Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 22

1
PanARGA
A Python Tool for Pan Antibiotic Resistance Genome Analysis
Yichen He
December 14, 2018

2
Contents
1. Introduction .................................................................................................... 3
2. What can PanARGA do ................................................................................ 3
3. Installation ...................................................................................................... 3
[1] Install Python3 v3.6+ for Win/Mac/Linux ............................................. 3
[2] Install Python3 accessory packages: ...................................................... 3
[3] Install blast v2.7.1+ for Win/Mac/Linux ............................................... 4
4. Modules of PanARGA ................................................................................... 4
[1] Preprocessing module .................................................................................. 4
[2] Gene identification modules ..................................................................... 4
[3] Analysis modules.......................................................................................... 4
5. Input files ....................................................................................................... 5
[1] Sequence files: ............................................................................................. 5
[2] Setting file ................................................................................................... 6
[3] Phenotype file: ............................................................................................. 7
[4] Annotation csv files: ................................................................................... 8
6. Usage and output files ................................................................................ 9
[1] Before running .............................................................................................. 9
[2] Running PanARGA ........................................................................................ 9
[3] Output files ................................................................................................. 12

3
1. Introduction
PanARGA is a platform independent Python3 tool used to analyze pan-
antibiotic resistance genome characteristics for multiple genomes. It
accepts various format of sequence files as input files. The identification
of antibiotic resistance genes is mainly based on the Comprehensive
Antibiotic Resistance Database (CARD) and ResFinder database.
2. What can PanARGA do
1) Antibiotic resistance genes (ARGs) identification
2) Pan-antibiotic resistance genome feature analysis
3) Classifying and counting for identified ARGs
4) Analysis of ARGs associated with given antibiotics
3. Installation
[1] Install Python3 v3.6+ for Win/Mac/Linux
(https://www.python.org/)
[2] Install Python3 accessory packages:
For PanARGA, several Python3 packages are required. Therefore, PIP,
(https://pypi.org/project/pip/) the PyPA tool for installing and managing
Python packages, is recommended to install firstly if you don’t have other
packages management tools. For details of pip documentations, please see
https://pip.pypa.io/en/stable/.
The packages required:
a) Biopython v1.7+ (https://biopython.org/)
b) NumPy v1.15+ (http://www.numpy.org/)
c) Pandas v0.23+ (http://pandas.pydata.org/)
d) SciPy v1.1+ (https://www.scipy.org/)
e) Matplotlib v3.0+ (https://matplotlib.org/)
f) Seaborn v0.9+ (http://seaborn.pydata.org/)
If you choose pip to install these packages, use “pip install XXX” for
each module to install in command line interface. For example, to install
Biopython module, try:
You can view all packages and their versions by:
pip install biopython
pip list

4
[3] Install blast v2.7.1+ for Win/Mac/Linux
Blast+ is available at https://blast.ncbi.nlm.nih.gov/Blast.cgi, you need
to modify the directory of where you install blast. For example, if the
blastn, blastp and makeblastdb programs are in C:/blast+/bin, please
change the directory of “blast+” in “settings.txt” like:
4. Modules of PanARGA
[1] Preprocessing module
1) CDSex.py: extracts coding sequences from GenBank files (only for files
with a “.gb” extension), forming both protein and nucleotide fasta files
(files with a “.faa” and a “.fna” extension).
[2] Gene identification modules
#these modules accept sequence files as input files
1) ArKmer.py/ResKmer.py: find resistance genes from raw reads FASTQ
files (files with a “.fastq” extension).
2) ArBlastn.py/ResBlastn.py: find resistance genes from nucleotide
sequence FASTA nucleic acid files (files with a “.fna” extension)
3) ArBlastp.py/ResBlastp.py: find resistance genes from protein sequence
FASTA amino acid files (files with a “.faa” extension)
[3] Analysis modules
#these modules accept annotation csv files genetated by “gene
identification modules” as input files.
1) Pangenome.py: analyze pan-ARgenome features, including analysis of
ARGs distribution and pan-ARgenome curve fitting.
2) PanAccess.py: classification and statistical analysis for all ARGs
3) ArMatrix.py/ResMatrix.py: analyze associated ARGs for each kind of
antibiotics which are given in the input file “ar_phenotype.csv” or
“res_phenotype.csv” for different database.
[blast+=C:/blast+/bin/] #please use “/” to separate the directory
Note: Module named with a prefix of “Ar” (example: ArKmer) refers to
analyzing based on CARD database, and named with a prefix of “Res” (example:
ResKmer) refers to analyzing based on ResFinder database.
Abbreviations: pan-ARgenome (pan-antibiotic resistance genome)
ARGs (antibiotic resistance genes)
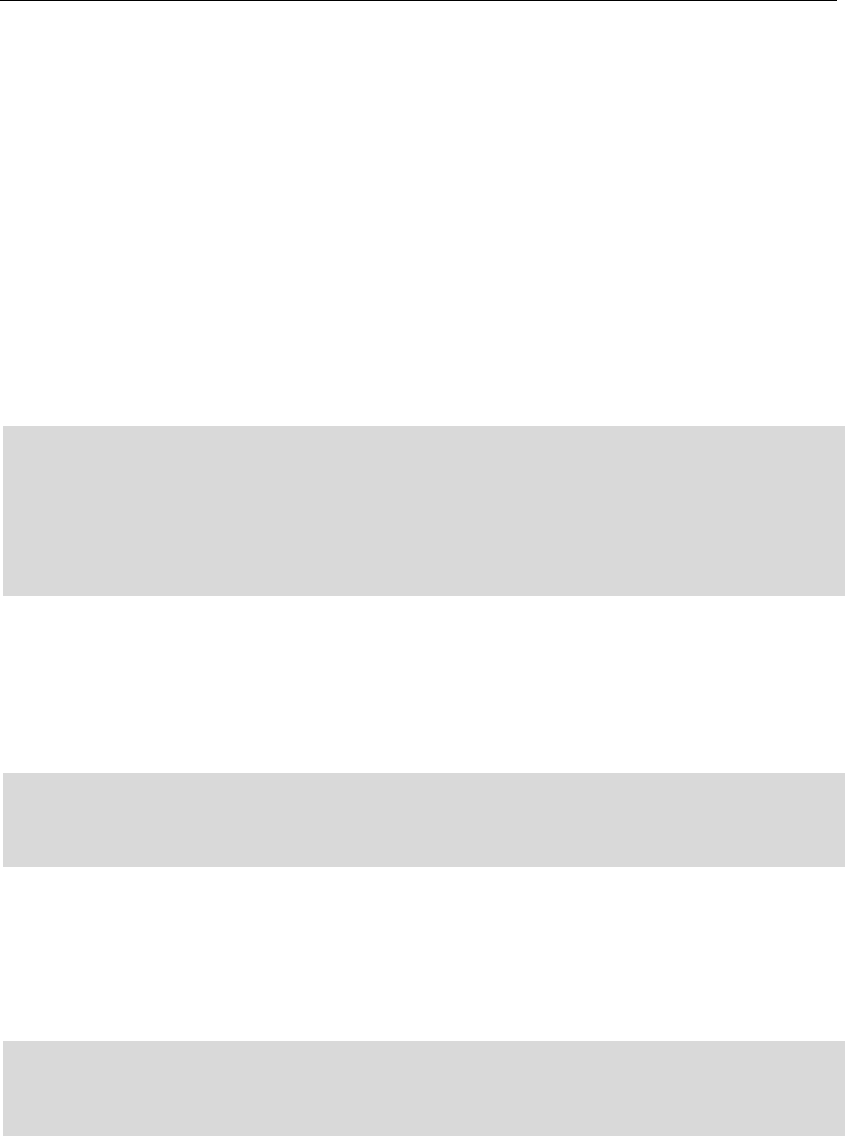
5
5. Input files
[1] Sequence files:
