The_SeqHound_Admin_Manual The Seq Hound Admin Manual
User Manual:
Open the PDF directly: View PDF ![]() .
.
Page Count: 421 [warning: Documents this large are best viewed by clicking the View PDF Link!]
- About this manual.
- Conventions
- How to contact us.
- Who is SeqHound?
- 4. Setting up SeqHound locally.
- 5. Description of the SeqHound parsers and data tables by module
- What are modules?
- How to use this section.
- Parser descriptions
- Table descriptions
- An overview of the SeqHound data table structure
- Parsers and resource files needed to build and update modules of SeqHound.
- core module
- Redundant protein sequences (redundb) module
- Complete genomes tracking (gendb) module
- Taxonomy hierarchy (taxdb) module
- Structural databases (strucdb) module
- Protein sequence neighbours (neighdb) module
- Locus link functional annotations (lldb) module
- GENE module
- Gene Ontology hierarchy (godb) module
- Gene Ontology Association (GOA) module
- dbxref module
- How to update the DBXref and GO Annotation modules using a cluster.
- Understanding the dbxref.ini file
- RPS-BLAST domains (rpsdb) module
- Molecular Interaction (MI) module
- Text mining module
- 6. Developing for SeqHound.
- 7. Appendices
The SeqHound Manual
Part II: Sections 4-7
For Administrators and Developers
Release 3.3
(April 20th, 2005)
Authors
Ian Donaldson, Katerina Michalickova, Hao Lieu, Renan Cavero, Michel Dumontier,
Doron Betel, Ruth Isserlin, Marc Dumontier, Michael Matan, Rong Yao, Zhe Wang,
Victor Gu, Elizabeth Burgess, Kai Zheng, Rachel Farrall
Edited by
Rachel Farrall and Ian Donaldson
© 2005 Mount Sinai Hospital
The SeqHound Manual 2 of 421 18/04/2005
Table of Contents
About this manual............................................................................................................ 7
Conventions .....................................................................................................................8
How to contact us. ........................................................................................................... 8
Who is SeqHound?........................................................................................................... 9
4. Setting up SeqHound locally. ....................................................................................... 10
4.1 Overview.................................................................................................................. 10
4.2 SeqHound system requirements............................................................................... 11
OS and hardware architecture .................................................................................... 11
Memory (RAM) ......................................................................................................... 11
Hard Disk ................................................................................................................... 12
Source code and executables .................................................................................. 12
Database.................................................................................................................. 12
Other Software ........................................................................................................... 12
Compiling SeqHound Code yourself. ........................................................................ 13
ODBC compliant database engines............................................................................ 13
Library dependencies ................................................................................................. 13
4.3 Obtaining precompiled SeqHound executables....................................................... 14
4.3.1 Obtaining SeqHound Source Code...................................................................... 16
4.4 Compiling SeqHound executables on Solaris.......................................................... 18
4.5 Building the SeqHound system on Solaris............................................................... 26
Catch up on SeqHound daily updates ........................................................................ 45
Setting up daily sequence updates.............................................................................. 47
Setting up SeqHound servers. Overview................................................................... 53
Trouble-shooting notes............................................................................................... 57
Error logs ................................................................................................................ 57
Recompiling SeqHound.......................................................................................... 57
Restarting the Apache server .................................................................................. 57
Other useful links.................................................................................................... 58
Parser schedule........................................................................................................ 58
MySQL errors ......................................................................................................... 58
5. Description of the SeqHound parsers and data tables by module................................. 59
What are modules? ........................................................................................................ 59
How to use this section. ................................................................................................. 59
Parser descriptions........................................................................................................ 59
Table descriptions.......................................................................................................... 60
An overview of the SeqHound data table structure ....................................................... 63
Parsers and resource files needed to build and update modules of SeqHound. ........... 64
seqhound@blueprint.org Version 3.3
The SeqHound Manual 3 of 421 18/04/2005
core module................................................................................................................ 66
mother parser .......................................................................................................... 66
update parser ........................................................................................................... 71
postcomgen parser .................................................................................................. 72
asndb table .............................................................................................................. 75
parti table ................................................................................................................ 78
nucprot table............................................................................................................ 80
accdb table .............................................................................................................. 82
histdb table.............................................................................................................. 88
pubseq table ............................................................................................................ 91
taxgi table................................................................................................................ 94
sengi table ............................................................................................................... 97
sendb table .............................................................................................................. 99
chrom table............................................................................................................ 101
gichromid table ..................................................................................................... 105
contigchromid table .............................................................................................. 107
gichromosome table.............................................................................................. 109
contigchromosome table ....................................................................................... 111
Redundant protein sequences (redundb) module ..................................................... 113
redund parser......................................................................................................... 113
redund table........................................................................................................... 115
Complete genomes tracking (gendb) module........................................................... 119
Taxonomy hierarchy (taxdb) module....................................................................... 120
importtaxdb parser ................................................................................................ 120
taxdb table............................................................................................................. 122
gcodedb table ........................................................................................................ 127
divdb table............................................................................................................. 132
del table................................................................................................................. 135
merge table............................................................................................................ 137
Structural databases (strucdb) module ..................................................................... 139
cbmmdb parser...................................................................................................... 139
vastblst parser........................................................................................................ 144
pdbrep parser......................................................................................................... 146
mmdb table............................................................................................................ 148
mmgi table ............................................................................................................ 154
domdb table........................................................................................................... 156
Protein sequence neighbours (neighdb) module ...................................................... 162
Installing nblast:.................................................................................................... 162
Configuration of nblast environment:................................................................... 163
Running NBLAST ................................................................................................ 164
NBLAST Update Procedure ................................................................................. 166
nbraccess program* .............................................................................................. 168
BLASTDB table................................................................................................... 169
NBLASTDB table................................................................................................. 172
Locus link functional annotations (lldb) module ..................................................... 177
llparser................................................................................................................... 177
seqhound@blueprint.org Version 3.3
The SeqHound Manual 4 of 421 18/04/2005
addgoid parser....................................................................................................... 179
ll_omim table ........................................................................................................ 181
ll_go table.............................................................................................................. 183
ll_llink table .......................................................................................................... 186
ll_cdd table............................................................................................................ 188
GENE module .......................................................................................................... 191
parse_gene_files.pl parser..................................................................................... 191
gene_dbxref table.................................................................................................. 193
gene_genomicgi table ........................................................................................... 195
gene_history table ................................................................................................. 198
gene_info table...................................................................................................... 201
gene_object table .................................................................................................. 204
gene_productgi table............................................................................................. 206
gene_pubmed table ............................................................................................... 208
gene_synonyms table............................................................................................ 210
Gene Ontology hierarchy (godb) module................................................................. 212
goparser................................................................................................................. 212
go_parent table...................................................................................................... 214
go_name table ....................................................................................................... 216
go_reference table................................................................................................. 219
go_synonym table ................................................................................................. 221
Gene Ontology Association (GOA) module ............................................................ 223
Table summarizing input files, parsers and command line parameters for GOA
module................................................................................................................... 225
Gene Ontology Module Diagram.......................................................................... 228
goa_seq_dbxref table ............................................................................................ 230
goa_association table ............................................................................................ 234
goa_reference table ............................................................................................... 237
goa_with table....................................................................................................... 239
goa_xdb table........................................................................................................ 242
goa_gigo table....................................................................................................... 245
dbxref module .......................................................................................................... 248
Who Cross-references who? ................................................................................. 249
Explanation of the data table structure: ................................................................ 249
How to update the DBXref and GO Annotation modules using a cluster. .............. 256
Understanding the dbxref.ini file ............................................................................. 257
Table summarizing input files, parsers and command line parameters for dbxref
module................................................................................................................... 262
dbxref table ........................................................................................................... 265
dbxrefsourcedb table............................................................................................. 268
Contents of dbxrefsourcedb table ......................................................................... 270
RPS-BLAST domains (rpsdb) module..................................................................... 272
domname parser.................................................................................................... 272
Rpsdb parser.......................................................................................................... 273
domname table...................................................................................................... 274
rpsdb table............................................................................................................. 278
seqhound@blueprint.org Version 3.3
The SeqHound Manual 5 of 421 18/04/2005
Molecular Interaction (MI) module.......................................................................... 285
MI-BIND parser.................................................................................................... 285
MI_source table .................................................................................................... 289
MI_ints table ......................................................................................................... 291
MI_objects table.................................................................................................... 292
MI_obj_dbases table ............................................................................................. 294
MI_mol_types table .............................................................................................. 295
MI_dbases table .................................................................................................... 296
MI_record_types table .......................................................................................... 297
MI_complexes table.............................................................................................. 298
MI_complex2ints table ......................................................................................... 299
MI_complex2subunits table.................................................................................. 300
MI_complex2subunits table.................................................................................. 301
MI_refs table......................................................................................................... 302
MI_refs_db table................................................................................................... 304
MI_exp_methods table.......................................................................................... 305
MI_obj_labels table .............................................................................................. 306
Text mining module ................................................................................................. 307
mother parser ........................................................................................................ 307
text searcher parser ............................................................................................... 308
yeastnameparser.pl parser..................................................................................... 312
text_bioentity table................................................................................................ 314
text_bioname table................................................................................................ 317
text_secondrefs table............................................................................................. 321
text_bioentitytype table......................................................................................... 324
text_fieldtype table................................................................................................ 325
text_nametype table .............................................................................................. 326
text_rules table...................................................................................................... 327
text_db table.......................................................................................................... 328
text_doc table........................................................................................................ 329
text_docscore table................................................................................................ 331
text_evidencescore table ....................................................................................... 336
text_method table.................................................................................................. 338
text_point table...................................................................................................... 341
text_pointscore table ............................................................................................. 342
text_result table..................................................................................................... 344
text_resultscore table ............................................................................................ 346
text_search table.................................................................................................... 348
text_searchscore table ........................................................................................... 351
text_rng table ........................................................................................................ 353
text_rngresult table................................................................................................ 355
text_doctax table ................................................................................................... 357
text_organism table............................................................................................... 359
text_englishdict table ............................................................................................ 361
text_bncorpus table ............................................................................................... 363
text_pattern table................................................................................................... 365
seqhound@blueprint.org Version 3.3
The SeqHound Manual 6 of 421 18/04/2005
text_stopword table............................................................................................... 367
6. Developing for SeqHound. ......................................................................................... 369
Open source development............................................................................................ 369
Code organization. ...................................................................................................... 370
Adding/Modifying a remote API function to SeqHound.............................................. 373
Overall architecture of the SeqHound system.......................................................... 374
Adding a new module to SeqHound............................................................................. 380
Database layer .......................................................................................................... 381
Parser layer............................................................................................................... 382
Local API layer (Query layer).................................................................................. 383
CGI layer.................................................................................................................. 383
Remote API layer..................................................................................................... 384
7. Appendices.................................................................................................................. 387
Example GenBank record ........................................................................................ 388
Example SwissProt record ....................................................................................... 393
Example EMBL record ............................................................................................ 400
Example PDB record................................................................................................ 406
Example Biostruc ..................................................................................................... 411
GO background material .......................................................................................... 421
* not available at time of writing
seqhound@blueprint.org Version 3.3
The SeqHound Manual 7 of 421 18/04/2005
About this manual.
This manual contains everything that has been documented about SeqHound. It is
distributed in two Parts (Part I: For Users and Part II: For Administrators and
Developers).
If you can’t find the answer here then please contact us. This manual was written and
reviewed by the persons listed under “Who is SeqHound”. Any errors should be reported
to seqhound@blueprint.org.
You can find out more about the general architecture of SeqHound by reading the
SeqHound paper that is freely available from BioMed Central. This paper is included in
the supplementary material distributed with this manual. See:
Michalickova K, Bader GD, Dumontier M, Lieu H, Betel D, Isserlin R, Hogue CW.
SeqHound: biological sequence and structure database as a platform for bioinformatics
research. BMC Bioinformatics. 2002 Oct 25;3(1):32.
PMID: 12401134
The SeqHound Manual (Part I: Sections 1-3) For Users.
Section1 and Section 2 is a one page description that tells you what to read first to get
started depending on what kind of user you are.
Section 3 is of interest to programmers who want to use the remote API to access
information in the SeqHound database maintained by the Blueprint Initiative.
The SeqHound Manual (Part II: Sections 4-7) For Administrators and Developers
Section 4 is of interest to programmers and system administrators who want to set up
SeqHound themselves so they can use the local API.
Section 5 is an in-depth description of everything that’s in the SeqHound database and
how it gets there (table by table). This section will be of interest to all users.
Section 6 describes how programmers can add to SeqHound. This section also describes
our internal development process at Blueprint.
Section 7 includes Appendices of background and reference material.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 8 of 421 18/04/2005
Conventions
The following section describes the conventions used in this manual.
Italic
is used for filenames, file extensions, URLs, and email addresses.
Constant Width
is used for code examples, function names and system output.
Constant Bold
is used in examples for user input.
Constant Italic
is used in examples to show variables for which a context-specific substitution should be
made.
How to contact us.
General enquiries or comments can be posted to the SeqHound usergroup mailing list
seqhound.usergroup@blueprint.org. You may also subscribe to this list to receive
regular updates about SeqHound developments by going to
http://lists.blueprint.org/mailman/listinfo/seqhound.usergroup .
Private enquiries, bug reports from external users, questions about SeqHound or errors
found in this manual may be sent to seqhound@blueprint.org.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 9 of 421 18/04/2005
Who is SeqHound?
Chronologically ordered according to when the person first started work on SeqHound.
Chris Hogue
Katerina Michalickova
Gary Bader
Ian Donaldson
Ruth Isserlin
Michel Dumontier
Hao Lieu
Marc Dumontier
Doron Betel
Renan Cavero
Ivy Lu
Rong Yao
Volodya Grytsan
Zhe Wang
Victor Gu
Rachel Farrall
Michael Matan
Elizabeth Burgess
Kai Zheng
seqhound@blueprint.org Version 3.3

The SeqHound Manual 10 of 421 18/04/2005
4. Setting up SeqHound locally.
4.1 Overview.
This section describes how one can set up the SeqHound system on your own hardware
using freely available SeqHound executables. These executables will allow you to build
and update the SeqHound database as well as run a web-interface and a remote API
server.
Section 4.2 should be reviewed first for system requirements before attempting to install
the SeqHound system.
Section 4.3 tells you how to download executables from the SeqHound ftp site for your
platform and operating system. SeqHound code may also be downloaded from this site.
Section 4.4 describes how SeqHound code may be compiled on your own hardware using
the freely available code available on the SeqHound ftp site. This step is only required if
SeqHound executables are not available for your platform or if you want to make use of
the local programming API. If you obtain SeqHound executables from the ftp site and
want to build your local SeqHound database, you still need to go through Steps 8, 9, 10,
11 and 13 in this section which describe how to install the MySQL server and ODBC
driver.
Section 4.5 contains detailed instructions for using the executables to build the SeqHound
data tables and for setting up the SeqHound web-interface and remote API server.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 11 of 421 18/04/2005
4.2 SeqHound system requirements.
Before attempting to set up SeqHound yourself, you should review the system
requirements listed below. The SeqHound system is able to run on a number of operating
systems (we recommend and can best support a UNIX operating system like Sun Solaris
or Red Hat Linux). Setting up SeqHound will require approximately 700 GB of disk
space (see below).
Questions about system requirements, compilation, setup and maintenance can be
addressed to seqhound@blueprint.org. We will do our best to address all inquiries but
resources may not allow us to solve all problems arising on all possible set ups.
OS and hardware architecture
SeqHound code is compiled on the following platforms based on release version code.
Blueprint production SeqHound is compiled and run on Sun-Fire-880 - Sun Solaris
(version 9). We have also compiled and tested SeqHound on the Fedora Core 2.0 and
the MacOS X operating systems.
Release versions of SeqHound executables are available for.
x86 architecture (Fedora Core 2.0)
Sun-Fire-880 Sun Solaris (version 9)
PowerPC architecture MacOS X
We have also successfully built executables on the following platforms.
x86 architecture FreeBSD
x86 architecture QNX
x86 architecture Windows NT
PowerPC architecture PPC Linux
SGI Irix 6
Alpha architecture Compaq Alpha OS
HPPA 2.0 architecture HPUX 11.0
HPPA 1.1 architecture PA-RISC Linux
Memory (RAM)
We recommend a minimum of 1 GB of RAM to run the SeqHound executables.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 12 of 421 18/04/2005
Hard Disk
Source code and executables
Component Image Size
SeqHound Source and compiled 220.0 MB
NCBI Toolkit 560.0 MB
NCBI C++ Toolkit 12GB
bzip2 Library 4.5 MB
slri lib 7.3MB
slri lib_cxx 9.4 MB
Source code and executables (total) 13GB approx
Database
Component Image Size
data tables 300 GB
data tables backup 300 GB
Database (total) 700 GB*
*700GB includes 300 GB for a single copy of the SeqHound data tables. The SeqHound
system includes a second copy of the data tables used for back up and updating. We
suggest a minimum of 700 GB for SeqHound installation. This allows for yearly growth
of the data tables as well as for a RAID5 disk configuration.
We are using the MySQL database storage engine InnoDB, which provides transaction
support and automatic recovery in the event of database server outage. There is no need
to keep a separate instance of the database when the InnoDB storage engine is used. To
prevent deadlock during data insertion and update, you should not run SeqHound parsers
in parallel against the InnoDB database server. As a result, it takes up to three extra days
for the initial build of SeqHound database using the InnoDB storage engine. If you wish
to use the MyISM storage engine, you can run parallel parsers to speed up the initial
build of SeqHound. However, you will need to keep a separate database instance for
database update and backup as the storage engine MyISM does not support transaction
and automatic recovery.
Other Software
Apache
Webserver(version 1.3)
See http://www.apache.org/ for software installation for you
platform.
Apache Jakarta Tocat
JSP/Servlet Container
(version 4.1)
See http://jakarta.apache.org/tomcat/ for software installation for
you platform.
Perl (version 5.8.3) See http://www.cpan.org/ for installation for your platform.
Requiredmodules include Net/FTP.pm, sun4-solaris-64/DBI.pm
seqhound@blueprint.org Version 3.3
The SeqHound Manual 13 of 421 18/04/2005
Compiling SeqHound Code yourself.
It is not necessary to compile SeqHound executables yourself; the system may be set up
using the executables provided on the ftp site for selected Operating Systems. However,
if you wish to make use of the local API then you must compile SeqHound yourself.
ODBC compliant database engines
Blueprint uses the ODBC compliant MySQL database engine. We are using version
4.1.10 in production; this version supports nested SQL queries and internationalization.
We have not tested SeqHound on other ODBC compliant RDBMS such as Oracle, DB2
and PostgreSQL.
Library dependencies
Library Source
NCBI Toolkit from ftp://ftp.ncbi.nih.gov/toolbox/ncbi_tools/
NCBI C++ Toolkit (optional*) from ftp://ftp.ncbi.nih.gov/toolbox/ncbi_tools/
bzip2 Library from http://sourceforge.net/projects/slritools/
slri lib from http://sourceforge.net/projects/slritools/
slri lib_cxx (optional*) from http://sourceforge.net/projects/slritools/
* This library is only required if you plan to use the SeqHound remote C++ API.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 14 of 421 18/04/2005
4.3 Obtaining precompiled SeqHound executables.
It is not necessary to compile SeqHound executables yourself; the system may be set up
using the precompiled executables provided on the ftp site for selected Operating
Systems. If you choose to compile the executables yourself, skip to step 4.3.1.
You will require about 220 MB of disk space to store the SeqHound compiled
executables. These instructions assume you are logged in as user “seqhound” on a UNIX
system running the bash shell and you have perl installed on your system.
1. Decide the location to install the SeqHound binary executables. For example, if you
want to install in the directory /home/seqhound/execs, do the following:
mkdir execs
cd execs
2. Download the SeqHound installation utility script installseqhound.pl from the FTP
site: ftp.blueprint.org
ftp ftp.blueprint.org
When prompted for a name enter
anonymous
When prompted for a password type your email address:
myemail@home.com
cd pub/SeqHound/script
get installseqhound.pl
Close the ftp session by typing:
bye
3. Run the perl script to download and install SeqHound executables. The perl script
will download SeqHound binary executables based on the specified platform (linux or
solaris), unpack the tar ball, modify the configurations files .odbc.ini and .intrezrc (for
ODBC database access) and deploy the configuration files. It requires two
commandline arguments: platform (linux or solaris) and installation path (e.g.
/home/seqhound/execs). Enter the path to the ODBC driver (e.g.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 15 of 421 18/04/2005
/usr/lib/libmyodbc3.so, please refer to step 10 in section 4.4 for ODBC driver path),
database server name, port number, user id, password and database instance name
when prompted by the perl script.
./installseqhound.pl [linux OR solaris] [/home/seqhound/execs]
Upon successful execution of the perl script, you should see the following directories
in the directory execs:
build
config
example
include
lib
sql
test
updates
www
The configuration file .odbc.ini can be found in the home
directory (e.g. /home/seqhound).
seqhound@blueprint.org Version 3.3

The SeqHound Manual 16 of 421 18/04/2005
4.3.1 Obtaining SeqHound Source Code.
Follow the instructions below to download SeqHound source code . If you downloaded
and unpacked the executables, you can skip section 4.3.1 and 4.4 and continue with
section 4.5.
1. In your home directory, make a new directory where you will store the new
SeqHound code.
mkdir compile
Move into this directory and set an environment variable called COMPILE to point to
this directory.
cd compile
export COMPILE=`pwd`
(where (`) is a single back-quote)
2. Download the perl utility seqhoundsrcdownload from the SeqHound ftp site
Note: We no longer support SeqHound download from the Sourceforge
FTP site. Please download SeqHound from
ftp://ftp.blueprint.org/pub/SeqHound/
From the compile directory, type:
ftp ftp.blueprint.org
When prompted for a name enter
anonymous
When prompted for a password type your email address:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 17 of 421 18/04/2005
myemail@home.com
cd pub/SeqHound/script
get seqhoundsrcdownload.pl
Close the ftp session by typing:
bye
3. Download SeqHound source code by running the perl script seqhoundsrcdownload.pl.
The script will download the source code tar file and unpack the tar file into two
directories slri and bzip2. You will also see a release note file Release_notes_x.x.txt
in the same directory compile.
./seqhoundsrcdownload.pl
4. Set the SLRI environment variable
Move to the slri directory and set the environment variable “SLRI” to point to this
directory.
cd $COMPILE/slri
export SLRI=`pwd`
seqhound@blueprint.org Version 3.3

The SeqHound Manual 18 of 421 18/04/2005
4.4 Compiling SeqHound executables on Solaris
These instructions describe how to compile SeqHound running on the Solaris platform.
They may be used as a guide for compiling SeqHound code on other platforms.
Instructions are similar for Linux and differences are noted.
Using these instructions
These instructions assume that:
You have downloaded the SeqHound code from the ftp server and you have set
environment variables called COMPILE and SLRI. See section 4.3.1
You are using the bash shell.
Note: On Linux platforms, to compile SeqHound libs with ODBC support you also need
unixODBC-devel package which contains the sql.h + other libs/headers required to
compile SeqHound libs with ODBC support. This is not needed to run SeqHound, just to
compile it.
These instructions were tested on a Sun-Fire-880 architecture running a Sun Solaris OS
(version 9). The system information for the test-box (results of a “uname –a” call)
were:
SunOS machine_name 5.9 Generic_117171-15 sun4u sparc
SUNW,Sun-Fire-880
1. Download the NCBI toolkit
SeqHound is dependent on code in the NCBI toolkit
Move to the compile directory and ftp to the NCBI ftp site:
cd $COMPILE
ftp ftp.ncbi.nlm.nih.gov
When prompted for a name enter anonymous
When prompted for a password type myemail@home.com
cd toolbox/CURRENT
Make a note of the FAQ.html and the readme.htm files.
Change your transfer type to binary and get the zipped directory called ncbi.tar.gz
bin
get ncbi.tar.gz
Close the ftp session by typing:
bye
seqhound@blueprint.org Version 3.3
The SeqHound Manual 19 of 421 18/04/2005
Uncompress the toolkit.
gunzip ncbi.tar.gz
tar xvf ncbi.tar
2. Edit the platform make file.
Go to the platform directory and locate the file with a “.mk” extension that applies to
your platform. For 64-bit Solaris system the file is “solaris64.ncbi.mk” and in Linux
the file is linux-x86.ncbi.mk.
cd $COMPILE/ncbi
cd platform
In Linux linux-x86.ncbi.mk replace the line /home/coremake/ncbi with
${NCBI}
Use the following line (a Perl command) to replace the string in the Solaris file
/netopt/ncbi_tools/ncbi64/ncbi with the string ${NCBI}
in the solaris64.ncbi.mk file:
perl -p -i.bak -e 's|/netopt/ncbi_tools/
ncbi64/ncbi|\${NCBI}|g' solaris64.ncbi.mk
so for instance, the line
NCBI_INCDIR = /netopt/ncbi_tools/ncbi64/ncbi/include
Will become:
NCBI_INCDIR = ${NCBI}/include
You could also edit this file in hand using a text editor if you don’t have Perl
installed.
Copy the file up one level to the ncbi directory and rename it “ncbi.mk”
cp solaris64.ncbi.mk ../ncbi.mk
3. Set environment variables in preparation for the toolkit build.
Move back to the ncbi directory and set the environment variable NCBI to point to
that directory
cd $COMPILE/ncbi
export NCBI=`pwd`
check this by typing
echo $NCBI
the value shown will replace ${NCBI} in the “solaris64.ncbi.mk” file that you
modified in the above step when the make file is run.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 20 of 421 18/04/2005
Note: The make file in the NCBI toolkit will use the C compiler from Sun
instead of the compiler gcc. We do not recommend using gcc as it
generates seqhound parsers that lead to segmentation fault at run time.
Finally, paths to the compiler and the archive executable ar should be added to your
PATH variable:
export
PATH=/usr/local/bin:/opt/SUNWspro/prod/bin:/usr/ccs/bin:$
PATH
You can check all of your environment variables by typing
set | sort
At this point, the relevant environment variables should be something like this:
COMPILE=/export/home/your_user_name/compile
NCBI=/export/home/your_user_name/compile/ncbi
OSTYPE=solaris2.9
PATH=/opt/SUNWspro/prod/bin:/usr/local/bin:/usr/ccs/bin:/
usr/bin:/usr/ucb:/etc:.
If you want, you can read the readme file in the make directory.
cd make
more readme.unx
seqhound@blueprint.org Version 3.3

The SeqHound Manual 21 of 421 18/04/2005
Note: For the Solaris UNIX OS only, the SeqHound API functions
SHoundGetGenBankff and SHoundGetGenBankffList breaks
due to a bug in the NCBI library file ncbistr.c (in directory ncbi/corelib
and ncbi/build). To fix the problem, replace all the code inside the
function Nlm_TrimSpacesAroundString() in the file ncbistr.c
with the following text
char *ptr, *dst, *revPtr;
int spaceCounter = 0;
ptr = dst = revPtr = str;
if ( !str || str[0] == '\0' )
return str;
while ( *revPtr != '\0' )
if ( *revPtr++ <= ' ' )
spaceCounter++;
if ( (revPtr - str) <= spaceCounter )
{
*str = '\0';
return str;
}
while ( revPtr > str && *revPtr <= ' ' )
revPtr--;
while ( ptr < revPtr && *ptr <= ' ' ) ptr++;
while ( ptr <= revPtr ) *dst++ = *ptr++;
*dst = '\0';
return str;
seqhound@blueprint.org Version 3.3

The SeqHound Manual 22 of 421 18/04/2005
4. Build the NCBI toolkit
Move back up to the compile directory and run the make command.
cd $COMPILE
./ncbi/make/makedis.csh |& tee out.makedis.txt
Note: to build Solaris 64 bit binaries add the following to the command
line:
SOLARIS_MODE=64 ./ncbi/make/makedis.csh
This runs a c-shell script to make the toolkit and tees the output to the screen and a
log file “out.makedis.txt”. It is safe to ignore the multiple error messages that you
may see.
At the end of a successful build you will see
*********************************************************
*The new binaries are located in ./ncbi/build/ directory*
*********************************************************
The ncbi.tar file can be removed from the “compile” directory after the successful build
process has been completed.
5. Make the bzip2 library
The bzip2 code was downloaded as part of the seqhound code in step 4.3.1 above.
Move to the bzip2 directory and run the make file.
cd $COMPILE/bzip2
make –f make.bzlib
6. Set the BZDIR environment variable.
cd $COMPILE/bzip2
export BZDIR=`pwd`
7. In your home directory, add the following environment parameters to the appropriate
configuration file such as .bashrc or .bash_profile. Text in italics should be changed
to the correct path on your machine that points to directory having DBI.pm:
export NCBI=$COMPILE/ncbi
export BZDIR=$COMPILE/bzip2
export SLRI=$COMPILE/slri
export VIBLIBS="-L/usr/X11R6/lib -lXm -lXpm -lXmu -lXp -
lXt -X11 -lXext"
seqhound@blueprint.org Version 3.3

The SeqHound Manual 23 of 421 18/04/2005
export
PERL5LIB=/usr/local/lib/perl5/site_perl/5.8.3/sun4-
solaris-64
8. Install MySQL server and create database “seqhound”.
SeqHound is built and tested in MySQL version 4.1.10. You can download MySQL
from http://dev.mysql.com/downloads/mysql/4.1.html and follow the manual at
http://dev.mysql.com/doc/mysql/en/index.html to install MySQL on your server. The
data directory where the MySQL server points to should have 700 GB for a full
SeqHound database. After MySQL is installed, you need to log into MySQL and
create database “seqhound”:
create database seqhound;
Note that ";" must be used at the end of all MySQL statements.
9. Install ODBC driver:
Note that for Linux platforms, the unixODBC package needs to be
installed prior to the ODBC driver otherwise the following error will
occur:
error: Failed dependencies:
libodbcinst.so.1 is needed by MyODBC-3.51.09-1
a) Go to web site: http://dev.mysql.com/doc/connector/odbc/en/faq_2.html
b) Find and download RPM distribution of ODBC driver MyODBC-3.51.07-
1.i586.rpm.
c) As user "root", install the driver.
For first time installation
rpm -ivh MyODBC-3.51.01.i386-1.rpm
For upgrade
rpm -Uvh MyODBC-3.51.01.i386-1.rpm
d) The library file libmyodbc3. will be installed in directory /usr/lib or
/usr/local/lib.
10. Set up the configuration file for ODBC driver.
Create a configuration file called .odbc.ini in your home directory with the following
content:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 24 of 421 18/04/2005
Edit the file called .intrezrc in directory slri/seqhound/config/.
[mysqlsh]
Description = MySQL ODBC 3.51 Driver DSN
Trace = Off
TraceFile = stderr
Driver = /usr/lib/libmyodbc3.so
DSN = mysqlsh
SERVER = my_server
header must not be used for other sections
your library path
same as the header name
PORT = my_port
USER = my_id
PASSWORD = my_pwd
DATABASE = seqhound
Text in italics should be changed. Text /usr in the value of variable Driver
should be changed to the path where unixodbc resides. Text my_server should be
changed to the IP address or the server name of the MySQL server. Text my_port
should be changed to port number of the MySQL instance. Text my_id and my_pwd
should be replaced by your user id and password to the MySQL database.
database name
Note that the values for the headers such as DSN, USER, PASSWORD and
DATABASE must be less than 9 characters.
11. Set up ODBC related variables:
export ODBC=path_to_unixodbc
Where path_to_unixodbc should be replaced by the path of the UnixODBC
driver on your machine.
In your home directory, add parameter “LD_LIBRARY_PATH” to the appropriate
configuration file such as .bashrc or .bash_profile:
export LD_LIBRARY_PATH =
/usr/local/unixodbc/lib:/usr/local/unixodbc/odbc/lib:/usr
/local/mysql/lib/mysql:/usr/local/mysql/lib/mysql/lib
The value of variable “LD_LIBRARY_PATH” should have all the paths that have the
library files libodbc*, libmyodbc*, and libmysqlclient*
12. Build the SeqHound executables
Move to the compile directory and list all the files in the directory:
cd $COMPILE
ls
You should see:
> ls
bzip2
ncbi
slri
out.makedis.txt
seqhound@blueprint.org Version 3.3
The SeqHound Manual 25 of 421 18/04/2005
Before proceeding you should check your environment variables
set | sort
to ensure that correct paths have been specified for each of the following variables:
NCBI
SLRI
ODBC
BZDIR
Compile the SLRI libraries using the following commands:
cd $SLRI/lib
make -f make.slrilib
make -f make.slrilib odbc
The above commands will build the SLRI libraries needed by SeqHound.
The make files which you are about to invoke call on these variables therefore the
paths must be correct. Move to the make directory for SeqHound and run the makeall
script. The script requires two command line arguments. The first parameter indicates
what database backend is to be used for the build (currently the only valid target is
odbc). The second parameter indicates what SeqHound programs are to be made (a
choice of all, cgi, domains, examples, genomes, go,
locuslink, parsers,scripts, taxon, updates). The output of the
build script will be captured in the text file out.makeseqhound.txt.
cd $SLRI/seqhound
./makeallsh odbc all 2>&1 | tee out.makeseqhound.txt
It is safe to ignore the multiple warning messages that you may see.
After this has finished running, move to the directory slri/seqhound/build/odbc/
where you will find the executables for SeqHound.
cd build/odbc
ls -1
You will see
>ls –1
addgoid
cbmmdb
chrom
clustmask
clustmasklist
comgen
fastadom
gen2fasta
gen2struc
goparser
goquery
seqhound@blueprint.org Version 3.3

The SeqHound Manual 26 of 421 18/04/2005
histparser
importtaxdb
isshoundon
llgoa
llparser
llquery
mother
pdbrep
precompute
redund
seqrem
sh_nbhrs
shunittest_odbc_local
shunittest_odbc_rem
shtest
update
vastblst
wwwseekgi
13. Set up the SQL files that create tables.
cd $SLRI/seqhound/sql
In each of files core.sql, redund.sql, ll.sql, taxdb.sql, gendb.sql,
strucdb.sql, cddb.sql, godb.sql, rps.sql, nbr.sql, there is a line close to
the beginning of each file:
#use testsql;
This line should be changed to
use seqhound;
4.5 Building the SeqHound system on Solaris
Using these instructions
These instructions show how the SeqHound executables may be used to build the
SeqHound system under a Solaris 8 OS. These instructions may also be used as a guide
for setting up SeqHound under other operating systems. These instructions assume that:
• You have downloaded the latest release version of the SeqHound code (see step
4.3.3)
• You have successfully installed MySQL
• You have successfully compiled the SeqHound code yourself (section 4.4)
OR
you have downloaded the SeqHound executables for your platform and operating
system (section 4.3.4).
• You have set environment variables called COMPILE and SLRI (see steps 4.3.1 and
4.3.6).
seqhound@blueprint.org Version 3.3

The SeqHound Manual 27 of 421 18/04/2005
• You have a default install of an Apache server running. See http://www.apache.org/
for freely available software and instructions for your platform.
• You have installed Perl. See http://www.cpan.org/ for freely available software and
installation instructions.
• You have at least 300 MB space available in a directory where you can check out
code and compile it.
• You have at least 600 GB available for the SeqHound executables and data tables.
See section 4.2.
These instructions were tested on a Sun Ultra machine running the Sun-Solaris 8 OS. The
system information for the test-box (results of a “uname –a” call) were:
SunOS machine_name 5.8 Generic_108528-01 sun4u sparc
SUNW,Ultra-4
These instructions assume that you are using the c shell. Syntax may differ for some
commands in other shells.
Note: These instructions begin with ‘step 14’.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 28 of 421 18/04/2005
14. Prepare to build the SeqHound database.
Create a new directory where you will set up SeqHound.
mkdir seqhound
Set the environment variable SEQH to point to this directory.
cd seqhound
setenv SEQH `pwd`
Move to this directory and create new directories
cd seqhound
mkdir 1.core.files
mkdir 2.redund.files
mkdir 3.taxdb.files
mkdir 4.godb.files
mkdir 5.lldb.files
mkdir 6.comgenome.files
mkdir 7.mmdb.files
mkdir 8.hist.files
mkdir 9.neighbours.files
mkdir 10.rpsdb.files
mkdir precompute
The numbered directories will hold parsers and files required for the build of the
SeqHound data tables. Directory “precompute” will hold the precomputed data of the
database.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 29 of 421 18/04/2005
Move to each of the numbered directories and copy all of the scripts and executables
required for the build.
cd $SEQH/1.core.files
cp $SLRI/seqhound/sql/core.sql .
cp $SLRI/seqhound/scripts/asnftp.pl .
cp $SLRI/seqhound/scripts/seqhound_build.sh .
cp $SLRI/seqhound/build/odbc/mother .
cp $SLRI/seqhound/build/odbc/update .
cp $SLRI/seqhound/config/.intrezrc .
cd $SEQH/2.redund.files
cp $SLRI/seqhound/sql/redund.sql .
cp $SLRI/seqhound/scripts/nrftp.pl .
cp $SLRI/seqhound/build/odbc/redund .
cd $SEQH/3.taxdb.files
cp $SLRI/seqhound/sql/taxdb.sql .
cp $SLRI/seqhound/scripts/taxftp.pl .
cp $SLRI/seqhound/build/odbc/importtaxdb .
cd $SEQH/4.godb.files
cp $SLRI/seqhound/sql/godb.sql .
cp $SLRI/seqhound/scripts/goftp.pl .
cp $SLRI/seqhound/build/odbc/goparser .
cd $SEQH/5.lldb.files
cp $SLRI/seqhound/sql/ll.sql .
cp $SLRI/seqhound/scripts/llftp.pl .
cp $SLRI/seqhound/build/odbc/llparser .
cp $SLRI/seqhound/build/odbc/addgoid .
cd $SEQH/6.comgenomes.files
cp $SLRI/seqhound/sql/gendb.sql .
cp $SLRI/seqhound/scripts/genftp.pl .
cp $SLRI/seqhound/scripts/humoasn.pl .
cp $SLRI/seqhound/scripts/humouse_build.sh .
cp $SLRI/seqhound/scripts/comgencron_odbc.pl .
cp $SLRI/seqhound/scripts/shconfig.pm .
cp $SLRI/seqhound/genomes/gen_cxx .
cp $SLRI/seqhound/genomes/pregen.pl .
seqhound@blueprint.org Version 3.3

The SeqHound Manual 30 of 421 18/04/2005
cp $SLRI/seqhound/genomes/gen.pl .
cp $SLRI/seqhound/genomes/ncbi.bacteria.pl .
cp $SLRI/seqhound/build/odbc/chrom .
cp $SLRI/seqhound/build/odbc/comgen .
cp $SLRI/seqhound/build/odbc/mother .
cd $SEQH/7.mmdb.files
cp $SLRI/seqhound/sql/strucdb.sql .
cp $SLRI/seqhound/scripts/mmdbftp.pl .
cp $SLRI/seqhound/config/.mmdbrc .
cp $SLRI/seqhound/config/.ncbirc .
cp $SLRI/seqhound/build/odbc/cbmmdb .
cd $SEQH/8.hist.files
cp $SLRI/seqhound/build/odbc/histparser .
Open the .intrezrc file with a text editor like pico and edit.
cd $SEQH/1.core.files
pico .intrezrc
An example .intrezrc file follows. Lines preceded by a semi-colon are comments that
explain what the settings are used for and their possible values.
Text in italics must be changed for the .intrezrc file to function correctly with
your SeqHound set-up. Variables username, password, dsn, database in
section [datab] should have the same values as USER, PASSWORD, DSN and
DATABASE respectively in the .odbc.ini file you set up in Step 10 in section 4.4. For
variable path and indexfile in section [precompute], replace the text in
italics with the absolute path of directory “precompute” you just created.
Warning: This file may have wrapped lines. Take care when editing this
file that you do not break any of the lines (i.e. introduce any unwanted
carriage returns).
seqhound@blueprint.org Version 3.3
The SeqHound Manual 31 of 421 18/04/2005
-------------------------------example .intrezrc begins--------------------------------
[datab]
;seqhound database that you are connecting
username=your_user_name
password=your_pass_word
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
[config]
;the executable the cgi runs off of.
CGI=wwwseekgi
[precompute]
;precomputed taxonomy queries
MaxQueries = 100
MaxQueryTime = 10
QueryCount = 50
path = /seqhound/precompute/
indexfile = /seqhound/precompute/index
[sections]
;indicated what modules are available in SeqHound
;1 for available, 0 for not available
;gene ontology hierarchy
godb = 1
;locus link functional annotations
lldb = 1
;taxonomy hierarchy
taxdb = 1
;protein sequence neighbours
neigdb = 1
;structural databases
strucdb = 1
;complete genomes tracking
gendb = 1
;redundant protein sequences
redundb = 1
;open reading frame database
;currently not exported to outside users of SeqHound
cddb = 0
;RPS-BLAST domains
rpsdb = 1
;DBXref Database Cross_Reference
dbxref = 0
[crons]
;customizable variables in cron jobs
;NOTE: all paths must end in '/'
pathupdates=./
pathinputfiles=./
pathinputfilescomgen=./
mail=user\@host.org
defaultrelease=141
pathflags=./
-------------------------------example .intrezrc ends----------------------------------
This file should be copied to other directories used during the build process:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 32 of 421 18/04/2005
cp .intrezrc $SEQH/2.redund.files/.
cp .intrezrc $SEQH/3.taxdb.files/.
cp .intrezrc $SEQH/4.godb.files/.
cp .intrezrc $SEQH/5.lldb.files/.
cp .intrezrc $SEQH/6.comgenome.files/.
cp .intrezrc $SEQH/7.mmdb.files/.
cp .intrezrc $SEQH/8.hist.files/.
cp .intrezrc $SEQH/9.neighbours.files/.
cp .intrezrc $SEQH/10.rpsdb.files/.
15. Build the core module of SeqHound.
Building the core module (basically all of the sequence data tables) is not optional.
The rest of the modules are optional if there is a need to spare resources or
administrative efforts but the corresponding API functionality will not be present.
cd $SEQH/1.core.files
Create the core tables in the database
Make sure file core.sql has line use seqhound close to the beginning of the file.
mysql –u my_id –p –P my_port –h my_server < core.sql
Where my_id, my_port and my_server should be replaced by your userid for
the database, the port of the database and the IP address or the server name of the
database server respectively. You will be prompted to enter your password.
This creates core tables accdb, asndb, nucprot, parti, pubseq, sendb, sengi, taxgi,
bioentity, bioname, secondrefs, bioentitytype, nametype, rules, fieldtype and histdb.
If you are building a full-instance of the SeqHound database then run the asnftp.pl
script while in the build directory:
./asnftp.pl
Note that any command in these instructions can be run as a ‘nohup’ to
prevent the process from ending if your connection to the machine should
be lost. For example:
nohup ./asnftp.pl &
If you only want to build a small test version of the database then manually download
a single file. For example:
ftp ftp.ncbi.nih.gov
When prompted for a name enter anonymous
When prompted for a password type myemail@home.com
cd refseq/cumulative
bin
get rscu.bna.Z (do not uncompress this file)
bye
seqhound@blueprint.org Version 3.3

The SeqHound Manual 33 of 421 18/04/2005
The asnftp.pl script downloads all of the GenBank sequence records (in binary ASN.1
format) required to make an initial build of the SeqHound core module. This script
will take approximately 24 hours to run and will consume 14 GB of disk space.
Note that all scripts are described in detail in section 5.
Two other files are generated by this script:
asn.list is a list of the sequence files that the script intends to download.
asnftp.log is where the script logs error messages during execution time.
If you open another session with the machine where you are building SeqHound, you
can check how far along asnftp.pl is by comparing the number of lines in the asn.list
file
grep “.aso.gz” asn.list | wc –l
to the number of lines in the build directory (number of files actually downloaded so
far)
ls *.aso.gz | wc -l
Once asnftp has finished, these two numbers should be the same.
Run the seqhound build script. Before running this script, make certain that the
.intrezrc file, in the same directory, and .odbc.ini, in your home directory, have
correct configuration values. (see steps 10 in section 4.4 and step 14 in the current
section). This parser MUST be given a single parameter that represents the release
version of GenBank. You can find the release number in the file:
ftp://ftp.ncbi.nih.gov/ncbi-asn1/daily/Last.Release.
./seqhound_build.sh 141
seqhound_build.sh executes the mother parser over all source files and populates
tables accdb, asndb, nucprot, parti, pubseq, sendb, sengi, taxgi, bioentity, bioname,
secondrefs, bioentitytype, nametype, rules and fieldtype. This will take about 75
hours. Table histdb is still empty at this stage. It is populated in Step 25.
Parser mother creates a log file for every *.aso file that it parses. These log files are
located in a subdirectory called “logs” and are named “rsnc0506run” where
“rsnc0506” is the name of the file that was being processed.
While seqhound_build.sh is running, you can move on to steps 16-18.
Once seqhound_build.sh has finished you can test that all of the files were properly
processed by showing that the results of
cd logs
grep “Done” | wc –l
is the same as
ls *run | wc –l
is the same as
cd ..
ls *aso.gz | wc -l
seqhound@blueprint.org Version 3.3
The SeqHound Manual 34 of 421 18/04/2005
The seqhound_build.sh script unzips .aso.gz files before feeding them as input to the
mother program. seqhound_build.sh then rezips the file after mother is done with it.
If for some reason, the build should crash part way through, you have to
a) recreate core tables using core.sql (see above) and
b) search for any unzipped (*.aso files) in the build directory and rezip them
c) restart seqhound_build.sh.
Once the seqhound_build.sh script has finished, you should move all of the *.aso.gz
files into a directory where they will be out of the way:
mkdir asofiles
mv *.aso.gz asofiles/.
16. Build the redundb module.
cd $SEQH/2.redund.files
Create table redund in the database.
Make sure file redund.sql has the line use seqhound close to the beginning of the
file.
mysql –u my_id –p –P my_port –h my_server < redund.sql
Where my_id”, “my_port” and “my_server” should be replaced by your userid
for the database, the port of the database and the IP address or the server name of the
database server respectively. You will be prompted to enter your password.
This creates table redund in the database.
Run the nrftp.pl script to download the FASTA nr database of proteins
(ftp://ftp.ncbi.nlm.nih.gov/blast/db).
./nrftp.pl
nrftp.pl generates a log file “nrftp.log” that informs you what happened. If everything
went ok, the last two lines should read:
Getting nr.gz
closing connection
A new file should appear in the build directory called “nr.Z”. You will have to
unpack this file by typing:
gunzip nr.gz
Run the redund parser to make the redund table of identical protein sequences.
Before running this script, make certain that the .intrezrc file in the same directory
and .odbc.ini in your home directory have correct configuration values (see step 10 in
section 4.4 and step 14 in the current section).
./redund -i nr -n F
redund generates the log file “redundlog”. If everything went ok, the only line in this
file should be:
NOTE: [000.000] {redund.c, line 259} Done.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 35 of 421 18/04/2005
And about 3 millions records will be inserted into table redund.
17. Build the taxdb module
Create tables of the taxdb module in the database.
cd $SEQH/3.taxdb.files
Make sure file taxdb.sql has line use seqhound close to the beginning of the file.
mysql –u my_id –p –P my_port –h my_server < taxdb.sql
Where my_id, my_port and my_server should be replaced by your userid for
the database, the port of the database and the IP address or the server name of the
database server respectively. You will be prompted to enter your password.
This creates tables taxdb, gcodedb, divdb, del, merge in the database.
Run the taxftp.pl script to download taxonomy info from the NCBI
(ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz).
taxftp.pl
taxftp.pl generates a log file taxftp.log that informs you what happened. If everything
went ok, the last two lines should read:
Getting taxdump.tar.gz
closing connection
A new file should appear in the build directory called taxdump.tar.gz. You will have
to unpack this file by typing:
gzip –d taxdump.tar.gz
tar -xvf taxdump.tar
There will be seven new files:
delnodes.dmp
division.dmp
gc.prt
gencode.dmp
merged.dmp
names.dmp
nodes.dmp
Run the importtaxdb parser to make the taxonomy data tables. Taxdump must be in
the same directory as this parser.
./importtaxdb
importtaxdb has no command line parameters. importtaxdb generates the log file
importtaxdb_log.txt. If everything went ok, the output of this file should be
something like:
Program start at Thu Sep 4 13:47:51 2003
Number of Tax ID records parsed: 191647
Number of Tax ID Name records parsed: 246263
Number of Division records parsed: 11
Number of Genetic Code records parsed: 18
seqhound@blueprint.org Version 3.3
The SeqHound Manual 36 of 421 18/04/2005
Number of Deleted Node records parsed: 25475
Number of Merged Node records parsed: 4607
Program end at Thu Aug 12 13:49:43 2004
And records will be inserted into tables taxdb, gcodedb, divdb, del and merge.
18. Build the GODB module
Create tables of the godb module in the database.
cd $SEQH/4.godb.files
Make sure file godb.sql has line use seqhound close to the beginning of the file.
mysql –u my_id –p –P my_port –h my_server < godb.sql
Where my_id, my_port and my_server should be replaced by your userid for
the database, the port of the database and the IP address or the server name of the
database server respectively. You will be prompted to enter your password.
This creates tables go_parent, go_name, go_reference, go_synonym in the database.
Run the goftp.pl script to download the gene ontology files
(ftp://ftp.geneontology.org/pub/go/gene-associations and
ftp://ftp.geneontology.org/pub/go/ontology).
goftp.pl
There is a log file for this script called goftp.log that indicates that it got all of these
files. Three new files should appear in the build directory called
component.ontology
function.ontology
process.ontology
Two other files also appear called
gene_association.Compugen.GenBank.gz
gene_association.Compugen.UnitProt.gz
but these are used as input files by addgoid in the next step.
Run the goparser to make the hierarchical gene ontology data tables. The three input
files must be in the same directory as this parser.
./goparser
goparser has no command line parameters. goparser generates the log file
goparserlog. If everything went ok, the output of this file should have only one
NOTE line:
NOTE: [000.000] {goparser.c, line 101} Main: Done!
And records will be inserted into tables go_parent, go_name, go_reference,
go_synonym.
19. Build the LLDB module
Create tables of the locus link module in the database.
cd $SEQH/5.lldb.files
Make sure file ll.sql has line use seqhound close to the beginning of the file.
mysql –u my_id –p –P my_port –h my_server < ll.sql
seqhound@blueprint.org Version 3.3
The SeqHound Manual 37 of 421 18/04/2005
Where my_id, my_port and my_server should be replaced by your userid for
the database, the port of the database and the IP address or the server name of the
database server respectively. You will be prompted to enter your password.
This creates tables ll_omim, ll_go, ll_llink, ll_cdd in the database.
Run the llftp.pl script to download the locus link template file (LL_tmpl) which is the
source for function annotation tables
(ftp://ftp.ncbi.nih.gov/refseq/LocusLink/LL_tmpl.gz).
llftp.pl
This script generates the llftp.log file. If everything executes correctly, the last two
lines of the file should read:
Getting LL_tmpl.gz
closing connection
And a new file should appear in the build directory called LL_tmpl.gz which you will
have to unpack using the commands
gzip –d LL_tmpl.gz
Run the llparser to create the set of functional annotation data tables. The input file
must be in the same directory as this parser.
./llparser
llparser has no command line parameters. llparser generates the log file
“llparserlog”. At the time of writing, the output of this file will have thousands of
lines like:
NOTE: [000.000] {ll_cb.c, line 654} LL_AppendRecord: No
NP id. Record skipped.
(these lines are expected since many LocusLink records are not linked to specific
sequence records)
followed by the last line of the file:
NOTE: [000.000] {llparser.c, line 90} Main: Done!
Records will be inserted into tables ll_omim, ll_go, ll_llink and ll_cdd. Run the
addgoid parser to populate the go annotation table. This parser uses input files that
were downloaded in the previous step 13. Copy those files to this directory:
cp ../4.godb.files/gene_association.Compugen.GenBank.gz
./
cp ../4.godb.files/gene_association.Compugen.UniProt.gz
./
The files need to be unpacked.
gunzip gene_association.Compugen.GenBank.gz
gunzip gene_association.Compugen.UnitProt.gz
The input files must be in the same directory as addgoid
./addgoid –i gene_association.Compugen.GenBank
after this parser has finished, use it to parse the other input file
./addgoid –i gene_association.Compugen.UniProt
seqhound@blueprint.org Version 3.3

The SeqHound Manual 38 of 421 18/04/2005
At the time of writing, this second input file is not parsed since cross references
between Swissprot and GenBank ids are not available. This is being corrected by the
dbxref module project.
addgoid MUST BE EXECUTED AFTER ALL CORE TABLES AND
LLDB TABLES HAVE BEEN BUILT; the llparser makes the ll_go table
into which the addgoid script writes. This program is dependent on tables
asndb, parti, accdb and nucprot..
addgoid generates the log file addgoidlog. The output of this file will look like:
=========[ Sep 5, 2003 10:28 AM ]========================
ERROR: [000.000] {addgoid.c, line 235} No GI from 100K_RAT.
ERROR: [000.000] {addgoid.c, line 235} No GI from 100K_RAT.
ERROR: [000.000] {addgoid.c, line 235} No GI from 100K_RAT.
ERROR: [000.000] {addgoid.c, line 235} No GI from 100K_RAT.
This is normal. These errors are caused by the inability to find GI’s for names of
proteins/loci that are annotated in the GO input file. This problem is being addressed
by the dbxref module.dir
This program writes to the existing ll_go table that was generated by llparser.
20. Build the GENDB module
Change directories to the Complete Genomes directory (comgenomes).
cd $SEQH/6.comgenomes.files
Create tables of the GENDB module in the database.
Make sure file gendb.sql has line use seqhound close to the beginning of the file.
mysql –u my_id –p –P my_port –h my_server < gendb.sql
Where my_id, my_port and my_server should be replaced by your userid for
the database, the port of the database and the IP address or the server name of the
database server respectively. You will be prompted to enter your password.
This creates table chrom in the database.
Building the GENDB module involves several steps. To simplify the process, a perl
script, comgencron_odbc.pl groups together all of the necessary scripts or binaries for
each individual step. These scripts and binaries must be present in this directory.
They are:
comgencron_odbc.pl
shconfig.pm
gen_cxx
pregen.pl
gen.pl
ncbi.bacteria.pl
genftp.pl
humoasn.pl
chrom
seqhound@blueprint.org Version 3.3
The SeqHound Manual 39 of 421 18/04/2005
iterateparti
humouse_build.sh
mother
comgen
Before building the GENDB module, the [crons] section in configuration file
.intrezrc should be set up properly. It should look like the following. Text in
italics must be changed. Variable mail should have the e-mail address where
you want the message to be sent to. Variable defaultrelease should have the
release number of the GenBank files you use to build the core tables of SeqHound
database (see Step 15):
[crons]
;customizable variables in cron jobs
;NOTE: all paths must end in '/'
pathupdates=./
pathinputfiles=./
pathinputfilescomgen=./genfiles/
mail=your_email_addr
defaultrelease=141
pathflags=./flag/
Make a subdirectory flag where the flag file comgen_complete.flg will be saved.
mkdir flag
Run the script to build the GENDB module:
./comgencron_odbc.pl
comgencron_odbc.pl generates flat file genff, log files bacteria.log, chromlog,
comgenlog, gen.log, iteratapartilog, a subdirectory genfiles and a lot of logs file with
postfix run which will be moved to a subdirectory logs. It also downloads many .asn
files which will be moved to subdirectory genfiles. During the process, temporary file
comff and directory asn are created. They are deleted before the end of the build
process. If the build process fails in the middle, they should be removed along with
file genff manually.
There are several lines printed on the screen during the build like:
mail = your_email_addr
pathupdates = ./
pathinputfilescomgen = ./genfiles/
defaultrelease = 141
pathflags = ./flag/
No source or subsource Plasmpdium falciparum NC_03043.
Update 1 chromosome type by hand.
It is OK to see above line.
An e-mail will be sent to the address you provide to inform if the process succeeds or
fails. If everything went ok, you will see the last line in file comgenlog as:
NOTE: [000.000] {comgen.c, line 504} Main: Done.
The last line in file iteratepartilog as:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 40 of 421 18/04/2005
NOTE: [000.000] {iterateparti.c, line 170} Done.
The last line in file chromlog as:
NOTE: [000.000] {chrom.c, line 173} Done.
The last two lines in file bacteria.log as:
deleteing asn
See bacteria.results for changes to ./genff
The last two lines in file gen.log as:
Removing asn
Deleting comff
The following is a detailed explanation of the script comgencron_odbc.pl. You may skip
it.
21. Generate flat file genff.
genff is a tab-delimited text file where each line in this file represents one "DNA unit"
(chromosome, plasmid, extrachromosomal element etc.) belonging to a complete
genome.
Column Description
1 Taxonomy identifier for the genome
2 Unique integer identifier for a given chromosome
3 Type of molecule (1 or chromosome, 8 for plasmid, …)
4 FTP file name for the genome without the .asn
extension)
5 Full name of the organism
Here is an example of several rows from genff:
305 286 8 NC_003296 Ralstonia solanacearum plasmid pGMI1000MP
258594 287 1 NC_005296 Rhodopseudomonas palustris CGA009 chromosome
781 288 1 NC_003103 Rickettsia conorii chromosome
782 289 1 NC_000963 Rickettsia prowazekii chromosome
90370 290 1 NC_003198 Salmonella typhi chromosome
90370 291 8 NC_003384 Salmonella typhi plasmid pHCM1
90370 292 8 NC_003385 Salmonella typhi plasmid pHCM2
209261 293 1 NC_004631 Salmonella typhi Ty2 chromosome
The genff flat file is generated in two steps.
a) gen.pl which will CREATE genff using the eukaryotic complete genomes.
b) ncbi.bacteria.pl which will UPDATE genff with bacteria complete genomes.
* both gen.pl and ncbi.bacteria.pl are dependent on pregen.pl so this must be in the
same directory as gen.pl and ncbi.bacteria.pl when you run it.
gen.pl will backup the current (if it exists) genff as genff.backup and then create a new
genff file. gen.pl will download asn files from NCBI’s ftp site and then extract the
relevant fields (as described above) and store them as records in genff.
The data of bacteria complete genome is written to genff by running ncbi.bacteria.pl.
This perl utility will compare the data in genff to the contents of the
/genomes/bacteria directory in NCBI’s ftp site and then automatically update genff.
ncbi.bacteria.pl will save the names of the bacteria that have been newly added to
genff in a separate file called bacteria.results. You can use this file to quickly verify
the results.
A sample output of bacteria.results.pl
seqhound@blueprint.org Version 3.3

