by pdbGetDouble Optimizer min.abs, which
defaults to 1.e-10 for all targets.
min.rel
Lists the minimum relative error for each target. If specified, the list must be the same
length as the number of targets. It is the minimum target ratio for which relative error is
used to calculate the target error during optimization. This value is compared with the ratio
A: Commands
optimize
Sentaurus™ Process User Guide 1043
I-2013.12
of the absolute target value to the maximum absolute target value. Default is given by
pdbGetDouble Optimizer min.rel, which defaults to 1.e-10 for all targets.
model.function
Provides the name of a user-defined Tcl procedure. The arguments of the procedure will be
those specified in model.parameters and be in the same order as in
model.parameters.
model.parameters
Lists names for the parameters that need to be optimized.
param.init
Lists initial values for the parameters that need to be optimized. Default value is 0.5, if not
specified.
param.lower
Lists lower bound values for the parameters that need to be optimized. Default value is 0.0,
if not specified.
param.upper
Lists upper bound values for the parameters that need to be optimized. Default value is 1.0,
if not specified.
param.log
Lists names for any parameters that you want its value varied logarithmically during
optimization.
target
Lists desired target data that the optimization attempted to achieve by varying the values of
parameters defined in model.parameters.
Note: If you use the target option for a fitting problem, you need to set up the independent
variable data through a Tcl global variable opt.independent.var. And you need to
ensure that the user-defined Tcl procedure returns a list of the results corresponding to the
independent variable data.
target.file
Specifies a text file (SIMS data, for example) which can be used instead of the target
parameter to set target values. The file has two columns of data: the first column contains
independent variable data; the second column contains corresponding target data. The
A: Commands
optimize
1044 Sentaurus™ Process User Guide
I-2013.12
independent variable and target data must be in pairs. You can either specify
target.file or target; you cannot have both.
tolerance
The root-mean-square (RMS) error for convergence criterion. The optimization terminates
when the RMS error of targets is smaller than tolerance. Default is given by
pdbGetDouble Optimizer Tolerance, which defaults to 0.1.
weight
Lists weighting factors for targets. If specified, the list must be the same length as the
number of targets. The weights are used to control the importance of individual targets in
calculations of the error during optimization. For a fitting problem since, you do not need
to specify weight since all targets have the same importance. Default is given by
pdbGetDouble Optimizer Weight, which defaults to 1.0 for all targets.
Description
This command allows users to optimize specified parameters to achieve desired target values.
Before using the optimize command, you must define a Tcl procedure which computes the
model’s results given a set of valid model parameters. The result of the command is a Tcl list
with the optimized parameters in the following format:
{c1 <n> c2 <n> ...}
Examples
This following example is a procedure to optimize the process conditions for the given oxide
thickness:
proc optFunc { temp dose}
{init tdr=init
implant Phosphorus dose=$dose energy=80
diffuse temperature=$temp time=20 O2
set z=NetActive name=NetActive store
set tox [interface Silicon /Oxide y=0.1]
set lox [interface Oxide /Gas y=0.1]
set thi [expr $tox -$lox]
return $thi
}
The optimize command with the above procedure would be:
optimize \
model.function = optFunc \
model.parameters = {temp dose} \
param.init = {temp=1000 dose=1E14} \
A: Commands
optimize
Sentaurus™ Process User Guide 1045
I-2013.12
param.lower = {temp=900 dose=1E12} \
param.upper = {temp=1200 dose=1E15} \
param.log = {dose} \
target = {0.06} \
max.iter = 500 \
tolerance = 0.1 \
min.abs = {1.e-10} \
min.rel = {1.e-10} \
history = test.dat
Following is an example procedure for a fitting problem. For fitting problem, we use
opt.independent.var global Tcl variable to store the independent variable data:
proc optFunc { cja vja mja} {
global {opt.independent.var}
foreach value ${opt.independent.var} {
lappend z [expr $cja/(1+$value/$vja)^mja]
}
return $z
}
The optimize command with the above procedure would be:
optimize \
model.function = optFunc \
model.parameters = {cja vja mja} \
param.init = {cja=0.2 vja=5. mja=0.5} \
param.lower = {cja=0.1 vja=2. mja=0.1} \
param.upper = {cja=0.4 vja=7. mja=0.9} \
target.file = model.dat \
max.iter = 500 \
tolerance = 0.1 \
history = test.dat

A: Commands
paste
1046 Sentaurus™ Process User Guide
I-2013.12
paste
Assembles 2D or 3D simulations by incorporating fragments from a TDR file.
Syntax
paste
tdr=<c>
direction= "left" | "right" | "front" | "back"
[Adaptive]
Arguments
Adaptive
If specified, Adaptive switches on adaptive meshing. Parameters for adaptive meshing
are described in Adaptive Refinement on page 699. The default is the return value of
pdbGet Grid Adaptive.
direction
Selects the side where to paste the incoming simulation. In 3D, the new structure can be
pasted on the left, right, front, or back side. In 2D, only left or right sides are used.
tdr
Name of the file to be imported and pasted into the current simulation.
Description
This command reads a TDR file containing valid geometry and appends it (pastes it) to the
current structure. The new structure is displaced automatically by the correct amount to
properly fit at the specified side, but the structures are not stretched automatically. If the
dimensions of nongas materials at the pasting sides are not the same, the command fails and
quits.
The dimension of all parts much match; 2D and 3D cannot be mixed. Also, in 2D, only the
direction values left and right are supported.
The paste command allows for the assembly of complex 2D or 3D structures by reading the
different pieces from TDR files and putting all of them together.
A: Commands
paste
Sentaurus™ Process User Guide 1047
I-2013.12
Examples
Append the structure in the file structure1_fps.tdr to the right side (maximum
y-coordinate) of the current structure:
paste tdr= "propertyx" direction= "right"
See Also
Inserting Polygons in Two Dimensions on page 785
struct on page 1158

A: Commands
pdbDelayDouble
1048 Sentaurus™ Process User Guide
I-2013.12
pdbDelayDouble
Retrieves an expression for a double parameter that will be evaluated at each time step during
diffusion.
Syntax
pdbDelayDouble <c> <c> ...
Arguments
<c>
This argument can be any double parameter declared in the parameter database and any
double parameter declared by the user in user-defined models. In all cases, the argument
must be specified with the full hierarchical path for the double parameter.
Description
This command is typically called from Alagator to retrieve a parameter expression. Since
among other things, the temperature can change during a diffusion step, the evaluation of
Arrhenius expressions must be delayed until the temperature is known. This command
provides this functionality.
Examples
Return an expression for D0 (not a value):
pdbDelayDouble Si B D0

A: Commands
pdbdiff
Sentaurus™ Process User Guide 1049
I-2013.12
pdbdiff
Compares the current structure with one from a TDR file.
Syntax
pdbdiff <c> <c>
Arguments
<c>
Specifies the full path or prefix of the TDR files to be compared. The prefix is the file name
without _fps.tdr.
Description
The command reports any differences between the parameters stored and any differences in
value.
Examples
Compare the pdb differences between n1_fps.tdr and n2_fps.tdr:
pdbdiff n1 n2

A: Commands
pdbDopantLike
1050 Sentaurus™ Process User Guide
I-2013.12
pdbDopantLike
Creates new dopants in materials.
Syntax
pdbDopantLike <c> <c>
Arguments
<c>
The first argument must be the name of the material.
The second argument must be the name of the new dopant.
Description
If dopants are not present in a material, an error message is displayed.
Examples
Create a new dopant called MyDopant in silicon. You can select dopant-related diffusion
switches (such as DiffModel and ActiveModel) for MyDopant:
pdbDopantLike Silicon MyDopant

A: Commands
pdbExprDouble
Sentaurus™ Process User Guide 1051
I-2013.12
pdbExprDouble
Retrieves an expression for a double parameter without evaluating.
Syntax
pdbExprDouble <c> <c> ...
Arguments
<c>
This argument can be any double parameter declared in the parameter database and any
double parameter declared by the user in user-defined models. In all cases, the argument
must be specified with the full hierarchical path for the double parameter.
Description
This command is typically called from Alagator to retrieve a parameter expression. If the
parameter depends on solution names, data fields, and so on, the evaluation of the expression
must be delayed until the solution time. This command provides this functionality.
Examples
Return an expression for Bulk (not a value):
pdbExprDouble Si Mechanics BulkModulus

A: Commands
pdbGet and Related Commands
1052 Sentaurus™ Process User Guide
I-2013.12
pdbGet and Related Commands
All these commands retrieve database parameters:
■pdbGet
■pdbGetArray
■pdbGetBoolean
■pdbGetDouble
■pdbDelayDouble
■pdbGetDoubleArray
■pdbGetElement
■pdbGetFunction
■pdbGetString
■pdbGetSwitch
■pdbGetSwitchString
Only pdbGet has syntax checking.
Syntax
pdbGet <c> <c> ...
Arguments
<c>
This argument can be any double parameter declared in the parameter database and any
double parameter declared by the user in user-defined models. In all cases, the argument
must be specified with the full hierarchical path for the double parameter.
Description
These commands are used to obtain parameters that reside in the property database, which is
hierarchical and is indicated by passing a series of strings to the command. In the command
file, the command pdbGet must replace all other pdbGet* commands because the type of the
parameter and the syntax are checked automatically.
If a parameter does not exist in the directory, the tool exits and prints a list of parameters that
can be found. Normal aliasing is applied to each string before the parameter is retrieved from
the database.
A: Commands
pdbGet and Related Commands
Sentaurus™ Process User Guide 1053
I-2013.12
The following commands all return 0 if the parameter is not found:
■pdbGetArray
■pdbGetBoolean
■pdbGetDouble
■pdbDelayDouble
■pdbGetDoubleArray
■pdbGetElement
■pdbGetFunction
■pdbGetString
■pdbGetSwitch
The command pdbGetSwitchString returns nothing if the parameter is not found.
These commands have a slight performance advantage and will not exit if a parameter has not
been defined, so they are preferred for Alagator scripting.
The command pdbGetSwitch returns an integer value of a switch, and the command
pdbGetSwitchString returns the string value. For example, if a switch has the choices a, b,
or c, and a is chosen, pdbGetSwitch returns 0, and pdbGetSwitchString returns a.
Examples
Retrieve the current value of StressHistory. The parameter StressHistory is known, but
if it is spelled incorrectly, Sentaurus Process exits and prints a list of known parameters at the
Mechanics level:
pdbGet Mechanics StressHistory
Retrieve StressHistory without syntax-checking. The command returns 0 if not found.
Sentaurus Process exits if there is a type mismatch between StressHistory and Boolean
(which is not the case in this example):
pdbGetBoolean Mechanics StressHistory

A: Commands
pdbIsAvailable
1054 Sentaurus™ Process User Guide
I-2013.12
pdbIsAvailable
Checks whether the given pdb command is available.
Syntax
pdbIsAvailable <c> <c> ...
Arguments
<c>
Specifies the pdb command.
Description
If the pdb command exists, the pdbIsAvailable command returns 1; otherwise, it returns 0.
Examples
Return 1 if the command "Silicon MyData" is available:
pdbIsAvailable Silicon MyData

A: Commands
pdbLike
Sentaurus™ Process User Guide 1055
I-2013.12
pdbLike
Creates new pdb parameter like an existing one in materials.
Syntax
pdbLike <c> <c> <c>Arguments
<c>
The first argument must be the name of the material.
The second argument must be the name of the new pdb parameter.
The third argument must be the name of the existing like pdb parameter.
Description
This command creates a new pdb parameter like an existing one in a material. It is usually used
to inherit all the parameters of a solution variable in a material for the new parameter (in other
words, new solution variable). If the like parameter is not present in a material, an error
message is displayed.
Examples
Create a new parameter called MyBoron in silicon:
pdbLike Silicon
MyBoron
Boron
MyBoron will inherit all the parameters defined for Boron in silicon including the user defined
ones. If MyBoron was defined as a solution name, all the inherited callback procedures names
and parameters will be used to build the diffusion equations. This is a fast way of introducing
a new dopant which is like an existing one.
See Also
solution on page 1145

A: Commands
pdbSet and Related Commands
1056 Sentaurus™ Process User Guide
I-2013.12
pdbSet and Related Commands
All of the following commands set database parameters:
■pdbSet
■pdbSetArray
■pdbSetBoolean
■pdbSetDouble
■pdbSetDoubleArray
■pdbSetElement
■pdbSetFunction
■pdbSetString
■pdbSetSwitch
Only pdbSet has syntax checking.
Syntax
pdbSet <c> <c> ... <value>
Arguments
<c>
This argument can be any parameter declared in the parameter database and any parameter
declared by the user in user-defined models. In all cases, the argument must be specified
with the full hierarchical path for the parameter.
<value>
The value associated with the type of the parameter. For example, a double value must be
given for the Double parameter type.
Description
These commands are used to set parameters that reside in the property database, which is
hierarchical and is indicated by passing a series of strings to the command. In the command
file, the command pdbSet must replace all other pdbSet* commands because the type of the
parameter and the syntax are checked automatically. If a parameter does not exist in the
directory, the tool exits and prints a list of parameters that can be found. Normal aliasing is
applied to each string before the parameter is retrieved from the database for all these
commands.
A: Commands
pdbSet and Related Commands
Sentaurus™ Process User Guide 1057
I-2013.12
The following commands all create a new parameter if one does not already exist:
■pdbSetArray
■pdbSetBoolean
■pdbSetDouble
■pdbSetDoubleArray
■pdbSetFunction
■pdbSetString
These commands have a slight performance advantage, and Sentaurus Process does not exit if
a parameter has not been defined, so they are preferred for Alagator scripting. The property
database uses the centimeter-gram-second (CGS) system of units [s, cm, g, , poise,
], except for activation energies [eV].
The command pdbSet checks the type of variable trying to be set and checks that type against
the <value> passed. The command pdbSetDouble takes a double for a value and, similarly,
pdbSetString takes a string and pdbSetBoolean takes a Boolean (1 or 0). The command
pdbSetSwitch will set a value for existing switches. If a switch is not found, a new one will
be created.
The command pdbSetArray defines the array for string data.
The data type DoubleArray has a special format and can be modified in several different ways
depending on the changes required.
This type is usually used for charge state–dependent parameters, in which case, the array index
refers to the charge state. For example, the database entry for Silicon Interstitial
ChargeStates is a list of length 10; the first entry is -2, the second entry is 0 (which means
ChargeStates[-2]=0), the third entry is -1, and the fourth is {[Arrhenius 5.68
0.48]}, which means ChargeStates[-1]=[Arrhenius 5.68 0.48] and so on.
There are also arrays that are intended for double sums. In this case, the array index entries have
a comma-separated field. For example, with Silicon Boron Interstitial kfKickOut,
the first entry (which corresponds to an array index) is {-2,-2}. The following examples show
how to set and change these types.
The command pdbSetElement modifies the value of one element in an array.
NOTE The arguments for pdbSetDouble, pdbSetDoubleArray, and
pdbSetBoolean must evaluate to numeric data. Calls to procedures or
to the pdbDelayDouble command in the arguments may cause errors
if they are not constructed correctly.
dyn/cm2
cm2/s
A: Commands
pdbSet and Related Commands
1058 Sentaurus™ Process User Guide
I-2013.12
Examples
Set the Dstar parameter for boron in silicon; exit with list if not found:
pdbSet Si B Dstar 1e-7
Set index 0 of ChargeStates to 0.1:
pdbSet Si Int ChargeStates 0 0.1
Set all members of ChargeStates (index 0 = 0.1, and so on):
pdbSet Si Int ChargeStates {0 0.1 1 0.2 3 0.3}
Set one index of a double array meant for double charge state indexing:
pdbSet Si B Int KfKickOut -2,1 {[expr 4.0*3.14159*([Arrhenius 0.1 0.2]+
[Arrhenius 0.3 0.4])]}
Now for the non-syntax-checked versions:
Set Dstar; create Dstar if it does not already exist (in this example, Dstar would exist):
pdbSetDouble Si B Dstar 1e-7
Set the DiffModel in silicon to Pair:
pdbSetSwitch Si Dopant DiffModel Pair
Create a new DoubleArray, index 0 = 0.1, and so on:
pdbSetDoubleArray Si MyVar MyArray {0 0.1 1 0.2 3 0.3}
pdbSetArray MyArray {0 abc 1 def 2 ghi}
pdbGetArray MyArray ;# print "0 abc 1 def 2 ghi"
pdbGetElement MyArray 1 ;# print "def"
pdbSetElement MyArray 1 jkl ;# modifies A[1] data from "def" to "jkl"

A: Commands
pdbUnSet-related Commands
Sentaurus™ Process User Guide 1059
I-2013.12
pdbUnSet-related Commands
All these commands unset database parameters:
■pdbUnSetBoolean
■pdbUnSetDouble
■pdbUnSetDoubleArray
■pdbUnSetString
Syntax
pdbUnSetBoolean <c> <c> ...
pdbUnSetDouble <c> <c> ...
pdbUnSetDoubleArray <c> <c> ...
pdbUnSetString <c> <c> ...
Arguments
<c>
This argument can be any parameter declared in the parameter database and any parameter
declared by the user in user-defined models. In all cases, the argument must be specified
with the full hierarchical path for the parameter.
Description
This command temporarily removes parameters from the parameter database during the
simulation.
Examples
The following command removes the list of Derived.Materials of InGaAs material:
pdbUnSetString InGaAs Derived.Materials

A: Commands
PDE2KMC
1060 Sentaurus™ Process User Guide
I-2013.12
PDE2KMC
Translates and transfers Sentaurus Process fields to Sentaurus Process KMC.
Syntax
PDE2KMC
Description
This command is called automatically when a switch from the PDE solver to the Sentaurus
Process KMC solver is detected. It translates the continuum concentrations into suitable
particle distributions to be used by Sentaurus Process KMC.
The atomization is performed using the select command with the appropriate KMC species
names.
The translation between PDE fields and KMC species is performed with a mapping that
translates the field names into their atomistic counterparts. This translation is made as
accurately as possible, but sometimes a perfect one-to-one mapping is not possible. In that
case, meaningful approximations are used. For example, ICluster is translated into I4.
A complete list of these translations is available in the file KMC.tcl.
See Also
SetAtomistic on page 1121

A: Commands
photo
Sentaurus™ Process User Guide 1061
I-2013.12
photo
Creates a photoresist layer of the specified thickness outside the mask.
Syntax
photo
[Adaptive] [mask=<c>] [repair] [sde= {<c>}]
[thickness=<n>][<m>|<cm>|<um>|<nm>]
Arguments
Adaptive
If specified, Adaptive switches on adaptive meshing. Parameters for adaptive meshing
are described in Adaptive Refinement on page 699. The default is the return value of
pdbGet Grid Adaptive.
mask
Name of the mask to be used to create the photoresist. The photoresist is deposited in the
openings of the mask.
repair
In MGOALS3D mode, small regions are removed automatically by default. Sometimes,
this causes small gas bubbles in the structure or other problems. Use !repair to switch
off the small region removal.
sde
String used to specify parameters and algorithms for 3D Sentaurus Structure Editor. By
default, mask and thickness are translated into appropriate Sentaurus Structure Editor
commands. If an algorithm is specified using sde, it overwrites the algorithm used by
default for isotropic or anisotropic etching, for example:
photo thickness= 2<um> mask= mask1 sde= {"algorithm" "lopx"}
thickness
Specifies the thickness of the photoresist. Default value and unit: .
Description
The mask must have been defined using a mask command. If the photoresist must be deposited
inside of the mask, the negative argument must be defined in the mask command.
2.0 µm
A: Commands
photo
1062 Sentaurus™ Process User Guide
I-2013.12
Examples
Create a resist layer 1-µm thick. The resist layer material will appear in open areas of mask
mask1. In other words, it will be the negative of mask1:
photo thickness= 2<um> mask= mask1
Create a resist layer 2-µm thick. The resist layer will have the same polarity as mask2:
photo mask= mask2
See Also
mask on page 1020