1) raw reads sequence: FASTQ files (example: A.fastq)
a) single-end reads, the filenames should end with “.fastq”
(If one of the input files is “A.fastq”, the output filename of this
genome will be written with a prefix “A”)
b) pair-end reads, the filenames end with “.1.fastq” and “.2.fastq”
(The input filenames of strain A should be “A.1.fastq” and
“A.2.fastq”, the output filename of this genome will be written
with a prefix “A”)
Data format (a unit of a fastq file contains four lines):
2) protein sequence: FASTA amino acid files (example: A.faa)
(If one of the input files is “A.faa”, the output filename of this genome
will be written with a prefix “A”)
Data format (a unit of a fasta amino acid file):
3) nucleotide sequence: FASTA nucleic acid files (example: A.fna)
(If one of the input files is “A.fna”, the output filename of this genome
will be written with a prefix “A”)
Data format (a unit of a fasta nuleic acid file):
4) GenBank annotation files (example: A.gb)
(If one of the input files is “A.gb”, the output filename of this genome
will be written with a prefix “A”)
Data format (a unit of a GenBank file):
@READ_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTT
+ READ_INFO
!''*((((***+))%%%++)(%%%%).1***-+*''))**55CCF>>>>>>CCCCCCC6
>PROTEIN_ID
MKAYFIAILTLFTCIATVVRAQQMSELENRIDSLLNGKKATVGIAVWTDKGDMLRYNDH
>NUCLEOTIDE_ID
GATTTGGGGTTCAAAGCAGTATCGATCAAATAGTAAATCCATTTGTTCAACTCACAGTT

6
[2] Setting file
The setting file in PanARGA package (/PanARGA/settings.txt) is used
to set parameters for individual modules.
1) Settings for identification modules
a) blast+ directory (e.g. [blast+=C:/blast+/bin])
b) blast identity threshold (e.g. [identity=95])
c) blast coverage=query length/subject length
(e.g. [query_coverage=0.80])
d) k value for kmer analysis (e.g. [k=25])
#k value must be an odd number
e) number of kernels for kmer analysis (e.g. [kernel=2])
f) depth of k bp length reads (e.g. [depth=20])
#only time repeats higher than depth will be calculated
g) threshold of score of area under curve (e.g. [area_score=100])
2) Settings for analysis modules
Graph parameters:
#different graphs have individual parameters
a) length of the graph (e.g. [page_length=15])
b) width of the graph (e.g. [page_width=15])
c) dots per inch (DPI) of the graph (e.g. [dpi=200])
d) the font type (e.g. [font_type=Times New Roman])
e) the font size (e.g. [font_size=20])
f) the size of dots in graph (e.g. [dot_size=30])
#only for the correlation matrix graph
g) the font size of labels of genomes (e.g. [genomelabel_fsize=auto])
#because the number of genomes may change, “auto” can change the
font size of labels of genomes according to genome amounts, and you
can also use an itegre to modify the parameter.
gene complement(<1..102)
/locus_tag="DXN26_00005"
CDS complement(<1..102)
/locus_tag="DXN26_00005"
/inference="COORDINATES: similar to AA
sequence:RefSeq:NP_460822.1"
/product="phage virulence factor"
/protein_id="PRJNA484101:DXN26_00005"
/translation="MKHVKSVFLAMVLILPSSLYPALTIAADSQDHKK"

7
Pangenome parameters:
h) fitting coverage of pan-ARgenome curve (e.g. [fit_coverage=0.8])
#only the use-specified portion will be fitted, because some models
exhibit better fitting performance for latter part of the curve.
i) fitting model of the curve (e.g. [fit_model=False])
#three models are provided: power_law (power law model), polyfit
(polynomial model), pangp (a model used by tool “PanGP”).
j) fitting order of polynomial model (e.g. [fit_order=6])
# the highest order of the polynomial model
Cluster graph parameters:
k) cluster for data in each row (e.g. [row_cluster=True])
#if “True”, cluster each row; if “False”, no clustering
l) cluster for data in each column (e.g. [column_cluster=True])
#if “True”, cluster each column; if “False”, no clustering
m) cluster method (e.g. [cluster_method=average])
#the method of cluster, please see:
https://docs.scipy.org/doc/scipy/reference/generated/scipy.clust
er.hierarchy.linkage.html
Phenotype analysis parameters:
n) the name of column (e.g. [column_name=allele])
#if “allele”, clustering for each antibiotic associated genes is based
on gene allele; if “detail”, it is based on each gene.
o) whether to show phenotype (e.g. [show_phenotype=False])
#require “res_phenotype.csv” or “ar_phenotype.csv” as an input file.
#if “True”, phenotype will be shown in the front of the first column;
if “False”, it will not be shown.
[3] Phenotype file:
This file is needed when you want to show associated ARGs for each
kind of antibiotics given in this file.
And if you want to show your experimental results of antibiotic
phenotypic traits together with their associated ARGs, more information
needs to be added. See below for details.
1) “ar_phenotype.csv” is needed if you want to analyze based on the CARD
database. An example file is given in PanARGA package. The first row
of the input file list all the antibiotics in the database. You can only
remain the antibiotics you want to analyze and then copy the file to the
8
directory of your sequence files.
An example of “ar_phenotype.csv”:
(if open the file using a text document, each item is separated by a
comma “,”; if using Microsoft Excel, each item will in an individual cell):
Using a text document:
Using Excel:
#name
glycopeptide antibiotic
fluoroquinolone antibiotic
tetracycline antibiotic
penam
If you want to add phenotypic traits, just write down the number of
antibiotics which the strain is resistant to for each kind. If a cell is left
blank, PanARGA will recognize it as value “0”. An example:
Using a text document:
Using Excel:
#name
glycopeptide antibiotic
fluoroquinolone antibiotic
tetracycline antibiotic
penam
A
1
0
2
B
0
1
3
C
1
1
2
2) “res_phenotype.csv” is needed if you want to analyze based on the
ResFinder database. The main differences are different drug classes.