The SeqHound Manual 41 of 421 18/04/2005
***********PERFECT MATCH***********
Aeropyrum pernix
***********SEMI MATCHED NCBI BACTERIA*************
NCBI BACTERIA CHROMFF
----------------------------------------
Buchnera aphidicola Buchnera sp
Buchnera aphidicola Sg Buchnera sp
***********UNMATCHED NCBI BACTERIA*************
Agrobacterium tumefaciens C58 Cereon
Agrobacterium tumefaciens C58 UWash
Perfectly matched bacteria are already present in genff. Semi matched bacteria means
that there is an organism that is closely related to a new organism. For the above
example, Buchnera aphidocola Sg and Buchnera aphidocola were newly released and
closely related to the Buchnera sp. The newly released data will have been added to
genff. Unmatched bacteria are completely new organism and will be added to genff.
Both gen.pl and ncbi.bacteria.pl will create an intermediate file called comff, and a
temporary directory asn. These are temporary and are critical to the functionality of
the perl scripts. Both gen.pl and ncbi.bacteria.pl will delete comff and asn after
execution.
While running gen.pl and ncbi.bacteria.pl you may see the following on the screen.
No source or subsource Plasmodium falciparum NC_03043.
Update 1 chromosome type by hand.
It means that for the specified organism, the asn file is missing the chromosome type.
In such a scenario, the chromosome type will default to 1 (chromosome
Once you have generated file genff, you will likely need to run it again periodically,
in case some of the data in genff has changed, for example if an organism taxid
changes, in which case it is crucial to rerun gen.pl.
Script genftp.pl downloads complete genome files from
ftp://ftp.ncbi.nih.gov/genomes/*.
A script called humoasn.pl must be in the same directory as genftp.pl since genftp.pl
calls the script.
humoasn.pl is a misnomer because the script actually processes files for
human, mouse AND rat genomes.
Each of these genomes has two files called rna.asn and protein.asn (these files are
called the same thing regardless of the organism that they refer to: the only way you
can tell which organism the file refers to is by looking at the directory name that it
came from or by looking at the contents. genftp.pl renames rna.asn and protein.asn
files to more specific names so they can be processed with the humoasn.pl script.
rna.asn and protein.asn files mostly contain XM’s and XP’s sequences: see for
example genomes/H_sapiens/protein. The sequences in these files are “loose”
bioseqs that have to be “stitched” together into bioseq sets by humoasn.pl. This
allows these sequences to be processed by the mother parser in the next step.
Many new *.asn files will appear in the comgenomes directory after this is run. There
is no log file for this script.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 42 of 421 18/04/2005
a) Populate table chrom
Binary chrom is used to populate table chrom from the list of complete genomes
found in genff. Chrom generates the log file “chromlog”. This log will look
something like:
============[ Sep 5, 2003 2:30 PM ]====================
NOTE: [000.000] {chrom.c, line 130} Assigned TaxId 56636.
NOTE: [000.000] {chrom.c, line 137} Assigned Kloodge 1.
NOTE: [000.000] {chrom.c, line 144} Assigned Chromfl 1.
NOTE: [000.000] {chrom.c, line 149} Assigned Access NC_000854
NOTE: [000.000] {chrom.c, line 152} Assigned Name Aeropyrum pernix.
…
NOTE: [000.000] {chrom.c, line 167} Done.
b) Delete all records from division gbchm from the tables of the core module.
This step is carried out for data integrity purpose. All the records that are inserted
into the core module tables are labeled as from division gbchm. Before they are
inserted, it needs to ensure no such record exists in the database. This is
accomplished using binary iterateparti. iterateparti takes the division name as one
parameter and deletes all GI’s that are part of that division from all of the tables in
the core module.
c) Set kloodge to 0 in table taxgi
This step is also carried out for data integrity purpose. The field “kloodge” in
table taxgi for all records should be set to 0 before they are updated in a later step
by binary comgen.
d) Move all Apis mellifera related files to a subdirectory.
The chromosome, rna and protein files of Apis mellifera are not processed at the
time of writing. They are moved to a subdirectory.
e) Add records to the core module tables.
Since the human, mouse and rat sequences from this source (the “Complete
Genomes” directory) are not a part of the GenBank release, the records are added
to the core module tables by script humouse_build.sh. 141
This script feeds all chromosome, rna and protein files downloaded by genftp.pl to
the mother parser. The mother parser makes a new division called “gbchm”
(GenBank Chromosome Human and Mouse) and touches all core module tables.
Log files will be created by mother for every chromosome file processed (called
*run).
f) Update field kloodge in table taxgi and field name in table accdb
Parser comgen is used to label sequences as belonging to a complete genome.
This program uses the files downloaded by genftp.pl and marks the complete
genomes in table taxgi. This program also adds loci names into table accdb (if
they are not present). comgen is dependent on the chrom table and writes to
accdb and taxgi. The comgen program has to be executed after all databases are
built.
Comgen writes to the log file comgenlog in the same directory where it is run.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 43 of 421 18/04/2005
22. Build the Strucdb module
Change to the mmdbdata directory.
cd $SEQH/7.mmdb.files
Create tables of the Strucdb module in the database.
Make sure file strucdb. sql has line use seqhound close to the beginning of the file.
mysql –u my_id –p –P my_port –h my_server < gendb.sql
Where my_id, my_port and my_server should be replaced by your userid for
the database, the port of the database and the IP address or the server name of the
database server repectively. You will be prompted to enter your password.
This creates tables mmdb, mmgi and domdb in the database.
Make certain that the configuration files have been properly set up. These include:
.mmdbrc, .ncbirc and .intrezrc.
In file .mmdbrc, variable “Gunzip” should have a value which is the path of
gunzip on the machine (change text in italics). File .mmdbrc looks like:
[MMDB]
;Database and Index required when local MMDB database is used
Database = ./
Index = mmdb.idx
Gunzip = /bin/gunzip
; [VAST]
;Database required for local VAST fetches.
; Database = .
In file .ncbirc, variable DATA should have a value which is the path of directory
ncbi/data on your machine. File .ncbirc looks like (change text in italics):
[NCBI]
ROOT=/
DATA=/my_home/compile/ncbi/data/
Copy file bstdt.val from the ncbi/data directory:
cp ~/compile/ncbi/data/bstdt.val ./
Run the mmdbftp.pl script to download the mmdb (Molecular Model Database)
ASN.1 files from ftp://ftp.ncbi.nih.gov/mmdb/mmdbdata. This will take
approximately 10 hours..
./mmdbftp.pl
This script writes to the mmdb.log file and records the files downloaded.
Approximately 20000 *.val.gz files will appear in the mmdbdata directory after
running this. Look at the first line in the mmdb.idx index file and this states the
number of files that should have been downloaded.
Run the cbmmdb parser to make the MMDB and MMGI datafiles. Use:
./cbmmdb –n F -m F
This program takes about 12 hours to run and writes errors to the cbmmdblog file.
After a typical run this file will contain:
============[ Nov 3, 2003 1:21 AM ]======================
ERROR: [004.001] {cbmmdb.c, line 125} Error opening MMDB id 22339
seqhound@blueprint.org Version 3.3

The SeqHound Manual 44 of 421 18/04/2005
WARNING: [011.001] {cbmmdb.c, line 240} Total elapsed time: 41857 seconds
NOTE: [000.000] {cbmmdb.c, line 245} Main: Done!
And records are inserted into tables mmdb and mmgi.
Run the vastblst parser to make the DOMDB datafile.
./vastblst –n F
This program writes errors to the vastblstlog file. After a typical run this file will
contain no messages and records are inserted into table domdb
In addition, vastblst makes a FASTA datafile of domains called mmdbdom.fas in the
directory where it is run.
Get the most recent nrpdb.* file from the NCBI ftp site in hand
(ftp://ftp.ncbi.nih.gov/mmdb/nrtable/nrpdb
Run the pdbrep parser to label representatives of nr chain sets in the domdb datatable.
This parser writes to the domdb table. Use:
uncompress nrpdb*.Z
pdbrep –i nrpdb.*
Where nrpdb.* is the name of the input file set. pdbrep will write errors to the
pdbreplog file in the same directory where it is run.
23. Build the Neighdb module
The sequence neighbours tables can be downloaded from
ftp://ftp.blueprint.org/pub/SeqHound/NBLAST/ as MySql database table files, as well
as mysqldump output, which should be adaptable to most SQL database systems. See
the readme on the ftp site for information on these files. To incorporate the mysql
database table files into your instance of seqhound, simply copy the files extracted
from the nblastdb and blastdb archives, downloaded from the ftp site, into your
seqhound database directory in your mysql instance. To incorporate the mysql dumps
of these tables into your seqhound instance, you need only pipe the contents of the
dump(which are SQL statements) to your database server. In the case of mysql,
simply execute:
gunzip -c seqhound.blastdb.SQLdump.YYYYMMDD.gz | mysql seqhound
gunzip -c seqhound.nblastdb.SQLdump.YYYYMMDD.gz | mysql seqhound
Be sure to fill in any required mysql options, such as username, hostname and
port number.
24. Build the Rpsdb and Domname modules
The pre-computed rps-blast table and the domname table can be downloaded from
ftp://ftp.blueprint.org/pub/SeqHound/RPS/ as MySQL database table files, as well as
mysqldump output, which should be adaptable to most SQL database systems. To
incorporate the mysql database table files into your instance of seqhound, simply
copy the files extracted from the rpsdb and domname archive, downloaded from the
ftp site, into your seqhound database directory in your mysql instance. To
incorporate the mysql dumps of these tables into your seqhound instance, you need
only pipe the contents of the dump(which are SQL statements) to your database
server. In the case of mysql, simply execute:
gunzip -c seqhound.rpsdb.SQLdump.YYYYMMDD.gz | mysql seqhound
gunzip -c seqhound.domname.SQLdump.YYYYMMDD.gz | mysql seqhound
seqhound@blueprint.org Version 3.3

The SeqHound Manual 45 of 421 18/04/2005
Be sure to fill in any required mysql options, such as username, hostname and port
number.
25. Build the histdb table.
cd $SEQH/8.hist.files
./histparser –n F
This parser populates table histdb. An entry will be generated for each of the
sequences that have valid accessions in table accdb that indicates that the sequence
was added on this day (when you ran histparser). This parser writes to the
histparserlog. This parser requires the accdb table and will take about 15 hours to
run.
26. You are done with the initial build of SeqHound.
If you did not build any of the optional modules, you will have to
remember this when setting up the .intrezrc configuration file for any
SeqHound application.
Set module values to zero if you did not build them. See the following section of the
.intrezrc configuration file.
example:
[sections]
;indicate what modules are available in SeqHound
;1 for available, 0 for not available
;gene ontology hierarchy (did you run goparser?)
godb = 1
;locus link functional annotations (did you run llparser and addgoid?)
lldb = 1
;taxonomy hierarchy (did you run importtaxdb?)
taxdb = 1
;protein sequence neighbours (did you download neighbours tables?)
neigdb = 1
;structural databases (did you run cbmmdb, vastblst and pdbrep?)
strucdb = 1
;complete genomes tracking (did you run chrom and comgen?)
gendb = 1
;redundant protein sequences (did you run redund?)
redundb = 1
;open reading frame database (currently not exported at all)
cddb = 0
;RPS-BLAST tables (did you download RPS-BLAST tables?)
rpsdb = 1
Catch up on SeqHound daily updates
27. Download all daily update files for genbank
Warning: There might have been a new GenBank release while you were
building SeqHound, in this case you cannot get updates from
ftp://ftp.ncbi.nih.gov/ncbi-asn1/daily-nc/ any more. You have to rebuild
SeqHound with a fresh GenBank release. You should check the file
ftp://ftp.ncbi.nih.gov/ncbi-asn1/daily/Last.Release to make certain that it
contains the same release number that was present when you started step
15.
cd $SEQH/
mkdir seqsync
seqhound@blueprint.org Version 3.3
The SeqHound Manual 46 of 421 18/04/2005
cd seqsync
ftp ftp.ncbi.nih.gov
When prompted for a name enter anonymous
When prompted for a password type myemail@home.com cd ncbi-asn1
cd daily-nc
bin
prompt
mget nc*.aso.gz
bye
Do not download the con_nc*.aso.gz files from this directory. SeqHound does not
use them.
28. Download all daily update files for refseq.
From ftp://ftp.ncbi.nih.gov/refseq/daily/ download all files past the date stamp on
gbrscu.aso.gz. gbrscu.aso.gz is the latest cumulative RefSeq division which was
downloaded by asnftp.pl and is located (in this example) in seqhound/build/asofiles.
cd $SEQH/seqsync
ftp ftp.ncbi.nih.gov
enter anonymous and your email address when prompted
cd refseq
cd daily
bin
get rsnc.****.2003.bna.Z
(where **** are files with timestamps greater than gbrscu.aso.gz)
bye
You must uncompress all of these files and rezip them so they can be processed by
the mother parser.
compress –d *.Z
gzip *.bna
29. Run update and mother on all downloaded files (excluding today's one; crons will do
it in the evening).
You can use the scripts all_update.sh and all_update_rs.sh. You will also need
mother, update and a properly configured .intrezrc file in the same directory as all of
the daily update files.
cd $SEQH/seqsync
cp $COMPILE/slri/seqhound/scripts/all_update.sh .
cp $COMPILE/slri/seqhound/scripts/all_update_rs.sh .
cp $SEQH/1.core.files/.intrezrc .
cp $SEQH/1.core.files/mother .
cp $SEQH/1.core.files/update .
Run all_update.sh first
seqhound@blueprint.org Version 3.3
The SeqHound Manual 47 of 421 18/04/2005
./all_update.sh 141
where 141 is the release number.
Run all_update_rs.sh second.
./all_update_rs.sh 141
These scripts will run update and mother executables (consecutively) on all
downloaded files present in the current directory.
All daily updates in SeqHound are stored in one division called gbupd regardless
how long SeqHound runs without a core rebuild.
mother will make a log file called “*run” for every file that it processes
update will make two log files called “*gis” and “*log” for every file that it processes
You can check that the two parsers have completed successfully. Each of the
following queries should return the same number (the number of starting input files):
ls *aso.gz | wc –l
ls *gis | wc –l
ls nc*log | wc –l
ls nc*run | wc –l
grep Done nc*run |wc -l
Setting up daily sequence updates
30. Make a new directory from where you will run daily sequence updates.
Populate this with the necessary scripts and programs.
cd $SEQH
mkdir updates
cd updates
cp $SLRI/seqhound/scripts/*cron_odbc.pl .
cp $SLRI/seqhound/scripts/shconfig.pm .
cp $SLRI/seqhound/build/odbc/redund .
cp $SLRI/seqhound/build/odbc/mother .
cp $SLRI/seqhound/build/odbc/update .
cp $SLRI/seqhound/build/odbc/precompute .
cp $SLRI/seqhound/build/odbc/isshoundon .
cp $SLRI/seqhound/build/odbc/importaxdb .
cp $SLRI/seqhound/build/odbc/goparser .
cp $SLRI/seqhound/build/odbc/llparser .
cp $SLRI/seqhound/build/odbc/addgoid .
cp $SLRI/seqhound/build/odbc/comgen .
seqhound@blueprint.org Version 3.3
The SeqHound Manual 48 of 421 18/04/2005
cp $SLRI/seqhound/build/odbc/chrom .
cp $SLRI/seqhound/scripts/genftp.pl .
cp $SLRI/seqhound/scripts/humoasn.pl .
cp $SLRI/seqhound/scripts/humouse_build.sh .
cp $SLRI/seqhound/genomes/gen_cxx .
cp $SLRI/seqhound/genomes/pregen.pl .
cp $SLRI/seqhound/genomes/gen.pl .
cp $SLRI/seqhound/genomes/ncbi.bacteria.pl .
mkdir logs
mkdir asofiles
mkdir inputfiles
mkdir genfiles
mkdir flags
31. Copy the .intrezrc config file to the updates directory and edit it.
cd $SEQH/updates
cp $SLRI/seqhound/config/.intrezrc .
cp $SEQH/1.core.files/.intrezrc .
Text in italics must be changed. in [crons] section, variable pathupdates
points to the path where the update jobs will be set up; variable pathinputfiles
points to the path that saves the input files (other than *.aso.gz and *.bna.gz files from
the core module and *.asn files from the gendb module); variable
pathinputfilescomgen points to the path that saves the input files *.asn for the
gendb module; variable mail indicates your e-mail address; variable
defaultrelease is the GenBank release you build SeqHound database with;
variable pathflags points to the path that save the flag files generated by each
updating job.
[crons]
;customizable variables in cron jobs
;NOTE: all paths must end in '/'
pathupdates=./
pathinputfiles=./inputfiles/
pathinputfilescomgen=./genfiles/
mail=my_email
defaultrelease=141
pathflags=./flags/
The cron daemon may consider your home directory to be the “current directory”.
For this reason, the .intrezrc file should be copied to your home directory too.
cd $SEQH/updates
cp .intrezrc ~/.
32. Set up the dupdcron_odbc.pl cron job.
dupdcron_odbc.pl (daily update cron) is a PERL script that retrieves the latest
GenBank and RefSeq update files from the NCBI ftp site and then passes them to
seqhound@blueprint.org Version 3.3

The SeqHound Manual 49 of 421 18/04/2005
“update” and “mother” where they are used to update the SeqHound data tables.
Specifically, it
a) downloads update files with today's date (from ftp://ftp.ncbi.nih.gov/ncbi-
asn1/daily-nc/ nc*.aso.gz and ftp://ftp.ncbi.nih.gov/refseq/daily/ rsnc*.bna.Z
b) runs update
(update -i nc*.aso.gz)
and then
c) runs mother
(mother -i nc*.aso.gz -r version# -n F -m F -u T).
You need to know this because if you miss a few updates before setting up
the cron job (and after completing the seqsync steps above) you have to
run update and mother in hand using the above commands.
All scripts (like dupdcron_odbc.pl) report success or failure via email. The mailto
address is set in the shconfig.pm script which you have just edited.
dupdcron_odbc.pl is the first cron job that has to be set up. Make a new text file
called list_crontab where you will list the cron jobs.
cd $SEQH/updates
pico list_crontabs
This file should have the single line
30 22 * * * cd /seqhound/update; LD_LIBRARY_PATH=libpath ./dupdcron_odbc.pl
where libpath should be replaced by the correct path you set up in Step 11 for
environment variable LD_LIBRARY_PATH. You can find it out by:
echo $LD_LIBRARY_PATH
This line specifies the time to run a job on a recurring basis. It consists of 6 fields
separated by spaces. The fields and allowable values are of the form:
minute (0-59) in this case 30
hour (0-23) in this case 22
day of the month (1-31) in this case *
month (1-12) in this case *
day-of-week (0-6 where 0 is Sunday) in this case *
command to run
The above line indicates that dupdcron_odbc.pl is to be run at 10:30 PM every day of
the month, every month, regardless of the day of the week. The * character is a wild-
card. The actual command consists of changing to the directory where
dupdcron_odbc.pl exists (this path will have to be modified depending on your set
up)
cd /seqhound/update;
and then executing the perl script
./dupdcron_odbc.pl
seqhound@blueprint.org Version 3.3
The SeqHound Manual 50 of 421 18/04/2005
After adding the above line and editing it to match your setup, close the file.
To activate this crontab file, type
crontab list_crontabs
If for some reason, you want to deactivate the cron job, type:
crontab –r list_crontabs
To find out what cron jobs you have activated, type
crontab -l
For more information on setting up cron jobs on UNIX type:
man crontab
33. Set up redundcron_odbc.pl to run daily.
cd $SEQH/updates
pico list_crontabs
Add the following line:
30 23 * * * cd /seqhound/update; LD_LIBRARY_PATH=libpath ./redundcron_odbc.pl
See Step 32 for the explanation of libpath.
After adding the above line, edit it to match your setup and close the file.
To activate this crontab file, type
crontab list_crontabs
This script basically does three things:
a) checks if file “nr” is updated on the ftp site ftp://ftp.ncbi.nlm.nih.gov/blast/db. If it
is, retrieves it
b) drops table redund from the database and recreate it.
c) rebuilds table redund using the downloaded nr file and the redund parser.
34. Run precompute for the first time.
First set up the configuration file
cd $SEQH/updates
pico .intrezrc
Edit the section under [precompute] to make it look like:
[precompute]
;precomputed taxonomy queries
MaxQueries = 0
MaxQueryTime = 10
QueryCount = 0
#path to precomputed searches has to have "/" at the end !!
path = /seqhound/precompute/
indexfile = /seqhound/precompute/index
Make sure the value of path is the absolute path of directory precompute you
make in Step 14 and the value of indexfile is the value of path plus index.
Variable path is the directory that holds results of the precompute executable.
indexfile is a path to the index that will be created by precompute.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 51 of 421 18/04/2005
Finally, run the precompute executable:
cd $SEQH/updates
./precompute –a redo
Where –a redo specifies that the program is being run for the first time.
This program basically precomputes the number of proteins and nucleic acids (and
their GI values) for each taxon in the taxgi table. The results of this query are stored
and indexed in text files (in the directory specified by path) if this query takes
longer than x seconds (where x is defined by MaxQueryTime in the above .intrezrc
file). These text files are used by SeqHound API calls such as
SHoundProteinsFromTaxIDIII(taxid)
35. Set up precomcron_odbc.pl to run daily.
cd $SEQH/updates
pico list_crontabs
Add the following line:
30 1 * * * cd /seqhound/update; LD_LIBRARY_PATH=libpath ./precomcron_odbc.pl
See Step 32 for the explanation of libpath.
After adding the above line and editing it to match your setup, close the file.
To activate this crontab file, type
crontab list_crontabs
This script basically runs the command
precompute -a update
and updates the precomputed search results.
36. Set up isshoundoncron_odbc.pl to run daily.
cd $SEQH/updates
pico list_crontabs
Add the following line:
30 7 * * * cd /seqhound/update; LD_LIBRARY_PATH=libpath ./isshoundoncron_odbc.pl
See Step 32 for the explanation of “libpath”.
After adding the above line and editing it to match your setup, close the file.
To activate this crontab file, type
crontab list_crontabs
This script basically does two things:
a) runs the executable called isshoundon. This program makes a single call to
the local SeqHound API to ensure that it is working.
b) moves all log, run and gis log files into a directory called logs
37. Set up llcron_odbc.pl to run daily.
cd $SEQH/updates
pico list_crontabs
seqhound@blueprint.org Version 3.3
The SeqHound Manual 52 of 421 18/04/2005
Add the following line:
30 21 * * * cd /seqhound/update; LD_LIBRARY_PATH=libpath ./llcron_odbc.pl
See Step 32 for the explanation of libpath.
After adding the above line and editing it to match your setup, close the file.
To activate this crontab file, type
crontab list_crontabs
This script basically repeats the actions listed in step 14 above and re-creates the
locus link tables in SeqHound. This includes:
a) getting the latest LL_tmpl.gz file from the NCBI ftp site.
b) removing the locus link tables from SeqHound
c) running llparser
d) getting 2 GO annotation files from GO ftp site
e) running the addgoid parser on these two files
38. Set up comgencron_odbc.pl to run daily.
cd $SEQH/updates
pico list_crontabs
Add the following line:
30 21 * * * cd /seqhound/update; LD_LIBRARY_PATH=libpath ./comgencron_odbc.pl
See Step 32 for the explanation of libpath.
After adding the above line and editing it to match your setup, close the file.
To activate this crontab file, type
crontab list_crontabs
This script basically repeats the actions listed in step 15 above and re-creates the
chrom table in SeqHound and updates the complete genome information in the core
tables. This includes:
a) generating a list of “DNA units” that belongs to a complete genome,
b) downloading complete genome files from NCBI ftp site,
c) rebuilding table chrom
d) removing all records in the core tables that belongs to division “gbchm”,
e) running script humous_build.sh to insert records into core tables
f) resetting the kloodge field in table taxgi for all records to 0
g) updating kloodge by running parser comgen
seqhound@blueprint.org Version 3.3
The SeqHound Manual 53 of 421 18/04/2005
Setting up SeqHound servers. Overview
39. Setting up SeqHound servers. Overview.
There are two web server applications that make up the SeqHound system:
a) wwwseekgi produces html pages for the SeqHound web interface and
b) seqrem processes requests to the SeqHound remote API.
Step 40 shows you how to find the two directories where you will set up these two
applications (assuming that you are using a default installation of Apache). The two
directories are called:
cgi-bin
htdocs
Step 40 may be skipped if you already know or have already been told where these two
directories are.
Steps 41 - describe the files that must be placed into these two sub-directories in order to
start the wwwseekgi and seqrem servers.
40. Examining the httpd.conf file for Apache.
These instructions assume that you already have an Apache server running. In order
to proceed further you must locate the directory where executables will be run from
(called “cgi-bin” in a default set-up of Apache) and a directory that contains html
documents (called “htdocs” in a default set-up of Apache). You can find (and reset)
the location of these two directories in an Apache configuration file called
“httpd.conf”. In a default set-up of Apache, the httpd.conf file can be accessed by
changing to the directory:
cd /etc/apache
and then opening the httpd.conf file found in this directory using a text editor such as
pico:
pico httpd.conf
To find the cgi-bin directory location, look for the line beginning with
“ScriptAlias”. In the default set-up, this line looks like this:
ScriptAlias /cgi-bin/ “/var/apache/cgi-bin/”
In this example, the path to the cgi-bin directory is /var/apache/cgi-bin/.
Write this path down, whatever it is.
To find the htdocs directory, look for the line beginning with “DocumentRoot”. In
the default set-up, this line looks like this:`
DocumentRoot “/var/apache/htdocs/”
In this example, the path to the cgi-bin directory is /var/apache/htdocs/.
Write this path down, whatever it is.
Also make a note of the line beginning with “User” and “Group” (who has
ownership to the server). In a default Apache set-up, these lines are likely
User nobody
Group nobody
seqhound@blueprint.org Version 3.3

The SeqHound Manual 54 of 421 18/04/2005
Make a note of this, whatever it is.
Exit from the httpd.conf file and save your changes. If you made changes to the file,
you must restart the Apache server using the command:
/usr/apache/bin/apachectl restart
See the Trouble Shooting section at the end for more information on this.
In the steps below you will set up the SeqHound server by adding to these two
directories
Contents of the cgi-bin and htdocs directories
directory contents
cgi-bin the SeqHound wwwseekgi and seqrem server applications will placed
here
htdocs all of the static html pages used by the SeqHound interface will be placed
here
41. Set up the cgi-bin directory.
Move to the cgi-bin directory you found in the step above. For the default set-up:
cd /var/apache/cgi-bin/
make a new subdirectory here called SeqHound
mkdir seqhound
cd seqhound
copy the SeqHound server applications here:
cp $COMPILE/slri/seqhound/build/odbc/seqrem .
cp $COMPILE/slri/seqhound/build/odbc/wwwseekgi .
also copy the following files to this directory:
cp $COMPILE/slri/seqhound/html/seekhead.txt .
cp $COMPILE/slri/seqhound/html/seektail.txt .
cp $COMPILE/slri/seqhound/html/seekhead.txt pics/.
cp $COMPILE/slri/seqhound/config/.intrezrc .
cp $COMPILE/slri/seqhound/config/.ncbirc .
42. Edit the .ncbirc configuration file.
Open the file with a text editor such as pico.
The setting for Data should contain a path to the ncbi/data directory. This directory
was downloaded as part of the ncbi toolkit in step 2.
--------------------example .ncbirc file begins-----------------------
[NCBI]
Data=/home/ncbi/data
--------------------example .ncbirc file ends-------------------------
43. Edit the .intrezrc configuration file.
Refer to step 14 in the current section for setting up of the .intrezrc file. The settings
for username, password, dsn and database in section [datab] should be
valid for the SeqHound database you have just built, and the setting for path and
seqhound@blueprint.org Version 3.3
The SeqHound Manual 55 of 421 18/04/2005
indexfile in section [precompute] should point to the valid path as in step 34
in the current section. Set up the index.html file for the web interface.
Move to the htdocs directory for your web-server. In the default case:
cd /var/apache/htdocs/
Make a SeqHound directory here:
mkdir seqhound
cd seqhound
Copy the index.html page to this directory:
cp $COMPILE/slri/seqhound/html/index.html .
Open the file in a text editor like pico and edit it so that its action points to the
wwwseekgi server.
pico index.html
then edit the line
<FORM ACTION="/cgi-bin/seqhound/wwwseekgi" METHOD="GET">
where "/cgi-bin/seqhound/wwwseekgi" should specify the path to the
wwwseekgi executable.
44. Set up ODBC configuration file .odbc.ini:.
Move to the home directory of the owner of the binary seqrem in directory
/var/apache/cgi-bin/. Text in italics should be changed (see Step 10):
cd /homedir
Set up file .odbc.ini as the following (text in italics should be changed):
[mysqlsh]
Description = MySQL ODBC 3.51 Driver DSN
Trace = On
TraceFile = stderr
Driver = /software/64/unixodbc/odbc/lib/libmyodbc3.so
DSN = mysqlsh
SERVER = my_server
PORT = my_port
USER = user_id
PASSWORD = my_pwd
DATABASE = seqhound
45. Set permissions on the cgi-bin directory.
Move to the cgi-bin directory.
cd /var/apache/cgi-bin/
Change the user and group ownership to nobody (or whatever the values of “User”
and “Group” were set to in step 40).
chown –R nobody:nobody seqhound
46. Set permissions on the htdocs directory.
Move to the htdocs directory.
cd /var/apache/htdocs/
seqhound@blueprint.org Version 3.3
The SeqHound Manual 56 of 421 18/04/2005
Change the user and group ownership to nobody (or whatever the values of “User”
and “Group” were set to in step 40).
chown –R nobody:nobody seqhound
47. Test the SeqHound web interface.
Open an internet browser and, in this example, go to the url
http://yourmachinename/cgi-bin/seqhound/
You should see the front page of the SeqHound wwwseekgi interface.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 57 of 421 18/04/2005
Trouble-shooting notes
Error logs
Error logs for each of the SeqHound parsers are described in the steps above where the
parser is used to initially build a given SeqHound module.
Error logs for the SeqHound wwwseekgi server software is located in the same directory
as the executable; see wwwseekgilog.
Error logs for the seqrem server are located in the same directory as the executable; see
seqremlogs.
Error logs for the Apache server software are located (in a default set-up) in
/var/apache/logs.
Recompiling SeqHound
If you make changes to and recompile SeqHound, you should first do a clean of the
existing object files and executables. If you are still in a super-user shell, you may wish
to exit this shell and return to the shell where you had set all of your environment
variables. These variables are required by the clean and make scripts.
COMPILE compile directory
SLRI slri directory
NCBI ncbi directory
CC gcc
PATH /usr/local/bin:/usr/ccs/bin:${PATH}
EXTRAOPT –D_FILE_OFFSET_BITS=64
ODBC (path to unixodbc)
LD_LIBRARY_PATH (path to mysql and odbc libraries)
The first three variables refer to directories that are created during the above instructions.
To clean, run any make file with the ‘clean’ target. For example, to clean the cgi
executables, type:
cd $COMPILE/slri/seqhound/cgi
make –f make.seqrem clean
make –f make.wwwseekgi clean
Restarting the Apache server
If changes are made to the httpd.conf file, the Apache server must be restarted for the
changes to take effect. Use the apachectl script to do this
For a default install of Apache this script is in the directory
cd /usr/apache/bin
and to start the server, type
./apachectl restart
or to get a list of apachectl script commands type
./apachectl
seqhound@blueprint.org Version 3.3
The SeqHound Manual 58 of 421 18/04/2005
Other useful links
SLRI on Sourceforge http://sourceforge.net/projects/slritools/
NCBI Info Engineering branch: http://www.ncbi.nlm.nih.gov/IEB/
Concurrent Versioning System (CVS) http://www.cvshome.org/
MySQL http://www.mysql.com/
Parser schedule
Parsers are run on a periodic basis as “cron” jobs on Unix platforms and as “Schedules
tasks” on Windows platforms.
The cron job schedule is set up in the file “seqhound/update/list_crontabs” on UNIX
platforms
The setup for the current production version of SeqHound described in these instructions
is shown below:
0 19 * * * /arena/seqhound/update/llcron_odbc.pl
0 21 * * * /arena/seqhound/update/redundcron_odbc.pl
30 22 * * * /arena/seqhound/update/dupdcron_odbc.pl
30 24 * * * /arena/seqhound/update/precomcron_odbc.pl
0 7 * * * /arena/seqhound/update/isshoundoncron_odbc.pl
MySQL errors
ERROR 1153 at line X: Got a packet bigger than
'max_allowed_packet'
When MySQL receives a packet bigger than max_allowed_packet bytes, it issues a
Packet too large error and closes the connection. For example, when a single SQL
statement from a mysqldump being imported exceeds the value for
"max_allowed_packet" configured on the MySQL server. Increasing this value to
64MB from the default 16MB should resolve the error. This value may be changed in the
global config or setting this on the running server via:
set global max_allowed_packet=67108864;
Please see http://dev.mysql.com/doc/mysql/en/Packet_too_large.html for more
information
seqhound@blueprint.org Version 3.3

The SeqHound Manual 59 of 421 18/04/2005
5. Description of the SeqHound parsers and data tables by module
What are modules?
The SeqHound system is divided into one required “core” module and several optionally configured modules. Modules are groups of
tables and API calls that are filled using a common data resource, for example the 3D structures. The purpose of this division is to give
the user an option to control hardware resources and complexity of system administration when parts of the SeqHound system are not
required. The list of SeqHound modules and their data resources is contained in the table below.
After a system build, the module information is recorded in the configuration file which is then utilized by the API to determine if
certain operations can be achieved with the current setup. The configuration file is called .intrezrc (Unix platforms) The relevant
section of this file is under the heading “[sections]”. Consult Section 4 under “Building the SeqHound system” for more information
on specifying the available modules in this file.
How to use this section.
This section describes the SeqHound system in detail module by module.
Parser and data tables associated with a given module are described under the section for that module.
A brief description of all of the parsers can be found below and in the Table 2 of the SeqHound paper. The table is repeated below
and will be updated here.
Note: It is assumed that you have read the material in section 3 and section 4 before delving into this material. These
two sections include everything you need to know to start using the SeqHound remote API or to install you own local
version of SeqHound. This section is intended for users who may want more details about how SeqHound is
constructed and exactly what it is doing behind the scenes. This section is also meant as background material for
developers who want to further develop the SeqHound system
Parser descriptions
Parser descriptions contain the following headings and information:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 60 of 421 18/04/2005
purpose: a brief description of what the parser is for
logic: more details on the parser – see update parser for example
module: what module the parser belongs to
input files: input files required by the parser (also available in Table 2)
tables altered: tables in SeqHound that are modified or created by the parser (also
available in Table 2)
source code
location:
location of the parser source code in the slri development tree
config file
dependencies:
what configuration file parameters must be set for the parser to work
command line
parameters:
used by the parser
example use: of the parser from the command line
associated scripts: that are used to run the parser
error and run-time
logs:
where they are located and what is in them
troubleshooting: problems that may occur with this parser
additional info: where to find it
Table descriptions
Each table that is relevant to a module is described under that module.
Here is an example with comments in brackets. A data table description consists of the following sections and content:
Database: that the table belongs to (almost always SeqHound)
Table: name of the table like “accdb”
Definition: a brief description of the table's purpose (for example, “This table
seqhound@blueprint.org Version 3.3

The SeqHound Manual 61 of 421 18/04/2005
correlates gi’s to accession identifiers).
Observation: notes about the table
Source db: where does this table’s information come from
Source file: the source file (used by the parser to fill this table) location is listed here
Parser: the name of parser if a single parser is responsible for filling this entire
table.
This is followed by a summary of the table’s definition (for example):
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto number row identifier
gi int(11)
N
o 0 GenInfo Identifier
asn1 longblob Yes
N
ULL BioSeqs: Sequences
and indexes
Keyname Type Field
PRIMARY PRIMARY gi
iasndb_rowid INDEX rowid
iasndb_gi INDEX gi
(each field is then described...only one field description is shown for this example)
***gi***
description: definition of the column (for example, GenInfo sequence record identifier”)
example: of a column entry (for example, “1232452”)
default value: if the value has a default value, it is listed here
ASN.1 structure: If the value in this column is derived from an NCBI data structure, this gives
you a quick idea of where to locate it--you can find more info by searching
for this data structure at:
http://www.ncbi.nlm.nih.gov/IEB/ToolBox/SB/hbr.html.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 62 of 421 18/04/2005
for example, “Bioseq->Seq-id (choice 12)
Alternatively, the column may store a binary object that is an NCBI or SLRI
ASN.1 object. Descriptions of SLRI ASN.1 objects may be found at
http://cvs.sourceforge.net/cgi-bin/viewcvs.cgi/slritools/slri/seqhound/asn.
source: If the value in this column is derived from a text file, this describes how to
find the information present in this column in the source file (for example in
the fourth column)
parser: this is the parser(s) that retrieves the value from some other db (for example,
mother)
function: this is the parser function that retrieves the value
API: if a SHound API function retrieves this value from this table it is listed here
(for example, ShoundFindAcc)
more info: (other notes)
seqhound@blueprint.org Version 3.3
The SeqHound Manual 63 of 421 18/04/2005
An overview of the SeqHound data table structure
An overview of the SeqHound data table structure is available as a separate document in pdf format. See
http://www.blueprint.org/seqhound/api_help/docs/SeqHound_Schema_Prod.pdf.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 64 of 421 18/04/2005
Parsers and resource files needed to build and update modules of SeqHound.
This table will be updated shortly
Input File Resource Parser Tables
Modified Module
ASN.1
sequences
f
tp://ftp.ncbi.nih.gov/ncbi-asn1/*.aso
f
tp://ftp.ncbi.nih.gov/
r
efseq/cumulative
/*.bna
mother
asndb, parti,
nucprot, accdb,
pubseq, taxgi,
sendb, sengi
core
ASN.1
sequences
f
tp://ftp.ncbi.nih.gov/ncbi-asn1/daily-
nc/*.aso
f
tp://ftp.ncbi.nih.gov/refseq/daily/*bna
update
asndb, parti,
nucprot, accdb,
pubseq, taxgi,
sendb, sengi
core
FASTA nr
database
f
tp://ftp.ncbi.nih.gov/blast/db/nr redund redund redundb
List of complete
genomes (flat
file)
http://cvs.sourceforge.net/cgi-
bin/viewcvs.cgi/slritools/slri/seqhound
/genomes/chromff
chrom chrom gendb
ASN.1 for
complete
genomes
f
tp://ftp.ncbi.nih.gov/genomes/*/*.asn comgen taxgi, accdb gendb
Taxonomy
release (flat file)
f
tp://ftp.ncbi.nih.gov/pub/taxonomy/tax
dump.tar importtaxdb TAX, GCODE,
DIV, del, merge taxdb
ASN.1 MMDB
release
f
tp://ftp.ncbi.nih.gov/mmdb/mmdbdata
/*.val cbmmdb mmdb, mmgi strucdb
MMDB
(database table) mmdb table vastblst domdb strucdb
seqhound@blueprint.org Version 3.3
The SeqHound Manual 65 of 421 18/04/2005
3-D chain
BLAST sets
(flat file)
f
tp://ftp.ncbi.nih.gov/mmdb/nrtable/nr
p
db.* pdbrep domdb strucdb
FASTA nr
database
f
tp://ftp.ncbi.nih.gov/blast/db/nr nblast nrB neigdb
BLAST ASN.1
results
nrB table available nrB and nrN tables
available at
f
tp://ftp.blueprint.org/pub/SeqHound/
NBLAST/
nbraccess nrN neigdb
LL_tmpl (flat
file)
f
tp://ftp.ncbi.nih.gov/refseq/LocusLink
/
LL_tmpl llparser ll_omim, ll_go,
ll_llink, ll_cdd lldb
gene_associaton
.com
pugen.GenBank
/Swissprot (flat
files)
http://www.geneontology.org addgoid ll_go lldb
function.ontolog
y
process.ontolog
y
component.onto
logy (flat files)
http://www.geneontology.org goparser
go_parent,
go_name,
go_reference,
go_synonym
godb
CDD database
f
tp://ftp.ncbi.nih.gov/pub/mmdb/cdd/ domname domname rpsdb
FASTA nr
database and
CDD database
f
tp://ftp.ncbi.nih.gov/blast/db/nr;
f
tp://ftp.ncbi.nih.gov/pub/mmdb/cdd/
DOMNAME and RPSDB tables
available at
f
tp://ftp.blueprint.org/pub/SeqHound/
RPS/
rpsdb rpsdb rpsdb
seqhound@blueprint.org Version 3.3
The SeqHound Manual 66 of 421 18/04/2005
core module
Last updated April 11, 2005. This section is maintained bt Elizabeth Burgess.
mother parser
Last updated April 11, 2005
purpose:
The mother parser is the first parser that is used to initially build the SeqHound “core” set of data tables. The input files consist of the
latest release of GenBank and RefSeq in binary ASN.1 format available on the NCBI ftp site. The resulting SeqHound data tables
hold DNA, RNA and protein sequence record information.
As of release 4.0, the mother parser is also used with the GenBank and RefSeq daily updates to update SeqHound so that that
sequence information is synchronized with that of NCBI. Previously, updates were handled by a separate parser called “update”.
logic:
The mother parser is run on a daily basis in update mode (-u T) to update with two input files. One input file is the daily update from
GenBank and the other is the daily update from the RefSeq database.
For each bioseq in the input file, mother retrieves the GI and accession number.
Mother then looks for this pair of GI and accession identifiers in the SeqHound accdb table.
If neither the accession (nor the GI) is found, then the record in the daily update represents a GI that is to be ADDED to SeqHound.
If the accession in the update file is found in SeqHound and is associated with the same GI (as listed in the update file) then the record
in the daily update represents a GI that has been CHANGED. This means that the sequence record was resubmitted to GenBank with
the same GI; the sequence remains the same but the associated annotation has changed.
If the accession in the update file is found in SeqHound but the GI associated with this accession differs between the update file and
SeqHound, then the GI that is newly associated with the accession represents a change in the sequence. The accession and updated GI
seqhound@blueprint.org Version 3.3

The SeqHound Manual 67 of 421 18/04/2005
pair point to a sequence record that will be ADDED to SeqHound. The accession and old GI (currently in SeqHound) point to a
record in SeqHound that will be KILLED (deleted).
Mother then records in the SeqHound history table whether the GI and accession in the update file represent an ADDED or
CHANGED record. The previous gi associated with the accession is also recorded, if there is one. Mother also records in the history
table those sequence records currently in SeqHound that will be KILLED (see above).
This process is completed for every bioseq in the update file.
A list of ADDED, CHANGED and KILLED GI’s is written to the file rsncmmddgis where rsncmmdd refers to the update file that was
being processed.
Mother then deletes from the following tables: parti, accdb, sendb,sengi, taxgi, pubseq, nucprot asndb. If the record is a complete
genome record, then the gi will be deleted from gichromid, contigchromid, gichromosome and contigchromosome. The strategy for
complete genome deletions is as follows:
Complete genome information in NCBI may be stored in several different places in the record.
1. Records from organisms other than those for certain higher eukaryotes ( e.g. human, mouse, rat, chicken and bee). These
records contain the flag NCBI_GENOMES and list the RNA and protein gis in the annotation. The RNA and protein gis will
be written to gichromid. The gi of the contig that contains the annotation is also written to this record if it is known.
2. Records from human, mouse, rat etc. The top level contig contains the NCBI_GENOMES flag, but no annotation. Instead, it
lists the gis for contigs that make up this chromosome. The annotation of these contig records contains the gis and proteins that
belong to this chromosome. The lower level contig gis are written to contigchromid so that the protein and RNA gis can be
parsed later from the contig bioseqs by the postcomgen parser. The gi of the top level contig is also written to this record.
3. Some records exist that only contain a chromosome number in the description of the bioseq. These records can be a contig
record or a record for an individual protein or RNA. These records are first written to gichromosome or contigchromosome and
later moved to gichromid and contigchromid by postcomgen.
For updating, we check to see if the accession for that gi is in the chrom table. The only records that are written to this table are the top
level gis that contain the NCBI_GENOMES flag. If the gi belongs to a top level contig, then all gis in contigchromid that belong to
that contig are retrieved and all gis in gichromid that belong to each low level contig gi are deleted. The low level contig gis are then
deleted from contigchromid.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 68 of 421 18/04/2005
If the record is present in contigchromid, then all gis in gichromid from that contig are deleted before the record is deleted from
contigchromid.
If the gi is present in only gichromid and contains a contig gi, then the appropriate record in contigchromid is marked as changed.
That way, after mother finishes, postcomgen will only process those contigs that have changed.
Mother writes errors and messages to a log file called rsncmmddrun where rsncmmdd refers to the update file that was being
processed.
module: core
input files:
latest GenBank release (ftp://ftp.ncbi.nih.gov/ncbi-asn1/*.aso)
latest RefSeq release (ftp://ftp.ncbi.nih.gov/refseq/cumulative/*.bna)
daily GenBank release (ftp://ftp.ncbi.nih.gov/ncbi-asn1/daily-nc/*.aso)
daily RefSeq release (ftp://ftp.ncbi.nih.gov/refseq/daily/*.bna)
tables altered:
asndb, parti, nucprot, accdb, pubseq, taxgi, sendb, sengi, bioentity, bioname, chrom, gichromid, contigchromid, gichromosome,
contigchromosome
source code location:
slri/seqhound/parsers/mother.c
config file dependencies:
The relevant configuration file is:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 69 of 421 18/04/2005
slri/seqhound/config/.intrezrc
The relevant section of the configuration file is:
[datab]
;seqhound database that you are connecting
username=your_user_name
password=your_pass_word
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
Text in italics must be changed for the .intrezrc file to function correctly with your SeqHound set-up. Variables username,
password, dsn, database in section [datab] should have the same values as USER, PASSWORD, DSN and DATABASE
respectively in your .odbc.ini (see section 4).
command line parameters:
Typing “./mother –“ at the command line while in the directory where mother resides will return a list of command line
parameters and default settings.
Note that -n and -m are listed for historical Codebase purposes. These values are always F for ODBC.
For example:
> ./mother –
mother arguments:
-i Filename for asn.1 input [File In]
-r Release [String]
-n Initialize the ASNDB database file [T/F] Optional
default = F
-m Initialize the remaing database files [T/F] Optional
default = F
-u Is this an update [T/F] Optional
default = F
-c Is this a file for human/mouse complete genome [T/F] Optional
default = F
-t Read input file in text mode [T/F] Optional
default = F
seqhound@blueprint.org Version 3.3
The SeqHound Manual 70 of 421 18/04/2005
example use:
mother –i nc1227.aso -r 135 -n F -m F
mother –I nc1227.aso –r 135
Note that mother is normally run under the control of a script (see below).
associated scripts:
The initial build of SeqHound is executed using the script called “seqhound_build.sh”
See “slri/seqhound/scripts/seqhound_build.sh”
This script cycles through all the GenBank and RefSeq release files downloaded by asn_ftp.pl unzips them, processes them with
mother and then zips them up again.
The script must be run in the directory containing the GenBank and RefSeq release files and a copy of the mother parser The script
takes one argument (a release number).
./seqhound_build.sh 135
mother is also called by the scripts that generate the daily updates (see “associated scripts” under update parser).
error and run-time logs:
mother writes to a log file called “rsncmmddrun” where rsncmmdd refers to the GenBank release or update file that was being
processed. When run in update mode, it also writes the gis to a file called “rsncmmddgis”. The log files are created in the directory
logs.
troubleshooting:
additional info:
See readme files that accompany the input files on the GenBank ftp site.
See data table descriptions for each of the tables that are listed under “tables altered”.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 71 of 421 18/04/2005
update parser
Last updated April 11, 2005
note:
As of Release 4.0, update functionality has been moved to the mother parser and associated scripts. See above.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 72 of 421 18/04/2005
postcomgen parser
Last updated April 11, 2005
purpose:
The postcomgen parser is used in conjunction with the mother parser. It is used to update the taxgi table with complete genome
information using the chrom, contigchromid, gichromid, gichromosome and contigchromosome tables. It is run after the initial build
and after any updates.
logic:
Complete genome information in NCBI may be stored in several different places in different records.
1.Records from organisms other than those for certain higher eukaryotes ( e.g. human, mouse, rat, chicken and bee) contain the flag
NCBI_GENOMES and list the RNA and protein gis in the annotation.
2.Records from certain higher eukaryotes (human, mouse, rat etc) are different. The top level contig contains the NCBI_GENOMES
flag, but no annotation. Instead, it lists gis for the contigs that make up this chromosome. The annotation of these records contains the
gis and proteins that belong to this chromosome.
3. Some records exist that only contain a chromosome number in the description of the bioseq. These records can be contig records or
records for an individual protein or RNA.
At the time that mother is run, gis in the annotation may not yet have been entered into accdb and taxgi. For this reason, mother writes
these gis out to the table gichromid. When mother encounters a record that contains only contig gis, these gis are written out to
contigchromid. The records that contain only a chromosome number are written to gichromosome or contigchromosome.
After mother is finished, postcomgen is run to process the data in these tables and write the kloodge to the taxgi table for each gi:
1. Postcomgen first writes every gi in gichromosome to gichromid with the appropriate chromid. After this is done, all records are
deleted from gichromosome.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 73 of 421 18/04/2005
2. Next, all contigchromosome gis are written to contigchromid with the appropriate chromid and all records are deleted from
contigchromosome.
3. For each gi in contigchromid, the bioseq is obtained from asndb and any protein or RNA gis in the annotation are written to
gichromid. In addition to the chromid, the contiggi is stored so that the appropriate contigchromid record can be reread on update if
necessary.
4. Each record in contigchromid is marked as read, so that upon update only records that have been modified need be read.
5. Finally, taxgi is updated for each gi in gichromid with the appropriate kloodge.
module: core
input files:
There are no input files. The parser uses the tables gichromid, contigchromid, gichromosome and contigchromosome, which are filled
by mother, to update the taxgi table.
tables altered:
taxgi, gichromid, contigchromid, gichromosome, contigchromosome
source code location:
slri/seqhound/genomes/postcomgen.c
config file dependencies:
The relevant configuration file is:
slri/seqhound/config/.intrezrc
The relevant section of the configuration file is:
[datab]
;seqhound database that you are connecting
username=your_user_name
seqhound@blueprint.org Version 3.3
The SeqHound Manual 74 of 421 18/04/2005
password=your_pass_word
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
Text in italics must be changed for the .intrezrc file to function correctly with your SeqHound set-up. Variables username, password,
dsn, database in section [datab] should have the same values as USER, PASSWORD, DSN and DATABASE respectively in your
.odbc.ini (see 4).
command line parameters:
None.
example use:
postcomgen
associated scripts:
error and run-time logs:
Postcomgen writes to a log file called “postcomgen.log”.
troubleshooting:
additional info:
See data table descriptions for each of the tables that are listed under “tables altered”.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 75 of 421 18/04/2005
asndb table
Last updated April 11, 2005
Database: SeqHound
Module: core
Table: asndb
Definition: Central table that stores sequence records in binary ASN.1 format and indexed by gi.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 76 of 421 18/04/2005
asndb table
Field Type Null Default Column_Definition Example Source API
rowid int(11)
N
o
N
ULL Mysql autoincrement
column
ts timestamp Yes
CURRENT_TIMES
TAMP timestamp 2005-03-23 12:32:16
gi int(11)
N
o 0
GenInfo primary
sequence record
identifier
4001923 FillASNDB calls GetGi.
See Bioseq->seqid->gi
asn1 longblob Yes
N
ULL
bioseq in binary ASN.1 An example of a bioseq
is included in the
Appendix (Example of
GenBank record).
ToBioseqSEQENTRY
See Bioseq->seqid->gi
SHoundGetBioseq
SHoundSequenceLength
division varchar(25)
N
o
N
CBI division PRI GetDivisionFromGBBlock.
See
Bioseq->descr->source
->org->orgname->div
release varchar(10)
N
o
The name of the release
given as a command
line input to the mother
parser
135
type varchar(15)
N
o The type of molecule
that this record refers to.
protein FillASNDB calls GetType.
See Bioseq-> mol
SHoundMoleculeType
seqhound@blueprint.org Version 3.3
The SeqHound Manual 77 of 421 18/04/2005
asndb indices
Keyname Type Field
PRIMARY PRIMARY gi
iasndb_rowid INDEX rowid
iasndb_ts INDEX ts
iasndb_gi INDEX gi
seqhound@blueprint.org Version 3.3
The SeqHound Manual 78 of 421 18/04/2005
parti table
Last updated April 11, 2005
Note: This table is documented for historical purposes and may be deprecated in a future release. PARTI was required
by codebase as an index to the ASNDB table.
Database: seqhound
Table: parti
Module: core
Definition: This table maps the gi to the NCBI division.
parti table
Field Type Null Default Column_Definition Example Source API
rowid int(11)
N
o MySQL autoincrement
column.
ts timestamp Yes CURRENT_TI
MESTAMP
MySQL timestamp
column
2005-03-23 12:32:16
gi int(11)
N
o 0
GenInfo Identifier 21676275 FillASNDB calls
AppendRecordPARTI. See
Bioseq->Seqid->gi (choice 12)
division char(15) Yes
N
ULL
N
CBI Division PRI GetDivisionFromGBBlock. See
Bioseq->descr->source
->org->orgname->div
seqhound@blueprint.org Version 3.3