A: Commands
plot.1d
Sentaurus™ Process User Guide 1063
I-2013.12
plot.1d
Plots a 1D cross section.
Syntax
plot.1d
[boundary] [clear] [close] [color=<c>] [fix.ratio] [label=<c>]
[max= <list>] [min= <list>]
[name=<c>] [rescale] [symb=<c>] [title=<c>]
[x=<n>][<m>|<cm>|<um>|<nm>]
[y=<n>][<m>|<cm>|<um>|<nm>]
[z=<n>][<m>|<cm>|<um>|<nm>]
Arguments
boundary
Specifies that any material boundaries that are crossed must be drawn in as vertical lines
on the plot. Default: false.
clear
Specifies whether the graphics screen must be cleared before the graph is drawn. Default:
true (the screen is cleared).
close
Closes the plot window.
color
Specifies the line color for the plot. It can be any color supported by X11 hardware and
named in the color database.
fix.ratio
Specifies the x-, y-axis ratio to be fixed. Default: false.
label
Specifies the name of the line in the legend of the plot window. The default is the name of
the current dataset.
A: Commands
plot.1d
1064 Sentaurus™ Process User Guide
I-2013.12
max
List of numeric values that will be the ends of the x- and y-axis. The first argument is the
x-value and the second is the y-value. A single value is always interpreted as the x-value.
The default is the maximum extent of the current structure.
min
List of numeric values that will be the ends of the x- and y-axis. The first argument is the
x-value and the second is the y-value. A single value is always interpreted as the x-value.
The default is the minimum extent of the current structure.
name
Name of a data field. This allows plots to be created without using the select command.
Default: Z_Plot_Var.
rescale
Rescales the plot to fit the entire simulation domain.
symb
Specifies a symbol type to be drawn on the cross-sectional line. Each point is drawn with
the specified symbol. It defaults to no symbol. Whatever character is entered is placed at
each data point on the plot.
title
Specifies the title of the plot window.
x, y, z
Specify the constant values of a line along which sectioning is performed. In one
dimension, these arguments are not necessary. In two dimensions, only one of x or y can
be specified for a given device. Specifying x produces a horizontal slice through the device
and y specifies a vertical slice. An easy way to remember is that the cross section is taken
at the constant value specified. For a 3D simulation, two of these three values must be
specified. Default unit: .
Description
This command plots cross sections vertically or horizontally through the device with
arguments to provide for initialization of the graphics device and plotting of axes. This
command can optionally draw vertical lines whenever a material boundary is crossed.
µm
A: Commands
plot.1d
Sentaurus™ Process User Guide 1065
I-2013.12
Examples
Clear the screen, draw a set of axes, and draw the data along a horizontal cross section at
x = . Each point is drawn with symbol 1:
plot.1d x= 1.0 symb= 1 clear
Draw a horizontal cross section at x = on the previous set of axis. The line is labeled
Lateral in the legend:
plot.1d x= 2.0 clear label= Lateral
See Also
select on page 1117
slice on page 1141
1.0 µm
2.0 µm

A: Commands
plot.2d
1066 Sentaurus™ Process User Guide
I-2013.12
plot.2d
Plots a 2D xy graphic.
Syntax
plot.2d
[boundary] [clear] [close] [col.bound=<c>] [col.grid=<c>]
[edges] [faces] [fill] [fix.ratio] [gas] [grid] [kmc] [label.bound]
[max= <list>] [min= <list>]
[nodes] [rescale] [title=<c>]
[vector=<c>]
[vlength=<n>][<m>|<cm>|<um>|<nm>]
[vmax=<n>][<m>|<cm>|<um>|<nm>]
[x=<n> | y=<n> | z=<n>][<m>|<cm>|<um>|<nm>]
Arguments
boundary
Specifies that the device outline and material interfaces must be drawn. Default: false.
clear
Specifies that the graphics screen must be cleared before the graph is drawn. Default: true
(the screen is cleared).
close
Closes the plot window.
col.bound
Specifies the color of the boundary. Any valid X11 color can be specified.
col.grid
Specifies the color of the grid. Any valid X11 color can be specified.
edges
Prints the edge indices on the plot. Default: false.
faces
Prints the face indices on the plot. Default: false.
A: Commands
plot.2d
Sentaurus™ Process User Guide 1067
I-2013.12
fill
Specifies that the device must be drawn with the proper aspect ratio. If fill is false, the
device is drawn with the proper aspect ratio. If fill is true, the device is expanded to fill
the screen. Default: false.
fix.ratio
By default, the x to y ratio is now fixed. This can be switched off using !fix.ratio.
gas
Specifies that the grid in the gas must also be plotted. Default: false (no gas grid is shown).
grid
Specifies that the numeric grid on which the problem was solved must be drawn. Default:
false.
kmc
Plots particles in an atomistic KMC simulations as dots.
label.bound
Name of the material in the lower-left corner of the material region.
max
List of numeric values that will be the ends of the x- and y-axis, respectively. The first
argument is the x-value and the second is the y-value. A single value is always interpreted
as the x-value. The default is the maximum extent of the current structure. Default unit:
.
min
List of numeric values that will be the ends of the x- and y-axis, respectively. The first
argument is the x-value and the second is the y-value. A single value is always interpreted
as the x-value. The default is the minimum extent of the current structure. Default unit:
.
nodes
Prints the node indices on the plot. Default: false.
rescale
Rescales the plot to fit the whole simulation domain.
µm
µm
A: Commands
plot.2d
1068 Sentaurus™ Process User Guide
I-2013.12
title
Specifies the title of the plot window.
vector
Name of a vector field. This indicates arrows proportional to the size of the vector and in
the direction of the vector at each node. This argument does not work in 3D simulations.
vlength
Scales the length of the vectors so that the maximum vector has length vlength. Default
value and unit: .
vmax
Use this as the maximum velocity instead of searching for it. Default unit: .
x, y, z
For 2D simulations, these arguments are unnecessary. In three dimensions, one of these
three must be specified to indicate the cutline through the structure. Default unit: .
Description
Usually, this command is used to look at material boundaries and grids; however, it also can be
used to plot a flow field. This command can be executed immediately before a contour
command to allow isoconcentration lines to be plotted in context with the structure.
To obtain standard color and other settings for the plot.2d window, use the following
command from the command line:
unix:> xrdb -merge ${STROOT}/tcad/${STRELEASE}/lib/score/XFloops
Examples
Draw the triangular grid and axis. Each material is plotted in a different color:
plot.2d grid
Draw the material interfaces with the minimum x- and y-values of and :
plot.2d boundary min= {2 5}
Draw the material interfaces and place symbols at each coordinate in the mesh:
plot.2d boundary diamonds
0.1 µm
µm
µm
2.0 µm
5.0 µm
A: Commands
plot.2d
Sentaurus™ Process User Guide 1069
I-2013.12
Plot the Velocity vector field. The maximum arrow drawn will have a length of . The
plot surface will not be cleared:
plot.2d vector= Velocity vlength= 0.1 !clear
See Also
bound on page 887
Compatibility on page 888
contour on page 894
select on page 1117
slice on page 1141
0.1 µm

A: Commands
plot.tec
1070 Sentaurus™ Process User Guide
I-2013.12
plot.tec
Updates or initiates Sentaurus Process–Tecplot SV 1D, 2D, and 3D graphics.
Syntax
plot.tec
[autofit] [autorange] [command=<c>] [connect] [contourvar=<c>] [create.abs]
[data] [data.bool] [data.int] [data.real] [data.symtensor] [data.vector]
[data.element] [data.face] [data.node]
[delete.frames] [detach] [display] [double_prec]
[framebg] [framebgname] [framecolor] [framecolorname] [frameheader]
[frameheight] [framewidth] [frameposx] [frameposy]
[frameshiftx] [frameshifty] [frametransparent]
[framezoom] [framezoomx] [framezoomy]
[Grid] [host=<c>] [interfaces]
[launch.timeout=<n>] [legend] [loadfile=<c>]
[macro=<c>]
[port=<n>]
[reset.display] [reset.xyaxes]
[scale= lin | log | ash]
[set.variables= <list>] [start]
[suppressmat=<c>] [unsuppressmat=<c>]
[suppressvar=<c>] [unsuppressvar=<c>]
[terms]
[view.fit]
[x1auto] [x2auto] [x3auto] [x4auto] [x5auto] [xauto]
[y1auto] [y2auto] [y3auto] [y4auto] [y5auto] [yauto]
[x1log] [x2log] [x3log] [x4log] [x5log] [xlog]
[y1log] [y2log] [y3log] [y4log] [y5log] [ylog]
[x1max=<n>] [x1min=<n>] [x2max=<n>] [x2min=<n>] [x3max=<n>] [x3min=<n>]
[x4max=<n>] [x4min=<n>] [x5max=<n>] [x5min=<n>] [xmax=<n>] [xmin=<n>]
[y1max=<n>] [y1min=<n>] [y2max=<n>] [y2min=<n>] [y3max=<n>] [y3min=<n>]
[y4max=<n>] [y4min=<n>] [y5max=<n>] [y5min=<n>] [ymax=<n>] [ymin=<n>]
[xyautofit] [xyshow=<c>]
[y1axisvar=<n>] [y2axisvar=<n>] [y3axisvar=<n>] [y4axisvar=<n>]
[y5axisvar=<n>]
Arguments
autofit
Automatically fits the view for 2D and 3D modes after each update.
A: Commands
plot.tec
Sentaurus™ Process User Guide 1071
I-2013.12
autorange
Automatically resets the minimum and maximum data range for contour plots after each
update.
command
Specifies the command string used to launch the Tecplot SV process. Default:
"tecplot_sv -s:ipc".
connect
Permits connection to a running Tecplot SV process. Default: true.
contourvar
Selects the specified variable as the contour variable. By default, the first variable is
selected as the contour variable.
create.abs
Automatically creates an abs() dataset for each vector variable.
data
Sends new values of all variables to Tecplot SV.
data.bool, data.int, data.real, data.symtensor, data.vector
Enables data of the corresponding value type. The default is true for data.real,
data.symtensor, and data.vector.
data.element, data.face, data.node
Enables data of the corresponding location type. The default is true for all types.
delete.frames
Deletes the old frame when a new frame is created in Tecplot SV, due to switching from
1D to 2D mode, or from 2D to 3D mode. Default: true.
detach
Detaches the display from a process.
display
Specifies the host name and sequence number for display.
A: Commands
plot.tec
1072 Sentaurus™ Process User Guide
I-2013.12
double_prec
Uses double precision for all data transfers to Tecplot SV. This causes slower data transfer
and higher memory consumption in Tecplot SV. You must specify double_prec before
or together with start. The default is to use single precision.
framebg
Specifies the background color of the frame.
framebgname
Specifies the background color name of the frame.
framecolor
Specifies the header color of the frame.
framecolorname
Specifies the header color name of the frame.
frameheader
Enables display of the frame header.
frameheight, framewidth
Specify the height and width of the frame.
frameposx, frameposy
Specify the horizontal or vertical display position of the frame.
frameshiftx, frameshifty
Specify the horizontal or vertical distance that the frame shifts.
frametransparent
Sets the transparency display mode of the frame.
framezoom
Specifies the scaling factor of the frame.
framezoomx, framezoomy
Specify the horizontal or vertical scaling factor of the frame.
A: Commands
plot.tec
Sentaurus™ Process User Guide 1073
I-2013.12
Grid
Updates the grid (vertices and elements) in Tecplot SV.
host
Specifies the name of the host where Tecplot SV must be started or connected to.
interfaces
Enables interface regions to be displayed. Default: false.
launch.timeout
Specifies how many seconds Sentaurus Process must wait for a Tecplot SV response after
trying to start it. Default: 10 s.
legend
Displays the contour legend.
loadfile
Name of the file to load in to Tecplot SV.
macro
Sends a macro command to Tecplot SV (see examples). The macro language is
documented in the Tecplot 360™ Scr ipting Guide.
port
Specifies the port number for the Tecplot SV socket connection. Default: 2203.
reset.display
Resets to the display mode to the default.
reset.xyaxes
Resets the axis scale and the axis range for all axes.
scale
Sets the contouring scale to either lin, log, or ash for the variables specified by
set.variables. The set.variables argument must be specified with scale.
set.variables
Specifies a list of variables to be used with another argument such as scale. The variable
names can contain wildcards, for example, Stress*.
A: Commands
plot.tec
1074 Sentaurus™ Process User Guide
I-2013.12
start
Tries to connect to a running Tecplot SV process or to launch a new process. See
Tecplot SV User Guide, Chapter 5 on page 13 for detailed information on the behavior of
the start procedure.
suppressmat, unsuppressmat
Specifies the name of a material to be excluded or not excluded from the display.
suppressvar, unsuppressvar
Specifies the name of a variable to be excluded or not excluded from the display. A pattern
can be specified to exclude a set of variables, for example, *Interstitial*. Patterns are
matched using the Tcl_StringMatch() function.
terms
Enables term fields.
view.fit
Fits the view.
x1auto, x2auto, x3auto, x4auto, x5auto, xauto
y1auto, y2auto, y3auto, y4auto, y5auto, yauto
Enables automatic range resetting for the specified axis. Default: true.
x1log, x2log, x3log, x4log, x5log, xlog
y1log, y2log, y3log, y4log, y5log, ylog
Sets or unsets the logarithmic mode for the specified axis. The default is true for y-axes.
x1max, x1min, x2max, x2min, x3max, x3min, x4max, x4min, x5max, x5min,
xmax, xmin
y1max, y1min, y2max, y2min, y3max, y3min, y4max, y4min, y5max, y5min,
ymax, ymin
Sets the lower and upper range limits for the specified axis. This disables automatic range
resetting for the corresponding axis.
xyautofit
Resets all xy axes ranges to preset values after each update.
xyshow
Specifies the variables that must be displayed as xy mappings. Wildcards can be used in
the variable names.
A: Commands
plot.tec
Sentaurus™ Process User Guide 1075
I-2013.12
y1axisvar, y2axisvar, y3axisvar, y4axisvar, y5axisvar
Specifies the variable name to be displayed on the y-axis.
Description
This command starts Tecplot SV, connects to a running Tecplot SV process, or sends
commands or data to a running Tecplot SV process. After an instance of Tecplot SV with
interprocess communication enabled (that is, the command-line option tecplot_sv -
s:ipc) is running, subsequent Sentaurus Process runs will send graphics information to the
same (already running) Tecplot SV to avoid a delay in Tecplot SV startup. The plot.tec
command is called automatically by the graphics command.
Examples
Open Tecplot SV if a structure has already been created:
plot.tec start
Update graphics:
plot.tec grd data
Allow independent scaling of axes and fit the view:
plot.tec macro= "TWODAXIS AXISMODE = INDEPENDENT"
plot.tec view.fit
Save a Tecplot SV package file:
plot.tec macro= "SAVELAYOUT 'example.lpk' INCLUDEDATA = YES"
Prepare to display boron and stress data on separate y-axes, with the legend enabled, with linear
scaling for the stress variables, and with boron as the contouring variable for 2D mode:
plot.tec xyshow= "Boron* Stress*" \
y1axisvar= Boron* y2axisvar= Stress*!y2log y2min= -1e-6 y2max =1e-5 \
contourvar= Boron legend
plot.tec set.variables= Boron* scale= log
plot.tec set.variables= Stress* scale= lin
See Also
graphics on page 938

A: Commands
plot.xy
1076 Sentaurus™ Process User Guide
I-2013.12
plot.xy
Prepares an xy plot to draw on.
Syntax
plot.xy
[clear]
[max= <list>] [min= <list>]
[x.axis=<c>] [y.axis=<c>] [x.log] [y.log]
Arguments
clear
Clears the existing plot surface. Default: true.
max
List of numeric values that will be the ends of the x- and y-axis, respectively. The first
argument is the x-value and the second is the y-value. A single value is always interpreted
as the x-value. The default is the maximum extent of the current structure.
min
List of numeric values that will be the ends of the x- and y-axis, respectively. The first
argument is the x-value and the second is the y-value. A single value is always interpreted
as the x-value. The default is the minimum extent of the current structure.
x.axis, y.axis
Specify the labels for the x-axis and y-axis.
x.log, y.log
Specify whether there is a linear or log axis. If x.log or y.log is selected, the logarithm
of the values on the point.xy command are taken. The axis also will have log scale form.
Description
This command configures a 2D plot surface for use with the point.xy command. This
prepares the axis scaling and labels, and controls the log axes.
Using this command and the point.xy command could simulate all other commands in this
section.

A: Commands
point
1078 Sentaurus™ Process User Guide
I-2013.12
point
Creates a point, for example, for a mask polygon.
Syntax
point
[clear]
[coord= {
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]}]
[list] [name=<c>]
Arguments
clear
Clears the list of all points. If name is specified, it clears only this point information.
coord
Defines the coordinates of the point. For a 3D point, all three coordinates must be specified.
If only two are defined, a 2D layout point in the yz plane is assumed. Default unit: .
list
Returns the list of currently defined points. If name is given, it prints the information for
this point only.
name
Name of the point.
Description
This command defines a point in three dimensions or a 2D point in the yz plane. It can be used
to construct polygons to define masks.
Examples
Define a 2D point with the coordinates y = 0 and z = –1.5:
point name= p1 coord= {0 -1.5}
µm

A: Commands
point.xy
1080 Sentaurus™ Process User Guide
I-2013.12
point.xy
Adds a line segment to a plot.
Syntax
point.xy
x=<n> y=<n>
[color=<c>] [move] [name=<c>] [symb=<c>]
Arguments
color
Specifies the color for the line. It can be any color supported by X11 hardware and named
in the color database.
move
Instead of drawing from the last point, the graphics pen is placed at this point without
moving. Using move with symb draws scatter plots.
name
Name of a line, so that points can be added to the line at a later time. The name can be any
valid character string and is used in the plot legend. If the named line does not exist, it is
created.
symb
The first character of this string is used to mark the line. If no symbol is specified, none will
be used. If a symbol is specified once for a line, it is used for all lines. Default: x.
x, y
Specify the values to be added to the plot.
Description
This command adds segments to a specified line on a plot surface using X-windows-based
plotting (plot.1d or plot.2d). The command is used to plot calculated values, data, or direct
outputs from the simulation. The values can be added to any named line.