Modification method of it is similar to “ar_phenoty.csv”.
[4] Annotation csv files:
These files are output files of “gene identification modules”, which are
name with a suffix “_ar.csv”, and they are also input files of “analysis
modules”.
For example, if you input a genome file “A.fasta”, and use “gene
identification module-ArBlastn.py, an output file named “A_ar.csv” will be
generated, and it is the input file for three analysis modules.
#name,glycopeptide antibiotic,fluoroquinolone antibiotic, tetracycline antibiotic,penam
#name,glycopeptide antibiotic,fluoroquinolone antibiotic, tetracycline antibiotic,penam
A,1,,0,2
B,0,1,,3
C,,1,1,2
Please note that the proper input file is used for each module!

9
6. Usage and output files
[1] Before running
1) All sequence files should be put in the same folder.
2) If you want to analyze associate genes and their antibiotic phenotypic
traits, “ar_phenotype.csv” or “res_phenotype.csv” is also needed to be put
in the same folder with sequence files. Please see “5. Input files-[3]
Phenotype file” for more details.
3) Modify the settings. Especially for blast directory. Please see “5. Input
files-[2] Setting file” for more details.
[2] Running PanARGA
1) Move to the installation directory. e.g.:
2) Run PanARGA
And then it will print:
cd C:/PanARGA/
python PanARGA.py
Note:
1) For one run, only files with the same extension will be analyzed. For
example, if “.gb” files and “.fna” files are in one folder simultaneously, and
you choose “G1-1” to analyze them, only the “.gb” files will be analyzed and
“.fna” files will be ignored.
2) If you have different type of input files, and want to analyze for all of
them, please select corresponding “gene identification modules” to
generate all annotation csv files. And after processing all the input
sequence files, select “analysis modules” to analyze all the annotation csv
files (all the annotation files should also be put in the same folder).
==================================================================
============Pan Antibiotic Resistance Genome Analyzer=============
==================================================================
If you choose one of RUN ALL MODULES,
you don't need to RUN SEPARATE MODULE;
If you just want to run one module in all modules,
please see RUN SEPARATE MODULE
------------------------------------------------------------------

10
==================================================================
RUN ALL MODULES:
------------------------------------------------------------------
# Raw reads as input files (files with a ".fastq" extension):
------------------------------------------------------------------
[R1] analysis with CARD nucleotide database
[R2] analysis with ResFinder nucleotide database
------------------------------------------------------------------
# Genbank files as input files (files with a ".gb" extension):
------------------------------------------------------------------
[G1-1] analysis with CARD nucleotide database
[G1-2] analysis with CARD protein database
[G2-1] analysis with ResFinder nucleotide database
[G2-2] analysis with ResFinder protein database
------------------------------------------------------------------
# Nucleotide seq as input files (files with a ".fna" extension):
------------------------------------------------------------------
[N1] analysis with CARD nucleotide database
[N2] analysis with ResFinder nucleotide database
------------------------------------------------------------------
# Protein seq as input files (files with a ".faa" extension):
------------------------------------------------------------------
[P1] analysis with CARD protein database
[P2] analysis with ResFinder protein database
------------------------------------------------------------------
==================================================================
==================================================================
RUN SEPARATE MODULE:
------------------------------------------------------------------
# Preprocessing module:
------------------------------------------------------------------
[a] "CDSex.py": extract coding sequence from genbank files;
form both protein and nucleotide fasta files;
------------------------------------------------------------------
# Gene identification modules:
------------------------------------------------------------------
## Modules using CARD database------------------------------------
[b-1] "ArKmer.py": find resistance genes from raw reads;
[b-2] "ArBlastn.py": find resistance genes from ".fna" files;
[b-3] "ArBlastp.py": find resistance genes from ".faa" files;

11
3) run all modules
If you choose to run all modules, for example, there are dozens of
GenBank annotation files in C:/data/, and you want to analyze them based
on ResFinder nucleotide database, just type “G2-1” after “Your choice:”,
like:
And then type the directory of input files:
All the annotation results and analysis output files will be generated in
the input directory C:/data/.
4) run separate module
If you choose to run just one module, for example, to identify ARGs
from dozens of raw reads files in C:/data/, and you want to analyze them
based on CARD nucleotide databases, just type “b-1” after “Your choice:”,
like:
Your choice: G2-1
please input the genebank files directory: C:/data/
## Modules using ResFinder database-------------------------------
[c-1] "ResKmer.py": find resistance genes from raw reads;
[c-2] "ResBlastn.py": find resistance genes from ".fna" files;
[c-3] "ResBlastn.py": find resistance genes from ".fna" files;
------------------------------------------------------------------
# Analysis modules:
# Input files of Analysis modules are annotation files (files
# with "_ar.csv" suffixes) formed by gene identification modules
------------------------------------------------------------------
[d] "Pangenome.py": pan-antibiotic resistance genome analysis
(mainly analyze for pan-genome features)
[e] "PanAccess.py": pan & accessory anti-resis genome analysis
(mainly classify and statistical analysis for ARGs)
## Module using CARD database-------------------------------------
[f-1] "ArMatrix.py": analysis associated genes for each kind of
antibiotics in CARD database
## Module using ResFinder database--------------------------------
[f-2] "ResMatrix.py": analysis associated ARGs for each kind of
antibiotics in ResFinder database
==================================================================
Your choice: b-1

12
And then type the directory of input files:
Only the annotation csv files will be generated in the same folder
C:/data, and for further analysis, for example, to study pan-ARgenome
features, just run PanARGA again and type “d” after “Your choice:”, like:
And then type the directory of annotation csv files:
The pangenome output files will be generated in C:/data/pangenome/.
[3] Output files
1) Outputs of gene identification modules.
a) ArKmer/ResKmer:
Input: C:/data/A.fastq or C:/data/A.1.fastq+C:/data/A.2.fastq
Output_1: C:/data/kmer/A_25mer_countVSar_nucl.csv
or C:/data/kmer/A_25mer_countVSres_nucl.csv
“25” in “A_25mer_count” is the k value in the setting file, all the
genes in the database beyond the threshold would be given in the file,
including the gene name, the score (area under the curve of the
corresponding gene in Output_2) and the coverage. The table below
shows a part of the output file (Using CARD database).