The SeqHound Manual 79 of 421 18/04/2005
parti indices
Keyname Type Field
PRIMARY PRIMARY gi
iparti_rowid INDEX rowid
iparti_ts INDEX ts
iparti_gi INDEX gi
iparti_div INDEX division
seqhound@blueprint.org Version 3.3
The SeqHound Manual 80 of 421 18/04/2005
nucprot table
Last updated April 11, 2005
Database: seqhound
Table: nucprot
Module: core
Definition: This table maps the gi of a protein to the gi of its encoding DNA.
Observation: A DNA may map to more than one protein. A protein can only map to one DNA. Not every DNA may
map to a protein, eg synthetic DNA fragments typically do not have a protein. In a bioseqset structure of
type nucprot, the first bioseq record is a nucleic acid, subsequent bioseq records correspond to proteins.
In such a case, the entries added to the nucprot database would be:
record 1) gi_1 gi_2
record 2) gi_1 gi_3
record 3) gi_1 gi_4
Each record would be a mapping of the nucleic acid (gi_1) to each protein (gi_2 ... gi_N)
seqhound@blueprint.org Version 3.3

The SeqHound Manual 81 of 421 18/04/2005
nucprot table
Field Type Null Default Column_Definition Source API
rowid int(11) MySQL autoincrement
column.
Example
N
o
gin
N
o 0 27464927 FillNUCPROT. See SHoundDNAFromProtein
timestamp Yes MySQL timestamp column
2005-03-23 12:32:16
int(11)
N
o
int(11) DNA GenInfo Identifier Bioseq->Seqid->gi (choice 12)
CURRENT_
TIMESTAM
P
ts
0 Protein GenInfo Identifier 27464928 FillNUCPROT. See
Bioseq->Seqid->gi (choice 12)
SHoundProteinFromDNA
gia
nucprot indices
Keyname Type Field
PRIMARY PRIMARY gia
inucprot_rowid INDEX rowid
inucprot_ts INDEX ts
inuc_gin INDEX gin
inuc_gia gia INDEX
seqhound@blueprint.org Version 3.3
The SeqHound Manual 82 of 421 18/04/2005
accdb table
Last updated April 11, 2005
Database: seqhound
Table: accdb
Definition: This table maps gi's to accession numbers and other identifiers (a combination of a database name and
identifier) found in GenBank sequence records.
Observation Note that gi is not unique in ACCDB. Also note that a gi does not always have an accession associated
with it. There are 2 kinds of records in ACCDB. In one case the gi is associated with an accession and in
the other case, the gi is associated with a database name and some identifier from that database (name
column). Any given gi may have both record types and may be associated with more than one name.
Note that internal NCBI databases may be stored in ACCDB, for example NCBI_GENOMES. In this
case the record will not have an accession, but it will have a name.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 83 of 421 18/04/2005
accdb table
Field Type Null Default Column_Definition Example Source API
rowid int(10)
N
o MySQL autoincrement
column.
ts timestamp Yes
CURRENT_TI
MESTAMP MySQL timestamp column 2005-03-23 12:32:16
gi int(11)
N
o
0 GenInfo Identifier 2313082 FillACCDB calls GetGi. See
Bioseq->seqid->gi
SHoundGiFromGBAcc
(ShoundFindAcc)
db varchar(15)
N
o
The source database of the
sequence record.
See note 1 below.
pdb FillACCDB. See
Bioseq->Seq-Id->dbtag->db
SHoundDbNameAndIdListFrom
GBAcc
name varchar(30)
N
o This is an accession number
from a foreign database.
1AAP See note 2 below.
SHoundDbNameAndIdListFrom
GBAcc
(ShoundFindName)
(ShoundGetNameByGi)
namelow varchar(30) Yes
N
ULL
Same as name but in lower
case. Present for historical
reasons and may probably be
removed in the future.
1aap See note 2 below.
access varchar(20)
N
o
Genbank Accession. The
primary sequence record
identifier.
n/a WriteACCDB. See Bioseq-
>Seq-id->Textseq-id-
>accession. This field is n/a
for sequences from PDB and
other databases. This field is
only relevant when name field
is n/a.
SHoundGBAccFromGi
(ShoundAccFromGi)
chain varchar(20) Yes
N
ULL
This describes which one of
(possibly) many chains in a
structure a sequence refers to.
This only refers to PDB
sequences.
A FillACCDB. See
Bioseq->Seq-id->PDB-seq-id-
>chain. See note 5 below.
SHoundGiFromPDBchain
seqhound@blueprint.org Version 3.3

The SeqHound Manual 84 of 421 18/04/2005
release varchar(20) Yes
N
ULL release date of the record Sep 14, 1990 See note 3 below.
SHoundSeqIdFromGi
version int(11) Yes
N
ULL version of the record
0 See note 4 below.
This value cannot be directly
retrieved from the table by the
API but a Seq-id can be retrieved
using ShoundSeqIdFromGi
given a gi.
title text Yes
N
ULL a brief description of the
sequence
Chain A, Protease
Inhibitor Domain Of
Alzheimer's Amyloid
Beta-Protein Precursor
(APPI)
FillACCDB calls
CreateDefline.
Notes
1.
A complete list of db names that may appear in this column is listed under API supplementary material at
http://www.blueprint.org/seqhound/apisupplement.html. Some common db name abbreviations are listed below.
gb means GenBank
embl means EMBL
pir means Protein Information Resource (PIR)
sp means Swiss-Prot
pbs means other database
ref means RefSeq
dbj means DNA Database of Japan (DDBJ)
prf means PRF
pdb means Protein Data Bank (PDB)
tpe means third party annotation from embl
seqhound@blueprint.org Version 3.3
The SeqHound Manual 85 of 421 18/04/2005
tpg means third party annotation from genbank
tpd means third party annotation from ddbj
other means some other database
2.
WriteAccdb.
For all db’s except PDB and ‘other’:
Bioseq->Seq-id->Textseq-id->name
For PDB sequences:
Bioseq->Seq-id->PDB->seq-id->PDB-mol-id (4 characters)
For other databases:
Bioseq->Seq-id->Dbtag->Object-id->string or integer
3.
FillACCDB, given a Bioseq pointer, retrieves a Seq-id pointer. If the Seq-id is of the type, pdb (protein data bank), then a PDB-seq-
id ptr is passed to WriteACCDB which retrieves the value of rel. If the Seq-id is of the type, genbank, embl, pir, swissprot, other,
ddbj, prf, tpg, tpd or tpe then a Textseq-id pointer is passed to WriteACCDB which retrieves the value of release. If the Seq-id is of
the type, giimport then a Giimport-id ptr is passed to WriteACCDB which retrieves the value of release. In all cases, the formatting
of the date is attempted with NCBI's function DatePrint (like this 07-OCT-2004). If the Formatting cannot be done, then the string
is just copied.
Bioseq->Seq-id->Textseq-id->release (an INTEGER) for all databases besides PDB, Giimport and ‘other’
Bioseq->Seq-id->PDB->seq-id->rel (a string or an NCBI data type) for PDB sequences
Bioseq->Seq-id->Giimport-id->release (a string) for Giimport
n/a for all ‘other’ databases.
4.
FillACCDB, given a Bioseq pointer, retrieves a Seq-id pointer. If the Seq-id is of the type, genbank, embl, pir, swissprot, other,
ddbj, prf, tpg, tpd or tpe then a Textseq-id pointer is passed to WriteACCDB which retrieves the value of version. For all other
databases, this value is n/a. See
seqhound@blueprint.org Version 3.3
The SeqHound Manual 86 of 421 18/04/2005
5.
Sequences that are part of structures will have a PDB identifier. Since there may be more than one chain (sequence) in a structure, the
PDB identifier must be accompanied by a chain identifier to uniquely identifiy a chain within the structure. A PDB identifier
supplemented with a chain identifier will be associated with a single sequence record (GI). For example, see PDB id “9XIM” and
chain A; this corresponds to GI sequence 443580. See example ASN.1 record for 9XIM_A in the Appendix.
Chain in the ASN.1 annotation is of type character, not of type string so it appears in its decimal form. So 65 is A and so on.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 87 of 421 18/04/2005
accdb indices
Keyname Type Field
PRIMARY PRIMARY gi
db
access
iaccdb_rowid INDEX rowid
iaccdb_ts INDEX ts
iaccdb_gi INDEX gi
iaccdb_db INDEX db
iaccdb_name INDEX name
iaccdb_namelow INDEX namelow
iaccdb_acc INDEX access
seqhound@blueprint.org Version 3.3
The SeqHound Manual 88 of 421 18/04/2005
histdb table
Last updated April 11, 2005
Database: seqhound
Table: histdb
Module: hist
Definition: A history table of gis. Includes the date and any action taken on them.
Observation: As of release 4.0, histdb is no longer populated by the histparser and update. Instead, mother populates
this table, both during the initial build and the update. During the initial build, gi-accession number pairs,
the version number, filename and date are stored in histdb with an action ACTION_ADDED.
During updates, mother extracts the gi-accession number pairs from ACCDB. If neither the gi nor the
accession are found, then this is a new record (ACTION_ADDED). If the accession is found in
SeqHound but is associated with a different gi, then the record represents a changed record
(ACTION_CHANGED), and if the gi-accession pair is present in SeqHound, then the record represents a
deleted record (ACTION_KILLED). The gi, accession, action along with the version, filename and date
(current date update executed) are logged in histdb. Gi's with ACTION_ADDED will get added to accdb,
nucprot, taxgi, pubseq, sengi, sendb, parti by mother. Gi's with ACTION_CHANGED will be deleted and
the new updated information will be added to the core databases. For gi's with ACTION_KILLED, the
old record information in the core databases is now obsolete and will be deleted from the core databases
and the new information will be added to the databases . For any gi that is killed, there will be another gi
(with the same accession) that is added.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 89 of 421 18/04/2005
histdb table
Field Type Null Default Column_Definition Example Source API
rowid int(11)
N
o MySQL autoincrement column.
ts timestamp Yes
CURRENT_
TIMESTAM
P
MySQL timestamp column
2005-03-23 12:32:16
gi int(11)
N
o 0 GenInfo Identifier
21676275 GetGi is called by ToBioseqUp in the
initial build and by ToBioseqUp in
updates. See:
Bioseq-->Seqid->id (choice 12)
oldgi int(11) Yes
N
ULL
The gi previously associated with this
accession. Not used during the initial
build.
10954454 ToBioseqUp retrieves the gi from ACCDB
using GetACCDBRecord.
access char(20)
N
o
The genbank accession from ACCDB
for the gi. If the accession is ‘n/a’,
then the ‘name’ is written here.
See details in accdb table description.
N
C_001911 ToBioseqUp retrieves the accession or
name from ACCDB using
GetACCDBRecord.
version int(11) Yes
N
ULL Version of the record. See details in
accdb table description.
1 ToBioseq and ToBioseqUp call
GetSeqIdInfoFromBioseq. See Bioseq-
>Seq-id->Textseq-id->version.
date date
N
o 0000-00-00 current date when histdb is updated in
the form (YYYYMMDD)
20040128 Mother uses the current date.
action int(11)
N
o 0
the action taken on the record:
ACTION_ADDED,
ACTION_KILLED,
ACTION_CHANGED
ACTION_CHANGED ToBioseq and ToBioseqUp calls
LogHistory.
filename Varchar(80)
N
o The name of the file that contains this
gi for debugging purposes.
rsnc0311.bna Main passes this to ToBioseq and
ToBioseqUp.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 90 of 421 18/04/2005
histdb indices
Keyname Type Field
ihistdb_rowid INDEX rowid
ihistdb_ts INDEX ts
ihistdb_gi INDEX gi
ihistdb_date INDEX date
ihistdb_action INDEX action
ihistdb_acc INDEX access
ihistdb_filename INDEX filename
ihistdb_oldgi INDEX oldgi
seqhound@blueprint.org Version 3.3
The SeqHound Manual 91 of 421 18/04/2005
pubseq table
Last updated April 11, 2005
Database: seqhound
Table: pubseq
Definition: This database maps a geninfo identifier to the medline/pubmed article(s) found in the sequence record
indicating who first published the sequence.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 92 of 421 18/04/2005
pubseq table
Field Type Null Default Column_Definition Example Source API
rowid int(11)
N
o MySQL autoincrement column.
ts timestamp Yes
CURRENT_
TIMESTAM
P
MySQL timestamp column
2005-03-23 12:32:16
ts
gi int(11)
N
o 0 GenInfo Identifier
25992759 ToBioseq calls GetGi.
See Bioseq-->Seqid
(choice 12)
SHoundGiFromReferenceID
SHoundGiFromReferenceList
muid int(11)
N
o
0
Medline identifier found in this
record (points to same article as
pmid (if listed)).
99091706 FillPUBSEQ calls
GetMuid. See Seq-
entry->Bioseq-
>Seqdescr->Pubdesc-
>Pub-equiv->Pub-
>muid (choice 4)
SHoundGetReferenceIDFromGi
SHoundGetReferenceIDFromGiList
SHoundMuidFrom3D
SHoundMuidFrom3Dlist
(ShoundMuidFromGi)
(ShoundMuidFromGiList)
pmid int(11)
N
o
0
Pubmed identifier found in this
record (points to same article as
muid (if listed))
9873079 FillPUBSEQ calls
GetMuid. See Seq-
entry->Bioseq-
>Seqdescr->Pubdesc-
>Pub-equiv->Pub-
>pmid (choice 13)
seqhound@blueprint.org Version 3.3

The SeqHound Manual 93 of 421 18/04/2005
pubseq indices
Keyname Type Field
PRIMARY PRIMARY gi
p
mi
d
ipubseq_rowid INDEX rowid
ipubseq_ts INDEX ts
ipubseq_gi INDEX gi
ipubseq_muid INDEX muid
ipubseq_pmid INDEX
p
mid
seqhound@blueprint.org Version 3.3
The SeqHound Manual 94 of 421 18/04/2005
taxgi table
Last updated April 11, 2005
Database: seqhound
Table: taxgi
Module: core
Definition: This table associates a gi with a taxon identifier.
Observation.: Gis are stored in taxgi even if they are not associated with any taxonomy id. This occurs, for example, for
some patent gis.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 95 of 421 18/04/2005
taxgi table
Field Type Null Default Column_Definition Example Source API
rowid int(11)
N
o MySQL autoincrement
colulmn.
ts timestamp Yes
CURRENT_T
IMESTAMP MySQL timestamp column 2005-03-23
16:50:42
gi int(11)
N
o 0
GenInfo Identifier 42565179 WriteTAXGI calls GetGi.
See Bioseq-->Seq-id (choice 12)
SHoundDNAFromTaxID()
SHoundDNAFromTaxIDList()
SHoundProteinsFromTaxID()
SHoundProteinsFromTaxIDList()
SHoundProteinsFromChromosome()
SHoundProteinsFromChromosomeList()
SHoundDNAFromChromosome()
SHoundDNAFromChromosomeList()
taxid int(11)
N
o 0 Taxonomy Identifier from
N
CBI’s taxonomy database.
3702 See note 1 below. SHoundTaxIDFromGi()
SHoundTaxIDFromGiList()
kloodge int(11) Yes
N
ULL Blueprint chromosome id from
the chrom table.
392 See note 2 below.
type varchar(50) Yes
N
ULL
Type of macromolecule
DNA, RNA, protein, NA,
other, "not specified".
protein WriteTaxgi calls GetType. See
Bioseq->mol (see note 3 below).
SHoundDNAFromChromosome()
SHoundDNAFromChromosomeList()
SHoundProteinsFromChromosome()
ShoundProteinsFromChromosomeList()
Notes:
1.
FillTAXGI calls GetTaxId. See Bioseq-->Seq-desc->BioSource->Org-ref->Dbtag.db == "taxon"
Bioseq-->Seq-desc->BioSource->Org-ref->Dbtag.tag
2.
Mother: ToBioseq calls SearchCHROMByChromNum.
Postcomgen: GetAllRecordsFromGICHROMID and UpdateTaxgiWithKloodge
seqhound@blueprint.org Version 3.3

The SeqHound Manual 96 of 421 18/04/2005
3.
DNA = 1
RNA = 2
Protein = 3
NA = 4
Other = 255
taxgi indices
Keyname Type Field
PRIMARY PRIMARY gi
itaxgi_rowid INDEX rowid
itaxgi_ts INDEX timestamp
itaxgi_gi INDEX gi
itaxgi_taxid INDEX taxid
itaxgi_kl INDEX kloodge
itaxgi_type INDEX type
seqhound@blueprint.org Version 3.3
The SeqHound Manual 97 of 421 18/04/2005
sengi table
Last updated April 11, 2005
Database: seqhound
Table: sengi
Module: core
Definition: Seqentry to 'gi' conversion.
Observation: Seqhound keeps track of which Bioseqs come from which Bioseq-sets using the sengi table. In the sengi
table, each Bioseq is represented by a GI (a unique identifier for the sequence record) and each Bioseq-set
is represented by the first GI in the set of sequences, the seid.
Sengi also stores the division for historical purposes.
sengi table
Field Type Null Default Column_Definition Example Source API
rowid int(10)
N
o MySQL autoincrement column
ts timestamp Yes
CURRENT_TI
MESTAMP MySQL timestamp column 2005-03-23 12:32:16
gi int(11)
N
o
0 GenInfo Identifier 2313082 ToBioseqSEQENTRY calls GetGi. See Seq-entry->Bioseq-
set->Bioseq-->Seqid (choice 12)
seid int(11)
N
o 0
GenInfo Identifier: the first
one in the bioseq-set. seid
means seqentry id.
231308 ToBioseq calls GetGi for the first Bioseq in a SeqEntry See
Seq-entry->Bioseq-set->Bioseq-->Seqid (choice 12).
division char(15) Yes
N
ULL Genbank division. PRI ToBioseqSEQENTRY calls GetDivisionFromPARTI.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 98 of 421 18/04/2005
sengi indices
Keyname Type Field
PRIMARY PRIMARY gi
isengi_rowid INDEX rowid
isengi_ts INDEX ts
isengi_gi INDEX gi
isengi_seid INDEX seid
seqhound@blueprint.org Version 3.3
The SeqHound Manual 99 of 421 18/04/2005
sendb table
Last updated April 11, 2005
Database: seqhound
Table: sendb
Module: core
Definition: SeqEntry Data Base
Observation: Sequences are distributed by GenBank in packages. Each package is referred to as a seq-entry.
A seq-entry may contain either a single sequence record (called a Bioseq) or a set of sequence records
(called a Bioseq-set). If a Seq-entry contains a Bioseq-set then that Bioseq-set contains a list of Seq-entry
packages (yes, this data structure is recursive). Each of these Seq-entry packages contains a single
sequence record (a Bioseq). There is annotation that is associated with single sequence records (Bioseqs).
An example of annotation is a list of authors who are responsible for submitting a sequence record. There
is also annotation associated with sets of sequence records (Bioseq-sets). This type of annotation applies
to all of the sequence records that are in the set. For example a set of authors may be responsible for all
of the sequence records in the set.
Seqhound stores Bioseqs and their associated GenInfo id's in one central table (called asndb). Since each
of these Bioseqs may have come from a Bioseq-set, Seqhound needs a way to store the Bioseq-set
associated annotation (that applies to each of the Bioseqs in the set). To accomplish this, Seqhound takes
the Bioseq-set, removes the Bioseqs that it contains, and stores the remainder of the Bioseq-set
(annotation) in the sendb table. Seqhound keeps track of which Bioseqs come from which Bioseq-sets
using the sengi table. In the sengi table, each Bioseq is represented by a GI (a unique identifier for the
sequence record) and each Bioseq-set is represented by the first GI in the set of sequences (the seid).
seqhound@blueprint.org Version 3.3
The SeqHound Manual 100 of 421 18/04/2005
sendb table
Field Type Null Default Column_Definition Example Source API
rowid int(10)
N
o Auto number row identifier
ts timestamp Yes
CURRENT_TI
MESTAMP MySQL timestamp column 2005-03-23 12:32:16
seid int(11)
N
o 0 Gene Info identifier for the first
bioseq in the SeqEntry.
2313082 ToBioseq calls GetGi for the
first Bioseq in a SeqEntry.
Main calls
AppendRecordSENDB. See
Seq-entry->Bioseq-set-
>Seq-entry->Bioseq->Seqid
(choice 12)
asn1 mediumblob
N
o
ASN1 Binary SeqEntry. Note
that the bioseqs themselves have
been removed to save space.
binary data ToBioseqSeqEntry removes
the bioseqs. Main calls
AppendRecordSENDB.
These functions return the Seqentry
or BioseqSet stored in this field
after first replacing all of the
Bioseqs that belong in it.
SHoundGetBioseqSet()
SHoundGetSeqEntry()
sendb indices
Keyname Type Field
PRIMARY PRIMARY seid
isendb_rowid INDEX rowid
isendb_ts INDEX ts
isendb_seid INDEX seid
seqhound@blueprint.org Version 3.3
The SeqHound Manual 101 of 421 18/04/2005
chrom table
Database: seqhound
Table: chrom
Module: core
Definition: This table maps taxonomy ids with their complete genome information (chromid, chromflag, accession, and name)
Observation: This table keeps track of all completely sequenced chromosomes of a particular organism (taxid). We also store the NCBI
chromid (chromid), the type of sequence (chromflag), the name (name) and accession number (access).
Every chromid will have a corresponding kloodge identifier (this is Blueprint’s internal version of the NCBI chromid. In
addition, every taxon will have one kloodge identifier that has no corresponding chromid. This kloodge id is assigned to any
proteins or RNAs that are have no chromid; i.e. they have not been assigned to a chromosome yet. The kloodge is the value of
an autoincrement column and is written to taxgi by postcomgen.
We also store the chromosome number (chromnum) as text. This is used so that we can assign records which have no complete
genome information other than taxid and chromosome number to a chromosome identifier.
For example, let’s say that chromosome 4 (circular plasmid, named chr_4, access XIV) of species “species X” (taxid 123) is
completely sequenced and that NCBI has assigned this chromosome a chromid of 191. Let us say our internal kloodge id for
this same chromosome is 1254. Then a record is added to the chrom table with fields:
kloodge: 1254
taxid: 123
chromid: 191
chromflag: plasmid
accession: XIV
name: species X chr_4
chromnum: chr 4
See more example entries in the notes section below.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 102 of 421 18/04/2005
chrom table
Field Type Null Default Column_Definition Example1Source API
kloodge int(11)
N
o
Blueprint chromosome
id (used internally).
Mysql Autoincrement
column.
568
ts timestamp
N
o CURRENT_T
IMESTAMP
2005-03-23
16:17:11
taxid int(11)
N
o 0
Taxonomy identifier 9606 ToBioseq calls GetTaxid See
Bioseq-->Seq-desc->BioSource->Org-
ref->Dbtag->tag
SHoundAllGenomes
chromid int(11)
N
o 0
N
CBI chromsome
identifier (see note 2
below)
1 ToBioseq calls
GetChromIdFromBioseq. See Bioseq-
>SeqId->Dbtag->tag
chromflag int(11)
N
o 0
the type of chromosome
(chromosome, plasmid,
mitochrondria etc)
defined as byte macros
in intrez.h.
(see note 3 below)
1 ToBioseq calls GetGenomeInfo, See
Bioseq->descr->source->subtype-
>subtype
SHoundChromosomeFromGenome
access varchar(20)
N
o NULL
Accession for the
sequence (see note 2
below).
N
C_000001 GetGenomeInfo calls
Misc_GetAccession. See Bioseq->Seq-
id->Textseq-id->accession
name text
N
o
The scientific name of
the organism with the
chromosome name.
Homo sapiens
Chromosome 1
GetGenomeFromBiosource constructs
this from various places in the
Biosource. See Bioseq->descr->source
chromnum varchar(10) Yes
N
ULL
Chromosome number as
text.
1 GetGenomeFromBiosource constructs
this from various places in the
Biosource subtype. See Bioseq->descr-
>source->subtype
seqhound@blueprint.org Version 3.3
The SeqHound Manual 103 of 421 18/04/2005
Notes.
1.
Example entries from the chrom table.
kloodge ts taxid chromid chromflag access name chromnum
1 '2005-03-23 12:33:24' 9 13723 9 'NC_001911' 'Buchnera aphidicola Plasmid pLeu-Dn' 'pLeu-Dn'
2 '2005-03-23 12:33:24' 9 17119 9 'NC_004843' 'Buchnera aphidicola Plasmid pBPS1' 'pBPS1'
567 '2005-03-23 16:06:41' 9606 -9606 1 'Homo sapiens Chromosome Un' 'Un'
568 '2005-03-23 16:17:11' 9606 1 1 'NC_000001' 'Homo sapiens Chromosome 1' '1'
569 '2005-03-23 16:17:12' 9606 2 1 'NC_000002' 'Homo sapiens Chromosome 2' '2'
570 '2005-03-23 16:17:12' 9606 3 1 'NC_000003' 'Homo sapiens Chromosome 3' '3'
592 '2005-03-23 16:35:05' 9606 2000021 5 'AC_000021' 'Homo sapiens Mitochondria'
4289 '2005-04-05 09:29:55' 287944 18301 5 'NC_006916' 'Harpiosquilla harpax Mitochondria'
4290 '2005-04-05 09:29:55' 302098 18316 5 'NC_006931' 'Eubalaena japonica Mitochondria'
Note that the third entry down represents an entry corresponding to an “unknown” chromosome. Proteins or RNAs that have not been
mapped to a chromosome will be assigned this kloodge identifier (567) in the accdb and table.
2.
Kloodge identifiers representing unknown chromosmomes will not have a corresponding chromid from NCBI. The negative value of
the taxid is used instead. See third entry in example table above. These entries will also have no accession associated with them.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 104 of 421 18/04/2005
notes continued…
3.
CHROM_PHAGE (phage sequence)
CHROM_NR
CHROM_ECE
CHROM_PLMD (plasmid sequence)
CHROM_CHLO (chloroplast sequence)
CHROM_MITO (mitochrondrial sequence)
CHROM_CHROM (chromosome sequence)
CHROM_ALL
chrom indices
Keyname Type Field
PRIMARY PRIMARY kloodge
ichrom_chromid INDEX chromid
ichrom_taxid INDEX taxid , chromnum
ichrom_kl INDEX kloodge
ichrom_acc INDEX access
Ichrom_chromnum INDEX chromnum
seqhound@blueprint.org Version 3.3

The SeqHound Manual 105 of 421 18/04/2005
gichromid table
Last updated April 11, 2005
Database: SeqHound
Table: gichromid
Module: core
Definition: Stores RNA and protein gis that are part of a complete genome. Used to
update taxgi with the kloodge.
Observation: This table is used internally to store RNA and Protein gis parsed from
contigs that are part of a complete genome. After a complete build is
done, the gis and chromids from this table are used by postcomgen to
update taxgi with the kloodge.
gichromid table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o Primary key. Mysql auto-
increment column.
ts timestamp Yes CURRENT_TIMESTAMP
MySQL timestamp
column
2005-03-23 12:32:16
gi int(11)
N
o 0 Gene Info identifier
231308 ToBioseq calls
FillGiChromid. See Bioseq--
>Seqid (choice 12)
chromid int(11)
N
o 0
N
CBI chromosome id 3144
N
CBI chromid
contiggi int(11) Yes
N
ULL Gi of the contig that
contains this gi.
231308 ToBioseq calls
GetChromidFromBioseq.
See Bioseq->Seqid(choice
11)->tag->id
gichromid indices
Keyname Type Field
seqhound@blueprint.org Version 3.3

The SeqHound Manual 106 of 421 18/04/2005
id PRIMARY gi
gi INDEX gi
igits INDEX ts
igichromid INDEX chromid
igicontiggi INDEX contiggi
seqhound@blueprint.org Version 3.3

The SeqHound Manual 107 of 421 18/04/2005
contigchromid table
Last updated April 11, 2005
Database: SeqHound
Table: contigchromid
Module: core
Definition: Stores contig gis that are part of a complete genome. Used to update
taxgi with the kloodge.
Observation: This table is used internally to store lists of GI’s that make up a contig
sequence for a complete genome. After a complete build is done, the gis
and chromids from these contigs are used by postcomgen to update
taxgi with the kloodge.
contigchromid table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o Mysql auto-increment column.
ts timestamp Yes
CURRENT_TI
MESTAMP MySQL timestamp column 2005-03-23 12:32:16
contiggi int(11)
N
o 0 Gene Info identifier for this sequence record
that is part of a contig.
50754263 ToBioseq calls GetGi. See Bioseq--
>Seqid (choice 12)
topgi int(11) Yes
N
ULL
Genbank gi of the contig that contains
multiple sequences (i.e. multiple
contiggi’s).
51039201 ToBioseq calls GetGi. See Bioseq--
>Seqid (choice 12)
chromid int(11)
N
o 0
N
CBI chromosome id.
461 ToBioseq calls
GetChromidFromBioseq. See Bioseq-
>Seqid(choice 11)->tag->id
changed Int(11) Yes
N
ULL
Indicates whether the record needs to be
processed by postcomgen. Used by mother
in update mode.
1 = process
2 = do not process
ToBioseq calls FillContigChromid. See
Bioseq->Seqid(choice 11)->tag->id
contigchromid indices
seqhound@blueprint.org Version 3.3

The SeqHound Manual 108 of 421 18/04/2005
Keyname Type Field
contiggi PRIMARY id
icontigts INDEX ts
contigi UNIQUE contiggi
iccontigi INDEX contiggi
ictopgi INDEX topgi
seqhound@blueprint.org Version 3.3
The SeqHound Manual 109 of 421 18/04/2005
gichromosome table
Last updated April 11, 2005
Database: SeqHound
Table: gichromosome
Module: core
Definition: Stores RNA and protein gis that have a chromosome name, but no other
complete genome info.
Observation: This table is used internally to store RNA and protein gis that have a
chromosome name, but no other complete genome info. After a
complete build is done, postcomgen will write these gis to gichromid
with the appropriate chromid.
gichromosome table
Field Type Null Default Column
_
Definition Example Source API
id int(11)
N
o
Primary key Mysql
auto-increment
column.
ts timestamp Yes
CURRENT_TIM
ESTAMP
MySQL timestamp
column
2005-03-23 12:32:16
gi int(11)
N
o 0 GenInfo Identifier 231308 ToBioseq calls GetGi. See Bioseq-->Seqid
(choice 12)
chromnum varchar(10)
N
o Chromosome
number
1 ToBioseq calls GetGenomeInfo. See Bioseq-
>descr->source->subtype->name
seqhound@blueprint.org Version 3.3

The SeqHound Manual 110 of 421 18/04/2005
gichromosome indices
Keyname Type Field
id PRIMARY id
igichrom_ts INDEX ts
gi UNIQUE gi
igichrom_gi INDEX gi
igichrom_num INDEX chromnum
seqhound@blueprint.org Version 3.3
The SeqHound Manual 111 of 421 18/04/2005
contigchromosome table
Last updated April 11, 2005
Database: SeqHound
Table: gichromosome
Module: core
Definition: Stores contig gis that have a chromosome name, but no other complete
genome info.
Observation: This table is used internally to store contig gis that have a chromosome
name, but no other complete genome info. After a complete build is
done, postcomgen will writethese gis to contigchromid with the
appropriate chromid.
contigchromosome table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o Primary key Mysql auto-
increment column.
ts timestamp Yes
CURREN
T_TIMES
TAMP
MySQL timestamp column
2005-03-23 12:32:16
contiggi int(11)
N
o GenInfo gi 231308 ToBioseq calls GetGi. See Bioseq-->Seqid
(choice 12)
chromnum Varchar(10)
N
o Chromosome number 1 ToBioseq calls GetGenomeInfo. See Bioseq--
>Seqid (choice 12)
seqhound@blueprint.org Version 3.3

The SeqHound Manual 112 of 421 18/04/2005
contigchromosome indices
Keyname Type Field
id PRIMARY id
igichrom_ts INDEX ts
contiggi UNIQUE contiggi
icontigchrom_gi INDEX contiggi
icontigchrom_num INDEX chromnum
seqhound@blueprint.org Version 3.3

The SeqHound Manual 113 of 421 18/04/2005
Redundant protein sequences (redundb) module
Last updated: August 4, 2004
redund parser
purpose:
The redund parser builds the redundb module, which consists of the redund table. The input file consists of information pertaining to
redundant GIs, accession numbers and the sequence information which the GIs refer to. The resulting data table contain information
on the GIs, redundant GI groups and the ranking of each GI within their redundant group.
module:
redundb
input files:
nr.gz from ftp://ftp.ncbi.nih.gov/blast/db/FASTA/
table created:
redund
source code location:
slri/seqhound/parsers/redund.c
config file dependencies:
slri/seqhound/config/.intrezrc (UNIX platform)
The relevant section of the configuration file is:
[datab]
;seqhound database that you are connecting
username=your_user_name
password=your_pass_word
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
[sections]
seqhound@blueprint.org Version 3.3
The SeqHound Manual 114 of 421 18/04/2005
;this should be set to 1 to allow usuage of the redundb
;redundant protein sequences
redundb = 1
Text in italics must be changed for the .intrezrc file to function correctly with your SeqHound set-up. Variables username,
password, dsn, database in section [datab] should have the same values as USER, PASSWORD, DSN and DATABASE
respectively in your .odbc.ini (see Step 10 in section 4.4.) In section [sections] redundb should be 1.
commandline parameters
Typing ./redund at the command line will return a list of command line parameters and default settings. For example:
>./redund
redund arguments:
-i Input non-redundant database fasta file [File In]
-n Initialize database file [T/F] Optional
default = F
example use:
./redund -i nr -n T
associated scripts:
nrftp.pl: retrieves the relevant data file from NCBI's ftp site
error & run-time logs:
redund writes to a log file called redundlog
additional information:
ftp://ftp.ncbi.nih.gov/blast/db/FASTA/
See data table descriptions for each of the tables that are listed under “tables altered”
seqhound@blueprint.org Version 3.3
The SeqHound Manual 115 of 421 18/04/2005
redund table
Last updated: August 4, 2004
Database: seqhound
Table: redund
Module: redundb
Definition: Partitions GIs into groups based on sequence redundancy, and ranks
each GI within their group.
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto number row identifier
gi int(11)
N
o 0 GenInfo Identifier
rordinal int(11)
N
o 0 Position in redundant group
rgroup int(11) Yes
N
ULL ID of redundant group(not static)
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY gi
iredund_rowid INDEX rowid
iredund_gi INDEX gi
iredund_ordinal INDEX rordinal
iredund_rgroup INDEX rgroup
seqhound@blueprint.org Version 3.3
The SeqHound Manual 116 of 421 18/04/2005
Observation: redund is a database table that places GIs with equivalent sequences
into redundant groups. The same sequence may be worked on by
independent researchers. Each discovery is submitted and assigned
a different gi. When the discoveries concern a common sequence,
the GIs point to sequence records that describe redundant sequences,
i.e. a single sequence has multiple GIs. These GIs then belong to a
common group (rgroup). In such cases, it is common for one
particular GI to be used more than the others because it is better
annotated. The ordinal (rordinal) is a ranking of the GIs, 1 for the
b
est annotated GI, 2 for the second, 3 for the third etc. This ordering
is determined by the ordering of the sequence headers in the nr
source file. These 3 pieces of information form the redund database
table.
Source org: NCBI
Source file: nr.gz from ftp://ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz
parser: redund
***gi***
description: GenInfo Identifier for a sequence record.
example: 7228451
default value: 0
source: parsed from nr (line 1 of record) as token(|) delimited fields
parser: redund
function: LabelGI, AssignRedund (redund.c)
seqhound@blueprint.org Version 3.3
The SeqHound Manual 117 of 421 18/04/2005
API: SHoundRedundantGroup (returns all GIs in the same group as
the input GI)
SHoundRedundantGroupFromID (returns GIs from the group
with input group ID)
SHoundFirstOfRedundantGroupFromID (GI of rank 1,
given a group)
SHoundIsNRFirst (true/false, is this GI is ranked 1 in it's
group)
***rordinal***
description: rank position in redundant set
example: 1
default value: n/a
source: based on the position of the gi within the record, the first gi has
rordinal 1, the 2nd has ordinal 2
parser: redund
function: LabelGI, AssignRedund (redund.c)
API: SHoundIsNRFirst (tells if a gi has rordinal = 1)
***rgroup***
description: redundant group the gi is a member of
example: 1
default value: n/a
source: rgroup IDs are assigned to GIs as the input file is parsed. Each GI
encountered is assigned the rgroup ID of those GIs already
encountered which have an identical sequence. If no such GIs have
been encountered, the GI is assigned a new rgroup ID one number
higher than the largest current rgroup ID in the database. The first
GI encountered is given an rgroup ID of 1.
parser: redund
seqhound@blueprint.org Version 3.3
The SeqHound Manual 118 of 421 18/04/2005
function: LabelGI, AssignRedund (redund.c)
API: SHoundRedundantGroupIDFromGI[List] (returns the ID of
the rgroup that the input GI is a part of)
seqhound@blueprint.org Version 3.3
The SeqHound Manual 119 of 421 18/04/2005
Complete genomes tracking (gendb) module
As of release 4.0 of SeqHound, the complete genomes module has been moved into the core module. The initial build and updates to
these tables are handled by the mother and postcomgen parsers. See the description of the core module above for more details.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 120 of 421 18/04/2005
Taxonomy hierarchy (taxdb) module
Last updated August 18, 2004
importtaxdb parser
purpose:
The importtaxdb parser builds the taxonomy module, which consist of the taxdb, gcodedb, divdb, del and merge table. The input file
consists of taxonomic information such as the taxonomy nodes, names, division, genetic codes, deleted nodes and merged nodes.
Table taxdb holds taxonomy ids and a binary file associated with each id. Table gcodedb holds genetic code ids and a binary file
associated with each id. Table divdb holds division ids and a binary file associated with each id. Table del holds the taxonomy id of
the deleted nodes. Table merge holds the taxonomy ids of nodes which has been merged and which is result of merging.
module:
taxdb
input files:
taxdump.tar.gz from ftp://ftp.ncbi.nih.gov/pub/taxonomy/
tables created:
taxdb
gcodedb
divdb
del
merge
source code location:
slri/seqhound/taxon/importtaxdb.c
config file dependencies:
slri/seqhound/config/.intrezrc (UNIX platform)
seqhound@blueprint.org Version 3.3

The SeqHound Manual 121 of 421 18/04/2005
The relevant section of the configuration file is:
[datab]
;seqhound database that you are connecting
username=your_user_name
password=your_pass_word
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
[sections]
;taxonomy hierarchy
taxdb = 1
Text in italics must be changed for the .intrezrc file to function correctly with your SeqHound set-up. Variables username,
password, dsn, database in section [datab] should have the same values as USER, PASSWORD, DSN and DATABASE
respectively in your .odbc.ini (see Step 10 in section 4.4.) In section [sections] taxdb should be 1.
commandline parameters
There are no command line parameters. Typing ./importtaxdb will run the program:
example use:
./importtaxdb
associated scripts:
taxftp.pl: retrieves the relevant data file from NCBI's ftp site
error & run-time logs:
importtaxdb writes to a log file called importtaxdb_log.txt
additional information:
readme files at ftp://ftp.ncbi.nih.gov/pub/taxnomy/
See data table descriptions for each of the tables that are listed under “tables altered”
seqhound@blueprint.org Version 3.3

The SeqHound Manual 122 of 421 18/04/2005
taxdb table
Database: seqhound
Table: taxdb
Module: taxdb
Definition: Maps a taxid to information about the taxid. The information about the
taxid is stored as an ASN blob.
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto number row identifier
tid int(11)
N
o 0 Taxonomy identifier
asn mediumblob Yes
N
ULL Information about tid
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY tid
itax_rowid INDEX rowid
itax_tid INDEX tid
Observation: The entire taxonomy can be viewed as a hierarchical taxonomy tree.
The tree expresses the relationships between the nodes within the tree.
At the root of the tree is a generic node that provides no information.
Descendents of the root node include superkingdoms such as Archea,
Eubacteria, Eukaryota, Viroids, Viruses. etc. Each of these
superkingdoms have corresponding taxonomy children. Also included
in the taxonomy tree are artificial sequences and unclassified
taxonomies such as the prions, unidentified agents, etc. taxdb maps the
taxonomy identifiers to the relevant information describing the
taxonomies. Organizing the taxonomy as a tree allows users to request
seqhound@blueprint.org Version 3.3

The SeqHound Manual 123 of 421 18/04/2005
complete lineages, ancestors, children and permits the user to browse
the tree exploiting its tree structure.
taxdb contains information from both nodes.dmp and names.dmp
Source org: NCBI
Source files: nodes.dmp from ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
names.dmp from ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
parser: importtaxdb
***tid***
description: the unique taxonomy identifier.
example: 9606 (Human)
default value: n/a
source: nodes.dmp: column 1
parser: importtaxdb
function: Parser_TaxDBNodeRecord
API: SHoundGetTaxChildNodes
SHoundGetTaxChildNodesList
SHoundGetAllTaxProgeny
SHoundGetTaxParent
ShoundGetAllTaxAncestors
Note that although relationships (parent, child) are not
stored in the database, it is a part of the asn field, and as
such it makes it possible to retrieve parent, children,
ancestors and lineages (see asn blob).
seqhound@blueprint.org Version 3.3
The SeqHound Manual 124 of 421 18/04/2005
***asn***
description: ASN blob containing information about the taxonomy.
Fields in the ASN blob include:
1) parent taxonomy of node
2) rank of node
3) embl code for node
4) division of node (see divdb)
5) inherited div flag (1 if this node inherits its division from its parent)
6) genetic code (see gcodedb)
7) inherited genetic code (1 if this nodes inherits its genetic code from
parent)
8) mitochondrial genetic code
9) inherited mit. genetic code (1 if nodes inherits mitochondrial gencode
from parent)
10) genbank hidden flag (1 if name is suppressed in GenBank entry
lineage)
11) subtree root flag (1 if this subtree has no sequence data yet)
12) comments
13) name of taxid (from names.dmp)
Possible rankings of node (there may be more):
superkingdom, kingdom
genus, subgenus
subspecies, species
subfamily, family, superfamily
phylum, subphylum,
subtribe, tribe
varietas
seqhound@blueprint.org Version 3.3
The SeqHound Manual 125 of 421 18/04/2005
infraorder, order, suborder
infraclass, class, subclass
no rank
example: A textual representation of the blob for taxid 9606:
SLRI-taxon ::= {
taxId 9606 ,
parent-taxId 9605 ,
children-taxId {
63221 } ,
names {
{
name "Homo sapiens" ,
name-class scientific-name } ,
{
name "human" ,
name-class other ,
other-class "genbank common name" } ,
{
name "man" ,
name-class common-name } } ,
rank {
rank species ,
premod none ,
postmod none } ,
embl-code "HS" ,
division 5 ,
inherited-div TRUE ,
gencode 1 ,
inherited-gencode TRUE ,
mito-gencode 2 ,
inherited-mito-gencode TRUE ,
genbank-hidden FALSE ,
hidden-subtree-root FALSE }
default value: n/a
source: nodes.dmp: column 2 - 13 and names.dmp: column 2, 3
parser: importtaxdb
function: Parser_TaxDBNodeRecord
API: The ASN object is not directly accessible through the API. Instead,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 126 of 421 18/04/2005
fields in the ASN object may be retrieved and be accessible from the
API. eg.
SHoundGetTaxNameFromTaxIDByClass
SHoundGetTaxNameFromTaxID
SHoundGetTaxLineageFromTaxID
You can also directly retrieve the ASN SLRITaxon by:
#include <taxdb_db.h>
AsnIoPtr aip = NULL;
SLRITaxonPtr ptax = NULL;
SHoundInit(FALSE, "name");
ptax = DB_GetTaxRec(9606);
aip = AsnIoNex(ASNIO_TEXT_OUT, stdout, NULL, NULL, NULL);
SLRITaxonAsnWrite(ptax, aip, NULL);
AsnIoClose(aip);
This will produce the text in the example above.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 127 of 421 18/04/2005
gcodedb table
database: seqhound
table: gcodedb
Module: taxdb
definition: Maps a genetic code identifier to the corresponding ASN taxonomy
genecode record.
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto number row identifier
gcid int(11)
N
o 0 genetic code
asn mediumblob Yes
N
ULL ASN taxon-gencode record
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY gcid
igcode_rowid INDEX rowid
igcode_gcid INDEX gcid
Observation: gcodedb is part of a group of databases (taxdb, gcodedb, divdb) that can
be used to form a taxonomy hierarchy tree. In the case of gcode, the
records map a genetic code identifier to an ASN object that holds
information about that genetic code. taxdb (see taxdb later) will specify
the type of genetic material each taxonomy uses, eg if the taxonomy has
a plasmid as a genetic code, mitochrondria DNA, or standard
chromosomal DNA etc. gcodedb houses information concerning each of
the genetic code. Relevant information include the translation table (the
mapping of nucleic acid codons to amino acids), & the start codon.
Further information at: http://www.ncbi.nlm.nih.gov/Taxonomy
seqhound@blueprint.org Version 3.3
The SeqHound Manual 128 of 421 18/04/2005
Source org: NCBI
Source file: gencode.dmp from ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
Parser: importtaxdb
***gcid***
description: The genetic code identifier. The identifiers may not be consecutive
because historically codes may have merged or new ones were created.
The code and their corresponding names:
0 Unspecified
1 Standard
2 Vertebrate Mitochondrial
3 Yeast Mitochondrial
4 Mold Mitochondrial; Protozoan Mitochondrial; Coelenterate
Mitochondrial; Mycoplasma; Spiroplasma
5 Invertebrate Mitochondrial
6 Ciliate Nuclear; Dasycladacean Nuclear; Hexamita Nuclear
9 Echinoderm Mitochondrial; Flatworm Mitochondrial
10 Euplotid Nuclear
11 Bacterial and Plant Plastid
12 Alternative Yeast Nuclear
13 Ascidian Mitochondrial
14 Alternative Flatworm Mitochondrial
15 Blepharisma Macronuclear
16 Chlorophycean Mitochondrial
21 Trematode Mitochondrial
22 Scenedesmus obliquus mitochondrial
seqhound@blueprint.org Version 3.3
The SeqHound Manual 129 of 421 18/04/2005
23 Thraustochytrium mitochondrial code
example: Because many of the taxonomy groups are bacterial, 11 is a common
genetic code.
default value: n/a
source: gencode.dmp: column 1
parser: importtaxdb
function: Parse_TaxDBGenCodeRecord
API: n/a
***asn***
description: An ASN blob for the genetic code information corresponding to the
genetic code identifiers. The individual fields in the blob are:
1) an optional abbreviation
2) name of genetic code (see gcid for names)
3) translation table
4) start codon
The translation table maps the nucleic acid codon to amino acids:
In the source file, the map is represented as:
FFLLSSSSYY**CC*WLLLLPPPPHHQQRRRRIIIMTTTTNNKKSSRRVVVVAAAADDEEGGGG
There are 64 characters in this map. Each position in the map
corresponds to a codon (see below) and the letter at that position
corresponds to the resulting amino acid that will be translated from that
codon. The same can map be better expressed as a direct mapping from
codon to amino acid where
1) first 3 letters represent a nucleic acid sequence (codon),
2) an amino acid letter identifier
3) an amino acid abbreviation
seqhound@blueprint.org Version 3.3
The SeqHound Manual 130 of 421 18/04/2005
TTT F Phe TCT S Ser TAT Y Tyr TGT C Cys
TTC F Phe TCC S Ser TAC Y Tyr TGC C Cys
TTA L Leu TCA S Ser TAA * Ter TGA * Ter
TTG L Leu TCG S Ser TAG * Ter TGG W Trp
CTT L Leu CCT P Pro CAT H His CGT R Arg
CTC L Leu CCC P Pro CAC H His CGC R Arg
CTA L Leu CCA P Pro CAA Q Gln CGA R Arg
CTG L Leu CCG P Pro CAG Q Gln CGG R Arg
ATT I Ile ACT T Thr AAT N Asn AGT S Ser
ATC I Ile ACC T Thr AAC N Asn AGC S Ser
ATA I Ile ACA T Thr AAA K Lys AGA R Arg
ATG M Met ACG T Thr AAG K Lys AGG R Arg
GTT V Val GCT A Ala GAT D Asp GGT G Gly
GTC V Val GCC A Ala GAC D Asp GGC G Gly
GTA V Val GCA A Ala GAA E Glu GGA G Gly
GTG V Val GCG A Ala GAG E Glu GGG G Gly
The start codon is the particular codon that signals the start of protein
translation for that particular genetic molecular (i.e. a standard
chromosome starts translation at a different codon than a protozoan
mitochrondrial DNA).
The start codon is shown in the same manner as the translation table:
----------------------------------MM----------------------------
This indicates that M (methionine) is the start codon.
Further information can be found at:
htpp://www.ncbi.nlm.nih.gov/Taxonomy in Genetic codes link.
example: A textually representation of a blob for genetic code 11:
SLRI-taxon-gencode ::= {
gencode-id 11 ,
name "Bacterial and Plant Plastid" ,
trans-table
"FFLLSSSSYY**CC*WLLLLPPPPHHQQRRRRIIIMTTTTNNKKSSRRVVVVAAAADDEEGGG
G " ,
start-codons "---M---------------M------------MMMM--------------
seqhound@blueprint.org Version 3.3
The SeqHound Manual 131 of 421 18/04/2005
-M------------ " }
default value: n/a
source: gencode.dmp: column 2, 3, 4, 5 are stored in the database as an ASN
SLRITaxonGencode structure.
parser importtaxdb
function: Parse_TaxDBGenCodeRecord
API: not directly accessible from the API, but you can retrieve the
SLRITaxonGencode object using:
#include <taxdb_db.h>
AsnIoPtr aip = NULL;
SLRITaxGencodePtr ptax = NULL;
SHoundInit(FALSE, "name");
ptax = DB_GetTaxGencodeRec(11);
aip = AsnIoNex(ASNIO_TEXT_OUT, stdout, NULL, NULL, NULL);
SLRITaxGencodeAsnWrite(ptax, aip, NULL);
AsnIoClose(aip);
This will produce the text in the example above.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 132 of 421 18/04/2005
divdb table
Database: seqhound
Table: divdb
Module: taxdb
Definition: Maps taxonomy division ID to an ASN SLRI-taxon-div object
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto number row identifier
did int(11)
N
o 0 Taxonomy Division ID
asn mediumblob Yes
N
ULL ASN taxonomy division record
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY did
idiv_rowid INDEX rowid
idiv_did INDEX did
Observation: divdb is part of a group of databases (taxdb, gcodedb, divdb) that can be
used to form a taxonomy hierarchy tree. In the case of divdb, the
records map a division identifier to an ASN object that holds
information about that division. This information include a 3-character
GenBank division code, a division name and an optional comment.
There are 10 divisions (see below).
Source org: NCBI
Source file: division.dmp from ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
Parser: importtaxdb
seqhound@blueprint.org Version 3.3
The SeqHound Manual 133 of 421 18/04/2005
***did***
description: The division identifier (0-11, see asn for the names of division).
example: 0
source: division.dmp: column 1
parser: importtaxdb
function: Parse_TaxDBDivRecord
API: n/a
***asn***
description: The ASN blob for the taxonomy division. The individual fields in the
ASN blob are:
1) a 3 character division code (see below)
2) a division name (see below)
3) an optional comment
Possible division codes and corresponding division names are:
BCT Bacteria
INV Invertebrates
MAM Mammals
PHG Phages
PLN Plants
PRI Primates
ROD Rodents
SYN Synthetic
UNA Unassigned
seqhound@blueprint.org Version 3.3
The SeqHound Manual 134 of 421 18/04/2005
VRL Viruses
VRT Vertebrates
example: the entire field is one ASN blob. A possible blob could be represented
textually as:
SLRI-taxon-div ::= {
div-id 1 ,
div-code "INV" ,
div-name "Invertebrates" }
source: Column 2, 3, and 4 of division.dmp are stored as one ASN blob
(SLRITaxonDiv) created in-house.
parser: importtaxdb
function: Parse_TaxDBDivRecord (importtaxdb)
API: there is no API function that allows you to retrieve the entire ASN blob.
The information is used to construct information about a taxid, (see
taxdb). You can retrieve a TaxonDivRec using:
#include <taxdb_db.h>
AsnIoPtr aip = NULL;
SLRITaxonDivPtr ptax = NULL;
SHoundInit(FALSE, "name");
ptax = DB_GetTaxDivRec(1);
aip = AsnIoNex(ASNIO_TEXT_OUT, stdout, NULL, NULL, NULL);
SLRITaxonDivAsnWrite(ptax, aip, NULL);
AsnIoClose(aip);
This will produce the text in the example above.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 135 of 421 18/04/2005
del table
Database: seqhound
Table: del
Module: taxdb
Definition: Contains all the deleted taxonomy identifiers.
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto number row identifier
tid int(11)
N
o 0 Taxonomy ID
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY tid
idel_rowid INDEX rowid
idel_tid INDEX tid
Observation:
Source org: NCBI
Source files: delnodes.dmp from ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
parser: importtaxdb
***tid***
description: The taxonomy identifier
example: 15
seqhound@blueprint.org Version 3.3
The SeqHound Manual 136 of 421 18/04/2005
default value: n/a
source: column 1 of delnodes.dmp
parser: importtaxdb
function: main of importtaxdb
API: SHoundIsTaxDeleted (true if taxid parameter is deleted)
seqhound@blueprint.org Version 3.3
The SeqHound Manual 137 of 421 18/04/2005
merge table
Database: seqhound
Table: merge
Module: taxdb
Definition: Contains all tax nodes that have merged. Maps an old taxid before
merging with its new taxid after the merge. A taxonomy may be
deleted because initially it was considered species X, but when its DNA
is analyzed, it is deemed to be insignificantly different from species Y,
in which case one of the species gets merged.
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto number row identifier
otid int(11)
N
o 0 old taxonomy id
ntid int(11)
N
o 0 new taxonomy id
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY otid
imerge_rowid INDEX rowid
imerge_ntid INDEX ntid
imerge_otid INDEX otid
Observation:
Source org: NCBI
Source files: merged.dmp from ftp://ftp.ncbi.nih.gov/pub/taxonomy/taxdump.tar.gz
parser: importtaxdb
seqhound@blueprint.org Version 3.3
The SeqHound Manual 138 of 421 18/04/2005
***otid***
description: the old taxid
example: 12
default value: n/a
source: merged.dmp: column 1
parser: importtaxdb
function: main of importtaxdb
API: SHoundIsTaxMerged (takes an old taxid and checks if it has been
merged into a new taxid)
***ntid***
description: the new taxid
example: 74109
default value:
source: merged.dmp: column 2
parser: importtaxdb
main of importtaxdb
API:
n/a
function:
SHoundIsTaxMerged (takes an old taxid and checks if it's been
merged into a new taxid)
seqhound@blueprint.org Version 3.3
The SeqHound Manual 139 of 421 18/04/2005
Structural databases (strucdb) module
Last updated October 6, 2004
cbmmdb parser
purpose:
The cbmmdb parser builds the strucdb module databases. The input files to cbmmdb contain data for experimentally determined
macromolecular 3D structures. The data files are in ASN.1 format available from the NCBI ftp site.
Module:
Build the Strucdb module
Change to the mmdbdata directory.
cd $SEQH/7.mmdb.files
Create tables of the Strucdb module in the database.
Make sure file strucdb.sql has line use seqhound close to the beginning of the file.
mysql –u my_id –p –P my_port –h my_server < strucdb.sql
Where my_id, my_port and my_server should be replaced by your userid for the database, the port of the database and the IP
address or the server name of the database server repectively. You will be prompted to enter your password.
This creates tables mmdb, mmgi and domdb in the database.
strucdb
input files:
*.val.gz (ftp://ftp.ncbi.nih.gov/mmdb/mmdbdata)
tables altered:
mmdb, mmgi
source code location:
slri/seqhound/parsers/cbmmdb.c
config file & other dependencies:
The relevant configuration files are:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 140 of 421 18/04/2005
slri/seqhound/config/.intrezrc (UNIX platform)
slri/seqhound/config/.mmdbrc (UNIX platform
ncbi/config/.ncbirc (UNIX platform)
ncbi/data/bstdt.val (optional)
* The requirement that bstdt.val be in the same directory as the parser is not strictly enforced. As long as .ncbirc (see below) is
properly configured, bstdt.val will be located. However, if bstdt.val is not in the same directory as parser, a warning will be issued in
cbmmdb's log file. Despite the warning, the parser should work properly, granted the configurations files are all set properly.
The .mmdbrc file should have at least 1 section [MMDB] with 3 fields:
1) Database: the path where the data source files (*.val.gz) are located
2) Index: the index file for the data source files (mmdb.idx: this is also downloaded by mmdbftp.pl)
3) Gunzip: path to gunzip
seqhound@blueprint.org Version 3.3
The SeqHound Manual 141 of 421 18/04/2005
sample .mmdbrc file:
[MMDB]
;Where the data source files are located
Database = ./
;Index of all data source files
Index = mmdb.idx
;Path to gunzip
Gunzip = /bin/gunzip
The .ncbirc file should have 1 section [NCBI] with 1 field:
1) Data: the path to bstdt.val
sample .ncbirc file:
In file .ncbirc, variable DATA should have a value which is the path of directory ncbi/data on your machine. File .ncbirc looks like
(change text in italics):
[NCBI]
ROOT=/
DATA=/my_home/compile/ncbi/data/
[NCBI]
;where bstdt.val is located
Data=/ncbi/data
The .intrezrc file should have the pathmm set to where you want the databases to be stored.
sample .intrezrc file:
[datab]
;seqhound database that you are connecting
username=your_user_name
password=your_pass_word
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
[sections]
;structural databases
strucdb = 1
seqhound@blueprint.org Version 3.3
The SeqHound Manual 142 of 421 18/04/2005
Text in italics must be changed for the .intrezrc file to function correctly with your SeqHound set-up. Variables username,
password, dsn, database in section [datab] should have the same values as USER, PASSWORD, DSN and DATABASE
respectively in your .odbc.ini (see Step 10 in section 4.4.) In section [sections] strucdb should be 1.
mmdb.idx should be stored in the same directory as the data sources (*.val.gz files). It is an index, containing a list of MMDB ID and
PDB code pairs. The first line is the number of Biostrucs in MMDB in the current release. The * in *.val.gz is the MMDB ID. The
cbmmdb parser will initially parse the content of mmdb.idx to ensure its integrity.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 143 of 421 18/04/2005
command line parameters:
Typing "./cbmmdb" at the command line will return a list of parameters
and default settings. For example:
> ./cbmmdb
cbmmdb arguments:
-n Initialize Database File for Biostrucs [T/F]
-m Initialize Database File for MMDB Id and GI pairs [T/F]
example use:
./cbmmdb -i T -m T
associated scripts:
mmdbftp.pl retrieves all the data files for input to cbmmdb.
error and run-time logs:
cbmmdb writes to cbmmdblog
additional info:
See additional readme files on the NCBI ftp site (ftp://ftp.ncbi.nih.gov/mmdb)
See data table descriptions for each of the tables that are listed under “tables altered”
seqhound@blueprint.org Version 3.3
The SeqHound Manual 144 of 421 18/04/2005
vastblst parser
purpose:
The vastblst parser constructs the 3D domain information that is extracted from the MMDB 3D structures. vastblst builds the domdb
table to store this information.
module:
strucdb
input files:
mmdb table
tables altered:
domdb
source code location:
slri/seqhound/domains/vastblst.c
config file dependencies:
slri/seqhound/config/.intrezrc (UNIX platform)
The relevant section in the configuration file is:
[datab]
;seqhound database that you are connecting
username=your_user_name
password=your_pass_word
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
[sections]
;structural databases
strucdb = 1
seqhound@blueprint.org Version 3.3
The SeqHound Manual 145 of 421 18/04/2005
Text in italics must be changed for the .intrezrc file to function correctly with your SeqHound set-up. Variables username,
password, dsn, database in section [datab] should have the same values as USER, PASSWORD, DSN and DATABASE
respectively in your .odbc.ini (see Step 10 in section 4.4.) In section [sections] strucdb should be 1.
command line parameters:
Typing ./vastblst at the command line will return a list of the command line
parameters and default setting. For example:
> ./vastblst
vastblst arguments:
-n Initialize Database File for Domains [T/F]
example use
./vastblst -n T
associated scripts:
n/a
additional information:
See data table description for each table listed under “tables altered”
seqhound@blueprint.org Version 3.3
The SeqHound Manual 146 of 421 18/04/2005
pdbrep parser
purpose:
The pdbrep parser fills in the 'rep' field in the domdb table. 'rep' is the best representative domain for that blast set. The input files
consist of non-redundant PDB chain set, BLAST rankings for the set and the representative for that chain set. pdbrep stores the
representative into the domdb table.
module:
strucdb
input files:
nrpdb.* from ftp://ftp.ncbi.nih.gov/mmdb/nrtable
tables altered:
domdb
source code location:
slri/seqhound/domains/pdbrep.c
config file dependencies:
The relevant configuration file is:
slri/seqhound/config/.intrezrc (UNIX platform)
The relevant sections are:
[datab]
;seqhound database that you are connecting
username=your_user_name
password=your_pass_word
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
[sections]
;structural databases
seqhound@blueprint.org Version 3.3
The SeqHound Manual 147 of 421 18/04/2005
strucdb = 1
Text in italics must be changed for the .intrezrc file to function correctly with your SeqHound set-up. Variables username,
password, dsn, database in section [datab] should have the same values as USER, PASSWORD, DSN and DATABASE
respectively in your .odbc.ini (see Step 10 in section 4.4.) In section [sections] strucdb should be 1.
command line parameters:
Typing ./pdbrep at the commandline will generate a listing of commandline
parameters. For example:
>./pdbrep
pdbrep arguments:
-i Input nrpdb table [File In]
example use:
./pdbrep -i nrpdb.010400
associated scripts:
n/a
error & run-time log:
pdbrep writes to pdbreplog
additional information:
README's at ftp://ftp.ncbi.nih.gov/mmdb/nrtable
see data table descriptions for each of the tables that are listed under "tables altered"
seqhound@blueprint.org Version 3.3
The SeqHound Manual 148 of 421 18/04/2005
mmdb table
Database: seqhound
Table: mmdb
Module: strucdb
Definition: Is a database of experimentally determined macromolecular 3D
structures (Molecular Modeling DB).
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto number row identifier
mmdbid int(11)
N
o 0 Molecular modeling database
identifier
asn1 mediumblob
N
o Biostruc blob
p
dbi
d
varchar(20)
N
o PDB id
bwhat int(11) Yes
N
ULL types of molecules in the biostruc
models int(11) Yes
N
ULL number of models in record
molecules int(11) Yes
N
ULL number of molecules in record
size int(11) Yes
N
ULL size of uncompressed biostruc
bzsize int(11) Yes
N
ULL size of compressed biostruc
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY mmdbid
immdb_rowid INDEX rowid
immdb_mmdbid INDEX mmdbid
immdb_pdbid INDEX
p
dbid
seqhound@blueprint.org Version 3.3
The SeqHound Manual 149 of 421 18/04/2005
Observation: Most 3D structures are obtained through X-ray crystallography and
NMR-spectroscopy. MMDB is a subset of Protein Data Bank (PDB),
excluding theoretical models. 3D structures can provide information
pertaining to the biological function, mechanism of function,
evolutionary history and relationships of a biomolecule.
Source org: NCBI
Source file: *.val.gz from ftp://ftp.ncbi.nih.gov/mmdb/mmdbdata
FTP script: slri/seqhound/scripts/mmdbftp.pl
parser: cbmmdb
***mmdbid***
description: a unique, stable numerical identifier. MMDB ID are assigned when a new
structure enters MMDB. If an entry is deleted from PDB, then its MMDB entry
is obsolete. Obsolete structures get archived.
example: 10
default value: n/a
ASN.1 struct: Biostruc->Biostruc-id (choice 1)
parser: cbmmdb
function: Biostruc2Modelstruc (ncbi/biostruc/mmdbapi1.c)
API: SHound3DExists
***asn1***
description: the Biostruct blob (see ASN structure below). Contains information about the
3D structure of a molecule including information about the researchers,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 150 of 421 18/04/2005
molecule bond information, etc.
example: see below
default value: n/a
source: *.val.gz
parser: cbmmdb
function: MMDBBiostrucGet
API: SHoundGet3DfromPdbId
SHoundGetPDB3D
SHoundGet3D
**pdbid
***
description: a 4 character identifier from the Protein Data Bank.
example: 100D
default: n/a
ASN.1 struct: Biostruc->Biostruc-id(choice 2)
parser: cbmmdb
function: Biostruc2Modelstruc (ncbi/biostruc/mmdbapi1.c)
API: no functions return the pdbid from mmdb, it is used as a key to retrieve a 3D
structure, see asn1 API.
***bwhat
***
description: an integer that represents the type of molecule.
The possible flags are (defined in ncbi/biostruc/mmdbapi1.h) and their
corresponding byte values are:
1) AM_ION 0x80
seqhound@blueprint.org Version 3.3
The SeqHound Manual 151 of 421 18/04/2005
2) AM_RNA 0x40
3) AM_WAT 0x20
4) AM_SOL 0x10
5) AM_HET 0x08
6) AM_DNA 0x04
7) AM_PROT 0x0
8) AM_POLY 0x01
9) AM_UNK 0x00
example: AM_ION
default: AM_UNK
ASN.1 stuct: Biostruc->Biostruc-graph->Molecule-graph->Bio-mol-descr->molecule-type
parser: cbmmdb
function: Biostruc2Modelstruc (ncbi/biostruc/mmdbapi1.c)
API: SHound3DbWhat
***models***
description: an enumeration of the different models for this structure
example: 3
default: n/a
source: this is enumerated by NCBI code, increment for EACH Biostruc->Biostruc-
model in the ASN structure
parser: cbmmdb
function: Biostruc2Modelstruc (ncbi/biostruc/mmdbapi1.c)
API: n/a, no public interface provided to this field
seqhound@blueprint.org Version 3.3
The SeqHound Manual 152 of 421 18/04/2005
***molecules***
description: an enumeration of the number of molecules in the structure
example: 3
default: n/a
source: this is enumerated by NCBI code, increment for EACH Biostruc->Biostruc-
graph->Molecule-graph in the ASN structure
parser: cbmmdb
function: Biostruc2Modelstruc (ncbi/biostruc/mmdbapi1.c)
API: n/a, no public interface provided to this field
***size***
description: the size of the uncompressed biostruc
example: 7691
default: n/a
source: the size of the uncompressed biostruc is determined just before it is compressed
and appended
parser: cbmmdb
function: AssignASNMemBZMemo (intrez_cb.c)
API: n/a, no public interface provided to this field
***bzsize***
the size of the compressed biostruc
description:
example: 31
default:
source:
n/a
the compressed size is determined after it is compressed
seqhound@blueprint.org Version 3.3
The SeqHound Manual 153 of 421 18/04/2005
parser: cbmmdb
n/a, no public interface provided to this field
function: AssignASNMemBZMemo (intrez_cb.c)
API:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 154 of 421 18/04/2005
mmgi table
Database: seqhound
Table: mmgi
Module:
Definition:
strucdb
A mapping of mmdbid to gi.
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto number row identifier
mmdbid int(11)
N
o mmdb identifier 0
gi int(11)
N
o 0 GenInfo Identifier
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY mmdbid
gi
immgi_rowid INDEX rowid
immgi_mmdbid INDEX mmdbid
immgi_gi INDEX gi
Observation:
Source org: NCBI
Source file: *.val.gz from ftp://ftp.ncbi.nih.gov/mmdb/mmdbdata
FTP script: slri/seqhound/scripts/mmdbftp.pl
parser: cbmmdb (see mmdb for initialization info)
seqhound@blueprint.org Version 3.3
The SeqHound Manual 155 of 421 18/04/2005
***mmdbid***
description: 3D molecular model unique identifier
example: 1
ASN.1 struct:
parser:
Biostruc->Biostruc-id (choice 1)
cbmmdb
function: Main
API: SHound3DExists
SHound3DFromGi
SHound3DFromGiList
***gi***
description: GenInfo Identifier
example: 1420979
ASN.1 struct: Biostruc->Biostruc-id
parser: cbmmdb
function: Main
API: SHoundGiFrom3D
SHoundGiFrom3DList
seqhound@blueprint.org Version 3.3