A: Commands
polygon
1082 Sentaurus™ Process User Guide
I-2013.12
polygon
Creates a polygon, for example, for a mask.
Syntax
polygon
clear |
list |
(name=<c>
[tdr=<c>] [materials= {<mat1> ... matn}] |
[max= {
<y>[<m>|<cm>|<um>|<nm>]
<z>[<m>|<cm>|<um>|<nm>]}]
[min= {
<y>[<m>|<cm>|<um>|<nm>]
<z>[<m>|<cm>|<um>|<nm>]}]
[rectangle] |
[points= {<point1> <point2> ... <pointn>}] [rectangle] |
[tdr=<c>] [regions= {<reg1> ... <regn>}] |
[segments= {
<y_1>[<m>|<cm>|<um>|<nm>]
<z_1>[<m>|<cm>|<um>|<nm>]
<y_2>[<m>|<cm>|<um>|<nm>]
<z_2>[<m>|<cm>|<um>|<nm>]
...
<y_n>[<m>|<cm>|<um>|<nm>]
<z_n>[<m>|<cm>|<um>|<nm>]}]
)
[xy]
Arguments
clear
Clears the list of all polygons. If name is specified, it clears only the named polygon.
list
Returns a list of all polygons. If name is given, it returns the information for this polygon
only.
materials
Specifies a material or list of materials that will be read when using tdr.
A: Commands
polygon
Sentaurus™ Process User Guide 1083
I-2013.12
max
Maximum point for a rectangular box. It must be used with rectangle to create a
rectangular polygon. The default is the structure bounding box maximum. Default unit:
.
min
Minimum point for a rectangular box. It must be used with rectangle to create a
rectangular polygon. The default is the structure bounding box minimum. Default unit:
.
name
Name of the polygon.
points
Lists the point names used to specify the polygon. A minimum of three must be specified.
The points must have been specified using the point command. The polygon is closed
implicitly by connecting the first and last points.
points also can be used with rectangle to specify a rectangular polygon. In this case,
two points must be given: the minimum and maximum points of the rectangle.
rectangle
Must be specified with max and min to define a rectangular box. Alternatively, two named
points can be given (using points) corresponding to the minimum and maximum of the
rectangle.
regions
Specifies a region or list of regions to be used when reading the polygon when using tdr.
segments
Lists the line segments in the yz plane (or the xy plane when xy is specified) used to specify
a polygon in three dimensions. The polygon is closed implicitly by connecting the first and
last points. A minimum of three segments must be given. Default unit: .
tdr
Name of the file from which to read the polygon. If you use tdr, you must specify xy. It
allows you to use materials and regions to further specify which polygon to be read from
the TDR file.
µm
µm
µm
A: Commands
polygon
1084 Sentaurus™ Process User Guide
I-2013.12
xy
Defines the polygon in the xy plane instead of the default yz plane. When using xy, the
segments are defined as {x_1 y_1 ... x_n y_n}, and min and max as xy. You must
use xy when specifying tdr. Specifying xy typically means that the polygon will be used
for insertion rather than for masking.
Description
This command defines a mask or uses the polygon during an insertion. One of the following
must be used to create a polygon:
■points
■rectangle
■segments
■tdr
If named points are not given explicitly when forming polygons, they are generated
automatically during the creation of the polygon.
Examples
Create three identical rectangles using points and coordinates:
point name= p1 coord= {0.0 0.0}
point name= p2 coord= {0.0 -0.5}
point name= p3 coord= {0.5 -0.5}
point name= p4 coord= {0.5 0.0}
polygon name= Box1 points= {p1 p2 p3 p4}
polygon name= Box2 points= {p2 p4} rectangle
polygon name= Box3 min= {0.0 -0.5} max= {0.5 0.0} rectangle
Define an L-shaped polygon using 1D line segments:
polygon name= LShape \
segments= {0.0 -1.5 0.0 -0.5 0.5 -0.5 0.5 1.5 1.5 1.5 1.5 -1.5}
Read the aluminum material structure in the file points_bnd.tdr as a polygon called box:
polygon name= "box" xy tdr= "points_bnd.tdr" materials= "Aluminum"
Print the list of polygons that have been defined:
LogFile [polygon list]
Delete Box3:
polygon name= Box3 clear

A: Commands
polyhedron
1086 Sentaurus™ Process User Guide
I-2013.12
polyhedron
Creates and stores 3D polyhedra, mainly for later insertion.
Syntax
polyhedron
clear |
list |
(name=<c>
(external.sde
(tdr=<c> [regions= {<reg1> ... regn}]) |
[materials= {mat1 mat2 ... matn}] [rotate]) |
brick= {
minx[<m>|<cm>|<um>|<nm>]
miny[<m>|<cm>|<um>|<nm>]
minz[<m>|<cm>|<um>|<nm>]
maxx[<m>|<cm>|<um>|<nm>]
maxy[<m>|<cm>|<um>|<nm>]
maxz[<m>|<cm>|<um>|<nm>]} |
polygons= {<pol1> <pol2> ... <poln>} |
(polygons= <list> |
min= min_x[<m>|<cm>|<um>|<nm>]
max= max_x[<m>|<cm>|<um>|<nm>])
)
Arguments
brick
Creates a rectangular prism, given its two corners as minx miny minz and maxx maxy
maxz. Default unit: .
clear
Removes all the previously defined polyhedra from memory.
external.sde
Creates a polyhedron from an external Sentaurus Structure Editor structure (see Sentaurus
Structure Editor Interface on page 791).
list
Lists the currently defined polyhedra.
µm
A: Commands
polyhedron
Sentaurus™ Process User Guide 1087
I-2013.12
materials
It is used only with tdr and chooses which materials will be included in the file. In addition
to explicit material names, bulk.materials is available to specify all nongas materials.
max
Maximum x-coordinate for extrusion (see polygons). Default unit: .
min
Minimum x-coordinate for extrusion (see polygons). Default unit: .
name
Name of the polyhedron to be created.
polygons
This argument can be used in two contexts:
• When specifying a list of polygons that form a polyhedron, it builds such a polyhedron.
• When specifying one planar axis-oriented polyhedron, it extrudes (using
min and max)
that polygon in the x-direction to form a polyhedron.
regions
It is used only with tdr and chooses which regions of the TDR boundary file are included.
rotate
It is used with tdr and avoids the automatic rotation that Sentaurus Process performs when
reading polyhedra to transfer them from a TDR boundary file (assumed to be in DF–ISE
coordinates) to a Sentaurus Process structure (in Sentaurus Process coordinates).
tdr
Name of the TDR boundary file from which to read all the polyhedra.
Description
This command creates a polyhedron and stores it under the name specified. Different
mechanisms can be used to create the polyhedron. It can be read from a TDR boundary file,
defined as a brick, defined from the beginning using polygonal faces, or created as an extruded
polygon. When a polyhedron is defined, it can be used to perform polyhedron insertion using
the insert command.
µm
µm
A: Commands
polyhedron
1088 Sentaurus™ Process User Guide
I-2013.12
Examples
Load the polyhedra containing silicon and gas from the boundary file sphere_bnd.tdr with
the name sphere:
polyhedron name= sphere tdr= sphere_bnd.tdr materials= {Silicon Gas}
Create a polyhedron named prism and extrude from x = to x = an already
existing polygon called triangle:
polyhedron name= prism polygons= {triangle} min= -6 max= 2
Using the polygons face1, face2, face3, and face4 (they must be correctly defined), build
a polyhedron called tetrahedron:
polyhedron name= tetrahedron polygons= {face1 face2 face3 face4}
Define a rectangular prism (brick shape) called smallPrism by using its two corners, that is,
minx = , miny = , minz = , and maxx = , maxy = ,
maxz =:
polyhedron name= smallPrism brick= {-6 -4 -2 -1 4.5 1}
See Also
Inserting Polyhedra in Three Dimensions on page 786
insert on page 982
6–µm
2µm
6–µm
4–µm
2–µm
1–µm
4.5 µm
1µm

A: Commands
PowerDeviceMode
Sentaurus™ Process User Guide 1089
I-2013.12
PowerDeviceMode
Sets diffusion models to match the pd.fermi model of TSUPREM-4 for power-device
applications. It also relaxes time-step controls and reduces mesh refinement around the
interfaces.
Syntax
PowerDeviceMode
Description
Sentaurus Process and TSUPREM-4 use different code and, sometimes, have different
assumptions or algorithms for diffusion. This command tries to minimize these differences by
setting appropriate switches that make the results of a Sentaurus Process simulation as close as
possible to those produced by TSUPREM-4 with the pd.fermi model for boron, phosphorus,
arsenic, antimony, and indium in silicon. This includes:
■Switch on Fermi model.
■Switch on solid solubility model.
■Switch on DopantOnly charge model.
■Switch on equilibrium activation model for arsenic.
■Switch on segregation model at oxide–silicon interface.
■Switch off dopant and defect clusters.
■Switch off point-defect equations.
■Relax time-step controls by modifying InitTimeStep, delT, delTox, delNT,
IncreaseRatio, ReduceRatio, and MaxGrowthStep.
■Switch on TSUPREM-4-style time-step controls.
■Reduce mesh refinement around the interfaces.
■Relax meshing criteria during boundary movement.

A: Commands
print.1d
1090 Sentaurus™ Process User Guide
I-2013.12
print.1d
Prints values along a 1D cross section.
Syntax
print.1d
[gas] [interfaces] [<material>] [name=<c>] [region=<c>]
[syntax.check.value=<c>]
[x=<n>][<m>|<cm>|<um>|<nm>]
[y=<n>][<m>|<cm>|<um>|<nm>]
[z=<n>][<m>|<cm>|<um>|<nm>]
Arguments
gas
By default, gas values are not reported. This argument allows the gas mesh to be included
in the extracted data.
interfaces
Prints interface data from the field specified by name. Values from all interfaces are
displayed on the screen and are organized by interface. For each point on the interface, a
set of numbers is displayed as follows:
•{x value} in one dimension
•{x y value} in two dimensions
•{x y z value} in three dimensions
where x, y, and z are the coordinates of the interface point, and value is the value of the
specified field.
<material>
Name of the material for which the data fields are printed.
name
Name of a data field. This allows printing without using the select command. The default
is to use the field specified in the most recent select command.
region
Name of the region for which the data fields are printed.
A: Commands
print.1d
Sentaurus™ Process User Guide 1091
I-2013.12
syntax.check.value
Sets a value to be returned only during syntax-checking mode. Sometimes, the value
returned by a command can cause a false syntax-check error because the value returned by
the command would not be the value during the normal run mode. Setting this value avoids
such problems.
x, y, z
Specify the constant values of a line along which sectioning is performed. In one
dimension, these parameters are not necessary. In two dimensions, only one of x or y can
be specified for a given device. Specifying x produces a horizontal slice through the device
and y specifies a vertical slice. An easy way to remember this is that the cross section is
taken at the constant value specified. For a 3D simulation, two of these three arguments
must be specified. Default unit: .
Description
This command is particularly useful for creating input for another xy plot. A Tcl list is returned
for all values. This allows subsequent processing (for example, integration) of the resulting
profile.
Examples
Print the selected value at x equal to :
print.1d x= 1.0
Print the data field named Arsenic along a vertical line at a lateral position of :
print.1d y= 1.0 name= Arsenic
See Also
plot.1d on page 1063
select on page 1117
tclsel on page 1164
µm
1.0 µm
1.0 µm

A: Commands
print.data
1092 Sentaurus™ Process User Guide
I-2013.12
print.data
Writes data in x-, y-, and z-format.
Syntax
print.data
[name=<c>] [NODE | EDGE] [outfile=<c>]
[xlo=<n>][<m>|<cm>|<um>|<nm>] [xhi=<n>][<m>|<cm>|<um>|<nm>]
[ylo=<n>][<m>|<cm>|<um>|<nm>] [yhi=<n>][<m>|<cm>|<um>|<nm>]
[zlo=<n>][<m>|<cm>|<um>|<nm>] [zhi=<n>][<m>|<cm>|<um>|<nm>]
Arguments
name
Name of a data field. This allows printing without using the select command. Default:
Z_Plot_Var.
NODE, EDGE
Specifies either a node-based field or an edge-based field. Default: NODE.
outfile
Name of output file. The file is opened for writing, and any previous content is destroyed.
xlo, ylo, zlo, xhi, yhi, zhi
Specify a 3D bounding box. Only data within these limits is printed. Default value and unit:
.
Description
The file format is the x-position, y-position, and z-position. This command is used primarily to
write a data field for use with more sophisticated 3D plotting tools.
Examples
Print the data field named Boron:
print.data outfile= foo name= Boron
See Also
select on page 1117
0µm

A: Commands
profile
Sentaurus™ Process User Guide 1093
I-2013.12
profile
Reads a data file and constructs a data field.
Syntax
profile
infile=<c> name=<c>
[concentration=<n>] [logarithmic | linear] [<material>]
[max= {<n> <n> <n>}] [min= {<n> <n> <n>}]
[offset= {<n> <n> <n>}][<m>|<cm>|<um>|<nm>]
[region=<c>]
[x.sigma=<n>] [xcoord=<n>] [xscale=<n>]
[y.sigma=<n>] [ymin=<n>] [z.sigma=<n>]
Arguments
concentration
Specifies the concentration of the field at the specified xcoord.
infile
Name of the file to be read. If it is an ASCII data file, the file must be in a two-column
format with depth (in ) in column 1 and the variable in column 2. It also will read the
output of the print.1d command, which includes Tcl braces for list processing and the
material name. If it is a TDR file, it must be in the same dimension as the simulated device
structure and requires .tdr as the file extension.
logarithmic, linear
Interpolates data using either logarithmic or linear interpolation. The default is
logarithmic, which is usually more accurate for concentration profiles.
<material>
Name of the material to which the field profile is applied. For information about specifying
materials, see Material Specification on page 52.
max
List of numeric values defining the x-, y-, and z- coordinates of the lower-right front corner
of the 1D, 2D, or 3D rectangular box in the internal coordinate system into which the
profile is imported. For 1D, 2D, and 3D structures, a list of one, two, or three numbers is
required, respectively. The possible maximum number is used for missing numbers.
µm
A: Commands
profile
1094 Sentaurus™ Process User Guide
I-2013.12
min
List of numeric values defining the x-, y-, and z- coordinates of the upper-left back corner
of the 1D, 2D, or 3D rectangular box in the internal coordinate system into which the
profile is imported. For 1D, 2D, and 3D structures, a list of one, two, or three numbers is
required, respectively. The possible minimum number is used for missing numbers.
name
Name of the data field. This argument allows for the creation of arbitrary fields, for
example, a field called Measured.
offset
List of numeric values that specify the offsets in the x-, y-, and z-direction, respectively.
The missing values are treated as 0. These values will be subtracted from the x-, y-, and z-
coordinate, respectively, when creating the data field from the imported field. This
argument allows a profile to be shifted. Default value and unit: .
region
Name of the region to which the profile is applied.
x.sigma
Standard deviation of erfc falloff from a rectangular box in the x-direction. It must be
specified if a rectangular box is specified.
xcoord
Coordinate in the x-direction where the concentration will be defined.
xscale
The profile command assumes the x-dimension is in micrometers. This argument allows
you to scale the depth dimension if necessary. For example, if the depth is in ångströms,
must be specified. Default: 1.0.
y.sigma
Standard deviation of erfc falloff from a rectangular box in the y-direction. If it is not
specified, it takes the value of x.sigma.
ymin
Minimum-acceptable value of the data field. Values less than ymin in the data field are set
to ymin. This is useful for data that may approach zero when using logarithmic
interpolation.
0µm
14–
×10
A: Commands
profile
Sentaurus™ Process User Guide 1095
I-2013.12
z.sigma
Standard deviation of erfc falloff from a rectangular box in the z-direction. If it is not
specified, it takes the value of y.sigma.
Description
This command reads data fields from an ASCII data file or a TDR file, and adds them to the
structure. It allows you to read a doping profile from a SIMS measurement. In this case, if the
simulated structure is 2D or 3D, the data field is created uniformly in the lateral direction. This
command also allows you to read a field from a TDR file with the same dimension as the
simulated structure. You also can limit the extent of the imported profile within a rectangular
box by specifying min or max, or both. Outside this box, the profile falls off with a
complementary error function (erfc) with standard deviations given by x.sigma, y.sigma,
and z.sigma in the x-, y-, and z-direction, respectively.
Examples
Read a boron profile (this could be from a Monte Carlo implant code – UT MARLOWE). Scale
the depth by to convert the ångström of MARLOWE to micrometer. Since the output
of MARLOWE is sometimes zero, specify a minimum value of :
profile name= Boron infile= utmar.bor xscale= 1.0e-4 ymin= 1.0e14
Read a file named SIMS into a data field called Data. This can be performed to initialize a
device doping profile or to read in a measured profile that is the target of a diffusion extraction:
profile name= Data infile= SIMS
See Also
print.1d on page 1090
14–
×10
114
×10

A: Commands
RangeRefineboxes
1096 Sentaurus™ Process User Guide
I-2013.12
RangeRefineboxes
Creates a set of refinement boxes based on a mask, and a set of range and extent parameters.
All boxes share a set of global refinement settings, but each box can have additional local
refinement settings.
Syntax
RangeRefineboxes
boxes= {
drange=<drange1> [<box-specific_arguments>]
[drange=<drange2> [<box-specific_arguments>]]
...
}
mask=<c>
name=<c>
range=<c>
[<other_arguments>]
Arguments
<box-specific_arguments>
Any argument of the refinebox command. These arguments are applied only to the
individual refinement box. They can overwrite global parameters defined in the
<other_arguments> section.
boxes
A Tcl list containing a set of Tcl lists. Each Tcl list contains specific parameter settings for
one individual refinement box.
drange
Specifies the primary extent of an individual refinement box, which extends from xmin =
range – drange to xmax = range + drange.
mask
Specifies the mask under which the refinement is to be applied.
name
Specifies the root name for the set of refinement boxes. Each individual refinement box
inherits a name of the form <c>_<n>, where <c> is the value of the name argument and
<n> is a counter.
A: Commands
RangeRefineboxes
Sentaurus™ Process User Guide 1097
I-2013.12
<other_arguments>
Any valid argument of the refinebox command.
range
Specifies the center of the primary extent for all refinement boxes in the set. For example,
you can use range from an implant table to define a set of refinement boxes to resolve the
area that dopants penetrate in a subsequent implantation step.
Description
The refinement is applied to the area under the specified mask. The lateral extent is controlled
by the same arguments as in the refinebox command, for example,
mask.edge.refine.extent and mask.edge.mns.
The primary extent is defined by range and drange. The range argument is common to all
refinement boxes and may be taken as the range argument for a given implantation. The
drange argument can be set for each related refinement box separately.
As optional global default arguments, any argument from the refinebox command is
allowed. These arguments are applied to all refinement boxes of the set. For each individual
refinement box, an additional refinebox command can be set, which can overwrite globally
defined arguments or add new arguments. There are no limits on how many refinement boxes
can be in the set. The individual refinement boxes inherit the root name (specified by the name
argument) with a numeric suffix counter.
NOTE The RangeRefineboxes command makes one call to the refinebox
command per individual refinement box.
NOTE Yo u ca n us e the DeleteRefinementboxes command to remove the
entire set of refinement boxes created with the RangeRefineboxes
command.
Examples
Create a set of refinement boxes:
mask name= M1_p segments= {6 10}
array set moments [implant Arsenic dose= 5e13<cm-2> energy= 1000<keV> \
tilt= 0 rotation= 0 get.moments]
set range $moments(rp)
set sigma $moments(stdev)
eval RangeRefineboxes name= "RM1" mask= "M1_p" range= $range \
boxes= \{ \
{drange= $sigma xrefine= [expr $sigma/4.0] yrefine= [expr $sigma/4.0] \
extend= 0} \
A: Commands
RangeRefineboxes
1098 Sentaurus™ Process User Guide
I-2013.12
{drange= [expr 4*$sigma] xrefine= $sigma yrefine= $sigma \
extend= [expr 2*$sigma]} \
{drange= [expr 2*$sigma] mask.edge.mns= [expr $sigma/8.0] \
mask.edge.refine.extent= [expr 2*$sigma] mask.edge.ngr= 1} } \
extend= 1.0 xrefine= 0.5 yrefine= 0.5 info= 2
See Also
DeleteRefinementboxes on page 900
refinebox on page 1101

A: Commands
reaction
Sentaurus™ Process User Guide 1099
I-2013.12
reaction
Defines reacting materials and the new material that forms as the product of the reaction.
Syntax
reaction
(list | mat.l=<c> mat.r=<c> mat.new=<c> name=<c>)
[ambient.name] [clear] [delete]
[diffusing.species= <list>]
[mat.final=<c> new.like=<c>]
Arguments
ambient.name
Specifies an ambient-type reaction and which ambient must be present for this reaction to
occur.
clear
If a reaction is named, this argument clears the diffusing species list from that reaction. If
no reaction is named, it deletes all reactions (may only be useful in special situations).
delete
Deletes the named reaction.
diffusing.species
List of reactants for material growth reactions either ambient type or nonambient type. For
ambient-type reactions, the (one only) default diffusing species name is the ambient name,
but it can be changed using this argument. For nonambient reactions (such as silicidation),
multiple diffusing species can be listed. Reactants are added automatically to the global
solution list in the SetReactantSolutions procedure.
list
Lists the names of the already defined reactions.
mat.final
For reactions that use a temporary material during growth, mat.final can be set to
convert the temporary material to a final material after the diffuse step is finished. This is
usually used with epitaxial reactions to keep regions separated during growth and only
merged afterwards.
A: Commands
reaction
1100 Sentaurus™ Process User Guide
I-2013.12
mat.l, mat.r
Specify the material names for each side (left and right) of the interface before the reaction.
mat.new
Specifies a valid material name as the product of the reaction.
name
Name of the reaction. The argument name is used to identify the reactions during the
growth process.
new.like
Name of the existing material that the new material is behaving like. This includes the
existing material and other material interfaces. It is performed in the ReactantLike
procedure.
Description
The convention for interface materials is mat1_mat2 where the materials are ordered
alphabetically. For the purpose of this command, left refers to mat1 and right refers to mat2.
Both materials must be specified when using this command. Silicidation and oxidation rely on
this information.
Examples
Define a reaction named MyDryOx. The reaction will occur at the gas–silicon interface and the
new material will be oxide. For the reaction to occur, O2 must be present in the structure. It is
expected that you will provide the actual reaction equation for the interface using the Alagator
language:
reaction name= MyDryOx mat.l= Silicon mat.r= Gas mat.new= oxide \
diffusing.species= O2
In this example, silicon and titanium are the reacting materials. The product of the reaction is
titanium silicide. Two species, silicon and titanium, are needed for the reaction. It is expected
that you will provide the actual reaction equations for the interface using the Alagator
language:
reaction name= silicidation mat.l= Silicon mat.r= Titanium \
mat.new= TiSilicide diffusing.species= {Silicon Titanium} new.like= oxide
See Also
Alagator for Generic Growth on page 596