A_25mer_countVSar_nucl.csv
#gene_name
area_score
coverage
gb|AM261837|+|73-865|ARO:3002619|aadA22
97763
100.00%
gb|AF047479|+|1295-2087|ARO:3002603|aadA3
118409
100.00%
gb|DQ677333|+|0-780|ARO:3002621|aadA24
106672
100.00%
Output_2: C:/data/kmer/A/ar_nucl_ 25/***(allele name).png
“25” in “ar_nucl_25” is the k value in the setting file, and for each
possible gene allele in Output_1, a kmer count graph will be drawn. In
each allele, if the number of genes exceeds four, only the four genes
with higher scores will be drawn, and the highest one will be drawn in
black thickening line. The picture below shows several typical examples
(allele of oqxA, aadA and aac(6’)).
please input the fastq files directory: C:/data/
Your choice: d
please input the annotation csv files directory: C:/data/
13
***(allele name).png

14
Output_3: C:/data/A_ar.csv
This is the final output file. Only one gene in each allele with highest
score and coverage will be present in the final annotation file. Together
with information in CARD/ResFinder database, the final elements for
each gene in the annotation file includes: gene_num (the gene name
formed by panARGA), gene_name (the gene name in database),
coverage, area_score, ARO_num (the ARO accession in CARD),
accession num (the accession of related gene in NCBI), ar_gene_allele
(allele of the gene), drug_class (the kind of antibiotics the gene
conferring according to the database) and ar_machanism (the
mechanism of the gene recorded in the database). Note that only CARD
has information of AR0_num and ar_mechanism. The table below shows
a part of the annotation csv file.
A_ar.csv
#gene_
num
gene_na
me
cove
rage
area_sc
ore
ARO_num
accessio
n_num
ar_gene
_allele
drug_class
ar_mechanism
A_AMR
gene_0
OXA-1
100
113192
ARO:3001
396
JN420
336.1
OXA
cephalosporin;
penam
antibiotic
inactivation
A_AMR
gene_1
APH(3')
-Ia
99.4
7849
68005
ARO:3002
641
BX6640
15.1
APH(3')
aminoglycosid
e antibiotic
antibiotic
inactivation
A_AMR
gene_2
AAC(3)-
IV
100
90553
ARO:3002
539
DQ2413
80.1
AAC(3)
aminoglycosid
e antibiotic
antibiotic
inactivation
b) ArBlastn/ResBlastn
Input: C:/data/A.fna
Output_1: C:/data/AVSar_nucl.xls or C:/data/AVSres_nucl.xls
The output of blast+ using command “blastn -query A.fna -db
ar_nucl/res_nucl -out AVSar_nucl/res_nucl.xls -outfmt “6 std slen” -
evalue 1e-20 -perc_identity identity (in the setting file)”.
Output_2: C:/data/arg/A_ar.fna
The sequence of ARGs identified from the input files will be written
into a new fasta nucleic acid file. The below shows part of the file.
>A_AMRgene_1_from_Scaffold32 [oqxA|identity:100.000|ARO:3003922|
NC_010378.1|position:1-1176]
TCAGTTAAGGGTGGCGCTG……CCAGGTTTTTTGCAGGCTCAT
>A_AMRgene_2_from_ Scaffold45 [AAC(6')-Ib-cr|identity:100.000|ARO:3002547|
DQ303918|position:1-555]
GTGACCAACAGCAACGATT……GAACACGCAGTGATGCCTAA

15
Output_3: C:/data/A_ar.csv
A similar annotation csv file to kmer output_3. The differences are
replacing “coverage” with “identity” and “area_score” with “e_value”. In
order to locate the gene in the scaffolds, the origin of the gene
together with start and end positions are added to the annotation file.
The table below shows a part of the annotation csv file.
A_ar.csv
#gene_num
gene_
name
ident
ity
e_val
ue
ARO_
num
access
ion_nu
m
ar_ge
ne_all
ele
drug_
class
ar_me
chanis
m
origin
start
end
A_AMRgen
e_1_from_
Scaffold32
oqxA
100
0
ARO:
3003
922
NC_0
10378
.1
oqxA
fluoro
quinol
one…
antibi
otic
efflux
Scaff
old32
1
1176
A_AMRgen
e_2_from_
Scaffold45
AAC(
6')-
Ib-cr
100
0
ARO:
3002
547
DQ30
3918
AAC(
6')
Fluoro
quinol
one…
antibi
otic
inacti
vation
Scaff
old45
1
555
c) ArBlastp/ResBlastp
Input: C:/data/A.faa
Output_1: C:/data/AVSar_prot.xls or C:/data/AVSres_prot.xls
The output of blast+ using command “blastp -query A.faa -db
ar_prot/res_prot -out AVSar_prot/res_prot.xls -outfmt “6 std slen”
-evalue 1e-20 -num_alignments 6”.
Output_2: C:/data/arg/A_ar.faa
The sequence of ARGs identified from the input files will be written
into a new fasta amino acid file. The below shows part of the file.
Output_3: C:/data/A_ar.csv
A similar annotation csv file to kmer output_3. The differences are
replacing “coverage” with “identity” and “area_score” with “e_value”.
Because there is no need to intercept amino acid sequences, so the
origin, start and end positions are not added comparing with
Ar/ResBlastn results, the original name of protein will remain. The
table below shows a part of the annotation csv file.