The SeqHound Manual 156 of 421 18/04/2005
domdb table
Database: seqhound
Table: domdb
Module: strucdb
Definition: Stores the information of structural domains.
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto number row identifier
mmdbid int(11)
N
o 0 mmdb identifier
asn1 mediumblob
N
o asn blob
p
dbi
d
varchar(20)
N
o
p
db identifie
r
p
dbchain varchar(10) Yes
N
ULL
p
db chain
gi int(11)
N
o 0 geninfo identifier
domno int(11) Yes
N
ULL domain number
domall int(11)
N
o 0 number of domains in the whole
structure
domid int(11)
N
o 0 vast domain id
rep int(11) Yes
N
ULL representative of blast set
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY domid
idomdb_rowid INDEX rowid
idomdb_mmdbid INDEX mmdbid
idomdb_pdbid INDEX
p
dbid
idomdb_gi INDEX gi
seqhound@blueprint.org Version 3.3

The SeqHound Manual 157 of 421 18/04/2005
idomdb_domall INDEX domall
idomdb_domid INDEX domid
Observation: VAST (vector alignment search tool) is an NCBI-developed algorithm
used to identify similar 3-D structures that may not possess sequence
similarities. Structural information is taken from the mmdb database.
3D structures are converted into vectors (which corresponding to
structurally significant regions of the protein). Adjacent vectors are
grouped into domains. Domain information is used to determine distant
homologs and provide structural information. The domains of proteins
can be compared with other protein domains. Those with similar
domain overlaps are likely to be distant homologs. The domdb database
stores the domain information.
Source org: NCBI
Source file: MMDB database & nrpdb.* from ftp://ftp.ncbi.nih.gov/mmdb/nrtable
Parser: vastblst & pdbrep
Note: vastblst parser uses SHoundGet3D to retrieve the MMDB information..
***mmdbid***
description: the 3D structure id from which the domain was computed.
example: 3446
default value: n/a
source: retrieved from mmdbid by calling SHoundAllMMDBID.
parser: vastblst
function: Main calls SHoundAllMMDBID and MakeAModelstruc
seqhound@blueprint.org Version 3.3
The SeqHound Manual 158 of 421 18/04/2005
API: not used by the API, there are other tables with more complete
mmdbid's.
***asn1***
description: the asn structure of the domain. The structure gives the gi of the domain
and the starting and ending point of the domain.
example: a text output of the SLRIValNode:
SLRIValNode ::= {
domain {
gi 443581 ,
from 2 ,
to 393 } }
default value: n/a
source: extracted from the biostruc reconstructed from what is stored in mmdb.
parser: vastblst
API:
function: WriteFASTAByDomain calls MakeDomain
SHoundGetDomain to actually print out the ASN structure, after
calling SHoundGetDomain, the return value (a SLRIValNodePtr
object) must be opened and streamed into an asn IO stream. eg.
{ // start of program
SLRIValNodePtr pdom = NULL;
AsnIoPtr aip = NULL;
pdom = SHoundGetDomain(443581, 0);
aip = AsnIoNew(ASNIO_TEXT_OUT, stdout, NULL, NULL, NULL);
SLRIValNodeAsnWrite(pdom, aip, NULL);
AsnIoClose(aip);
} // end of program, text output as in example
SHoundGetFastaDomain (to retrieve the text, follow code above,
replace SLRIValNode with SLRIFasta
seqhound@blueprint.org Version 3.3
The SeqHound Manual 159 of 421 18/04/2005
***pdbid***
description: the PDB id of the 3D structure from which the domain was computed
example: 9XIM
default value: n/a
ASN.1 struct: n/a
parser: vastblst
function: WriteFASTAByDomain
API: SHoundGetDomain
***gi***
description: the gi of the chain from which the domain was computed
example: 443581
default value: n/a
source: extracted from the biostruc object reconstructed from data in mmdb.
Biostruc->Biostruc-id (choice 1)
parser: vastblst
function: WriteFASTAByDomain
API: SHoundGiFromPDBchain
This field can also be parsed from the SLRIValNode and
SLRIFasta ASN structures (above).
***domno***
description: a protein chain may have several domains, each domain is enumerated,
eg if a protein X has 10 domains, the first domain has domno 0, the
seqhound@blueprint.org Version 3.3
The SeqHound Manual 160 of 421 18/04/2005
second domain has domno 1, ...etc
example: 0
default value: n/a
source: this is enumerated by NCBI code
parser: vastblast
function: WriteFASTAByDomain
API: n/a, by itself, the domno provides little information, so it is not
provided in the API
***domall***
description: The total number of domains in the 3D structure. In the above example
protein X, domall is 10
example: 1
default value: n/a
source: this is enumerated by NCBI code
parser: vastblast
function: WriteFASTAByDomain
API: n/a
***domid***
description: the id for the domain. domid is computed by hashing various id's,
including (but not limited to) mmdbid, domno, etc.
example: 34460200
default value: n/a
source: hashed in WriteFASTAByDomain
seqhound@blueprint.org Version 3.3
The SeqHound Manual 161 of 421 18/04/2005
parser: vastblast
function: WriteFASTAByDomain
API: n/a
***rep***
description: domains get grouped based on properties. The domains are then
compared to the other domains in their group and then the best over
domain representative of that group is chosen. This field reflects that
representative.
example: 1
default: 0
source: nrpdb.* from ftp://ftp.ncbi.nih.gov/mmdb/nrtable columns 6, 9 & A
parser: pdbrep
function: Main
API: n/a
seqhound@blueprint.org Version 3.3

The SeqHound Manual 162 of 421 18/04/2005
Protein sequence neighbours (neighdb) module
Last updated: August 18, 2004
Note: The neighbours tables are precalculated on a cluster and the resulting tables are distributed in MySQL format on
our ftp site. Therefore, this section is provided for informational purposes only, or for those who would like to build
neighbours tables from there own sequence data; it is not necessary if one wishes simply to include the neighbours
module into their own seqhound instance, in which case they should simply download the precomputed tables.
Note: the nblast development tree is in the slri directory on the same level as SeqHound (i.e.; slri/nblast).
This documentation includes a description of the nblast program.
The NBLAST paper is freely available from BioMed Central and is provided in the supplementary documents directory distributed
with this manual. See:
Dumontier M, Hogue CW. NBLAST: a cluster variant of BLAST for NxN comparisons.
BMC Bioinformatics. 2002 May 8;3(1):13. Epub 2002 May 08.
PMID: 12019022
Purpose: This section describes how to build and use nblast, a program for generating a database of sequence neighbours.
Installing nblast:
There are two ways to install nblast:
* Download nblast binaries for your platform from the sourceforge website:
http://sourceforge.net/project/showfiles.php?group_id=17918
or
* Compile nblast on your system: In order to compile nblast, two pieces of third party software are prerequisite: NCBI's C toolkit
and Sequiter software's CodeBase database library. Nblast currently only supports CodeBase as its database backend, but it may
be ported to other databases in the future. If your system meets these requirements, download the platform independent source
code for nblast from the link above and proceed.
The NCBI C toolkit can be downloaded from NCBI's website at
ftp://ftp.ncbi.nih.gov/toolbox/ncbi_tools/ncbi.tar.gz
CodeBase is a commercial database software library available from Sequiter software.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 163 of 421 18/04/2005
A description of how to compile and set up the nblast software follows. If you installed the binaries, or already have compiled the
software, skip to the Configuration section. The installation procedure for CodeBase and the NCBI C toolkit are not detailed here, but
are assumed to have been successful.
Once you have unpacked the source code tree, change directories into the topmost nblast directory and then exercise one of the
following sets of instructions, depending on whether you are using a Unix or Windows system.
Unix:
• Modify the nblast.mk file for different source and library paths.
• Modify the make.nblast file or project settings to create an executable that incorporates NBLAST (-D NBLAST_API), logging
(-D NBLAST_LOG), and/or MoBiDiCK(-D MOBIDICK_API) (The MoBiDick library is currently not publicly available).
• From the src directory type 'make -f make.nblast', if the nblast binary is built then build nblastcleanup with 'make -f
make.nblastcleanup' The binaries will be placed in the build/ subdirectory of the source tree.
Windows:
• Open the nblast.dsw workspace in MSVC.
Build NBLAST and NBLASTCLEANUP
The two nblast executables, nblast and nblastcleanup, should now be present in the slri/nblast/build/ directory. Set your PATH
environment variable appropriately so that you can execute them. Before you can use nblast, you'll have to do some more
configuration, described in the next section.
Configuration of nblast environment:
At least one configuration file is required to use nblast and it's associated applications, more if you wish to have some of the relevant
files outside of the current directory. At the very least, you must have an nblast configuration file, called .nblastrc on Unix, and
nblast.ini on Windows. This file must be present either in your current directory or your home directory[may not apply on windows]
and has the following formats:
.nblastrc on Unix:
; NBLAST configuration file
[NBLAST]
writepath = /home/nblast/build/
nblast.ini on Windows:
[NBLAST]
writepath = g:\code\slri\nblast\build
seqhound@blueprint.org Version 3.3
The SeqHound Manual 164 of 421 18/04/2005
where the directory path named after “writepath = “ specifies where the files generated by nblast will be written to. A trailing
slash(unix) or backslash(windows) is necessary for the directory path to work.
If you want to place the scoring matrix (used by the blast algorithm) and/or the formatted fasta database in any directory but the
current working one, you must also have an ncbi configuration file:
.ncbirc on Unix:
[NCBI]
DATA = g:\code\ncbi\data
[BLAST]
BLASTDB = g:\blastdb\
ncbi.ini on Windows:
[NCBI]
DATA = /code/ncbi/data
[BLAST]
BLASTDB = /blastdb/
Where the path following “DATA =” specifies the path to find the scoring matrix file, and the path following “BLASTDB” specifies
the path to find the formatted fasta database.
The BLAST algorithm uses the BLOSUM62 scoring matrix. The file containing this matrix is required for nblast to function. The file
is named BLOSUM62, and comes with the NCBI C toolkit. It should also come with the binary distribution of nblast.
Once the configuration files have been properly set, you are ready to run nblast, detailed in the next section.
Running NBLAST
* Format the fasta database using formatdb: Before Nblast can process the protein sequences, they must be properly formatted.
This is done using the formatdb program, available from NCBI, which takes a fasta formatted database of protein sequences
and processes them into a form which can be BLASTed. Once you have downloaded formatdb, format your fasta database
using the following command:
formatdb -oT -i <dbname>
Where <dbname> is the file name of your fasta formatted protein sequence database. Generally this is nr, the non-redundant
protein sequence database, which can be downloaded from ftp://ftp.ncbi.nlm.nih.gov/blast/db/nr.tar.gz file (at the time of this
writing, that file is ~500 MB compressed). <dbname> will be used throughout the rest of this document to mean the filename of
your original fasta formatted sequence database.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 165 of 421 18/04/2005
* Use NBLAST to build the skeleton database: Before nblast can compare sequences, it needs to build a skeleton of the nblastdb
table with information on how the sequences will be ordered and indexed. This is done by running nblast with the -N1
command line argument as follows:
nblast -i<dbname> -d<dbname> -N1
* Use Nblast to do the blast comparisons. Before the neighbour tables can be built, nblast must do the pairwise blast
comparisons. In addition to the nblast specific command line options mentioned here, you may also apply options related to
NCBI's blastall to augment the BLAST results. For small databases, it is feasible to do this step on a single computer. Single
computer execution:
nblast -i<dbname> -d<dbname> -e<eval_cutoff> -N2
Where <eval_cutoff> is the maximum evalue allowed for BLAST comparisons between sequences. BLAST comparisons
which result in evalues higher than this are not saved.
Parallel Computer Execution: Since these comparisons are computationally intensive for large databases (like nr), nblast is
capable of splitting up the task across multiple compute nodes. This is done by distributing the <dbname>N.* files generated by
the last step, and the <dbname>.p* files generated by formatdb, to the compute nodes, and then running nblast on each compute
node with node specific options:
nblast -i<dbname> -d<dbname> -e<eval_cutoff> -N2 -C<node_#> -D<total_#of_nodes>
Where <total_#of_nodes> is the total number of compute nodes in your parallel computing system, and <node_#> is the
index (ranging from 1 to <total_#of_nodes>) of the current compute node with respect to the set of nodes which the nblast
task is divided across. <eval_cutoff> has the same meaning as in single computer usage.
Both single and parallel computer execution of the N2 mode of nblast results in creation of files entitled
<dbname>B<node_#>.* (for single computers, <node_#> defaults to 1). These contain the set of blast results which that
particular compute node generated.
* Build the nblastdb table of sequence neighbours: Now the blastdb table of pairwise blast results can be used to build/fill the
nblastdb table of neighbouring sequences. The pairwise BLAST results generated during the last step are processed and
consolidated to generate the records for the neighbouring sequences table, nblastdb. Single Computer Execution:
nblast -i<dbname> -d<dbname> -e<eval_cutoff> -N4
Parallel Computer Execution: You must first collect the database files generated on the compute nodes during the last step onto
the head node before running this step. Then run the following on the head node:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 166 of 421 18/04/2005
nblast -i<dbname> -d<dbname> -e<eval_cutoff> -N4 -C<node_#> -D<total_#of_nodes>
* Cleanup the database and generate number of neighbours fields: The neighbours table, nblastdb, is nearly complete. All that
needs to be done is to fill in the #Neighbours fields of the nblastdb table. This is done using the program nblastcleanup with
the following arguments:
nblastcleanup -bT -pT -aT -qT -d<dbname> -n <num_blastdbs>
Where <num_blastdbs> is the number of blastdb's to check/build from, generally equal to the number of compute nodes you
used (1 in the single computer case).
Your Neighbours database should now be completely built.
NBLAST Update Procedure
Nblast was designed to process NCBI's non-redundant sequence database, named nr. This database contains a non-redundant list of
protein sequences and their associated GenInfo identifier numbers (GIs). The NCBI's nr database is updated on a regular basis, with
some GI's being removed, and others being added. It is desirable to keep the NeighDB module's nblastdb and blastdb tables updated
with the nr database, without having to recompute all the neighbours. This is the purpose of nblast's update procedure, which removes
GI's which have been “killed” from any entries in the nblast tables which contain them, and BLASTs and inserts “new” GI's where
they are appropriate. This process involves multiple steps, which are outlined and explained here.
* Format new version of the fasta database using formatdb: Use formatdb to format your new version of the fasta database in the
same way you did before. Note that your new fasta database should have the same name as the one you initially used to build the
nblast tables.
formatdb -oT -i <dbname>
* Run Nblast in Update mode N5: This step compares the GIs in the out of date nblastdb with those in the updated FASTA
database. It removes entries of the killed Gis from the neighbour lists of all gi's, deletes records which only exist because of the
killed GI, and inserts empty records for the newly added GIs in the nblastdb. It also creates an update file called
NBLAST_UPDATE.val which lists which GI's have been killed and which are to be added, for the next stages to use. On the head
compute node, execute:
nblast -i<dbname> -N5
* Run Nblast in Update mode N6: This step performs the pairwise BLAST comparisons between the newly added sequences and all
the old sequences that haven't been killed, as well as comparing the new sequences with each other. Single Computer Execution:
nblast -i<dbname> -e<eval_cutoff> -N6
seqhound@blueprint.org Version 3.3
The SeqHound Manual 167 of 421 18/04/2005
Parallel Computer Execution:
nblast -i<dbname> -e<eval_cutoff> -N6 -C<node_#> -D<total_#of_nodes>
Where <eval_cutoff> is the maximum allowed evalue for comparison results, as described earlier.
* Rebuild the Neighbouring Sequences Table: Now rebuild the nblast database using the same command you used to build it
previously. Single Computer Execution:
nblast -i<dbname> -d<dbname> -e<eval_cutoff> -N4
Parallel Computer Execution: First collect all the <dbname>B<node#>.* files from the compute nodes onto the head node.
Then run the following command:
nblast -i<dbname> -d<dbname> -e<eval_cutoff> -N4 -C<node_#> -D<total_#of_nodes>
* Cleanup: The cleanup procedure is done in the same manner as for the initial build of the neighbour tables. Single Computer
Execution:
nblastcleanup -bT -pT -aT -qT -d<dbname> -n <num_blastdbs>
Where <num_blastdbs> is the number of blastdb's to check/build from, generally equal to the number of compute nodes you
used (1 in the single computer case).
Your Neighbours database should now be properly updated.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 168 of 421 18/04/2005
nbraccess program*
Note: Not available at time of release.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 169 of 421 18/04/2005
BLASTDB table
Last updated: August 4, 2004
Database: SeqHound
Module: (Sequence) Neighbours
Source: NBLAST
Source File: Derived from NxN comparison of sequences from the NR database
from ftp://ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz
Parser/Application: NBLAST http://www.sourceforge.net/projects/slritools (source &
binaries)
Published
Documentation:
BMC Bioinformatics http://www.biomedcentral.com/1471-
2105/3/13/
Definition: Pairwise sequence alignments from NBLAST computation
Note: The neighbours tables are precalculated on a cluster and the resulting tables are distributed in MySQL format on
our ftp site. Therefore, this section is provided for informational purposes only, or for those who would like to build
neighbours tables from there own sequence data; it is not necessary if one wishes simply to include the neighbours
module into their own seqhound instance, in which case they should simply download the precomputed tables.
Codebase
Column_Name Index NULL Data_Type Size Column_Definition
UID P
N
INTEGER/Hash
Key
14 Perfect Hash for 2
Ordinal Numbers
EVAL
N
N
FLOAT E-Value 12.7 Alignment Evalue
ASNSA
N
N
MEMO ASN.1 Modified SeqAlign for
p
airwise sequence
alignment
MySQL
Field Type Null Default Column_Definition
rowid int(11) Yes
N
ULL Auto incremented id
uid bigint(20) Yes
N
ULL Perfect Hash for 2 Ordinal
N
umbers
seqhound@blueprint.org Version 3.3
The SeqHound Manual 170 of 421 18/04/2005
eval decimal(12,7) Yes
N
ULL 12.7 Alignment Evalue
asnsa mediumblob Yes
N
ULL Modified SeqAlign for pairwise
sequence alignment
MySQL Indexes:
Keyname Type Field
iblastdb_rowid INDEX rowid
iblastdb_uid INDEX uid
*** uid ***
Description: 64 bit Perfect Hash Key generated from 2 32 bit Ordinal Numbers in
nblastdb
example:
default value: none
source: NBLAST - a cluster computer extension of BLAST to compute NxN
sequence comparisons
functions: ShoundGetBlastResult: Given two GIs, checks if alignment
exists, returns NBlast-result-set else NULL
SHoundGetBlastSeqAlign : Given two GIs, calls
SHoundGetBlastResult and formats the NBlast-result-set to a
SeqAlign
*** eval ***
Description: The BLAST e-value reported for this alignment
example: 0.0001
default value: none
function: Used by NBLAST to create the NBLASTDB table using a user-
specified minimum evalue
seqhound@blueprint.org Version 3.3
The SeqHound Manual 171 of 421 18/04/2005
***asnsa***
Description: NBlast-Result-Set (ASN.1 definition is described in
slri/nblast/asn/NBlastasn.asn)
- Stores the useful parts of a seqalign from the BLAST computation
including the alignment, bitscore, evalue
seqhound@blueprint.org Version 3.3
The SeqHound Manual 172 of 421 18/04/2005
NBLASTDB table
Last updated: August 4, 2004
Database: SeqHound
Module: (Sequence) Neighbours
Source: NBLAST
Source File: Derived from NxN comparison of the NR database from
f
tp://ftp.ncbi.nih.gov/blast/db/FASTA/nr.gz
Parser/Application: NBLAST http://www.sourceforge.net/projects/slritools (source &
binaries)
Published
Documentation:
BMC Bioinformatics http://www.biomedcentral.com/1471-
2105/3/13/
Definition: Sequence neighbour lists from NBLAST computation
Codebase
Column_Name Index NULL Data_Type Size Column_Definition
ORD P
N
INTEGER
Ordinal
N
umbe
r
10 The ordinal number of
this entry (auto-
increment)
GI I
N
INTEGER
GenInfo Id
10 The GenInfo Identifier
for the sequence for
which there are
neighbours in the list
N
UM I
N
INTEGER#
N
eighbours
10
N
umber of neighbours
in ASN.1 structure
ASNNBR
N
N
MEMO ASN.1 ASN.1 structure
containing the list of GIs
whose sequences
neighbour that of this
entries GI, and the
corresponding evalues
of the alignments.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 173 of 421 18/04/2005
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto incremented id
ord int(11)
N
o 0 The ordinal number of this entry
gi int(11)
N
o 0
The GenInfo Identifier for the
sequence for which there are
neighbours in the list
num int(11)
N
o 0
N
umber of neighbours in ASN.1
structure
asnnbr mediumblob
N
o
ASN.1 structure containing the
list of GIs whose sequences
neighbour that of this entries GI,
and the corresponding evalues of
the alignments.
MySQL Indexes:
Keyname Type Field
inblastdb_rowid INDEX rowid
inblastdb_ord INDEX ord
inblastdb_gi INDEX gi
inblastdb_num INDEX num
***ord***
Description: The ordinal value of the entry in the database (starts with 1). The
ordinal values from two GIs in a pairwise alignment are perfectly
and reversibly hashed into/from the BLASTDB UID value. This
approach saves on space required to store two 32 bit integers.
Example: 1
Default Value: NA
Function:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 174 of 421 18/04/2005
***gi***
Description: The GenInfo Identifier of the query sequence
Example: 2495000
Default Value: NA
seqhound@blueprint.org Version 3.3
The SeqHound Manual 175 of 421 18/04/2005
API Function: SHoundNeighboursFromGiEx: Given GI, an evalue cutoff and
a limit of 100 returned results, returns FLinkSet containing list of
neighbour GIs and their alignment e-value
Other functions that use SeqHound Functionality (Redundant
Groups & Taxonomy Protein Lists)
SHoundNeighboursFromGi: Calls
SHoundNeighboursFromGiEx, and if the GI is not found,
searches through the list of redundant GIs for the respective
sequence to find an equivalent GI for which there is neighbour
information, returning the ShoundNeighboursFromGiEx results for
that GI.
SHoundNeighboursFromGiList: Calls
SHoundNeighboursFromGi with each GI in a valnode list
SHoundNeighboursFromTaxID: Calls
ShoundNeighboursFromGiList for each GI in a given
taxonomy (SHoundProteinsFromOrganism)
SHoundNeighboursOfNeighbours: Fetches the neighbours o
f
supplied GI and each of their neighbours (limit 100)
SHoundNeighboursOfNeighboursList: Fetches the
neighbours of supplied GI list and each of their neighbours
SHoundGiAndNumNeighboursList: returns a list of all the
GIs with more than 0 neighbours
SHoundNumNeighboursInDB: Fetches the number of
neighbours in the nblastdb
***num***
seqhound@blueprint.org Version 3.3
The SeqHound Manual 176 of 421 18/04/2005
Description: Quick lookup value to find out how many neighbours are in the
ASN.1 structure. Can also be used to sort the GIs with most/least
neighbours
Example: range of integer values
Default Value: 0
Function:
***asnnbr***
Description: NBlast-GiAndEval-set (ASN.1 definition is described in
slri/nblast/asn/NBlastasn.asn)
stores the GI and it's list of sequence neighbours (as GI, evalue
pairs)
Functions: see above for GI
seqhound@blueprint.org Version 3.3
The SeqHound Manual 177 of 421 18/04/2005
Locus link functional annotations (lldb) module
llparser
Last updated: August 5, 2004
purpose:
The llparser parses the LocusLink data files to create and update a set of tables that
correlate curated sequence data and descriptive information about genetic loci. It retrieves
information on official nomenclature, aliases, sequence accessions, phenotypes, EC
numbers, MIM numbers, UniGene clusters, homology and map locations.
module: lldb
input files:
LL_tmpl
From ftp://ftp.ncbi.nih.gov/refseq/LocusLink/
tables altered:
ll_cdd, ll_go, ll_llink, ll_omim
source code location:
slri/seqhound/locuslink/llparser.c
config file dependencies:
The relevant configuration file is:
slri/seqhound/parsers/.intrezrc (for Unix platforms) or
The relevant section of the configuration file is:
[datab]
;seqhound database that you are connecting
username=your_user_name
password=your_pass_word
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
[sections]
;locus link functional annotations
lldb = 1
Text in italics must be changed for the .intrezrc file to function correctly with your
SeqHound set-up. Variables username, password, dsn, database in section
[datab] should have the same values as USER, PASSWORD, DSN and DATABASE
respectively in your .odbc.ini (see Step 10 in section 4.4.) In section [sections]
lldb should be 1.
command line parameters:
This parser does not have any command line parameter, Just type “./llparser” and
it will parse the LL_tmpl file. This input file must be in the same directory as the llparser
executable.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 178 of 421 18/04/2005
associated scripts:
see “slri/seqhound/scripts/llftp.pl”
This script retrieves the input file or this parser.
error and run-time logs:
llparser writes to a log file called “llparserlog” where it writes Time Stamp, Error #,
goparser.c line # and cause of the problem.
e.g.
=================[ May 21, 2003 3:49 PM ]==================
ERROR: [000.000] {llparser.c, line 81} Main: Cannot find LL_tmpl file.
troubleshooting:
additional info:
The LocusLink web page is at NCBI http://www.ncbi.nlm.nih.gov/LocusLink/
The LocusLink README file describes the input file for this parser
ftp://ftp.ncbi.nih.gov/refseq/LocusLink/
See data table descriptions for each of the tables in the lldb module.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 179 of 421 18/04/2005
addgoid parser
Last updated: August 5, 2004
purpose:
Correlates sequence record gi's with GO annotation identifiers.
The Gene Ontology flat file parser adds information to the supplements ll_go table. This
information correlates sequence records GI’s with GO annotation identifiers. This
information is retrieved from the gene_association.compugen.Genbank and
gene_association.compugen.Swissprot files from www.geneontology.org.
Other GO annotation data is available at this site but it is not currently incorporated into
SeqHound. This is actively being worked on at the time of writing.
Note that the input files used here do not contain PubMed Identifiers.
This parser is dependent on the following tables, asndb, parti, accdb and nucprot. This
parser is also dependent on SeqHound API.
For this reason, the mother parser must be run before using the addgoid parser.
module: lldb
input files:
gene_ association.compugen.Genbank
gene_association.compugen.Swissprot
from ftp://ftp.geneontology.org/pub/go/gene-associations/
tables altered:
ll_go
source code location:
slri/seqhound/locuslink/addgoid.c
config file dependencies:
The relevant configuration file is:
slri/seqhound/parsers/.intrezrc (for Unix platforms) or
The relevant section of the configuration file is:
[datab]
;seqhound database that you are connecting
username=your_user_name
password=your_pass_word
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
[sections]
;locus link functional annotations
lldb = 1
seqhound@blueprint.org Version 3.3
The SeqHound Manual 180 of 421 18/04/2005
Text in italics must be changed for the .intrezrc file to function correctly with your
SeqHound set-up. Variables username, password, dsn, database in section
[datab] should have the same values as USER, PASSWORD, DSN and DATABASE
respectively in your .odbc.ini (see Step 10 in section 4.4.) In section [sections]
lldb should be 1.
command line parameters:
Typing “./addgoid –“ at the command line while in the directory where addgoid
resides will return a list of command line parameters and default settings. For example:
> ./addgoid –
pdbrep arguments:
-i Input file [File In]
associated scripts:
see /slri/seqhound/goftp.pl
The script retrieves the files used as input by the addgoid parser. This script also
retrieves the three input files required by the goparser (see godb module).
error and run-time logs:
addgoid parser writes to a log file called addgoidlog where it writes Time Stamp, Error #,
goparser.c line # and cause of the problem. For example
==================[ May 21, 2003 4:14 PM ]==================
NOTE: CoreLib [002.003] {ncbifile.c, line 624} FileOpen("gene_association.compugen.Swissprot","r")
failed
troubleshooting:
additional info:
The NCBI LocusLink web page is at
http://www.ncbi.nlm.nih.gov/LocusLink/.
The Gene Ontology Consortium documentation at is at :http://www.geneontology.org/..
See data table descriptions for each of the tables that are listed under the lldb module.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 181 of 421 18/04/2005
ll_omim table
Last updated: August 5, 2004
Database seqhound
Table ll_omim
Module lldb
Definition OMIM Online Mendelian Inheritance in Man.
Online Mendelian Inheritance in Man (OMIMTM) is a continuously
updated catalog of human genes and genetic disorders. OMIM focuses
primarily on inherited, or heritable, genetic diseases. It is also
considered to be a phenotypic companion to the human genome project.
OMIM is based upon the text Mendelian Inheritance in Man, authored
and edited by Dr. Victor A. McKusick and a team of science writers and
editors at Johns Hopkins University and elsewhere.
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto incremented id
ll_id int(11)
N
o 0 Locus Link Identifier
omim_id int(11)
N
o 0 OMIM Identifier.
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY ll_id
omim_id
illomim_rowid INDEX rowid
illomim_llid INDEX ll_id
illomim_omimid INDEX omim_id
Observation:
Organization: NCBI
Source db: ftp://ftp.ncbi.nih.gov/refseq/LocusLink/LL_tmpl.gz
*** ll_id ***
description: Locus link record identifier.
example: 1
default value: 0
source: First field in each locus link record. Eg:
>>1
is the record with locus link id 1.
parser: llparser.c --> ll_parser.c
seqhound@blueprint.org Version 3.3
The SeqHound Manual 182 of 421 18/04/2005
function: LL_ParseFile() --> LL_LineParser
db_layer: LL_Append2OMIM_DB()
API: SHoundLLIDFromOMIM()
*** omim_id ***
description: OMIM Online Mendelian
example: 138670
default value: n/a
source: After tag 'OMIM: '
parser: llparser.c --> ll_parser.c
function: LL_LineParser() --> LL_ParseOMIM()
db_layer: LL_AppendRecord() --> LL_Append2OMIM_DB()
API: SHoundOMIMFromLLID()
SHoundOMIMFromGi()
SHoundOMIMFromGiList()
seqhound@blueprint.org Version 3.3
The SeqHound Manual 183 of 421 18/04/2005
ll_go table
Last updated: August 5, 2004
Database: seqhound
Table: ll_go
Module: lldb
Definition: Associates a locus link identifier with the gene ontology (GO)
annotation.
Field Type Default
MySQL
Null Column_Definition
int(11)
N
o Auto incremented id
int(11)
N
o 0 Locus Link Identifier
go_id int(11) Yes
N
ULL Gene Ontology ID.
p
mi
d
int(11) Yes
N
ULL Pub Med ID.
evidence varchar(50) Yes
N
ULL
rowid
ll_id
Evidence Code.
MySQL Indexes
Keyname Type Field
illgo_rowid INDEX rowid
illgo_llid INDEX ll_id
illgo_goid INDEX go_id
illgo_pmid INDEX
p
mi
d
Observation.:
Organization: NCBI
Source file: ftp://ftp.ncbi.nih.gov/refseq/LocusLink/LL_tmpl.gz
Parser: ll_parser.c
*** ll_id ***
description: Locus link identifier
example: 2
default value: n/a
source: see above
parser: ll_parser.c -> ll_parser.c
function: LL_LineParser() -> LL_ParseNPUnit
SHoundLLIDFromGOIDAndECode ()
***go_id***
description:
API:
Gene Ontology Identifier.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 184 of 421 18/04/2005
example: 5717
default value: n/a
source: after tag GO, 4th element
e.g.:
GO: cellular
component|extracellular|IDA|GO:0005574|GOA|na
parser: ll_parser.c or ll_parser.c
function: LL_LineParser() -> LL_ParseGOUnit
API: SHoundGOIDFromLLID()
SHoundGOIDFromGi()
SHoundGOIDFromRedundantGi
SHoundGOIDFromGiList
SHoundGOIDFromRedundantGiList
example:
GO: cellular component|extracellular|IDA|GO:0005574|GOA|345201
eg:
***pmid***
description: PubMed Identification.
3458201
default value: n/a
source: after tag GO, 6th element
e.g.:
parser: llparser.c -> ll_parser.c
function: LL_LineParser() -> LL_ParseGOUnit
API: SHoundGOPMIDFromGiAndGOID()
***evidence***
description: Every GO annotation must indicate the type of evidence that supports
it; these evidence codes correspond to broad categories of experimental
or other support. http://www.geneontology.org/GO.evidence.html
example: IC: Inferred by Curator.
ND: No biological Data available.
TAS: Traceable Author Statement.
IEA: Inferred from Electronic Annotation.
default value: n/a
source: After a 'GO:' line, it's the 3rd. field element
GO: cellular
component|extracellular|IDA|GO:0005574|GOA|34520
1
parser: llparser.c -> ll_parser.c
seqhound@blueprint.org Version 3.3
The SeqHound Manual 185 of 421 18/04/2005
function: LL_LineParser()-> LL_ParseGOUnit()
API: SHoundGOECodeFromGiAndGOID()
seqhound@blueprint.org Version 3.3
The SeqHound Manual 186 of 421 18/04/2005
ll_llink table
Last updated: August 5, 2004
Database: seqhound
Table:
Module:
ll_llink
lldb
Definition: Associates a locus link id with a sequence identifier
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto incremented id
ll_id int(11)
N
o 0 Locus Link Identifier.
gi int(11)
N
o 0 Gene Info Identifier
map text Yes
N
ULL Gene location in chromosome.
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY ll_id
gi
illll_rowid INDEX rowid
illll_llid INDEX ll_id
illll_gi INDEX gi
Observation.: Maps a locus link id with a gi and its location in the chromosome. Not
all locus link ids will have a gi. A gi may be specified as an NP gi or a
XP gi ( experimentally determined ). If an NP gi is available, it will be
used. If an NP gi is not available, then the XP gi will be used. If neither
the NP nor XP gi is available, then no gi will be used.
Organization: NCBI
Source file: ftp://ftp.ncbi.nih.gov/refseq/LocusLink/LL_tmpl.gz
*** ll_id ***
description: Locus Link Identifier.
example: 2
default value: n/a
source: start of record
eg.
>>2
parser: llparser.c --> ll_parser.c
function: LL_AppendLLID_DB()
LL_ParseFile() -> LL_LineParser()
seqhound@blueprint.org Version 3.3
The SeqHound Manual 187 of 421 18/04/2005
API: SHoundLLIDFromGi()
SHoundLLIDFromGiList()
***gi***
description: Gene Info Identifier
example: 21071030
default value: n/a
source: in the NP tag, 2nd element
eg
NP: NP_570602|21071030
parser: llparser.c --> ll_parser.c
function: LL_ParseFile() --> LL_LineParser() -->
LL_Append2LLID_DB()
API: SHoundGiFromLLID()
SHoundGiFromLLIDList()
***map***
description: Location of the gene in the Chromosome
example: 19q13.4
default value: n/a
source: in the MAP tag: 1st element
MAP: 19q13.4|RefSeq|C|
parser: llparser.c --> ll_parser.c
function: LL_ParseFile() --> LL_LineParser()
LL_Append2LLID_DB()
API: SHoundLocusFromGi()
seqhound@blueprint.org Version 3.3
The SeqHound Manual 188 of 421 18/04/2005
ll_cdd table
Last updated: August 5, 2004
Database: seqhound
Table: ll_cdd
Module: lldb
Definition: Conserved Domain Database.
Maps a locus link id to a Conserved Domain ID.
Proteins often contain several modules or domains, each with a distinct
evolutionary origin and function. The CDD database may be used to
identify the conserved domains present in a protein sequence.
Conserved Domains can be summarized with multiple local sequence
alignments. Computational biologists have compiled collections of
such alignments representing conserved domains, and LocusLink
imports them from three major sources:
* Smart, the Simple Modular Architecture Research Tool
* Pfam (UK), Pfam-A seed alignments
* COG (Clusters of Orthologous Groups)
Smart and Pfam are public domain databases, which are offered in
combination with HMM-based search engines and alignment
visualization services.
COG is an NCBI-curated protein classification resource. Sequence
alignments corresponding to COGs are created automatically from
constituent sequences and have not been validated manually for import
in CDD.
The default e-value cutoff of for data in this table is 0.01
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto incremented id
ll_id int(11)
N
o 0 Locus Link Identifier
cdd_id varchar(50) Yes
N
ULL CDD Source Database.
evalue decimal(20,10)Yes
N
ULL Descriptive match.
MySQL Indexes
Keyname Type Field
illcdd_rowid INDEX rowid
illcdd_llid INDEX ll_id
illcdd_cddid INDEX cdd_id
Observation.: Domains can be thought of as distinct functional and/or structural units
of a protein. These two classifications coincide rather often, as a matter
of fact, and what is found as an independently folding unit of a
seqhound@blueprint.org Version 3.3
The SeqHound Manual 189 of 421 18/04/2005
polypeptide chain also carries specific function. Domains are often
identified as recurring (sequence or structure) units, which may exist in
various contexts. e.g.: of the CDD Record.
CDD: pfam00207: Alpha-2-macroglobulin
family|5952|2318|na|8.970050e+02
CDDID: pfam00207
Evalue: 8.970050e+02
SHoundLLIDFromCDDID()
after CDD tag: 1st element
function:
***evalue***
Organization: NCBI
Source db: ftp://ftp.ncbi.nih.gov/refseq/LocusLink/LL_tmpl.gz
*** ll_id ***
description: Locus link identifier
example: 2
default value: n/a
source: start of record
eg.
>>2
parser: llparser.c --> ll_parser.c
function: LL_AppendLLID_DB()
LL_ParseFile() -> LL_LineParser()
API:
SHoundLLIDFromGi()
SHoundLLIDFromGiList()
***cdd_id***
description: Conserved Domain Database Identifier
example: pfam00207
default value: n/a
source:
CDD: pfam00207: Alpha-2-macroglobulin
family|5952|2318|na|8.970050e+02
parser: llparser.c --> ll_parser.c
LL_ParseNPUnit()
LL_ParseFile() --> LL_LineParser()
API: SHoundCDDIDFromGi()
SHoundCDDIDFromGiList()
SHoundCDDIDFromLLID()
description: Describes match between CDD and the sequence
seqhound@blueprint.org Version 3.3
The SeqHound Manual 190 of 421 18/04/2005
example: 8.970050e+02
default value: 0
source: after CDD tag: last element
CDD: pfam00207: Alpha-2-macroglobulin
family|5952|2318|na|8.970050e+02
parser: llparser.c --> ll_parser.c
function: LL_ParseNPUnit()
LL_ParseFile() --> LL_LineParser()
API: SHoundCDDScoreFromGi()
seqhound@blueprint.org Version 3.3
The SeqHound Manual 191 of 421 18/04/2005
GENE module
parse_gene_files.pl parser
Last updated September 27, 2004
purpose:
parse_gene_files.pl parses 4 files from the NCBI Gene database and populates gene_*
tables in SeqHound.
logic:
parse_gene_files.pl first drops the gene_* tables and then recreates them. It then reads the
files generated by gene_cron.pl to populate the new tables. In the future, the tables will
not be dropped upon update.
module: gene
gene_infoUniq
tables altered: gene_dbxref, gene_genomicgi, gene_history, gene_info, gene_productgi,
gene_pubmed, gene_object, gene_synonyms.
[datab]
password=your_pass_word
input files:
ftp://ftp.ncbi.nih.gov/gene/DATA
gene2refseqUniq
gene_historyUniq
gene2pubmedUniq
source code location:
/seqhound/gene/parse_gene_files.pl
/seqhound/scripts/gene_cron.pl
config file dependencies:
The relevant configuration file is:
slri/seqhound/config/.intrezrc (for Unix platforms)
The relevant section of the configuration file is:
;seqhound database that you are connecting
username=your_user_name
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
Text in italics must be changed for the .intrezrc file to function correctly with your
SeqHound set-up. Variables username, password, dsn, database in section
[datab] should have the same values as USER, PASSWORD, DSN and DATABASE
respectively in your .odbc.ini (see Step 10 in section 4.4.) .intrezrc must reside in the
directory where the parser is running and .odbc.ini should be in your home directory.
You should also have a copy of shconfig.pm in the directory where the parser is running
to read .intrezrc and .odbc.ini.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 192 of 421 18/04/2005
command line parameters:
example use:
perl parse_gene_files.pl
associated scripts:
Four files are downloaded from ftp://ftp.ncbi.nih.gov/gene/DATA by gene_cron.pl:
gene2refseq.gz
gene_info.gz
gene_history.gz
gene2pubmed.gz.
gene_cron.pl unzips the files and makes sure that each file only contains unique records
generating the following files:
gene2refseqUniq
gene_infoUniq
gene_historyUniq
error and run-time logs:
parse_gene_files.log
gene2pubmedUniq
troubleshooting:
additional info:
http://www.ncbi.nlm.nih.gov/entrez/query/static/help/genehelp.html
Note that the NCBI gene database is experimental. This means that the
input files and the SeqHound tables may change.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 193 of 421 18/04/2005
gene_dbxref table
Last updated September 28, 2004
Database: SeqHound
Table: gene_dbxref
Module: GENE
Definition: The table holds cross references to other databases for GeneIds and their
associated gis.
MySQL
Field Type Null Default Column_Definition
id Int(11)
N
o Identifier for a cross reference.
geneinfoid Int(11)
N
o Id of the gene_info record that
this cross reference refers to.
dbname Varchar(255)
N
o Database name
Varchar(30)
N
o Accession. dbaccession
MySQL Indexes
Keyname Type Field
igenedbxrefs_id INDEX id
igenedbxrefs_infoid INDEX geneinfoid
igenedbxrefs_db_name INDEX dbname
igenedbxrefs_db_access INDEX dbaccess
Observation:
Source org: NCBI
Source file: gene_info.gz
FTP script: gene_cron.pl
Parser: parse_gene_files.pl
***id***
Identifier for the cross reference. Mysql auto-increment table.
function:
description:
example: 1
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
API: not available yet
seqhound@blueprint.org Version 3.3
The SeqHound Manual 194 of 421 18/04/2005
***geneinfoid***
description: Identifier for the geneinfo record where this cross reference was found.
example: 53577
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
not available yet
***dbname***
description: Name of the database that contains the second reference.
example: SGD
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
***dbaccess***
description: Accession for the record that contains the name.
example: S0000572
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 195 of 421 18/04/2005
gene_genomicgi table
Last updated September 29, 2004
Module: GENE
Database: SeqHound
Table: gene_genomicgi
Definition: Table that contains the gis of genomic DNAs that contain genes. The
start, stop location and orientation on the genomic DNA are also
included.
MySQL
Field Type Null Default Column_Definition
id int(11)
N
o Identifier of a product gi. Mysql
auto-increment column.
geneobjectid int(11)
N
o Identifier of the geneobject that
this genomic gi refers to.
gi int(11)
N
o The genomic gi.
int(11) Yes
The start location on the genomic
DNA.
end int(11) Yes
The start location on the genomic
DNA.
orientation char(1) Yes The orientation on the genomic
DNA.
start
MySQL Indexes
Keyname Type Field
id
igenomic_objectid geneobjectid
igenomic_gi gi
igenomic_id
Observation:
Source org: NCBI
Source file: gene_info.gz
FTP script: gene_cron.pl
Parser: parse_gene_files.pl
seqhound@blueprint.org Version 3.3
The SeqHound Manual 196 of 421 18/04/2005
***id***
description: Identifier of a genomic gi.
example: 1
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
***geneobjectid***
description: The gene object that this gi refers to.
example:
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
***gi***
description: The genomic gi.
example: 10954454
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
***start***
description: The start position of the gene on the genomic DNA.
example: 348
default value:
ASN.1 struct:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 197 of 421 18/04/2005
source:
parser: parse_gene_files.pl
function:
API: not available yet
***end***
description: The end position of the gene on the genomic DNA.
example: 1190
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
***orientation***
description: The orientation of the gene on the genomic DNA.
- meaning "minus" strand or
+ meaning "plus" strand
example: -
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
seqhound@blueprint.org Version 3.3