A: Commands
refinebox
Sentaurus™ Process User Guide 1101
I-2013.12
refinebox
Sets the local grid parameters and performs a grid refinement using the MGOALS library.
Syntax
refinebox
[3d] [Adaptive] [clear] [double.side]
[kmc] [list]
[(mask=<c> extrusion.min=<n> extrusion.max=<n>) [extend=<n>]
[mask.edge.mns=<n>] [mask.edge.ngr=<n>] [mask.edge.refine.extent=<n>]]
[<material>] [materials= <list>]
[max= <list>] [min= <list>]
[name=<c>]
[print]
[regions= <list>]
[xrefine= <list>]
[yrefine= <list>]
[zrefine= <list>]
[interface.mat.pairs= <list>]
[interface.materials= <list>]
[interface.region.pairs= <list>]
[interface.regions= <list>]
[max.lateral.size=<n>][<um>]
[min.normal.size=<n>][<um>]
[normal.growth.ratio=<n>]
[offsetting]
[offsetting.maxlevel=<i>]
[abs.error= {<field1>=<n> <field2>=<n> ...}]
[def.abs.error=<n>]
[def.max.asinhdiff=<n>]
[def.max.difference=<n>]
[def.max.dose.error= n>]
[def.max.gradient=<n>]
[def.max.logdiff=<n>]
[def.rel.error=<n>]
[max.asinhdiff= {<field1>=<n> <field2>=<n> ...}]
[max.difference= {<field1>=<n> <field2>=<n> ...}]
[max.dose.error= {<field1>=<n> <field2>=<n> ...}]
[max.gradient= {<field1>=<n> <field2>=<n> ...}]
[max.logdiff= {<field1>=<n> <field2>=<n> ...}]
[max.value=<n>] [min.value=<n>]
[refine.add.fields= <list>]
[refine.dir.factor= <list>]
[refine.expr=<c>]
A: Commands
refinebox
1102 Sentaurus™ Process User Guide
I-2013.12
[refine.field.expr= {<field1>=<c> <field2>=<c> ...}
[refine.fields= <list>]
[refine.max.edge= <list>]
[refine.min.edge= <list>]
[refine.rm.fields= <list>]
[refine.type=<c>]
[rel.error= {<field1>=<n> <field2>=<n> ...}]
[target.length=<n>] [target.length.scaling=<n>]
Arguments
3d
Specifies the refinement box for only three dimensions, or for only one dimension and two
dimensions. The default behavior is to always apply the refinement box. If 3d is specified,
the refinement box only applies to three dimensions. If !3d is specified, the refinement box
only applies to one dimension and two dimensions.
Adaptive
Specifies an adaptive refinement box. Adaptive refinement boxes are used during all
MGOALS remeshing operations (such as deposit, etch, photo, transform) but will
not be used during solve unless adaptive meshing is switched on (by using pdbSet Grid
Adaptive 1).
clear
When used without other arguments, clear deletes all previously defined refinement
boxes. When used with name, only the named refinement box is deleted.
double.side
If !double.side is specified with offsetting, interface.mat.pairs and
interface.region.pairs are interpreted in a nonsymmetric fashion by Sentaurus
Mesh. The default is double.side, that is, the specification of a material or region pair
is interpreted by Sentaurus Mesh as if the parameters were defined symmetrically
for both and .
extend
Optional extension when using a mask-driven refinement. This value can be positive (or
negative) and extends (shrinks) the refinement isotropically in y and z. The original mask
remains unchanged.
extrusion.max, extrusion.min
Maximum and minimum coordinates in the x-axis where the refinement will be applied
when using mask.
x1x2
⁄
x1x2
⁄
x2x1
⁄
A: Commands
refinebox
Sentaurus™ Process User Guide 1103
I-2013.12
kmc
Refines the internal KMC boxes only.
list
Lists the defined refinement boxes.
mask
Uses an existing mask name as an extra constraint to where the refinement will be applied.
If the refinement contains another spatial constraint (for example, using min and max), the
final application region is the intersection of the other constraints and the specified
extruded mask.
This argument requires specifying the box length in x (lacked by the mask) using
extrusion.min and extrusion.max, and optionally allows the use of extend, which
allows for the definition of layout (mask)-driven refinements.
mask.edge.mns
Specifies the minimum mesh size near the mask edge (actual edge length may be up to two
times smaller than this setting). This argument must be used with
mask.edge.refine.extent to have an effect.
mask.edge.ngr
Specifies the growth rate of refinement away from the mask edge This argument must be
used with mask.edge.refine.extent to have an effect. Default: 1.0 (no growth).
mask.edge.refine.extent
Specifies the distance from the mask edge over which edge-based refinement occurs. It
must be specified to obtain mask edge–based refinement.
<material>
Limits the refinement box to a particular material. By default, the refinement box applies
to all materials. For more information about specifying materials, see Material
Specification on page 52.
materials
Limits the refinement box to a list of materials. By default, the refinement box applies to
all materials. For more information about specifying materials, see Material Specification
on page 52.
A: Commands
refinebox
1104 Sentaurus™ Process User Guide
I-2013.12
max, min
Limits the extent of the refinement box. Both arguments take a Tcl list of numbers defining
the refinement box extent in the x-, y-, and z-axes. You can specify either one or both min
and max with a Tcl list of one, two, or three numbers for each argument. If one number is
specified, it is taken to be the limit in the x-axis. If two numbers are specified, they set limits
for the x-axis and y-axis. Similarly, three numbers specify a limit in all three axes. Default
unit: .
name
Name of the refinement box.
print
Prints information for all refinement boxes. If name is specified, only the named refinement
box information is printed.
regions
Limits the refinement box to a list of regions. By default, the refinement box applies to all
regions.
xrefine
List of three numbers defining the element sizes in the x-direction at the top, middle, and
bottom of the box. Default unit: .
yrefine
List of three numbers defining the element sizes in the y-direction at the left, middle, and
right of the box. Default unit: .
zrefine
List of three numbers defining the element sizes in the z-direction at the front, middle, and
back of the box. Default unit: .
Arguments: Interface Refinement Control
interface.mat.pairs
A set of pairs of materials where interface meshing will be switched on (1st and 2nd, 3rd
and 4th, and so on).
interface.materials
All interfaces that contain any of the materials listed here are refined using the
min.normal.size criterion. By default, in two dimensions, interface refinement is
µm
µm
µm
µm
A: Commands
refinebox
Sentaurus™ Process User Guide 1105
I-2013.12
applied to all interfaces of Silicon, Polysilicon, or Oxide. In three dimensions,
interface refinement is only by default applied to interfaces of Silicon.
interface.region.pairs
A set of pairs of regions where interface meshing will be switched on (1st and 2nd, 3rd and
4th, and so on). This region-based interface specification is supported only for Sentaurus
Mesh offsetting, that is, when offsetting also is given.
interface.regions
Used only in conjunction with offsetting or offsetting.maxlevel to switch on
Sentaurus Mesh offsetting or to specify offsetting.maxlevel on a regionwise basis for
Sentaurus Mesh offsetting.
max.lateral.size
Specifies the maximum lateral spacing at the interface.
min.normal.size
Specifies the minimum edge spacing at interfaces for this box.
normal.growth.ratio
Specifies the edge-to-edge growth ratio moving away from an interface.
offsetting
When offsetting is specified along with interface.materials,
interface.mat.pairs, interface.regions, or interface.region.pairs, the
Sentaurus Mesh offsetting algorithm is used to generate offsetting layers at the given
interface.
offsetting.maxlevel
Specifies the number of offsetting layers at the interface when Sentaurus Mesh offsetting
is used, specified either by material or region using interface.materials or
interface.regions, respectively.
Arguments: Adaptive Meshing
abs.error
Sets a field-dependent value of the minimum significant field value.
def.abs.error
Sets the field-independent default value of the minimum significant field value.
A: Commands
refinebox
1106 Sentaurus™ Process User Guide
I-2013.12
def.max.asinhdiff
Sets the field-independent default value of the maximum inverse hyperbolic sine (asinh)
difference criteria.
def.max.difference
Sets the field-independent default value of the maximum absolute difference criteria.
def.max.dose.error
Sets the field-independent default value of the maximum local dose error criteria.
def.max.gradient
Sets the field-independent default value of the maximum gradient criteria.
def.max.logdiff
Sets the field-independent default value of the maximum logarithmic difference criteria.
def.rel.error
Sets the field-independent default value of the required relative change of a field across an
edge.
max.asinhdiff
Sets a field-dependent value of the inverse hyperbolic sine difference criteria.
max.difference
Sets a field-dependent value of the maximum absolute difference criteria.
max.dose.error
Sets a field-dependent value of the maximum local dose error criteria.
max.gradient
Sets a field-dependent value of the maximum gradient criteria.
max.logdiff
Sets a field-dependent value of the maximum logarithmic difference criteria.
max.value
Maximum interval value for interval refinement.
A: Commands
refinebox
Sentaurus™ Process User Guide 1107
I-2013.12
min.value
Minimum interval value for interval refinement.
refine.add.fields
List of fields to be added to the default list of fields considered for adaptive refinement.
refine.dir.factor
Applies adaptive refinement more strongly in one direction than another. A factor of 1 has
no effect. A factor less than 1 causes smaller edges in that direction.
For example, refine.dir.factor= {0.1 1.0} requests that, for a given adaptive
refinement expression value, edges in the x-direction be 10 times smaller than edges in the
y-direction.
refine.expr
Specifies a refinement expression. It takes any valid Alagator expression that produces a
node-based result.
NOTE Earlier releases required the diff() operator, but now, the diff
operator must not be used. Similar results can be obtained for earlier
releases by removing the diff operator.
refine.field.expr
Sets a field-dependent refinement expression.
refine.fields
Replaces the default list of fields considered for adaptive refinement. Solution variables
and terms can appear in the refine.fields list. (For a description of a term, see term on
page 1173.) The default list includes all dopants and point defects.
refine.max.edge
Sets the direction-dependent maximum edge length.
refine.min.edge
Sets the direction-dependent minimum edge length.
refine.rm.fields
Removes the specified fields from the default list of fields considered for adaptive meshing.
A: Commands
refinebox
1108 Sentaurus™ Process User Guide
I-2013.12
refine.type
Specifies the type of criteria to apply for adaptive refinement. Allowed values are
interval and error (default).
rel.error
Sets a field-dependent value of the required relative change of the refined field across an
edge.
target.length
Target length (in micrometers) for interval refinement.
target.length.scaling
Scaling factor used in the calculation of the effective target length for interval refinement.
Description
This command specifies mesh refinement. The following types of refinement box are available:
■Standard: Independent xrefine, yrefine, zrefine settings.
■Interface: Refinement near one or more interfaces.
■Adaptive: Adaptive refinement on fields.
■Plane: Planar refinement for crystal boundaries.
■Bulk mask: Confine refinement to an extruded boundary defined by a mask.
■Mask edge: Confine refinement to a specified distance from a specified mask.
All refinement boxes can be limited by material or spatially by specifying x-, y-, or z- minimum
or maximum limits.
Examples
Define two refinement boxes:
refinebox min= {-0.25 0.4} max= {0.4 0.6} xrefine= {0.1 0.06 0.1} \
yrefine= {0.1 0.01 0.1} oxide
refinebox min= {0.6 0.6} max= {0.8 0.8} xrefine= {0.1 0.03 0.1} \
yrefine= {0.1 0.03 0.1} silicon
Create an adaptive refinement box that applies maximum dose error criteria to the default list
of adaptive species, and effectively switch off relative error criteria, which is on by default:
refinebox adaptive def.rel.error= 100 def.max.dose.error= 5e9
A: Commands
refinebox
Sentaurus™ Process User Guide 1109
I-2013.12
Form the boundary for the refinement by extending (by ) an existing mask called
Mask1 extruded from to :
polygon name= pol segments= {-.5 -.5 .5 -.5 .5 .5 0 .5 0 0 -.5 0}
mask name= Mask1 polygons= {pol}
refinebox name= "ref1" mask= Mask1 yrefine= {0.05 0.075 0.075} \
extrusion.min= 0 extrusion.max= 0.05 extend= 0.2
See Also
Mesh Refinement on page 694
mask on page 1020
mgoals on page 1037
0.2 µm
0µm
0.05 µm

A: Commands
region
1110 Sentaurus™ Process User Guide
I-2013.12
region
Creates regions, marks substrates, and changes region materials.
Syntax
region
<material>
xlo=<c> [ylo=<c>] [zlo=<c>]
xhi=<c> [yhi=<c>] [zhi=<c>]
[alt.matername] [bbox | bbox.cm | bbox.um]
[change.material] [cropped.bbox] [exact.name]
[field=<c> (resistivity=<n>[<ohm-cm>] | concentration=<n>)]
[list | list.bulk | list.gas | list.interface]
[material]
[max= {<n> <n> <n>} min= {<n> <n> <n>}]
[name=<c>] [new.name=<c> point= {<n> <n> <n>}]
[substrate] [syntax.check.value=<c>] [volume] [zero.data]
Arguments
alt.matername
Specifies an alternative material to be used when saving a structure. When using the TDR
format, regions that are converted using alt.matername are correctly converted back to
the simulation material when the TDR file is read in from the init command. Although
common materials such as SiGe and III–Vs are by default handled this way, special
situations may require additional conversions when transferring to device simulation.
bbox, bbox.cm, bbox.um
If specified, the region command returns the maximum extents of the region in two
points. If bbox.cm is specified, it returns the maximum extents of the region in centimeter.
If bbox.um is specified, it returns the maximum extents of the region in micrometer.
change.material
Changes the material of an existing region (must be used with name). Changing the
material of selected regions (to and from gas) can be used to change the structure without
remeshing. Meshes of material gas are ignored in most process steps: implantation and
oxidation.
concentration
Specifies the value of the field directly.
A: Commands
region
Sentaurus™ Process User Guide 1111
I-2013.12
cropped.bbox
If specified, returns the cropped bounding box of a region that lies within a user-specified
bounding box.
exact.name
Usually when changing the material of a region, all ancestors of the named region (if there
are any) are converted as well as the named region if it exists (see Regionwise Parameters
and Region Name-handling on page 58).
If exact.name is switched on, only a region whose name exactly matches the name
argument will have its material changed. Default: false.
field
Name of a field to be initialized within this region.
list, list.bulk, list.gas, list.interface
Used to obtain a Tcl list of regions:
•list returns a list of all regions.
•list.bulk returns a list of nongas, noninterface regions (that is, all bulk regions).
•list.gas returns a list of gas regions.
•list.interface returns a list of interface regions.
<material>
Material of the region. For more information about specifying materials, see Material
Specification on page 52.
material
If specified, it returns the material name of the named region.
max, min
User-specified bounding box.
name
Name of the region. The name must not contain an underscore (_) or a period (.) because
these characters have special meaning.
A: Commands
region
1112 Sentaurus™ Process User Guide
I-2013.12
new.name, point
Used together to change the name of a region, where point must specify a point (a list of
doubles) within a region. The point must not be on or very near a border. The argument
new.name specifies the new name of the region.
resistivity
Sets the value of the field by requesting a resistivity. This argument only works for fields
that have the resistivity parameters in the PDB (which by default is only As, B, P, Sb, and
In in silicon).
substrate
Tags a named region as the substrate for subsequent analysis. Setting !substrate clears
the substrate tag. If no region name is specified and !substrate is set, all substrate tags
are cleared.
syntax.check.value
Sets a value to be returned only during syntax-checking mode. Sometimes, the value
returned by a command can cause a false syntax-check error because the value returned by
the command would not be the value during the normal run mode. Setting this value avoids
such problems.
xlo, ylo, zlo, xhi, yhi, zhi
Specify the bounds of the region. The <c> value must be one of the tags created in a
preceding line command.
volume
If specified, the region command returns the volume of the named region. The units will
be in cmD, where D is the simulation dimension.
zero.data
Usually when the material of a region is changed using change.material, all data in that
region is set to 0. Setting !zero.data leaves the data untouched. The default value for this
parameter is taken from pdbGet Grid default.zero.data, which allows a global
setting for this argument.
A: Commands
region
Sentaurus™ Process User Guide 1113
I-2013.12
Description
The region command has different applications:
■At the beginning of a simulation, the initial regions are created with the region command
in concert with the line command and the init command. The line command defines
where mesh lines go.
■The region command specifies between which mesh lines the regions are created and
what material the regions will be, and whether this region will be a substrate. It is used to
change the material of a region at any point in the simulation after the structure has been
initialized.
■The region command can be used to return a cropped bounding box of a region, specified
within a user-specified bounding box, defined by min and max along with a region name.
Examples
Create a new material MySilicon, and then change the material of a region named bulk to
MySilicon without changing the data:
mater name= MySilicon new.like= Silicon add
region name= bulk MySilicon change.material !zero.data
Change the region Gate to Gas before setting all fields to zero in Gate (zero.data defaults
to true):
region name= Gate Gas change.material
Create a 2D silicon region using the statements from the example for the line command:
region silicon ylo= left yhi= right xlo= surf xhi= back
Return a cropped bounding box of the region bulk that lies within the specified bounding box
defined by min and max:
region name= bulk min= {-5.0 0.0 0.0} max= {5.0 1.0 1.0} cropped.bbox
See Also
integrate on page 985
line on page 1010

A: Commands
sde
1114 Sentaurus™ Process User Guide
I-2013.12
sde
Dispatches commands to Sentaurus Structure Editor (only available for 3D simulations).
Syntax
sde
{<Sentaurus Structure Editor commands>}
[Adaptive] [external] [logfile=<c>] [off] [on]
[polyhedron=<c>] [polyhedron.material=<c>]
[remesh] [SdeCheck]
Arguments
Adaptive
If specified, Adaptive switches on adaptive meshing if remesh is given. Parameters for
adaptive meshing are described in Adaptive Refinement on page 699. The default is the
return value of pdbGet Grid Adaptive.
external
Puts the Sentaurus Structure Editor interface in external mode, which can be used to create
polyhedra to be inserted into a Sentaurus Process structure using MGOALS3D. When the
external mode is switched on, all geometry transformations such as etching and deposition
are applied to the external Sentaurus Structure Editor structure (see Sentaurus Structure
Editor Interface: External Mode on page 788).
logfile
Name of the file that will log all the Scheme commands dispatched to Sentaurus Structure
Editor. The recommended file extension is .scm. The file will contain both the Scheme
commands translated from Sentaurus Process etch, deposit, strip, photo, and
transform commands, and the user-specified Scheme commands inside the sde
command.
The log file can be used for fine-tuning and debugging in a stand-alone run of Sentaurus
Structure Editor such as:
sde -l mylogfile.scm
off
Switches off Sentaurus Structure Editor mode. Operations will be performed by the
MGOALS library instead.
A: Commands
sde
Sentaurus™ Process User Guide 1115
I-2013.12
on
Switches on Sentaurus Structure Editor for 3D geometry modeling. Even when Sentaurus
Structure Editor is the default engine for 3D etching and deposition, the sde on command
must always be specified to ensure that future simulations are performed using the same
algorithms.
polyhedron
Used for external mode only (see external). This polyhedron is used to initialize the
external Sentaurus Structure Editor interface. The material to be used for this polygon is
chosen using polyhedron.material.
polyhedron.material
Selects the material of the polyhedron that is used to initialize the external mode (see
external and polyhedron). Default: Silicon.
remesh
Forces a remesh at the end of the sde command.
<Sentaurus Structure Editor commands>
Any number of sde commands in the Scheme scripting language. You must enclose the
Scheme commands in a pair of braces to protect them from the Tcl command interpreter.
The opening brace must be on the same line as the sde command, for example:
sde {
(sdepe:depo "thickness" 0.01 "type" "iso" "algorithm" "pt"
"max-chamfer-angle" 30 "steps" 1 "material" "Oxide")
(sdeio:save-dfise-bnd "all" "out1_sde.bnd")
}
SdeCheck
Performs a geometry check of every boundary file that is created by Sentaurus Structure
Editor. This helps to detect failures in the geometry-modeling part and prevents the
Sentaurus Process simulation from continuing after an incorrect boundary representation
is found.
Description
This command enables and configures the interface between Sentaurus Process and Sentaurus
Structure Editor. When sde on is specified, all 3D geometry modeling is performed using
Sentaurus Structure Editor. Sentaurus Process will translate geometry-modifying commands to
the Sentaurus Structure Editor language and retrieve the resulting modified structure when
necessary. The following commands are supported: etch, deposit, photo, strip, and
transform.
A: Commands
sde
1116 Sentaurus™ Process User Guide
I-2013.12
NOTE Scheme commands can be sent directly to Sentaurus Structure Editor
using the sde command, but they must be enclosed in a pair of braces
to prevent syntax errors in the Tcl interpreter. Several Scheme
commands can be specified inside one sde command; each of them
must start on a new line.
Examples
Enable the use of Sentaurus Structure Editor or geometry modeling, specify the log file for the
Scheme commands, and check all boundary files written by Sentaurus Structure Editor:
sde logfile= depo.scm on SdeCheck
pdbSet InfoDefault 1
Create a cuboid in Sentaurus Structure Editor with tapered sidewalls and save the structure to
a.
bnd file:
sde {
(sdegeo:set-default-boolean "ABA")
(define r1 (sdegeo:create-cuboid (position 0 0.6 0)
(position 0.2 0.3 0.5) "Silicon" "Silicon_2"))
(define facelist (list (car (find-face-id (position 0.1 0.3 0.25)))
(car (find-face-id (position 0.2 0.5 0.25)))))
(sdegeo:taper-faces facelist (position 0.2 0.3 0.5) (gvector 0 0 1) 5)
(sdeio:save-dfise-bnd "all" "out1_sde.bnd")
}
NOTE The coordinates in the position vectors must be specified in DF–ISE
coordinates: x, y, z in the position vectors correspond to Sentaurus
Process z-, y-, and -x-coordinates.
See Also
For details about Scheme commands, refer to the Sentaurus™ Structure Editor User Guide.