>Protein_id1 [oqxA|identity:100.000|ARO:3003922|YP_001693237.1]
MSLQKTWGNIHLTALGAMM……GMPVNAKTVAMTSSATLN
>Protein_id2 [AAC(6')-Ib-cr|identity:99.457|ARO:3002547|ABC17627.1]
MTNSNDSVTLRLMTEHDLAM……AVYMVQTRQAFERTRSDA
16
A_ar.csv
#gene_num
gene_
name
ident
ity
e_val
ue
ARO_
num
access
ion_nu
m
ar_ge
ne_all
ele
drug_
class
ar_me
chanis
m
Protein_id1
oqxA
100
0
ARO:3
00392
2
YP_00
16932
37.1
oqxA
fluoro
quinolo
ne…
antibi
otic
efflux
Protein_id2
AAC(6
')-Ib-
cr
100
0
ARO:3
00254
7
ABC17
627.1
AAC(6
')
Fluoro
quinolo
ne…
antibi
otic
inactiv
ation
2) Outputs of analysis modules
a) Pangenome
Input: A batch of annotation csv files
(C:/data/A_ar.csv+B_ar.csv+C_ar.csv+……)
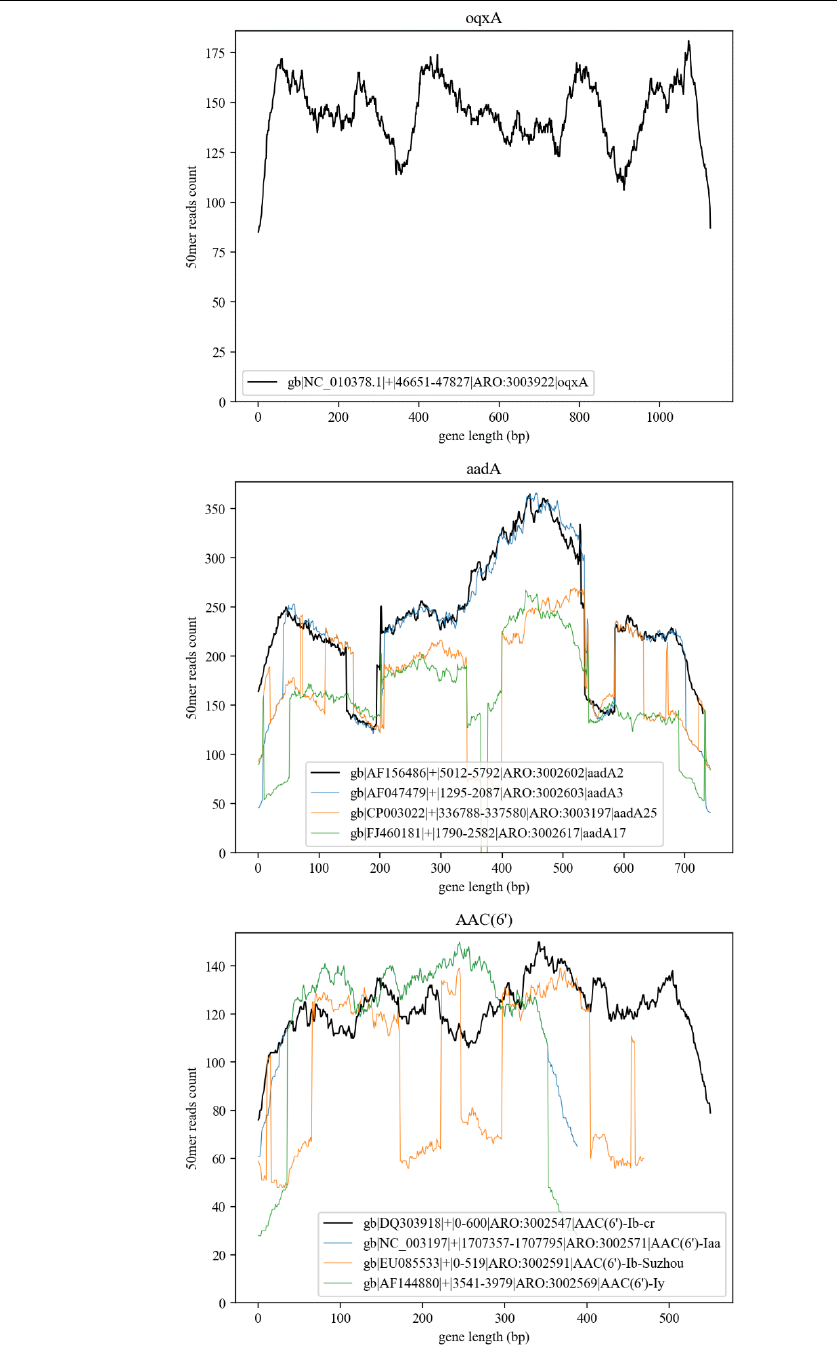
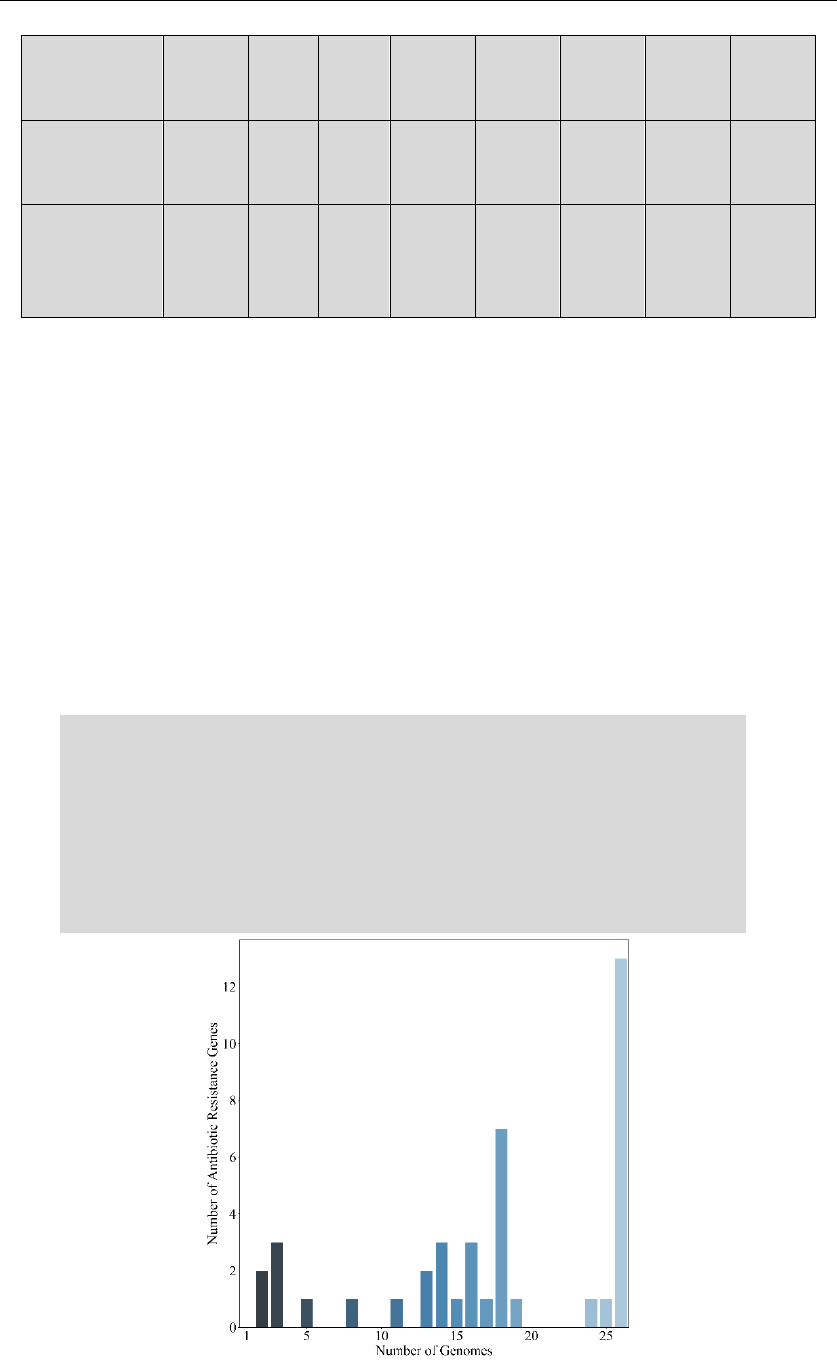
Output_1: C:/data/pangenome/ 4_ar_distribution.txt
& C:/data/pangenome/ 4_ar_distribution.png
Count the number of occurrences of each gene allele in each genome.