The SeqHound Manual 198 of 421 18/04/2005
gene_history table
Last updated September 29,2004
Database: SeqHound
Table: gene_history
Module: GENE
Definition: This table has information about geneids that are no longer current.
MySQL
Field Type Null Default Column_Definition
id int(11)
N
o Identifier for a history record.
Mysql auto-increment column.
taxid int(11) Yes
N
CBI taxonomy identifier.
currentgeneid int(11) Yes The current NCBI Gene Id for
this gene.
oldgeneid int(11) Yes The discontinued Gene Id.
oldsymbol varchar(20) Yes
The symbol assigned to the
discontinued Gene Id, if the
discontinued record was not
replaced with another.
MySQL Indexes
Keyname Type Field
igenehistory_id Index id
current_gene_id Index currentgeneid
discont_gene_id Index oldgeneid
discont_symbol Index oldsymbol
Observation: Note that all fields except for id are optional.
Source org: NCBI
Source file: gene_history.gz
FTP script: gene_cron.pl
Parser: parse_gene_files.pl
seqhound@blueprint.org Version 3.3
The SeqHound Manual 199 of 421 18/04/2005
***id***
description: Identifier for the history record. Mysql auto-increment column.
example: 212815
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
***taxid***
description: The NCBI taxonomy identifier for this gene..
example: 10116
default value:
ASN.1 struct:
***currentgeneid**
*
ASN.1 struct:
The discontinued NCBI gene id.
source:
parser: parse_gene_files.pl
function:
API: not available yet
description: The current NCBI gene id.
example: 29666
default value:
source:
parser: parse_gene_files.pl
function:
API: not available yet
***oldgeneid***
description:
example:
default value:
ASN.1 struct:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 200 of 421 18/04/2005
source:
parser: parse_gene_files.pl
function:
API: not available yet
***oldsymbol***
description: The symbol associated with the discontinued gene id, if no new record
replaced the discontinued record.
example: hlyB
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
seqhound@blueprint.org Version 3.3
The SeqHound Manual 201 of 421 18/04/2005
gene_info table
Last updated September 29, 2004
Database: SeqHound
Table: gene_info
Module: GENE
Definition: Information about a gene.
MySQL
Field Type Null Default Column_Definition
id int(11)
N
o Identifier for this record.
geneobjectid int(11)
N
o geneobjectid association with this
information.
symbol varchar(255) Yes The default symbol for this gene.
locustag varchar(255) Yes The LocusTag for this gene.
varchar(32) Yes
maplocation varchar(255) Yes The map location of this gene on
the chromosome.
description mediumtext Yes A description of this gene.
chromosome The chromosome where this gene
is found.
MySQL Indexes
Keyname Type Field
igeneinfo_id Index
geneobjectid
symbol Index symbol
locus_tag Index locustag
chr Index chromosome
id
object_id Index
Observation:
FTP script:
Source org: NCBI
Source file: gene_info.gz
gene_cron.pl
Parser: parse_gene_files.pl
seqhound@blueprint.org Version 3.3
The SeqHound Manual 202 of 421 18/04/2005
***id***
description: Identifier for this record.
example: 74058
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
***geneobjectid**
description: The geneobjectid for the gene that this information refers to.
example: 73870
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
***symbol***
description: The symbol for this gene.
example: 1A981
default value:
ASN.1 struct: gene->locus (NCBI says this is where the data comes from).
source:
parser: parse_gene_files.pl
function:
API:
***locustag***
description: The locus tag for this gene.
example: 1A981
default value:
ASN.1 struct: gene->locus-tag (NCBI says this is where the data comes from).
seqhound@blueprint.org Version 3.3
The SeqHound Manual 203 of 421 18/04/2005
source:
parser: parse_gene_files.pl
function:
API:
***chromosome***
description: The chromosome where this gene is found.
example: I
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
***maplocation***
description: The map location of this gene on the chromosome.
example: I;-19.13 cM (interpolated genetic position)
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
***description***
description: A description of this gene.
example: putative protein (69.3 kD) (1A981)
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 204 of 421 18/04/2005
gene_object table
Last updated September 29,2004
Database: SeqHound
Table: gene_object
Module: GENE
Definition: This table contains information on the status of gene records.
MySQL
Field Type Null Default Column_Definition
id int(11)
N
o Identifier for this gene.
geneid int(11)
N
o
N
CBI's Gene Id.
status varchar(64)) Yes
Status of the RefSeq for this gene.
May be provisional, INFERRED,
MODEL, PREDICTED,
Reviewed or VALIDATED
MySQL Indexes
Keyname Type Field
igeneobject_id Index id
igeneobject_geneid Index geneid
igeneobject_status Index status
Observation:
Source file:
Source org: NCBI
gene2refseq.gz
FTP script: gene_cron.pl
Parser: parse_gene_files.pl
seqhound@blueprint.org Version 3.3
The SeqHound Manual 205 of 421 18/04/2005
***id***
description: Identifier for this gene.
example: 8
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
***geneid**
description: NCBI's Gene Id.
example: 1246510
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
***status***
description: The status of the RefSeq record that contains this gene.
NULL
INFERRED
MODEL
PREDICTED
Provisional
Reviewed
VALIDATED
example: Provisional
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
seqhound@blueprint.org Version 3.3
The SeqHound Manual 206 of 421 18/04/2005
gene_productgi table
Last updated September 29, 2004
Database: SeqHound
Table: gene_productgi
Module: GENE
Definition: Stores the gis of the RNA and Proteins produced by a gene.
MySQL
Field Type Null Default Column_Definition
id int(11)
N
o Internal identifier for the product
gi. Mysql auto-increment column.
geneobjectid int(11)
N
o The geneobjectid for the gene that
this gi belongs to.
gi int(11)
N
o The gi.
MySQL Indexes
Keyname Type Field
iproduct_id Index id
iproduct_objected Index geneobjectid
iproduct_gi Index gi
Observation:
Source org: NCBI
Source file: gene2refseq.gz
FTP script: gene_cron.pl
Parser: parse_gene_files.pl
***id***
description: Internal identifier for this gi. Mysql auto-increment column.
example: 9
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
***geneobjectid***
seqhound@blueprint.org Version 3.3
The SeqHound Manual 207 of 421 18/04/2005
description: NCBI's identifier for the gene that this gi belongs to.
example: 10
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
***gi***
description: The gi. May be a protein or an RNA gi.
example: 32455275
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API: not available yet
seqhound@blueprint.org Version 3.3

The SeqHound Manual 208 of 421 18/04/2005
gene_pubmed table
Last updated September 29, 2004
Database: SeqHound
Table: gene_pubmed
Module: GENE
Definition: This table stores publications that refer to genes.
MySQL
Field Type Null Default Column_Definition
id int(11)
N
o Internal identifier for this
reference.
geneobjectid int(11) Yes The geneobjectid that this
reference is about.
p
mi
d
int(11) Yes The pmid of the reference.
MySQL Indexes
Keyname Type Field
ipubmed_id Index id
ipubmed_geneid Index geneobjectid
ipmid Index
p
mi
d
Observation:
Source org: NCBI
Source file: gene2pubmed.gz
FTP script: gene_cron.pl
Parser: parse_gene_files.pl
***id***
description: Internal identifier of this reference or of this gene objectid-pmid pair
example: 112394
default value:
ASN.1 struct:
source:
source:
parser: parse_gene_files.pl
function:
API: not available yet
seqhound@blueprint.org Version 3.3
The SeqHound Manual 209 of 421 18/04/2005
***geneobjectid***
description: The geneobjectid of the gene.
example: 173392
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
***pmid***
description: The pmid of the reference.
example: 8889548
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 210 of 421 18/04/2005
gene_synonyms table
Last updated September 28, 2004
Database: SeqHound
Table: gene_synonyms table
Module: GENE
Definition: Stores synonyms for genes.
MySQL
Field Type Null Default Column_Definition
id int(11)
N
o Identifier of this synonym, Mysql
auto-increment column
geneinfoid int(11) Yes The geneinfoid for this synonym.
synonym text Yes The synonym.
MySQL Indexes
Keyname Type Field
igenesyn_id Index id
igeneinfoid Index geneinfoid
isynonym Index synonym
Observation:
Source org: NCBI
Source file: gene_info.gz
FTP script: gene_cron.pl
Parser: parse_gene_files.pl
***id*
description: Internal identifier for this synonym geneinfoid pair. Basically a rowid.
Mysql auto-increment column.
example: 11179
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 211 of 421 18/04/2005
***geneinfoid
description: The geneinfoid for the record where this synonym was found.
example: 27686
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
***synonym
description: The synonym.
example: T1J24.19
default value:
ASN.1 struct:
source:
parser: parse_gene_files.pl
function:
API:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 212 of 421 18/04/2005
Gene Ontology hierarchy (godb) module
goparser
Last updated: August 5, 2004
purpose:
Go data files contain information from the Gene Ontology Consortium that describe
controlled vocabularies for the description of the molecular function, biological process
and cellular component of gene products.
The GO parser is responsible for generating the hierarchical gene ontology datafiles:
go_name, go_parent, go_synonym and go_reference. This is accomplished by parsing
the flat files component.ontology, function.ontology and process.ontology.
module: godb
input files:
component.ontology
function.ontology
process.ontology.
From ftp://ftp.geneontology.org/pub/go/ontology/
tables altered:
go_name, go_parent, GO_REF, GO_SYN
source code location:
slri/seqhound/go/goparser.c
config file dependencies:
The relevant configuration file is:
slri/seqhound/parsers/.intrezrc (for Unix platforms) or
The relevant section of the configuration file is:
[datab]
;seqhound database that you are connecting
username=your_user_name
password=your_pass_word
dsn=dsn_in_.odbc.ini_file
database=seqhound
local=
[sections]
;gene ontology hierarchy
godb = 1
seqhound@blueprint.org Version 3.3
The SeqHound Manual 213 of 421 18/04/2005
Text in italics must be changed for the .intrezrc file to function correctly with your
SeqHound set-up. Variables username, password, dsn, database in section
[datab] should have the same values as USER, PASSWORD, DSN and DATABASE
respectively in your .odbc.ini (see Step 10 in section 4.4.) In section [sections]
godb should be 1.
command line parameters:
This parser does not have any command line parameters. Typing “./goparser” will
process the three Gene Ontology files.
These files (component.ontology, function.ontology and process.ontology) must be
present in the same directory as the compiled goparser executable
associated scripts:
see “/slri/seqhound/goftp.pl”
This script retrieves the three input files required by the goparser. The script also
retrieves the files used as input by the addgoid parser (see lldb module).
error and run-time logs:
goparser writes to a log file called “goparserlog” where it writes Time Stamp, Error #,
goparser.c line # and cause of the problem.
e.g.
================[ May 21, 2003 2:36 PM====================
ERROR: [000.000] {goparser.c, line 74} Main: Cannot find function.ontology file
in current directory.
troubleshooting:
additional info:
The Gene Ontology Consortium documentation is located at
http://www.geneontology.org/
See data table descriptions for each of the tables that are listed under SeqHound's Data
Dictionary.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 214 of 421 18/04/2005
go_parent table
Last updated: August 5, 2004
Database: Seqhound
Table: go_parent
Module: Godb
Definition:
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto number row identifier
go_id int(11)
N
o 0 GeneOntology ID
p
arent_goi
d
int(11)
N
o 0 Parent Go Id..
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY go_id
p
arent_goi
d
igoparent_rowid INDEX rowid
igoparent_goid INDEX go_id
igoparent_pid INDEX
p
arent_goi
d
Observation.: Function, process and component are represented as directed acyclic
graphs (DAGs) or networks. The difference between a DAG and a
hierarchy is that in the latter each child can only have one parent; a
DAG allows a child to have more than one parent. A child term may be
an "instance" of its parent term(isa relationship) or a component of its
parent term (part-of relationship). A child term may have more than one
parent term and may have a different class of relationship with its
different parents.
Organization: Gene Ontology (ftp://ftp.geneontology.org/pub/go/ontology)
Source db: function.ontology (Molecular function)
process.ontology (Biological process)
component.ontology (Cellular component)
***go_id***
description: Gene Ontology identifier Child.
example: 0016172
%antifreeze activity ; GO:0016172 {Parent}
%ice nucleation inhibitor activity ; GO:0016173 {Child}
default value: n/a
source: related to the indentation in the file.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 215 of 421 18/04/2005
parser: goparser.c -> go_parser.c
function: GODB_ParseFile()->GO_LineParser()-
>GO_AppendRecord()
db_layer: GO_AppendRecord()->GO_Append_Name()->
GO_AppendParent()
GO_GetParentOf()
GO_GetChildrenOf()
GO_GetAllChildren()
GO_GetAllAncestors()
API: SHoundGODBGetChildrenOf()
***parent_goid***
description: Gene Ontology identifier Parent.
example: 0016173
%antifreeze activity ; GO:0016172 {Parent}
%ice nucleation inhibitor activity ; GO:0016173 {Child}
default value: n/a
source: Related to indentation in the file
parser: goparser.c -> go_parser.c
function: GODB_ParseFile()->GO_LineParser()-
>GO_AppendRecord()
db_layer: GO_AppendRecord()->GO_Append_Parent()
API:
GO_GetParentOf()
GO_GetChildrenOf()
GO_GetAllChildren()
GO_GetAllAncestors()
SHoundGODBGetParentOf()
seqhound@blueprint.org Version 3.3
The SeqHound Manual 216 of 421 18/04/2005
go_name table
Last updated: August 5, 2004
Database: seqhound
Table: go_name
Module: godb
Definition:
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto incremented id
go_id int(11)
N
o 0 GeneOntology ID.
go_name text
N
o Go_id Identifier name.
go_db int(11)
N
o 0 GO Database name.
go_level int(11)
N
o 0 Hierarchy level of the GO ID.
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY
go_id
go_name
go_db
go_level
igoname_rowid INDEX rowid
igoname_goid INDEX go_id
Observation.: This table contains a list of 'go_id' and it's molecular function name as
well the database where the go_id was parsed from and the level in the
Gene Ontology hierarchy.
Organization: GeneOntology (ftp://ftp.geneontology.org/pub/go/ontology)
Source db: function.ontology (Molecular function)
process.ontology (Biological process)
Gene Ontology identifier.
15643
component.ontology (Cellular component)
***go_id***
description:
example:
default value: n/a
source: See example Go File. The GO id follows 'GO:' tag.
parser: goparser.c -> go_parser.c
function: GODB_ParseFile()->GO_LineParser()-
>GO_AppendRecord()
db_layer: GO_AppendRecord()->GO_Append_Name()
seqhound@blueprint.org Version 3.3

The SeqHound Manual 217 of 421 18/04/2005
GO_GetRecordByID()
API: SHoundGOIDFromRedundantGi
SHoundGOIDFromRedundantGiList
SHoundGOIDFromGi
SHoundGOIDFromGiList
***go_name***
This is not used because the same GO ID can appear at
description: Gene Ontology identifier name.
example: anti-toxin;
default value: n/a
source: go_name follows, '%' and proceeds ';GO:'.
parser: goparser.c -> go_parser.c
function: GODB_ParseFile()->GO_LineParser()-
>GO_AppendRecord()
db_layer: GO_AppendRecord()->GO_Append_Name()
GO_GetNameByID()
API: SHoundGODBGetNameByID
***go_db***
description: Gene Ontology Database file. This is an integer 1, 2 or 3
example: 1
where:
1 GO_MOLFUNCTION
2 GO_BIOPROCESS
3 GO_CELLCOMPONEN
default value: n/a
source: NA
parser: goparser.c -> go_parser.c
function: GODB_ParseFile()->GO_LineParser()-
>GO_AppendRecord()
db_layer: GO_AppendRecord()->GO_Append_Name()
GO_GetClassification()
API: SHoundGODBGetClassification()
***go_level***
description: Hierarchy level of the GO ID.
different levels making the process to determine its level
irrelevant. This was intended to be used to indicate the
distance of a given GO node from the root node.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 218 of 421 18/04/2005
example: 3
default value: n/a
source: Related to the indentation of
'%{function name}'
parser: goparser.c -> go_parser.c
function: GODB_ParseFile()->GO_LineParser()-
>GO_AppendRecord()
db_layer: GO_AppendRecord()->GO_Append_Name()
GO_GetHierarchyLevel()
API: SHoundGODBGetHierarchyLevel() this function is deprecated.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 219 of 421 18/04/2005
go_reference table
Last updated: August 5, 2004
Database: seqhound
Table:
MySQL
go_reference
Module: godb
Definition:
Field Type Null Default Column_Definition
rowid int(11) Auto incremented id
go_id int(11)
N
o 0 GeneOntology ID
go_ref text
N
o GeneOntology Reference.
go_ref_db varchar(50) Yes
N
ULL Reference Data Base Name
N
o
MySQL Indexes
Keyname Type Field
PRIMARY PRIMARY go_id
go_ref
igoref_rowid INDEX rowid
igoref_go_ref_db INDEX go_ref_db
Observation.: This table stores the 'go_id' with its database cross-reference. The
cross-reference can be an external database identifier that points to
something that is equivalent to a given GO term.
Organization: GeneOntology (ftp://ftp.geneontology.org/pub/go/ontology)
Source db: function.ontology (Molecular function)
process.ontology (Biological process)
component.ontology (Cellular component)
***go_id***
description: Gene Ontology identifier.
example: 45174
default value: n/a
source: The go_id comes after '%{function name} ; GO:xxxxxx ; '
parser: goparser.c -> go_parser.c
function: GODB_ParseFile()->GO_LineParser()-
>GO_AppendRecord()
db_layer: GO_AppendRecord()-
>Go_Append_Reference()
GO_GetRecordByReference()
seqhound@blueprint.org Version 3.3
The SeqHound Manual 220 of 421 18/04/2005
API: SHoundGODBGetRecordByReference()
example:
***go_ref***
description: Gene Ontology Synonym Description.
example: ISBN: 0198506732
default value: n/a
source: go_ref comes after "GO:xxxxxx ; ISBN: 0198506732 ;"
parser: goparser.c -> go_parser.c
function: GODB_ParseFile()->GO_LineParser()-
>GO_AppendRecord()
db_layer: GO_AppendRecord()-
>GO_Append_Reference()
GO_GetRecordByReference()
API: NA
***go_ref_db***
description: Gene Ontology Reference Database.
EC
ISBN
TC
default value: n/a
source: After '%{function_name} ; GO:xxxxxxx ; {go_ref_db}
After '%{component_name} ; GO:xxxxxxx ; {go_ref_db}
parser: goparser.c -> go_parser.c
function: GODB_ParseFile()->GO_LineParser()-
>GO_AppendRecord()
db_layer: GO_AppendRecord()-
>GO_Append_Reference()
GO_GetRecordByReference()
API: NA
seqhound@blueprint.org Version 3.3
The SeqHound Manual 221 of 421 18/04/2005
go_synonym table
Last updated: August 5, 2004
Database: seqhound
Table: go_synonym
Module: godb
Definition:
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto incremented id
go_id int(11)
N
o 0 GeneOntology ID
go_syn text
N
o GeneOntology Synonym.
MySQL Indexes
Keyname Type Field
PRIMARY KEY INDEX go_id
go_syn(100)
igosynonym_rowid INDEX go_id
igosynonym_go_id INDEX go_syn
Observation.:
Organization: GeneOntology (ftp://ftp.geneontology.org/pub/go/ontology)
Source db: function.ontology (Molecular function)
process.ontology (Biological process)
component.ontology (Cellular component)
Example of source
file:
n/a
%glycine binding activity ; GO:0016594 ; synonym:Gly binding ;
synonym:aminoacetic acid binding ; synonym:aminoethanoic acid
binding
***go_id***
description: Gene Ontology identifier.
example: 45174
default value: n/a
source: The go_id follows '%{function name}; GO:'
parser: goparser.c -> go_parser.c
function: GODB_ParseFile()->GO_LineParser()-
>GO_AppendRecord()
db_layer: GO_AppendRecord()->Go_Append_Synonym()
API:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 222 of 421 18/04/2005
***go_syn***
description: Gene Ontology Synonym Description. This can be a 'synonym' of a
'process' or 'component' name.
example: 'Gly binding'
default value:
synonym:aminoacetic acid binding ;
n/a
source: After '%{function_name} ; GO:xxxxxxx ; synonym:{synonym name}'
After '%{component_name} ; GO:xxxxxxx ; synonym:{synonym name}'
After '%{process_name} ; GO:xxxxxxx ; synonym:{synonym name}'
glutathione dehydrogenase (ascorbate); glutamic acid binding
%glycine binding activity ; GO:0016594 ;
synonym:Gly binding ;
synonym:aminoethanoic acid binding
parser: goparser.c -> go_parser.c
function: GODB_ParseFile()->GO_LineParser()-
>GO_AppendRecord()
db_layer: GO_AppendRecord()->GO_Append_Synonym()
API: n/a
seqhound@blueprint.org Version 3.3

The SeqHound Manual 223 of 421 18/04/2005
Gene Ontology Association (GOA) module
Last updated April 5, 2005
This section is maintained by Renan Cavero.
purpose:
The GOA Module provides Gene Ontology (GO) information for all possible organisms that have GO terms. GO terms are controlled
vocabulary for molecular function, biological process and cellular component of gene products. GO Annotation assignments are
derived from ftp://ftp.geneontology.org/pub/go/gene-associations/ and from ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2go.gz.
The GO terms are linked to identifiers provided by the curating database (e.g. FlyBase) AND to the NCBI Gene Info identifier (GI)
using the SeqHound DBXref Module.
The GOA Module contains
• GO term assignments to genes or proteins
• Literature references like PubMed IDs
• Evidence codes between the gene product and the GO term
• Object types that get annotated such as gene, transcript, protein or protein structure
• Gene symbols or other associated text
• Taxonomic identifiers
• Date on which the annotation was made.
module:
GOA
seqhound@blueprint.org Version 3.3

The SeqHound Manual 224 of 421 18/04/2005
input files:
All files found under gene ontology FTP site
ftp://ftp.geneontology.org/pub/go/gene-associations/
NCBI EntrezGene
ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2go.gz
tables altered:
goa_association
goa_gigo
goa_reference
goa_seq_dbxref
goa_with
goa_xdb
seqhound@blueprint.org Version 3.3

The SeqHound Manual 225 of 421 18/04/2005
Table summarizing input files, parsers and command line parameters for GOA module.
Input file parser command line
parameters
All files from: ftp://ftp.geneontology.org/pub/go/gene-associations/ goa_parser_cluster.pl
See note below.
-d {Org.abbr.} (*)
–c {Cluster: T F}
-o {Offset: n}
-n {Num. Lines: n}
-f {Flag File}
ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2go.gz go_geneparser_cluster.pl
See note below.
-d GO_GENE (**)
-c {Cluster: T F}
-o {Offset: n}
-n {Num. Lines: n}
-f {Flag File}
Tables: goa_gigo, dbxref, goa_seq_dbxref, goa_association, accdb,
redund, taxgi.
Seqhound_gigo_cluster.pl
See note below.
-c {Cluster: T F}
-o {Offset: n}
-n {Num. Lines: n}
-f {Flag File}
Tables: goa_gigo, goa_seq_dbxref, goa_association, accdb, redund, taxgi. Seqhound_gigoPDB_cluster.pl
See note below.
-c {Cluster: T F}
-o {Offset: n}
-n {Num. Lines: n}
-f {Flag File}
Tables: goa_gigo, dbxref, goa_seq_dbxref, goa_association. Seqhound_gigoCGEN_cluster.pl
See note below.
-c {Cluster: T F}
-o {Offset: n}
-n {Num. Lines: n}
-f {Flag File}
Notes:
(*) Parser Command Line Parameters. (See dbxref.ini for details.)
goa_parser_cluster.pl: Parse Gene Ontology Association files and populate GOA module tables.
-d Organism Database File Abbreviation: Can be found under dbxref.ini Partition [GENE_ASSOCIATON_FILE] Ex.: GOA_DDB,
GOA_CGEN.
-c Cluster Option: T: True (run in a cluster environment); F: False: (run stand alone).
The following options are optional. Needs to be set-up only if running in a Cluster environment.
-o Offset: The offset in a flat file where a cluster node will start parsing.
-n Number of lines: The number of line-records to process by a cluster node after the Offset is reached.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 226 of 421 18/04/2005
-f Flag File: For synchronization purpose. A file name that will be generated by a cluster node when finished parsing, telling the
Cluster’s head node that the parsing was completed. (Cluster Head node will track completion of all nodes before continuing with the
next process in the queue.
(**)
goa_gene_parser: Same as above but this parser will only parse gene2go file.
seqhound_gigo_cluster.pl: Populates goa_gigo with GI’s (GenBank assessions) extracted from dbxref table and by looking-up Xref
in goa_seq_dbxref will get GO terms from goa_association. When looking up GI’s in accdb, redundancy and taxonomies are
considered.
seqhound_gigoPDB_cluster.pl: Looks up PDB records and chains from goa_seq_dbxref and by looking-up GIs in accdb populates
gi-go pairs in goa_gigo. When looking up GI’s in accdb, redundancy and taxonomies are considered.
seqhound_gigoCGEN_cluster.pl: Same as above but for GIs from Compugen records.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 227 of 421 18/04/2005
source code location:
The parsers for this module have been updated to work in a cluster configuration. Source code is unavailable at the time of this
manual release but will be released with the next code release.
/slri/seqhound/dbxrefgoa
config file dependencies:
The relevant configuration file is:
dbxref.ini (for Unix platforms)
see dbxref.ini documentation.
command line parameters:
See summary table above.
associated scripts:
The following shell scripts execute the parsers:
dbxrefgoa_cron.sh: Cron job that runs dbxrefgoa_updatecron.pl
dbxrefgoa_updatecron.pl: Program that automates deployment and runs parsers in a cluster. (see dbxref.ini documentation for details)
auxiliary scripts:
In a cluster environment the following scripts will help dbxrefgoa_updatecron.pl in the deployment and execution:
parsers.deploy.sh: deploy files to be parsed to the cluster nodes.
generate_goa_run.pl: Generate an instruction script that distributes processes in the cluster nodes.
run{organism database_file_abbreviation}: instruction script generated by generate_goa_run.pl. Example.: runGOA_MGI.sh
clean.sh: clean (remove) files previously deployed by script deploy.sh.
wait.pl: makes the cluster’s head node wait until all the nodes finish processing the parser that was deployed. When wait.pl receives a
signal from all the nodes, it will continue with the next parser.
See dbxref.ini for more details.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 228 of 421 18/04/2005
error and run-time logs:
at summarize number of records updates by each goa parser.
dbxrefgoa_updatecron.pl will execute instructions from the configuration file dbxref.ini. Any error generated by this process or by the
parsers will be recorded under dbxrefgoa_error.log. The final results will be summarized in dbxrefgoa_updatecron.log and
goa_parser.log. By following the error messages, the SeqHound administrator will be able to adjust configuration parameters under
dbxref.ini and run it again.
dbxrefgoa_errors.log: log that tracks run time error by the parsers.
dbxrefgoa_updatecron.log: log that indicate what parser was run, completed or failed . A copy of this log will be send by email to the
SeqHound administrator.
goa_parser.log: log th
troubleshooting:
additional info:
The Gene Ontology Consortium documentation is located at
http://www.geneontology.org/GO.contents.doc.html
ftp://ftp.geneontology.org/pub/go/doc/GO.xrf_abbs_spec
Gene Ontology Module Diagram
The tables also appears in http://www.blueprint.org/seqhound/api_help/docs/SeqHound_Schema_Prod.pdf
seqhound@blueprint.org Version 3.3

The SeqHound Manual 229 of 421 18/04/2005
goa_seq_dbxref
PK id
FK3 goa_xdb_id
I1 xref_key (2)
taxid_1 (13a)
taxid_2 (13b)
type_id (12)
symbol (3)
full_name (10)
synonym (11)
timestamp
goa_with
PK id
FK4 goa_association_id
FK5 goa_xdb_id (8)
xref_key (8a, 8b, 8x)
key_type
FK6 goa_seq_dbxref_id
timestamp
goa_xdb
PK id
abbreviation (1)
name
object
example
generic_url
url_syntax
url_example
timestamp
goa_reference
PK id
FK3 goa_association_id
goa_xdb_id
xref_key (6a, 6b, 6x)
timestamp
go_name
PK go_id
go_name
I2 go_db
I1 go_level
goa_association
PK,FK4 id
is_not (4)
FK8 go_id (5)
goa_seq_dbxref_id
assigned_by (15)
code (7)
date (14)
timestamp
Gene Ontology Flat File
DB (1)
DB_Object_ID (2)
DB_Object_Symbol (3)
NOT (4)
GOid (5)
DB_Reference (6)
Evidence (7)
With (8)
Aspect (9)
DB_Object_Name (10)
DB_Object_Synonym (11)
DB_Object_Type (12)
taxon (13)
Date (14)
Assigned_by (15)
Relational Data Base representation of Gene
Ontology Association module in SeqHound.
(Last updated March 23, 2005)
Numbers beside column names refer to columns in
the Gene Ontology flat file representation
(see http://www.geneontology.org/
GO.annotation.shtml#file and table below).
goa_gigo
gi
go
code
xdbid
xref_key
seqhound@blueprint.org Version 3.3
The SeqHound Manual 230 of 421 18/04/2005
goa_seq_dbxref table
Last updated April 12, 2005
Database:
Table:
Seqhound
goa_seq_dbxref
Module: GOA
Definition: The main table of the GOA Module. This table contains entries for a
database record ID (e.g.: Fly Base ID FBgn0013277) and the
information associated with the record that can be found in
gene_association files from Gene Ontology. The record is the base to
obtain other information like GO terms, reference and other annotation.
Source org: Gene Ontology ftp://ftp.geneontology.org/pub/go/gene-associations/
Source file: See module description above.
Parser: See module description above.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 231 of 421 18/04/2005
goa_seq_dbxref table
Field Type API Null Default Column_Definition Example Source
id int(10)
N
o Auto incremented
integer.
Internal primary record
identifier.
2099131 Assigned by source
document parser.
N
A
goa_xdb_id int(10)
N
o
0
Database identifier from
table goa_xdb. For
example, an xdb_id of 4
indicates the Candida
Genome Database.
5 See note below.
N
A
xref_key varchar(30)
N
o
Record ID (in the above-
mentioned database)
which points to the object
being annotated.
PrID1098818 See note below.
N
A
taxid_1 int(11)
N
o
0
Taxonomic identifier of
the species encoding the
gene product.
11676 See note below.
N
A
taxid_2
int(11) Yes 0
Taxonomic identifier of
the species where the gene
product acts (in the
manner described by the
GO annotation). For
example, if the protein is
from a virus, the host
organism might be listed
here.
0 See note below.
N
A
type_id varchar(20)
N
o
Indicates the type of
object being annotated
(gene, transcript, protein
etc.)
protein See note below.
N
A
seqhound@blueprint.org Version 3.3

The SeqHound Manual 232 of 421 18/04/2005
symbol varchar(30)
N
o
A unique and valid
symbol to which the
xref_key is matched. It
can be an ORF name,
gene product symbol or
any other identifier.
GI424263 See note below.
N
A
full_name varchar(255) Yes
N
ame of gene or gene
product.
cell surface glycoprotein
gp138
See note below.
N
A
synonym varchar(50) Yes
N
ULL Gene symbol or other text.fusA See note below.
N
A
lastupdate CURRENT_TIMES
TAMP
2005-04-01 17:25:44
N
A
N
ULL
timestamp
N
o When was this entry last
updated.
Note: columns in this table may be mapped to the GO Annotation flat file format described at
http://www.geneontology.org/GO.annotation.shtml#file. Also see the Figure entitled “GOA relationships” below.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 233 of 421 18/04/2005
goa_seq_dbxref indices
Keyname Type Cardinality Field
id_idx INDEX 2138717 id
goa_xdb_id_idx PRIMARY 91 goa_xdb_id
xref_key_idx PRIMARY VC0002 xref_key
taxid_1_idx INDEX 686 taxid_1
symbol_idx INDEX VC0002 symbol
synonym_idx INDEX mioC synonym
lastupdate_idx INDEX 2005-04-01 17:25:44 lastupdate
seqhound@blueprint.org Version 3.3
The SeqHound Manual 234 of 421 18/04/2005
goa_association table
Last updated April 12, 2005
Database: Seqhound
Table: goa_association
Module: GOA
Definition: This table contains GO term information associated with the Record ID
(goa_seq_xref.xref_key).
Source file: See module description above.
Parser: See module description above.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 235 of 421 18/04/2005
goa_association table
TField ype Null Default Column_Definition Example Source API
id int(10)
N
o Auto
increment
integer.
Internal unique identifier for this record. 33 This identifier is incremented
by the source file parser.
N
A
N
ULL Flags that modify the interpretation of an
annotation. A GO ID with a Not in this
field means that a particular gene product
is NOT associated with a particular GO
term.
For more information please read "Using
the Qualifier column" from
“T” indicates that “NOT was found in
this column.
“F” indicates that “NOT” was not found.
F See note below.
N
A
go_id int(10)
N
o 0 Gene Ontology identifier for the term
attributed to the object described by
goa_seq_xref.xref_key.
3677 See note below.
N
A
goa_seq_dbxref_id int(10)
N
o 0 Foreign key pointing to
goa_seq_dbxref.id
7 See note below.
N
A
assigned_by int(10)
N
o 0 database that made the annotation. 117 See note below.
code char(4)
N
o Evidence Code. One of IMP, IGI, IPI,
ISS, IDA, IEP, IEA, TAS, NAS, ND, IC.
IEA See note below.
N
A
date char(8) Yes
N
ULL Date on which the annotation was made. 20040107
N
A
lastupdate timestamp Yes
N
ULL Date when this record was last modified.
2005-04-01 17:25:44
See note below.
N
A
is_not char(1) Yes
http://www.geneontology.org/GO.annota
tion.html
See note below.
Note: columns in this table may be mapped to the GO Annotation flat file format described at
http://www.geneontology.org/GO.annotation.shtml#file. See the Figure entitled “GOA relationships” below.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 236 of 421 18/04/2005
goa_association indices
Keyname Type Cardinality Field
PRIMARY PRIMARY 8720064 id
go_id
goa_seq_dbxref_id
go_id UNIQUE 8720064
code
goa_id_idx INDEX 21268 go_id
goa_seq_dbxref_id_idx INDEX 8720064 goa_seq_dbxref_id
assigned_by_idx INDEX 19 assigned_by
code_idx INDEX code
lastupadate_idx INDEX 2005-04-01 17:25:44 lastupdate
seqhound@blueprint.org Version 3.3
The SeqHound Manual 237 of 421 18/04/2005
goa_reference table
Last updated April 12, 2005
Database: Seqhound
Table: goa_reference
Module: GOA
Definition: This table contains the reference identifier for the GO annotation.
Reference identifiers are unique identifiers appropriate to a database
authority for the attribution of the go_id to the Record ID. They may be
a literature reference or a database record.
Source file: See module description above.
Parser: See module description above.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 238 of 421 18/04/2005
goa_reference table
Field Type Null Default Column_Definition Example Source API
id int(10)
N
o Auto
increment
integer.
A unique identifier for this record.
1 This value is autoincremented
by the source file parser.
N
A
goa_association_id int(10)
N
o 0 Foreign key pointing to
goa_association.id.
530508 See note below.
N
A
goa_xdb_id int(10)
N
o 0 Database identifier that made the
reference. Foreign key pointing to
goa_xdb.id..
49 See note below.
N
A
xref_key varchar(20)
N
o Reference Identifier. For example, if
the reference is a published paper
that has a PubMed ID, the PubMed
ID number will be in this field
MGI:1354194 See note below.
N
A
lastupdate timestamp Yes
N
ULL When was this entry last updated? 2005-04-01 17:25:44
N
A
Note: columns in this table may be mapped to the GO Annotation flat file format described at
http://www.geneontology.org/GO.annotation.shtml#file. See the Figure entitled “GOA relationships” below.
goa_reference indices
Keyname Type Cardinality Field
PRIMARY PRIMARY 8424423 id
goa_association_id_idx INDEX 8424423 goa_association_id
goa_xdb_id_idx INDEX 18 goa_xdb_id
xref_key_idx INDEX 2864 xref_key
lastupdate_idx INDEX 17122 lastupdate
seqhound@blueprint.org Version 3.3
The SeqHound Manual 239 of 421 18/04/2005
goa_with table
Last updated April 12, 2005
Database: Seqhound
Table: goa_with
Module: GOA
Definition: This table is used to hold additional identifiers for annotations using
certain evidence codes. For more information please see "With (or)
From" section at http://www.geneontology.org/GO.annotation.html
Source files: See module description above.
Parsers: See module description above.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 240 of 421 18/04/2005
goa_with table
Field Type Null Default Column_Definition Example Source API
id int(10)
N
o Auto
increment
integer.
A unique identifier for this record. 1 This column is autoincremented
by the source file parser.
N
A
goa_association_id int(10)
N
o 0 Foreign key pointing to
goa_association.id
530509 See note below.
N
A
goa_xdb_id int(10)
N
o 0 Database Identifier that made the
'with' annotation.
37 See note below.
N
A
xref_key varchar(20)
N
o Reference identifier. IPR001601 See note below.
N
A
key_type int(10)
N
o 0 Type of the symbol annotated:
Type of the symbol annotated:
1=gene_symbol,
2=allele_symbol,
3=gene_id,
4=sequence_id,
5=go_id.
0 See note below.
N
A
goa_seq_dbxref_id int(10)
N
o 0 Foreign key pointing to
goa_seq_dbxref.id for quick look-
up from a ‘with’ annotation back
to the record id.
0 See note below.
This field is not currently
implemented.
N
A
lastupdate timestamp
N
o
N
ULL When was this entry last updated? 2005-04-01 17:25:44
N
A
Note: columns in this table may be mapped to the GO Annotation flat file format described at
http://www.geneontology.org/GO.annotation.shtml#file. See the Figure entitled “GOA relationships” below.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 241 of 421 18/04/2005
goa_with indices
Keyname Type Cardinality Field
PRIMARY PRIMARY 1234449 id
goa_association_id_idx INDEX 1234449 goa_association_id
goa_xdb_id_idx INDEX 17 goa_xdb_id
xref_key_idx INDEX xref_key
key_type_idx INDEX key_type
goa_seq_dbxref_id_idx INDEX goa_seq_dbxref_id
lastupdate_idx INDEX lastupdate
seqhound@blueprint.org Version 3.3
The SeqHound Manual 242 of 421 18/04/2005
goa_xdb table
Last updated April 12, 2005
Database: Seqhound
Table: goa_xdb
Module: GOA
Definition: The table goa_xdb contains metadata about the organizations which
contribute to the GO. There is a one to one relationship between
abbreviations and URLs where data can be retrieved. A single URL
which can be queried using database ids is referred to as a datasource.
Each organization may have multiple datasources. Each abbreviation
identifies one section of the file which provides the abbreviation and
full name of that data source, the object type which is retrieved, an
example database id, the generic url which identifies that data source
uniquely and globally and the syntax of an actual query request with
parameters filled in.
For more information please read:
ftp://ftp.geneontology.org/pub/go/doc/GO.xrf_abbs_spec
Source files: See module description above.
Parsers: See module description above.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 243 of 421 18/04/2005
goa_xdb table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o Auto
increment
integer.
A unique identifier for this record.
N
ote that this identifier is unstable
and may change from one release
to another.
76 This column is
autoincremented
by the source file
parser.
N
A
abbreviation varchar(50)
N
o Database abbreviation. SWISS-PROT See note below.
N
A
name varchar(255) Yes
N
ULL Database name or description. Swiss-Prot protein database. See note below.
N
A
object varchar(255) Yes
N
ULL type of identifier returned from the
data source:
Accession number
Locus identifier
call number
Gene symbol etc.
See note below.
N
A
example varchar(50) Yes
N
ULL An example database identifier. Swiss-Prot:P45867 See note below.
N
A
generic_url varchar(255) Yes
N
ULL The root or representative URL for
this data source.
http://ca.expasy.org/spro
t/
See note below.
N
A
url_syntax varchar(255) Yes
N
ULL A string to which one can append
a database ID and get a valid URL
query for the object referenced by
that id. There is no wild card to
represent the database ID, it is
simply appended to the end of the
string.
http://www.expasy.ch/cgi-
bin/sprot-search-ac?
See note below.
N
A
url_example varchar(255) Yes
N
ULL An example of what the
url_syntax will look like
http://www.expasy.ch/cgi-
bin/sprot-search-
ac?P45867
See note below.
N
A
lastupdate timestamp
N
o
N
ULL 2005-04-01 17:25:44
N
A
Note: columns in this table may be mapped to the GO Annotation flat file format described at
http://www.geneontology.org/GO.annotation.shtml#file. See the Figure entitled “GOA relationships” below.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 244 of 421 18/04/2005
goa_xdb indices
Keyname Type Cardinality Field
PRIMARY PRIMARY 117 id
abbreviation_idx INDEX abbreviation
name_idx INDEX name
seqhound@blueprint.org Version 3.3
The SeqHound Manual 245 of 421 18/04/2005
goa_gigo table
Pre-computed list of gi-go pairs.
Parsers:
Last updated April 5, 2005
Database: Seqhound
Table:
Module:
goa_gigo
GOA
Definition:
Since SeqHound database is NCBI GI Centric and GOA is organism
database ID centric; the process to get a GO term given a GI can be
complicated and time consuming, for this reason a pair list of gi-go
were pre-computed and stored in goa_gigo table. The SeqHound core
database (mainly accdb and redund tables) is required to run these
parsers.
Source files: See module description above.
See module description above.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 246 of 421 18/04/2005
goa_gigo table
Field API Type Null Default Column_Definition Example Source
gi INTEGER
N
o 0 Gene Info Identifier for an NCBI sequence
record
6552303 The record indicated by the last
two columns of this table are
converted to a matching Gene
Info Identifier using the
DBXref module.
N
A
go INTEGER
N
o 0 Gene Ontology Identifier. 6281 From table
goa_association:gi_id
N
A
varchar(4) Yes
N
ULL Evidence Code. One of IMP, IGI, IPI, ISS,
IDA, IEP, IEA, TAS, NAS, ND, IC.
TAS
For more information please read
http://www.geneontology.org/GO.evidenc
e.html
From table
goa_association:code
N
A
xdb_id INTEGER Yes
N
ULL Identifier for database. See also xref_key
below. From table goa_xdb.
103 From table goa_xdb.id
N
A
xref_key varchar(30) Yes
N
ULL Identifier in database (see previous
column) pointing to object that was
originally annotated.
P38398 From table
goa_seq_dbxref:xref_key.
N
A
lastupdate timestamp
N
o
N
ULL When was this entry last updated? 2005-04-01 17:25:44
N
A
code
seqhound@blueprint.org Version 3.3
The SeqHound Manual 247 of 421 18/04/2005
goa_gigo indices
Keyname Type Cardinality Field
PRIMARY PRIMARY 7936320
gi
go
code
gi_idx INDEX 3968160 gi
go_idx INDEX 7254 go
code_idx INDEX code
xdb_id_idx INDEX 22 xdb_id
xref_key_idx INDEX 7936320 xref_key
lastupdate_idx INDEX 22 lastupdate
seqhound@blueprint.org Version 3.3
The SeqHound Manual 248 of 421 18/04/2005
dbxref module
Last updated April 12, 2005
purpose:
The purpose of the SeqHound dbxref module is to have a centralized data source (cross references) where related information can be
found from a given ID. By using the dbxref module, it is possible to find one to "n" relationships between IDs from 3rd party
databases for DNA, Protein Sequences, Domains and Interactions, GenBank Accession Numbers, Swiss-Prot, LocusLink, SGD,
MGD, ZFIN, FB, PFAM, SMART, etc. See the “Explanation of the data table structure” below.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 249 of 421 18/04/2005
Who Cross-references who?
The following table indicates what database records we collect as cross-references from primary database records in the SeqHound
DBXref module.
This table was last updated April 15th and may change on a regular basis.
DB Cross references to:
Primary DBs GB SPTR CG TAIR UNIGENE IPI ENSEMBL OMIM
GENE X X X X X X X
SPTR X X X X X X X X
ENSEMBL X X X
AFCS X
TIGR_ATH X
MGI X X
RGD X
FB X
WB X X
SGD X
DDB X
ZFIN X
GRAMINE X X
GENEDB_SPOMBE X
TIGR_ATH X
TIGR_CMR X
UNIGENE X
VIDA X
Explanation of the data table structure:
The dbxref table is the core of the dbxref module. This explanation refers to that table.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 250 of 421 18/04/2005
Dbxref represents cross-references between 3rd party databases and GeneBank. This table is created by parsing 3rd party flat files and
creating records in "dbxref" for each record parsed. dbxref is a self-referencing table.
The value in the "record_id" field may represent one of two things:
1. A source record:
If the value of "parent_id" in the row is zero (0) it is referred to as an object id. Its "record_id" is the identifier/primary id for a
record in the database ("source_db") from which cross-references have been retrieved.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 251 of 421 18/04/2005
For example, from the dbxref table content example listed below.
P38903 is a Primary ID in SwissProt Database
S00055403 is a Primary ID in SGD Database.
2. A database cross-reference found in a source record. The retrieved cross-references are stored as a combination of
a) Source database ("source_db").
b) an identifier for a record in that database ("record_id").
c) the record that the cross-reference was found in. The field "parent_id" contains an integer. This integer refers to the row in this
table (see "id") that contains the identifier of the record from which this cross reference was retrieved.
d) cross references are retrieved from some field in a record. The name of this field is recorded in the "field" column or if there
are no field names then the column number is recorded. For example : Col1 or Col4.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 252 of 421 18/04/2005
Example entries
| id | source_db | record_id | parent_id | link | field | cv |
+----+-----------+-----------+-----------+------+-------+----+
| 1 | SP | P38903 | 0 | 0 | ID | 0 |
| 2 | GB | U06630 | 1 | 0 | DR | 1 |
| 3 | GB | AAB38372 | 1 | 2 | DR | 2 |
| 4 | GB | S79635 | 1 | 0 | DR | 1 |
| 5 | GB | AAB35312 | 1 | 4 | DR | 2 |
| 6 | GB | X87331 | 1 | 0 | DR | 1 |
| 7 | GB | CAA60763 | 1 | 6 | DR | 2 |
| 8 | GB | Z74922 | 1 | 0 | DR | 1 |
| 9 | GB | CAA99203 | 1 | 8 | DR | 2 |
| 10 | InterPro | IPR001757 | 1 | 0 | DR | 3 |
| 11 | Pfam | PF00689 | 1 | 0 | DR | 3 |
| 12 | SGD | S0005540 | 1 | 0 | DR | 0 |
| 13 | SP | P47096 | 0 | 0 | ID | 0 |
| 14 | GB | Z49525 | 13 | 0 | DR | 1 |
| 15 | GB | CAA89550 | 13 | 14 | DR | 2 |
| 16 | GB | X87297 | 13 | 0 | DR | 1 |
| 17 | IPR | IPR007113 | 13 | 0 | DR | 0 |
| 18 | SGD | S00055403 | 0 | 0 | Col4 | 0 |
| 19 | GB | 1420113 | 18 | 0 | GI | 0 |
| 20 | GermOnline| 143602 | 18 | 0 | Col1 | 0 |
| 21 | DIP | 4191 | 18 | 0 | Col1 | 4 |
| 22 | GB | 6324588 | 18 | 0 | GI | 0 |
| 23 | GB | AX596518 | 18 | 0 | Col1 | 1 |
| 24 | CandidaDB | CA1247 | 18 | 0 | Col1 | 0 |
| 25 | GB | CAA99203 | 18 | 0 | Col1 | 2 |
| 26 | InterPro | IPR002554 | 18 | 0 | Col1 | 0 |
| 27 | GB | NP_014656 | 18 | 0 | Col1 | 2 |
| 28 | SP | P38903 | 18 | 0 | Col1 | 0 |
| 29 | PIR | S54620 | 18 | 0 | Col1 | 0 |
| 30 | GB | S79635 | 18 | 0 | Col1 | 1 |
| 31 | GB | U06630 | 18 | 0 | Col1 | 1 |
| 32 | GB | X87331 | 18 | 0 | Col1 | 1 |
| 33 | MIPS | YOR014W | 18 | 0 | Col1 | 0 |
| 34 | GB | Z74922 | 18 | 0 | Col1 | 1 |
+----+-----------+-----------+-----------+------+-------+----+
The "link", "field" and "cv" fields in the dbxref table help specify where the information is coming from. These are described below.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 253 of 421 18/04/2005
link
The link field was created to support some databases (for example Swiss-Prot) which may store more than one cross-reference in one
field. For example a swiss-prot record for a protein may contain a "DR" field that lists two EMBL identifiers. ie.:
DR EMBL; U06630; AAB38372.1; -.
The first identifier is a cross-reference for a nucleotide record in EMBL that encodes a protein (second identifier).
The link field was created to capture this relationship between the two cross-references. The integer in the link field points to a row in
the dbxref table (see "id") and indicates that the current cross-reference is linked (or comes after) some other cross reference in the
same source record and field.
The exact meaning of the database cross-reference can be discerned from the "cv" (Controled Vocabulary) column of this table. In
this example, one cross-reference would be labeled as "nucleotide" and one would be labeled as "protein".
2: points to record "id=2" meaning that protein sequence identifier "AAB38372" comes after the identifier “U06630” in the DR
field.
0: if no relationship exists or if the dbxref is the first one in a list
field
This describes the field in the source record where the database cross-reference was found. For example, “DR” is a field name in
Swiss-Prot records indicating a Database Reference. Alternatively, a column number might be listed here is the source file was a tab-
delimited text file.
cv
cv is a controlled vocabulary term that is used to describe the type of record that the database cross-reference is pointing to. This
controlled vocabulary is simple at the moment and may be expanded in future. Briefly,
1 indicates a DNA sequence record
2 indicates a protein sequence record
3 RNA
100 Swiss-Prot record
101 Trembl record
110 Swiss-Prot secondary accession
111 Trembl secondary accession
seqhound@blueprint.org Version 3.3