A: Commands
select
Sentaurus™ Process User Guide 1117
I-2013.12
select
Selects the variable for display in all postprocessing commands.
Syntax
select
[delete] [edge.vector] [element] [interfaces] [list] [list.all]
[<material>]
([min | max] | [report.location])
[name=<c>] [node.evaluate] [permanent] [present]
[region=<c>] [store] [syntax.check.value=<c>] [value=<c>] [z=<c>]
Arguments
delete
Deletes the data field specified by name.
edge.vector
Computes the weighted field with respect to the edge orientation strongly favoring axis-
oriented edges. Used with adaptive meshing.
element
Computes the field on elements interpolating fields in the z expression if necessary.
If !element is specified and element fields appear in the z expression, those values are
interpolated to the nodes first.
interfaces
Computes the field or minimum or maximum on interfaces as well as bulk. Default: true
(include interfaces).
list
Returns a list of currently defined and named real data fields. This returns a full Tcl list for
use with those commands that require list variables.
list.all
Returns a list of currently defined and named data fields (for example, real data and vector
data). This returns a full Tcl list for use with commands that require list variables.
A: Commands
select
1118 Sentaurus™ Process User Guide
I-2013.12
<material>
Specifies the material to which the command applies. Different expressions for the data
field initialization in different materials can be used. For information about specifying
materials, see Material Specification on page 52.
min, max
Used with the name argument. When min or max is specified, the select command
returns the minimum or maximum of the field name. You can limit the query to either a
specific material using <material> or a specific region using region.
name
Name of the new data field. Default: Z_Plot_Var. This name is used by all the commands
when a plot name is not specified. This is a powerful feature, as solution fields also can be
created.
node.evaluate
Computes the divergence of a vector field at a node.
permanent
Returns 1 if the data field is written into permanent storage. If not, it returns 0.
present
Returns 1 if the data field with the name defined by name exists. If it does not exist, it
returns 0.
region
Name of the region. Specifies the region to which the command applies. Different
expressions for the data field initialization in different regions can be used.
report.location
Works with min and max. Reports the coordinate of the minimum or maximum value of
the selected field.
store
Sets the data field with the name defined by name to be written into permanent storage
when a structure file is output. Default: false.
syntax.check.value
Sets a value to be returned only during syntax-checking mode. Sometimes, the value
returned by a command can cause a false syntax-check error because the value returned by
A: Commands
select
Sentaurus™ Process User Guide 1119
I-2013.12
the command would not be the value during the normal run mode. Setting this value avoids
such problems.
value, z
Accepts an expression of data fields that are used to build a new data field. The operators
*, /, +, –, and ^ all work as expected. The vector variables are listed here. The data fields
available can be listed using the list argument. In addition to the listed data fields, the x-
and y-coordinates can be specified. Several functions also are available to operate on data
fields:
•abs: Absolute value
•erf: Error function
•erfc: Complementary error function
•exp: Exponential
•log: Logarithm
•log10: Logarithm base 10
•sqrt: Square root
Description
Data can be selected directly in most commands, but it is usually more effective to specify it
with the select command, which allows for manipulation of data fields and also will list all
currently defined data fields. The quantity can be computed on nodes (default) or elements
using the element argument. In either case, if necessary, interpolation will be performed to
obtain the proper value type (to obtain element values from nodal ones or vice versa).
NOTE The select command can be abbreviated to sel.
NOTE The select command always sets or retrieves data in internal units.
Internal units are CGS, for example, pressure is .
Examples
Select as the plot variable the base 10 logarithm of the MyData concentration:
select z= log10(MyData)
Select as the plot variable the MyData concentration minus a constant value of :
select z= (MyData - 5.0e14)
dyn/cm2
514
×10
A: Commands
select
1120 Sentaurus™ Process User Guide
I-2013.12
Select as the plot variable the difference between the MyData profile and an analytic profile.
This data field will be named MyField. The store argument indicates that the doping field
must be written into any saved structure files:
sel z= (MyData - 1.0e18 * exp ( y * y / 1.0e-8 )) name= MyField store
Set the value of the data field Pressure to (CGS units are used internally for
mechanics):
sel z= 1.0e9 name= Pressure store
Delete the MyField data field:
select name= MyField delete
Calculate the electric field in a new data field called ElectricField. The store argument
ensures that the new data field is stored to disk in subsequent calls of the struct command:
select z= "-diff(Potential)" edge.vector store name= ElectricField
List all available real data fields:
select list
Calculate the total heat field in a new data field called TotalHeat:
select z= "grad(Temperature)" node.evaluate store name= TotalHeat
See Also
All postprocessing commands
109dyn/cm2

A: Commands
SetAtomistic
Sentaurus™ Process User Guide 1121
I-2013.12
SetAtomistic
Sets the atomistic mode as the simulation mode.
Syntax
SetAtomistic
Description
This command switches the simulation domain to the atomistic mode. The following
commands are affected in this mode:
■deposit
■diffuse
■etch
■implant
■profile
■region
■select
■strip
■struct
If there are continuum fields, SetAtomistic automatically calls PDE2KMC to atomize the
fields and to make them available as an initial state for the KMC simulation.
See Also
Chapter 5 on page 381

A: Commands
SetDFISEList
1122 Sentaurus™ Process User Guide
I-2013.12
SetDFISEList
Sets a list of solution or term names to be included when saving DF–ISE format files.
Syntax
SetDFISEList
[Dopants]
[<solution/term names>]
[Solutions]
Arguments
Dopants
Dopants include the total and active dopant concentrations. Specifying !Dopants does not
save the total and active dopant concentrations, but still saves NetActive
(DopingConcentration). Default: true.
<solution/term names>
Any known fields listed on the command line are added to files saved with
struct dfise=<c>.
Solutions
Stores all solution variables (necessary for restarting a simulation). Using !Solutions
switches off all default savings (only fields specified by name will be saved to DF–ISE
files). Default: true.
Description
This command creates a solution or term name list that is passed to the struct command.
Depending on the arguments provided by users, solution names or dopant names can be
included or excluded from the fields that must be written to the DF–ISE files.
If the SetDFISEList command is executed without arguments, the default saving is used,
which includes all solutions, total and active dopant fields, and NetActive
(DopingConcentration).
Examples
Write only the VTotal field to the DF–ISE file:
SetDFISEList VTotal !Solutions
A: Commands
SetDFISEList
Sentaurus™ Process User Guide 1123
I-2013.12
Add the Vac and Temperature fields to those usually saved in DF–ISE files:
SetDFISEList Vac Temperature
Add the Int0, Intm, Vac0, and Vacpp fields to those usually saved in DF–ISE files:
SetDFISEList Int0 Intm Vac0 Vacpp
Save only NetActive, and the total and active dopants:
SetDFISEList !Solutions Dopants
See Also
SetTDRList on page 1129
struct on page 1158

A: Commands
SetDielectricOxidationMode
1124 Sentaurus™ Process User Guide
I-2013.12
SetDielectricOxidationMode
Sets the oxidation mode to grow oxide with dielectric on top.
Syntax
SetDielectricOxidationMode
<Dielectric> <Oxidant>
[Continuous | Segregation]
[Dirichlet | MassTransfer]
Arguments
Continuous, Segregation
Sets continuous boundary conditions at the dielectric–oxide interface.
Sets segregation boundary conditions at the dielectric–oxide interface.
<Dielectric>
Specifies the name of the dielectric material to grow oxide underneath.
Dirichlet, MassTransfer
Sets Dirichlet boundary conditions at the gas–dielectric interface.
Sets mass transfer boundary conditions at the gas–dielectric interface.
<Oxidant>
Specifies the name of the oxidant.
Description
This command sets the related parameters and models for oxidation with a dielectric on top.
Boundary conditions at the gas–dielectric interface and the dielectric–oxide interface default
to Dirichlet and Continuous, respectively.
In addition, these settings can be changed using:
pdbSetString <Dielectric> <Oxidant> DielectricOxTransportBC MassTransfer
pdbSetString <Dielectric> <Oxidant> DielectricOxInterfaceBC Segregation
A: Commands
SetDielectricOxidationMode
Sentaurus™ Process User Guide 1125
I-2013.12
Enables oxidation with a nitride layer on top for O2 ambient and uses the mass transfer
boundary condition at the gas-dielectric interface and the segregation boundary condition at the
dielectric-oxide interface, respectively:
SetDielectricOxidationMode Nitride O2 MassTransfer Segregation
See Also
UnsetDielectricOxidationMode on page 1189

A: Commands
SetFastMode
1126 Sentaurus™ Process User Guide
I-2013.12
SetFastMode
Omits diffusion and Monte Carlo implantation to simulate the device geometry quickly.
Syntax
SetFastMode
Description
This command runs the simulation quickly without simulating dopants and defects. This can
be useful when setting up a command file to confirm quickly that the geometry is satisfactory
before simulating more computationally expensive steps.
To switch off implantation, use:
pdbSet ImplantData NoImplant 1
To switch off oxidation or reaction, use:
pdbSet Diffuse NoDiffusionReaction 1
This may be useful for 3D geometry simulation.

A: Commands
setMobilityModel
Sentaurus™ Process User Guide 1127
I-2013.12
setMobilityModel
Sets the mobility model and mobility model parameters in silicon and polysilicon for antimony,
arsenic, boron, indium and phosphorus.
Syntax
setMobilityModel
[model]
Arguments
model
Specifies the name of the mobility model. model can be Model1, Antoniadis, Model2,
Masetti, Model3, or Thurber, where Model1=Antoniadis, Model2=Masetti, and
Model3=Thurber.
Description
This command sets the related parameters and switches for the mobility model given in silicon
and polysilicon for antimony, arsenic, boron, indium and phosphorus. Set values are printed
into the log file.
Examples
Set mobility model to Masetti:
setMobilityModel Masetti
or:
setMobilityModel Model12
See Also
Resistivity on page 857

A: Commands
SetPlxList
1128 Sentaurus™ Process User Guide
I-2013.12
SetPlxList
Sets a list of solution and term names to be passed to the WritePlx command.
Syntax
SetPlxList [<solution/term names>]
Arguments
<solution/term names>
Defines the name list to be passed to the WritePlx command.
Description
This command sets the list of fields to be saved in the next call to WritePlx. The list can
contain solutions or term names.
Examples
Write a .plx file with the data fields Temperature and Potential:
SetPlxList {Temperature Potential}
WritePlx T_and_P.plx
See Also
WritePlx on page 1191

A: Commands
SetTDRList
Sentaurus™ Process User Guide 1129
I-2013.12
SetTDRList
Sets a list of solution or term names to be included when saving TDR format files.
Syntax
SetTDRList
[Dopants]
[<solution/term names>]
[Solutions]
Arguments
Dopants
Dopants include the total and active dopant concentrations. Specifying !Dopants does not
save the total and active dopant concentrations, but does save NetActive
(DopingConcentration). Default: true.
<solution/term names>
Any known fields listed on the command line are added to files saved with
struct tdr=<c>.
Solutions
Stores all solution variables (necessary for restarting a simulation). Using !Solutions
switches off all default savings (only fields specified by name will be saved to TDR files).
Default: true.
Description
This command stores solution or term names in a TDR format file.

A: Commands
SetTemp
1130 Sentaurus™ Process User Guide
I-2013.12
SetTemp
Sets the temperature value.
Syntax
SetTemp
<n>[<C>|<K>]
Arguments
<n>
Specifies the temperature. Default unit: degree Celsius.
Description
This command sets the temperature value. The value is also saved in a TDR file.
Examples
Set the temperature to :
SetTemp 1000.0
1000°C

A: Commands
SetTS4ImplantMode
Sentaurus™ Process User Guide 1131
I-2013.12
SetTS4ImplantMode
Sets implant-related parameters and models to match those of TSUPREM-4.
Syntax
SetTS4ImplantMode [Native | Taurus]
Arguments
Native
Makes simulation results close to those of the TSUPREM-4 native implanter.
Taurus
Makes simulation results close to those of the TSUPREM-4 Taurus implanter.
Description
Sentaurus Process and TSUPREM-4 use different codes and, sometimes, have different
assumptions or algorithms for analytic implantations. The SetTS4ImplantMode command
tries to minimize this difference by setting appropriate switches that make Sentaurus Process
simulation results as close as possible to those produced by TSUPREM-4. This includes:
■Use the beam dose.
■Switch on the ts4.backscattering model.
■Switch off the Sentaurus Process backscattering model.
■Switch on TS4FluorineMode which computes fluorine profiles using fluorine tables for
BF2 implant.
■In Taurus mode, also switch on the PAI model with the TSUPREM-4-compatible PAI
mode.
The results may not be exactly the same due to differences in numeric methods for some cases.

A: Commands
SetTS4MechanicsMode
1132 Sentaurus™ Process User Guide
I-2013.12
SetTS4MechanicsMode
Sets mechanics-related parameters and models to match those of TSUPREM-4.
Syntax
SetTS4MechanicsMode [2008.09 | advanced]
Arguments
2008.09
Used for backward compatibility.
advanced
Sets TSUPREM-4 advanced settings.
Description
This command sets mechanics-related parameters and models in Sentaurus Process to match
TSUPREM-4 settings. This includes:
■Viscoelastic model and parameters
■Elastic moduli
■Stress relaxation factor setting
■Stress smoothing setting
■Thermal mismatch coefficients
■Some settings for oxidation used for backward compatibility (these settings are the same
as the defaults in the PDB)
The above parameters are set to match TSUPREM-4 defaults. The results may differ due to
different numeric methods.

A: Commands
SetTS4OxidationMode
Sentaurus™ Process User Guide 1133
I-2013.12
SetTS4OxidationMode
Sets oxidation-related parameters and models to match those of TSUPREM-4.
Syntax
SetTS4OxidationMode [2008.09 | advanced]
Arguments
2008.09
Sets oxidation and mechanics parameters to Version A-2008.09 default values.
advanced
Sets parameters to TSUPREM-4 advanced settings.
Description
This command sets oxidation-related parameters and models in Sentaurus Process to match
TSUPREM-4 settings. This includes:
■Settings from SetTS4MechanicsMode
■Activation volumes of stress-dependent oxidation (SDO) reaction rate and diffusivity
■Activation volumes of stress-dependent viscosity
■Native layer thickness
■Stress-dependent oxidation flag
Because TSUPREM-4 has different values for the activation volume of stress-dependent
viscosity during oxidation and nonoxidation steps, it is recommended to call
SetTS4OxidationMode immediately before the oxidation step and call
SetTS4MechanicsMode after it.
The above parameters are set to match TSUPREM-4 defaults. The results may not be very
close due to differences in numeric methods for some cases.

A: Commands
SetTS4PolyMode
1134 Sentaurus™ Process User Guide
I-2013.12
SetTS4PolyMode
Sets the polycrystalline model to match those of TSUPREM-4.
Syntax
SetTS4PolyMode
Description
This command sets the related parameters and models for the polycrystalline model in
Sentaurus Process to match TSUPREM-4 settings.