For example, the alleles behind number “26” refers to the allele which
has appeared in 26 genomes.
4_ar_distribution.png
1
2 rmtB,QepA
3 rpoB,mphA,Mrx
…
25 ramR
26 parC,parE,soxR,soxS,MdtK,mdsC,mdsB,mdsA,golS,gyrA,gyrB,sdiA
17
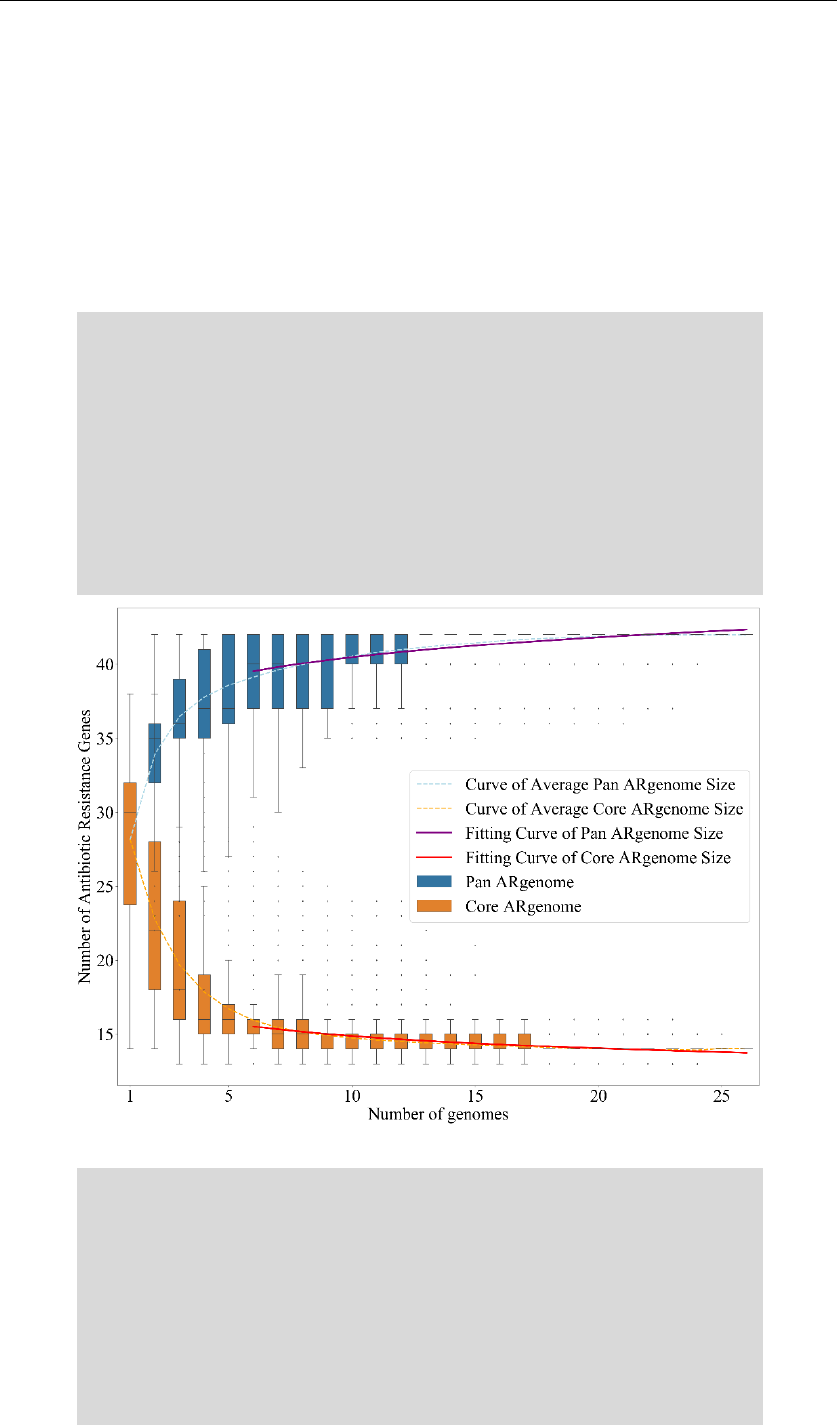
Output_2: C:/data/pangenome/5_pangenome.txt
& C:/data/pangenome/5_pangenome.png
& C:/data/pangenome/5_pangenome _fitmodel.txt
Count the pan genome size and core genome size for antibiotic gene
alleles. It traverses all combinations of given genomes and form a text
document. A boxplot is drawn according to the data and the model in
the setting file is applied for curve fitting. The final model together
with R^2 value is also written into a text document.
5_pangenome.png
Genome_Number Pan_Genome_Size Core_Genome_Size
1 30 30
1 34 34
1 31 31
… … …
25 41 14
25 41 15
26 41 14
Power Law Model of PanGenome:
P=(36.331034418648684)*x^(0.0469962382291344)
(R^2=0.9534322181666640470632029231)
Power Law Model of CoreGenome:
C=(18.021585517236634)*x^(-0.08327794834525747)
(R^2=0.9211436740795695974874930157)

18
b) PanAccess
Input: A batch of annotation csv files
(C:/data/A_ar.csv+B_ar.csv+C_ar.csv+……)
Output_1: C:/data/analysis/1_class_summary.csv
& C:/data/analysis/1_mech_summary.csv
& C:/data/analysis/1_class_summary.png
& C:/data/analysis/1_ar_cluster.png
& C:/data/analysis/1_ar_corr.png
Analyze all the ARGs for all genomes, and classify them into
different drug classes and mechanism classes. A stacked bar graph, a
cluster map and a comparison graph are drawn according to summary of
drug classes.
1_class_summary.csv
name
aminocoumar
in antibiotic
aminoglycosid
e antibiotic
carbapenem
cephalosporin
…
A
1
8
5
9
…
B
1
9
5
10
…
C
1
4
5
10
…
#Each number in the cell represent the number of antibiotic
associated ARGs in each genome.
1_mech_summary.csv
name
antibiotic
efflux
antibiotic
inactivation
antibiotic
target
alteration
antibiotic
target
replacement
…
A
12
12
7
4
…
B
15
13
8
4
…
C
15
10
8
4
…
#Each number in the cell represent the number of of ARGs
related to each kind of antibiotic resistance mechanisms in each
genome.