The SeqHound Manual 254 of 421 18/04/2005
0 means not defined.
source code location: /slri/seqhound/dbxref
input Files: See summary table.
parsers: See summary table.
config file dependencies:
dbxref.ini
command line parameters: See summary table.
example use: See summary table.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 255 of 421 18/04/2005
associated scripts: (See associated scripts under GOA Module.)
The following shell scripts execute the parsers.
dbxrefgoa_cron_monthly.sh: Script to create all tables and sub-directories when running dbxref-goa module for the first time.
dbxrefgoa_updatecron.pl: Program that automates deployment and runs parsers in a cluster. (see dbxref.ini documentation for details)
error and run-time logs:
dbxrefgoa_errors.log: log that tracks run time error by the parsers.
dbxrefgoa_updatecron.log: log that indicate what parser was run, completed or failed . A copy of this log will be sent by email to the
SeqHound administrator.
dbxref_parser.log: log that summarizes number of record updates by each dbxref parser.
Error messages are sent by email when dbxrefgoa_updatecron.pl runs.
troubleshooting:
Check the email sent by dbxrefgoa_updatecron.pl to find out if any parsers had problems. For more details consult the log files.
additional info:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 256 of 421 18/04/2005
How to update the DBXref and GO Annotation modules using a cluster.
Last updated April 15th, 2005.
This section is maintained by Zhe Wang and Renan Cavero.
Note for those installing there own local instance of SeqHound. The data sets generated by this process are made available on the
SeqHound ftp site at ftp://ftp.blueprint.org/pub/SeqHound/Data/ . These instructions are supplied as a description of our internal
process. The scripts described are provided as part of the SeqHound code release package for those who may wish to use them as a
guide for setting up their own internal cluster build. The framework presented here might also be useful to others for setting up cluster
jobs.
Dbxrefgoa_update.pl will generate scripts for distribution and execution of processes on a cluster. To accomplish this it uses clusterit
tools. More information about clusterit can be found at: http://www.garbled.net/clusterit.html.
The file dbxref.ini has all the configurations to build and update the DBXRef/GOA modules on a computer cluster. This configuration
file is read and executed by the script dbxrefgoa_update.pl.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 257 of 421 18/04/2005
Understanding the dbxref.ini file
Section [DBXREFDATA] describes the database to which the updated or new DBXRef/GOA data will be written. Variable
“database” should be an existing database. The text in italics should be modified. The user must have write privilege to the
database.
[DBXREFDATA]
# this should point to the server which has dbxref and goa modules
host = staging_box
port = 33306
user = user
password = passwd
database = dbxrefgoa
table = dbxref
tablegigo = goa_gigo
Section [SEQHOUND] describes the SeqHound database from which information will be read in order to update the DBXRef/GOA
modules specified in section [DBXREFDATA]. The user must have read privilege to the database.
[SEQHOUND]
# this should point to the server which has the most up-to-date tables of seqhound
hostshound = production_box
portshound = 3306
usersshound = user
passwordshound = passwd
databaseshound = seqhound
working_dir = /home/user123/dbxrefgoa/
data_dir = /scratch/dbxrefgoa/download
myemail = you@you.org
The variable “working_dir” specifies the directory in which to run script dbxref_updatecron.pl.
Directory /home/ is mapped to all cluster nodes.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 258 of 421 18/04/2005
Variable “data_dir” specifies where the input files are saved.
Directory /scratch/ is a local directory on each node.
Variable “myemail” should be the e-mail address of the SeqHound administrator who will be notified of the result of the
DBXRef/GOA update.
Section [LOG_FILES] has the name for three logs files:
[LOG_FILES]
results_log = dbxrefgoa_results.log
errors_log = dbxrefgoa_errors.log
update_log = dbxrefgoa_updatecron.log
Section [DBXREF_FILES] and [GENE_ASSOCIATION_FILES] specify the data files to be processed.
Each of sections [ORGANISM_DBXREF] and [ORGANISM_GOA] specifies the categories of data to be processed.
[ORGANISM_DBXREF]
ORGANISMS = “GENE;SP;TR”
The value of the variable “ORGANISMS” indicates that there are three groups of data that need to be processed. This particular order
of GENE, SP and TR is important for internal purposes. By looking at the value of the variable “ORGANISMS”, script
dbxrefgoa_update.pl goes to the proper section in this configuration file, dbxref.ini, to find information such as where to download the
data file and what commands to be executed on the data file.
For example, when dbxrefgoa_update.pl reads “GENE”, it looks for section [GENE] in the dbxref.ini file. Section [GENE] is shown
below. “GENE_URL” specifies the FTP path of the data file, “GENE_CMD” specifies the command to be run, and
“GENE_CMD2RUN” specifies the command to be run on the cluster nodes.
[GENE]
GENE_URL="ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/mim2gene"
GENE_CMD2RUN=perl dbxref_gene_cluster.pl
seqhound@blueprint.org Version 3.3
The SeqHound Manual 259 of 421 18/04/2005
GENE_CMD="./deploy.sh GENE gene2accession"
GENE_CMD="./generate_dbxref_run.pl XREF_GENE > runXREF_GENE.sh"
GENE_CMD="./runXREF_GENE.sh"
GENE_URL="ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2accession.gz"
GENE_OMIM_CMD2RUN=perl dbxref_gene_extra.pl -d OMIM
GENE_CMD="./deploy.sh GENE mim2gene"
GENE_CMD="./generate_dbxref_run.pl XREF_GENE_OMIM > runXREF_GENE_OMIM.sh"
GENE_CMD="./runXREF_GENE_OMIM.sh"
GENE_CMD="./clean.sh GENE"
GENE_URL="ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2unigene"
GENE_UNIGENE_CMD2RUN=perl dbxref_gene_extra.pl -d UNIGENE
GENE_CMD="./deploy.sh GENE gene2unigene"
GENE_CMD="./generate_dbxref_run.pl XREF_GENE_UNIGENE > runXREF_GENE_UNIGENE.sh"
GENE_CMD="./runXREF_GENE_UNIGENE.sh"
GENE_CMD="./clean.sh GENE"
Script dbxrefgoa_update.pl first creates a sub-directory called “GENE/wget/” in the directory specified by the variable “data_dir”.
The newly created directory would be /scratch/dbxrefgoa/download/GENEl/wget/. Script dbxrefgoa_update.pl then reads all
GENE_URLs sequentially and downloads the data files if they been updated. These files are placed into the directory
/scratch/dbxrefgoa/download/GENE/wget/, and then subsequently copied one level up to the directory
/scratch/dbxrefgoa/download/GENE/. If no up-to-date data files are downloaded, script dbxrefgoa_update.pl finishes with this section
and moves to the next according to the value of “ORGANISMS” in section [ORGANISM_DBXREF]. Once all the data files are
downloaded, the script dbxrefgoa_update.pl reads the values of “GENE_CMD” and executes one command at a time.
The first part of section “[GENE]” is used to explain how this works.
[GENE]
#GENE_URL="ftp://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2accession.gz"
GENE_CMD2RUN=perl dbxref_gene_cluster.pl
GENE_CMD="./deploy.sh GENE gene2accession"
GENE_CMD="./generate_dbxref_run.pl XREF_GENE > runXREF_GENE.sh"
GENE_CMD="./runXREF_GENE.sh"
seqhound@blueprint.org Version 3.3
The SeqHound Manual 260 of 421 18/04/2005
The first command to be executed is "./deploy.sh GENE gene2accession ". Script deploy.sh makes directory
/scratch/dbxrefgoa/download/GENE/ on each of the cluster nodes and copy the data file gene2accession into the newly created
directory on each node.
The second command to be executed is "./generate_dbxref_run.pl XREF_GENE > runXREF_GENE.sh". Script
generate_dbxref_run.pl takes in one parameter, XREF_GENE, which refers to the full path of the data file specified in variable
“data_dir” plus “XREF_GENE” in section [DBXREF_FILE] of the configuration file dbxref.ini. The data file has been copied to each
cluster node by executing command "./deploy.sh GENE gene2accession". Each of the cluster nodes will process a part of the data file.
The data file will be evenly divided into multiple segments each of which is processed by one node. In this way, the jobs on the nodes
can finish in approximately the same time.
The cluster nodes need to know where in the file to start and to end. Script generate_dbxref_run.pl reads the data file and
calculates the start and end points of the file for each cluster node to process. The script also constructs the command line for each
cluster node. The output of script generate_dbxref_run.pl is written to the file runXREF_GENE.sh.
An example of such a shell script is:
#! /usr/bin/bash
rsh an090 "cd /home/user123/dbxrefgoa; perl dbxref_gene_cluster.pl -c T -o 0 -n
269259 -f an090.dat >> /home/user123/dbxrefgoa/xref_parser.log 2>&1 &"
rsh an091 "cd /home/user123/dbxrefgoa; perl dbxref_gene_cluster.pl -c T -o 269259 -n 269259 -f an091.dat >>
/home/user123/dbxrefgoa/xref_parser.log 2>&1 &"
rsh an092 "cd /home/user123/dbxrefgoa; perl dbxref_gene_cluster.pl -c T -o 538518 -n 269259 -f an092.dat >>
/home/user123/dbxrefgoa/xref_parser.log 2>&1 &"
# Num of records: 807777
./wait.pl an090.dat an091.dat an092.dat
rm an090.dat an091.dat an092.dat
Where “perl dbxref_gene_cluster.pl” is the value of variable “GENE_CMD2RUN” in section [GENE]. In this example, the data file
has 807,777 records that are evenly divided into three segments each of which is processed on one cluster node.
Script dbxref_ll.pl takes four parameters:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 261 of 421 18/04/2005
–c indicates whether this is a run on a cluster node (-c F would make the script process the entire data file regardless of the values of –
o and –n),
–o indicates the starting point or offset in the file for this node,
–n indicates the number of records to be processed,
–f specifies the name of the flag file.
Since the next data file cannot be processed until the previous one is finished, it is necessary to know whether the process on every
cluster node is completed. On each node, a flag file named node.dat (e.g. an091.dat) is generated when the process on that particular
node is finished. The next data file won’t be processed until script wait.pl finds file node.dat on all of the nodes. After this condition is
met, wait.pl removes all flag files.
The last line in section [GENE] is:
GENE_CMD="./cleana.sh"
This command deletes all data files downloaded for this section from the cluster nodes in order to reclaim disk space.
All of the other sections in the dbxref.ini file have the same format as just described for [GENE]. These additional sections are:
[GENE], [SP], [TR], [FB]. [WB], [MGI], [SGD], [TIGR_ATH], [DDB], [RGD], [ZFIN], [XREFGOA], [GENEDB_SPOMBE],
[TAIR], [TIGR_CMR], [UNIGENE], [VIDA], [GOA_CGEN], [GOA_DDB], [GOA_FB], [GOA_GENEDB_GMORSITANS],
[GOA_GENEDB_LMAJOR], [GOA_GENEDB_PFALCIPARUM], [GOA_GENEDB_SPOMBE], [GOA_GENEDB_TBRUCEI],
[GOA_GRAMENE_ORYZA], [GOA_MGI], [GOA_GOA_PDB], [GOA_RGD], [GOA_SGD], [GOA_TAIR], [GOA_TIGR_ATH1],
[GOA_TIGR_CMR], [GOA_TIGR_TBRUCEI_CHR2], [GOA_TIGR_GENE_INDEX], [GOA_VIDA], [GOA_WB],
[GOA_GOA_UNIPROT], [GOA_ZFIN] and [GO_GENE].
seqhound@blueprint.org Version 3.3

The SeqHound Manual 262 of 421 18/04/2005
Table summarizing input files, parsers and command line parameters for dbxref module.
Input file parser 1command line parameters
ftp://expasy.org/databases/uniprot/knowledgebase/uniprot_sprot.dat.gz dbxref_sptr_cluster.pl -d XREF_SP
ftp://expasy.org/databases/uniprot/knowledgebase/uniprot_trembl.dat.gz dbxref_sptr_cluster.pl -d XREF_TR
http://flybase.bio.indiana.edu/allied-data/extdb/external-databases.txt dbxref_fb_cluster.pl none
ftp://ftp.sanger.ac.uk/pub/databases/wormpep/wormpep.table dbxref_wb_cluster.pl none
ftp://ftp.informatics.jax.org/pub/reports/MRK_Sequence.rpt dbxref_mgi_cluster.pl none
ftp://ftp.informatics.jax.org/pub/reports/MRK_SwissProt_TrEMBL.rpt dbxref_mgi_cluster.pl none
ftp://genome-
ftp.stanford.edu/pub/yeast/data_download/chromosomal_feature/dbxref.tab
dbxref_sgd_cluster.pl none
ftp://ftp.afcs.org/pub/mpdata/afcsflat.txt dbxref_AFCS_cluster.pl none
ftp://ftp.tigr.org/pub/data/a_thaliana/ath1/DATA_RELEASE_SUPPLEMENT/r
elease_5.genbank_accessions.txt.gz
dbxref_tigr_ath_cluster.pl
-d XREF_TIGR_ATH
ftp://ftp.geneontology.org/pub/go/gp2protein/gp2protein.tigr_ath dbxref_tigr_ath_cluster.pl -d XREF_TIGR_ATH_GP
ftp://ftp.blueprint.org/pub/SeqHound/Private/DDB/dictybaseid_gb_accession.t
xt.gz
dbxref_ddb_cluster.pl none
ftp://rgd.mcw.edu/pub/data_release/genbank_to_gene_ids.txt dbxref_rgd_cluster.pl none
http://zfin.org/data_transfer/Downloads/genbank.txt dbxref_zfin_cluster.pl none
http://zfin.org/data_transfer/Downloads/refseq.txt dbxref_zfin_cluster.pl none
ftp://ftp.geneontology.org/pub/go/gp2protein/gp2protein.zfin dbxref_zfin_cluster.pl none
ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/HUMAN/human.xrefs.gz dbxref_goa_xrefs_cluster.pl -d
XREF_XREFGOA_HUMAN
ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/MOUSE/mouse.xrefs.gz dbxref_goa_xrefs_cluster.pl –d
seqhound@blueprint.org Version 3.3
The SeqHound Manual 263 of 421 18/04/2005
Input file parser 1command line parameters
XREF_XREFGOA_MOUSE
ftp://ftp.ebi.ac.uk/pub/databases/GO/goa/RAT/rat.xrefs.gz dbxref_goa_xrefs_cluster.pl -d XREF_XREFGOA_RAT
ftp://ftp.sanger.ac.uk/pub/yeast/pombe/Mappings/gp2swiss.txt dbxref_DBs_SPTR_cluster.pl -d spombe
ftp://ftp.geneontology.org/pub/go/gp2protein/gp2protein.tair dbxref_DBs_SPTR_cluster.pl -d tair
ftp://ftp.geneontology.org/pub/go/gp2protein/gp2protein.tigr_cmr dbxref_DBs_SPTR_cluster.pl -d tigr_cmr
ftp://ftp.geneontology.org/pub/go/gp2protein/gp2protein.unigene dbxref_DBs_SPTR_cluster.pl -d unigene
ftp://ftp.geneontology.org/pub/go/gp2protein/gp2protein.vida dbxref_DBs_SPTR_cluster.pl -d vida
ftp://ftp.ncbi.nih.gov/gene/DATA/gene2accessions.gz dbxref_gene_cluster.pl
ftp://ftp.ncbi.nih.gov/gene/DATA/mim2gene.gz dbxref_gene_extra.pl -d OMIM
ftp://ftp.ncbi.nih.gov/gene/DATA/gene2unigene.gz dbxref_gene_extra.pl -d UNIGENE
1 command line parameters:
All DBXref parsers can run in a cluster environment setting the –c argument.
-d Organism Database File Abbreviation: Can be found under dbxref.ini Partition [ORGANISM_DBXREF] Ex.: SPTR; FB.
-c Cluster Option: T: True (run in a cluster environment); F: False: (run stand alone).
The following options are optional. Needs to be set-up only if running in a Cluster environment.
-o Offset: The offset in a flat file where a cluster node will start parsing.
-n Number of lines: The number of line-records to process by a cluster node after the Offset is reached.
-f Flag File: For synchronization purposes. A file name that will be generated when a cluster node finishes parsing telling the Cluster’s
head node that the parsing is finished. (The Cluster Head node will track completion of all nodes before it continues with the next
process in queue).
seqhound@blueprint.org Version 3.3
The SeqHound Manual 264 of 421 18/04/2005
seqhound@blueprint.org Version 3.3

The SeqHound Manual 265 of 421 18/04/2005
dbxref table
Last updated April 12, 2005
Database: SeqHound
Table: dbxref
Module: dbxref
Definition: The dbxref table is the core of the dbxref module.
It represents cross references between 3rd party databases and
GeneBank. This table is created by parsing 3rd party flat files and
creating records in "dbxref" for each record parsed. dbxref is a self-
referencing table. See “Explanation of data table structure” above.
Source org: multiple - see summary table
multiple - see summary table
Source file:
multiple - see summary table
FTP script:
multiple - see summary table
Parser:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 266 of 421 18/04/2005
dbxref table
Field Type Null Default Column_Definition Example Source1API
id int(11)
N
o Auto
increment Identifier for this entry. 2 All SQL insert statements will auto
increment this field.
N
A
source_id int(11)
N
o
0
Foreign key pointing to an identifier for a database, in table
dbxrefsourcedb. For example,
1 = Swiss-Prot,
2 = GenBank…etc.
2 See dbxrefsourcedb:source_id.
N
A
record_id char(30)
N
o
Record identifier in database mentioned in previous column.
The format of this identifier will be that of the source
database.
AAD12597
N
A
parent_id int(11)
N
o
0
Link pointing to "id" (first column in this table) to the
record that is the source id of the Cross-Reference. An
entry of “0” indicates that this is a source record from which
database cross-references are parsed.
1 See dbxref:id
N
A
link int(11) Yes
N
ULL
The link field was created to support some databases (for
example Swiss-Prot) which may store more than one cross-
reference in one field. See the section above “Explanation
of data table structure”.
0 SQL insert statements for proteins that
have dbXref to the nucleotide will
have the "link" field pointing to the
"id" record of the nucleotide.
If this relationship cannot be
established the "link" value will be set
to 0.
N
A
field char(20) Yes
N
ULL
Describes the field in the source record where the database
cross-reference was found. For example, “DR” is a field
name for Database Reference found in Swiss-Prot records.
Col6
N
A
cv int(11) Yes
N
ULL
Controled vocabulary. This will be used to describe the
type of record that the cross-reference is pointing to. See
the section above “Explanation of data table structure”.
2
N
A
lastupdate timestamp
N
o
N
ULL When was this entry last updated 2005-04-01
17:25:44
N
A
1. Multiple source files are parsed by multiple parsers. For details, see summary table.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 267 of 421 18/04/2005
dbxref indices
Keyname Type Cardinality Field
id_idx INDEX 16849002 id
source_id_idx PRIMARY 18 source_id
dbxref_id _idx PRIMARY 16849002 record_id
parent_id_idx PRIMARY 16849002 parent_id
link_idx INDEX 16849002 link
field_idx INDEX field
cv_idx INDEX 18 cv
lastupdate_idx INDEX 51843 lastupdate
seqhound@blueprint.org Version 3.3

The SeqHound Manual 268 of 421 18/04/2005
dbxrefsourcedb table
Last updated April 12, 2005
Database: seqhound
Table: dbxrefsourcedb
Module: dbxref
Definition: This table assigns an internal identifier to all data sources where cross-
references are found. These identifiers are used in the source_id
column of the dbxref table.
Source org: multiple - see summary table
multiple - see summary table
Source file:
multiple - see summary table
FTP script:
multiple - see summary table
Parser:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 269 of 421 18/04/2005
dbxrefsourcedb table1
Field Type Null Default Column_Definition Example Source1API
source_id int(11)
N
o
0
Database ID, primary key in table dbxrefsourcedb.
For example,
1 = Swiss-Prot
1
N
A
source_db char(50)
N
o
Abbreviated name of database.
Abbreviations for database names are the same as
those used by the Gene Ontology group see:
"GO.xrf_abbs"
f
tp://ftp.geneontology.org/pub/go/doc/GO.xrf_abbs
This value is mandatory.
SP
N
A
isprimary_db tinyint(4)
N
o
0
Are cross-references retrieved from this database?
1 = YES
0 = NO
1
N
A
lastupdate timestamp
N
o
2005-04-01 17:25:44
N
A
1. The contents of this table are hand-edited as part of the dbxrefgoa.sql file. The entire contents of this table as of April 5, 2005 are
listed below.
dbxrefsourcedb indices
Keyname Type Cardinality Field
PRIMARY PRIMARY 33 source_id
source_db_idx INDEX 33 source_db
lastupdate_idx INDEX lastupdate
seqhound@blueprint.org Version 3.3
The SeqHound Manual 270 of 421 18/04/2005
Contents of dbxrefsourcedb table
(last updated March 22, 2005).
source
_
id source_db isprimar
y_
db
1 SP 1
2 GB 0
3 PFAM 0
4 INTERPRO 0
5 MGI 1
6 SGD 1
7 SMART 0
8 ZFIN 1
9 FB 1
10 CG 0
11 TR 1
12 SPTR 1
13 WB 1
14 LL 0
15 CGEN 0
16 TIGR_ATH 1
17 REFSEQ 0
18 GENEDB_SPOMBE 1
19 DDB 1
GR 1
21 TAIR
22 TIGR_CMR 1
23 UNIGENE 1
24 VIDA 1
20
1
seqhound@blueprint.org Version 3.3
The SeqHound Manual 271 of 421 18/04/2005
25 RGD 1
26 IPI 1
27 ENSEMBL 1
28 AFCS 1
29 HUGO 0
30 OMIM 0
31 PIR 0
32 GENE 1
seqhound@blueprint.org Version 3.3

The SeqHound Manual 272 of 421 18/04/2005
RPS-BLAST domains (rpsdb) module
domname parser
Note: Not available at this time, as it has not been ported to an ODBC backend. Please go to our ftp site at
ftp://ftp.blueprint.org/pub/SeqHound/RPS/
to download precomputed domname tables.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 273 of 421 18/04/2005
Rpsdb parser
Last updated: August 4, 2004
Note: The rpsdb table is precalculated on a cluster and the resulting table is distributed in MySQL format on our ftp
site. Therefore, this section is provided for informational purposes only, or for those who would like to build rpsdb
tables from there own sequence/domain data; it is not necessary if one wishes simply to include the rpsdb module into
their own seqhound instance, in which case they should simply download the precomputed tables.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 274 of 421 18/04/2005
domname table
Last updated: August 4, 2004
Database: SeqHound
Table: domname
Definition: Domain information parsed from CDD database.
Parser: The table is populated by the program “DomNameToDB” whose
source file is seqhoun/rps/domname.c
The program traverses a directory that contains all the *.acd files
(obtained from NCBI ftp site
f
tp://ftp.ncbi.nih.gov/pub/mmdb/cdd/acd.tar.gz) which contains the
CDD ASN.1 records
Comments: Similarly to rpsdb, this table has two API’s. The CDD ASN.1
definition seems to be under constant flux and with every new
release there are new and revised fields. This in turn requires that we
modify the parser functions and update API functions accordingly.
Codebase (for historical purposes)
Column_name Indexed NULL Column_Definition Data_type Size
ACCESSION Yes
N
o String 15 CDD id
N
AME Yes
N
o String 25 Short domain label
PDB-ID Yes Yes String 10 ID of PDB structure that is
used as 3D representative
of the domain
ASN1
N
o Binary
object
The entire CDD ASN.1
record from the source file
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto incremented id
accession varchar(15)
N
o CDD id
name varchar(25)
N
o Short domain label
seqhound@blueprint.org Version 3.3
The SeqHound Manual 275 of 421 18/04/2005
p
dbi
d
varchar(12)
N
o
ID of PDB structure that is used
as 3D representative of the
domain
asn1 mediumblob
N
o The entire CDD ASN.1 record
from the source file
MySQL Indexes
Keyname Type Field
idom_rowid INDEX rowid
idom_acc INDEX accession
idom_name INDEX name
idom_pdbid INDEX
p
dbid
***accession***
Description: This is the domain’s Conserved Domain Database (CDD) unique
identifier assigned by the CDD group in NCBI. It can also be
SMART, Pfam, LOAD or CDD identifier.
Null
CddIdPtr,
DomNameToDB
Default Value:
Source (ASN.1) :
Parser :
Function: FillDomNameNode
More Info: In early versions of CDD the accession was the Pfam or SMART
identifier. Therefore, in some of the comments in the code this field
is referred to as the Pfam or SMART identifier. Currently, most of
the domains have a unique CDD id but some may not.
More info: The API functions return a list. The reason is that a domain label
such as SH3 may have a SMART and Pfam entry each stored as a
separate CDD entry. Both ID’s will be returned.
API: SHoundGetDomainIdFromLabel(CharPtr label);
seqhound@blueprint.org Version 3.3

The SeqHound Manual 276 of 421 18/04/2005
***name***
Description: Domain’s short label.
Default value: Null
source: This information is obtained from *.csq file that contains a FASTA
description of the domain. The label is parsed from the definition
line.
string GetDomainLabelFromDomainId(string s); CharPtr LIBCALL
SHoundGetDomainLabelFromDomainId(CharPtr accession)
Get3DStructureFromDomainId
Function: FillDomNameNode
More info:
API:
***pdbid***
Description:
Default value:
A 3D structure representative of the domain.
null
source: pCdd->master3d, part of Cdd record
FillDomNameNode
Function:
More info:
API: SHoundGetDomain3DStructure
***asn1***
Description: This is the entire NCBI CDD ASN.1 structure.
Default value: Null
source: Cdd
More info:
Important Note: The CDD structure contains place holders for describing the domains parent, sibling and child
domains. These are domains that are structurally or otherwise related to the domain on the sequence level. The program
DomNameToDB collects this information. In the latest release of CDD that was parsed these fields were left empty.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 277 of 421 18/04/2005
NCBI plans to (or may have) include this information in future releases. This is important information pertaining to the
domains that may be included in future versions of the DomName table. Currently, the table has not been expanded
with additional fields to hold this information.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 278 of 421 18/04/2005
rpsdb table
Last updated: October 07, 2004
Database: SeqHound
Table: rpsdb
Definition: Domain annotation of proteins derived from RPS-BLAST and
Conserved Domains Database (CDD).
Comments: The rpsdb is precalculated on a cluster and distributed in MySQL
format on our ftp site.
This table contains two local API versions (C and C++) defined in
rpsdbapi.hpp and rpsdbapi.c. For the most part the same information
can be accessed by both API albeit in different forms. However,
there may be cases where the two APIs are not identical.
The default e-value cutoff for data in rpsdb is 1
Note: The rpsdb table is precalculated on a cluster and the resulting table is distributed in MySQL format on our ftp
site. Therefore, this section is provided for informational purposes only, or for those who would like to build rpsdb
tables from there own sequence/domain data; it is not necessary if one wishes simply to include the rpsdb module into
their own seqhound instance, in which case they should simply download the precomputed tables.
Codebase (for historical purposes)
Column_name Indexed NULL Data_type Size Column_Definition
GI Yes
N
o Integer 10 Sequence identifier
CDDID Yes
N
o Integer 10 CDD ID (domain ID from
CDD)
DOMID Yes
N
o String 12 Domain ID from primary
database (Pfam, SMART,
COG, KOG or cd)
FROM
N
o
N
o Integer 6 First a.a. position aligned
to this domain
seqhound@blueprint.org Version 3.3
The SeqHound Manual 279 of 421 18/04/2005
ALIGN_LEN Yes
N
o Integer 6 Length of alignment b/w
p
rotein and domain
SCORE
N
o
N
o Integer 10 RPS-BLAST score
EVALUE
N
o
N
o Double 15,8 Base 10 log of RPS-
BLAST E- value
BITSCORE
N
o
N
o Double 15,8 RPS-BLAST bit score
MISSING_N
N
o
N
o Integer 6 The length of N-terminus
residues on the domain
that were not aligned
MISSING_C
N
o
N
o Integer 6 The length of C-terminus
residues on the domain
that were not aligned
N
UMDOM Yes
N
o Integer 4
N
umber of total domains
mapped to this protein
MySQL
Field Type Null Default Column_Definition
rowid int(11)
N
o Auto incremented id
gi int(11)
N
o 0 Sequence identifier
cddid int(11)
N
o 0 CDD ID (domain ID from CDD)
domid char(12)
N
o
Domain ID from primary
database (Pfam, SMART, COG,
KOG or cd)
rfrom int(11)
N
o 0 First a.a. position aligned to this
domain
align_len int(11)
N
o 0 Length of alignment b/w protein
and domain
score int(11) 0 RPS-BLAST score
evalue decimal(15,8) Yes
N
ULL Base 10 log of RPS-BLAST E-
value
N
o
seqhound@blueprint.org Version 3.3

The SeqHound Manual 280 of 421 18/04/2005
bitscore decimal(15,8) Yes
N
ULL RPS-BLAST bit score
missing_n int(11)
N
o 0
The length of N-terminus residues
on the domain that were not
aligned
missing_c int(11)
N
o 0
The length of C-terminus residues
on the domain that were not
aligned
numdom int(11)
N
o 0
N
umber of total domains mapped
to this protein
MySQL Indexes
Keyname Type Field
irps_rowid INDEX rowid
irps_gi INDEX gi
irps_cddid INDEX cddid
irps_domid INDEX domid
irps_len INDEX align_len
irps_numdom INDEX numdom
irps_gi_e INDEX gi
evalue
Source db: SeqHound redundant table
Source program:
Parser :
RPS-BLAST results
rpsdb.h/c in seqhound/rps
***gi***
description: Primary sequence identifier assigned at NCBI
Default value : 0
seqhound@blueprint.org Version 3.3
The SeqHound Manual 281 of 421 18/04/2005
source: GI’s are collected from Seqhound redund table. Proteins that were
not annotated in this table were the hypothetical ORF from RefSeq
(XP_xxxxxx) and SWISS-PROT proteins. These proteins are not
present in SeqHound (no Bioseq) although their GI's are in redund
table. In each redundant group the first GI was used for computing
RPS-BLAST (ordinal 1). These proteins are considered to be the
best representative of the redundant group. However, some
redundant groups may not have the first GI in SeqHound, in those
cases the program collects ordinal 2 or higher. The redundant list is
collected from redund table using “redundlist” program
(seqhound/rps).
API: SHoundGetGisByDomainIdAndEvalue
SHoundGetGisByDomainId
SHoundGetGisByNumberOfDomains
More info:
Comment: There are two functions in the C version that use Codebase relational
query. They should not be used and are there for experimental
purposes only.
seqhound/rps/rpsdb_README.txt
***cddid***
description: Conserved Domain Database unique identifier.
Default Value: none
Source (ASN.1) : CddIdPtr, can also be collected from CddHitPtr cdd_id field.
Parser : rpsdb
Function: RPSDBSHoundRedund2ResultsCallback
More info: seqhound/rps/rpsdb_README.txt
***domid***
Description: Domain identifier from the primary database of origin. These
correspond to either Pfam or SMART string identifiers.
Default Value: Null
seqhound@blueprint.org Version 3.3
The SeqHound Manual 282 of 421 18/04/2005
source: Definition field in CddHitPtr structure.
Function: RPSDBSHoundRedund2ResultsCallback
***rfrom***
Description: The index position of the first amino acid in the protein that is
aligned with this domain.
Default value: 0
source:
Function:
Start field in CddHitPtr structure
RPSDBSHoundRedund2ResultsCallback
***align_len***
Description: The length of sequence alignment between the protein and the
domain.
Default value : 0
source:
Function:
stop- start field values in CddHitPtr.
RPSDBSHoundRedund2ResultsCallback
***score***
Description : RPS-BLAST score parameter
Default value: 0
source: score field in CddHitPtr
Function: RPSDBSHoundRedund2ResultsCallback
***evalue***
Description : Base 10 log of RPS-BLAST evalue score
0
Default value:
Source: evalue field in CddHitPtr
Function:
RPSDBSHoundRedund2ResultsCallback
***bitscore***
seqhound@blueprint.org Version 3.3
The SeqHound Manual 283 of 421 18/04/2005
Description: RPS-BLAST bit score
0
Default:
Default:
source:
Default :
source: bitscore field in CddHitPtr
Function:
RPSDBSHoundRedund2ResultsCallback
***missing_n***
Description: The number of residues on the domain’s N-terminus that were not
aligned with the protein. The missing length on the N-terminus of
the domain.
0
source: It is collected from DenseDegPtr which is part of SeqAlign
structure. This structure is filled up by the RPS-BLAST engine. It
contains the collection of aligned segments between the domain and
the protein.
Function: RPSDBSHoundRedund2ResultsCallback
***missing_c***
Description: The number of missing residues not aligned in the domain’s C-
terminus.
0
Same as MISSING_N
Function: RPSDBSHoundRedund2ResultsCallback
***numdom***
Description: The total number of domains aligned with the protein.
Default: 0
source: The number of entries for the query GI in the rpsdb..
Function: SLRICddCountSeqAligns in rpsdb
seqhound@blueprint.org Version 3.3
The SeqHound Manual 284 of 421 18/04/2005
API: The above fields are all accessed through a set of calls that retrieve
the domain annotation based on different requirements.
SHoundGetDomainsFromGi
SHoundGetDomainsFromGiWithEvalue
SHoundGetDomainsFromGiListWithEvalue
seqhound@blueprint.org Version 3.3
The SeqHound Manual 285 of 421 18/04/2005
Molecular Interaction (MI) module
MI-BIND parser
Last updated October 1, 2004
purpose:
The Molecular Interaction module is meant to consolidate the interaction data, and associated annotation, from disparate source
interaction databases(e.g. BIND, IntAct, MINT, etc). The source data is parsed out of their own unique formats and placed into the
MI module tables' data model. This data model has been designed to provide maximum flexibility in terms of the complexity of
queries that can be made to it. Additionally, the module adds value to the data by cross-referencing distinct records, regardless of their
source databases, to provide information on molecular object redundancy and interaction similarity. The MI module's set of parsers
takes records from source interaction modules, parse out their data, and insert that data into the MI tables. Currently, there exists only
one such parser, for parsing BIND XML records; parsers for other interaction databases will be developed in the future.
Logic:
MI-BIND parser:
This parses the BIND XML records, available on the BIND ftp site. It uses the SAX XML parsing API, in order to achieve maximum
parsing speed. In order to foster code reusability, the parser is broken down into three components: The first component parses out
the BIND XML records interaction data and places it into a general MI data structure. This data structure is then passed to another
component, which is responsible for placing the contents into the MI tables in the database. Another component is then called to do
the cross-referencing and redundancy/similarity analysis on the data just processed, and places that additional annotation into the
database. This break-down is convenient, because it means that developers of MI parsers for other source interaction databases will
only need to replace the first component, the one which parses the data out of the source record, which they can then pass to the
already written data feed and cross-referencing components. The parser was written in the Java programming language to take
advantage of Java's mature XML processing capabilities, and because a great deal of java source code was available from the open
source BIND project for processing BIND XML records.
input files:
The MI-BIND parser processes BIND records in their native XML format. These are available from the BIND ftp site, in a number of
different partitions, each of which can be used interchangeable. In order to import all of BIND into the MI module, one would
download the “Divisions” partition from
ftp://ftp.blueprint.org/pub/BIND/data/divisions/xml/*.xml
seqhound@blueprint.org Version 3.3
The SeqHound Manual 286 of 421 18/04/2005
and then process this list of input files using the MI-BIND parser.
tables altered:
MI_complex2ints
MI_complex2subunits
MI_complexes
MI_dbases
MI_exp_methods
MI_ints
MI_mol_type
MI_obj_dbases
MI_obj_labels
MI_objects
MI_record_types
MI_refs
MI_refs_db
MI_source
source code location:
The source is contained in the following seqhound java packages:
org.bluprint.seqhound.parsers.mi
org.bluprint.seqhound.parsers.mi.bind
config file dependencies:
MI.properties file
This file must be present in the same directory as where the MI-BIND parser is being invoked, and must contain the entries:
dbDriverName=myDatabaseDriversName
dbUserName=myDatabaseUserName
dbPassWord=myDatabasePassword
dbURL=myDatabaseConnectionURL
These contain the settings, which the parser will use to connect to the database management system, which manages the MI data
tables. An example entry follows:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 287 of 421 18/04/2005
dbDriverName=com.mysql.jdbc.Driver
dbUserName=johnsmith
dbPassWord=johmsmithspassword
dbURL=jdbc:mysql://dbhostname:dbportnumber/MIdbname
seqhound@blueprint.org Version 3.3
The SeqHound Manual 288 of 421 18/04/2005
library dependencies:
Along with the standard java runtime environment, the MI-BIND parser requires one third party java library; this is the open source
xerces2 XML parser implementation, available for free from the apache foundation(see http://xml.apache.org/xerces2-j/download.cgi
for free download instructions).
The xerces jar files xercesImpl.jar and xmlParserAPIs.jar must be in classpath when executing the parser.
The MI-BIND parser has been tested only with xerces2 java version 2.6.2, but may work with other versions.
command line parameters:
The MI-BIND parser takes one command line argument: a file containing a list of filenames of files for the parser to process.
example use:
java org.blueprint.seqhound.parsers.mi.bind.bmdParse BINDXMLFileList.txt
Where BINDXMLFileList.txt is a text file containing a newline delimited list of the names of files containing the BIND XML
records to be processed; these BIND XML files must be in the same directory. Note that the compiled bind module parser files, as
well as the xerces jars, must be in classpath in order for the above command to function.
associated scripts:
none
error and run-time logs:
runtime and error information is printed to standard output and standard error, respectively. This will likely be changed in a future
release, to print to a standard log file.
troubleshooting:
additional info:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 289 of 421 18/04/2005
MI_source table
Last updated October 4, 2004
Database:
MI_source
MI
Table:
Module: Molecular Interaction(MI)
Definition: Contains information about the source of an interaction record
MySQL
Field Type Null Default Column Definition
uid Int(11)
N
o Internal MI identifier for record
intcompid Int(11)
N
o interaction, complex or pathway
this record refers to
0
Internal MI identifier of the
db Smallint(6) Yes
N
ULL
Internal MI identifier of the
source db which this record
comes room, corresponds to db
column of MI_dbases
acc Varchar(10) Yes
N
ull Source db accession of this
record, if it exists
id Int(11) Yes
N
ull Source db ID of this record, if it
exists
type Smallint(6)
N
ull
Type of this record, either
interaction(1), complex(2) or
p
athway(3)
descr Text Yes
N
ull Text description of this record
from the source db
data_blob Longblob Yes Binary version of original record
from source db, if it exists
data_clob Longtext Yes
N
ull Text version of original record
N
o
N
ull
seqhound@blueprint.org Version 3.3
The SeqHound Manual 290 of 421 18/04/2005
from source db, if it exists
MySQL Indexes
Keyname Field Type
Primary Primary uid
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script: N/A
Parser: MI-BIND
***uid***
function:
description:
example:
default value
ASN.1 structure:
parser:
API:
more info:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 291 of 421 18/04/2005
MI_ints table
Last updated October 4, 2004
Database: MI
Table:
Module:
MI_ints
MI
Definition: Contains information about individual interactions
MySQL
Field Type Null Default Column Definition
intid Int(11)
N
o Internal MI id of this interaction
objAid Int(11)
N
o 0 Internal MI id of the first object
in this interaction
objBid Int(11)
N
o 0 Internal MI id of the second
object in this interaction
rig Int(11)
N
o 0
Internal MI id of the redundant
interaction group to which this
interaction belongs
MySQL Indexes
Keyname Type Field
Primary Primary intid
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script: N/A
Parser: MI-BIND
seqhound@blueprint.org Version 3.3
The SeqHound Manual 292 of 421 18/04/2005
MI_objects table
Last updated October 4, 2004
Database: MI
Table: MI_objects
Module: MI
Definition: Contains information about the individual objects involved in
interactions, complexes and pathways
MySQL
Field Type Null Default Column Definition
objid Int(11)
N
o Internal MI identifier for the
object which this row describes
type Smallint(6)
N
o 0
Internal MI identifier for the
molecule type of this object;
corresponds to column type of
MI_mol_type
db Smallint(6)
N
o 0
Internal MI identifier for the
source db of this object;
corresponds to column db of
MI_obj_dbases
id Int(11) Yes
N
ull Source db id of this object
tax Int(11) Yes
N
ull
N
CBI taxonomy id of this object,
if applicable
acc Varchar(20) Yes
N
ull Source db accession of this object
rog Int(11)
N
o 0
Internal MI id of the redundant
object group to which this object
belongs
MySQL Indexes
Keyname Type Field
Primary Primary objid
seqhound@blueprint.org Version 3.3
The SeqHound Manual 293 of 421 18/04/2005
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script: N/A
Parser: MI-BIND
seqhound@blueprint.org Version 3.3
The SeqHound Manual 294 of 421 18/04/2005
MI_obj_dbases table
Last updated October 4, 2004
Database: MI
Table: MI_obj_dbases
Module: MI
Definition: Table of objects source databases
MySQL
Field Type Null Default Column Definition
db Smallint(6)
N
o Internal MI identifier for the
source db
db_name Varchar(30)
N
o The name of the object source db
MySQL Indexes
Keyname Type Field
Primary Primary db
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script: N/A
Parser: MI-BIND
seqhound@blueprint.org Version 3.3
The SeqHound Manual 295 of 421 18/04/2005
MI_mol_types table
Last updated October 4, 2004
Database: MI
Table: MI_mol_types
Module: MI
Definition: Molecular types of objects
MySQL
Field Type Null Default Column Definition
type Smallint(6)
N
o Internal MI identifier for
molecules of this type
type_name Varchar(15)
N
o
N
atural language name for this
molecule type
MySQL Indexes
Keyname Type Field
Primary Primary type
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script: N/A
Parser: MI-BIND
seqhound@blueprint.org Version 3.3
The SeqHound Manual 296 of 421 18/04/2005
MI_dbases table
Last updated October 4, 2004
Database: MI
Table: MI_dbases
Module: MI
Definition: Table of source databases for interaction records
MySQL
Field Type Null Default Column Definition
db Smallint(6)
N
o Internal MI identifier for this
source interaction database
db_name Varchar(30)
N
o
N
atural language name for this
source interaction database.
MySQL Indexes
Keyname Type Field
Primary Primary db
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script: N/A
Parser: MI-BIND
seqhound@blueprint.org Version 3.3
The SeqHound Manual 297 of 421 18/04/2005
MI_record_types table
Last updated October 4, 2004
Database: MI
Table: MI_record_types
Module: MI
Definition: Table of types for interaction records
MySQL
Field Type Null Default Column Definition
type Smallint(6)
N
o Internal MI identifier for this
record type
type_name Varchar(20)
N
o
N
atural language name for this
record type(by default, either
interaction, complex or pathway).
MySQL Indexes
Keyname Type Field
Primary Primary type
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script: N/A
Parser: MI-BIND
seqhound@blueprint.org Version 3.3
The SeqHound Manual 298 of 421 18/04/2005
MI_complexes table
Last updated October 4, 2004
Database: MI
Table: MI_complexes
Module: MI
Definition: Table of complex and pathway records
MySQL
Field Type Null Default Column Definition
compid Int(11) Internal MI identifier for this
complex record
numsubunits Int(11)
N
o 0
N
umber of subunits in this
complex
N
o
MySQL Indexes
Keyname Type Field
Primary Primary compid
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script: N/A
Parser: MI-BIND
seqhound@blueprint.org Version 3.3
The SeqHound Manual 299 of 421 18/04/2005
MI_complex2ints table
Last updated October 4, 2004
Database: MI
Table: MI_complex2ints
Module: MI
Definition: Mapping of complexes to their component interactions
MySQL
Field Type Null Default Column Definition
compid Int(11)
N
o 0Internal MI identifier for this
complex
intid Int(11)
N
o 0
Internal MI identifier for an
interaction which is a component
of this complex
MySQL Indexes
Keyname Type Field
Observation:
Source org: Blueprint
Source file:
FTP script:
BIND division files
N/A
Parser: MI-BIND
seqhound@blueprint.org Version 3.3

The SeqHound Manual 300 of 421 18/04/2005
MI_complex2subunits table
Last updated October 4, 2004
Database:
Table:
MI
MI_complex2subunits
MI
Module:
Definition: Mapping of complexes to subunits
MySQL
Field Type Null Default Column Definition
compid Int(11)
N
o 0Internal MI identifier for this
complex
objid Int(11)
N
o 0
Internal MI identifier for the
subunit object which belongs to
this complex
MySQL Indexes
Keyname Type Field
Observation:
Source org:
Blueprint
Source file: BIND division files
FTP script: N/A
Parser: MI-BIND
seqhound@blueprint.org Version 3.3

The SeqHound Manual 301 of 421 18/04/2005
MI_complex2subunits table
Last updated October 4, 2004
Database: MI
Table: MI_complex2subunits
Module: MI
Definition: Mapping of complexes to subunits
MySQL
Field Type Null Default Column Definition
compid Int(11)
N
o 0 Internal MI identifier for this
complex
objid Int(11)
N
o 0
Internal MI identifier for the
subunit object which belongs to
this complex
MySQL Indexes
Keyname Type Field
Observation:
Source org: Blueprint
Source file: BIND division files
N/A
FTP script:
Parser: MI-BIND
seqhound@blueprint.org Version 3.3

The SeqHound Manual 302 of 421 18/04/2005
MI_refs table
Last updated October 6, 2004
Database: MI
Table: MI_refs
Module: MI
Definition: Mapping of MI records to literature references which support it
MySQL
Field Type Null Default Column_Definition
uid Int(11)
N
o 0
Internal MI identifier for the
interaction record which this
reference is a part of
db Smallint(6)
N
o 0
Internal MI identifier for the
reference database which this
reference is from (eg pubmed or
medline)
acc Varchar(15) Yes
N
ull Accession of this reference from
source reference database
id Int(11) Yes
N
ull ID of this reference from the
source reference database
method Smallint(6)
N
o 0
Internal MI identifier for the
experimental method used in the
referenced experiment
MySQL Indexes
Keyname Type Field
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script: N/A
seqhound@blueprint.org Version 3.3
The SeqHound Manual 303 of 421 18/04/2005
Parser: MI-BIND
seqhound@blueprint.org Version 3.3
The SeqHound Manual 304 of 421 18/04/2005
MI_refs_db table
Last updated October 6, 2004
Database: MI
Table: MI_refs_db
Module: MI
Definition: Mapping of internal MI reference database Ids to source reference
databases
MySQL
Field Type Null Default Column_Definition
db Smallint(6)
N
o Internal MI identifier for this
reference database
db_name Varchar(15) Yes
N
ull The natural language name of the
reference source database
MySQL Indexes
Keyname Type Field
Primary Primary db
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script:
Parser:
N/A
MI-BIND
seqhound@blueprint.org Version 3.3
The SeqHound Manual 305 of 421 18/04/2005
MI_exp_methods table
Last updated October 6, 2004
Database: MI
Table: MI_exp_methods
Module: MI
Definition: Mapping of internal experimental method identifiers to their
descriptions
MySQL
Field Type Null Default Column_Definition
method Smallint(6)
N
o Internal MI identifier for this
experimental method
method_descr Varchar(40) Yes
N
ull
N
atural language na
m
e of the
experimental method
MySQL Indexes
Keyname Type Field
Primary Primary method
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script: N/A
Parser: MI-BIND
seqhound@blueprint.org Version 3.3
The SeqHound Manual 306 of 421 18/04/2005
MI_obj_labels table
Last updated October 6, 2004
Database: MI
Table: MI_obj_labels
Module: MI
Definition: Mapping of molecular objects to their free form labels
MySQL
Field Type Null Default Column_Definition
uid Int(11)
N
o 0
Internal MI identifier for the
interaction/complex/pathway
record which assigns this label to
this object
objid Int(11)
N
o 0 Internal MI identifier for the
molecular object being labeled
label Text Yes
N
ull
Free form label given to this
molecular object by this
interaction/complex/pathway
record
MySQL Indexes
Keyname Type Field
Observation:
Source org: Blueprint
Source file: BIND division files
FTP script: N/A
Parser: MI-BIND
seqhound@blueprint.org Version 3.3
The SeqHound Manual 307 of 421 18/04/2005
Text mining module
Overview:
The SeqHound text mining module helps researchers locate mentions and co-mentions of biologically related entities in the scientific
literature.
At the time of writing this is limited to finding protein mentions in PubMed abstracts. The module however has been designed to be
extensible to small molecules, complexes and even biological concepts in both abstract and full-text articles. Each of the steps in the
process may be scored by multiple methods that can be developed internally or by external developers.
mother parser
Note that a number of the tables in the text module are created at the same time as the creation of the other core tables (see core.sql)
and some of these tables are populated by the mother parser (see table descriptions below). Mother, is used to retrieve protein names
and synonyms from the RefSeq database, so these tables are created at the same time as other core module tables. The mother parser
is described under the core module section above. Parsers specific to the text module are described below.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 308 of 421 18/04/2005
text searcher parser
last updated February 25th, 2005
purpose:
The text update parser and related programs are used to collect bionames, search against a literature database, and then investigate co-
mentions of bioentities in the literatures. The co-mentions of bioentities are scored using pattern recognition and statistical machine
learning methods to look for potential biophysical interactions between bioentities. The text mining module tables are updated daily.
logic:
Most of the update logic is implemented in the Text.pm Perl module and other scoring programs. The text update parser calls the
functions in Text.pm and other Perl modules. The text mining module depends on the CORE module to generate and update names of
proteins and on the MyMED in house literature database. These resources are updated daily. The steps taken to update the text-mining
module are as follows:
Step 1: Collect bioentity names from the lexicon
Step 2: Formulate searches; collect search results and scores
Step 3: Collect co-occurrences of names and scores
Step 4: Summarize evidence for each pair of bioentities and scores
module: text
input files:
Latest Medline release: ftp://ftp.ncbi.nih.gov/nlmdata/.medlease/*.xml.gz
Latest PubMed Central release: ftp://ftp.ncbi.nlm.nih.gov/pub/pmc/
Latest Biomed Central release: ftp://ftp.biomedcentral.com/
Latest NRC LitMiner SVM release: http://ii200.iit.nrc.ca/~martinj/
British National Corpus: ftp://ftp.itri.bton.ac.uk/bnc/
seqhound@blueprint.org Version 3.3

The SeqHound Manual 309 of 421 18/04/2005
Moby project English word list: http://www.dcs.shef.ac.uk/research/ilash/Moby/
PubMed help stop word list: http://www.ncbi.nlm.nih.gov/entrez/query/static/help/pmhelp.html#Stopwords
Smart English stop word list: ftp://ftp.cs.cornell.edu/pub/smart/english.stop
tables altered:
text_bncorpus, text_db, text_doc, text_docscore, text_doctax, text_englishdict, text_evidence, text_evidencescore, text_method,
text_namepair, text_namepairresult, text_organism, text_pattern, text_point, text_pointscore, text_result, text_resultscore, text_rng,
text_rngscore, text_search, text_searchscore, text_stopword
source code location:
slri/seqhound/text/text_update.pl
config file dependencies:
slri/seqhound/text/text.ini
command line parameters:
Typing “./text_update.pl -“ or “perl text_update.pl -” at the command line while in the directory where
text_update.pl resides will return a list of command line parameters and default settings.
text_update.pl arguments:
-r redo all name searches against new insertions from MyMED using time stamp [T/F] optional
-t taxnomy ids for organism to be searched, seperated by comma. If specified in command line, only provided
organism(s) will be searched. Otherwise, a default list of taxids stored in text.ini file will be searched.
Optional
-u update target table only, default is all. Optional
seqhound@blueprint.org Version 3.3
The SeqHound Manual 310 of 421 18/04/2005
example use:
For example:
>./text_update.pl -r F –t 4932,10090,9606 -u text_searchscore
associated scripts:
slri/seqhound/text/text.sql Data Definition file for text mining module tables.
slri/seqhound/text/text_dump.sh Mysql dump script for portable tables.
slri/seqhound/text/text_create.sh Script used to create Text database, the starting point.
slri/seqhound/text/text_regex.pl Script used to score evidence using regex patterns.
slri/seqhound/text/Text.pm contains most functions that will be using for update.
slri/seqhound/text/Pattern.pm Perl module used to represent regular expression objects.
slri/seqhound/text/Tee.pm Perl module used to branch the output to different outputs.
slri/seqhound/text/text_updatecron.sh Text module daily update cron script.
slri/seqhound/text/myeutils.pl Entrez eutil script used for comparing results between MyMED and entrez searches.
error and run-time logs:
Errors and runtime logs will be directed to file specified in text.ini , current default log file name is text_update.log
Daily update log will also be send to email account specified in text.ini file
troubleshooting:
Check the email message sent by text_update.pl to see if there is any error during update. Consult update log file to look for detail
problem.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 311 of 421 18/04/2005
additional info:
All text mining module tables are in small cases, and are prefixed with "text_". Tables with auto incremented rowid have a primary
key "id" as default. When referencing primary key in other table, the field name in the referencing table will be the referenced table
name plus id.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 312 of 421 18/04/2005
yeastnameparser.pl parser
Last updated September 27, 2004
purpose:
The yeastnameparser extracts names from the SGD file SGD_features.tab. Only names that belong to yeast records that are already in
RefSeq are added, as determined using the DBXRef module. This means that this parser MUST be run after the DBXRef parsers.
It is not necessary to run this parser, if yeast names are not desired.
logic:
The yeastnameparser reads through each record in SGD_features.tab. It searches for a refseq cross reference for each record. If one is
found, then the parser gets the relevant bioentityid and checks all names for that bioentityid.
If the name does not already exist in the database, then it is added and the db field is set to “sgd” and the access field is set to the
SGDID. The action field is set to 1, ADD and the current data is written to actiondate.
Existing names in the database are also compared to the file. Names which are no longer present in the yeast file are marked as deleted
(action =2) and the current data is written to actiondate. If the name already exists in refseq, then a SECONDREFS record is filled out
for the yeast record.
module: names
input files:
SGD_features.tab from
ftp://genome-ftp.stanford.edu/pub/yeast/data_download/chromosomal_feature/
tables altered:
bioentity, bioname, secondrefs
source code location:
slri/seqhound/names/yeastnameparser.pl
config file dependencies:
The relevant configuration files are:
.intrezrc and .odbc.ini should be set up as described above for seqhound. The values will be read by shconfig.pm, which should be
located in the same directory as yeastnameparser.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 313 of 421 18/04/2005
command line parameters:
None.
example use:
perl yeastnameparser.pl
associated scripts:
The program yeastnamecron.pl can be used for both the initial read of SGD_features.tab and updates of the file. yeastnamecron.pl
checks whether SGD_features.tab needs updating, downloads it and calls yeastnameparser.pl. SGD_features.tab is updated weekly by
SGD.
error and run-time logs:
yeastnameparser writes errors to a file called yeastname.log. Updates are written to a file called yeastupdate.log as a tab delimited file
where the fields are: name, sgdid, bionameid and field. Additions are written to a file called yeastadd.log as a tab delimited file where
the fields are:name, sgdid bioentityid and field.
troubleshooting:
additional info:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 314 of 421 18/04/2005
text_bioentity table
Last updated Febuary 15, 2004
Database: SeqHound
Table: text_bioentity
Module: text (note mother parser is part of the core module)
Definition: A bioentity refers to any biological object with names that may be used in the literature to refer to these objects.
Currently, all bioentities are proteins from RefSeq. This table tells us which database contains the primary record
for this bioentity, the accession and the field which refers to this bioentity. At the moment, all bioentities are
obtained from RefSeq. RefSeq was chosen because it represents a high quality database of non-redundant records.
The intention of this part of the core is to collect a non-redundant list of biological objects and the names that are
used in written language to refer to them by.
Source file: *.bna.gz from ftp://ftp.ncbi.nih.gov/refseq/release/complete
FTP script: asnftp.pl
Parser: mother
seqhound@blueprint.org Version 3.3

The SeqHound Manual 315 of 421 18/04/2005
text_bioentity table
Field Type Null Default Column_Definition Example Source API
id
int(11) no
Auto incremented id. A
unique identifier for this
bioentity.
1 mother parser
autoincrements this
field
SHoundBioentityIdFromGi
SHoundBioentityIdFromAcc
SHoundBioentityIdListFromBionameAndTaxId
bioentitytypeid int(11)
N
o
0
Bioentity type (for
example, molecule type).
Id from the bioentitytype
table.
1 mother parser (see
GetType()) parsing
and see bioseq->mol
SHoundBioentityFromBioentityId
db varchar(15)
N
o
The primary database
where this bioentity was
found.
ref mother parser inserts
“ref”
SHoundBioentityFromBioentityId
access varchar(20)
N
o
Accession in the primary
database. Any
alphanumeric identifier
used by the primary
database.
N
P_858066 mother parser (see
FillBionameDB())
and see bioseq--
>seqid
SHoundBioentityFromBioentityId
identifier int(11) Yes
N
umeric identifier for this
bioentity in the primary
database; for example an
N
CBI Gene Info identifier
(GI). This field is not
required.
31982991 mother parser (see
GetGI) see bioseq--
>seqid (choice 12)).
SHoundBioentityFromBioentityId
fieldtypeid int(11)
N
o
A number that represents
the field from which this
bioentity was derived.
This is the id from the
fieldtype table. For
example, 1 indicates the
ASN.1 path
“seqentry/seqset/bioseq”
1 mother parser (see
ASN.1 path)
SHoundBioentityFromBioentityId
seqhound@blueprint.org Version 3.3

The SeqHound Manual 316 of 421 18/04/2005
text_bioentity indices
Keyname Type Field
PRIMARY PRIMARY access
ibioe_id INDEX id
ibioe_type INDEX bioentitytypeid
ibioe_identifier INDEX identifier
seqhound@blueprint.org Version 3.3

The SeqHound Manual 317 of 421 18/04/2005
text_bioname table
Last updated February 15, 2005
Database: SeqHound
Table:
Module:
text_bioname
text (note that the mother parser is part of the core module)
Definition: This table holds names for bioentities. A Bioname is some “name” applied to some bioentity.
Source org: NCBI
Source file: *.bna.gz from ftp://ftp.ncbi.nih.gov/ncbi/refseq/release/complete
FTP script: asnftp.pl
Parser: mother
seqhound@blueprint.org Version 3.3

The SeqHound Manual 318 of 421 18/04/2005
text_bioname table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o A unique identifier for this
bioentity-name pair.
1 mother parser
auto increments
this column
SHoundBionameListFromBioentityId
timestamp datetime Yes
N
ULL mother parser
yeastnameparse
r
bioentityid int(11)
N
o
0The id of the bioentity to which
this name refers.
1 mother parser
yeastnameparse
r
SHoundBionameListFromBioentityId
name text
N
o
mother parser
The name.
2-
isopropylmal
ate synthase
yeastnameparse
r
SHoundBionameListFromBioentityId
nametypeid int(11)
N
o
0
Type of name. For example,
protein name (1) or gene name
(2). This is the id from the
nametype table.
1 mother parser
yeastnameparse
r
SHoundBionameListFromBioentityId
db varchar(15)
N
o
The database in which this name
was found.
ref mother parser
yeastnameparse
r
SHoundBionameListFromBioentityId
access varchar(20)
N
o The accession of the record in
which this name was found.
NP_047187 mother parser
yeastnameparse
r
SHoundBionameListFromBioentityId
identifier int(11) Yes
N
ULL
The identifier of the record in
which this name was found; for
example a Gene Info identifier
(GI).
10954458 mother parser
yeastnameparse
r
SHoundBionameListFromBioentityId
fieldtypeid int(11)
N
o
0
The field of the record in which
this name was found. This is the
id from the fieldtype table; for
example, 5 indicates the
ASN.1 path
“seqentry/seqset/bioseq/descr-
title/”
5 mother parser
yeastnameparse
r
SHoundBionameListFromBioentityId
seqhound@blueprint.org Version 3.3