A: Commands
SheetResistance
Sentaurus™ Process User Guide 1135
I-2013.12
SheetResistance
Calculates the sheet resistance and the p-n junction depth.
Syntax
SheetResistance
[x=<n>] [y=<n>] [z=<n>]
Arguments
x, y, z
Specify the cut position. For 1D simulations, no cut specification is necessary. For 2D
simulations, either x or y must be specified. For 3D simulations, two axes must be
specified. Default unit: .
Description
This command calculates the sheet resistance and the p-n junction depth of a semiconductor
layer in the vertical direction. It can only be used after a diffusion step.
Examples
Calculate the sheet resistance and the p-n junction depth of a 3D structure using the cutplane
y= and z = :
SheetResistance y= 0.4 z= -0.1
See Also
Chapter 13 on page 849
µm
0.5 µm
0.1–µm

A: Commands
simDelayDouble
1136 Sentaurus™ Process User Guide
I-2013.12
simDelayDouble
Retrieves the Tcl expression used to evaluate a double-precision simulator state variable.
Syntax
simDelayDouble <c> [<c>]
Arguments
<c>
This argument can be any double parameter declared in the parameter database and any
double parameter declared by the user in user-defined models. In all cases, the argument
must be specified with the full hierarchical path for the double parameter.
Description
This command is very similar to the simGetDouble command except that the evaluation of
the returned expression is delayed.
Examples
Return [simGetDouble Diffuse tempC], which is the unevaluated expression itself:
simDelayDouble Diffuse tempC
See Also
simGetDouble on page 1138

A: Commands
simGetBoolean
Sentaurus™ Process User Guide 1137
I-2013.12
simGetBoolean
Reads a global simulator state variable.
Syntax
simGetBoolean <c> [<c>]
Arguments
<c>
This argument can be any double parameter declared in the parameter database and any
double parameter declared by the user in user-defined models. In all cases, the argument
must be specified with the full hierarchical path for the double parameter.
Description
This command returns one of the following global simulator state variables:
■AmbientReactions
■IsEpi
■IsGrowing
■laser
■MaterialReactions
Examples
Return true if laser annealing is switched on:
simGetBoolean Diffuse laser

A: Commands
simGetDouble
1138 Sentaurus™ Process User Guide
I-2013.12
simGetDouble
Reads a double-precision simulator state variable.
Syntax
simGetDouble <c> [<c>]
Arguments
<c>
This argument can be any double parameter declared in the parameter database and any
double parameter declared by the user in user-defined models. In all cases, the argument
must be specified with the full hierarchical path for the double parameter.
Description
This command returns a simulator set global variable. These variables are:
■EpiThick
■PH2O
■pO2
■pressure
■ramprate
■temp
■tempC
■tempK
■time
■Vt
■Vti
If the variable is not defined, it returns zero.
Examples
Return the last diffusion temperature [K]:
simGetDouble Diffuse tempK
Return the oxygen partial pressure used during the simulation:
simGetDouble Diffuse pO2

A: Commands
simSetBoolean
Sentaurus™ Process User Guide 1139
I-2013.12
simSetBoolean
Sets a global simulator state variable.
Syntax
simSetBoolean <c> [<c>] <n>
Arguments
<c>
This argument can be any double parameter declared in the parameter database and any
double parameter declared by the user in user-defined models. In all cases, the argument
must be specified with the full hierarchical path for the double parameter.
<n>
Specifies the value of the variable.
Description
This command sets one of the following global simulator state variables:
■AmbientReactions
■IsEpi
■IsGrowing
■laser
■MaterialReactions
NOTE Modifying global simulator state variables may cause errors in the
simulation.
Examples
Set the value of the global simulator state variable laser to true:
simSetBoolean Diffuse laser 1

A: Commands
simSetDouble
1140 Sentaurus™ Process User Guide
I-2013.12
simSetDouble
Sets a double-precision simulator state variable.
Syntax
simSetDouble <c> [<c>] <n>
Arguments
<c>
This argument can be any double parameter declared in the parameter database and any
double parameter declared by the user in user-defined models. In all cases, the argument
must be specified with the full hierarchical path for the double parameter.
<n>
Specifies the value of the variable.
Description
This command sets one of the following global double-precision simulator state variables:
■PH2O
■pO2
■temp
■tempC
■tempK
■Vti
NOTE Modifying global simulator state variables may cause errors in the
simulation.
Examples
Set the last diffusion temperature to :
simSetDouble Diffuse temp 900.0
900°C

A: Commands
slice
Sentaurus™ Process User Guide 1141
I-2013.12
slice
Extracts a 1D data slice through a 2D to 3D simulation object.
Syntax
slice
[<material>] [mdist] [mx] [my] [mz] [name=<c>]
[p1= <list>]
[p2= <list>]
[side=<c>] [syntax.check.value=<c>]
[value=<n>]
[x=<n>][<m>|<cm>|<um>|<nm>]
[y=<n>][<m>|<cm>|<um>|<nm>]
[z=<n>][<m>|<cm>|<um>|<nm>]
[include.interfaces]
[only.interfaces]
Arguments
include.interfaces
Includes interface values with the returned data. At an interface, the distance coordinate of
the 3 nodes (2 bulk and 1 interface) will be the same, and the interface value will be inserted
between the 2 neighboring bulk values.
<material>
Specifies the material. For information about specifying materials, see Material
Specification on page 52.
mdist, mx, my, mz
Changes the reporting information when interface materials are selected. The interface
distance can be reported as projected along one of the three primary axes (mx, my, mz).
Alternately, it can be reported as the distance along the extracted line (mdist).
name
Name of the data field. Default: Z_Plot_Var.
only.interfaces
Returns interface values exclusively in the returned data. When specified, no bulk values
are returned.
A: Commands
slice
1142 Sentaurus™ Process User Guide
I-2013.12
p1, p2
Specify the start point and endpoint for the cutline. Each argument takes a list of numeric
values.
The first, second, and third values in the list are taken as the x-, y-, and z-value, respectively.
The missing value will be treated as zero. These arguments allow the slice command to
extract data along an arbitrary line. The output from the slice command is a list of
(distance, value) pairs, where distance is measured from the p1 point, and value is the
extracted value of the selected quantity along the line.
NOTE Error messages will be generated if p1 and p2 are mixed with x, y, z, or
value.
side
Takes the value from one of the two bulk materials consisting of the interface or ‘interface’
(literally) itself. If side is not specified, ‘interface’ itself is assumed. If side is specified
as one of the bulk materials, the value of the selected quantity for the bulk material at the
interface is returned. This argument is effective only if an interface material is specified.
syntax.check.value
Sets a value to be returned only during syntax-checking mode. Sometimes, the value
returned by a command can cause a false syntax-check error because the value returned by
the command would not be the value during the normal run mode. Setting this value avoids
such problems.
value, x, y, z
Specify a cutline for up to a four-dimensional solid, so that a 2D return is provided. For 1D
simulations, none of these arguments is required. For 2D simulations, one is required. For
3D simulations, two are required. These requirements are reduced by one if an interface
material has been specified. The default unit for x, y, and z is .
Description
This is an extremely powerful data analysis command. It extracts xy data along a slice through
a specified material. It returns a Tcl list of coordinate–value pairs, where the coordinate is the
distance [ ] along the reference segment, and the value is the local value of the argument
specified either with the -name option or, if that is not provided, in the most recent select
command. For example:
select z= Boron
set sliceRet [slice y= 0.5]
foreach { x value } $sliceRet {
LogFile "$x\t$value\n"
}
µm
µm
A: Commands
slice
Sentaurus™ Process User Guide 1143
I-2013.12
This command will print and send to the log file the boron profile in x-coordinate value pairs
at y=0.5.
The command extracts the selected variables as a function of position along a constant line. In
one dimension, the command returns the concentration versus depth, for example. It also can
extract a constant contour of the data selected and returns the coordinates of the
isoconcentration line.
Examples
Return the selected variable as a function of depth at a constant lateral position of :
slice silicon y= 0.01
Return the x- and y-positions of a contour of the selected variable at 16.0:
slice silicon value= 16.0
Return the value of the selected quantity at the silicon side of the interface as a function of
distance from the start of the interface:
slice silicon /oxide mdist side= silicon
Return the boron concentration along a line passing through points (0, 0) and (1, 2):
slice name= Boron p1= {0 0} p2= {1. 2}
See Also
select on page 1117
tclsel on page 1164
0.01 µm

A: Commands
smooth
1144 Sentaurus™ Process User Guide
I-2013.12
smooth
Smooths a set of fields.
Syntax
smooth
smooth.field= <list>
[init=<n>][<hr>|<min>|<s>]
[smooth.distance= {<double array>}]
Arguments
init
Specifies the first time step for solving the smoothing equations. The default is 0.0001 s,
which is sometimes inappropriate for defect simulations, particularly in cases of damage.
Default unit: minute.
smooth.distance
Specifies the smoothing distance for each field as specified in smooth.field. Default:
2.0 nm.
smooth.field
List of fields to be smoothed. Any existing field can be specified.
Description
This command smoothes a set of fields with specified smooth distances.
Examples
Smooth the interstitial and vacancy implantation profiles with smooth distances of 2 nm and
3 nm, respectively:
smooth smooth.field= {Int_Implant Vac_Implant} smooth.distance= {2<nm> 3<nm>}

A: Commands
solution
Sentaurus™ Process User Guide 1145
I-2013.12
solution
Obtains and sets solution parameters for generic solutions using the Alagator language.
Syntax
solution
[add] [damp]
[DiffStep] [GrowthStep] [Heat] [InitStep] [list]
[material.list= <list>]
[name=<c>] [needsolution] [negative]
[nosolve | solve | ifpresent=<c>]
[present] [reset] [smooth] [store] [unit=<c>]
Arguments
add
Creates a new solution.
damp
Applies a damping algorithm to the Newton iteration.
DiffStep, GrowthStep, Heat, InitStep, smooth
Determines in which solver the solution variable will be solved:
•DiffStep variables are solved with the diffusion solver such as dopants and defects.
•GrowthStep variables are solved in the reaction step and are usually oxidants.
•Heat variables are solved during the laser annealing step.
•InitStep variables are solved during the initialization step.
•smooth variables are solved during smoothing steps (such as occurs when using the
smooth command).
list
Lists all the currently defined solutions.
material.list
List of materials where the solution variable will be solved. If the list is empty, the solution
variable will be solved only if the equations are set for a specific material.
A: Commands
solution
1146 Sentaurus™ Process User Guide
I-2013.12
name
The character string used for the solution. Capitalization is not ignored, for example,
Potential and potential are different. Abbreviations of names are not accepted.
needsolution
Returns true if the solution must be solved.
negative
Allows the specified solution to have negative values.
nosolve, solve, ifpresent
Only one of these options can be used at a time. They control the solution status for the next
command:
•nosolve means do not solve.
•solve switches on the solution status for the next command.
•ifpresent sets up a conditional solve.
If all the solutions in the specified list also are being solved, this solution is solved.
present
Returns true if the solution is defined and a data field matches the name.
reset
Allows reaction solution variables to be reset before the diffusion starts. Default: true.
store
Allows the solution command to be stored in a TDR file.
unit
Unit of the solution variable. Default: .
Description
This command creates and modifies solution names, and sets conditions for their inclusion in
the matrix assembly. Solutions also can be listed and checked.
cm 3–
A: Commands
solution
Sentaurus™ Process User Guide 1147
I-2013.12
Examples
Create a solution named Potential and always solve for it. Allow the solution to have
negative values and use damping on the Newton iteration updates:
solution name= Potential damp negative solve add
Create a solution name Vac and always solve for it. Do not use damping and do not allow
values to become negative:
solution name= Vac !damp !negative solve add
Create a solution named I2 and solve for it if Int and Vac are also present and being solved:
solution name= I2 !damp !negative ifpresent= {Int Vac} add
Return a list of all solutions:
solution list
Return a Boolean true if Vac has been defined and if there is a data field with the name Vac:
solution name= Vac present
See Also
term on page 1173

A: Commands
sptopo
1148 Sentaurus™ Process User Guide
I-2013.12
sptopo
Transfers structures and dispatches commands to Sentaurus Topography.
Syntax
sptopo {<Sentaurus Topography commands>}
Arguments
<Sentaurus Topography commands>
Any number of Sentaurus Topography commands. Enclose the commands in a pair of
braces to protect them from interpretation by the Tcl interpreter.
Description
This command transfers the boundary representation of the current structure and dispatches the
commands to Sentaurus Topography. After executing the commands in Sentaurus Topography,
the modified structure is retrieved and remeshed in Sentaurus Process.
NOTE A license for Sentaurus Topography must be available, and a version of
the Sentaurus Process binary with Sentaurus Topography included must
be installed.
Examples
Use Sentaurus Topography 3D to deposit 2 layers isotropically: Oxide thickness 0.005
(5 nm) and PolySilicon 0.18 :
sptopo {
deposit material= Oxide thickness= 0.005
deposit material= PolySilicon thickness= 0.180
}
See Also
Sentaurus™ Topography User Guide
For 3D operations, see topo on page 1176.
µm
µm

A: Commands
stdiff
Sentaurus™ Process User Guide 1149
I-2013.12
stdiff
Compares the current structure with one from a TDR format file.
Syntax
stdiff <c>
Arguments
<c>
Specifies the full path or the prefix of a TDR file. The prefix is the file name without
_fps.tdr.
Description
This command reads the external TDR file, interpolates the data onto the current structure,
compares data, and reports if data exceeds the relative error criteria (subject to the absolute
error minimum value).
Examples
Compare field values in the current structure in memory with those contained in file:
./
n1_fps.tdr
Current structure:
stdiff n1_fps.tdr

A: Commands
strain_profile
1150 Sentaurus™ Process User Guide
I-2013.12
strain_profile
Defines the strain introduced by an impurity as a piecewise linear function of the mole fraction
in a given substrate.
Syntax
strain_profile
<material> ratio= <list>
species=<c> strain= <list>
Arguments
<material>
Substrate material where the strains are defined.
ratio
List of numeric values of the mole fraction of the species; ranges from 0 to 1.
species
Species in the substrate that cause the strain.
strain
List of numeric values of the strain caused by the specified mole fraction; ranges from 0 to
1.
Description
The presence of certain materials such as germanium in silicon can modify the lattice spacing.
This command computes strains using the impurity mole fraction.
Examples
Defines the strain profile for germanium impurity in silicon:
strain_profile silicon species= Germanium ratio= {0 1} strain= {0 0.0425}
See Also
transform on page 1177

A: Commands
stressdata
Sentaurus™ Process User Guide 1151
I-2013.12
stressdata
This command:
■Defines the intrinsic stress of materials for use in stress calculations.
■Defines boundary conditions for stress analysis.
■Reports the maximum stress values and their locations.
■Prints anisotropic material matrix.
■Defines edge dislocation settings.
Syntax
stressdata
[<material> | region=<c>]
[sxxi=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[syyi=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[szzi=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[sxyi=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[syzi=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[szxi=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[base=<n>][<m>|<cm>|<um>|<nm>]
[sxx1=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[syy1=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[szz1=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[sxx2=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[syy2=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[szz2=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[bc.location= Left | Right | Front | Back | Bottom]
[bc.rotation.axis= {[xa=<n>] | [ya=<n>] | [za=<n>]}]
[bc.value= {
[dx=<n>] | [dy=<n>] | [dz=<n>] | [pressure=<n>] |
[pfx=<n>] | [pfy=<n>] | [pfz=<n>] |
[rx=<n>] | [ry=<n>] | [rz=<n>] }]
[point.coord= {<n> <n> <n>}]
[number=<n>]
[sxx] [syy] [szz] [sxy] [syz] [szx] [hs] [pr] [ps] [vms]
[print.anisotropic.matrix] [print.equiv.Poisson.matrix]
[print.equiv.YoungsMod.x] [print.equiv.YoungsMod.y]
[print.equiv.YoungsMod.z]
[print.equiv.ShearMod.xy] [print.equiv.ShearMod.yz]
[print.equiv.ShearMod.zx][
A: Commands
stressdata
1152 Sentaurus™ Process User Guide
I-2013.12
[apply.dislocation]
[name=<c>][saveTDR]
[dislocation.origin= {<n> <n> <n>}]
[para.orient= {<n> <n> <n>}]
[perp.orient= {<n> <n> <n>}]
[opt.maxiter=<n>] [opt.mindnrm=<n>] [opt.mindssq=<n>] [opt.tolerance=<n>]
[optimize.dislocation]
[origin.max= {<n> <n> <n>}]
[origin.min= {<n> <n> <n>}]
Arguments
<material>
Material in which the stress parameters are to be set.
region
Region where the stresses are to be applied.
sxxi, syyi, szzi, sxyi, syzi, szxi
Intrinsic stresses. Default unit: .
Arguments: Width-dependent Intrinsic Stress
base
Base width. Default unit: .
sxx1, syy1, szz1
Scale factors in linear width-dependent intrinsic stress.
sxx2, syy2, szz2
Scale factors in natural logarithmic width-dependent intrinsic stress.
Arguments: Boundary Conditions
bc.location
Specifies the area where the boundary conditions are applied.
Left | Right | Front | Back | Bottom refer to the outer boundary surfaces of
the simulation domain.
dyn/cm2
µm
A: Commands
stressdata
Sentaurus™ Process User Guide 1153
I-2013.12
bc.rotation.axis
Specifies the coordinates of the point around which rotational boundary conditions are
applied. Default unit: cm.
bc.value
Specifies the boundary condition types and values. The type can be:
•dx/dy/dz for the displacement rate (default unit: cm/s).
•pressure for pressure (default unit: ).
•pfx/pfy/pfz for point force (default unit: dyne).
•rx/ry/rz for rotational velocity (default unit: rad/s).
point.coord
Specifies the location where the point force is applied. Defualt unit: .
Arguments: Maximum Stress List
number
Specifies the number of the largest stress values to report.
sxx, syy, szz, sxy, syz, szx, hs, pr, ps, vms
Specifies from which stress component (sxx, syy, szz, sxy, syz, szx) or which derived
stress (vms is the von Mises stress, ps is the principal stress, hs is the hydrostatic stress,
and pr is the pressure) to extract the maximum stress values. Values for stress components
and principal stresses are computed at element centroid, while values for von Mises
stresses, hydrostatic stresses, and pressures are computed at nodes.
NOTE To extract maximum principal stresses, use:
pdbSet Mechanics Calculate.Principal.Stress 1
Arguments: Anisotropic Material Matrix
print.anisotropic.matrix
Prints the anisotropic material matrix.
print.equiv.Poisson.matrix
Prints equivalent Poisson’s ratio matrix.
dyn/cm2
µm
A: Commands
stressdata
1154 Sentaurus™ Process User Guide
I-2013.12
print.equiv.YoungsMod.x
Prints equivalent Young’s modulus in the x-direction.
print.equiv.YoungsMod.y
Prints equivalent Young’s modulus in the y-direction.
print.equiv.YoungsMod.z
Prints equivalent Young’s modulus in the z-direction.
print.equiv.ShearMod.xy
Prints equivalent shear modulus in the xy plane.
print.equiv.ShearMod.yz
Prints equivalent shear modulus in the yz plane.
print.equiv.ShearMod.zx
Prints equivalent shear modulus in the zx plane.
Arguments: Edge Dislocation
apply.dislocation
Indicates that an edge dislocation will be defined.
name
Specifies the name of the edge dislocation.
dislocation.origin
Specifies the location of the dislocation core.
para.orient
Specifies the direction of the edge dislocation.
perp.orient
Specifies the Burger’s vector in the perpendicular direction to the half plane. The
magnitude is the slip distance.
saveTDR
Saves the current defined edge dislocation to a TDR file for visualization. This should be
specified with the edge dislocation definition.
A: Commands
stressdata
Sentaurus™ Process User Guide 1155
I-2013.12
Arguments: Edge Dislocation–induced Strain Energy Minimization
opt.maxiter
Specifies the maximum number of iterations allowed in the optimization loop. Default:
500.
opt.mindnrm
Specifies the change in norm of the parameter vector for convergence. Default: 5e-3.
opt.mindssq
Specifies the relative change in the sum of the squares for convergence. Default:1e-5.
opt.tolerance
Specifies the tolerance of target errors. Default: 1e-3.
optimize.dislocation
Switches on the elastic strain energy minimization of edge dislocations defined
with !apply.dislocation.
origin.max
List of numbers defining the x-, y-, and z-coordinates of the lower-right front corner of the
bounding box for the location of the edge dislocation core.
origin.min
List of numbers defining the x-, y-, and z-coordinates of the upper-left back corner of the
bounding box for the location of the edge dislocation core.
Description
This command provides stress analysis parameters for input and output. Zero is the default
value for all intrinsic stress parameters. Wherever possible, use the deposit command with
specified stresses to apply intrinsic stresses.
Examples
Set the yy component of the intrinsic stress in nitride to :
stressdata nitride syyi= 1.4e10
1.4 10
×10 dyn/cm2

A: Commands
StressDependentSilicidation
1156 Sentaurus™ Process User Guide
I-2013.12
StressDependentSilicidation
Enables stress-dependent silicidation for a specified silicide.
Syntax
StressDependentSilicidation <silicide>
Arguments
<silicide>
Name of the silicide that will use the pressure-dependent model.
Description
In the model, reaction rates are normal stress dependent. The diffusivity of the reactant species,
silicon, (represented by the field SiliconReact) is pressure dependent, and the silicide is
allowed to relax.
Examples
Enables stress-dependent silicidation for nickel silicide:
StressDependentSilicidation NickelSilicide
See Also
Stress-dependent Silicidation on page 637

A: Commands
strip
Sentaurus™ Process User Guide 1157
I-2013.12
strip
Completely removes a layer exposed to the top gas region.
Syntax
strip <material> [remesh]
Arguments
<material>
The specified material, if exposed, is completely removed. For information about
specifying materials, see Material Specification on page 52.
remesh
By default, the structure is remeshed in two dimensions after strip. Setting !remesh
prevents remeshing. In three dimensions, the boundary is changed without generating a
mesh, so this argument has no effect in three dimensions. Preventing remeshing can save
time for very large structures.
Description
In two dimensions, the mesh is regenerated immediately. In three dimensions, only the
boundary is modified and the mesh is regenerated later when necessary.
Examples
Remove all oxide regions exposed to the top gas region:
strip oxide