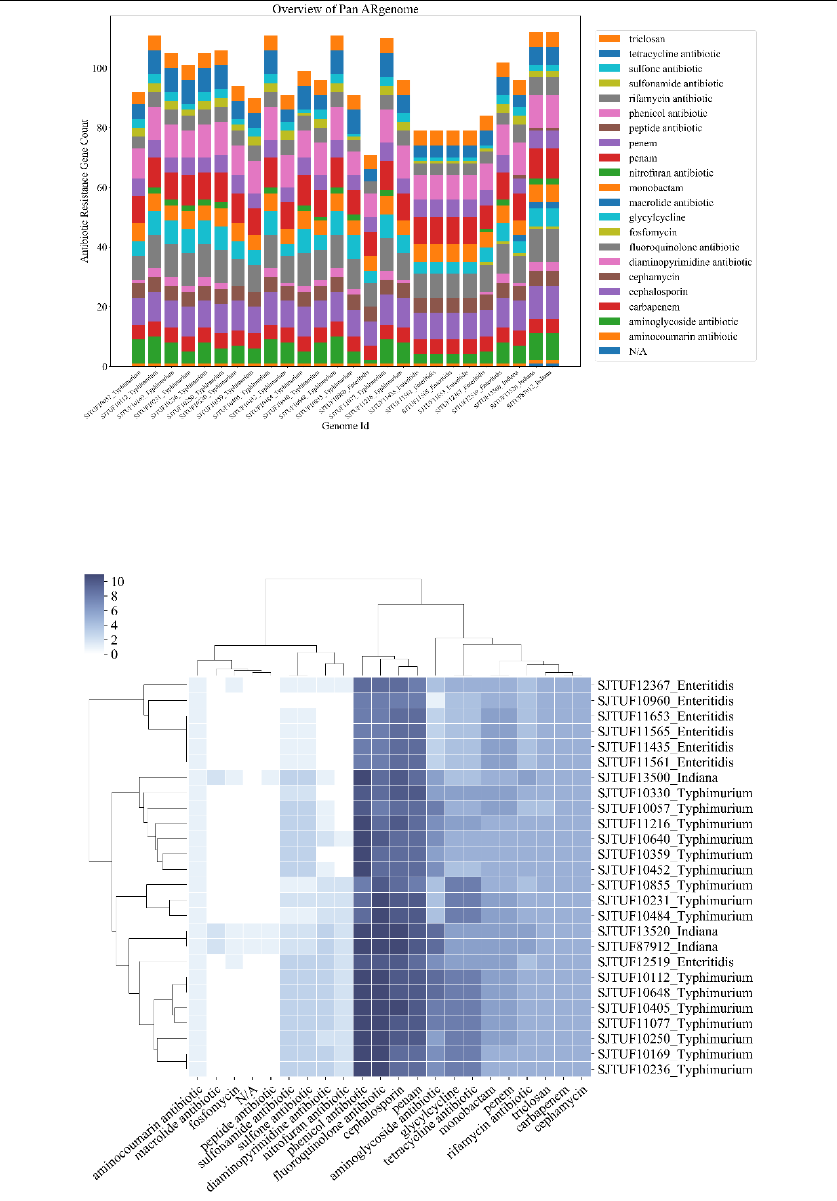
19
1_class_summary.png
#A stacked bar graph based on 1_class_summary.csv. Different colors
represent different kind of antibiotics
1_ar_cluster.png
#A cluster map based on 1_class_summary.csv. The darker the color, the
more the number of related genes
20
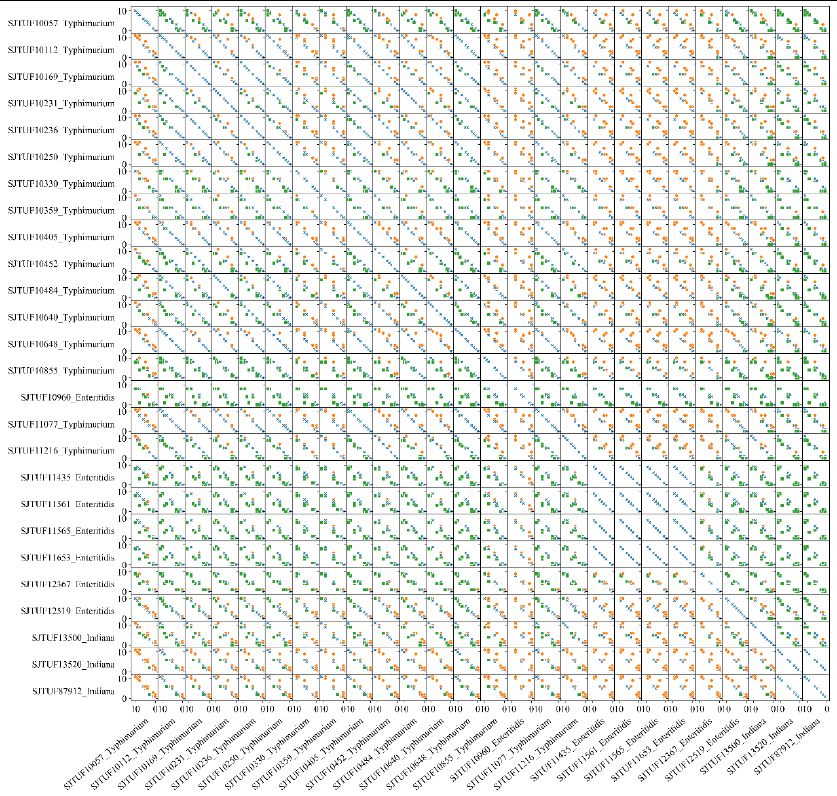
1_ar_corr.png
#A comparison matrix graph based on 1_class_summary.csv. Each
subgraph represents a comparison of resistance genes in two genomes.
Each dot represents an antibiotic, and dots in orange means the number
of associated ARGs of the antibiotic in the genome in y axis is higher
than that in x axis, while blue means equal and green mean lower.
Output_2: C:/data/A_ar_accessory.csv
& C:/data/analysis/2_accessory_class_summary.csv
& C:/data/analysis/2_accessory_class_summary.png
& C:/data/analysis/2_accessory_ar_cluster.png
& C:/data/analysis/2_accessory_ar_corr.png
The output_2 is similar to output_1, and the difference is that the
data used is accessory ARgenome instead of pan-ARgenome.
A_ar_accessory.csv is the annotation file that excludes all core ARGs
from A_ar.csv, and their elements are all the same.