The SeqHound Manual 319 of 421 18/04/2005
official
int(11) Yes 0 Is this an official name?
1=Yes, 2=N0
1 yeastnameparse
r
SHoundBionameListFromBioentityId
deprecated int(11) Yes 0 Has this name been deprecated?
N
ot used at present.
0 SHoundBionameListFromBioentityId
datedeprecated datetime Yes
000000000
00000
The date the name was
deprecated.
00000000000
000
mother parser
yeastna
m
eparser
SHoundBionameListFromBioentityId
ruleid int(11) Yes
N
ULL
What rule was used to construct
this name? This is the id from the
rules table. For example, 1
indicates that A gene name is
being used to refer to a protein.
1 mother parser SHoundBionameListFromBioentityId
yeastnameparser
action
char(1) Yes A
What was the last action taken on
this record. A=ADD,
D=DELETE.
mother parser
yeastnameparse
r
actiondate datetime Yes
000000000
00000 Date of the last action. 2004-08-20
01:30:08
mother parser
yeastnameparser
seqhound@blueprint.org Version 3.3
The SeqHound Manual 320 of 421 18/04/2005
text_bioname indices
Keyname Type Field
ibioname_id INDEX id
ibioname_identifier INDEX identifier
ibioname_access INDEX access
ibioname_bioentityid INDEX bioentityid
ibioname_nametypeid INDEX nametypeid
ibioname_official INDEX official
ibioname_deprecated INDEX deprecated
ibioname_ruleid INDEX ruleid
ibioname_action INDEX action
ibioname_actiondate INDEX actiondate
seqhound@blueprint.org Version 3.3

The SeqHound Manual 321 of 421 18/04/2005
text_secondrefs table
Last updated February 15, 2005
Database: SeqHound
Table: text_secondrefs
Module: core
Definition: Additional references for sources of bionames.
Source org: SGD
Source file: SGD_features.tab from ftp://genome-
ftp.stanford.edu/pub/yeast/data_download/chromosomal_feature/
FTP script: yeastnamecron.pl
Parser: yeastnameparser.pl
seqhound@blueprint.org Version 3.3

The SeqHound Manual 322 of 421 18/04/2005
text_secondrefs table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o
id of the second reference. Mysql
auto-increment column.
1 yeastnameparser
will Auto-
increment this
column
timestamp datetime Yes
N
ULL yeastnameparse
r
bionameid int(11)
N
o
0id of the name that this reference
refers to.
1 yeastnameparser
retrieves this from
text_bioname table
db varchar(15)
N
o
Database in which this reference is
found.
sgd yeastnameparser
(see
AddSeconfRef())
access varchar(20) Yes
N
ULL Accession of the record that refers
to the name.
S000033 yeastnameparser
(see
AddSeconfRef())
fieldtypeid int(11)
N
o
0Identifies the field in the record
where this name was found.
101 yeastnameparser
(see
AddSeconfRef())
seqhound@blueprint.org Version 3.3
The SeqHound Manual 323 of 421 18/04/2005
text_secondrefs indices
Keyname Type Field
isecondrefs_id INDEX id
isecondrefs_bionameid INDEX bionameid
isecondrefs_dbsearch INDEX access, db
isecondrefs_field INDEX field
seqhound@blueprint.org Version 3.3

The SeqHound Manual 324 of 421 18/04/2005
text_bioentitytype table
Last updated February 15, 2005
Database: SeqHound
Table: text_bioentitytype
Module: text (note that this table is currently defined when the core module is
created)
Definition: Look up table that stores bioentity types.
Source org: Blueprint
Source file: core.sql
FTP script: NA
Parser: NA
text_bioentitytype table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o Identifier for a bioentitytype.
Mysql auto-increment column.
1 core.sql
type varchar(80)
N
o The molecule type. protein core.sql
text_bioentitytype indices
NA
seqhound@blueprint.org Version 3.3

The SeqHound Manual 325 of 421 18/04/2005
text_fieldtype table
Last updated February 15, 2005
Database: SeqHound
Table: text_fieldtype
Module: text (note that this table is defined when the core module is created)
Definition: Look up table that stores the field that contains the name.
Source org: Blueprint
Source file: This is a look up table that is filled by core.sql
FTP script: NA
Parser: NA
text_fieldtype table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o Identifier for a fieldtype. 1 core.sql
pathtofield varchar(80)
N
o
The field that contains the name.
For an ASN.1 record, this is the
ASN.1 "path" to the field that
contains the name. For a flat file,
this is of the form "Column
#:Column Name".
seqentry/seq
set/bioseq/s
eq-
annot/seqfea
t-gene/syn
core.sql
text_fieldtype indices
NA
seqhound@blueprint.org Version 3.3
The SeqHound Manual 326 of 421 18/04/2005
text_nametype table
Last updated February 15, 2005
Database:
Table:
SeqHound
text_nametype
Module: text (note that this table is defined when the core module is created)
Definition:
Look up table that stores name types.
Source org: Blueprint
Source file: This is a look up table that is filled by core.sql
FTP script: NA
Parser: NA
text_nametype table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o
Identifier for a nametype.
Mysql auto-increment
column.
1 core.sql
type varchar(880)
N
o The type of name.
p
rotein core.sql
text_nametype indices
NA
seqhound@blueprint.org Version 3.3

The SeqHound Manual 327 of 421 18/04/2005
text_rules table
Last updated September 30, 2004
Database: SeqHound
Table: text_rules
Module: text (note that this table is defined when the core module is created)
Definition: Look up table that stores rules for generating names.
Source org: Blueprint
Source file: This is a look up table that is filled by core.sql
FTP script: NA
Parser: NA
text_rules table
Field Type Example Null Default Column_Definition Source API
id int(11) Identifier for a rule. Mysql auto-
increment column.
core.sql
type varchar(80) The rule. Use gene name
for protein.
core.sql
N
o 1
N
o
text_rules indices
NA
seqhound@blueprint.org Version 3.3

The SeqHound Manual 328 of 421 18/04/2005
text_db table
Last updated February 15, 2005
Database: seqhound
Table: text_db
Module: text
Definition: Lookup table that lists biomedical literature databases used by the text
module.
Source file:
Source org: Blueprint
FTP script: NA
Parser: NA
text_db table
Field Type Null Default Column Definition Example Source API
dbid int(11)
N
o Auto incremented id 1 text.sql
name varchar(80)
N
o name of database PubMed text.sql
text_db indices
Keyname Type Field
PRIMARY PRIMARY dbid
seqhound@blueprint.org Version 3.3

The SeqHound Manual 329 of 421 18/04/2005
text_doc table
Last updated February 16, 2005
Database: seqhound
Table: text_doc
Module: text
Definition: This table lists the accession ids from each literature database and
assigns them an internal document id.
Source file:
ftp://ftp.ncbi.nlm.nih.gov/pub/pmc/
Source org: NLM MEDLINE and other biomedical literature databases
FTP script: slri/medline/updates/updatecron.sh
slri/medline/pubmedcentral/pmc_updatecron.sh
Parser: text_update.pl
ftp://ftp.nlm.nih.gov/nlmdata/.medlease/medline*.xml.gz
seqhound@blueprint.org Version 3.3
The SeqHound Manual 330 of 421 18/04/2005
text_doc table
Field Type Null Default Column Definition Example Source API
docid int(11)
N
o auto incremented identifier 1077882
dbid int(11)
N
o 0 literature database id 1
accession int(11)
N
o
0
accession in literature
database such as a PubMed
identifier (PMID)
1077882 Medline Xpath:
“/MedlineCitati
on/PMID”
Or
PMC Xpath:
/art/ui[@type="
p
mid"]'
status char(10) Yes
N
ULL insert, delete or update
N
ULL
text_doc indices
Keyname Type Field
id PRIMARY id
dbid INDEX dbid
accession INDEX accession
seqhound@blueprint.org Version 3.3

The SeqHound Manual 331 of 421 18/04/2005
text_docscore table
Last updated September 28, 2004
Database: seqhound
Table:
text_docscore
Module: text
Definition: This table lists scores for documents. Multiple scores (from different
scoring methods) may be listed for each document.
Source file:
Source org: NRC
FTP script: wget –P ~/nrc -m http://ii200.iit.nrc.ca/~martinj/
slri/text/nrc.sh
Program:
LitMiner :http://textomy.iit.nrc.ca/cgi-
bin/bindpresent.cgi?qry=10747882
Notes: The current method for scoring a document is an SVM (support vector
machine) classifier implemented by National Research Council's Joel
Martin and Berry deBruijn. Detailed information can be found in the
prebind publication:
http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pubmed&pu
bmedid=12689350
seqhound@blueprint.org Version 3.3
The SeqHound Manual 332 of 421 18/04/2005
text_docscore table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o foreign key from doc table. See
docid
25
methodid int(11)
N
o
scoring method identifier. See the
text_method table. For example, 1
indicates an SVM trained to
recognize papers describing
interaction data.
1
score double
N
o
0 score -0.852876574 http://ii200.iit.nr
c.ca/~martinj/
text_docscore indices
Keyname Type Field
PRIMARY PRIMARY (docid, methodid, score)
methodid INDEX methodid
score INDEX score
seqhound@blueprint.org Version 3.3
The SeqHound Manual 333 of 421 18/04/2005
text_evidence table
Last updated February 16, 2004
Database: seqhound
Table: text_evidence
Module: text
Definition: This table assigns a unique identifier to each co-occurrence of two
bionames in the same document. A bioname points to a specific name
bioentity pair in the bioname table (this pair is repeated here). This is to
be distinguished from a pair of names that co-occur in a document; this
pair of names is identified by a namepairid and does not make any
reference to a specific pair of bioentities. This evidence is in support of
some point and is based on some name pair.
Program:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 334 of 421 18/04/2005
text_evidence table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o A unique identifier for this
p
iece of evidence.
1 Auto
incremented
identifier
docid int(11)
N
o
0the document identifier
where this evidence occurs
325387
int(11)
N
o
0search result identifier for
bioname A
2200
bioentityida int(11)
N
o 0 identifier for bioentity A 107564
nama char(80)
N
o name referring to A LEU1
resultidB int(11)
N
o
0search result identifier for
bioname B
9513
bioentityidb int(11)
N
o 0 identifier for bioentity B 109911
namb char(80)
N
o name referring to B HIS4
p
ointi
d
int(11) Yes
N
ULL
POtential INTeraction
between two bioentites that
this evidence supports
2
namepairid Yesint(11)
N
ULL
corresponding namepairid
on which this evidence is
based
10634
state smallint(6) Yes book keeping 0
resultidA
N
ULL
seqhound@blueprint.org Version 3.3

The SeqHound Manual 335 of 421 18/04/2005
text_evidence indices
Keyname Type Field
PRIMARY PRIMARY evidenceid
docid INDEX docid
resultida INDEX resultidA
resultidb INDEX resultidB
seqhound@blueprint.org Version 3.3
The SeqHound Manual 336 of 421 18/04/2005
text_evidencescore table
Last updated February 16, 2005
Database: seqhound
Table: text_evidencescore
Module: text
Definition: This table assigns a score to a piece of evidence. See evidence table.
Multiples cores may be assigned to one evidence if different methods
were used. Currently, this table stores potential protein-protein
interaction scores for each co-occurrence of bionames. The current
scoring method uses a set of manually collected regular expression
patterns to identity interactions. Detailed information can be found at:
ftp://ftp.blueprint.org/pub/BIND/PreBIND/README
Program:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 337 of 421 18/04/2005
text_evidencescore table
Field Type Null Default Column_Definition Example Source API
evidenceid int(11)
N
o 0 see text_evidence table 8
methodid int(11)
N
o 0
scoring method identifier
(see text_method table); for
example, 2 indicates a
method that uses regular
expressions to detect
bioentities that are
p
hysically interacting
2
score double
N
o 0.63 0 the score
text_evidencescore indices
Keyname Type Field
PRIMARY PRIMARY (evidenceid, methodid, score)
methodid INDEX methodid
score INDEX score
seqhound@blueprint.org Version 3.3
The SeqHound Manual 338 of 421 18/04/2005
text_method table
Last updated March 15, 2005
Database: seqhound
Table: text_method
Module: text
Definition: This table assigns a unique identifier to each of the multiple scoring
schemes used in the SeqHound text mining module.
Source: This file is hand-edited.
Source Org.
Blueprint
seqhound@blueprint.org Version 3.3

The SeqHound Manual 339 of 421 18/04/2005
text_method table
Field Type Default Source Null Column_Definition Example API
id int(11)
N
o 0 Auto incremented identifier 1
type varchar(30)
N
o
This field describes the type of
score that is generated by the
method. It will be one of
searchscore, docscore, resultscore,
evidencescore or pointscore.
searchscore
hypothid
int(11) Yes 0
corresponding hypothesis
identifier. See the text_hypoth
table.
23
hypoth text Yes
This document
describes
biophysical
interaction data for
some molecule(s).
method text Yes
N
ULL
a more detailed description of the
method with pointers to more
details about the method and its
performance.
A support vector
machine was
trained to recognize
abstracts containing
biophysical
interaction data.
See PubMed
Identifier 12689350
for more details.
positive text Yes
N
ULL
the value (range) of scores
corresponding to the hypothesis
being found TRUE
>0
negative text Yes
N
ULL
the value (range) of scores
corresponding to the hypothesis
being found FALSE
<0
N
ULL a hypothesis that this method
attempts to support or refute.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 340 of 421 18/04/2005
undecided text Yes
N
ULL
the value (range) of scores
corresponding to the hypothesis
being found UNDECIDED
0
implemented enumerated Yes
N
ULL has this method been implemented
N
O
assume text Yes
N
ULL
the score value that can be
assumed if the method has been
implemented but no score is found
>0
script text Yes
N
ULL the script or program that
implements the method
text_search.pl
text_method indices
Keyname Type Field
methodid PRIMARY methodid
seqhound@blueprint.org Version 3.3

The SeqHound Manual 341 of 421 18/04/2005
text_point table
Last updated February 16, 2005
seqhound
Database:
Table: text_point
Module: text
Definition: A POINT represents two bioentities for which some POtential
INTeraction may occur in the literature. Each POINT may be supported
by multiple pieces of evidence in the evidence table.
Program:
text_point table
Field Type Column_Definition Example API Null Default Source
id int(11)
N
o auto incremented identifier 1
bioentityidA int(11)
N
o 0 bioentity identifier 110070
bioentityidB int(11)
N
o 0 bioentity identifier 1268438
small int Yes
N
ULL book keeping 0state
text_point indices
Keyname Type Field
tid PRIMARY id
bioentityida INDEX bioentityida
bioentityidb INDEX bioentityidb
seqhound@blueprint.org Version 3.3
The SeqHound Manual 342 of 421 18/04/2005
text_pointscore table
Last updated February 16, 2005
Database: seqhound
Table: text_pointscore
Module: text
Definition: This table lists scores for potential interactions. These scores may be
viewed as a summary of the scores for all the pieces of evidence that
support this POINT. A POINT may have multiple scores that are
generated by multiple methods.
Program:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 343 of 421 18/04/2005
text_pointscore table
Field Type Null Default Column_Definition Example Source API
p
ointi
d
int(11)
N
o e
POINT (see text_point table)
0a unique identifier for som 11
methodid
an identifier for the method
used to generate this score
for the POINT (see
text_method table).
3
score double
N
o 0 the score 0.63
int(11)
N
o 0
text_pointscore indices
Ty Field
PRIMARY PRIMARY
Keyname
(pointid, methodid, score)
methodid
pe
INDEX methodid
score score INDEX
p
ointi
d
INDEX
p
ointi
d
seqhound@blueprint.org Version 3.3
The SeqHound Manual 344 of 421 18/04/2005
text_result table
Last updated February 16, 2004
Database: seqhound
Table: text_result
Module: text
Definition: This table stores all the search results (document ids) for performed
searches (see text_search table). Position default is 0, which means that
the bioname appears somewhere in the document without specifying
exactly where or how many times.
Program:
text_result table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o auto incremented identifier 1
searchid int(11)
N
o
0identifies the search that
generated this result
1
docid int(11)
N
o
0the document identifier
where the name was found
7874750
p
ositioni
d
int(11)
N
o
0
the postion id in the
document where the name
appears (0 indicates no
specified position)
0
state smallint(6) Yes
N
ULL book keeping 0
text_result indices
Keyname Type Field
tid PRIMARY id
seqhound@blueprint.org Version 3.3
The SeqHound Manual 345 of 421 18/04/2005
searchid INDEX searchid
docid INDEX docid
p
ositione
d
INDEX
p
ositioni
d
seqhound@blueprint.org Version 3.3
The SeqHound Manual 346 of 421 18/04/2005
text_resultscore table
Last updated February 16, 2005
Database: seqhound
Table: text_resultscore
Module: text
Definition: This table holds scores for search results; i.e., is the searched-for
bioentity really mentioned in the document. This table might be used to
store disambiguation scores for search results. Because a bioname can
refer to many different bioentities, an algorithm (some method) )may be
used to determine which bioentity a name occurrence refers to. This
table might also hold the results of methods that disambiguate
bioentities that have English words as names.
Program:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 347 of 421 18/04/2005
text_resultscore table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o 0 Auto incremented identifier 1
methodid int(11)
N
o
0
identifier for the method
used to score this result (see
text_method table)
3
score double
N
o 0 the score -1
text_resultscore indices
Keyname Type Field
PRIMARY PRIMARY (resultid, methodid, score)
resultid INDEX resultid
methodid INDEX methodid
score INDEX score
seqhound@blueprint.org Version 3.3
The SeqHound Manual 348 of 421 18/04/2005
text_search table
Last updated February 16, 2004
Database: seqhound
Table: text_search
Module: text
Definition: This search table is generated using the text_bioname table. A search is
minimally composed of a name that is used to look for some bioentity
(listed in this table) using some method. Currently all bioname items
with nametypeid=2 and a taxid that can be found in the taxgi table
and are inserted into this search table for MyMedline searching.
Program:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 349 of 421 18/04/2005
text_search table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o auto incremented identifier 1
bioentityid int(11)
N
o 0 bioentity being searched for 106945
bionameid int(11)
N
o 0
b
ionameid used to search for
bioentity
414737
name char(80)
N
o name used to find mention
of bioentity
AI1
taxid int(11)
N
o
0taxonomy identifier for
bioentity (if applicable)
4932
rngid int(11) Yes
N
ULL redundant name group
identifier
70513
methodid int(11)
N
o
method used to search for
the bioentity mention using
name
1
searched datetime
N
o
0000-00-
00
00:00:00
date and time that search
was last performed
2004-12-01 10:12:22
results int(11) Yes
N
ULL number of search results
returned
17
text_search indices
Keyname Type Field
PRIMARY PRIMARY searchid
bioentityid INDEX bioentityid
bionameid INDEX bionameid
taxid INDEX taxid
seqhound@blueprint.org Version 3.3

The SeqHound Manual 350 of 421 18/04/2005
searched INDEX searched
seqhound@blueprint.org Version 3.3
The SeqHound Manual 351 of 421 18/04/2005
text_searchscore table
Last updated February 16, 2004
Database: seqhound
Table: text_searchscore
Module: text
Definition: This table holds some score that may be used to determine if a given
search strategy WILL BE informative if performed or its results are
likely TO BE informative if examined. This score might also be used to
determine whether a search strategy is to be performed at all or if some
search strategy is best left until more informative searches have been
informed. For example, if the name to be used is an English word, the
search may be scored so as to skip this search. Multiple methods (and
their scores) may be applied to a single search.
Program:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 352 of 421 18/04/2005
text_searchscore table
Field Type Null Default Column_Definition Example Source API
searchid int(11)
N
o
0identifies a search strategy (see
text_search table)
1
methodid int(11)
N
o 0 identifies the method used to score
the search strategy
score double
N
o 0 the score
text_searchscore indices
Keyname Type Field
PRIMARY PRIMARY (searchid, methodid, score)
searchid resultid INDEX
methodid INDEX methodid
score INDEX score
seqhound@blueprint.org Version 3.3
The SeqHound Manual 353 of 421 18/04/2005
text_rng table
Last updated February 16, 2004
Database: seqhound
Table: text_rng (redundant name group)
Module: text
Definition: This table groups together bionames that have equivalent names (i.e.
homonyms). This table facilitates searching by reducing the eliminating
redundant searches for the same string in the document collection. It is
thus an intermediate table in the process of creating the text_result table.
Program:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 354 of 421 18/04/2005
text_rng table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o
auto incremented identifier. This
is a unique identifier for the
redundant name group identifier.
8
name char(80)
N
o the homonym represented by this
group
SRB8
searched datetime
N
o 0000-00-00
00:00:00
when this name was last used to
search
2004-12-01 12:02:23
results Yes
N
ULL the number of documents returned
by this search
0
status char(20) Yes
N
ULL searched book keeping
text_rng indices
Keyname Type Field
Id PRIMARY id
N
ame Unique
N
ame
Searched INDEX Searched
Results Results INDEX
Status status INDEX
seqhound@blueprint.org Version 3.3
The SeqHound Manual 355 of 421 18/04/2005
text_rngresult table
Last updated February 16, 2004
seqhound
Database:
Table: text_rngresult
Module: text
Definition: This table is an intermediate step in one method for searching for
mentions of protein names in text. The table stores search results for
redundant name groups. These results are combined with the
text_doctax table to generate the final text_results table.
Program:
Note: This is an intermediate table and is not distributed as part of SeqHound.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 356 of 421 18/04/2005
text_rngresults table
Field Type Default Example Null Column_Definition Source API
id int(11)
N
o Auto incremented identifier. 1
rngid int(11)
N
o 2
0a redundant name group identifier.
See the text_rng table.
docid int(11) Yes
N
ULL a document identifier where this
name appears
45549
pmid int(11) Yes
N
ULL the corresponding PubMed
identifier where this name appears
45549
state int(11)
N
o 0 book keeping 0
text_rngresults indices
Keyname Type Field
Id PRIMARY Id
Rngid INDEX Rngid
Docid INDEX Docid
Pmid INDEX Pmid
State INDEX State
seqhound@blueprint.org Version 3.3
The SeqHound Manual 357 of 421 18/04/2005
text_doctax table
Last updated February 16, 2004
Database: seqhound
Table: text_doctax
Module: text
Definition: This table keeps a list of the organisms (by taxon identifiers) that are
described in a document.
Program:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 358 of 421 18/04/2005
text_doctax table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o Auto incremented identifier. A
unique identifier.
1
docid int(11)
N
o
0document identifier (see text_doc
table).
taxid int(11)
N
o
0
organism described in this
document (listed by NCBI
taxonomy database identifier)
text_doctax indices
Keyname Type Field
Id PRIMARY Id
Docid INDEX Docid
Taxid Index taxid
seqhound@blueprint.org Version 3.3
The SeqHound Manual 359 of 421 18/04/2005
text_organism table
Last updated February 16, 2004
Database: seqhound
Table: text_organism
Module: text
Definition: This table keeps a list of the MESH terms that are used to identify the
presence of an organism in a PubMed abstract.
Program:
Source Org: Blueprint
Source file:
Note:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 360 of 421 18/04/2005
text_organism table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o auto incremented identifier 1
taxid int(11)
N
o 0
N
CBI taxonomy identifie
r
4932
mesh timestamp
N
o mesh term found in abstracts
that describe this organism
Saccharomyces cerevisiae
searched timestamp Yes
CURRENT_TIM
ESTAMP
time that this search was last
completed
2005-02-15 15:58:38
results int(11) Yes
N
ULL number of documents
returned
N
ULL
bioentities int(11) Yes
N
ULL number of bioentities for this
organism
N
ULL
bionames int(11) Yes
N
ULL number of names for this
organism
N
ULL
maxbionameid int(11) Yes
N
ULL maximum bioname identifier
N
ULL
lastupdate timestamp Yes
0000-00-00
00:00:00
time that all all updates were
last completed
0000-00-00 00:00:00
text_organism indices
Keyname Type Field
Id PRIMARY Id
seqhound@blueprint.org Version 3.3

The SeqHound Manual 361 of 421 18/04/2005
text_englishdict table
Last updated February 16, 2004
Database: seqhound
Table: text_englishdict
Module: text
Definition: This table holds an English Dictionary from Moby project
http://www.dcs.shef.ac.uk/research/ilash/Moby/
Program:
Organization: Oxford University
File:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 362 of 421 18/04/2005
text_englishdict table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o
Auto incremented identifier.
Aunique identifier for this word-
pos combination.
1
word char(16) Yes
N
ULL the word the
pos char(10) Yes
N
ULL part of speech Det
freq int(11) Yes
N
ULL frequency in the bnc per million
words
61847
count int(11) Yes
N
ULL how many bioname identifiers
does this word refer to
1
source char(10) Yes
N
ULL
pubmed = this word is in th
pubmed stopword list
stop = present in other stopword
lists
other possibilities
pubmed
text_englishdict indices
Keyname Type Field
Id PRIMARY Id
Word Index Word
Pos Index Pos
Freq Index Freq
Count Index count
seqhound@blueprint.org Version 3.3

The SeqHound Manual 363 of 421 18/04/2005
text_bncorpus table
Last updated February 16, 2004
Database: seqhound
Table: text_bncorpus
Module: text
Definition: This table holds the British National Corpus.
Parser:
Organization: Oxford University (http://www.natcorp.ox.ac.uk/)
File: ftp://ftp.itri.bton.ac.uk/bnc/
Note: this file is not distributed with SeqHound
seqhound@blueprint.org Version 3.3

The SeqHound Manual 364 of 421 18/04/2005
text_bncorpus table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o auto incremented identifier. A
unique identifier for this word.
1
word char(160)
N
o the word the
freq int(11)
N
o 0 frequency in the corpus 6187267
pos char(16)
N
o part of speech at0
files int(11)
N
o
0number of files (out of 4120) that
this word appears in
4120
text_bncorpus indices
Keyname Type Field
Id Primary Id
Word Index Word
Freq Index Freq
seqhound@blueprint.org Version 3.3
The SeqHound Manual 365 of 421 18/04/2005
text_pattern table
Last updated February 16, 2004
Database: seqhound
Table: text_pattern
Module: text
Definition: This table holds a list of regular expressions used to detect mentions of
biophysical interactions between two given names that appear in a
sentence.
Program:
Source Org: Blueprint
Source File:
Note:
seqhound@blueprint.org Version 3.3

The SeqHound Manual 366 of 421 18/04/2005
text_pattern table
Field Type Null Default Column_Definition Example Source API
id varchar(8)
N
o a unique identifier for this regular
expression.
9920
parentid varchar(8) Yes
N
ULL
the parent identifier if this
expression is derived from
another.
99
class int(11)
N
o
0
does this expression identify an
interaction (1) or the absence of an
interaction (-1).
1
score double Yes
N
ULL score for this regex 0.85
regex text Yes
N
ULL the regular expression A(\S*\s+){0,4}\S*B(\S*\s+){0,4}\S
*heterodimer
text_pattern indices
Keyname Type Field
Id Primary Id
Parentid Index Parentid
seqhound@blueprint.org Version 3.3

The SeqHound Manual 367 of 421 18/04/2005
text_stopword table
Last updated February 16, 2004
Database: seqhound
Table: text_stopword
Module: text
Definition: This table contains a complete list of stopwords.
1.http://www.ncbi.nlm.nih.gov/entrez/query/static/help/pmhelp.html#Sto
pwords
2.ftp://ftp.cs.cornell.edu/pub/smart/english.stop
3. Manually collected from bioname table
Program:
Source Org: Various (see definition)
Source file:
Note:
seqhound@blueprint.org Version 3.3
The SeqHound Manual 368 of 421 18/04/2005
text_stopword table
Field Type Null Default Column_Definition Example Source API
id int(11)
N
o Auto incremented identifier 1
word char(16)
N
o the stop word a
source char(16) Yes Source of this stop word PubMed help
text_ indices
Keyname Type Field
Id Primary Id
Word Index word
seqhound@blueprint.org Version 3.3
The SeqHound Manual 369 of 421 18/04/2005
6. Developing for SeqHound.
Open source development.
SeqHound code is developed on a cvs tree internal to the Samuel Lunenfeld Research
Institute by the members of the Blueprint Initiative Development team. The most stable
current release is available at
ftp://ftp.blueprint.org/pub/SeqHound/
External developers are encouraged to discuss major additions or modifications to the
system with the Project Manager at seqhound@blueprint.org.
Minor additions or corrections may also submitted to seqhound@blueprint.org.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 370 of 421 18/04/2005
Code organization.
Note: The following section is under revision and will be updated shortly.
This document summarizes the contents in the directories under slri/seqhound and
slri/nblast as of June of 2003. The contents of directories are first stated and then the
purpose of the contents is given.
Under the current directory hierarchy, the seqhound directory contains the following
directories (/) and files (*):
asn/
bioperl/
build/
cgi/
config/
db2/
domains/
examples/
genomes/
go/
html/
include/
include_cxx
java/
lib/
locuslink/
parsers/
perl/
rps/
scripts/
shreadme/
shreadme_cxx/
src/
src_cxx/
taxon/
tindex/
updates/
yeast/
seqhound .mk*
seqhound_cb.mk*
seqhound_db2.mk*
seqhound_odbc.mk*
seqhound_rem.mk*
shreadme*
shreadme_cxx*
seqhound@blueprint.org Version 3.3
The SeqHound Manual 371 of 421 18/04/2005
asn/ : contains the asn specifications (*.asn) for various objects used in
seqhound, auxiliary files used by datatool & asntool (*.def), and the
scripts used by asntool and datatool to autogenerate the objects in unix
(*.sh) and windows (*.bat) platforms.
bioperl/: contains the files used in the SeqHound bioperl module
build/: contains a directory structure used to store the executables. As
executables get compiled, they will be moved into their relevant
directories inside the build directory. Executables using Codebase will
be moved to cb/, db2 executables are moved to db2/, and odbc
executables to odbc/. The appropriate directories will get created as the
need arises.
cgi/: contains the source code for the cgi and web services.
config/: the configuration files used in seqhound remote & local applications.
db2/: scripts to create the tables in db2 and redund for db2.
domains/: source code for the domain module.
examples/: source code showing how to use some of the code in seqhound, ex the
asn structures and functions, the C++ remote library.
genomes/: source code for the complete genome module.
go/: source code for the gene ontology module.
html/: documentation for SeqHound API, and some scripts for converting the
documentation to html
include/: the *.h files for SeqHound
include_cxx/: the include files for the C++ remote library
java/: source code for the SeqHound Java remote library
lib/: libraries for SeqHound are copied here once compiled.
locuslink/: source code for the locuslink module.
parsers/: source code for various parsers (mother, cbmmdb, cddb, redund,
mmdbloc)
perl/: the SeqHound perl module (deprecated in favor of bioperl)
rps/: source code for the rps module (domname, rpsdb)
scripts/: various scripts to retrieve flatfiles from NCBI, and to build SeqHound
src/: source code for SeqHound (includes db layers for GO, HIST,
LOCUSLINK, NBR, rpsdb, taxdb& core modules), the C remote API
src_cxx/: source code for the remote C++ library.
taxon/: source code for the taxon module parser.
tindex/: source code for the text indexer.
updates/: source code for the daily updates & histparser.
yeast/: source code for importing yeast GO into Seqhound (never completed).
seqhound@blueprint.org Version 3.3
The SeqHound Manual 372 of 421 18/04/2005
*.mk: various auxiliary files used by SeqHound makefiles.
shreadme*: readme files for C & C++ SeqHound
Under the current directory hierarchy, the nblast directory contains the following
directories(/) and files(*):
asn/
db/
docs/
lib/
msvc/
scripts/
src/
nblast.mk*
nblastflags.mk*
asn/: contains the ASN specifications for nblast.
db/: source code for nblast Codebase layer.
docs/: instructions for compiling nblast.
lib/: source code for the ASN nblast object.
msvc/: files for Microsoft Visual C++ project.
scripts/
:
various scripts for retrieving NCBI Blast flatfiles & setting up NBlast.
src/: core source code for nblast.
*.mk: auxiliary makefile for nblast applications.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 373 of 421 18/04/2005
Adding/Modifying a remote API function to SeqHound.
Note: This section is included for historical purposes and is being
rewritten for a future release (5.0)
Overall steps:
1. open a new Bugzilla report
2. create the database search functionality
3. create the local API call
4. create the CGI call
5. add the remote calls (C/C++/Java/Bioperl/Perl)
6. test the new functionality in each of the layers
7. write/update documentation
8. inform technical writer
9. inform tester
10. update seqrem and local library in production server
11. In some rare cases, modifications may have to be made to the underlying data table
structure, then SeqHound must be rebuilt in test and production environments before
code is checked into cvs.
12. check in the new source code
13. update API website
14. close Bugzilla report
This document only goes over steps 2-5 in detail.
seqhound@blueprint.org Version 3.3
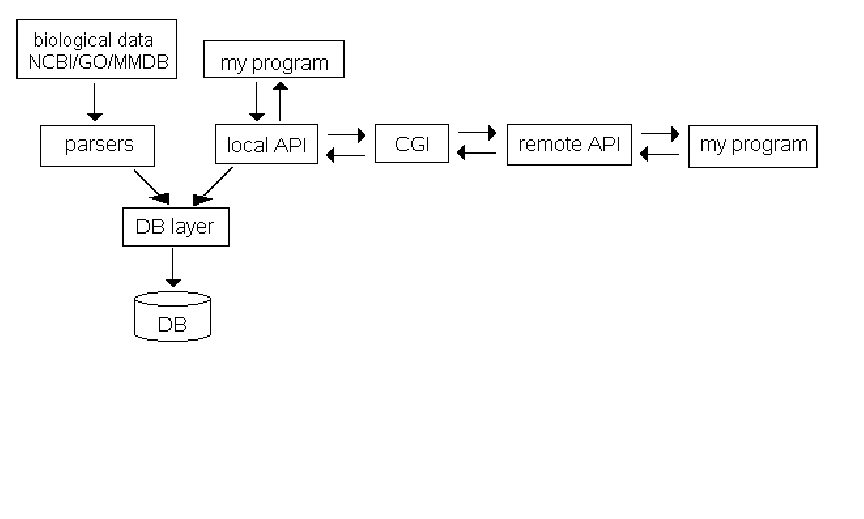
The SeqHound Manual 374 of 421 18/04/2005
Overall architecture of the SeqHound system.
The diagram shows how the various code layers in SeqHound.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 375 of 421 18/04/2005
2. Create the Database Search Functionality
If adding a new data file, you will need to write the database search functions, one for
each data file. Database Search functions are found in ‘slri/seqhound/src/*_cb.c’
files.
‘*’ is the name of the module that the searched table belongs to like ‘GO’ or ‘intrez’.
‘cb’ refers to the database engine that the function talks to like CodeBase (cb) or
ODBC compliant databases (odbc).
Functions that search data tables are typically called SearchXXX. New data files
require you to define the ASN structure, typically each field in the data file
corresponds to an ASN field.
Most of the existing databases already have search functions so you should not have
to write these yourself. If they don’t exist, then you should consult the instructions
under Creating a new SeqHound module first. The details below are here to help you
understand the database layer and to use Search functions in your API function.
Boolean LIBCALL SearchACCDB (StAccdbPtr PNTR ppac)
The search functions should find ALL instances of a key in the database and return
them through the ASN pointer. Each ASN structure is a linked list, so multiple
records can be retrieved. Pseudo code for the typical search function:
Boolean SearchXXX(ASNLinkList Pointer)
{
foreach record that matches key in Pointer
create a new ASNLinkList Node;
fill node with record fields;
join node to Pointer;
end foreach
return TRUE if key found
else return FALSE;
}
As an example, the function
Boolean LIBCALL SearchACCDB (StAccdbPtr PNTR ppac)
finds records in the accdb table for an ODBC compliant database engine. This
function is located in slri/seqhound/src/intrez_odbc.c. The corresponding function
for a CodeBase database engine has the same name but is found in
slri/seqhound/src/intrez_cb.c. The ASN structure that StAccdbPtr points to is defined
in slri/seqhound/asn/slristruc.asn (search for StAccdb) and the corresponding C
structure is defined in slri/seqhound/include/objslristruc.h. You need to know this
when you use a database search function in your local API function.
3. Create The Local API Function
You will need to change one of three files in order to add new functionality to
SeqHound.
a) go_query.c : to add functionality to gene ontology module API functions
b) ll_query.c : to add to locuslink module API functions
c) intrez.c : to add new functionality to the other data tables.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 376 of 421 18/04/2005
Declare function prototype in slri/seqhound/include/seqhound.h. Try to group the
functions logically in the .c and .h files.
The API functions follow a general logic. You should try to stick to that logic.
Typical example of an API function:
Int4 LIBCALL SHoundFindAcc(CharPtr pcAcc)
{
Int4 gi = 0;
StAccdbPtr pac = NULL, pachead = NULL;
Int2 res = 0;
if ((pcAcc == NULL) || (strcmp(pcAcc, "n/a") == 0))
{
ErrPostEx(SEV_ERROR,0,0,"SHoundFindAcc: No accession.");
Input integrity
check
Each data table has specific asn structure.
return 0;
}
pac = StAccdbNew();
pachead = pac;
pac->access = StrSave(pcAcc);
res = SearchACCDB(&pac);
if (res == FALSE)
{
ErrPostEx(SEV_ERROR,0,0,"SHoundFindAcc: Search failed.");
StAccdbFree(pachead);
Failed search.
Free structure
and return
Successful search - extract what
you need and return it.
Set the key in the asn structure
database layer to search for the key.
The asn structure will hold all the
records that match the key.
return 0;
}else if(res == TRUE){
gi = pac->gi;
pac = pac->next;
}
StAccdbFree(pachead);
return gi;
}
4. Step 4. Create the CGI Function
Definitions:
CGI (Common Gateway Interface):
a program that allows 2 computers connected to the internet to communicate with
each other.
HTTP (Hypertext Transfer Protocol):
a way of passing messages between the 2 computers. Is used for web services, eg
CGI, servlets … Format of HTTP:
http://server_name/path/to/cgi/cgi_name?key1=value1&key2=value2&….
Query string:
The portion of the HTTP call that is used to pass parameters to the CGI. Format of
query strings: ?param1=value1¶m2=value2….
SeqHound’s CGI
is slri/seqhound/cgi/seqrem.c.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 377 of 421 18/04/2005
Remote users
can call seqrem on our servers using HTTP calls. Format of a HTTP call:
http://seqhound.blueprint.org/cgi-
bin/seqrem?fnct=SeqHoundFindAcc&acc=AA73235
2 parts to edit in seqrem.c
1. if statement in function: ProcessFnctRequest(CharPtr pfnct);
2. Add a corresponding CGI function call that calls the local API
There is currently an if-else statement in the
ProcessFnctRequest function. It needs to be modified to check if
the first field of the query string contains your new function name. If so, it
will call a CGI function that calls your local API function.
SLRI_ERR ProcessFnctRequest(CharPtr pfnct)
{
if(SeqHoundInit() == SLRI_FAIL) {
MemFree(pfnct);
return 1;
} else if (strcmp(pfnct, "SeqHoundFindAcc") == 0) {
if(SeqHoundFindAcc() == SLRI_FAIL) {
MemFree(pfnct);
Calling internal CGI function
… see below
return 1;
}
} ...
} else if(strcmp(pfnc, “YourNewFunction” ..
if(SeqHoundYourNewFunction() ...
}
}
Next you need to define a function with the prototype
SLRI_ERR SeqHoundYourNewFunction(void)
This function will extract the remaining keys in the query string portion of the HTTP
call, and call the local API function you defined in step 3, then output the HTTP
response to be sent back to the user.
http://seqhound.blueprint.org/cgi-bin/seqrem?fnct=SeqHoundFindAcc&acc=AA73235
SLRI_ERR SeqHoundFindAcc(void)
{
Int4 gi = 0;
Int4 IndexArgs = -1;
printf("Content-type: text/html\r\n\r\n");
if ((IndexArgs = WWWFindName(ginfo,"acc")) >= 0)
{
pcThis = WWWGetValueByIndex(ginfo,IndexArgs);
pacc = StringSave(pcThis);
}
if ((pacc == NULL) || (strlen(pacc) == 0))
Output the HTTP
headers
Extract the remaining
fields from the CGI
ti
that calls Internal CGI function
the local API
Your new function gets
called here
{
ErrPostEx(SEV_ERROR,0,0, " Failed to get parameter value.");
fprintf(stdout, "SEQHOUND_ERROR Failed to get parameters.");
return SLRI_FAIL;
seqhound@blueprint.org Version 3.3
}
gi = SHoundFindAcc(pacc); Calling the local API
fti
The SeqHound Manual 378 of 421 18/04/2005
fprintf(stdout, "SEQHOUND_OK\n");
Send output
back to remote
users using
HTTP
fprintf(stdout, "%ld\n", (long) gi);
MemFree(pacc);
return SLRI_SUCCESS;
}
seqhound@blueprint.org Version 3.3
The SeqHound Manual 379 of 421 18/04/2005
5. Step 5. Create the Remote Calls
We have programs available in most of the widely used languages that allow SeqHound
users to write programs in their favorite language, accessing SeqHound data without
having to understand how everything works. Our remote programs in effect constructs
the HTTP calls (described above), sends the HTTP calls to the server and then parses the
server’s return value and sends this back to the user’s program.
Our remote interfaces are:
1. slri/seqhound/src/seqhoundapi.c
2. slri/seqhound/src_cxx/Seqhound.cpp
3. slri/seqhound/java/SeqHound.java
4. slri/seqhound/perl/SeqHound.pm
5. slri/seqhound/bioperl/SeqHound.pm
Int4 LIBCALL SHoundFindAcc(CharPtr pcAcc)
{
Char fpath[PATH_MAX];
Int4 gi = 0;
if(pcAcc == NULL)
Making the HTTP
call.
Contains the server
name
,
p
ath to CGI
,
{
ErrPostEx(SEV_ERROR,0,0, "Invalid parameter.");
return 0;
}
if(SHoundWWWGetfile(slri_sername, fpath) == 0)
sprintf(fpath,"%s?fnct=SeqHoundFindAcc&acc=%s", slri_cgipath, pcAcc);
ErrPostEx(SEV_INFO,0,0, "SeqHoundFindAcc request: %s.\n", fpath);
{
ErrPostEx(SEV_ERROR,0,0, "SHoundWWWGetfile failed.");
Send the HTTP
call
return 0;
}
gi = ReplyBSGetInteger();
if (gi == 0)
{
ErrPostEx(SEV_INFO,0,0, "SeqHoundFindAcc returned zero.");
Get the return
value.
A family of
ReplyBSGetXX
X exists for
return 0;
}
return gi;
}
Similar logic is used in the remote C++, Java, and Perl libraries.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 380 of 421 18/04/2005
Adding a new module to SeqHound
Note: This section is included for historical purposes and is being
rewritten for a future release (5.0)
This section describes how to go about adding a new module to SeqHound from the
developer’s point of view. It is basically a description of the different files that have to
be written for a new module and where they should go in the code tree. So this section
may be used as a guide for looking at existing modules to find out what files are expected
and where they are. However, historically, several programmers have added modules to
SeqHound and have used different code organization schemes; therefore, historical
modules may be organized differently. Going forward (as of November 2003) all
modules will have the components that are organized in the way described below.
1. Start a new module project plan
Creating a new module begins with starting a new project plan (see Format for project
reports). This should contain a clearly stated “Need” and “Objective” as well as a
“General approach” to developing the module; this may include background
information on the data resource being incorporated into SeqHound. The “Detailed
planning” section will contain a section on “Data table design” and “Code
Organization” that will be described further. See the DBXREF (Database cross-
reference) module Project Plan as an example
(./slri/seqhound_priv/dbxref/dbxref_module_desc.txt).
The components of a new module are summarized in the diagram below. The rest of
this section describes the creation of each of these components and their organization.
File names in bold italics indicate files that may be looked at as examples.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 381 of 421 18/04/2005
Database layer
2. Design the data table structure
The module’s project description should contain a description of the final table
design. See the example in the DBXREF (Database cross-reference) module Project
Plan as an example.
3. Write the script file that creates the data table.
You must modify the existing script file that creates the SeqHound data tables (this
step is only for ODBC SeqHound). This file is called seqhound.sql and is located in
./slri/seqhound/sql/seqhound.sql
Add the line
DROP TABLE SEQHOUND.DBXREF_tablename;
near the top of the file. Every time this script is run, it will destroy any pre-existing
data tables that belong to the module.
Add the lines that describe the table(s) that are a part of the module so the script
creates the new tables belonging to the module. See the example file “seqhound.sql”.
Note that CodeBase tables to not require a script to be made; these tables are created
by the function InitCodeBase in the <module_name>_cb.c file.
4. Write ASN.1 structure(s) that corresponds to data table descriptions.
The file ./slri/seqhound/asn/slristruc.asn must be modified to contain a description
of tables in the module. See the example structure StDbXref in the slristruc.asn file.
The shell script “./slri/seqhound/asn/makeasn” calls asntool which auto generates
slristruc.h, objsslristruc.h and objslristruc.c files. These files contain functions that
allow one to allocate and free memory for a structure corresponding to a database
record. See the makeasn file.
5. Step 5: Design new functions for the Database code layer
The DB layer contains C functions that create and retrieve records from tables in the
module. In the case of the DBXREF module, the tables are populated by a parser
written in PERL so there are no functions listed that write to the tables. The
requirement for a certain Perl modules is however noted in the project plan; these
modules allow Perl to converse with a database.
The DB layer will have three files that were auto generated from the preceding step.
a) ./slri/seqhound/include/slristruc.h
b) ./slri/seqhound/include/objsslristruc.h
c) ./slri/seqhound/src/objsslristruc.c
These files will be placed in this location by the makeasn script.
In addition, DB layer will require that two new files be made.
a) ./slri/seqhound/include/<module_name>_odbc.h
This will contain function prototypes and comments for functions in
seqhound@blueprint.org Version 3.3
The SeqHound Manual 382 of 421 18/04/2005
b) ./slri/seqhound/src/<module_name>_odbc.c
This will contain at least one functions called “Search<module_name> that
retrieves records from the database. Other functions that read or write to the database
may be included in this file.
The example function “SearchDBXREF” (see dbxref_odbc.h and dbxref_odbc.c)
takes as input a pointer to a structure called StDbXref. This structure has the same
fields as the DBXREF table. This structure is described in ASN.1 and functions to
allocate and free memory for this structure are auto generated using ASNtool. Any
row in a DBXREF table that contains field values matching anyone of the field values
passed to “SearchDBXREF” will be returned in a linked list of Valnodes that
contain pointers to StDBXref structures.
6. Make changes to existing code to accommodate changes to the database code layer.
You must modify the existing function called InitCodeBase so that it handles
tables that are new to the module. This function is found in two locations:
a) ./slri/seqhound/src/intrez_cb.c is the file that supports a CodeBase database
backend to SeqHound. InitCodeBase() needs to be changed to open the
code base data files that belong to the new module. See the example intrez_cb.c
file.
b) ./slri/seqhound/src/intrez_odbc.c is the file that supports a supports an ODBC
database backend to SeqHound. InitCodeBase() need to contain the
necessary code to establish a connection to the Database Server. See the example
intrez_odbc.c file.
The function InitCodeBase() under intrez_odbc.c has a function call:
GetAppParam(intrez", "datab", "db2alias", NULL,
(Char*)dsn, sizeof(char) * 10) <= 0 )
that retrieves database connection information from the
./slri/seqhound/config/.intrezrc configuration file.
Parser layer
7. Design the parser layer.
Parsers are generally written in C or Perl. A separate script is written to download
some file from an external ftp site. The parser takes this file as input and uses it to
populate a set of data tables belonging to the module.
Pseudocode for parsers should be documented in the project plan.
Parser layer code is located in
./slri/seqhound/<module_name>/[parser_name]
for example
./slri/seqhound/dbxref/dbxref_parser_sp.pl
Finally, by project end, all parsers must be documented according to the examples
given in the SeqHound manual. See the example parser description for “mother”.
seqhound@blueprint.org Version 3.3

The SeqHound Manual 383 of 421 18/04/2005
Local API layer (Query layer)
8. Design the Local API layer
Design the local API local query layer. This layer will consist of three files
a) ./slri/seqhound/src /[Module name]_query.c
This file contains all of the API functions that query the module’s tables as well
as auxiliary functions (if any). Note that this naming convention is not followed
by API calls that belong to the core module of SeqHound; code for these local
API calls that query core module tables is in ./slri/seqhound/src/intrez.c.
b) ./slri/seqhound/include/[Module name]_query.h
This file contains function prototypes and comments for auxiliary functions (if
any) that may be used by the module’s parsers, API functions or other
applications specific to the module.
c) ./slri/seqhound/include/seqhound.h
This is where all publicly available API functions are defined. Note that the local
and the remote API’s use the same header file. ALL API functions for ALL
modules are defined in this header. This file already exists and must be simply
modified.
Examples are:
./slri/seqhound/src /dbxref_query.c
./slri/seqhound/include/dbxref_query.h
./slri/seqhound/include/seqhound.h
An example of an API function is a function(s) to retrieve data base cross-references
given a source record (SHoundDBXREFGetDBXrefListBySourceRecord).
See the example in the dbxref_query.c file. This is a local API call. Notice the
naming convention: ‘Shound’ followed by the module name ‘GODB’ followed by the
actual API function name.
In the example function note the line
Note that this function calls a database layer function called
if(!SHoundModule("godb"))
This checks the SeqHound configuration file to make certain that the build of
SeqHound actually includes this module. All API function calls must have an
analogous check.
SearchDBXREF.
CGI layer
9. Design the CGI layer
The SeqHound cgi layer that supports the remote API is contained in only one file
./slri/seqhound/cgi/seqrem.c
There is no header file for seqrem.c since all functions are defined before “Main()”
and Main only calls functions in this file.
seqhound@blueprint.org Version 3.3
The SeqHound Manual 384 of 421 18/04/2005
This file must be edited to include cgi support for the remote API calls for the new
module. See the examples in the seqrem.c file. See the example function
SeqHoundDBXREFGetDBXrefListBySourceRecord.
10. Design the remote API layer
For C:
SEQH_SRC_ODBC
Note the naming convention: SeqHound followed by the module name followed by
the same function name used by the local API function.
Remote API layer
The remote API for SeqHound supports 4 languages. So there are four files that must
be modified to include new API functions for the new module.
./slri/seqhound/src/seqhoundapi.c
For C++: ./slri/seqhound/src_cxx/SeqHound.c
For Java: ./slri/seqhound/java/SeqHound.java
For Perl: ./slri/seqhound/perl/SeqHound.pm
The function names will be exactly the same as those listed in the local API layer (for
example; SHoundDBXREFGetDBXrefListBySourceRecord).
Examples for the C remote API are given in the seqhoundapi.c file.
11. Modify the seqhound config file.
New modules require that another entry be made in the .intrezrc file. This setting
allows the SeqHound administrator to indicate whether any given SeqHound module
has been built. The function SHoundModule() will look at these config file
settings every time a local API call is made to determine if the module is present.
Modify the ./slri/seqhound/config/intrezrc according to the example. Analogous
additions must be made to the ./slri/seqhound/config/intrez.ini file for Windows
platforms.
Modify the ShoundModule() function in ./slri/seqhound/src/intrez_cfg.c file to
support the new module. Follow the example in this file.
12. Modify/create make files to support the new module.
Modify the following files.
./slri/seqhound/seqhound_odbc.mk
SEQH_OBJ_OODBC
./slri/seqhound/seqhound.mk
SEQH_SRC_COM
SEQH_OBJ_COM
SEQH_ODBC_COM
seqhound@blueprint.org Version 3.3
The SeqHound Manual 385 of 421 18/04/2005
./slri/seqhound/seqhound_rem.mk
./slri/seqhound/seqhoundrem.mk??
./slri/seqhound/src/make.shoundlocllib
./slri/seqhound/src/make.shoundremlib
Create the following make files for any parsers written in C.
./slri/seqhound/<module_name>/make.[parser_name]
see the example in
./slri/seqhound/locuslink/make.llparser
13. Design regression tests
Regression tests are based on the CuTest C Unit Testing Framework.
(see http://cutest.sourceforge.net/).
One file must be modified to support tests for the new module. Follow the examples
in:
./slri/seqhound/test/regresion/main.c
And a new file must be created that contains the actual test functions. See the
examples in the test driver for the database layer for the DBXREF module:
./slri/seqhound/test/regresion/dbxref_odbc_driver.c
Examples of some test cases Function calls.
void testDBXREF_GetObjectIDbyAcc(CuTest *tc){...}
Another driver contains test functions for the
./slri/seqhound/test/regresion/dbxref_query_driver.c
Finally, the following file must be modified to accommodate the new module test
drivers. Follow the examples in:
./slri/seqhound/test/regresion/make.test_driver
14. Design test cases
These test cases refer to tests of the local and remote API functions relevant to the
module.
15. Code test and debug
16. Finish documentation
17. Compile SeqHound code in test environment
18. Build SeqHound db in test environment
19. Check in code
20. Pass the project on to delivery team (Test Dev/Systems Dev/Software Training)
21. Build module in production (SEQHOUND ADMIN)
22. Update data tables (SEQHOUND ADMIN)
seqhound@blueprint.org Version 3.3
The SeqHound Manual 386 of 421 18/04/2005
23. Update seqrem (SEQHOUND ADMIN)
24. Update docs (SEQHOUND ADMIN)
25. Update website (SEQHOUND ADMIN)
seqhound@blueprint.org Version 3.3