A: Commands
struct
1158 Sentaurus™ Process User Guide
I-2013.12
struct
Writes files containing the structure or the mesh and solutions.
Syntax
struct
[Adaptive] [alt.maternames] [binary] [bnd]
[bndfile=<c>] [compress] [contacts] [datfile=<c>]
[dfise=<c> | tdr=<c>]
[FullD] [Gas] [grdfile=<c>] [interfaces]
[mshcmd] [pdb] [pdb.only]
[sat] [satfile=<c>] [scale=<n>]
[simplify=<n>] [tdr.bnd]
[visual.1D]
([x=<n>][<m>|<cm>|<um>|<nm>]
[y=<n>][<m>|<cm>|<um>|<nm>]
[z=<n>][<m>|<cm>|<um>|<nm>])
Arguments
Adaptive
In three dimensions, meshing is delayed until it is needed; to save a file, a mesh may need
to be created. Adaptive controls whether adaptive meshing is used. The default value is
the return of pdbGet Grid Adaptive.
alt.maternames
Saves alternative material names to DF–ISE or TDR format files. If you choose an
alternative name for a material using mater alt.matername or region
alt.matername, the alternative material name is used in the file when this argument is
specified. When writing DF–ISE files, the original material name is not available upon
loading. In TDR files, the material name also is stored in the file, so both the proper
material names and the alternative names are restored when loading a TDR file.
binary
Uses binary (not the default compressed ASCII) format when writing DF–ISE files.
bnd
Saves a boundary file in two dimensions and three dimensions along with the DF–ISE or
TDR file. Default: false.
A: Commands
struct
Sentaurus™ Process User Guide 1159
I-2013.12
bndfile, datfile, grdfile
Specify the corresponding file names separately.
compress
Writes compressed or uncompressed DF–ISE files. Default: true.
contacts
Writes contacts defined in the contact command into the boundary file. Default: true.
dfise
Saves files in DF–ISE format. The extensions _fps.grd and _fps.dat are added
automatically. Therefore, if dfise=filename is specified, Sentaurus Process saves the
files filename_fps.grd and filename_fps.dat.
The coordinate system for Sentaurus Process differs from the coordinate system in the
DF–ISE files. In Sentaurus Process, the x-direction is always perpendicular to the substrate
surface and the positive direction increases with depth into the substrate (the negative
direction is up). The y-direction and z-direction are parallel to the initial substrate surface
in two dimensions and three dimensions, respectively. In DF–ISE files, different coordinate
systems are used for 1D, 2D, and 3D simulations. In two dimensions, x is parallel to the
initial substrate surface and negative-y points up. In the 3D DF–ISE coordinate system,
positive-z is up, and x and y are parallel to the initial substrate surface. The appropriate
coordinate transformation is applied by default. To change the coordinate rotation, use the
math command.
FullD
If FullD is specified, the mesh is extruded to the maximum dimension allowed in the
simulation temporarily before saving the file. After saving the file, the simulation continues
in the same dimension as before.
If !FullD is specified, the saved files contain mesh and data in the dimension currently
used in the simulation.
NOTE When TDR restart files are saved, by default, no extrusion is performed.
Gas
By default, Sentaurus Process writes regions of material gas to DF–ISE, .bnd, and TDR
files. If !Gas is specified, regions of material gas are not saved.
interfaces
Saves interface data in DF–ISE and TDR files. Specify !interfaces to prevent storing
interface data. Default: true.
A: Commands
struct
1160 Sentaurus™ Process User Guide
I-2013.12
mshcmd
When specified with tdr, mshcmd writes a .cmd file with refinement information readable
by Sentaurus Mesh.
pdb
Saves pdb parameters along with the geometry and data in a TDR file.
pdb.only
Saves only pdb parameters (without geometry and data) in a TDR file.
sat
Enforces or prevents the saving of a Sentaurus Structure Editor restart file.
satfile
When using Sentaurus Structure Editor for 3D geometry-modeling steps, a Sentaurus
Structure Editor restart file is saved by default. The argument satfile defines the file
name. The default file extension is .sat.
When saving a 2D or 3D boundary file in .bnd or.tdr files, the extracted geometry is
simplified before saving to file. The double parameter simplify defines the maximum
deviation of the simplified boundary from the extracted geometry.
scale
The coordinates are multiplied by the specified value before writing them to the file. The
default is 1.0e4, which converts Sentaurus Process internal standard units (cm) to DF–ISE
units ( ).
simplify
When saving a 2D or 3D boundary file in .bnd or.tdr files, the extracted geometry is
simplified before saving to file. The double parameter simplify defines the maximum
deviation of the simplified boundary from the extracted geometry.
tdr
Saves a file in TDR format. The extension _fps.tdr is added automatically. By default,
all modifications to the parameter database are written to the TDR file to support splitting
and restarting simulations.
The coordinate system in TDR files is the same as DF–ISE files.
By default, TDR files can be used to split and restart simulations. Coordinates and field
values are stored with their unscaled internal values.
µm
A: Commands
struct
Sentaurus™ Process User Guide 1161
I-2013.12
If !Gas or!interfaces is specified, coordinates and field values are scaled to the
DF–ISE units, and information required for restarting is omitted.
For information about the TDR format, refer to the Sentaurus™ Data Explorer User
Guide.
tdr.bnd
Writes a TDR file that contains only the boundary representation.
visual.1D
Applies only to 1D simulations. If specified, Sentaurus Process orders the nodes when
writing them in a TDR file, so that the file can be easily visualized with Tecplot SV.
x, y, z
Specify a cutline for up to a 3D solid, so that a 1D TDR file is stored. For 1D simulations,
none of these arguments is required. For 2D simulations, one is required. For 3D
simulations, two are needed. Since the file is stored in TDR format, the tdr argument must
be used together with these arguments.
Description
This command writes the structure and the simulation mesh and field data to one or several
files. The data saved is from the current set of solution values.
Examples
Save the DF–ISE files output_fps.grd.gz and output_fps.dat.gz:
struct dfise= output
Write a file for device simulation (output_fps.tdr):
struct tdr= output !Gas
Write a TDR file with the current simulation mesh and data. By default, a restart file is written:
struct tdr= output
Write two files (output_fps.tdr and output_bnd.tdr):
struct tdr= output bnd
Write one file output_fps.bnd with the boundary representation in DF–ISE format:
struct bndfile= output

A: Commands
substrate_profile
Sentaurus™ Process User Guide 1163
I-2013.12
substrate_profile
Defines the impurity profile in the substrate.
Syntax
substrate_profile
<material> species=<c>
xcoord= <list>
concentration= <list>
Arguments
concentration
Numeric list of concentrations of the impurity at the specified xcoord.
<material>
Substrate material where the impurities will to be defined.
species
Name of the impurity.
xcoord
Numeric list of coordinates in x-direction where the concentration will be defined.
Description
This command defines the profile of a species in a substrate in a piecewise linear manner. The
piecewise linear function is given by the concentration corresponding to xcoord.
Examples
Defines the germanium profile in silicon substrate as a piecewise linear function:
substrate_profile Silicon species= Germanium \
xcoord= {0 0.01 0.011 0.5 0.7 10} \
concentration= {1e10 1e10 2e22 2e22 1e10 1e10}
See Also
transform on page 1177

A: Commands
tclsel
1164 Sentaurus™ Process User Guide
I-2013.12
tclsel
Selects the plot variable for the postprocessing routines.
Syntax
tclsel
[list] [<material>] [name=<c>]
[store] [vec] [z=<c>]
Arguments
list
Returns a list of currently defined and named data fields. The real data fields are listed by
default. Vector data fields can be listed using vec. This returns a Tcl list for use with those
commands that require list variables.
<material>
Specifies the material to which the command applies. Different expressions for the data
field initialization in different materials can be used. For information about specifying
materials, see Material Specification on page 52.
name
Name of the new data field. Default: Z_Plot_Var.
This is used by all commands when a plot name is not specified. This is a powerful feature,
as solution fields also can be created.
store
Controls whether the data field is written into permanent storage when a structure file is
output. Default: false.
vec
Lists the vector data fields. Default: false.
z
Accepts a Tcl expression that are used to build a new data field. All valid Tcl expressions
can be used in the string. Existing data fields are defined as Tcl variables, and the
expression is evaluated node-by-node with the updated value of the variable. In general,
this argument must be enclosed in braces, so that variable substitution is performed when
the string is parsed.
A: Commands
tclsel
Sentaurus™ Process User Guide 1165
I-2013.12
Description
This command specifies the plot variable for almost all other plot commands. It is a companion
to the select command, but it differs from the select command in that it accepts any general
Tcl expression. Data fields are made into Tcl variables and can be accessed with standard Tcl
variable methods.
Examples
Select as the plot variable the base 10 logarithm of the arsenic concentration:
tclsel z= {log10($Arsenic)}
Select as the plot variable the phosphorus concentration minus a constant value of :
tclsel z= {($Phosphorus - 5.0e14)}
Select as the plot variable the difference between the phosphorus and an analytic profile. This
data field will be named Doping. The store argument indicates that the doping field must be
written into any saved structure files:
tclsel z= {($Phosphorus - 1.0e18 * exp ( $y * $y / 1.0e-8 ))} \
name= Doping store
List all available real and vector data fields:
tclsel list vec
See Also
All postprocessing commands
514
×10

A: Commands
temp_ramp
1166 Sentaurus™ Process User Guide
I-2013.12
temp_ramp
Defines a temperature profile for use with the diffuse command.
Syntax
temp_ramp
(clear | list | name=<c>)
[<ambient>]
[angles.factors= {
[<interface_mat1>= <list>]
[<interface_mat2>= <list>] }]
[auto.doping= <list>]
[coeffs= {<A0> <A1> <A2> ... <An>}]
[crystal.rate= {"<100>"=<n> "<110>"=<n> "<111>"=<n>}]
[current.time=<n>][<hr>|<min>|<s>]
[delNT=<n>[<C>|<K>] | delT=<n>[<C>|<K>]]
[density.increase= <regionName>=<n> | <material>=<n>]
[deposit.type=<c>]
[epi.doping= <list>] [epi.doping.final= <list>]
[epi.layers=<i>] [epi.model=<i>]
[epi.resist= { [<dopant1=<n>[<ohm-cm>]] [<dopant2=<n>[<ohm-cm>]] ... }]
[epi.thickness=<n>][<m>|<cm>|<um>|<nm>]
[flow<ambient>=<n>][<l/min>]
[flows= {
[<ambient1>=<n>][<l/min>]
[<ambient2>=<n>][<l/min>]
}]
[gas.flow=<c>]
[hold] [ISSG] [last]
[mat.coeffs= {
<mat1>= {<A0> <A1> <A2> ... <An>}
<mat2>= {<A0> <A1> <A2> ... <An>}
...
<matn>= {<A0> <A1> <A2> ... <An>} }]
[p<ambient>=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[partial.pressure= {
[<ambient1>=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa> |<dyn/cm2>]
[<ambient2>=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa> |<dyn/cm2>]
...}]
[pressure=<n>][<atm>|<GPa>|<MPa>|<KPa>|<Pa>|<dyn/cm2>]
[ramprate=<n>][<C/s>|<K/s>|<C/min>|<K/min>]
[read.temp.file=<c>]
[repair]
[reset.init.time]
[sources= {<beam1> <beam2> ... <beamn>}]
[t.final=<n>][<C>|<K>]
A: Commands
temp_ramp
Sentaurus™ Process User Guide 1167
I-2013.12
[temperature=<n>][<C>|<K>] |
[time=<n>][<hr>|<min>|<s>]
Arguments
<ambient>
Shorthand specification to set the ambient partial pressure the same as the total pressure. If
an ambient is specified this way, it must be the only ambient set in the temp_ramp
command.
angles.factors
Specifies interface-specific anisotropic epi growth rate factors. This argument specifies a
piecewise linear growth rate factor versus angle for each growing interface (the factors
must be between 0 and 1). For example, to create a silicon facet and a polysilicon
facet, specify:
angles.factors= {
EpiOnSilicon_Gas= {0.0 1.0 25.0 1.0 30 0.0}
EpiOnPolySilicon_Gas= {0.0 1.0 35.0 1.0 40 0.0}
}
auto.doping
String list of species for which the auto-doping model will be switched on during epitaxial
growth.
clear
Clears the global list of temperature ramps. When defining profiles, the action is to unite
the new definition with any prior profiles of the same name.
coeffs
List of single-material coefficients used in Fourier deposition when
epi.model=1 and deposit.type=fourier.
crystal.rate
List of etching rates defined per crystallographic direction in the format:
{"<100>"=<dep_rate> "<110>"=<dep_rate> "<111>"=<dep_rate>}
used for crystallographic deposition when epi.model=1 and deposit.type=crystal.
current.time
Returns the value of the ramp for the given time.
30°
40°
A0A1…An
,,,
A: Commands
temp_ramp
1168 Sentaurus™ Process User Guide
I-2013.12
delNT
Defines the maximum temperature step during a temperature ramp-down if specified.
Default unit: degree Celsius. It also can be defined globally with the command:
pdbSet Diffuse delNT {<n>}
delT
Defines the maximum temperature step during a temperature ramp-up if specified. Default
unit: degree Celsius. It also can be defined globally with the command:
pdbSet Diffuse delT {<n>}
density.increase
Applies densification model where the density increases in percentage. The increase value
can be specified per region <regionName>=<n> (where regionName is the name of an
existing region in the current structure) or per material <material>=<n> (where
material is the name of a material in the current structure).
deposit.type
When using epi.model=1, epitaxy is solved as a series of alternating deposition and
diffuse steps. This argument specifies the deposit type, and the allowed values are:
•isotropic (default)
•fourier (in which case, either coeffs or mat.coeffs must be specified as well)
•crystal (in which case, crystal.rate must be specified as well)
epi.doping
List of parameters where the parameter name is the name of the species to be initialized,
and the value is the initial value. A list of fields of any name can be initialized with this
argument. For solution variables, units are accepted, for example:
epi.doping= {boron= 1e18<cm-3> GSize= 1<nm> myfield= 1}
epi.doping.final
List of parameters where the parameter name is the name of the species to be initialized,
and the value is the final value. A list of fields of any name can be initialized with this
argument. For solution variables, units are accepted, for example:
epi.doping.final= {boron= 1e18<cm-3> GSize= 1<nm> myfield= 1}
epi.layers
Number of layers of mesh lines required during epitaxial growth (for epi.model=0).
Default: 40. You also can set globally a distance between mesh lines using:
A: Commands
temp_ramp
Sentaurus™ Process User Guide 1169
I-2013.12
pdbSet Silicon Grid epi.perp.add.dist <n>
If epi.perp.add.dist is set to a positive number, epi.layers is ignored.
epi.model
Select the method for epitaxial growth:
•epi.model=0 (default) applies a boundary-moving algorithm similar to oxidation.
•epi.model=1 uses alternating doped deposition and inert annealing steps.
epi.resist
List of parameters with dopant name and resistivity to calculate the background dopant
concentration. If more than one dopant name appears in the list, the doping concentration
is calculated individually for each dopant by ignoring the other ones.
epi.thickness
Sets the epitaxial layer thickness to be deposited. Default unit: .
flow<ambient>, flows
List of gas flows in the reaction chamber. The gas flows are used to compute the partial
pressures of the active ambients (those causing material growth). You can specify flows
using either a parameter name composed of flow + <ambient> (for example, flowO2 and
flowHCl where O2 and HCl are ambient names) or flows that takes a list of parameters
with the names of the ambients, for example:
flows= {O2= 1.0<l/min> HCl= 1.0<l/min>}
Table 64 on page 617 lists the available ambients, but this list can be extended by using the
ambient command. When a gas flow is specified as a combination of flows (and not when
using partial pressures), a complete reaction of the ambients is assumed to occur, for
example, O2 + 2H2 -> 2H2O. Besides gas reactions, the addition of inert gases will change
the partial pressure of the material-growing ambients. For example, if the flows of only N2
and O2 are specified and are equal, the partial pressure of O2 will be <total pressure>/
2.0 where <total pressure> is given by pressure.
NOTE Flows and partial pressures must not be specified in the same
temp_ramp command.
gas.flow
Specifies a gas flow to be used for this temp_ramp (must not be used with other gas_flow
arguments).
µm
A: Commands
temp_ramp
1170 Sentaurus™ Process User Guide
I-2013.12
hold
During this segment of the temperature ramp, hold gives the diffuse command the
opportunity to specify the time of the segment.
ISSG
Switches on in situ steam-generated (ISSG) oxidation.
last
Defines the final component of the temperature profile. There will be no more additions to
the ramp.
list
Generates a list of temperature profiles. It returns a Tcl list and can be operated on as such.
The default action for commands is to print the return, so if no handling is required, this
prints a list of names of defined temperature profiles. If a name is specified, the
temp_ramp command only is listed along with details about the ramps.
mat.coeffs
List of multimaterial coefficients used in Fourier deposition when
epi.model=1 and deposit.type=fourier.
name
Name used to identify the temperature ramp. Use this name in a subsequent diffuse
command.
p<ambient>, partial.pressure
List of the partial pressures of active ambients. Partial pressure specifications must not be
used with flows, flow<ambient>, or pressure specifications. Partial pressures can be
specified using either a parameter name composed of p + <ambient> (for example, pO2
and pN2O where O2 and N2O are active ambient names) or partial.pressure that takes
a list of parameters with the names of the ambients, for example:
partial.pressure= {O2= 1.0<atm> N2O= 1.0<atm>}
Table 64 on page 617 lists the available ambients, but this list can be extended by using the
ambient command. These partial pressures are assumed to contribute to the oxidation or
user-defined reaction processes. No reaction between the species is assumed. Default
unit: atm.
NOTE Only the partial pressures of the active ambients are used directly in the
oxidation reaction equations. Therefore, setting the partial pressure of
A0A1…An
,,,
A: Commands
temp_ramp
Sentaurus™ Process User Guide 1171
I-2013.12
inactive (in the sense that they cause a material growth reaction)
ambients, such as N2 and HCl, has no effect.
pressure
The (total) pressure of the ambient gas. Default value and unit: 1.0 atm. This setting takes
effect only if flows or flow<ambient> is defined explicitly. If gas.flow is specified,
the pressure is set in the corresponding gas_flow command.
ramprate
Temperature change during anneal. Default value and unit: .
read.temp.file
Reads a thermal profile from a file. It must not be used with any other thermal specification.
To create this profile file during laser annealing, use write.temp.file of the diffuse
command. The format of the file is two columns: time (in seconds) and temperature (in
degree Celsius). Lines beginning with a hash (#) are ignored.
repair
In MGOALS3D mode, small regions are removed automatically by default. Sometimes,
this causes small gas bubbles in the structure or other problems. Use !repair to switch
off removal of small regions.
reset.init.time
Starts each annealing step with the same initial time step.
sources
Defines deposition sources used in Fourier deposition when epi.model=1 and
deposit.type=fourier.
t.final
Final temperature for a temperature ramp-up or ramp-down. It is used if ramprate is not
given. The ramp time is calculated automatically.
temperature
Annealing temperature. Default unit: degree Celsius.
time
Annealing time. Default unit: minute.
0.0°C/s
A: Commands
temp_ramp
1172 Sentaurus™ Process User Guide
I-2013.12
Description
This command specifies multiple-step temperature ramps and holds. It can be used to construct
a complex temperature sequence to be simulated with the diffuse command (by specifying
temp.ramp of the diffuse command).
All gas_flow command arguments are available with the temp_ramp command. Table 64 on
page 617 lists the available ambients and includes O2, H2O, HCl, N2, H2, and N2O. To specify
epi, an epi-type ambient must be used. By default, two are available: Epi and LTE. For more
information, see Epitaxy on page 282.
Examples
Define the temperature profile named tr1 with a temperature rate of 10 K/s:
temp_ramp name= tr1 temp= 20 ramprate= 10<K/s> time= 100<s>
See Also
diffuse on page 908
gas_flow on page 935