21
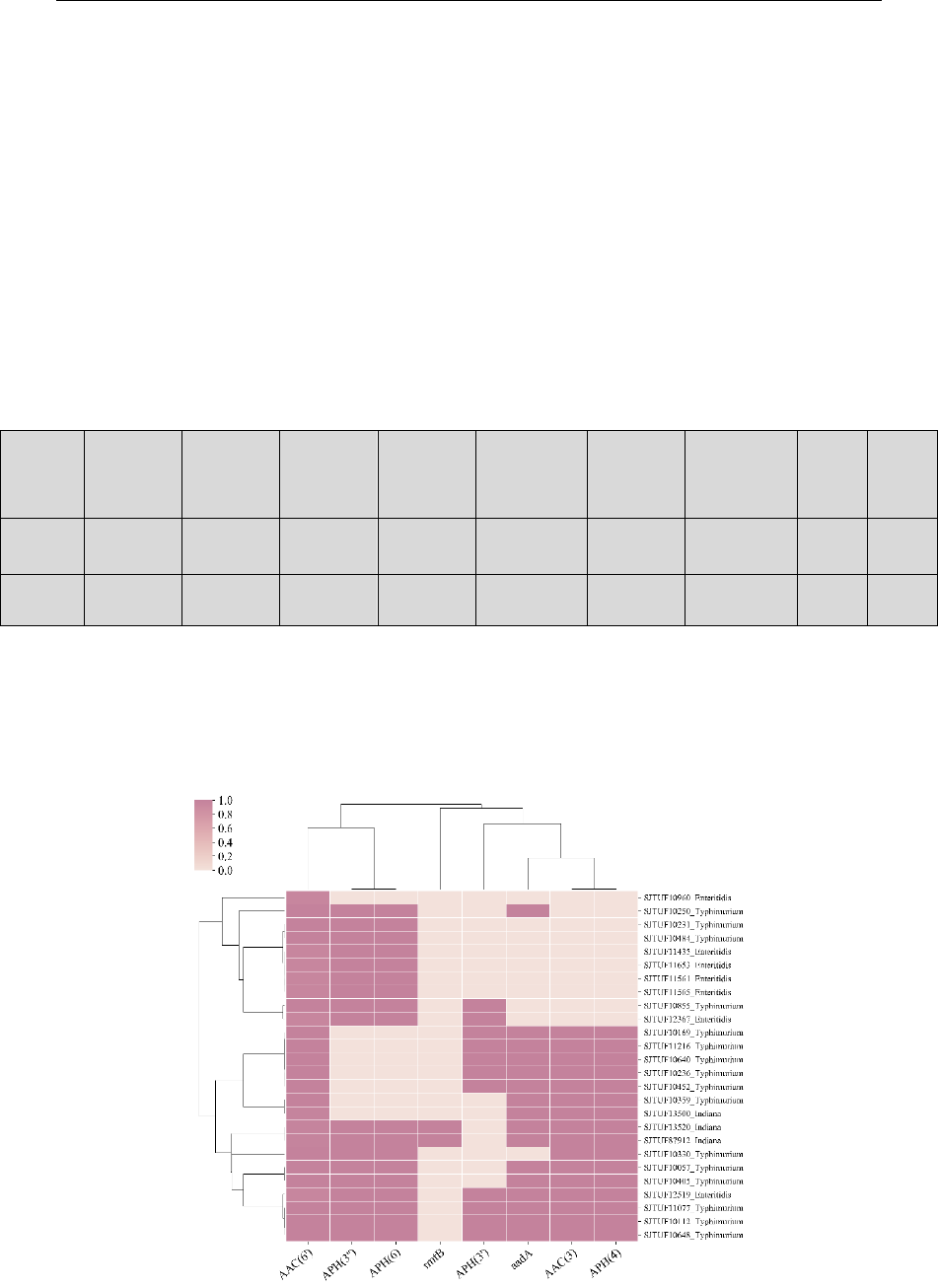
d) ArMatrix/ResMatrix
Input: A batch of annotation csv files
(C:/data/A_ar.csv+B_ar.csv+C_ar.csv+……)
& “C:/data/ar_phenotype.csv” or “C:/data/res_phenotype.csv”
Output: C:/data/analysis/3_***_matrix.csv
& C:/data/analysis/3_***_matrix.png
“***” is the name of antibiotics given in the first row of
“ar_phenotype.csv” or “res_phenotype.csv”. For example, if one of the
antibiotics given is “aminoglycoside antibiotic”, the output name will be
“3_aminoglycoside antibiotic_matrix”. The details are shown below:
3_aminoglycoside antibiotic_matrix.csv
name
AAC(6')
aadA
AAC(3)
APH(4)
APH(3'')
APH(6)
APH(3')
rmtB
PHE
NOT
YPE
A
1
0.9974
7
0.99871
1
0.99751
0.99881
0
0
N/A
B
1
0.99621
0.99871
1
0.99751
0.99881
0.99386
0
N/A
#Genes or alleles (according to settings [column_name=detali] or
[column_name=allele]) in the first row are all genes or alleles identified
from all genomes, which are related to the given antibiotic. The number
in each cell is the identity or coverage of the gene in annotation files.
3_aminoglycoside antibiotic_matrix.png
#A cluster map based on the matrix file above ([column_name=allele]).
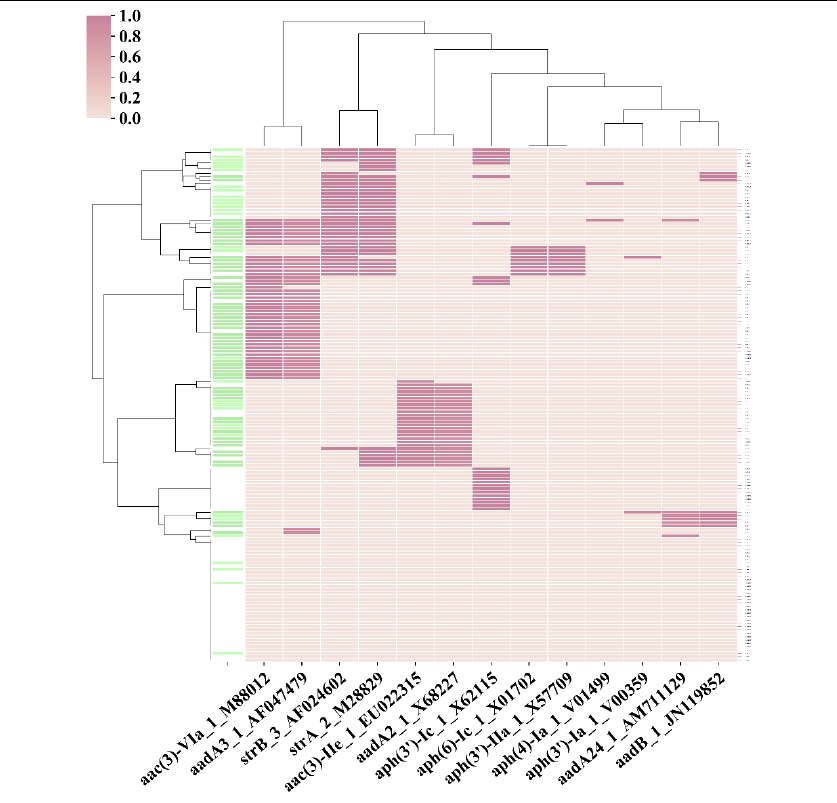
22
3_aminoglycoside antibiotic_matrix.png
#A cluster map for another hundreds of genomes using setting
[column_name=details]. The phenotypic triats information are
added (see “Input files for more details”), so a green column is
drawn to represent antibiotic resistance phenotype.