The SeqHound Manual 388 of 421 18/04/2005
Example GenBank record
Seq-entry ::= set {
title "Vairimorpha necatrix largest subunit of RNA polymerase II
source {
db {
std {
name {
country "UK" ,
month 4 ,
day 4 } ,
class nuc-prot ,
descr {
(RPB1)
gene, complete cds." ,
org {
taxname "Vairimorpha necatrix" ,
{
db "taxon" ,
tag
id 6039 } } , <==========================TAXGI/taxid
orgname {
name
binomial {
genus "Vairimorpha" ,
species "necatrix" } ,
lineage "Eukaryota; Fungi; Microsporidia; Burenellidae;
Vairimorpha" ,
gcode 1 ,
mgcode 1 ,
div "INV" } } } ,
create-date
std {
year 1998 ,
month 12 ,
day 10 } ,
pub {
pub {
sub {
authors {
names
{
name
name {
last "Hirt" ,
first "R" ,
initials "R.P." } } ,
{
name
last "Healy" ,
first "B" ,
initials "B." } } } ,
affil
std {
affil "The Natural History Museum" ,
div "Zoology" ,
city "London" ,
street "Cromwell Road" ,
postal-code "SW7 5BD" } } ,
medium email ,
date
std {
year 1998 ,
day 16 } } } } ,
update-date
std {
year 1999 ,
month 2 ,
pub {
pub {
article {
seqhound@blueprint.org Version 3.3
The SeqHound Manual 389 of 421 18/04/2005
title {
name "Microsporidia are related to Fungi: evidence from the
largest subunit of RNA polymerase II and other proteins." } ,
authors {
names
std {
{
name
name {
last "Hirt" ,
initials "R.P." } } ,
{
name
name {
last "Doolittle" ,
name
medline 99110933 } } ,
last "Logsdon" ,
initials "J.M." ,
suffix "Jr." } } ,
{
name
name {
last "Healy" ,
initials "B." } } ,
{
name
name {
last "Dorey" ,
initials "M.W." } } ,
{
name
name {
initials "W.F." } } ,
{
name {
last "Embley" ,
initials "T.M." } } } ,
affil
str "Department of Zoology, The Natural History Museum,
London
SW7 5BD, United Kingdom." } ,
from
journal {
title {
iso-jta "Proc. Natl. Acad. Sci. U.S.A." ,
ml-jta "Proc Natl Acad Sci U S A" ,
issn "0027-8424" ,
name "Proceedings of the National Academy of Sciences
of the
United States of America." } ,
imp {
date
std {
year 1999 ,
month 1 ,
day 19 } ,
volume "96" ,
issue "2" ,
pages "580-585" ,
language "eng" } } ,
ids {
pubmed 9892676 ,
muid 99110933 ,
pmid 9892676 } } } ,
seq-set {
<==================================beginning of bioseq
<===========================look down for end of bioseq(see ASNDB/asn1)
seq {
id {
genbank{<===============================ACCDB/DB
name "AF060234" ,<================== ======ACCDB/name
seqhound@blueprint.org Version 3.3
The SeqHound Manual 390 of 421 18/04/2005
accession "AF060234" ,<=====================ACCDB/accession
version 1 } ,<============================ACCDB/version
gi 4001823 } , <==========================Many tables/GI
descr {
molinfo {
biomol genomic } } ,
inst {
repr raw ,
mol dna ,
length 5019 ,
seq-data
ncbi2na
'171B6C0C434EFC0FBDC301F7E3FFFF3F396FF3FD7FFFF355C03BF8E03EC
10020DDB4350F6BF3FDD48203080352EDB5030D4578053A00EAF57037AEBF0C8FC00EA4
71202A7
FFCEF4D7B8008C3FF7E57AD3FE944F83C1C250EF53BEBC4E18034200DFA0EEFEFFCFBDC
87C03CB
1000DF0808FE0FFB3A0CFDC21C02EFE60AE03EA803ABF129EE80C2497B0C00082838BF8
C9FF380
A208202E3AC0B0FF83A2022F4C33DE0823EC0E08E7BBFFABFE3482FC70978BA7CFF87BD
FF2F5D7
5F7B0A5CB3ED3A0A0EF889208E3F874C0FA48CCB009C31F37C203383C82A917AD3B2C2A
3CE043C
750FD33E510E3E10E130BA5051291F4802EA657F0024FDE761E029020A98B5AA03F0EA0
02AC8FD
2E52B7B0F1978D60CFDEC820B68BD7D223E608D447F5480F3075FF0CF8597C701FB740E
A5038B1
56A983CEC3080E1A1020C87C0FD0CAA233C2F2082BCECB622113908688ECBFCFC32105F
7F1100E
24E3A74FFB58B4E829007FC8703FB7EEDD95CC19A3FE1AA180E0DF44E5902F104B0A683
C820F3B
F2C2C042CF0B510DC3014B0EA8CB1084BF049DC21FFC4F188F4FFF8DB22010E43CF33DE
C0CF0C3
CE2FC78F6D80F0F3874E3874FEB0C3F131E0837D033CE00CF03F549CFDF35C203EE87AC
040DF0B
C4FF140C43F30E80370E244182087E803B608F7C6D30F203A6037C2E83CF842027B6AD0
4429AFC
3D130F90387FAD7848B87EFDFE38E748038E0DFCFD9C430197FDCFA0FAE190C92102010
E2D0B10
20B32012702042C0E034CB4091000301C808F14ACED0E22227F80B42C0F13FE0C050884
FDE99B4
9F4037F8BFFB0C138A18EB5E928D00A74FCC0CFD10B449FBF2B4100EC828008357F6BFC
3CC8DFC
513FCB0268F3DEA037228FEC8032F3B00A0F1D7820F7DFD39CEA2B220ADC3E33E73C012
4804A33
3D020872C0243A08E71ED1DF8C8DEC228923A3F3F3433831A208EBFE3947FFC80E40038
74E386E
9C702386EDFF003F93F2C84EFC48FE0FD90C000803BC48D03F103CF871E3B03F1200FF3
38E0FE2
EBC0E00EC000CE000E0CF9CB53B0FD40830C0FC98FCC0FE3EC802884FD56CFC3FC8C7F0
00FC320
0FC50F083F3C3E033C300CC3F3630228FF0383303CD3C827CC3E8FF0080F03F00FF00D0
C3F7503
80EB291FE9A7437B2A81494540E1FC0C7F53F24A2CD240CF18EA2C508F0080F303BE600
33C21D7
ECE08FCFC02353F0C01FC80E9C200D40B83C83FD233002DDEE0FCB80FCF3857B8C8211F
7300823
023FEF4A0CFF8FD5E3813F23FCB00E5C0FCD308F003E120F03EB0B003F80F200CFB002F
C4E297F
5C33FD4CC3C8D238032D20FC30C20C23B3F4DFE0C30EC833CC3F10CC00CCF03F023CEAF
30C0342
02FFCC2E08C08408E2EB3F12123A2FECC22033DDD3503B20A13FAC1F70E3F0380CB20BF
CA0C827
9C888437E0E0F87F2F321A03A0BCED0D329134971E9E3B8E10E0A8C5F12B304213A2E0C
0BA8FEA
E47019934FE0807BA33FC721927F2C920033B0402BCC488330E3A845C957FAA12832805
E7FE8EC
2C0F230273147F70148330F3820BE3194FCC44B52ED2005E2CC24BA01E895A7CFCB20A0
3633994
0042FCCF75862561F32D4189504CCB5462541332D4109507C495842541F32D4189585C7
4504D94
DB32D610B58BC4B5042562F12D4109589CCB5042D4DF3254109537C4B504D16DF12D413
seqhound@blueprint.org Version 3.3
The SeqHound Manual 391 of 421 18/04/2005
45B7C4B
504256DF32D41095B7CCB504256DF32D41B18C38600801C3280600A024300CF0CB0000F
C00FFFF
FFEC03FFCCD035CCF8DCCBFC33C030304'H } ,
annot {
{
data
ftable {
{
data
gene {
locus "RPB1" } ,
location
int {
from 100 ,
to 4917 ,
id
gi 4001823 } } } } } } ,
<==============================the first bioseq ends here
see ASNDB/asn1
<==============================next bioseq begins here
seq {
id {
genbank {
accession "AAD12604" ,
NGPNEYP
version 1 } ,
gi 4001824 } ,
descr {
molinfo {
biomol peptide ,
tech concept-trans-a } ,
title "largest subunit of RNA polymerase II; RPO21 [Vairimorpha
necatrix]" } ,
inst {
repr raw ,
mol aa ,
length 1605 ,
topology not-set ,
seq-data
ncbieaa
"MFDEIVTKRISSIQFGLFSPEEIRKSSVVQIIHPETMENGFPKSGGLIDLKMGTTERAF
LCSSCEKDNFSCPGHFGHIELTKPMFHVGYMTKIKKILECVCFYCSRLKISTKNLKKDLNFVWNISKTKSV
CEGEIGE
NGFTGCGNKQPVIKKEGMSLIAFMKGEEESDGKVILNGERVHNILKKIVNEDAVFLGFDQKFTKPEWLILT
VLLVPPP
SVRPSIVMEGMLRAEDDLTHKLADIVKANTYLKKYELEGAPGHVVRDYEQLLQFHIATMIDNDISGQPQAL
QKSGRPL
KSISARLKGKEGRVRGNLMGKRVDFSARSVITPDPNISVEEVGVPSEIAKIHTFPEIITPFNIDRLTKLVS
GANYVIRNDGQRIDLNFNRGDIKLEEGYVVERHMQDGDVVLFNRQPSLHKMSMMAHFVRVMEGKTFRLNLS
CVSPYNA
DFDGDEMNLHMPQSYNSKAELEELCLVSKQVLSPQSNKPVMGIVQDSLTALRLFTLRDSFFDRRETMQLLY
SVNINNY
EFTDSSKLIMTHDDSFGNNLHTEESSNIMKILNFPAISYPKKLWTGKQILSYILPNTIYNGKSNEHNEEDL
ENVEDSY
VIIRNGEILSGIIDKKAVGSTQGGLIHIIANDFGPDRVTCFFDDAQKMMNLYFATINAFSIGIGDAIADKE
TMSQVQR
SIETAKEQVNEIIVKAQKNKLERLPGMSMRESFESQVNYILNKPRDISGASASKSLSFCNNMRTMVLAGSK
GSFINIS
QVTACLGQQNVEGKRIPFGFNYRSLPHFSKADYSGKSRGFVENSYVKGITPEEFFFHAMGGREGLIDIAIK
TAETGYI
QRRLVKAMEDATVTLDRSVRGADGFIYQYEYGEDGFDATFLEMQKMTHDDVATKDDVSFKNLHLVDMFTDL
NFAIKKE
NVTDQIYKLLTTDVNLQKILYDEFEWLNENVKKYEKMNIASPCNFQRIINLAIYKFDCRKGDISPYLILDT
LKNLIEN
LPIKNLLIEILIKYNLSIKRILNEYKLSLEAYNWILKEIKFKILKSIISPNEMVGTLAAQSVGEPATQMTL
NTFHLAG
VSANITMGVPRLKEIINVAKNIKTPCMKIYLKDPFNKTLEMAKKIQSELEFSDIKSLCEFSEIYYDPVIED
TSIKEDK
DFVQEYFDFPDEHLDFSKMPKFIIRLKIDRIKLVSKNLKLENIVKSLHEAFPNIFHIIRSDENSQNLIIRI
RCISSLN
seqhound@blueprint.org Version 3.3
The SeqHound Manual 392 of 421 18/04/2005
NNVEYYNLQYKNILNLKIMGYNKIKKVFISEDKDKDEWYLQTDGVCIREIFSHPNVEGHLVTSNDLNEIVE
VLGIEAA
RETILNELTLVIDGNGSYVNHRHISLLADVMTMKGYLTGITRHGVNKVGFGCTKRASFEETVDILLDAALV
AEKYVTK
GYTENIMMGHLAPLGTGIGNLLLDVSKLDKAIPLSKPEYNYEEVDTPFIHSPVSENLSISSGNWSPAYLVE
GNRYAPK
TSLYSPTSPTYSPTSPTYSPTSPTYSPTSPTYSPTSPTYSPTSPTYSPTSPSYSPTSPSYSPTSPSYSPTS
"RPO21" } } ,
data
PSYSPTS
PSYSPTSPSYSPTSPSYSPTSPSYSPTSPSYSPTSPSYSPTSPSYSPTYDNDEKKTNRKRKGKQ" } ,
annot {
{
data
ftable {
{
data
prot {
name {
"largest subunit of RNA polymerase II" ,
location
int {
from 0 ,
to 1604 ,
strand plus ,
id
gi 4001824 } } } } } } } ,
annot {
{
ftable {
{
data
cdregion {
frame one ,
code {
id 1 } } ,
product
whole
gi 4001824 ,
location
int {
from 100 ,
to 4917 ,
strand plus ,
id
gi 4001823 } } } } } }
seqhound@blueprint.org Version 3.3
The SeqHound Manual 393 of 421 18/04/2005
Example SwissProt record
Seq-entry ::= seq {
id {
swissprot {
POLYMERASE II FOR THE MRNA PRECURSOR, AND POLYMERASE III FOR 5S AND
FAMILY." ,
gi 886082 ,
str "L0001744" } ,
name "RPB1_YEAST" ,<============================ACCDB/name
accession "P04050" } ,<=========================ACCDB/accession
gi 2507347 } ,
descr { <======ACCDB/title
title "DNA-DIRECTED RNA POLYMERASE II LARGEST SUBUNIT (B220)." ,
comment "----------------------------------------------------------
-------
--~This SWISS-PROT entry is copyright. It is produced through
a~collaboration
between the Swiss Institute of Bioinformatics and~the EMBL outstation
- the
European Bioinformatics Institute.~The original entry is available
from
http://www.expasy.ch/sprot~and
http://www.ebi.ac.uk/sprot~-------------------------------------------
-------
----------------" ,
comment "[FUNCTION] DNA-DEPENDENT RNA POLYMERASE CATALYZES THE
TRANSCRIPTION OF DNA INTO RNA USING THE FOUR RIBONUCLEOSIDE
TRIPHOSPHATES AS
SUBSTRATES." ,
comment "[CATALYTIC ACTIVITY] N NUCLEOSIDE TRIPHOSPHATE = N
PYROPHOSPHATE
+ RNA(N)." ,
comment "[SUBUNIT] RNA POLYMERASE II CONSISTS OF 12 DIFFERENT
SUBUNITS.
THIS SUBUNIT IS THE LARGEST COMPONENT OF RNA POLYMERASE II." ,
comment "[SUBCELLULAR LOCATION] NUCLEAR." ,
comment "[PTM] THE TANDEM 7 RESIDUES REPEATS CAN BE HIGHLY
PHOSPHORYLATED.
THE PHOSPHORYLATION ACTIVATES POL2." ,
comment "[MISCELLANEOUS] THREE DISTINCT ZINC-CONTAINING RNA
POLYMERASES
ARE FOUND IN EUKARYOTIC NUCLEI: POLYMERASE I FOR THE RIBOSOMAL RNA
PRECURSOR,
TRNA
GENES." ,
comment "[SIMILARITY] BELONGS TO THE RNA POLYMERASE BETA' CHAIN
sp {
class standard ,
extra-acc {
"Q12364" ,
"Q92315" } ,
seqref {
gi 4397 ,
gi 4398 ,
gi 1419218 ,
gi 1419221 ,
gi 1431216 ,
gi 1431217 ,
gi 886080 ,
gi 2144431 } ,
dbref {
{
db "SGD" ,
tag
{
db "PFAM" ,
tag
str "PF00623" } ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 394 of 421 18/04/2005
{
db "PROSITE" ,
tag
str "PS00115" } } ,
keywords {
"Transferase" ,
month 11 ,
org {
db "taxon" ,
orgname {
gcode 1 ,
molinfo {
pub {
"DNA-directed RNA polymerase" ,
"Transcription" ,
"Zinc" ,
"Repeat" ,
"DNA-binding" ,
"Nuclear protein" ,
"Phosphorylation" ,
"Zinc-finger" } ,
created
std {
year 1986 ,
month 11 ,
day 1 } ,
sequpd
std {
year 1997 ,
month 11 ,
day 1 } ,
annotupd
std {
year 1999 ,
month 7 ,
day 15 } } ,
create-date
std {
year 1986 ,
day 1 } ,
update-date
std {
year 1999 ,
month 7 ,
day 15 } ,
source {
taxname "Saccharomyces cerevisiae" ,
common "baker's yeast" ,
db {
{
tag
id 4932 } } ,
name
binomial {
genus "Saccharomyces" ,
species "cerevisiae" } ,
lineage "Eukaryota; Fungi; Ascomycota; Saccharomycotina;
Saccharomycetes; Saccharomycetales; Saccharomycetaceae; Saccharomyces"
,
mgcode 3 ,
div "PLN" } } } ,
biomol peptide ,
completeness complete } ,
pub {
gen {
serial-number 1 } ,
muid 85282617 ,
article {
title {
name "Extensive homology among the largest subunits of
eukaryotic
and prokaryotic RNA polymerases." } ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 395 of 421 18/04/2005
authors {
names
std {
{
name
name {
last "Allison" ,
initials "L.A." } } ,
name
name {
issn "0092-8674" ,
language "eng" } } ,
muid 97127826 ,
{
{
last "Moyle" ,
initials "M." } } ,
{
name
name {
last "Shales" ,
initials "M." } } ,
{
name
name {
last "Ingles" ,
initials "C.J." } } } } ,
from
journal {
title {
iso-jta "Cell" ,
ml-jta "Cell" ,
name "Cell." } ,
imp {
date
std {
year 1985 ,
month 9 } ,
volume "42" ,
issue "2" ,
pages "599-610" ,
ids {
pubmed 3896517 ,
medline 85282617 } } ,
pmid 3896517 } ,
comment "SEQUENCE FROM N.A.~STRAIN=A364A" } ,
pub {
pub {
gen {
serial-number 2 } ,
article {
title {
name "Analysis of a 26,756 bp segment from the left arm of
yeast
chromosome IV." } ,
authors {
names
std {
name
name {
last "Wolfl" ,
initials "S." } } ,
{
name
name {
last "Hanemann" ,
initials "V." } } ,
{
name
name {
last "Saluz" ,
initials "H.P." } } } ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 396 of 421 18/04/2005
affil
str "Hans-Knoll-Institut fur Naturstoff-Forschung,
Department of
Cell and Molecular Biology, Jena, Germany." } ,
from
journal {
title {
iso-jta "Yeast" ,
ml-jta "Yeast" ,
issn "0749-503X" ,
name "Yeast (Chichester, England)" } ,
imp {
date
std {
year 1996 ,
month 12 } ,
volume "12" ,
issue "15" ,
pages "1549-1554" ,
language "eng" } } ,
ids {
pubmed 8972577 ,
medline 97127826 } } ,
pmid 8972577 } ,
comment "SEQUENCE FROM N.A.~STRAIN=S288C / FY1679" } ,
pub {
pub {
gen {
serial-number 3 } ,
muid 95377607 ,
article {
authors {
Urbana
6180, USA." } ,
journal {
date
title {
name "The gene encoding the biotin-apoprotein ligase of
Saccharomyces cerevisiae." } ,
names
std {
{
name
name {
last "Cronan" ,
initials "J.E." ,
suffix "Jr." } } ,
{
name
name {
last "Wallace" ,
initials "J.C." } } } ,
affil
str "Department of Microbiology, University of Illinois,
from
title {
iso-jta "FEMS Microbiol. Lett." ,
ml-jta "FEMS Microbiol Lett" ,
issn "0378-1097" ,
name "FEMS microbiology letters." } ,
imp {
std {
year 1995 ,
month 8 ,
day 1 } ,
volume "130" ,
issue "2-3" ,
pages "221-229" ,
language "eng" } } ,
ids {
pubmed 7649444 ,
medline 95377607 } } ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 397 of 421 18/04/2005
pmid 7649444 } ,
comment "SEQUENCE OF 1669-1733 FROM N.A.~STRAIN=S288C" } } ,
inst {
repr raw ,
mol aa ,
length 1733 ,
seq-data
ncbieaa
"MVGQQYSSAPLRTVKEVQFGLFSPEEVRAISVAKIRFPETMDETQTRAKIGGLNDPRLGSIDR
NLKCQTCQEGMNECPGHFGHIDLAKPVFHVGFIAKIKKVCECVCMHCGKLLLDEHNELMRQALAIKDSKKR
FAAIWTL
SIAKTLT
ETIAEAK
ASEIYYD
ids {
int {
CKTKMVCETDVPSEDDPTQLVSRGGCGNTQPTIRKDGLKLVGSWKKDRATGDADEPELRVLSTEEILNIFK
HISVKDF
TSLGFNEVFSRPEWMILTCLPVPPPPVRPSISFNESQRGEDDLTFKLADILKANISLETLEHNGAPHHAIE
EAESLLQ
FHVATYMDNDIAGQPQALQKSGRPVKSIRARLKGKEGRIRGNLMGKRVDFSARTVISGDPNLELDQVGVPK
YPEVVTPYNIDRLTQLVRNGPNEHPGAKYVIRDSGDRIDLRYSKRAGDIQLQYGWKVERHIMDNDPVLFNR
QPSLHKM
SMMAHRVKVIPYSTFRLNLSVTSPYNADFDGDEMNLHVPQSEETRAELSQLCAVPLQIVSPQSNKPCMGIV
QDTLCGI
RKLTLRDTFIELDQVLNMLYWVPDWDGVIPTPAIIKPKPLWSGKQILSVAIPNGIHLQRFDEGTTLLSPKD
NGMLIID
GQIIFGVVEKKTVGSSNGGLIHVVTREKGPQVCAKLFGNIQKVVNFWLLHNGFSTGIGDTIADGPTMREIT
KKVLDVTKEAQANLLTAKHGMTLRESFEDNVVRFLNEARDKAGRLAEVNLKDLNNVKQMVMAGSKGSFINI
AQMSACV
GQQSVEGKRIAFGFVDRTLPHFSKDDYSPESKGFVENSYLRGLTPQEFFFHAMGGREGLIDTAVKTAETGY
IQRRLVK
ALEDIMVHYDNTTRNSLGNVIQFIYGEDGMDAAHIEKQSLDTIGGSDAAFEKRYRVDLLNTDHTLDPSLLE
SGSEILG
DLKLQVLLDEEYKQLVKDRKFLREVFVDGEANWPLPVNIRRIIQNAQQTFHIDHTKPSDLTIKDIVLGVKD
LQENLLV
LRGKNEIIQNAQRDAVTLFCCLLRSRLATRRVLQEYRLTKQAFDWVLSNIEAQFLRSVVHPGEMVGVLAAQ
SIGEPAT
QMTLNTFHFAGVASKKVTSGVPRLKEILNVAKNMKTPSLTVYLEPGHAADQEQAKLIRSAIEHTTLKSVTI
PDPRSTVIPEDEEIIQLHFSLLDEEAEQSFDQQSPWLLRLELDRAAMNDKDLTMGQVGERIKQTFKNDLFV
IWSEDND
EKLIIRCRVVRPKSLDAETEAEEDHMLKKIENTMLENITLRGVENIERVVMMKYDRKVPSPTGEYVKEPEW
VLETDGV
NLSEVMTVPGIDPTRIYTNSFIDIMEVLGIEAGRAALYKEVYNVIASDGSYVNYRHMALLVDVMTTQGGLT
SVTRHGF
NRSNTGALMRCSFEETVEILFEAGASAELDDCRGVSENVILGQMAPIGTGAFDVMIDEESLVKYMPEQKIT
EIEDGQD
GGVTPYSNESGLVNADLDVKDELMFSPLVDSGSNDAMAGGFTAYGGADYGEATSPFGAYGEAPTSPGFGVS
SPGFSPT
SPTYSPTSPAYSPTSPSYSPTSPSYSPTSPSYSPTSPSYSPTSPSYSPTSPSYSPTSPSYSPTSPSYSPTS
PSYSPTS
PSYSPTSPSYSPTSPSYSPTSPSYSPTSPSYSPTSPAYSPTSPSYSPTSPSYSPTSPSYSPTSPSYSPTSP
NYSPTSP
SYSPTSPGYSPGSPAYSPKQDEQKHNENENSR" ,
hist {
replaces {
date
std {
year 1997 ,
month 10 ,
day 9 } ,
gi 133330 } } } } ,
annot {
{
data
ftable {
{
data
region "Zinc finger region" ,
comment "C2H2-TYPE (POTENTIAL)." ,
location
from 66 ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 398 of 421 18/04/2005
to 82 ,
id
gi 2507347 } ,
exp-ev not-experimental } ,
{
data
region "Domain" ,
gi 2507347 } ,
data
{
comment "CARBOXYL-TERMINAL 7-RESIDUE REPEATS." ,
location
int {
from 1543 ,
to 1718 ,
id
gi 2507347 } ,
exp-ev experimental } ,
{
data
region "Variant" ,
comment "MISSING (IN STRAIN A364A)." ,
location
int {
from 1652 ,
to 1658 ,
id
exp-ev experimental } ,
{
data
region "Conflict" ,
comment "A -> V (IN REF. 1)." ,
location
pnt {
point 1513 ,
id
gi 2507347 } ,
exp-ev experimental } ,
{
region "Conflict" ,
comment "G -> A (IN REF. 1)." ,
location
pnt {
point 1523 ,
id
gi 2507347 } ,
exp-ev experimental } ,
data
region "Conflict" ,
comment "T -> M (IN REF. 1)." ,
location
pnt {
point 1600 ,
id
gi 2507347 } ,
exp-ev experimental } ,
{
data
gene {
locus "RPB1" ,
syn {
"RPO21" ,
"RPB220" ,
"SUA8" ,
"YDL140C" ,
"D2150" } } ,
location
int {
from 0 ,
to 1732 ,
id
gi 2507347 } } ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 399 of 421 18/04/2005
{
data
prot {
name { <=============ACCDB/title
"DNA-DIRECTED RNA POLYMERASE II LARGEST SUBUNIT" } ,
ec {
"2.7.7.6" } } ,
location
int {
from 0 ,
to 1732 ,
id
gi 2507347 } } } } } }
seqhound@blueprint.org Version 3.3
The SeqHound Manual 400 of 421 18/04/2005
Example EMBL record
Seq-entry ::= set {
level 1 ,
class nuc-prot ,
descr {
pub {
pub {
gen {
cit "Unpublished" ,
authors {
names
std {
{
name
name {
last "Drebot" ,
initials "M.A." } } ,
{
name
name {
last "Jansma" ,
initials "D." } } ,
{
name
name {
last "Himmelfarb" ,
initials "H.J." } } ,
{
name
name {
last "Friesen" ,
initials "J.D." } } } } ,
title "Suppressors of yeast RNA polymerase II mutations
belong to a
family of gene products that interact with a protein kinase" } } } ,
pub {
pub {
sub {
authors {
names
std {
{
name
name {
last "Jansma" ,
initials "D." } } } ,
affil
str "David Jansma, Genetics, Hospital for Sick Children,
555
University, Avenue, Toronto, Ontario, M5G 1X8, Canada" } ,
medium other ,
date
std {
year 1992 ,
month 7 ,
day 28 } } } } ,
create-date
std {
year 1992 ,
month 12 ,
day 11 } ,
update-date
std {
year 1993 ,
month 3 ,
day 12 } ,
source {
org {
taxname "Saccharomyces cerevisiae" ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 401 of 421 18/04/2005
common "baker's yeast" ,
db {
{
db "taxon" ,
tag
id 4932 } } ,
orgname {
name
binomial {
genus "Saccharomyces" ,
species "cerevisiae" } ,
mod {
{
subtype isolate ,
subname "Mating type a" } } ,
lineage "Eukaryota; Fungi; Ascomycota; Saccharomycotina;
Saccharomycetes; Saccharomycetales; Saccharomycetaceae; Saccharomyces"
,
gcode 1 ,
mgcode 3 ,
div "PLN" } } ,
subtype {
{
subtype chromosome ,
name "7" } } } } ,
seq-set {
seq {
id {
embl {
name "SCSPM2" ,<==============================ACCDB/name
accession "Z14128" ,<========================ACCDB/accession
version 1 } ,<==================================ACCDB/version
gi 287914 } ,
descr {
title "S.cerevisiae spm2+ gene for spm2+ protein" ,
embl {
div fun ,
creation-date
std {
year 1992 ,
month 12 ,
day 11 } ,
update-date
std {
year 1993 ,
month 3 ,
day 12 } ,
keywords {
"CDC68/SPT16 protein" ,
"spm2+ gene" ,
"spm2+ protein" } ,
xref {
{
dbname
name "SGD" ,
id {
str "L0001891" ,
str "SIP2" } } ,
{
dbname
code swissprot ,
id {
str "P34164" ,
str "SIP2_YEAST" } } } } ,
molinfo {
biomol genomic } } ,
inst {
repr raw ,
mol dna ,
length 2032 ,
seq-data
ncbi2na
seqhound@blueprint.org Version 3.3
The SeqHound Manual 402 of 421 18/04/2005
'00F965EC6C171E0103E3BFD06B54022FC17F9A55FFCDD14FFD770607B4E
0107C933066C7C4C8C8F0323053933470724DB4E0C91BF973183E69693ADFF33E757E85
8B023F1
7E6FF68DB4037FCA80A303114423D7A80E00B3780C3EE7FE82AC78892E0BF73AB1C610B
4D49500
024041000BB6E45CD38B86F2200152439D0A798164203A39EE24202F1E0789F0303BD23
D50866A
925F42A2A373C50000361BCF9E6E38A18951CE502737BCE8079EE847877AB7DB71C98E3
822A233
CF9521848950908E52D78E38D9D294F7D952228A841240C6E50209F6A2B50B80D089D3C
3AF56E8
8C2BA490AEBD00BF1B84A74F4503A28038DAFE3178F78430E9D3F4EE09C29E7D4A444C8
F48FCCB
A30E27C8BCB8FD7951241E348EA01F6D0F1320B4A4148000541C180230AD428248F5398
5D51498
D8D743E77903EB0A35218FFAE1A3310AFD38237B45485D6FA0CC41213D596EFC7856DED
3A0631C
F317E8DB4902431E31B4E9E114550F175D2FA20EF37E0C0C739464214BD062041D299DE
54F5814
EEB9F051FAF1CB24F0910C47FBB25D4DBD8CC0480CED1523DFCC654C8B5DB0D6A3D71FF
C0A6CFB
333FFA49CE911C3F73F50FCBB0922332093304C38FC70EA74B1DD0E7CFBCD1014631C31
5A0B66E
37FFFFF6803883651FF706020CF80DF0CFAE208A0C02C9C222D1A8700AC00BD273D41FC
75FDE33
E45FDB81E9F6ECFC2E0CE'H ,
hist {
replaces {
date
std {
year 1993 ,
month 6 ,
day 11 } ,
ids {
gi 4525 } } } } ,
annot {
{
data
ftable {
{
data
pub {
pub {
muid 92017853 ,
article {
title {
name "CDC68, a yeast gene that affects
regulation of
cell proliferation and transcription, encodes a protein with a highly
acidic
carboxyl terminus." } ,
authors {
names
std {
{
name
name {
last "Rowley" ,
initials "A." } } ,
{
name
name {
last "Singer" ,
initials "R.A." } } ,
{
name
name {
last "Johnston" ,
initials "G.C." } } } ,
affil
str "Department of Microbiology, Dalhousie
University, Halifax, Nova Scotia, Canada." } ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 403 of 421 18/04/2005
from
journal {
title {
iso-jta "Mol. Cell. Biol." ,
ml-jta "Mol Cell Biol" ,
issn "0270-7306" ,
name "Molecular and cellular biology." }
,
imp {
date
std {
year 1991 ,
month 11 } ,
volume "11" ,
issue "11" ,
pages "5718-5726" ,
language "eng" } } ,
ids {
pubmed 1833637 ,
medline 92017853 } } ,
pmid 1833637 } } ,
location
int {
from 1282 ,
to 2031 ,
id
gi 287914 } } ,
{
data
gene {
locus "spm2+" } ,
location
int {
from 398 ,
to 1645 ,
id
gi 287914 } } } } } } ,
seq {
id {
embl {
accession "CAA78503" ,
version 1 } ,
gi 287915 } ,
descr {
title "spm2+ [Saccharomyces cerevisiae]" ,
molinfo {
biomol peptide } } ,
inst {
repr raw ,
mol aa ,
length 415 ,
seq-data
ncbieaa
"MGTTTSHPAQKKQTTKKCRAPIMSDVREKPSNAQGCEPQEMDAVSKKVTELSLNKCSDS
QDAGQPSREGSITKKKSTLLLRDEDEPTMPKLSVMETAVDTDSGSSSTSDDEEGDIIAQTTEPKQDASPDD
DRSGHSS
PREEGQQQIRAKEASGGPSEIKSSLMVPVEIRWQQGGSKVYVTGSFTKWRKMIGLIPDSDNNGSFHVKLRL
LPGTHRF
RFIVDNELRVSDFLPTATDQMGNFVNYIEVRQPEKNPTNEKIRSKEADSMRPPTSDRSSIALQIGKDPDDF
GDGYTRF
HEDLSPRPPLEYTTDIPAVFTDPSVMERYYYTLDRQQSNTDTSWLTPPQLPPQLENVILNKYYATQDQFNE
NNSGALP
IPNHVVLNHLVTSSIKHNTLCVASIVRYKQKYVTQILYTPIESS" ,
hist {
replaces {
date
std {
year 1993 ,
month 6 ,
day 11 } ,
ids {
gi 4526 } } } } ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 404 of 421 18/04/2005
annot {
{
data
ftable {
{
data
prot {
name {
"spm2+" } ,
activity {
"Wild-type version of SPM2,a dominant extragenic
suppressor of some temperature-sensitve mutations in RPO21 and PRP4."
} } ,
location
whole
gi 287915 } } } } } ,
seq {
id {
embl {
accession "CAA78504" ,
version 1 } ,
gi 4388554 } ,
descr {
title "CDC68 /SPT16 [Saccharomyces cerevisiae]" ,
annot {
prot {
data
data
molinfo {
biomol peptide ,
completeness partial } } ,
inst {
repr raw ,
mol aa ,
length 1 ,
seq-data
ncbieaa "M" } ,
{
data
ftable {
{
data
name {
"CDC68 /SPT16" } } ,
partial TRUE ,
location
whole
gi 4388554 } } } } } } ,
annot {
{
data
ftable {
{
cdregion {
frame one ,
code {
id 1 } } ,
product
whole
gi 287915 ,
location
int {
from 398 ,
to 1645 ,
id
gi 287914 } ,
dbxref {
{
db "SWISS-PROT" ,
tag
str "P34164" } } } ,
{
seqhound@blueprint.org Version 3.3
The SeqHound Manual 405 of 421 18/04/2005
cdregion {
frame one ,
code {
id 1 } } ,
partial TRUE ,
product
whole
gi 4388554 ,
location
int {
from 2029 ,
to 2031 ,
id
gi 287914 ,
fuzz-to
lim gt } ,
cit
pub {
muid 92017853 } } } } } }
seqhound@blueprint.org Version 3.3
The SeqHound Manual 406 of 421 18/04/2005
Example PDB record
Seq-entry ::= set {
class pdb-entry ,
descr {
pdb {
deposition
std {
year 1992 ,
month 4 ,
day 3 } ,
class "Isomerase(Intramolecular Oxidoreductse)" ,
compound {
"D-Xylose Isomerase (E.C.5.3.1.5) Mutant With Glu 186 Replaced
By Gln
(E186Q) Complex With Xylose And Mn" } ,
source {
"(Actinoplanes missouriensis) E186Q Mutant Gene Expressed In
(Escherichia coli)" } ,
exp-method "X-Ray Diffraction" } ,
comment "Revision History:~JUL 15 93 Initial Entry" ,
pub {
pub {
sub {
authors {
names
std {
{
name
name {
last "Janin" ,
full "J.Janin" ,
initials "J." } } } } ,
date
std {
year 1992 ,
month 4 ,
day 3 } } } } ,
pub {
pub {
muid 92304915 ,
article {
title {
name "Protein engineering of xylose (glucose) isomerase
from
title {
Actinoplanes missouriensis. 1. Crystallography and site-directed
mutagenesis
of metal binding sites." } ,
authors {
names
str {
"J.Jenkins" ,
"J.Janin" ,
"F.Rey" ,
"M.Chiadmi" ,
"H.van Tilbeurgh" ,
"I.Lasters" ,
"M.De Maeyer" ,
"D.Van Belle" ,
"S.J.Wodak" ,
"M.Lauwereys" ,
"P.Stanssens" ,
"N.T.Mrabet" ,
"J.Snauwaert" ,
"G.Matthyssens" ,
"A.-M.Lambeir" } } ,
from
journal {
seqhound@blueprint.org Version 3.3
The SeqHound Manual 407 of 421 18/04/2005
iso-jta "Biochemistry" ,
ml-jta "Biochemistry" ,
issn "0006-2960" ,
name "Biochemistry." } ,
year 1992 ,
issue "24" ,
pubmed 1610791 ,
article {
"N.T.Mrabet" ,
"H.van Tilbeurgh" ,
year 1992 ,
pubmed 1610792 ,
imp {
date
std {
month 6 ,
day 23 } ,
volume "31" ,
pages "5449-5458" ,
language "eng" } } ,
ids {
medline 92304915 } } ,
pmid 1610791 } } ,
pub {
pub {
muid 92304916 ,
title {
name "Protein engineering of xylose (glucose) isomerase
from
Actinoplanes missouriensis. 2. Site-directed mutagenesis of the xylose
binding site." } ,
authors {
names
str {
"A.-M.Lambeir" ,
"M.Lauwereys" ,
"P.Stanssens" ,
"J.Snauwaert" ,
"G.Matthyssens" ,
"I.Lasters" ,
"M.De Maeyer" ,
"S.J.Wodak" ,
"J.Jenkins" ,
"M.Chiadmi" ,
"J.Janin" } } ,
from
journal {
title {
iso-jta "Biochemistry" ,
ml-jta "Biochemistry" ,
issn "0006-2960" ,
name "Biochemistry." } ,
imp {
date
std {
month 6 ,
day 23 } ,
volume "31" ,
issue "24" ,
pages "5459-5466" ,
language "eng" } } ,
ids {
medline 92304916 } } ,
pmid 1610792 } } ,
pub {
pub {
muid 92304917 ,
article {
title {
name "Protein engineering of xylose (glucose) isomerase
from
Actinoplanes missouriensis. 3. Changing metal specificity and the pH
seqhound@blueprint.org Version 3.3
The SeqHound Manual 408 of 421 18/04/2005
profile
by site-directed mutagenesis." } ,
authors {
names
std {
{
name
name {
last "van Tilbeurgh" ,
initials "H." } } ,
{
name
name {
last "Jenkins" ,
initials "J." } } ,
{
name
name {
last "Chiadmi" ,
initials "M." } } ,
{
name
name {
last "Janin" ,
initials "J." } } ,
{
name
name {
last "Wodak" ,
initials "S.J." } } ,
{
name
name {
last "Mrabet" ,
initials "N.T." } } ,
{
name
name {
last "Lambeir" ,
initials "A.M." } } } ,
affil
str "Plant Genetic Systems N.V., Gent, Belgium." } ,
from
name "Biochemistry." } ,
journal {
title {
iso-jta "Biochemistry" ,
ml-jta "Biochemistry" ,
issn "0006-2960" ,
imp {
date
std {
year 1992 ,
month 6 ,
day 23 } ,
volume "31" ,
issue "24" ,
pages "5467-5471" ,
language "eng" } } ,
ids {
pubmed 1610793 ,
medline 92304917 } } ,
pmid 1610793 } } ,
pub {
pub {
muid 92172844 ,
article {
title {
name "Arginine residues as stabilizing elements in
proteins." } ,
authors {
names
seqhound@blueprint.org Version 3.3
The SeqHound Manual 409 of 421 18/04/2005
str {
"N.T.Mrabet" ,
"A.Van Den Broek" ,
"I.Van Den Brande" ,
"P.Stanssens" ,
"Y.Laroche" ,
"A.-M.Lambeir" ,
"G.Matthijssens" ,
"J.Jenkins" ,
"M.Chiadmi" ,
"H.van Tilbeurgh" ,
"F.Rey" ,
"J.Janin" ,
"W.J.Quax" ,
"I.Lasters" ,
"M.De Maeyer" ,
"S.J.Wodak" } } ,
from
journal {
title {
iso-jta "Biochemistry" ,
ml-jta "Biochemistry" ,
issn "0006-2960" ,
name "Biochemistry." } ,
imp {
date
std {
year 1992 ,
month 3 ,
day 3 } ,
volume "31" ,
issue "8" ,
pages "2239-2253" ,
language "eng" } } ,
ids {
pubmed 1540579 ,
medline 92172844 } } ,
pmid 1540579 } } ,
pub {
pub {
muid 89184498 ,
article {
title {
name "Structural analysis of the 2.8 A model of Xylose
isomerase
from Actinoplanes missouriensis." } ,
authors {
names
std {
{
name
name {
last "Rey" ,
initials "F." } } ,
{
name
name {
last "Jenkins" ,
initials "J." } } ,
{
name
name {
last "Janin" ,
initials "J." } } ,
{
name
name {
last "Lasters" ,
initials "I." } } ,
{
name
name {
seqhound@blueprint.org Version 3.3
The SeqHound Manual 410 of 421 18/04/2005
last "Alard" ,
initials "P." } } ,
{
name
name {
last "Claessens" ,
initials "M." } } ,
{
name
name {
last "Matthyssens" ,
initials "G." } } ,
{
name
name {
last "Wodak" ,
initials "S." } } } ,
affil
str "Laboratoire de Biologie Physicochimique, Universite
Paris
Sud, Orsay, France." } ,
from
journal {
title {
iso-jta "Proteins" ,
ml-jta "Proteins" ,
issn "0887-3585" ,
name "Proteins." } ,
imp {
date
std {
year 1988 } ,
volume "4" ,
issue "3" ,
pages "165-172" ,
language "eng" } } ,
ids {
pubmed 3237716 ,
medline 89184498 } } ,
pmid 3237716 } } } ,
seq-set {
seq {
id {
pdb {
mol "9XIM" ,<===================================ACCDB/name
chain 65 ,<====================================ACCDB/chain
rel
std {
year 1992 , ,<===========================ACCDB/release
month 4 ,
day 3 } } ,
gi 443580 } ,
descr {
// record truncated
seqhound@blueprint.org Version 3.3
The SeqHound Manual 411 of 421 18/04/2005
Example Biostruc
An example of the ASN biostruc. Some data has been removed for
the sake of brevity.
Biostruc ::= {
id {
mmdb-id 2 } ,
descr {
name "101D" ,
pdb-comment "remark 3: Refinement." ,
pdb-comment "remark 3: Program Nuclsq" ,
pdb-comment "remark 3: Authors Westhof,Dumas,Moras" ,
pdb-comment "remark 3: R Value 0.163" ,
pdb-comment "remark 3: Free R Value 0.252" ,
pdb-comment "remark 3: Number Of Reflections 2430" ,
pdb-comment "remark 3: Resolution Range 8.0 - 2.25 Angstroms" ,
pdb-comment "remark 3: Data Cutoff 2.0 Sigma(F)" ,
pdb-comment "remark 3: Number Of Protein Atoms
0" ,
pdb-comment "remark 3: Number Of Nucleic Acid Atoms
488" ,
pdb-comment "remark 3: Number Of Solvent Atoms
33" ,
pdb-comment "remark 3: Rms Deviations From Ideal Values (The Values Of" ,
pdb-comment "remark 3: Sigma, In Parentheses, Are The Input Estimated" ,
pdb-comment "remark 3: Standard Deviations That Determine The Relative" ,
pdb-comment "remark 3: Weights Of The Corresponding Restraints)" ,
pdb-comment "remark 3: Distance Restraints (Angstroms)" ,
pdb-comment "remark 3: Sugar-Base Bond Distance
0.024(0.030)" ,
pdb-comment "remark 3: Sugar-Base Bond Angle Distance
0.040(0.040)" ,
pdb-comment "remark 3: Phosphate Bond Distance
0.026(0.040)" ,
pdb-comment "remark 3: Phosphate Bond Angle Distance, H-Bond
0.057(0.050)" ,
pdb-comment "remark 3: Plane Restraint (Angstroms)
0.014(0.020)" ,
pdb-comment "remark 3: Chiral-Center Restraint (Angstroms3)
0.161(0.150)" ,
pdb-comment "remark 3: Non-Bonded Contact Restraints (Angstroms)" ,
pdb-comment "remark 3: Single Torsion Contact
0.093(0.100)" ,
pdb-comment "remark 3: Multiple Torsion Contact
0.097(0.100)" ,
pdb-comment "remark 3: Isotropic Thermal Factor Restraints (Angstroms2)" ,
pdb-comment "remark 3: Sugar-Base Bond
4.282(6.000)" ,
pdb-comment "remark 3: Sugar-Base Angle
4.990(6.000)" ,
pdb-comment "remark 3: Phosphate Bond
5.693(6.000)" ,
pdb-comment "remark 3: Phosphate Bond Angle, H-Bond
5.227(6.000)" ,
pdb-comment "remark 105: Nomenclature For Atoms Of The DeoxyriboseRIBOSE
pdb-comment "remark 106: The Hydrogen Bonds Between Base Pairs In This
pdb-comment "remark 101: Residue +c A 9 Has Br Bonded To C5." ,
pdb-comment "remark 101: Residue +c B 21 Has Br Bonded To C5." ,
pdb-comment "remark 105: The Protein Data Bank Has Adopted The Saccharide
Chemists" ,
MOIETY" ,
pdb-comment "remark 105: Rather Than That Of The Nucleoside Chemists. The
Ring" ,
pdb-comment "remark 105: Oxygen Atom Is Labelled O4 Instead Of O1." ,
Entry Follow" ,
pdb-comment "remark 106: The Conventional Watson-Crick Hydrogen Bonding
Pattern." ,
pdb-comment "remark 106: They Have Not Been Presented On Conect Records In
This" ,
pdb-comment "remark 106: Entry." ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 412 of 421 18/04/2005
history {
data-source {
name-of-database "Protein Data Bank" ,
version-of-database
release-date
std {
year 1995 ,
month 2 ,
day 28 } ,
database-entry-id
other-database {
db "PDB" ,
tag
str "101D" } ,
database-entry-date
std {
year 1994 ,
month 12 ,
day 14 } } } ,
attribution
sub {
authors {
names
std {
{
name
name {
last "Goodsell" ,
full "D.S.Goodsell" ,
initials "D.S." } } ,
{
name
name {
last "Kopka" ,
full "M.L.Kopka" ,
initials "M.L." } } ,
{
name
name {
last "Dickerson" ,
full "R.E.Dickerson" ,
initials "R.E." } } } } ,
imp {
date
std {
year 1994 ,
month 12 ,
day 14 } } } ,
attribution
gen {
cit "To Be Published" ,
authors {
names
std {
{
name
name {
last "Goodsell" ,
full "D.S.Goodsell" ,
initials "D.S." } } ,
{
name
name {
last "Kopka" ,
full "M.L.Kopka" ,
initials "M.L." } } ,
{
name
name {
last "Dickerson" ,
full "R.E.Dickerson" ,
initials "R.E." } } } } ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 413 of 421 18/04/2005
title "Refinement Of Netropsin Bound To Dna: Bias And Feedback In
Electron Density Map Interpretation" } ,
attribution
equiv {
muid 85264810 ,
article {
title {
name "Binding of an antitumor drug to DNA, Netropsin and
C-G-C-G-A-A-T-T-BrC-G-C-G." } ,
authors {
names
std {
{
name
name {
last "Kopka" ,
initials "M.L." } } ,
{
name
name {
last "Yoon" ,
initials "C." } } ,
{
name
name {
last "Goodsell" ,
initials "D." } } ,
{
name
name {
last "Pjura" ,
initials "P." } } ,
{
name
name {
last "Dickerson" ,
initials "R.E." } } } } ,
from
journal {
title {
iso-jta "J. Mol. Biol." ,
ml-jta "J Mol Biol" ,
issn "0022-2836" ,
jta "J6V" } ,
imp {
date
std {
year 1985 ,
month 6 ,
day 25 } ,
volume "183" ,
issue "4" ,
pages "553-563" } } } } } ,
chemical-graph {
descr {
name "Dna (5'-D(CpGpCpGpApApTpTp(Br)cpGpCpG)-3') Complexed With
Netropsin, Re-Refinement" ,
pdb-class "Deoxyribonucleic Acid" ,
pdb-source "Synthetic" ,
assembly-type other } ,
molecule-graphs {
{
id 1 ,
descr {
name "A" ,
pdb-comment "SEQRES" ,
molecule-type dna ,
organism {
org {
taxname "synthetic construct" ,
db {
{
seqhound@blueprint.org Version 3.3
The SeqHound Manual 414 of 421 18/04/2005
db "taxon" ,
tag
id 32630 } } ,
orgname {
name
partial {
{
fixed-level other ,
level "species" ,
name "synthetic construct" } } ,
lineage "artificial sequence" ,
gcode 11 ,
div "SYN" } } } } ,
seq-id
gi 996094 ,
residue-sequence {
{
id 1 ,
name " 1 " ,
residue-graph
standard {
biostruc-residue-graph-set-id
other-database {
db "Standard residue dictionary" ,
tag
id 1 } ,
residue-graph-id 66 } } ,
{
id 5 ,
name " 5 " ,
residue-graph
standard {
biostruc-residue-graph-set-id
other-database {
db "Standard residue dictionary" ,
tag
id 1 } ,
residue-graph-id 61 } } ,
{
id 8 ,
name " 8 " ,
residue-graph
standard {
biostruc-residue-graph-set-id
other-database {
db "Standard residue dictionary" ,
tag
id 1 } ,
residue-graph-id 70 } } ,
{
id 9 ,
name " 9 " ,
residue-graph
local 1 } ,
{
id 10 ,
name " 10 " ,
residue-graph
standard {
biostruc-residue-graph-set-id
other-database {
db "Standard residue dictionary" ,
tag
id 1 } ,
residue-graph-id 67 } } ,
{
id 11 ,
name " 11 " ,
residue-graph
standard {
biostruc-residue-graph-set-id
other-database {
seqhound@blueprint.org Version 3.3
The SeqHound Manual 415 of 421 18/04/2005
db "Standard residue dictionary" ,
tag
id 1 } ,
residue-graph-id 64 } } ,
{
id 12 ,
name " 12 " ,
residue-graph
standard {
biostruc-residue-graph-set-id
other-database {
db "Standard residue dictionary" ,
tag
id 1 } ,
residue-graph-id 68 } } } ,
inter-residue-bonds {
{
atom-id-1 {
molecule-id 1 ,
residue-id 11 ,
atom-id 9 } ,
atom-id-2 {
molecule-id 1 ,
residue-id 12 ,
atom-id 1 } } } } ,
{
id 2 ,
descr {
name "B" ,
pdb-comment "SEQRES" ,
molecule-type dna ,
organism {
org {
taxname "synthetic construct" ,
db {
{
db "taxon" ,
tag
id 32630 } } ,
orgname {
name
partial {
{
fixed-level other ,
level "species" ,
name "synthetic construct" } } ,
lineage "artificial sequence" ,
gcode 11 ,
div "SYN" } } } } ,
seq-id
gi 996095 ,
residue-sequence {
{
id 9 ,
name " 21 " ,
residue-graph
local 1 } ,
{
id 10 ,
name " 22 " ,
residue-graph
standard {
biostruc-residue-graph-set-id
other-database {
db "Standard residue dictionary" ,
tag
id 1 } ,
residue-graph-id 67 } } ,
{
id 12 ,
name " 24 " ,
residue-graph
seqhound@blueprint.org Version 3.3
The SeqHound Manual 416 of 421 18/04/2005
standard {
biostruc-residue-graph-set-id
other-database {
db "Standard residue dictionary" ,
tag
id 1 } ,
inter-molecule-bonds {
residue-graph-id 68 } } } ,
inter-residue-bonds {
{
atom-id-1 {
molecule-id 2 ,
residue-id 1 ,
atom-id 6 } ,
atom-id-2 {
molecule-id 2 ,
residue-id 2 ,
atom-id 1 } } ,
{
atom-id-1 {
molecule-id 2 ,
residue-id 11 ,
atom-id 9 } ,
atom-id-2 {
molecule-id 2 ,
residue-id 12 ,
atom-id 1 } } } } ,
{
id 3 ,
descr {
name "1" ,
molecule-type other-nonpolymer } ,
residue-sequence {
{
id 1 ,
name " 9 " ,
residue-graph
local 2 } } } ,
{
id 4 ,
descr {
name "2" ,
molecule-type other-nonpolymer } ,
residue-sequence {
{
id 1 ,
name " 21 " ,
residue-graph
local 2 } } } ,
{
id 38 ,
descr {
name "3" ,
molecule-type solvent } ,
residue-sequence {
{
id 1 ,
name " 58 " ,
residue-graph
local 5 } } } ,
{
id 39 ,
descr {
name "3" ,
molecule-type solvent } ,
residue-sequence {
{
id 1 ,
name " 59 " ,
residue-graph
local 5 } } } } ,
{
seqhound@blueprint.org Version 3.3
The SeqHound Manual 417 of 421 18/04/2005
atom-id-1 {
molecule-id 1 ,
residue-id 9 ,
atom-id 18 } ,
atom-id-2 {
molecule-id 3 ,
residue-id 1 ,
atom-id 1 } } ,
{
atom-id-1 {
molecule-id 2 ,
residue-id 9 ,
atom-id 18 } ,
atom-id-2 {
molecule-id 4 ,
residue-id 1 ,
atom-id 1 } } } ,
residue-graphs {
{
id 1 ,
descr {
name " +C DNA" ,
pdb-comment "" } ,
residue-type deoxyribonucleotide ,
iupac-code {
"N" } ,
atoms {
{
id 1 ,
name " P " ,
iupac-code {
" P " } ,
element p } ,
{
id 18 ,
name " C5 " ,
iupac-code {
" C5 " } ,
element c } ,
{
id 19 ,
name " C6 " ,
iupac-code {
" C6 " } ,
element c } } ,
bonds {
{
atom-id-1 16 ,
atom-id-2 17 ,
bond-order unknown } ,
{
atom-id-1 16 ,
atom-id-2 18 ,
bond-order unknown } } } ,
{
id 2 ,
descr {
name " BR" ,
pdb-comment "Bromine" } ,
residue-type other ,
iupac-code {
"X" } ,
atoms {
{
id 1 ,
name "BR " ,
iupac-code {
"BR " } ,
element br } } ,
bonds {
} } ,
{
seqhound@blueprint.org Version 3.3
The SeqHound Manual 418 of 421 18/04/2005
id 3 ,
descr {
name " NT" ,
pdb-comment "Netropsin" } ,
residue-type other ,
iupac-code {
"X" } ,
atoms {
{
id 1 ,
name " C1 " ,
iupac-code {
" C1 " } ,
element c } ,
{
id 5 ,
name " C2 " ,
iupac-code {
" C2 " } ,
element c } ,
{
id 31 ,
name " N10" ,
iupac-code {
" N10" } ,
element n } } ,
bonds {
{
atom-id-1 1 ,
atom-id-2 2 ,
bond-order unknown } ,
{
atom-id-1 1 ,
atom-id-2 3 ,
bond-order unknown } ,
{
atom-id-1 12 ,
atom-id-2 14 ,
bond-order unknown } ,
{
atom-id-1 29 ,
atom-id-2 31 ,
bond-order unknown } } } ,
{
id 4 ,
descr {
name "MO3" ,
pdb-comment "Magnesium Ion, 3 Waters Coordinated" } ,
residue-type other ,
iupac-code {
"X" } ,
atoms {
{
id 1 ,
name "MG " ,
iupac-code {
"MG " } ,
element mg } ,
{
id 2 ,
name " O1 " ,
iupac-code {
" O1 " } ,
element o } ,
{
id 3 ,
name " O2 " ,
iupac-code {
" O2 " } ,
element o } ,
{
id 4 ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 419 of 421 18/04/2005
name " O3 " ,
iupac-code {
" O3 " } ,
element o } } ,
bonds {
{
atom-id-1 1 ,
atom-id-2 2 ,
bond-order unknown } ,
{
atom-id-1 1 ,
atom-id-2 3 ,
bond-order unknown } ,
{
atom-id-1 1 ,
atom-id-2 4 ,
bond-order unknown } } } ,
{
id 5 ,
descr {
name "HOH" ,
pdb-comment "" } ,
residue-type other ,
iupac-code {
"X" } ,
atoms {
{
id 1 ,
name " O " ,
iupac-code {
" O " } ,
element o } } ,
bonds {
} } } } ,
model {
{
id 3 ,
type pdb-model ,
descr {
name "Model 1 from PDB entry 101D" ,
pdb-reso "Resolution: 2.25" ,
pdb-method "X-Ray Diffraction" ,
pdb-comment "FEB 27 95 Initial Entry" } ,
model-space {
coordinate-units angstroms ,
thermal-factor-units b } ,
model-coordinates {
{
id 1 ,
coordinates
literal
atomic {
number-of-points 556 ,
atoms {
number-of-ptrs 556 ,
molecule-ids {
1 ,
1 ,
1 ,
1 ,
2 ,
2 ,
2 ,
3 ,
4 ,
5 ,
5 ,
5 ,
5 ,
6 ,
6 ,
6 ,
seqhound@blueprint.org Version 3.3
The SeqHound Manual 420 of 421 18/04/2005
6 ,
7 ,
8 ,
9 ,
10 ,
11 ,
12 ,
13 ,
14 ,
15 ,
16 ,
17 ,
18 ,
19 ,
20 ,
12 ,
12 ,
12 ,
12 ,
12 ,
1 ,
1 ,
1 } } ,
sites {
scale-factor 1000 ,
x {
18598 ,
19853 ,
20375 ,
16812 } ,
y {
34469 ,
34632 ,
33233 ,
30694 } ,
z {
24672 ,
22605 ,
8518 ,
-2033 ,
26343 } } ,
temperature-factors
isotropic {
scale-factor 1000 ,
b {
23239 ,
24930 } } } } } } } }
seqhound@blueprint.org Version 3.3