A: Commands
term
Sentaurus™ Process User Guide 1173
I-2013.12
term
Defines a new subexpression for use in the equation specification of the Alagator language.
Syntax
term
[add] [clear] [delete] [eqn=<c>] [list]
[<material>]
[name=<c>] [print] [store]
Arguments
add
Creates a new term. A term with that name will be overwritten.
clear
Removes a term from the current set if the term exists. Otherwise, it clears the content of
all terms.
delete
Removes a term from the current set.
eqn
The string defines the equation part of the term. The equation must conform to all the
standard constraints of the Alagator language. Terms can be nested; the equation specified
here can refer to other terms. Parsing of the equation is performed during diffusion, so there
is no need for everything to be predefined.
list
Lists all the names of the current terms. This is returned as a Tcl list, so it can be used in
conjunction with all the list functionality.
<material>
If a material is specified, the term becomes specific for this material only. This allows the
same name to have different equations in different materials. For information about
specifying materials, see Material Specification on page 52.
name
Reference name for the term. This name is defined and is compared to strings found in the
equation parsing. Capitalization is important, and only exact matches are allowed.
A: Commands
term
1174 Sentaurus™ Process User Guide
I-2013.12
print
Prints the equation for the term matching the name specified. If no term matches, 0.0 is
returned. If the material name is not given, the first term with the matching name is returned
(for example, you may obtain VTotal in oxide instead of silicon).
store
Allows the term command to be stored in a TDR file.
Description
Terms are never required but can offer substantial computational benefit. Each term is
evaluated only once during assembly, and the results are cached. If multiple equations refer to
a term, the first equation to use it evaluates the expression and the remainder use the cached
values. Terms are usually used for expressions that need to appear in several partial differential
equations.
For example, a recombination term between vacancies and interstitials must appear in both the
vacancy and interstitial equation. A term can be used for the recombination and can be placed
in both partial differential equations. The recombination is then evaluated only once during the
assembly process.
Terms can be created, searched, and printed, which allow inquiries about terms to be made in
the various callback procedures. For example, the charge term in the Poisson equation can be
accumulated by obtaining the current charge and adding new pieces to the term.
Examples
Create a term named VTotal in silicon only. The keyword VTotal will be replaced with the
subexpression Vacancy+VacancyGbc:
term name= VTotal add silicon eqn= "Vacancy+VacancyGbc"
Create a term named Noni in silicon only. The equation will be the exponential of Potential
multiplied by $Vti. The normal rules for Tcl string variables and executions apply, so that Vti
must be a currently defined variable. The value will be replaced when the term command is
executed:
term name= Noni add silicon eqn= exp(Potential*$Vti)
This is the same as the previous example. The difference is the braces around the equation,
which delay variable expansion. The variable will not be expanded until the diffuse
command is executed. This is the more normal form. You want the Vti variable to be replaced
A: Commands
term
Sentaurus™ Process User Guide 1175
I-2013.12
with the value of the current temperature of the diffuse command, not the temperature at the
time of the term command execution:
term name= Noni add silicon eqn= {exp(Potential*$Vti)}
Return a list of all the current term names defined:
term list
Return the currently defined equation corresponding to the name Charge:
term name= Charge print
See Also
solution on page 1145

A: Commands
topo
1176 Sentaurus™ Process User Guide
I-2013.12
topo
Performs 3D etching and deposition using Sentaurus Topography 3D.
Syntax
topo {<Sentaurus Topography 3D commands>}
Arguments
<Sentaurus Topography 3D commands>
All arguments of the topo command are described in the Sentaurus™ Topography 3D
User Guide.
Description
Physical etching and deposition are available through the interface to Sentaurus
Topography 3D and are executed using the topo command.
Commands entered into the topo command are passed directly to the Sentaurus
Topography 3D library. The exchange of the boundary between Sentaurus Process and
Sentaurus Topography 3D is handled automatically and only when required.
NOTE A licence for Sentaurus Topography 3D must be available, and a version
of the Sentaurus Process binary with Sentaurus Topography 3D
included must be installed.

A: Commands
transform
Sentaurus™ Process User Guide 1177
I-2013.12
transform
Reflects, stretches, cuts, flips, rotates, or translates a structure.
Syntax
transform
(cut | flip | reflect | rotate | stretch)
[Adaptive] [keep.original] [mesh.align] [remesh]
[angle=<n> axis= "X" | "Y" | "Z"]
[length=<n>][<m>|<cm>|<um>|<nm>]
[[location=<n>][<m>|<cm>|<um>|<nm>]
(left | right | front | back | up | down) | (ymin | ymax | zmin | zmax)]
[max= {
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]}
min= {
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]}]
[translate= {
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]}]
Arguments: cut
Adaptive
If specified, Adaptive switches on adaptive meshing if remesh is given. Parameters for
adaptive meshing are described in Adaptive Refinement on page 699. The default is the
return value of pdbGet Grid Adaptive.
cut
Crops the structure to a new bounding box (using max and min) or crops half the structure
(using left, right, front, back, up, or down).
left, right, front, back, up, down
Indicates a cut at a location given by the location argument, and specifies which half is
to be removed. These arguments must not be used with max and min.
A: Commands
transform
1178 Sentaurus™ Process User Guide
I-2013.12
location
Specifies the x-, y-, or z-coordinate (in the internal coordinate system) where the cut will
be made. Default: . The location argument is used with left, right, front,
back, up, or down to indicate which direction and side to cut.
max, min
The cut box can be specified by either:
•Both the
max and min arguments:
max= {maxx maxy maxz} min= {minx miny minz}
•One of left, right, front, back, up, down to specify an axis-aligned cut at the
coordinate given by location.
NOTE The max and min arguments must be used together. Do not use them
with any of the left, right, front, back, up, or down arguments.
mesh.align
By default, MGOALS cuts the structure at the nearest mesh line and does not perform a
remesh. If !mesh.align is specified, MGOALS cuts precisely at the specified
coordinates and remeshes the structure.
remesh
Available only for two dimensions. Forces a remesh after the transformation. However,
remeshing is always possible using the grid remesh command if required.
Arguments: flip
Adaptive
If specified, Adaptive switches on adaptive meshing. Parameters for adaptive meshing
are described in Adaptive Refinement on page 699. The default is the return value of
pdbGet Grid Adaptive.
flip
Selects the flip operation (flips from top to bottom). See The transform flip Command and
Backside Processing on page 776.
location
Selects the x-coordinate about which the structure will be flipped. By default, the middle
of the structure is chosen. Subsequent transform flip commands will, by default, use
the same location for flipping whether the default is used or a chosen location is used.
In three dimensions, the z-coordinate of the rotation is the middle of the structure in the
0.0 µm
A: Commands
transform
Sentaurus™ Process User Guide 1179
I-2013.12
z-direction. The location of the flip is also the fixed coordinate for mechanics simulations,
which is otherwise at the bottom of the structure when no flip has occurred.
Arguments: reflect
Adaptive
If specified, Adaptive switches on adaptive meshing. Parameters for adaptive meshing
are described in Adaptive Refinement on page 699. The default is the return value of
pdbGet Grid Adaptive.
keep.original
Retains the original structure after reflection (having both the original and the reflected
structure), or stores only the reflected structure when disabled with !keep.original.
Default: true.
left, right, front, back
Selects the side of the simulation domain at which the reflection is performed.
reflect
Indicates that a reflection will be performed.
ymin, ymax, zmin, zmax
Specify the location where the reflection is performed:
•ymin is the same as left.
•ymax is the same as right.
•zmin is the same as back.
•zmax is the same as front.
Arguments: rotate
Adaptive
If specified, Adaptive switches on adaptive meshing. Parameters for adaptive meshing
are described in Adaptive Refinement on page 699. The default is the return value of
pdbGet Grid Adaptive.
angle
Rotation angle. It must be one of 90, 180, or 270. Angles leading to structures having gas
in a side are not allowed. This means that only is allowed for y- and z-axes. Two-
dimensional simulations are extruded into three dimensions and are then rotated.
180°
A: Commands
transform
1180 Sentaurus™ Process User Guide
I-2013.12
axis
Rotation axis. It must be x, y, or z.
rotate
Indicates that a rotation will be performed.
Arguments: stretch
Adaptive
If specified, Adaptive switches on adaptive meshing. Parameters for adaptive meshing
are described in Adaptive Refinement on page 699. The default is the return value of
pdbGet Grid Adaptive.
left, right, front, back
Indicates which side of the structure will be moved.
length
Length of stretching. Default value and unit: .
location
The y- or z-coordinate (in the internal coordinate system) where the structure will be
stretched. Default value and unit: .
remesh
Specifies that a remesh will be performed. Default: true.
stretch
Indicates that a stretch operation will be performed.
Arguments: translate
translate
Translates the entire structure by specifying a translation vector:
translate= {translate_x translate_y translate_z}
Description
Previously, the cut and clip commands had slightly different behavior. Now, they are the
same and are referred to as the cut command. All operations work in both two and three
dimensions.
0µm
0µm
A: Commands
transform
Sentaurus™ Process User Guide 1181
I-2013.12
All these transformations, except flip and stretch, are also available in the KMC mode.
Examples
Both commands reflect the structure to the right side:
transform reflect right
transform reflect ymax
Reflect the structure to the right side and keep the reflected part only:
transform reflect right !keep.original
Stretch the structure to the right side. The structure left of 0.7 remains unchanged; the structure
to the right of 0.7 will be moved by 20 nm:
transform stretch location= 0.7 length= 0.02 right remesh
Cut the structure at y=0.7. The left part will be removed without remeshing:
transform cut location= 0.7 left !remesh
Cut the structure at x between and , and y between and :
transform cut min= {0<um> 0<um>} max= {1<um> 3<um>}
Shift the structure up in the x-direction by :
transform translate= {-1 0 0}
Flip the structure from top to bottom about its midpoint if it is the first flip, or store the flip
location for subsequent flips:
transform flip
Rotate the structure in the x-axis:
transform rotate axis= X angle= 90
See Also
Stress Handling on page 757
mgoals on page 1037
0µm
1µm
0µm
3µm
1µm
90°

A: Commands
transform.refinement
1182 Sentaurus™ Process User Guide
I-2013.12
transform.refinement
Reflects, stretches, cuts, flips, rotates, or translates a given refinement box or all refinement
boxes.
Syntax
transform.refinement
(cut | flip | reflect | rotate | stretch)
[angle=<n> axis= "X" | "Y" | "Z"]
[keep.original]
[length=<n>][<m>|<cm>|<um>|<nm>]
[name=<c>] [name.new=<c>]
[[location=<n>][<m>|<cm>|<um>|<nm>]
(left | right | front | back | up | down) | (ymin | ymax | zmin | zmax)]
[max= {
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]}
min= {
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]}]
[translate=
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]
<n>[<m>|<cm>|<um>|<nm>]}]
Arguments: General
keep.original
Specifies whether to keep the original after the transformation. When keeping the original
refinement, the original is untouched, and a new one is created by transforming the original
refinement. Otherwise, the refinement itself is transformed.
name
Name of the refinement to apply the transformation.
name.new
Name of the transformed refinement. If not specified, a default name is given.
A: Commands
transform.refinement
Sentaurus™ Process User Guide 1183
I-2013.12
Arguments: cut
cut
Crops the refinement to a new bounding box (using max and min) or crops half of it (using
the left, right, front, back, up, or down arguments).
left, right, front, back, up, down
Indicates a cut at a location given by location, and specifies which half is to be removed.
These arguments must not be used with max and min.
location
Specifies the x-, y-, or z-coordinate where the cut is to be performed. The location
argument is used with left, right, front, or back to indicate which direction and side
to cut. Default: .
max, min
The cut box can be specified by either:
•Both the
max and min arguments:
max= {maxx maxy maxz} min= {minx miny minz}
•One of left, right, front, back, up, down to specify an axis-aligned cut at the
coordinate given by the location argument.
NOTE The max and min arguments must be used together. Do not use them
with any of the left, right, front, back, up, down arguments.
ymin, ymax, zmin, zmax
Specify the location where the refinement reflection is performed:
•ymin is the same as left.
•ymax is the same as right.
•zmin is the same as back.
•zmin is the same as front.
Arguments: flip
flip
Flips a refinement (top to bottom).
0.0 µm
A: Commands
transform.refinement
1184 Sentaurus™ Process User Guide
I-2013.12
location
Selects the x-coordinate about which the refinement will be flipped. By default, the middle
of the structure is chosen. Subsequent transform flip commands will, by default, use
the same location for flipping whether the default is used or a chosen location is used.
In three dimensions, the z-coordinate of the rotation is the middle of the structure in the z-
direction.
Arguments: reflect
left, right, front, back
Selects the side of the simulation domain at which the reflection is performed.
reflect
Indicates that a reflection will be performed.
Arguments: rotate
angle
Rotation angle. It must be 90, 180, or 270.
axis
Rotation axis. It must be x, y, or z.
rotate
Indicates that a rotation will be performed.
Arguments: stretch
left, right, front, back
Indicates which side of the refinement will be moved.
length
Length of stretching. Default value and unit: .
location
Specifies the y- or z-coordinate (in the internal coordinate system) where the refinement
will be stretched. Default value and unit: .
stretch
Indicates that a stretch operation will be performed.
0µm
0µm
A: Commands
transform.refinement
Sentaurus™ Process User Guide 1185
I-2013.12
Arguments: translate
translate
Translates the refinement by specifying a translation vector:
translate= {translate_x translate_y translate_z}
Description
A new transformed refinement box is created by default, while the old one is kept. This can be
overridden with !keep.original. The transformation applies to all existing refinements,
except if a name is specified. In this case, a transformed refinement name also can be specified
by using name.new.
Examples
Create a set of new refinements as reflections of all the current refinements to the right side:
transform.refinement reflect right
Create a new refinement called sbox1 by stretching the existing refinement box1 to the right
side. The area left of 0.7 remains unchanged; the structure to the right of 0.7 will be moved by
20 nm:
transform stretch location= 0.7 length= 0.02 right name= box1 name.new= sbox1
Cut all the existing refinements at y = 0.7. The left part will be removed:
transform cut location= 0.7 !keep.original
Create a new refinement new2 by copying and shifting r1 up in the x-direction by :
transform translate= {-1 0 0} name= r1 name.new= new2
Rotate the refinement refbox around the y-axis without changing its name:
transform rotate axis= "Y" angle= 270 name= refbox \
name.new= refbox !keep.original
See Also
Mesh Refinement on page 694
Stress Handling on page 757
refinebox on page 1101
transform on page 1177
1µm
270°

A: Commands
translate
1186 Sentaurus™ Process User Guide
I-2013.12
translate
Translates a named dataset with the specified offset.
Syntax
translate
[<material>]
[min=<n>]
[name=<c>]
[offset= {<n> <n> <n>}]
Arguments
<material>
If a material is specified, the dataset is translated in the specified material only. Otherwise,
the dataset is translated in all materials. For information about specifying materials, see
Material Specification on page 52.
min
Minimum value to fill the points with undefined value. Default: 0.0.
name
Name of a dataset. Default: Z_Plot_Var.
offset
List of numeric values, where the first, second, and third values in the list are taken as the
x-, y-, and z-value, respectively. The missing value is treated as zero.
Description
This command spatially shifts a profile (dataset) with the specified offset. If a material is
specified, the profile is shifted in the specified material only. Otherwise, the profile is shifted
in all materials. When a profile is shifted, the value at some points may become undefined, in
which case, these points are filled with a minimum value as specified by the min argument.
Examples
Shift the Boron data field with a shifting vector ( , , ):
translate name= Boron offset= {0.01 0.02}
0.01 µm
0.02 µm
0.0 µm

A: Commands
UnsetAtomistic
Sentaurus™ Process User Guide 1187
I-2013.12
UnsetAtomistic
Disables the atomistic KMC diffusion model and continues the simulation using the PDE
solver.
Syntax
UnsetAtomistic
[sano] [sano.list] [sano.materials]
Arguments
sano
Remeshes the Sentaurus Process finite-element mesh and converts KMC particles to finite-
element fields. To adaptively remesh on Sano fields and Sano-smoothed NetActive
(DopingConcentration), you must specify adaptive meshing parameters before
UnsetAtomistic.
Any adaptive criteria specified for a field that is in the Sano list will be applied to the Sano-
smoothed value of the field, and any criteria specified for NetActive will be applied by
default to NetActive computed from Sano-smoothed active fields. To set the list of Sano
fields, use sano.list, but by default the list contains the active dopants. The field
NetActive is updated automatically using Sano fields and does not need to be included
explicitly.
sano.list
Sets the list of Sano fields. These fields are converted from KMC particle distributions
using the Sano method and are used for adaptive remeshing, and subsequently converted to
finite-element fields on the newly created mesh. By default, the Sano list includes all active
dopants that are present. The field NetActive is updated automatically using Sano fields
and does not need to be included explicitly.
sano.materials
Sets the list of materials in which the Sano method is applied. By default, the list contains
only Silicon because this is the only material that by default has nontrivial KMC
diffusion models.
Description
This command transfers all information into the Sentaurus Process standard mesh (by calling
KMC2PDE), sets the atomistic mode to false, and deletes all atomistic-related information.
A: Commands
UnsetAtomistic
1188 Sentaurus™ Process User Guide
I-2013.12
Examples
Use the default conversion (closes mesh point) to convert discrete particles to continuum field
values:
UnsetAtomistic
Use the Sano method to convert phosphorus, arsenic, and boron particles to active
concentrations:
UnsetAtomistic sano sano.list= {PActive AsActive BActive}

A: Commands
UnsetDielectricOxidationMode
Sentaurus™ Process User Guide 1189
I-2013.12
UnsetDielectricOxidationMode
Disables the oxidation mode to grow oxide with dielectric on top.
Syntax
UnsetDielectricOxidationMode
<Dielectric> <Oxidant>
Arguments
<Dielectric>
Specifies the name of the dielectric material to grow oxide underneath.
<Oxidant>
Specifies the name of the oxidant.
Description
This command disables the dielectric oxidation mode of material <Dielectric> and ambient
<Oxidant>. It deletes all the dielectric oxidation-related callback settings, the boundary
conditions, and the parameter settings.
Examples
Disables oxide growth with nitride on top for O2 ambient:
UnsetDielectricOxidationMode Nitride O2
See Also
SetDielectricOxidationMode on page 1124

A: Commands
update_substrate
1190 Sentaurus™ Process User Guide
I-2013.12
update_substrate
Sets up the substrate with impurities, strains, and modified lattice constants for analyses that
involve strained silicon layers.
NOTE This command is deprecated.
Syntax
update_substrate
top.relaxed.coord=<n>
Arguments
top.relaxed.coord
Top of the region that is totally relaxed from the lattice strains. It can be considered to be
the top point above which no dislocations can be found. The top.relaxed.coord is the
lowest x-coordinate below which the wafer is totally relaxed from the impurity-related
strains.
Description
This command takes into account the strains that certain impurities introduce to the wafer as
defined by the strain_profile command or in the PDB, and sets up the lattice constants
and strains in the substrate as defined in the region command. This command applies to the
impurity profile of the substrate as defined by the profile command or the
substrate_profile command.
This command must be called only once for initialization. It is recommended to replace it with
a short solve step and set:
pdbSet Silicon Mechanics UpdateStrain 1

A: Commands
WritePlx
Sentaurus™ Process User Guide 1191
I-2013.12
WritePlx
Writes a 1D .plx file.
Syntax
WritePlx
<filename>
[<material>]
[x=<n>] [y=<n>] [z=<n>]
[include.interfaces]
[only.interfaces]
Arguments
<filename>
Name of the output file.
NOTE The <filename> argument must be the first argument on the
WritePlx command line.
include.interfaces
Includes interface values with the returned data.At an interface, the distance coordinate of
the 3 nodes (2 bulk and 1 interface) will be the same, and the interface value will be inserted
between the 2 neighboring bulk values.
<material>
If a material is specified, only the plot from the given material is created. For information
about specifying materials, see Material Specification on page 52.
only.interfaces
Returns interface values exclusively in the returned data. When specified, no bulk values
are returned.
x, y, z
Specify the cut position. For 1D simulations, no cut specification is necessary. For 2D
simulations, either x or y must be specified. For 3D simulations, two axes must be
specified. It is also possible to shift .plx output files by specifying PlxShift variables.
Default unit: .
µm
A: Commands
WritePlx
1192 Sentaurus™ Process User Guide
I-2013.12
Description
This command makes a 1D profile along a given cutline and writes a .plx file of the solutions
and terms given in the list provided by the SetPlxList command. If the list is not provided,
only present solution names are written. If a material is specified, only data from the given
material is used to create the plot.
NOTE The <filename> argument must be the first argument on the
WritePlx command line.
Examples
Write a 1D .plx file at the y = cutline:
WritePlx 1.5.plx y= 1.5
Shift the axis by and write a 1D .plx file:
set PlxShift 0.2
WritePlx test.plx
See Also
SetPlxList on page 1128
1.5 µm
0.2 µm
